 Jie Liang1†
Jie Liang1† Jacqueline D. Ziegler1
Jacqueline D. Ziegler1 Beate Jahraus1
Beate Jahraus1 Christian Orlik1
Christian Orlik1 Renata Blatnik2Norbert Blank3Beate Niesler4,5
Renata Blatnik2Norbert Blank3Beate Niesler4,5 Guido Wabnitz1
Guido Wabnitz1 Thomas Ruppert2Katrin Hübner1
Thomas Ruppert2Katrin Hübner1 Emre Balta1
Emre Balta1 Yvonne Samstag1*
Yvonne Samstag1*- 1Section Molecular Immunology, Institute of Immunology, Heidelberg University, Heidelberg, Germany
- 2Mass Spectrometry Core Facility, Center for Molecular Biology (ZMBH), Heidelberg University, Heidelberg, Germany
- 3Division of Rheumatology, Department of Internal Medicine V, Heidelberg University, Heidelberg, Germany
- 4Department of Human Molecular Genetics, Heidelberg University, Heidelberg, Germany
- 5nCounter Core Facility, Department of Human Molecular Genetics, Heidelberg University, Heidelberg, Germany
Piperlongumine (PL), a natural small molecule derived from the Piper longum Linn plant, has received growing interest as a prooxidative drug with promising anticancer properties. Yet, the influence of PL on primary human T cells remained elusive. Knowledge of this is of crucial importance, however, since T cells in particular play a critical role in tumor control. Therefore, we investigated the effects of PL on the survival and function of primary human peripheral blood T cells (PBTs). While PL was not cytotoxic to PBTs, it interfered with several stages of T cell activation as it inhibited T cell/APC immune synapse formation, co-stimulation-induced upregulation of CD69 and CD25, T cell proliferation and the secretion of proinflammatory cytokines. PL-induced immune suppression was prevented in the presence of thiol-containing antioxidants. In line with this finding, PL increased the levels of intracellular reactive oxygen species and decreased glutathione in PBTs. Diminished intracellular glutathione was accompanied by a decrease in S-glutathionylation on actin suggesting a global alteration of the antioxidant response. Gene expression analysis demonstrated that TH17-related genes were predominantly inhibited by PL. Consistently, the polarization of primary human naïve CD4+ T cells into TH17 subsets was significantly diminished while differentiation into Treg cells was substantially increased upon PL treatment. This opposed consequence for TH17 and Treg cells was again abolished by thiol-containing antioxidants. Taken together, PL may act as a promising agent for therapeutic immunosuppression by exerting prooxidative effects in human T cells resulting in a diminished TH17 but enhanced Treg cell differentiation.
Introduction
PL is a compound that can be isolated from different piper species, especially from Piper longum Linn (P. longum, Indian long pepper). It has been traditionally used in Asia for the treatment of gastrointestinal complaints, respiratory diseases and infectious diseases, such as malaria (1). In recent years, PL has gained increasing interest as a potential drug against cancer. The anticancer effects were mainly shown by an inhibition of the proliferation of various human cancer cell lines under in vitro conditions and an increased cancer cell death in the presence of PL (2–5). Anticancer effects of PL have also been found in vivo in mouse xenograft tumor models (4, 6, 7). Based on that, several patents have been filed for the treatment of cancer using PL and PL analogs (8). Mechanistically, PL is a prooxidative compound that increases the amount of reactive oxygen species (ROS) in cancer cells, which is directly linked to PL-associated anticancer activities (9, 10). Further studies could identify several ROS-dependent signals, e.g., PI3K/AKT/mTOR (4) and NF-κB pathways (11–13), signal transducer and activator of transcription (STAT) 3 (7) and p38 (3, 14) as targets of PL in cancer cells. PL also interacts with biologically important small molecules, e.g., it is considered as a direct inhibitor of the thioredoxin reductase 1 in human gastric cancer cells, which leads to ROS accumulation and ROS-dependent cell death (15). Moreover, in cancer cells PL was reported to deplete the reduced glutathione (GSH) stores (16) and to inactivate other thiol-containing proteins involved in maintaining cellular redox homeostasis through thiol modification (5). Besides, PL induces endoplasmic reticulum stress (17) and inhibits the ubiquitin-proteasome system (18), which are also linked to increased ROS levels.
Despite intensive research on its anticancer properties, potential effects of PL on human immune cells, especially T cells, were disregarded in initial studies in this field. In tumor patients potent anti-tumor immune responses are extremely important, e.g., for successful immunotherapy. We, therefore, asked whether treatment with PL affected the function of human T cells. Earlier studies, in which the effects of PL in chronic inflammatory diseases were analyzed, provided first insights into the immunomodulatory properties of PL in the mouse system. Xiao et al. demonstrated, under in vitro conditions, that PL inhibits the LPS-induced maturation of mouse bone marrow-derived dendritic cells (DCs). This observation was confirmed by in vivo experiments showing decreased maturation of splenic DCs in mice with collagen-induced arthritis (CIA) (19). Furthermore, Sun et al. have shown in a mouse model of CIA that PL expanded myeloid-derived suppressor cells (MDSC) and reduced the arthritis score and histopathologic lesions (20). Another study reported that PL improved the symptoms of lupus nephritis in MRL-Fas (lpr) mice by decreasing the levels of proinflammatory cytokines and the frequency of TH17 cells while increasing the frequency of Treg cells (21). In line with the shifted TH17/Treg ratio, PL ameliorated MOG-induced experimental autoimmune encephalomyelitis (EAE) in mice due to dampened NF-κB signaling (22). PL also inhibited the activation and function of human fibroblast-like synoviocytes (FLS) that were derived from rheumatoid arthritis (RA) patients (20, 23). However, a potential direct influence of PL on human T cells has not been investigated. Given the crucial role of T cells in the immune system, it is, however, important to know whether and how PL affects T cell immunity in the human system in order to assess its potential clinical benefit.
The molecular mode of action of PL on various immune cell types is only partially known. In contrast to the situation in cancer cells, in splenic DCs PL diminished the intracellular ROS levels, thereby suppressing the maturation of DCs (19). Likewise, PL decreased TNFα-induced intracellular ROS production in RA FLS (23). Overall, a confusing picture emerged with regard to the effects of PL on the intracellular redox homeostasis in different cell types.
T cell functions can be strongly modulated by influencing the actin cytoskeleton via the redox balance (24). In addition, lowered ROS levels in T cells of RA patients were reported to promote their differentiation into IL-17 and IFNγ-producing inflammatory cells (25). Vice versa, a prooxidative microenvironment reduced the cytokine response of terminally differentiated TH1 and TH17 cells (26). Furthermore, Fu et al. reported that accumulation of ROS active misshapen/NIK-related kinase 1 (MINK1) limited the generation of TH17 cells. This phenotype was prevented in the presence of the antioxidant N-acetyl-cysteine (NAC) (27). Along the same line, inhibition of pyruvate dehydrogenase kinase (PDHK) specifically impaired TH17 cells, while sparing TH1 and promoting Treg cells. Again, this was mediated in part through ROS since NAC treatment restored TH17 cell generation (28). Based on the reported redox regulation by PL, our goal was to investigate whether PL serves as a novel means to modulate the redox balance and thus the function of primary human T cells.
In the present study, we found that PL was not toxic to primary human T cells, as opposed to the malignant T leukemia line Jurkat. However, PL inhibited the activation and proliferation of primary human T cells after costimulation through CD3 and CD28. In particular, the development of proinflammatory TH17 cells was significantly diminished, while anti-inflammatory Treg cells were upregulated. We were able to attribute these results to the prooxidative activity of PL in primary human T cells, because PL increased intracellular ROS levels and decreased GSH levels, and the PL-mediated immunosuppressive effects were weakened by thiol-containing antioxidants. Furthermore, we identified redox-regulated targets of PL, e.g., hypoxia-inducible factor (HIF)-1α.
Together, our data demonstrate that PL acts as an immunomodulating agent for primary human T cells. By altering the redox balance toward a prooxidative milieu PL shifts T cell immunity toward an immunosuppressive phenotype. Thus, PL may represent a novel option to control autoimmune disorders.
Materials and Methods
Materials
Piperlongumine (#SML0221), DMSO, NAC, Paraformaldehyde (PFA), phorbol 12-myristate 13-acetate (PMA), Ionomycin and Brefeldin A were purchased from Sigma-Aldrich (St. Louis, USA). GSH, 5-(and-6) chloromethyl-2′, 7′-dichlorodihydro-fluorescein diacetate, acetyl ester (CM-H2DCFDA), RPMI 1640 and ThiolTrackerTM Violet were purchased from Thermo Fisher Scientific. Carboxyfluorescein diacetate succinimidyl ester (CFSE) was bought from Invitrogen (Eugene, USA) and fetal bovine serum (FBS) from PAN-Biotech (Aidenbach, Germany). nCounter® GX Human Immunology v2 panel was purchased from NanoString Technologies and Direct-zolTM RNA MiniPrep kit from QIAGEN. Ficoll-Hypaque (FicoLite H) was obtained from Linarisblue (Wertheim-Bettingen, Germany) and BD FACSTM lysing solution from BD Bioscience. Recombinant human IL-12, IL-4, IL-1β, IL-23, IL-6, TGF-β, neutralizing antibodies against IFNγ and IL-4, True-Nuclear™ transcription factor buffer set, LEGEND MAX™ Human IL-17A, IL-17F and LEGEND MAX™ Free Active TGF-β ELISA kit were obtained from BioLegend (San Diego, USA).
Antibodies used in this study were specific for the following molecules: CD3 (clone OKT3, mouse mAb), CD28 (clone 28.2, mouse mAb) and isotype control antibodies IgG1, κ and IgG2a, κ (mouse mAb, BD Biosciences, Heidelberg, Germany), GAPDH (clone 6C5, mouse mAb, Invitrogen; Eugene, USA), actin (rabbit polyclonal, Sigma-Aldrich, Hamburg, Germany), GSH (clone D8, mouse mAb, Santa Cruz Biotechnology), HIF-1α (rabbit polyclonal, Cayman chemical, USA). For the secondary antibodies, IRDye® 680CW donkey anti-mouse and IRDye® 800CW donkey anti-rabbit were purchased from LICOR Biosciences (Lincoln, USA). Goat anti-mouse IgG+IgM used as coating antibody was from Jackson ImmunoResearch (West Grove, USA). 7-AAD and all fluorescently-labeled antibodies were obtained from BD Biosciences.
Primary Human T Cell Preparation and Cell Culture
Human peripheral blood mononuclear cells (PBMCs) were obtained from heparinized blood of voluntary healthy donors by density-gradient centrifugation using Ficoll-Hypaque. Pan T cells or naïve CD4+ T cells were isolated by negative selection using the pan T cell isolation kit or naïve CD4+ T cells isolation kit from Miltenyi Biotec (Bergisch Gladbach, Germany), respectively, according to the manufacturer's instructions. The purity of Pan T cells reached more than 99.5% (Supplementary Figure 1a), of naïve CD4+ T cells 99% (Supplementary Figure 1b). The purified T cells were adjusted to 3 x 106 cells/ml in RPMI complete medium (RPMI 1640+10% FBS) and cultured in incubator at 5% CO2 and 37°C until use. To costimulate human peripheral blood T cells (PBTs), goat anti-mouse IgG+IgM antibody was used to pre-coat microplates (Nunc, Wiesbaden, Germany), followed by blocking with RPMI complete medium and coating with anti-CD3 (20 ng/mL)/anti-CD28 (5 μg/mL) antibodies or the respective isotype controls. T cells were spun down on the antibodies and incubated at 5% CO2 and 37°C for the indicated time points. This study was approved by the Ethics Committee of the Heidelberg University (S-269/2015). The human T cell leukemia cell line Jurkat ACC282 and Raji cells, which served as antigen-presenting cells (APC) were cultured in RPMI complete medium at 5% CO2 and 37°C, and cells were split 1:5 every second day by replacing the medium.
Sampling of Blood From Patients
Peripheral blood from patients with different diseases was collected into sterile heparinized tubes under aseptic conditions. Informed consent for use of the cells was obtained from all patients included in this study. To test the effects of PL on the lymphocytes in whole blood, 100 μl of blood were diluted 1:1 with RPMI medium, blood was treated with DMSO or with 5 μM PL and activated with 1 μg/ml staphylococcal enterotoxin B (SEB) for 20 hours (h). Afterwards, Brefeldin A (10 μg/ml) was used to prevent the secretion of cytokines (29). After 4 h incubation, red blood cells were lysed with BD FACS Lysis Solution for 10 min at room temperature (RT) and simultaneously, white blood cells were fixed. Samples were then washed with FACS wash buffer (FW, PBS containing 0.5% BSA, 0.5% FBS, and 0.07% NaN3), permeabilized with FWS (FW containing 0.1% Saponin) and stained with fluorescent-labeled antibodies against CD3, CD69, CD25, IFNγ, and IL-2 in FWS for 20 min at RT. Thereafter, samples were washed and assessed by flow cytometry (LSRII, BD Bioscience, Heidelberg, Germany). Data were analyzed with FlowJo X (FlowJo LLC, Ashland, OR, USA). This study was approved by the Ethics Committee of the Heidelberg University (S-119/2017).
Cell Viability Assay
Primary human T cells and Jurkat cells were cultured in 200 μL RPMI complete medium with or without 5 μM PL for the indicated time points (5% CO2 and 37°C). PL was solved in DMSO, therefore cells were treated with equal concentrations of DMSO as a control. The cells were then washed once with PBS, stained with AnnexinV and 7-AAD in Annexin binding buffer for 20 min at RT. Thereafter, cells were washed once with Annexin binding buffer and analyzed by flow cytometry.
Measurement of Intracellular ROS and GSH Levels
ROS detection reagent CM-H2DCFDA was used for the measurement of intracellular ROS levels and ThiolTrackerTM violet dye was used for the measurement of intracellular GSH levels as described before (30). Briefly, for the intracellular ROS measurement, T cells (1 × 106 cells/ml) or Jurkat cells (3 × 105 cells/ml) were washed with PBS and stained with CM-H2DCFDA (5 μM) in PBS for 15 min at 37°C. Cells were then washed with PBS, resuspended in RPMI complete medium and treated with DMSO, PL or H2O2. After the indicated time points, the mean fluorescence intensity (MFI) of the CM-H2DCFDA signal was determined by flow cytometry. For the intracellular GSH measurement, T cells (1 × 106 cells/ml) or Jurkat cells (3 × 105 cells/ml) were incubated with DMSO or PL in RPMI complete medium for 1 h or 24 h. Afterwards, cells were rinsed with PBS and stained with 1 μM ThiolTrackerTM violet dye (diluted in PBS) for 15 min at 37°C. After washing with PBS, cells were resuspended in PBS and measured immediately by flow cytometry.
Detection of S-glutathionylation
Western blotting was used for the detection of S-glutathionylated proteins. Control or PL-treated T cells or Jurkat cells were washed with PBS and postnuclear lysates (cytoplasm) were generated. Therefore, T cells were lysed with TKM lysis buffer on ice for 20 min, the nuclei and the cell debris were removed by centrifugation at 14,000 g for 10 min. Proteins in the cytoplasmic fraction were separated by electrophoresis in denaturing SDS polyacrylamide gels (SDS-PAGE), proteins were transferred onto PVDF-membranes and the membranes were blocked in blocking buffer for 1 h. Afterwards, membranes were probed with primary antibodies against GSH (1:200), Actin (1:1,000), GAPDH (1:10,000), and respective IRDye fluorescent-labeled secondary antibodies (1:10,000). The membranes were scanned by a Licor infrared scanner (LI-COR Biosciences, Germany).
The S-glutathionylated protein was further analyzed by mass spectrometry. To this end, postnuclear cell lysates from control or PL-treated T cells (3 × 106 cells/sample), were run on SDS-PAGE and subjected to Coomassie-staining. The bands corresponding to the 42-kDa protein were cut out and analyzed by mass spectrometry. Protein digestion and LC-MS measurement were done as described elsewhere (31). Briefly, gel pieces were washed, dehydrated and incubated with trypsin solution (Thermo-Fisher, Rockford, USA) for 4 h at 37°C. The reaction was quenched by adding of 20 μL of 0.1% trifluoroacetic acid (TFA; Biosolve, Netherlands). The supernatant was dried in a vacuum concentrator before LC-MS analysis. Nanoflow LC-MS analysis was performed with a NanoAcquity UPLC liquid chromatography (Waters, Eschborn, Germany) system coupled to an Orbitrap XL or with an Ultimate 3000 liquid chromatography system coupled to an Orbitrap Q Exactive mass spectrometer (Thermo-Fischer, Bremen, Germany). Detailed information for the LC-MS measurements is described in the Supplementary Material. For the protein identification, raw files were analyzed using Proteome Discoverer with the Sequest version 2.3 (Thermo Fisher Scientific, USA). Sequest was set up to search against Uniprot human databases (retrieved in June, 2017) with trypsin as the digestion enzyme with maximum two missed cleavages. A parent ion mass tolerance was set to 10 ppm and a fragment ion mass tolerance was set to 0.50 Da or 0.02 Da for measurements on the Orbitrap XL or the Q Exactive mass spectrometer, respectively. Proteome Discoverer results were used for a spectral library generation in the Skyline (32) using BiblioSpec algorithm for MS1 full-scan quantification of glutathionylated peptides and their unmodified counterparts as previously described (33). After raw file data import into Skyline, chromatographic traces for top three isotopic peaks were manually inspected for proper peak picking of MS1 filtered peptides and peak area determination. The summed area of three isotopic peaks was used to calculate peptide intensities. Target peptide areas were normalized to summed area of selected actin peptides (AVFPSIVGRPR; RGILTLK; VAPEEHPVLLTEAPLNPK; SYELPDGQVITIGNER).
Measurement of Intracellular Calcium
Control or PL-treated T cells (3.5 × 106 cells/sample, 1 × 106 cells/ml) were centrifuged and re-suspended in 250 μL RPMI medium (+2% FBS) with the calcium (Ca2+) -binding dye, Indo-1, at a concentration of 2 μg/ml. Cells were then stained at 37°C for 45 min. The dye changes its emission wavelength when binding to Ca2+, so a ratio between bound and unbound dye can be calculated. The samples were washed twice with PBS, then resuspended in 500 μL RPMI complete medium. The kinetics of the Ca2+ signal was measured by flow cytometry (Fortessa, BD Bioscience, Heidelberg, Germany). To this end, the baseline Ca2+ signal was acquired for 1 min, followed by adding anti-CD3 antibody to a final concentration of 20 ng/ml. The Ca2+ signal was acquired for another 8 min, until goat-anti-mouse antibody was added (7.2 μg/mL) for crosslinking of the anti-CD3 antibody to induce activation and Ca2+ influx to the cytosol. Thereafter, the Ca2+ signal was monitored for up to 18 min and then ionomycin was added to induce a maximum Ca2+ signal as a positive control and the measurement was stopped after 20 min.
T Cell/APC Conjugate and Immune Synapse Formation
Conjugates were formed between T cells and Raji cells as described below. T cells (1 × 106 cells/ml) were preincubated with or without 5 μM PL for 1 h or overnight and Raji cells were loaded with 5 μg/ml SEB or kept unloaded. Pretreated T cells and Raji cells were then coincubated for 45 min at 37°C at a ratio of 1:1. Afterwards, 1.5% PFA was added while vortexing the sample for 10 s in order to separate non-specifically bound cells. After 10 min fixation, cells were washed twice with FW buffer followed by staining with anti-CD19 PE-Cy5 and anti-CD3 PE conjugated antibodies. 1 × 104 T cells per sample of three independent experiments were acquired to quantify the proportion of T cell/APC conjugates. CD3 and CD19 double positive events were counted as cell couples.
Immune synapse formation was assessed by multispectral imaging flow cytometry (MIFC, IS100, Amnis Corp., Seattle, WA, USA). To this end, T cell/APC conjugates were formed and fixed as described above. The samples were then stained with anti-CD3 PE-Cy7 and anti-CD18 PE. After permeabilization with FWS buffer, samples were further stained with SiR-actin and DAPI. Thereafter, cells were subjected to MIFC and as many as 15,000 images were acquired per sample. The IDEAS 6.0 software (Amnis, Seattle, WA, USA) was used to analyze the subcellular localization of proteins (34).
T Cell Activation
T cells (1 × 106 cells/ml) were preincubated with DMSO or different concentrations of PL for 30 min and were seeded in 96-well microplates coated with anti-CD3/CD28 antibodies. In parallel, mouse anti-human IgG1, κ, and IgG2a, κ were coated as isotype control. Then the cells were incubated at 37°C for 24 h and the activation state of the T cells was determined by measuring the expression of CD69 and CD25. Briefly, T cells were collected after plate stimulation and washed twice with FW buffer, fixed with 1.5% PFA for 10 min and stained at RT with anti-CD69 PE-Cy7 and anti-CD25 APC antibodies in FW buffer. After 20 min of staining, cells were washed with FW and measured by flow cytometry. For the detection of the total extra- and intracellular CD69 and CD25, T cells were permeabilized by FWS buffer prior to staining with antibodies in FWS buffer.
T Cell Proliferation
T cells were washed with PBS, resuspended in PBS containing 1 μM CFSE and incubated at 37°C for 15 min. The unbound CFSE was then removed by washing twice with PBS. T cells were then resuspended in RPMI complete medium and treated with DMSO or PL for 30 min. Afterwards, the cells were seeded on a 96-well microplate coated with anti-CD3/CD28 antibodies and incubated for 72 h. The CFSE signal was determined using flow cytometry and data were analyzed with FlowJo X. The proliferation index was calculated as described previously (30).
Co-stimulation Induced Cytokine Production
For the assessment of costimulation induced intracellular cytokines, Pan T cells were treated with various concentrations of PL in the presence and absence of exogenously added NAC and then costimulated for 48 h. Afterwards, T cells were restimulated with 10 nM PMA and 1 μg/ml ionomycin for 4 h to enhance the cytokine production. Simultaneously, 10 μg/ml Brefeldin A was used to prevent the secretion of cytokines. Then, T cells were collected and washed twice with FW, fixed in 1.5% PFA for 10 min and permeabilized with FWS for 20 min. Afterwards, T cells were stained with anti-IL-2, anti-IFNγ and anti-TNFα (50 μL/sample) antibodies diluted by FWS for 30 min. Florescence Minus One (FMO) staining was included in control samples to detect unspecific staining. Cells were then washed twice with FWS and measured by flow cytometry. Data were analyzed with FlowJo X.
Gene Expression Profiling
T cells (1 × 106 cells/sample) were pretreated with DMSO or 5 μM PL for 30 min and then stimulated for 4 h in anti-CD3/CD28 antibodies coated plates. Total RNA was extracted from T cells by trizol. The RNA yield from each sample was detected using a NanoDropTM 2000 spectrophotometer (Thermo Fisher Scientific, Wilmington, DE), RNA integrity was confirmed using a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). The gene expression profiling was performed with 25 ng total RNA per sample and all RNA samples were analyzed using the Nanostring nCounter GX Human Immunology v2 kit. The gene expression profiling and data analyses were performed as previously described (30). Briefly, the hybridization reaction was carried out at 65°C overnight by mixing RNA samples with nCounter reporter probes and capture probes in hybridization buffer. After hybridization of the probes with the targets of interest, samples were purified and immobilized on a cartridge and data assessed on the nCounter SPRINT Profiler. The data were then collected with an automated fluorescence microscope and nCounter digital analyzer, and subsequently assessed with the help of the nSolver Analysis Software Version 4.0 (NanoString Technologies). For the data analysis, sample counts were initially normalized by scaling all the values by the ratios of geometric mean (GM) of sample controls to the overall GM of control gene counts across all samples. This was done for both spike-in positive controls and housekeeping genes. The final counts of the mRNA transcripts were used for the further evaluation.
Naïve CD4+ T Cell Differentiation
Naïve CD4+ T cells were isolated as described above. Cells (2 × 105 cells/ml) were treated with DMSO or 1 μM PL and seeded in a 12 well plate coated with anti-CD3/CD28 antibodies. Naïve CD4+ T cells differentiation was induced by adding the following cytokines and antibodies. TH1: IL-12 (2 ng/ml), anti-IL-4 antibody (2 μg/ml); TH2: IL-4 (2.5 ng/ml), anti-IFNγ antibody (2 μg/ml); TH17: IL-6 (10 ng/ml), TGF-β (2.5 ng/ml), IL-1β (10 ng/ml), IL-23 (20 ng/ml), anti-IFNγ (2 μg/ml), and anti-IL-4 antibody (2 μg/ml); Treg: IL-2 (20 U/ml), TGF-β (5 ng/ml), anti-IFNγ (2 μg/ml) and anti-IL-4 antibody (2 μg/ml). After stimulation and differentiation for 6 days, cells were collected and washed twice with FW buffer, then stained with antibodies specific for the surface molecules CXCR3 (CD183), CCR4 (CD194), and CCR6 (CD196) as mentioned above. The TH subtypes were identified according to the surface staining as TH1 cells are CXCR3+CCR4−CCR6−, TH2 cells are CXCR3−CCR4+CCR6− and TH17 cells are CXCR3−CCR4+CCR6+ according to standard flow cytometry procedures (35). The Treg cell population was determined by the expression of transcription factor FOXP3. For the FOXP3 staining, cells (1 × 106 cells/sample) were fixed with 100 μL 1 × True-Nuclear™ fixation buffer for 1 h, followed by three times washing with 200 μL 1 × True-Nuclear™ Perm buffer. Afterwards, cells were resuspended in 50 μL 1 × True-Nuclear™ Perm buffer with anti-FOXP3 (1:20) antibody. After incubation for 30 min at RT, cells were washed three times with 200 μL of 1 × True-Nuclear™ Perm buffer, resuspended in 200 μL PBS and measured with flow cytometry.
For the measurement of cytokine production after 6 days of T cell differentiation, cells were centrifuged, cell free supernatants were collected and immediately aliquoted and stored at −80°C. Secreted cytokines in undiluted cell culture supernatants were detected by ELISA kits specific for IL-17A, IL-17F, and TGF-β as per the manufacturer's instructions.
HIF-1α Detection
Western blot was used to detect HIF-1α. Briefly, T cells (1 × 106 cells/ml) were treated with DMSO or PL as indicated, and activated in anti-CD3/CD28 antibodies coated plates for 24 h. Then, the cells were collected, washed with PBS and total lysate was prepared using PBS with 1 × reducing sample buffer. Proteins were detected by western blotting as described above with the following antibodies: HIF-1α (1:200), Actin (1:1,000) and respective IRDye® conjugated secondary antibodies (1:10,000).
Statistics
Statistical tests were performed with GraphPad Prism 6.0 (GraphPad; San Diego, USA). Two data sets were analyzed using t-test or paired t-test for matched observations. Multiple groups were compared using ANOVA. All data are presented as mean ± standard error of the mean (SEM). P < 0.05 were considered statistically significant.
Results
PL Induces a Prooxidative Milieu in Primary Human T Cells
First, we have determined whether PL is toxic to human T cells. To this end, we prepared freshly isolated human PBTs and assessed T cell viability after treatment with 10 μM PL up to 24 h. Since, it was reported that 1511.9 ng/ml (4.8 μM) of PL could be found in rat plasma after 1 dose of 50 mg/kg PL intraperitoneal injection (36), in our study we considered concentrations of PL up to 10 μM as physiologically relevant. Even the highest PL concentration used in our experiments (10 μM) showed no significant cytotoxicity on primary human T cells (Supplementary Figure 2a). Interestingly, this concentration of PL induced cell death in malignant Jurkat T leukemia cells over the same time period (Supplementary Figure 2b) which is in line with previous studies showing cytotoxic effects of 10 μM PL on myeloid and B cell leukemia cells (37, 38).
PL exhibits potent anticancer activity in multiple cancer types by increasing the cellular ROS levels (39). ROS are also important regulators of T cell activation and function (24, 40, 41). The impact of PL on the redox system of primary human T cells, however, is unknown. We therefore evaluated the intracellular ROS levels in primary human T cells after PL treatment. To this end, the ROS-sensitive probe, CM-H2DCFDA, was used to stain T cells, followed by T cell treatment with 1–10 μM PL for 1 h prior to flow cytometric analysis. Supplementary Figure 3a shows the gating of lymphocytes and the staining with CM-H2DCFDA. PL treatment induced a significant increase of ROS in a concentration-dependent manner (Figure 1A). The intracellular ROS levels in primary human T cells after treatment with 5–10 μM PL were comparable to those observed after treatment with 50 μM H2O2. Similar to primary human T cells, the ROS levels in Jurkat leukemia cells also increased significantly after PL treatment (Figure 1A).
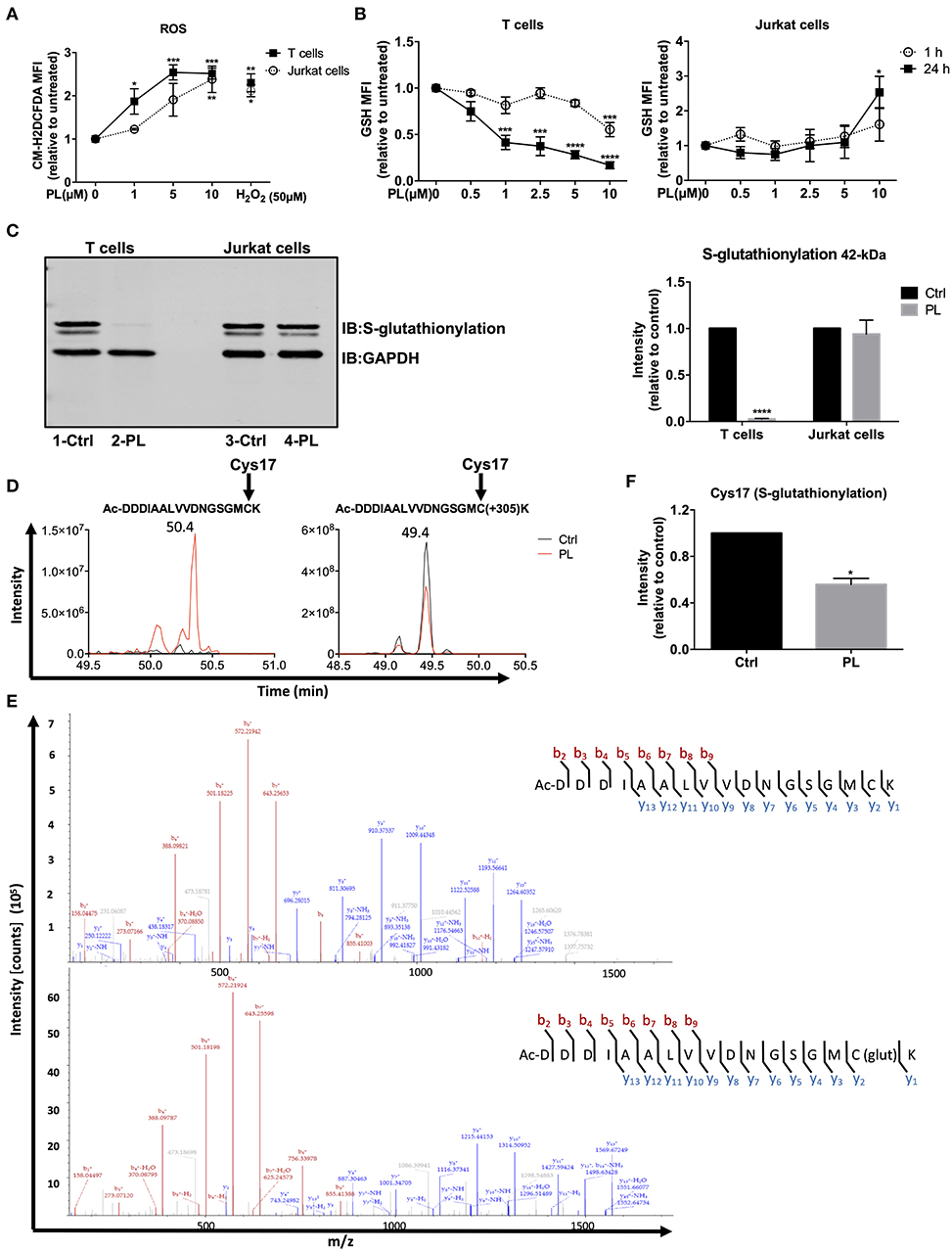
Figure 1. PL induces a prooxidative state in primary human T cells. (A) The intracellular ROS levels were assessed by flow cytometry. Primary human T cells (black squares) and Jurkat cells (open circles) were preloaded with CM-H2DCFDA and, thereafter, treated for 1 h with or without PL as indicated (n = 3; mean; SEM; *p < 0.05, **p < 0.01, ***p < 0.001). (B) Intracellular GSH levels of T cells (left panel) and Jurkat cells (right panel) were assessed by flow cytometry. Cells were treated with or without PL as indicated for 1 h (open circle) or 24 h (black square) and stained with ThiolTrackerTM violet dye for 15 min (n = 3; mean; SEM; *p < 0.05, ***p < 0.001, ****p < 0.0001). (C) Protein S-glutathionylation was detected by western blotting. Representative immunoblot (left) shows the S-glutathionylated 42-kDa protein and GAPDH in control or 5 μM PL-treated T cells (lane 1, 2) and Jurkat cells (lane 3, 4). Bar graph (right) shows the quantitative analysis of three different experiments. S-glutathionylated 42-kDa protein levels were related to the expression levels of GAPDH. Values were normalized to the control, which was set as 1 (n = 3; mean; SEM; ****p < 0.0001). (D) Elution profile of acetylated N-terminal peptide Ac-DDDIAALVVDNGSGMCK of actin in control (black) or PL-treated (red) T cells in its unmodified (left) and glutathionylated (+305) form (right). Shown are the representative extracted ion chromatograms from three biological replicates. (E) MS2 spectrum of unmodified (upper graph) and glutathionylated (lower graph) showing N-terminal acetylated Cys17 identified at 50.4 min and 49.4 min, respectively. (F) Quantification of S-glutathionylated Cys17 under control and PL treatment conditions. Values were normalized to the control, which was set as 1 (n = 3; mean; SEM; *p < 0.05).
The response of cells to prooxidative substances is antagonized by antioxidant systems, primarily by GSH. Therefore, we next determined the intracellular levels of GSH, an important ROS-scavenger, using ThiolTrackerTM violet dye in both primary human T cells and Jurkat cells. Supplementary Figure 3b shows the gating of lymphocytes and the staining with ThiolTrackerTM. After 1 h treatment, 10 μM PL was able to significantly diminish GSH levels, whereas already 1 μM PL was sufficient to significantly decrease the amount of GSH in T cells after 24 h treatment (Figure 1B, left). In contrast, the GSH levels in Jurkat cells were even increased after 24 h treatment with 10 μM PL (Figure 1B, right).
GSH can form mixed disulfides with oxidized cysteine residues in proteins in response to mild oxidative stress, which is named S-glutathionylation. This process is considered as a defense mechanism to protect proteins from irreversible oxidative states (42). Since PL shifted the cellular redox equilibrium toward prooxidative conditions, we next investigated whether PL interfered with the S-glutathionylation of cellular proteins. For this purpose, we treated the cells with 5 μM PL for up to 4 h. Subsequently, cell lysates were loaded on SDS-PAGE and immunostained for S-glutathionylation using a monoclonal anti-GSH antibody. We observed a strong glutathionylation signal of a 42-kDa protein which decreased strongly (deglutathionylation) in T cells treated with PL (Figure 1C, lane 1-Ctrl and 2-PL). Notably, treatment of Jurkat cells with PL did not change the S-glutathionylation of the same protein band (Figure 1C, lane 3-Ctrl and 4-PL). This finding is consistent with the strongly diminished intracellular levels of GSH, the essential material for S-glutathionylation, only in PL-treated T cells but not in Jurkat cells.
The 42-kDa protein was further analyzed by liquid chromatography-tandem mass spectrometry (LC-MS2). LC-MS2 analysis revealed that the highly S-glutathionylated 42-kDa band was beta-actin. To further elaborate this finding, we inspected which cysteines in actin were S-glutathionylated and whether PL treatment alters the relative levels of this modification. To this end, we searched for unmodified, S-glutathionylated (+305), sulfenylated (SOH), sulfinylated (SO2H), and sulfonylated (SO3H) forms of the cysteine-containing peptides. The peptide Ac-DDDIAALVVDNGSGMCK was detected as unmodified, S-glutathionylated (+305), sulfinylated (SO2H, +32) and sulfonylated (SO3H, +48) on Cys17 with MS2 spectra at 50.4, 49.4, 50, and 50 min, respectively (Figure 1D and Supplementary Figures 4a,c). The MS2 spectrum fingerprint for all peptides is comparable. b ions have the same masses and relative intensities in all spectra. Relative intensities of y ions are comparable in all spectra while their masses are (except for y1) 305.0681, 31.990 or 47.986 m/z higher in the spectrum of the S-glutathionylated, sulfinylated or sulfonylated peptide, respectively (Figure 1E and Supplementary Figures 4b,d). These MS2 spectra show that Cys17 is present as unmodified, S-glutathionylated (+305), sulfinylated (SO2H) and sulfonylated (SO3H) residue in the peptide Ac-DDDIAALVVDNGSGMCK.
For relative quantification of the peptides, peak areas were calculated with the Skyline software (32) and non-cysteine-containing peptides were used for normalization in each sample. The unmodified peptide Ac-DDDIAALVVDNGSGMCK was detected in the PL-treated sample, whereas it was absent in the control (Figure 1D, left). Cys17 glutathionylated peptide Ac-DDDIAALVVDNGSGMC(+305)K was detected with intense signals in both control and PL-treated samples (Figure 1D, right). Figure 1F shows that the intensity of the S-glutathionylated peptide (Cys17) decreased significantly upon PL treatment compared to the control sample. Moreover, lower levels of S-glutathionylation on Cys217 upon PL treatment were also found in two experiments, while it was not detected in the third experiment in both control and PL-treated samples (data not shown). Reasonably, sulfinylated (SO2H) and sulfonylated (SO3H) forms of the Cys17 in peptide Ac-DDDIAALVVDNGSGMCK in control and PL-treated sample were detected, and the signal intensities increased for both peptides with PL treatment (Supplementary Figures 4a,c,e). This indicates oxidation of the cysteine to sulfinic or sulfonic acid.
Taken together, PL exerts prooxidative activity on primary human T cells, based on an increase in ROS levels and a decrease in GSH levels. In Jurkat cells, PL also enhanced the intracellular ROS levels, but the levels of GSH remained stable or rather increased. In line with these observations, PL diminished the S-glutathionylation of actin in T cells but not in Jurkat cells.
PL Inhibits Immune Synapse Formation Between APC and T Cells
Since excessive ROS production or prolonged exposure to high ROS levels impair T cell functions (24), we investigated whether PL influences T cell activation and T cell-dependent immune responses.
In a physiological setting, one of the first steps of T cell activation is the formation of a contact zone with an APC and the maturation of the immunological synapse (IS). To investigate this, control or PL-pretreated T cells (1 h or overnight pretreatment with 5 μM PL) were cultured with SEB-loaded Raji cells which served as APCs. First, T cell/APC conjugates were identified by flow cytometry (Figures 2A,B). As expected, in the absence of SEB, only 2.5 ± 0.25% T cell/APC conjugates were found, whereas this number of conjugates increased to 9.4 ± 0.8% in the presence of SEB. No effect on cell couple formation was observed after 1 h preincubation with 5 μM PL (data not shown), but overnight pretreatment with PL significantly decreased the formation of T cell/APC conjugates in the presence of SEB to 3.0 ± 0.79% (Figure 2B). In order to clarify whether this result is due to the prooxidative capacity of PL described above (compare Figure 1), we made use of the antioxidant NAC. As a synthetic precursor of intracellular cysteine and GSH, NAC can lead to replenishment of GSH stores (43). Intriguingly, we observed that NAC (3 mM) could prevent the PL-induced downregulation of T cell/APC conjugates formation (Figure 2B).
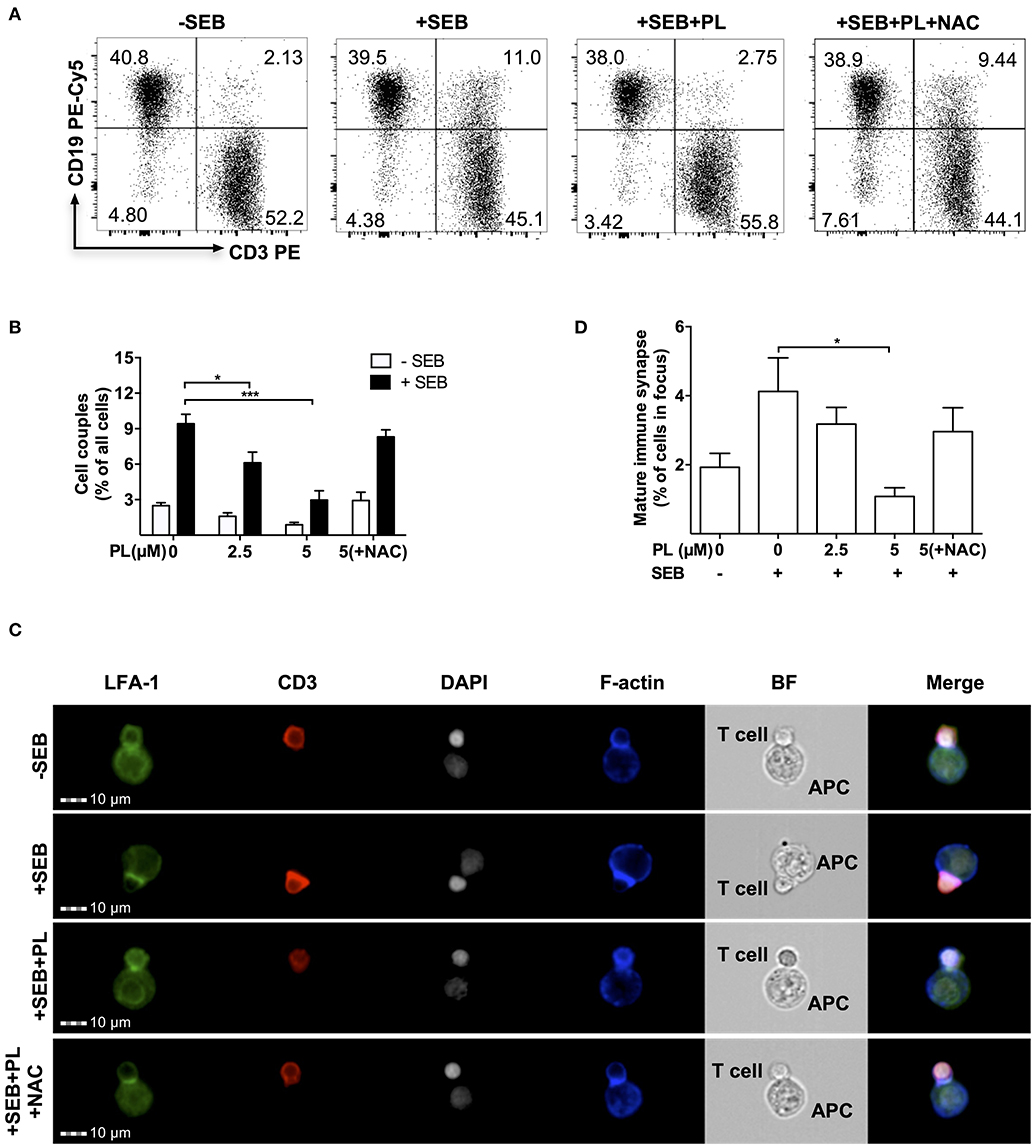
Figure 2. PL interferes with T cell/APC couple formation and immune synapse maturation. (A) Dot plots of T cell/APC couples. Formation of couples was assessed by flow cytometry via CD3 and CD19 staining. T cells were treated overnight without or with 5 μM PL or 5 μM PL +NAC (3 mM) prior to incubation with APCs in the absence or presence of SEB. The dot plots are representative for three experiments. (B) Quantification of the couple formation between T cells and APCs (n = 3; mean; SEM; *p < 0.05, ***p < 0.001). (C) Formation of mature immune synapses was analyzed by multispectral imaging flow cytometry (MIFC). Cells were stained for LFA-1 (green), CD3 (red), nuclei (DAPI, white), and F-actin (blue). BF represents the Bright Field image. The merged images show the digital overlay of all four colors. Images are representative for three experiments. (D) Quantification of mature immune synapses between T cells and APCs (n = 3; mean; SEM; *p < 0.05).
The impact of PL on formation of a mature immune synapse was investigated by multi-spectral imaging flow cytometry (MIFC). By defining regions of interest, MIFC allows the spatial quantification of the accumulation of receptors at the IS. T cell/APC couples were identified according to staining of nuclei with DAPI and detection of CD3 expression in T cells. The mature IS was then defined by accumulation of LFA-1 (green) and CD3 (red) in the T cell/APC contact area. As expected, while most T cells did not show LFA-1 and CD3 enrichment in the contact area without SEB treatment, a clear receptor enrichment -hence IS maturation- was observed in the presence of SEB. In samples pretreated overnight with PL, the enrichment of LFA-1 and CD3 in the contact zone was very low but it occurred normally when T cells were preincubated with PL and NAC (Figures 2C,D). Together, overnight pretreatment of T cells with PL impairs the formation of a mature IS between T cells and APCs, most likely due to depletion of GSH.
PL Inhibits T Cell Calcium Signaling
IS formation initiates and controls Ca2+ signals in T cells (44). We asked whether PL also influences the anti-TCR/CD3-induced Ca2+ signaling which occurs independently of T cell/APC conjugate formation. Figure 3A shows the Ca2+ flux signal monitored by flow cytometry. The parameter AUC (area under curve) was used to evaluate the Ca2+ signal in our experiments, which refers to the total amount of cytosolic Ca2+ during the whole recorded period of Ca2+ flux. These data revealed that the Ca2+ signal in control T cells increased steeply after CD3 cross-linking while overnight pretreatment of T cells with 5 μM PL inhibited the TCR/CD3-mediated Ca2+ flux. This inhibition was completely prevented by NAC (Figures 3A,B). Together, these results imply that PL also interferes with Ca2+ signaling in T cells by depleting GSH.
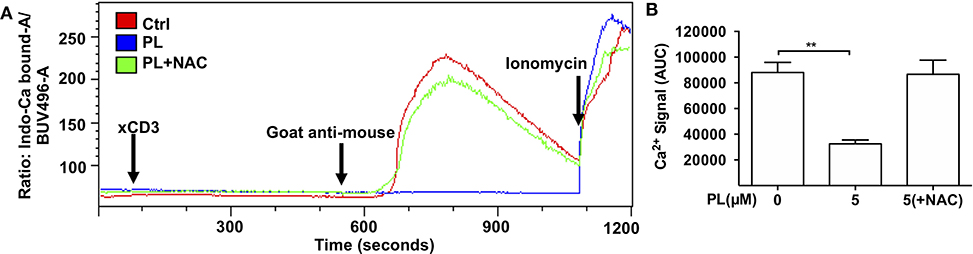
Figure 3. PL inhibits TCR-mediated Ca2+ flux. (A) Ca2+ flux in control (red), 5 μM PL (blue) or 5 μM PL+NAC (green) pretreated (overnight) T cells was measured using flow cytometry. Cells were stimulated by adding CD3 antibodies (xCD3) followed by crosslinking with goat anti-mouse antibodies as indicated. Ionomycin was added as a positive control at the end of each measurement. The depicted graphs are representative for three independent experiments. (B) Quantification of Ca2+ signal as the area under the curve (AUC) (n = 3; mean; SEM; **p < 0.01).
PL Inhibits Costimulation-Induced CD69 and CD25 Expression in Primary Human T Cells
To investigate whether pretreatment with PL for 30 mins interferes with later T cell activation events, expression of the T cell activation markers CD69 and CD25 in primary human T cells was assessed after costimulation with anti-CD3/CD28 antibodies or isotype control antibodies. The flow cytometry analysis procedure is shown in Supplementary Figure 5a. T cells seeded in isotype control antibody-coated plates are similar to resting T cells according to the CD69 and CD25 surface staining (Supplementary Figure 5b). After costimulation with anti-CD3/CD28 antibodies for 24 h, CD69 and CD25 were highly expressed on the T cell surface in the absence of PL, while in the presence of PL, their expression was suppressed in a dose-dependent manner (Figure 4A and Supplementary Figure 5c). Intriguingly, while the MFI value of CD69 was decreased after PL treatment, the percentage of CD69-positive cells was not altered under the same conditions. Moreover, not only the surface expression, but also the total amount of CD69 and CD25 within the cells was decreased upon PL treatment (Supplementary Figure 5d). This indicates that PL inhibited the expression of both proteins rather than impairing the receptor transport to the surface. Again, treatment with NAC prevented the PL-induced downregulation of CD69 and CD25 (Figure 4A).
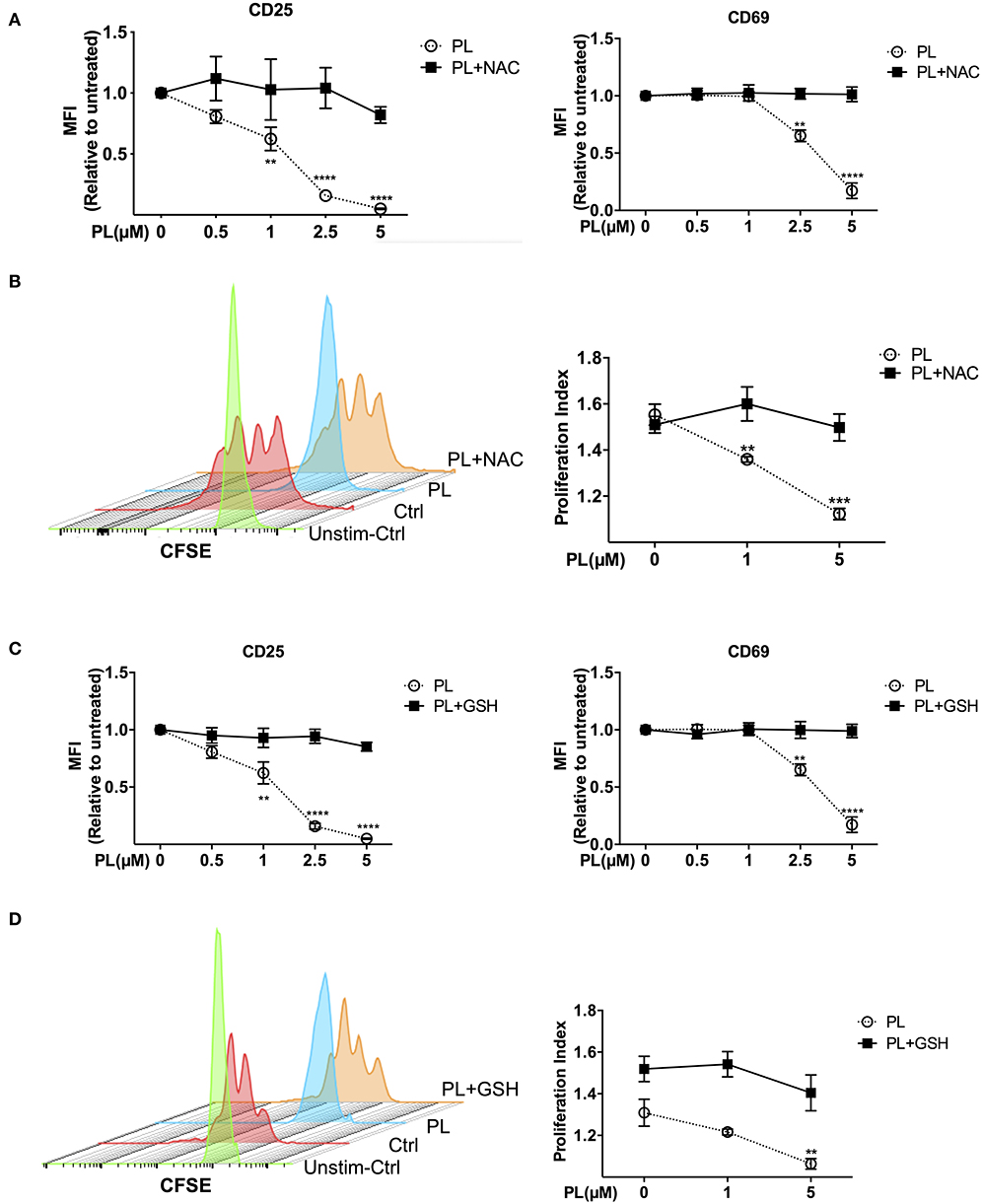
Figure 4. Immunosuppressive effects of PL were abrogated by thiol-containing antioxidants. (A,C) Expression of CD25 (left) and CD69 (right) in T cells was analyzed by flow cytometry. T cells in the presence/absence of PL and (A) NAC or (C) GSH were costimulated with anti-CD3/CD28 antibodies for 24 h. Shown are the MFI ratios of PL-treated to untreated samples. (B,D) T cell proliferation was detected via staining of CFSE. T cells were loaded with CFSE, thereafter costimulated with anti-CD3/CD28 antibodies in the absence/presence of PL without or with addition of exogenous (B) NAC or (D) GSH. CFSE signal was measured after 72 h of costimulation by flow cytometry. Shown are representative histograms (left) and the proliferation index (right) (n = 3; mean; SEM; **p < 0.01, ***p < 0.001, ****p < 0.0001).
PL Inhibits Costimulation-Induced Proliferation of Primary Human T Cells
T cell proliferation over time can be monitored via labeling T cells with CFSE. To this end, we pretreated CFSE-labeled T cells with or without PL for 30 mins, and determined T cell proliferation by flow cytometry after 72 h of costimulation. As shown in Figure 4B, compared to untreated control cells, PL treatment significantly inhibited T cell proliferation. Again, NAC abolished the suppressive effect of PL on T cell proliferation. In order to determine whether these immunosuppressive effects are due to the depleted GSH stores, we repeated the experiments with exogenously added GSH. Indeed, inhibition of the expression of CD69 and CD25 (Figure 4C), and T cell proliferation (Figure 4D) were completely prevented in the presence of GSH. All together, these findings demonstrate that PL acts as an immunosuppressive agent on primary human T cells through depletion of intracellular GSH. PL induced immunosuppression can be prevented by the antioxidants GSH or NAC.
PL Suppresses Expression of Genes Important for T Cell Activation and Proliferation
Next, we took advantage of the NanoString nCounter® Technology and the GX Human Immunology v2 panel to gain an unbiased view on the expression of differential mRNA profiles. The genes evaluated were related to leukocyte functions including major classes of cytokines and their receptors. The final counts of the mRNA transcripts were visualized by hierarchical clustering (Figure 5A). For these experiments, primary human T cells were again pretreated for 30 min with PL or solvent control and then costimulated with anti-CD3/CD28 antibodies for 4 h. Upon PL treatment, 81 genes were significantly downregulated and 27 genes were significantly upregulated (fold change >2, p < 0.05) compared to the control. Notably, consistent with our finding of diminished CD25 (IL-2RA) at the protein level (compare Figure 4A), the mRNA level of IL-2RA was also substantially decreased by PL treatment (Figure 5B, red dot). Intriguingly, other genes that are important for T cell activation and proliferation were also inhibited upon PL treatment, e.g., CD2 and the TNF receptor superfamily member 9 [TNFRSF9, a TNF receptor that possesses costimulatory activity for activated T cells (45)] and JAK2, JAK3, and STAT5A [an essential mediator of IL-2 signaling in T cells (46)] (Figure 5B, red dots), as well as the proinflammatory cytokines IFNγ and TNFα (Figure 5B, green dots). Moreover, the expression of the key proinflammatory cytokine IL-1B (47), the chemokine (C-C motif) ligands (CCL) 3 and CCL4, which can be produced by T cells following nonspecific activation or T cell receptor engagement (48) were inhibited by PL treatment (Figure 5B, green dots). In addition, PL was able to suppress the expression of cytolytic enzymes like Granzyme (Grz) A, GrzB and Perforin, which are critical mediators of anti-viral and anti-tumor immunity (Figure 5B, blue dots). These results, together with the aforementioned findings, demonstrate that PL is an inhibitor for T cell activation and possesses anti-inflammatory properties. Furthermore, our data imply that PL is a potential suppressor of T cell-mediated cytotoxicity.
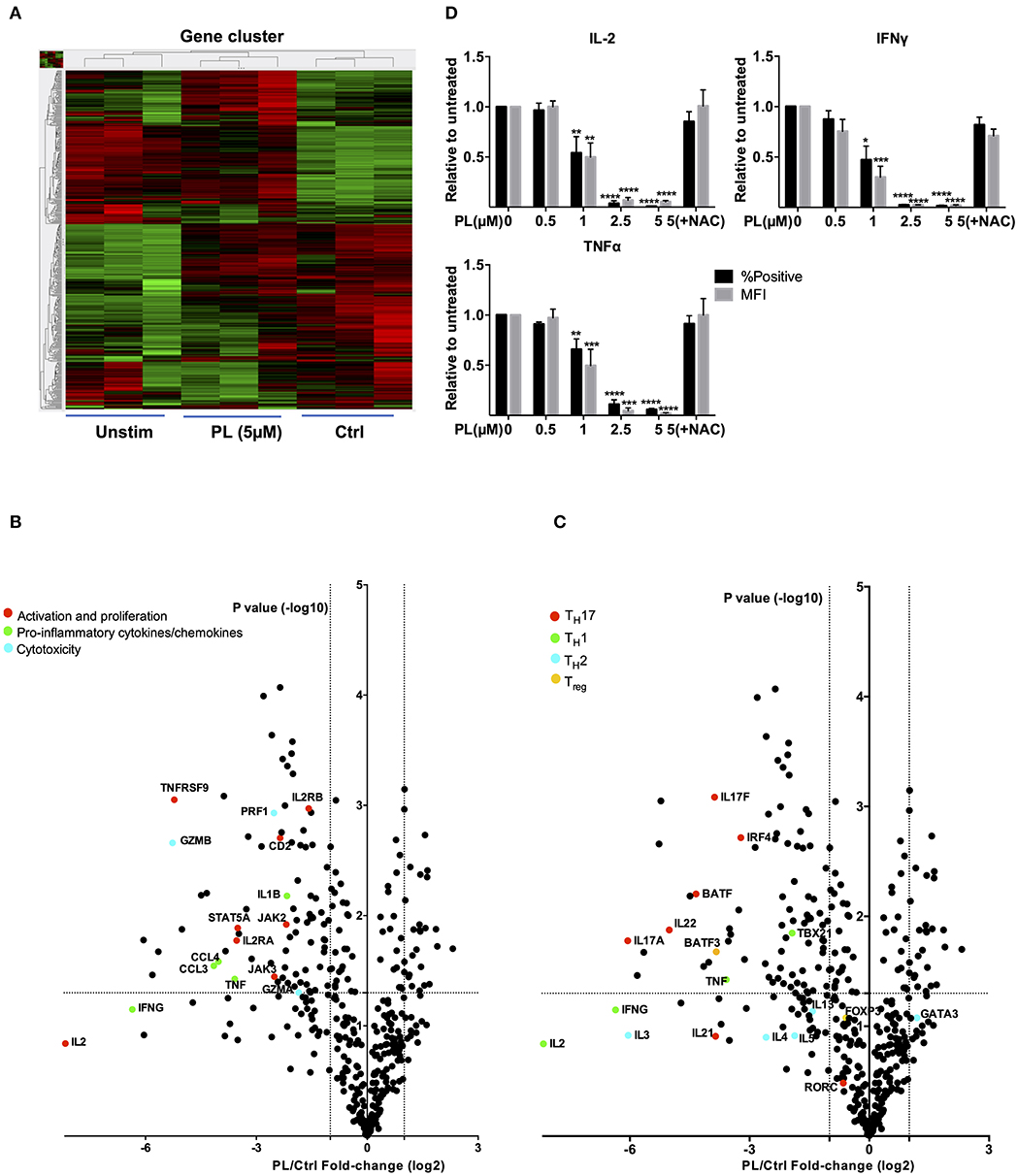
Figure 5. Gene expression analysis (nCounter®) and intracellular cytokine staining. (A) Hierarchical clustering of differentially expressed mRNAs in T cells which were unstimulated (Unstim), anti-CD3/CD28 antibodies stimulated with PL (PL 5 μM) or without PL (Ctrl) treatment. Each column represents an individual sample (n = 3). Relative change is indicated by the color scale (red: high; green: low). (B,C) Volcano plots show differentially expressed genes (B) important for T cell activation and cytokines and (C) related to TH1, TH2, Treg, and TH17 cells. Log2 transformed fold-change in expression (PL vs. Control) on the x-axis is plotted against significance [–log10 (p-value)] on the y-axis. The horizontal and vertical dashed lines indicate cutoff for significance p < 0.05 (−log10 p-value >1.3) and for fold-change ≥2/ ≤ −2, respectively. (D) Protein levels of IFNγ, TNFα and IL-2 were detected by intracellular cytokine staining. Shown are ratios of PL-treated cells to the control with regard to the % positive cells (black bars) and MFI (gray bars) (n = 3; mean; SEM; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Intriguingly, the expression of TH17-specific cytokines, e.g., IL-17A, IL-17F, and IL-22 as well as expression of B cell activating transcription factor (BATF) (49) and interferon regulatory factor 4 (IRF4) (50), which together initiate a transcriptional program for TH17 cell development (51), were inhibited by PL treatment (Figure 5C, red dots). Expression of RORC gene was, however, unaffected. We have then further checked genes associated with other T cell subpopulations, i.e., TH1, TH2, and Treg cells. In addition to TNFα, the expression of IL-2 and the TH1 cytokine IFNγ were inhibited by PL although the differences to untreated control cells were not statistically significant. PL significantly inhibited the TH1-specific transcription factor TBX21 (Figure 5C, green dots). The expression of TH2-associated cytokines, e.g., IL-3, IL-4, IL-5, and IL-13 was in part suppressed by PL, but not significantly and to a lower extent compared to TH1 cytokines (Figure 5C, sky blue dots). The TH2-specific transcription factor GATA3 was even increased by tendency upon PL treatment (Figure 5C, sky blue dot). Following costimulation for 4 h, the Treg-related transcription factor FOXP3 remained unaffected by PL, but BATF3, a transcription factor that prevents the differentiation of Treg cells (52), was inhibited significantly by PL (Figure 5C, yellow dots). These results together provided first hints that PL acts differently on various T cell populations: At the gene expression level, TH17-associated cytokines were more sensitive to PL treatment compared to TH1 and TH2 cytokines (Figure 5C).
To test these findings at the protein level, the cellular content of IL-2, IFNγ, and TNFα was determined by intracellular staining. Consistent with our findings of diminished mRNA levels of IL-2, IFNγ, and TNFα (Figures 5B,C), also the intracellular protein levels of these cytokines were substantially decreased by PL treatment (Figure 5D). Note that FMO samples showed that there is no nonspecific staining for these cytokines (Supplementary Figure 6a). Also, at the protein level, the PL-mediated decrease of IL-2, IFNγ, and TNFα was prevented when the cells were coincubated with NAC, which is consistent with the hypothesis that depletion of GSH plays a crucial role in PL-mediated T cell immunomodulation. Of note, NAC alone did not upregulate expression of IL-2, IFNγ, and TNFα (Supplementary Figure 6b).
PL Inhibits the Differentiation of Naïve CD4+ T Cells Into TH17 Cells While Promoting the Development of Treg Cells
The results from the nCounter gene analysis suggested that PL differentially affects T cell subpopulations. To further investigate this finding, we have next measured the effect of PL on the differentiation of naïve CD4+ T cells into TH1, TH2, TH17, and Treg cells. Naïve CD4+ T cells were cultured under appropriate polarizing differentiation conditions (+cytokines) in the absence or presence of 1 μM PL (+cytokines+PL) as described in Materials and Methods. After 6 days, the differentiation into TH1, TH2, and TH17 cells was assessed by surface marker staining (Figure 6A and supplementary Figure 7a) and cytokine detection (Figures 6C–E). Nuclear FOXP3 staining was used for identifying Treg cells (Figure 6B and Supplementary Figure 7b). 7-AAD staining confirmed that cells did not die after 6 days of 1 μM PL treatment (data not shown). Quantification of the T cell subpopulations (Figures 6A,B) confirmed that the percentage of TH1, TH2, TH17, and Treg cells in the respective polarizing conditions (+cytokines) was much higher than with anti-CD3/28 costimulation alone (none), which proved that the polarizing conditions were optimal. PL inhibited the differentiation of naïve CD4+ T cells into TH17 cells while it had no clear effect on the differentiation into TH1 and TH2 cells, as assessed by staining of the respective T cell surface markers (Figure 6A). Interestingly, FOXP3 staining showed that PL significantly promoted the differentiation of naïve CD4+ T cells into Treg cells (Figure 6B). This notion holds true not only for the proportion of FOXP3+ Treg cells, but also for the absolute number of FOXP3+ Treg cells. Importantly, NAC treatment prevented the PL mediated decrease in the TH17 population and the PL mediated increase in the Treg population (+cytokines+PL+NAC) (Figures 6A,B).
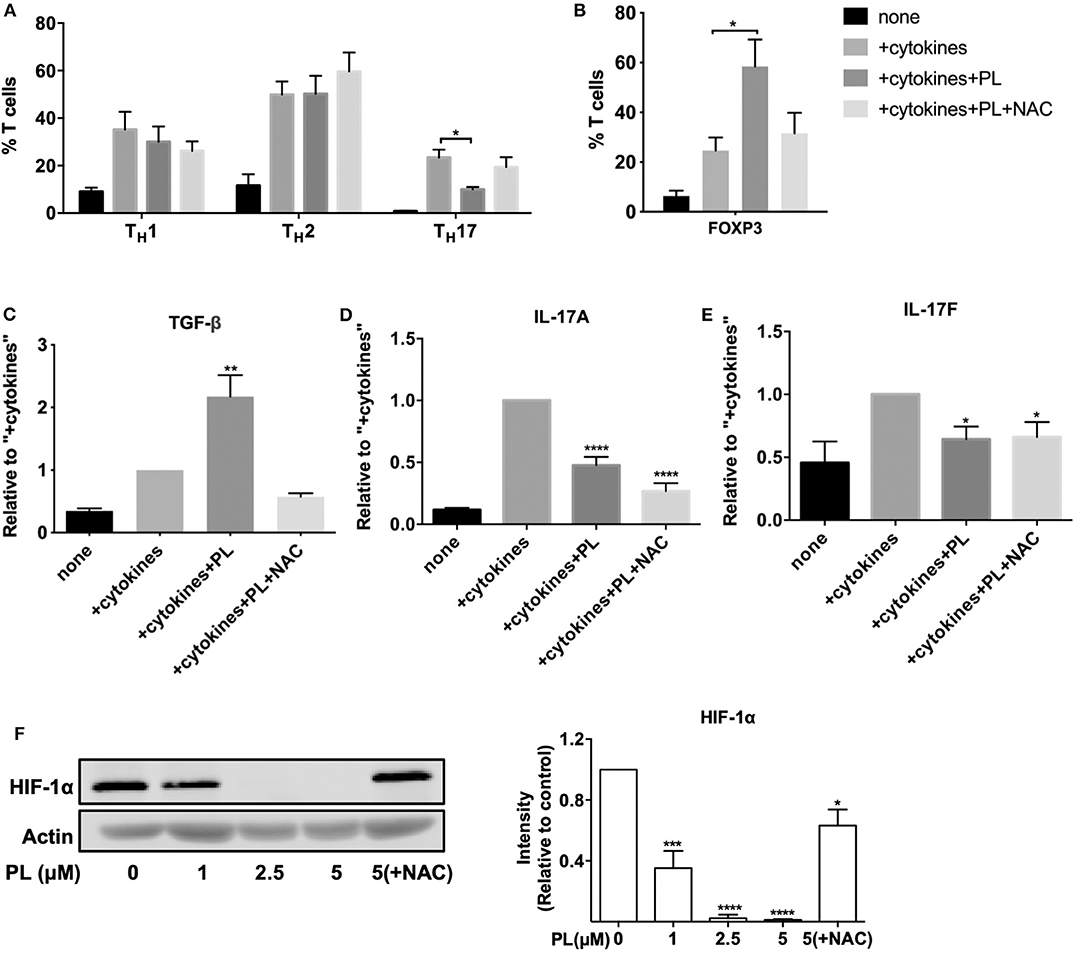
Figure 6. PL inhibits the differentiation of naïve CD4+ T cells into TH17 cells while promoting the development of Treg cells. (A–C) Naïve CD4+ T cells were costimulated by anti-CD3/CD28 antibodies either alone (none) or in the presence of TH1, TH17, or TH2 polarizing cytokines without (+cytokines) or with 1 μM PL treatment (+cytokines +PL), or with both PL and NAC treatment (+cytokines +PL +NAC). (A) The differentiation of TH1, TH2, and TH17 cells was assessed by surface staining of CD183, CD194, and CD196 and was analyzed by flow cytometry. Shown are the percentage of the respective TH subset in the total T cell population (n = 5; mean; SEM; *p < 0.05). (B) Treg cells were identified by nuclear staining of FOXP3 (n = 3; mean; SEM; *p < 0.05). (C) TGF-β levels after Treg polarizing differentiation conditions, (D) IL-17A and (E) IL-17F levels after TH17 polarizing differentiation conditions were detected by ELISA (n = 3; mean; SEM; *p < 0.05, **p < 0.01, ****p < 0.0001). (F) HIF-1α was detected by western blot (left). T cells were pretreated with or without PL and NAC as indicated for 30 min and seeded into anti-CD3/CD28-coated plates for 24 h. Cell lysates were loaded on SDS-PAGE gel and immunoblotted for HIF-1α and actin. The expression levels of HIF-1α were normalized to the expression levels of actin. The bar graph (right) shows the quantitative analysis of three different experiments (n = 3; mean; SEM; *p < 0.05, ***p < 0.001, ****p < 0.0001).
To get a more detailed picture about the consequences of PL for the differentiation of naïve CD4+ T cells into TH17 and Treg cells, we analyzed the levels of the anti-inflammatory Treg cell cytokine TGF-β, and the proinflammatory TH17 cell cytokines IL-17A and IL-17F. Indeed, in line with the increased proportion of FOXP3+ cells (Figure 6B), under Treg polarizing differentiation conditions the concentration of TGF-β also increased significantly upon treatment with PL (+cytokines+PL) compared to the control without PL (+cytokines). And again, NAC prevented this effect (Figure 6C). As expected, PL treatment (+cytokines+PL) significantly diminished the levels of the proinflammatory TH17 cell cytokines IL-17A and IL-17F (Figures 6D,E). Surprisingly, these cytokine levels remained unaffected by NAC.
It was reported that HIF-1α, a subunit of the heterodimeric transcription factor HIF-1, enhances TH17 development and attenuates Treg development (53). Intriguingly, we could show by western blot analysis that PL strongly inhibits the expression of HIF-1α in a dose-dependent manner starting already at a concentration of 1 μM PL. This inhibition was prevented by NAC treatment (Figure 6F). Thus, the diminished expression of HIF-1α may provide one molecular explanation for the PL-induced downregulation of TH17-related genes. Taken together, PL affects T cell subpopulations differently. It inhibits the differentiation of naïve CD4+ T cells into proinflammatory TH17 cells, while promoting the differentiation into Treg cells.
PL Shows Immunosuppressive Effects on Whole Blood Lymphocytes Derived From Patients With Autoimmune Diseases
Given the newly discovered immunomodulatory role of PL on primary human T cells, it was tempting to speculate that PL could be beneficial for patients with autoimmune diseases. Therefore, we analyzed the impact of PL on whole blood samples from patients with RA and systemic lupus erythematosus (SLE), which are prototypical autoimmune rheumatic diseases (54), as well as from patients with TNF receptor-associated periodic syndrome (TRAPS), a very rare autoinflammatory disease. For these analyses, 100 μl of heparin blood were pretreated with 5 μM PL and activated with SEB overnight. Erythrocytes were then lysed and leukocytes were permeabilized, stained with fluorescently-labeled antibodies specific for CD3, CD69, CD25, IFNγ, and IL-2 and assessed by flow cytometry. In the presence of PL, the expression of the T cell activation markers CD25 (Figure 7A, and Supplementary Figure 8) and CD69 (Figure 7B, and Supplementary Figure 8) after SEB stimulation was significantly downregulated in the lymphocyte population (CD3+). Consistently, a PL-mediated inhibition was also observed on the production of IL-2 (Figure 7C) and the proinflammatory cytokine IFNγ (Figure 7D). Taken together, this experiment confirmed the immunosuppressive activity of PL on human T cells in clinically relevant samples. This implies that PL might be a promising substance for the treatment of autoimmune diseases.
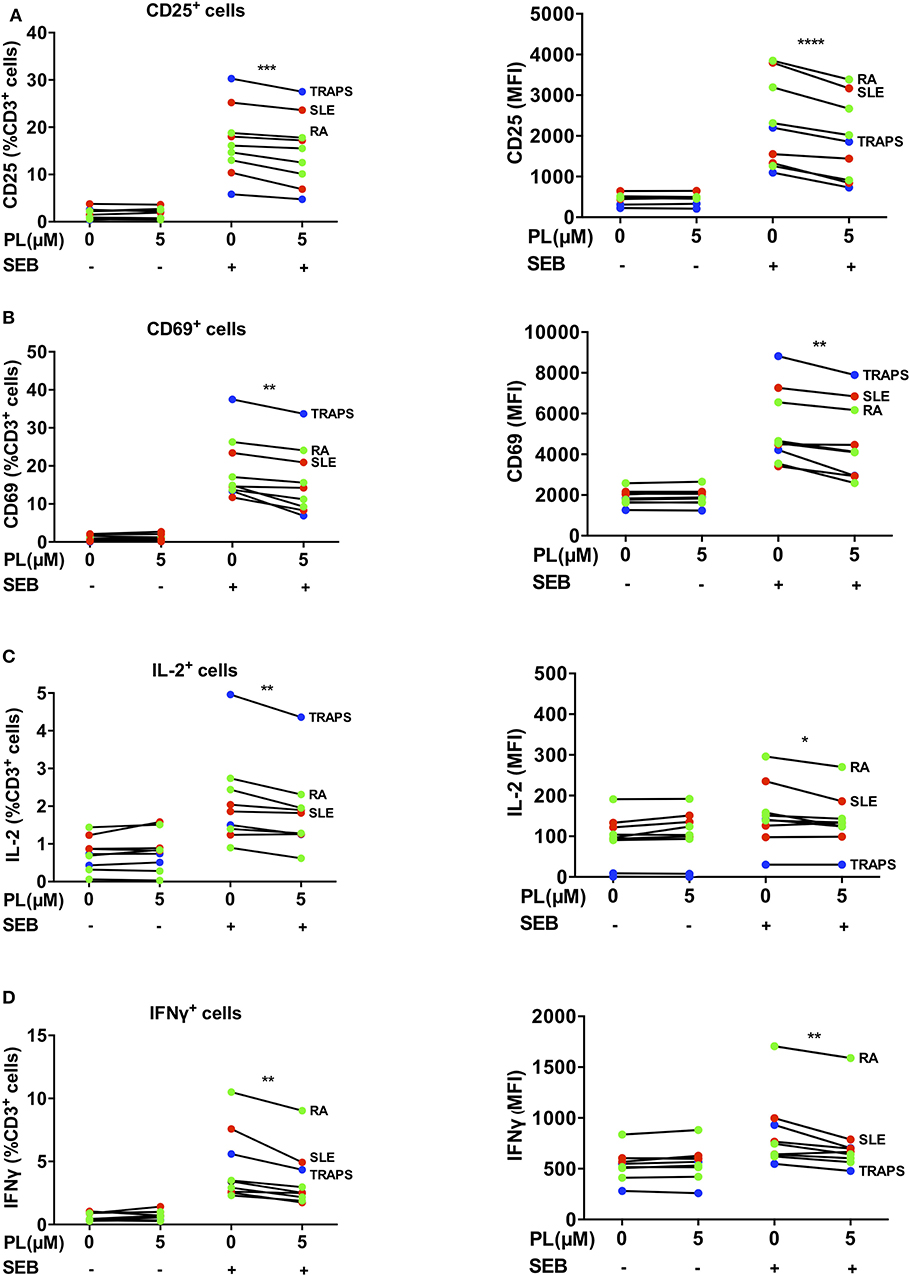
Figure 7. PL downregulates CD69, CD25, IL-2 and IFNγ in lymphocytes of patient-derived whole blood samples. (A–D) Whole blood samples from patients with RA (green), SLE (red) and TRAPS (blue) were treated with or without PL and activated with SEB overnight followed by erythrocytes lysis and subsequent immunofluorescence staining and flow cytometry. The CD3 positive cells within the white blood cells were analyzed for expression of (A) CD25, (B) CD69, (C) IL-2, and (D) IFNγ. Shown are percent positive T cells (left) and MFI (right). Statistical analysis was conducted using paired t-test (n = 8; mean; SEM; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Discussion
PL has been of interest as a potential anticancer drug, while its impact on primary human T cells, which are responsible for anti-tumor immune responses, remained unknown. We therefore examined in our study whether PL interferes with the function and differentiation of primary human T cells. While PL decreased ROS in mouse DCs (19) and in human TNFα stimulated FLS of RA patients (23), we have found that PL induces a prooxidative state in primary human PBTs. This manifested as an increased amount of ROS and largely depleted intracellular GSH pools. Moreover, we have revealed here for the first time that PL inhibits the activation and the proliferation of primary human PBTs. Even more important, PL is able to attenuate the development of human proinflammatory TH17 cells and to enhance the development of Treg. These data uncover a potential of PL to act as a cell type-specific immunomodulator in human and suggest that PL might be a promising substance for treating autoimmune diseases.
The effects of PL on T cell functions can at least partially be explained by the depletion of GSH, since a precursor for GSH biosynthesis, namely NAC, or exogenously added GSH were able to restore T cell functions in the presence of PL. Maintaining optimal GSH levels in T cells is critical for regulating the redox state of proteins, thus keeping the cells in a normal functional state. Depletion of GSH could potentially interfere with the S-glutathionylation, thereby the oxidation state and function of various proteins. Indeed, analysis of protein S-glutathionylation by western blotting and mass spectrometry demonstrated that PL decreased S-glutathionylation of beta-actin. In this context, we have shown that in human T cells Cys17 and Cys217 were highly glutathionylated in the absence of PL. So far, only S-glutathionylation on Cys217 of actin had been described (55). S-glutathionylation on Cys17 of actin is shown for the first time in this study. S-glutathionylation of proteins can change their function in several ways. Intriguingly, we found that when Cys17 was not glutathionylated in PL treated T cells, it existed in higher oxidation states, i.e., sulfinylated or sulfonylated states. This is in line with the notion that GSH protects cysteines from irreversible oxidation (42). Since diminished GSH and diminished protein S-glutathionylation resulting in hyperoxidation of proteins can alter the function of many proteins, these findings provide a potential general mechanistical explanation for the loss of cellular functions upon PL treatment.
While high levels of ROS lead to irreversible protein oxidation, thus loss of cellular function (24), low levels of ROS with readily reversible protein oxidation even promote T cell functions (40). The sensitivity of different proteins and T cell subsets to oxidative stress varies (56). In this context, different T cell subsets may react differentially to the same level of oxidative stress. Immune cell-specific gene expression analysis revealed that the most dampened genes upon PL treatment were TH17 cell-related, followed by TH1-related genes. Importantly, while PL impaired the development of TH17 cells, at the same time it increased the development of Treg cells. This is consistent with the finding that ROS limit TH17 cell differentiation (27) but increase Treg cell differentiation (57). HIF-1α is another key factor that controls the TH17/Treg balance. Lack of HIF-1α in mice resulted in enhanced Treg development but diminished TH17 development and protected the mice from autoimmune neuroinflammation (53, 58). In this context, we found that in human T cells PL inhibited the expression of HIF-1α in a dose-dependent way. This effect was in turn prevented by NAC treatment. Thus, inhibition of HIF-1α may offer an additional explanation for the altered TH17/Treg balance under PL treatment.
TH1 and TH17 cells are involved in the progression of many autoimmune diseases (59, 60). Due to the inhibitory effect on TH1 and TH17 cells, PL could be helpful for the treatment of these diseases. In line with this assumption, we showed that also in T cells from patients with autoimmune diseases, PL led to reduced expression of the T cell activation markers CD69 and CD25 and diminished production of IFNγ and IL-2. These results are in line with previous findings, which showed that PL inhibits TH17 cells in mouse CIA models and reduces the activation of human FLS in RA patients (20). Yang et al. found that RA T cells with diminished ROS production are spontaneously biased to differentiate into IFNγ and IL-17-producing proinflammatory T cells, which play a central role for disease progression (61). Thus, strategies that upregulate ROS levels in RA T cells and rebalance the ROS signaling systems might be promising for therapeutic purposes. This discovery strengthens the assumption that PL might be a promising agent for treating autoimmune diseases such as RA, SLE or EAE. However, one hindrance in the application of PL is its low water solubility. Therefore, water soluble derivates of PL that possess similar biological functions are currently under investigation (62, 63).
Also, of high clinical importance may be our second finding: PL differentially affects malignant Jurkat T leukemia cells (increase of GSH and tumor cell killing) and primary T cells (decrease of GSH and immunosuppression). This may be explained by the fact that tumor cells usually contain a higher antioxidant capacity than untransformed cells (e.g., Trx1 and GSH systems). Thereby, they can keep their redox balance more stable. For example, the GSH synthesis in tumor cells can be increased significantly during oxidative stress by upregulating the cystine/glutamate antiporter SLC7A11 and glutamate cysteine ligase modifier subunit (GCLM). Aside from the biosynthesis, tumor cells can also regenerate GSH by upregulating the production of nicotinamide adenine dinucleotide phosphate (NADPH) (64). A functional immune response plays a decisive role for the outcome of cancer. Therefore, when discussing the efficacy of PL against cancer, potential T cell immunosuppression by PL should be considered. Given that PL has a prooxidative effect on T cells, and that T cells are more susceptible to ROS than tumor cells, it is likely that the immunosuppressive effects of PL are more pronounced than the cytotoxic effects on tumor cells. This may be particularly important if cancer patients receive T cell-based immunotherapies (e.g., CAR T cells or checkpoint inhibitors). The sensitivity of T cells to low concentrations of PL further aggravates this situation, as the publications on anticancer effects of PL often show significant inhibition only with concentrations of 5 μM PL or higher (2, 5). In the future, it should also be investigated whether PL has a differential impact on other immune cell types and whether this involves similar molecular mechanisms as observed in T cells.
In summary, we have shown for the first time that PL differentially influences the fate of human primary T cells. Moreover, we have uncovered a molecular mechanism by which PL controls human T cell activation: PL induces a prooxidative milieu in primary human T cells by increasing ROS and depleting GSH. Thereby, it inhibits costimulation-induced T cell activation and proliferation. Importantly, PL diminishes TH17 differentiation while it enhances Treg differentiation potentially by inhibiting HIF-1α. Furthermore, in T cells from patients with different autoimmune diseases PL inhibits the expression of the T cell activation markers CD69 and CD25 as well as production of IFNγ and IL-2. Together, these data suggest that PL may provide a new therapeutic option for chronic TH17-related inflammatory diseases. Simultaneously, it should be considered that during immunotherapies of cancer (e.g., CAR T cells or checkpoint inhibitors), in which T cell-mediated anti-tumor responses play a decisive role for the outcome of the disease, a treatment with PL may be harmful.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee of the Heidelberg University. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
JL and YS: conceptualization. JL, JZ, BJ, EB, BN, and GW: methodology. JL, JZ, BJ, EB, CO, RB, and TR: investigation. YS and NB: resources. JL, JZ, and YS: drafting of the article. JL, EB, KH, and YS: critical revision of the article. YS: supervision and funding acquisition. All authors have approved the final version of the manuscript.
Funding
This study was supported by the Ministry of Baden-Württemberg, Germany, to YS (AZKIM, www.azkim.de); JL was financially supported by the Chinese Scholarship Council (No. 201408370079).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Ralph Röth for his support in the nCounter analysis, and Jan-Christoph Schumacher for the organization of the clinical samples.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.01172/full#supplementary-material
References
1. Bezerra DP, Pessoa C, de Moraes MO, Saker-Neto N, Silveira ER, Costa-Lotufo LV. overview of the therapeutic potential of piplartine (piperlongumine). Eur J Pharm Sci. (2013) 48:453–63. doi: 10.1016/j.ejps.2012.12.003
2. Golovine KV, Makhov PB, Teper E, Kutikov A, Canter D, Uzzo RG, et al. Piperlongumine induces rapid depletion of the androgen receptor in human prostate cancer cells. Prostate. (2013) 73:23–30. doi: 10.1002/pros.22535
3. Liu JM, Pan F, Li L, Liu QR, Chen Y, Xiong XX, et al. Piperlongumine selectively kills glioblastoma multiforme cells via reactive oxygen species accumulation dependent JNK and p38 activation. Biochem Res Commun. (2013) 437:87–93. doi: 10.1016/j.bbrc.2013.06.042
4. Wang F, Mao Y, You Q, Hua D, Cai D. Piperlongumine induces apoptosis and autophagy in human lung cancer cells through inhibition of PI3K/Akt/mTOR pathway. Int J Immunopathol Pharmacol. (2015) 28:362–73. doi: 10.1177/0394632015598849
5. Lee HN, Jin HO, Park JA, Kim JH, Kim JY, Kim B, et al. Heme oxygenase-1 determines the differential response of breast cancer and normal cells to piperlongumine. Mol Cells. (2015) 38:327–35. doi: 10.14348/molcells.2015.2235
6. Dhillon H, Chikara S, Reindl KM. Piperlongumine induces pancreatic cancer cell death by enhancing reactive oxygen species and DNA damage. Toxicol Rep. (2014) 1:309–18. doi: 10.1016/j.toxrep.2014.05.011
7. Bharadwaj U, Eckols TK, Kolosov M, Kasembeli MM, Adam A, Torres D, et al. Drug-repositioning screening identified piperlongumine as a direct STAT3 inhibitor with potent activity against breast cancer. Oncogene. (2015) 34:1341–53. doi: 10.1038/onc.2014.72
8. de Lima Moreira F, Habenschus MD, Barth T, Marques LM, Pilon AC, da Silva Bolzani V, et al. Metabolic profile and safety of piperlongumine. Sci Rep. (2016) 6:33646. doi: 10.1038/srep33646
9. Jin H-O, Y-Lee H, J-Park A, H-Lee N, J-Kim H, Kim J-Y, et al. Piperlongumine induces cell death through ROS-mediated CHOP activation and potentiates TRAIL-induced cell death in breast cancer cells. J Cancer Res Clin Oncol. (2014) 140:2039–46. doi: 10.1007/s00432-014-1777-1
10. Chen W, Lian W, Yuan Y, Li M. The synergistic effects of oxaliplatin and piperlongumine on colorectal cancer are mediated by oxidative stress. Cell Death Dis. (2019) 10:1–12. doi: 10.1038/s41419-019-1824-6
11. Ginzburg S, Golovine KV, Makhov PB, Uzzo RG, Kutikov A, Kolenko VM. Piperlongumine inhibits NF-kappaB activity and attenuates aggressive growth characteristics of prostate cancer cells. Prostate. (2014) 74:177–86. doi: 10.1002/pros.22739
12. Han JG, Gupta SC, Prasad S, Aggarwal BB. Piperlongumine chemosensitizes tumor cells through interaction with cysteine 179 of IκBα kinase, leading to suppression of NF-κB-regulated gene products. Mol Cancer Ther. (2014) 13:2422–35. doi: 10.1158/1535-7163.MCT-14-0171
13. Zheng J, Son DJ, Gu SM, Woo JR, Ham YW, Lee HP, et al. Piperlongumine inhibits lung tumor growth via inhibition of nuclear factor kappa B signaling pathway. Sci Rep. (2016) 6:26357. doi: 10.1038/srep26357
14. Liu QR, Liu JM, Chen Y, Xie XQ, Xiong XX, Qiu XY, et al. Piperlongumine inhibits migration of glioblastoma cells via activation of ROS-dependent p38 and JNK signaling pathways. Oxid Med Cell Longev. (2014) 2014:653732. doi: 10.1155/2014/653732
15. Zou P, Xia Y, Ji J, Chen W, Zhang J, Chen X, et al. Piperlongumine as a direct TrxR1 inhibitor with suppressive activity against gastric cancer. Cancer Lett. (2016) 375:114–26. doi: 10.1016/j.canlet.2016.02.058
16. Harshbarger W, Gondi S, Ficarro SB, Hunter J, Udayakumar D, Gurbani D, et al. Structural and biochemical analyses reveal the mechanism of glutathione s-transferase pi 1 inhibition by the anti-cancer compound piperlongumine. J Biol Chem. (2017) 292:112–20. doi: 10.1074/jbc.M116.750299
17. Kim TH, Song J, Kim SH, Parikh AK, Mo X, Palanichamy K, et al. Piperlongumine treatment inactivates peroxiredoxin 4, exacerbates endoplasmic reticulum stress, and preferentially kills high-grade glioma cells. Neuro Oncol. (2014) 16:1354–64. doi: 10.1093/neuonc/nou088
18. Jarvius M, Fryknas M, D'Arcy P, Sun C, Rickardson L, Gullbo J, et al. Piperlongumine induces inhibition of the ubiquitin-proteasome system in cancer cells. Biochem Biophys Res Commun. (2013) 431:117–23. doi: 10.1016/j.bbrc.2013.01.017
19. Xiao Y, Shi M, Qiu Q, Huang M, Zeng S, Zou Y, et al. Piperlongumine suppresses dendritic cell maturation by reducing production of reactive oxygen species and has therapeutic potential for rheumatoid arthritis. J Immunol. (2016) 196:4925–34. doi: 10.4049/jimmunol.1501281
20. Sun J, Xu P, Du X, Zhang Q, Zhu Y. Piperlongumine attenuates collagen-induced arthritis via expansion of myeloid-derived suppressor cells and inhibition of the activation of fibroblast-like synoviocytes. Mol Med Rep. (2015) 11:2689–94. doi: 10.3892/mmr.2014.3001
21. Yao L, Chen HP, Ma Q. Piperlongumine alleviates lupus nephritis in MRL-Fas(lpr) mice by regulating the frequency of Th17 and regulatory T cells. Immunol Lett. (2014) 161:76–80. doi: 10.1016/j.imlet.2014.05.001
22. Gu SM, Yun J, Son DJ, Kim HY, Nam KT, Kim HD, et al. Piperlongumine attenuates experimental autoimmune encephalomyelitis through inhibition of NF-kappaB activity. Free Radic Biol Med. (2017) 103:133–145. doi: 10.1016/j.freeradbiomed.2016.12.027
23. Xu S, Xiao Y, Zeng S, Zou Y, Qiu Q, Huang M, et al. Piperlongumine inhibits the proliferation, migration and invasion of fibroblast-like synoviocytes from patients with rheumatoid arthritis. Inflamm Res. (2018) 67:233–243. doi: 10.1007/s00011-017-1112-9
24. Klemke M, Wabnitz GH, Funke F, Funk B, Kirchgessner H, Samstag Y. Oxidation of cofilin mediates T cell hyporesponsiveness under oxidative stress conditions. Immunity. (2008) 29:404–13. doi: 10.1016/j.immuni.2008.06.016
25. Weyand CM, Shen Y, Goronzy JJ. Redox-sensitive signaling in inflammatory T cells and in autoimmune disease. Free Radic Biol Med. (2018) 125:36–43. doi: 10.1016/j.freeradbiomed.2018.03.004
26. Abimannan T, Peroumal D, Parida JR, Barik PK, Padhan P, Devadas S. Oxidative stress modulates the cytokine response of differentiated Th17 and Th1 cells. Free Radic Biol Med. (2016) 99:352–63. doi: 10.1016/j.freeradbiomed.2016.08.026
27. Fu G, Xu Q, Qiu Y, Jin X, Xu T, Dong S, et al. Suppression of Th17 cell differentiation by misshapen/NIK-related kinase MINK1. J Exp Med. (2017) 214:1453–69. doi: 10.1084/jem.20161120
28. Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J Clin Invest. (2015) 125:194–207. doi: 10.1172/JCI76012
29. Dinter A EG. Berger: golgi-disturbing agents. Histochem Cell Biol. (1998) 109:571–90. doi: 10.1007/s004180050256
30. Liang J, Jahraus B, Balta E, Ziegler JD, Hubner K, Blank N, et al. Sulforaphane inhibits inflammatory responses of primary human T-cells by increasing ros and depleting glutathione. Front Immunol. (2018) 9:2584. doi: 10.3389/fimmu.2018.02584
31. Fecher-Trost C, Wissenbach U, Beck A, Schalkowsky P, Stoerger C, Doerr J, et al. The in vivo TRPV6 protein starts at a non-AUG triplet, decoded as methionine, upstream of canonical initiation at AUG. J Biol Chem. (2013) 288:16629–44. doi: 10.1074/jbc.M113.469726
32. MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. (2010) 26:966–8. doi: 10.1093/bioinformatics/btq054
33. Schilling B, Rardin MJ, MacLean BX, Zawadzka AM, Frewen BE, Cusack MP, et al. Platform-independent and label-free quantitation of proteomic data using MS1 extracted ion chromatograms in skyline: application to protein acetylation and phosphorylation. Mol Cell Proteomics. (2012) 11:202–14. doi: 10.1074/mcp.M112.017707
34. Wabnitz GH, Nessmann A, Kirchgessner H, Samstag Y. InFlow microscopy of human leukocytes: a tool for quantitative analysis of actin rearrangements in the immune synapse. J Immunol Methods. (2015) 423:29–39. doi: 10.1016/j.jim.2015.03.003
35. Cossarizza A, H-Chang D, Radbruch A, Acs A, Adam D, Adam-Klages S, et al. Guidelines for the use of flow cytometry and cell sorting in immunological studies (second edition). Eur J Immunol. (2019) 49:1457–1973. doi: 10.1002/eji.201970107
36. Bezerra DP, Pessoa C, Moraes MO, Costa-Lotufo LV, Rubio Gouvea D, Jabor VAP, et al. Sensitive method for determination of piplartine, an alkaloid amide from piper species, in rat plasma samples by liquid chromatography-tandem mass spectrometry. Química Nova. (2012) 35:460–5. doi: 10.1590/S0100-40422012000300004
37. Pei S, Minhajuddin M, Callahan KP, Balys M, Ashton JM, Neering SJ, et al. Targeting aberrant glutathione metabolism to eradicate human acute myelogenous leukemia cells. J Biol Chem. (2013) 288:33542–58. doi: 10.1074/jbc.M113.511170
38. Han SS, Han S, Kamberos NL. Piperlongumine inhibits the proliferation and survival of B-cell acute lymphoblastic leukemia cell lines irrespective of glucocorticoid resistance. Biochem Biophys Res Commun. (2014) 452:669–75. doi: 10.1016/j.bbrc.2014.08.131
39. Raj L, Ide T, Gurkar AU, Foley M, Schenone M, Li X, et al. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature. (2011) 475(7355), 231-4. doi: 10.1038/nature10167
40. Franchina DG, Dostert C, Brenner D. Reactive oxygen species: involvement in T cell signaling and metabolism. Trends Immunol. (2018) 39:489–502. doi: 10.1016/j.it.2018.01.005
41. Samstag Y, John I, Wabnitz GH. Cofilin: a redox sensitive mediator of actin dynamics during T-cell activation and migration. Immunol Rev. (2013) 256:30–47. doi: 10.1111/imr.12115
42. Klatt P, Lamas S. Regulation of protein function by S-glutathiolation in response to oxidative and nitrosative stress. Eur J Biochem. (2000) 267:4928–44. doi: 10.1046/j.1432-1327.2000.01601.x
43. Zafarullah M, Li WQ, Sylvester J, Ahmad M. Molecular mechanisms of N-acetylcysteine actions. Cell Mol Life Sci. (2003) 60:6–20. doi: 10.1007/s000180300001
44. Kummerow C, Junker C, Kruse K, Rieger H, Quintana A, Hoth M. The immunological synapse controls local and global calcium signals in T lymphocytes. Immunol Rev. (2009) 231:132–47. doi: 10.1111/j.1600-065X.2009.00811.x
45. Shao Z, Schwarz H. CD137 ligand, a member of the tumor necrosis factor family, regulates immune responses via reverse signal transduction. J Leukoc Biol. (2011) 89:21–9. doi: 10.1189/jlb.0510315
46. Moriggl R, Topham DJ, Teglund S, Sexl V, McKay C, Wang D, et al. Stat5 is required for IL-2-induced cell cycle progression of peripheral T cells. Immunity. (1999) 10:249–59. doi: 10.1016/S1074-7613(00)80025-4
47. Satoh T, Otsuka A, Contassot E, French LE. The inflammasome and IL-1β: implications for the treatment of inflammatory diseases. Immunotherapy. (2015) 7:243–54. doi: 10.2217/imt.14.106
48. Menten P, Wuyts A, Van Damme J. Macrophage inflammatory protein-1. Cytokine Growth Factor Rev. (2002) 13:455–81. doi: 10.1016/S1359-6101(02)00045-X
49. Martinez GJ, Dong C. BATF: bringing (in) another Th17-regulating factor. J Mol Cell Biol. (2009) 1:66–68. doi: 10.1093/jmcb/mjp016
50. Brustle A, Heink S, Huber M, Rosenplanter C, Stadelmann C, Yu P, et al. The development of inflammatory T(H)-17 cells requires interferon-regulatory factor 4. Nat Immunol. (2007) 8:958–66. doi: 10.1038/ni1500
51. Ciofani M, Madar A, Galan C, Sellars M, Mace K, Pauli F, et al. A validated regulatory network for Th17 cell specification. Cell. (2012) 151:289–303. doi: 10.1016/j.cell.2012.09.016
52. Lee W, Kim HS, Hwang SS, Lee GR. The transcription factor Batf3 inhibits the differentiation of regulatory T cells in the periphery. Exp Mol Med. (2017) 49:e393. doi: 10.1038/emm.2017.157
53. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell. (2011) 146:772–84. doi: 10.1016/j.cell.2011.07.033
54. Pentony P, Duquenne L, Dutton K, Mankia K, Gul H, Vital E, et al. The initiation of autoimmunity at epithelial surfaces: a focus on rheumatoid arthritis and systemic lupus erythematosus. Discov Med. (2017) 24:191–200.
55. Terman JR, Kashina A. Post-translational modification and regulation of actin. Curr Opin Cell Biol. (2013) 25:30–8. doi: 10.1016/j.ceb.2012.10.009
56. Yarosz EL, Chang CH. The role of reactive oxygen species in regulating T cell-mediated immunity and disease. Immune Netw. (2018) 18:e14. doi: 10.4110/in.2018.18.e14
57. Kraaij MD, Savage NDL, van der Kooij SW, Koekkoek K, Wang J, van den Berg JM, et al. Induction of regulatory T cells by macrophages is dependent on production of reactive oxygen species. Proc Natl Acad Sci USA. (2010) 107:17686–91. doi: 10.1073/pnas.1012016107
58. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, Chi H. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. (2011) 208:1367–76. doi: 10.1084/jem.20110278
59. Dardalhon V, Korn T, Kuchroo VK, Anderson AC. Role of Th1 and Th17 cells in organ-specific autoimmunity. J Autoimmun. (2008) 31:252–6. doi: 10.1016/j.jaut.2008.04.017
60. Leung S, Liu X, Fang L, Chen X, Guo T, Zhang J. The cytokine milieu in the interplay of pathogenic Th1/Th17 cells and regulatory T cells in autoimmune disease. Cell Mol Immunol. (2010) 7:182–9. doi: 10.1038/cmi.2010.22
61. Yang Z, Shen Y, Oishi H, Matteson EL, Tian L, Goronzy JJ, Weyand CM. Restoring oxidant signaling suppresses proarthritogenic T cell effector functions in rheumatoid arthritis. Sci Transl Med. (2016) 8:331ra38. doi: 10.1126/scitranslmed.aad7151
62. Zou Y, Zhao D, Yan C, Ji Y, Liu J, Xu J, et al. Novel ligustrazine-based analogs of piperlongumine potently suppress proliferation and metastasis of colorectal cancer cells in vitro and in vivo. J Med Chem. (2018) 61:1821–32. doi: 10.1021/acs.jmedchem.7b01096
63. Zou Y, Yan C, Zhang H, Xu J, Zhang D, Huang Z, Zhang Y. Synthesis and evaluation of N-heteroaromatic ring-based analogs of piperlongumine as potent anticancer agents. Eur J Med Chem. (2017) 138:313–9. doi: 10.1016/j.ejmech.2017.06.046
Keywords: piperlongumine, primary human T cells, reactive oxygen species, glutathione, TH17 cells, Treg cells
Citation: Liang J, Ziegler JD, Jahraus B, Orlik C, Blatnik R, Blank N, Niesler B, Wabnitz G, Ruppert T, Hübner K, Balta E and Samstag Y (2020) Piperlongumine Acts as an Immunosuppressant by Exerting Prooxidative Effects in Human T Cells Resulting in Diminished TH17 but Enhanced Treg Differentiation. Front. Immunol. 11:1172. doi: 10.3389/fimmu.2020.01172
Received: 18 January 2020; Accepted: 12 May 2020;
Published: 12 June 2020.
Edited by:
Thomas Herrmann, Julius Maximilian University of Würzburg, GermanyReviewed by:
Xin Chen, University of Macau, ChinaBhalchandra Mirlekar, UNC School of Medicine, United States
Copyright © 2020 Liang, Ziegler, Jahraus, Orlik, Blatnik, Blank, Niesler, Wabnitz, Ruppert, Hübner, Balta and Samstag. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yvonne Samstag, eXZvbm5lLnNhbXN0YWdAdXJ6LnVuaS1oZWlkZWxiZXJnLmRl
†Present address: Jie Liang, Department of Immunology, Medical College of Qingdao University, Qingdao, China