 Lindsay M. Snyder
Lindsay M. Snyder Juhi Arora
Juhi Arora Mary J. Kennett1
Mary J. Kennett1 Margherita T. Cantorna
Margherita T. Cantorna- 1Department of Veterinary and Biomedical Sciences, The Pennsylvania State University, University Park, PA, United States
- 2Center for Evolutionary & Theoretical Immunology, University of New Mexico, Albuquerque, NM, United States
Vitamin A deficiency (A–) increases morbidity and mortality to gastrointestinal (GI) infection. Blocking retinoid signaling (dominant negative retinoic acid receptor, dnRAR) in intestinal epithelial cells (IEC, IECdnRAR) had no effect on vitamin A absorption, the expression of tight junction proteins or the integrity of the barrier. Immune cells in the gut were present in normal frequencies in the IECdnRAR mice, with the exception of the T cell receptor (TCR)αβ+/CD8αα cells, which were significantly lower than in wildtype littermates. Challenging the IECdnRAR mice with dextran sodium sulfate to induce colitis or Citrobacter rodentium infection resulted in similar disease to wildtype littermates. Feeding mice vitamin A deficient diets reduced vitamin A status and the A– IECdnRAR mice developed more severe colitis and C. rodentium infection. In particular, retinoid signaling in the IEC was crucial for the A– host to survive early infection following C. rodentium. Treating A– mice with retinoic acid (RA) beginning on the day of infection protects most mice from early lethality. However, RA treatment of the A– IECdnRAR mice was ineffective for preventing lethality following C. rodentium infection. Retionid signaling in IEC is critical, especially when there are reduced levels of dietary vitamin A. IEC are direct targets of vitamin A for mounting early defense against infection.
Introduction
Vitamin A is a micronutrient that is essential for embryonic development, vision, and immune function. Vitamin A deficiency (A–) is a persistent problem in resource limited countries worldwide. It is estimated that over 250 million preschool-age children are vitamin A deficient (1, 2). Vitamin A deficient children develop night blindness and have increased susceptibility to enteric infections (1). Longitudinal studies have shown that supplementing children with vitamin A deficiency with one or two bolus doses of vitamin A reduced infection rates and lessened the severity of enteric infections (3). Vitamin A deficiency is prevalent in developing countries and vitamin A protects the gastrointestinal (GI) tract from infection.
The GI tract is composed of a population of heterogeneous cells whose role is to maintain ignorance of the large number of antigens present in food as well as the abundant microbiota. The mechanisms by which tolerance in the gut is controlled includes complex interactions between the microbiota, the intestinal epithelial cells (IEC) and the immune system. The IEC express tight junction proteins that maintain intestinal integrity and pathogen recognition receptors that function as part of the innate immune response (4, 5). When the barrier becomes compromised following infection, bacteria breach the barrier which can elicit local and systemic inflammation (6, 7). IEC regulate and activate intestinal immunity in the GI tract and are important for resistance to infection and development of inflammatory bowel disease (IBD) (6–8). The IEC of the GI tract are important for the regulation and maintenance of homeostasis.
Vitamin A status regulates intestinal barrier function and is important for the production of the mucous layer that lines the gut. Vitamin A deficiency resulted in keratinization of the mucosa (9, 10). The active metabolite of vitamin A (retinoic acid, RA) induced the expression of several tight junction proteins including: ZO-1, occludin, claudin-6, and claudin-7 (11, 12). Supplementing vitamin A deficient children with vitamin A, decreased urine lactulose/ mannitol levels, suggesting that vitamin A enhances the intestinal barrier integrity in humans (13, 14). Vitamin A is an important regulator of IEC and GI barrier function.
Vitamin A is an essential regulator of the development and function of the immune system. In the GI tract, dendritic cells metabolize vitamin A to produce RA locally (15). RA upregulates the expression of gut-homing receptors α4β7 and CCR9 receptors on T and B cells (16, 17). Mice with a dominant negative retinoic acid receptor (dnRAR) expressed in T cells failed to clear Citrobacter rodentium infection (18). RA treatment suppressed experimental colitis by inhibiting IL-17 and inducing IL-10 and T regs in vivo (19). RA induced IL-22 by innate lymphoid cells and γδ T cells that was critical for resolution of inflammation (20). IL-22 induced the production of anti-bacterial peptides by IEC and protected the gut from injury (20, 21). Vitamin A regulates the immune response in the gastrointestinal tract.
We hypothesized that vitamin A was a direct regulator of IEC function and that mice with IEC that were refractory to retinoids would be more susceptible to infectious or chemical injury. To determine the effects of vitamin A on IEC function, mice were generated in which retinoid signaling was inhibited in IEC by expressing the dnRAR in villin expressing cells (IECdnRAR). The dnRAR inhibits retinoid signaling through all 3 RAR (α, β, and γ) receptors (22, 23). The IECdnRAR mice were not different than the wildtype (WT) littermates in growth rate, vitamin A status, barrier function, and expression of other markers of IEC function. The IECdnRAR mice had reduced frequencies of T cell receptor (TCR)αβ+/CD8αα+ cells in the gut compared to the WT littermates. Challenging the IECdnRAR mice with dextran sodium sulfate (DSS) or C. rodentium resulted in colitis that was not different than WT. Feeding, the IECdnRAR mice, vitamin A deficient diets resulted in the increased susceptibility to colitis and severe C. rodentium infection. The A– IECdnRAR mice developed a lethal infection with C. rodentium that was refractory to RA treatment. We concluded that IEC are direct targets of vitamin A that are especially critical when the amount of dietary vitamin A is low. In the A+ IECdnRAR, the role of RA in IEC was compensated for by the effects of vitamin A on other targets in the GI tract.
Materials and Methods
Animals
C57BL/6J villinCre+, LckCre+, or LysMCre+ mice were originally from Jackson Laboratories (Bar Harbor, ME) and were bred and maintained at the Pennsylvania State University (University Park, PA) according to IACUC and university guidelines. Mice expressing dnRARfl/fl were generously provided by Dr. Randolph J. Noelle (Dartmouth Medical School, Lebanon, NH). VillinCre+ mice were crossed with dnRARfl/fl mice to generate IECdnRAR mice with blocked retinoid signaling through all 3 RAR (α, β, and γ) isoforms in intestinal epithelial cells (22). dnRARfl/fl mice were crossed with LckCre+ to make TdnRAR and LysMCre+ to make LysMdnRAR mice. dnRARfl/fl Cre- (WT) littermates were used as controls. A+ and A– mice were generated by feeding pregnant females on purified diets with and without 25 μg/d retinyl acetate as previously described (24). Serum was collected and pooled from each batch of experimental mice to confirm vitamin A status. By 8 wks of age there is a significant difference in serum retinol among the A+ and A– mice (Figure 1A). Vitamin A status was determined by quantifying serum retinol levels using ultra-highpressure liquid chromatography (UPLC). Vitamin A deficiency was defined as serum retinol concentrations <0.7 μmol/L.
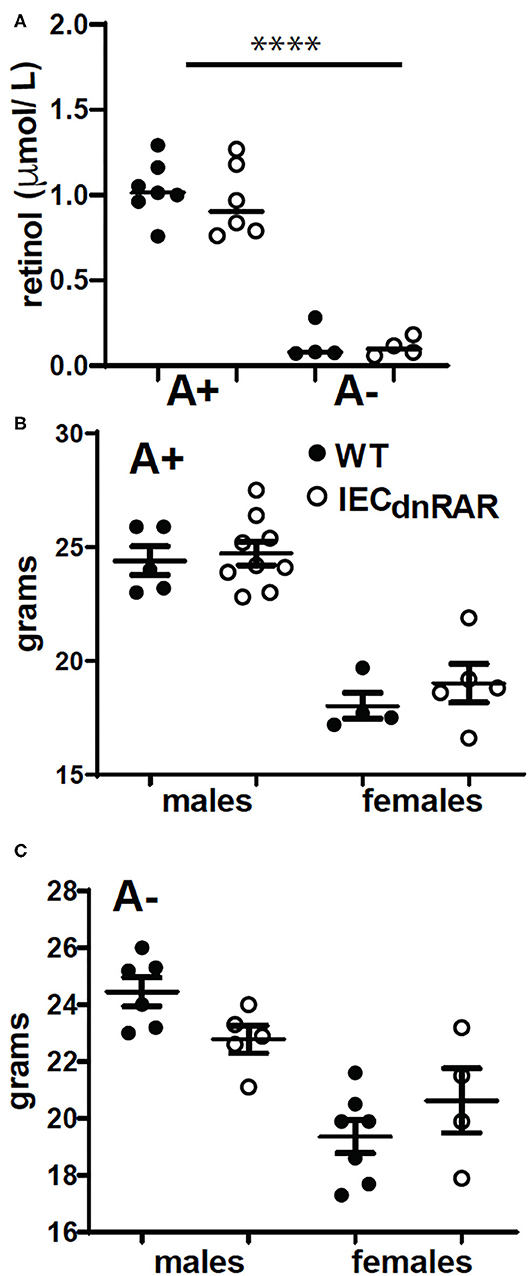
Figure 1. IECdnRAR mice were phenotypically similar to WT mice. For each experiment serum was collected and pooled to monitor vitamin A status. (A) Serum retinol values. Values are the mean ± SEM of 4–7 pooled experiments. WT and IECdnRAR (B) A+ and (C) A– mice were weighed. Values are the mean ± SEM of n = 4–9 mice of each gender. One-way ANOVA with Bonferroni post-test (A), and unpaired student's T-test (B,C). ****P < 0.0001.
Dextran Sodium Sulfate (DSS)
3.75–4.5% DSS (MP Biomedicals, Solon, OH) was administered orally in the drinking water for 5 days followed by 5 days on water for recovery exactly as described (25–28). The amount of DSS used in the experiments was based on the starting weight of the mice and the use of purified diets that require higher amounts of DSS to induce injury (25–28). None of the mice bled rectally and therefore blood scores were not included in the analyses. Body weight changes were monitored for 10 days. Mice were euthanized at d8 or d10 post DSS exposure. Colon lengths were measured, and distal colon tissue was collected for histological analysis (Supplementary Methods).
Citrobacter rodentium
Nalidixic acid resistant C. rodentium strain ICC169 was kindly provided by Gad Frankel (London School of Medicine and Dentistry, London, UK). Bacteria were cultured in Luria-Bertani broth (LB; Becton, Dickinson, & Co, Franklin Lakes, NJ) or LB agar for 18–24 h at 37°C. Overnight log phase bacterial cultures in LB broth were used to prepare inoculums. Adult mice 8–10 wks of age were individually housed, fasted overnight, and then orally gavaged with 100 μl of sterile PBS containing 5 × 109 CFU C. rodentium. Fecal pellets, spleens, and livers were collected, homogenized and plated in serial dilutions on LB agar plates containing nalidixic acid to quantify bacterial burdens and track infection kinetics as we described (18, 24).
Flow Cytometry
Single cell suspensions of spleen, meseneteric lymph nodes (MLN) and thymus were made exactly as previously described (29–31). The Peyer's patches were removed and the entire small intestine (SI) IEL and SI or colon IEC were isolated as we have described previously (30, 32, 33). SI and colon tissues were cut longitudinally to increase surface area and incubated twice (20 min at 37°C) with 1 mM 1,4 dithiothreitol (DTT, Sigma Aldrich) and 10 mM EDTA to release IEC and IEL (30, 32, 33). A 25/40% discontinuous Percoll (Sigma Aldrich) gradient was used to purify IEC while a 40/80% discontinuous gradient was used to purify IEL. 1–2 million cells were stained with fluorescein isothiocyanate (FITC) CD8β, FITC or phycoerythrin (PE) GL3 (γδ TCR), PE-CF594 CD4, PE-Cy 5 TCRβ, Brilliant violet (BV) 421 TLA (BD Biosciences, San Jose, CA), PE IL15Rα (Thermo Fisher Scientific, Waltham, MA), PE-Cy7 TLR4, or PE-Cy7 CD8α (BioLegend, San Diego, CA). CD8αα was detected in the thymus with PE-labeled TL-tetramer (T3b) (34). The tetramers were a gift from Dr. Hilde Cheroutre (La Jolla Institute for Allergy and Immunology, La Jolla, CA). Single positive and fluorescence minus one (FMO) controls were used to set gates. Cells were analyzed on an FC500 benchtop cytometer (Beckman Coulter, Brea, CA) or a Becton Dickinson LSR Fortessa cytometer (Becton, Dickinson and Company, Franklin Lakes, NJ), and data was analyzed using FlowJo 7.6.5 software (Tree Star, Ashland, OR).
FITC Dextran Permeability Assay
4 kDa FITC dextran (Sigma Aldrich, St. Louis, MO) permeability assay was performed as previously described (26). Briefly, mice were fasted for 4 h and then gavaged with an 80 mg/kg dose of FITC dextran. 4 h later, mice were bled to obtain serum and a FITC dextran standard curve (20 to 0.3125 μg/ml) was prepared by diluting the stock solution with PBS. The standard and serum samples from naïve and DSS treated mice were transferred to black bottom 96 well plates and fluorescence was read at 525 nm on a Perkin Elmer Wallace Microplate Reader (GMI, Ramsey, MN). A linear curve was fitted to the standard and used to quantify serum FITC dextran levels.
Histology
Distal colons from naïve and C. rodentium infected mice were fixed in 4% formalin, embedded in paraffin, sectioned, and stained with hematoxylin and eosin at the Pennsylvania State University Animal Diagnostics Laboratory. C. rodentium tissue sections were coded and scored by a board-certified laboratory animal veterinarian with pathology training (Dr. Mary Kennett, University Park, PA). Tissue samples were scored for inflammatory cell infiltrate cell infiltrates and the severity of mucosal damage, edema, and crypt loss (35). The five longest crypts were identified visually from each sample and measured to determine an average crypt length.
RT-PCR
Tissues were snap frozen and stored at −80°C until RNA isolation with TriZOL (Invitrogen, Carlsbad, CA). Cells (2 × 106) were suspended in 0.5 mL TriZOL and stored at −80°C until processing. RNA isolation was performed according to TriZOL manufacturer's protocol. Complementary DNA (cDNA) was created by reverse transcribing 2–4 ug RNA using TaqMan reverse transcription kit (Applied Biosystems, Carlsbad, CA). qPCR was performed using SYBR green mix (BioRad, Hercules, CA) and the MyiQ Single-Color Real Time PCR machine (BioRad). Relative standards were prepared by serially diluting DNA products of the genes of interest. A standard curve was generated to quantify relative expression levels in samples. Relative expression levels were normalized to a housekeeping gene (HPRT) and fold change values were reported relative to WT or untreated control tissues. Primers are listed in Supplementary Table 1.
Statistical Analyses
Statistical analyses were performed using GraphPad Prism (GraphPad, La Jolla, CA). Two-tailed student t-tests were used to compare gene expression fold change, histological scores, and DSS parameters. A student's t-test with Welch's correction was also used to compare data sets with unequal variances and a Mann Whitney test was used to compare data sets that were not normally distributed. One-way analysis of variance (ANOVA) with Bonferroni's post hoc test was used to analyze histology scores. Two-way ANOVA with Bonferroni's post hoc test was used to analyze IEL/thymocyte populations through time and fecal shedding and weight loss curves. Log rank Mantel-Cox test was used to assess survival rates between genotypes and treatments. A P < 0.05 was used as the threshold to determine statistically significant changes.
Results
Normal Intestinal Epithelial Cells in IECdnRAR Mice
Genotype had no effect on body weight in male or female mice that were either A+ (Figure 1B) or A– (Figure 1C). A+ IECdnRAR and A+ WT mice had significantly higher serum retinol than age matched A– IECdnRAR and A– WT mice (Figure 1A). There were no differences in serum retinol as a result of genotype (Figure 1A). The total number of lymphocytes in thymus, spleen, MLN and SI IEL were not different in the IECdnRAR and WT mice (Supplementary Figure 1). The frequencies of CD4+ cells in the MLN and the frequencies of CD19+ cells in the spleen were slightly but significantly lower in the IECdnRAR as compared to the WT mice (Supplementary Figure 1). The number of IEC cells from IECdnRAR and WT mice were not different in either the SI or colon (Supplementary Figure 2A). The frequency of IEC that express TLR-4, IL-15Rα, and TLA were the same in IECdnRAR and WT SI and colon (Supplementary Figure 2). In addition, mean fluorescence intensities (MFI) for TLR4, MHCI, IL15R-α, and TLA were the same on the WT and IECdnRAR IEC (Supplementary Figure 2 and data not shown). The expression of mRNA for madcam1, ccl25, occludin, claudin 6, and claudin 7 were not different in IECdnRAR and WT mice (Supplementary Figure 3). The IEC from IECdnRAR mice were phenotypically similar to WT IEC.
Fewer CD8αα Expressing T Cells in IECdnRAR Mice
IECs are important in the development of intestinal T cell populations. The total number of cells isolated from the SI IEL were the same in A+ IECdnRAR and WT mice (Supplementary Figure 1). The frequencies of TCRαβ+ T cells, CD4+, and TCRγδ+ T cells in the IEL of IECdnRAR and WT were the same in adult mice (Figures 2A–C). There were some significant shifts in CD4+ and CD8+ T cell frequencies in SI IEL as the mice aged (Figures 2C,D). At 8 wks of age there were no differences in CD4+ or CD8+ frequencies but at 12 wks the CD8+ T cells in IECdnRAR mice were significantly higher compared to WT mice (Figure 2D). The TCRαβ+/CD8αα+ cells were lower in the IECdnRAR mice beginning at 5 wks of age and continuing to adulthood (Figure 2E), while the TCRγδ+/CD8αα+ cells were not different in WT and IECdnRAR mice (Figure 2F). All TCRαβ+ T cells, including the TCRαβ+/CD8αα+ cells, develop in the thymus. In order to determine whether the IECdnRAR had a defect in thymocyte development, thymic precusors were measured in IECdnRAR and WT mice. Double negative (DN, CD4–/CD8–) cells become double positive (DP, CD4+/CD8+) cells that then develop into single CD8+ or CD4+ T cells (30, 34, 36). A subset of the DP thymocytes become triple positive (TP, CD4+/CD8+/CD8αα) and then downregulate all three receptors as they exit the thymus (34). TP cells are the thymic precursors of TCRαβ+/CD8αα+ cells in the SI (34). The frequencies of the DN, DP, and TP cells in the thymus were not different in IECdnRAR and WT mice (Figure 3), ruling out a thymus development effect of the IECdnRAR. IL-15, IL-15 receptor (R), TLA and TGF-β are important in the induction of CD8αα on TCRγδ+ and TCRαβ+ T cells in the gut (37). Il15rα, Tla, and Tgfβ mRNA expression was the same in IECdnRAR and WT SI (Supplementary Figure 4). Il15 mRNA expression was significantly higher in IECdnRAR SI than in WT (Supplementary Figure 4). The reduced frequency of TCRαβ+/CD8αα+ in IECdnRAR SI was not due to changes in IL-15, IL-15Rα, TLA, or TGF-β. Expression of the dnRAR in IEC resulted in fewer TCRαβ+/CD8αα+ in the SI as compared to WT mice.
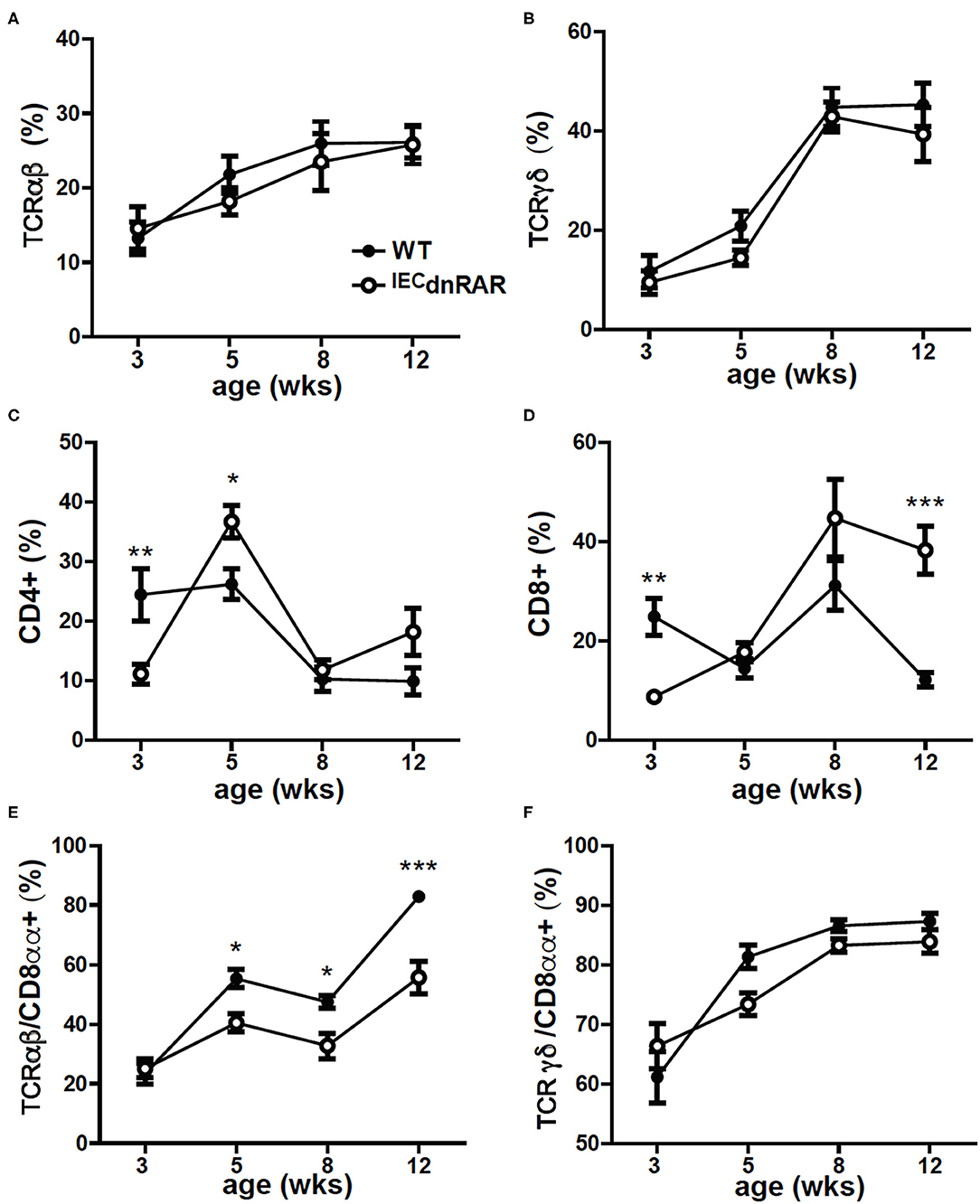
Figure 2. Reduced frequencies of CD8αα+ T cells in the SI of A+ IECdnRAR mice. The A+ WT and A+ IECdnRAR frequencies of (A) TCRαβ+, (B) TCRγδ+, (C) CD4+, (D) CD8+, (E) TCRαβ+/CD8αα+, and (F) TCRγδ+/CD8αα+ T cells in the SI IEL. Values are the mean ± SEM of two combined experiments with n = 7–11 mice at each time point. Two-way ANOVA with Bonferroni post-test, *P < 0.05, **P < 0.01, ***P < 0.001.
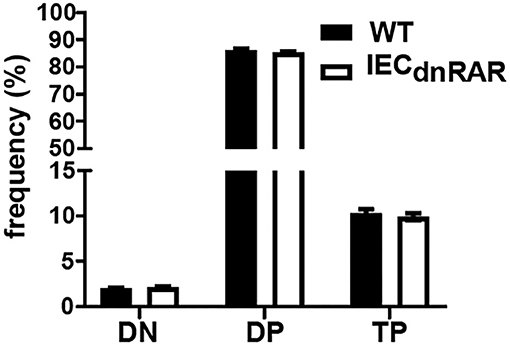
Figure 3. Normal thymocyte development in IECdnRAR mice. The frequencies of DN, DP, and TP cells in the thymus of WT and IECdnRAR mice. Values are mean ± SEM of two combined experiments, n = 6–11 mice of each genotype. Statistical significance was evaluated using two-way ANOVA with Bonferroni post-test.
WT and IECdnRAR Mice Are Resistant to DSS Colitis
TCRαβ+/CD8αα+ T cells protect the host from GI injury (38). Vitamin A sufficient (A+) WT and IECdnRAR mice were treated with DSS to induce colitis. DSS induced weight loss in A+ WT and IECdnRAR mice, when the males and females were analyzed separately (significant time effect, Supplementary Figure 5A), but not when they were combined together (Figure 4A). There was no genotype effect on weight loss following DSS treatment in males, females or combined males and females (Supplementary Figure 5A and Figure 4A). After 8 days of DSS treatment A+ WT and IECdnRAR treated mice had significantly shortened colon lengths compared to day 0 colon length (Figure 4B). There were no differences between the colon lengths of A+ WT and IECdnRAR mice at day 0 or day 8 post-DSS (Figure 4B). There was a small but insignificant increase in intestinal permeability that occurred following DSS treatment of A+ WT and IECdnRAR mice (Figure 4C). Histopathology sections were not different in the A+ WT and IECdnRAR mice at day 10 post-DSS (Supplementary Figures 5C, 6A). There was no effect of IECdnRAR expression on the susceptibility of mice to DSS.
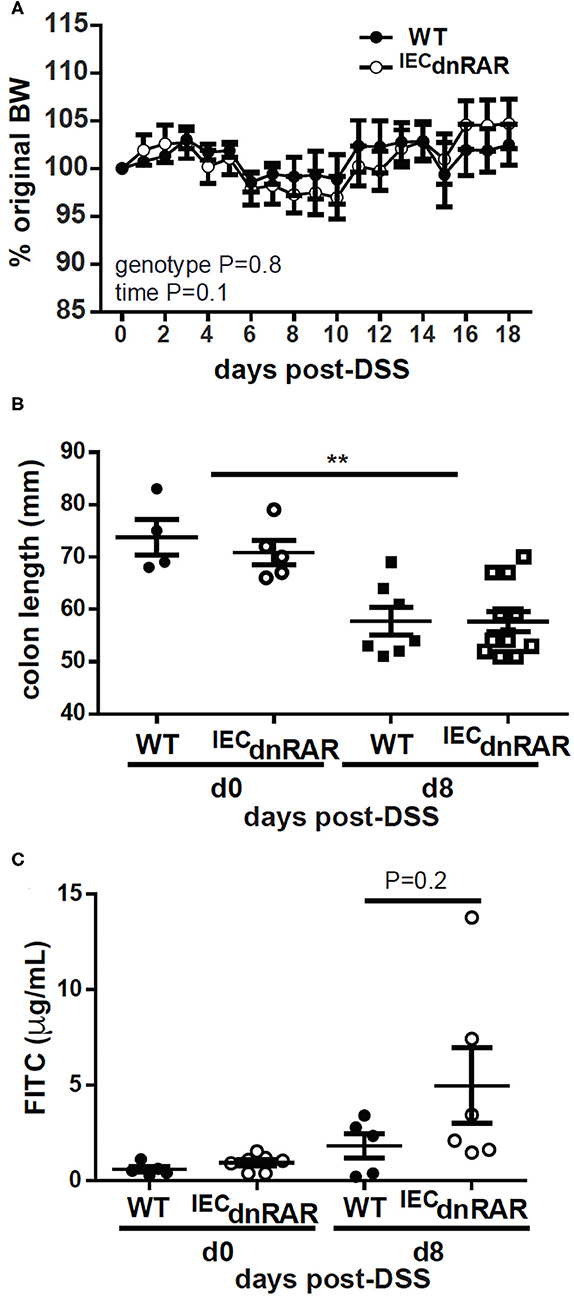
Figure 4. No change in susceptibility to DSS in A+ IECdnRAR mice. (A) Weight loss, (B) colon length, and (C) intestinal permeability following DSS administration. Values are the mean ± SEM of two experiments with n = 6–12 mice her group and time point. Two-way ANOVA with Bonferroni post-test (A), and one-way ANOVA (B,C), **P < 0.01.
Vitamin A deficient (A–) WT and IECdnRAR mice were also challenged with DSS. A– IECdnRAR mice lost significantly more weight following DSS treatment than their A– WT counterparts (Figure 5A). The effect of genotype on A– weight loss was largely due to the males and not the females since there was no effect of genotype on weight loss in the A– females (Supplementary Figure 5B). All other parameters of colitis severity were unaffected by sex. As expected colon lengths were shorter at day 8 post-DSS than at day 0 (Figure 5B). There was no effect of genotype on A– colon length either at day 0 or day 8 post-DSS (Figure 5B). Colonic histopathology at d10 post-DSS was not different in A– WT and A– IECdnRAR mice (Supplementary Figures 5C, 6A). Intestinal permeability was significantly higher in A– IECdnRAR mice compared to A– WT mice at both day 0 and day 8 post-DSS (Figure 5C). Expression of the dnRAR in IEC cells, with vitamin A deficiency, resulted in the increased permeability of the GI tract, and the increased weight loss following injury of the GI tract with DSS.
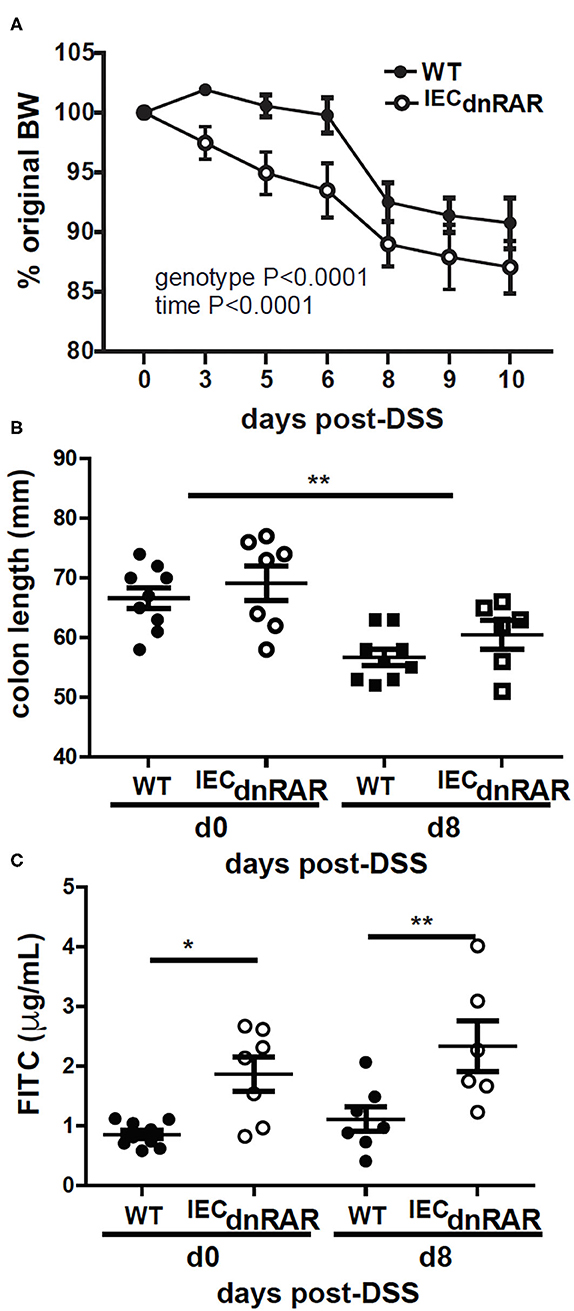
Figure 5. Increased susceptibility to DSS in A– IECdnRAR mice. (A) Weight loss, (B) colon length, and (C) intestinal permeability following DSS treatment of A– WT and A– IECdnRAR mice. Values are mean ± SEM of two experiments with n = 6–9 mice per group. Two-way ANOVA with Bonferroni post-test (A) or one-way ANOVA (C,D) was used to determine significance, *P < 0.05, **P < 0.01.
A– IECdnRAR Develop a Severe Early Infection With Citrobacter rodentium
A– mice had reduced survival following C. rodentium infection (24). The surviving A– mice were unable to clear the infection (24). Treating A– mice with RA on the day of infection or at the peak of infection completely protected the mice and resulted in clearance of C. rodentium (18). Three groups of IECdnRAR mice were infected with C. rodentium, A+, A–, and A– +RA treated (Figure 6). The A+ IECdnRAR mice had C. rodentium shedding curves (Figure 6A) that were the same as A+ WT (18, 24). A– IECdnRAR mice had decreased survival following C. rodentium infection (Figure 6A and Table 1). Surviving A– IECdnRAR mice became chronic shedders of C. rodentium and had very high numbers of bacteria in their feces on day 30 post-infection, when A+ IECdnRAR mice had already cleared the infection (Figure 6B). RA treatment of A– IECdnRAR mice significantly reduced the fecal shedding of C. rodentium and the kinetics of the clearance were similar to the A+ IECdnRAR mice (Figure 6B). The chronically infected A– IECdnRAR mice had short, visibly thickened colons. At day 30 post-infection the A– IECdnRAR mice had significantly longer crypt lengths than all other groups (Figure 6C). The histopathology scores showed increased damage of the A+ and A– colons at day 10 post-infection that resolved by day 30 post-infection (Figure 6C and Supplementary Figure 6B). There were no differences in histopathology scores when comparing A+ IECdnRAR and A– IECdnRAR mice (Figure 6C and Supplementary Figure 6B). Expression of the IECdnRAR exacerbated the effects of vitamin A deficiency on GI infection.
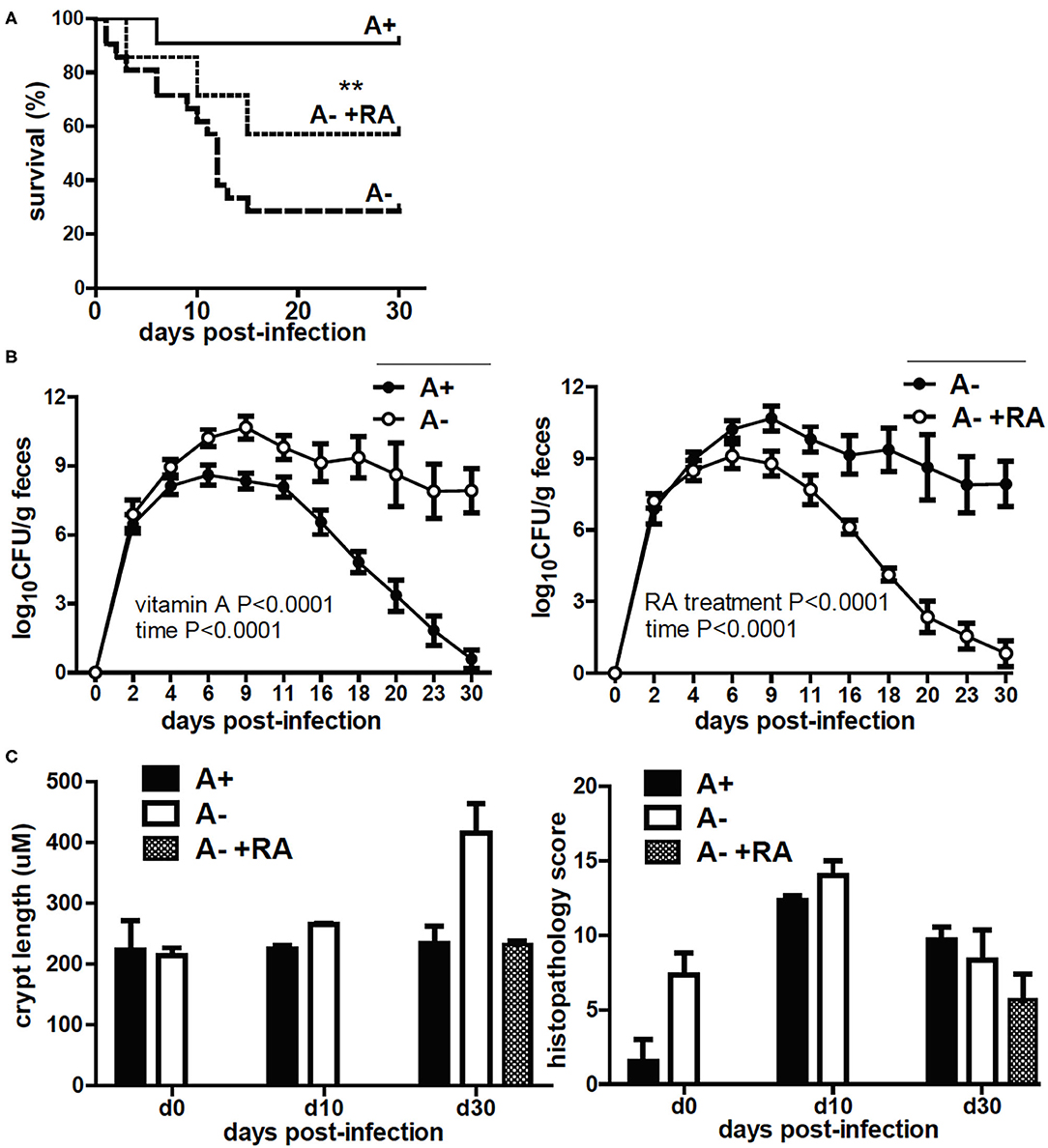
Figure 6. C. rodentium infection in A+ IECdnRAR, A– IECdnRAR mice and A– +RA IECdnRAR mice. A+ IECdnRAR, A– IECdnRAR and A– +RA IECdnRAR mice were infected with C. rodentium and (A) survival, (B) fecal shedding, and (C) crypt length/histopathology score of the distal colon were measured. Values are the mean ± SEM of n = 4–10 mice per group and time-point. Significance was determined using the Log rank Mantel-Cox test (A), two-way ANOVA with Bonferroni post-test (B), and one-way ANOVA (C), **P < 0.01.
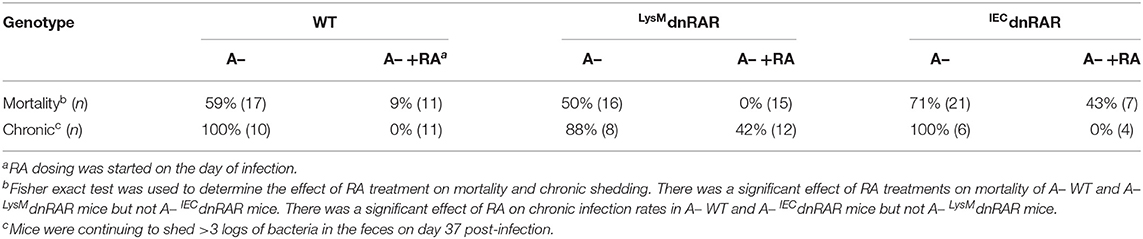
Table 1. A– IECdnRAR are refractory to the early protective effects of RA.
IEC Are Critical Retinoid Targets for Protection From Early Lethality Following C. rodentium Infection
Treating A– WT mice with RA at d0 protected mice from lethality and resulted in the complete clearance of C. rodentium infection (24) (Table 1). Virulence of C. rodentium is regulated by the LEE pathogencity island that includes the transcriptional regulator ler (39). Ler expression is highest at the peak of infection (day 5–7) and then is downregulated (40). C. rodentium from A– mice expressed more ler than the C. rodentium from RA treated A– mice (Supplementary Figure 7). At day 5 post-infection when both the A– and A– +RA LysMdnRAR had very high infections, ler was lower in the RA treated C. rodentium (Supplementary Figure 7). RA treatment of mice that express the dnRAR in macrophage and neutrophil (LysMdnRAR) protected the mice from lethality but resulted in chronically C. rodentium infected mice (18) (Table 1). At d37 post-infection the A– LysMdnRAR still had C. rodentium that expressed ler (Supplementary Figure 7). RA dosing of A– IECdnRAR mice starting at d0 of infection failed to significantly improve survival rates (Table 1). However, the surviving IECdnRAR A– +RA mice cleared the C. rodentium infection (Table 1 and Figure 6). Functional retinoid signaling in macrophage/neutrophil is not required to survive the early infection but is required for clearance of the infection. Conversely, IEC are critical retinoid targets that prevent early lethality following C. rodentium infection.
Discussion
Mice with a dnRAR expressed in IEC were phenotypically normal and had IEC that expressed similar levels of several tight junction proteins as their WT littermates. Conversely, the IECdnRAR mice had reduced frequencies of TCRαβ+/CD8αα+ T cells in the IEL. The thymic precursors of the CD8αα+/TCRαβ+ were unaffected by the IECdnRAR expression. IL-15 and trans-presentation of IL-15 is required for CD8αα maturation and survival (41). IL15R knockout (KO) mice had reduced TCRαβ+/CD8αα+ frequencies and restoration of IL15-Rα expression on IEC restored TCRαβ+/CD8αα populations (41, 42). IL15-Rα expression was normal in IECdnRAR mice. Other potential regulators of IEL include RA, TLA, TGF-β, IFN-γ, and IL-27 (43, 44). TGF-β and TLA expression were normal in the IECdnRAR GI tract. IEC have been shown to be required for the production of serum amyloid A (SAA) proteins that are hypothesized to deliver retinol to T cells and IEC important for GI immunity (45). It is possible that the IEC require functional RAR to induce SAA delivery of retinol for the RA mediated induction of CD8αα+ on TCRαβ+ T cells in the gut. There is a cell intrinsic requirement for functional RARs in the IEC to indirectly induce CD8αα+ on TCRαβ+ T cells in the gut.
Expression of the dnRAR in IEC affects the intestinal barrier. Intestinal barrier function, susceptibility to DSS colitis, and resistance to C. rodentium infection was not affected by expression of the IECdnRAR in vitamin A suffient mice. Expression of mRNA for tight junction proteins and intestinal permeability were not different in A+ IECdnRAR and WT mice (Supplementary Figure 3 and Figure 4). A limitation of the study is that protein levels for the tight junction proteins were not measured. Gattu et.al showed that A+ IECdnRAR had normal barrier function (45). However, Salmonella infection of A+ IECdnRAR mice resulted in more translocation of bacteria to the spleen and liver than in the WT littermates (45). Blocking retinoid signaling in IEC had mild effects on susceptibility to colitis or protection from GI infection. When the IECdnRAR mice were vitamin A deficient, blocking retinoid signaling in IEC had severe consequences. Intestinal permeability and weight loss were significantly higher in A– IECdnRAR mice compared to A– WT mice (Figure 5). Vitamin A is a critical regulator of the GI barrier and blocking the effects of retinoids in IEC cells resulted in increased permeability even when the mice were unchallenged (Figure 5). A– IECdnRAR mice were more susceptible to DSS colitis (Figure 5) and extremely susceptible to C. rodentium (72% mortality rate, Figure 6). Treating A– WT and A–LysMdnRAR mice with RA on the day of infection eliminated the lethality of a C. rodentium infection [Table 1 (18, 24)]. RA treatments were ineffective for preventing mortality in the C. rodentium infected A– IECdnRAR mice (Table 1). There was no lethality in the A+ IECdnRAR or the A+ LysMdnRAR infected with C. rodentium [Figure 6 (18)]. When vitamin A is low IEC must be able to respond to RA, which helps to maintain the barrier and protect the host from early lethality following GI infection. The early lethality following C. rodentium infection in A– mice requires retinoid signaling in IEC but not macrophage/neutrophils.
The inflammation following C. rodentium infection has been shown to be greatest at the peak of infection, followed by resolution of inflammation as the infection is cleared. Infection of germfree mice with C. rodentium resulted in inflammation that was highest at 10–14 days post-infection and then resolution of inflammation; although the mice remained colonized with high numbers of organisms (40). Ler was downregulated in the chronically C. rodentium monocolonized mice without inflammation (40). Chronically infected A– and A– +RA LysMdnRAR mice shed high numbers of C. rodentium in the feces 30 days post-infection [Table 1 (18, 24)]. RA treatment of the A– LysMdnRAR on the day of C. rodentium infection, protected the mice from early lethality but 42% of the mice developed chronic infection (Table 1). RA treated A– LysMdnRAR mice had C. rodentium with lower expression of ler at day 5 and day 37 post-infection. Ler expression did not correspond with the ability of the host to eliminate C. rodentium, since the RA treated A– LysMdnRAR developed chronic infections (Table 1). RA treatments inhibit the expression of the C. rodentium ler pathogenicity genes in otherwise A– hosts perhaps by reducing inflammation (39).
Vitamin A deficiency remains a public health concern in resource limited countries. The GI tract needs vitamin A for mucous production and the regulation of the barrier. During vitamin A sufficiency other cells in the GI tract can compensate for the IEC that express dnRAR to regulate the barrier. Conversely, the development of CD8αα+/TCRαβ+ T cells require IEC with functional retinoid receptors. RA in the IEC protects the vitamin A deficient host from severe GI disease including lethal bacterial infections. It would be important for developing countries to continue to work toward strategies to improve vitamin A nutrition. Even in the vitamin A deficient host, short-term RA treatments restored IEC function protecting the host from the most serious consequences of vitamin A deficiency.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by The Pennsylvania State University IACUC committee.
Author Contributions
LS, JA, and MC conceptualized and designed the experimental studies. LS, JA, MK, and VW performed the experiments and acquired and analyzed the data. LS and JA drafted the manuscript with the help of MC. All authors approved the publication of the manuscript.
Funding
This work was supported by the National Institutes of Health (R01AT005378), (R56 AI114972), and (T32GM108563) and United State Department of Agriculture (2914-38420-21822).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank A. Catharine Ross and Cheng-Hsin Wei for the serum retinol analyses. We thank the Center for Molecular Immunology and Infectious Diseases for lively discussion and critical feedback.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.559635/full#supplementary-material
References
1. West KpJ. Extent of vitamin A deficiency among preschool children and women of reproductive age. J Nutr. (2002) 132:2857–66S. doi: 10.1093/jn/132.9.2857S
2. WHO. “Global Prevalence of Vitamin A Deficiency in Populations at Risk 1995-2005”. WHO Global Database on Vitamin A Deficiency. Geneva: World Health Organization (2009).
3. Villamor E, Fawzi WW. Vitamin A supplementation: implications for morbidity and mortality in children. J Infect Dis. (2000) 182(Suppl. 1):S122–33. doi: 10.1086/315921
4. Deplancke B, Gaskins HR. Microbial modulation of innate defense: goblet cells and the intestinal mucus layer. Am J Clin Nutr. (2001) 73:1131–41S. doi: 10.1093/ajcn/73.6.1131S
5. Peterson LW, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol. (2014) 14:141–53. doi: 10.1038/nri3608
6. Fasano A, Shea-Donohue T. Mechanisms of disease: the role of intestinal barrier function in the pathogenesis of gastrointestinal autoimmune diseases. Nat Clin Pract Gastroenterol Hepatol. (2005) 2:416–22. doi: 10.1038/ncpgasthep0259
7. Curciarello R, Canziani KE, Docena GH, Muglia CI. Contribution of non-immune cells to activation and modulation of the intestinal inflammation. Front Immunol. (2019) 10:647. doi: 10.3389/fimmu.2019.00647
8. Mundy R, Macdonald TT, Dougan G, Frankel G, Wiles S. Citrobacter rodentium of mice and man. Cell Microbiol. (2005) 7:1697–706. doi: 10.1111/j.1462-5822.2005.00625.x
9. Fan X, Liu S, Liu G, Zhao J, Jiao H, Wang X, et al. Vitamin A deficiency impairs mucin expression and suppresses the mucosal immune function of the respiratory tract in chicks. PLoS ONE. (2015) 10:e0139131. doi: 10.1371/journal.pone.0139131
10. Polcz ME, Barbul A. The role of vitamin A in wound healing. Nutr Clin Pract. (2019) 34:695–700. doi: 10.1002/ncp.10376
11. Kubota H, Chiba H, Takakuwa Y, Osanai M, Tobioka H, Kohama G, et al. Retinoid X receptor alpha and retinoic acid receptor gamma mediate expression of genes encoding tight-junction proteins and barrier function in F9 cells during visceral endodermal differentiation. Exp Cell Res. (2001) 263:163–72. doi: 10.1006/excr.2000.5113
12. Osanai M, Nishikiori N, Murata M, Chiba H, Kojima T, Sawada N. Cellular retinoic acid bioavailability determines epithelial integrity: role of retinoic acid receptor alpha agonists in colitis. Mol Pharmacol. (2007) 71:250–8. doi: 10.1124/mol.106.029579
13. Thurnham DI, Northrop-Clewes CA, Mccullough FS, Das BS, Lunn PG. Innate immunity, gut integrity, and vitamin A in Gambian and Indian infants. J Infect Dis. (2000) 182(Suppl. 1):S23–8. doi: 10.1086/315912
14. Lima AA, Soares AM, Lima NL, Mota RM, Maciel BL, Kvalsund MP, et al. Effects of vitamin A supplementation on intestinal barrier function, growth, total parasitic, and specific Giardia spp infections in Brazilian children: a prospective randomized, double-blind, placebo-controlled trial. J Pediatr Gastroenterol Nutr. (2010) 50:309–15. doi: 10.1097/MPG.0b013e3181a96489
15. Iwata M, Yokota A. Retinoic acid production by intestinal dendritic cells. Vitam Horm. (2011) 86:127–52. doi: 10.1016/B978-0-12-386960-9.00006-X
16. Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C, Song SY. Retinoic acid imprints gut-homing specificity on T cells. Immunity. (2004) 21:527–38. doi: 10.1016/j.immuni.2004.08.011
17. Hammerschmidt SI, Friedrichsen M, Boelter J, Lyszkiewicz M, Kremmer E, Pabst O, et al. Retinoic acid induces homing of protective T and B cells to the gut after subcutaneous immunization in mice. J Clin Invest. (2011) 121:3051–61. doi: 10.1172/JCI44262
18. Snyder LM, Mcdaniel KL, Tian Y, Wei CH, Kennett MJ, Patterson AD, et al. Retinoic acid mediated clearance of Citrobacter rodentium in vitamin A deficient mice requires CD11b+ and T cells. Front Immunol. (2018) 9:3090. doi: 10.3389/fimmu.2018.03090
19. Bai A, Lu N, Guo Y, Liu Z, Chen J, Peng Z. All-trans retinoic acid down-regulates inflammatory responses by shifting the Treg/Th17 profile in human ulcerative and murine colitis. J Leukoc Biol. (2009) 86:959–69. doi: 10.1189/jlb.0109006
20. Mielke LA, Jones SA, Raverdeau M, Higgs R, Stefanska A, Groom JR, et al. Retinoic acid expression associates with enhanced IL-22 production by gammadelta T cells and innate lymphoid cells and attenuation of intestinal inflammation. J Exp Med. (2013) 210:1117–24. doi: 10.1084/jem.20121588
21. Cantorna MT, Snyder L, Arora J. Vitamin A and vitamin D regulate the microbial complexity, barrier function, and the mucosal immune responses to ensure intestinal homeostasis. Crit Rev Biochem Mol Biol. (2019) 54:184–92. doi: 10.1080/10409238.2019.1611734
22. Damm K, Heyman RA, Umesono K, Evans RM. Functional inhibition of retinoic acid response by dominant negative retinoic acid receptor mutants. Proc Natl Acad Sci USA. (1993) 90:2989–93. doi: 10.1073/pnas.90.7.2989
23. Rajaii F, Bitzer ZT, Xu Q, Sockanathan S. Expression of the dominant negative retinoid receptor, RAR403, alters telencephalic progenitor proliferation, survival, and cell fate specification. Dev Biol. (2008) 316:371–82. doi: 10.1016/j.ydbio.2008.01.041
24. Mcdaniel KL, Restori KH, Dodds JW, Kennett MJ, Ross AC, Cantorna MT. Vitamin A-deficient hosts become nonsymptomatic reservoirs of Escherichia coli-like enteric infections. Infect Immun. (2015) 83:2984–91. doi: 10.1128/IAI.00201-15
25. Yu S, Weaver V, Martin K, Cantorna MT. The effects of whole mushrooms during inflammation. BMC Immunol. (2009) 10:12. doi: 10.1186/1471-2172-10-12
26. Ooi JH, Li Y, Rogers CJ, Cantorna MT. Vitamin D regulates the gut microbiome and protects mice from dextran sodium sulfate-induced colitis. J Nutr. (2013) 143:1679–86. doi: 10.3945/jn.113.180794
27. Ooi JH, Waddell A, Lin YD, Albert I, Rust LT, Holden V, et al. Dominant effects of the diet on the microbiome and the local and systemic immune response in mice. PLoS ONE. (2014) 9:e86366. doi: 10.1371/journal.pone.0086366
28. Cantorna MT, Lin YD, Arora J, Bora S, Tian Y, Nichols RG, et al. Vitamin D regulates the microbiota to control the numbers of RORgammat/FoxP3+ regulatory T cells in the colon. Front Immunol. (2019) 10:1772. doi: 10.3389/fimmu.2019.01772
29. Froicu M, Weaver V, Wynn TA, Mcdowell MA, Welsh JE, Cantorna MT. A crucial role for the vitamin D receptor in experimental inflammatory bowel diseases. Mol Endocrinol. (2003) 17:2386–92. doi: 10.1210/me.2003-0281
30. Bruce D, Cantorna MT. Intrinsic requirement for the vitamin D receptor in the development of CD8alphaalpha-expressing T cells. J Immunol. (2011) 186:2819–25. doi: 10.4049/jimmunol.1003444
31. Yu S, Cantorna MT. Epigenetic reduction in invariant NKT cells following in utero vitamin D deficiency in mice. J Immunol. (2011) 186:1384–90. doi: 10.4049/jimmunol.1002545
32. Yu S, Bruce D, Froicu M, Weaver V, Cantorna MT. Failure of T cell homing, reduced CD4/CD8alphaalpha intraepithelial lymphocytes, and inflammation in the gut of vitamin D receptor KO mice. Proc Natl Acad Sci USA. (2008) 105:20834–9. doi: 10.1073/pnas.0808700106
33. Chen J, Waddell A, Lin YD, Cantorna MT. Dysbiosis caused by vitamin D receptor deficiency confers colonization resistance to Citrobacter rodentium through modulation of innate lymphoid cells. Mucosal Immunol. (2015) 8:618–26. doi: 10.1038/mi.2014.94
34. Gangadharan D, Lambolez F, Attinger A, Wang-Zhu Y, Sullivan BA, Cheroutre H. Identification of pre- and postselection TCRalphabeta+ intraepithelial lymphocyte precursors in the thymus. Immunity. (2006) 25:631–41. doi: 10.1016/j.immuni.2006.08.018
35. Erben U, Loddenkemper C, Doerfel K, Spieckermann S, Haller D, Heimesaat MM, et al. A guide to histomorphological evaluation of intestinal inflammation in mouse models. Int J Clin Exp Pathol. (2014) 7:4557–76.
36. Qiu Y, Yang H. Effects of intraepithelial lymphocyte-derived cytokines on intestinal mucosal barrier function. J Interferon Cytokine Res. (2013) 33:551–62. doi: 10.1089/jir.2012.0162
37. Cheroutre H, Lambolez F. The thymus chapter in the life of gut-specific intra epithelial lymphocytes. Curr Opin Immunol. (2008) 20:185–91. doi: 10.1016/j.coi.2008.03.009
38. Das G, Augustine MM, Das J, Bottomly K, Ray P, Ray A. An important regulatory role for CD4+CD8 alpha alpha T cells in the intestinal epithelial layer in the prevention of inflammatory bowel disease. Proc Natl Acad Sci USA. (2003) 100:5324–9. doi: 10.1073/pnas.0831037100
39. Collins JW, Keeney KM, Crepin VF, Rathinam VA, Fitzgerald KA, Finlay BB, et al. Citrobacter rodentium: infection, inflammation and the microbiota. Nat Rev Microbiol. (2014) 12:612–23. doi: 10.1038/nrmicro3315
40. Kamada N, Kim YG, Sham HP, Vallance BA, Puente JL, Martens EC, et al. Regulated virulence controls the ability of a pathogen to compete with the gut microbiota. Science. (2012) 336:1325–9. doi: 10.1126/science.1222195
41. Ma LJ, Acero LF, Zal T, Schluns KS. Trans-presentation of IL-15 by intestinal epithelial cells drives development of CD8alphaalpha IELs. J Immunol. (2009) 183:1044–54. doi: 10.4049/jimmunol.0900420
42. Lodolce JP, Boone DL, Chai S, Swain RE, Dassopoulos T, Trettin S, et al. IL-15 receptor maintains lymphoid homeostasis by supporting lymphocyte homing and proliferation. Immunity. (1998) 9:669–76. doi: 10.1016/S1074-7613(00)80664-0
43. Reis BS, Rogoz A, Costa-Pinto FA, Taniuchi I, Mucida D. Mutual expression of the transcription factors Runx3 and ThPOK regulates intestinal CD4(+) T cell immunity. Nat Immunol. (2013) 14:271–80. doi: 10.1038/ni.2518
44. Kwong B, Lazarevic V. T-bet orchestrates CD8alphaalpha IEL differentiation. Immunity. (2014) 41:169–71. doi: 10.1016/j.immuni.2014.08.003
Keywords: vitamin A deficiency, infection, barrier, gastrointestinal tract, retinoic acid receptor
Citation: Snyder LM, Arora J, Kennett MJ, Weaver V and Cantorna MT (2020) Retinoid Signaling in Intestinal Epithelial Cells Is Essential for Early Survival From Gastrointestinal Infection. Front. Immunol. 11:559635. doi: 10.3389/fimmu.2020.559635
Received: 06 May 2020; Accepted: 07 September 2020;
Published: 08 October 2020.
Edited by:
Raquel Hontecillas, Virginia Tech, United StatesReviewed by:
Hairong Wei, University of Alabama at Birmingham, United StatesShubha Priyamvada, University of Illinois at Chicago, United States
Copyright © 2020 Snyder, Arora, Kennett, Weaver and Cantorna. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Margherita T. Cantorna, bXhjNjlAcHN1LmVkdQ==