 Matthew J. Elder1,2
Matthew J. Elder1,2 Steve J. Webster1,3
Steve J. Webster1,3 Timothy J. Fitzmaurice1
Timothy J. Fitzmaurice1 Aran S. D. Shaunak1Martin Steinmetz4Ronnie Chee5
Aran S. D. Shaunak1Martin Steinmetz4Ronnie Chee5 Ziad Mallat6
Ziad Mallat6 E. Suzanne Cohen7David L. Williams8
E. Suzanne Cohen7David L. Williams8 J. S. Hill Gaston1
J. S. Hill Gaston1 Jane C. Goodall1*
Jane C. Goodall1*- 1Department of Medicine, School of Clinical Medicine, Addenbrookes Hospital, University of Cambridge, Cambridge, United Kingdom
- 2Early Oncology R&D Division, AstraZeneca, Cambridge, United Kingdom
- 3Department of Veterinary Medicine, University of Cambridge, Cambridge, United Kingdom
- 4Unit 970, INSERM, Paris Cardiovascular Research Center, Paris, France
- 5Department of Immunology, Royal Free Hospital, London, United Kingdom
- 6Division of Cardiovascular Medicine, Department of Medicine, University of Cambridge, Cambridge, United Kingdom
- 7Biopharmaceutical Research Division, AstraZeneca, Cambridge, United Kingdom
- 8Department of Surgery, Center for Inflammation, Infectious Disease and Immunity, James H. Quillen College of Medicine, East Tennessee State University, Johnson City, TN, United States
Thymic stromal lymphopoietin (TSLP) is a functionally pleotropic cytokine important in immune regulation, and TSLP dysregulation is associated with numerous diseases. TSLP is produced by many cell types, but has predominantly been characterized as a secreted factor from epithelial cells which activates dendritic cells (DC) that subsequently prime T helper (TH) 2 immunity. However, DC themselves make significant amounts of TSLP in response to microbial products, but the functional role of DC-derived TSLP remains unclear. We show that TSLPR signaling negatively regulates IL-1β production during dectin-1 stimulation of human DC. This regulatory mechanism functions by dampening Syk phosphorylation and is mediated via NADPH oxidase-derived ROS, HIF-1α and pro-IL-1β expression. Considering the profound effect TSLPR signaling has on the metabolic status and the secretome of dectin-1 stimulated DC, these data suggest that autocrine TSLPR signaling could have a fundamental role in modulating immunological effector responses at sites removed from epithelial cell production of TSLP.
Introduction
Thymic Stromal Lymphopoietin (TSLP) is a four-helix bundle cytokine belonging to the IL-2 family that was initially described as a lymphocyte growth factor (1). Since this initial report, it has been shown to be produced by a plethora of cell types (2–8). Functionally TSLP is pleotropic; TSLP is described to have an important role in maintaining tolerance within the gut (9, 10) yet it is implicated in asthma (11) and in the skin in both the development of itch (12) and atopic inflammation (12–14). TSLP binds to its unique receptor, called the TSLP receptor (TSLPR) composed of a unique TSLPR chain and the IL-7 receptor alpha (15) which initiates JAK-STAT mediated activation of downstream target genes (16–18).
Dendritic cells (DC) are immunologically important TSLP responsive cells (19). DC activated with TSLP can induce naïve CD4+ T cell proliferation (20) and T helper (TH) 2-cell differentiation (21) which requires the up-regulation of OX40L on the DC (22). DC can also produce TSLP in response to pattern recognition receptor (PRR) engagement (3, 4, 8, 23). Therefore, DC are the only cell population known to both produce and respond to TSLP by altering their effector responses. However, the functional role of DC-derived TSLP remains unclear; this work addresses this issue.
Amongst PRRs, dectin-1 stimulation induces TSLP production by DC (8, 23). Dectin-1 recognizes exposed β-1,3 glucan residues on the cell surface of fungi and studies utilizing dectin-1 gene knockout (−/−) mice emphasize the importance of this PRR to anti-fungal immunity (24). Activation of dectin-1 signaling induces immunological effector responses including phagocytosis (25), oxidative burst (26, 27) and the secretion of inflammatory cytokines including IL-1β, IL-6, and IL-23 (27–32). IL-1β production plays a critical role in the generation of protective anti-fungal immunity (33, 34). However, IL-1β dysregulation is associated with numerous diseases including inflammatory bowel disease (IBD) and auto-inflammatory conditions such as the cryopyrin associated periodic syndromes (CAPS) (35, 36). Therefore, IL-1β production is tightly regulated in DC, requiring two independent signals for its production. An initial priming signal (signal 1) is generated from ligation of PRRs, activating the inflammatory transcription factor NF-κB required for the up-regulated transcription of pro-IL-1β (36). A second activatory signal (signal 2) then causes inflammasome-mediated cleavage of pro-IL-1β into its active form (26, 27, 30, 32, 36–39). Recent work has augmented the understanding of IL-1β regulation, describing how changes to cellular metabolism after PRR stimulation regulate IL-1β expression (40, 41).
We report here that autocrine TSLPR signaling in human DC negatively regulates IL-1β production in response to dectin-1 stimulation. It likely does this through limiting a metabolic switch to glycolysis in DC which is required for IL-1β expression.
Results
Inhibition of TSLPR Signaling in mDC Modulates IL-1β Production
DC secretion of TSLP can be readily induced following dectin-1 stimulation (4, 8, 23). We investigated the functional relevance of DC-derived TSLP by neutralizing TSLP signaling in human monocyte-derived dendritic cells (mDC) which had been stimulated with heat-killed C. albicans or β-glucan purified from either S. cerevisiae (SC glucan) or C. albicans (CA glucan). We blocked TSLP activity or TSLPR signaling using neutralizing antibodies, and evaluated IL-1β, IL-6, IL-23, TSLP, and CCL22 secretion. Inhibition of either TSLP or TSLPR on mDC augmented the production of IL-1β, IL-6, IL-23 (Figures 1A–C), and TSLPR inhibition augmented TSLP itself (Figure 1G) from mDC in response to dectin-1 stimulation. These effects were not observed using control antibodies. CCL22 is a known TSLP-responsive chemokine in epithelial cells (21), and as expected inhibition of TSLPR signaling also reduced DC CCL22 production in response to dectin-1 stimulation (Figure 1H). Augmented IL-1β, IL-6, and IL-23 were all dependent on signaling through dectin-1 via Syk (Supplemental Figures 1A–F). To ensure that this effect was not an artifact caused by antibody binding and subsequent mDC activation, we generated bone marrow-derived dendritic cells (BMDC) from wildtype TSLPR+/+ and knockout TSLPR−/− BALB/c mice. In agreement with the results using TSLPR blocking antibodies, TSLPR−/− BMDC stimulated with either C. albicans or β-glucans produced more IL-1β, IL-6, and IL-23 compared to TSLPR+/+ BMDC (Figures 1D–F). Furthermore, inhibiting TSLPR signaling on human blood-derived CD1c+ DC also increased IL-1β secretion confirming that these findings were not an artifact of in vitro differentiation of monocytes (Figure 1I).
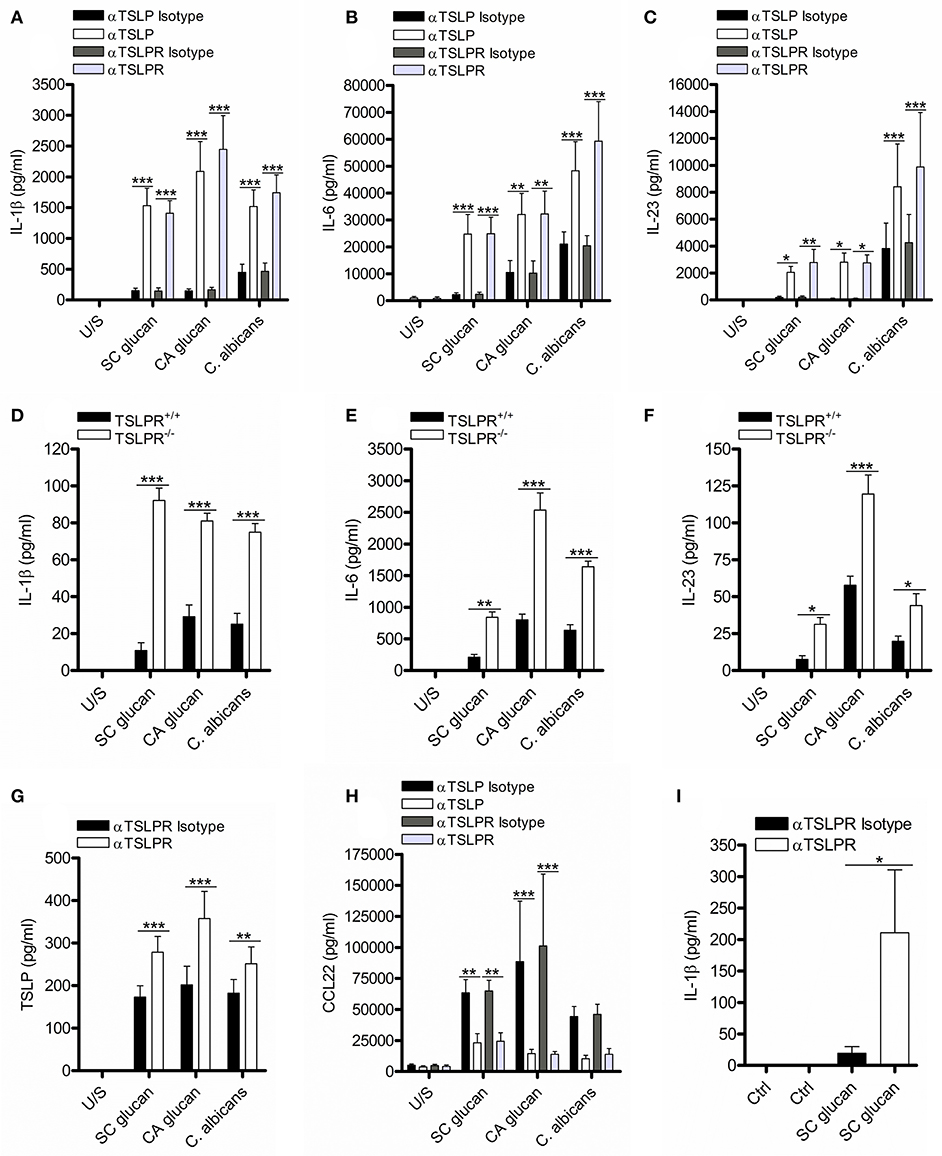
Figure 1. Inhibition of TSLPR signaling in mDC modulates IL-1β production. (A–C,G,H) Human mDC stimulated with SC glucan, CA glucan or heat killed C. albicans hyphae with anti-TSLP, anti-TSLPR or IgG isotype control antibodies for 24 h (n = 14 independent donors, presented as pooled data for SC glucan stimulated mDC), (n = 6 independent donors, presented as pooled data for CA glucan stimulated mDC) and (n = 10 independent donors, presented as pooled data for heat killed C. albicans hyphae stimulated mDC). (D–F) Wildtype TSLPR+/+ or TSLPR−/− BMDC derived from BALB/c mice were stimulated with, SC glucan, CA glucan or C. albicans hyphae for 24 h (n = 4 independent animals from a representative experiment, presented as pooled data. Experiment was repeated four times). (I) Human ex vivo CD1c+ DC stimulated with SC glucan with anti-TSLPR or IgG isotype control antibodies for 24 h (n = 3 independent donors, presented as pooled data). IL-1β, IL-6, IL-23, TSLP and CCL22 was measured in 24-h cell culture supernatants by ELISA. Cumulative data displayed as mean ±SEM. Statistical analysis calculated using one-way ANOVA with Bonferroni post-tests (***p = 0.001, **p = 0.01, *p = 0.05).
TSLPR Signaling Negatively Regulates IL-6 and IL-23 Secretion by Controlling IL-1β
The importance of IL-1β for generating effective anti-fungal immunity is well-established (31). Therefore, we wanted to determine whether the increase in IL-6 and IL-23 secretion from mDC was a direct effect of inhibiting TSLPR signaling on production of these cytokines or due to the effects of increased IL-1β. We have showed that IL-1β mRNA expression precedes that of IL-6 and IL-23p40 (Supplemental Figures 2A–D) and inhibition of IL-1 receptor signaling with IL-1 receptor antagonist (IL-1RA) significantly inhibits IL-6 and IL-23 secretion from SC glucan-stimulated mDC (Supplemental Figures 2E, F). However, to directly address this, we stimulated mDC with C. albicans or β-glucans and neutralized TSLPR signaling in the presence or absence of IL-1RA, or caspase-1 and caspase-8 inhibitors to prevent inflammasome-mediated processing of IL-1β which our group has previously demonstrated (8). This showed that the increased IL-6 and IL-23 observed when TSLPR was blocked, was reduced in the presence of IL-1RA or caspase-1 and caspase-8 inhibitors (Figures 2A–F). Therefore, autocrine TSLPR signaling indirectly regulated IL-6 and IL-23 secretion by its effect on IL-1β production.
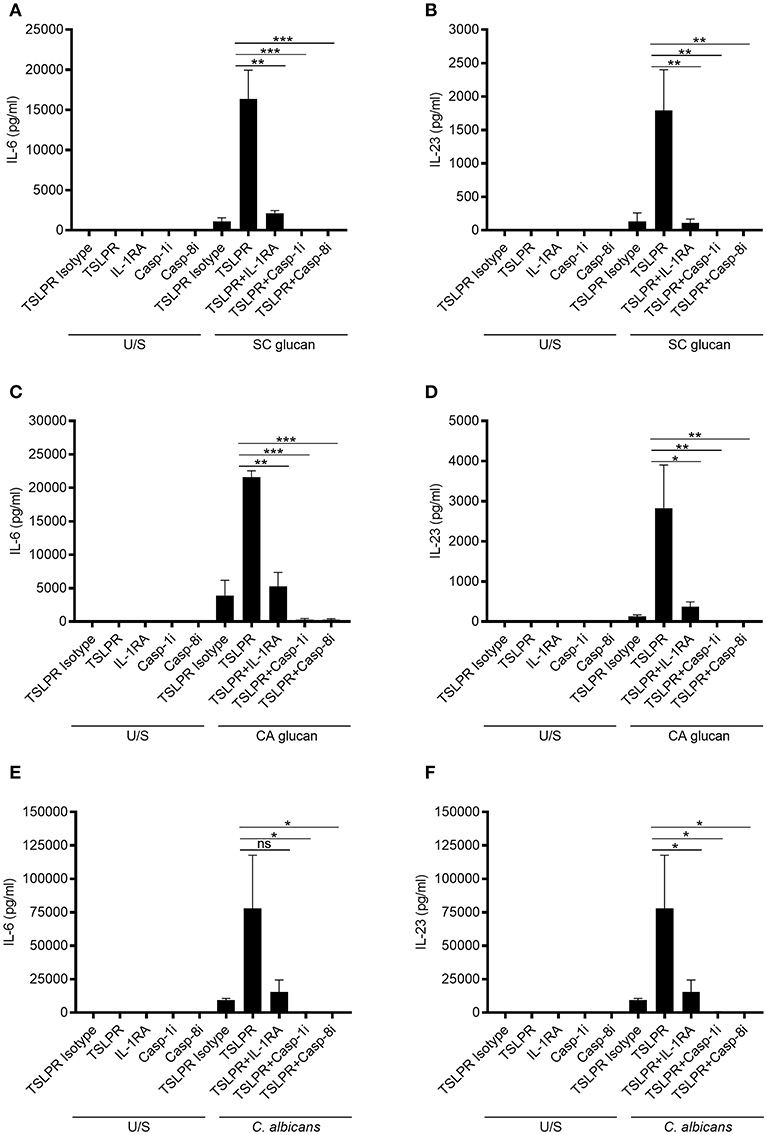
Figure 2. mDC-derived TSLP indirectly regulates IL-6 and IL-23 secretion by controlling IL-1β production. Human mDC stimulated with, (A,B) SC glucan, (C,D) CA glucan or (E,F) heat killed C. albicans hyphae with anti-TSLPR or IgG isotype control antibodies in the presence or absence of IL-RA, caspase-1 inhibitor or caspase-8 inhibitor for 24 h (n = 3 independent donors, presented as pooled data). IL-6 and IL-23 was measured in 24-h cell culture supernatants by ELISA. Cumulative data displayed as mean ±SEM. Statistical analysis calculated using one-way ANOVA with Bonferroni post-tests (ns = not significant, ***p = 0.001, **p = 0.01, *p = 0.05).
Dectin-1-Induced TSLP Negatively Regulates Pro-IL-1β and HIF-1α
Recent work has described how LPS-treated macrophages and β-glucan-stimulated monocytes undergo a metabolic switch toward glycolysis and away from oxidative phosphorylation (40). This is a process which has many parallels with the Warburg effect observed in tumors. We observed that mDC culture media was more acidic when TSLPR signaling was neutralized during dectin-1 stimulation. Therefore, we measured lactate production to determine whether increased lactic acid production accounted for the pH change. Significantly higher concentrations of lactate were indeed detected in cell culture supernatants from mDC when TSLPR signaling was neutralized (Figure 3A) and from TSLPR−/− BMDC (Supplemental Figure 3A), suggesting that autocrine mDC-derived TSLP negatively regulates this metabolic shift to lactate production. Tannahill et al reported that this cellular metabolic switch was crucial for pro-IL-1β expression via the induction of the transcription factor, hypoxia-inducible factor 1-alpha (HIF-1α), and in agreement with this report TSLP or TSLPR neutralization in mDC during dectin-1 signaling augmented both HIF-1α and pro-IL-1β protein expression (Figures 3B–E). An identical augmentation was observed in TSLPR−/− BMDC (Supplemental Figure 3B). The enhancement in HIF-1α expression could not be explained by changes in gene expression since HIF-1α mRNA expression was not significantly modulated by inhibition of TSLPR signaling. In contrast pro-IL-1β mRNA expression was augmented in mDC when TSLPR activity was neutralized (Supplemental Figures 4A,B). These effects were specific and not as a result of a general increase in expression of effector molecules downstream of dectin-1 signaling; for instance p38 mitogen-activated protein kinase (MAPK) activation (Thr 180/Tyr 182) was not modulated by TSLP or TSLPR neutralization (Figures 3B,F). It has previously been shown that chemicals which induce AMP-activated protein kinase (AMPK) activation can oppose the metabolic switch in DCs and macrophages which is induced by PRR stimulation (41). Accordingly, neutralization of TSLPR signaling reduced phosphorylation of Thr 172 on the catalytic subunit of AMPK, a key modification required for AMPK activation (Figures 3B,G). To determine whether the modulation of HIF-1α expression and AMPK phosphorylation occurred as a result of the increased IL-1β expression, IL-1β activity was neutralized in combination with inhibition of TSLPR signaling. This showed that the modulation of AMPK activation by inhibition of TSLPR activity was dependent on IL-1β signaling, but this did not apply to HIF-1α expression (Figures 4A–C).
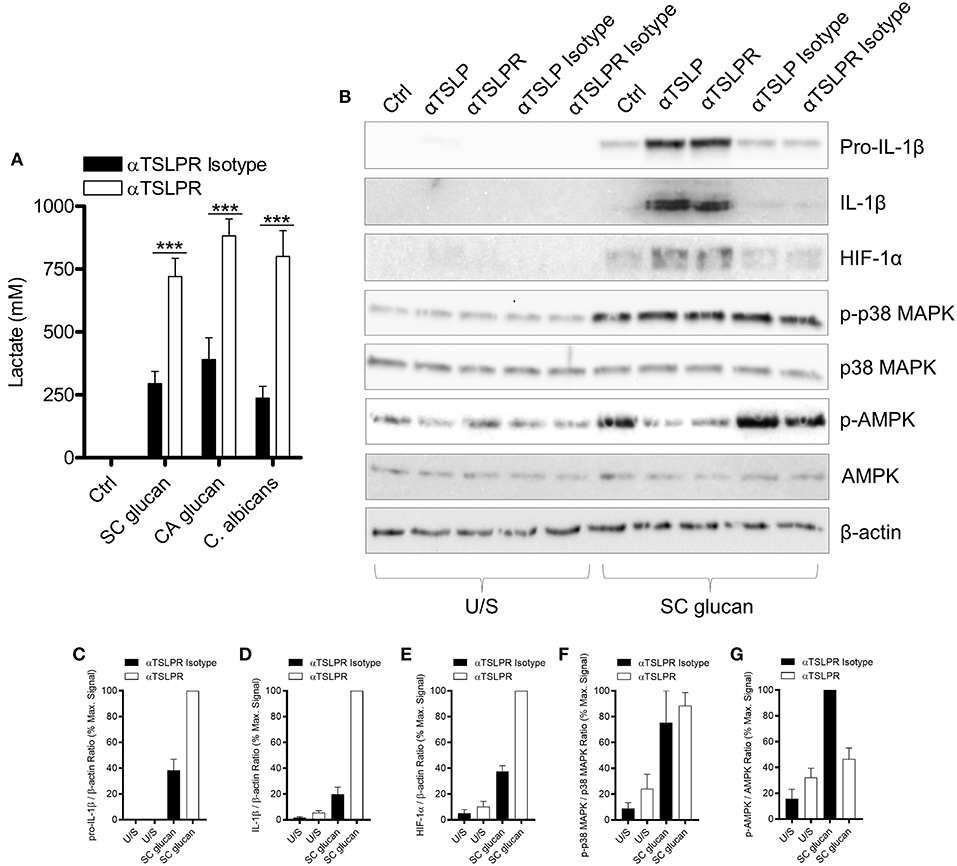
Figure 3. Dectin-1-induced TSLP negatively regulates pro-IL-1β and HIF-1α. (A) Human mDC were stimulated with SC glucan, CA glucan or heat killed C. albicans hyphae with anti-TSLPR antibodies or IgG isotype control for 24 h (n = 6 independent donors, presented as pooled data). Lactate production was measured in cell-culture supernatants using colourmetric L-lactate detection kit. (B) Human mDC were stimulated SC glucan with either anti-TSLP, anti-TSLPR or IgG isotype control antibodies for 8 h (n = 1 representative donor presented, three separate experiments performed). Pro-IL-1β, IL-1β, HIF-1α, phospho-p38 MAPK, p38 MAPK, phospho-AMPK, AMPK and β-actin were measured by immunoblot. (C–G) Densitometry of cumulative data was performed using Image Studio Lite software with pro-IL-1β, IL-1β and HIF-1α normalized to β-actin and phospho-p38 MAPK and phospho-AMPK normalized to total p38 MAPK and AMPK respectively. Data is reported as percentage of maximal signal observed within each donor (n = 3 independent donors, presented as pooled data). Cumulative data displayed as mean +SEM. Statistical analysis calculated using one-way ANOVA with Bonferroni post-tests (***p = 0.001).
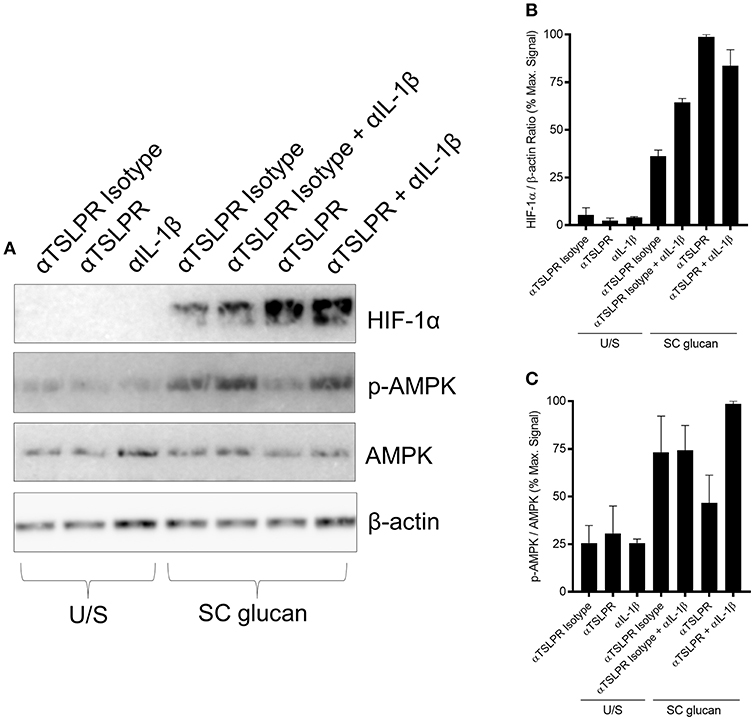
Figure 4. Increased HIF-1α expression is regulated by TSLPR inhibition and not IL-1β secretion (A) Human mDC were stimulated with SC glucan with either anti-TSLPR or IgG isotype control antibodies for 8 h in the presence or absence of IL-1β neutralization antibodies (n = 1 representative donor presented, three separate experiments performed). Pro-IL-1β, IL-1β, HIF-1α, phospho-p38 MAPK, p38 MAPK, phospho-AMPK, AMPK and β-actin were measured by immunoblot. (B,C) Densitometry of cumulative data was performed using Image Studio Lite software with HIF-1α normalized to β-actin and phospho-AMPK normalized to AMPK. Data is reported as percentage of maximal signal observed within each donor (n = 3 independent donors, presented as pooled data). Cumulative data displayed as mean +SEM.
Dectin-1-Induced TSLP Limits IL-1β, HIF-1α Expression, Syk Phosphorylation and the Activation of NADPH Oxidase-Derived ROS
It has previously been shown that reactive oxygen species (ROS) are induced in macrophages during dectin-1 signaling and are important for IL-1β production (26, 27). Chronic granulomatous disease (CGD) patients possess mutations in genes encoding proteins that form the nicotinamide adenine dinucleotide phosphate-oxidase (NADPH) complex; thus these patients are unable to generate NADPH oxidase-derived ROS (Supplemental Figure 5) and TSLP from mDC in response to dectin-1 agonists (23). mDC from CGD donors did not show induction of HIF-1α and pro-IL-1β in response to dectin-1 signaling, highlighting the critical role of ROS in the expression of HIF-1α and pro-IL1β by DCs (Figures 5A–D). In contrast, induction of AMPK phosphorylation was unaffected by the absence of a functional NADPH oxidase (Figures 5A,E), indicating that AMPK activation has distinct signaling from that required for HIF-1α expression. Given the important role of ROS in HIF-1α protein expression, we investigated whether inhibition of TSLPR signaling in CGD patients modulated IL-1β production. TSLPR neutralization during dectin-1 stimulation did not alter the minimal amount of IL-1β secretion seen in CGD donors (Figures 5F–H). Our data suggest NADPH oxidase-derived ROS contributes to the enhancement of HIF-1α and IL-1β expression observed with the loss of TSLPR signaling.
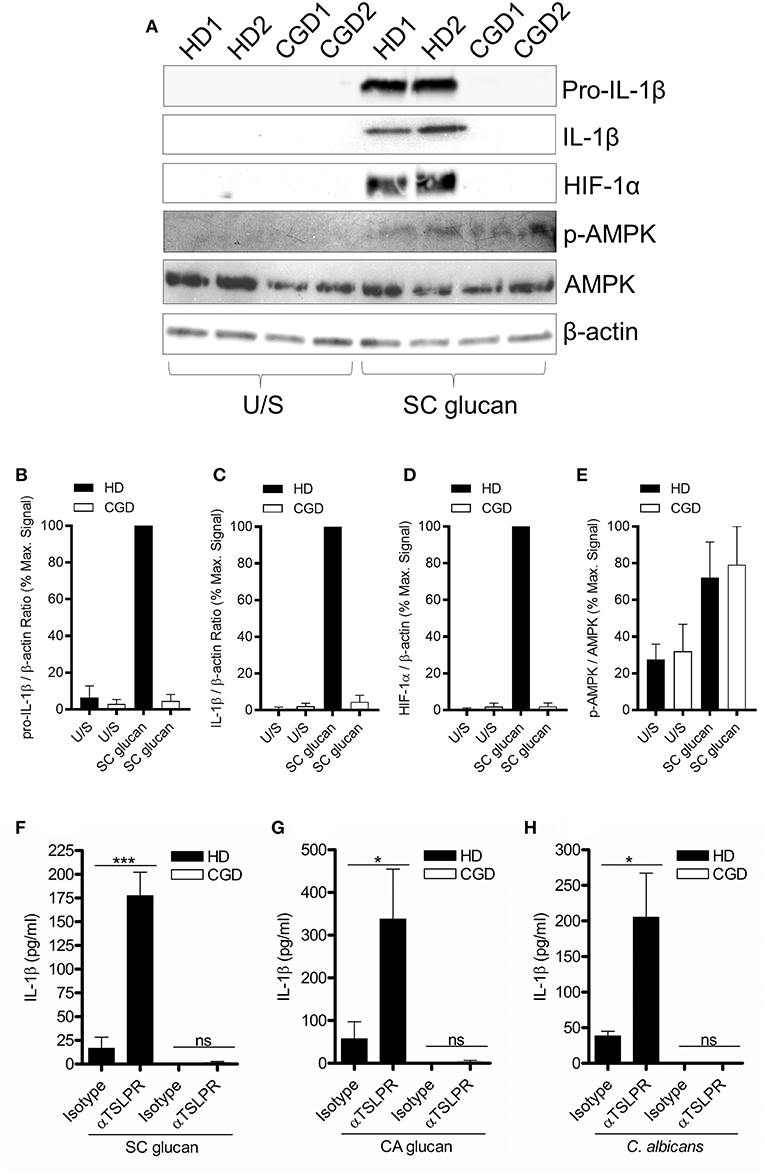
Figure 5. Dectin-1-induced NADPH oxidase-derived ROS is required for augmented IL-1β during TSLPR inhibition. (A) Human mDC derived from healthy donors (HD) or CGD patients were stimulated with SC glucan for 8 h (n = 2 representative donors presented, three separate experiments performed). Pro-IL-1β, IL-1β, HIF-1α, phospho-AMPK, AMPK and β-actin were measured by immunoblot. (B–E) Densitometry of cumulative data was performed using Image Studio Lite software with pro-IL-1β, IL-1β and HIF-1α normalized to β-actin and phospho-AMPK normalized to total AMPK. Data is reported as percentage of maximal signal observed within each donor (n = 3 independent donors, presented as pooled data). (F–H) Human mDC derived from HD or CGD patients were stimulated with SC glucan, CA glucan or heat killed C. albicans hyphae with anti-TSLPR or IgG isotype control antibodies for 24 h (n = 3 independent donors, presented as pooled data). IL-1β was measured in 24-h cell culture supernatants by ELISA. Cumulative data displayed as mean +SEM. Statistical analysis calculated using one-way ANOVA with Bonferroni post-tests (***p = 0.001,*p = 0.05).
Dectin-1-Induced TSLP Negatively Regulates Syk Activation
Dectin-1-mediated effector responses are controlled by the recruitment and activation of Syk (28), and we have previously shown that inhibition of Syk signaling in mDC stimulated with dectin-1 agonists substantially reduces TSLP and IL-1β expression (8). Furthermore, inhibition of Syk activity in DCs with the Syk inhibitor R406 reduced SC glucan-induced HIF-1α and pro-IL-1β expression (Supplemental Figure 6A). We hypothesized that TSLPR signaling may directly modulate Syk activation and therefore examined the phosphorylation status of critical Tyr residues in Syk which are associated with its activation and interaction with downstream signaling pathways. As expected, SC glucan-induced Syk phosphorylation (Tyr 525/526) was dectin-1 dependent (Supplemental Figure 6B). Neutralization of autrocrine TSLPR signaling resulted in enhanced Syk phosphorylation at this residue (Figures 6A–D). Furthermore, analysis of the phosphorylation state of other Tyr residues associated with Syk activation was also enhanced (Figures 6A,C). These data suggest that autocrine mDC-derived TSLP limits Syk-mediated activation. This in turn may negatively regulate the metabolic shift to glycolysis, the production of HIF-1α and hence expression of pro-IL-1β.
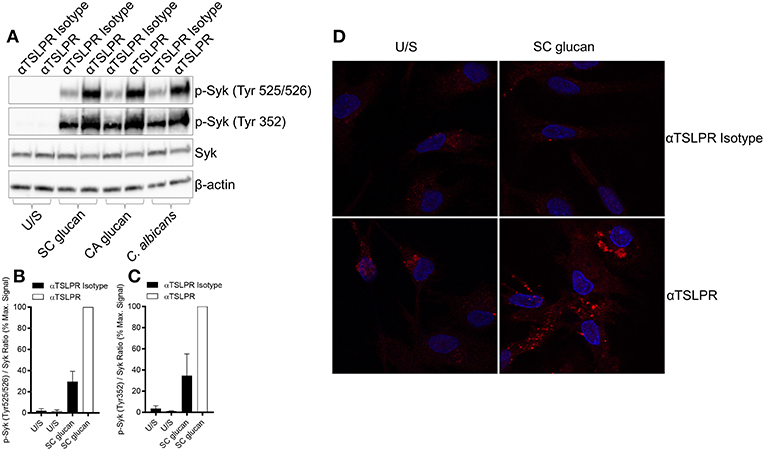
Figure 6. Dectin-1-induced TSLP negatively regulates Syk activation. (A–D) Human mDC were stimulated with SC glucan with anti-TSLPR or IgG isotype control antibodies for 2 h (n = 1 representative donor presented, three separate experiments performed). (B,C) Densitometry of cumulative data was performed using Image Studio Lite software with phospho-Syk normalized to total Syk. Data is reported as percentage of maximal signal observed within each donor (n = 3 independent donors, presented as pooled data). Phospho-Syk measured by (A) immunoblot and (D) confocal microscopy (Red represents p-Syk and Blue represents nuclear DAPI staining).
Discussion
We have previously demonstrated that human mDC and murine BMDC generate TSLP in response to C. albicans or β-glucans (8, 23). In this study, we showed that this TSLP acts in an mDC autocrine fashion to regulate IL-1β, and hence IL-6 and IL-23 production. We propose that this increase in inflammatory cytokine expression which is seen when DC responses to TSLP are blocked is a result of enhanced HIF-1α expression and a more marked glycolytic shift in the metabolism of the DC. Furthermore, we showed that TSLPR signaling dampens Syk phosphorylation likely acting to decrease HIF-1α and pro-IL-1β production.
TSLPR signaling negatively regulates IL-1β production, which in turn modulates the expression of IL-6 and IL-23. It is well-established that IL-1β production plays a critical role in the generation of protective anti-fungal immunity (31); however, IL-1β dysregulation is associated with IBD and CAPS such as Muckle-Wells syndrome (35, 36). Given the importance of IL-1β regulation, we speculate that DC-derived TSLP acts as an important molecular checkpoint to limit IL-1β-mediated effector responses. Furthermore, the differentiation of naïve CD4+ T cells to TH1- and TH17-cells is important for protective anti-fungal immunity and the inflammatory cytokines IL-1β, IL-6, and IL-23 are important in generating these T cell phenotypes (28, 31, 42–44). TSLPR−/− mice have been shown to produce more IFN-γ in an experimental modal of Trypanosoma congolense infection (45), more IFN-γ and IL-17 in an inducible modal of colitis (46) compared to TSLPR+/+ mice and IL-1β is crucial for the generation of inflammatory IFN-γ/IL-17 double producing T cells during C. albicans infection (31, 42). Given that autocrine TSLPR signaling negatively regulated IL-1β production during C. albicans and β-glucan stimulation: TSLPR signaling might also function to regulate TH1- and TH17-cell differentiation.
Recent work has established that myeloid-derived cells stimulated with activators of PRRs switch from oxidative phosphorylation to aerobic glycolysis (40). Similarly, we observed that C. albicans and β-glucan stimulated mDC also induced HIF-1α and increased the production of lactate. Furthermore, both HIF-1α and lactate production were further augmented when TSLPR signaling was neutralized on dectin-1-stimulated mDC. Given that Tannahill et al identified that HIF-1α expression was crucial for pro-IL-1β induction in LPS-treated macrophages (40) and β-glucan stimulated monocytes induce HIF-1α expression required for this glycolytic switch (41): these data are compatible with the idea that autocrine TSLPR signaling controls pro-IL-1β expression in mDC by regulating HIF-1α. Further work will be required to confirm that HIF-1α is a key factor that promotes increased IL-1β expression when TSLPR signaling is inhibited.
IL-1β has been shown to directly induce HIF-1α expression (47–49), but despite an increase in IL-1β when inhibiting TSLPR signaling, our data provide evidence that the enhanced HIF-1α expression induced by dectin-1 occurred independently of this cytokine. In contrast to the regulation of HIF-1α, modulation of AMPK phosphorylation was shown to be dependent on the secretion of IL-1β and to our knowledge this is the first report that suggests that IL-1β may negatively regulate AMPK activation. Our data also highlighted the differences in the signaling requirements for ROS in AMPK activation and HIF-1α expression. We showed that following dectin-1 stimulation, phosphorylation of the AMPK catalytic subunit was unaffected by the absence of ROS but in contrast, HIF-1α expression was completely dependent on ROS production. Given that AMPK activation has been shown to antagonize HIF-1α expression, it may not be surprising that the signaling requirements for these factors have shared and distinct arms, highlighting the potential for independent regulation of these factors in the dectin-1 signaling pathway. Most importantly, the modulation of HIF-1α and AMPK in the presence of TSLPR neutralizing antibodies, could be interpreted as complimentary responses, since enhanced HIF-1α or a reduction in AMPK activation have been shown to contribute to a metabolic shift toward aerobic glycolysis and the increased production of inflammatory cytokines in monocytes and DC (41, 50).
Our data also showed that inhibition of TSLPR signaling on mDC enhanced Syk activation. We show that CGD patients cannot generate ROS, HIF-1α or IL-1β secretion; and unlike in healthy donors, mDC-derived from CGD patients do not augment HIF-1α expression or IL-1β secretion when autocrine TSLPR signaling is neutralized. These data are in agreement with published work showing that both Syk and ROS regulate IL-1β cleavage during dectin-1 stimulation (27) and that ROS can activate HIF-1α expression (51). Therefore, autocrine TSLP production may directly regulate HIF-1α and pro-IL-1β, via Syk-mediated activation of NADPH oxidase-derived ROS: a regulatory mechanism that does not exist in CGD patients.
Therefore, dysregulation of TSLP production or TSLPR signaling might be a feature of diseases associated with Syk and IL-1β overproduction. Opportunistic invasive fungal infections present serious clinical complications particularly in immunosuppressed individuals. Given the importance of IL-1β to anti-fungal immunity these findings describe an important regulatory mechanism of IL-1β that could ultimately lead to the development of approaches to boost resistance.
Materials and Methods
Ethics Statement
Human blood was sourced from apheresis cones derived from healthy donors (HD) (Addenbrooke's Hospital, Cambridge) and age- and sex-matched CGD patients (Royal Free Hospital, London). Appropriate consent to use blood-derived cells for research was obtained. These studies were approved by the Joint UCL/UCLH Committee for the Ethics of Human Research, project number 04/Q0501/119.
Cell Isolation and Generation of Dendritic Cells
Human monocyte-derived dendritic cells (mDC) were generated from CD14+ monocytes isolated from human PBMC by magnetic bead separation (Miltenyi) and were differentiated by culturing for 6-days in RPMI1640 (Lonza) 5% FCS (Biosera) supplemented with 20 ng/ml GM-CSF (Life Technologies) and 4 ng/ml IL-4 (BD Biosciences) as described previously (8). CGD donors were recruited at the Royal Free Hospital, London and age and sex matched to healthy donors recruited at the University of Cambridge, in accordance with ethical rules set out by each institution. Human CD1c+ DC were isolated from PBMC by magnetic bead separation (Miltenyi). Murine bone marrow-derived dendritic cells (BMDC) were generated by culturing cells isolated from bone marrow of wildtype (TSLPR+/+) and TSLPR knockout (TSLPR−/−) BALB/c mice for 7-days in RPMI1640 10% FCS supplemented with 5% X63 conditioned media and 10 ng/ml IL-4 (Peprotech).
Cell Stimulations
mDCs were stimulated with 50 μg/ml of either β-1,3 glucan (SC glucan) derived from Saccharomyces cerevisiae (S. cerevisiae) isolated by David. L. Williams, East Tennessee State University as previously described (52), β-1,3 glucan (CA glucan) derived from hyphal Candida albicans (C. albicans) isolated by David. L. Williams as previously described (53) or heat-killed hyphal C. albicans (MOI 2:1) gifted from John Trowsdale, University of Cambridge. C. albicans was grown in sabouraud dextrose broth for 8 h at 37°C to an optical density of 0.2. C. albicans was killed by heating for 1 h at 70°C.
Reagents
10 μg/ml sheep anti-TSLP blocking antibody (R&D Systems), 10 μg/ml sheep IgG isotype control (R&D Systems), 10 μg/ml goat anti-TSLPR blocking antibody (R&D Systems), 10 μg/ml goat IgG isotype control (R&D Systems), 2 μg/ml IL-1β blocking antibody (R&D Systems), 10 μg/ml mouse IgG2B dectin-1 blocking antibody (clone-259931 R&D Systems), 10ug/ml mouse IgG2B isotype control (clone-20116 R&D Systems), 1 μM Syk inhibitor, R406 (Selleckchem), 1 μg/ml IL-1 receptor antagonist (IL-1RA) (R&D Systems), 50 μM caspase-1 inhibitor (Z-YVAD-FMK) (Calbiochem), 50 μM caspase-8 inhibitor (Z-IE(OMe)TD(OMe)-FMK) (Calbiochem). Where inhibitors, blocking antibodies and modifiers were used, mDCs were pre-treated 1 h prior to cell stimulation. Repeated experiments were performed on independent donors unless otherwise stated.
Cytokine Production
IL-1β, IL-6, IL-23 (eBioscience), TSLP, and CCL22 (R&D) were measured in 24 h mDC, CD1c+ DC or BMDC culture supernatants by ELISA according to manufacturer's protocols.
Quantitative Real-Time PCR
mDC were stimulated for indicated time period and IL-1β, HIF-1α, IL-6, IL-23p19 and IL-23p40 mRNA expression was measured by quantitative real-time PCR from isolated RNA (Norgen) using TaqMan Gene Expression Assays (Applied Biosystems). Gene expression was normalized to HPRT and calculated as relative expression (2-dCT).
Immunoblot
mDC were stimulated for indicated time period and protein lystates were generated, quantified by Bradford assay (Thermo) and resolved using SDS-PAGE. IL-1β (R&D, AB-201-AB), HIF-1α (Novus, NB100-449), phospho-AMPK (Thr 172) (Cell Signaling, 2535), AMPK (Cell Signaling, 5831), phospho-Syk (Tyr 525/526) (Cell Signaling, 2710), phospho-Syk (Tyr 352) (Cell Signaling, 2701), Syk (Cell Signaling, 13198), phospho-p38 MAPK (Thr 180/ Tyr 182) (Cell Signaling, 4511), p38 MAPK (Cell Signaling, 8690) and β-actin (Abcam, 8226) protein expression were measured by immunoblot, by incubation with indicated primary antibodies followed by incubation with HRP-conjugated secondary antibodies, ECL detection (PerkinElmer) and visualized using GBox (Syngene). Densitometry of cumulative data was performed using Image Studio Lite software. Pro-IL-1β, IL-1β and HIF-1α expression was normalized to β-actin and phospho-p38 MAPK, phospho-AMPK and phospho-Syk was normalized to total p38 MAPK, AMPK and Syk respectively. Cumulative data is reported as percentage of maximal signal observed within each donor.
Quantification of Reactive Oxygen Species (ROS) Production
mDC derived from HD or CGD patients were stimulated with SC glucan and ROS production was measured by fluorescence of the luminol-based chemiluminescent probe L-012 (WAKO) over 30 min using a luminometer (Centro LB960, Berthold).
Confocal Microscopy
mDC were stimulated for indicated time period on poly-D-lysine coated coverslips (BD), washed with cold PBS and stained for phospho-Syk (Tyr 525/526) (Cell Signaling, 2710). Coverslips were then mounted on slides with DAPI fluoromount G (Southern Biotech) and analyzed by confocal microscopy (Leica SP5).
Lactate Detection
mDC were stimulated and lactate production was measured instantly from 24 h cell-culture supernatants using colourmetric L-lactate detection kit (Abcam) according to manufacturer's protocols.
Data Analysis
Data were analyzed using GraphPad Prism statistical package. Cumulative data are displayed as mean ±SEM. Statistical analysis using either t test (p-values stated in figures legends) or one-way ANOVAs with Bonferroni post-tests ns = not significant, *p < 0.05, **p < 0.01, ***p < 0.001.
Ethics Statement
Human blood was sourced from apheresis cones derived from healthy donors (Addenbrooke's Hospital, Cambridge) and age and sex-matched CGD patients (Royal Free Hospital, London). Appropriate consent to use blood-derived cells for research was obtained. Ethics Reference Number: 04/Q0501/119.
Author Contributions
ME designed, performed and analyzed all experimental data and drafted the manuscript. SW, ZM, EC, JSG, and JCG were key to experimental design, data interpretation, and reviewed manuscript. TF, AS, and MS performed and analyzed experimental data. RC facilitated access to patient blood and aided data interpretation and reviewed manuscript. DW facilitated access to β-glucan agonists and aided experimental design, data interpretation, and reviewed manuscript.
Funding
This research was supported by Arthritis Research UK 19962 to JSG and Arthritis Research UK RG61798 to JCG and in part, by NIH R01GM53522, RO1GM119197, and RO1GM083016 to DW and NIH C06RR0306551 to ES.
Conflict of Interest Statement
ME and EC are employed by AstraZeneca.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Sarita Workman and Professor John Trowsdale for providing us with key reagents; Lou Ellis and Sarah Gibbs for some of the sample processing and Sam Strickson for some of the data analysis.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.00921/full#supplementary-material
Abbreviations
TSLP, Thymic stromal lymphopoietin; mDC, monocyte-derived DC; BMDC, bone marrow-derived dendritic cell.
References
1. Friend SL, Hosier S, Nelson A, Foxworthe D, Williams DE, Farr A. A thymic stromal cell line supports in vitro development of surface IgM+ B cells and produces a novel growth factor affecting B and T lineage cells. Exp Hematol. (1994) 22:321–8.
2. Lee HC, Ziegler SF. Inducible expression of the proallergic cytokine thymic stromal lymphopoietin in airway epithelial cells is controlled by NFkappaB. Proc Natl Acad Sci USA. (2007) 104:914–9. doi: 10.1073/pnas.0607305104
3. Goodall JC, Wu C, Zhang Y, McNeill L, Ellis L, Saudek V, et al. Endoplasmic reticulum stress-induced transcription factor, CHOP, is crucial for dendritic cell IL-23 expression. Proc Natl Acad Sci USA. (2010) 107:17698–703. doi: 10.1073/pnas.1011736107
4. Kashyap M, Rochman Y, Spolski R, Samsel L, Leonard WJ. Thymic stromal lymphopoietin is produced by dendritic cells. J Immunol. (2011) 187:1207–11. doi: 10.4049/jimmunol.1100355
5. Urata Y, Osuga Y, Izumi G, Takamura M, Koga K, Nagai M, et al. Interleukin-1beta stimulates the secretion of thymic stromal lymphopoietin (TSLP) from endometrioma stromal cells: possible involvement of TSLP in endometriosis. Hum Reprod. (2012) 27:3028–35. doi: 10.1093/humrep/des291
6. Zhang Y, Zhou X, Zhou B. DC-derived TSLP promotes Th2 polarization in LPS-primed allergic airway inflammation. Eur J Immunol. (2012) 42:1735–43. doi: 10.1002/eji.201142123
7. Dewas C, Chen X, Honda T, Junttila I, Linton J, Udey MC, et al. TSLP expression: analysis with a ZsGreen TSLP reporter mouse. J Immunol. (2015) 194:1372–80. doi: 10.4049/jimmunol.1400519
8. Elder MJ, Webster SJ, Williams DL, Gaston JS, Goodall JC. TSLP production by dendritic cells is modulated by IL-1β and components of the endoplasmic reticulum stress response. Eur J Immunol. (2016) 46:455–63. doi: 10.1002/eji.201545537
9. Iliev ID, Spadoni I, Mileti E, Matteoli G, Sonzogni A, Sampietro GM, et al. Human intestinal epithelial cells promote the differentiation of tolerogenic dendritic cells. Gut. (2009) 58:1481–9. doi: 10.1136/gut.2008.175166
10. Taylor BC, Zaph C, Troy AE, Du Y, Guild KJ, Comeau MR, et al. TSLP regulates intestinal immunity and inflammation in mouse models of helminth infection and colitis. J Exp Med. (2009) 206:655–67. doi: 10.1084/jem.20081499
11. Corren J, Parnes JR, Wang L, Mo M, Roseti SL, Griffiths JM, et al. Tezepelumab in adults with uncontrolled asthma. N Engl J Med. (2017) 377:936–46. doi: 10.1056/NEJMoa1704064
12. Wilson SR, The L, Batia LM, Beattie K, Katibah GE, McClain SP, et al. The epithelial cell-derived atopic dermatitis cytokine TSLP activates neurons to induce itch. Cell. (2013) 155:285–95. doi: 10.1016/j.cell.2013.08.057
13. Zhou B, Comeau MR, De Smedt T, Liggitt HD, Dahl ME, Lewis DB, et al. Thymic stromal lymphopoietin as a key initiator of allergic airway inflammation in mice. Nat Immunol. (2005) 6:1047–53. doi: 10.1038/ni1247
14. Volpe E, Pattarini L, Martinez-Cingolani C, Meller S, Donnadieu MH, Bogiatzi SI, et al. Thymic stromal lymphopoietin links keratinocytes and dendritic cell-derived IL-23 in patients with psoriasis. J Allergy Clin Immunol. (2014) 134:373–81. doi: 10.1016/j.jaci.2014.04.022
15. Park LS, Martin U, Garka K, Gliniak B, Di Santo JP, Muller W, et al. Cloning of the murine thymic stromal lymphopoietin (TSLP) receptor: formation of a functional heteromeric complex requires interleukin 7 receptor. J Exp Med. (2000) 192:659–70. doi: 10.1084/jem.192.5.659
16. Arima K, Watanabe N, Hanabuchi S, Chang M, Sun SC, Liu YJ. Distinct signal codes generate dendritic cell functional plasticity. Sci Signal. (2010) 3:ra4. doi: 10.1126/scisignal.2000567
17. Rochman Y, Kashyap M, Robinson GW, Sakamoto K, Gomez-Rodriguez J, Wagner KU, et al. Thymic stromal lymphopoietin-mediated STAT5 phosphorylation via kinases JAK1 and JAK2 reveals a key difference from IL-7-induced signaling. Proc Natl Acad Sci USA. (2010) 107:19455–60. doi: 10.1073/pnas.1008271107
18. Bell BD, Kitajima M, Larson RP, Stoklasek TA, Dang K, Sakamoto K, et al. The transcription factor STAT5 is critical in dendritic cells for the development of TH2 but not TH1 responses. Nat Immunol. (2013) 14:364–71. doi: 10.1038/ni.2541
19. Ziegler SF, Artis D. Sensing the outside world: TSLP regulates barrier immunity. Nat Immunol. (2010) 11:289–93. doi: 10.1038/ni.1852
20. Watanabe N, Hanabuchi S, Soumelis V, Yuan W, Ho S, de Waal Malefyt R, et al. Human thymic stromal lymphopoietin promotes dendritic cell-mediated CD4+ T cell homeostatic expansion. Nat Immunol. (2004) 5:426–34. doi: 10.1038/ni1048
21. Soumelis V, Reche PA, Kanzler H, Yuan W, Edward G, Homey B, et al. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat Immunol. (2002) 3:673–80. doi: 10.1038/ni805
22. Ito T, Wang YH, Duramad O, Hori T, Delespesse GJ, Watanabe N, et al. TSLP-activated dendritic cells induce an inflammatory T helper type 2 cell response through OX40 ligand. J Exp Med. (2005) 202:1213–23. doi: 10.1084/jem.20051135
23. Elder MJ, Webster SJ, Chee R, Williams DL, Hill Gaston JS, Goodall JC. Beta-glucan size controls dectin-1-mediated immune responses in human dendritic cells by regulating Il-1beta production. Front Immunol. (2017) 8:791. doi: 10.3389/fimmu.2017.00791
24. Taylor PR, Tsoni SV, Willment JA, Dennehy KM, Rosas M, Findon H, et al. Dectin-1 is required for beta-glucan recognition and control of fungal infection. Nat Immunol. (2007) 8:31–8. doi: 10.1038/ni1408
25. Brown GD, Taylor PR, Reid DM, Willment JA, Williams DL, Martinez-Pomares L, et al. Dectin-1 is a major beta-glucan receptor on macrophages. J Exp Med. (2002) 196:407–12. doi: 10.1084/jem.20020470
26. Underhill DM, Rossnagle E, Lowell CA, Simmons RM. Dectin-1 activates Syk tyrosine kinase in a dynamic subset of macrophages for reactive oxygen production. Blood. (2005) 106:2543–50. doi: 10.1182/blood-2005-03-1239
27. Gross O, Poeck H, Bscheider M, Dostert C, Hannesschlager N, Endres S, et al. Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature. (2009) 459:433–6. doi: 10.1038/nature07965
28. Goodridge HS, Wolf AJ, Underhill DM. Beta-glucan recognition by the innate immune system. Immunol Rev. (2009) 230:38–50. doi: 10.1111/j.1600-065X.2009.00793.x
29. Hise AG, Tomalka J, Ganesan S, Patel K, Hall BA, Brown GD, et al. An essential role for the NLRP3 inflammasome in host defense against the human fungal pathogen Candida albicans. Cell Host Microbe. (2009) 5:487–97. doi: 10.1016/j.chom.2009.05.002
30. Gringhuis SI, Kaptein TM, Wevers BA, Theelen B, van der Vlist M, Boekhout T, et al. Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1beta via a noncanonical caspase-8 inflammasome. Nat Immunol. (2012) 13:246–54. doi: 10.1038/ni.2222
31. Zielinski CE, Mele F, Aschenbrenner D, Jarrossay D, Ronchi F, Gattorno M, et al. Pathogen-induced human TH17 cells produce IFN-gamma or IL-10 and are regulated by IL-1beta. Nature. (2012) 484:514–8. doi: 10.1038/nature10957
32. Ganesan S, Rathinam VA, Bossaller L, Army K, Kaiser WJ, Mocarski ES, et al. Caspase-8 modulates Dectin-1 and complement receptor 3-driven il-1beta production in response to beta-glucans and the fungal pathogen, Candida albicans. J Immunol. (2014) 193:2519–30. doi: 10.4049/jimmunol.1400276
33. Van't Wout JW, Van der Meer JW, Barza M, Dinarello CA. Protection of neutropenic mice from lethal Candida albicans infection by recombinant interleukin 1. Eur J Immunol. (1988) 18:1143–6.
34. Vonk AG, Netea MG, van Krieken JH, Iwakura Y, van der Meer JW, Kullberg BJ. Endogenous interleukin (IL)-1 alpha and IL-1 beta are crucial for host defense against disseminated candidiasis. J Infect Dis. (2006) 193:1419–26. doi: 10.1086/503363
35. Kuemmerle-Deschner JB, Wittkowski H, Tyrrell PN, Koetter I, Lohse P, Ummenhofer K, et al. Treatment of Muckle-Wells syndrome: analysis of two IL-1-blocking regimens. Arthritis Res Ther. (2013) 15:R64. doi: 10.1186/ar4237
36. Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. (2013) 13:397–411. doi: 10.1038/nri3452
37. Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose-Girma M, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. (2006) 440:228–32. doi: 10.1038/nature04515
38. Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. (2008) 9:847–56. doi: 10.1038/ni.1631
39. Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM, Nunez G. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. (2013) 38:1142–53. doi: 10.1016/j.immuni.2013.05.016
40. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature. (2013) 496:238–42. doi: 10.1038/nature11986
41. Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. (2014) 345:1250684. doi: 10.1126/science.1250684
42. van de Veerdonk FL, Joosten LA, Shaw PJ, Smeekens SP, Malireddi RK, van der Meer JW, et al. The inflammasome drives protective Th1 and Th17 cellular responses in disseminated candidiasis. Eur J Immunol. (2011) 41:2260–8. doi: 10.1002/eji.201041226
43. Lemoine S, Jaron B, Tabka S, Ettreiki C, Deriaud E, Zhivaki D, et al. Dectin-1 activation unlocks IL12A expression and reveals the TH1 potency of neonatal dendritic cells. J Allergy Clin Immunol. (2015) 136:1355–68 e1315. doi: 10.1016/j.jaci.2015.02.030
44. Li J, Leyva-Castillo JM, Hener P, Eisenmann A, Zaafouri S, Jonca N, et al. Counterregulation between thymic stromal lymphopoietin- and IL-23-driven immune axes shapes skin inflammation in mice with epidermal barrier defects. J Allergy Clin Immunol. (2016) 138:150–61 e113. doi: 10.1016/j.jaci.2016.01.013
45. Onyilagha C, Singh R, Gounni AS, Uzonna JE. Thymic stromal lymphopoietin is critical for regulation of proinflammatory cytokine response and resistance to experimental trypanosoma congolense infection. Front Immunol. (2017) 8:803. doi: 10.3389/fimmu.2017.00803
46. Spadoni I, Iliev ID, Rossi G, Rescigno M. Dendritic cells produce TSLP that limits the differentiation of Th17 cells, fosters Treg development, and protects against colitis. Mucosal Immunol. (2012) 5:184–93. doi: 10.1038/mi.2011.64
47. Hellwig-Burgel T, Rutkowski K, Metzen E, Fandrey J, Jelkmann W. Interleukin-1beta and tumor necrosis factor-alpha stimulate DNA binding of hypoxia-inducible factor-1. Blood. (1999) 94:1561–7.
48. Thornton RD, Lane P, Borghaei RC, Pease EA, Caro J, Mochan E. Interleukin 1 induces hypoxia-inducible factor 1 in human gingival and synovial fibroblasts. Biochem J. (2000) 350 Pt 1:307–12. doi: 10.1042/bj3500307
49. Jung YJ, Isaacs JS, Lee S, Trepel J, Neckers L. IL-1beta-mediated up-regulation of HIF-1alpha via an NFkappaB/COX-2 pathway identifies HIF-1 as a critical link between inflammation and oncogenesis. FASEB J. (2003) 17:2115–7. doi: 10.1096/fj.03-0329fje
50. Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood. (2010) 115:4742–9. doi: 10.1182/blood-2009-10-249540
51. Qutub AA, Popel AS. Reactive oxygen species regulate hypoxia-inducible factor 1alpha differentially in cancer and ischemia. Mol Cell Biol. (2008) 28:5106–19. doi: 10.1128/MCB.00060-08
52. Williams DL, McNamee RB, Jones EL, Pretus HA, Ensley HE, Browder IW, et al. A method for the solubilization of a (1–−3)-beta-D-glucan isolated from Saccharomyces cerevisiae. Carbohydr Res. (1991) 219:203–13. doi: 10.1016/0008-6215(91)89052-H
Keywords: TSLP, dectin-1, IL-1β, hypoxia, ROS, HIF-1α, Syk
Citation: Elder MJ, Webster SJ, Fitzmaurice TJ, Shaunak ASD, Steinmetz M, Chee R, Mallat Z, Cohen ES, Williams DL, Gaston JSH and Goodall JC (2019) Dendritic Cell-Derived TSLP Negatively Regulates HIF-1α and IL-1β During Dectin-1 Signaling. Front. Immunol. 10:921. doi: 10.3389/fimmu.2019.00921
Received: 22 November 2018; Accepted: 10 April 2019;
Published: 08 May 2019.
Edited by:
Catarina R. Almeida, University of Aveiro, PortugalReviewed by:
Elisabetta Volpe, Fondazione Santa Lucia (IRCCS), ItalyKushagra Bansal, Harvard Medical School, United States
Copyright © 2019 Elder, Webster, Fitzmaurice, Shaunak, Steinmetz, Chee, Mallat, Cohen, Williams, Gaston and Goodall. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jane C. Goodall, amNnMjNAbWVkc2NobC5jYW0uYWMudWs=