 Jasper F. Nies
Jasper F. Nies Ulf Panzer
Ulf Panzer- 1Translational Immunology, III. Department of Medicine, University Medical Center Hamburg-Eppendorf Hamburg, Hamburg, Germany
- 2Hamburg Center of Translational Immunology (HCTI), University Medical Center Hamburg-Eppendorf, Hamburg, Germany
Therapeutic targeting of IL-17A and its receptor IL-17RA with antibodies has turned out to be a tremendous success in the treatment of several autoimmune conditions. As the IL-17 cytokine family consists of six members (IL-17A to F), it is intriguing to elucidate the biological function of these five other molecules to identify more potential targets. In the past decade, IL-17C has emerged as quite a unique member of this pro-inflammatory cytokine group. In contrast to the well-described IL-17A and IL-17F, IL-17C is upregulated at very early timepoints of several disease settings. Also, the cellular source of the homodimeric cytokine differs from the other members of the family: Epithelial rather than hematopoietic cells were identified as the producers of IL-17C, while its receptor IL-17RE is expressed on TH17 cells as well as the epithelial cells themselves. Numerous investigations led to the current understanding that IL-17C (a) maintains an autocrine loop in the epithelium reinforcing innate immune barriers and (b) stimulates highly inflammatory TH17 cells. Functionally, the IL-17C/RE axis has been described to be involved in the pathogenesis of several diseases ranging from infectious and autoimmune conditions to cancer development and progression. This body of evidence has paved the way for the first clinical trials attempting to neutralize IL-17C in patients. Here, we review the latest knowledge about identification, regulation, and function of the IL-17C/IL-17receptor E pathway in inflammation and immunity, with a focus on the mechanisms underlying tissue injury. We also discuss the rationale for the translation of these findings into new therapeutic approaches in patients with immune-mediated disease.
Introduction
The discovery of TH17 cells as a novel subset of CD4+ T cells in 2005 (1) lead to a paradigm shift in the field of immunology. Our previously incomplete and inconsistent understanding of many diseases' pathogenesis was manifold enhanced thanks to rigorous examination of this new T cell lineage. These discoveries are not only important for basic immunological research, but drugs targeting TH17-related molecules have had a significant impact on the treatment of immunological diseases (2–4).
As the name of the TH17 cells was coined by their characteristic production of the highly inflammatory cytokine IL-17A upon activation, most scientific effort has been put into understanding the biological activity of this protein. However, five more cytokines with structural similarity to IL-17A have been identified (IL-17B-F). In this six-member cytokine family, IL-17A is best characterized, followed by the very closely related IL-17F. Structurally, all members of the IL-17 cytokine family are homodimers in their biologically active form, yet one heterodimer consisting of IL-17A and IL-17F (IL-17A/F) is described (5, 6). The proteins bind to heterodimeric receptor complexes to induce signaling in their target cells. Most of those complexes consist of the ubiquitously expressed subunit IL-17RA and a second, ligand-specific subunit (IL-17RB-RE) (7–11). IL-17D remains an orphan ligand in the cytokine family (Figure 1).
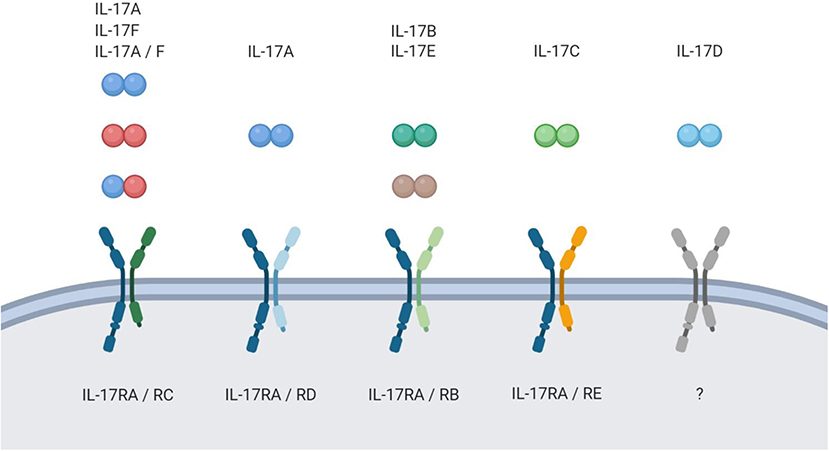
Figure 1. The IL-17 family. Schematic overview of the IL-17 family members and their respective receptor complexes.
In line with the current understanding of TH17 cells being a highly inflammatory lineage, IL-17A and F induce several inflammatory pathways. Most markedly, their binding to the receptor complex IL-17RA/RC, which is predominantly expressed on epithelial cells, leads to upregulation of cytokines, anti-bacterial peptides, and chemokines. The chemokines then recruit innate immune cells like neutrophils which potently enhance the inflammatory reaction. Thus, it is fair to say that by now we have got a good grasp of how IL-17A and F unfold their inflammatory effect.
The role of the remaining four IL-17 family members has long been considered rather elusive. However, the last years have shed a little more light on the IL-17C/RE axis, which unveiled some unique features.
In this review, we provide an overview of expression patterns and the functional importance of IL-17C and its receptor IL-17RE in immunological diseases, present a hypothesis of how the IL-17C/RE axis mediates its inflammatory effect, summarize intracellular signaling pathways, and give an outlook on translational approaches.
Identification of the IL-17C/RE Axis
First Characterization of IL-17C
In 2000, the cytokine IL-17C has first been identified by a homology-search for proteins similar to IL-17A (12). IL17C is located on chromosome 16q24, is 1.1 kb long, and the protein IL-17C shares roughly 27% amino acid identity with IL-17A. Interestingly, after stimulation no induction of IL17C mRNA was observed in CD4+ cells, which are the main source of IL-17A and F. This was the first evidence that IL-17C seems to assume a unique role in the IL-17 family. In an initial functional analysis of the protein, the authors showed that IL-17C stimulated the monocytic cell line THP-1 to release TNF-α and IL-1β.
IL-17C Is Expressed by Epithelial Cells and Not by Hematopoietic Cells
Unlike what is known about the other IL-17 family members, many studies suggest that IL17C is not expressed by leukocytes, but by non-hematopoietic cells.
The characteristic production of IL-17A by a subset of CD4+ cells has led to the name of TH17 cells, which emerged to be a distinct lineage apart from the classical dichotomy of TH1 and TH2 cells. However, not only CD4+ T cells produce IL-17A, but also CD8+ T cells (13), γδ T cells (14, 15), invariant natural killer T cells (iNKT) (16), group 3 innate lymphoid cells (ILCs) (17), and even B cells (18).
In contrast, IL17C is expressed by epithelial cells. In a model for psoriasis, keratinocytes are the main source of IL-17C (19). Several groups confirmed this IL17C expression in keratinocytes (20–23). Other epithelial cells producing the cytokine include colonic epithelial cells (9), resident kidney cells (24, 25), and respiratory epithelial cells (26–28).
Although this strong evidence points to epithelial cells as the main source of IL17C, its expression has also been found in leukocytes (29, 30) and smooth muscle cells (31).
IL-17C and the Microbiome
TH17 biology is closely linked to the microbiome as it influences TH17 cell development: Experiments with antibiotic treatment or germ-free mice drastically reduced intestinal TH17 cells (32, 33). However, specific bacteria are required for proper induction of this cell type. Segmented filamentous bacteria (SFB) can potently induce the TH17 cell development (34, 35), while Bacteroides fragilis suppresses this differentiation (36). Thus, changes in the gut flora influence the development of TH17 cells, which can both aggravate or ameliorate extra-intestinal TH17-driven autoimmunity (37).
Even though TH17 cells themselves are not the source of IL-17C, intestinal bacteria still seem to play a role for IL17C expression in the gut. Antibiotic treatment of mice blocked the induction of Il17c. IL-23 and IL-22 were reported to be dispensable, but TLR-MyD88 signaling in gut resident cells was essential for the induction of Il17c expression. In the same experiment, the authors identified MyD88 as being essential for proper induction of Il17a, but not Il17c, in hematopoietic cells (38). Co-culture of murine colonic epithelial cells with Citrobacter rodentium induces IL-17C production in those cells. Specifically, Lipopolysaccharide (LPS) and flagellin are two pattern-associated molecular patterns (PAMPs) that can be recognized by toll-like receptors (TLRs) and culturing the cells with those components alone resulted in strong IL-17C production (9). A change in Il17c expression was not observed in any of the analyzed leukocyte populations (T lymphocytes, B lymphocytes, intraepithelial lymphocytes, lamina propria mononuclear cells). This finding was validated by the fact that no difference in Il17c induction was seen between wildtype and recombination-activating gene 1 (Rag1) deficient mice. Within the non-leukocytic cell populations in the colon, Il17c induction was indeed limited to only the colonic epithelial cells since no mRNA upregulation Il17c was seen in colonic stromal cells after infection (9). Those findings indicate that TLR activation by microbiota in the gut is important for both IL-17A and IL-17C, albeit the source of those cytokines is located to different cell types: Hematopoietic cells and gut resident epithelial cells, respectively.
Thus, epithelial cells are the main source of the cytokine in different tissues. That stands in stark contrast to the cellular source of other cytokines of the IL-17 family, which are mainly expressed by leukocytes.
IL17C Is Upregulated Early in Disease
Regarding the temporal expression of IL17C, current data point to an early upregulation during disease. Il17c mRNA was strongly upregulated after 4 days in the colons of bacterially infected mice, while Il17a expression peaked at day 12 (9). Ramirez-Carrozzi et al. analyzed the kinetics of Il17c expression in detail: in vitro stimulation of HCT-15 cells with heat-killed E. coli lead to a rapid expression of the cytokine after 1 h and murine skin challenged with imiquimod showed strong Il17c expression after 2 days. In the DSS-colitis model, the authors found induction of Il17c expression after 2 days in colons and mesenteric lymph nodes, but upregulation of I17a and Il17f mRNA transcripts was not detected before day 6 (19). In the nephrotoxic nephritis (NTN) mouse model for crescentic glomerulonephritis, we showed that Il17c is upregulated as early as 12 h after induction of the disease, while Il17a and Il17f expression starts after a couple of days (24).
IL-17C Binds to the Receptor Complex IL-17RA/RE
The group that first described IL-17C also suggested that IL-17C does not bind to IL-17RA, but to another receptor. Expressing a His-tagged and metabolically labeled form of the extracellular domain of IL-17RA in 293T cells, precipitation with IL-17A was observed as expected, but no precipitation could be detected during incubation with IL-17C (12). Six years later, another group discovered this receptor, which has been named IL-17RE. Murine IL-17RE shares 40% DNA and 18% amino acid sequence with IL-17RC (39). However, the authors did not yet identify a ligand binding to this receptor subunit. The receptor was found to be expressed in lung, kidney, stomach, intestine, and testis of mice and to have six different isoforms.
Several groups described that IL-17C is the specific ligand for IL-17RE in 2011: Transfection of 293T cells with the receptor subunits IL-17RA-RE revealed that IL-17C seems to bind exclusively to IL-17RE (40). Another group used a similar approach by analyzing the binding of Flag-tagged human IL-17C to HEK293 cells overexpressing each of the five IL-17 receptor subunits. In contrast to the findings of the first characterization of IL-17C (no binding to IL-17RA) (12), flow cytometer examination of the cells after incubation with IL-17C showed binding to both IL-17RA and IL-17RE, but none of the other receptor subunits. Also, no interactions were found between IL-17RE and any of the other IL-17 cytokine family members (19).
Song and colleagues used glutathione S-transferase precipitation to demonstrate that IL-17C associates not only with IL-17RE but with a heterodimeric receptor complex consisting of IL-17RA and IL-17RE (9). Using a blocking antibody against IL-17RA during stimulation of keratinocytes with IL-17C, a dose-dependent inhibition of IL-17C-induced G-CSF and β-defensin-2 expression was observed, underlining the functional dependence on IL-17RA (19).
The IL-17RA/RE Receptor Complex Is Expressed on Both Epithelial and TH17 Cells
Interestingly, epithelial cells—the main source of IL-17C—express the specific receptor for the cytokine. Strong Il17re expression has been detected in keratinocytes and colon epithelial cells (19). Reynolds et al. report expression of the receptor subunit in the colonic epithelial cell line YAMC (41). IL17RE is also expressed in nerve fibers of human skin after HSV-2 reactivation (21).
Apart from epithelial cells, Chang et al. first described that Il17re is expressed on TH17 cells. While numerous tissues express an isoform of IL-17RE that lacks the transmembrane domain, TH17 cells expressed high amounts of full-length IL17RE. This expression is strongly enhanced when the cells are stimulated with a cytokine cocktail of IL-6, TGF-β, IL-1, and IL-23 and IL-17C also induced expression of the receptor on TH17 cells (40). Validating this finding, our group found strong Il17re expression IL-17A+ YFP+ cells from IL-17A YFP+ fate reporter mice and in TH17 polarized cells (compared to TH0, TH1, and Treg cells) (24).
Similar to IL17C, we reported that Il17re was upregulated 24 h after induction of NTN (24).
In summary, epithelial cells produce IL-17C at early timepoints in disease. The cytokine signals through the heterodimeric receptor complex IL-17RA/RE. This complex is expressed by several epithelial cells themselves. Secondly, TH17 cells express IL17RE which indicates that this T cell lineage is also responsive to IL-17C (Table 1).
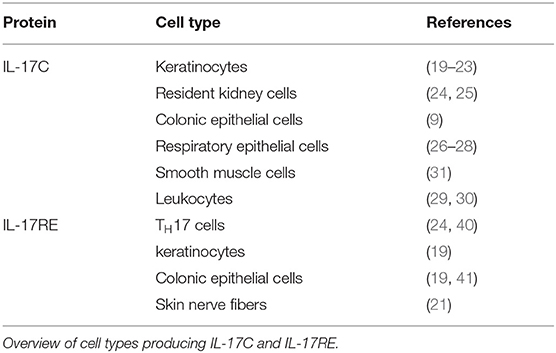
Table 1. Sources of IL-17C and IL-17RE.
Infection and Autoimmunity
Many studies report that IL17C expression is upregulated at an early stage in both infectious and autoimmune diseases. This suggests that it is involved in the innate first-line immunity in the pathogenesis of those conditions. Intriguingly, IL-17C also plays an important role in the initiation of the adaptive immune response later. First, we will take a closer look at the role of the IL-17C/RE immune axis in infectious diseases. Second, we will zoom in on autoimmune conditions.
IL-17C/RE Signaling Induces Innate Immune Functions in Bacterial, Fungal, and Viral Infections
Signaling through the IL-17C/RE is involved in host defense against foreign pathogenic microorganisms. In the following paragraphs, we will summarize the role of the axis in bacterial, fungal, and viral infections.
Bacterial Infections
The IL-17C/RE axis plays a significant role in several bacterial infection models. Mice infected with the intestinal pathogen Citrobacter rodentium showed upregulation of Il17c mRNA in the colon (9). Ex vivo cultured murine colon tissue and colonic epithelial cells showed marked mRNA expression of antibacterial peptides, inflammatory cytokines, and chemokines after stimulation with IL-17C. Clinically, lack of signaling through the IL-17C/RE axis modeled with Il17re−/− mice lead to decreased mRNA levels of said molecules and failure to clear the infection. This resulted in loss of body weight, higher intestinal and splenic weight, increased bacterial burden, and death. Interestingly, there was no difference when the cells were treated with IL-17A or F, which indicates that IL-17RE is dispensable for these two cytokines.
In a model of acute colitis, I17c−/− mice challenged with dextran sulfate sodium (DSS) had a significantly worse outcome than mice with physiological IL-17C production, which is reflected by earlier and more pronounced weight loss and colonic shortening. The authors explain this observation with the fact that IL-17C induced mRNA expression of tight-junction molecules, which are essential for the integrity of the colonic mucosal barrier (41).
Another group examined the role of IL-17RE in this model confirming those findings of the IL-17C/RE axis assuming a crucial role in protection against bacteria-driven DSS-induced colitis (19).
The immune axis also plays a role in the defense against airway infections with Pseudomonas aeruginosa and Haemophilus influenza (26, 27).
Fungal Infections
The impact of IL-17C has also been examined in fungal infections. Huang and colleagues reported that IL-17C is required for a lethal course of systemic infection with Candida albicans in mice since II17c−/− mice displayed increased survival and less severe functional and morphological kidney damage (25). This is in contrast to the function of IL-17A in this model: While Il17a overexpression protects the mice, lack of signaling through IL-17RA results in increased susceptibility to this fungal infection (42). Similarly, patients with Job's syndrome, a condition with TH17 cell defects, are also at great risk to suffer from such fungal infections (43, 44). Another study reported that IL-17C is not involved in immunity to systemic, oral and dermal candidiasis (45). Even though Il17c mRNA expression was upregulated 2 days after exposure to the fungus, the group did not observe a difference in clearance of the infection or gene expression profiles between mice lacking IL-17C or IL-17RE compared to a wildtype control group.
Viral Infections
Two studies investigated the role of IL-17C/RE in viral infections. Peng et al. showed that IL-17C was the only IL-17 family cytokine that was induced in keratinocytes from human genital skin biopsies during recurrent HSV-2 reactivation. Also, cultured human keratinocytes produced IL-17C in response to infection with HSV-2. Since cutaneous nerve fibers expressed IL17RE and ex vivo application of IL-17C reduced apoptosis in the nerve cells, the authors hypothesize that keratinocyte-derived IL-17C serves as a protective agent for nerve fibers during HSV-2 reactivation in the skin (21). Another group recently analyzed the effects of IL-17C in in vitro virus-bacteria coinfection of human bronchial epithelial cells to assess the cytokine's role in COPD exacerbations. A challenge with both pathogens resulted in a synergistic induction of IL-17C. Interestingly, tissue from healthy smokers released little IL-17C upon exposure to the pathogens, but epithelial cells from COPD patients released significantly more. Thus, the IL-17C/RE axis might be involved in the pathogenesis of COPD exacerbations of mixed upper airway infections (46).
Several TH17-Driven Autoimmune Diseases Are Exacerbated by IL-17C/RE
Inflammation orchestrated by TH17 cells is a hallmark of various autoimmune conditions like rheumatoid arthritis, psoriasis, multiple sclerosis, autoimmune kidney diseases, and autoimmune hepatitis.
In 2007, Yamaguchi et al. attributed IL-17C a role in the pathogenesis of collagen-induced arthritis (30). Mice adoptively transferred with CD4+ T cells, which were retrovirally transduced with either IL-17A, B, C, or F, had significantly higher arthritis scores than those that got cells transduced with an empty vector.
Several studies also evaluated the role of IL-17C in skin inflammation complementing the picture of IL-17C-induced auto-aggression. Johansen et al. first showed that IL17C mRNA and protein levels were increased in the skin of patients with psoriatic lesions compared to non-lesional skin (47). In fact, IL-17C is by far the most abundant IL-17 cytokine found in the skin of such skin lesions: Its protein levels were reported to be roughly 125-fold higher than those of IL-17A in the lesions (48). Transgenic mice lacking Il17c, Il17ra, or Il17re display a less severe course of imiquimod-induced psoriasis (19, 49) while an overexpression of Il17c in skin keratinocytes lead to spontaneous development of psoriasiform skin lesions (48). IL-17C also drives inflammation in atopic dermatitis as IL17C expression was increased in lesional skin of patients and blocking IL-17C with an antibody ameliorated skin inflammation in one mouse model for psoriasis and two models for atopic dermatitis (50).
Also, IL-17C/RE signaling aggravates the course of experimental autoimmune encephalitis (EAE) (40). Il17c−/− mice were less prone to develop the disease and those that did showed less pronounced clinical manifestations of the inflammation. Vice versa, increased signaling through the axis in transgenic mice overexpressing Il17re in CD4+ cells lead to a worse clinical situation of the animals.
We have recently described that the serum levels of IL-17C are significantly higher in patients with ANCA-associated glomerulonephritis compared to a healthy control group, which was not true for IL-17A, F, and B. We showed the pro-inflammatory role of IL-17C in established mouse models for lupus nephritis and crescentic glomerulonephritis. In accordance with the mentioned previous studies, our experiments showed expression of Il17re by TH17 cells and significantly less TH17 cells in inflamed kidneys of both Il17c−/− and Il17re−/− mice (24).
Two studies investigated the involvement of the IL-17C/RE axis in autoimmune hepatitis. One group found evidence that IL-17C stimulates intrahepatic CD4+ T cells to release IL-2 with subsequent NK-cell mediated liver damage. In this study, lesser levels of GOT and GPT in sera of Il17c−/− and Il17re−/− mice were found compared to wildtype mice (51). However, another group found no differences in GOT and GPT activities and granulocyte infiltration into the liver between Il17c−/− and wildtype mice in the same model (52).
Further diseases involving the IL-17C/RE axis include psoriasiform skin lesions in inflammatory bowel disease (IBD) patients under anti-TNF-α treatment (53), recurrent aphthous ulcers (20), LPS-induced endotoxin shock (52), and different forms of cancer (38, 54–56) (Tables 2, 3).
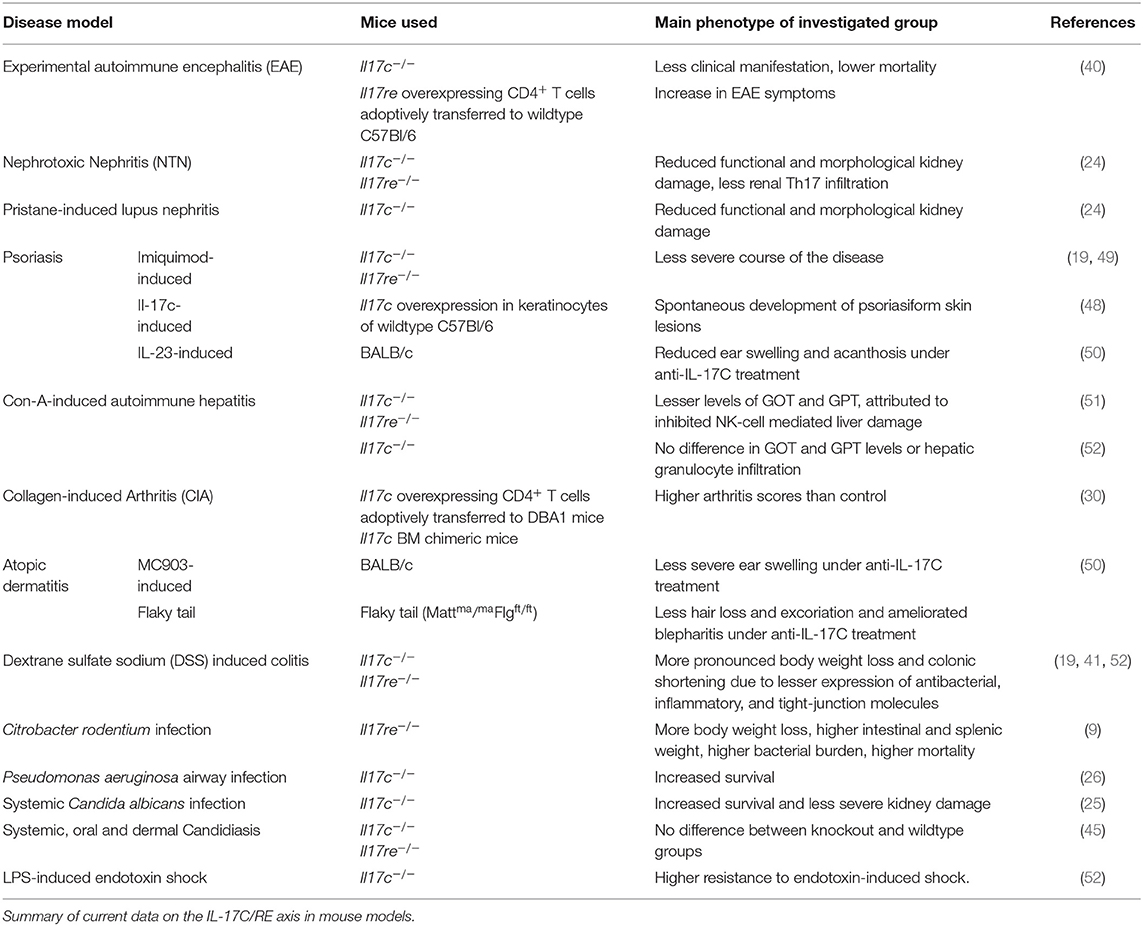
Table 2. Main findings of experimental data on IL-17C and IL-17RE.
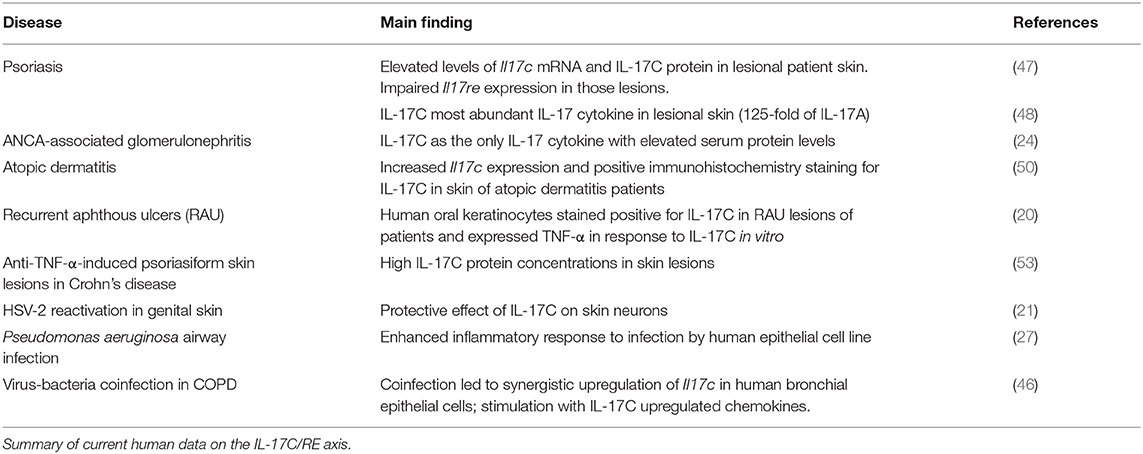
Table 3. IL-17C/RE data on human samples.
Mechanisms of IL-17C/RE Driven Inflammation
Mechanistically, a body of evidence suggests that IL-17C exerts two important immunological effects: (a) In an autocrine feedback loop with epithelial cells, IL-17C strengthens innate barriers against infectious agents. (b) Boosting TH17 cell function, IL-17C also stimulates the adaptive immune system to efficiently fight off infections. Yet, those pathways harbor the risk of TH17-driven autoimmunity.
As IL-17A has been studied much more extensively as IL-17C and acts on epithelial cells, it is worthwhile to recapitulate the signaling of IL-17A through IL-17RA.
The similar expression of fibroblast growth factor and IL-17R (SEFIR) domain is highly conserved within the IL-17 receptor family and structurally similar to the Toll/IL-1R (TIR) domain found in TLRs and the IL-1β receptor (57). Yet, IL-17 signaling employs an adaptor protein unique to IL-17 signaling called ACT1, which also carries the SEFIR domain. The adaptor protein can then bind several intracellular signaling proteins to induce several conserved signaling pathways. Pathways activated by IL-17 receptor signaling include nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) (58), inhibitor of NF-κB ζ (IκBζ) (59), mitogen-activated protein kinase (MAPK) (60–62), and CCAAT/enhancer-binding protein (C/EBP) (63, 64). Together, these pathways mediate mitogenic signals and induce expression of pro-inflammatory cytokines and chemokines.
Another domain called TIR-like loop (TILL) domain is crucial for IL-17 signaling but is only present in the IL-17RA subunit (65). However, most of the other subunits of this family heterodimerize with IL-17RA to form a functional complex, which suggests that IL-17RA is the domain necessary for intracellular signaling. Also, the C/EBPβ activation domain (CBAD) on IL-17RA stimulates signaling through the transcription factor C/EBPβ (65), initiating one of the few known inhibitory mechanisms of IL-17 signaling (66).
A very important aspect of IL-17 signaling is synergism: De novo gene expression by IL-17A in target cells does not fully account for the observed strong inflammatory effect of the cytokine. Signaling through IL-17RA stabilizes mRNA transcripts of genes expressed by other strong inflammatory stimuli like TNF-α (59, 67). Ligand binding to IL-17RA recruits the kinase IKKi to phosphorylate ACT1. TRAF2 and 5 then bind to form a complex that can inhibit cleavage of mRNA (61, 68).
Thus, the full biological activity of IL-17A becomes apparent only in concert with other factors of the inflammatory milieu. Such synergetic effects have also been described between IL-17C and three other cytokines: TNF-α (19, 24, 48), IL-22 (9, 24), and IL-1β (19). However, the underlying molecular mechanisms have not specifically been studied for IL-17C/RE signaling.
IL-17C and the Epithelial Cell
The first site of IL-17C immunity is the epithelial cell. Group-specific innate signaling pathways like the activation of TLRs in response to PAMPs induce expression of Il17c (9, 19, 38). Activation of TLR is one of the first responses of the immune system after contact with pathogens, which explains the early upregulation of IL17C in the various infectious diseases. Intracellular MyD88 signaling induced by TLR2 and 5 agonists or IL-1β stimulated the expression of IL17C in mucosal epithelial cells (19). Another intracellular mechanism for IL17C expression in response to pathogens is activation of nucleotide-binding oligomerization domain-containing protein 2 (NOD2) by Staphylococcus aureus (69).
There is strong evidence for a synergistic effect between TNF-α and IL-17A as IL-17A signaling stabilizes mRNA of target genes of TNF-α. Interestingly, one target gene that is synergistically induced by IL-17A and TNF-α is IL17C (70). However, stimulation of murine and human epithelial cells with TNF-α or IL-17A alone is also capable of upregulating IL17C expression (9, 19).
These findings are underlined by the fact that IL17C expression is decreased in skin biopsies of psoriasis patients under anti-TNF-α therapy (22). Likewise, IL-17RA blockade with Brodalumab lead to decreased levels of IL17C expression in psoriatic skin (71).
In terms of signaling cascades, TNF-α signaling seems to employ the p38 mitogen activated protein kinase (22) and the NF-κB pathway to enhance IL17C expression. Direct evidence of this are three bindings sites for NF-κB in the IL17C promotor (23).
Thus, both PAMPs and pro-inflammatory cytokines can induce IL17C expression. TNF-α and IL-17A are able to induce IL17C individually and a strong synergistic effect between the two cytokines drastically boosts the expression.
Binding of IL-17C to the IL-17RA/RE complex on the epithelial IL-17C-source cells forms an autocrine loop in the epithelium. Like IL-17A, IL-17C signaling through IL-17RA/RE employs the adaptor molecule ACT1 (40). The signaling cascade then activates the MAPK pathway by phosphorylation of p38, ERK, and JNK as well as the NF-κB pathway by phosphorylation of the p65 subunit and the NF-κB inhibitor IκBα (9). Also, signaling through L-17RA/RE on epithelial cells reinforces the mechanical epithelial barrier by expressing the tight-junction proteins occludin, claudin-1, and claudin-4 (41). Host defense mechanisms in epithelial cells induced by IL-17C include the expression of hBD2, S100A7/8/9, CXCL1/2/3, CCL20, TNFAIP6, and TNIP3 (19) as well as pro-inflammatory cytokines like IL-1β, IL-17A/F, IL-22, IL-6, IL-8, VEGF, and TNF-α (48). This expression profile is a potent response to actively fight off invading pathogens.
Thus, the autocrine loop of IL-17C in the epithelium is an early protective response against pathogenic alterations in the microbiome and other epithelial tissues.
IL-17C and the TH17 Cell
The second site of action of IL-17C is the TH17 cell. We have shown that the numbers of TH17 cells significantly decrease in the absence of IL-17C or IL-17RE in a murine models of autoimmune kidney diseases (24). This effect of IL-17C on TH17 cells might be due to increased proliferation or differentiation, inhibited apoptosis, or impeded exhaustion. Other groups have investigated these intracellular effects in more detail.
In the EAE model, TH17 differentiation was induced by IL-17C/RE signaling via IκBζ (40). Signaling through IL-17RE lead to increased production of IL-17A, IL-17F, and IL-22. Song et al. showed that IL-17C induces the expression of anti-apoptotic factors BCL2 and BCL2L1 in intestinal epithelial cells (38). This anti-apoptotic effect of the IL-17C/RE axis was also seen in nerve fibers during HSV-2 reactivation (21). As mentioned before, signaling pathways of IL-17C in epithelial cells involve NF-κB and MAPK (9), which might also be true for TH17 cells and would be indicative of an effect on proliferation of target cells.
Many groups have shown the pro-inflammatory role of IL-17C in disease settings that are known to be driven by a strong TH17 cell activity. As IL-17C induces the expression of IL-17A in TH17 cells (40), it may be that this effect of IL-17C is dependent on IL-17A. Indeed, blockade of IL-17A with an antibody abolished the difference in renal damage between wildtype and Il17c−/− mice in a model for crescentic glomerulonephritis (24). Thus, this stimulatory effect of IL-17C/RE on TH17 cells leads to higher levels of TH17 signature cytokines—above all IL-17A—which accounts for the strong inflammatory effect of IL-17C. As excessive TH17 cell activity is linked to many autoimmune diseases, IL-17C-mediated stimulation of the TH17 cell represents a cause for TH17 autoimmunity upstream of main effector cytokines like IL-17A and F.
In terms of regulating IL-17C signaling, Monin et al. identified the endoribonuclease MCP-1 induced protein 1 (MCPIP1) as a negative regulator of both IL-17A and C signaling: In a model of imiquimod-induced skin inflammation, mice deficient in MCPIP1 showed increased inflammation and upregulation of IL-17A- and IL-17C-dependent genes, but unaltered levels of IL-17A and C. This indicates that MCPIP1 influences intracellular pathways downstream of IL-17 receptor signaling as opposed to modulation of the expression of IL-17 cytokines (72). The exact mechanism of this negative regulation on IL-17A and C signaling has not been described. However, previous studies have shown that MCPIP1 hampers TLR signaling in response to LPS by degrading mRNA of Il6 (73) and interferes with MAPK and NF-κB signaling by deubiquitination of signaling molecules (74). Even more, MCPIP1 degrades Il17ra and Il17rc mRNA (75) and MCPIP1 deficiency boosts TH17 effector functions (76), which underlines its regulatory effect in IL-17 signaling.
Figure 2 summarizes intracellular signaling pathways of IL-17C.
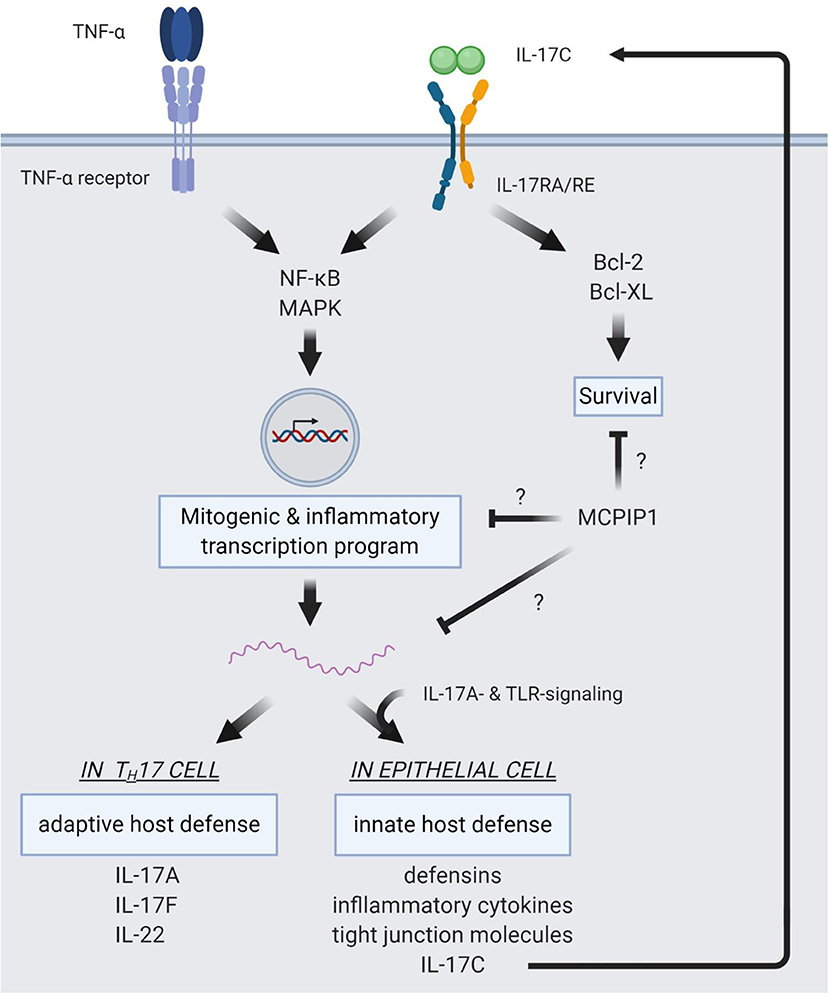
Figure 2. Intracellular pathways of IL-17C signaling. IL-17C signaling induces NF-κB and MAPK signaling pathways. This induction shows a synergistic effect with TNF-α signaling, resulting in strong induction of a mitogenic and pro-inflammatory expression profile. In epithelial cells, target genes of IL-17C/RE signaling are defensins, inflammatory cytokines, and tight junction molecules to reinforce innate host barriers in response to pathogens. Also, IL-17C expression is upregulated in the epithelium and subject to a synergism of IL-17A- and TLR-signaling. IL-17C is then released from the epithelial cell and binds to the IL-17RA/RE receptor complex expressed on the same cell, forming an autocrine loop. In TH17 cells, IL-17C induces expression of IL17A, IL17F, and IL22, boosting adaptive defense mechanisms. IL-17C also activates anti-apoptotic pathways via Bcl-2 and Bcl-XL. MCPIP1 is a regulator of IL-17C/RE signaling, but the distinct mechanisms of this negative regulation are not yet elucidated.
Taken together, IL-17C assumes a position at the interface of innate and adaptive immune system: It is upregulated during early stages of disease and reinforces innate defense lines in the epithelium via an autocrine loop. Its stimulatory action on the TH17 cells induces the adaptive immune response and can trigger autoimmune disease (Figure 3).
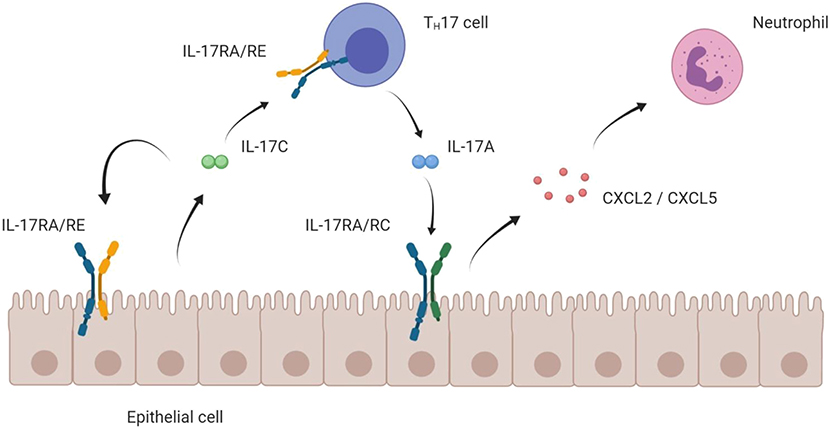
Figure 3. Mode of action of IL-17C on epithelial and TH17 cells. Schematic hypothesis of the pro-inflammatory mode of action of the IL-17C/RE axis. IL-17C is mainly expressed by epithelial cells. Expressing the IL-17RA/RE receptor complex, both the epithelial cell itself and TH17 cells are targets for IL-17C. Boosting expression of IL17A in TH17 cells, IL-17C indirectly enhances epithelial expression of chemokines that attract neutrophils, which ultimately cause a strong inflammatory reaction.
First Steps in Therapeutic Targeting of the IL-17C/RE Axis
Antibodies targeting TH17 cell functions are already in clinical use for a host of autoimmune disorders like psoriasis, psoriatic arthritis, and IBD.
Ustekinumab is a monoclonal antibody directed against the p40 subunit which is shared by the cytokines IL-12 and IL-23. It has been shown to be very successful in the treatment of psoriasis and psoriatic arthritis (77, 78) and is approved for Crohn's disease (79).
Other antibodies directly targeting IL-17A (Secukinumab, Ixekizumab) or the receptor IL-17RA (Brodalumab) show astonishing effects in psoriasis patients (4, 80, 81). Other indications are psoriatic arthritis (2, 3, 82) and ankylosing spondylitis (83).
Neutralizing IL-17C is an intriguing approach in the treatment of autoimmune diseases as it might hamper TH17 function in general and not only the impact signaling of the signature cytokine IL-17A. Indeed, the first clinical studies with an anti-IL-17C-neutralizing antibody have been started in patients with atopic dermatitis (84) after trials in murine models showed promising results (50).
Interestingly, targeting the cytokine IL-17A or its receptor IL-17RA aggravates symptoms in IBD patients (85, 86). This shows that intervening in those signaling pathways might not be as straightforward as initially thought. Thus, it might be possible that the protective role that IL-17C plays for the integrity of epithelial barrier function exceeds its pathological effect for TH17 stimulation in autoimmunity. Disrupting the autocrine loop of the epithelial cells with an antibody might lead to unwanted adverse effects like gastrointestinal or respiratory infections. Inhibiting the TH17 cell function obviously harbors the risk of a general susceptibility to infections with extracellular bacteria and fungi.
Discussion
In summary, IL-17C is a homodimeric cytokine that is expressed by non-hematopoietic—mainly epithelial—cells. It binds to its heterodimeric receptor complex IL-17RA/RE that is expressed on both a variety of epithelial cells and TH17 cells. Compared to other IL-17 cytokine family members, IL17C is upregulated at early stages of the diseases and plays two roles. (a) In an autocrine manner it sustains barrier integrity of epithelial cell layers and thus supports the innate immune system to keep infections in check. (b) By binding to IL-17RE on TH17 cells, IL-17C also stimulates the adaptive immune response to potently fight off invading pathogens. The downside of this mode of action is the risk of immunological derailment, leading to autoimmune conditions.
Intracellular signaling of IL-17C/RE involves anti-apoptotic Bcl-2 and Bcl-XL as well as the NF-κB and MAPK pathways to promote proliferation and host defense. The induction of IL17C has been shown to be dependent on TLR signaling and pro-inflammatory cytokines. IL17C expression is subject to a synergism between TNF-α and IL-17A, presumably due to mRNA stabilization by IL-17A. To date, the molecular mechanisms of described synergisms between IL-17C and other cytokines (TNF-α, IL-22, and IL-1β) have not specifically been investigated.
Being an inflammatory mediator upstream of TH17 effector cytokines, IL-17C represents an interesting target for pharmacological intervention. The first clinical trials have been started for atopic dermatitis and data from human samples suggest transferability of experimental data to the clinical setting for some diseases (24, 48, 53).
The first translational approaches to pharmacologically exploit the IL-17C/RE axis are on the way. We believe that the main potential of such interventions lies in the treatment of autoimmune disorders. Yet, a lot of experimental data on more disease settings requires further analyses of human samples to investigate potential patient populations for this kind of treatment.
Author Contributions
UP and JN wrote the manuscript and designed the figures.
Funding
UP and JN were supported by the Deutsche Forschungsgemeinschaft (SFB1192 project A1 and MGK).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors apologize to all colleagues whose work could not be cited due to space restrictions.
References
1. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. (2005) 201:233–40. doi: 10.1084/jem.20041257
2. Mease PJ, McInnes IB, Kirkham B, Kavanaugh A, Rahman P, van der Heijde D, et al. Secukinumab inhibition of interleukin-17A in patients with psoriatic arthritis. N Engl J Med. (2015) 373:1329–39. doi: 10.1056/NEJMoa1412679
3. McInnes IB, Mease PJ, Kirkham B, Kavanaugh A, Ritchlin CT, Rahman P, et al. Secukinumab, a human anti-interleukin-17A monoclonal antibody, in patients with psoriatic arthritis (FUTURE 2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. (2015) 386:1137–46. doi: 10.1016/S0140-6736(15)61134-5
4. Langley RG, Elewski BE, Lebwohl M, Reich K, Griffiths CE, Papp K, et al. Secukinumab in plaque psoriasis–results of two phase 3 trials. N Engl J Med. (2014) 371:326–38. doi: 10.1056/NEJMoa1314258
5. Wright JF, Guo Y, Quazi A, Luxenberg DP, Bennett F, Ross JF, et al. Identification of an interleukin 17F/17A heterodimer in activated human CD4+ T cells. J Biol Chem. (2007) 282:13447–55. doi: 10.1074/jbc.M700499200
6. Chang SH, Dong C. A novel heterodimeric cytokine consisting of IL-17 and IL-17F regulates inflammatory responses. Cell Res. (2007) 17:435–40. doi: 10.1038/cr.2007.35
7. Toy D, Kugler D, Wolfson M, Vanden Bos T, Gurgel J, Derry J, et al. Cutting edge: interleukin 17 signals through a heteromeric receptor complex. J Immunol. (2006) 177:36–9. doi: 10.4049/jimmunol.177.1.36
8. Rickel EA, Siegel LA, Yoon BR, Rottman JB, Kugler DG, Swart DA, et al. Identification of functional roles for both IL-17RB and IL-17RA in mediating IL-25-induced activities. J Immunol. (2008) 181:4299–310. doi: 10.4049/jimmunol.181.6.4299
9. Song X, Zhu S, Shi P, Liu Y, Shi Y, Levin SD, et al. IL-17RE is the functional receptor for IL-17C and mediates mucosal immunity to infection with intestinal pathogens. Nat Immunol. (2011) 12:1151–8. doi: 10.1038/ni.2155
10. Ramirez-Carrozzi V, Ota N, Sambandam A, Wong K, Hackney J, Martinez-Martin N, et al. Cutting edge: IL-17B uses IL-17RA and IL-17RB to induce Type 2 inflammation from human lymphocytes. J Immunol. (2019) 202:1935–41. doi: 10.4049/jimmunol.1800696
11. Su Y, Huang J, Zhao X, Lu H, Wang W, Yang XO, et al. Interleukin-17 receptor D constitutes an alternative receptor for interleukin-17A important in psoriasis-like skin inflammation. Sci Immunol. (2019) 4:eaau9657. doi: 10.1126/sciimmunol.aau9657
12. Li H, Chen J, Huang A, Stinson J, Heldens S, Foster J, et al. Cloning and characterization of IL-17B and IL-17C, two new members of the IL-17 cytokine family. Proc Natl Acad Sci USA. (2000) 97:773–8. doi: 10.1073/pnas.97.2.773
13. Huber M, Heink S, Pagenstecher A, Reinhard K, Ritter J, Visekruna A, et al. IL-17A secretion by CD8+ T cells supports Th17-mediated autoimmune encephalomyelitis. J Clin Invest. (2013) 123:247–60. doi: 10.1172/JCI63681
14. Martin B, Hirota K, Cua DJ, Stockinger B, Veldhoen M. Interleukin-17-producing gammadelta T cells selectively expand in response to pathogen products and environmental signals. Immunity. (2009) 31:321–30. doi: 10.1016/j.immuni.2009.06.020
15. Shibata K, Yamada H, Hara H, Kishihara K, Yoshikai Y. Resident Vdelta1+ gammadelta T cells control early infiltration of neutrophils after Escherichia coli infection via IL-17 production. J Immunol. (2007) 178:4466–72. doi: 10.4049/jimmunol.178.7.4466
16. Michel ML, Keller AC, Paget C, Fujio M, Trottein F, Savage PB, et al. Identification of an IL-17-producing NK1.1(neg) iNKT cell population involved in airway neutrophilia. J Exp Med. (2007) 204:995–1001. doi: 10.1084/jem.20061551
17. Takatori H, Kanno Y, Watford WT, Tato CM, Weiss G, Ivanov II, et al. Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. J Exp Med. (2009) 206:35–41. doi: 10.1084/jem.20072713
18. Bermejo DA, Jackson SW, Gorosito-Serran M, Acosta-Rodriguez EV, Amezcua-Vesely MC, Sather BD, et al. Trypanosoma cruzi trans-sialidase initiates a program independent of the transcription factors RORgammat and Ahr that leads to IL-17 production by activated B cells. Nat Immunol. (2013) 14:514–22. doi: 10.1038/ni.2569
19. Ramirez-Carrozzi V, Sambandam A, Luis E, Lin Z, Jeet S, Lesch J, et al. IL-17C regulates the innate immune function of epithelial cells in an autocrine manner. Nat Immunol. (2011) 12:1159–66. doi: 10.1038/ni.2156
20. Al-Samadi A, Kouri VP, Salem A, Ainola M, Kaivosoja E, Barreto G, et al. IL-17C and its receptor IL-17RA/IL-17RE identify human oral epithelial cell as an inflammatory cell in recurrent aphthous ulcer. J Oral Pathol Med. (2014) 43:117–24. doi: 10.1111/jop.12095
21. Peng T, Chanthaphavong RS, Sun S, Trigilio JA, Phasouk K, Jin L, et al. Keratinocytes produce IL-17c to protect peripheral nervous systems during human HSV-2 reactivation. J Exp Med. (2017) 214:2315–29. doi: 10.1084/jem.20160581
22. Johansen C, Vinter H, Soegaard-Madsen L, Olsen LR, Steiniche T, Iversen L, et al. Preferential inhibition of the mRNA expression of p38 mitogen-activated protein kinase regulated cytokines in psoriatic skin by anti-TNFα therapy. Br J Dermatol. (2010) 163:1194–204. doi: 10.1111/j.1365-2133.2010.10036.x
23. Johansen C, Riis JL, Gedebjerg A, Kragballe K, Iversen L. Tumor necrosis factor α-mediated induction of interleukin 17C in human keratinocytes is controlled by nuclear factor κB. J Biol Chem. (2011) 286:25487–94. doi: 10.1074/jbc.M111.240671
24. Krohn S, Nies JF, Kapffer S, Schmidt T, Riedel JH, Kaffke A, et al. IL-17C/IL-17 receptor E Signaling in CD4(+) T cells promotes TH17 cell-driven glomerular inflammation. J Am Soc Nephrol. (2018) 29:1210–22. doi: 10.1681/ASN.2017090949
25. Huang J, Meng S, Hong S, Lin X, Jin W, Dong C. IL-17C is required for lethal inflammation during systemic fungal infection. Cell Mol Immunol. (2016) 13:474–83. doi: 10.1038/cmi.2015.56
26. Wolf L, Sapich S, Honecker A, Jungnickel C, Seiler F, Bischoff M, et al. IL-17A-mediated expression of epithelial IL-17C promotes inflammation during acute Pseudomonas aeruginosa pneumonia. Am J Physiol Lung Cell Mol Physiol. (2016) 311:L1015–22. doi: 10.1152/ajplung.00158.2016
27. Pfeifer P, Voss M, Wonnenberg B, Hellberg J, Seiler F, Lepper PM, et al. IL-17C is a mediator of respiratory epithelial innate immune response. Am J Respir Cell Mol Biol. (2013) 48:415–21. doi: 10.1165/rcmb.2012-0232OC
28. Kusagaya H, Fujisawa T, Yamanaka K, Mori K, Hashimoto D, Enomoto N, et al. Toll-like receptor-mediated airway IL-17C enhances epithelial host defense in an autocrine/paracrine manner. Am J Respir Cell Mol Biol. (2014) 50:30–9. doi: 10.1165/rcmb.2013-0130OC
29. Hou C, Zhang Y, Yu S, Li Z, Zhai Q, Li Z, et al. Presence of interleukin-17C in the tissue around aseptic loosened implants. Int Orthop. (2013) 37:953–9. doi: 10.1007/s00264-013-1812-x
30. Yamaguchi Y, Fujio K, Shoda H, Okamoto A, Tsuno NH, Takahashi K, et al. IL-17B and IL-17C are associated with TNF-α production and contribute to the exacerbation of inflammatory arthritis. J Immunol. (2007) 179:7128–36. doi: 10.4049/jimmunol.179.10.7128
31. Butcher MJ, Waseem TC, Galkina EV. Smooth muscle cell-derived interleukin-17C plays an atherogenic role via the recruitment of proinflammatory interleukin-17A+ T cells to the aorta. Arterioscler Thromb Vasc Biol. (2016) 36:1496–506. doi: 10.1161/ATVBAHA.116.307892
32. Ivanov II, Frutos Rde L, Manel N, Yoshinaga K, Rifkin DB, Sartor RB, et al. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe. (2008) 4:337–49. doi: 10.1016/j.chom.2008.09.009
33. Atarashi K, Nishimura J, Shima T, Umesaki Y, Yamamoto M, Onoue M, et al. ATP drives lamina propria T(H)17 cell differentiation. Nature. (2008) 455:808–12. doi: 10.1038/nature07240
34. Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. (2009) 139:485–98. doi: 10.1016/j.cell.2009.09.033
35. Gaboriau-Routhiau V, Rakotobe S, Lecuyer E, Mulder I, Lan A, Bridonneau C, et al. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity. (2009) 31:677–89. doi: 10.1016/j.immuni.2009.08.020
36. Round JL, Lee SM, Li J, Tran G, Jabri B, Chatila TA, et al. The Toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science. (2011) 332:974–7. doi: 10.1126/science.1206095
37. Krebs CF, Paust HJ, Krohn S, Koyro T, Brix SR, Riedel JH, et al. Autoimmune renal disease is exacerbated by S1P-receptor-1-dependent intestinal Th17 cell migration to the kidney. Immunity. (2016) 45:1078–92. doi: 10.1016/j.immuni.2016.10.020
38. Song X, Gao H, Lin Y, Yao Y, Zhu S, Wang J, et al. Alterations in the microbiota drive interleukin-17C production from intestinal epithelial cells to promote tumorigenesis. Immunity. (2014) 40:140–52. doi: 10.1016/j.immuni.2013.11.018
39. Li TS, Li XN, Chang ZJ, Fu XY, Liu L. Identification and functional characterization of a novel interleukin 17 receptor: a possible mitogenic activation through ras/mitogen-activated protein kinase signaling pathway. Cell Signal. (2006) 18:1287–98. doi: 10.1016/j.cellsig.2005.10.010
40. Chang SH, Reynolds JM, Pappu BP, Chen G, Martinez GJ, Dong C. Interleukin-17C promotes Th17 cell responses and autoimmune disease via interleukin-17 receptor E. Immunity. (2011) 35:611–21. doi: 10.1016/j.immuni.2011.09.010
41. Reynolds JM, Martinez GJ, Nallaparaju KC, Chang SH, Wang YH, Dong C. Cutting edge: regulation of intestinal inflammation and barrier function by IL-17C. J Immunol. (2012) 189:4226–30. doi: 10.4049/jimmunol.1103014
42. Huang W, Na L, Fidel PL, Schwarzenberger P. Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J Infect Dis. (2004) 190:624–31. doi: 10.1086/422329
43. Ma CS, Chew GY, Simpson N, Priyadarshi A, Wong M, Grimbacher B, et al. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J Exp Med. (2008) 205:1551–7. doi: 10.1084/jem.20080218
44. Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, et al. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature. (2008) 452:773–6. doi: 10.1038/nature06764
45. Conti HR, Whibley N, Coleman BM, Garg AV, Jaycox JR, Gaffen SL. Signaling through IL-17C/IL-17RE is dispensable for immunity to systemic, oral and cutaneous candidiasis. PLoS ONE. (2015) 10:e0122807. doi: 10.1371/journal.pone.0122807
46. Jamieson KC, Traves SL, Kooi C, Wiehler S, Dumonceaux CJ, Maciejewski BA, et al. Rhinovirus and bacteria synergistically induce IL-17C release from human airway epithelial cells to promote neutrophil recruitment. J Immunol. (2019) 202:160–70. doi: 10.4049/jimmunol.1800547
47. Johansen C, Usher PA, Kjellerup RB, Lundsgaard D, Iversen L, Kragballe K. Characterization of the interleukin-17 isoforms and receptors in lesional psoriatic skin. Br J Dermatol. (2009) 160:319–24. doi: 10.1111/j.1365-2133.2008.08902.x
48. Johnston A, Fritz Y, Dawes SM, Diaconu D, Al-Attar PM, Guzman AM, et al. Keratinocyte overexpression of IL-17C promotes psoriasiform skin inflammation. J Immunol. (2013) 190:2252–62. doi: 10.4049/jimmunol.1201505
49. El Malki K, Karbach SH, Huppert J, Zayoud M, Reissig S, Schuler R, et al. An alternative pathway of imiquimod-induced psoriasis-like skin inflammation in the absence of interleukin-17 receptor a signaling. J Invest Dermatol. (2013) 133:441–51. doi: 10.1038/jid.2012.318
50. Vandeghinste N, Klattig J, Jagerschmidt C, Lavazais S, Marsais F, Haas JD, et al. Neutralization of IL-17C reduces skin inflammation in mouse models of psoriasis and atopic dermatitis. J Invest Dermatol. (2018) 138:1555–63. doi: 10.1016/j.jid.2018.01.036
51. Huang J, Yuan Q, Zhu H, Yin L, Hong S, Dong Z, et al. IL-17C/IL-17RE augments T cell function in autoimmune hepatitis. J Immunol. (2017) 198:669–80. doi: 10.4049/jimmunol.1600977
52. Yamaguchi S, Nambu A, Numata T, Yoshizaki T, Narushima S, Shimura E, et al. The roles of IL-17C in T cell-dependent and -independent inflammatory diseases. Sci Rep. (2018) 8:15750. doi: 10.1038/s41598-018-34054-x
53. Friedrich M, Tillack C, Wollenberg A, Schauber J, Brand S. IL-36gamma sustains a proinflammatory self-amplifying loop with IL-17C in anti-TNF-induced psoriasiform skin lesions of patients with Crohn's disease. Inflamm Bowel Dis. (2014) 20:1891–901. doi: 10.1097/MIB.0000000000000198
54. Jungnickel C, Schmidt LH, Bittigkoffer L, Wolf L, Wolf A, Ritzmann F, et al. IL-17C mediates the recruitment of tumor-associated neutrophils and lung tumor growth. Oncogene. (2017) 36:4182–90. doi: 10.1038/onc.2017.28
55. Liao R, Sun J, Wu H, Yi Y, Wang JX, He HW, et al. High expression of IL-17 and IL-17RE associate with poor prognosis of hepatocellular carcinoma. J Exp Clin Cancer Res. (2013) 32:3. doi: 10.1186/1756-9966-32-3
56. Ritzmann F, Jungnickel C, Vella G, Kamyschnikow A, Herr C, Li D, et al. IL-17C-mediated innate inflammation decreases the response to PD-1 blockade in a model of Kras-driven lung cancer. Sci Rep. (2019) 9:10353. doi: 10.1038/s41598-019-46759-8
57. Novatchkova M, Leibbrandt A, Werzowa J, Neubuser A, Eisenhaber F. The STIR-domain superfamily in signal transduction, development and immunity. Trends Biochem Sci. (2003) 28:226–9. doi: 10.1016/S0968-0004(03)00067-7
58. Yao Z, Fanslow WC, Seldin MF, Rousseau AM, Painter SL, Comeau MR, et al. Herpesvirus Saimiri encodes a new cytokine, IL-17, which binds to a novel cytokine receptor. Immunity. (1995) 3:811–21. doi: 10.1016/1074-7613(95)90070-5
59. Karlsen JR, Borregaard N, Cowland JB. Induction of neutrophil gelatinase-associated lipocalin expression by co-stimulation with interleukin-17 and tumor necrosis factor-α is controlled by IκB-zeta but neither by C/EBP-beta nor C/EBP-delta. J Biol Chem. (2010) 285:14088–100. doi: 10.1074/jbc.M109.017129
60. Qian Y, Liu C, Hartupee J, Altuntas CZ, Gulen MF, Jane-Wit D, et al. The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nat Immunol. (2007) 8:247–56. doi: 10.1038/ni1439
61. Bulek K, Liu C, Swaidani S, Wang L, Page RC, Gulen MF, et al. The inducible kinase IKKi is required for IL-17-dependent signaling associated with neutrophilia and pulmonary inflammation. Nat Immunol. (2011) 12:844–52. doi: 10.1038/ni.2080
62. Huang G, Wang Y, Vogel P, Chi H. Control of IL-17 receptor signaling and tissue inflammation by the p38α-MKP-1 signaling axis in a mouse model of multiple sclerosis. Sci Signal. (2015) 8:ra24. doi: 10.1126/scisignal.aaa2147
63. Chang SH, Park H, Dong C. Act1 adaptor protein is an immediate and essential signaling component of interleukin-17 receptor. J Biol Chem. (2006) 281:35603–7. doi: 10.1074/jbc.C600256200
64. Ruddy MJ, Wong GC, Liu XK, Yamamoto H, Kasayama S, Kirkwood KL, et al. Functional cooperation between interleukin-17 and tumor necrosis factor-α is mediated by CCAAT/enhancer-binding protein family members. J Biol Chem. (2004) 279:2559–67. doi: 10.1074/jbc.M308809200
65. Maitra A, Shen F, Hanel W, Mossman K, Tocker J, Swart D, et al. Distinct functional motifs within the IL-17 receptor regulate signal transduction and target gene expression. Proc Natl Acad Sci USA. (2007) 104:7506–11. doi: 10.1073/pnas.0611589104
66. Shen F, Li N, Gade P, Kalvakolanu DV, Weibley T, Doble B, et al. IL-17 receptor signaling inhibits C/EBPbeta by sequential phosphorylation of the regulatory 2 domain. Sci Signal. (2009) 2:ra8. doi: 10.1126/scisignal.2000066
67. Hartupee J, Liu C, Novotny M, Li X, Hamilton T. IL-17 enhances chemokine gene expression through mRNA stabilization. J Immunol. (2007) 179:4135–41. doi: 10.4049/jimmunol.179.6.4135
68. Sun D, Novotny M, Bulek K, Liu C, Li X, Hamilton T. Treatment with IL-17 prolongs the half-life of chemokine CXCL1 mRNA via the adaptor TRAF5 and the splicing-regulatory factor SF2 (ASF). Nat Immunol. (2011) 12:853–60. doi: 10.1038/ni.2081
69. Roth SA, Simanski M, Rademacher F, Schroder L, Harder J. The pattern recognition receptor NOD2 mediates Staphylococcus aureus-induced IL-17C expression in keratinocytes. J Invest Dermatol. (2014) 134:374–80. doi: 10.1038/jid.2013.313
70. Chiricozzi A, Guttman-Yassky E, Suarez-Farinas M, Nograles KE, Tian S, Cardinale I, et al. Integrative responses to IL-17 and TNF-α in human keratinocytes account for key inflammatory pathogenic circuits in psoriasis. J Invest Dermatol. (2011) 131:677–87. doi: 10.1038/jid.2010.340
71. Russell CB, Rand H, Bigler J, Kerkof K, Timour M, Bautista E, et al. Gene expression profiles normalized in psoriatic skin by treatment with brodalumab, a human anti-IL-17 receptor monoclonal antibody. J Immunol. (2014) 192:3828–36. doi: 10.4049/jimmunol.1301737
72. Monin L, Gudjonsson JE, Childs EE, Amatya N, Xing X, Verma AH, et al. MCPIP1/regnase-1 restricts IL-17A- and IL-17C-dependent skin inflammation. J Immunol. (2017) 198:767–75. doi: 10.4049/jimmunol.1601551
73. Matsushita K, Takeuchi O, Standley DM, Kumagai Y, Kawagoe T, Miyake T, et al. Zc3h12a is an RNase essential for controlling immune responses by regulating mRNA decay. Nature. (2009) 458:1185–90. doi: 10.1038/nature07924
74. Liang J, Saad Y, Lei T, Wang J, Qi D, Yang Q, et al. MCP-induced protein 1 deubiquitinates TRAF proteins and negatively regulates JNK and NF-κB signaling. J Exp Med. (2010) 207:2959–73. doi: 10.1084/jem.20092641
75. Garg AV, Amatya N, Chen K, Cruz JA, Grover P, Whibley N, et al. MCPIP1 endoribonuclease activity negatively regulates interleukin-17-mediated signaling and inflammation. Immunity. (2015) 43:475–87. doi: 10.1016/j.immuni.2015.07.021
76. Uehata T, Iwasaki H, Vandenbon A, Matsushita K, Hernandez-Cuellar E, Kuniyoshi K, et al. Malt1-induced cleavage of regnase-1 in CD4(+) helper T cells regulates immune activation. Cell. (2013) 153:1036–49. doi: 10.1016/j.cell.2013.04.034
77. Croxtall JD. Ustekinumab: a review of its use in the management of moderate to severe plaque psoriasis. Drugs. (2011) 71:1733–53. doi: 10.2165/11207530-000000000-00000
78. McInnes IB, Kavanaugh A, Gottlieb AB, Puig L, Rahman P, Ritchlin C, et al. Efficacy and safety of ustekinumab in patients with active psoriatic arthritis: 1 year results of the phase 3, multicentre, double-blind, placebo-controlled PSUMMIT 1 trial. Lancet. (2013) 382:780–9. doi: 10.1016/S0140-6736(13)60594-2
79. Feagan BG, Sandborn WJ, Gasink C, Jacobstein D, Lang Y, Friedman JR, et al. Ustekinumab as induction and maintenance therapy for Crohn's disease. N Engl J Med. (2016) 375:1946–60. doi: 10.1056/NEJMoa1602773
80. Griffiths CE, Reich K, Lebwohl M, van de Kerkhof P, Paul C, Menter A, et al. Comparison of ixekizumab with etanercept or placebo in moderate-to-severe psoriasis (UNCOVER-2 and UNCOVER-3): results from two phase 3 randomised trials. Lancet. (2015) 386:541–51. doi: 10.1016/S0140-6736(15)60125-8
81. Lebwohl M, Strober B, Menter A, Gordon K, Weglowska J, Puig L, et al. Phase 3 studies comparing brodalumab with ustekinumab in psoriasis. N Engl J Med. (2015) 373:1318–28. doi: 10.1056/NEJMoa1503824
82. van der Heijde D, Cheng-Chung Wei J, Dougados M, Mease P, Deodhar A, Maksymowych WP, et al. Ixekizumab, an interleukin-17A antagonist in the treatment of ankylosing spondylitis or radiographic axial spondyloarthritis in patients previously untreated with biological disease-modifying anti-rheumatic drugs (COAST-V): 16 week results of a phase 3 randomised, double-blind, active-controlled and placebo-controlled trial. Lancet. (2018) 392:2441–51. doi: 10.1016/S0140-6736(18)31946-9
83. Baeten D, Sieper J, Braun J, Baraliakos X, Dougados M, Emery P, et al. Secukinumab, an interleukin-17A inhibitor, in ankylosing spondylitis. N Engl J Med. (2015) 373:2534–48. doi: 10.1056/NEJMoa1505066
84. GlobeNewswire Inc. Galapagos and Morphosys Report First Promising Signs of Clinical Activity in a Phase 1 Study with IL-17c Antibody Mor106 in Atopic Dermatitis Patients. (2017). Available online at: https://www.globenewswire.com/news-release/2017/09/27/1133502/0/en/Galapagos-and-MorphoSys-report-first-promising-signs-of-clinical-activity-in-a-Phase-1-study-with-IL-17C-antibody-MOR106-in-atopic-dermatitis-patients.html
85. Hueber W, Sands BE, Lewitzky S, Vandemeulebroecke M, Reinisch W, Higgins PD, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn's disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut. (2012) 61:1693–700. doi: 10.1136/gutjnl-2011-301668
Keywords: IL-17C, IL-17RE, immunity, inflammation, Th17
Citation: Nies JF and Panzer U (2020) IL-17C/IL-17RE: Emergence of a Unique Axis in TH17 Biology. Front. Immunol. 11:341. doi: 10.3389/fimmu.2020.00341
Received: 05 December 2019; Accepted: 12 February 2020;
Published: 26 February 2020.
Edited by:
Katarzyna Bulek, Jagiellonian University, PolandReviewed by:
Peter A. Ward, University of Michigan, United StatesPiergiuseppe De Berardinis, Istituto di Biochimica delle Proteine (IBP), Italy
Copyright © 2020 Nies and Panzer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ulf Panzer, cGFuemVyQHVrZS5kZQ==