 Lili Chen
Lili Chen Keiko Ozato
Keiko Ozato- Division of Developmental Biology, National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD, United States
Innate immune memory was first described for monocytes and other myeloid cells. This memory is designated Immune Training, in which the host animals that had experienced pathogen infection earlier acquire improved resistance to a second infection. Innate immune memory is mediated by an epigenetic mechanism traced to transcriptional memory that is conserved throughout evolution and has been selected for the ability to mount an adaptive response to shifting environments. Accumulating evidence shows that not only peripheral myeloid cells but hematopoietic stem/progenitor cells (HSCs/HSPCs) can acquire epigenetic memory upon pathogen exposure. Systemic pathogen infection causes HSCs to exit from quiescence and facilitate myeloid-biased differentiation that leads to efficient host defense. This sequence of events is common in HSC memory generation, which is triggered by different stimuli. Recent studies show that not only pathogens but other stimuli such as metabolic stress can generate memory in HSCs. This review summarizes recent publications relevant to HSC memory. We discuss the current understanding of initial sensors, soluble mediators/cytokines involved in memory formation, including Type I and Type II interferons along with future implications.
Introduction
Epigenetic traits, such as histone modifications and certain gene expression programs are inherited through somatic cell divisions, allowing for the maintenance of phenotypic attributes across cell generations. The inheritance of epigenetic traits is largely attributed to transcriptional memory, an evolutionarily conserved mechanism known from bacteria to plants, and mammals (1). Typically, in transcriptional memory, certain sets of genes that had been expressed earlier in response to external cues, mount a faster and greater transcriptional response when these genes are induced again. Enhanced transcriptional response in turn provides the capacity to adapt to a shifting environment which can improve survival (1–4). Faster and greater response, however, does not represent the entire range of transcriptional memory, as in some cases, a previous induction renders the gene(s) unresponsive to the subsequent stimulus, illustrating a dual feature of memory. Innate immune memory/trained immunity shares these features.
Hematopoietic Stem Cells (HSCs)
Adult hematopoietic stem cells (HSCs) reside in the bone marrow (BM) and hierarchically give rise to all lineages of immune cells, which subsequently migrate into peripheral blood and tissues to perform various physiological functions (5–7). HSCs are heterogeneous with respect to self-renewing and differentiation activity (8, 9). Long-term HSCs (LT-HSCs) are capable of self-renewal and full-range lineage differentiation. Short-term HSCs (ST-HSCs) and multipotent progenitors (MPPs) are generated from LT-HSCs. While they maintain multipotency, these progenitors no longer have the self-renewal capacity. MPPs give rise to downstream progenitors, i.e., common lymphoid progenitors (CLPs), common myeloid progenitors (CMPs), megakaryocyte-erythroid progenitors (MEPs), which generate functional lymphocytes and myeloid cells (10, 11).
When encountered with systemic infection, inflammation, blood loss, or other forms of hematopoietic stress, HSCs exit from a dormant state, undergo proliferation, and then differentiation to facilitate efficient myelopoiesis (12, 13). This process is accompanied by peripheral production of hematopoietic growth factors and cytokines, such as granulocyte-macrophage colony-stimulating factor (GM-CSF), Interleukin-6 (IL-6), IL-1, and Type I and Type II interferons (IFNs), which activates new signaling pathways (14–20). Furthermore, HSCs/HSPCs express pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs), and recognize pathogen components, which could then induce cytokines themselves to facilitate emergency myelopoiesis (21–24).
After acute HSC proliferation and myeloid cell differentiation subside, a new homeostasis is established in HSCs which possess a new chromatin landscape and epigenetic traits. This epigenetic modification is thought to provide a basis of innate immune memory/trained immunity, which typically confers enhanced myelopoiesis and greater pathogen clearance (25). Conversely, in other cases, initial priming causes an unresponsive state, resulting in a reduced response upon secondary stimulus, as typified by bacterial lipopolysaccharides (LPS) from gram-negative bacteria (26–29). In either case, innate immune memory is dependent upon epigenetic mechanisms, and as such differs from the classical immunological memory in B and T lymphocytes, which involves genetic changes in the immunoglobulin, and T cell receptor genes, respectively. Unlike adaptive immune memory, innate immune memory created in peripheral myeloid cells is thought to be short-lived, since these cells are turned over relatively rapidly. However, memory in HSCs/HSPCs, if formed, could persist longer, and produce greater downstream consequences.
Pathogen Component-Induced Innate Immunity
A broad range of microbial infections results in alterations in the BM compartment, involving rapid proliferation and differentiation of HSCs as well as progenitor cells, and the subsequent mobilization to the site of infections (30). Escherichia coli infection leads to enhanced granulopoiesis and mobilization of progenitor LK (Lin-ckit+Sca-1-) cells into the peripheral circulation (31). In addition, in the Pseudomonas aeruginosa induced sepsis model, the infection causes HSC expansion that permits rapid compensation to cover the loss of mature immune cells (32). Extensive alterations in the HSPCs compartment have also been observed after other forms of systemic infection (33–35). As summarized in Figure 1, systemic pathogen exposure can afford improved protection against secondary infection by related or unrelated pathogens (25, 36–40).
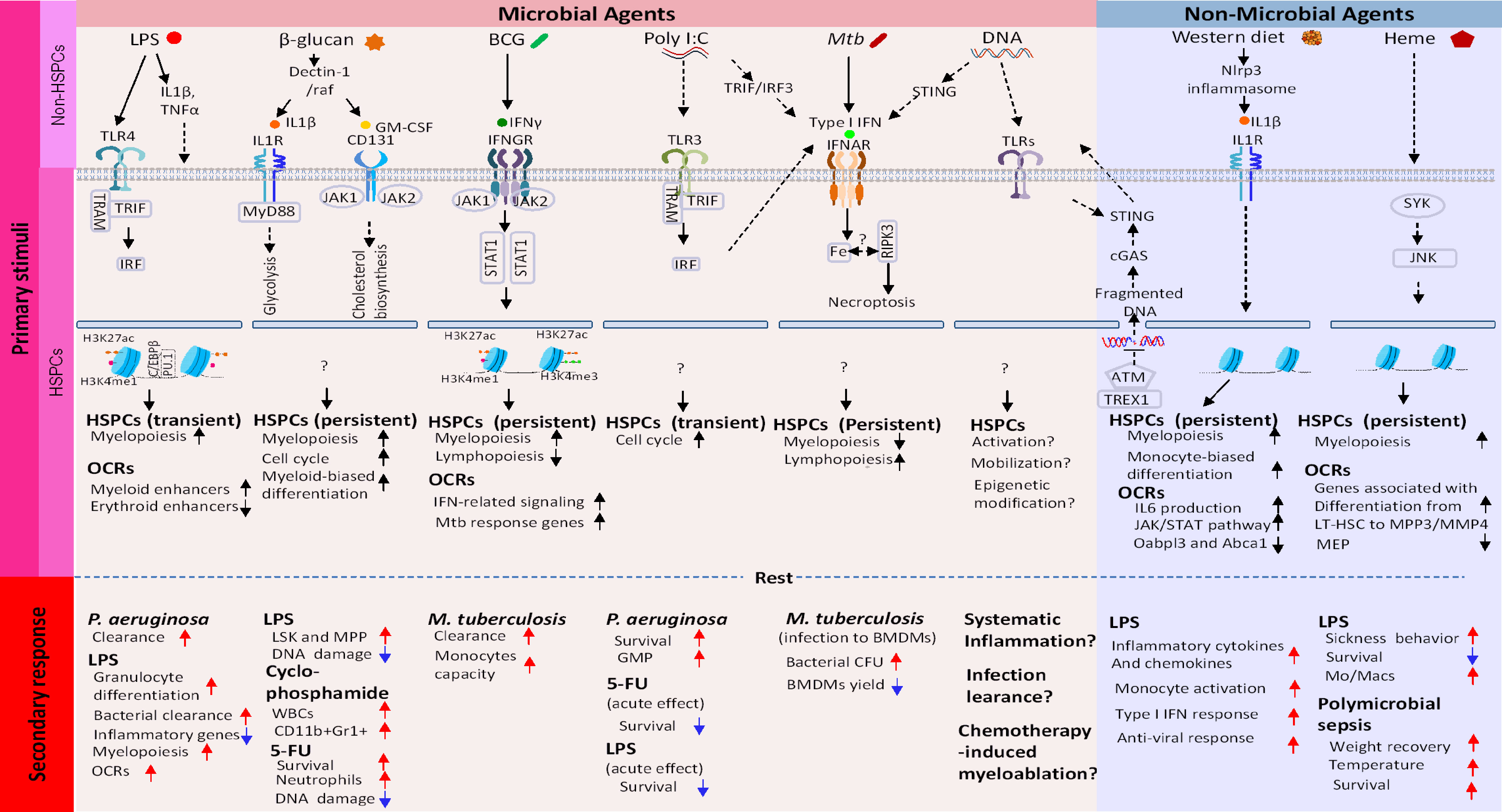
Figure 1 Molecular cascades that create epigenetic memory in HSCs. Top row: Microbial Training Agents and Non-Microbial Training Agents recognized by PRRs and other sensors. Images underneath are subsequent events occurring in descending order. (1): Activation of signaling pathway involving transcription factors and kinases. (2): This then globally alters chromatin accessibility, which leads to building new transcriptome profiles. Open chromatin regions (OCR) can persist longer than transcriptome changes, providing a basis of lasting epigenetic marks. Shown in the bottom two rows are (3): Duration of memory and (4): Phenotypic manifestation of memory. In all cases, HSC memory acquisition involves exit from quiescence, proliferation, and myeloid-biased differentiation of LT-HSC and progenitor cells.
Toll-Like Receptor 4 (TLR4) -Induced HSC Activation and Acquisition of Innate Immune Memory
TLRs (10 in humans, and 13 in mice known) detect pathogen-associated molecular patterns (PAMPs) from invading microbes (23, 24). HSCs/HSPCs express a number of TLRs, including TLR1-4, and TLR 6-9 (41), allowing the cells to recognize various forms of PAMPs and to stimulate proliferation and differentiation into myeloid cells. It is reported that HSPCs (Lin-IL-7Rα-ckit+Sca-1+ (LKS+) cells, LKS+Flk2− long-term stem cells (LT-HSCs), LKS+Flk-2+ multipotent progenitors (MPPs) are capable of responding to LPS through TLR4 or Pam3CSK 4 via TLR2. The downstream adaptor, MyD88 is shown to be required for HSPC activation (21). Another study, on the other hand, reported that LPS induced TLR4 activation depends on TRIF, an alternate adaptor in the TLR signaling cascade (22). Although seemingly inconsistent, these results may not be contradictory, since TLR4 employs both MyD88 and TRIF (23, 24).
Recently, de Laval et al. reported that upon LPS exposure, HSCs undergo expansion and myeloid differentiation and gaining epigenetic memory, which provided an increased protective response to Gram-negative bacteria, Pseudomonas aeruaginosa by reducing bacterial burden and increasing survival rate (38). Although LT-HSC populations returned to a steady-state (cell number) 4 weeks following LPS priming, the LT-HSCs, conferred protection against p. aeruaginosa infection when transferred into naïve mice. The LT-HSCs retained the self-renewal and lineage differentiation capacity along with the transcriptome profile of quiescent HSCs, in which LPS induced inflammatory gene expression was transiently seen earlier. LPS induced a number of transcription factors known to promote myelopoiesis, including members of the C/EBP, ATF, and IRF families, which correlated with a sustained change in chromatin accessibility with an increase at PU.1 and RUNX1 motifs. Consistent with this, open chromatin regions correlated with enhancer marks such as H3K3me1 and H3K27ac and are linked to genes involved in myeloid cell development and activity. These observations indicate that LPS induced transcription factors set a new epigenetic mark in chromatin that leads to the establishment of innate immune memory. Accordingly, HSCs without C/EBPβ were unable to alter chromatin accessibility and failed to provide memory. Other transcription factors expressed in HSCs and regulated by LPS may also modulate these processes (42, 43). Thus, persistent alteration of epigenetic landscape is likely to reflect the state and duration of HSC innate immune memory. In line with this study, another paper reported that LPS priming improved bacterial clearance and survival of mice when challenged with P. aeruginosa (37). In addition, increased granulocyte monocyte progenitors (GMP) were also found in an LPS mediated sepsis model (38, 44).
Besides these studies, LPS is known to cause a profound unresponsive state known as LPS tolerance after a single administration (26–28). Thus, LPS tolerance can leave the host more vulnerable to a secondary infection in some cases. Tolerance is the opposite side of innate immune memory/trained immunity, in which many proinflammatory cytokines, including IL-1, TNFα, and IL-6 remain uninduced after a second LPS stimulation as observed in vivo and in vitro (45, 46).
The Role for TLR3 in HSC Training
Poly (I: C), synthetic ds RNA, used as an RNA virus mimic is a ligand of TLR3 (31–34). de Laval showed that when injected into mice, Poly (I: C), like LPS, led to increased resistance to P. aeruginosa, showing that TLR3 signaling activated following RNA virus infection could give rise to trained immunity (38). In addition, Ribes et al. showed that intraperitoneal pre-injection of Poly (I: C) protects mice from the intracranial E.coli infection, which is known to cause meningoencephalitis (47). Although this study does not present data for HSCs, it indicates that Poly (I: C) is capable of generating some forms of innate immune memory, as it produced broad effects, including those on NK cell mobility and microglia phagocytic activity. Taken together, given that many RNA viruses are major pathogens that afflict all animals, further investigations are warranted to elucidate TLR3 mediated innate immune training, not only in HSCs but peripheral myeloid cells. In this context, RIG-I and MDA5 that also sense viral dsRNA may also play a role in training (48).
β-glucan Induced Trained Immunity in HSCs
β-glucans are a group of polysaccharides that represent key components of the skeletal cell wall of fungi (such as Candida albicans), bacteria, and some plants (such as grain and seaweed) (49, 50). The ability of β-glucans in modulating immune response has been well established. The first evidence for the role of β-glucan in trained immunity was shown in a study of C. albicans infection where preinfection with the fungus protected mice from the second, lethal C. albicans infection (36). This protection was dependent on monocytes, not lymphocytes, unraveling a novel training effect in monocyte in vivo (36). Cellular response to β-glucans is initiated mostly by the binding to dectin-1, studied extensively in macrophages (50–52). In these studies, the long-term epigenetic reprogramming afforded by C. albicans or β-glucans exposure was shown to be mediated by dectin-1 and through the noncanonical Raf-1 pathway (36, 53).
Mitroulis et al. showed that β-glucan, when injected intraperitoneally, induces a dynamic change in the proportion of HSCs and MPPs (39). Gene expression analysis illustrated induction of proliferation and differentiation of LT-HSCs towards myeloid lineage-biased CD41+ LT-HSCs subsets, along with an increase in myeloid-biased MPPs. β-glucan injection also led to an increase in CMP, GMP, and granulocytes (Gr1+CD11b+). The authors performed adoptive transfer of LT-HSCs from β-glucan injected mice into naïve mice and showed that LT-HCSs from β-glucan injected mice provides sustained myelopoiesis. In addition, β-glucan training afforded a protective response to LPS induced DNA damage in HSCs. Also, β-glucan training led to improved resistance to cytotoxic drugs, 5-fluoracil (5-FU), and cyclophosphamide, resulting in a marked increase in the survival rate (54–56). Importantly, the authors found that IL-1β is produced upon β-glucan injection, and this cytokine is responsible for HSC expansion and myeloid biased progenitor differentiation. IL-1 was also responsible for a metabolic shift towards glycolysis. Verifying these results, pharmacological inhibition of IL-1 by IL1RA (anakinra) abrogated HSC expansion, myelopoiesis, metabolic change and immune training. Presumably relevant to these findings, it is reported that SHIP1 signaling is involved in β-glucan induced myeloid cell training, suggesting the role for the phosphatase in this process (57).
Extending the observations of Mitroulis et al., Moorlag et al. recently demonstrated that β-glucan dependent immune training offers a broad anti-pathogen protection, not only in mice, but in human against virulent Mycobacterium. tuberculosis (M.tb) (58). Human monocytes pre-exposed to β-glucan in vitro followed by M.tb infection had lower bacterial load than those without β-glucan preexposure. RNA-seq and ChIP seq analyses showed that some IL-1 family cytokines/chemokines were upregulated in β-glucan trained monocytes, which correlated with increased the H3K27ac mark that indicates enhancers. Preinjection of β-glucan in mice conferred longer survival in mice in response to the secondary M.tb infection. As reported by Mitroulis et al, β-glucan increased LT-HSCs and myelopoiesis in an IL-1 dependent manner. Corroborating the critical requirement of IL-1 signaling, IL-1RA treatment increased M.tb burden in the lung. These reports provide substantive evidence that β-glucan educates HSCs through IL-1 pathways.
Bacillus Calmette-Guérin (BCG) Induced Trained Immunity in HSCs
BCG vaccine is a live, attenuated strain of Mycobacterium bovis, used for protection against M. tb. Epidemiological studies on BCG vaccination support its efficacy and the role of innate immunity (59). BCG vaccines are also shown to give cross-protection against different pathogens, even cancers (60). Based on the cross-protective activity of BCG, O’Neill and Netea proposed the possibility that BCG vaccination may be beneficial for boosting host resistance against coronavirus, including Covid-19, pandemic at the time of this writing (61). It is reported that peripheral monocytes acquire trained immunity in volunteers who received BCG vaccine. These monocytes expressed higher levels of proinflammatory cytokines, including IFNγ, TNFα, and IL-6 than those without BCG (62, 63). Also, BCG is reported to provide increased resistance against experimental yellow fever in human monocytes (64), which coincides with a shift towards glycolytic metabolism, important for BCG induced training (65). It is known that HSCs are refractory to direct bacterial infection, including BCG and Mycobacterium avium, as HSCs do not take up the bacteria (19, 40, 66, 67).
Kaufmann et al. showed that i.v. injection of BCG in mice causes long term innate immune memory in HSCs, conferring improved resistance to second infection by the virulent M.tb (40). The authors found that BCG injection facilitates HSC expansion and development of myeloid lineage dominant multipotent progenitor (MPP3) (19, 40). BM derived macrophages from BCG injected mice gave enhanced protection against M. tb compared to those from naïve mice. Moreover, in cell transfer experiments, naive mice given BM HSCs (LKS) from BCG injected mice demonstrated lasting protection against M.tb, verifying that memory took place in the HSCs. BCG education of HSCs and enhanced resistance to M. tb was dependent on IFNγ (Type II IFN), in which Ifngr-/- mice lacking IFNγ signaling failed to provide anti-M.tb protection. Bulk and single-cell (sc) RNA-seq revealed that this memory correlated with changes in the transcriptome programs in HSCs and MPPs. At the epigenome-level, the transcriptome profiles were associated with the appearance of key enhancer elements marked by acetylation of H3K27.
More recently, Khan et al. asked if the virulent M.tb strain, H37Rv generates trained immunity and report the results startlingly different from those observed with BCG (67). M.tb infection by i.v. injection or aerosol, weakened host’s ability to mount resistance against the subsequent M.tb infection. The weakened resistance was mediated by Type I IFN signaling, and lasted at least a year. While M.tb and BCG both expanded LT-HSCs and MPPs, unlike BCG, M.tb suppressed myelopoiesis leading to a dramatic reduction of neutrophils and Ly6Chi monocytes in periphery. RIPK1 dependent necroptosis accounted for the neutrophil deficiency. BM derived macrophages from naïve mice which adoptively received HSCs from M.tb infected mice were lower in cell yield and deficient in clearing M.tb in vitro. Type I IFN signaling was found critical for the increased susceptibility, as Ifnar1-/- mice (lacking type I IFN receptor), but not Ifngr-/- mice showed better survival after M.tb infection than WT mice and displayed reduced phenotypes. The inhibitory role of type I IFNs in M.tb infection is partly in line with some of previous clinical/epidemiological studies. Together, M.tb trains HSCs somewhat paradoxically to diminish host’s own innate resistance.
Non-Microbial Agents Induced Trained Immunity
PRRs recognize not only pathogen-derived molecular patterns, but non-pathogen derived patterns, produced endogenously or exogenously. Some of these can lead to trained immunity with extensive phenotypic changes.
Western Diet-Induced Trained Immunity
Western-style diet (high calorie, high cholesterol), combined with a sedentary lifestyle are prone to cause obesity, type II diabetes, and other health risks. Inflammation in myeloid cells is a major factor for these health problems. Christ et al. reported that Western diet leads to the generation of trained immunity similar to that produced by pathogens described above (68, 69). The authors examined an atherosclerosis model with Ldlr-/- mice, and found that Western diet prompted the expansion of HSPCs and increased myeloid cell outputs as well as the recruitment of myeloid cells to the site of inflammation. In BM, the proportion of HSPCs, MPPs, and GMPs was increased after consumption of the Western diet. Prolonged myeloid prone changes are associated with low-grade inflammation, also seen in aging. In LPS rechallenge experiments, Western diet-fed mice displayed increased monocyte activation and hyper-inflammation, similar to the reported impacts of innate immune training. Analogous effects were previously reported for rabbits fed with cholesterol-rich diets (70). Supporting an epigenetic mechanism, the Western diet altered overall chromatin accessibility with open chromatin regions associated with IL-6 gene expression and the JAK/STAT pathway activity. Moreover, the NLRP3 inflammasome pathway was involved in Western diet-induced trained immunity, in that the diet-induced effects were reversed in Ldlr-/-/Nlrp3-/- double knock-out mice, which exhibited a reduction in systemic inflammation, excessive hematopoiesis, and reprogramming of GMPs. A prolonged Western diet is known to cause cholesterol overloading in HSCs, leading to an increase in the production of growth factors/cytokines, such as GM-CSF and IL-3. Together these studies suggest that prolonged Western diet promotes formation of immune memory in HSCs. However, underlying processes by which Western diet regulates transcription and epigenome programs in HSCs remain to be elucidated.
Heme-Induced Trained Immunity in HSCs
Heme, a key prosthetic group of hemoproteins or enzymes, is composed of protoporphyrin IX and a ferrous ion (71). Free heme can accumulate excessively during sterile and infectious hemolysis, including hemolytic anemias, ischemia-reperfusion, and malaria, once heme scavengers are over-saturated (72, 73). Heme accumulation increases oxidative stress and systemic inflammatory response (72). Somewhat paradoxically, sickle-caused heme accumulation provides protective effects against Plasmodium Infection, partially through the NR2 2/heme oxygenase-1 (HO-1) pathway (74). Moreover, heme can induce IL-1β production in LPS primed macrophages through activation of NLRP3 inflammasomes (72, 75).
Jentho et al. reported that heme administration increases myeloid-biased LT-HSCs (CD41+LT-HSC), and myeloid-biased MPPs (MPP3,Flt3−CD48+CD150−LSK) with a concomitant decrease in lymphoid-biased MPP4 cells (Flt3+CD48+CD150− LSK) (76). In addition, heme-primed mice were more sensitive to LPS induced acute inflammation, leading to an increase in mortality. Conversely, heme-primed mice showed a protective response to smoldering bacterial sepsis induced by peritoneal contamination and infection. ATAC-seq analysis revealed that heme induces a dramatic change in chromatin accessibility, consistent with myeloid cell-prone development. Heme mediated immune training shared common features with β-glucan driven training, such as upregulated glycolytic metabolism, and enrichment of AP-1 motif in accessible chromatin sites. These findings indicate that labile heme mediates training in LT-HSCs facilitating long-term myelopoiesis with varying outcomes in host defense. It remains to be explored how HSCs sense heme and then reprogram myeloid-biased training in vivo.
Role of IFNs in Innate Immune Memory: IFN Action in LT-HSCs
IL-1 and GM-CSF, cytokines produced by β-glucan priming play a role in HSC immune training (39). IFNs are another class of cytokines that take part in generating innate immune memory in HSCs and peripheral myeloid cells.
Expression and Function of IFNs
There are three types of IFNs, Type I (IFNα/β), Type II (IFNγ), and Type III (IFNλ). Type I and Type II IFNs are shown to be involved in innate immune memory (see below). However, to date, the role of Type III IFN in memory formation has yet to be deciphered. Type I IFNs are encoded by a cluster of related genes (one Ifnb gene, many Ifna genes), and synthesized mostly in DCs and macrophages in response to PRR signaling, but other non-immune cells such as fibroblasts and epithelial cells also produce Type I IFNs. Type II IFN is encoded by a single gene and synthesized in NK and T cells in response to cytokines such as IL-12 and TCR activation (77, 78). Type I IFNbinds to the surface receptor, IFNAR, and signals through a JAK-STAT pathway, leading to activation of the STA1/STAT2/IRF9 complex. This prompts transcriptional induction of more than 2,000 IFN stimulated genes (ISGs), which collectively confer anti-viral and anti-microbial activity on the host cells (79). Type II IFN binds IFNGR and signals through a similar, but distinct JAK-STAT pathway which activates STAT1 dimers. Type II IFN also induces over 2,000 ISGs, many overlapping with ISGs induced by Type I IFN (79, 80). Together, these IFNs provide innate resistance against all types of pathogens, from viruses (DNA and RNA viruses) to bacteria, fungi, and even parasites, a trait that distinguishes them from other cytokines (81). There is an extensive crosstalk between IFNs and NFκB induced inflammatory responses. For example, IFNβ is activated not only by IRFs but by NFκB, which in turn creates an IFNβ feedback loop. Thus, ISGs and NFκB-induced factors are often co-expressed during infection and inflammation.
Both Type I and Type II IFNs are involved in regulating HSC activity and play a role in forming innate immune memory (18, 38, 40). It is reported that the injection of IFNα and Poly (I: C), a Type I IFN inducer, prompts LT- HSCs to exit quiescence prompting their proliferation (18). This process is dependent on IFNAR and JAK-STAT1 signaling.
Convincing evidence has been presented for the requirement of IFNγ in BCG mediated HSC immune training: Kaufmann et al. showed that priming mice with BCG trains HSCs to form memory, which provided enhanced protection against M.tb and that this training was dependent on IFNGR (40). In addition, IFNγ creates innate immune memory in peripheral myeloid cells (82, 83).
Baldridge et al. showed that injection of recombinant IFNγ activates LT-HSCs, triggering the cell cycle entry, and the subsequent mobilization to the spleen (19, 84). Infection with Mycobacterium avium which induces IFNγ also stimulated LT-HSC expansion. A later study by Matatall et al. showed that LCMV infection, also inducing IFNγ, triggered LT-HSC proliferation, and directed myeloid-biased differentiation along with an increased expression of C/EBPβ (85). Furthermore, myeloid-biased HSCs expressed IFNGR at higher levels than lymphoid biased HSCs, thus were more sensitive to IFNγ signaling than lymphoid biased HSCs. The differential IFNGR expression reinforced the selective expansion of myeloid-biased progenitors and their differentiation. Furthermore, IFNγ primed myeloid-biased HSC were preferentially mobilized to periphery upon Mycobacterium. avium infection (85). These observations support a significant role of IFNγ for HSC memory and provide a clue to the mechanism of IFNγ action.
IFN Stimulation Creates Classical Transcriptional Memory
In a separate line of approach, our group reported that Type I and Type II IFNs generate transcriptional memory in somatic cells (83). When NIH 3T3 cells, mouse embryonic fibroblasts, and BM macrophages were treated with IFNβ or IFNγ, respectively in advance, the cells mounted a faster and greater ISG response upon the second IFN stimulation, a typical feature of transcriptional memory. Supporting the biological significance of this memory, IFN pretreatment led to improved protection against EMCV viral infection. This memory was inherited through generations, as the memory response was retained after cycles of fibroblast proliferation, another hallmark of transcriptional memory. Transcriptome analysis revealed that memory has a dual quality. While some ISGs exhibited enhanced transcription, other ISGs became unresponsive (or less responsive) to the second IFN stimulation. A similar dual feature has been documented for LPS induced memory, in that LPS pre-administration enhanced expression of some LPS response genes, but repressed other genes (26–28, 38, 45). GO analysis indicated that this duality has a functional meaning, since ISGs showing enhanced expression in the second response were associated with anti-viral, anti-pathogen responses, whereas ISGs with reduced second response were enriched with terms for cell growth, metabolic regulation, etc, unrelated to host defense. The memory response was accounted for by accelerated recruitment of STAT1 and RNA polymerase II to ISGs for ISGs with the enhanced second response. On the other hand, a block in transcriptional elongation was observed for ISGs tolerized in the second response. Epigenome analysis showed that memory coincides with the deposition of the histone H3.3, a conserved histone variant implicated in memory (86). H3.3 and its specific chaperon HIRA, which is responsible for genic H3.3 deposition are expressed highly in murine adult BM HSCs. Our subsequent study with conditional Hira-/- mice demonstrated that HIRA is essential for the generation and maintenance of BM LT-HSCs. In the absence of HIRA, the number of BM LT-HSCs were dramatically reduced, along with the reduction in immediate (MPPs) and downstream progenitors (CMPs, GMPs), leading to a marked paucity in mature, functional myeloid and lymphoid cells. These observations support the possibility that the histone H3.3 and its chaperon HIRA play a substantial role in shaping the development and function of HSCs, and may contribute to their memory formation.
Trained Immunity and DNA Damage in HSCs: Unsolved Questions
Although a limited number of IFN stimulation can generate trained immunity, repeated IFN exposure is shown to exhaust HSC pools by an internally controlled process, not fully understood (84, 87, 88). Transcription factor families including the IRF family appear to play a role (84, 87). HSC attrition is presumably a result of DNA damage that occurs during HSC proliferation and associated replication stress (88). In addition to IFNs, Poly I: C and LPS are shown to cause DNA damage in HSCs even after a single exposure, as evidenced by phosphorylation of H2AX and nuclear foci formation (39, 88). Similarly, chronic exposure to IL-1, a proinflammatory cytokine involved in β-glucan mediated HSC training is shown to exhaust HSC pools (17). Since HSC activation and resultant proliferation leads to DNA damage, it is possible that HSC training is in some way linked to DNA damage. On the other hand, excessive HSC activation/proliferation may have a negative consequence on HSC’s self-renewal capacity and lifespan. It remains unclear how HSC exhaustion affects immune training and vice versa.
There is evidence suggesting that DNA damage activates another signaling pathway, STING, and influences innate immune memory in HSCs. STING is a cytoplasmic adaptor for a DNA sensing signaling pathway (89). Canonical STING ligands, cyclic di-GMP/AMP are produced by various pathogens, which activate TBK and IRF3, resulting in Type I IFN and ISG induction (86). The STING pathway is functional in LT-HSC since they are activated and mobilized by a canonical STING ligand (90). It is now evident that not only cyclic di-GMP/AMP, but DNA breaks produced by genotoxic, chemotherapy drugs activate the STING pathway (91). STING is also activated in mice defective in DNA repair (92). DNA damage-induced STING pathway is reported to chronically activate ISGs and NFκB mediated inflammatory cytokines in some cell types (91, 92).
It is noteworthy that chronic ISG expression and STING activation is a hallmark of Aicardi-Goutieres Syndromes (AGS) and related retinal vasculopathy with cerebral leukodystrophy (RVCL), which produce complex inflammatory diseases often involving neurological defects (93, 94). It may be of interest to study how HSC DNA damage and immune training intersect with AGS and related chronic inflammatory disorders.
Concluding Remarks
Innate immune memory is an emerging concept that opened a radically new perspective on infection and inflammation. Convincing evidence has been presented demonstrating that HSCs form epigenetic memory in response to pathogens and other stress, which confers adaptive responses to the subsequent stress upon the host. HSC memory coincides with the induction of proliferation and myeloid-biased progenitor differentiation, the process driven by IFN, IL-1 and other inflammatory cytokines. Many questions regarding molecular mechanisms, signaling pathways, and epigenome landscapes leading to HSC innate immune memory remain to be elucidated further. In addition, relationships between HSC immune training, DNA damage, and hematopoietic aging are subjects of future investigation.
Author Contributions
KO proposed LC to write a minireview on innate immune memory. The two authors discussed the scope of the task, undertook literature surveys, and outlined the structure of the review. Both authors wrote the manuscript and performed editing. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the intramural program of the National Institute of Child Health and Human Development, National Institutes of Health (Project designation: ZIA HD 001310-33).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Xue Y, Acar M. Mechanisms for the epigenetic inheritance of stress response in single cells. Curr Genet (2018) 64(6):1221–8. doi: 10.1007/s00294-018-0849-1
2. Brickner JH. Transcriptional memory: staying in the loop. Curr Biol (2010) 20(1):R20–1. doi: 10.1016/j.cub.2009.11.013
3. D’Urso A, Brickner JH. Epigenetic transcriptional memory. Curr Genet (2017) 63(3):435–9. doi: 10.1007/s00294-016-0661-8
4. Avramova Z. Transcriptional ‘memory’ of a stress: transient chromatin and memory (epigenetic) marks at stress-response genes. Plant J (2015) 83(1):149–59. doi: 10.1111/tpj.12832
5. Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell (2008) 132(4):631–44. doi: 10.1016/j.cell.2008.01.025
6. Mendelson A, Frenette PS. Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nat Med (2014) 20(8):833–46. doi: 10.1038/nm.3647
7. Warr MR, Pietras EM, Passegué E. Mechanisms controlling hematopoietic stem cell functions during normal hematopoiesis and hematological malignancies. Wiley Interdiscip Rev Syst Biol Med (2011) 3(6):681–701. doi: 10.1002/wsbm.145
8. Haas S, Trumpp A, Milsom MD. Causes and Consequences of Hematopoietic Stem Cell Heterogeneity. Cell Stem Cell (2018) 22(5):627–38. doi: 10.1016/j.stem.2018.04.003
9. Sawai CM, Babovic S, Upadhaya S, Knapp D, Lavin Y, Lau CM, et al. Hematopoietic Stem Cells Are the Major Source of Multilineage Hematopoiesis in Adult Animals. Immunity (2016) 45(3):597–609. doi: 10.1016/j.immuni.2016.08.007
10. Chavakis T, Mitroulis I, Hajishengallis G. Hematopoietic progenitor cells as integrative hubs for adaptation to and fine-tuning of inflammation. Nat Immunol (2019) 20(7):802–11. doi: 10.1038/s41590-019-0402-5
11. Yamamoto R, Wilkinson AC, Nakauchi H. Changing concepts in hematopoietic stem cells. Science (2018) 362(6417):895–6. doi: 10.1126/science.aat7873
12. Schultze JL, Mass E, Schlitzer A. Emerging Principles in Myelopoiesis at Homeostasis and during Infection and Inflammation. Immunity (2019) 50(2):288–301. doi: 10.1016/j.immuni.2019.01.019
13. Mitroulis I, Kalafati L, Hajishengallis G, Chavakis T. Myelopoiesis in the Context of Innate Immunity. J Innate Immun (2018) 10(5-6):365–72. doi: 10.1159/000489406
14. Boettcher S, Manz MG. Regulation of Inflammation- and Infection-Driven Hematopoiesis. Trends Immunol (2017) 38(5):345–57. doi: 10.1016/j.it.2017.01.004
15. Burgess AW, Metcalf D. The nature and action of granulocyte-macrophage colony stimulating factors. Blood (1980) 56(6):947–58. doi: 10.1182/blood.V56.6.947.947
16. Bernad A, Kopf M, Kulbacki R, Weich N, Koehler G, Gutierrez-Ramos JC. Interleukin-6 is required in vivo for the regulation of stem cells and committed progenitors of the hematopoietic system. Immunity (1994) 1(9):725–31. doi: 10.1016/s1074-7613(94)80014-6
17. Pietras EM, Mirantes-Barbeito C, Fong S, Loeffler D, Kovtonyuk LV, Zhang S, et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat Cell Biol (2016) 18(6):607–18. doi: 10.1038/ncb3346
18. Essers MA, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, et al. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature (2009) 458(7240):904–8. doi: 10.1038/nature07815
19. Baldridge MT, King KY, Boles NC, Weksberg DC, Goodell MA. Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature (2010) 465(7299):793–7. doi: 10.1038/nature09135
20. Pietras EM, Lakshminarasimhan R, Techner JM, Fong S, Flach J, Binnewies M, et al. Re-entry into quiescence protects hematopoietic stem cells from the killing effect of chronic exposure to type I interferons. J Exp Med (2014) 211(2):245–62. doi: 10.1084/jem.20131043
21. Nagai Y, Garrett KP, Ohta S, Bahrun U, Kouro T, Akira S, et al. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity (2006) 24(6):801–12. doi: 10.1016/j.immuni.2006.04.008
22. Takizawa H, Fritsch K, Kovtonyuk LV, Saito Y, Yakkala C, Jacobs K, et al. Pathogen-Induced TLR4-TRIF Innate Immune Signaling in Hematopoietic Stem Cells Promotes Proliferation but Reduces Competitive Fitness. Cell Stem Cell (2017) 21(2):225–40. doi: 10.1016/j.stem.2017.06.013
23. O’Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol (2007) 7(5):353–64. doi: 10.1038/nri2079
24. Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science (2003) 301(5633):640–3. doi: 10.1126/science.1087262
25. Netea MG, Joosten LA, Latz E, Mills KH, Natoli G, Stunnenberg HG, et al. Trained immunity: A program of innate immune memory in health and disease. Science (2016) 352(6284):aaf1098. doi: 10.1126/science.aaf1098
26. Lu M, Varley AW, Ohta S, Hardwick J, Munford RS. Host inactivation of bacterial lipopolysaccharide prevents prolonged tolerance following gram-negative bacterial infection. Cell Host Microbe (2008) 4(3):293–302. doi: 10.1016/j.chom.2008.06.009
27. Munford RS. Murine responses to endotoxin: another dirty little secret? J Infect Dis (2010) 201(2):175–7. doi: 10.1086/649558
28. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol (2010) 11(10):889–96. doi: 10.1038/ni.1937
29. Xing Z, Afkhami S, Bavananthasivam J, Fritz DK, D’Agostino MR, Vaseghi-Shanjani M, et al. Innate immune memory of tissue-resident macrophages and trained innate immunity: Re-vamping vaccine concept and strategies. J Leukoc Biol (2020) 108(3):825–34. doi: 10.1002/jlb.4mr0220-446r
30. Glatman Zaretsky A, Engiles JB, Hunter CA. Infection-induced changes in hematopoiesis. J Immunol (2014) 192(1):27–33. doi: 10.4049/jimmunol.1302061
31. Shahbazian LM, Quinton LJ, Bagby GJ, Nelson S, Wang G, Zhang P. Escherichia coli pneumonia enhances granulopoiesis and the mobilization of myeloid progenitor cells into the systemic circulation. Crit Care Med (2004) 32(8):1740–6. doi: 10.1097/01.ccm.0000132900.84627.90
32. Rodriguez S, Chora A, Goumnerov B, Mumaw C, Goebel WS, Fernandez L, et al. Dysfunctional expansion of hematopoietic stem cells and block of myeloid differentiation in lethal sepsis. Blood (2009) 114(19):4064–76. doi: 10.1182/blood-2009-04-214916
33. Yáñez A, Murciano C, O’Connor JE, Gozalbo D, Gil ML. Candida albicans triggers proliferation and differentiation of hematopoietic stem and progenitor cells by a MyD88-dependent signaling. Microbes Infect (2009) 11(4):531–5. doi: 10.1016/j.micinf.2009.01.011
34. Singh P, Yao Y, Weliver A, Broxmeyer HE, Hong SC, Chang CH. Vaccinia virus infection modulates the hematopoietic cell compartments in the bone marrow. Stem Cells (2008) 26(4):1009–16. doi: 10.1634/stemcells.2007-0461
35. Shi X, Zhang P, Sempowski GD, Shellito JE. Thymopoietic and bone marrow response to murine Pneumocystis pneumonia. Infect Immun (2011) 79(5):2031–42. doi: 10.1128/iai.01213-10
36. Quintin J, Saeed S, Martens JHA, Giamarellos-Bourboulis EJ, Ifrim DC, Logie C, et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe (2012) 12(2):223–32. doi: 10.1016/j.chom.2012.06.006
37. Murphey ED, Fang G, Varma TK, Sherwood ER. Improved bacterial clearance and decreased mortality can be induced by LPS tolerance and is not dependent upon IFN-gamma. Shock (2007) 27(3):289–95. doi: 10.1097/01.shk.0000245024.93740.28
38. de Laval B, Maurizio J, Kandalla PK, Brisou G, Simonnet L, Huber C, et al. C/EBPβ-Dependent Epigenetic Memory Induces Trained Immunity in Hematopoietic Stem Cells. Cell Stem Cell (2020) 26(5):657–74. doi: 10.1016/j.stem.2020.01.017
39. Mitroulis I, Ruppova K, Wang B, Chen LS, Grzybek M, Grinenko T, et al. Modulation of Myelopoiesis Progenitors Is an Integral Component of Trained Immunity. Cell (2018) 172(1-2):147–61. doi: 10.1016/j.cell.2017.11.034
40. Kaufmann E, Sanz J, Dunn JL, Khan N, Mendonça LE, Pacis A, et al. BCG Educates Hematopoietic Stem Cells to Generate Protective Innate Immunity against Tuberculosis. Cell (2018) 172(1-2):176–90. doi: 10.1016/j.cell.2017.12.031
41. Cannova J, Breslin SJP, Zhang J. Toll-like receptor signaling in hematopoietic homeostasis and the pathogenesis of hematologic diseases. Front Med (2015) 9(3):288–303. doi: 10.1007/s11684-015-0412-0
42. Wilson NK, Foster SD, Wang X, Knezevic K, Schutte J, Kaimakis P, et al. Combinatorial transcriptional control in blood stem/progenitor cells: genome-wide analysis of ten major transcriptional regulators. Cell Stem Cell (2010) 7(4):532–44. doi: 10.1016/j.stem.2010.07.016
43. Olsson A, Venkatasubramanian M, Chaudhri VK, Aronow BJ, Salomonis N, Singh H, et al. Single-cell analysis of mixed-lineage states leading to a binary cell fate choice. Nature (2016) 537(7622):698–702. doi: 10.1038/nature19348
44. Bomans K, Schenz J, Sztwiertnia I, Schaack D, Weigand MA, Uhle F. Sepsis Induces a Long-Lasting State of Trained Immunity in Bone Marrow Monocytes. Front Immunol (2018) 9:2685. doi: 10.3389/fimmu.2018.02685
45. Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature (2007) 447(7147):972–8. doi: 10.1038/nature05836
46. Wendeln AC, Degenhardt K, Kaurani L, Gertig M, Ulas T, Jain G, et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature (2018) 556(7701):332–8. doi: 10.1038/s41586-018-0023-4
47. Ribes S, Adam N, Ebert S, Regen T, Bunkowski S, Hanisch UK, et al. The viral TLR3 agonist poly(I:C) stimulates phagocytosis and intracellular killing of Escherichia coli by microglial cells. Neurosci Lett (2010) 482(1):17–20. doi: 10.1016/j.neulet.2010.06.078
48. Reikine S, Nguyen JB, Modis Y. Pattern Recognition and Signaling Mechanisms of RIG-I and MDA5. Front Immunol (2014) 5:342. doi: 10.3389/fimmu.2014.00342
49. Vetvicka V. Glucan-immunostimulant, adjuvant, potential drug. World J Clin Oncol (2011) 2(2):115–9. doi: 10.5306/wjco.v2.i2.115
50. Brown GD, Gordon S. Fungal beta-glucans and mammalian immunity. Immunity (2003) 19(3):311–5. doi: 10.1016/s1074-7613(03)00233-4
51. Brown GD, Herre J, Williams DL, Willment JA, Marshall AS, Gordon S. Dectin-1 mediates the biological effects of beta-glucans. J Exp Med (2003) 197(9):1119–24. doi: 10.1084/jem.20021890
52. Brown GD, Gordon S. Immune recognition of fungal beta-glucans. Cell Microbiol (2005) 7(4):471–9. doi: 10.1111/j.1462-5822.2005.00505.x
53. Saeed S, Quintin J, Kerstens HH, Rao NA, Aghajanirefah A, Matarese F, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science (2014) 345(6204):1251086. doi: 10.1126/science.1251086
54. Karigane D, Takubo K. Metabolic regulation of hematopoietic and leukemic stem/progenitor cells under homeostatic and stress conditions. Int J Hematol (2017) 106(1):18–26. doi: 10.1007/s12185-017-2261-x
55. Takubo K, Goda N, Yamada W, Iriuchishima H, Ikeda E, Kubota Y, et al. Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell (2010) 7(3):391–402. doi: 10.1016/j.stem.2010.06.020
56. Kocabas F, Zheng J, Thet S, Copeland NG, Jenkins NA, DeBerardinis RJ, et al. Meis1 regulates the metabolic phenotype and oxidant defense of hematopoietic stem cells. Blood (2012) 120(25):4963–72. doi: 10.1182/blood-2012-05-432260
57. Saz-Leal P, Del Fresno C, Brandi P, Martinez-Cano S, Dungan OM, Chisholm JD, et al. Targeting SHIP-1 in Myeloid Cells Enhances Trained Immunity and Boosts Response to Infection. Cell Rep (2018) 25(5):1118–26. doi: 10.1016/j.celrep.2018.09.092
58. Moorlag S, Khan N, Novakovic B, Kaufmann E, Jansen T, van Crevel R, et al. beta-Glucan Induces Protective Trained Immunity against Mycobacterium tuberculosis Infection: A Key Role for IL-1. Cell Rep (2020) 31(7):107634. doi: 10.1016/j.celrep.2020.107634
59. Koeken V, Verrall AJ, Netea MG, Hill PC, van Crevel R. Trained innate immunity and resistance to Mycobacterium tuberculosis infection. Clin Microbiol Infect (2019) 25(12):1468–72. doi: 10.1016/j.cmi.2019.02.015
60. Kleinnijenhuis J, van Crevel R, Netea MG. Trained immunity: consequences for the heterologous effects of BCG vaccination. Trans R Soc Trop Med Hyg (2015) 109(1):29–35. doi: 10.1093/trstmh/tru168
61. O’Neill LAJ, Netea MG. BCG-induced trained immunity: can it offer protection against COVID-19? Nat Rev Immunol (2020) 20(6):335–7. doi: 10.1038/s41577-020-0337-y
62. Kleinnijenhuis J, Quintin J, Preijers F, Joosten LA, Ifrim DC, Saeed S, et al. Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc Natl Acad Sci USA (2012) 109(43):17537–42. doi: 10.1073/pnas.1202870109
63. Cirovic B, de Bree LCJ, Groh L, Blok BA, Chan J, van der Velden W, et al. BCG Vaccination in Humans Elicits Trained Immunity via the Hematopoietic Progenitor Compartment. Cell Host Microbe (2020) 28(2):322–4.e5. doi: 10.1016/j.chom.2020.05.014
64. Arts RJW, Moorlag S, Novakovic B, Li Y, Wang SY, Oosting M, et al. BCG Vaccination Protects against Experimental Viral Infection in Humans through the Induction of Cytokines Associated with Trained Immunity. Cell Host Microbe (2018) 23(1):89–100. doi: 10.1016/j.chom.2017.12.010
65. Arts RJW, Carvalho A, La Rocca C, Palma C, Rodrigues F, Silvestre R, et al. Immunometabolic Pathways in BCG-Induced Trained Immunity. Cell Rep (2016) 17(10):2562–71. doi: 10.1016/j.celrep.2016.11.011
66. Kolb-Mäurer A, Wilhelm M, Weissinger F, Bröcker EB, Goebel W. Interaction of human hematopoietic stem cells with bacterial pathogens. Blood (2002) 100(10):3703–9. doi: 10.1182/blood-2002-03-0898
67. Khan N, Downey J, Sanz J, Kaufmann E, Blankenhaus B, Pacis A, et al. M. tuberculosis Reprograms Hematopoietic Stem Cells to Limit Myelopoiesis and Impair Trained Immunity. Cell (2020) 183(3):752–70. doi: 10.1016/j.cell.2020.09.062
68. Christ A, Günther P, Lauterbach MAR, Duewell P, Biswas D, Pelka K, et al. Western Diet Triggers NLRP3-Dependent Innate Immune Reprogramming. Cell (2018) 172(1-2):162–75. doi: 10.1016/j.cell.2017.12.013
69. Christ A, Latz E. The Western lifestyle has lasting effects on metaflammation. Nat Rev Immunol (2019) 19(5):267–8. doi: 10.1038/s41577-019-0156-1
70. Fleet JC, Clinton SK, Salomon RN, Loppnow H, Libby P. Atherogenic diets enhance endotoxin-stimulated interleukin-1 and tumor necrosis factor gene expression in rabbit aortae. J Nutr (1992) 122(2):294–305. doi: 10.1093/jn/122.2.294
71. Ponka P. Cell biology of heme. Am J Med Sci (1999) 318(4):241–56. doi: 10.1097/00000441-199910000-00004
72. Dutra FF, Bozza MT. Heme on innate immunity and inflammation. Front Pharmacol (2014) 5:115. doi: 10.3389/fphar.2014.00115
73. Gozzelino R, Jeney V, Soares MP. Mechanisms of cell protection by heme oxygenase-1. Annu Rev Pharmacol Toxicol (2010) 50:323–54. doi: 10.1146/annurev.pharmtox.010909.105600
74. Ferreira A, Marguti I, Bechmann I, Jeney V, Chora A, Palha NR, et al. Sickle hemoglobin confers tolerance to Plasmodium infection. Cell (2011) 145(3):398–409. doi: 10.1016/j.cell.2011.03.049
75. Li Q, Fu W, Yao J, Ji Z, Wang Y, Zhou Z, et al. Heme induces IL-1β secretion through activating NLRP3 in kidney inflammation. Cell Biochem Biophys (2014) 69(3):495–502. doi: 10.1007/s12013-014-9823-9
76. Jentho E, Novakovic B, Ruiz-Moreno C, Kourtzelis I, Martins R, Chavakis T, et al. Heme induces innate immune memory. bioRxiv (2019). doi: 10.1101/2019.12.12.874578
77. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol (2014) 14(1):36–49. doi: 10.1038/nri3581
78. González-Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I interferons. Nat Rev Immunol (2012) 12(2):125–35. doi: 10.1038/nri3133
79. Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol (2014) 32:513–45. doi: 10.1146/annurev-immunol-032713-120231
80. Kawai T, Akira S. TLR signaling. Cell Death Differ (2006) 13(5):816–25. doi: 10.1038/sj.cdd.4401850
81. MacMicking JD. Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat Rev Immunol (2012) 12(5):367–82. doi: 10.1038/nri3210
82. Yao Y, Jeyanathan M, Haddadi S, Barra NG, Vaseghi-Shanjani M, Damjanovic D, et al. Induction of Autonomous Memory Alveolar Macrophages Requires T Cell Help and Is Critical to Trained Immunity. Cell (2018) 175(6):1634–50. doi: 10.1016/j.cell.2018.09.042
83. Kamada R, Yang W, Zhang Y, Patel MC, Yang Y, Ouda R, et al. Interferon stimulation creates chromatin marks and establishes transcriptional memory. Proc Natl Acad Sci USA (2018) 115(39):E9162–e71. doi: 10.1073/pnas.1720930115
84. Baldridge MT, King KY, Goodell MA. Inflammatory signals regulate hematopoietic stem cells. Trends Immunol (2011) 32(2):57–65. doi: 10.1016/j.it.2010.12.003
85. Matatall KA, Shen CC, Challen GA, King KY. Type II interferon promotes differentiation of myeloid-biased hematopoietic stem cells. Stem Cells (2014) 32(11):3023–30. doi: 10.1002/stem.1799
86. Ng RK, Gurdon JB. Epigenetic memory of an active gene state depends on histone H3.3 incorporation into chromatin in the absence of transcription. Nat Cell Biol (2008) 10(1):102–9. doi: 10.1038/ncb1674
87. Sato T, Onai N, Yoshihara H, Arai F, Suda T, Ohteki T. Interferon regulatory factor-2 protects quiescent hematopoietic stem cells from type I interferon-dependent exhaustion. Nat Med (2009) 15(6):696–700. doi: 10.1038/nm.1973
88. Walter D, Lier A, Geiselhart A, Thalheimer FB, Huntscha S, Sobotta MC, et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature (2015) 520(7548):549–52. doi: 10.1038/nature14131
89. Barber GN. STING: infection, inflammation and cancer. Nat Rev Immunol (2015) 15(12):760–70. doi: 10.1038/nri3921
90. Kobayashi H, Kobayashi CI, Nakamura-Ishizu A, Karigane D, Haeno H, Yamamoto KN, et al. Bacterial c-di-GMP affects hematopoietic stem/progenitors and their niches through STING. Cell Rep (2015) 11(1):71–84. doi: 10.1016/j.celrep.2015.02.066
91. Dunphy G, Flannery SM, Almine JF, Connolly DJ, Paulus C, Jønsson KL, et al. Non-canonical Activation of the DNA Sensing Adaptor STING by ATM and IFI16 Mediates NF-κB Signaling after Nuclear DNA Damage. Mol Cell (2018) 71(5):745–60. doi: 10.1016/j.molcel.2018.07.034
92. Härtlova A, Erttmann SF, Raffi FA, Schmalz AM, Resch U, Anugula S, et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity (2015) 42(2):332–43. doi: 10.1016/j.immuni.2015.01.012
93. Crow YJ, Livingston JH. Aicardi-Goutieres syndrome: an important Mendelian mimic of congenital infection. Dev Med Child Neurol (2008) 50(6):410–6. doi: 10.1111/j.1469-8749.2008.02062.x
Keywords: HSC, myeloid-bias, trained immunity, epigenetic memory, interferon
Citation: Chen L and Ozato K (2021) Innate Immune Memory in Hematopoietic Stem/Progenitor Cells: Myeloid-Biased Differentiation and the Role of Interferon. Front. Immunol. 12:621333. doi: 10.3389/fimmu.2021.621333
Received: 26 October 2020; Accepted: 15 March 2021;
Published: 29 March 2021.
Edited by:
Jose Luis Subiza, Inmunotek SL, SpainCopyright © 2021 Chen and Ozato. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Keiko Ozato, b3phdG9rQGRpcjYubmljaGQubmloLmdvdg==
†Present address: Lili Chen, Center for Stem Cell and Regenerative Disease, Brown Foundation Institute of Molecular Medicine for the Prevention of Human Diseases (IMM), The University of Texas Health Science Center at Houston, Houston, TX, United States