 Carlos Zamora
Carlos Zamora Elisabet Cantó
Elisabet Cantó Sílvia Vidal
Sílvia Vidal- Inflammatory Diseases, Institut de Recerca de l'Hospital de la Santa Creu i Sant Pau, Biomedical Research Institute Sant Pau (IIB Sant Pau), Barcelona, Spain
Patients with chronic inflammatory diseases often exhibit cardiovascular risk. This risk is associated with the systemic inflammation that persists in these patients, causing a sustained endothelial activation. Different mechanisms have been considered responsible for this systemic inflammation, among which activated platelets have been regarded as a major player. However, in recent years, the role of platelets has become controversial. Not only can this subcellular component release pro- and anti-inflammatory mediators, but it can also bind to different subsets of circulating lymphocytes, monocytes and neutrophils modulating their function in either direction. How platelets exert this dual role is not yet fully understood.
Introduction
Systemic inflammation (SI) has been described as a consequence of increased levels of the circulating pro-inflammatory mediators that activate endothelial cells (EC). Endothelial activation is part of a normal immune system defense, but a prolonged inflammatory stimulus induces a sustained endothelial activation/dysfunction that is often associated with atherogenesis and cardiovascular events. The role of SI on cardiovascular (CV) risk has been explored in autoimmune diseases. Systemic lupus erythematosus (SLE)-like mouse models display endothelial dysfunction and cardiac hypertrophy, mediated through IL-6 and IL-1α (1). In these patients, it has been shown that B lymphocyte stimulator induced apoptosis of endothelial progenitors cells and EC (2). In addition, it has been suggested that autoantibodies play a role in endothelial dysfunction, possibly by modulating the adhesion of neutrophils (3, 4). In a model of arthritis, endothelial dysfunction was only observed in rats with a persisting imbalance between NOS and COX-2 pathways, higher plasma levels of IL-1β and tumor necrosis factor-α (TNF-α) (5). The presence of diabetes mellitus type 2 in patients with metabolic syndrome impairs the endothelial function (6). In a model of SI, the levels of endothelin-1 and endocan are related to endothelial dysfunction (7). However, there are some circumstances in which the relationship between SI and endothelial dysfunction is less clear. In Systemic Inflammatory Response syndrome, the levels of dysfunctional EC were associated with mortality and organ dysfunction independently of inflammatory markers (8).
The link between inflammation and endothelial dysfunction has been confirmed by the inhibition of molecules related to SI. Anti-TNFα antibodies reduces sE-selectin and sVCAM expression (9) and decreases endothelium-dependent relaxation (10, 11). EC treated with etanercept revert the apoptosis induced by TNF-α (12). JAK inhibitors improve endothelium dependent vasorelaxation, endothelial cell differentiation and lipoprotein profiles, while decreasing pro-inflammatory cytokines in SLE-like syndrome (13). Glucocorticoids decrease IL-1β and TNF-α levels, improving the function of endothelium in rheumatoid arthritis (RA) (14). Patients with CV risk factors had increased levels of IL-1β and its gene expression signature and blocking IL-1β with canakinumab was observed to prevent recurrent cardiovascular disease (CVD) (15). Agents with nucleoside triphosphate hydrolase activity decrease platelet-leukocyte-endothelium interaction, the transcription of pro-inflammatory cytokines, microvascular platelet-neutrophil aggregate sequestration, activation marker expression on platelets (PLTs) and neutrophils contained in these aggregates, leukocyte extravasation, and organ damage (16). Furthermore, dihydroartemisin inhibits the occludin downregulation induced by TNF-α, improving the permeability of EC (17) and the inhibition of cannabinoid receptors reduces leukocyte-adhesion and improves microvascular blood flow (18). The pre-treatment of primary cultured human umbilical vein endothelial cells (HUVECs) with sevoflurane reduces ICAM-1 (intercellular adhesion molecule-1) and VCAM-1 (vascular cell adhesion molecule-1) IκBα, and NF-κB activation, and blocks the adhesion of leukocytes (19).
Products with anti-inflammatory properties can improve endothelial function. Lactobacillus plantarum 299v supplements decreased inflammatory markers (20), while the active form of vitamin D diminishes IL-6 secretion and increases the angiogenic capacity of myeloid angiogenic cells via CXCL10 down-regulation (21).
Features of Endothelial Dysfunction
The inflammatory phase that leads to endothelial dysfunction is initiated by TNF-α and subsequently amplified by IL-1, IL-6 and downstream mediators. Endothelial dysfunction refers to the failure by ECs to perform their physiological functions, often due to a maladaptive response to pathological stimuli. The phenotypic features of endothelial dysfunction include the upregulated expression of endothelial leukocyte adhesion molecules (ELAMs) [E-selectin, ICAM-1 and VCAM-1]. On leukocytes, activated ECs induce the affinity of counter-receptors for ELAMs and secrete and display chemokines on the luminal surface. Endothelial dysfunction also includes a compromised barrier function, the secretion of microvesicles, an increased vascular smooth muscle tone, and the increased production of vasoconstrictor substances, the reduced resistance to thrombosis via platelet aggregation and oxidative stress upregulation (22–24). It was described that high levels of IL-8 and TNF-α up-regulate CX3CR1 expression on platelet-monocyte aggregates, increasing adhesion to activated endothelium (25). In mouse models, increased IL-17 was associated with reactive oxygen species formation, circulating inflammatory leukocytes and endothelial dysfunction (26), while higher levels of resistin, TNF-α, IL-1β, and MMP-9 expression were associated with the levels of inflammatory infiltrates in artery walls (27).
Beyond the activation of the well-known signaling cascades, the stimulation of EC induces gene expression via microRNAs (miRNA) and epigenetic modifications. The overexpression of miR100 in ECs attenuates leukocyte-endothelial interaction, represses the mammalian target of rapamycin complex 1 signaling, stimulating endothelial autophagy, and attenuates NF-κB signaling. Local miR100 expression is inversely correlated with an inflammatory cell content (28). miR181b inhibits downstream and upstream NF-κB signaling in response to activation (29), while the NF-κB target genes (VCAM-1, ICAM-1, E-selectin, and tissue factor) (30) and miR223 are associated with HUVEC dysfunction (31). Additionally, IFN-α, through miR155, promotes an endothelial dysfunction signature in HUVECs characterized by transcription suppression and the mRNA instability of eNOS and by the upregulation of MCP-1 and VCAM-1 and enhanced neutrophil adhesion (32).
Endothelial microparticles (MPs), shed as a result of the activation of EC are considered a source of important information on the status of ECs and vascular function (33, 34). Circulating levels of endothelial MPs reflect a balance between cell stimulation, proliferation, apoptosis, and cell death (35) and are increased by inflammatory stimuli, mediated by the activation of NF-κB and associated with oxidative stress intensity (36, 37). Endothelial MPs are increased in autoimmune diseases (38) and serve as markers for vascular dysfunction and their effects depend on their cargo and on the surface molecules. Recently, levels of endothelial MPs have been associated with disease activity in SLE patients and CV risk (39).
MPs from RA patients had higher expression of TNF-α on the surface compared to healthy donors (HD), increasing apoptosis and autophagy levels on EC and correlating with clinical RA activity (40).
Platelets and Systemic Inflammation
PLTs have come to be recognized as active players in SI. After activation, PLTs participate in the vasculature inflammation and damage, atherogenesis and thrombosis (41–45). Wide ranges of stimulus are able to activate PLTs. The strong PLT activation was achieved with the ligation of the agonist thrombin, collagen and ADP to the PLTs receptors: protease-activated receptor 1, GPVI and P2Y1 or P2Y12 respectively (46–48). Other non-classical pathways are able to activate PLTs due to the expression of Toll-like receptors (TLR), TNF-α receptor, IL-1β receptors and C-type lectin-like receptors (49–53). Some autoantibodies presents in autoimmune disease patients such as anti-citrullinated protein, anti-β2 glycoprotein I and anti-D4GDI have also the ability to induce PLT activation through FCγRIIa (54–56).
However, PLTs also participate in the resolution of inflammation as anti-inflammatory elements. How PLTs sense the signal to exert pro- or anti-inflammatory functions is not yet fully known. However, it is known that PLTs exert their functions by releasing soluble factors and interacting with cells. The dual role of PLTs in inflammation (57, 58) may be the result of differences in the PLT packing of molecules, activated-dependent release by different stimuli, the kinetics of release and the de novo synthesis of soluble factors and their binding to certain molecules on the surface of leukocytes.
Pro-inflammatory and Anti-inflammatory Soluble Factors Released by PLTs
Some of the released factors of PLTs are synthesized de novo, whereas others are stored and are secreted from granules as pro-thrombotic, immunoregulatory molecules and growth factors immediately after activation. Molecules from dense granule components contribute to hemostasis and coagulation. Molecules from α-granules contain multiple cytokines, mitogens, pro- and anti-inflammatory factors and other bioactive molecules that are essential regulators in the complex microenvironment (Table 1).

Table 1. Evidences of dual role of expressed/secreted platelet factors.
Platelet Factor 4 (PF4, also called CXCL4) is the most abundant protein secreted by activated PLTs and is deposited on endothelium. Higher levels of circulating PF4 have been observed in patients with chronic inflammation (78–80). PF4 increases phagocytosis, respiratory burst, survival and the secretion of inflammatory cytokines in monocytes (59). PF4 blocking reduces the inflammation of vasculature and CV events by reducing leukocyte recruitment and the generation of neutrophil extracellular traps (NETs) by neutrophils (60, 81, 82).
PF4 also acts as an anti-inflammatory factor on T lymphocytes (83), limiting Th17 differentiation by suppressing RORγ expression (62). A lack of PF4 induces the rejection of cardiac transplantation by increasing levels of IL-17 and T cell mediated inflammation. We has observed that PF4 decreases T lymphocyte proliferation and granzyme B expression in CD8+ T lymphocytes (61), explaining how higher levels of PF4 in a malignant context can limit T lymphocyte stimulation (61).
Stimulated PLTs are able to secrete and store IL-1β (63). PLT levels were closely associated with plasmatic IL-1β levels (15). This cytokine activates HUVECs, inducing neutrophil adhesion and endothelium damage (63). The co-culture of PLTs from SLE with HUVECs increased EC damage and inflammatory marker expression in an IL-1β dependent manner (64). In experimental models of inflammation, IL-1α secreted by activated PLTs also played a crucial role in SI (84–86).
The soluble P-selectin secreted from α-granules is also implicated in inflammatory responses. P-selectin from activated PLTs induces the release of 3-10 folds of inflammatory cytokines by monocytes (65) and also promotes NETs formation (66). Elevated levels of circulating soluble P-selectin may contribute to early vascular disease by promoting the adhesion of leukocytes to the endothelium (87). However, soluble P-selectin can prevent in vitro adhesion of neutrophils to activated endothelium (67).
Circulating soluble CD40L (sCD40L) is secreted mainly by activated PLTs. In human immunodeficiency virus (HIV) patients, sCD40L levels correlated with pro-inflammatory cytokines (68). Moreover, PLTs support B cell isotype switching through CD40L-CD40 binding (70). In patients with an increased CV risk plasmatic sCD40L was increased and correlated with disease activity and with pro-inflammatory cytokines (69). The addition of thrombin-activated PLTs to TLR-stimulated monocytes has been seen to reduce TNF-α and IL-6 secretion and induce IL-10 production, and were abolished by blocking sCD40L (71). In HIV, sCD40L levels were correlated with IDO enzymatic activity and Treg frequency, in addition to induced Treg expansion and differentiation (68).
TGF-β is a potent anti-inflammatory factor, produced mainly by PLTs, which suppresses T lymphocytes function and is involved in Treg differentiation. The culture of CD4+ T lymphocytes with PLTs enhances Th1 and Th17 cytokine production but the TGF-β secreted by PLTs activates Treg suppressing Th1 and Th17 response (72). PLTs inhibit CD4+ and CD8+IFN-γ production, proliferation and granzyme B and perforin expression in a TGF-β dependent manner (73).
PLTs also release the damage-associated molecular pattern molecule high-mobility group box 1 (HMGB1) contributing to thrombosis process (87) promoting monocytes migration, suppressing monocyte apoptosis via TLR4-ligation and the monocyte accumulation at the site of vascular thrombosis (74, 75) and promote NETs formation (76, 77).
Binding of PLTs to Leukocytes and Endothelial Cells
The interaction of PLTs with leukocytes and EC contributes to SI by favoring the arrest of leukocytes on endothelium, the production of inflammatory cytokines and NETs formation. Under certain circumstances, the binding of PLTs to leukocytes decreases the inflammatory response, participating in the resolution of thrombo-inflammation (Table 2).
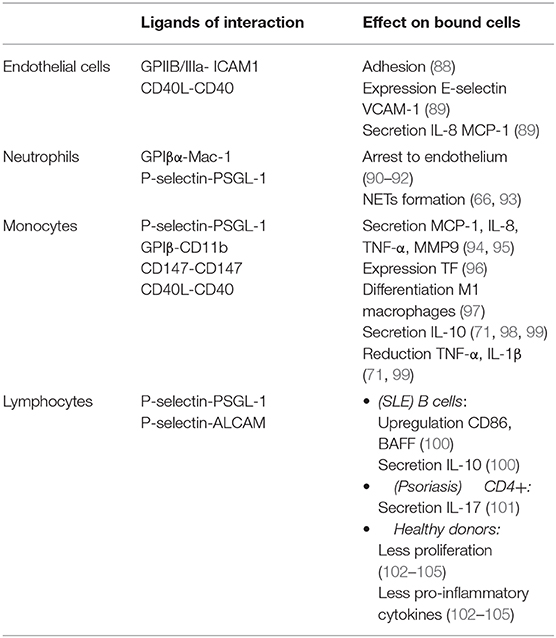
Table 2. Effects of PLT binding to other cells.
Under inflammatory stress, PLTs have a firm adhesion to endothelium via GPIIB/IIIa-ICAM-1 in a fibrinogen dependent manner (88). CD40L expressed by PLTs induces the expression of E-selectin, VCAM-1 and ICAM-1 on endothelium and the secretion of IL-8 and MCP-1 (89), increasing the recruitment of leukocytes.
Although the P-selectin-PSGL-1 (P-selectin glycoprotein 1) axis is essential to the binding of PLTs to leukocytes, other pathways are involved in this process: P-selectin-ALCAM, GPIβα-Mac1, CD40L-CD40, P-selectin-CD15, JAM-C-Mac1, TREM1L-TREM1, CD36-trombospondin-CD36, and CD147 pathway (94, 106, 107).
The interaction of PLTs with neutrophils through GPIβα-Mac-1 and P-selectin-PSGL-1 is crucial for the development of thrombo-inflammation and vascular damage by arresting neutrophils to endothelium and the induction of NETs formation (66, 90–93). The neutrophil-platelet aggregates can also be seen in tissues in acute coronary syndrome and the skin of psoriatic patients (108, 109). Stimulated TLR4 PLTs, through the binding with neutrophils, induced robust neutrophil activation and formation of NETs (93). Induced NETs formation by PLTs was abolished blocking their binding with neutrophils (110). The increase of neutrophil-platelet aggregates in the circulation of autoimmune disease patients correlates with neutrophil activation (111) and vascular abnormalities (112) which was abolished with intravenous immunoglobulins plus prednisolone treatment (112). However, even all the current evidences supports that interaction of PLTs with neutrophils are involved in inflammatory process and vasculature damage, this interaction could have also anti-inflammatory consequences depending on the neutrophil status. The addition of PLTs to previously stimulated TLR-neutrophils downregulates degranulation markers expression and the secretion of elastase (113).
Although monocyte-PLT aggregates are increased in CVD (114), the interaction of PLTs with monocytes have pro- or anti-inflammatory consequences depending on the experimental assay and the activation status of monocytes. As pro-inflammatory consequence, thrombin-activated PLTs through P-selectin-PSGL-1 binding induces the expression of MCP-1 and IL-8 in resting monocytes (95). P-selectin expressed on the surface of PLTs induced a rapid tissue factor expression by monocytes (96). The binding of PLTs to monocytes through GPIβ-CD11b induces a M1 macrophage phenotype that produce TNF-α (97). Via CD147 axis, RA patients had more circulating intermediate monocytes-PLTs aggregates, increasing the TNF-α and MMP-9 secretion (94). Autoimmune patients with a higher CV risk have more monocyte-PLT aggregates and its associated with the activated state of monocytes (115). As anti-inflammatory consequences, it has been observed that the binding of PLTs to activated monocytes induces IL-10 production and decreases TNF-α and IL-1β production, and were abolished by the blocking of P-selectin-PSGL-1, CD40L-CD40 axis and Ca2+ chelator (71, 98, 99).
Although the binding of PLTs to lymphocytes has preferably anti-inflammatory consequences, it was demonstrated a contribution in inflammatory process. High levels of lymphocytes-PLTs aggregates were observed in SLE and psoriasis. In SLE, lymphocytes-PLTs aggregates had an up-regulation of CD86, B cell activation factor receptor and IL-10 production and correlated positively with plasmatic levels of IgG, IgA, IL-10, sCD40L and renal manifestation, and correlated negatively with IgM levels (100). In psoriasis, the IL-17+ CD4+ had higher levels of bound PLTs and anti-TNF-α drugs normalize the numbers (101), while in HIV there are more lymphocytes-PLTs aggregates and are associated to D-dimer levels, increasing the CV risk (116).
We observed that CD4+ T lymphocytes-PLT aggregates had a reduced proliferation and production of pro-inflammatory cytokines. A less severe phenotype and a decreased CV risk was observed in RA patients with higher levels of circulating CD4+T lymphocytes-PLTs aggregates (102). The addition of PLTs to lymphocytes from RA synovial fluids decreased their proliferation and the secretion of IFN-γ, IL-17, and increased IL-10 production (103). PLTs MPs cultured with Tregs prevented the differentiation into IL-17– and IFN-γ-producing cells in a P-selectin dependent manner (104). The co-cultures of CD4+ CD25- T cells with PLTs induced Tregs and this effect was abolished by the blocking of glycoprotein A repetitions predominant (105). In HIV, the aberrant function of CD8+ T lymphocytes was abolished when these cells were co-cultured with PLTs from HD, implying that direct contact with PLTs and TGF-β secretion contributed to this functional improvement (117). In later stages of experimental autoimmune encephalomyelitis (EAE), there was an increase CD4+ T lymphocytes-PLTs aggregates through the interaction of P-selectin-ALCAM, down-regulating their activation, proliferation and the production of IFN-γ, crucial for the spontaneous resolution of EAE. The blocking of CD4+-PLT aggregates exacerbate EAE (118).
Platelet to Lymphocyte Ratio in Systemic Inflammation
The platelet to lymphocyte ratio (PLR) has emerged as a reliable marker of SI. Although elevated counts of PLTs and low counts of lymphocytes per se has been associated with worse prognosis of CVD, increase CV mortality and morbidity and SI (119, 120), PLR predicts better the outcomes of CVD. The role of PLR as an independent marker in CVD and SI has been extensively reviewed (121, 122). In patients with chronic inflammatory diseases, PLR is elevated and it correlates with markers of SI (122–125).
However, there are confounding factors that alter PLR. Sex and ethnic origin also modulates PLR (126) as well as drugs that affect blood cell maturation in the bone marrow (127, 128). Other confounding factors of PLR may be the technical limitations of PLR measurements, such as EDTA dependent agglutination (129, 130).
Conclusions
PLTs have been considered to play a pro-inflammatory role in SI. However, their binding to leukocytes and EC and the secretion of immunomodulatory molecules during activation also have anti-inflammatory consequences. Different effects were observed with platelets from healthy donors or chronic inflammatory patients. In addition, the binding to each subpopulation of leukocytes has distinctive consequences. Further research is necessary to reveal how platelets exert their dual function.
Author Contributions
CZ and EC: literature review, manuscript preparation. SV: oversight, editing, and planning. All authors contributed to the article and approved the submitted version.
Funding
This study is supported by Instituto de Salud Carlos III and Fondos FEDER (PI17/00072 and PI20/00184). SV was supported by Fondo Investigaciones Sanitarias and a participant in the Program for Stabilization of Investigators the Direcció i d'Estrategia i Coordinació del Departament Salut de la Generalitat de Catalunya.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Akbar N, Nanda S, Belch J, Cohen P, Khan F. An important role for A20-binding inhibitor of nuclear factor-kB-1 (ABIN1) in inflammation-mediated endothelial dysfunction: an in vivo study in ABIN1 (D485N) mice. Arthritis Res Ther. (2015) 17:1–10. doi: 10.1186/s13075-015-0543-3
2. Spinelli FR, Barbati C, Cecarelli F, Morello F, Colasanti T, Vomero M, et al. B lymphocyte stimulator modulates number and function of endothelial progenitor cells in systemic lupus erythematosus. Arthritis Res Ther. (2019) 21:1–11. doi: 10.1186/s13075-019-2015-7
3. Khawaja AA, Pericleous C, Ripoll VM, Porter JC, Giles IP. Autoimmune rheumatic disease IgG has differential effects upon neutrophil integrin activation that is modulated by the endothelium. Sci Rep. (2019) 9:1–13. doi: 10.1038/s41598-018-37852-5
4. Bugała K, Mazurek A, Gryga K, Komar M, Kopeć G, Musiał J, et al. Influence of autoimmunity and inflammation on endothelial function and thrombosis in systemic lupus erythematosus patients. Clin Rheumatol. (2018) 37:2087–93. doi: 10.1007/s10067-018-4104-4
5. Totoson P, Maguin-Gaté K, Nappey M, Wendling D, Demougeot C. Endothelial dysfunction in rheumatoid arthritis: mechanistic insights and correlation with circulating markers of SI. PLoS ONE. (2016) 11:e146744. doi: 10.1371/journal.pone.0146744
6. Walther G, Obert P, Dutheil F, Chapier R, Lesourd B, Naughton G, et al. Metabolic syndrome individuals with and without type 2 diabetes mellitus present generalized vascular dysfunction: cross-sectional study. Arterioscler Thromb Vasc Biol. (2015) 35:1022–9. doi: 10.1161/ATVBAHA.114.304591
7. Cox LAE, Van Eijk LT, Ramakers BPC, Dorresteijn MJ, Gerretsen J, Kox M, et al. Inflammation-induced increases in plasma endocan levels are associated with endothelial dysfunction in humans in vivo. Shock. (2015) 43:322–6. doi: 10.1097/SHK.0000000000000320
8. Mikacenic C, Hahn WO, Price BL, Harju-Baker S, Katz R, Kain KC, et al. Biomarkers of endothelial activation are associated with poor outcome in critical illness. PLoS One. (2015) 10:e141251. doi: 10.1371/journal.pone.0141251
9. Benhamou Y, Miranda S, Armengol G, Harouki N, Drouot L, Zahr N, et al. Infliximab improves endothelial dysfunction in a mouse model of antiphospholipid syndrome: Role of reduced oxidative stress. Vascul Pharmacol. (2015) 71:93–101. doi: 10.1016/j.vph.2015.03.014
10. Genre F, López-Mejías R, Miranda-Filloy JA, Ubilla B, Mijares V, Carnero-López B, et al. Anti-TNF-α therapy reduces endothelial cell activation in non-diabetic ankylosing spondylitis patients. Rheumatol Int. (2015) 35:2069–78. doi: 10.1007/s00296-015-3314-1
11. Pérez-Sánchez C, Cecchi I, Barbarroja N, Patiño-Trives AM, Luque-Tévar M, Pérez-Sánchez L, et al. Early restoration of immune and vascular phenotypes in systemic lupus erythematosus and rheumatoid arthritis patients after B cell depletion. J Cell Mol Med. (2019) 23:6308–18. doi: 10.1111/jcmm.14517
12. Barbati C, Colasanti T, Vomero M, Ceccarelli F, Celia AI, Perricone C, et al. Up-regulation of autophagy by etanercept treatment results in TNF-induced apoptosis reduction in EA.hy926 endothelial cell line. Clin Exp Rheumatol. (in press).
13. Thacker SG, Abdalrahman Z, Sciumè G, Tsai WL, Anna M. Tofacitinib ameliorates murine lupus and its associated vascular. Arthritis Rheumatol. (2017) 69:148–160. doi: 10.1002/art.39818
14. Verhoeven F, Totoson P, Maguin-Gaté K, Prigent-Tessier A, Marie C, Wendling D, et al. Glucocorticoids improve endothelial function in rheumatoid arthritis: a study in rats with adjuvant-induced arthritis. Clin Exp Immunol. (2017) 188:208–18. doi: 10.1111/cei.12938
15. Tunjungputri RN, Li Y, De Groot PG, Dinarello CA, Smeekens SP, Jaeger M, et al. The inter-relationship of platelets with interleukin-1β-mediated inflammation in humans. Thromb Haemost. (2018) 118:2112–25. doi: 10.1055/s-0038-1675603
16. Granja T, Körner A, Glück C, Hohmann JD, Wang X, Köhler D, et al. Targeting CD39 toward activated platelets reduces SI and improves survival in sepsis: a preclinical pilot study. Crit Care Med. (2019) 47:e420–e7. doi: 10.1097/CCM.0000000000003682
17. Cheng Z, Qi R, Li L, Liu Q, Zhang W, Zhou X, et al. Dihydroartemisinin ameliorates sepsis-induced hyperpermeability of glomerular endothelium via up-regulation of occludin expression. Biomed Pharmacother. (2018) 99:313–8. doi: 10.1016/j.biopha.2018.01.078
18. Toguri JT, Moxsom R, Szczesniak AM, Zhou J, Kelly MEM, Lehmann C. Cannabinoid 2 receptor activation reduces leukocyte adhesion and improves capillary perfusion in the iridial microvasculature during SI. Clin Hemorheol Microcirc. (2015) 61:237–49. doi: 10.3233/CH-151996
19. Li S, Xu J, Yao W, Li H, Liu Q, Xiao F, et al. Sevoflurane pretreatment attenuates TNF-α-induced human endothelial cell dysfunction through activating eNOS/NO pathway. Biochem Biophys Res Commun. (2015) 460:879–86. doi: 10.1016/j.bbrc.2015.03.126
20. Malik M, Suboc TM, Tyagi S, Salzman N, Wang J, Ying R, et al. Lactobacillus plantarum 299v supplementation improves vascular endothelial function and reduces inflammatory biomarkers in men with stable coronary artery disease. Circ Res. (2018) 123:1091–102. doi: 10.1161/CIRCRESAHA.118.313565
21. Reynolds JA, Haque S, Williamson K, Ray DW, Alexander MY, Bruce IN. Vitamin D improves endothelial dysfunction and restores myeloid angiogenic cell function via reduced CXCL-10 expression in systemic lupus erythematosus. Sci Rep. (2016) 6:1–11. doi: 10.1038/srep22341
22. Zhang H, Park Y, Wu J, Chen XP, Lee S, Yang J, et al. Role of TNF-α in vascular dysfunction. Clin Sci. (2009) 116:219–30. doi: 10.1042/CS20080196
23. Pober JS. Endothelial activation: intracellular signaling pathways. Arthritis Res. (2002) 4:S109–S16. doi: 10.1186/ar576
24. Jimenez JJ, Jy W, Mauro LM, Soderland C, Horstman LL, Ahn YS. Endothelial cells release phenotypically and quantitatively distinct microparticles in activation and apoptosis. Thromb Res. (2003) 109:175–80. doi: 10.1016/S0049-3848(03)00064-1
25. Marques P, Collado A, Martinez-Hervás S, Domingo E, Benito E, Piqueras L, et al. SI in metabolic syndrome: increased platelet and leukocyte activation, and key role of CX3CL1/CX3CR1 and CCL2/CCR2 axes in arterial platelet-proinflammatory monocyte adhesion. J Clin Med. (2019) 8:708. doi: 10.3390/jcm8050708
26. Karbach S, Croxford AL, Oelze M, Schüler R, Minwegen D, Wegner J, et al. Interleukin 17 drives vascular inflammation, endothelial dysfunction, and arterial hypertension in psoriasis-like skin disease. Arterioscler Thromb Vasc Biol. (2014) 34:2658–68. doi: 10.1161/ATVBAHA.114.304108
27. Gao F, Si F, Feng S, Yi Q, Liu R. Resistin enhances inflammatory cytokine production in coronary artery tissues by activating the NF-κB signaling. Biomed Res Int. (2016) 2016:3296437. doi: 10.1155/2016/3296437
28. Pankratz F, Hohnloser C, Bemtgen X, Jaenich C, Kreuzaler S, Hoefer I, et al. MicroRNA-100 suppresses chronic vascular inflammation by stimulation of endothelial autophagy. Circ Res. (2018) 122:417–32. doi: 10.1161/CIRCRESAHA.117.311428
29. Sun X, Baron RM, Feinberg MW, Sun X, Icli B, Wara AK, et al. MicroRNA-181b regulates NF- k B – mediated vascular inflammation Find the latest version : MicroRNA-181b regulates NF- κ B – mediated vascular inflammation. J Clin Invest. (2012) 122:1973–90. doi: 10.1172/JCI61495
30. Lin J, He S, Sun X, Franck G, Deng Y, Yang D, et al. MicroRNA-181b inhibits thrombin-mediated endothelial activation and arterial thrombosis by targeting caspase recruitment domain family member 10. FASEB J. (2016) 30:3216–26. doi: 10.1096/fj.201500163R
31. Cavallari C, Dellepiane S, Fonsato V, Medica D, Marengo M, Migliori M, et al. Online hemodiafiltration inhibits inflammation-related endothelial dysfunction and vascular calcification of uremic patients modulating miR-223 expression in plasma extracellular vesicles. J Immunol. (2019) 202:2372–83. doi: 10.4049/jimmunol.1800747
32. Buie JJ, Renaud LL, Muise-Helmericks R, Oates JC. IFN-α negatively regulates the expression of endothelial nitric oxide synthase and nitric oxide production: implications for systemic lupus erythematosus. J Immunol. (2017) 199:1979–88. doi: 10.4049/jimmunol.1600108
33. Boulanger CM, Leroyer AS, Amabile N, Tedgui A. Circulating endothelial microparticles: A new marker of vascular injury. Ann Cardiol Angeiol (Paris). (2008) 57:149–54. doi: 10.1016/j.ancard.2008.02.016
34. Boyle LJ, Credeur DP, Jenkins NT, Padilla J, Leidy HJ, Thyfault JP, et al. Impact of reduced daily physical activity on conduit artery flow-mediated dilation and circulating endothelial microparticles. J Appl Physiol. (2013) 115:1519–25. doi: 10.1152/japplphysiol.00837.2013
35. Spencer DM, Mobarrez F, Wallén H, Pisetsky DS. The expression of HMGB1 on microparticles from jurkat and HL-60 cells undergoing apoptosis in vitro. Scand J Immunol. (2014) 80:101–10. doi: 10.1111/sji.12191
36. Winner M, Koong AC, Rendon BE, Zundel W, Mitchell RA. Amplification of tumor hypoxic responses by macrophage migration inhibitory factor-dependent hypoxia-inducible factor stabilization. Cancer Res. (2007) 67:186–93. doi: 10.1158/0008-5472.CAN-06-3292
37. Mezentsev A, Merks RMH, O'Riordan E, Chen J, Mendelev N, Goligorsky MS, et al. Endothelial microparticles affect angiogenesis in vitro: role of oxidative stress. Am J Physiol - Hear Circ Physiol. (2005) 289:1106–14. doi: 10.1152/ajpheart.00265.2005
38. Wu YJJ, Hua CC, Chen JY, Chang YW, Tseng JC. The role of endothelial microparticles in autoimmune disease patients with Raynaud's phenomenon. J Microbiol Immunol Infect. (2017) 50:857–62. doi: 10.1016/j.jmii.2015.12.010
39. Tydén H, Lood C, Gullstrand B, Nielsen CT, Heegaard NHH, Kahn R, et al. Endothelial dysfunction is associated with activation of the type i interferon system and platelets in patients with systemic lupus erythematosus. RMD Open. (2017) 3:508. doi: 10.1136/rmdopen-2017-000508
40. Barbati C, Vomero M, Colasanti T, Diociaiuti M, Ceccarelli F, Ferrigno S, et al. TNFα expressed on the surface of microparticles modulates endothelial cell fate in rheumatoid arthritis. Arthritis Res Ther. (2018) 20:1–12. doi: 10.1186/s13075-018-1768-8
41. Smith TL, Weyrich AS. Platelets as central mediators of systemic inflammatory responses. Thromb Res. (2011) 127:391–4. doi: 10.1016/j.thromres.2010.10.013
42. Koupenova M, Clancy L, Corkrey HA, Freedman JE. Circulating platelets as mediators of immunity, inflammation, and thrombosis. Circ Res. (2018) 122:337–51. doi: 10.1161/CIRCRESAHA.117.310795
43. Langer HF, Gawaz M. Platelet-vessel wall interactions in atherosclerotic disease. Thromb Haemost. (2008) 99:480–6. doi: 10.1160/TH07-11-0685
44. Smyth SS, Mcever RP, Weyrich AS, Morrell CN, Hoffman MR, Arepally GM, et al. Platelet functions beyond hemostasis. J Thromb Haemost. (2009) 7:1759–66. doi: 10.1111/j.1538-7836.2009.03586.x
45. Page CP. Platelets as inflammatory cells. Immunopharmacology. (1989) 17:51–9. doi: 10.1016/0162-3109(89)90008-8
46. Murugappan S, Kunapuli SP. The role of ADP receptors in platelet function. Front Biosci. (2006) 11:1977–86. doi: 10.2741/1939
47. Surin WR, Barthwal MK, Dikshit M. Platelet collagen receptors, signaling and antagonism: Emerging approaches for the prevention of intravascular thrombosis. Thromb Res. (2008) 122:786–803. doi: 10.1016/j.thromres.2007.10.005
48. Ofosu FA, Nyarko KA. Human platelet thrombin receptors: Roles in platelet activation. Hematol Oncol Clin North Am. (2000) 14:1185–98. doi: 10.1016/S0889-8588(05)70178-7
49. May F, Hagedorn I, Pleines I, Bender M, Vögtle T, Eble J, et al. CLEC-2 is an essential platelet-activating receptor in hemostasis and thrombosis. Blood. (2009) 114:3464–72. doi: 10.1182/blood-2009-05-222273
50. Xia L. Platelet CLEC-2: A molecule with 2 faces. Blood. (2017) 130:2158–60. doi: 10.1182/blood-2017-09-804088
51. Brown GT, Narayanan P, Li W, Silverstein RL, McIntyre TM. Lipopolysaccharide stimulates platelets through an IL-1β autocrine loop. J Immunol. (2013) 191:5196–203. doi: 10.4049/jimmunol.1300354
52. Pignatelli P, De Biase L, Lenti L, Tocci G, Brunelli A, Cangemi R, et al. Tumor necrosis factor-α as trigger of platelet activation in patients with heart failure. Blood. (2005) 106:1992–4. doi: 10.1182/blood-2005-03-1247
53. Hally K, Fauteux-Daniel S, Hamzeh-Cognasse H, Larsen P, Cognasse F. Revisiting platelets and toll-like receptors (TLRS): at the interface of vascular immunity and thrombosis. Int J Mol Sci. (2020) 21:1–28. doi: 10.3390/ijms21176150
54. Barbati C, Stefanini L, Colasanti T, Cipriano E, Celia A, Gabriele G, et al. Anti-D4GDI antibodies activate platelets in vitro: a possible link with thrombocytopenia in primary antiphospholipid syndrome. Arthritis Res Ther. (2019) 21:2. doi: 10.1186/s13075-019-1947-2
55. Hollerbach A, Müller-Calleja N, Ritter S, Häuser F, Canisius A, Orning C, et al. Platelet Activation by Antiphospholipid Antibodies Depends on Epitope Specificity and is Prevented by mTOR Inhibitors. Thromb Haemost. (2019) 119:1147–53. doi: 10.1055/s-0039-1685453
56. Habets KLL, Trouw LA, Levarht EWN, Korporaal SJA, Habets PAM, de Groot P, et al. Anti-citrullinated protein antibodies contribute to platelet activation in rheumatoid arthritis. Arthritis Res Ther. (2015) 17: doi: 10.1186/s13075-015-0665-7
57. Heijnen H, van der Sluijs P. Platelet secretory behaviour: As diverse as the granules or not? J Thromb Haemost. (2015) 13:2141–51. doi: 10.1111/jth.13147
58. Chatterjee M, Huang Z, Zhang W, Jiang L, Hultenby K, Zhu L, et al. Distinct platelet packaging, release, and surface expression of proangiogenic and antiangiogenic factors on different platelet stimuli. Blood. (2011) 117:3907–11. doi: 10.1182/blood-2010-12-327007
59. Kasper B, Brandt E, Brandau S, Petersen F. Platelet factor 4 (CXC chemokine ligand 4) differentially regulates respiratory burst, survival, and cytokine expression of human monocytes by using distinct signaling pathways. J Immunol. (2007) 179:2584–91. doi: 10.4049/jimmunol.179.4.2584
60. Vajen T, Koenen RR, Werner I, Staudt M, Projahn D, Curaj A, et al. Blocking CCL5-CXCL4 heteromerization preserves heart function after myocardial infarction by attenuating leukocyte recruitment and NETosis. Sci Rep. (2018) 8:1–11. doi: 10.1038/s41598-018-29026-0
61. Mulet M, Zamora C, Porcel JM, Nieto JC, Pajares V, Muñoz-Fernandez AM, et al. Platelet factor 4 regulates T cell effector functions in malignant pleural effusions. Cancer Lett. (2020) 491:78–86. doi: 10.1016/j.canlet.2020.06.014
62. Shi G, Field DJ, Ko KA, Ture S, Srivastava K, Levy S, et al. Platelet factor 4 limits Th17 differentiation and cardiac allograft rejection. J Clin Invest. (2014) 124:543–52. doi: 10.1172/JCI71858
63. Lindemann S, Tolley ND, Dixon DA, McIntyre TM, Prescott SM, Zimmerman GA, et al. Activated platelets mediate inflammatory signaling by regulated interleukin 1β synthesis. J Cell Biol. (2001) 154:485–90. doi: 10.1083/jcb.200105058
64. Nhek S, Clancy R, Lee KA, Allen NM, Barrett TJ, Marcantoni E, et al. Activated platelets induce endothelial cell activation via an interleukin-1β pathway in systemic lupus erythematosus. Arterioscler Thromb Vasc Biol. (2017) 37:707–16. doi: 10.1161/ATVBAHA.116.308126
65. Suzuki J, Hamada E, Shodai T, Kamoshida G, Kudo S, Itoh S, et al. Cytokine secretion from human monocytes potentiated by P-selectin-mediated cell adhesion. Int Arch Allergy Immunol. (2013) 160:152–60. doi: 10.1159/000339857
66. Etulain J, Martinod K, Wong SL, Cifuni SM, Schattner M, Wagner DD. P-selectin promotes neutrophil extracellular trap formation in mice. Blood. (2015) 126:242–6. doi: 10.1182/blood-2015-01-624023
67. Gamble JR, Skinner MP, Berndt MC, Vadas MA. Prevention of activated neutrophil adhesion to endothelium by soluble adhesion protein GMP140. Science. (1990) 249:414–7. doi: 10.1126/science.1696029
68. Jenabian MA, Patel M, Kema I, Vyboh K, Kanagaratham C, Radzioch D, et al. Soluble CD40-ligand (sCD40L, sCD154) plays an immunosuppressive role via regulatory T cell expansion in HIV infection. Clin Exp Immunol. (2014) 178:102–11. doi: 10.1111/cei.12396
69. Goules A, Tzioufas AG, Manousakis MN, Kirou KA, Crow MK, Routsias JG. Elevated levels of soluble CD40 ligand (sCD40L) in serum of patients with systemic autoimmune diseases. J Autoimmun. (2006) 26:165–71. doi: 10.1016/j.jaut.2006.02.002
70. Cognasse F, Hamzeh-Cognasse H, Lafarge S, Chavarin P, Cogné M, Richard Y, et al. Human platelets can activate peripheral blood B cells and increase production of immunoglobulins. Exp Hematol. (2007) 35:1376–87. doi: 10.1016/j.exphem.2007.05.021
71. Gudbrandsdottir S, Hasselbalch HC, Nielsen CH. Activated platelets enhance IL-10 secretion and reduce TNF-α secretion by monocytes. J Immunol. (2013) 191:4059–67. doi: 10.4049/jimmunol.1201103
72. Zhu L, Huang Z, Stålesen R, Hansson GK, Li N. Platelets provoke distinct dynamics of immune responses by differentially regulating CD4 + T-cell proliferation. J Thromb Haemost. (2014) 12:1156–65. doi: 10.1111/jth.12612
73. Rachidi S, Metelli A, Riesenberg B, Wu BX, Nelson MH, Wallace C, et al. Platelets subvert T cell immunity against cancer via GARP-TGF axis. Sci Immunol. (2017) 2:7911. doi: 10.1126/sciimmunol.aai7911
74. Vogel S, Rath D, Borst O, Mack A, Loughran P, Lotze MT, et al. Platelet-derived high-mobility group box 1 promotes recruitment and suppresses apoptosis of monocytes. Biochem Biophys Res Commun. (2016) 478:143–8. doi: 10.1016/j.bbrc.2016.07.078
75. Vogel S, Bodenstein R, Chen Q, Feil S, Feil R, Rheinlaender J, et al. Platelet-derived HMGB1 is a critical mediator of thrombosis. J Clin Invest. (2015) 125:4638–54. doi: 10.1172/JCI81660
76. Kim SW, Lee JK. Role of HMGB1 in the interplay between NETosis and thrombosis in ischemic stroke: a review. Cells. (2020) 9:1794. doi: 10.3390/cells9081794
77. Dyer MR, Chen Q, Haldeman S, Yazdani H, Hoffman R, Loughran P, et al. Deep vein thrombosis in mice is regulated by platelet HMGB1 through release of neutrophil-extracellular traps and DNA. Sci Rep. (2018) 8:2068. doi: 10.1038/s41598-018-20479-x
78. Vettori S, Irace R, Riccardi A, Iacono D, Pellecchia L, Vicedomini L, et al. Serum CXCL4 increase in primary Sjögren's syndrome characterizes patients with microvascular involvement and reduced salivary gland infiltration and lymph node involvement. Clin Rheumatol. (2016) 35:2591–6. doi: 10.1007/s10067-016-3386-7
79. Ye L, Zhang YP, Yu N, Jia YX, Wan SJ, Wang FY. Serum platelet factor 4 is a reliable activity parameter in adult patients with inflammatory bowel disease. Medince. (2017) 96:6323. doi: 10.1097/MD.0000000000006323
80. Patsouras MD, Sikara MP, Grika EP, Moutsopoulos HM, Tzioufas AG, Vlachoyiannopoulos PG. Elevated expression of platelet-derived chemokines in patients with antiphospholipid syndrome. J Autoimmun. (2015) 65:30–7. doi: 10.1016/j.jaut.2015.08.001
81. Rossaint J, Herter JM, Van Aken H, Napirei M, Döring Y, Weber C, et al. Synchronized integrin engagement and chemokine activation is crucial in neutrophil extracellular trap-mediated sterile inflammation. Blood. (2014) 123:2573–84. doi: 10.1182/blood-2013-07-516484
82. Koenen RR, Von Hundelshausen P, Nesmelova IV, Zernecke A, Liehn EA, Sarabi A, et al. Disrupting functional interactions between platelet chemokines inhibits atherosclerosis in hyperlipidemic mice. Nat Med. (2009) 15:97–103. doi: 10.1038/nm.1898
83. Liu CY, Battaglia M, Lee SH, Sun Q-H, Aster RH, Visentin GP. Platelet factor 4 differentially modulates CD4 + CD25 + (Regulatory) versus CD4 + CD25 – (Nonregulatory) T Cells. J Immunol. (2005) 174:2680–6. doi: 10.4049/jimmunol.174.5.2680
84. Burzynski LC, Humphry M, Pyrillou K, Wiggins KA, Chan JNE, Figg N, et al. The coagulation and immune systems are directly linked through the activation of interleukin-1α by thrombin. Immunity. (2019) 50:1033–42.e6. doi: 10.1016/j.immuni.2019.03.003
85. Thornton P, McColl BW, Greenhalgh A, Denes A, Allan SM, Rothwell NJ. Platelet interleukin-1α drives cerebrovascular inflammation. Blood. (2010) 115:3632–9. doi: 10.1182/blood-2009-11-252643
86. Boilard E, Nigrovic PA, Larabee K, Watts GFM, Coblyn JS, Weinblatt ME, et al. Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science. (2010) 327:580–3. doi: 10.1126/science.1181928
87. Woollard KJ, Suhartoyo A, Harris EE, Eisenhardt SU, Jackson SP, Peter K, et al. Pathophysiological levels of soluble P-selectin mediate adhesion of leukocytes to the endothelium through mac-1 activation. Circ Res. (2008) 103:1128–38. doi: 10.1161/CIRCRESAHA.108.180273
88. Khlgatian M, Nassar H, Chou HH, Gibson FC, Genco CA. Fimbria-dependent activation of cell adhesion molecule expression in Porphyromonas gingivalis-infected endothelial cells. Infect Immun. (2002) 70:257–67. doi: 10.1128/IAI.70.1.257-267.2002
89. Henn V, Slupsky JR, Gräfe M, Anagnostopoulos I, Förster R, Müller-Berghaus G, et al. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature. (1998) 391:591–4. doi: 10.1038/35393
90. Hidalgo A, Chang J, Jang JE, Peired AJ, Chiang EY, Frenette PS. Heterotypic interactions enabled by polarized neutrophil microdomains mediate thromboinflammatory injury. Nat Med. (2009) 15:384–91. doi: 10.1038/nm.1939
91. Kim K, Li J, Tseng A, Andrews RK, Cho J. NOX2 is critical for heterotypic neutrophil-platelet interactions during vascular inflammation. Blood. (2015) 126:1952–64. doi: 10.1182/blood-2014-10-605261
92. von Brühl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. (2012) 209:819–35. doi: 10.1084/jem.20112322
93. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. (2007) 13:463–9. doi: 10.1038/nm1565
94. Rong MY, Wang CH, Wu ZB, Zeng W, Zheng ZH, Han Q, et al. Platelets induce a proinflammatory phenotype in monocytes via the CD147 pathway in rheumatoid arthritis. Arthritis Res Ther. (2014) 16:478. doi: 10.1186/s13075-014-0478-0
95. Weyrich AS, Elstad MR, McEver RP, McIntyre TM, Moore KL, Morrissey JH, et al. Activated platelets signal chemokine synthesis by human monocytes. J Clin Invest. (1996) 97:1525–34. doi: 10.1172/JCI118575
96. Ivanov II, Apta BHR, Bonna AM, Harper MT. Platelet P-selectin triggers rapid surface exposure of tissue factor in monocytes. Sci Rep. (2019) 9:13397. doi: 10.1038/s41598-019-49635-7
97. Carestia A, Mena HA, Olexen CM, Ortiz Wilczyñski JM, Negrotto S, Errasti AE, et al. Platelets promote macrophage polarization toward pro-inflammatory phenotype and increase survival of septic mice. Cell Rep. (2019) 28:896–908.e5. doi: 10.1016/j.celrep.2019.06.062
98. Takeda Y, Marumo M, Nara H, Feng ZG, Asao H, Wakabayashi I. Selective induction of anti-inflammatory monocyte-platelet aggregates in a model of pulsatile blood flow at low shear rates. Platelets. (2016) 27:583–92. doi: 10.3109/09537104.2016.1153616
99. Zamora C, Canto E, Nieto JC, Garcia-Planella E, Gordillo J, Ortiz MA, et al. Inverse association between circulating monocyte-platelet complexes and inflammation in ulcerative colitis patients. Inflamm Bowel Dis. (2018) 24:818–28. doi: 10.1093/ibd/izx106
100. Zamora C, Toniolo E, Diaz-Torné C, Cantó E, Magallares B, Ortiz MA, et al. Association of platelet binding to lymphocytes with B cell abnormalities and clinical manifestations in systemic lupus erythematosus. Mediators Inflamm. (2019) 2019:2473164. doi: 10.1155/2019/2473164
101. Sanz-Martínez MT, Moga E, Sánchez Martínez MA, Zamora Atenza C, Vidal S, Juárez C, et al. High levels of platelet-lymphocyte complexes in patients with psoriasis are associated with a better response to anti–TNF-α therapy. J Invest Dermatol. (2020) 140:1176–83. doi: 10.1016/j.jid.2019.08.457
102. Zamora C, Cantó E, Nieto JC, Ortiz MA, Diaz-Torné C, Diaz-Lopez C, et al. Functional consequences of platelet binding to T lymphocytes in inflammation. J Leukoc Biol. (2013) 94:521–9. doi: 10.1189/jlb.0213074
103. Zamora C, Cantó E, Nieto JC, Bardina J, Diaz-Torné C, Moya P, et al. Binding of platelets to lymphocytes: a potential anti-inflammatory therapy in rheumatoid arthritis. J Immunol. (2017) 198:3099–108. doi: 10.4049/jimmunol.1601708
104. Dinkla S, Van Cranenbroek B, Van Der Heijden WA, He X, Wallbrecher R, Dumitriu IE, et al. Platelet microparticles inhibit IL-17 production by regulatory T cells through P-selectin. Blood. (2016) 127:1976–86. doi: 10.1182/blood-2015-04-640300
105. Zimmer N, Krebs FK, Zimmer S, Mitzel-Rink H, Kumm EJ, Jurk K, et al. Platelet-derived GARP induces peripheral regulatory t cells—potential impact on t cell suppression in patients with melanoma-associated thrombocytosis. Cancers (Basel). (2020) 12:1–24. doi: 10.3390/cancers12123653
106. Li N. Platelet-lymphocyte cross-talk. J Leukoc Biol. (2008) 83:1069–78. doi: 10.1189/jlb.0907615
107. van Gils JM, Zwaginga JJ, Hordijk PL. Molecular and functional interactions among monocytes, platelets, and endothelial cells and their relevance for cardiovascular diseases. J Leukoc Biol. (2009) 85:195–204. doi: 10.1189/jlb.0708400
108. Herster F, Bittner Z, Codrea MC, Archer NK, Heister M, Löffler MW, et al. Platelets aggregate with neutrophils and promote skin pathology in psoriasis. Front Immunol. (2019) 10:1867. doi: 10.3389/fimmu.2019.01867
109. Adrover JM, Hidalgo A. Activated platelets jam up the plaque. Circ Res. (2015) 116:557–559. doi: 10.1161/CIRCRESAHA.115.305823
110. Carestia A, Kaufman T, Rivadeneyra L, Landoni VI, Pozner RG, Negrotto S, et al. Mediators and molecular pathways involved in the regulation of neutrophil extracellular trap formation mediated by activated platelets. J Leukoc Biol. (2016) 99:153–62. doi: 10.1189/jlb.3a0415-161r
111. Ott I, Neumann F-J, Gawaz M, Schmitt M, Schömig A. Increased neutrophil-platelet adhesion in patients with unstable angina. Circulation. (1996) 94:1239–46. doi: 10.1161/01.CIR.94.6.1239
112. Ueno K, Nomura Y, Morita Y, Eguchi T, Masuda K, Kawano Y. Circulating platelet-neutrophil aggregates play a significant role in Kawasaki disease. Circ J. (2015) 79:1349–56. doi: 10.1253/circj.CJ-14-1323
113. Hally KE, Bird GK, la Flamme AC, Harding SA, Larsen PD. Platelets modulate multiple markers of neutrophil function in response to in vitro Toll-like receptor stimulation. PLoS One. (2019) 14: doi: 10.1371/journal.pone.0223444
114. Furman MI, Barnard MR, Krueger LA, Fox ML, Shilale EA, Lessard DM, et al. Circulating monocyte-platelet aggregates are an early marker of acute myocardial infarction. J Am Coll Cardiol. (2001) 38:1002–6. doi: 10.1016/S0735-1097(01)01485-1
115. Allen N, Barrett TJ, Guo Y, Nardi M, Ramkhelawon B, Rockman CB, et al. Circulating monocyte-platelet aggregates are a robust marker of platelet activity in cardiovascular disease. Atherosclerosis. (2019) 282:11–8. doi: 10.1016/j.atherosclerosis.2018.12.029
116. Green SA, Smith M, Hasley RB, Stephany D, Harned A, Nagashima K, et al. Activated platelet-T-cell conjugates in peripheral blood of patients with HIV infection: coupling coagulation/inflammation and T cells. AIDS. (2015) 29:1297–308. doi: 10.1097/QAD.0000000000000701
117. Mudd JC, Panigrahi S, Kyi B, Moon SH, Manion MM, Younes SA, et al. Inflammatory function of cx3cr1+ cd8+ t cells in treated HIV infection is modulated by platelet interactions. J Infect Dis. (2016) 214:1808–16. doi: 10.1093/infdis/jiw463
118. Starossom SC, Veremeyko T, Yung AWY, Dukhinova M, Au C, Lau AY, et al. Platelets play differential role during the initiation and progression of autoimmune neuroinflammation. Circ Res. (2015) 117:779–92. doi: 10.1161/CIRCRESAHA.115.306847
119. Ly HQ, Kirtane AJ, Murphy SA, Buros J, Cannon CP, Braunwald E, et al. Association of platelet counts on presentation and clinical outcomes in ST-elevation myocardial infarction (from the TIMI Trials). Am J Cardiol. (2006) 98:1–5. doi: 10.1016/j.amjcard.2006.01.046
120. Nunez J, Minana G, Bodi V, Nunez E, Sanchis J, Husser O, et al. Low lymphocyte count and cardiovascular diseases. Curr Med Chem. (2011) 18:3226–3233. doi: 10.2174/092986711796391633
121. Kurtul A, Ornek E. Platelet to lymphocyte ratio in cardiovascular diseases: a systematic review. Angiology. (2019) 70:802–18. doi: 10.1177/0003319719845186
122. Gasparyan AY, Ayvazyan L, Mukanova U, Yessirkepov M, Kitas GD. The platelet-to-lymphocyte ratio as an inflammatory marker in rheumatic diseases. Ann Lab Med. (2019) 39:345–57. doi: 10.3343/alm.2019.39.4.345
123. Akpinar MY, Ozin YO, Kaplan M, Ates I, Kalkan IH, Kilic ZMY, et al. Platelet-to-lymphocyte ratio and neutrophil-to-lymphocyte ratio predict mucosal disease severity in ulcerative colitis. J Med Biochem. (2018) 37:155–62. doi: 10.1515/jomb-2017-0050
124. Abdulrahman MA, Afifi N, El-Ashry M. Neutrophil/lymphocyte and platelet/lymphocyte ratios are useful predictors comparable to serum IL6 for disease activity and damage in naive and relapsing patients with lupus nephritis. Egypt Rheumatol. (2020) 42:107–12. doi: 10.1016/j.ejr.2019.08.002
125. Peng YF, Cao L, Zeng YH, Zhang ZX, Chen D, Zhang Q, et al. Platelet to lymphocyte ratio and neutrophil to lymphocyte ratio in patients with rheumatoid arthritis. Open Med. (2015) 10:249–53. doi: 10.1515/med-2015-0037
126. Wu L, Zou S, Wang C, Tan X, Yu M. Neutrophil-to-lymphocyte and platelet-to-lymphocyte ratio in Chinese Han population from Chaoshan region in South China. BMC Cardiovasc Disord. (2019) 19:7. doi: 10.1186/s12872-019-1110-7
127. Bromberg L, Roufosse F, Pradier O, Delporte C, Van Antwerpen P, De Maertelaer V, et al. Methylprednisolone-induced lymphocytosis in patients with immune-mediated inflammatory disorders. Am J Med. (2016) 129:746–52.e3. doi: 10.1016/j.amjmed.2016.02.013
128. Taillan B, Garnier G, Castanet J, Ferrari E, Pesce A, Dujardin P. Lymphoma developing in a patient with rheumatoid arthritis taking methotrexate. Clin Rheumatol. (1993) 12:93–4. doi: 10.1007/BF02231567
129. Ohashi-Fukuda N, Inokuchi R, Sato H, Nakamura K, Iwagami M, Wada T, et al. Poorer prognosis with ethylenediaminetetraacetic acid-dependent pseudothrombocytopenia. Med (United States). (2015) 94:674. doi: 10.1097/MD.0000000000000674
Keywords: platelet, chronic inflammation, cardiovascular risk, leukocytes, systemic inflammation
Citation: Zamora C, Cantó E and Vidal S (2021) The Dual Role of Platelets in the Cardiovascular Risk of Chronic Inflammation. Front. Immunol. 12:625181. doi: 10.3389/fimmu.2021.625181
Received: 02 November 2020; Accepted: 15 February 2021;
Published: 01 April 2021.
Edited by:
Eva Reali, University of Milano-Bicocca, ItalyReviewed by:
Cristiana Barbati, Sapienza University of Rome, ItalyAngelo A. Manfredi, Vita-Salute San Raffaele University, Italy
Copyright © 2021 Zamora, Cantó and Vidal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sílvia Vidal, c3ZpZGFsQHNhbnRwYXUuY2F0