 Amani Abraheem Alsadiq Alyaseer
Amani Abraheem Alsadiq Alyaseer Murilo Henrique Saturnino de Lima
Murilo Henrique Saturnino de Lima Tarcio Teodoro Braga
Tarcio Teodoro Braga- 1Department of Pathology, Federal University of Parana, Curitiba, Brazil
- 2Instituto Carlos Chagas, Fiocruz-Parana, Curitiba, Brazil
Fibrosis is considered a complex form of tissue damage commonly present in the end stage of many diseases. It is also related to a high percentage of death, whose predominant characteristics are an excessive and abnormal deposition of fibroblasts and myofibroblasts -derived extracellular matrix (ECM) components. Epithelial-to-mesenchymal transition (EMT), a process in which epithelial cells gradually change to mesenchymal ones, is a major contributor in the pathogenesis of fibrosis. The key mediator of EMT is a multifunctional cytokine called transforming growth factor-β (TGF-β) that acts as the main inducer of the ECM assembly and remodeling through the phosphorylation of Smad2/3, which ultimately forms a complex with Smad4 and translocates into the nucleus. On the other hand, the bone morphogenic protein-7 (BMP-7), a member of the TGF family, reverses EMT by directly counteracting TGF-β induced Smad-dependent cell signaling. NLRP3 (NACHT, LRR, and PYD domains-containing protein 3), in turn, acts as cytosolic sensors of microbial and self-derived molecules and forms an immune complex called inflammasome in the context of inflammatory commitments. NLRP3 inflammasome assembly is triggered by extracellular ATP, reactive oxygen species (ROS), potassium efflux, calcium misbalance, and lysosome disruption. Due to its involvement in multiple diseases, NLRP3 has become one of the most studied pattern-recognition receptors (PRRs). Nevertheless, the role of NLRP3 in fibrosis development has not been completely elucidated. In this review, we described the relation of the previously mentioned fibrosis pathway with the NLRP3 inflammasome complex formation, especially EMT-related pathways. For now, it is suggested that the EMT happens independently from the oligomerization of the whole inflammasome complex, requiring just the presence of the NLRP3 receptor and the ASC protein to trigger the EMT events, and we will present different pieces of research that give controversial point of views.
Fibrosis
The inflammatory response is triggered in an attempt to remove an aggressor agent and ultimately leads to the wound healing process as a result of reparative or reactive steps in different organs concomitant with the elimination of the agent. This agent can be a pathogen, in which the inflammatory response is initiated after the recognition of a PAMP (Pathogen Associated Molecular Pattern), or the agent can be an endogenous product, named DAMP (Damage Associated Molecular Pattern), both recognized by PRRs (Pattern Recognition Receptors), which are receptors of innate immune system (1). Although it is considered a normal process for maintaining the architecture and functional integrity of the affected organ, if the injurious stimuli persist, such a response will result in the deregulation of normal processes and scar formation.
Fibrosis can be defined as an accumulation of fibrous connective tissue, in particular extracellular matrix (ECM) components such as type I collagen and fibronectin (2), the accumulation of which can cause the malfunction of the organ and its failure. This process is the end result of chronic inflammatory diseases such as rheumatoid arthritis (3), idiopathic pulmonary fibrosis (4, 5), Chron's Disease (5), and Chronic Kidney Disease (CKD) (6, 7) besides autoimmune reactions, allergic responses, chemical insults, radiation, and tissue injury (6), all of which progress to an advanced state characterized by a poor quality of life of the patient, mainly due to the lack of specific treatments for the reversibility of the fibrotic process (2). The origin of the fibrosis process is, therefore, an unresolved inflammatory response (8) or the improper removal of stimulus, which culminates in a chronic inflammatory response (9). The strategies to deal with fibrotic diseases rely on an attempt to neutralize the aggressor but not all of them are easily diffusible or with a known origin, which forces clinicians to try to find ways to control and reverse the fibrosis (2). Inflammatory mediators are responsible for controlling the response triggered against aggressor agents and for starting the tissue repair process. In chronic inflammatory diseases, macrophages are the main characters in expressing these mediators (10). These cells are differentiated, mainly, from monocytes when they leave the circulatory system and invade the tissue to become phagocytes and are able to produce inflammatory cytokines (11). Besides bone marrow hematopoietic cells, macrophages are also derived from different sources, including local proliferation of embryonic progenitor cells, which is responsible for the generation of tissue-resident macrophages (12). They are plastic cells and functionally subdivided into profiles that vary from an inflammatory subpopulation (usually called M1 macrophages) to an anti-inflammatory one (usually referred as M2 macrophages) (13). It is noteworthy to state that anti-inflammatory macrophages can be derived from inflammatory ones or resident cells, in contrast to inflammatory cells which are derived mainly from circulating monocytes. Additionally, inflammatory macrophages express IL-1β, IL-23, TNF-α, and lipid-derived mediators (14, 15). Another important characteristic of the inflammatory profile is the production of reactive oxygen species (ROS), which is necessary to kill the invader and therefore responsible for the injury during inflammation (14, 15). The anti-inflammatory cells, on the other hand, appear to exert the opposite effect, i.e., controlling the inflammatory process. This subpopulation is responsible for producing IL-10, IL-4, and transformation growth factor-β (TGF-β) (15), playing, therefore, a role in the clearance of apoptotic cells and the formation of wound healing, mainly due to the expression of TGF-β, although it has not been shown that the presence of anti-inflammatory macrophages in the absence of an inflammatory context leads to the formation of fibrotic tissue.
Epithelial to Mesenchymal Transition
Epithelial to mesenchymal transition (EMT) is a biological process, found in the context of fibrosis, that is characterized by the changing in the phenotype of polarized epithelial cells into mobile mesenchymal cells through a switch of the expressed proteins (16). During EMT it has been observed that there is an up-regulation of α-sma (α-smooth muscle actin), a marker of mesenchymal cells, and a downregulation of e-cadherin, the key marker of epithelial cells responsible for the lateral adherent junctions and adhesion (17). Usually, epithelial cells have a square-shaped morphology and they are strongly adherent to their basement membrane and the neighbor cells, ultimately being responsible for maintaining the integrity of cell junctions (18). However, after a process of EMT, induced by a constellation of signaling pathways, whether intrinsic (such as genetic mutations) or extrinsic (such as growth factors including TGF-β, the WNT, and Sonic Hedgehog), those cells will gradually start losing their ability to express their known adhesion proteins, structure, and function to end up turning to the mesenchymal cells. The EMT process converges in an increase of transcription factors such as snail, ERK, and slug which initiate and maintain the process of EMT (19). The outcome of the EMT process is the changing morphology of epithelial cells into spindle-shaped form cells that do not adhere to other cells, but instead present an enhanced migratory capacity, the ability of invasion, and a high resistance to apoptosis (18). The EMT process does not represent a bipolarity between epithelial and mesenchymal cells, but instead is a spectrum of different protein expression throughout the time that culminates in a complete transition. Both EMT and the opposite process, known as mesenchymal to epithelial transition (MET), are considered integral stages of many physiological processes that are tightly coordinated by several molecular regulators (20–22). The down-regulation of the epithelial markers that have been observed in the EMT process is also mediated by mesenchymal markers up-regulation such as vimentin, n-cadherin, integrin, fibronectin, and matrix metalloproteinase, which will result in loss of the cytoskeleton polarization and development of lamellipodia and, thus, an increase in its cell motility. Additionally, the EMT process is also responsible for the increase in extracellular matrix deposition (18, 23).
EMT plays a main role in diverse biological processes other than the fibrotic process, such as embryogenesis and cancer metastasis. It is separated into three types, each of them presenting a different function in the mesenchymal cells at the end of the process (16, 18, 24). The type I EMT is present in embryogenesis implantation, during the initiation of the placenta development and organ development; the type II EMT, on the other hand, occurs during fibrotic diseases in organs such as the liver, kidney, lung, and intestine, and it is also associated with tissue regeneration and wound healing; the type III EMT is conceptually present in the progression of cancer and metastasis (18, 25). In the early days of a mammal's embryo development, a structure called blastocyst, composed of a blastocoele and a trophoblast, triggers type I EMT to invade and adhere to the endometrium to subsequently form the placenta (26). In cancer, the cells can migrate to another tissue via vessels. This ability is attributed to the type III EMT that promotes a loss of cell adhesion and helps the posterior invasion. Both type I and type III EMT processes are reversible by the MET process after a tissue invasion, a reversion that does not happen in type II EMT (27). During fibrotic diseases, the main triggered mechanism is the type II EMT. Here, the mesenchymal type of cells formed after complete transitions are myofibroblasts and fibroblasts, responsible for expressing ECM components in tissue repair after an injury (16). In the present review, we will give special attention to type II EMT due to its importance in fibrosis.
Type II EMT
In inflammation, after damage is caused by infectious agents, toxins, allergens, or trauma, a release of DAMPs, mainly due to cellular death, which are latter recognized by PRRs such as like Toll-Like receptors (TLR) and Nod-like receptors (NLR), become the major element of the inflammasome complex (28). This recognition promotes the assembly of an inflammatory response that aims to destroy the aggressor agent in order to control and repair the tissue damage. To repair this damaged tissue, myofibroblasts are recruited to the lesion site and start to release metalloproteinases and digestive enzymes. After the clearance, myofibroblasts produce new ECM proteins to promote the wound healing (29, 30). The origin of myofibroblasts is due to a high expression of TGF-β, responsible for triggering EMT in epithelial cells through its receptor (31). Any small flaw in this mechanism might result in the promotion of a large amount of ECM components synthesis, leading to an exacerbated scar formation and compromising the tissue function (29, 30).
The intracellular signaling pathways involved in EMT are complex and can cross-talk between them. TGF-β was first described as an inducer of EMT in normal mammary epithelial cells by signals through the serine/threonine kinase complexes receptor (32). The signaling pathway better described is the TGF-β/Smad pathway. As EMT represents a gradual transition from an epithelial cell type to a mesenchymal one, the first signals are responsible for forming a hybrid cell type that is partially epithelial but already starts the expression of mesenchymal markers, a process mediated by ERK, snail, and slug (19). This process can be reversible once the damage stimuli are removed and the hybrid return to an epithelial form (19). The TGF-β pathway is a complex signaling mechanism involving different proteins, and, additionally, may be a Smad-independent pathway (19), but Rhoa-dependent, a GTPase-related enzyme. This enzyme is related to the cytoskeletal modeling as it activates the phosphorylation of the downstream ROCK I/II, a serine/threonine kinase-specific protein that acts in actin protein (33, 34). Another important signal is MAPK/ERK. Once TGF-β binds into its receptor, it activates an upstream GTPase RAS responsible for triggering a set of mitogen-protein kinases, the last of which is the MAPK/ERK (35). This pathway was shown to establish a relationship with EMT once an inhibition of this signal interrupts EMT (34). Phosphatidylinositol 3-Kinase (PI3K) is also a signal activated by TGF-β (36) and it has been linked to EMT as its inhibition also stops the transition processes in the HK-2 cell model, a renal epithelial cell line (37). But the exact mechanism remains unknown in tubulointerstitial fibrosis (34).
In vitro models of EMT have been used in the past few years to elucidate the mechanism and therapeutics targets of EMT in several diseases that compromise vital organs like the lungs, heart, and kidneys. In pulmonary fibrosis, for instance, the induction of EMT can be done by different inducers such as bleomycin, silica, and the atmospheric particular matter (38–40). All of them are responsible for triggering EMT as observed by the increase of mesenchymal markers such as FSP-1 and α-sma and a decrease of e-cadherin. In other tissues, other inducers are shown to be successful in modulating the EMT markers as TGF-β in kidney diseases (41–44) and heart diseases (45). In the next sections, we will give special attention to some EMT modulators.
TGF-β
Transforming growth factor-β (TGF-β) is a cytokine member of a large family which carries the same name and is linked with several essential cellular functions, including embryological development, fibrosis, and cancer (46). Early research studying cancer biology found out the mechanisms by which TGF-β acts (47) and its name was termed due to the incorrect belief that this cytokine was only expressed in transformed cells during the establishment of cancer (47). Other members that belong to this family are bone morphogenetic protein-7 (BMP-7), activin, nodal, and anti-mullerian hormone, which are all structurally related molecules and are all involved in critical cellular functions such as proliferation, differentiation, motility, extracellular matrix production, angiogenesis, and apoptosis (48, 49).
In addition to the known anti-inflammatory effect of TGF-β, it has also shown to have the ability to act as a profibrotic agent, through the increase of ECM synthesis and the expression of proteinase inhibitors, including plasminogen activator inhibitor-1 (PAI-1), and by decreasing the ECM-degrading proteins, such as collagenase (50). Moreover, according to both Li et al. and Strutz et al. this profibrotic activity is a potent inducer of EMT and part of the TGF-β-induced EMT program which promotes the remodeling of cell contacts with the basal membrane (51, 52). Besides, such cellular contact remodeling is triggered by the activation of the matrix metalloproteases MMP2 and MMP9 which also results in the degradation of type IV collagen, a component of the basement membrane (51, 52). In humans, TGF-β exists in three main isoforms (TGF-β1, TGF-β2, and TGF-β3) with TGF-β1 being the most common (53) and, although they are encoded by distinct genes, they share a similar homology and activate the same intracellular signaling pathway (54). TGF-β1 is also responsible for the formation of myofibroblasts through the activation of resident fibroblasts and induction of both EMT and endothelial to mesenchymal transition (EndMT) (55, 56). Mice overexpressing an active form of the TGF-β1 in the liver, for instance, have shown to develop progressive liver and renal fibrosis (57, 58). Other experimental models using knockout mice led to a lethal multifocal inflammatory disease, which shows that they also play different roles in maintaining homeostasis and in embryogenic development (59–61). Moreover, the blockage of TGF-β 1 signaling either in T cells or in bone marrow results in a similar multifocal inflammatory responses (62, 63). The three isoforms of TGF-β are also secreted as inactive latent precursors that require activation before binding to the TGF-β receptors (64).
All isoforms of TGF-β interact with two receptors in the cell membrane, the type I receptor (TGF-βRI) and the type II receptor (TGF-βRII), which are both threonine/serine kinase receptors. When TGF-β link to TGF-βRI, this promotes the phosphorylation of TGF-βRII, which will posteriorly recruit downstream signals in a Smad-dependent pathway (65), a signaling pathway better explored in the next section. The complex Smad2 and Smad3 form a trimer with the common Smad4 to reach the nucleus and start the transcription of genes related to different cellular functions (65). Additionally, TGF-β can activate a number of non-Smad signaling pathways where the activated receptors also signal through other transducers, such as the mitogen-activated protein kinase (MAPK) pathways, that includes each of the extracellular signal-regulated kinases (ERKs), AKT, Rho family GTPases, c-Jun amino-terminal kinase (JNK), and p38 MAPK, as well as the IkB kinase (IKK) and phosphatidylinositol-3 kinase (PI3K) (66, 67), as stated above. These non-Smad signals play different roles: they are involved in TGF-β-mediated biological responses, but can also regulate the canonical Smad pathway (68).
Smad
Smad, named due to the members Mad (the Drosophila gene Mothers Against Decapentaplegic) and Sma (the Mad homologs observed in the nematode Caenorhabditis elegans) (69) have shown to be activated by numerous upstream molecules with pleiotropic functions and are thought to constitute the core-point for transcriptional regulation of multiple genes with diverse activities (70). Smad are proteins with a transcription factor function, responding to TGF-β signaling after phosphorylation of their receptors' activities (70). They are categorized into two subgroups according to their functions, i.e., R-Smad, the receptor-activated Smad, and I-Smad, the inhibitory Smad. R-smad are phosphorylated by either TGF-βRII (include Smad-2 and Smad-3) or by BMP receptor (BMPR) (and include Smad1, Smad-5, and Smad-8). These R-Smad bind to the Smad-4, also known as the Co-Smad, which form a heterodimer with the activated phosphorylated R-Smad, and, together, move from the cytoplasm to the nucleus, initiating the transcriptional activity (71). I-Smad (which includes Smad-6 and Smad-7), on the other hand, inhibit the signaling activity of the R-Smads and the formation of the Co-Smad/R-Smad complex, important for creating a balance during non-fibrotic functions (70, 72).
It has been described that both Smad2 and Smad3 promote EMT in the context of fibrosis after TGF-β signaling pathway activation. Moreover, based on knockout studies, the specific role of Smad2 or Smad3 has been investigated (73). TGF-β1-mediated induction of MMP-2 was selectively dependent on Smad2, whereas activation of the activin response was strongly suppressed in Smad2 KO fibroblasts but enhanced in Smad3 KO cells (73). The Smad proteins consist of a highly conserved amino-terminal MH1 domain and a carboxyl-terminal MH2 domain, which are separated by a proline-rich linker region (74). Both Smad2 MH1 and MH2 domains' knockout approaches led to lethality in early stages of mice embryonic development, due to a misormation of an anterior-posterior axis, gastrulation, and mesoderm (75, 76), suggesting that Smad2 is more closely related to embryogenic events than others following the TGF-β/Smad pathway. Smad3 KO mice showed viability in newborn mice and guaranteed survival for a few months but led to systemic chronic inflammation and death when left for a longer time (73). Therefore, Smad3 is not important to embryological development but it is crucial in the pathobiology of fibrosis development (77). Although both Smad2 and Smad3 are strongly activated in liver fibrosis (78), Smad3 appears to be the key element in the signal transduction pathways that are responsible for fibrosis (79). Smad3 activation leads to an increased expression of fibrogenic genes, including α-sma and e-cadherin (80, 81). Besides, Smad3 activation through TGF-β signaling triggers the induction of tissue inhibitor of metalloproteinases (TIMP) (82) and thus prevents the degradation of ECM (77). Therefore, Smad3 knockdown results in the blocking of EMT and attenuation of renal fibrosis, inflammation, and apoptosis in a model of unilateral ureteral obstruction (UUO) (83).
Additionally, some inactivating mutation into Smad genes were described in the cancer context. DPC4 (deleted in pancreatic carcinoma locus 4), for instance, was identified as a tumor suppressor gene in carcinoma, implicated in Smad4 signaling (84). Early colorectal carcinoma was found to be associated with Smad2 and Smad4 inactivating mutations (85, 86). Despite some reports pointing to an absence of acquired mutations, insertions, or microdeletions in Smad3 in the skin and parathyroid tumors (87), its somatic mutation associated with Smad2 and Smad4 mutations is a cause of sporadic colorectal carcinoma (88). Smad3 also promotes cancer progression by inhibiting E4BP4-mediated NK cell development (89). Accordingly, Smad3 functions as both a negative and positive regulator of carcinogenesis depending on cell type and clinical stage of the tumor (90).
BMP-7
Bone morphogenic protein 7 (Bmp-7), also known as osteogenic protein 1, is a member of the transforming growth factor-β proteins superfamily (48, 49). The Bmp-7 compromises an anti-inflammatory, anti-fibrotic, anti-apoptotic, and a proliferating stimulating function (91–93). It also exerts a crucial role in kidney development, whereas Bmp-7 KO mice die after birth due to renal failure (94, 95). Like TGF-β, Bmp-7 signaling is also mediated by the heterodimeric receptors with serine/threonine receptor kinases and the cytoplasmic proteins Smads (96), but while the TGF-β acts on the TGF-βR to phosphorylate certain types of Smad, the Bmp-7 binds to the BMPIIRs to activate R-Smad1/5/8 (96, 97) and the activin-like kinase (ALK) receptors ALK2, ALK6, and ALK3. Moreover, BMP-7 can signal through non-canonical pathways to exert its biological activity (65, 98). Although the precise function of Bmp-7 is not well-understood, researches have proven that Bmp-7 acts as an endogenous regulator of organ homeostasis and regeneration in the kidney and liver (91, 99). Hence, recombinant Bmp-7 reduces the severity of injury in acute and chronic organ failure by counteracting profibrogenic TGF-β activities (100).
Studies have also shown that Bmp-7 is effective in reversing both EMT and the fibrotic effect that is induced by TGF-β; this was proved by Ziesberg's lab team in 2003, who focused on chronic renal injury, and by Ying and colleagues in 2017, where they studied the inhibition of TGF-β -induced EMT using BMP-7 in breast cancer (93, 101). In other words, Bmp-7 acts as an opposing factor to TGF-β, thereby reducing inflammation and ECM-related products, such as type I and type III collagen, that are hallmarks of organ fibrosis (102). Moreover, Higgings and colleagues proved in an experimental study both in vivo and in vitro that this inhibition is done through the decreased activation of PI3K signaling via Akt and also the inhibition of Smad3 phosphorylation without any effect reported on Smad2 or the Smad2/3/4 complex (103). Another study by Luo et al. showed that this inhibition was caused by interference with Smad3 DNA binding site and the enhancement of SnoN expression (104). Other studies that were concerned with the function of BMP-7 showed that it played a role in preserving the blood flow, preventing tubular atrophy, and reducing tubular interstitial fibrosis in obstructive neuropathy in rats (105). Additionally, in an acute tubular necrosis model performed by Vukicevic's team, it was proved that BMP-7 preserves the kidney function and increases the patient's survival rate (100). Zeisberg et al. using mice with acute glomerular nephritis, showed that BMP-7 induces EMT reversion by increasing the expression of e-cadherin and leading to ECM degradation (93).
BMP-7 was also studied in hepatic fibrosis using mice models where the fibrosis was induced by an injection of carbon tetrachloride (CCL4) (106). After the administration of recombinant BMP-7, there was a suppressed progression of liver fibrosis and an improved liver function, partly due to downregulation of type I collagen, α-sma, and TIMP2 and up-regulation of MMP-2 with the suppression of expression of TGF-β (106). Most studies aiming to understand the BMP-7 function were performed on kidney and liver diseases. Meanwhile, rat dermal papilla cells (DPC) treated with BMP-7 demonstrated a counteractive function to the TGF-β profibrotic effect by preventing and reversing the fibrosis (107). We, therefore, strongly relate the opposing relation between BMP-7 and TGF-β in fibrosis and EMT, as schematized in Figure 1. However, whether there is a direct relation between TGF-β or BMP-7 pathways with inflammasome still remains a mystery.
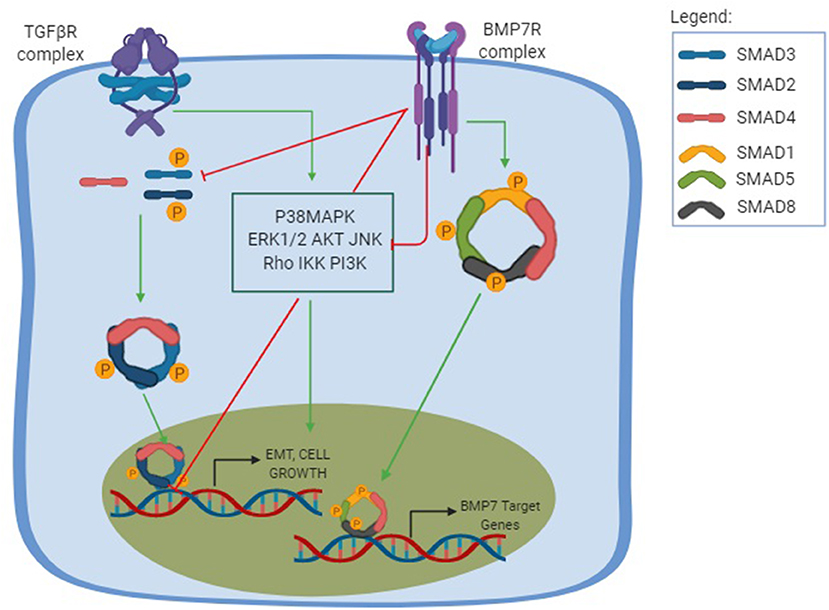
Figure 1. TGF-β and BMP-7 pathways. The TGF-β pathway is shown in its canonical and non-canonical forms. The canonical pathway is Smad-dependent, in which the TGF-β binds to the TGF-βRI and promotes a phosphorylation of TGF-βRII, which will posteriorly recruit downstream signals that results in the phosphorylation of both Smad2 and Smad3 to form a trimer complex with the Smad 4, a common Smad. This results in the trimer translocation into the nucleus and the activation of a number of genes related to cellular functions and profibrotic effect including EMT, cell growth, and apoptosis. The non-canonical pathway, also known as the non-Smad signaling pathways, is mediated by other transducers, such as the p38 mitogen-activated protein kinase (P38MAPK) pathways, that includes each of the extracellular signal regulated kinases (ERKs), AKT, c-Jun amino terminal kinase (JNK), Rho family GTPases, as well as the IkB kinase (IKK), and phosphatidylinositol-3 kinase (PI3K). The non-canonical pathway results in different cellular functions and can amplify the Smad pathway. The BMP-7 pathway is activated after the binding of BMP-7 to the BMP-7R complex and results in the phosphorylation of the Smad1/5/8 to form a complex with the common Smad4 and translocate in the nucleus to activate the opposing function to TGF-β, creating a normal physiological balance between the TGF-β pathway, ultimately leading to an antifibrotic effect, increasing ECM degradation and inducing MET.
NLRP3 Inflammasome
In 2002, the research group headed by Jurg Tschopp coined the term “Inflammasome,” defined as a protein complex composed of a nucleotide-binding oligomerization (Nod) receptor, able to activate an inflammatory caspase and, ultimately, lead to IL-1β production (108). Members of the Nod receptor family, which are known as Nod-like receptors (NLRs), have three domains: a carboxy-terminal leucine-rich repeat (LRR), which was first established as the one responsible for sensing the function of the NLRP3 protein (109), but Hafner-Bratkovič and colleagues demonstrated that even with the LRR domain truncation, the inflammasome assembly is still viable, suggesting that its sensor function is probably executed by another domain (110); a central NACHT domain (named after NAIP, CIITA, HET-E, and TP1); and an effector amino-terminal domain, that recruits other proteins with the same domain to exert the signaling function of the oligomer recently formed (111). These domains guarantee the functions of PAMPs and DAMPs recognition, the conformational change, and the formation of oligomers (112).
There are subfamilies of NLRs that differ from each other in the effector domains, with these being the acidic domain (NLRA), BIR domain (NLRB), CARD domain (NLRC), and Pyrin domain (NLRP) (113). Some members of NLRC and NLRP form inflammasome complexes, such as NLRC4, NLRP1, and NLRP3, as well as two other protein receptors, absent in melanoma-2 (Aim-2) and Pyrin (114). An inflammasome complex consisting of NLRC4 does not bind to the ASC adapter protein but instead binds directly to caspase-1 in a card-card interaction since both proteins contain this domain (115). The results from Professor Tschopp's group first brought the notion of a complex containing the cytosolic receptor NALP1 (nowadays renamed as NLRP1) linked to the ASC adapter protein and the inflammatory caspase-1, functioning as a complex that is able to cleave the inactive form of pro IL-1β into its active form (108).
The subfamilies whose amino-terminal domain are Pyrin constitute the major forms of the inflammasome: NLRP1, NLRP3, Aim-2, and Pyrin (116, 117). When activated by an endogenous or exogenous ligand, the proteins bind to each other and interact with the ASC adapter protein, which in turn binds to the inactivated caspase-1. Once cleaved, caspase-1 becomes active and able to cleave protein precursors in aspartate residues, whose main representatives are the cleavage of inactive forms of the pro IL-β and pro IL-18 cytokines, making them active (118). In addition to Pyrin, the ASC protein also contains a CARD domain, responsible for the interaction with the inactive form of caspase-1 (117).
The most studied inflammasome complex to date is the NLRP3 inflammasome. Activation of the NLRP3 inflammasome is divided into canonical and non-canonical. Both pathways present, as a first stage, a response to either TLR activation or some cytokine receptors which are able to promote the transcription of the pro IL-1β and pro IL-18 forms as well as the NLRP3, all via NF-κB, translocation into the nucleus (119). The second step in the canonical pathway is the oligomerization phase of the complex, in which there is recruitment of the adapter protein ASC and pro caspase-1 (119). Subsequently, the complex is activated and performs its function of converting pro IL-1β and pro IL-18 forms into their active forms (120). Besides the cytokine cleavage, the assembly of the NLRP3 inflammasome after its activation and consequent caspase-1 autoclavation, results in the cleavage of a pore forming protein called Gasdermin D (GSMD), resulting in a programmed type of cell death called pyroptosis (121). Pyroptosis is a programmed cell death that is characterized by cell swelling followed by lysis and ending with the release of the intracellular contents.
Once the GSMD is cleaved, it results in the oligomerization of the N terminal domain of the GSDMD which will translocate to the inner cell membrane and bind to the phospholipids (phosphatidylserine and phosphatidylinositol), promoting pore formation and the release of the proinflammatory cytokines IL1β and IL18. As a result, the cell swells and the cytosol content overflows, causing the death of the cell due to a disruption in its osmotic potential. (122). Another phospholipid that GSMD binds to is the cardiolipin, a member of both the bacterial membrane and the mitochondrial outer membrane (122). Rogers and colleagues showed that GSMD-N binding to mitochondrial cardiolipin releases cytochrome C and enhances the apoptosis by the formation of the apoptosome with Apaf-1 and caspase-9 (123, 124).
As mentioned, another result of the pore formation trough GSMD action is the leakage of IL-1β and IL18. Shi and colleagues demonstrated that the release of the IL-1β is increased during pyroptosis. They used gsmd−/− Bone Marrow Derived Macrophages (BMDM) and observed a conserved caspase-1 action downstream the NLRP3 activation. Moreover, the secretion of IL-1β in these knockout cells was reduced when compared to wild type ones (121). This IL-1β secretion was later confirmed by Li et al. in an arsenic-induced liver fibrosis model. They showed that arsenate increases GSMD levels and cleavages caspase-1 (125). On the other hand, high expression of miR-379-5p restrained the GSMD expression and the IL-1β expression. Additionally, the EMT appears together with GSMD expression and IL-1β is released by hepatocyte cells (125). However, it is noteworthy that an GSDMD-independent IL1β release does also exist (126, 127).
The non-canonical pathway is a caspase-1 independent pathway and caspase-11 (for mice) or caspase-4/5 -dependent (for humans). Such a pathway, as mentioned before, is initiated after a TLR recognition, specifically, the recognition of a gram-negative bacteria, a group of pathogens that can bind and activate the TLR4-Myd88 and TRIF (Toll-IL-1 receptor homology domain-containing adapter-inducing interferon-β) pathways (128). The consequence of such activation is the translocation of the NF-κB transcription factor into the nucleus, promoting the transcription of NLRP3, pro IL-1β, and pro IL-18, as well the transcription of regulatory factors of interferons 3 and 7 (IRF3 and IRF7). The IRF3 and IRF7 complexes enhance the expression of type I interferons (IFNα/β) which, when binding to its respective receptors, activate the JAK/STAT signaling pathway, leading to the transcription of capsase-11 genes (or caspase-4/5) (120). In different pieces of research it was noted that it is not possible to cleave pro-IL1β and pro-IL18 by caspase-11 but caspase-11 does increase the activity of caspase-1 (129). Moreover, although it is not yet fully known, many data suggest that the pores formed by gasdermin are the predominant point in this non-canonical process (130). In this sense, caspase 11 is capable of initiating the pore forming pyroptosis in a similar way to caspase 1 and thus resulting in the release in the IL1β (131, 132). However, it has been well-noticed that active caspase-11 amplifies the canonical pathway, possibly by processing pannexin 1 and causing potassium efflux (133, 134).
A recent study showed a new integrant of the inflammasome complex, the Nek7 (NIMA-related kinase 7), a known protein involved in mitosis. Schmid-Burgk et al. performed a genomic experiment based on a CRISPR/Cas9 technology that promoted a misfunction of some proteins that were speculated to be involved in the NLRP3 inflammasome activation and regulation. One of these proteins was the Nek7 (135). He et al. demonstrated that Nek7 interacts with NLRP3 but not with any other inflammasome component. They generated Nek7-deficient macrophages and showed no caspase-1 and IL-1β activity (131). More interestingly, the absence of caspase-1 and IL-1β activity happened only in the NLRP3 inflammasome but not in either a NLRC4 or AIM2 inflammasomes activation context (131). Therefore, it was demonstrated that Nek7 interacts with the LRR region of the NLRP3 after ROS inflammasome oligomerization induction (136).
Despite the vast amount of literature concerning the NLRP3 inflammasome, there are still controversial aspects. For example, it was just recently found that the LRR region of the NLRP3 was described as not being required for its activation (110). As the Nek7 binds to the LRR region, this conclusion puts in check the role of this NIMA protein in the inflammasome activation. Another recent description supports a stochastic model for NLRP3 activation, depending on its isoform expressed into the cell, the full-length NLRP3, or the NLRP3 lacking exon 5, that ultimately loses the interaction with Nek7 (137).
IL-1β and IL-18
IL-1β corresponds to a multifunctional pro-inflammatory cytokine that is secreted by different cell types as a result of the host defense against both infection and injury, whether it is working alone or together with other cytokines (138). It is considered the most popular and the most studied among the family of interleukins (139). IL-1β is released in response to many PAMPs and DAMPs which can activate a variety, or in some cases multiple, PRRs to form inflammasome complexes and is known for its ability to cause fever, hypotension, and the induction of other cytokines such as IL-6 (139). The IL-1β is initially produced in its inactive precursor with 31 KDa in response to different molecular triggers and it is posteriorly cleaved to its 17.3 KDa active form by the pro-inflammatory protease caspase-1 (140), and, once activated, it can be involved in its autocrine and paracrine signaling (138).
There are 10 members of the IL-1 receptors family and of them the IL-1R1 is responsible for binding to IL-1β (141). This recognition happens with the help of a co-receptor IL-1R3, which does not have an IL-1β binding site (141). The IL-1R1 contains a cytosolic functional domain similar to TLRs called Toll-Interleukin receptor (TIR). After the IL-1β binding to IL-1R1, structural changes happen and allow the IL-1R3 to bind and form a trimeric structure (IL-1β, IL-1R1, and IL-1R3) (141). Thereafter, the cytosolic functional domain TIR recruits MyD88 which enables a signaling cascade that promotes NF-κB activation (141).
Inflammasomes and IL-1β are involved in the pathogenesis of many inflammatory disorders, yet the mechanism by which IL-1β is involved in fibrosis is complex. The main hypothesis lies in the relationship between the inflammatory cytokine and TGF-β. According to Luo et al. the short exposure of IL-1β inhibits the action of TGF-β via the NF-κB pathway, and consequently, the Smad pathway, responsible for the transcription of the TGF-β -encoding genes (142). However, this situation is transient due to a change in the NF-κB heterodimer from p65/p50 to p50/p50 at 24 h after exposure to IL-1β (142). The newly formed heterodimer is not able to inactivate Smad and thus, prolonged IL-1β exposure ultimately promotes the expression of TGF-β via Smad (142, 143). Such data represent the first finding regarding the potential of IL-1β in activating TGF-β in vascular muscle cells (144).
Another less discussed cytokine is the IL18 which, like IL1β, is a pro inflammatory cytokine implicated in the pathogenesis of many inflammatory renal diseases (145) and shares a structural homology, produced as a 24-kD inactive precursor lacking a signal peptide (pro-IL-18). This proinflammatory cytokine has proved its relation to EMT in a study performed by Bani-Hani and colleagues, using transgenic mice overexpressing human IL-18-binding protein in HK2 cells, where fibrosis was enhanced by ureteral obstruction, and led to a significant increase in IL-18 production, collagen deposition, α-sma, TGF-β1, and TNF-α production, whereas E-cadherin expression was simultaneously decreased (146). Further analysis revealed that neutralization of IL18 led to reductions in these indicators of obstruction-induced renal fibrosis and EMT, without any alterations in TGF-β1 or TNF-α activity (146).
Another study done by Zhang and colleagues showed that the IL-18 is highly expressed in bleomycin-induced pulmonary fibrosis (147). They used an IL-18 binding protein to block IL-18, and, as result, it prevented the EMT induction by reducing α-sma and reestablishing E-cadherin. The downstream signaling pathway of IL-18 inducing EMT is believed to be the transcriptor factor snail-1, supported by the finding that this factor was upregulated after IL-18 treatment. Also, the snail-1 knockout approach reduces the α-sma and increases E-cadherin with the same IL-18 treatment (147).
The Role of NLRP3 Activation in the Onset of EMT
As discussed previously, inflammasome are intracellular complexes formed by receptors of the Nod family, mostly belonging to cells of the innate immune system such as macrophages and dendritic cells. However, few studies show the activation of the complex in epithelium cells of certain tissues, such as the intestine (148), kidneys (149), and spleen (150). In contrast, IL-1β and IL-18 expression has been widely demonstrated to be expressed in tubular epithelial cells (151). Despite the complete relationship between inflammasomes and the onset of fibrosis not being fully known, the inflammasome -dependent and -independent pathways are summarized in Figure 2. The role of NLRP3 inflammasome into the EMT process was questioned by Wang and colleagues in a set of experiments, in which they demonstrated that the EMT process is independent of the NLRP3 inflammasome complex formation, but requires the presence of the NLRP3 protein (152). The levels of nlrp3 mRNA in mice primary tubular epithelial cells (pTECs) and human proximal tubular cells (HPTC) were found to be increased in response to TGF-β1 (152). Moreover, knockdown of NLRP3 retained an epithelial spindle-like morphology and reversed the mesenchymal characteristics of the cells. Additionally, the TGF-β1-induced MMP-9 and α-sma expression were significantly decreased in NLRP3−/− pTECs. When IL-1β, IL-18, MyD88, and casapse-1 are inhibited, MMP-9 and α-sma expression remains at comparable levels to WT cells (152). These data are also supported by another study demonstrating that the NLRP3 expression, but not the NLRP3 inflammasome complex activation, was required for EMT in colorectal cancer cells, in a model in which cancer cells were transfected with inflammasome-related genes, such as NLRP3. To elucidate the independence of NLRP3 inflammasome complex activation, western blotting analysis was performed and the cleaved form of caspase-1 was not observed after the induction of the EMT process, but only the pro-form of caspase-1 (153).
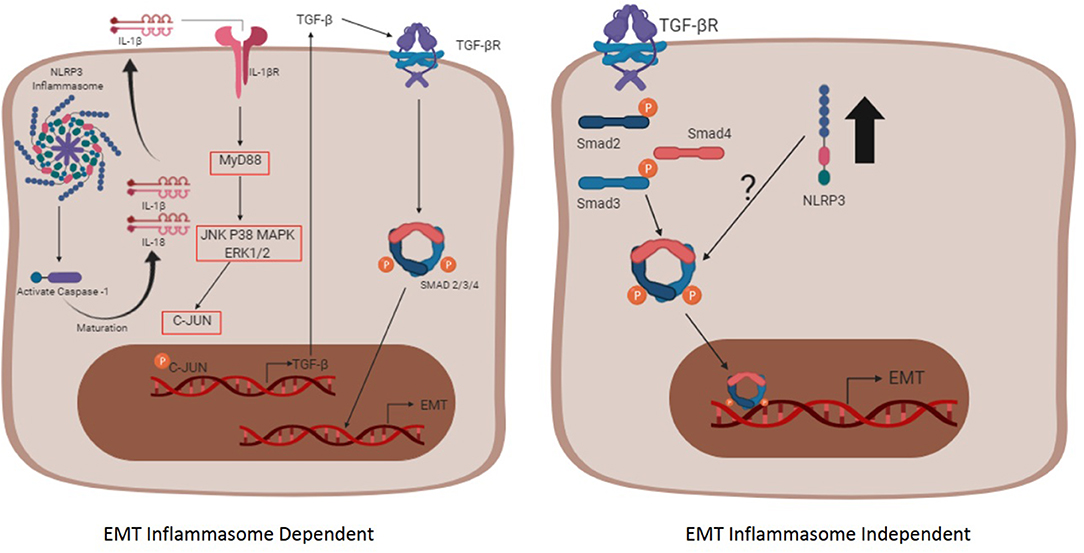
Figure 2. The inflammasome complex -dependent and -independent pathways involved in EMT. The inflammasome complex is activated after two signals (not shown), the first one (priming phase) that promotes the transcription of inflammasome-related proteins (NLRP3, Pro IL-1β, and Pro IL-18) through NF-κB, and the second one (activating phase) that promotes the oligomerization of the complex itself, contributing for the maturation of the cytokines IL-1β and IL-18 through caspase-1. Afterwards, the mature form of IL-1β reaches its receptor in an autocrine, paracrine, or endocrine way and promotes the transcription of the TGF-β gene. The secreted TGF-β binds to its receptor, triggering the pathway dependent of the Smad and promoting the EMT process (Left schematic cell). The pathway independent of the inflammasome oligomerization (right schematic cell) initiates when TGF-β reaches its receptor and, upon the Smad signaling, the nlrp3 expression is enhanced (black arrow). The interaction between the NLRP3 and the Smad3 is not fully understood and requires more studies.
Recently, some studies have also been elucidating the upstream inflammasome signaling pathways and their role in EMT. One of these studies showed that a medicinal herb called Huangkui is able to reduce the mRNA expression of nlrp3 through diminishing TLR4 and NF-κB protein levels (154). Additionally, another compound, a drug called paricalcitol, was suggested to decrease the nuclear translocation of p65 subunit of NF-κB, although the study did not evaluate the inflammasome pathway (155). Years later, another group used the paricalcitol in a mesothelial model to investigate the role of NLRP3 in the EMT process. They treated human peritoneal mesothelial cells with TGF-β1 and the addition of paricalcitol led to a reduction of nlrp3 transcription followed by reduced oxidative stress (156). Taken together, these results suggested that paricalcitol regulates the EMT process through modulating NLRP3 pathway signaling in an upstream manner, due its action in blocking the NF-κB translocation.
Just like the relationship between fibrosis and NLRP3 inflammasome, the relationship between TGF-β and the NLRP3 inflammasome is not well-understood (152). The understanding that inflammasome complex activation has the function of activating IL-1β and, in turn, IL-1β leads to increased TGF-β expression, brings the idea that the inflammasome complex triggers EMT via TGF-β derived from IL-1β production (157), as shown in Figure 2. It was reported that nanomaterials used in some biosensors, such as nanorods, nanowires, carbonaceous, and rare earth oxides (REOs) are able to activate the NLRP3 inflammasome, and, therefore, trigger EMT in the context of chronic lung fibrosis (158). The relationship between TGF-β and the NLRP3 inflammasome was also demonstrated in an in vitro model induced by angiotensin II, a protein able to activate the inflammasome via ROS (157). In this scenario, EMT occurs through the IL-1β signaling since it binds to its receptor and triggers a cascade of signals that promotes the transcription of TGF-β via MyD88 and c-JUN transcription factors. The TGF-β can act in an autocrine and paracrine way by binding to the TGF-βRII that ultimately leads to phosphorylation of Smad2/3 and binding to Smad4, acting as a transcription factor for EMT-related genes (157, 159).
Additionally, there is some evidence showing that IL-1β can enhance TGF-β action and, consequently, lead to EMT in an in vitro model using bronchial epithelial cells (160). Dorner and Zuraw treated bronchial epithelial cells with TGF-β alone, or combined with IL-1β, and they observed that when cells were treated with both cytokines, the levels of e-cadherin considerably decreased in comparison with the TGF-β-treated group. Also, the combined treatment raised the levels of tenascin C compared to individual treatment (160). Romero et al. suggests that there is a physical interaction between the NLRP3 and the Smad2/3 transcription factor in the cytosol in order to regulate the expression of genes responsible for EMT (161), which corroborate the results previously described regarding the participation of the NLRP3 inflammasome and the dependence of Smad to lead to EMT (157). It was demonstrated that the involvement of the NLRP3 inflammasome in inducing EMT may be related to the action of TGF-β in renal, pulmonary, and hepatic models of fibrosis (38, 152).
Moreover, Tian et al. demonstrated, in a model of bleomycin-induced pulmonary fibrosis, that the activation of the NLRP3 inflammasome leads to EMT via TGF-β, and once NLRP3 is silenced there are reduced TGF-β levels and, thus, the EMT does not occur (38). The same result was reached by Song et al. in a high glucose induced renal tubular fibrosis (162). Also, a more recent study performed by Li et al. in a silicosis model used the clinically approved drug for treating idiopathic pulmonary fibrosis, pirefenidone, and showed it effectively suppressed the EMT process and caused NLRP3 inhibition through phosphorylation of TAK1-MAPK-Snail/NF-κB pathway (40). The relationship between inflammasome and EMT is summarized in Table 1, and, due its controversial consensus, more studies are required in order to clarify results, especially the dependence of the NLRP3 complex formation for the EMT process to occur.

Table 1. The relationship between inflammasome and EMT.
Conclusion Remarks
The full understanding of the inflammasome complex in the context of the EMT triggering pathway, especially in its relationship with the TGF-β/Smad pathway, remains unclear and requires further study. According to the collected data from the literature, it is suggested that the EMT process might happen in two ways: one is dependent on the inflammasome activation when the IL-1β executes its pro-inflammatory action by up-regulating the TGF-β pathway; another is independent of the inflammasome activation, being necessary only in the presence of the NLRP3 receptor which enhances R-Smads in the TGF-β/Smad pathway. Hopefully, these findings might provide more opportunity for future targeting through blocking fibrosis, or at least retarding it in therapeutic approaches aimed at inflammatory diseases, chronic kidney disease, pulmonary silicosis, or any other disease that is end staged by fibrosis.
Author Contributions
All authors conceived the ideas present in the paper and wrote the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Chen L, Deng H, Cui H, Fang J, Zuo Z, Deng J, et al. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget. (2018) 9:7204–18. doi: 10.18632/oncotarget.23208
2. Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. (2012) 18:1028–40. doi: 10.1038/nm.2807
3. Scott DL, Wolfe F, Huizinga TW. Rheumatoid arthritis. Lancet. (2010) 376:1094–108. doi: 10.1016/S0140-6736(10)60826-4
4. Martinez FJ, Collard HR, Pardo A, Raghu G, Richeldi L, Selman M, et al. Idiopathic pulmonary fibrosis. Nat Rev Dis Primers. (2017) 3:17074. doi: 10.1038/nrdp.2017.74
5. Torres J, Mehandru S, Colombel J-F, Biroulet L. Crohn's disease. Lancet. (2017) 389:1741–55. doi: 10.1016/S0140-6736(16)31711-1
6. Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. (2008) 214:199–210. doi: 10.1002/path.2277
7. Liu Y. Renal fibrosis: new insights into the pathogenesis and therapeutics. Kidney Int. (2006) 69:213–7. doi: 10.1038/sj.ki.5000054
8. Meng XM, Nikolic-Paterson DJ, Lan HY. Inflammatory processes in renal fibrosis. Nat Rev Nephrol. (2014) 10:493–503. doi: 10.1038/nrneph.2014.114
9. Gallo J, Raska M, Kriegova E, Goodman SB. Inflammation and its resolution and the musculoskeletal system. J Orthop Translat. (2017) 10:52–67. doi: 10.1016/j.jot.2017.05.007
10. Fenyo IM, Gafencu AV. The involvement of the monocytes/macrophages in chronic inflammation associated with atherosclerosis. Immunobiology. (2013) 218:1376–84. doi: 10.1016/j.imbio.2013.06.005
11. Yang J, Zhang L, Yu C, Yang XF, Wang H. Monocyte and macrophage differentiation: circulation inflammatory monocyte as biomarker for inflammatory diseases. Biomark Res. (2014) 2:1. doi: 10.1186/2050-7771-2-1
12. Braga TT, Agudelo JS, Camara NO. Macrophages during the fibrotic process: M2 as friend and foe. Front Immunol. (2015) 6:602. doi: 10.3389/fimmu.2015.00602
13. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. (2008) 8:958–69. doi: 10.1038/nri2448
14. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. (2014) 6:13. doi: 10.12703/P6-13
15. Atri C, Guerfali FZ, Laouini D. Role of human macrophage polarization in inflammation during infectious diseases. Int J Mol Sci. (2018) 19:1801. doi: 10.3390/ijms19061801
16. Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. (2003) 112:1776–84. doi: 10.1172/JCI200320530
17. Liu F, Gu LN, Shan BE, Geng CZ, Sang MX. Biomarkers for EMT and MET in breast cancer: an update. Oncol Lett. (2016) 12:4869–76. doi: 10.3892/ol.2016.5369
18. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. (2009) 119:1420–8. doi: 10.1172/JCI39104
19. Zhang J, Tian XJ, Xing J. Signal transduction pathways of EMT induced by TGF-β, SHH, and WNT and their crosstalks. J Clin Med. (2016) 5:41. doi: 10.3390/jcm5040041
20. Lipschutz JH, Kissil JL. Expression of beta-catenin and gamma-catenin in epithelial tumor cell lines and characterization of a unique cell line. Cancer Lett. (1998) 126:33–41. doi: 10.1016/S0304-3835(97)00522-3
21. Rothenpieler UW, Dressler GR. Pax-2 is required for mesenchyme-to-epithelium conversion during kidney development. Development. (1993) 119:711–20.
22. Hogan BL, Kolodziej PA. Organogenesis: molecular mechanisms of tubulogenesis. Nat Rev Genet. (2002) 3:513–23. doi: 10.1038/nrg840
23. Zeisberg M, Bonner G, Maeshima Y, Colorado P, Müller GA, Strutz F, et al. Renal fibrosis: collagen composition and assembly regulates epithelial-mesenchymal transdifferentiation. Am J Pathol. (2001) 159:1313–21. doi: 10.1016/S0002-9440(10)62518-7
24. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. (2009) 139:871–90. doi: 10.1016/j.cell.2009.11.007
25. Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest. (2009) 119:1429–37. doi: 10.1172/JCI36183
26. Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. (2008) 14:818–29. doi: 10.1016/j.devcel.2008.05.009
27. Campbell K. Contribution of epithelial-mesenchymal transitions to organogenesis and cancer metastasis. Curr Opin Cell Biol. (2018) 55:30–5. doi: 10.1016/j.ceb.2018.06.008
28. Mack M. Inflammation and fibrosis. Matrix Biol. (2018) 68–9:106–21. doi: 10.1016/j.matbio.2017.11.010
29. Kothari AN, Mi Z, Zapf M, Kuo PC. Novel clinical therapeutics targeting the epithelial to mesenchymal transition. Clin Transl Med. (2014) 3:35. doi: 10.1186/s40169-014-0035-0
30. Nisticò P, Bissell MJ, Radisky DC. Epithelial-mesenchymal transition: general principles and pathological relevance with special emphasis on the role of matrix metalloproteinases. Cold Spring Harb Perspect Biol. (2012) 4:a011908. doi: 10.1101/cshperspect.a011908
31. Willis BC, duBois RM, Borok Z. Epithelial origin of myofibroblasts during fibrosis in the lung. Proc Am Thorac Soc. (2006) 3:377–82. doi: 10.1513/pats.200601-004TK
32. Miettinen PJ, Ebner R, Lopez AR, Derynck R. TGF-beta induced transdifferentiation of mammary epithelial cells to mesenchymal cells: involvement of type I receptors. J Cell Biol. (1994) 127(6 Pt 2):2021–36. doi: 10.1083/jcb.127.6.2021
33. Bhowmick NA, Ghiassi M, Bakin A, Aakre M, Lundquist CA, Engel ME, et al. Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol Biol Cell. (2001) 12:27–36. doi: 10.1091/mbc.12.1.27
34. Burns WC, Thomas MC. The molecular mediators of type 2 epithelial to mesenchymal transition (EMT) and their role in renal pathophysiology. Expert Rev Mol Med. (2010) 12:e17. doi: 10.1017/S1462399410001481
35. McCain J. The MAPK (ERK) pathway: investigational combinations for the treatment of BRAF-mutated metastatic melanoma. P T. (2013) 38:96–108.
36. Bakin AV, Tomlinson AK, Bhowmick NA, Moses HL, Arteaga CL. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J Biol Chem. (2000) 275:36803–10. doi: 10.1074/jbc.M005912200
37. Docherty NG, O'Sullivan OE, Healy DA, Murphy M, O'neill AJ, Fitzpatrick JM, et al. TGF-beta1-induced EMT can occur independently of its proapoptotic effects and is aided by EGF receptor activation. Am J Physiol Renal Physiol. (2006) 290:F1202–12. doi: 10.1152/ajprenal.00406.2005
38. Tian R, Zhu Y, Yao J, Meng X, Wang J, Xie H, et al. NLRP3 participates in the regulation of EMT in bleomycin-induced pulmonary fibrosis. Exp Cell Res. (2017) 357:328–34. doi: 10.1016/j.yexcr.2017.05.028
39. Xu Z, Li Z, Liao Z, Gao S, Hua L, Ye X, et al. PM2.5 induced pulmonary fibrosis in vivo and in vitro. Ecotoxicol Environ Saf. (2019) 171:112–21. doi: 10.1016/j.ecoenv.2018.12.061
40. Li X, Yan X, Wang Y, Wang J, Zhou F, Wang H, et al. NLRP3 inflammasome inhibition attenuates silica-induced epithelial to mesenchymal transition (EMT) in human bronchial epithelial cells. Exp Cell Res. (2018) 362:489–97. doi: 10.1016/j.yexcr.2017.12.013
41. Zhou T, Luo M, Cai W, Zhou S, Feng D, Xu C, et al. Runt-related transcription factor 1 (RUNX1) promotes TGF-β-induced renal tubular epithelial-to-mesenchymal transition (EMT) and renal fibrosis through the PI3K subunit p110δ. EBioMedicine. (2018) 31:217–25. doi: 10.1016/j.ebiom.2018.04.023
42. Sun Z, Ma Y, Chen F, Wang S, Chen B, Shi J. miR-133b and miR-199b knockdown attenuate TGF-β1-induced epithelial to mesenchymal transition and renal fibrosis by targeting SIRT1 in diabetic nephropathy. Eur J Pharmacol. (2018) 837:96–104. doi: 10.1016/j.ejphar.2018.08.022
43. Wang S, Zhou Y, Zhang Y, He X, Zhao X, Zhao H, et al. Roscovitine attenuates renal interstitial fibrosis in diabetic mice through the TGF-β1/p38 MAPK pathway. Biomed Pharmacother. (2019) 115:108895. doi: 10.1016/j.biopha.2019.108895
44. Gu TT, Chen TY, Yang YZ, Zhao XJ, Sun Y, Li TS, et al. Pterostilbene alleviates fructose-induced renal fibrosis by suppressing TGF-β1/TGF-β type I receptor/Smads signaling in proximal tubular epithelial cells. Eur J Pharmacol. (2019) 842:70–8. doi: 10.1016/j.ejphar.2018.10.008
45. Song S, Liu L, Yu Y, Zhang R, Li Y, Cao W, et al. Inhibition of BRD4 attenuates transverse aortic constriction- and TGF-β-induced endothelial-mesenchymal transition and cardiac fibrosis. J Mol Cell Cardiol. (2019) 127:83–96. doi: 10.1016/j.yjmcc.2018.12.002
46. Clark DA, Coker R. Transforming growth factor-beta (TGF-beta). Int J Biochem Cell Biol. (1998) 30:293–8. doi: 10.1016/S1357-2725(97)00128-3
47. Moses HL, Roberts AB, Derynck R. The discovery and early days of TGF-β: a historical perspective. Cold Spring Harb Perspect Biol. (2016) 8:a021865. doi: 10.1101/cshperspect.a021865
48. Kingsley DM. The TGF-beta superfamily: new members, new receptors, and new genetic tests of function in different organisms. Genes Dev. (1994) 8:133–46. doi: 10.1101/gad.8.2.133
49. Chang H, Brown CW, Matzuk MM. Genetic analysis of the mammalian transforming growth factor-beta superfamily. Endocr Rev. (2002) 23:787–823. doi: 10.1210/er.2002-0003
50. Chevalier RL. Specific molecular targeting of renal injury in obstructive nephropathy. Kidney Int. (2006) 70:1200–1. doi: 10.1038/sj.ki.5001815
51. Li Y, Yang J, Dai C, Wu C, Liu Y. Role for integrin-linked kinase in mediating tubular epithelial to mesenchymal transition and renal interstitial fibrogenesis. J Clin Invest. (2003) 112:503–16. doi: 10.1172/JCI200317913
52. Strutz F, Zeisberg M, Ziyadeh FN, Yang CQ, Kalluri R, Müller GA, et al. Role of basic fibroblast growth factor-2 in epithelial-mesenchymal transformation. Kidney Int. (2002) 61:1714–28. doi: 10.1046/j.1523-1755.2002.00333.x
53. Hata A, Lo RS, Wotton D, Lagna G, Massagué J. Mutations increasing autoinhibition inactivate tumour suppressors Smad2 and Smad4. Nature. (1997) 388:82–7. doi: 10.1038/40424
54. Barton DE, Foellmer BE, Du J, Tamm J, Derynck R, Francke U. Chromosomal mapping of genes for transforming growth factors beta 2 and beta 3 in man and mouse: dispersion of TGF-beta gene family. Oncogene Res. (1988) 3:323–31.
55. Zavadil J, Böttinger EP. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene. (2005) 24:5764–74. doi: 10.1038/sj.onc.1208927
56. Zeisberg EM, Potenta SE, Sugimoto H, Zeisberg M, Kalluri R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J Am Soc Nephrol. (2008) 19:2282–7. doi: 10.1681/ASN.2008050513
57. Kopp JB, Factor VM, Mozes M, Nagy P, Sanderson N, Böttinger EP, et al. Transgenic mice with increased plasma levels of TGF-beta 1 develop progressive renal disease. Lab Invest. (1996) 74:991–1003.
58. Sanderson N, Factor V, Nagy P, Kopp J, Kondaiah P, Wakefield L, et al. Hepatic expression of mature transforming growth factor beta 1 in transgenic mice results in multiple tissue lesions. Proc Natl Acad Sci USA. (1995) 92:2572–6. doi: 10.1073/pnas.92.7.2572
59. Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, et al. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci USA. (1993) 90:770–4. doi: 10.1073/pnas.90.2.770
60. Sanford LP, Ormsby I, Gittenberger-de Groot AC, Sariola H, Friedman R, Boivin GP, et al. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development. (1997) 124:2659–70.
61. Kaartinen V, Voncken JW, Shuler C, Warburton D, Bu D, Heisterkamp N, et al. Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat Genet. (1995) 11:415–21. doi: 10.1038/ng1295-415
62. Gorelik L, Flavell RA. Abrogation of TGFbeta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity. (2000) 12:171–81. doi: 10.1016/S1074-7613(00)80170-3
63. Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. (1992) 359:693–9. doi: 10.1038/359693a0
64. Miyazono K, Heldin CH. Latent forms of TGF-beta: molecular structure and mechanisms of activation. Ciba Found Symp. (1991) 157:81–9; discussion: 89–92. doi: 10.1002/9780470514061.ch6
65. Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. (2003) 425:577–84. doi: 10.1038/nature02006
66. Moustakas A, Heldin CH. Non-Smad TGF-beta signals. J Cell Sci. (2005) 118(Pt 16):3573–84. doi: 10.1242/jcs.02554
67. Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Res. (2009) 19:128–39. doi: 10.1038/cr.2008.328
68. Hong M, Wilkes MC, Penheiter SG, Gupta SK, Edens M, Leof EB. Non-Smad transforming growth factor-β signaling regulated by focal adhesion kinase binding the p85 subunit of phosphatidylinositol 3-kinase. J Biol Chem. (2011) 286:17841–50. doi: 10.1074/jbc.M111.233676
69. Savage C, Das P, Finelli AL, Townsend SR, Sun CY, Baird SE, et al. Caenorhabditis elegans genes sma-2, sma-3, and sma-4 define a conserved family of transforming growth factor beta pathway components. Proc Natl Acad Sci USA. (1996) 93:790–4. doi: 10.1073/pnas.93.2.790
70. Massagué J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. (2005) 19:2783–810. doi: 10.1101/gad.1350705
71. Howell M, Itoh F, Pierreux CE, Valgeirsdottir S, Itoh S, ten Dijke P, et al. Xenopus Smad4beta is the co-Smad component of developmentally regulated transcription factor complexes responsible for induction of early mesodermal genes. Dev Biol. (1999) 214:354–69. doi: 10.1006/dbio.1999.9430
72. Macias MJ, Martin-Malpartida P, Massagué J. Structural determinants of Smad function in TGF-β signaling. Trends Biochem Sci. (2015) 40:296–308. doi: 10.1016/j.tibs.2015.03.012
73. Piek E, Ju WJ, Heyer J, Escalante-Alcalde D, Stewart CL, Weinstein M, et al. Functional characterization of transforming growth factor beta signaling in Smad2- and Smad3-deficient fibroblasts. J Biol Chem. (2001) 276:19945–53. doi: 10.1074/jbc.M102382200
74. Liu F, Hata A, Baker JC, Doody J, Cárcamo J, Harland RM, et al. A human Mad protein acting as a BMP-regulated transcriptional activator. Nature. (1996) 381:620–3. doi: 10.1038/381620a0
75. Nomura M, Li E. Smad2 role in mesoderm formation, left-right patterning and craniofacial development. Nature. (1998) 393:786–90. doi: 10.1038/31693
76. Weinstein M, Yang X, Li C, Xu X, Gotay J, Deng CX. Failure of egg cylinder elongation and mesoderm induction in mouse embryos lacking the tumor suppressor smad2. Proc Natl Acad Sci USA. (1998) 95:9378–83. doi: 10.1073/pnas.95.16.9378
77. Xu F, Liu C, Zhou D, Zhang L. TGF-β/SMAD pathway and its regulation in hepatic fibrosis. J Histochem Cytochem. (2016) 64:157–67. doi: 10.1369/0022155415627681
78. Yao QY, Xu BL, Wang JY, Liu HC, Zhang SC, Tu CT. Inhibition by curcumin of multiple sites of the transforming growth factor-beta1 signalling pathway ameliorates the progression of liver fibrosis induced by carbon tetrachloride in rats. BMC Complement Altern Med. (2012) 12:156. doi: 10.1186/1472-6882-12-156
79. Medeiros AI, Sá-Nunes A, Soares EG, Peres CM, Silva CL, Faccioli LH. Blockade of endogenous leukotrienes exacerbates pulmonary histoplasmosis. Infect Immun. (2004) 72:1637–44. doi: 10.1128/IAI.72.3.1637-1644.2004
80. Latella G, Vetuschi A, Sferra R, Zanninelli G, D'Angelo A, Catitti V, et al. Smad3 loss confers resistance to the development of trinitrobenzene sulfonic acid-induced colorectal fibrosis. Eur J Clin Invest. (2009) 39:145–56. doi: 10.1111/j.1365-2362.2008.02076.x
81. Masszi A, Kapus A. Smaddening complexity: the role of Smad3 in epithelial-myofibroblast transition. Cells Tissues Organs. (2011) 193:41–52. doi: 10.1159/000320180
82. Leivonen SK, Lazaridis K, Decock J, Chantry A, Edwards DR, Kähäri VM. TGF-β-elicited induction of tissue inhibitor of metalloproteinases (TIMP)-3 expression in fibroblasts involves complex interplay between Smad3, p38α, and ERK1/2. PLoS ONE. (2013) 8:e57474. doi: 10.1371/journal.pone.0057474
83. Meng XM, Huang XR, Chung AC, Qin W, Shao X, Igarashi P, et al. Smad2 protects against TGF-beta/Smad3-mediated renal fibrosis. J Am Soc Nephrol. (2010) 21:1477–87. doi: 10.1681/ASN.2009121244
84. Hahn SA, Schutte M, Hoque AT, Moskaluk CA, da Costa LT, Rozenblum E, et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science. (1996) 271:350–3. doi: 10.1126/science.271.5247.350
85. Eppert K, Scherer SW, Ozcelik H, Pirone R, Hoodless P, Kim H, et al. MADR2 maps to 18q21 and encodes a TGFbeta-regulated MAD-related protein that is functionally mutated in colorectal carcinoma. Cell. (1996) 86:543–52. doi: 10.1016/S0092-8674(00)80128-2
86. Hata A, Lagna G, Massagué J, Hemmati-Brivanlou A. Smad6 inhibits BMP/Smad1 signaling by specifically competing with the Smad4 tumor suppressor. Genes Dev. (1998) 12:186–97. doi: 10.1101/gad.12.2.186
87. Shattuck TM, Costa J, Bernstein M, Jensen RT, Chung DC, Arnold A. Mutational analysis of Smad3, a candidate tumor suppressor implicated in TGF-beta and menin pathways, in parathyroid adenomas and enteropancreatic endocrine tumors. J Clin Endocrinol Metab. (2002) 87:3911–4. doi: 10.1210/jc.87.8.3911
88. Fleming NI, Jorissen RN, Mouradov D, Christie M, Sakthianandeswaren A, Palmieri M, et al. SMAD2, SMAD3 and SMAD4 mutations in colorectal cancer. Cancer Res. (2013) 73:725–35. doi: 10.1158/0008-5472.CAN-12-2706
89. Tang PM, Zhou S, Meng XM, Wang QM, Li CJ, Lian GY, et al. Smad3 promotes cancer progression by inhibiting E4BP4-mediated NK cell development. Nat Commun. (2017) 8:14677. doi: 10.1038/ncomms14677
90. Millet C, Zhang YE. Roles of Smad3 in TGF-beta signaling during carcinogenesis. Crit Rev Eukaryot Gene Expr. (2007) 17:281–93. doi: 10.1615/CritRevEukarGeneExpr.v17.i4.30
91. Sugimoto H, Yang C, LeBleu VS, Soubasakos MA, Giraldo M, Zeisberg M, et al. BMP-7 functions as a novel hormone to facilitate liver regeneration. FASEB J. (2007) 21:256–64. doi: 10.1096/fj.06-6837com
92. Boon MR, van der Horst G, van der Pluijm G, Tamsma JT, Smit JW, Rensen PC. Bone morphogenetic protein 7: a broad-spectrum growth factor with multiple target therapeutic potency. Cytokine Growth Factor Rev. (2011) 22:221–9. doi: 10.1016/j.cytogfr.2011.08.001
93. Zeisberg M, Hanai J, Sugimoto H, Mammoto T, Charytan D, Strutz F, et al. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med. (2003) 9:964–8. doi: 10.1038/nm888
94. Dudley AT, Lyons KM, Robertson EJ. A requirement for bone morphogenetic protein-7 during development of the mammalian kidney and eye. Genes Dev. (1995) 9:2795–807. doi: 10.1101/gad.9.22.2795
95. Luo G, Hofmann C, Bronckers AL, Sohocki M, Bradley A, Karsenty G. BMP-7 is an inducer of nephrogenesis, and is also required for eye development and skeletal patterning. Genes Dev. (1995) 9:2808–20. doi: 10.1101/gad.9.22.2808
96. López-Rovira T, Chalaux E, Massagué J, Rosa JL, Ventura F. Direct binding of Smad1 and Smad4 to two distinct motifs mediates bone morphogenetic protein-specific transcriptional activation of Id1 gene. J Biol Chem. (2002) 277:3176–85. doi: 10.1074/jbc.M106826200
97. Meng XM, Chung AC, Lan HY. Role of the TGF-β/BMP-7/Smad pathways in renal diseases. Clin Sci. (2013) 124:243–54. doi: 10.1042/CS20120252
98. Hu MC, Wasserman D, Hartwig S, Rosenblum ND. p38MAPK acts in the BMP7-dependent stimulatory pathway during epithelial cell morphogenesis and is regulated by Smad1. J Biol Chem. (2004) 279:12051–9. doi: 10.1074/jbc.M310526200
99. Zeisberg M, Kalluri R. The role of epithelial-to-mesenchymal transition in renal fibrosis. J Mol Med. (2004) 82:175–81. doi: 10.1007/s00109-003-0517-9
100. Vukicevic S, Basic V, Rogic D, Basic N, Shih MS, Shepard A, et al. Osteogenic protein-1 (bone morphogenetic protein-7) reduces severity of injury after ischemic acute renal failure in rat. J Clin Invest. (1998) 102:202–14. doi: 10.1172/JCI2237
101. Ying X, Sun Y, He P. MicroRNA-137 inhibits BMP7 to enhance the epithelial-mesenchymal transition of breast cancer cells. Oncotarget. (2017) 8:18348–58. doi: 10.18632/oncotarget.15442
102. Weiskirchen R, Meurer SK. BMP-7 counteracting TGF-beta1 activities in organ fibrosis. Front Biosci. (2013) 18:1407–34. doi: 10.2741/4189
103. Higgins DF, Ewart LM, Masterson E, Tennant S, Grebnev G, Prunotto M, et al. BMP7-induced-Pten inhibits Akt and prevents renal fibrosis. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:3095–104. doi: 10.1016/j.bbadis.2017.09.011
104. Luo DD, Phillips A, Fraser D. Bone morphogenetic protein-7 inhibits proximal tubular epithelial cell Smad3 signaling via increased SnoN expression. Am J Pathol. (2010) 176:1139–47. doi: 10.2353/ajpath.2010.090459
105. Hruska KA, Guo G, Wozniak M, Martin D, Miller S, Liapis H, et al. Osteogenic protein-1 prevents renal fibrogenesis associated with ureteral obstruction. Am J Physiol Renal Physiol. (2000) 279:F130–43. doi: 10.1152/ajprenal.2000.279.1.F130
106. Yang T, Chen SL, Lu XJ, Shen CY, Liu Y, Chen YP. Bone morphogenetic protein 7 suppresses the progression of hepatic fibrosis and regulates the expression of gremlin and transforming growth factor β1. Mol Med Rep. (2012) 6:246–52. doi: 10.3892/mmr.2012.892
107. Bin S, Li HD, Xu YB, Qi SH, Li TZ, Liu XS, et al. BMP-7 attenuates TGF-β1-induced fibroblast-like differentiation of rat dermal papilla cells. Wound Repair Regen. (2013) 21:275–81. doi: 10.1111/wrr.12015
108. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. (2002) 10:417–26. doi: 10.1016/S1097-2765(02)00599-3
109. Ng A, Xavier RJ. Leucine-rich repeat (LRR) proteins: integrators of pattern recognition and signaling in immunity. Autophagy. (2011) 7:1082–4. doi: 10.4161/auto.7.9.16464
110. Hafner-Bratkovič I, Sušjan P, Lainscek D, Tapia A, Cerović K, Kadunc L, et al. NLRP3 lacking the leucine-rich repeat domain can be fully activated via the canonical inflammasome pathway. Nat Commun. (2018) 9:5182. doi: 10.1038/s41467-018-07573-4
111. Proell M, Riedl SJ, Fritz JH, Rojas AM, Schwarzenbacher R. The Nod-like receptor (NLR) family: a tale of similarities and differences. PLoS ONE. (2008) 3:e2119. doi: 10.1371/journal.pone.0002119
112. Kim YK, Shin JS, Nahm MH. NOD-like receptors in infection, immunity, and diseases. Yonsei Med J. (2016) 57:5–14. doi: 10.3349/ymj.2016.57.1.5
113. Negroni A, Pierdomenico M, Cucchiara S, Stronati L. NOD2 and inflammation: current insights. J Inflamm Res. (2018) 11:49–60. doi: 10.2147/JIR.S137606
114. Kanneganti TD. The inflammasome: firing up innate immunity. Immunol Rev. (2015) 265:1–5. doi: 10.1111/imr.12297
115. Jha S, Ting JP. Inflammasome-associated nucleotide-binding domain, leucine-rich repeat proteins and inflammatory diseases. J Immunol. (2009) 183:7623–9. doi: 10.4049/jimmunol.0902425
116. Schattgen SA, Fitzgerald KA. The PYHIN protein family as mediators of host defenses. Immunol Rev. (2011) 243:109–18. doi: 10.1111/j.1600-065X.2011.01053.x
117. de Zoete MR, Palm NW, Zhu S, Flavell RA. Inflammasomes. Cold Spring Harb Perspect Biol. (2014) 6:a016287. doi: 10.1101/cshperspect.a016287
118. Schroder K, Tschopp J. The inflammasomes. Cell. (2010) 140:821–32. doi: 10.1016/j.cell.2010.01.040
119. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. (2019) 19:477–89. doi: 10.1038/s41577-019-0165-0
120. Pellegrini C, Antonioli L, Lopez-Castejon G, Blandizzi C, Fornai M. Canonical and non-canonical activation of NLRP3 inflammasome at the crossroad between immune tolerance and intestinal inflammation. Front Immunol. (2017) 8:36. doi: 10.3389/fimmu.2017.00036
121. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. (2015) 526:660–5. doi: 10.1038/nature15514
122. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. (2016) 535:153–8. doi: 10.1038/nature18629
123. Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat Commun. (2019) 10:1689. doi: 10.1038/s41467-019-09397-2
124. Brown GC, Borutaite V. Regulation of apoptosis by the redox state of cytochrome c. Biochim Biophys Acta. (2008) 1777:877–81. doi: 10.1016/j.bbabio.2008.03.024
125. Li J, Xue J, Wang D, Dai X, Sun Q, Xiao T, et al. Regulation of gasdermin D by miR-379-5p is involved in arsenite-induced activation of hepatic stellate cells and in fibrosis via secretion of IL-1β from human hepatic cells. Metallomics. (2019) 11:483–95. doi: 10.1039/C8MT00321A
126. Baroja-Mazo A, Compan V, Martín-Sánchez F, Tapia-Abellán A, Couillin I, Pelegrín P. Early endosome autoantigen 1 regulates IL-1β release upon caspase-1 activation independently of gasdermin D membrane permeabilization. Sci Rep. (2019) 9:5788. doi: 10.1038/s41598-019-42298-4
127. Monteleone M, Stanley AC, Chen KW, Brown DL, Bezbradica JS, von Pein JB, et al. Interleukin-1β maturation triggers its relocation to the plasma membrane for gasdermin-D-dependent and -independent secretion. Cell Rep. (2018) 24:1425–33. doi: 10.1016/j.celrep.2018.07.027
128. Broz P, Monack DM. Measuring inflammasome activation in response to bacterial infection. Methods Mol Biol. (2013) 1040:65–84. doi: 10.1007/978-1-62703-523-1_6
129. Viganò E, Mortellaro A. Caspase-11: the driving factor for noncanonical inflammasomes. Eur J Immunol. (2013) 43:2240–5. doi: 10.1002/eji.201343800
130. Platnich JM, Muruve DA. NOD-like receptors and inflammasomes: a review of their canonical and non-canonical signaling pathways. Arch Biochem Biophys. (2019) 670:4–14. doi: 10.1016/j.abb.2019.02.008
131. He Y, Hara H, Núñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci. (2016) 41:1012–21. doi: 10.1016/j.tibs.2016.09.002
132. He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. (2015) 25:1285–98. doi: 10.1038/cr.2015.139
133. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. (2016) 16:407–20. doi: 10.1038/nri.2016.58
134. Yang D, He Y, Muñoz-Planillo R, Liu Q, Núñez G. Caspase-11 requires the pannexin-1 channel and the purinergic P2X7 pore to mediate pyroptosis and endotoxic shock. Immunity. (2015) 43:923–32. doi: 10.1016/j.immuni.2015.10.009
135. Schmid-Burgk JL, Chauhan D, Schmidt T, Ebert TS, Reinhardt J, Endl E, et al. A genome-wide CRISPR (clustered regularly interspaced short palindromic repeats) screen identifies NEK7 as an essential component of NLRP3 inflammasome activation. J Biol Chem. (2016) 291:103–9. doi: 10.1074/jbc.C115.700492
136. Shi H, Wang Y, Li X, Zhan X, Tang M, Fina M, et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat Immunol. (2016) 17:250–8. doi: 10.1038/ni.3333
137. Hoss F, Mueller JL, Rojas Ringeling F, Rodriguez-Alcazar JF, Brinkschulte R, Seifert G, et al. Alternative splicing regulates stochastic NLRP3 activity. Nat Commun. (2019) 10:3238. doi: 10.1038/s41467-019-11076-1
138. Dinarello CA. Interleukin-1. Cytokine Growth Factor Rev. (1997) 8:253–65. doi: 10.1016/S1359-6101(97)00023-3
139. Lopez-Castejon G, Brough D. Understanding the mechanism of IL-1β secretion. Cytokine Growth Factor Rev. (2011) 22:189–95. doi: 10.1016/j.cytogfr.2011.10.001
140. Thornberry NA, Bull HG, Chapman KT, Howard AD, Kostura MJ, Miller DK, et al. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature. (1992) 356:768–74. doi: 10.1038/356768a0
141. Dinarello CA. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol Rev. (2018) 281:8–27. doi: 10.1111/imr.12621
142. Luo DD, Fielding C, Phillips A, Fraser D. Interleukin-1 beta regulates proximal tubular cell transforming growth factor beta-1 signalling. Nephrol Dial Transplant. (2009) 24:2655–65. doi: 10.1093/ndt/gfp208
143. Lee KY, Ito K, Hayashi R, Jazrawi EP, Barnes PJ, Adcock IM. NF-kappaB and activator protein 1 response elements and the role of histone modifications in IL-1beta-induced TGF-beta1 gene transcription. J Immunol. (2006) 176:603–15. doi: 10.4049/jimmunol.176.1.603
144. Yue TL, Wang XK, Olson B, Feuerstein G. Interleukin-1 beta (IL-1 beta) induces transforming growth factor-beta (TGF-beta 1) production by rat aortic smooth muscle cells. Biochem Biophys Res Commun. (1994) 204:1186–92. doi: 10.1006/bbrc.1994.2588
145. Dinarello CA. Interleukin 1 and interleukin 18 as mediators of inflammation and the aging process. Am J Clin Nutr. (2006) 83:447S−55S. doi: 10.1093/ajcn/83.2.447S
146. Bani-Hani AH, Leslie JA, Asanuma H, Dinarello CA, Campbell MT, Meldrum DR, et al. IL-18 neutralization ameliorates obstruction-induced epithelial-mesenchymal transition and renal fibrosis. Kidney Int. (2009) 76:500–11. doi: 10.1038/ki.2009.216
147. Zhang LM, Zhang Y, Fei C, Zhang J, Wang L, Yi ZW, et al. Neutralization of IL-18 by IL-18 binding protein ameliorates bleomycin-induced pulmonary fibrosis via inhibition of epithelial-mesenchymal transition. Biochem Biophys Res Commun. (2019) 508:660–6. doi: 10.1016/j.bbrc.2018.11.129
148. Nakata A, Nadatani Y, Watanabe T, Matsumoto Y, Kosaka S, Higashimori A, et al. NOD-like receptor family pyrin domain-containing 3 inflammasome activation exacerbates 5-fluorouracil-induced small intestinal mucositis via interleukin-1β activation. Digestion. (2019). doi: 10.1159/000504974. [Epub ahead of print].
149. Kim YG, Kim SM, Kim KP, Lee SH, Moon JY. The role of inflammasome-dependent and inflammasome-independent NLRP3 in the kidney. Cells. (2019) 8:1389. doi: 10.3390/cells8111389
150. Lech M, Avila-Ferrufino A, Skuginna V, Susanti HE, Anders HJ. Quantitative expression of RIG-like helicase, NOD-like receptor and inflammasome-related mRNAs in humans and mice. Int Immunol. (2010) 22:717–28. doi: 10.1093/intimm/dxq058
151. Homsi E, Janino P, de Faria JB. Role of caspases on cell death, inflammation, and cell cycle in glycerol-induced acute renal failure. Kidney Int. (2006) 69:1385–92. doi: 10.1038/sj.ki.5000315
152. Wang W, Wang X, Chun J, Vilaysane A, Clark S, French G, et al. Inflammasome-independent NLRP3 augments TGF-β signaling in kidney epithelium. J Immunol. (2013) 190:1239–49. doi: 10.4049/jimmunol.1201959
153. Wang H, Wang Y, Du Q, Lu P, Fan H, Lu J, et al. Inflammasome-independent NLRP3 is required for epithelial-mesenchymal transition in colon cancer cells. Exp Cell Res. (2016) 342:184–92. doi: 10.1016/j.yexcr.2016.03.009
154. Han W, Ma Q, Liu Y, Wu W, Tu Y, Huang L, et al. Huangkui capsule alleviates renal tubular epithelial-mesenchymal transition in diabetic nephropathy via inhibiting NLRP3 inflammasome activation and TLR4/NF-κB signaling. Phytomedicine. (2019) 57:203–14. doi: 10.1016/j.phymed.2018.12.021
155. Lee JW, Kim SC, Ko YS, Lee HY, Cho E, Kim MG, et al. Renoprotective effect of paricalcitol via a modulation of the TLR4-NF-κB pathway in ischemia/reperfusion-induced acute kidney injury. Biochem Biophys Res Commun. (2014) 444:121–7. doi: 10.1016/j.bbrc.2014.01.005
156. Ko J, Kang HJ, Kim DA, Ryu ES, Yu M, Lee H, et al. Paricalcitol attenuates TGF-β1-induced phenotype transition of human peritoneal mesothelial cells (HPMCs) via modulation of oxidative stress and NLRP3 inflammasome. FASEB J. (2019) 33:3035–50. doi: 10.1096/fj.201800292RR
157. Zhang LL, Huang S, Ma XX, Zhang WY, Wang D, Jin SY, et al. Angiotensin(1-7) attenuated Angiotensin II-induced hepatocyte EMT by inhibiting NOX-derived H2O2-activated NLRP3 inflammasome/IL-1β/Smad circuit. Free Radic Biol Med. (2016) 97:531–43. doi: 10.1016/j.freeradbiomed.2016.07.014
158. Wang X, Sun B, Liu S, Xia T. Structure activity relationships of engineered nanomaterials in inducing NLRP3 inflammasome activation and chronic lung fibrosis. NanoImpact. (2017) 6:99–108. doi: 10.1016/j.impact.2016.08.002
159. Artlett CM. The role of the NLRP3 inflammasome in fibrosis. Open Rheumatol J. (2012) 6:80–6. doi: 10.2174/1874312901206010080
160. Doerner AM, Zuraw BL. TGF-beta1 induced epithelial to mesenchymal transition (EMT) in human bronchial epithelial cells is enhanced by IL-1beta but not abrogated by corticosteroids. Respir Res. (2009) 10:100. doi: 10.1186/1465-9921-10-100
161. Romero CA, Remor A, Latini A, De Paul AL, Torres AI, Mukdsi JH. Uric acid activates NRLP3 inflammasome in an in-vivo model of epithelial to mesenchymal transition in the kidney. J Mol Histol. (2017) 48:209–18. doi: 10.1007/s10735-017-9720-9
Keywords: NLRP3, EMT—epithelial to mesenchymal transition, fibrosis, inflammasome, TGF-β
Citation: Alyaseer AAA, de Lima MHS and Braga TT (2020) The Role of NLRP3 Inflammasome Activation in the Epithelial to Mesenchymal Transition Process During the Fibrosis. Front. Immunol. 11:883. doi: 10.3389/fimmu.2020.00883
Received: 04 February 2020; Accepted: 16 April 2020;
Published: 20 May 2020.
Edited by:
Robson Coutinho-Silva, Federal University of Rio de Janeiro, BrazilReviewed by:
Alberto Baroja-Mazo, Biomedical Research Institute of Murcia (IMIB), SpainJunya Masumoto, Ehime University Graduate School of Medicine, Japan
Copyright © 2020 Alyaseer, de Lima and Braga. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tarcio Teodoro Braga, dGFyY2lvX3RiQHlhaG9vLmNvbS5icg==