 Kim Ohl
Kim Ohl Klaus Tenbrock
Klaus Tenbrock- Department of Pediatrics, Medical Faculty, RWTH Aachen, Aachen, Germany
Reactive oxygen species (ROS) molecules are implicated in signal transduction pathways and thereby control a range of biological activities. Immune cells are constantly confronted with ROS molecules under both physiologic and pathogenic conditions. Myeloid-derived suppressor cells (MDSCs) are immunosuppressive, immature myeloid cells and serve as major regulators of pathogenic and inflammatory immune responses. In addition to their own release of ROS, MDSCs often arise in oxidative-stress prone environments such as in tumors or during inflammation and infection. This evidently close relationship between MDSCs and ROS prompted us to summarize what is currently known about ROS signaling within MDSCs and to elucidate how MDSCs use ROS to modulate other immune cells. ROS not only activate anti-oxidative pathways but also induce transcriptional programs that regulate the fate and function of MDSCs. Furthermore, MDSCs release ROS molecules as part of a major mechanism to suppress T cell responses. Targeting redox-regulation of MDSCs thus presents a promising approach to cancer therapy and the role of redox-signaling in MDSCs in other disease states such as infection, inflammation and autoimmunity would appear to be well worth investigating.
Introduction
Reactive oxygen species (ROS) appear to have harmful as well as beneficial effects(1, 2). Their harmful effects include oxidation-induced damage to cellular contents, such as lipids, proteins, carbohydrates and nucleic acids, which subsequently induce cell pathologies and cell death. Damaged and oxidized molecules contribute to a number of alterations including atherosclerosis, neurodegenerative diseases and aging. Beyond this, ROS molecules are implicated in signal transduction pathways and redox-dependent regulations controlling a range of biological activities. In this regard, it is interesting to examine myeloid-derived suppressor cells (MDSCs). These heterogenic myeloid cells are controlled by ROS but they also use ROS to fulfill suppressive functions. Pathological conditions such as chronic inflammation, infection and cancer, induce MDSCs, which consist of a heterogeneous population of immature myeloid cells (3, 4). A hallmark of these immunosuppressive cells is their capability to suppress T cell responses, which contributes to cancer immune evasion on the one hand but suppression of exaggerated T cell responses during inflammation on the other. In mice, MDSCs are broadly characterized by the surface expression of CD11b and Gr-1 and are further grouped into monocytic (CD11b+Ly6ChighLy6G−) and polymorphonuclear (CD11b+Ly6ClowLy6G+) MDSCs (5). The polymorphonuclear (PMN-MDSC) subset displays increased STAT3 and NADPH oxidase (Nox) activity, which results in high release of ROS but low NO release. The monocytic subset (M-MDSC) express high levels of STAT1 and iNOS and enhanced level of NO but show low ROS production. Both of them express arginase 1 (3). Although ROS have toxic effects on most cells, MDSCs survive despite elevated levels and continuous production of ROS (6). ROS production is not only central to the immunosuppressive properties of MDSCs but also seems to maintain them in an undifferentiated state. Furthermore, steady-state production of ROS by MDSCs is upregulated in a variety of murine tumor models and in human cancer, and also after activation in inflammatory and autoimmune conditions (4, 7). In addition to their own ROS release, MDSCs often arise in oxidative-stress prone environments such as in tumors or during inflammation and infection. This evidently close relationship between MDSCs and ROS prompted us to summarize what is currently known about ROS signaling in MDSCs itself and to elucidate how MDSCs use ROS to modulate other immune cells.
Main Text
Regulation of MDSCs by ROS
A state of “oxidative stress” describes a situation where high levels of ROS -derived from cellular metabolism, toxic insults, or oxidative burst- outbalance the anti-oxidative system (8). This breakdown of cellular homeostasis results from mitochondrial dysfunction or increased metabolic activity, oncogene activity or infiltrating immune cells (8) and induces damage to lipids, proteins, carbohydrates and nucleic acids and can even lead to cell death (9). Excessive production of ROS molecules is associated with several inflammatory and pathologic conditions. For example, oxidative stress within the intestinal epithelium is thought to be involved in the pathogenesis of intestinal inflammation (10) and oxidative stress is also associated with neurodegenerative diseases (8). Furthermore, elevated rates of ROS can be observed in almost all cancers and are involved in tumor metastasis (11). On the other hand, emerging evidence suggests that ROS molecules serve as signaling intermediates that play central roles in several molecular pathways and also serve as central mediators of immune cells (12). Low levels of ROS are continuously generated under healthy cellular conditions, and are neutralized by the endogenous antioxidant machinery that is regulated by nuclear factor (erythroid-derived 2)-like 2 (Nrf2). Nrf2 is retained and degraded in the cytosol by Kelch ECH associating protein 1 (Keap1) under basal conditions (13). Cellular stimuli such as oxidative stress lead to conformational changes in Keap1, which are followed by the release of Nrf2 from Keap1. Afterwards, Nrf2 translocates into the nucleus, where transactivation of genes containing an antioxidant response element (ARE) in their promoter regions takes place (14). Thereby, Nrf2 up-regulates phase II detoxifying enzymes and antioxidant proteins. These processes play a vital role in maintaining cellular homeostasis and are of major relevance upon exposure of cells to chemical or oxidative stress and inflammation. Particulary enzymes mediating gluathione (GSH) synthesis, the thioredoxin enzyme system, and detoxifying enzymes such as heme oxygenases, or NAD(P)H: quinone oxidoreductase 1 are part of the Nrf2 induced enzymatic machinery.
The most prevalent intracellular sources of ROS are mitochondria and NADPH oxidases (Nox) but beyond this, the ER and also the peroxisome (organelle that metabolizes long chain fatty acids) generate ROS molecules. Nox-mediated release of ROS induces the so-called oxidative burst and eliminates invading microorganisms (15). The relevance of Nox-derived ROS in host immunity is best demonstrated by the disease pattern of chronic granulomatous disease (GCD), which is caused by NOX2 defects, and results in hypersensitivity to common infections and accumulation of bacteria-containing phagocytes with subsequent granulome development (15, 16).
Mitochondrial ROS are central regulators of the innate immune system. They are indispensable for Toll-like receptor (TLR)-initiated pathways (17). In detail TLR1, TLR2, and TLR4 signaling leads to recruitment of mitochondria to phagosomes and enhances ROS production in macrophages, indicating that mitochondrial ROS form an important component of antibacterial responses and are necessary for activation of NLRP3 inflammasome (18). In addition to this, mitochondrial ROS are involved in NLRP3 activation. Accumulation of damaged ROS-generating mitochondria leads to NLRP3 activation (19), and increased levels of mitochondrial ROS resulting from NLRP3 activation serve as a feedback mechanism to sustain activation (20).
Furthermore, mitochondrial ROS and ROS derived from other sources and cellular metabolism are intimately linked. Oxidative phosphorylation (OXPHOS) is a major cellular source of ROS and requires adequate availability of antioxidants to prevent apoptosis. One advantage of glycolysis over OXPHOS lies therefore in a better maintenance of the redox balance. Lian et al. recently observed that MDSCs counteract OXPHOS-derived ROS by upregulation of glycolysis, thereby protecting MDSCs from apoptosis (Figure 1) (23). We observed high OXPHOS in MDSCs of mice with a constitutive Nrf2 activation and subsequently low levels of intracellular ROS (22). The constitutive activation and availability of antioxidant enzymes regulated by Nrf2 activation in these cells might be a central mechanism enabling the cells to increase mitochondrial ATP production by simultaneously counteracting subsequent high ROS levels. High oxygen consumption rate (OCR) levels were associated with a highly suppressive and tolerizing phenotype. Recent studies have shown that aerobic glycolysis constitutes the metabolic basis for trained immunity (24). The metabolism of tolerant myeloid cells, particularly of MDSCs, is less clearly understood and was one focus of our study (22). It is generally assumed that naïve or tolerant cells primarily use OXPHOS as an energy source, but activated cells, e.g., after LPS stimulation, undergo a shift toward aerobic glycolysis (25). However, metabolic characteristics of MDSCs seem to differ within this quite heterogeneous cell population and might also depend on disease context. In comparison to splenic MDSCs, tumor-infiltrating MDSCs enhance fatty acid oxidation (26). However, rapamycin, which specifically inhibis mTOR, reduces M-MDSC in mice with allografts or tumors (27). Flux of glucose down the pentose phosphate pathway (PPP) is essential for redox buffering as PPP produces NADPH. This is required to maintain GSH, the most important cellular antioxidant, in the reduced state (Figure 1). Again, we observed a high expression of PPP enzymes in Nrf2-activated MDSCs, which suggests that Nrf2 is critically involved in redox and in the metabolic signaling of MDSCs, and acts either by mediating ROS signaling or possibly also by targeting other genes (22).
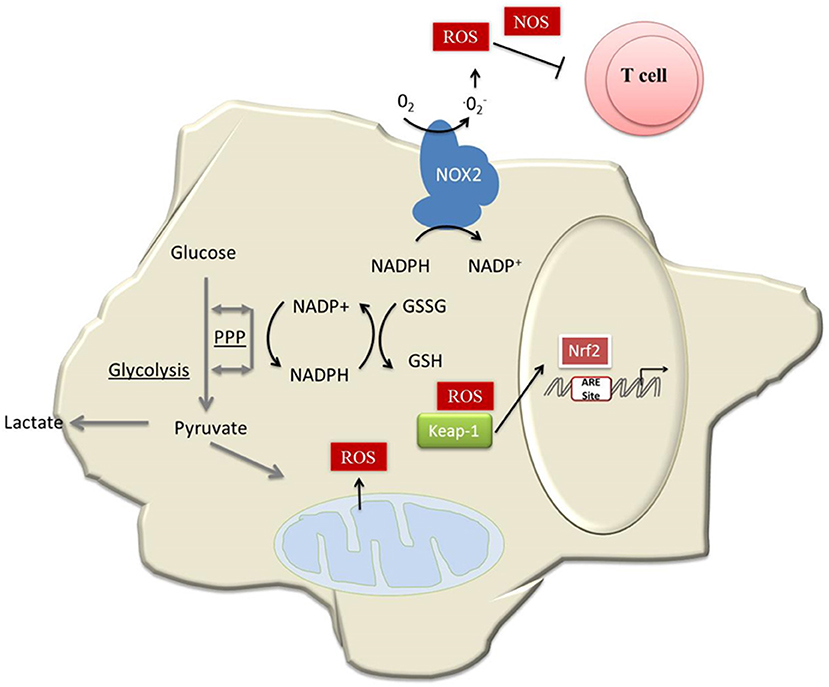
Figure 1. Model of how MDSCs maintain redox homeostasis. Activated MDSCs produce high amounts of ROS molecules by the action of NOX2 (4). This, in addition to mitochondrial ROS and ROS derived from cancer cells, compromises redox homeostasis in MDSCs and most likely induces apoptosis in the absence of Nrf2 (21). Another consequence of Nrf2 activation, besides expression of antioxidative genes, is a metabolic reprogramming of MDSCs. This leads to enhanced expression of the PPP (22), which provides GSH. GSH not only serves as a major antioxidant, but is essential for MDSC differentiation (22). In addition, MDSCs counteract OXPHOS-derived ROS by upregulation of glycolysis (23).
Redox signaling is moreover involved in several signal transduction pathways. In most cases cystein (Cys) residues serve as redox-dependent switches and the oxidation/reduction of specific amino acids, that bear reactive Cys residues, induces activation, or inactivation of target proteins such as phosphatases. Moreover, ROS modulate antioxidant enzymes that not only serve as scavengers but also transduce redox-dependent signals (28). GSH is not only the most important antioxidant in cells in general but also mediates specific effects in immune cells. For instance, GSH is involved in reprogramming of effector T cells during inflammation (29). With regard to MDSCs, increased levels of GSH are especially important for MDSC differentiation (30). Probably, GSH affects MDSCs differentiation by neutralization of ROS but other direct effects of GSH on MDSCs are conceivable.
ROS molecules are essential for maintainance of MDSCs in their undifferentiated state. Scavenging of H2O2 with catalase induces differentiation of immature myeloid cells into macrophages in mice bearing tumors (31), while in the absence of Nox activity, MDSCs differentiate into macrophages and DCs in tumor-bearing mice (4). Increased levels of endogenous H202 might thereby present a mechanism by which tumors prevent the differentiation of MDSCs. The precise molecular mechanism maintaining MDSCs in their undifferentiated state in the presence of ROS remains to be elucidated.
Regulation of Cellular Immune Responses by MDSC-Derived ROS
Release of ROS molecules is one of the major mechanisms that MDSCs use to suppress T cells in mice and humans (4, 32, 33). Administration of ROS inhibitors was found to counteract the suppressive effect of human MDSCs on T cells (34). And, at least in tumor-bearing mice, suppression of T cells is dependent on NOX2 activity (4). Superoxide released by MDSCs rapidly reacts with a large number of molecules e.g., H2O2, hydroxyl radical, hypochlorous acid, and peroxynitrite to form ROS, which then damage proteins, lipids, and nucleic acids, enhance inflammation and promote apoptosis. ROS are even thought to enable Ag-specific suppression of T cell responses by MDSCs. Nagaraj et al. showed that MDSC-derived ROS molecules and peroxynitrite, which is the product of the reaction of ROS with NO, modify TCR and CD8 molecules. Through these modifications, CD8+ T cells lose their ability to bind phosphorylated MHC and induce antigen-specific tolerance of peripheral CD8+ T cells (35).
H202, formed from MDSC-derived superoxide, decreases T cellular CD3ζ expression, thereby limits the ability of the T cells to become activated (36) and reduces their expression of IFN-γ (4).
While MDSCs suppress effector T cells, they induce the expansion of regulatory T cells (Tregs) in cancer, and also in inflammatory conditions (37–42). Induction of Tregs is therefore one important mechanism of MDSC-mediated T cell inhibition. The role of ROS molecules in the interaction of MDSCs and Tregs is not clear. The induction of Treg cells by macrophages involves production of ROS and therefore ROS deficiency might lead to reduced Treg induction and might aggravate T-cell suppression (43). In addition, Tregs, are less susceptible to oxidative stress-induced cell death compared to other T cell populations (44). This is most likely caused by a greater secretion of redox proteins such as thioredoxin (44) or hemeoxygenase 1 (45). In addition to this, human Tregs have been shown to express high levels of cell surface thiols, that are important reducing agents, and facilitate enhanced intracellular anti-oxidative abilities (44). Nevertheless, a recent study claims that Treg cells are less resistant to oxidative stress in the tumor microenvironment compared to conventional T cells and even undergo ROS-induced apoptosis due to a weak Nrf2-associated antioxidant system (46). These apoptotic Treg cells suppress antitumor T cell immunity even more efficiently via the adenosine and A2A pathways. As a consequence, Tregs or at least Treg-mediated suppression seems to benefit from oxidative stress conditions and might therefore contribute to MDSC-mediated immune suppression.
Beyond direct effects on T cells, ROS molecules also indirectly modulate T cell responses. Peroxynitrite indirectly hinders T cells activation by modifying the antigen presenting structure on tumor cells. To this end, peroxynitrite reduces the binding of antigens to tumor cell-associated MHC and thereby generates tumor cells that are resistant to antigen-specific cytotoxic T cell responses (47). Furthermore reactive nitrogen species induce posttranslational modifications of T cell chemokines and thereby hinder antigen-specific T cells invasion of tumors (48).
Furthermore, not only T cell responses are targets of ROS mediated suppression by MDSCs. PMN-MDSCs also suppress NK cell responses to adenoviral vectors and to vaccinia virus infection by ROS release (49, 50). In addition, MDSCs also suppress NK cell toxicity in tumor bearing mice and might critically contribute to the attenuated NK cell activity and cytotoxicity in tumors (51), however the exact mechanism and involvement of ROS are not fully determined.
Recent research demonstrates that MDSCs also negatively regulate B cell- mediated immune responses using ROS. In a murine AIDS model (LP-BM5 reotroviral infection) M-MDSC suppressed B cell responses at least in part by ROS mediated suppression (52, 53). Astudy with human PMN-MDSCs demonstrates that MDSCs suppress B cell proliferation and antibody production in a cell contact manner by means of arginase, NO and ROS (54).
Redox-dependent Transcriptional Reprogramming of MDSCs in Cancer and Inflammation
It is of note that HIF-1α and Nrf2, which are both involved in redox-signaling and oxidative stress responses, emerge as critical regulators of MDSCs. Beyond redox regulation; both factors control other mechanisms and thereby regulate MDSC fate and function.
A critical role of HIF-1α signaling in MDSCs is described in murine cancer models, such as hepatocellular carcinoma (HCC) (21, 55, 56) and in patients with non-small cell lung cancer (57). Interestingly, HIF-1α controls the manner of MDSC-mediated suppression, depending on the hypoxic state of the environment. The dominant mechanism in peripheral lymphoid organs is mediated by ROS and results in antigen-specific T cell non-responsiveness. However, within the hypoxic tumor microenvironment, MDSCs bearing the same phenotype and morphology revealed low levels of ROS levels but significantly enhanced NO production as well as arginase activity and thereby suppressed T cells (21). Several mechanisms have been analyzed by which HIF-1α regulates the fate and function of MDSCs in a hypoxic tumor environment. Some of these studies come to contradictory conclusions, possibly due to the use of different tumor models or the heterogeneity of MDSC populations. Liu et al. showed that lineage differentiation of MDSCs to M1 cells requires glycolytic activity induced by mTOR- and HIF-1α, as brought about by SIRT1 in tumors (56), while Cocl2 (an HIF-1α activator) effectively promotes M1-MDSC differentiation, and potentiates tumor-killing and glycolytic activities. On the other hand, HIF-1α was found to upregulate PD-L1 on MDSCs and induce miR-210, both of which enhance MDSC-mediated T cell suppression (58, 59). In conclusion, these studies reveal that by regulating several pathways including metabolic reactions and miRNA expression, HIF-1α critically regulates the function and maintenance of MDSCs within the hypoxic tumor environment.
Nrf2 is involved in the regulation of various pathways in MDSCs as well. Through an analysis of Nrf2-deficient mice in mammary carcinoma and colon carcinoma models, Beury et al. initially showed that Nrf2 regulates numbers and function of MDSCs (6). Nrf2 deficient mice had increased survival rates and reduced tumor progression with reduced numbers of MDSCs and MDSCs from Nrf2-deficient mice had a reduced suppressive capacity and, surprisingly, a reduced H202 production. Intracellular oxidative stress and apoptosis were enhanced in the absence of Nrf2. However, myeloid-lineage specific Nrf2 deficiency enhances lung metastasis and has been shown to lead to an aberrant ROS accumulation in myeloid cells (60). Nrf2 is known to play dual roles in cancer prevention and progression, which depends on the cellular context and environment (61). However, the exact mechanisms involved remain to be elucidated. It is also not clear whether Nrf2 expression—like HIF-1α expression—is different in peripheral lymphoid organs and tumor MDSCs and whether it might therefore also influence local MDSC maintenance. We observed spontaneously enhanced numbers of MDSCs in mice with a constitutive activation of Nrf2 with intact suppressive functions in vitro. This was also found in a transfer colitis model and in a sepsis model in vivo (22). MDSCs with constitutive Nrf2 activation displayed low levels of intracellular ROS, but a high metabolic activity and high proliferation rates. This suggests that, beyond its anti-oxidative action, Nrf2 has several other effects that need to be taken into account and might contribute to a context-dependent regulation of MDSCs.
Conclusion
ROS signaling is without doubt a central mediator of MDSC function and fate. Furthermore, beyond their role in MDSC-mediated immune-suppression, ROS molecules are intrinsically involved in activation of transcription factors such as Nrf2 and HIF-1α, which can induce transcriptional and metabolic reprogramming of MDSCs and influence their differentiation and maintenance. Compounds that target ROS in MDSCs to enhance the effects of cancer immune therapy are promising therapeutic options. The synthetic triterpenoid C-28 methyl ester of 2-cyano-3,12-dioxooleana-1,9,-dien-28-oic acid (CDDO-Me, also referred to as bardoxolone methyl, RTA402, TP-155 and NSC713200) is a potent Nrf2 activator and has been found to reduce MDSC production of ROS and tumor growth in mouse tumor models (62). CDDO-Me shows a promising anticancer effect in a phase I trial (63). In addition, systemic treatment with all-trans-retinoic acid (ATRA) promotes maturation of human MDSCs and reverses their immune suppressor function. Accumulation of GSH in MDSCs by ATRA decreases levels of ROS and induces MDSC differentiation into mature myeloid cells (30, 64). Until now, most studies have focused on cancer models and suggest that inhibition of ROS production in MDSCs helps to enhance anti-tumor immune responses. Beyond their pathogenic role in cancer, expansion and activation of MDSCs also occurs in autoimmunity, infection and chronic inflammation, conditions that are associated with oxidative stress and hypoxic states (10, 65, 66). Thus, redox-signaling in MDSCs might be a promising therapeutic target in these diseases as well. However, the role of MDSCs here seems to be less clear here, and both positive and negative roles of MDSCs have been revealed with regard to progression of autoimmune diseases. Therefore, further studies are warranted to uncover the specific role of redox signaling in MDSCs in autoimmunity and infection.
Author Contributions
KO and KT both researched data for the article, contributed to discussion of the content as well as writing and reviewing of the manuscript.
Funding
This work was supported by the Interdisciplinary Center for Clinical Research at the Uniklinik RWTH Aachen (O3-10).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ATRA, All-trans retinoic acid; CDDO-Me, C-28 methyl ester of 2-cyano-3,12-dioxooleana-1,9,-dien-28-oic acid; Cys, cysteine; GSH, Glutathione; HCC, Hepatocellular carcinoma; HIF, Hypoxia-inducible factor; Keap1, Kelch ECH associating protein 1; MDSC, Myeloid-derived suppressor cell; M-MDSC, monocytic MDSC; NOX, NADPH oxidase; Nrf2, Nuclear factor (erythroid-derived 2)-like 2; OXPHOS, oxidative phosphorylation; PMN-MDSC, polymorphonuclear MDSC; PPP,pentose phosphate pathway; ROS, reactive oxygen species.
References
1. Kalyanaraman B, Cheng G, Hardy M, Ouari O, Bennett B, Zielonka J. Teaching the basics of reactive oxygen species and their relevance to cancer biology: Mitochondrial reactive oxygen species detection, redox signaling, and targeted therapies. Redox Biol. (2018) 15:347–62. doi: 10.1016/j.redox.2017.12.012
2. Sareila O, Kelkka T, Pizzolla A, Hultqvist M, Holmdahl R. NOX2 complex-derived ROS as immune regulators. Antioxid Redox Signal (2011) 15:2197–208. doi: 10.1089/ars.2010.3635
3. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. (2009) 9:162–74. doi: 10.1038/nri2506
4. Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, et al. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol. (2009) 182:5693–701. doi: 10.4049/jimmunol.0900092
5. Manjili MH, Wang XY, Abrams S. Evolution of our understanding of myeloid regulatory cells: from MDSCs to Mregs. Front Immunol. (2014) 5:303. doi: 10.3389/fimmu.2014.00303
6. Beury DW, Carter KA, Nelson C, Sinha P, Hanson E, Nyandjo M, et al. Myeloid-derived suppressor cell survival and function are regulated by the transcription factor Nrf2. J Immunol. (2016) 196:3470–8. doi: 10.4049/jimmunol.150178
7. Dietlin TA, Hofman FM, Lund BT, Gilmore W, Stohlman SA, van der Veen RC. Mycobacteria-induced Gr-1+ subsets from distinct myeloid lineages have opposite effects on T cell expansion. J Leukoc Biol. (2007) 81:1205–12. doi: 10.1189/jlb.1006640
8. Ohl K, Tenbrock K, Kipp M. Oxidative stress in multiple sclerosis: central and peripheral mode of action. Exp Neurol. (2016) 277:58–67. doi: 10.1016/j.expneurol.2015.11.010
9. Kaminski MM, Roth D, Krammer PH, Gulow K. Mitochondria as oxidative signaling organelles in T-cell activation: physiological role and pathological implications. Arch Immunol Ther Exp. (2013) 61:367–84. doi: 10.1007/s00005-013-0235-0
10. Novak EA, Mollen KP. Mitochondrial dysfunction in inflammatory bowel disease. Front Cell Dev Biol. (2015) 3:62. doi: 10.3389/fcell.2015.00062
11. Chen Y, Zhang H, Zhou HJ, Ji W, Min W. Mitochondrial redox signaling and tumor progression. Cancers (2016) 8:40. doi: 10.3390/cancers8040040
12. Nathan C, Cunningham-Bussel A. Beyond oxidative stress: an immunologist's guide to reactive oxygen species. Nat Rev Immunol. (2013) 13:349–61. doi: 10.1038/nri3423
13. Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol. (2003) 23:8137–51. doi: 10.1128/MCB.23.22.8137-8151.2003
14. Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. (2007) 47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046
15. Panday A, Sahoo MK, Osorio D, Batra S. NADPH oxidases: an overview from structure to innate immunity-associated pathologies. Cell Mol Immunol. (2015) 12:5–23. doi: 10.1038/cmi.2014.89
16. Quie PG, White JG, Holmes B, Good RA. In vitro bactericidal capacity of human polymorphonuclear leukocytes: diminished activity in chronic granulomatous disease of childhood. J Clin Invest. (1967) 46:668–79. doi: 10.1172/JCI105568
17. West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature (2011) 472:476–80. doi: 10.1038/nature09973
18. Gurung P, Lukens JR, Kanneganti TD. Mitochondria: diversity in the regulation of the NLRP3 inflammasome. Trends Mol Med. (2015) 21:193–201. doi: 10.1016/j.molmed.2014.11.008
19. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature (2011) 469:221–5. doi: 10.1038/nature09663
20. Dan Dunn J, Alvarez LA, Zhang X, Soldati T. Reactive oxygen species and mitochondria: a nexus of cellular homeostasis. Redox Biol. (2015) 6:472–85. doi: 10.1016/j.redox.2015.09.005
21. Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, et al. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. (2010) 207:2439–53. doi: 10.1084/jem.20100587
22. Ohl K, Fragoulis A, Klemm P, Baumeister J, Klock W, Verjans E, et al. Nrf2 Is a central regulator of metabolic reprogramming of myeloid-derived suppressor cells in steady state and sepsis. Front Immunol. (2018) 9:1552. doi: 10.3389/fimmu.2018.01552
23. Jian SL, Chen WW, Su YC, Su YW, Chuang TH, Hsu SC, et al. Glycolysis regulates the expansion of myeloid-derived suppressor cells in tumor-bearing hosts through prevention of ROS-mediated apoptosis. Cell Death Dis. (2017) 8:e2779. doi: 10.1038/cddis.2017.192
24. Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science (2014) 345:1250684. doi: 10.1126/science.1250684
25. O'Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. (2016) 16:553–65. doi: 10.1038/nri.2016.70
26. Hossain F, Al-Khami AA, Wyczechowska D, Hernandez C, Zheng L, Reiss K, et al. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol Res. (2015) 3:1236–47. doi: 10.1158/2326-6066.CIR-15-0036
27. Wu T, Zhao Y, Wang H, Li Y, Shao L, Wang R, et al. mTOR masters monocytic myeloid-derived suppressor cells in mice with allografts or tumors. Sci Rep. (2016) 6:20250. doi: 10.1038/srep20250
28. Holmstrom KM, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol. (2014) 15:411–21. doi: 10.1038/nrm3801
29. Mak TW, Grusdat M, Duncan GS, Dostert C, Nonnenmacher Y, Cox M, et al. Glutathione primes T cell metabolism for inflammation. Immunity (2017) 46:675–89. doi: 10.1016/j.immuni.2017.03.019
30. Nefedova Y, Fishman M, Sherman S, Wang X, Beg AA, Gabrilovich DI. Mechanism of all-trans retinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Res. (2007) 67:11021–8. doi: 10.1158/0008-5472.CAN-07-2593
31. Kusmartsev S, Gabrilovich DI. Inhibition of myeloid cell differentiation in cancer: the role of reactive oxygen species. J Leukoc Biol. (2003) 74:186–96. doi: 10.1189/jlb.0103010
32. Chen X, Song M, Zhang B, Zhang Y. Reactive oxygen species regulate T cell immune response in the tumor microenvironment. Oxid Med Cell Longev. (2016) 2016:1580967. doi: 10.1155/2016/1580967
33. Zhang H, Li ZL, Ye SB, Ouyang LY, Chen YS, He J, et al. Myeloid-derived suppressor cells inhibit T cell proliferation in human extranodal NK/T cell lymphoma: a novel prognostic indicator. Cancer Immunol Immunother. (2015) 64:1587–99. doi: 10.1007/s00262-015-1765-6
34. Wei J, Zhang M, Zhou J. Myeloid-derived suppressor cells in major depression patients suppress T-cell responses through the production of reactive oxygen species. Psychiatry Res. (2015) 228:695–701. doi: 10.1016/j.psychres.2015.06.002
35. Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med. (2007) 13:828–35. doi: 10.1038/nm1609
36. Schmielau J, Finn OJ. Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of t-cell function in advanced cancer patients. Cancer Res. (2001) 61:4756–60.
37. Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, et al. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. (2006) 66:1123–31. doi: 10.1158/0008-5472.CAN-05-1299
38. Garcia MR, Ledgerwood L, Yang Y, Xu J, Lal G, Burrell B, et al. Monocytic suppressive cells mediate cardiovascular transplantation tolerance in mice. J Clin Invest. (2010) 120:2486–96. doi: 10.1172/JCI41628
39. Park MJ, Lee SH, Kim EK, Lee EJ, Baek JA, Park SH, et al. Interleukin-10 produced by myeloid-derived suppressor cells is critical for the induction of Tregs and attenuation of rheumatoid inflammation in mice. Sci Rep. (2018) 8:3753. doi: 10.1038/s41598-018-21856-2
40. Nagaraj S, Youn JI, Gabrilovich DI. Reciprocal relationship between myeloid-derived suppressor cells and T cells. J Immunol. (2013) 191:17–23. doi: 10.4049/jimmunol.1300654
41. Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol. (2018) 19:108–19. doi: 10.1038/s41590-017-0022-x
42. Greten TF, Manns MP, Korangy F. Myeloid derived suppressor cells in human diseases. Int Immunopharmacol. (2011) 11:802–7. doi: 10.1016/j.intimp.2011.01.003
43. Kraaij MD, Savage ND, van der Kooij SW, Koekkoek K, Wang J, van den Berg JM, et al. Induction of regulatory T cells by macrophages is dependent on production of reactive oxygen species. Proc Natl Acad Sci USA. (2010) 107:17686–91. doi: 10.1073/pnas.1012016107
44. Mougiakakos D, Johansson CC, Kiessling R. Naturally occurring regulatory T cells show reduced sensitivity toward oxidative stress-induced cell death. Blood (2009) 113:3542–5. doi: 10.1182/blood-2008-09-181040
45. Pae HO, Oh GS, Choi BM, Chae SC, Chung HT. Differential expressions of heme oxygenase-1 gene in CD25- and CD25+ subsets of human CD4+ T cells. Biochem Biophys Res Commun. (2003) 306:701–5. doi: 10.1016/S0006-291X(03)01037-4
46. Maj T, Wang W, Crespo J, Zhang H, Wang W, Wei S, et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat Immunol. (2017) 18:1332–41. doi: 10.1038/ni.3868
47. Lu T, Ramakrishnan R, Altiok S, Youn JI, Cheng P, Celis E, et al. Tumor-infiltrating myeloid cells induce tumor cell resistance to cytotoxic T cells in mice. J Clin Invest. (2011) 121:4015–29. doi: 10.1172/JCI45862
48. Molon B, Ugel S, Del Pozzo F, Soldani C, Zilio S, Avella D, et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med. (2011) 208:1949–62. doi: 10.1084/jem.20101956
49. Fortin C, Huang X, Yang Y. NK cell response to vaccinia virus is regulated by myeloid-derived suppressor cells. J Immunol. (2012) 189:1843–9. doi: 10.4049/jimmunol.1200584
50. Zhu J, Huang X, Yang Y. Myeloid-derived suppressor cells regulate natural killer cell response to adenovirus-mediated gene transfer. J Virol. (2012) 86:13689–96. doi: 10.1128/JVI.01595-12
51. Liu C, Yu S, Kappes J, Wang J, Grizzle WE, Zinn KR, et al. Expansion of spleen myeloid suppressor cells represses NK cell cytotoxicity in tumor-bearing host. Blood (2007) 109:4336–42. doi: 10.1182/blood-2006-09-046201
52. Rastad JL, Green WR. Myeloid-derived suppressor cells in murine AIDS inhibit B-cell responses in part via soluble mediators including reactive oxygen and nitrogen species, and TGF-beta. Virology (2016) 499:9–22. doi: 10.1016/j.virol.2016.08.031
53. Green KA, Cook WJ, Green WR. Myeloid-derived suppressor cells in murine retrovirus-induced AIDS inhibit T- and B-cell responses in vitro that are used to define the immunodeficiency. J Virol. (2013) 87:2058–71. doi: 10.1128/JVI.01547-12
54. Lelis FJN, Jaufmann J, Singh A, Fromm K, Teschner AC, Poschel S, et al. Myeloid-derived suppressor cells modulate B-cell responses. Immunol Lett. (2017) 188:108–15. doi: 10.1016/j.imlet.2017.07.003
55. Chiu DK, Tse AP, Xu IM, Di Cui J, Lai RK, Li LL, et al. Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat Commun. (2017) 8:517. doi: 10.1038/s41467-017-00530-7
56. Liu G, Bi Y, Shen B, Yang H, Zhang Y, Wang X, et al. SIRT1 limits the function and fate of myeloid-derived suppressor cells in tumors by orchestrating HIF-1alpha-dependent glycolysis. Cancer Res. (2014) 74:727–37. doi: 10.1158/0008-5472.CAN-13-2584
57. Li J, Wang L, Chen X, Li L, Li Y, Ping Y, et al. CD39/CD73 upregulation on myeloid-derived suppressor cells via TGF-beta-mTOR-HIF-1 signaling in patients with non-small cell lung cancer. Oncoimmunology (2017) 6:e1320011. doi: 10.1080/2162402X.2017.1320011
58. Noman MZ, Janji B, Hu S, Wu JC, Martelli F, Bronte V, et al. Tumor-promoting effects of myeloid-derived suppressor cells are potentiated by hypoxia-induced expression of miR-210. Cancer Res. (2015) 75:3771–87. doi: 10.1158/0008-5472.CAN-15-0405
59. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. (2014) 211:781–90. doi: 10.1084/jem.20131916
60. Satoh H, Moriguchi T, Taguchi K, Takai J, Maher JM, Suzuki T, et al. Nrf2-deficiency creates a responsive microenvironment for metastasis to the lung. Carcinogenesis (2010) 31:1833–43. doi: 10.1093/carcin/bgq105
61. Moon EJ, Giaccia A. Dual roles of NRF2 in tumor prevention and progression: possible implications in cancer treatment. Free Rad Biol Med. (2015) 79:292–9. doi: 10.1016/j.freeradbiomed.2014.11.009
62. Nagaraj S, Youn JI, Weber H, Iclozan C, Lu L, Cotter MJ, et al. Anti-inflammatory triterpenoid blocks immune suppressive function of MDSCs and improves immune response in cancer. Clin Cancer Res. (2010) 16:1812–23. doi: 10.1158/1078-0432.CCR-09-3272
63. Wang YY, Yang YX, Zhe H, He ZX, Zhou SF. Bardoxolone methyl (CDDO-Me) as a therapeutic agent: an update on its pharmacokinetic and pharmacodynamic properties. Drug Des Dev Therapy (2014) 8:2075–88. doi: 10.2147/DDDT.S68872
64. Mirza N, Fishman M, Fricke I, Dunn M, Neuger AM, Frost TJ, et al. All-trans-retinoic acid improves differentiation of myeloid cells and immune response in cancer patients. Cancer Res. (2006) 66:9299–307. doi: 10.1158/0008-5472.CAN-06-1690
65. Mateen S, Moin S, Khan AQ, Zafar A, Fatima N. Increased reactive oxygen species formation and oxidative stress in rheumatoid arthritis. PloS ONE (2016) 11:e0152925. doi: 10.1371/journal.pone.0152925
Keywords: ROS, MDSC, Nrf2, redox regulation, metabolism
Citation: Ohl K and Tenbrock K (2018) Reactive Oxygen Species as Regulators of MDSC-Mediated Immune Suppression. Front. Immunol. 9:2499. doi: 10.3389/fimmu.2018.02499
Received: 20 August 2018; Accepted: 10 October 2018;
Published: 30 October 2018.
Edited by:
Olivera J. Finn, University of Pittsburgh, United StatesReviewed by:
Rolf Kiessling, Karolinska Institutet (KI), SwedenGiacomo Desantis, San Raffaele Hospital (IRCCS), Italy
Copyright © 2018 Ohl and Tenbrock. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kim Ohl, a29obEB1a2FhY2hlbi5kZQ==