 Tugce Karaderi1,2†
Tugce Karaderi1,2† Halin Bareke3,4†
Halin Bareke3,4† Imge Kunter5
Imge Kunter5 Adil Seytanoglu3
Adil Seytanoglu3 Ilgin Cagnan3
Ilgin Cagnan3 Deniz Balci3
Deniz Balci3 Burc Barin6
Burc Barin6 Mevhibe B. Hocaoglu7,8
Mevhibe B. Hocaoglu7,8 Nilufer Rahmioglu9,10
Nilufer Rahmioglu9,10 Esra Asilmaz11
Esra Asilmaz11 Bahar Taneri3,12*
Bahar Taneri3,12*- 1Center for Health Data Science, Faculty of Medical and Health Sciences, University of Copenhagen, Copenhagen, Denmark
- 2Disease Systems Biology Program, Novo Nordisk Foundation Center for Protein Research, Faculty of Medical and Health Sciences, University of Copenhagen, Copenhagen, Denmark
- 3Department of Biological Sciences, Faculty of Arts and Sciences, Eastern Mediterranean University, Famagusta, Cyprus
- 4Department of Pharmaceutical Biotechnology, Faculty of Pharmacy, Institute of Health Sciences, Marmara University, Istanbul, Turkey
- 5Faculty of Pharmacy, Eastern Mediterranean University, Famagusta, Cyprus
- 6Vaccines and Infectious Diseases Therapeutic Research Area, The Emmes Company, Rockville, MD, United States
- 7Cicely Saunders Institute of Palliative Care, Policy & Rehabilitation, Florence Nightingale Faculty of Nursing, Midwifery & Palliative Care, King’s College London, London, United Kingdom
- 8Dr Fazil Kucuk Faculty of Medicine, Eastern Mediterranean University, Famagusta, Cyprus
- 9Wellcome Centre for Human Genetics, University of Oxford, Oxford, United Kingdom
- 10Nuffield Department of Women’s and Reproductive Health, University of Oxford, Oxford, United Kingdom
- 11Department of Gastroenterology, Homerton University Hospital, Clapton, United Kingdom
- 12Department of Genetics and Cell Biology, Faculty of Health, Medicine & Life Sciences, Institute for Public Health Genomics, Maastricht University, Maastricht, Netherlands
COVID-19 presentation is very heterogeneous across cases, and host factors are at the forefront for the variables affecting the disease manifestation. The immune system has emerged as a key determinant in shaping the outcome of SARS-CoV-2 infection. It is mainly the deleterious unconstrained immune response, rather than the virus itself, which leads to severe cases of COVID-19 and the associated mortality. Genetic susceptibility to dysregulated immune response is highly likely to be among the host factors for adverse disease outcome. Given that such genetic susceptibility has also been observed in autoimmune diseases (ADs), a number of critical questions remain unanswered; whether individuals with ADs have a significantly different risk for COVID-19–related complications compared to the general population, and whether studies on the genetics of ADs can shed some light on the host factors in COVID-19. In this perspective, we discuss the host genetic factors, which have been under investigation in association with COVID-19 severity. We touch upon the intricate link between autoimmunity and COVID-19 pathophysiology. We put forth a number of autoimmune susceptibility genes, which have the potential to be additional host genetic factors for modifying the severity of COVID-19 presentation. In summary, host genetics at the intersection of ADs and COVID-19 may serve as a source for understanding the heterogeneity of COVID-19 severity, and hence, potentially holds a key in achieving effective strategies in risk group identification, as well as effective treatments.
Introduction
Coronaviruses have been a source of global alarm for the last two decades (1). The most recent emerging coronavirus SARS-CoV-2 has led to a pandemic, as declared by the World Health Organization (WHO) in March 2020. There have been more than 40 million confirmed cases of COVID-19 worldwide, and it has claimed more than 1,000,000 lives (2). Considering the risk of healthcare systems being overwhelmed, it is of utmost importance to pinpoint the risk factors for adverse disease outcomes to implement informed preventive measures limiting the global burden of the current pandemic.
Epidemiological reports from different countries highlighted COVID-19 mortality risk factors; comorbidities such as hypertension, diabetes, obesity, cardiovascular disease, chronic respiratory disease and cancer (3), as well as older age and male sex regardless of any comorbidities (2, 4). Discrepancies in testing availability, differences in testing algorithms and the presence of asymptomatic cases confound cross-country comparisons of the actual infection and mortality rates. These challenges are also relevant to research studies on host susceptibility factors for COVID-19 severity. Therefore, additional host susceptibility factors are likely to transpire as further epidemiological data emerge over time.
symptomatic and mild cases are estimated to comprise the majority of COVID-19 cases (5). As a multisystem disease, severe and critical cases may have acute respiratory distress syndrome (ARDS), and organ injuries due to cytokine storm and coagulopathies potentially leading to death (5–7). The striking interpersonal differences in clinical presentation have led to questions on the role of genetic factors. For instance, angiotensin-converting enzyme 2 (ACE2) was one of the first genes that attracted a lot of interest due to ACE2 being the viral portal of entry to the host cells, and the high mortality rate observed among cases with hypertension receiving ACE-inhibitor treatment.
Exaggerated inflammatory response is the culprit of the majority of the COVID-19 deaths. Hence, genetic susceptibility to dysregulated immune response is potentially among the host factors for adverse disease outcome. Such genetic susceptibility has also been observed in autoimmune diseases (ADs) (8, 9). Therefore, a significant question remains to be answered; does genetic susceptibility to ADs affect the risk for COVID-19–related complications and mortality?
ADs are complex diseases due to both genetic and environmental factors. They are characterized by an aberrant immune response to self-antigens due to the presence of autoreactive lymphocytes and loss of immune tolerance (10). Uncovering any potential genetic, and possibly, biological link between ADs and immune response to SARS-CoV-2 may in turn help to (1) identify individuals at risk, (2) shed light on COVID-19 immunopathology, (3) explain the broad heterogeneity in the disease progression and treatment responses, and (4) guide the vaccine development to prevent any vaccine-induced destructive immune response.
Herein, we briefly discuss the host genetic factors that have been under investigation with regards to association with COVID-19 disease severity, and also suggest a number of AD susceptibility genes, with the potential to be additional host genetic factors for heterogeneous COVID-19 presentation.
Progress in the Investigation of COVID-19 Host Genetics
From the very start of the COVID-19 outbreak, protein members of the biological pathway essential for the entry of the virus into the host cells have become a focus of attention as candidate host susceptibility genes. Epidemiological findings on chronic conditions such as hypertension having more severe COVID-19 disease and a higher mortality risk, have also pointed out specific proteins functioning in the viral entry pathway (11). Of main interest were two proteins, ACE2 and transmembrane serine protease 2 (TMPRSS2), but the latter received significantly less attention with regards to host genetics studies (12–16). ACE2, a transmembrane protein, mainly found in airway ciliated epithelial cells with different enzymatic activities related to the renin-angiotensin-aldosterone system, has been shown to serve as a functional receptor for SARS-CoV-2 to infect nasal and alveolar epithelial cells in the lungs (17). The spike glycoprotein (S-protein) on the viral envelope of SARS-CoV-2 binds to the host ACE2 via its receptor-binding domain. Upon binding, the S-protein is activated by the TMPRSS2, which is a cellular protease that co-localizes with ACE2. This interaction assists the virus to fuse with the plasma membrane and facilitates the viral invasion of the host cell (18) (Table 1).
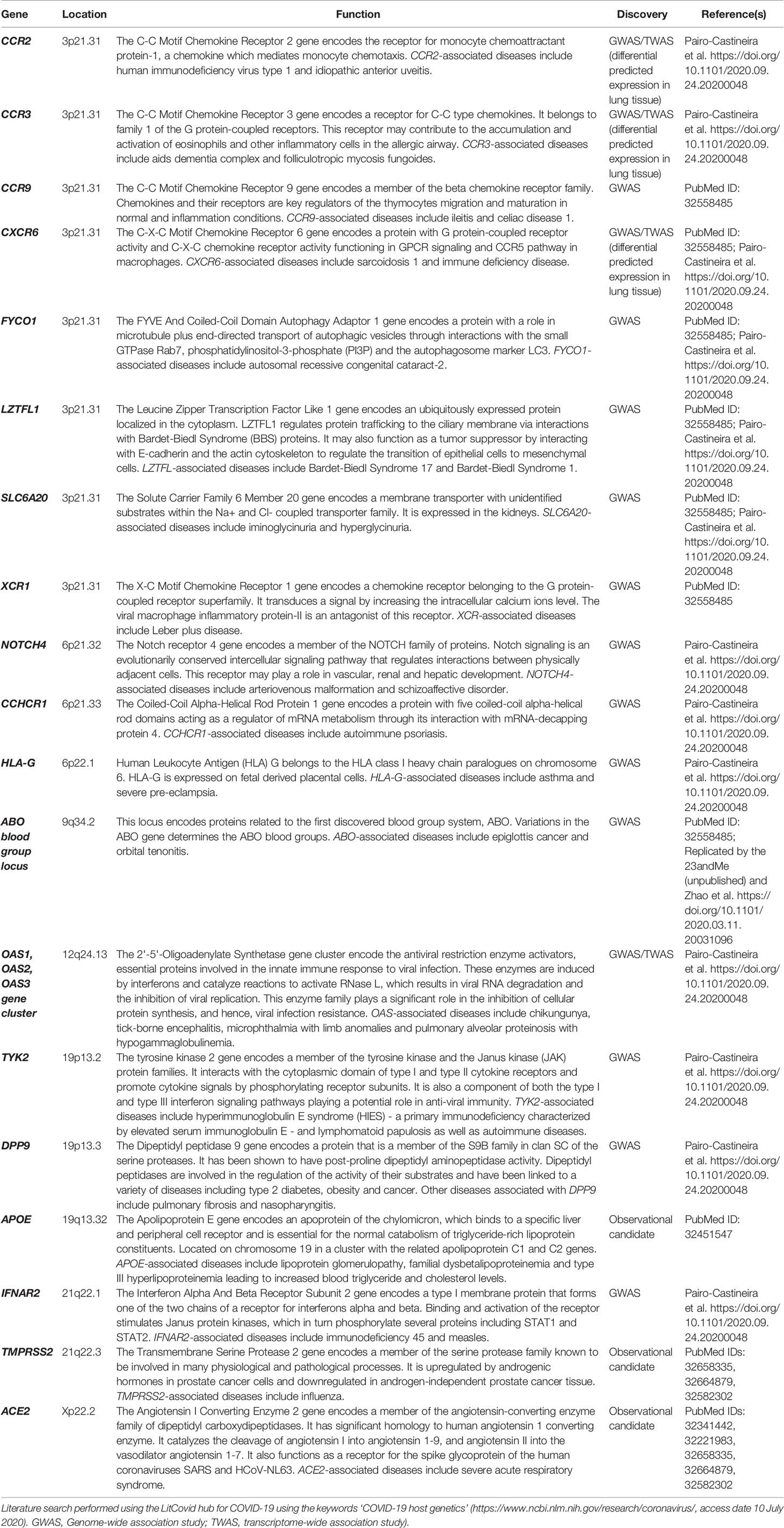
Table 1 Descriptions of selected host genes and their functions relevant to COVID-19 susceptibility and severity.
ACE inhibitors and angiotensin-receptor blockers (ARBs), which are used to balance blood pressure and vascular complications in chronic diseases (e.g. cardiovascular diseases and diabetes), provide various clinical benefits by increasing the expression of ACE2 while blocking ACE. Given that ACE2 facilitates viral invasion of human cells, there have been concerns that upregulation of ACE2, via use of ACE inhibitors and ARBs, may increase COVID-19 susceptibility and severity (19). However, it was also suggested that increasing ACE2 by the same intervention might be beneficial for a subset of cases, mainly because of its anti-inflammatory effects (19, 20). Supporting this perspective, a population-based case-control study reported that the use of ACE inhibitors or ARBs does not directly correlate with COVID-19 susceptibility or outcome severity (11). Studies in mice also suggested a potential protective role for ACE2 as its downregulation resulted in more severe respiratory failure (21). Furthermore, ACE2 deficiency has been shown to increase inflammatory response via increased expression of cytokines, promoting vascular inflammation (22). Hence, ACE2 may potentially play contrasting roles at different stages of the disease, affecting COVID-19 susceptibility and severity in multiple ways; at early stages, enabling viral entry to the cell, and hence, increasing disease susceptibility, and later, down-regulating cytokines/inflammatory response, and therefore, decreasing severity of the disease.
So far, studies on population genetics and genetic epidemiology of ACE2 variants have been inconclusive, not showing a significant global pattern. A number of potentially functional variants (such as missense variants rs758278442, rs759134032, and rs763395248) have been shown to have varying frequencies in populations of European vs. Asian descent, but convincing evidence for biological effects of these on disease susceptibility and severity require further functional experiments (23–27).
Another gene of interest has been apolipoprotein E (APOE), due to the observation of pre-existing dementia as a risk factor for COVID-19 severity and mortality in the older UK Biobank population (age range ~ 40–69 years; UKBB; Table 1). The UKBB study concluded that ApoE e4 allele increases the risk of severe COVID-19 infection, independent of pre-existing dementia, cardiovascular disease, and type-2 diabetes. Biologically, ApoE e4 plays a role both in lipoprotein function and in regulation of macrophage pro-/anti-inflammatory phenotypes (28). ApoE is highly expressed in the lungs (29). The precise biological mechanisms linking ApoE variants to COVID-19 severity require further investigation (30).
Further approaches to exploring COVID-19 host genetics involve international collaborations to perform hypothesis-free association analysis across the human genome. The global COVID-19 Host Genetics Initiative (HGI) has been formed to bring together international human genetics and epidemiology experts, and to gather and analyze scientific data on genetic determinants of COVID-19 susceptibility (31) (https://www.covid19hg.org/). Their main focus is to comprehensively investigate the human genome to obtain insights into disease susceptibility, as well as severity and outcome. As part of the Initiative, a genome-wide association study (GWAS) including 1980 cases from Italy and Spain was conducted by the Severe COVID-19 GWAS Group (32). Two loci, 3p21.31 gene cluster and 9q34.2 ABO blood group locus, were associated with severe COVID-19, defined by respiratory failure (Table 1). Individuals with the blood group A had a higher risk of severe COVID-19, whereas there was a protective effect for those with the blood group O. The gene cluster on chromosome 3 contains six genes (CCR9, CXCR6, FYCO1, LZTFL1, SLC6A20, XCR1) with potential roles in COVID-19 severity, such as those involved in immune response (CCR9 and CXCR6), as well as in amino acid transport interacting with ACE2 (SLC6A20). A very recent GWAS preprint by Genetics of Mortality in Clinical Care (GenOMICC) collaborators including HGI (https://genomicc.org) reported additional novel loci (DPP9, TYK2, OAS gene cluster, IFNAR2, CCR2, CCR3, HLA-G, CCHCR1, and NOTCH4) associated with COVID-19 severity, all of which contain genes with roles in immune response and/or immune-mediate diseases (Table 1) (33). Although identifying actual causal gene variant(s) requires further association analyses in larger and more diverse sample populations as well as functional experiments, these results provide important insights into the potential factors affecting COVID-19 severity.
Autoimmunity-Associated Genes Providing Potential Insights Into Susceptibility to Severe COVID-19
Immune system is key in shaping the outcome of SARS-CoV-2 infection. An appropriate immune response to SARS-CoV-2 is dependent not only on mounting the right type of response at the right time, but also at the right intensity. Exaggerated immune response to SARS-CoV-2, involving increased pro-inflammatory serum cytokines [e.g. interleukin-1B (IL-1B), IL-1RA, IL-7, IL-9, IFNγ, CXCL10, TNFα, and especially, IL-6 and IL-8], C-reactive protein, and lung inflammatory mononuclear infiltrates, may cause lung fibrosis and lead to life-threatening ARDS (34–37). Likewise, cytokine storm increases the risk for disseminated intravascular coagulation (DIC) and multiple organ failure that may result in death (38). Thus, it is mainly the deleterious unconstrained immune response, rather than the virus itself, that leads to COVID-19–associated mortality (39).
Several factors may cause the hyper-inflammation observed in severe COVID-19. One of them might be the weak early interferon response that leads to excessive viral replication, which then triggers an exaggerated inflammatory response (40, 41). Other factors promoting this aggressive immune response might be the initial dose of exposure and previous infections of the host. An important host factor is highly likely to be the host’s genetic predisposition to a dysregulated immune response, similar to those seen in ADs.
An intricate link is emerging between autoimmunity and COVID-19 pathophysiology. The presence of autoantibodies is associated with an increased need for respiratory support (42), and furthermore, anti-cardiolipin IgA antibody detected frequently in anti-phospholipid syndrome (APS) has also been observed in a number of COVID-19 cases, with thrombotic events (43, 44). Auto-antibodies against type I interferons have also recently been identified in some of the severe COVID-19 cases (45). Several groups have also put forth that the molecular mimicry by SARS-CoV-2 may induce a disseminated autoimmune reaction in the body (46–48). Interestingly, lymphopenia is both a prognostic factor in COVID-19 and a trigger in multiple ADs (35, 49–51).
Exacerbated immune response in COVID-19 is overall akin to hemophagocytic lymphohistiocytosis (HLH), with its hyperferritinemia, cytopenia, and increased cytokine levels (52). Interestingly, secondary HLH can be seen in rheumatological diseases, such as systemic lupus erythematosus (SLE), juvenile idiopathic arthritis, and rheumatoid arthritis (RA), which are all autoimmune conditions (53). Besides HLH, hyperferritinemia is a finding in a number of ADs such as adult-onset Still’s disease, and catastrophic APS (54). As per the observed link between autoimmunity and COVID-19, drugs commonly used in AD, such as corticosteroids, and IL-6R and IL-1 antagonists, are being tested and used in COVID-19 cases (55, 56).
Given the aforementioned link between autoimmunity and COVID-19, we propose a connection between genetic susceptibility to ADs and to COVID-19 severity. We put forth candidates for shared genetics, among severe COVID-19 and AD susceptibility, by considering the well-known and replicated genetic variants shared across multiple ADs (Table 2). As shown in Table 2 and further discussed below, these genes code for proteins involved in antigen sensing, T cell activation and in cytokine signaling, hence they functionally participate in shaping the intensity of the immune response; a paramount factor in COVID-19 severity.
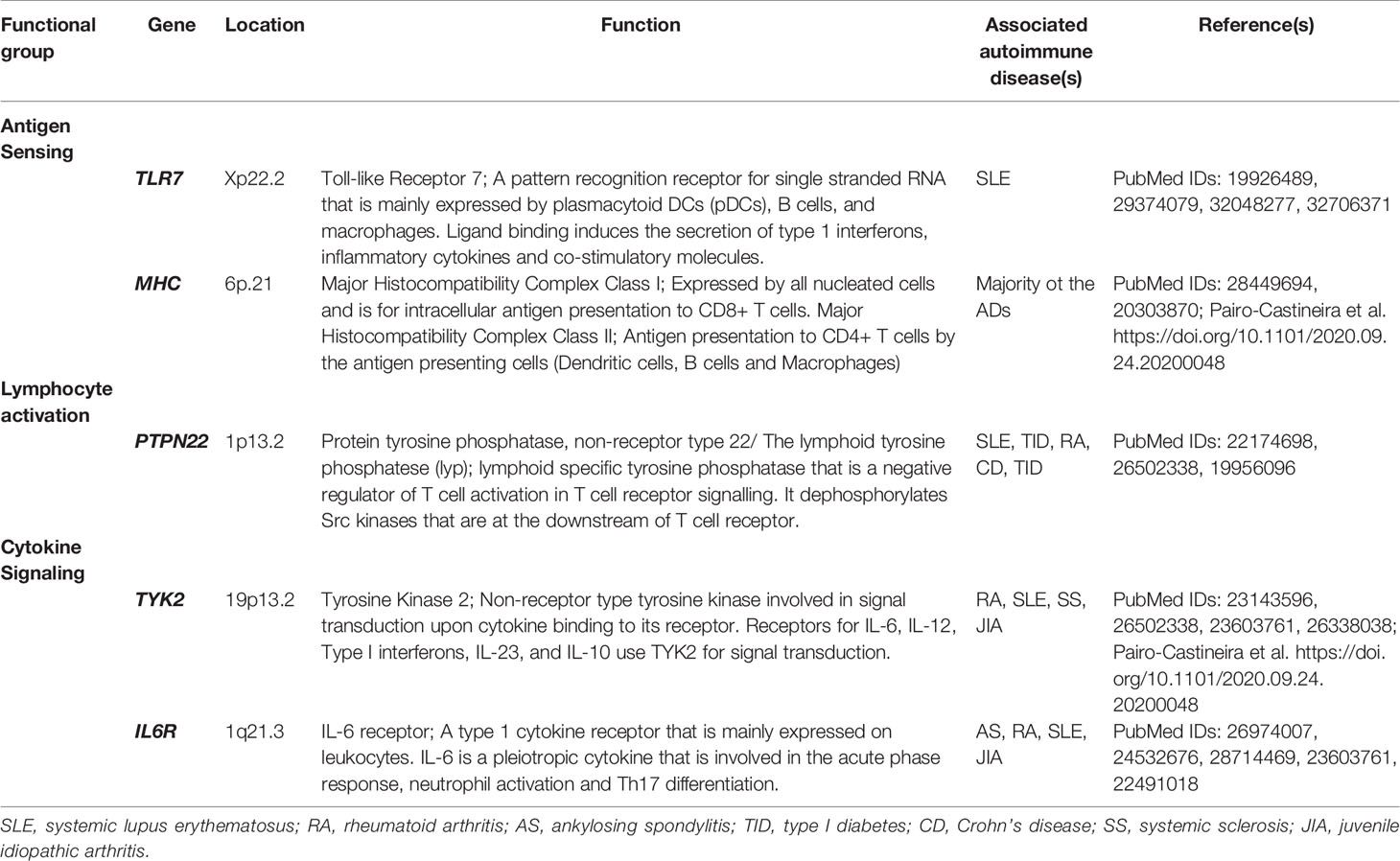
Table 2 Putative candidates for shared genes between severe COVID-19 and autoimmune disease (AD) susceptibility by considering the overlapping genetic associations across multiple ADs in genes that shape the immune response intensity.
In pathogen sensing by the innate immune system, toll-like receptor 7 (TLR7), an endosomal pattern recognition receptor (PRR), recognizes single-stranded RNA, and thus, it is among the initial innate immune cell receptors that sense SARS-CoV-2. TLR signaling leads to expression of pro-inflammatory cytokines and interferon genes (10). ADs, such as SLE, RA, and systemic sclerosis, have associations with TLR variants (10, 57, 58). Even though the early interferon response is delayed in SARS-CoV-2, upon increased viral load, excessive signaling due to genetic variants in TLR signal transduction pathways may lead to exacerbated macrophage and neutrophil activation, and subsequent, cytokine secretion (59). Interestingly, a recent case-series has shown that four young males with severe COVID-19 harbored rare TLR7 variants, which hampered interferon responses upon TLR7 engagement (60). Since TLR7 is located on the X-chromosome and is expressed bi-allelically, it has been suggested as one of the reasons for the sex bias, albeit in opposite direction, seen in AD and COVID-19 (61). These claims are yet to be proved, but it is becoming more apparent that a coordinated immune response is key for successfully controlling SARS-CoV-2 infection (62). Therefore, it is probable that any change in TLR7 signaling might increase the risk for a dysregulated immune response seen in severe COVID-19. The role of genetic variants in other players for type I interferon response, including TLR3 in severe COVID-19 is also emerging (63). In the case of antigen sensing in the adaptive immune system, T cells are dependent on antigens presented on human leukocyte antigen (HLA) molecules, and HLA loci are strongly associated with ADs (64). However, the mechanistic link between HLA loci and autoimmunity is complex, hence it is challenging to put forth a specific candidate variant that might also be present in severe COVID-19 cases. The GWAS in Italian and Spanish populations did not find any link between the HLA locus and respiratory failure in COVID-19 (32). However, a recent small-scale Han Chinese study has shown that HLA-C*07:29 and B*15:27 genotypes were more common in 82 recovered COVID-19 cases (65). As a highly polymorphic region, any HLA association is also bound to be population-dependent, similar to the previously observed HLA associations with various ADs (66).
Upon antigen recognition, signal transduction through the T cell receptor (TCR) leads to the activation of T cells. Among the proteins involved in T cell activation, protein tyrosine phosphatase non-receptor type 22 (PTPN22) is a negative regulator of T cell signaling. PTPN22R620W variant is very frequently detected in a variety of ADs (Table 2) (67, 68). Interestingly, PTPN22R620W is a gain of function variant, where TCR signaling is more inhibited (69). Increased activity of PTPN22 might tip the balance in regulatory T (Treg) cell and effector T cell homeostasis against Tregs (70). Such a variant may also contribute to the T cell depletion and immunoparalysis seen in severe COVID-19 cases (51, 71).
Cytokines play a pleiotropic role in modulating leukocyte activity, differentiation and intensity of the immune response. Given the deleterious effects of hypercytokinemia in severe COVID-19 and association of IL-6 with disease severity, genetic variants that cause altered cytokine signaling might be among the host genetic factors for severe COVID-19 susceptibility (35). Janus kinase-signal transducer and activator of transcription (JAK-STAT) pathway is involved in signal transduction from cytokine receptors. Baricitinib, a selective JAK1/JAK2 inhibitor that is approved for RA treatment, is currently being tested for COVID-19 management (72). Tyrosine kinase 2 (TYK2) is a non-receptor tyrosine kinase from the JAK family that functions together with different JAK molecules, for signal transduction from IL6R, type I interferon receptors, IL-12, IL-23 and IL-10 receptors. TYK2 variants have been shown to affect disease susceptibility in a variety of ADs (Table 2) (73, 74). Similarly, these TYK2 variants may be associated with disease manifestation, as well as the response to Jak inhibitors (Jakinibs) in COVID-19. Response to Jak inhibitors is especially important as Jakinibs are among the prime drugs that are being tested for drug repurposing in COVID-19 (75) (clinical trials: NCT04320277, NCT04338958). Of great interest, during the peer review process of this perspective, the GenOMICC study identified TYK2 association with critical illness in COVID-19 (Table 1) (33).
Furthermore, the IL6R gene variants have been associated with heterogeneous response to anti-IL6R (Tocilizumab) therapy in RA (76–78) (Table 2). Hence, in terms of inferring potential host genetic factors affecting treatment responses in COVID-19 from the genetic studies of ADs, the genetic variants of IL6R might potentially be promising biomarkers of response to tocilizumab in COVID-19 (78).
Discussion and Future Directions
COVID-19 pandemic has had significant healthcare, socio-economic and personal implications, and led to extreme protective measures worldwide, such as lockdowns and border closures. Significantly increased mortality rates for the high-risk groups such as the elderly and those with chronic diseases, as well as concerns of overwhelmed healthcare systems, necessitated use of such drastic measures (3). Accumulating epidemiological data across countries have also been revealing potential new risk groups and post-infection effects of COVID-19 (e.g. acute thyroiditis in young individuals, an inflammatory condition similar to Kawasaki disease in children, and onset or worsening of diabetes (79–82)). There is a plethora of global research currently ongoing in this rapidly progressing field. Therefore, more factors and outcomes related to COVID-19 susceptibility and severity will become evident in the coming months.
Individuals with ADs and those predisposed to ADs (e.g. with family history for one or more ADs, and/or with clinically/genetically determined higher risk for ADs) are of special interest given the immune-related pathways and immunomodulatory treatments shared with COVID-19. In a number of epidemiological studies conducted, ADs have not been indicated as major risk-modulating co-morbidities for severe COVID-19 (83). However, new evidence from a large recent study from the United Kingdom has revealed RA, lupus and psoriasis, as risk factors for COVID-19–related deaths (3). Given the current contrasting epidemiological evidence, it should be considered whether a possible association might be masked by the use of some immunomodulatory drugs in ADs, or because underlying autoimmunity has not yet manifested (84). Investigating the family history of ADs and/or estimating the risk of individuals for having ADs, using available clinical and/or genetic variables in cases with severe and critical COVID-19, could be an approach to reveal a possible link between predisposition to ADs and COVID-19 severity. Even though consistent epidemiological connection between severe COVID-19 and AD is currently lacking, the shared biological pathways and genetic variants related to those pathways may still aid in deciphering the dysregulated immune response to the infection and identifying additional targets for treatment and drug repurposing.
Moreover, candidate gene studies and GWAS including larger samples of COVID-19 cases of diverse ethnic backgrounds may shed more light on the host genetic factors in severe COVID-19. Furthermore, performing sex-stratified genetic analyses in these larger sample populations is necessary for investigating the observed epidemiological sex difference in the COVID-19 susceptibility and severity. Although environmental/behavioral factors are likely to play a role, at least partially, in the observed sex differences, genetic and biological factors may also contribute to these observations (4). Sex differences observed in ADs and COVID-19 severity may also share a number of these factors.
Besides host genetic factors, it has been shown that cases with severe COVID-19 has a higher viral load and sustain this load for a longer period (85). Although it remains to be determined whether the high viral load is the direct cause of the severe disease, or it is due to a dysregulated immune response, viral load has emerged as a potential key player affecting COVID-19 severity. Additionally, mutations detected in the virus, especially in the spike protein, have a potential to alter its virulence (86). Overall, the current scientific evidence shows that COVID-19 severity is determined by a combination and interplay of host and viral factors. Thus, multidisciplinary and comprehensive studies are required to tackle the problem of severe COVID-19 and to unravel the biological mechanisms involved to reveal rational drug targets for better treatment.
The genetic underpinnings of ADs and the biological relevance of many autoimmune-associated variants are yet to be clearly elucidated. Thus, in this perspective, we focused on the immune-related genetic factors, frequently observed across different ADs, which may also be functionally relevant in COVID-19 severity and treatment. To contribute towards an effective response to the pandemic, we put forth a number of gene candidates such as TLR7, the MHC region, PTPN22, TYK2 and IL6R (Table 2), albeit not exhaustive, that may affect the risk of having ADs, and modulate COVID-19 severity, as well as treatment response via common biological pathways. After preparation of this manuscript, supportive evidence for the role of the two of the gene candidates, TLR7 and TYK2, in severe COVID-9 has already started to surface, underscoring the importance and relevance of using the knowledge gained on ADs to shed light on SARS-CoV-2 (33, 60).
Data Availability Statement
Publicly available datasets were analyzed in this study. This data can be found here: Via literature search performed using the LitCovid hub for COVID-19 using the keywords ‘COVID-19 host genetics’ (https://www.ncbi.nlm.nih.gov/research/coronavirus/).
Author Contributions
Performed the main literature search and wrote the first draft: TK, HB (contributed equally). Provided substantial literature search and writing: IK, AS, IC, DB. Critically evaluated and revised the manuscript: BB, MBH, NR, EA, BT. Conceptualization: TK, HB, EA, BT. Supervised the work: BT. All authors contributed to the article and approved the submitted version.
Funding
TK was supported by the Novo Nordisk Foundation Center for Protein Research (grant NNF14CC0001). MBH was supported by the National Institute for Health Research (NIHR) Applied Research Collaboration South London (NIHR ARC South London) at King’s College Hospital NHS Foundation Trust. The views expressed are those of the author and not necessarily those of the NIHR or the Department of Health and Social Care.
Conflict of Interest
BB was employed by The Emmes Company.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Helmy YA, Fawzy M, Elaswad A, Sobieh A, Kenney SP, Shehata AA, et al. The COVID-19 Pandemic: A Comprehensive Review of Taxonomy, Genetics, Epidemiology, Diagnosis, Treatment, and Control. J Clin Med (2020) 9(4). doi: 10.3390/jcm9041225
2. World Health Organization. https://covid19.who.int/.
3. Williamson EJ, Walker AJ, Bhaskaran K, Bacon S, Bates C, Morton CE, et al. OpenSAFELY: factors associated with COVID-19 death in 17 million patients. Nature (2020). doi: 10.1038/s41586-020-2521-4
4. Chakravarty D, Nair SS, Hammouda N, Ratnani S, Gharib Y, Wagaskar V, et al. Sex differences in SARS-CoV-2 infection rates and the potential link to prostate cancer. Commun Biol (2020) 3(1):374. doi: 10.1038/s42003-020-1088-9
5. Borges do Nascimento IJ, Cacic N, Abdulazeem HM, von Groote TC, Jayarajah U, Weerasekara I, et al. Novel Coronavirus Infection (COVID-19) in Humans: A Scoping Review and Meta-Analysis. J Clin Med (2020) 9(4). doi: 10.2139/ssrn.3550028
6. Rodriguez-Morales AJ, Cardona-Ospina JA, Gutierrez-Ocampo E, Villamizar-Pena R, Holguin-Rivera Y, Escalera-Antezana JP, et al. Clinical, laboratory and imaging features of COVID-19: A systematic review and meta-analysis. Travel Med Infect Dis (2020) 34:101623. doi: 10.1016/j.tmaid.2020.101623
7. Behzad S, Aghaghazvini L, Radmard AR, Gholamrezanezhad A Extrapulmonary manifestations of COVID-19: Radiologic and clinical overview. Clin Imaging (2020) 66:35–41. doi: 10.1016/j.clinimag.2020.05.013
8. Pascual V, Chaussabel D, Banchereau J. A genomic approach to human autoimmune diseases. Annu Rev Immunol (2010) 28:535–71. doi: 10.1146/annurev-immunol-030409-101221
9. Ramos PS, Criswell LA, Moser KL, Comeau ME, Williams AH, Pajewski NA, et al. A comprehensive analysis of shared loci between systemic lupus erythematosus (SLE) and sixteen autoimmune diseases reveals limited genetic overlap. PloS Genet (2011) 7(12):e1002406. doi: 10.1371/journal.pgen.1002406
10. Farrugia M, Baron B. The Role of Toll-Like Receptors in Autoimmune Diseases through Failure of the Self-Recognition Mechanism. Int J Inflam (2017) 2017:8391230. doi: 10.1155/2017/8391230
11. Mancia G, Rea F, Ludergnani M, Apolone G, Corrao G. Renin-Angiotensin-Aldosterone System Blockers and the Risk of Covid-19. N Engl J Med (2020) 382(25):2431–40. doi: 10.1056/NEJMoa2006923
12. Russo R, Andolfo I, Lasorsa VA, Iolascon A, Capasso M. Genetic analysis of the novel SARS-CoV-2 host receptor TMPRSS2 in different populations. bioRxiv (2020). doi: 10.1101/2020.04.23.057190
13. Asselta R, Paraboschi EM, Mantovani A, Duga S. ACE2 and TMPRSS2 variants and expression as candidates to sex and country differences in COVID-19 severity in Italy. medRxiv (2020). doi: 10.2139/ssrn.3559608
14. Ovsyannikova IG, Haralambieva IH, Crooke SN, Poland GA, Kennedy RB. The role of host genetics in the immune response to SARS-CoV-2 and COVID-19 susceptibility and severity. Immunol Rev (2020) 296(1):205–19. doi: 10.1111/imr.12897
15. Hou Y, Zhao J, Martin W, Kallianpur A, Chung MK, Jehi L, et al. New insights into genetic susceptibility of COVID-19: an ACE2 and TMPRSS2 polymorphism analysis. BMC Med (2020) 18(1):216. doi: 10.1186/s12916-020-01673-z
16. Lopera Maya EA, van der Graaf A, Lanting P, van der Geest M, Fu J, Swertz M, et al. Lack of Association Between Genetic Variants at ACE2 and TMPRSS2 Genes Involved in SARS-CoV-2 Infection and Human Quantitative Phenotypes. Front Genet (2020) 11:613. doi: 10.3389/fgene.2020.00613
17. Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q, et al. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science (2020) 367(6485):1444–8. doi: 10.1126/science.abb2762
18. Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S, et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell (2020) 181(2):271–280 e8. doi: 10.1016/j.cell.2020.02.052
19. Kai H, Kai M. Interactions of coronaviruses with ACE2, angiotensin II, and RAS inhibitors-lessons from available evidence and insights into COVID-19. Hypertens Res (2020) 43(7):648–54. doi: 10.1038/s41440-020-0455-8
20. Cheng H, Wang Y, Wang GQ. Organ-protective effect of angiotensin-converting enzyme 2 and its effect on the prognosis of COVID-19. J Med Virol (2020) 92(7):726–30. doi: 10.1002/jmv.25785
21. Gagliardi MC, Tieri P, Ortona E, Ruggieri A, et al. ACE2 expression and sex disparity in COVID-19. Cell Death Discovery (2020) 6:37. doi: 10.1038/s41420-020-0276-1
22. Thomas MC, Pickering RJ, Tsorotes D, Koitka A, Sheehy K, Bernardi S, et al. Genetic Ace2 deficiency accentuates vascular inflammation and atherosclerosis in the ApoE knockout mouse. Circ Res (2010) 107(7):888–97. doi: 10.1161/CIRCRESAHA.110.219279
23. Cao Y, Li L, Feng Z, Wan S, Huang P, Sun X, et al. Comparative genetic analysis of the novel coronavirus (2019-nCoV/SARS-CoV-2) receptor ACE2 in different populations. Cell Discovery (2020) 6:11. doi: 10.1038/s41421-020-0147-1
24. Hussain M, Jabeen N, Raza F, Shabbir S, Baig AA, Amanullah A, et al. Structural variations in human ACE2 may influence its binding with SARS-CoV-2 spike protein. J Med Virol (2020). doi: 10.1002/jmv.25832
25. Stawiski EW, Diwanji D, Suryamohan K, Gupta R, Fellouse FA, Sathirapongsasuti JF, et al. Human ACE2 receptor polymorphisms predict SARS-CoV-2 susceptibility. bioRxiv (2020). doi: 10.1101/2020.04.07.024752
26. Li Q, Rahman P. Genetic Variability of Human Angiotensin-Converting Enzyme 2 (hACE2) Among Various Ethnic Populations. bioRxiv (2020). doi: 10.1101/2020.04.14.041434
27. Chen Y, Shan K, Qian W. Asians and Other Races Express Similar Levels of and Share the Same Genetic Polymorphisms of the SARS-CoV-2 Cell-Entry Receptor. Preprints (2020). doi: 10.20944/preprints202002.0258.v1
28. Tudorache IF, Trusca VG, Gafencu AV. Apolipoprotein E - A Multifunctional Protein with Implications in Various Pathologies as a Result of Its Structural Features. Comput Struct Biotechnol J (2017) 15:359–65. doi: 10.1016/j.csbj.2017.05.003
29. Zhao Y, Zhao Z, Wang Y, Zhou Y, Ma Y, Zuo W, et al. Single-cell RNA expression profiling of ACE2, the receptor of SARS-CoV-2. bioRxiv (2020). doi: 10.1101/2020.01.26.919985
30. Kuo CL, Pilling LC, Atkins JL, Masoli JAH, Delgado J, Kuchel GA, et al. APOE e4 genotype predicts severe COVID-19 in the UK Biobank community cohort. J Gerontol A Biol Sci Med Sci (2020). doi: 10.1101/2020.05.07.20094409
31. Initiative C-HG. The COVID-19 Host Genetics Initiative, a global initiative to elucidate the role of host genetic factors in susceptibility and severity of the SARS-CoV-2 virus pandemic. Eur J Hum Genet (2020) 28(6):715–8. doi: 10.1038/s41431-020-0636-6
32. Ellinghaus D, Degenhardt F, Bujanda L, Buti M, Albillos A, Invernizzi P, et al. Genomewide Association Study of Severe Covid-19 with Respiratory Failure. N Engl J Med (2020). doi: 10.1056/NEJMoa2020283
33. Pairo-Castineira E, Clohisey S, Klaric L, Bretherick A, Rawlik K, Parkinson N, et al. Genetic mechanisms of critical illness in Covid-19. medRxiv (2020).
34. Prete M, Favoino E, Catacchio G, Racanelli V, Perosa F. SARS-CoV-2 Inflammatory Syndrome. Clinical Features and Rationale for Immunological Treatment. Int J Mol Sci (2020) 21(9). doi: 10.3390/ijms21093377
35. Zhang X, Tan Y, Ling Y, Lu G, Liu F, Yi Z, et al. Viral and host factors related to the clinical outcome of COVID-19. Nature (2020) 583(7816):437–40. doi: 10.1038/s41586-020-2355-0
36. Han H, Ma Q, Li C, Liu R, Zhao L, Wang W, et al. Profiling serum cytokines in COVID-19 patients reveals IL-6 and IL-10 are disease severity predictors. Emerg Microbes Infect (2020) 9(1):1123–30. doi: 10.1080/22221751.2020.1770129
37. Lee JS, Park S, Jeong HW, Ahn JY, Choi SJ, Lee H, et al. Immunophenotyping of COVID-19 and influenza highlights the role of type I interferons in development of severe COVID-19. Sci Immunol (2020) 5(49). doi: 10.1126/sciimmunol.abd1554
38. Soy M, Keser G, Atagunduz P, Tabak F, Atagunduz I, Kayhan S, et al. Cytokine storm in COVID-19: pathogenesis and overview of anti-inflammatory agents used in treatment. Clin Rheumatol (2020) 39(7):2085–94. doi: 10.1007/s10067-020-05190-5
39. Tay MZ, Poh CM, Renia L, MacAry PA, Ng LFP. The trinity of COVID-19: immunity, inflammation and intervention. Nat Rev Immunol (2020) 20(6):363–74. doi: 10.1038/s41577-020-0311-8
40. Prompetchara E, Ketloy C, Palaga T. Immune responses in COVID-19 and potential vaccines: Lessons learned from SARS and MERS epidemic. Asian Pac J Allergy Immunol (2020) 38(1):1–9. doi: 10.12932/AP-200220-0772
41. Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Moller R, et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell (2020) 181(5):1036–1045 e9. doi: 10.1016/j.cell.2020.04.026
42. Gagiannis D, Steinestel J, Hackenbroch C, Hannemann M, Umathum V, Gebauer N, et al. COVID-19-induced acute respiratory failure: an exacerbation of organ-specific autoimmunity? medRxiv (2020). doi: 10.1101/2020.04.27.20077180
43. Zhang Y, Xiao M, Zhang S, Xia P, Cao W, Jiang W, et al. Coagulopathy and Antiphospholipid Antibodies in Patients with Covid-19. N Engl J Med (2020) 382(17):e38. doi: 10.1056/NEJMc2007575
44. Tan YK, Goh C, Leow AST, Tambyah PA, Ang A, Yap ES, et al. COVID-19 and ischemic stroke: a systematic review and meta-summary of the literature. J Thromb Thrombolysis (2020). doi: 10.1007/s11239-020-02228-y
45. Bastard P, Rosen LB. Auto-antibodies against type I IFNs in patients with life-threatening COVID-19. Science (2020).
46. Lyons-Weiler J. Pathogenic Priming Likely Contributes to Serious and Critical Illness and Mortality in COVID-19 via Autoimmunity. J Transl Autoimmun (2020), 100051. doi: 10.1016/j.jtauto.2020.100051
47. Kanduc D, Shoenfeld Y. On the molecular determinants of the SARS-CoV-2 attack. Clin Immunol (2020) 215:108426. doi: 10.1016/j.clim.2020.108426
48. Cappello F. Is COVID-19 a proteiform disease inducing also molecular mimicry phenomena? Cell Stress Chaperones (2020) 25(3):381–2. doi: 10.1007/s12192-020-01112-1
49. Chevalier N, Thorburn AN, Macia L, Tan J, Juglair L, Yagita H, et al. Inflammation and lymphopenia trigger autoimmunity by suppression of IL-2-controlled regulatory T cell and increase of IL-21-mediated effector T cell expansion. J Immunol (2014) 193(10):4845–58. doi: 10.4049/jimmunol.1302966
50. L’Huillier A, Ren G, Shi Y, Zhang J. A two-hit model of autoimmunity: lymphopenia and unresponsiveness to TGF-beta signaling. Cell Mol Immunol (2012) 9(5):369–70. doi: 10.1038/cmi.2012.25
51. Diao B, Wang C, Tan Y, Chen X, Liu Y, Ning L, et al. Reduction and Functional Exhaustion of T Cells in Patients With Coronavirus Disease 2019 (COVID-19). Front Immunol (2020) 11:827. doi: 10.3389/fimmu.2020.00827
52. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet (2020) 395(10229):1054–62. doi: 10.1016/S0140-6736(20)30566-3
53. Kleynberg RL, Schiller GJ. Secondary hemophagocytic lymphohistiocytosis in adults: an update on diagnosis and therapy. Clin Adv Hematol Oncol (2012) 10(11):726–32.
54. Giemza-Stoklosa J, Islam MA, Kotyla PJ. Hyperferritinaemia: An Iron Sword of Autoimmunity. Curr Pharm Des (2019) 25(27):2909–18. doi: 10.2174/1381612825666190709202804
55. Huet T, Beaussier H, Voisin O, Jouveshomme S, Dauriat G, Lazareth I, et al. Anakinra for severe forms of COVID-19: a cohort study. Lancet Rheumatol (2020) 2(7):e393–400. doi: 10.1016/S2665-9913(20)30164-8
56. Guaraldi G, Meschiari M, Cozzi-Lepri A, Milic J, Tonelli R, Menozzi M, et al. Tocilizumab in patients with severe COVID-19: a retrospective cohort study. Lancet Rheumatol (2020) 2(8):e474–84.
57. Souyris M, Cenac C, Azar P, Daviaud D, Canivet A, Grunenwald S, et al. TLR7 escapes X chromosome inactivation in immune cells. Sci Immunol (2018) 3(19). doi: 10.1126/sciimmunol.aap8855
58. Frasca L, Lande R. Toll-like receptors in mediating pathogenesis in systemic sclerosis. Clin Exp Immunol (2020) 201(1):14–24. doi: 10.1111/cei.13426
59. Medvedev AE. Toll-like receptor polymorphisms, inflammatory and infectious diseases, allergies, and cancer. J Interferon Cytokine Res (2013) 33(9):467–84. doi: 10.1089/jir.2012.0140
60. van der Made CI, Simons A, Schuurs-Hoeijmakers J, van den Heuvel G, Mantere T, Kersten S, et al. Presence of Genetic Variants Among Young Men With Severe COVID-19. JAMA (2020). doi: 10.1001/jama.2020.13719
61. Salvati L, Biagioni B, Vivarelli E, Parronchi P. A gendered magnifying glass on COVID-19. Clin Mol Allergy (2020) 18:14. doi: 10.1186/s12948-020-00129-2
62. Moderbacher CR, Ramirez SI, Dan JM, Grifoni A, Hastie KM, Weiskopf D, et al. Antigen-specific adaptive immunity to SARS-CoV-2 in acute COVID-19 and associations with age and disease severity. Cell (2020). doi: 10.1016/j.cell.2020.09.038
63. Zhang Q, Bastard P. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science (2020). doi: 10.1126/science.abd4570
64. Zenewicz LA, Abraham C, Flavell RA, Cho JH. Unraveling the genetics of autoimmunity. Cell (2010) 140(6):791–7. doi: 10.1016/j.cell.2010.03.003
65. Wang W, Zhang W, Zhang J, He J, Zhu F. Distribution of HLA allele frequencies in 82 Chinese individuals with coronavirus disease-2019 (COVID-19). HLA (2020) 96(2):194–6. doi: 10.1111/tan.13941
66. Matzaraki V, Kumar V, Wijmenga C, Zhernakova A. The MHC locus and genetic susceptibility to autoimmune and infectious diseases. Genome Biol (2017) 18(1):76. doi: 10.1186/s13059-017-1207-1
67. Steck AK, Baschal EE, Jasinski JM, Boehm BO, Bottini N, Concannon P, et al. rs2476601 T allele (R620W) defines high-risk PTPN22 type I diabetes-associated haplotypes with preliminary evidence for an additional protective haplotype. Genes Immun (2009) 10(Suppl 1):S21–6. doi: 10.1038/gene.2009.87
68. Bentham J, Morris DL, Graham DSC, Pinder CL, Tombleson P, Behrens TW, et al. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat Genet (2015) 47(12):1457–64. doi: 10.1038/ng.3434
69. Vang T, Congia M, Macis MD, Musumeci L, Orru V, Zavattari P, et al. Autoimmune-associated lymphoid tyrosine phosphatase is a gain-of-function variant. Nat Genet (2005) 37(12):1317–9. doi: 10.1038/ng1673
70. Fousteri G, Liossis SN, Battaglia M. Roles of the protein tyrosine phosphatase PTPN22 in immunity and autoimmunity. Clin Immunol (2013) 149(3):556–65. doi: 10.1016/j.clim.2013.10.006
71. Qin C, Zhou L, Hu Z, Zhang S, Yang S, Tao Y, et al. Dysregulation of immune response in patients with COVID-19 in Wuhan, China. Clin Infect Dis (2020). doi: 10.1093/cid/ciaa248
72. Cantini F, Niccoli L, Matarrese D, Nicastri E, Stobbione P, Goletti D, et al. Baricitinib therapy in COVID-19: A pilot study on safety and clinical impact. J Infect (2020). doi: 10.1016/j.jinf.2020.04.017
73. Eyre S, Bowes J, Diogo D, Lee A, Barton A, Martin P, et al. High-density genetic mapping identifies new susceptibility loci for rheumatoid arthritis. Nat Genet (2012) 44(12):1336–40. doi: 10.1038/ng.2462
74. Dendrou CA, Cortes A, Shipman L, Evans HG, Attfield KE, Jostins L, et al. Resolving TYK2 locus genotype-to-phenotype differences in autoimmunity. Sci Transl Med (2016) 8(363):363ra149. doi: 10.1126/scitranslmed.aag1974
75. Spinelli FR, Conti F, Gadina M. HiJAKing SARS-CoV-2? The potential role of JAK inhibitors in the management of COVID-19. Sci Immunol (2020) 5(47). doi: 10.1126/sciimmunol.abc5367
76. Wang J, Bansal AT, Martin M, Germer S, Benayed R, Essioux L, et al. Genome-wide association analysis implicates the involvement of eight loci with response to tocilizumab for the treatment of rheumatoid arthritis. Pharmacogenomics J (2013) 13(3):235–41. doi: 10.1038/tpj.2012.8
77. Maldonado-Montoro M, Canadas-Garre M, Gonzalez-Utrilla A, Angel Calleja- Hernandez M. Influence of IL6R gene polymorphisms in the effectiveness to treatment with tocilizumab in rheumatoid arthritis. Pharmacogenomics J (2018) 18(1):167–72. doi: 10.1038/tpj.2016.88
78. Perricone C, Conigliaro P, Ciccacci C, Marcucci E, Cafaro G, Bartoloni E, et al. The differential response to anti IL-6 treatment in COVID-19: the genetic counterpart. Clin Exp Rheumatol (2020) 38(3):580.
79. Caruso P, Longo M, Esposito K, Maiorino MI. Type 1 diabetes triggered by covid-19 pandemic: A potential outbreak? Diabetes Res Clin Pract (2020) 164:108219. doi: 10.1016/j.diabres.2020.108219
81. Cheung EW, Zachariah P, Gorelik M, Boneparth A, Kernie SG, Orange JS, et al. Multisystem Inflammatory Syndrome Related to COVID-19 in Previously Healthy Children and Adolescents in New York City. JAMA (2020). doi: 10.1001/jama.2020.10374
82. Brancatella A, Ricci D, Viola N, Sgro D, Santini F, Latrofa F, et al. Subacute Thyroiditis After Sars-COV-2 Infection. J Clin Endocrinol Metab (2020) 105(7). doi: 10.1210/clinem/dgaa276
83. Ehrenfeld M, Tincani A, Andreoli L, Cattalini M, Greenbaum A, Kanduc D, et al. Covid-19 and autoimmunity. Autoimmun Rev (2020) 19(8):102597. doi: 10.1016/j.autrev.2020.102597
84. Gianfrancesco M, Hyrich KL, Al-Adely S, Carmona L, Danila MI, Gossec L, et al. Characteristics associated with hospitalisation for COVID-19 in people with rheumatic disease: data from the COVID-19 Global Rheumatology Alliance physician-reported registry. Ann Rheum Dis (2020) 79(7):859–66. doi: 10.1136/annrheumdis-2020-217871
85. Liu Y, Yan LM, Wan L, Xiang TX, Le A, Liu JM, et al. Viral dynamics in mild and severe cases of COVID-19. Lancet Infect Dis (2020) 20(6):656–7. doi: 10.1016/S1473-3099(20)30232-2
Keywords: COVID-19, SARS-CoV-2, immune response, autoimmunity, host genetics, susceptibility, cytokine, polymorphism
Citation: Karaderi T, Bareke H, Kunter I, Seytanoglu A, Cagnan I, Balci D, Barin B, Hocaoglu MB, Rahmioglu N, Asilmaz E and Taneri B (2020) Host Genetics at the Intersection of Autoimmunity and COVID-19: A Potential Key for Heterogeneous COVID-19 Severity. Front. Immunol. 11:586111. doi: 10.3389/fimmu.2020.586111
Received: 22 July 2020; Accepted: 17 November 2020;
Published: 22 December 2020.
Edited by:
Pier Luigi Meroni, Istituto Auxologico Italiano (IRCCS), ItalyReviewed by:
Danilo Malandrino, University of Florence, ItalyKuender D. Yang, Mackay Memorial Hospital, Taiwan
Copyright © 2020 Karaderi, Bareke, Kunter, Seytanoglu, Cagnan, Balci, Barin, Hocaoglu, Rahmioglu, Asilmaz and Taneri. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bahar Taneri, YmFoYXIudGFuZXJpQGVtdS5lZHUudHI=
†These authors have contributed equally to this work