Abstract
Natural killer (NK) cells are lymphocytes primarily involved in innate immunity and possess important functional properties in anti-viral and anti-tumor responses; thus, these cells have broad potential for clinical utilization. NK cells originate from hematopoietic stem cells (HSCs) through the following two independent and continuous processes: early commitment from HSCs to IL-15-responsive NK cell progenitors (NKPs) and subsequent differentiation into mature NK cells in response to IL-15. IL-15 is the most important cytokine for NK cell development, is produced by both hematopoietic and nonhematopoietic cells, and functions through a distinct delivery process termed transpresentation. Upon being transpresented to NK cells, IL-15 contributes to NK cell development via the activation of several downstream signaling pathways, including the Ras–MEK–MAPK, JAK–STAT5, and PI3K–ATK–mTOR pathways. Nonetheless, the exact role of IL-15 in NK cell development has not been discussed in a consecutive and comprehensive manner. Here, we review current knowledge about the indispensable role of IL-15 in NK cell development and address which cells produce IL-15 to support NK cell development and when IL-15 exerts its function during multiple developmental stages. Specifically, we highlight how IL-15 supports NK cell development by elucidating the distinct transpresentation of IL-15 to NK cells and revealing the downstream target of IL-15 signaling during NK cell development.
Introduction
NK cells constitute the third most abundant lineage of lymphocytes in the peripheral blood after B and T cells, accounting for approximately 8–15% of circulating cells in humans or 2–6% in mice (1). Similar to CD8+ cytotoxic T lymphocytes, NK cells effectively eliminate virus-infected cells and malignant cells by producing proinflammatory cytokines and directly lysing target cells. NK cell activation is determined by the balance between signals transduced from multiple activating receptors and inhibitory receptors, which interact with their cognate ligand on target cells (2, 3).
Interleukin (IL)-15, a member of the common gamma chain cytokine family, was first described as a T cell growth factor, like IL-2 (4). IL-15 signals through a heterotrimeric receptor consisting of IL-15Rα (CD215), IL-2/IL-15Rβ (CD122) and the common γ chain (γc, CD132) (5). Similar to IL-2, IL-15 requires the receptors IL-2/IL-15Rβ and γc to transduce signaling but differs from IL-2 by virtue of its private binding receptor IL-15Rα, which is incapable of transducing signaling but has high affinity for IL-15 and forms a complex (IL-15–IL-15Rα) in IL-15-expressing cells (4, 6, 7). These IL-15/IL-15Rα complexes have the potential to stimulate neighboring cells that express IL-2/IL-15Rβ and γc via a unique mechanism referred to as tanspresentation (8, 9). Since the discovery of transpresentation, increasing evidence has suggested that IL-15 responses are largely mediated by transpresentation at steady state (10, 11).
NK cells primarily develop in the bone marrow (BM), which contains abundant hematopoietic stem cells (HSCs) capable of differentiating toward NK cells through common lymphoid progenitor (CLP) and lineage-restricted progenitor (NKP) cells (12). Multiple internal pathways and external factors contribute to the development of NK cells from HSCs (13). Most importantly, the pleiotropic cytokine IL-15 is indispensable for the development and homeostasis of NK cells as highlighted by their significant deficiency in IL-15-deficient mice. Correspondingly, deficiency in IL-15 or any one of the IL-15 receptor subunits, such as the IL-15Rα, IL-15Rβ, and γc in mice, results in a dramatic paucity of mature NK cells (14–17). Parallel with the role of IL-15 in mice, several studies have demonstrated that the early commitment of NK cells from human CD34+ hemopoietic progenitor cells into NKP cells is dependent on the coordinated function of IL-3, IL-7, c-kit ligand (KL), and flt3 ligand (FL) but not IL-15, whereas IL-15 is involved in the emergence of CD56+ NK cells (18, 19). Furthermore, Huntington et al. demonstrated that human NK cell differentiation that occurs in a linear fashion from CD56hiCD16−KIR− to CD56loCD16+KIR− and finally to CD56loCD16+KIR+ requires IL-15 in a humanized model (20). Collectively, these findings illustrate that IL-15 signaling is essential for NK cell development and homeostasis in both mice and humans. Interestingly, Sun et al. recently reported that the requirement of IL-15 for NK cell development could be partially overcome by acute mouse cytomegalovirus (MCMV) infection, as IL-12 but not IL-15 primarily drives the anti-viral response of NK cells even in mice lacking the γc (21). Further studies are required to investigate whether this represents an IL-15-independent NK cell development manner.
Due to the critical role of IL-15 in NK cell development, dissecting the signaling pathways that allow IL-15 to control the development and homeostasis of NK cells is fundamental to determine the molecular details of immune regulation. In this review, we provide an overview of the specific IL-15 signaling that transcriptionally regulates NK cell development and maturation.
When Does IL-15 Promote NK Cell Development?
IL-15 Is Dispensable for NK Cell Commitment but Promotes Later Development
Mice and human NK cells are generated from HSCs through multiple but sophisticated stages in specific developmental niches with internal and external regulatory pathways governing NK cell development. In brief, NK cell development primarily involves the following two independent and continuous processes: early NK cell commitment to IL-15-responsive NKPs and subsequent phenotypical and functional maturation of NK cells in response to IL-15. Early NK cell commitment to NKP cells is characterized by the acquisition of CD122 (IL-15Rβ), which is a critical subunit of the IL-15 receptor and dimerizes with γc to transduce IL-15 signaling (22, 23). However, IL-15 is not involved in the generation of IL-15-responsive NKPs because the IL-15 receptor is not expressed prior to the NKP stage (24). Recently, pre-NKP cells were identified as the earliest committed NK cell progenitors in murine BM, and these cells reside downstream of CLP and differentiate into NKPs (23). Although pre-NKP cells express undetectable levels of CD122, they are fully committed to the NK lineage both in vitro and in vivo. Therefore, IL-15 is not necessary for NK cell lineage commitment. Furthermore, mice deficient in γc exhibit an intact NKP compartment (16), and IL-3, IL-7, KL, and FL synergistically drive the differentiation of NKP cells from human HSCs in vitro in the absence of IL-15 (25). Conversely, IL-15 is indispensable for the later development of NK cells. The expression of CD122 endows NK cells with the capacity to be responsive to IL-15; thus, these cells can become phenotypically and functionally mature and exhibit survival in response to IL-15 (16).
IL-15 Receptor Expression Varies in Different Stages of NK Cell Development
Intriguingly, the expression of CD122 on NK cells is not static but dynamically changes with NK cell maturation. It has been previously demonstrated that CD56bright NK cells express higher levels of CD122 as well as elevated CD122 transcripts compared with CD56dim NK cells, and thus are intrinsically more responsive to IL-15 (26–29). This observation explains the decreased proliferation capacity in response to IL-15 or dendritic cell (DC) stimulation during NK cell maturation (30, 31) and is consistent with the fact that cytokines, such as IL-2 and IL-15, fail to reverse the proliferation defects of CD57+ terminally matured NK cells (32). Consistent with the observation in human NK cells, CD122 expression is significantly decreased during maturation from mice CD11b+CD27+ NK cells to CD11b+CD27− NK cells and concomitant with decreased proliferation capacity (33). Despite the vitally important role of IL-15 in NK cell maturation, the exact role of decreased CD122 expression during NK cell terminal maturation needs to be further elucidated.
Transcriptional Regulation of IL-15 Receptor Expression at Different Stages During NK Cell Development
Although CD122 (encoded by Il2rb) is critical for NK cell development by transducing IL-15 signaling, the coordinated regulation of CD122 expression by various transcription factors remains elusive. Previous studies have demonstrated that RUNX3 (one of the Runx family transcription factors), T-bet, and Eomesodermin (Eomes) directly bind to the promoter region of Il2rb and induce CD122 expression (34, 35). However, these transcription factors are not simultaneously functional, but rather function at different stages of NK cell development. In the NK cell development pathway, RUNX3 expression is initiated at the NKP stage. The inactivation of RUNX3 in HSCs partially disturbed the generation of CD122+NKP cells in vitro but not completely, indicating that other unknown transcription factors contribute to the expression of CD122 during NK cell commitment. In addition, the deletion of RUNX3 in immature NK cells in mice only slightly reduced CD122 expression on NK cells, and the absolute number of NK cells was not significantly affected. These results confirmed that RUNX3 is necessary for the acquisition of CD122 during NK cell lineage commitment but is not essential for the maintenance of CD122 at the later maturation stages of NK cell development (34).
In contrast, T-bet and Eomes are weakly expressed at the NKp stage but highly expressed during NK cell maturation; therefore we speculated that T-bet and Eomes are not firmly involved in the induction of CD122 at the NKp stage but may contribute to the maintenance of CD122 expression during maturation (35, 36). Consistently, mice harboring genomic deletions of T-bet and Eomes lack NK cells, but CD122hi precursors of NK cells were observed (36). In addition, the deletion of Eomes in mice results in significantly decreased CD122 expression at different stages of NK cell maturation (33, 35, 37). Moreover, Eomes+ NK cells express more CD122 and proliferate better than Eomes− NK cells, which are called Innate Lymphoid Cells (ILCs) 1 now (38, 39). However, CD122 expression is upregulated in T-bet-deficient NK cells, and this finding may be attributed to increased Eomes expression, which is repressed by T-bet (40). These results indicate that Eomes but not T-bet plays a dominant role in the maintenance of CD122 expression during NK cell maturation. Consistently, although T-bet expression is upregulated during the NK cell transition from the CD11b+CD27+ to CD11b+CD27− stage, CD122 expression is progressively decreased, accompanied by a reduction in Eomes expression (33).
In conclusion, the induction of CD122 during early NK cell commitment is dependent on RUNX3, whereas Eomes but not T-bet maintains the expression of CD122 to promote NK cell maturation (Figure 1).
Figure 1

Transcriptional regulation of CD122 expression during different stages of NK cell development. RUNX3 coordinates with T-bet and Eomes to control CD122 expression during NK cell development. Therein, the induction of CD122 expression during early NK cell commitment is determined by RUNX3 and other unknown transcription factors. Moreover, both T-bet and Eomes contribute to CD122 expression during NK cell maturation. However, Eomes but not T-bet plays a predominant role in the maintenance of CD122 expression to promote NK cell maturation.
Which Cells Produce IL-15 to Promote NK Cell Development?
Isolated Expression of IL-15 mRNA and Protein
IL-15 mRNA is constitutively expressed in a broad range of tissues, including hematopoietic cells [monocytes, macrophages, and dendritic cells (DCs)] and non-hematopoietic cells (epithelial cells, fibroblasts, nerve cells, skeletal muscle and keratinocytes) (4). In contrast to the widespread expression of IL-15 mRNA, IL-15 protein is only detectable in a more restricted population at steady state. This discrepancy between widespread IL-15 transcript expression and restricted protein expression is attributed to extensive checkpoints at transcription, translation, and intracellular trafficking, particularly post-transcriptional checkpoints. Multiple 5′-untranslated region (UTR) AUG sequences, a long signal peptide (LSP) (48 amino acid) and a negative regulatory element in the C-terminus of the coding sequence and mature protein all contribute to impede translation (41–44). Surprisingly, a 250-fold increase in IL-15 expression is observed after the removal of those three predominant restraints, further demonstrating the contribution of multiple post-transcriptional mechanisms in limiting IL-15 translation (42). Additionally, there are two isoforms of IL-15 mRNA, differing in their signal peptide, which result in distinct intracellular trafficking, localization, and secretion patterns (44–46). Both isoforms produce mature IL-15 protein. IL-15 with LSP is primarily located in the Golgi apparatus, early endosomes, and endoplasmic reticulum and functions as a secretory signal peptide, whereas IL-15 with a short signal peptide (SSP) (21 amino acid) is not secreted, appears to reside in the nucleus and cytoplasmic components (44). Tight regulation of IL-15 expression is important because of the potent capacity of IL-15 to promote inflammation.
The Production of IL-15 by Hematopoietic and Non-Hematopoietic Cells
Due to the extremely low level of IL-15 protein expression at steady state, even after stimulation, IL-15 is barely detectable by antibodies. However, the establishment of an IL-15 reporter mouse line allows IL-15-producing cells to be visualized by flow cytometry or fluorescence microscopy as well as immunohistochemistry in vivo (47–49). Among hematopoietic cells, IL-15 is predominantly produced by monocytes, macrophages, DCs, myeloid cells, and some early hematopoietic cells (Table 1) (4, 47). Therein, CD8+conventional DCs are the major DC subsets responsible for IL-15 expression rather than plasmacytoid DCs (47, 48). Moreover, myeloid cells, including neutrophils, basophils, and eosinophils, express high levels of IL-15 in vivo, whereas lymphoid lineages, such as T cells, B cells, NKT cells, and NK cells, express minimal to undetectable IL-15 levels. Interestingly, LSK cells (Lineage−Sca-1+c-kit+), which constitute a heterogeneous population of both long-term and short-term HSCs in BM, uniformly express high levels of IL-15.
Table 1
| Hematopoietic cells | Non-hematopoietic cells |
|---|---|
| Monocytes Macrophages DCs: CD8+conventional DCs Myeloid cells: neutrophils, basophils and eosinophils Early hematopoietic cells: LSK cells |
In the BM: CXCL12-abundant reticular (CAR) cells In the thymus: thymic medulla and medullary thymic epithelial cells with high MHC class II expression In the lymph nodes: fibroblastic reticular cells (FRCs), gp38−CD31− stromal cells and blood endothelial cells (BECs) In the spleen: VCAM-1+ stromal cells |
The production of IL-15 by hematopoietic cells and non-hematopoietic cells.
Among nonhematopoietic cells, a distinct category of stromal cells together with epithelial cells directs IL-15 expression in primary and secondary lymphoid organs (Table 1) (49). In BM, IL-15 is predominantly expressed by VCAM1+PDGFRβ+CD31−Sca-1− mesenchymal stromal cells, which correspond to a distinct subset of CXC chemokine ligand-12 (CXCL12)-abundant reticular (CAR) cells and may function as a developmental niche for NK cells (50, 51). In the thymus, IL-15 is highly expressed in the thymic medulla and medullary thymic epithelial cells with high MHC class II expression, providing a major source of IL-15. In the lymph nodes, IL-15-expressing cells, which include some fibroblastic reticular cells (FRCs) and gp38−CD31− stromal cells, primarily reside in the T-cell zone and medulla. In addition, in the lymph nodes, blood endothelial cells (BECs) also express high IL-15 levels. In the spleen, VCAM-1+ stromal cells are responsible for IL-15 expression.
In contrast to the low expression of IL-15 at steady state, its expression capacity is further strengthened by several inflammatory stimuli, including Toll-like receptor (TLR) ligands and cytokines (47, 52–54). Previously, studies have proven that bacterial lipopolysaccharide (LPS) or the double-stranded RNA mimic Poly I:C initiates TLR signaling to induce IL-15 expression (55). Similarly, IL-15 induction is interferon (IFN)-α receptor (IFNAR)-dependent after viral infection (47). Although IL-15 mRNA is elevated in all DC subsets after inflammatory stimuli, only CD8α+ DCs upregulated IL-15 protein expression, further specifying a DC subset for IL-15 production (47, 55, 56). Moreover, it has been demonstrated that upregulated IL-15 expression also exists in monocytes, macrophages, and tumor-associated neutrophils in inflammatory environments (57–59). In addition, LPS-induced inflammation also greatly increases IL-15 expression in stromal cells, including BECs and lymphatic endothelial cells (LECs), whereas this effect is not significant in other stromal cells (49).
Both Hematopoietic and Non-Hematopoietic Cells Promote NK Cell Development by Producing IL-15
The diverse subsets of IL-15-expressing cells play different but overlapping roles in the development of NK cells in BM and peripherally by producing and transpresenting IL-15 (Figure 2). Overall, hematopoietic cells were found to override the importance of non-hematopoietic cells in promoting NK cell development (10, 11). Correspondingly, restricting IL-15Rα or IL-15 expression to hematopoietic cells completely recovered NK cell development at all stages in BM with a slight defect in peripheral mature NK cells, whereas the development of NK cells was only partially rescued in all tissues when IL-15Rα or IL-15 expression was specifically limited to non-hematopoietic cells.
Figure 2

NK cell commitment is represented by the transition from CLPs to NKPs that acquire the most representative hallmark CD122. With the expression of NK1.1 and NKp46, immature NK (iNK) cells are originated from NKPs. According to the expression of CD27 and CD11b, NK cell maturation can be distinguished into four stages: CD11b−CD27−(iNK) → CD11b−CD27+(M1) → CD11b+CD27+(M2) → CD11b+CD27−(M3). Stromal cells expressing IL-15α are sufficient for the generation of immature NK cells in the BM. Moreover, some unrecognized hematopoietic cells contribute to differentiate into M1 and M2 NK cells by supplying IL-15. After migration from BM to the periphery, DCs and macrophages transpresent IL-15 for M2 to promote NK cell terminal maturation. Both hematopoietic and non-hematopoietic cells promote NK cell development by producing IL-15 at different stages.
As a critical component of hematopoietic cells, monocytes, DCs, and macrophages contribute to NK cell development by producing IL-15. The indispensable role of monocytes in NK cell development and homeostasis was exemplified by the observation that the interaction between NK cells and spleen monocytes promotes CD11b+CD27+ NK cell differentiation into CD11b+CD27− NK cell in an IL-15Rα- and IL-15-dependent and cell–cell contact-dependent manner (60). Consistently, immunobiological studies revealed that monocytes and NK cells reside in close proximity of the red pulp of the spleen (60). Additionally, although IL-15Rα expression on DCs and macrophages is dispensable for NK cell differentiation in BM, it is required for the maintenance of mature NK cells in the periphery given that specific knockdown of IL-15Rα on DCs or macrophages results in a substantial reduction in NK cells in the periphery (61). Moreover, NK cell homeostasis is not exacerbated when IL-15Rα is conditionally deleted from both DCs and macrophages, indicating that DCs and macrophages maintain NK cell populations in the peripheral blood and organs in a similar manner (61). Furthermore, mice with conditional deletion of IL-15Rα in DCs or macrophages exhibit significant deficits in terminally differentiated CD27−CD11b+ NK cells, although the subsets of peripheral CD27+CD11b− and CD27+CD11b+ NK cells remain intact. Thus, DCs and macrophages were dispensable for NK cell development in the BM and necessary for NK cells’ terminal differentiation in the periphery. However, using CD11c/IL-15Rα Tg mice with an IL-15Rα−/− background, Castillo et al. revealed that DCs contribute to the development of NK cells in both the BM and peripheral blood and organs. Mice that exclusively expressed IL-15Rα on DCs exhibited partial recovery of NK cells in all tissues, and the greatest reconstitution was noted in the BM (10). Furthermore, IL-15 exclusive transpresentation via DCs is insufficient for the maturation of CD27−CD11b+ NK cells, which preferentially reside in the peripheral blood and organs. These discrepancies in the function of DCs during NK cell development may be attributed to divergent models. Nonetheless, transpresentation of IL-15 by DCs and macrophages is not responsible for the all IL-15 events attributed to IL-15Rα+ hematopoietic cells, as NK cell deficiency in the BM of mice with IL-15Rα deletion in DCs or macrophages is less apparent than that observed in IL-15Rα-deficient mice (10, 61). Therefore, besides DCs and macrophages, other unrecognized hematopoietic cells in the BM that contribute to NK cell development have not been identified.
Moreover, IL-15 expression by non-hematopoietic cells is more important for NK cell development in BM other than in the periphery, as limiting IL-15Rα expression to non-hematopoietic cells results in more evident NK cell recovery in BM, and this effect is virtually non-existent in the spleen or liver (10). Non-hematopoietic cells expressing IL-15α are sufficient for the generation of immature NK cells but are incapable of NK cell maturation (10). This finding may be attributed to the high expression of IL-15 in CXCL12-abundant reticular (CAR) cells, which are in close contact with NK cells in BM (49–51). In addition to transducing the downstream signaling of CXC chemokine receptor (CXCR4), the engagement of CXCL-12 on CAR cells via CXCR4 expressed by NK cells also contributes to NK cell retention in BM, which provides a special IL-15-sufficient niche for NK cell development. In vivo and in vitro studies demonstrated that the CXCL-12/CXCR4 axis is essential for NK cell maturation and proliferation (50, 51). However, the exact role of IL-15 expression in CAR cells is unidentified. Additionally, consistent with the high expression of IL-15 in fibroblastic reticular cells (FRCs) of lymphoid nodes, the specific ablation of IL-15 in FRCs results in almost complete abrogation of NK cells in Peyer’s patches (PPs) and gut-associated secondary lymphoid organs (SLOs), indicating that FRCs promote NK cell homeostasis via the establishment of an IL-15-dependent niche (62).
Furthermore, human spleen-derived fibroblasts are sufficient for the development of functional CD56brightCD3− NK cells in vitro, and neutralizing IL-15 signaling or disturbing direct contact significantly abrogates CD56dimCD3− NK cell generation, indicating that fibroblasts express and transpresent IL-15 to support NK cell development (63). However, no in vivo studies have demonstrated the role of fibroblasts in NK cell development. In conclusion, although previous studies have uncovered the distinct function of DCs and macrophages in NK cell development, the exact biological role of IL-15 expression in other hematopoietic cells (myeloid cells and early HSCs) and diverse stromal cells that reside in the BM or peripheral blood and organs during NK cell development remains poorly described.
How Does IL-15 Transpresentation Support NK Cell Development?
Although IL-15 is critical for NK cell development, IL-15 alone only weakly activates its downstream signaling. In fact, the exertion of IL-15 function is dependent on IL-15Rα, which has high affinity to IL-15 (64). With the aid of IL-15Rα, IL-15 is protected from degradation, accumulates on the membrane and in the circulation of mice, and exhibits increased biological activity (65). Accordingly, IL-15-expressing cells must simultaneously express IL-15Rα to supply IL-15 to IL-15-responsive NK cells bearing IL-15Rβ and γc (66, 67). The distinct requirement is further unveiled by the discovery that IL-15 is preassembled with IL-15Rα in a complex in the endoplasmic reticulum/Golgi and subsequently shuttled to the cell surface (8, 68). This cell surface complex is called membrane-associated IL-15-IL-15Rα complex (mIL-15 complex) (Figure 3).
Figure 3

The production of mIL-15 complex and sIL-15 complex. IL-15 preassembles with IL-15Rα in a complex in the endoplasmic reticulum/Golgi and subsequently shuttled to the cell surface, becoming membrane-associated IL-15–IL-15Rα complex (mIL-15 complex). After immune stimulating, the ADAM17 translocates to the cell surface with increased activity. The mIL-15 is cleaved by ADAM17 at the ectodomain of IL-15Rα to induce the formation of soluble IL-15–IL-15Rα complex (sIL-15 complex).
Nonetheless, the mIL-15 complex could be cleaved from the surface to form soluble IL-15–IL-15Rα complex (sIL-15 complex) in response to several immune stimuli, including type I interferons (type I IFNs), Poly I:C stimulation, total body irradiation (TBI), Toll-like receptor (TLR) stimulation, virus infections, and activation of the stimulator of IFN genes (STING) pathway (57, 69, 70). It is reported that this process is mediated by A Disintegrin and Metalloprotease (ADAM) 17 protease, whose expression is upregulated on the surface of IL-15 expressing cells after immune stimulus (69). Consistently, in vivo evidence demonstrated that the IL-15–IL-15Rα complex exists in two forms, mIL-15 complex and sIL-15 complex, in humans and mice (65, 70). Although the sIL-15 complex was identified several years ago, its biological significance remains controversial. Interestingly, in vivo studies revealed that the sIL-15 complex serves as a potent agonist and is approximately 50–100 times more potent at promoting NK cell proliferation than recombinant IL-15 alone (71, 72). Thus, the sIL-15 complex may play an important role in stimulating IL-15 responses. Consistently, Anton et al. (73) demonstrated that low doses of sIL-15 contribute to the phosphorylation of Stat5 in NK cells, whereas higher concentrations of sIL-15 are required to stimulate S6 phosphorylation in vitro. However, other studies discovered that the mIL-15 complex mediates NK cell activation rather than the sIL-15 complex present in the supernatants of IL-15-expressing cells cultured in vitro or in the serum of mice ex vivo (68, 74). This contradiction may be attributed to the different experimental methods and low concentration of sIL-15 complex in the supernatants and serum. Considering the rare detection of sIL-15 complex at steady states and substantial sIL-15 complex produced after immune stimulation, we hypothesize that the sIL-15 complex mediates IL-15 responses during immune activation but not during steady states. However, due to the technological limitation, it is hard to distinguish IL-15 responses mediated by sIL-15 complex from mIL-15 complex.
In contrast to the sIL-15 complex, which functions independently of cell–cell interactions, the mIL-15 complex functions through a distinct delivery mechanism termed transpresentation during cell–cell contact to transduce IL-15 signaling to NK cells via the IL-12/IL-15Rβ and γc complex (8, 64). Consistently, although IL-15Rα knockout mice exhibit dramatic defects in NK cell development (15), the specific deletion of IL-15Rα in NK cells has no detrimental effect on NK cell development. However, adoptive transfer of normal NK cells into IL-15Rα-deficient mice results in the abrupt loss of these cells, indicating that IL-15Rα expressed by non-NK cells but not NK cells is required to mediate IL-15 signaling for NK cell development (75, 76).
During transpresentation, the IL-15Rα–IL-15 complex functions through three different mechanisms to transduce IL-15 signaling in NK cells (73, 77) (Figure 4). First, presenting cells can directly interact with NK cells via the formation of an immunologic synapse where the membrane-associated IL-15Rα–IL-15 complex on presenting cells interacts with the IL-15Rβ-γc receptor at the plasma membrane of NK cells to transduce IL-15 signaling. Consistently, the mIL-15 complex expressed by DCs accumulates at the synapse with NK cells, and the use of an antibody to block IL-15Rα promotes NK cell apoptosis and significantly reduces NK cell survival (78). In addition to the IL-15/IL-15Rα-β/γc interaction, many other receptor–ligand interactions may simultaneously occur at NK cell immunologic synapses, such as interactions between activating receptors or inhibitory receptors and their ligands, separately (78, 79). Interestingly, using a confocal microscopy assay, the mIL-15 complex accumulated in the periphery of activating synapses, whereas the mIL-15 complex was evenly distributed along the entire contact area when the NK cell line made contact with IL-15-expressing cells (79). Nonetheless, the regulatory role of these receptor–ligand interactions in IL-15 signaling remains elusive. In vitro studies have demonstrated that the interaction between inhibitory receptors, such as KIR2DL1, KIR2DL2/3, or CD94-NKG2A, and their cognate ligands selectively inhibited the phosphorylation of AKT and S6 but not Stat5, and this effect was concomitant with reduced proliferation induced by the mIL-15 complex but not the sIL-15 complex (79).
Figure 4

The transpresentation of IL-15 to NK cells during NK cell development. (A) The sIL-15 complex (soluble IL-15–IL-15Rα complex), which exists as a soluble extracellular complex in serum, directly interacts with NK cells that express IL-15Rβ–γc chains without transpresentation by presenting cells. (B) Presenting cells directly interact with NK cells via the formation of an immunologic synapse. Then, the mIL-15 complex (membrane-associated IL-15–IL-15Rα complex) on presenting cells interacts with the IL-15Rβ–γc receptor at the plasma membrane of NK cells to transduce IL-15 signaling. (C) During synapse formation, the mIL-15 complex is cleaved from the plasma membrane and then endocytosed by NK cells. The internalized mIL-15 complex co-localizes with the IL-15Rβ–γc receptor in the cytoplasm and contributes to IL-15 signaling activation. (D) The intact mIL-15 complex together with the plasma membrane of presenting cells is internalized into NK cells through a distinct process termed transendocytosis without being cleaved. Subsequently, the transendocytosed IL-15Rα–IL-15 complex colocalizes with the IL-15Rβ/γc chain in intracellular NK cell compartments and contributes to IL-15 signaling activation.
During cell-to-cell contact, the membrane-bound IL-15Rα–IL-15 complex is internalized by NK cells and contributes to the activation of IL-15 signaling (77). This process is dependent on the proteolytic cleavage of IL-15Rα, which allows the IL-15Rα–IL-15 complex to separate from the presenting cells. In addition, the IL-15Rα–IL-15 complex gradually accumulates in NK cells during the interaction between IL-15-presenting cells and NK cells. After separation from the presenting cells, the previously restored IL-15 complex contributes to the survival and residual proliferation of NK cells in a time-limited manner. In contrast, abrogation of IL-15Rα cleavage results in enhanced and prolonged Stat5 phosphorylation concomitant with increased IL-15 expression in the synapse. This observation further demonstrated that the mIL-15 complex on presenting cells also contributes to the activation of IL-15 signaling during cell-to-cell contact (77). Therefore, mIL-15 complex cleavage and internalization could represent a negative regulatory mechanism that reduces the availability of transpresented-IL-15 and protects NK cell from excessive IL-15 signaling.
However, inhibition of IL-15Rα cleavage did not completely abrogate the entry of the IL-15Rα–IL-15 complex into NK cells, indicating that the IL-15 entry is not exclusively dependent on the shedding of the membrane-associated IL-15Rα–IL-15 complex (77). Indeed, the intact membrane-associated IL-15Rα–IL-15 complex from the presenting cells together with the plasma membrane of presenting cells is internalized into NK cells through a distinct process termed transendocytosis without being cleaved (73). Subsequently, the transendocytosed IL-15Rα–IL-15 complex colocalizes with the IL-15Rβ/γc chain in intracellular NK cell compartments to promote ribosomal protein S6 phosphorylation and NK cell proliferation. Consistently, interference of transendocytosis by silencing the small GTPase TC21, which is a critical component of transendocytosis, substantially inhibits S6 phosphorylation but not Stat5 phosphorylation in NK cells.
What is the Downstream Target of IL-15 Signaling During NK Cell Development?
IL-15-JAK-STAT5 Signaling for NK Cell Development
Upon the engagement of the IL-15Rα–IL-15 complex with the IL-15Rβ/γc receptor, three distinct signaling pathways, including Ras–MEK–MAPK, JAK–STAT5 and PI3K–ATK–mTOR are activated and contribute to NK cell development. The IL-15Rα–IL-15 complex primarily induces the activation of the JAK–STAT5 pathway via recruiting Janus kinase 1 (JAK1) and JAK3 (Figure 5). Interestingly, JAK1 binding to the IL-2/IL-15Rβ and JAK3 combining with γc is crucial for signal transduction by activating JAK1 and JAK3, which induce the phosphorylation of tyrosine residues in IL-2/IL-15 Rβ (80–82). This model has been further confirmed by the discovery that humans with deletion of JAK3 exhibited similar phenotypes of severe combined immunodeficiency (SCID) as γc-deficient patients (83). Although the specific function of JAK1 and JAK3 varies considerably, genetically engineered mice provide the possibility to determine the distinct roles of individual proteins. While deficiency of Jak1 in mice leads to perinatal lethality (84), a remarkable decrease of immature B220+ NK cells was observed in adult mice with inducible loss of Jak1, indicating that Jak1 is essential for NK cell development (85). These observations were recently validated in mice with conditional deletion of Jak1 in Ncr1-expressing cells (Jak1fl/fl Ncr1Cre), displaying blockade of NK cell development at the NKp and iNK stages in a dose-dependent manner (86). Not surprisingly, JAK3 also plays an important role in NK cell development, coinciding with the finding that Jak3-deficient mice suffer from differentiation block of NK cells at the pre-NKP stage (87). Despite the cooperation of Jak1 and Jak3 in NK cell development, accumulating evidence has proposed that Jak1 plays a dominant role overriding Jak3 during the signal transduction (88, 89). The specific inactivity of Jak3 in human cells lines fails to attenuate STAT5 phosphorylation as anticipated, as two Jak kinases have an equivalent function in signal transduction, whereas remarkable abrogated downstream signaling was found in Jak1-inactive cells lines (88). Furthermore, the knockdown experiments suggested that Jak1 is responsible for the phosphorylation of Jak3 and STAT5 after cytokine receptor activation, and Jak3 contributes to enhance Jak1 activity by phosphorylating it. Likewise, quantitative mass spectrometry analysis also revealed that Jak1 is more important that Jak3 in mature NK cells (90). Although, many studies have addressed the vital roles of Jak1 and Jak3 in signal transduction, molecular interactions between the two Jak kinases and their individual contributions in NK cells remain to be determined.
Figure 5
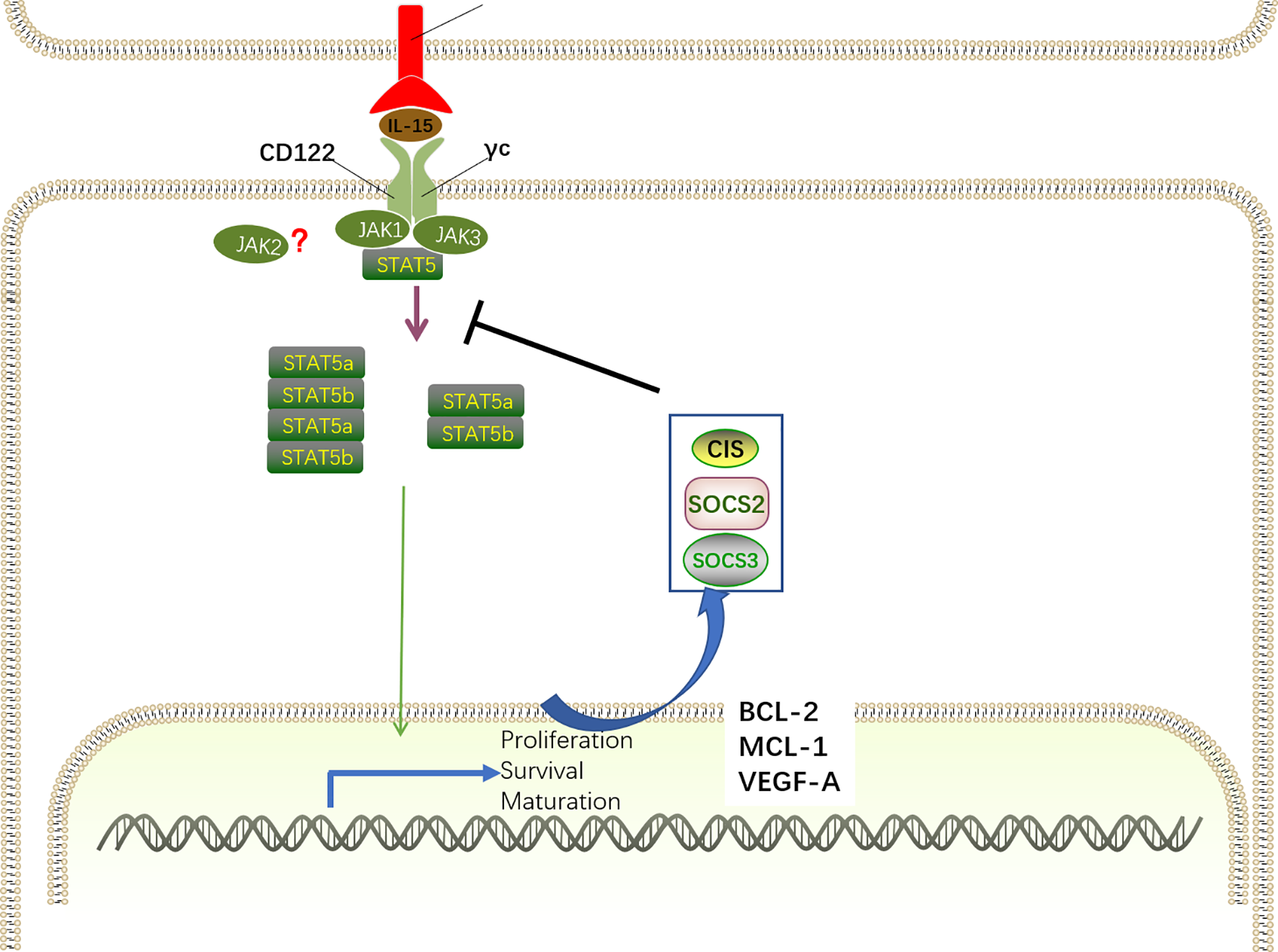
IL-15-JAK-STAT5 signaling for NK cell development. The IL-15Rα–IL-15 complex primarily induces the activation of the JAK–STAT5 pathway via recruiting JAK1 and JAK3. For signal transduction, JAK1 binds to the IL-2/IL-15Rβ and JAK3 combines with γc, inducing the recruitment and phosphorylation of STAT5. By oligomerizing into dimers and tetramers, phosphorylated STAT5a and STAT5b translocate into the nucleus to drive the expression of STAT-target genes encoding proteins related with NK cell development, survival, proliferation, and function, including MCL-1, BCL-2, and VEGF-A. Specifically, IL-15–JAK–STAT5 signaling also promotes the transcription of SOCS family members. SOCS proteins comprise a negative feedback loop to retrain the JAK–STAT5-mediated pathway in NK cell development.
Although it has long been believed that IL-15 signaling is exclusively mediated by JAK1/3, the role of JAK2 in IL-15 signaling is controversial. Notably, a recent study described that JAK2 phosphorylates STAT5 downstream of IL-15 during NK cell differentiation in vitro (91). Accordingly, mice with conditional deletion of Jak2 in HSC exhibited impaired NK cell maturation (92). However, it has been shown that JAK2 is intrinsically dispensable for NK cell development as mice with conditional deletion of JAK2 in NKp46+ cells exhibited intact NK cell numbers and maturation (86). These discrepancies indicate that the absence of JAK2 may extrinsically interfere in NK cell maturation by altering the cytokine milieu.
The discovery of the IL-15-JAK association has contributed to the finding that members of the signal transducer and activation of transcription (STAT) family directly bind to phosphor-tyrosine docking site(s) in the IL-2/IL-15Rβ chain and are then phosphorylated by JAK1 and JAK3 on their tyrosine residues (80). Similar with IL-2, IL-15 predominantly induces STAT5 activation, despite the finding that STAT3 and STAT1 can also be activated to a lesser extent (93). STAT5 is comprised by two distinct transcription factors, STAT5a and STAT5b, that have a remarkable degree of sequence homology (approximately 96%) (94). By oligomerizing into dimers and tetramers, phosphorylated STAT5a and STAT5b translocate into the nucleus to drive the expression of STAT-target genes, which is critical for NK cell development, survival, proliferation, and function (95–97). STAT5 dimers preferentially bind to γ-interferon-activated sequence (GAS) motifs, whereas STAT5 tetramers are more flexible given the capacity for various non-consensus GAS motifs (98). Interestingly, Lin and colleagues revealed that STAT5 dimers are sufficient for early NK cell development, proliferation and cytotoxic capacity, whereas STAT5 tetramers are necessary for NK cell maturation and survival through the induction of the anti-apoptotic protein Bcl2 (99).
It is indisputable that STAT5-related transcriptional programs mediated by IL-15 activation are essential for the biological functions of IL-15. The indispensable role of STAT5-mediated transcriptional regulation in NK cell development has been highlighted by the finding that NK cell differentiation was abrogated at the NKp stage in Ncr1-iCreTg mice with conditionally deleted STAT5 (100). Consistently, disrupted NK cell maturation and impaired lytic function were observed in humans with STAT5b mutations (101). Therefore, STAT5 downstream of Jak kinases is essential for transducing IL-15 signaling. Despite the largely redundant functions of STAT5a and STAT5b, their distinct roles have been verified in single knockout mice for STAT5a or STAT5b (102, 103). Deficiency of STAT5b results in more dramatic defects in NK cell development than deletion of STAT5a, indicating that STAT5b plays a dominant role in NK cell development (104, 105). Consistently, transcriptional analysis revealed that the transcripts mediated by STAT5b are more abundant (104). Furthermore, only Stat5b knockout mice exhibit elevated transcription of VEGFA, an angiogenic factor that is transcriptionally repressed by STAT5.
Moreover, chromatin immunoprecipitation (ChIP) analysis of STAT5 binding sites revealed that STAT5 directly targets a large number of genes encoding proteins related with NK cell development and function, including ID2, EOMES, T-BET, perforin, granzymes, and IFN-γ. Additionally, STAT5 can also bind to Mcl-1 and Bcl-2, correlating with the ability of IL-15 to induce the expression of these genes and sustain NK cell survival (97, 106). Overexpression of BCL-2 enables the survival of STAT5-deficient NK cells but has no influence on proliferation, maturation, or effector functions. However, it seems that Mcl1 is more important in promoting NK cell survival than Bcl-2, as IL-15 stimulation maintains NK cell survival when Bcl-2 was inhibited but not when Mcl1 was inactivated (96).
However, STAT5 is not only correlated with transcriptional activation of gene expression, as the repressive effect of STAT5 binding is also present in NK cells. STAT5 has been shown to bind the Vegf-a gene promoter in NK cells, correlating with suppressed expression of the pro-angiogenic factor VEGF-A in mice and humans (106). In vitro studies revealed that STAT5-inactive NK cells showed abundant VEGFA expression, and this effect was also confirmed in vivo by increased tumor formation in the absence of STAT5 (106). In line with the observations in mice, tumor-infiltrating NK cells with VEFGA secretion properties promote tumor progression and are associated with poor outcomes in patients (107–109). According to the repressive effects of STAT5 on IL-17 and Bcl6 mRNA expression in T cells (110, 111), further research is essential to deepen our understanding of the distinct roles of STAT5 in NK cells.
IL-15 signaling contributes to the induction of suppressor of cytokine signaling (SOCS) family members, including cytokine inducible SH2-containing protein (CIS), SOCS2, and SOCS3, which comprise a negative feedback loop to retrain the IL-15–JAK–STAT5-mediated pathway in NK cell development (90, 112). Several studies have demonstrated that STAT5 directly targets the genes of these SOCS proteins (99, 113). SH2-containing protein (CIS, encoded by Cish gene) directly interacts with JAK1 to mediate the inhibition of its enzymatic activity and proteasomal degradation, thereby constraining JAK–STAT5 signaling. Consistently, mice with CIS ablation exhibit accumulation of terminally differentiated CD27−CD11b+ NK cells in the BM and spleen, which is associated with the hyper-responsive nature of NK cells to IL-15 (114). By directly interacting with JAK2, SOCS2 attenuated JAK2 activity and the corresponding JAK2–STAT5 signaling to negatively regulate NK cell differentiation (91). Increased NK cell differentiation has been observed in the absence of SOCS2 in vivo and in vitro, whereas the development advantage is reversed after the addition of a JAK2 inhibitor in vitro. In contrast to its effect on murine NK cells, SOCS2 has no influence on IL-15-mediated human NK cell differentiation in vitro but is essential for human NK cell effector function via the regulation of phosphorylated proline-rich tyrosine kinase 2 (Pyk2) (112). These discrepancies may be attributed to different protocols for mouse and human NK cell development in vitro or species differences. Although knockdown of SOCS3 in mice has no impact on NK cell development and maturation (90), a recent study revealed that SOCS3 suppressed IL-15-mediated STAT5 phosphorylation, correlating with the desensitization of NK cells to IL-15 simulation, resulting in disrupted NK cell terminal differentiation (115).
IL-15–PI3K–AKT–mTOR Signaling for NK Cell Development
The interaction of IL-15 with its receptor on NK cells also activates the canonical downstream PI3K–AKT–mTOR pathway (Figure 6). Phosphoinositide 3-kinases (PI3Ks) are comprised of three subclasses, including class I, class II, and class III (116). The class I PI3Ks, which predominantly transduce signaling triggered by cytokine receptor, are heterodimeric enzymes that include a regulatory subunit (p85α, p50α, p55α, p85β, p55γ, and p101) and a catalytic subunit (p110α, p110β, p110γ, and p110δ). Mice exclusively or simultaneously lacking the PI3K subunits P110 γ and δ exhibit severely defective NK cell maturation and total numbers (116–119). Consistently, p110 δ mutations in patients impair the development and cytotoxic function of NK cells, leading to severe viremia, whereas rapamycin treatment partially rescues defective NK cells (117). Despite the multiple membranes of PI3Ks, it is unknown which subtypes are required for IL-15 signaling in NK cell development.
Figure 6

IL-15–PI3K–AKT-mTOR signaling for NK cell development. IL-15 complex interacts with its receptor IL-15Rβ/γc on NK cells to trigger PI3K/AKT pathway autophosphorylation and activation and subsequent activation of mTORC1 and mTORC2. mTORC1 and mTORC2 differentially promote NK cell development in a cooperative and non-redundant manner primarily by divergent induction of corresponding transcription factor Eomes and T-bet. Eomes binds to the il2rb promoter and drives CD122 expression to maintain IL-15 responsiveness, generating a positive feedback loop to amplify the IL-15 signaling. Despite the negative regulation of T-bet expression by FoxO1, several transcription factors, including GATA3, TOX2, and ETS-1, promote T-bet expression. However, the activation of mTOR signaling is tightly modulated by cooperation of TSC1 and PTEN.
PI3K phosphorylates the three positions of the inositol ring of plasma membrane-associated phosphatidylinositol-4,5-bisphosphate [PI(4,5)P2] to generate PI(3,4,5)P3, which interacts with proteins containing pleckstrin homology (PH) domains, including the serine/threonine kinases phosphoinositide-dependent kinase (PDK1) and protein kinase B (PKB; also known as AKT), and localizes these proteins to membranes (120). The interaction between PI(3,4,5)P3 and AKT initiates conformational changes in AKT, allowing PDK1 to phosphorylate AKT at threonine 308 for AKT activation (121).
Subsequently, as an important downstream effector of PI3K/AKT signaling, the mammalian target of rapamycin (mTOR) is activated. mTOR, a serine/threonine protein kinase, includes two components, namely, mTOR complex1 (mTORC1) and mTORC2. Genetic studies have revealed that Raptor and Rictor are important components of mTORC1 and mTORC2, respectively, by defining their downstream substrates (122).
It was proposed that the activation of the IL-15R–PI3K–AKT–mTOR signaling cascade is dose-dependent. Specifically, low IL-15 concentrations only activate the phosphorylation of JAK/STAT5 signaling molecules, whereas the PI3K–AKT–mTOR pathway is further activated after exposure to high IL-15 concentrations (123). PI3K–AKT–mTOR signaling primarily regulates proliferation, differentiation, and maturation as well as NK cell effector function (124). The indispensable role of mTOR in controlling NK cell development was validated in mice with a specific deficiency in mTOR in NK cells in which NK cells almost disappeared in the peripheral organs, and the remaining NK cells in BM were severely blocked at the CD11b−CD27+ stage (123). Recent studies have demonstrated that mTORC1 and mTORC2 differentially promote NK cell development in a cooperative and non-redundant manner primarily by divergent induction of corresponding transcription factors, namely, T-bet and Eomes (33, 125). Intriguingly, mTORC1 and mTORC2 also positively or negatively regulate NK cell effector function, respectively. Ncr1iCre-mediated ablation of Raptor in mice results in disrupted mTORC1 function, leading to the impaired transition from CD27+CD11b− to CD27+CD11b+ NK cells and reduced NK cell function. Conversely, terminal maturation from CD27+CD11b+ to CD27−CD11b+ NK cells is impeded in mice in the absence of Rictor, which is essential for mTORC2 metabolic signaling. However, Rictor-deficient NK cells display enhanced effector function.
E4 promoter-binding protein 4 (E4BP4), encoded by Nfil3 (nuclear factor interleukin-3), is the predominant target downstream of mTORC1 (33, 126). Mechanistically, PDK1, a kinase downstream of PI3K, is thought to mediate IL-15-triggered mTORC1 and AKT phosphorylation to drive E4BP4 expression during NK cell development (127). Ectopic expression of E4BP4 rescued NK cell developmental defects in mTORC1-inactiavted and PDK1-deficient mice (126–128). Meanwhile, the absence of PDK1 in NK cells results in attenuated IL-15-triggered mTORC1 activation and significantly decreased E4BP4 expression (127). Moreover, the inactivation of mTORC1 diminishes the IL-15-mediated E4BP4 expression. These results suggest that the IL-15–PI3K–PDK1–mTORC1 signaling pathway is essential for E4BP4 induction. E4BP4 expression is initiated as early as the CLP stage and highly expressed in the iNK and mNK stages. Nfil3−/–mice display intact CLP compartment, and the population of NKPs, iNK cells, and mNK cells significantly reduced in the BM, indicating E4BP4 acts as early as CLP stage via an IL-15-independent manner and is essential for NK cell commitment. However, Ncr1iCre-mediated deletion of Nfil3 has no effect on NK cell development (129), indicating that Nfil3 is dispensable for NK cell maturation, and other unknown signaling pathways compensate for the absence of Nfil3.
The induction of E4BP4 promotes the expression of Eomes, which binds to the il2rb promoter and drives CD122 expression to maintain IL-15 responsiveness (35). Mice with depletion of PDK1 or Eomes exhibit significant accumulation of CD27+CD11b− NK cells but are devoid of terminally mature CD27−CD11b+ NK cells, and these findings resemble the findings in Raptor-deficient mice (33, 130, 131). Collectively, the IL-15–PI3K–PDK1–mTORC1–E4BP4–Eomes–CD122 pathway generates a positive feedback loop to amplify IL-15 signaling. However, mTORC1 activation is tightly modulated by Tuberous sclerosis 1 (Tsc1), which exhibits significantly increased expression after long-term IL-15 stimulation and forms a complex with Tsc2 with the aid of AKT (132).
Contrary to the indispensable role of mTORC1 in the early maturation of NK cells, mTORC2 is essential for the terminal maturation of NK cells from the CD27+CD11b+ to the CD27−CD11b+ stage. Previous studies have demonstrated that mTORC2 phosphorylates Akt at Serine 473 and augments its kinase activity, leading to the phosphorylation of FoxO1 by Akt (133, 134). Akt-triggered phosphorylation promotes modulator protein to interact with FoxO1, thereby inactivating it by blocking DNA binding and accelerating translocation from the nucleus to the cytosol (33, 135). This model has been further validated in NK cells through the discovery that mTORC2-inactivated NK cells display reduced phosphorylation of AktS473 and FoxO1 (33). In addition, Ingenuity Pathway Analysis (IPA) found a remarkable enrichment of FoxO1 targets in mTORC2-inactivated NK cells. Furthermore, in vitro studies reported that IL-15 efficiently induces phosphorylation, and hence inactivation of FoxO1 in developing NK cells, together with the activation of mTOR signaling (136, 137). Based on these results, we speculate that FoxO1 is a direct target of the mTORC2–AktS473 signaling axis that exists downstream of IL-15 signaling in NK cells. In contrast to the high expression of FoxO1 in NKp and iNKs, the level of FoxO1 is significantly decreased in mNKs (136). Apart from FoxO1, FoxO3 is also expressed by NK cells, although it is maintained at relatively low levels throughout NK cell development. Both Ncr1-Cre -FoxO1fl/fl and -FoxO1fl/fl mice exhibit accumulation of terminally differentiated CD27−CD11b+ NK cells, indicating FoxO1 and FoxO3 redundantly suppress NK cell maturation (137), contradicting the promoting effects of mTORC2. Owing to the weak expression of FoxO3, it is believed that FoxO1 plays a prominent role in NK cell development. However, in the same Ncr1-Cre -FoxO1fl/fl mice, Wang et al. reported a remarkable deficiency of iNK and mNK cells that is attributed to impaired FoxO1-mediated autophagy in iNK cells (136). Therefore, the distinct role of FoxO1 in NK cell development remains to be clearly clarified.
Several studies have demonstrated that the negative regulation of NK cell development by FoxO1 is associated with suppressed T-bet expression (33, 137). In humans, ChIP experiments showed that FoxO1 directly binds to the Tbx21 promoter, promoting decreased T-bet expression. However, in mice, the recruitment of FoxO1 to the Tbx21 proximal promoter region by Sp1, which is a FoxO1 protein binding partner, resulted in impaired transactivation of Tbx21, leading to disrupted T-bet expression. Consistently, the absence of FoxO1 in NK cells promotes T-bet mRNA and protein expression, whereas T-bet expression is decreased in NK cells with overexpression of FoxO1. In further support of this, Foxo1 and T-bet expression inversely correlate with each other during NK cell maturation. Immature NK cells express high levels of Foxo1, whereas T-bet is present in high amounts in terminally mature NK cells. Furthermore, in contrast to the accelerated maturation of NK cells in FoxO1−/− mice, T-bet deficiency abrogated NK cell terminal maturation (40). Taken together, these results demonstrate that decreased levels of FoxO1 are necessary for NK cell maturation by releasing the negative regulation of T-bet.
Despite the negative regulation of T-bet expression by FoxO1, several transcription factors, including GATA binding protein 3 (GATA3), thymocyte selection-associated HMG box 2 (TOX2) and Ets proto-oncogene 1 (ETS-1), promote T-bet expression (138–141). Thus, inactivation of FoxO1 mediated by IL-15−PI3K−mTORC2 signaling coordinated with several transcription factors to promote T-bet expression. T-bet−/− mice exhibited remarkably decreased NK cell populations in the periphery, but the NK cell number was modestly elevated in BM (142). This defect is attributed to the decreased expression of S1P5, which is induced by T-bet and responsible for NK cell egress from BM (143). In the absence of T-bet, NK cell maturation is specifically arrested at CD27+CD11b+stage, suggesting that T-bet is essential for NK cell terminal maturation (60). It has been proposed that T-bet promotes NK cell maturation by transiently inhibiting Eomes expression (36, 142). Consistent with this, T-bet levels are gradually increased during NK cell maturation, accompanied by the decreased expression of Eomes. T-bet also contributes to the induction of Zinc Finger E-box Binding Homeobox 2 (Zeb2) and B lymphocyte-induced maturation protein 1 (Blimp-1), which are critical for NK cell maturation (36, 40, 144). Thus, the IL-15R–PI3K–mTORC2–AKT–FoxO1–T-bet pathway determines the terminal maturation of NK cells.
In addition, mTORC2 suppresses mTORC1-mediated NK cell effector function by mainly downregulating SLC7A5 expression, which is downstream of STAT5 and regulates mTORC1 activity independent of AKT signaling (125, 145). Therefore, mTORC2 counteracts IL-15-mediated mTORC1 hyperactivation to prevent activation-induced NK cell apoptosis. Inversely, mTORC1 maintains IL-15–CD122–IL-15 signaling to sustain mTORC2 activity.
Phosphatase and tensin homolog (PTEN) directly antagonizes the PI3K–AKT pathway by specifically dephosphorylating PI(3,4,5)P3, which is downstream of PI3K and functions as an activator for downstream signaling proteins, including Vav, Akt, PDK1, and PI(4,5)P2 (146). Consistently, PTEN suppresses PI3K–AKT signaling and MAPK activation in humans, leading to compromised cytotoxic function (147). Conversely, the PTEN signaling pathway is impaired in Rictor-deficient NK cells with an inactive mTORC2 pathway, indicating that mTORC2 promotes PTEN expression to antagonize the PI(3,4,5)P3-mediated activation of mTORC2 (33, 148). Thus, a negative feedback exists between mTORC2 signaling and PTEN expression.
Perspectives
NK cell development is tightly regulated by the interplay between intracellular transcription factors and extracellular signals, such as cognate ligands, chemokines, and cytokines. Notably, pleiotropic cytokine IL-15 is indispensable for the development of NK cells. Recently, immunotherapy has been applied in anti-cancer and anti-infection treatments. As an important component of immune cell, NK cells have potent cytotoxicity and cytokine production capacity, which allows effective eradication of malignant and infected cells in the absence of graft versus host disease (GVHD). Therefore, NK cells are promising for therapeutic utilization. The prerequisite of clinical application is to substantially expand mature NK cells in vitro. Correspondingly, understanding of the molecular mechanisms by which IL-15 promotes NK cell development and manipulation of IL-15 for proper NK cell expansion in vitro will improve NK cell-based therapeutic strategies (18).
NK cells are the earliest donor-derived lymphocytes recovering after HSCT, whose populations quickly reach donor levels within 1 month (149, 150). The well-established reconstitution of NK cells exhibits a protective effect against leukemia relapse and is associated with improved disease-free survival after HSCT (151–153). Although the level of IL-15 is remarkably high after HSCT, the immature CD56brightKIR−NK cells dominate the early reconstruction (149, 150). Investigating the role of IL-15 in NK cell development after HSCT contributes to better prognosis by intervening NK cell maturation.
Given the formidable efficacy in enhancing NK cell development, IL-15 is much more promising than other cytokines in controlling tumor progression and viral infections. Several murine immunotherapy trials have demonstrated that the administration of IL-15 efficiently drove the expansion and activation of NK cells and CD8+T cells in vivo, without stimulating the expansion of regulatory T cells which exert an immunosuppressive effect (154–156). However, due to its adverse effects, such as toxicities, hypotension, thrombocytopenia, IL-15 application was constrained (157). More basic research is required to optimize the structure of IL-15 before it can extended to clinical practice.
Funding
This study was supported by the National Key Research and Development Program of China (No. 2017YFA0104500), the National Natural Science Foundation of China (Nos. 81870140, 82070184), the Innovative Research Groups of the National Natural Science Foundation of China (No. 81621001), and Clinical Medicine Plus X - Young Scholars Project of Peking University (No. PKU2020LCXQ015) supported by “the Fundamental Research Funds for the Central Universities”, and Peking University People’s Hospital Research and Development Funds (No. RDX2019-14).
Statements
Author contributions
XW wrote the manuscript. X-YZ outlined the manuscript and made a deep intellectual contribution to the work. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
Caligiuri MA . Human natural killer cells. Blood (2008) 112(3):461–9. doi: 10.1182/blood-2007-09-077438
2
Koch J Steinle A Watzl C Mandelboim O . Activating natural cytotoxicity receptors of natural killer cells in cancer and infection. Trends Immunol (2013) 34(4):182–91. doi: 10.1016/j.it.2013.01.003
3
Raulet DH Vance RE . Self-tolerance of natural killer cells. Nat Rev Immunol (2006) 6(7):520–31. doi: 10.1038/nri1863
4
Grabstein KH Eisenman J Shanebeck K Rauch C Srinivasan S Fung V et al . Cloning of a T-Cell Growth-Factor That Interacts with the Beta-Chain of the Interleukin-2 Receptor. Science (1994) 264(5161):965–8. doi: 10.1126/science.8178155
5
Fehniger TA Caligiuri MA . Interleukin 15: biology and relevance to human disease. Blood (2001) 97(1):14–32. doi: 10.1182/blood.v97.1.14
6
Giri JG Anderson DM Kumaki S Park LS Grabstein KH Cosman D . Il-15, a Novel T-Cell Growth-Factor That Shares Activities and Receptor Components with Il-2. J Leukocyte Biol (1995) 57(5):763–6. doi: 10.1002/jlb.57.5.763
7
Giri JG Kumaki S Ahdieh M Friend DJ Loomis A Shanebeck K et al . Identification and Cloning of a Novel Il-15 Binding-Protein That Is Structurally Related to the Alpha-Chain of the Il-2 Receptor. EMBO J (1995) 14(15):3654–63. doi: 10.1002/j.1460-2075.1995.tb00035.x
8
Dubois S Mariner J Waldmann TA Tagaya Y . IL-15Ralpha recycles and presents IL-15 In trans to neighboring cells. Immunity (2002) 17(5):537–47. doi: 10.1016/s1074-7613(02)00429-6
9
Lodolce JP Burkett PR Boone DL Chien M Ma A . T cell-independent interleukin 15Ralpha signals are required for bystander proliferation. J Exp Med (2001) 194(8):1187–94. doi: 10.1084/jem.194.8.1187
10
Castillo EF Stonier SW Frasca L Schluns KS . Dendritic cells support the in vivo development and maintenance of NK cells via IL-15 trans-presentation. J Immunol (2009) 183(8):4948–56. doi: 10.4049/jimmunol.0900719
11
Schluns KS Nowak EC Cabrera-Hernandez A Puddington L Lefrancois L Aguila HL . Distinct cell types control lymphoid subset development by means of IL-15 and IL-15 receptor alpha expression. Proc Natl Acad Sci USA (2004) 101(15):5616–21. doi: 10.1073/pnas.0307442101
12
Di Santo JP . NATURAL KILLER CELL DEVELOPMENTAL PATHWAYS: A Question of Balance. Annu Rev Immunol (2006) 24(1):257–86. doi: 10.1146/annurev.immunol.24.021605.090700
13
Stabile H Fionda C Santoni A Gismondi A . Impact of bone marrow-derived signals on NK cell development and functional maturation. Cytokine Growth Factor Rev (2018) 42:13–9. doi: 10.1016/j.cytogfr.2018.03.008
14
Kennedy MK Glaccum M Brown SN Butz EA Viney JL Embers M et al . Reversible defects in natural killer and memory CD8 T cell lineages in interleukin 15-deficient mice. J Exp Med (2000) 191(5):771–80. doi: 10.1084/jem.191.5.771
15
Lodolce JP Boone DL Chai S Swain RE Dassopoulos T Trettin S et al . IL-15 receptor maintains lymphoid homeostasis by supporting lymphocyte homing and proliferation. Immunity (1998) 9(5):669–76. doi: 10.1016/s1074-7613(00)80664-0
16
Vosshenrich CA Ranson T Samson SI Corcuff E Colucci F Rosmaraki EE et al . Roles for common cytokine receptor gamma-chain-dependent cytokines in the generation, differentiation, and maturation of NK cell precursors and peripheral NK cells in vivo. J Immunol (2005) 174(3):1213–21. doi: 10.4049/jimmunol.174.3.1213
17
Suzuki H Duncan GS Takimoto H Mak TW . Abnormal development of intestinal intraepithelial lymphocytes and peripheral natural killer cells in mice lacking the IL-2 receptor beta chain. J Exp Med (1997) 185(3):499–505. doi: 10.1084/jem.185.3.499
18
Mrózek E Anderson P Caligiuri MA . Role of interleukin-15 in the development of human CD56+ natural killer cells from CD34+ hematopoietic progenitor cells. Blood (1996) 87(7):2632–40. doi: 10.1182/blood.V87.7.2632.bloodjournal8772632
19
Yu H Fehniger TA Fuchshuber P Thiel KS Vivier E Carson WE et al . Flt3 ligand promotes the generation of a distinct CD34(+) human natural killer cell progenitor that responds to interleukin-15. Blood (1998) 92(10):3647–57. doi: 10.1182/blood.V92.10.3647.422k43_3647_3657
20
Huntington ND Legrand N Alves NL Jaron B Weijer K Plet A et al . IL-15 trans-presentation promotes human NK cell development and differentiation in vivo. J Exp Med (2009) 206(1):25–34. doi: 10.1084/jem.20082013
21
Sun JC Ma A Lanier LL . Cutting edge: IL-15-independent NK cell response to mouse cytomegalovirus infection. J Immunol (2009) 183(5):2911–4. doi: 10.4049/jimmunol.0901872
22
Vosshenrich CAJ Di Santo JP . Developmental programming of natural killer and innate lymphoid cells. Curr Opin Immunol (2013) 25(2):130–8. doi: 10.1016/j.coi.2013.02.002
23
Fathman JW Bhattacharya D Inlay MA Seita J Karsunky H Weissman IL . Identification of the earliest natural killer cell–committed progenitor in murine bone marrow. Blood (2011) 118(20):5439. doi: 10.1182/blood-2011-04-348912
24
Rosmaraki EE Douagi I Roth C Colucci F Cumano A Di Santo JP . Identification of committed NK cell progenitors in adult murine bone marrow. Eur J Immunol (2001) 31(6):1900–9. doi: 10.1002/1521-4141(200106)31:6<1900::aid-immu1900>3.0.co;2-m
25
McCullar V Oostendorp R Panoskaltsis-Mortari A Yun G Lutz CT Wagner JE et al . Mouse fetal and embryonic liver cells differentiate human umbilical cord blood progenitors into CD56-negative natural killer cell precursors in the absence of interleukin-15. Exp Hematol (2008) 36(5):598–608. doi: 10.1016/j.exphem.2008.01.001
26
Rautela J Huntington ND . IL-15 signaling in NK cell cancer immunotherapy. Curr Opin Immunol (2017) 44:1–6. doi: 10.1016/j.coi.2016.10.004
27
Poli A Michel T Thérésine M Andrès E Hentges F Zimmer J . CD56bright natural killer (NK) cells: an important NK cell subset. Immunology (2009) 126(4):458–65. doi: 10.1111/j.1365-2567.2008.03027.x
28
Michel T Poli A Cuapio A Briquemont B Iserentant G Ollert M et al . Human CD56bright NK Cells: An Update. J Immunol (2016) 196(7):2923–31. doi: 10.4049/jimmunol.1502570
29
Lima M Teixeira MA Queirós ML Leite M Santos AH Justiça B et al . Immunophenotypic characterization of normal blood CD56+lo versus CD56+hi NK-cell subsets and its impact on the understanding of their tissue distribution and functional properties. Blood Cells Mol Dis (2001) 27(4):731–43. doi: 10.1006/bcmd.2001.0443
30
Cooper MA Fehniger TA Turner SC Chen KS Ghaheri BA Ghayur T et al . Human natural killer cells: a unique innate immunoregulatory role for the CD56(bright) subset. Blood (2001) 97(10):3146–51. doi: 10.1182/blood.v97.10.3146
31
Vitale M Della Chiesa M Carlomagno S Romagnani C Thiel A Moretta L et al . The small subset of CD56brightCD16- natural killer cells is selectively responsible for both cell proliferation and interferon-gamma production upon interaction with dendritic cells. Eur J Immunol (2004) 34(6):1715–22. doi: 10.1002/eji.200425100
32
Lopez-Vergès S Milush JM Pandey S York VA Arakawa-Hoyt J Pircher H et al . CD57 defines a functionally distinct population of mature NK cells in the human CD56dimCD16+ NK-cell subset. Blood (2010) 116(19):3865–74. doi: 10.1182/blood-2010-04-282301
33
Yang C Tsaih SW Lemke A Flister MJ Thakar MS Malarkannan S . mTORC1 and mTORC2 differentially promote natural killer cell development. Elife (2018) 7:e35619. doi: 10.7554/eLife.35619
34
Ohno S-i Sato T Kohu K Takeda K Okumura K Satake M et al . Runx proteins are involved in regulation of CD122, Ly49 family and IFN-gamma expression during NK cell differentiation. Int Immunol (2008) 20(1):71–9. doi: 10.1093/intimm/dxm120
35
Intlekofer AM Takemoto N Wherry EJ Longworth SA Northrup JT Palanivel VR et al . Effector and memory CD8(+) T cell fate coupled by T-bet and eomesodermin. Nat Immunol (2005) 6(12):1236–44. doi: 10.1038/ni1268
36
Gordon SM Chaix J Rupp LJ Wu J Madera S Sun JC et al . The transcription factors T-bet and Eomes control key checkpoints of natural killer cell maturation. Immunity (2012) 36(1):55–67. doi: 10.1016/j.immuni.2011.11.016
37
Wagner JA Wong P Schappe T Berrien-Elliott MM Cubitt C Jaeger N et al . Stage-Specific Requirement for Eomes in Mature NK Cell Homeostasis and Cytotoxicity. Cell Rep (2020) 31(9):107720. doi: 10.1016/j.celrep.2020.107720
38
Daussy C Faure F Mayol K Viel S Gasteiger G Charrier E et al . T-bet and Eomes instruct the development of two distinct natural killer cell lineages in the liver and in the bone marrow. J Exp Med (2014) 211(3):563–77. doi: 10.1084/jem.20131560
39
Colonna M . Innate Lymphoid Cells: Diversity, Plasticity, and Unique Functions in Immunity. Immunity (2018) 48(6):1104–17. doi: 10.1016/j.immuni.2018.05.013
40
van Helden MJ Goossens S Daussy C Mathieu AL Faure F Marçais A et al . Terminal NK cell maturation is controlled by concerted actions of T-bet and Zeb2 and is essential for melanoma rejection. J Exp Med (2015) 212(12):2015–25. doi: 10.1084/jem.20150809
41
Bamford RN Battiata AP Burton JD Sharma H Waldmann TA . Interleukin (IL) 15/IL-T production by the adult T-cell leukemia cell line HuT-102 is associated with a human T-cell lymphotrophic virus type I R region/IL-15 fusion message that lacks many upstream AUGs that normally attenuate IL-15 mRNA translation. Proc Natl Acad Sci USA (1996) 93(7):2897–902. doi: 10.1073/pnas.93.7.2897
42
Bamford RN DeFilippis AP Azimi N Kurys G Waldmann TA . The 5‘ untranslated region, signal peptide, and the coding sequence of the carboxyl terminus of IL-15 participate in its multifaceted translational control. J Immunol (1998) 160(9):4418–26.
43
Gaggero A Azzarone B Andrei C Mishal Z Meazza R Zappia E et al . Differential intracellular trafficking, secretion and endosomal localization of two IL-15 isoforms. Eur J Immunol (1999) 29(4):1265–74. doi: 10.1002/(sici)1521-4141(199904)29:04<1265::Aid-immu1265>3.0.Co;2-v
44
Tagaya Y Kurys G Thies TA Losi JM Azimi N Hanover JA et al . Generation of secretable and nonsecretable interleukin 15 isoforms through alternate usage of signal peptides. Proc Natl Acad Sci USA (1997) 94(26):14444–9. doi: 10.1073/pnas.94.26.14444
45
Meazza R Gaggero A Neglia F Basso S Sforzini S Pereno R et al . Expression of two interleukin-15 mRNA isoforms in human tumors does not correlate with secretion: role of different signal peptides. Eur J Immunol (1997) 27(5):1049–54. doi: 10.1002/eji.1830270502
46
Kurys G Tagaya Y Bamford R Hanover JA Waldmann TA . The long signal peptide isoform and its alternative processing direct the intracellular trafficking of interleukin-15. J Biol Chem (2000) 275(39):30653–9. doi: 10.1074/jbc.M002373200
47
Colpitts SL Stoklasek TA Plumlee CR Obar JJ Guo C Lefrançois L . Cutting edge: the role of IFN-α receptor and MyD88 signaling in induction of IL-15 expression in vivo. J Immunol (2012) 188(6):2483–7. doi: 10.4049/jimmunol.1103609
48
Colpitts SL Stonier SW Stoklasek TA Root SH Aguila HL Schluns KS et al . Transcriptional regulation of IL-15 expression during hematopoiesis. J Immunol (2013) 191(6):3017–24. doi: 10.4049/jimmunol.1301389
49
Cui G Hara T Simmons S Wagatsuma K Abe A Miyachi H et al . Characterization of the IL-15 niche in primary and secondary lymphoid organs in vivo. Proc Natl Acad Sci USA (2014) 111(5):1915–20. doi: 10.1073/pnas.1318281111
50
Sugiyama T Kohara H Noda M Nagasawa T . Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity (2006) 25(6):977–88. doi: 10.1016/j.immuni.2006.10.016
51
Noda M Omatsu Y Sugiyama T Oishi S Fujii N Nagasawa T . CXCL12-CXCR4 chemokine signaling is essential for NK-cell development in adult mice. Blood (2011) 117(2):451–8. doi: 10.1182/blood-2010-04-277897
52
Zhang XH Sun SQ Hwang IK Tough DF Sprent J . Potent and selective stimulation of memory-phenotype CD8(+) T cells in vivo by IL-15. Immunity (1998) 8(5):591–9. doi: 10.1016/S1074-7613(00)80564-6
53
Xie CB Jiang B Qin L Tellides G Kirkiles-Smith NC Jane-Wit D et al . Complement-activated interferon-γ-primed human endothelium transpresents interleukin-15 to CD8+ T cells. J Clin Invest (2020) 130(7):3437–52. doi: 10.1172/jci135060
54
Oppenheimer-Marks N Brezinschek RI Mohamadzadeh M Vita R Lipsky PE . Interleukin 15 is produced by endothelial cells and increases the transendothelial migration of T cells In vitro and in the SCID mouse-human rheumatoid arthritis model In vivo. J Clin Invest (1998) 101(6):1261–72. doi: 10.1172/jci1986
55
Mattei F Schiavoni G Belardelli F Tough DF . IL-15 is expressed by dendritic cells in response to type IIFN, double-stranded RNA, or lipopolysaccharide and promotes dendritic cell activation. J Immunol (2001) 167(3):1179–87. doi: 10.4049/jimmunol.167.3.1179
56
Nolz JC Richer MJ . Control of memory CD8(+) T cell longevity and effector functions by IL-15. Mol Immunol (2020) 117:180–8. doi: 10.1016/j.molimm.2019.11.011
57
Anthony SM Rivas SC Colpitts SL Howard ME Stonier SW Schluns KS . Inflammatory Signals Regulate IL-15 in Response to Lymphodepletion. J Immunol (2016) 196(11):4544–52. doi: 10.4049/jimmunol.1600219
58
Soudja SM Ruiz AL Marie JC Lauvau G . Inflammatory monocytes activate memory CD8(+) T and innate NK lymphocytes independent of cognate antigen during microbial pathogen invasion. Immunity (2012) 37(3):549–62. doi: 10.1016/j.immuni.2012.05.029
59
Carrero RMS Beceren-Braun F Rivas SC Hegde SM Gangadharan A Plote D et al . IL-15 is a component of the inflammatory milieu in the tumor microenvironment promoting antitumor responses. Proc Natl Acad Sci USA (2019) 116(2):599–608. doi: 10.1073/pnas.1814642116
60
Soderquest K Powell N Luci C van Rooijen N Hidalgo A Geissmann F et al . Monocytes control natural killer cell differentiation to effector phenotypes. Blood (2011) 117(17):4511–8. doi: 10.1182/blood-2010-10-312264
61
Mortier E Advincula R Kim L Chmura S Barrera J Reizis B et al . Macrophage- and dendritic-cell-derived interleukin-15 receptor alpha supports homeostasis of distinct CD8+ T cell subsets. Immunity (2009) 31(5):811–22. doi: 10.1016/j.immuni.2009.09.017
62
Gil-Cruz C Perez-Shibayama C Onder L Chai Q Cupovic J Cheng HW et al . Fibroblastic reticular cells regulate intestinal inflammation via IL-15-mediated control of group 1 ILCs. Nat Immunol (2016) 17(12):1388–96. doi: 10.1038/ni.3566
63
Briard D Brouty-Boyé D Azzarone B Jasmin C . Fibroblasts from human spleen regulate NK cell differentiation from blood CD34(+) progenitors via cell surface IL-15. J Immunol (2002) 168(9):4326–32. doi: 10.4049/jimmunol.168.9.4326
64
Stonier SW Schluns KS . Trans-presentation: a novel mechanism regulating IL-15 delivery and responses. Immunol Lett (2010) 127(2):85–92. doi: 10.1016/j.imlet.2009.09.009
65
Bergamaschi C Rosati M Jalah R Valentin A Kulkarni V Alicea C et al . Intracellular interaction of interleukin-15 with its receptor alpha during production leads to mutual stabilization and increased bioactivity. J Biol Chem (2008) 283(7):4189–99. doi: 10.1074/jbc.M705725200
66
Sandau MM Schluns KS Lefrancois L Jameson SC . Cutting edge: transpresentation of IL-15 by bone marrow-derived cells necessitates expression of IL-15 and IL-15R alpha by the same cells. J Immunol (2004) 173(11):6537–41. doi: 10.4049/jimmunol.173.11.6537
67
Burkett PR Koka R Chien M Chai S Boone DL Ma A . Coordinate expression and trans presentation of interleukin (IL)-15Ralpha and IL-15 supports natural killer cell and memory CD8+ T cell homeostasis. J Exp Med (2004) 200(7):825–34. doi: 10.1084/jem.20041389
68
Mortier E Woo T Advincula R Gozalo S Ma A . IL-15Ralpha chaperones IL-15 to stable dendritic cell membrane complexes that activate NK cells via trans presentation. J Exp Med (2008) 205(5):1213–25. doi: 10.1084/jem.20071913
69
Anthony SM Howard ME Hailemichael Y Overwijk WW Schluns KS . Soluble interleukin-15 complexes are generated in vivo by type I interferon dependent and independent pathways. PloS One (2015) 10(3):e0120274. doi: 10.1371/journal.pone.0120274
70
Bergamaschi C Bear J Rosati M Beach RK Alicea C Sowder R et al . Circulating IL-15 exists as heterodimeric complex with soluble IL-15Rα in human and mouse serum. Blood (2012) 120(1):e1–8. doi: 10.1182/blood-2011-10-384362
71
Stoklasek TA Schluns KS Lefrançois L . Combined IL-15/IL-15Ralpha immunotherapy maximizes IL-15 activity in vivo. J Immunol (2006) 177(9):6072–80. doi: 10.4049/jimmunol.177.9.6072
72
Rubinstein MP Kovar M Purton JF Cho JH Boyman O Surh CD et al . Converting IL-15 to a superagonist by binding to soluble IL-15R{alpha}. Proc Natl Acad Sci USA (2006) 103(24):9166–71. doi: 10.1073/pnas.0600240103
73
Anton OM Peterson ME Hollander MJ Dorward DW Arora G Traba J et al . Trans-endocytosis of intact IL-15Rα-IL-15 complex from presenting cells into NK cells favors signaling for proliferation. Proc Natl Acad Sci USA (2020) 117(1):522–31. doi: 10.1073/pnas.1911678117
74
Sato N Patel HJ Waldmann TA Tagaya Y . The IL-15/IL-15Ralpha on cell surfaces enables sustained IL-15 activity and contributes to the long survival of CD8 memory T cells. Proc Natl Acad Sci USA (2007) 104(2):588–93. doi: 10.1073/pnas.0610115104
75
Koka R Burkett PR Chien M Chai S Chan F Lodolce JP et al . Interleukin (IL)-15R[alpha]-deficient natural killer cells survive in normal but not IL-15R[alpha]-deficient mice. J Exp Med (2003) 197(8):977–84. doi: 10.1084/jem.20021836
76
Kawamura T Koka R Ma A Kumar V . Differential roles for IL-15R alpha-chain in NK cell development and Ly-49 induction. J Immunol (2003) 171(10):5085–90. doi: 10.4049/jimmunol.171.10.5085
77
Tamzalit F Barbieux I Plet A Heim J Nedellec S Morisseau S et al . IL-15.IL-15Rα complex shedding following trans-presentation is essential for the survival of IL-15 responding NK and T cells. Proc Natl Acad Sci USA (2014) 111(23):8565–70. doi: 10.1073/pnas.1405514111
78
Brilot F Strowig T Roberts SM Arrey F Münz C . NK cell survival mediated through the regulatory synapse with human DCs requires IL-15Ralpha. J Clin Invest (2007) 117(11):3316–29. doi: 10.1172/jci31751
79
Anton OM Vielkind S Peterson ME Tagaya Y Long EO . NK Cell Proliferation Induced by IL-15 Transpresentation Is Negatively Regulated by Inhibitory Receptors. J Immunol (2015) 195(10):4810–21. doi: 10.4049/jimmunol.1500414
80
Leonard WJ O’Shea JJ . JAKS AND STATS: Biological implications. Annu Rev Immunol (1998) 16:293–322. doi: 10.1146/annurev.immunol.16.1.293
81
Nakamura Y Russell SM Mess SA Friedmann M Erdos M Francois C et al . Heterodimerization of the Il-2 Receptor Beta-Chain and Gamma-Chain Cytoplasmic Domains Is Required for Signaling. Nature (1994) 369(6478):330–3. doi: 10.1038/369330a0
82
Nelson BH Lord JD Greenberg PD . Cytoplasmic Domains of the Interleukin-2 Receptor Beta-Chain and Gamma-Chain Mediate the Signal for T-Cell Proliferation. Nature (1994) 369(6478):333–6. doi: 10.1038/369333a0
83
Macchi P Villa A Giliani S Sacco MG Frattini A Porta F et al . Mutations of Jak-3 gene in patients with autosomal severe combined immune deficiency (SCID). Nature (1995) 377(6544):65–8. doi: 10.1038/377065a0
84
Rodig SJ Meraz MA White JM Lampe PA Riley JK Arthur CD et al . Disruption of the Jak1 gene demonstrates obligatory and nonredundant roles of the Jaks in cytokine-induced biologic responses. Cell (1998) 93(3):373–83. doi: 10.1016/s0092-8674(00)81166-6
85
Kleppe M Spitzer MH Li S Hill CE Dong L Papalexi E et al . Jak1 Integrates Cytokine Sensing to Regulate Hematopoietic Stem Cell Function and Stress Hematopoiesis. Cell Stem Cell (2017) 21(4):489–+. doi: 10.1016/j.stem.2017.08.011
86
Witalisz-Siepracka A Klein K Prinz D Leidenfrost N Schabbauer G Dohnal A et al . Loss of JAK1 Drives Innate Immune Deficiency. Front Immunol (2018) 9:3108. doi: 10.3389/fimmu.2018.03108
87
Robinette ML Cella M Telliez JB Ulland TK Barrow AD Capuder K et al . Jak3 deficiency blocks innate lymphoid cell development. Mucosal Immunol (2018) 11(1):50–60. doi: 10.1038/mi.2017.38
88
Haan C Rolvering C Raulf F Kapp M Druckes P Thoma G et al . Jak1 Has a Dominant Role over Jak3 in Signal Transduction through gamma c-Containing Cytokine Receptors. Chem Biol (2011) 18(3):314–23. doi: 10.1016/j.chembiol.2011.01.012
89
Liu KD Gaffen SL Goldsmith MA Greene WC . Janus kinases in interleukin-2-mediated signaling: JAK1 and JAK3 are differentially regulated by tyrosine phosphorylation. Curr Biol (1997) 7(11):817–26. doi: 10.1016/s0960-9822(06)00369-1
90
Delconte RB Kolesnik TB Dagley LF Rautela J Shi W Putz EM et al . CIS is a potent checkpoint in NK cell-mediated tumor immunity. Nat Immunol (2016) 17(7):816–24. doi: 10.1038/ni.3470
91
Kim WS Kim MJ Kim DO Byun JE Huy H Song HY et al . Suppressor of Cytokine Signaling 2 Negatively Regulates NK Cell Differentiation by Inhibiting JAK2 Activity. Sci Rep (2017) 7:46153. doi: 10.1038/srep46153 doi: ARTN 46153.
92
Bottos A Gotthardt D Gill JW Gattelli A Frei A Tzankov A et al . Decreased NK-cell tumour immunosurveillance consequent to JAK inhibition enhances metastasis in breast cancer models. Nat Commun (2016) 7:12258. doi: 10.1038/ncomms12258 doi: ARTN 12258.
93
Lin JX Migone TS Tsang M Friedmann M Weatherbee JA Zhou L et al . The role of shared receptor motifs and common Stat proteins in the generation of cytokine pleiotropy and redundancy by IL-2, IL-4, IL-7, IL-13, and IL-15. Immunity (1995) 2(4):331–9. doi: 10.1016/1074-7613(95)90141-8
94
Lin JX Leonard WJ . The role of Stat5a and Stat5b in signaling by IL-2 family cytokines. Oncogene (2000) 19(21):2566–76. doi: 10.1038/sj.onc.1203523
95
Huntington ND . The unconventional expression of IL-15 and its role in NK cell homeostasis. Immunol Cell Biol (2014) 92(3):210–3. doi: 10.1038/icb.2014.1
96
Huntington ND Puthalakath H Gunn P Naik E Michalak EM Smyth MJ et al . Interleukin 15-mediated survival of natural killer cells is determined by interactions among Bim, Noxa and Mcl-1. Nat Immunol (2007) 8(8):856–63. doi: 10.1038/ni1487
97
Sathe P Delconte RB Souza-Fonseca-Guimaraes F Seillet C Chopin M Vandenberg CJ et al . Innate immunodeficiency following genetic ablation of Mcl1 in natural killer cells. Nat Commun (2014) 5:4539. doi: 10.1038/ncomms5539
98
Soldaini E John S Moro S Bollenbacher J Schindler U Leonard WJ . DNA binding site selection of dimeric and tetrameric Stat5 proteins reveals a large repertoire of divergent tetrameric Stat5a binding sites. Mol Cell Biol (2000) 20(1):389–401. doi: 10.1128/mcb.20.1.389-401.2000
99
Lin JX Du N Li P Kazemian M Gebregiorgis T Spolski R et al . Critical functions for STAT5 tetramers in the maturation and survival of natural killer cells. Nat Commun (2017) 8(1):1320. doi: 10.1038/s41467-017-01477-5
100
Eckelhart E Warsch W Zebedin E Simma O Stoiber D Kolbe T et al . A novel Ncr1-Cre mouse reveals the essential role of STAT5 for NK-cell survival and development. Blood (2011) 117(5):1565–73. doi: 10.1182/blood-2010-06-291633
101
Vargas-Hernández A Witalisz-Siepracka A Prchal-Murphy M Klein K Mahapatra S Al-Herz W et al . Human signal transducer and activator of transcription 5b (STAT5b) mutation causes dysregulated human natural killer cell maturation and impaired lytic function. J Allergy Clin Immunol (2020) 145(1):345–357.e349. doi: 10.1016/j.jaci.2019.09.016
102
Liu XW Robinson GW Wagner KU Garrett L WynshawBoris A Hennighausen L . Stat5a is mandatory for adult mammary gland development and lactogenesis. Gene Dev (1997) 11(2):179–86. doi: 10.1101/gad.11.2.179
103
Wakao H Gouilleux F Groner B . Mammary-Gland Factor (Mgf) Is a Novel Member of the Cytokine Regulated Transcription Factor Gene Family and Confers the Prolactin Response. EMBO J (1994) 13(9):2182–91. doi: 10.1002/j.1460-2075.1994.tb06495.x
104
Villarino AV Sciume G Davis FP Iwata S Zitti B Robinson GW et al . Subset-and tissue-defined STAT5 thresholds control homeostasis and function of innate lymphoid cells. J Exp Med (2017) 214(10):2999–3014. doi: 10.1084/jem.20150907
105
Imada K Bloom ET Nakajima H Horvath-Arcidiacono JA Udy GB Davey HW et al . Stat5b is essential for natural killer cell-mediated proliferation and cytolytic activity. J Exp Med (1998) 188(11):2067–74. doi: 10.1084/jem.188.11.2067
106
Gotthardt D Putz EM Grundschober E Prchal-Murphy M Straka E Kudweis P et al . STAT5 Is a Key Regulator in NK Cells and Acts as a Molecular Switch from Tumor Surveillance to Tumor Promotion. Cancer Discovery (2016) 6(4):414–29. doi: 10.1158/2159-8290.Cd-15-0732
107
Bruno A Focaccetti C Pagani A Imperatori AS Spagnoletti M Rotolo N et al . The proangiogenic phenotype of natural killer cells in patients with non-small cell lung cancer. Neoplasia (2013) 15(2):133–42. doi: 10.1593/neo.121758
108
Levi I Amsalem H Nissan A Darash-Yahana M Peretz T Mandelboim O et al . Characterization of tumor infiltrating natural killer cell subset. Oncotarget (2015) 6(15):13835–43. doi: 10.18632/oncotarget.3453
109
Bruno A Ferlazzo G Albini A Noonan DM . A think tank of TINK/TANKs: tumor-infiltrating/tumor-associated natural killer cells in tumor progression and angiogenesis. J Natl Cancer Inst (2014) 106(8):dju200. doi: 10.1093/jnci/dju200
110
Yang XP Ghoreschi K Steward-Tharp SM Rodriguez-Canales J Zhu J Grainger JR et al . Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat Immunol (2011) 12(3):247–54. doi: 10.1038/ni.1995
111
Villarino A Laurence A Robinson GW Bonelli M Dema B Afzali B et al . Signal transducer and activator of transcription 5 (STAT5) paralog dose governs T cell effector and regulatory functions. Elife (2016) 5:e08384. doi: 10.7554/eLife.08384
112
Lee SH Yun S Piao ZH Jeong M Kim DO Jung H et al . Suppressor of cytokine signaling 2 regulates IL-15-primed human NK cell function via control of phosphorylated Pyk2. J Immunol (2010) 185(2):917–28. doi: 10.4049/jimmunol.1000784
113
Lin JX Li P Liu D Jin HT He J Ata Ur Rasheed M et al . Critical Role of STAT5 transcription factor tetramerization for cytokine responses and normal immune function. Immunity (2012) 36(4):586–99. doi: 10.1016/j.immuni.2012.02.017
114
Delconte RB Guittard G Goh W Hediyeh-Zadeh S Hennessy RJ Rautela J et al . NK Cell Priming From Endogenous Homeostatic Signals Is Modulated by CIS. Front Immunol (2020) 11:75. doi: 10.3389/fimmu.2020.00075
115
Wang XF Sun R Hao XL Lian ZX Wei HM Tian ZG . IL-17 constrains natural killer cell activity by restraining IL-15-driven cell maturation via SOCS3. Proc Natl Acad Sci USA (2019) 116(35):17409–18. doi: 10.1073/pnas.1904125116
116
Kim N Saudemont A Webb L Camps M Ruckle T Hirsch E et al . The p110delta catalytic isoform of PI3K is a key player in NK-cell development and cytokine secretion. Blood (2007) 110(9):3202–8. doi: 10.1182/blood-2007-02-075366
117
Ruiz-García R Vargas-Hernández A Chinn IK Angelo LS Cao TN Coban-Akdemir Z et al . Mutations in PI3K110δ cause impaired natural killer cell function partially rescued by rapamycin treatment. J Allergy Clin Immunol (2018) 142(2):605–17. doi: 10.1016/j.jaci.2017.11.042
118
Tassi I Cella M Gilfillan S Turnbull I Diacovo TG Penninger JM et al . p110 gamma and p110 delta phosphoinositide 3-kinase signaling pathways synergize to control development and functions of murine NK cells. Immunity (2007) 27(2):214–27. doi: 10.1016/j.immuni.2007.07.014
119
Guo HL Samarakoon A Vanhaesebroeck B Malarkannan S . The p110 delta of PI3K plays a critical role in NK cell terminal maturation and cytokine/chemokine generation. J Exp Med (2008) 205(10):2419–35. doi: 10.1084/jem.20072327
120
Cantrell DA . Phosphoinositide 3-kinase signalling pathways. J Cell Sci (2001) 114(8):1439–45.
121
Pearce LR Komander D Alessi DR . The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol (2010) 11(1):9–22. doi: 10.1038/nrm2822
122
Zoncu R Efeyan A Sabatini DM . mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol (2011) 12(1):21–35. doi: 10.1038/nrm3025
123
Marais A Cherfils-Vicini J Viant C Degouve S Viel S Fenis A et al . The metabolic checkpoint kinase mTOR is essential for IL-15 signaling during the development and activation of NK cells. Nat Immunol (2014) 15(8):749–+. doi: 10.1038/ni.2936
124
Mao Y van Hoef V Zhang X Wennerberg E Lorent J Witt K et al . IL-15 activates mTOR and primes stress-activated gene expression leading to prolonged antitumor capacity of NK cells. Blood (2016) 128(11):1475–89. doi: 10.1182/blood-2016-02-698027
125
Wang FJ Meng M Mo BH Yang Y Ji Y Huang P et al . Crosstalks between mTORC1 and mTORC2 variagate cytokine signaling to control NK maturation and effector function. Nat Commun (2018) 9(1):4874. doi: 10.1038/s41467-018-07277-9
126
Li D Wang Y Yang M Dong Z . mTORC1 and mTORC2 coordinate early NK cell development by differentially inducing E4BP4 and T-bet. Cell Death Differ (2021). doi: 10.1038/s41418-020-00715-6
127
Yang M Li D Chang Z Yang Z Tian Z Dong Z . PDK1 orchestrates early NK cell development through induction of E4BP4 expression and maintenance of IL-15 responsiveness. J Exp Med (2015) 212(2):253–65. doi: 10.1084/jem.20141703
128
Gascoyne DM Long E Veiga-Fernandes H de Boer J Williams O Seddon B et al . The basic leucine zipper transcription factor E4BP4 is essential for natural killer cell development. Nat Immunol (2009) 10(10):1118–U1199. doi: 10.1038/ni.1787
129
Firth MA Madera S Beaulieu AM Gasteiger G Castillo EF Schluns KS et al . Nfil3-independent lineage maintenance and antiviral response of natural killer cells. J Exp Med (2013) 210(13):2981–90. doi: 10.1084/jem.20130417
130
He J Wang Y Liu T Liu G Chen S Li Q et al . Stage-specific requirement of kinase PDK1 for NK cells development and activation. Cell Death Differ (2019) 26(10):1918–28. doi: 10.1038/s41418-018-0263-8
131
Kamizono S Duncan GS Seidel MG Morimoto A Hamada K Grosveld G et al . Nfil3/E4bp4 is required for the development and maturation of NK cells in vivo. J Exp Med (2009) 206(13):2977–86. doi: 10.1084/jem.20092176
132
Yang MX Chen SS Du J He JM Wang YD Li ZH et al . NK cell development requires Tsc1-dependent negative regulation of IL-15-triggered mTORC1 activation. Nat Commun (2016) 7:12. doi: 10.1038/ncomms12730
133
Brunet A Bonni A Zigmond MJ Lin MZ Juo P Hu LS et al . Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell (1999) 96(6):857–68. doi: 10.1016/s0092-8674(00)80595-4
134
Guertin DA Stevens DM Thoreen CC Burds AA Kalaany NY Moffat J et al . Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKC alpha but not S6K1. Dev Cell (2006) 11(6):859–71. doi: 10.1016/j.devcel.2006.10.007
135
Cahill CM Tzivion G Nasrin N Ogg S Dore J Ruvkun G et al . Phosphatidylinositol 3-kinase signaling inhibits DAF-16 DNA binding and function via 14-3-3-dependent and 14-3-3-independent pathways. J Biol Chem (2001) 276(16):13402–10. doi: 10.1074/jbc.M010042200
136
Wang S Xia PY Huang GL Zhu PP Liu J Ye BQ et al . FoxO1-mediated autophagy is required for NK cell development and innate immunity. Nat Commun (2016) 7:11023. doi: 10.1038/ncomms11023 doi: ARTN 11023.
137
Deng Y Kerdiles Y Chu J Yuan S Wang Y Chen X et al . Transcription factor Foxo1 is a negative regulator of natural killer cell maturation and function. Immunity (2015) 42(3):457–70. doi: 10.1016/j.immuni.2015.02.006
138
Samson SI Richard O Tavian M Ranson T Vosshenrich CA Colucci F et al . GATA-3 promotes maturation, IFN-gamma production, and liver-specific homing of NK cells. Immunity (2003) 19(5):701–11. doi: 10.1016/s1074-7613(03)00294-2
139
Yun S Lee SH Yoon SR Kim MS Piao ZH Myung PK et al . TOX regulates the differentiation of human natural killer cells from hematopoietic stem cells in vitro. Immunol Lett (2011) 136(1):29–36. doi: 10.1016/j.imlet.2010.11.008
140
Vong QP Leung W-H Houston J Li Y Rooney B Holladay M et al . TOX2 regulates human natural killer cell development by controlling T-BET expression. Blood (2014) 124(26):3905–13. doi: 10.1182/blood-2014-06-582965
141
Taveirne S Wahlen S Van Loocke W Kiekens L Persyn E Van Ammel E et al . The transcription factor ETS1 is an important regulator of human NK cell development and terminal differentiation. Blood (2020)136(3):288–98. doi: 10.1182/blood.2020005204
142
Townsend MJ Weinmann AS Matsuda JL Salomon R Farnham PJ Biron CA et al . T-bet regulates the terminal maturation and homeostasis of NK and Valpha14i NKT cells. Immunity (2004) 20(4):477–94. doi: 10.1016/s1074-7613(04)00076-7
143
Jenne CN Enders A Rivera R Watson SR Bankovich AJ Pereira JP et al . T-bet-dependent S1P5 expression in NK cells promotes egress from lymph nodes and bone marrow. J Exp Med (2009) 206(11):2469–81. doi: 10.1084/jem.20090525
144
Kallies A Carotta S Huntington ND Bernard NJ Tarlinton DM Smyth MJ et al . A role for Blimp1 in the transcriptional network controlling natural killer cell maturation. Blood (2011) 117(6):1869–79. doi: 10.1182/blood-2010-08-303123
145
Loftus RM Assmann N Kedia-Mehta N O’Brien KL Garcia A Gillespie C et al . Amino acid-dependent cMyc expression is essential for NK cell metabolic and functional responses in mice. Nat Commun (2018) 9(1):2341. doi: 10.1038/s41467-018-04719-2
146
Leong JW Schneider SE Sullivan RP Parikh BA Anthony BA Singh A et al . PTEN regulates natural killer cell trafficking in vivo. Proc Natl Acad Sci USA (2015) 112(7):E700–9. doi: 10.1073/pnas.1413886112
147
Briercheck EL Trotta R Chen L Hartlage AS Cole JP Cole TD et al . PTEN is a negative regulator of NK cell cytolytic function. J Immunol (Baltimore Md 1950) (2015) 194(4):1832–40. doi: 10.4049/jimmunol.1401224
148
Liu P Gan W Chin YR Ogura K Guo J Zhang J et al . PtdIns(3,4,5)P-3-Dependent Activation of the mTORC2 Kinase Complex. Cancer Discovery (2015) 5(11):1194–209. doi: 10.1158/2159-8290.Cd-15-0460
149
Seggewiss R Einsele H . Immune reconstitution after allogeneic transplantation and expanding options for immunomodulation: an update. Blood (2010) 115(19):3861–8. doi: 10.1182/blood-2009-12-234096
150
Dulphy N Haas P Busson M Belhadj S Peffault de Latour R Robin M et al . An unusual CD56(bright) CD16(low) NK cell subset dominates the early posttransplant period following HLA-matched hematopoietic stem cell transplantation. J Immunol (Baltimore Md 1950) (2008) 181(3):2227–37. doi: 10.4049/jimmunol.181.3.2227
151
Cichocki F Cooley S Davis Z DeFor TE Schlums H Zhang B et al . CD56dimCD57+NKG2C+ NK cell expansion is associated with reduced leukemia relapse after reduced intensity HCT. Leukemia (2016) 30(2):456–63. doi: 10.1038/leu.2015.260
152
Foley B Cooley S Verneris MR Pitt M Curtsinger J Luo X et al . Cytomegalovirus reactivation after allogeneic transplantation promotes a lasting increase in educated NKG2C+ natural killer cells with potent function. Blood (2012) 119(11):2665–74. doi: 10.1182/blood-2011-10-386995
153
Cichocki F Taras E Chiuppesi F Wagner JE Blazar BR Brunstein C et al . Adaptive NK cell reconstitution is associated with better clinical outcomes. JCI Insight (2019) 4(2):e125553. doi: 10.1172/jci.insight.125553
154
Klebanoff CA Finkelstein SE Surman DR Lichtman MK Gattinoni L Theoret MR et al . IL-15 enhances the in vivo antitumor activity of tumor-reactive CD8+ T cells. Proc Natl Acad Sci USA (2004) 101(7):1969–74. doi: 10.1073/pnas.0307298101
155
Desbois M Beal C Charrier M Besse B Meurice G Cagnard N et al . IL-15 superagonist RLI has potent immunostimulatory properties on NK cells: implications for antimetastatic treatment. J Immunother Cancer (2020) 8(1):e000632. doi: 10.1136/jitc-2020-000632
156
Waldmann TA . Interleukin-15 in the treatment of cancer. Expert Rev Clin Immunol (2014) 10(12):1689–701. doi: 10.1586/1744666X.2014.973856
157
Conlon KC Lugli E Welles HC Rosenberg SA Fojo AT Morris JC et al . Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J Clin Oncol (2015) 33(1):74–82. doi: 10.1200/JCO.2014.57.3329
Summary
Keywords
IL-15, signaling/signaling pathways, natural killer cell, development, transcription factor
Citation
Wang X and Zhao X-Y (2021) Transcription Factors Associated With IL-15 Cytokine Signaling During NK Cell Development. Front. Immunol. 12:610789. doi: 10.3389/fimmu.2021.610789
Received
27 September 2020
Accepted
01 March 2021
Published
18 March 2021
Volume
12 - 2021
Edited by
William Ying Khee Hwang, National Cancer Centre Singapore, Singapore
Reviewed by
Stanislaw Stepkowski, University of Toledo, United States; Barbara L. Kee, University of Chicago, United States
Updates
Copyright
© 2021 Wang and Zhao.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiang-Yu Zhao, zhao_xy@bjmu.edu.cn
This article was submitted to Alloimmunity and Transplantation, a section of the journal Frontiers in Immunology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.