 Richard J. Jones1*
Richard J. Jones1* Ram K. Singh1
Ram K. Singh1 Fazal Shirazi1
Fazal Shirazi1 Jie Wan1Hua Wang1Xiaobin Wang2
Jie Wan1Hua Wang1Xiaobin Wang2 Min Jin Ha3
Min Jin Ha3 Muhamed Baljevic4Isere Kuiatse1
Muhamed Baljevic4Isere Kuiatse1 Richard E. Davis1
Richard E. Davis1 Robert Z. Orlowski1,5
Robert Z. Orlowski1,5- 1Department of Lymphoma and Myeloma, The University of Texas MD Anderson Cancer Center, Houston, TX, United States
- 2The Urology Department, ShengJing Hospital, China Medical University, ShenYang, China
- 3Department of Biostatistics, The University of Texas MD Anderson Cancer Center, Houston, TX, United States
- 4Department of Internal Medicine, University of Nebraska Medical Center, Omaha, NE, United States
- 5Department of Experimental Therapeutics, The University of Texas MD Anderson Cancer Center, Houston, TX, United States
Intravenous immunoglobulin G (IVIgG) is approved for primary immunodeficiency syndromes but may induce anti-cancer effects, and while this has been attributed to its anti-inflammatory properties, IgG against specific tumor targets may play a role. We evaluated IVIgG alone, and with a Heat shock protein (HSP)-90 or proteasome inhibitor, using multiple myeloma and mantle cell lymphoma (MCL) cells in vitro, and with the proteasome inhibitor bortezomib in vivo. IVIgG inhibited the growth of all cell lines tested, induced G1 cell cycle arrest, and suppressed pro-tumor cytokines including Interleukin (IL)-6, IL-8, and IL-10. Genomic and proteomic studies showed that IVIgG reduced tumor cell HSP70-1 levels by suppressing the ability of extracellular HSP70-1 to stimulate endogenous HSP70-1 promoter activity, and reduced extracellular vesicle uptake. Preparations of IVIgG were found to contain high titers of anti-HSP70-1 IgG, and recombinant HSP70-1 reduced the efficacy of IVIgG to suppress HSP70-1 levels. Combining IVIgG with the HSP90 inhibitor AUY922 produced superior cell growth inhibition and correlated with HSP70-1 suppression. Also, IVIgG with bortezomib or carfilzomib was superior to each single agent, and enhanced bortezomib's activity in bortezomib-resistant myeloma cells. Moreover, IVIgG reduced transfer of extracellular vesicles (EVs) to cells, and blocked transfer of bortezomib resistance through EVs. Finally, IVIgG with bortezomib were superior to the single agents in an in vivo myeloma model. These studies support the possibility that anti-HSP70-1 IgG contained in IVIgG can inhibit myeloma and MCL growth by interfering with a novel mechanism involving uptake of exogenous HSP70-1 which then induces its own promoter.
Introduction
Immunoparesis with suppression of uninvolved immunoglobulins is a well-recognized feature of multiple myeloma (MM) and has been associated with an inferior prognosis. For example, studies have linked immunoparesis with an increased risk of progression to MM from precursor states (1–6). Similarly, several studies suggest immunoparesis in newly diagnosed patients is connected with a worse progression-free and overall survival (7, 8), while immunoglobulin recovery after high-dose therapy confers a good prognosis (9, 10). These relationships may be due to the association of immunoparesis with disease burden and an increased risk of infection (11–13). The latter has prompted consideration of immunoglobulin replacement, and two older randomized studies found this reduced serious infections (14, 15). However, as antibiotic prophylaxis was not used in these trials and myeloma care has changed since then, current consensus guidelines do not routinely recommend prophylactic gamma-globulin (16).
Beyond its use to replace endogenous gamma-globulins in primary immune deficiency patients (17), intravenous gamma-globulin (IVIgG) is used in auto-immune and inflammatory conditions (18). Its immune modulating properties may be due to several mechanisms, including modifying complement activation, suppressing idiotypic antibodies, saturating Fc receptors, and quelling inflammatory mediators (18). At the molecular level, IVIgG may inhibit Nuclear factor kappa-B (NF-κB) activation (19–22), and impact on Mitogen-activated protein kinase (MAPK) pathways (21, 23), Tumor necrosis factor-α (TNF-α) (23), and Toll-like receptor signaling (24–26).
The ability of IVIgG to suppress inflammation suggests it has a role in suppressing malignant cell growth given the relationship between inflammation and cancer. In case reports, patients with Kaposi's sarcoma, thyroid cancer, peripheral nerve sheath tumors, and pancreatic cancer experienced remissions or tumor growth delay after receiving IVIgG (27, 28). Furthermore, a trial in patients with advanced melanoma reported a therapeutic effect of the anti-inflammatory, 2 g/kg IVIgG dose (29). Moreover, some reports suggested IVIgG may enhance endogenous immunoglobulin production (30), and we therefore hypothesized that this could increase proteotoxic stress, a key mechanism of action for proteasome inhibitors in myeloma (31).
Given this rationale, we examined the potential anti-myeloma effect of IVIgG, a polyclonal product pooled from 1,000s to 100,000 blood and plasma donors (32) containing 1,000 of antibodies against an array of targets (33). IVIgG induced an anti-proliferative and pro-apoptotic effect in myeloma and mantle cell lymphoma (MCL) cell lines. This was accompanied by a reduction of the abundance of Heat shock protein (HSP)-70 protein, and HSP70 promoter activity inhibition. The latter occurred in association with blockade of uptake of extracellular vesicles by the IVIgG preparation, and interruption of an autocatalytic loop whereby extracellular HSP70 stimulated its own intracellular transcription. Finally, IVIgG enhanced the anti-myeloma activity of an HSP90 and a proteasome inhibitor, both of which strongly induce anti-apoptotic HSP70, and an IVIgG/bortezomib combination enhanced in vivo anti-myeloma efficacy. These studies shed new light on the mechanisms of action of IVIgG, and on its potential applicability to myeloma patients in particular.
Materials and Methods
Reagents
IVIgG and rituximab (RTX) were from The University of Texas MD Anderson Cancer Center Pharmacy (Houston, TX), and IVIgG was dialyzed against phosphate-buffered saline (PBS) to remove L-proline. AUY922, bortezomib (BZB), and carfilzomib (CFZ) were from Selleck Chemicals (Houston, TX), recombinant human HSP70-1 protein was from StressMarq Biosciences (British Columbia, Canada), and bovine serum albumin (BSA) was from Sigma-Aldrich (St. Louis, MO).
Cell Culture
Cell lines were from the American Type Culture Collection (Manassas, VA) or the Leibniz-Institute DSMZ Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (Braunschweig, Germany), except for MUTU-I, an EBV+ Burkitt's lymphoma (BL) line, which was a gift from Dr. Alan Rickinson (University of Birmingham). These were validated through our Characterized Cell Line Core Facility, and grown in RPMI 1640 or Dulbecco's modified Eagle medium (Thermo Fisher Scientific; Waltham, MA) with 10% fetal bovine serum, penicillin, and streptomycin (Sigma-Aldrich).
Immunoblotting, Enzyme-Linked Immunosorbent Assays, Immunoprecipitations
Protein expression was determined by immunoblot analysis as previously described (34). Antibodies used were sourced as follows: anti-HSP70-1 (C92F3A-5) and -HSF-1 were from Enzo Life Sciences, Inc. (Farmingdale, NY); anti-HSP70-1 (MA3009) was from Thermo Fisher Scientific; anti-td-Tomato was from MyBioSource, Inc. (San Diego, CA); and anti-p53, -HSP90, -HSP40, -CD9, -CD63, -LAMP-2, and -β-actin were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). The anti-HSP70 IgG ELISA kit from Enzo Life Sciences, Inc., and the TeloTAGGGTM Telomerase PCR ELISA from Roche (Basel, Switzerland) were used according to the manufacturer's instructions. RPMI 8226 cells were treated overnight with RTX or IVIgG (both at 5 mg/ml), and the culture supernatant was collected. Cells were then washed in PBS to remove these antibodies, lysed in IP Lysis buffer (Thermo Fisher Scientific), and both the harvested supernatant and the cell lysate was incubated with protein A/G agarose (Sigma-Aldrich). The agarose beads were incubated overnight at 4°C and washed in IP lysis buffer and immunoblotting was performed for HSP70-1.
Plasmids and Quantitative Real-Time Polymerase Chain Reaction
Total RNA from treated cells was reverse transcribed as previously described (35). cDNA was subsequently used in quantitative real-time polymerase chain reaction (qPCR) for HSP70-1 using inventoried TaqMan human HSP70 probes and primers with GAPDH as a control (Thermo Fisher Scientific). The HSP70 promoter construct was generated from MM1.S cell genomic DNA using the forward and reverse primers: TCGGGGTACCGCCTTTCAGGTTCACAATCAATCAG, which contains a KpnI restriction site and binds 695 base pairs (bps) upstream of the HSP70 transcription initiation site; and GGCAAGATCTCAGGTTCGCTCTGGGAAGCCTTGG, which binds 87 bps downstream and has a BglII site. The resulting product was cloned into the pGL3 luciferase vector (Promega Corporation; Madison, WI). The palmitoylation (PALM) sequence tag coupled to the tdTomato fluorophore (tdT) vector was a gift from Xandra Breakefield (Massachusetts General Hospital).
Cell Viability Assay and Flow Cytometry
The WST-1 reagent from Roche was used to determine cell viability, while apoptosis was determined by Annexin V Pacific Blue and TO-PRO-3 (Life Technologies; Grand Island, NY) staining, along with Count Bright Beads (Thermo Fisher Scientific), and analyzed by flow using FlowJo, version 10 (Tree Star, Inc.; Ashland, OR). Cell cycle analysis was performed using the NPE AnalyzerTM reagent (NPE Systems, Inc.; Pembrook Pines, FL) on a Gallios flow cytometer (Beckman Coulter, Inc.; Brea, CA), and analyzed using Multicycle Software (Phoenix Flow Systems; San Diego, CA). Live cells were treated with 1 μM ER-Tracker Green (Thermo Fisher Scientific) for 1 h at 37°C and washed in PBS prior to analysis with flow using the FITC channel.
Gene Expression Profiling
The Illumina TotalPrep RNA Amplification Kit (Thermo Fisher Scientific) was used to generate amplified, biotinylated cRNA from total RNA isolated from OPM-2 cells treated with BSA or IVIgG in triplicate. The cRNA was hybridized overnight to Illumina HT-12 BeadArrays (San Diego, CA), stained with streptavidin-Cy3 (GE Healthcare; Amersham, PI), and scanned on a BeadArray Reader (Illumina). Bead-level data were extracted from GenomeStudio (Illumina) files and processed using open-source and custom software, following the approach of Dunning et al. (36). Values were corrected by the model-based background correction method that uses values for negative control probes to estimate and remove the non-specific signal component for each transcript probe, and the quantile distribution was normalized. We excluded the ~20% of probes that were not good matches to actual transcripts, and log2-transformed the probe-level data, which are the median values for each probe after discarding outliers. The relative gene expression in BSA and IVIgG samples were used to generate fold-change data, and p-values and heat maps were generated.
Cytokine Analysis
The Bio-Plex ProTM human cytokine 48-plex screening panel (Bio-Rad; Hercules, CA) was used to analyze BSA- and IVIgG-treated culture media supernatants and analyzed on a Bio-Plex® 200/LX200.
Extracellular Vesicle (EV) Isolation
Normal growth medium from 50 × 106 cells was collected, washed, and re-plated in serum-free RPMI containing 1x insulin-transferrin-selenium-ethanolamine (Thermo Fisher Scientific). EVs were then isolated from the culture media using the ExoQuick-TC reagent (System Biosciences; Palo Alto, CA), and particle size and concentration were measured using the ZetaView® Nanoparticle Tracking Analyzer (Particle Metrix; Meerbusch, Germany). CD63 and CD9 were used for validation, as was the presence of HSP70-1 and LAMP-2. The RPMI 8226 drug-naïve cells (WT) and RPMI 8226 BZB-resistant (V10R) myeloma cells (37) were engineered to express a plasmid carrying the PALM tdT tag (38).
Ethics Statement and Xenograft Studies
Experiments were performed under procedures and protocols approved by our Institutional Animal Care and Use Facility. Six- to 8-week-old C.B-17 severe combined immunodeficiency (SCID) mice (Envigo; Indianapolis, IN) (5 per group) were injected intravenously with 1 × 106 luciferase-labeled MM1.S cells, and tumor burden was monitored by bioluminescent imaging using the IVIS Spectrum (PerkinElmer. Inc.; Waltham, MA). At 3 weeks post-cell injection, mice were randomized to once-weekly PBS or IVIgG (500 mg/kg) for 2 weeks, or intraperitoneal BZB (0.5 mg/kg) twice weekly for 2 weeks in 30% poly-(ethylene-glycol). Mice were sacrificed by CO2 asphyxiation when they displayed distress.
Statistical Analyses
Data were analyzed by calculation of the standard error of the mean (SEM) and t-tests were used with a significance level of <0.05. The percentage change of tumor growth was plotted over time and the overall anti-tumor activity was the adjusted area under the curve (AUC) computed as an average AUC over time per mouse (39). Using both measurements, pairwise comparisons of the treatment groups were performed using the two-sample t-test and Wilcoxon rank sum test.
Results
IVIgG Induces Apoptosis and Cell Cycle Arrest
To begin to evaluate IVIgG's effects on myeloma and MCL cell lines, we exposed them to IVIgG or BSA, and used IVIgG at 10 mg/mL, which is equivalent to the IVIgG serum level achieved at the 2 g/kg dose for autoimmune disorders (40). Exposure of myeloma (Figure 1A) and MCL (Figure 1B) cell lines to IVIgG led to an anti-proliferative effect compared to the BSA control, with cell viability decreases ranging from 60% in MM1.S to 35% in ANBL-6 cells. Testing of marrow-derived HS-5 stromal cells and the gastric cancer cell line AGS, which do not express Fc receptors, demonstrated that IVIgG also reduced their viability (Figures 1A,B). Studies of RPMI 8226 and MOLP-8 myeloma, and of the JeKo-1 and MAVER-1 MCL cell lines, indicated IVIgG induced apoptosis based on the appearance of Annexin V-positivity (Figure 1C). Cell death occurred in 20% of RPMI 8226 and 30% of MOLP-8 cells, and 50% of JeKo-1 and 32% of MAVER-1 cells (Figure 1D). Cell cycle analysis in MOLP-8, U266, and HS-5 cells indicated IVIgG induced a G1S growth arrest (Supplementary Figure 1), though this was limited to a slight increase in the S phase fraction in MM1.S cells.
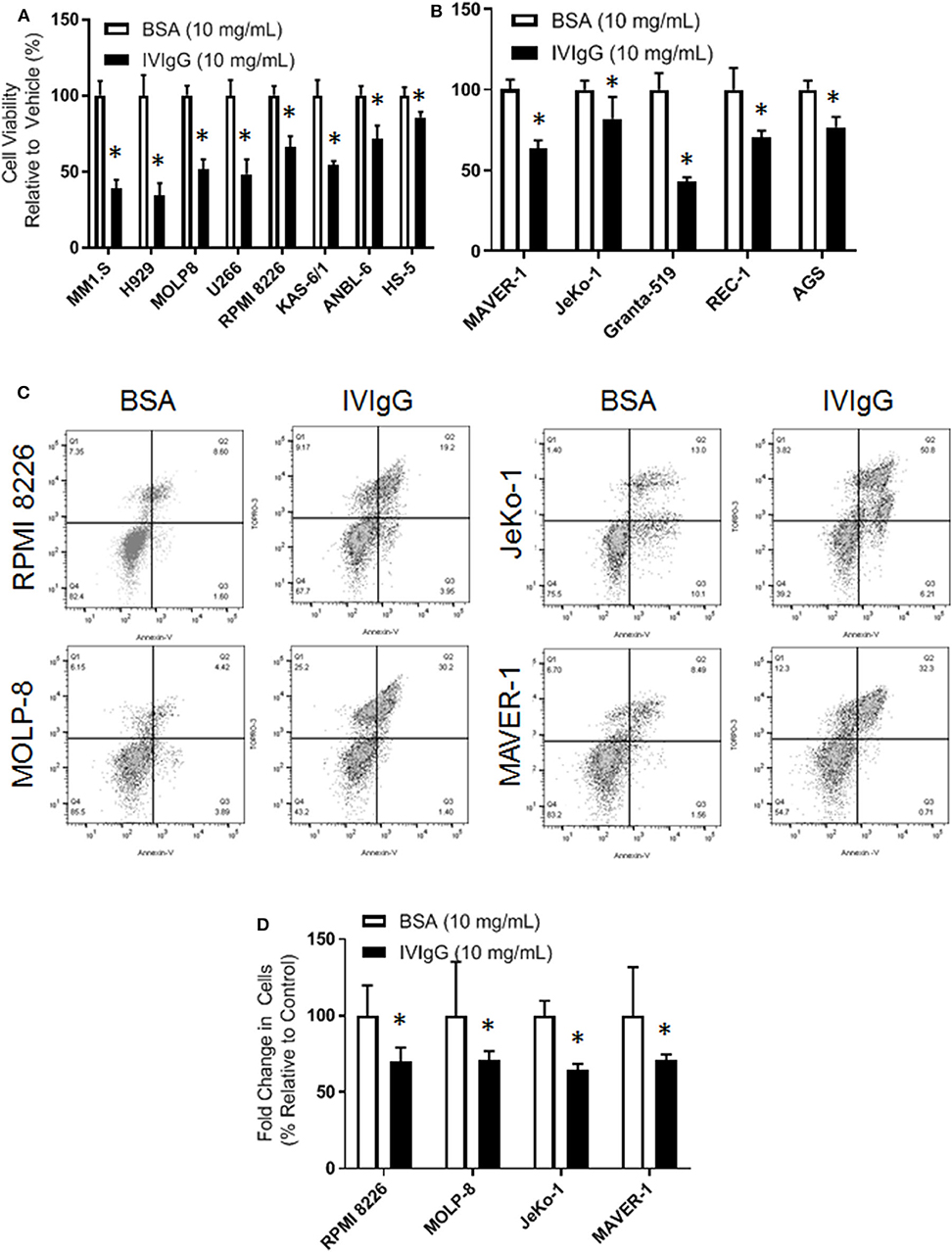
Figure 1. IVIgG inhibits growth of multiple myeloma and lymphoma cell lines. (A) Myeloma cell lines and the stromal cell line HS-5 were exposed for 72 h to IVIgG or bovine serum albumin (BSA), and cell viability was determined using the WST-1 assay. Results are expressed as the percentage viability relative to the vehicle control, which was arbitrarily set as 100%. (B) Cell viability of MCL cell lines and the Fc-gamma receptor-negative gastric cell line AGS after exposure to BSA vs. IVIgG was determined as above. (C) Flow cytometric analysis of RPMI 8226 and MOLP-8 cells, as well as of JeKo-1 and MAVER-1 cells exposed to BSA or IVIgG (10 mg/mL) for 72 h was performed after staining with Annexin V/TO-PRO-3. (D) Annexin V/TO-PRO-3 and Count Bright Bead flow was used to determine the change in viable cell number in response to BSA versus IVIgG. The values shown represent the mean ± standard error of the mean from three independent experiments, and an unpaired t-test was used to evaluate significance, where “*” denotes p < 0.05.
Impact on Cytokines and Endoplasmic Reticulum (ER) Abundance
Given the known impact of IVIgG on immune-modulating cytokines, we evaluated its effect on MM and MCL cell line cytokine production. IVIgG induced pleiotropic changes in cytokines involved in immune regulation and growth (Figure 2; Supplementary Table 1). Most cells had decreases in Interleukin (IL)-6, Macrophage inflammatory protein (MIP)-1α, and five myeloma cell lines had a decrease in IL-8. In contrast, IVIgG stimulated the active heterodimer of IL-12 (IL-12-p70) in three myeloma and both MCL cell lines. Uptake of large amounts of IgG may lead to protein overload and expansion of ER abundance to cope with this excess protein (41). Myeloma and MCL cells treated with BSA vs. IVIgG, and stained with ER-Tracker Green, showed increased fluorescence, indicating an increase in the ER abundance (Figure 2B). Similar results were seen in BL cell lines (Supplementary Figure 2).
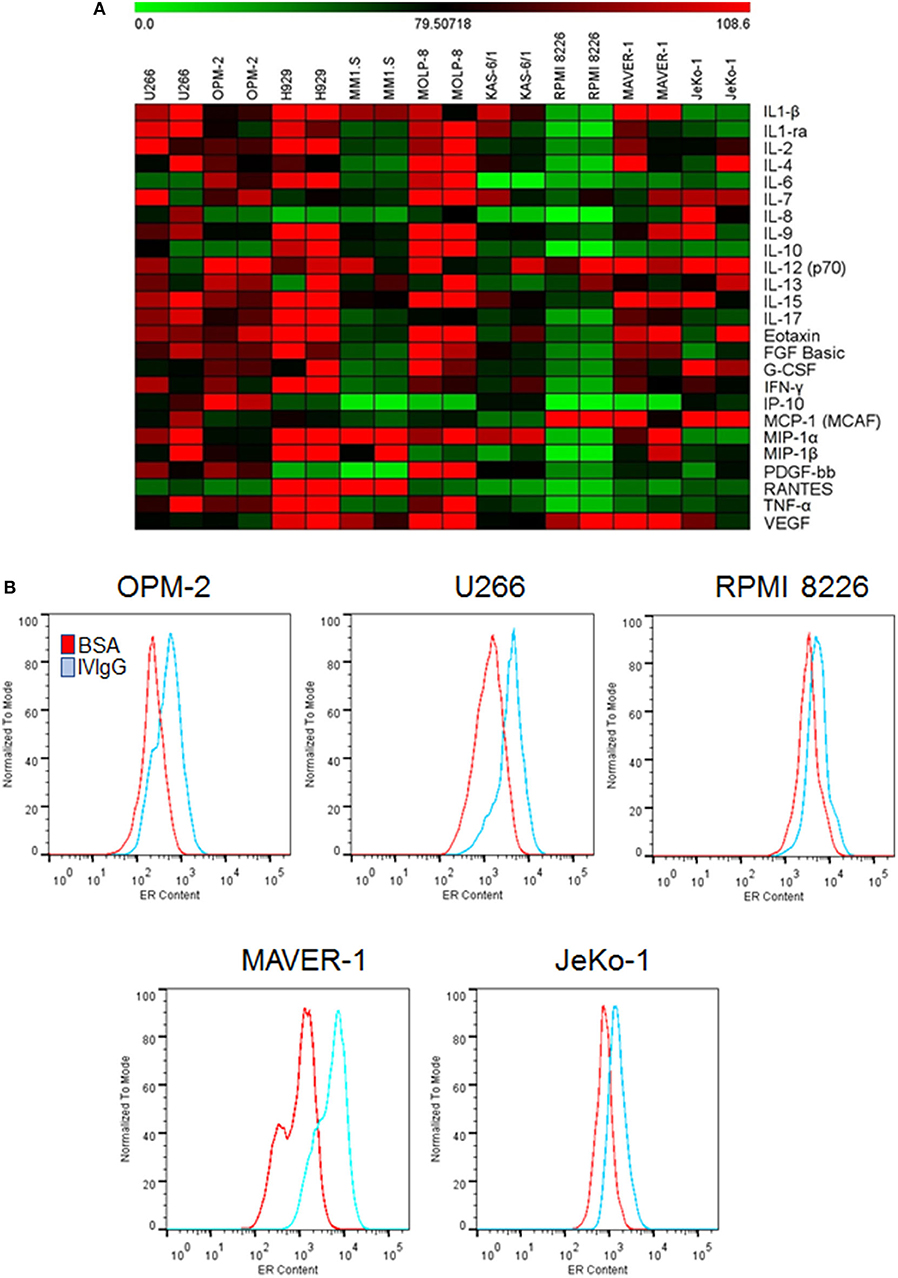
Figure 2. IVIgG-suppresses pro-myeloma and lymphoma cytokines and increases ER abundance. (A) The supernatant from IVIgG- and BSA-treated (10 mg/mL) MM or MCL cells was analyzed using the Bioplex human cytokine array in duplicate. Fold changes in expression were determined using BSA as a normalizing factor for the IVIgG-exposed samples. (B) MM and MCL cells were treated as above for 72 h, and live cells stained with ER-Tracker Green and analyzed by flow cytometry.
IVIgG Suppresses HSP70-1 Expression
To gain a broader understanding of IVIgG's effects, we performed gene expression profiling of OPM-2 myeloma cells exposed to IVIgG or BSA. A 2-fold expression variance cut-off with a p < 0.03 identified 15 genes with increased expression, and 33 that decreased after IVIgG (Figure 3; Supplementary Table 2). The genes most stimulated included the early B-cell marker V-Set Pre-B cell surrogate light chain 1 (VPREB1; 3.67-fold change) and the tumor suppressor Cadherin 1 (CDH1; 2.88-fold). HSP70 family members were among the genes most suppressed, including HSPA1B (HSP70-1; 7.87-fold), its paralog HSPA6 (HSP70B; 7.78-fold), and DNAJB1 (HSP40; 6.85-fold). This coincided with decreased expression of DNA damage inducible transcript 3 (DDIT3; 6.85-fold), which is stimulated by HSP70-1 (42), and a decrease (4.14-fold) in Telomerase reverse transcriptase (TERT), another HSP70-1 associate (43, 44).
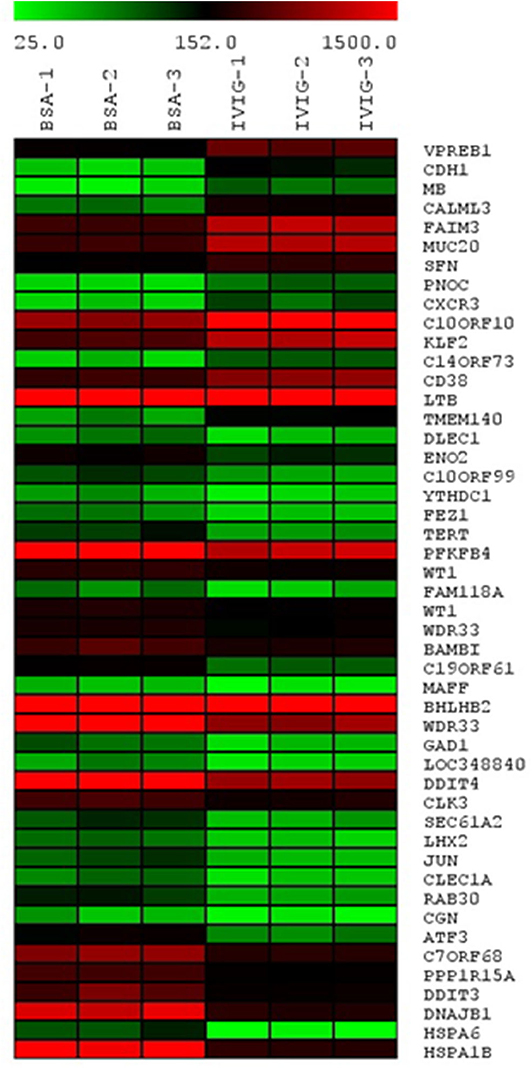
Figure 3. Transcription of HSP70 family members HSPA1B (HSP70-1), HSPA6, and DNAJB1 is reduced by IVIgG. The OPM-2 myeloma cell line was exposed in triplicate to BSA or IVIgG (10 mg/mL) for 48 h, subjected to gene expression profiling (GEP), and the relative gene expression levels were organized in a heat map based on fold change compared to the BSA control. These cells were selected for GEP because they had a less robust cell death response to IVIgG at the time point used, allowing a focus on early transcriptional changes of IVIgG in the absence of later changes due to induction of apoptosis.
Next, we evaluated the effect of IVIgG on HSP family members at the protein level. In all cell lines tested, HSP70-1 showed the largest change, and was lower with IVIgG in myeloma, MCL, and the BL models (Figure 4A; Supplementary Figure 3). Total levels of Heat shock transcription factor (HSF)-1, which induces HSP70-1, did not correlate with HSP70-1 since it was decreased in some cells but stable or increased in others, and HSP40 changes were similarly not consistent. The expression of p53, an HSP70 client (45), was also depleted in the majority, while HSP90 showed no change.
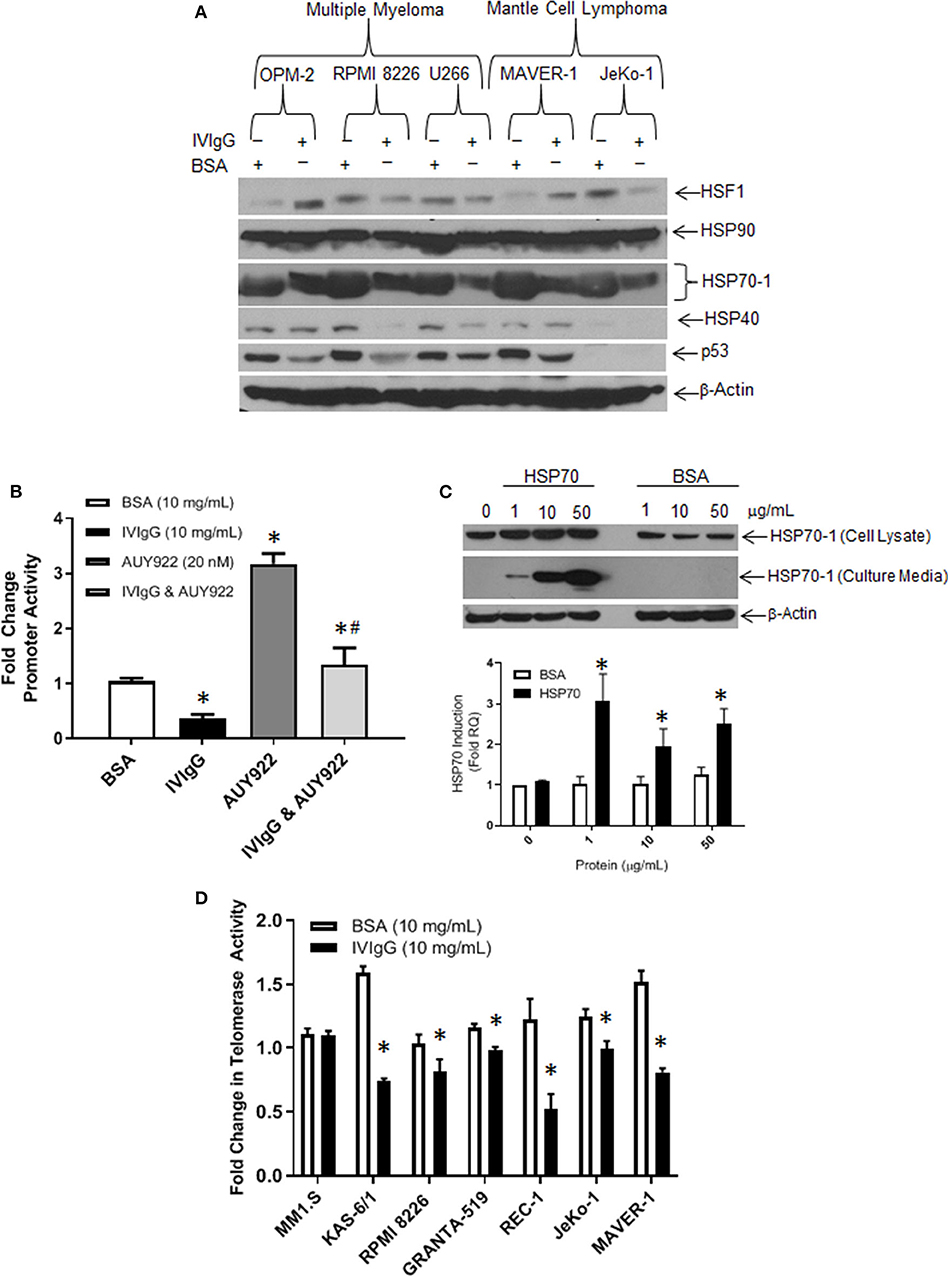
Figure 4. IVIgG suppresses expression of HSP70 and its co-chaperone protein targets. (A) MM and MCL cell lines were exposed to BSA or IVIgG for 48 h, and cell lysates were immunoblotted as indicated. (B) HEK 293T cells were transfected with an HSP70-1 promoter luciferase reporter construct, and promoter activity was monitored following treatment with BSA, IVIgG, AUY922, or IVIgG and AUY922 for 24 h. Values were normalized to the BSA control (* denotes p < 0.05 compared to BSA and #denotes p < 0.05 compared to AUY922). (C) RPMI 8226 cells were exposed to recombinant HSP70-1 for 24 h in serum-free media, and cell lysates and supernatants were evaluated with the indicated sera, while qPCR was performed on RNA extracted from cells and HSP70-1 transcription measured. (D) Telomerase activity in myeloma and MCL cell lines was evaluated using the TeloTAGGGTM Telomerase PCR ELISA following 48 h of exposure to BSA or IVIgG, and fold change was calculated in relation to the BSA control. The values shown represent the mean ± standard error from three independent experiments, and an unpaired t-test was used to evaluate significance, where “*” denotes p < 0.05 compared to the BSA control.
To further confirm the effect of IVIgG on HSP70-1 transcription, we generated a luciferase reporter containing the HSP70-1 promoter element. Transfected HEK 293T cells exposed to IVIgG vs. BSA showed a 50% decrease in basal promoter activity (Figure 4B). Addition of the HSP90 inhibitor AUY922 as a positive control increased HSP70 promoter activity, while IVIgG suppressed this by 50%. Since HSP70-1 is secreted and can be taken up into a variety of cell types (46), we next used exogenous recombinant HSP70-1. Addition of 1 μg of HSP70-1 increased HSP70-1 levels in cell lysates (Figure 4C, top panel), and led to a significant (>3-fold) increase in HSP70-1 transcription (Figure 4C, bottom). Given the known interaction between HSP70-1 and TERT (43, 44), we measured telomerase activity after IVIgG exposure, and five of six cell lines showed a decreased activity, with REC-1 and KAS-6/1 cells displaying the largest decreases of 50% (Figure 4D).
Antibodies to HSP70-1 Are Present in IVIgG
In that extracellular HSP70-1 stimulated intracellular HSP70A1A transcription and enhanced protein abundance, we next evaluated if IVIgG could bind extracellular HSP70-1 shed from cells. Whole RPMI 8226 cells in culture were incubated with IVIgG or RTX as a control, and both the supernatant and intact cells were collected. Cells were then washed to remove IVIgG or RTX, and then the original culture supernatant and lysates of the intact cells unexposed to IVIgG or RTX were subjected to immunoprecipitation. IVIgG precipitated HSP70-1 from the cell culture supernatant, indicating both that IVIgG contains IgG against extracellular HSP70-1 and that the latter is shed into the media (Figure 5A). Recombinant HSP70-1 protein was then titrated onto a Western blot and immunoblotted using single IVIgG lots or a pooled mixture of lots, and IVIgG detected HSP70-1 (Figure 5B). Notably, screening of four different IVIgG lots using a quantitative anti-HSP70-IgG ELISA indicated the presence of anti-HSP70-1 IgG at ~3 μg/mL of IVIgG across the tested lots (Figure 5C; Supplementary Table 3).
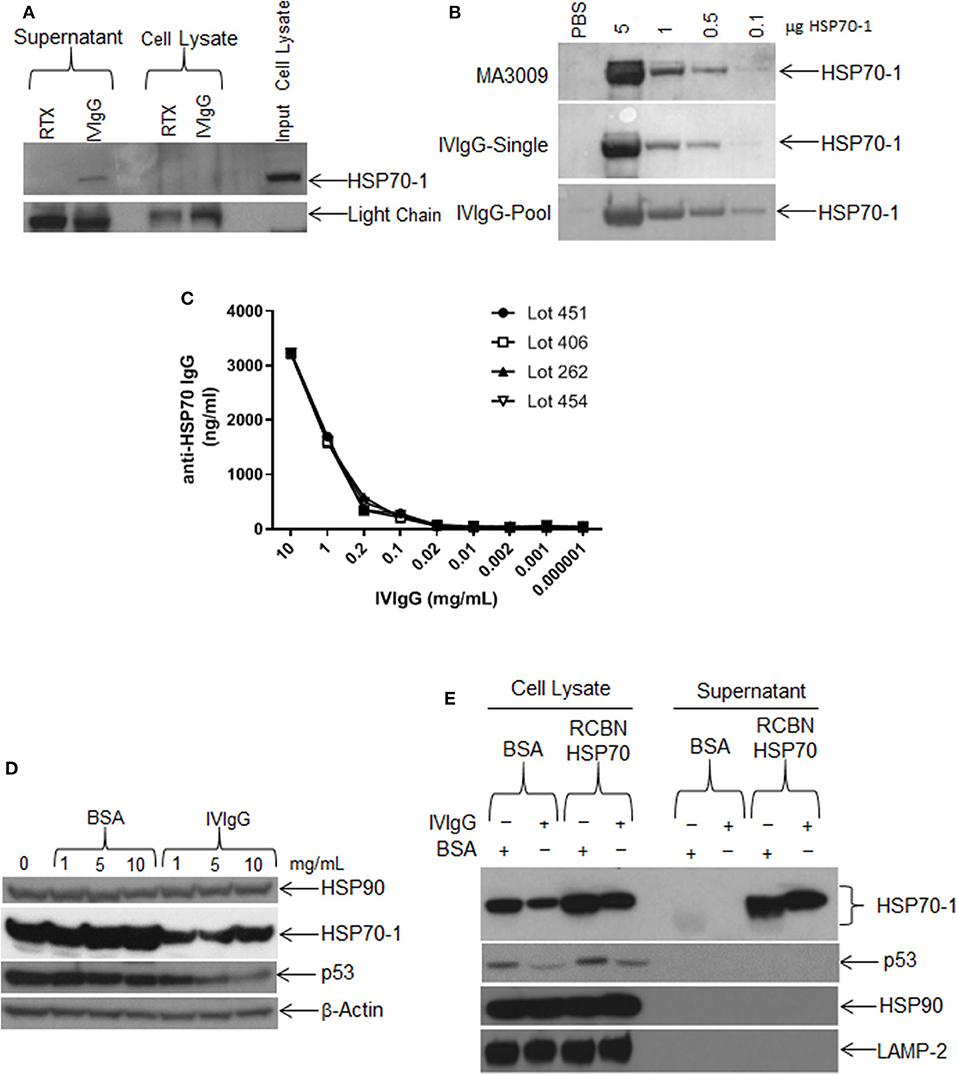
Figure 5. Single IVIgG lots contain high titers of anti-HSP70 IgG. (A) RPMI 8226 cells were treated for 24 h with rituximab (RTX) or IVIgG and culture media was immunoprecipitated with protein A/G agarose and immunoblotted for HSP70-1. (B) A single lot of IVIgG or multiple pooled lots were screened against increasing concentrations of recombinant HSP70-1 and compared to a commercially available anti-HSP70 IgG (MA3009). (C) Individual IVIgG lots were screened for anti-HSP70 IgG titers using a commercial ELISA and the concentration calculated for each lot. (D) RPMI 8226 cells were treated for 48 h with increasing concentrations of BSA vs. IVIgG and immunoblotted for the indicated targets. (E) RPMI 8226 cells were incubated for 24 h with BSA or IVIgG (10 mg/mL) in the presence of 50 μg of recombinant (RCBN) HSP70-1 and cell lysates of culture supernatant were immunoblotted as indicated.
We next titrated IVIgG onto cells to determine the dose effect on HSP70-1 levels compared to BSA. HSP70-1 decreased at the lowest IVIgG level used (1 mg/mL), and increasing this concentration did not suppress HSP70-1 further. In contrast, p53 levels were decreased at 5, and further decreased at 10 mg/mL, while there was no change in HSP90 or β-actin (Figure 5D). To see if the effect of IVIgG on HSP70-1 could be blocked, we added excess recombinant HSP70-1 to RPMI 8226 cells in the presence of IVIgG and evaluated cell lysates and supernatant. IVIgG reduced intracellular HSP70-1 to a lower level in the presence of BSA in the media than it could in the presence of recombinant HSP70 (Figure 5E). Moreover, the excess did not rescue the decrease in proliferation (data not shown), supporting the possibility that anti-HSP70 IgG within the IVIgG preparation bound exogenous HSP70-1 and modulated intracellular HSP70-1 levels.
IVIgG Prevents Compensatory HSP70-1 Upregulation by HSP90 Inhibition
HSP90 inhibitors are in trials for various malignances, and one mechanism of inducible chemoresistance is through upregulation of anti-apoptotic HSP70-1 (47). Treatment of OPM-2, U266, and RPMI 8226 cells with AUY922 decreased cell viability to ~30%, which was further decreased to 9, 15, and 13%, respectively, with IVIgG (Figure 6A), consistent with the possibility of an additive effect. The RPMI 8226 V10R BZB-resistant cell line exhibited resistance to IVIgG alone (87.25% viability) compared to drug-naïve RPMI 8226 cells (78% viability), and this was mirrored by a slight increase in resistance to AUY922, with viability dropping to 35% in V10R cells compared to 26% in the naïve cells. AUY922 and IVIgG showed limited benefit in V10R cells, with viability decreasing from 35% with the single agent to 30% with the combination (Figure 6A). Evaluation of two MCL cell lines revealed similar results: AUY922 decreased viability in JeKo-1 and MAVER-1 cells to 36 and 41%, respectively, which was further decreased to 15 and 19%, respectively, with IVIgG (Figure 6B).
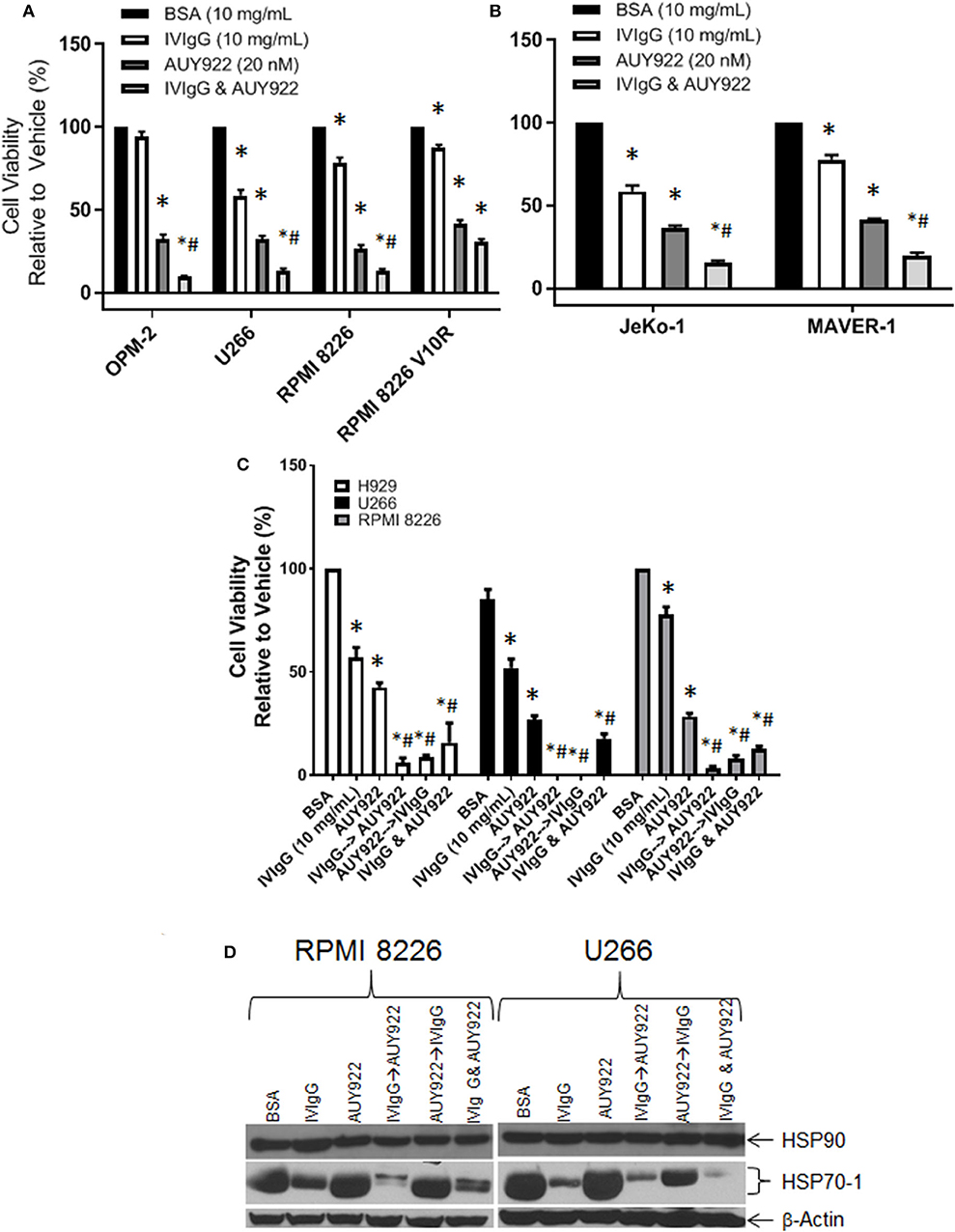
Figure 6. IVIgG suppresses AUY-922-mediated induction of HSP70-1 leading to enhanced cell death when used in sequence-specific combinations. Myeloma (A) and MCL (B) cell lines were treated with BSA, IVIgG, or AUY922 alone, or IVIgG with AUY922 for 72 h and cell viability was determined using the WST-1 assay. The BZB-resistant RPMI 8226 V10R cell line was also evaluated, along with its drug-naïve RPMI 8226 counterpart. (C) Cell viability measuring the sequence-specific effects of administration of IVIgG vs. AUY922 was then performed in myeloma cell lines, where IVIgG or AUY922 was added followed by the other agent 24 h later and then the two drugs were incubated for an additional 48 h. As a control, AUY922 and IVIgG were also added simultaneously. * denotes p < 0.05 to the BSA control and #denotes p < 0.05 to both single agents. (D) Cell lysates from cells treated in (C) were evaluated for the indicated targets by immunoblotting.
Next, we evaluated whether adding the agents in a particular sequence optimized their effects. H929, U266, and RPMI 8226 cells all responded to IVIgG or AUY922, and simultaneous addition enhanced cell killing (Figure 6C). Using either IVIgG followed by AUY922, or the reverse, showed that IVIgG added first and AUY922 later generally produced a greater effect than AUY922 followed by IVIgG in H929 and RPMI 8226 cells. AUY922 stimulated HSP70-1 in U266 and RPMI 8226 cells (Figure 6D), while IVIgG followed by AUY922 precluded the AUY922-mediated HSP70-1 upregulation in RPMI 8226 cells, whereas AUY922 followed by IVIgG did not (Figure 6D). In U266 cells, IVIgG followed by AUY922, or with both agents simultaneously, led to suppression of AUY922-mediated HSP70-1 upregulation, whereas AUY922 followed by IVIgG did so to a lesser extent.
IVIgG Enhances BZB Activity and Suppresses EV-Mediated Drug Resistance
BZB is a key clinically relevant drug that is used for myeloma treatment in multiple settings but resistance frequently arises, and HSP70-1 expression has been associated with BZB resistance (48). We therefore evaluated the effects of IVIgG and BZB, and IVIgG alone reduced H929, MM1.S, and U266 cell viability by 28, 21, and 37%, respectively, BZB reduced viability by 66, 68, and 63%, respectively, and adding both led to virtually 100% cell killing (Figure 7A). Next, we examined the effect of IVIgG on RPMI 8226 wild-type (WT) and V10R cells alone and with BZB or CFZ. Wild-type RPMI 8226 cells were sensitive to IVIgG, BZB, and CFZ, with viability decreasing by 38, 50, and 35%, respectively, while IVIgG combinations with BZB or CFZ led to near complete cell viability loss (Figure 7B). Combining IVIgG with BZB or CFZ in the V10R cells led to enhanced loss of viability over the single agents alone, indicating that IVIgG enhanced BZB activity in cells previously resistant to the drug (Figure 7B).
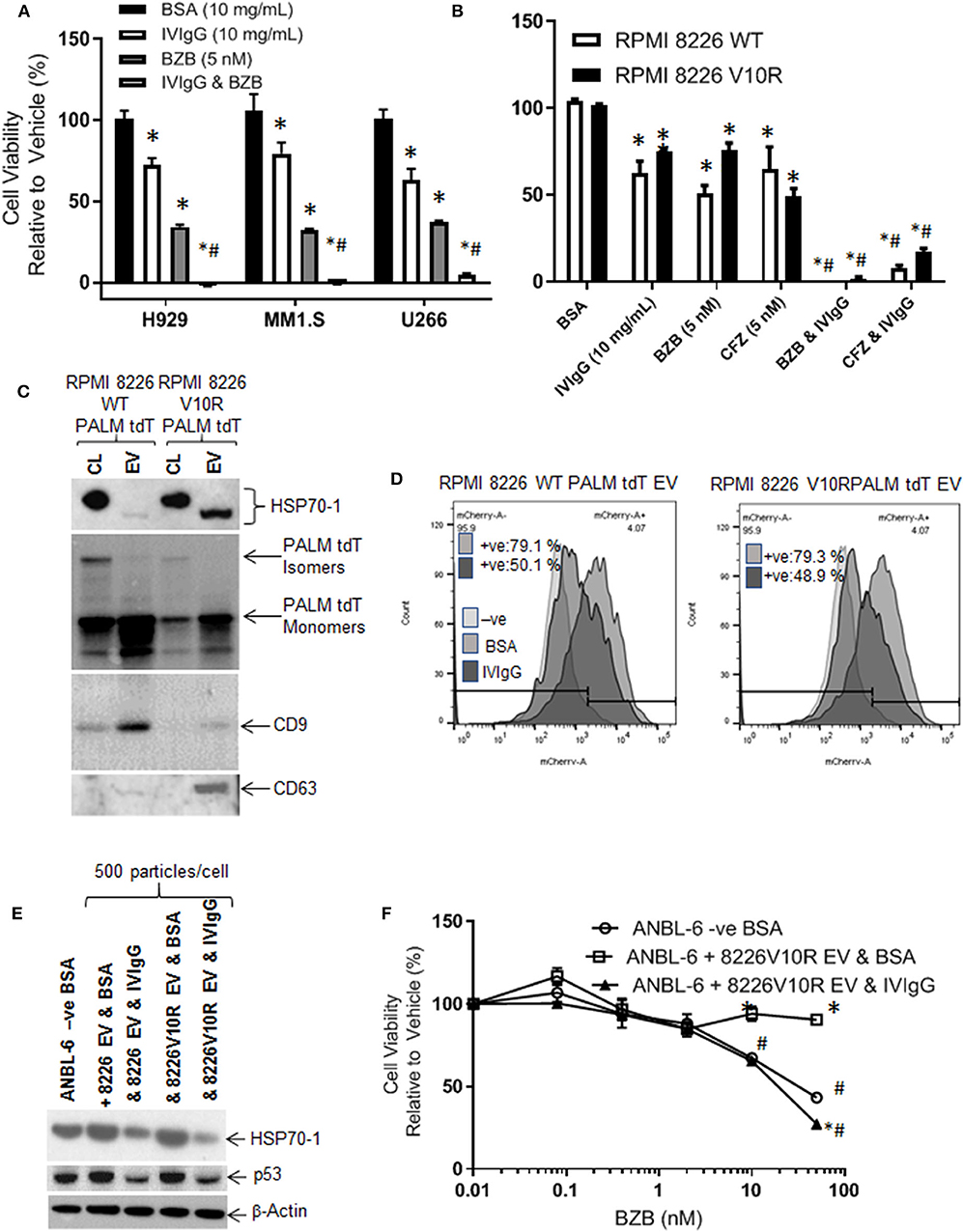
Figure 7. IVIgG prevents EV-mediated transfer of BZB resistance. (A) The anti-proliferative effect of IVIgG and BZB was evaluated in a panel of myeloma cell lines exposed to single agents or the combinations for 72 h followed by a WST-1 assay. (B) BZB-naïve RPMI 8826 and BZB-resistant RPMI 8226 V10R cells were exposed to single-agent BSA, IVIgG, CFZ, and BZB or a combination of IVIgG and either CFZ or BZB for 72 h, and the cell viability was measured by WST-1 assay. * denotes p < 0.05 compared to the BSA control and #denotes p < 0.05 compared to both single agents. (C) EVs were isolated from BZB-naïve RPMI 8226 or BZB-resistant RPMI 8226 V10R cells carrying a PALM tdT tag to allow EV tracking, and EV lysates or the corresponding cell lysates (CL) were analyzed by immunoblot with the indicated antibodies. (D) ANBL-6 cells were exposed to EVs as described in (C) at 500 particles per cell for 48 h in the presence of BSA or IVIgG and analyzed for the percentage of PALM tdT tag-positive cells by flow cytometry. (E) Immunoblotting is shown of cell lysates from (D) probed as indicated. (F) Cell viability assay of ANBL-6 cells treated as in (D) with increasing concentrations of BZB for 48 h. ANBL-6 cells were utilized because they readily accepted EVs from RPMI 8226 cells in preliminary studies (not shown). * denotes p < 0.05 compared to ANBL-6 –ve BSA, whilst #denotes p < 0.05 to ANBL-6 cells receiving 8226 V10R EV & IVIgG.
HSP70-1 is found on the surface and inside EVs (49, 50), which may be drug resistance mediators. We therefore assessed IVIgG's effect on EVs from RPMI 8226 WT and V10R cells engineered to release PALM tdT-tagged EVs (38). EVs from WT and V10R cells displayed a 150 nM mean particle size for the WT and 110 nM for the V10R EVs (range for both, 50–800 nM; Supplementary Figures 4A,B). These EVs contained CD9 and CD63 tetraspanins (Figure 7C), while PALM tdT was detected in the cell lysates and EVs. Cell lysates had both smaller PALM tdT monomers and larger PALM tdT multimers, while EVs had enrichment of PALM tdT monomers only (Figure 7C). Interestingly, the V10R EVs showed enrichment of HSP70-1, whereas the WT EVs did not, and the HSP70-1 variant in cell lysates migrated as a slower form, while that found in EVs had an apparent lower mass.
As EVs could transfer HSP70-1 between cells, we were interested to see if IVIgG blocked this mechanism. We titrated 50–500 particles/cell onto ANBL-6 myeloma cells and found that 500 particles/cell gave 100% uptake (Supplementary Figure 4C). Next, we added WT- or V10R-derived EVs to ANBL-6 cells in the presence of BSA or IVIgG. EV uptake was highly efficient, with 79% of the ANBL-6 cells bearing the PALM tdT fluorophore from both the WT and V10R EVs. IVIgG addition suppressed this uptake to 50.1% in WT EV-treated cells and 48.9% in V10R EV-treated cells (Figure 7D). Lysates from the treated cells demonstrated that addition of WT and V10R EVs increased HSP70-1 and p53 levels compared to untreated cells, while IVIgG suppressed these to levels below the control cells (Figure 7E).
We also evaluated whether the V10R EVs could transfer BZB resistance to ANBL-6 cells. When these were incubated with 500 particles/cell and BZB was added with BSA or IVIgG, ANBL-6 control cells were sensitive to BZB but, with the addition of V10R EVs, they were resistant to the highest BZB concentration. Addition of IVIgG to the V10R EVs prevented the increase in resistance and restored BZB sensitivity to a level similar to the control cells (Figure 7F).
Modeling in vivo Shows IVIgG Enhances BZB's Effects
To evaluate whether IVIgG enhanced BZB's anti-tumor activity, we utilized a systemic model of myeloma in which human-derived, luciferase-labeled MM1.S myeloma cells hone to bone in immunodeficient SCID mice, which were treated with vehicle, IVIgG, BZB, or both. IVIgG or BZB suppressed MM1.S tumor growth (p < 0.05; t-test and Wilcoxon rank sum test) from days 39–49 (Figure 8A). Notably, IVIgG with BZB inhibited tumor growth beyond the single agents from day 40 to the conclusion of the experiment (day 56). In order to assess the overall anti-tumor activity, we calculated the average adjusted AUC (aAUC) over time per mouse. Both IVIgG and BZB as single agents reduced the aAUC, and the effect was greater with the two combined, with all treatments being significant (p < 0.05) (Figure 8B). Application of the Wilcoxon rank sum test demonstrated that only the combination had a significant effect when measured by the aAUC. Analysis of the serum from treated mice indicated that the IVIgG-containing regimens led to high anti-HSP70 IgG titers in the serum (~45 μg/mL at day 39), which was not seen with PBS or BZB (Figure 8C).
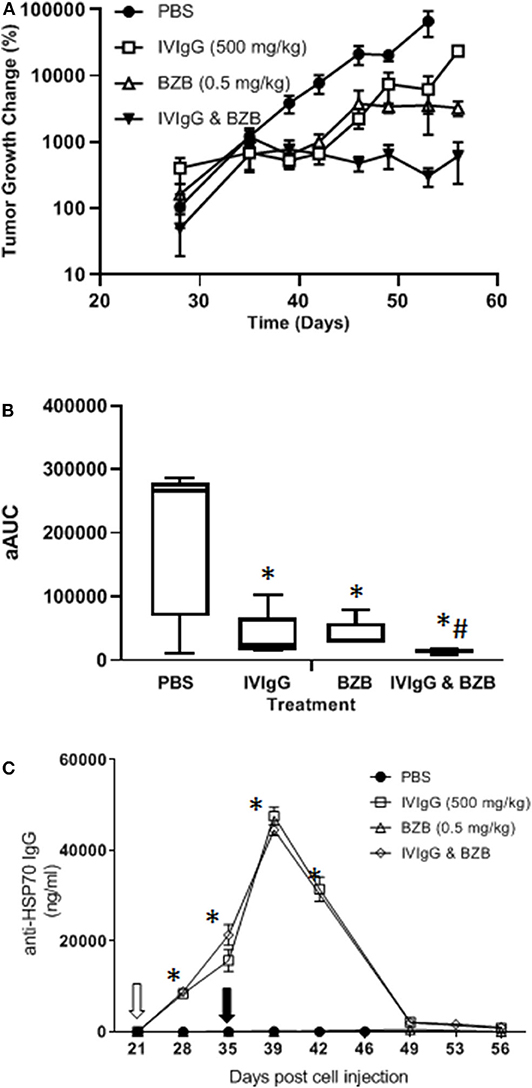
Figure 8. The IVIgG and BZB combination reduces myeloma disease burden and leads to high titers of anti-HSP70 IgG in mouse serum. (A) SCID mice injected with MM1.S cells carrying a luciferase expression cassette were monitored by IVIS imaging following a weekly dose of IVIgG for 3 weeks alone or IVIgG in combination with BZB twice per week for 2 weeks. The difference in tumor growth change (%) is shown. (B) Overall anti-tumor activity measured as the area under the curve (aAUC) over time is shown. (C) Serum titers of anti-HSP70 IgG in mice treated as above. * denotes p < 0.05 compared to the PBS control using the T-test in the above, while #denotes p < 0.05 compared to the PBS control using the Wilcoxon rank sum test.
Discussion
IVIgG has been used in cancer patients for immune maintenance following immune-suppressive therapies, but anecdotal reports detail remissions in a variety of malignancies. We evaluated the anti-cancer effects of IVIgG in myeloma and MCL cell lines, and found it decreased their proliferation and induced apoptosis, which coincided with cell cycle arrest (Figure 1; Supplementary Figure 1). Genomic analyses indicated a significant decrease in HSP70-1 transcription and decreased HSP70-1 protein expression in multiple cell lines with decreased telomerase activity (Figures 3, 4D; Supplementary Table 2). While these two factors do not always correlate quantitatively, there is a qualitative concordance, and differences may be due to the varying half-lives of the corresponding mRNA and protein. Indeed, the half-life of HSP70 mRNA is ~50 min (51), while that for HSP70 protein can be from 2 h (52) to up to 48 h or more (53) depending on the model and conditions studied. Interestingly, IVIgG suppressed activity of the endogenous HSP70 promoter (Figure 4B), while exogenous HSP70 activated HSP70 promoter activity (Figure 4C). It is tempting to speculate that these two are linked, and since extracellular HSP70 has been reported to induce Nuclear factor kappa B (NF-κB) in at least some models (54), and NF-κB can in turn up-regulate HSP70 (55), this may be the basis for such a positive feedback loop. However, additional studies will be needed to better understand this link, and it could be that IVIgG and HSP70 work on different pathways altogether.
Testing of multiple different IVIgG lots demonstrated a high titer of anti-HSP70 IgG, which recognized HSP70-1 by immunoblot and ELISA (Figure 5), and its effect on HSP70-1 was inhibited by exogenous HSP70-1 (Figure 5). A variety of chemotherapeutics induce HSP70-1, and this mechanism can help tumors circumvent pro-apoptotic effects (56). Notably, IVIgG suppressed HSP70-1 up-regulation by AUY922 when it was administered first, or with both together (Figure 6). Also, IVIgG with AUY922 or BZB enhanced anti-myeloma activity, and IVIgG enhanced BZB's activity against a BZB-resistant cell line (Figure 7). This sensitization occurred in association with an inhibition of transfer of EVs which themselves contained HSP70 (Figure 7). Finally, in an in vivo model, IVIgG with BZB were superior to the single agents, and increased anti-HSP70 IgG titers in mouse serum (Figure 8). It is possible that these in vivo effects of IVIgG could have been mediated in part through antibody-dependent cellular cytotoxicity by natural killer (NK) cells in the C.B17-SCID model. However, the mechanistic in vitro studies demonstrate that IVIgG can influence tumor cell sensitivity by influencing HSP70-1 levels in the absence of immune effector cells. It would also be interesting in future studies to examine cell surface HSP70-1 levels and how these are changed, if at all, by IVIgG, since tumor cells do express HSP70 (57, 58), and this can be a target for NK-mediated immune responses (59, 60).
One interesting aspect of our combination studies was that cells exposed to IVIgG first, and then an HSP90 inhibitor, suffered a greater loss in viability than if AUY922 was given first. IVIgG down-regulates HSP70-1 both by a direct mechanism, since it reduces cellular uptake of HSP70-1-containing EVs (Figure 7E), and an indirect mechanism by reducing HSP70-1 transcription (Figure 4B). The latter is likely to be a slower mechanism since the half-life of HSP70 mRNA is ~50 min (51). Therefore, if AUY922 is given first and strongly induces HSP70 mRNA levels, then IVIgG is unable to as fully suppress HSP70-1 protein translation, leaving higher intracellular HSP70-1 levels (Figure 6D). This in turn allows cells to be more resistant to AUY922, producing a less robust reduction in cell viability, and arguing that, if this were ever to be a therapeutic strategy, pre-treatment with IVIgG should be prioritized.
IVIgG has previously been shown to have an array of effects in cancer, ranging from anti-inflammatory effects to the presence of specific antibodies that target proteins which impact tumor pathobiology. The spectrum of antibodies within IVIgG is reflective of normal serum from healthy individuals, and includes anti-idiotype antibodies to DNA, antibodies against TNF family members, Metalloproteinase-9 (61, 62), IL-6, IL-8, and Vascular endothelial growth factor (63). Indeed, in our experiments, IVIgG modulated several cytokines important for tumor cell growth (Figure 2; Supplementary Table 1). Several cancers, including pancreatic cancer, melanoma, sarcoma, and colon cancer, have been reported to respond to IVIgG in preclinical and clinical studies (27, 28, 64). Specific effects within each tumor type may depend on their antigenic burden or cytokine-dependence, and on the amount of anti-cancer IgG in the preparation. Immune modulatory effects of IVIgG may be mediated through Fc-gamma receptors, which are abundantly expressed on immune cells (65), and the in vitro effects of IVIgG often require a cross-linking antibody. However, we found that IVIgG's effect could be independent of the Fc-gamma receptor, as HS-5 stromal and AGS gastric cancer cell lines do not express these receptors (Figure 1).
Because HSP70-1 is abundantly expressed in many cancer types, our findings of high anti-HSP70 IgG titers within IVIgG is novel and potentially relevant. The presence of anti-HSP70-1 IgG in IVIgG indicates that healthy donors generate these antibodies to potentially modulate HSP70-1 function. Notably, HSP70-1 levels increase with pathogenic and normal health conditions, including after exercise and with cancer, preeclampsia, sickle cell disease, and septic shock (66, 67). Presence of anti-HSP70-1 IgG in these states represents a response to stress, while HSP70-1 protein levels in the circulation are decreased during normal pregnancy (68), which may decrease the mother's immune response to the fetus. Also of interest, anti-HSP70 IgG levels in expectant mothers were reportedly elevated in those whose developing children had fetal abnormalities and lower in those with normal fetuses (69). High HSP70-1 and anti-HSP70-1 IgG levels have also been reported in chronic spontaneous urticaria, which is associated with autoimmunity. Healthy donors had titers of anti-HSP70-1 IgG, IgM, and IgA at 53.36 μg/mL compared to 188.67 in the urticaria patients (70). The ability of the anti-HSP70 IgG in IVIgG to suppress HSP70-1 in vitro indicates that this could occur in vivo to control HSP70-1 levels, and the presence of anti-HSP70 IgG in IVIgG indicates that this could be a normal physiologic response. Higher levels of anti-HSP70 IgG in lung cancer patients have been shown to not correlate with increased HSP70 in the serum (67), and this could represent a breakdown in the immune response and a failure to elicit an increase in humoral immunity.
HSP70-1 is associated with drug resistance and is commonly found on EVs, including exosomes, microvesicles/ectosomes, oncosomes, and apoptotic bodies (50). This makes HSP70-1 especially interesting, particularly as its location on the EV surface can be used for purification (71) and therapeutically with antibodies or peptides (72). Our comparison of HSP70-1 expression in cell lysates and EVs indicated that the EVs contained a faster migrating HSP70-1 form (Figure 6C), suggesting that these EVs carry different cargo. Indeed, EVs may carry different/altered amounts of microRNA (73) compared to their producer cells and have different EV membrane protein cargo (74). The function of this faster migrating form remains to be determined, but its presence suggests it may have potential exocrine/paracrine effects. Our data showing that IVIgG reduced uptake of EVs into cells, and prevented transfer of BZB resistance, suggests this effect could be a result of the anti-HSP70 IgG in the preparation. We have not evaluated which mediators the EVs convey to recipient cells to transfer BZB resistance, but the proteasome subunits PSMA3 and PSMA3-S1 may transfer such resistance in vitro, and can be found in BZB-resistant myeloma patient EVs (75).
Our preclinical data may provide another potential facet to the growing list of therapeutic effects of IVIgG, and may help explain its activities against various tumors. The broad spectrum of IgG against various antigens in IVIgG precludes us from narrowing down the exact contribution of anti-HSP70 IgG, but we did demonstrate that exogenous HSP70-1 decreased suppression of HSP70-1 in treated cells (Figure 5E). Addition of IVIgG to the cancer armamentarium is an interesting idea, but may be precluded by its availability and cost, especially since these effects typically require the high, 1–2 g/kg dosing used in inflammatory disease. Use of HSP70-1 monoclonal antibodies (76) could be an alternative, since their greater specificity could allow for lower dosing and cost, though further studies would be needed to identify which epitopes are exposed and best targeted. Indeed, the recent interest in cancer immune therapy makes IVIgG a potential road map to identify new therapies that could be generated as monoclonal antibodies. It also suggests that a potential anti-HSP70 IgG-based therapy is unlikely to have significant side effects, given that high titers of such antibodies are already found in humans.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics Statement
The animal study was reviewed and approved by MD Anderson Cancer Center IACUC.
Author Contributions
RJ designed and performed most of the research and wrote the manuscript. RS designed and performed research and data analysis. FS performed the animal studies. JW and XW performed research. IK performed research, animal work, and cell studies. RD performed GEP. RO designed and supervised all the research completed herein and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported in part by funding from the US National Institutes of Health by grants P50 CA142509 (RO), R01 CA184464 (RO), and 194264 (RO). Flow cytometry services were provided by the MD Anderson Flow Cytometry Core Facility, which was supported by the MD Anderson Cancer Center Support Grant (P30 CA016672). Additional support came from the National Cancer Institute (R01s CA184464 and CA194264), the Leukemia & Lymphoma Society Specialized Center of Research (SCOR-12206-17), the Florence Maude Thomas Cancer Research Professorship, and the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation.
Conflict of Interest
RO declares research funding from BioTheryX, and has served on advisory boards for Amgen, Inc., Bristol-Myers Squibb, Celgene, EcoR1 Capital LLC, Forma Therapeutics, Genzyme, GSK Biologicals, Ionis Pharmaceuticals, Inc., Janssen Biotech, Juno Therapeutics, Kite Pharma, Legend Biotech USA, Molecular Partners, Sanofi-Aventis, Servier, and Takeda Pharmaceuticals North America, Inc., and is a Founder of Asylia Therapeutics, Inc., with an equity interest. RJ is a Founder of Asylia Therapeutics, Inc., with an equity interest.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.01816/full#supplementary-material
References
1. Landgren O, Hofmann JN, McShane CM, Santo L, Hultcrantz M, Korde N, et al. Association of immune marker changes with progression of monoclonal gammopathy of undetermined significance to multiple myeloma. JAMA Oncol. (2019) 5:1293–301. doi: 10.1001/jamaoncol.2019.1568
2. Sandecka V, Hajek R, Pour L, Spicka I, Scudla V, Gregora E, et al. A first czech analysis of 1887 cases with monoclonal gammopathy of undetermined significance. Eur J Haematol. (2017) 99:80–90. doi: 10.1111/ejh.12894
3. Kuwabara C, Sakuma Y, Kume A, Tamura Y, Shimizu R, Iwai R, et al. Characteristics and prognosis of patients with non-immunoglobulin-M monoclonal gammopathy of undetermined significance: a retrospective study. Int J Hematol. (2019) 109:154–61. doi: 10.1007/s12185-018-2555-7
4. Sorrig R, Klausen TW, Salomo M, Vangsted AJ, Ostergaard B, Gregersen H, et al. Smoldering multiple myeloma risk factors for progression: a Danish population-based cohort study. Eur J Haematol. (2016) 97:303–9. doi: 10.1111/ejh.12728
5. Fernandez de Larrea C, Isola I, Pereira A, Cibeira MT, Magnano L, Tovar N, et al. Evolving M-protein pattern in patients with smoldering multiple myeloma: impact on early progression. Leukemia. (2018) 32:1427–34. doi: 10.1038/s41375-018-0013-4
6. Wu V, Moshier E, Leng S, Barlogie B, Cho HJ, Jagannath S, et al. Risk stratification of smoldering multiple myeloma: predictive value of free light chains and group-based trajectory modeling. Blood Adv. (2018) 2:1470–9. doi: 10.1182/bloodadvances.2018016998
7. Sorrig R, Klausen TW, Salomo M, Vangsted AJ, Frolund UC, Andersen KT, et al. Immunoparesis in newly diagnosed multiple myeloma patients: effects on overall survival and progression free survival in the Danish population. PloS ONE. (2017) 12:e0188988. doi: 10.1371/journal.pone.0188988
8. Heaney JLJ, Campbell JP, Iqbal G, Cairns D, Richter A, Child JA, et al. Characterisation of immunoparesis in newly diagnosed myeloma and its impact on progression-free and overall survival in both old and recent myeloma trials. Leukemia. (2018) 32:1727–38. doi: 10.1038/s41375-018-0163-4
9. Jimenez-Zepeda VH, Duggan P, Neri P, Chaudhry A, Tay J, Bahlis N. Immunoparesis and polyclonal immunoglobulin recovery after auto-SCT for patients with multiple myeloma treated at a single institution. Leukem Lymphom. (2018) 59:1920–6. doi: 10.1080/10428194.2017.1403026
10. Gonzalez-Calle V, Cerda S, Labrador J, Sobejano E, Gonzalez-Mena B, Aguilera C, et al. Recovery of polyclonal immunoglobulins one year after autologous stem cell transplantation as a long-term predictor marker of progression and survival in multiple myeloma. Haematologica. (2017) 102:922–31. doi: 10.3324/haematol.2016.158345
11. Sorrig R, Klausen TW, Salomo M, Vangsted A, Gimsing P. Risk factors for blood stream infections in multiple myeloma: a population-based study of 1154 patients in Denmark. Eur J Haematol. (2018) 101:21–7. doi: 10.1111/ejh.13066
12. Valkovic T, Gacic V, Nacinovic-Duletic A. Multiple myeloma index for risk of infection. J Cancer. (2018) 9:2211–4. doi: 10.7150/jca.24288
13. Cesana C, Nosari AM, Klersy C, Miqueleiz S, Rossi V, Ferrando P, et al. Risk factors for the development of bacterial infections in multiple myeloma treated with two different vincristine-adriamycin-dexamethasone schedules. Haematologica. (2003) 88:1022−8.
14. Chapel HM, Lee M, Hargreaves R, Pamphilon DH, Prentice AG. Randomised trial of intravenous immunoglobulin as prophylaxis against infection in plateau-phase multiple myeloma. The UK group for immunoglobulin replacement therapy in multiple myeloma. Lancet. (1994) 343:1059–63. doi: 10.1016/S0140-6736(94)90180-5
15. Musto P, Brugiatelli M, Carotenuto M. Prophylaxis against infections with intravenous immunoglobulins in multiple myeloma. Br J Haematol. (1995) 89:945–6. doi: 10.1111/j.1365-2141.1995.tb08447.x
16. Girmenia C, Cavo M, Offidani M, Scaglione F, Corso A, Di Raimondo F, et al. Management of infectious complications in multiple myeloma patients: expert panel consensus-based recommendations. Blood Rev. (2019) 34:84–94. doi: 10.1016/j.blre.2019.01.001
17. Krivan G, Jolles S, Granados EL, Paolantonacci P, Ouaja R, Cisse OA, et al. New insights in the use of immunoglobulins for the management of immune deficiency (PID) patients. Am J Clin Exp Immunol. (2017) 6:76–83.
18. Galeotti C, Kaveri SV, Bayry J. IVIG-mediated effector functions in autoimmune and inflammatory diseases. Int Immunol. (2017) 29:491–8. doi: 10.1093/intimm/dxx039
19. Ichiyama T, Ueno Y, Hasegawa M, Niimi A, Matsubara T, Furukawa S. Intravenous immunoglobulin inhibits NF-kappaB activation and affects Fcgamma receptor expression in monocytes/macrophages. Naunyn Schmiedebergs Arch Pharmacol. (2004) 369:428–33. doi: 10.1007/s00210-004-0877-x
20. Ichiyama T, Ueno Y, Isumi H, Niimi A, Matsubara T, Furukawa S. An immunoglobulin agent (IVIG) inhibits NF-kappaB activation in cultured endothelial cells of coronary arteries in vitro. Inflamm Res. (2004) 53:253–6. doi: 10.1007/s00011-004-1255-3
21. Murakami K, Suzuki C, Fujii A, Kobayashi F, Nakano A, Kamizono A. Intravenous immunoglobulin preparation prevents the production of pro-inflammatory cytokines by modulating NFkappaB and MAPKs pathways in the human monocytic THP-1 cells stimulated with procalcitonin. Inflamm Res. (2014) 63:711–8. doi: 10.1007/s00011-014-0744-2
22. Liu SY, Yu XL, Zhu J, Liu XM, Zhang Y, Dong QX, et al. Intravenous immunoglobulin ameliorates motor and cognitive deficits and neuropathology in R6/2 mouse model of Huntington's disease by decreasing mutant huntingtin protein level and normalizing NF-kappaB signaling pathway. Brain Res. (2018) 1697:21–33. doi: 10.1016/j.brainres.2018.06.009
23. Zhou C, Huang M, Xie L, Shen J, Xiao T, Wang R. IVIG inhibits TNF-alpha-induced MMP9 expression and activity in monocytes by suppressing NF-kappaB and P38 MAPK activation. Int J Clin Exp Pathol. (2015) 8:15879–86.
24. Loubaki L, Chabot D, Pare I, Drouin M, Bazin R. MiR-146a potentially promotes IVIg-mediated inhibition of TLR4 signaling in LPS-activated human monocytes. Immunol Lett. (2017) 185:64–73. doi: 10.1016/j.imlet.2017.02.015
25. Murakami K, Suzuki C, Kobayashi F, Nakano A, Fujii A, Sakai K, et al. Intravenous immunoglobulin preparation attenuates LPS-induced production of pro-inflammatory cytokines in human monocytic cells by modulating TLR4-mediated signaling pathways. Naunyn Schmiedebergs Arch Pharmacol. (2012) 385:891–8. doi: 10.1007/s00210-012-0765-8
26. Kessel A, Peri R, Haj T, Snir A, Slobodin G, Sabo E, et al. IVIg attenuates TLR-9 activation in B cells from SLE patients. J Clin Immunol. (2011) 31:30–8. doi: 10.1007/s10875-010-9469-3
27. Shoenfeld Y, Krause I. i.v.IG for autoimmune, fibrosis, and malignant conditions: our experience with 200 patients. J Clin Immunol. (2004) 24:107–14. doi: 10.1023/B:JOCI.0000019809.55787.ec
28. Danieli MG, Gelardi C, Pedini V, Gabrielli A. Potential anti-tumor activity of intravenous and subcutaneous immunoglobulin. Isr Med Assoc J. (2018) 20:782–3.
29. Schachter J, Katz U, Mahrer A, Barak D, David LZ, Nusbacher J, et al. Efficacy and safety of intravenous immunoglobulin in patients with metastatic melanoma. Ann N Y Acad Sci. (2007) 1110:305–14. doi: 10.1196/annals.1423.032
30. Muller F, Aukrust P, Nordoy I, Froland SS. Possible role of interleukin-10 (IL-10) and CD40 ligand expression in the pathogenesis of hypergammaglobulinemia in human immunodeficiency virus infection: modulation of IL-10 and Ig production after intravenous Ig infusion. Blood. (1998) 92:3721–9. doi: 10.1182/blood.V92.10.3721
31. Manasanch EE, Orlowski RZ. Proteasome inhibitors in cancer therapy. Nat Rev Clin Oncol. (2017) 14:417–33. doi: 10.1038/nrclinonc.2016.206
32. Barahona Afonso AF, Joao CM. The Production Processes and Biological Effects of Intravenous Immunoglobulin. Biomolecules. (2016) 6:15. doi: 10.3390/biom6010015
33. Sapir T, Shoenfeld Y. Uncovering the hidden potential of intravenous immunoglobulin as an anticancer therapy. Clin Rev Allergy Immunol. (2005) 29:307–10. doi: 10.1385/CRIAI:29:3:307
34. Jones RJ, Dickerson S, Bhende PM, Delecluse HJ, Kenney SC. Epstein-Barr virus lytic infection induces retinoic acid-responsive genes through induction of a retinol-metabolizing enzyme, DHRS9. J Bio Chem. (2007) 282:8317–24. doi: 10.1074/jbc.M608667200
35. Hong GK, Gulley ML, Feng WH, Delecluse HJ, Holley-Guthrie E, Kenney SC. Epstein-Barr virus lytic infection contributes to lymphoproliferative disease in a SCID mouse model. J Virol. (2005) 79:13993–4003. doi: 10.1128/JVI.79.22.13993-14003.2005
36. Dunning MJ, Barbosa-Morais NL, Lynch AG, Tavare S, Ritchie ME. Statistical issues in the analysis of illumina data. BMC Bioinform. (2008) 9:85. doi: 10.1186/1471-2105-9-85
37. Kuhn DJ, Berkova Z, Jones RJ, Woessner R, Bjorklund CC, Ma W, et al. Targeting the insulin-like growth factor-1 receptor to overcome bortezomib resistance in preclinical models of multiple myeloma. Blood. (2012) 120:3260–70. doi: 10.1182/blood-2011-10-386789
38. Lai CP, Kim EY, Badr CE, Weissleder R, Mempel TR, Tannous BA, et al. Visualization and tracking of tumour extracellular vesicle delivery and RNA translation using multiplexed reporters. Nat Commun. (2015) 6:7029. doi: 10.1038/ncomms8029
39. Wu J, Houghton PJ. Interval approach to assessing antitumor activity for tumor xenograft studies. Pharm Stat. (2010) 9:46–54. doi: 10.1002/pst.369
40. Morell. A. Pharmacokinetics of intravenous immunoglobulin preparations. In: Lee M, Strand V, editor. Intravenous Immunoglobulins in Clinical Practice. New York, NY: Marcel Dekker, Inc. (1997). p. 1.
41. Schuck S, Prinz WA, Thorn KS, Voss C, Walter P. Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. J Cell Biol. (2009) 187:525–36. doi: 10.1083/jcb.200907074
42. Sabnis AJ, Guerriero CJ, Olivas V, Sayana A, Shue J, Flanagan J, et al. Combined chemical-genetic approach identifies cytosolic HSP70 dependence in rhabdomyosarcoma. Proc Natl Acad Sci USA. (2016) 113:9015–20. doi: 10.1073/pnas.1603883113
43. Forsythe HL, Jarvis JL, Turner JW, Elmore LW, Holt SE. Stable association of hsp90 and p23, but Not hsp70, with active human telomerase. J Biol Chem. (2001) 276:15571–4. doi: 10.1074/jbc.C100055200
44. Perera ON, Sobinoff AP, Teber ET, Harman A, Maritz MF, Yang SF, et al. Telomerase promotes formation of a telomere protective complex in cancer cells. Sci Adv. (2019) 5:eaav4409. doi: 10.1126/sciadv.aav4409
45. Walerych D, Olszewski MB, Gutkowska M, Helwak A, Zylicz M, Zylicz A. Hsp70 molecular chaperones are required to support p53 tumor suppressor activity under stress conditions. Oncogene. (2009) 28:4284–94. doi: 10.1038/onc.2009.281
46. Theriault JR, Mambula SS, Sawamura T, Stevenson MA, Calderwood SK. Extracellular HSP70 binding to surface receptors present on antigen presenting cells and endothelial/epithelial cells. FEBS Lett. (2005) 579:1951–60. doi: 10.1016/j.febslet.2005.02.046
47. Yuno A, Lee MJ, Lee S, Tomita Y, Rekhtman D, Moore B, et al. Clinical evaluation and biomarker profiling of hsp90 inhibitors. Methods Mol Biol. (2018) 1709:423–41. doi: 10.1007/978-1-4939-7477-1_29
48. Mitsiades N, Mitsiades CS, Poulaki V, Chauhan D, Fanourakis G, Gu X, et al. Molecular sequelae of proteasome inhibition in human multiple myeloma cells. Proc Natl Acad Sci USA. (2002) 99:14374–9. doi: 10.1073/pnas.202445099
49. Multhoff G, Hightower LE. Distinguishing integral and receptor-bound heat shock protein 70 (Hsp70) on the cell surface by Hsp70-specific antibodies. Cell Stress Chaperones. (2011) 16:251–5. doi: 10.1007/s12192-010-0247-1
50. Kowal J, Arras G, Colombo M, Jouve M, Morath JP, Primdal-Bengtson B, et al. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci USA. (2016) 113:E968–77. doi: 10.1073/pnas.1521230113
51. Theodorakis NG, Morimoto RI. Posttranscriptional regulation of hsp70 expression in human cells: effects of heat shock, inhibition of protein synthesis, and adenovirus infection on translation and mRNA stability. Mol Cell Biol. (1987) 7:4357–68. doi: 10.1128/MCB.7.12.4357
52. Li D, Duncan RF. Transient acquired thermotolerance in drosophila, correlated with rapid degradation of Hsp70 during recovery. Eur J Biochem. (1995) 231:454–65. doi: 10.1111/j.1432-1033.1995.tb20719.x
53. Turturici G, Geraci F, Candela ME, Cossu G, Giudice G, Sconzo G. Hsp70 is required for optimal cell proliferation in mouse A6 mesoangioblast stem cells. Biochem J. (2009) 421:193–200. doi: 10.1042/BJ20082309
54. Somensi N, Brum PO, de Miranda Ramos V, Gasparotto J, Zanotto-Filho A, Rostirolla DC, et al. Extracellular HSP70 Activates ERK1/2, NF-kB and pro-inflammatory gene transcription through binding with RAGE in A549 human lung cancer cells. Cell Physiol Biochem. (2017) 42:2507–22. doi: 10.1159/000480213
55. Stuhlmeier KM. Activation and regulation of Hsp32 and Hsp70. Eur J Biochem. (2000) 267:1161–7. doi: 10.1046/j.1432-1327.2000.01112.x
56. Murphy ME. The HSP70 family and cancer. Carcinogenesis. (2013) 34:1181–8. doi: 10.1093/carcin/bgt111
57. Stangl S, Gehrmann M, Riegger J, Kuhs K, Riederer I, Sievert W, et al. Targeting membrane heat-shock protein 70 (Hsp70) on tumors by cmHsp70.1 antibody. Proc Natl Acad Sci USA. (2011) 108:733–8. doi: 10.1073/pnas.1016065108
58. Shevtsov M, Huile G, Multhoff G. Membrane heat shock protein 70: a theranostic target for cancer therapy. Philos Trans R Soc Lond B Biol Sci. (2018) 373:20160526. doi: 10.1098/rstb.2016.0526
59. Botzler C, Li G, Issels RD, Multhoff G. Definition of extracellular localized epitopes of Hsp70 involved in an NK immune response. Cell Stress Chaperones. (1998) 3:6–11. doi: 10.1379/1466-1268(1998)003<0006:DOELEO>2.3.CO;2
60. Multhoff G, Mizzen L, Winchester CC, Milner CM, Wenk S, Eissner G, et al. Heat shock protein 70 (Hsp70) stimulates proliferation and cytolytic activity of natural killer cells. Exp Hematol. (1999) 27:1627–36. doi: 10.1016/S0301-472X(99)00104-6
61. Shoenfeld Y, Fishman P. Gamma-globulin inhibits tumor spread in mice. Int Immunol. (1999) 11:1247–52. doi: 10.1093/intimm/11.8.1247
62. Shapiro S, Shoenfeld Y, Gilburd B, Sobel E, Lahat N. Intravenous gamma globulin inhibits the production of matrix metalloproteinase-9 in macrophages. Cancer. (2002) 95:2032–7. doi: 10.1002/cncr.10905
63. Schwartz-Albiez R, Monteiro RC, Rodriguez M, Binder CJ, Shoenfeld Y. Natural antibodies, intravenous immunoglobulin and their role in autoimmunity, cancer and inflammation. Clin Exp Immunol. (2009) 158(Suppl. 1):43–50. doi: 10.1111/j.1365-2249.2009.04026.x
64. Shoenfeld Y, Levy Y, Fishman P. Shrinkage of melanoma metastases following high dose intravenous immunoglobulin treatment. Isr Med Assoc J. (2001) 3:698–9.
65. Nagelkerke SQ, Kuijpers TW. Immunomodulation by Ivig and the role of fc-gamma receptors: classic mechanisms of action after all? Front Immunol. (2014) 5:674. doi: 10.3389/fimmu.2014.00674
66. Molvarec A, Rigo J Jr, Lazar L, Balogh K, Mako V, Cervenak L, et al. Increased serum heat-shock protein 70 levels reflect systemic inflammation, oxidative stress and hepatocellular injury in preeclampsia. Cell Stress Chaperones. (2009) 14:151–9. doi: 10.1007/s12192-008-0067-8
67. Ren B, Luo S, Xu F, Zou G, Xu G, He J, et al. The expression of DAMP proteins HSP70 and cancer-testis antigen SPAG9 in peripheral blood of patients with HCC and lung cancer. Cell Stress Chaperones. (2017) 22:237–44. doi: 10.1007/s12192-016-0758-5
68. Molvarec A, Tamasi L, Losonczy G, Madach K, Prohaszka Z, Rigo J Jr. Circulating heat shock protein 70 (HSPA1A) in normal and pathological pregnancies. Cell Stress Chaperones. (2010) 15:237–47. doi: 10.1007/s12192-009-0146-5
69. Child DF, Hudson PR, Hunter-Lavin C, Mukhergee S, China S, Williams CP, et al. Birth defects and anti-heat shock protein 70 antibodies in early pregnancy. Cell Stress Chaperones. (2006) 11:101–5. doi: 10.1379/CSC-130R1.1
70. Kasperska-Zajac A, Damasiewicz-Bodzek A, Bieniek K, Skrzypulec-Frankel A, Tyrpien-Golder K, Grzanka A. Elevated circulating heat shock protein 70 and its antibody concentrations in chronic spontaneous urticaria. Int J Immunopathol Pharmacol. (2018) 31:394632017750440. doi: 10.1177/0394632017750440
71. Ghosh A, Davey M, Chute IC, Griffiths SG, Lewis S, Chacko S, et al. Rapid isolation of extracellular vesicles from cell culture and biological fluids using a synthetic peptide with specific affinity for heat shock proteins. PLoS ONE. (2014) 9:e110443. doi: 10.1371/journal.pone.0110443
72. Gobbo J, Marcion G, Cordonnier M, Dias AMM, Pernet N, Hammann A, et al. Restoring anticancer immune response by targeting tumor-derived exosomes with a hsp70 peptide aptamer. J Natl Cancer Inst. (2016) 108:330. doi: 10.1093/jnci/djv330
73. Balbi C, Piccoli M, Barile L, Papait A, Armirotti A, Principi E, et al. First characterization of human amniotic fluid stem cell extracellular vesicles as a powerful paracrine tool endowed with regenerative potential. Stem Cell Transl Med. (2017) 6:1340–55. doi: 10.1002/sctm.16-0297
74. Moreno-Gonzalo O, Fernandez-Delgado I, Sanchez-Madrid F. Post-translational add-ons mark the path in exosomal protein sorting. Cell Mol Life Sci. (2018) 75:1–19. doi: 10.1007/s00018-017-2690-y
75. Xu H, Han H, Song S, Yi N, Qian C, Qiu Y, et al. Exosome-Transmitted PSMA3 and PSMA3-AS1 promote proteasome inhibitor resistance in multiple myeloma. Clin Cancer Res. (2019) 25:1923–35. doi: 10.1158/1078-0432.CCR-18-2363
Keywords: IVIgG, HSP70, heat shock response, bortezomib, extracellular vesicles
Citation: Jones RJ, Singh RK, Shirazi F, Wan J, Wang H, Wang X, Ha MJ, Baljevic M, Kuiatse I, Davis RE and Orlowski RZ (2020) Intravenous Immunoglobulin G Suppresses Heat Shock Protein (HSP)-70 Expression and Enhances the Activity of HSP90 and Proteasome Inhibitors. Front. Immunol. 11:1816. doi: 10.3389/fimmu.2020.01816
Received: 26 April 2020; Accepted: 07 July 2020;
Published: 13 August 2020.
Edited by:
Nurit Hollander, Tel Aviv University, IsraelReviewed by:
Jason C. Young, McGill University, CanadaGabriele Multhoff, Technical University of Munich, Germany
Copyright © 2020 Jones, Singh, Shirazi, Wan, Wang, Wang, Ha, Baljevic, Kuiatse, Davis and Orlowski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Richard J. Jones, cmpvbmVzQG1kYW5kZXJzb24ub3Jn