 Roberto Amadio
Roberto Amadio Giulia Maria Piperno1
Giulia Maria Piperno1 Federica Benvenuti
Federica Benvenuti- 1Cellular Immunology, International Centre for Genetic Engineering and Biotechnology (ICGEB), Trieste, Italy
- 2Department of Biomedical Sciences, Venetian Institute of Molecular Medicine, University of Padova, Padova, Italy
Modified or misplaced DNA can be recognized as a danger signal by mammalian cells. Activation of cellular responses to DNA has evolved as a defense mechanism to microbial infections, cellular stress, and tissue damage, yet failure to control this mechanism can lead to autoimmune diseases. Several monogenic and multifactorial autoimmune diseases have been associated with type-I interferons and interferon-stimulated genes (ISGs) induced by deregulated recognition of self-DNA. Hence, understanding how cellular mechanism controls the pathogenic responses to self-nucleic acid has important clinical implications. Fine-tuned membrane trafficking and cellular compartmentalization are two major factors that balance activation of DNA sensors and availability of self-DNA ligands. Intracellular transport and organelle architecture are in turn regulated by cytoskeletal dynamics, yet the precise impact of actin remodeling on DNA sensing remains elusive. This review proposes a critical analysis of the established and hypothetical connections between self-DNA recognition and actin dynamics. As a paradigm of this concept, we discuss recent evidence of deregulated self-DNA sensing in the prototypical actin-related primary immune deficiency (Wiskott-Aldrich syndrome). We anticipate a broader impact of actin-dependent processes on tolerance to self-DNA in autoimmune disorders.
Introduction
TLR9 and cGAS-STING are the two major DNA sensors that have been implicated in recognition of self-DNA and induction of pathogenic IFN-I responses in monogenic and systemic autoimmunity (1–4). TLR9 is localized in endosomes, and it is preferentially expressed in immune cells, such as B cells and phagocytes, where it scrutinizes extracellular material taken up by receptor-mediated endo-phagocytosis. cGAS-STING is ubiquitously distributed in every cell type, patrolling the cytosol for the presence of viral or self-DNA extruded from inner compartments such as mitochondria and nuclei. The activity of TLR9 and cGAS-STING is intimately linked to membrane trafficking. Both TLR9 and STING translocate from a compartment in which they are inactive to a different one where signaling occurs and both need tight control of DNA localization to modulate their activity. Similarly, they share a common pathway for signal extinction in late degradative compartments. One critical and still poorly understood aspect of DNA sensors regulation is their relationship with cytoskeletal structures and the associated actin regulatory proteins. Triggering of TLR9 actively induces actin remodeling as part of the response, impacting signal intensity. Sharing of some of the downstream signaling components suggests that cytoskeletal remodeling may occur as well during activation of cGAS-STING, for instance during autophagy induction, through molecular mediators, though this remains to be defined. On the other hand, actin dynamics support translocation events required for signal modulation and impact the availability of ligands by controlling compartmentalization and accessibility to DNA.
Here, we will discuss the emerging evidence of the role played by cytoskeletal remodeling and actin regulatory proteins during activation of DNA sensors. Inborn errors of immunity caused by defects in actin regulators are beginning to shed light on this connection and will likely boost additional studies to identify the impact of actin dynamics on the balance between immunity and tolerance to self-DNA.
Self-DNA and Autoimmunity
The concept of immunogenicity of self-DNA is well illustrated by the presence of antibodies reacting to it, which is a hallmark of several systemic autoimmune diseases and especially well characterized in systemic lupus erythematosus (SLE) (5). Later studies established the contribution of endogenous nucleic acids as triggers of innate activation, notably type-I interferon, in SLE and other autoimmune diseases (6, 7). The structural determinants and cellular conditions that promote the immunogenic properties of DNA have been widely investigated and associated with the development of various autoinflammatory and autoimmune conditions (7–9). Monogenic disorders due to deficiencies in DNA metabolizing enzymes provide a further formal demonstration of the immunogenicity of extracellular and intracellular self-DNA. Mutations in the cytosolic DNA degrading enzyme TREX1 are linked to the rare type I interferonopathy called Aicardi-Goutières Syndrome or AGS (10). Human and mouse models lacking extracellular DNAse-I develop a SLE-like phenotype (11, 12). Mouse models and the recent identification of human mutations in lysosomal DNAse-II confirmed the importance of DNA degradation in lysosomes to avoid IFN-I activation (13, 14). DNAse1L3, a DNAse specifically secreted by dendritic cells and macrophages is required to digest circulating chromatin associated with microparticles, and its deficiency is associated with autoinflammation (15, 16). In the following chapters, we will focus on the regulation of the two main receptors implicated in endosomal and cytosolic self-DNA recognition and autoimmunity. The triggering of RNA innate receptors by endogenous molecules is another important pathway to autoimmunity that has been discussed elsewhere (17, 18).
TLR9 Signaling
The endosomal DNA receptor Toll-like receptor 9 (TLR9) was discovered in 2000 (19) and extensively reviewed in (4, 20). TLR9 is a transmembrane receptor preferentially expressed in immune cells, such phagocytes and B cells. It detects double-stranded DNA (dsDNA) via a leucine-rich luminal region and mediates downstream signaling by recruiting MyD88 to its cytosolic C-terminal TIR domain to promote NF-kβ and IRF7 nuclear translocation and cytokines/interferon type I production (21, 22). At the steady state, inactive TLR9 resides in the endoplasmic reticulum (ER). TLR9 trafficking involves exit from the ER assisted by chaperone Unc93b1, transit through the Golgi, and delivery to endolysosomes (23, 24). Upon arrival into the acidic environment of endolysosomes, TLR9 undergoes stepwise processing that makes it competent to bind ligand, multimerize and initiate signaling (25, 26). Collectively these mechanisms restrict signaling to endosomes, preventing ectopic activation and excessive inflammation. TLR9 binds preferentially CpG-rich sequences in DNA of bacterial origin (19, 27), however the receptor can be activated by endogenous self-DNA under specific circumstances. Engulfed self-DNA becomes immunogenic when combined with antimicrobial peptides, chromatin proteins or when release by mitochondria in an oxidized state, as it occurs during extrusion of neutrophil extracellular traps (28–30). These modifications protect DNA from degradation and increase the time of retention in endosomes, facilitating TLR9 activation, and they have been directly linked to the development of autoimmune diseases (31).
Trafficking and Cytoskeletal Remodeling During TLR9 Signaling
In parallel to activation of inflammatory genes transcription, TLR9 triggering induces rapid cytoskeletal changes through a cross-talk with actin regulatory proteins. Earlier studies had identified a link between TLR signaling and actin remodeling that leads to increased endocytosis in dendritic cells (32). In macrophages, CpG DNA induces actin reorganization and cell spreading by Src mediated phosphorylation of Vav1, an exchange factor for Rac1, and by activation of paxillin (33, 34). Cell spreading, adhesion, and motility induced by the synthetic ligand CpG-B were shown to depend on Src and to occur before acidification of TLR9 in endosomes, suggesting that cytoskeletal remodeling is an early event establishing before full activation of the signaling cascade (35). In pDCs, phosphorylation of IKK and nuclear translocation of IRF-7 to induce interferon production is blunted in DOCK-2 deficient cells. Although the precise mechanism was not elucidated in this case, it was proposed that DOCK-2-mediated Rac1 activation controls IKK-α through NAPDH and generation of oxygen species (36). In line with this report, recent evidence showed that elevation of the B cell adaptor for PI3K (BCAP) in SLE contributes to heightened IFN-I production via the DOCK2-Rac1 axis (37). Extracellular matrix stiffness may also control DNA receptor activity. For instance, actin-dependent mechano-transduction via Rho-associated coiled-coil kinase (ROCK) was found to dampen TLR9 signaling in macrophages (38). BCR signaling in B cells is intimately linked to actin dynamics. A network of cortical actin and ezrin confines BCR diffusion at the steady state, preventing spontaneous activation. Actin depolymerization by cofilin and ezrin dephosphorylation free BCR mobility inducing assembly of BCR microclusters and signaling (39). Intriguingly, TLR9 signaling was shown to increase cofilin activity inducing BCR mobility and lowering the threshold of BCR receptor activation. The exact mechanism for cofilin activation was not described, yet this study provides an important link between TLR9 signaling and actin dissolution to favor receptor mobility (40). A very interesting extension to this concept is that actin may also control the distribution and mobility of TLR9 in endosomal membranes, preventing collision and spurious activation. Technical challenges inherent to visualization of TLR9 and actin dynamics on endosomes of immune cells prevent direct analysis, yet some of our observations in cells mutated for actin regulators provided some clues in this direction (see below).
A further poorly unexplored area relates to the control of translocation from the ER to signaling endosomes by actin dynamics. In this direction, one report documented that the cytoskeletal protein FODH2, which is linked to SLE predisposition, is critical to retain TLR9 into IRAP+ pre-signaling compartments, preventing activation (41, 42). Budding from the ER, transport to the Golgi and post-Golgi trafficking events are all connected to cytoskeletal remodeling in other cellular systems (43–46), therefore the link between actin and TLR9 signaling in immune cells is likely underestimated. This is in part explained by limitations in applying cutting-edge cell biology approaches to primary immune cells.
cGAS-STING Signaling
The structure and function of the cGAS-STING axis have been extensively reviewed recently (47, 48). Here we will briefly summarize the major features to focus on self-DNA recognition. The cGAS-STING pathway relies on two independent receptors. cGAS is an enzyme with a positively charged surface that recognizes, preferentially, B-form dsDNA to synthesize cGAMP. The newly formed cGAMP molecules bind to STING, an ER-resident protein in the resting state. cGAMP binding induces a conformational change in the inactive dimer of this protein and translocation to the Golgi/ER–Golgi intermediate compartment (ERGIC) where STING oligomerize forming a hub for recruitment of TBK1 and IKK, resulting in IRF3 and NF-kβ-mediated transcription of IFN-I and inflammatory cytokines. cGAMP can also be transferred to bystander cells in a process that involves the transporter Slc19a1, ATP-gated P2X7R channels, and the LRRCA voltage-regulated anion channels (49–51). This signal amplification strategy is extremely powerful in the case of viral infection but may become detrimental during self-DNA recognition. Mechanisms of cGAS negative regulation include low expression at a steady state and sequestration of inactive forms at the plasma membrane and in protected nuclear structures. Outside the scope of the present review, it has to be mentioned that some evidence points to the cytokine-independent output of cGAS-STING activation (52, 53).
cGAS-STING can be triggered by cytosolic self-DNA originated from mitochondria (mtDNA) or nucleus (genomic DNA, gDNA). Oxidized mtDNA spills from damaged mitochondria because of defective mitophagy or formation of VDAC voltage-dependent anion channel or gasdermin-D-dependent pores in the Mt membrane (54–56). Once in the cytosol, oxidized mtDNA counteracts inhibitory mechanisms launching innate activation (57). Nucleoid instability and compromised mitochondrial structure by TFAM deficiency are further examples of aberrant mtDNA release that cause pathogenic TLR9 and cGAS-STING activation (58–60). There is evidence that also genomic DNA can become immunogenic as exemplified in rare genetic disorders. In Ataxia Telangiectasia or Bloom syndrome, the predisposition to genomic instability causes DNA leakage and cGAS/STING activation (61). Mutations in the cytosolic DNA degrading enzyme TREX1 cause Aicardi-Goutières Syndrome or AGS a type I interferonopathy (10). In this group of diseases, and in familiar SLE, gDNA accumulates in the cytosol because of basal S-phase by-products generation or retroelement transposition. Prominent nuclear damage and genomic instability in cancer cells are further examples where the formation of micronuclei or mobilization of transposable genetic elements can feed the cytosol with DNA leading to cGAS activation and IFN-I production (62, 63).
cGAS-STING Trafficking and the Connection With the Cytoskeleton
Proteins and mechanisms involved in movements of cGAS-STING across intracellular compartments are being increasingly elucidated and illustrate the critical role of trafficking for signal regulation [reviewed in (64)]. STIM1 is an ER-resident Ca2+ binding protein implicated in replenishing Ca2+ stores at ER-plasma membrane (ER-PM) contact sites. STIM1 was recently reported to interact with STING and to retain it in the ER. Loss of STIM1 in cellular models causes spontaneous IFN-I production and mutations in STIM1 are linked to a severe immune deficiency with autoimmune complications (65, 66). A specular disease mechanism was identified for COPA syndrome, an interferon-mediated disease caused by STING gain of function. COPA interacts with STING and the mutant form inhibits retrograde transport leading to enhanced type-I interferon signaling (67, 68). Another elegant mechanistic insight on the trafficking process of STING has been recently provided by the characterization of STEEP, the “STING ER exit protein”. STEEP was found to interact with STING and to recruit VPS34 to increase the local concentration of phosphatidylinositol 3-phosphate (PtdIns(3)P), facilitating ER membrane curvature, packaging into COPII vesicles and budding. Accordingly, in STEEP mutant cells, STING is retained in the ER, and IFN-I induction is suppressed (69). TMEM203 is a further adaptor that helps STING exit from the ER and regulates its activation (70). Hence, fine-tuned control of STING retention/exit in and from the ER is key to regulate signaling. Intriguingly, it was recently reported that STING controls the accumulation of F-actin via WASP and PI3K in B cells, impacting BCR signaling (71). There is a strong rationale to predict more connections between actin and STING trafficking. For instance, translocation of STIM1 to ER-PM contact sites to refill Ca2+ stores was shown to be assisted by F-actin assembly (72). Second, membrane phosphoinositides recruited during STING exit from the ER are likely to influence downstream actin regulatory proteins. Moreover, cytoskeletal remodeling at the Golgi provides an interesting area to understand STING regulation (73, 74).
The second outcome of STING signaling, independent of TBK1 and cytokine activation, is induction of autophagy, which helps signal termination by clearance of cytosolic DNA. Mechanistically, cGAMP-induced LC3 lipidation is proposed to be dependent on WIPI2 and ATG5 and ERGIC membranes as a source of LC3 lipidation (52). As widely documented, actin provides an essential scaffold in each step of autophagosomal biogenesis, through a complex balance of Rho GTPAses and downstream nucleation promoting factors (NPFs). Interaction between WASH and components of the membrane trafficking machinery coordinates initial phases of autophagy, such as sorting of ATG9-positive vesicles to early sites of phagophore biogenesis (75). WHAMM orchestrates actin-comet tails generation to sustain phagophore propulsion and invagination (76) and JMY instead shapes autophagosomes by direct interaction with LC3 (77). To note, luminal actin has been spotted by super-resolution microscopy (78), suggesting that polymerization may continue in the lumen of phagosomes. Some evidence suggests that actin may also be important in the late stage of phagosomal closure, as shown by the requirement for Arp2/3 during selective autophagy (79). More recently, Wiskott-Aldrich syndrome protein (WASp) has been linked to the global orchestration of both general and selective autophagy in myeloid cells (80, 81). Even if a detailed mechanism of WASp-autophagy connections remains to be explored, it can be extrapolated that deregulation of actin dynamics (see below) will impact cGAS-STING signaling in phagocytes.
The Role of Actin in Maintaining Compartmentalization of Self-DNA
Actin dynamics play essential roles in mitochondrial and nuclear stability and therefore impact DNA confinement and prevention of unwanted cGAS-STING triggering. Transient actin polymerization induced by Arp2/3, cortactin, and formins contributes to mitochondria fission and conversely, actin relaxation allows mitochondria fusion (82, 83). In addition, actin flows were recently shown to ensure mitochondria redistribution to daughter cells during mitosis (84), ensuring a proper metabolic balance and mitochondrial turnover. Importantly, mitochondrial disposal by mitophagy is mediated by Arp2/3-dependent actin polymerization downstream of phosphatidylinositol 4,5-bisphosphate (PtdIns(4,5)P2) (85). Failure to degrade damaged mitochondria or problems during fission/fusion processes have been directly associated with mtDNA leakage and unwanted inflammation in SLE (54).
A second important level of actin-mediated regulation of DNA sensing is through the control of nuclear integrity and genomic stability (86). WASp and Arp2/3 have been implicated in preserving genomic stability via homology-directed repair of DNA double-strand breaks (DSB) during replication stress and by counteracting nuclear deformation (87, 88). On the other hand, genomic instability has been clearly linked to cGAS activation (63). It is thus reasonable to expect an interplay between perturbed actin dynamics, genomic stability, and cGAS activation. In this direction, uncoupling the cytoskeleton from the nuclear envelope in ATR deficient cells was recently shown to cause accumulation of perinuclear cGAS in conditions of mechanical stress (62, 89, 90). The ESCRT machinery involved in endosomal sealing is also recruited at sites of the damaged nuclear envelope for repair (91, 92). Interestingly, cGAS interacts with multiple ESCRT components, suggesting an interplay between cGAS, ESCRTs, and actin, in determining whether nuclear ruptures and DSB can be repaired or an inflammatory response should be triggered. The cross-talk between nuclear integrity, actin polymerization, and excessive responses to self-DNA has emerged as well in neurodegenerative and aging-related diseases. Increased availability of cytosolic self-DNA can occur in laminopathies and increased cytoskeletal stiffness was found to reduce nuclear plasticity, promoting nuclear blebbing and stimulating cGAS-driven senescence in a model of Hutchinson-Gilford progeria (93, 94).
Crosstalk Between Endosomal and Cytosolic Sensing in Phagocytes
The conventional model of TLR9 activation by ingested DNA in endosomes and cGAS activation by mt/gDNA in the cytosol is challenged in phagocytes. In fact, the two systems may connect in these cells, a process that we propose to call “cross-sensing”. Early evidence already suggested that phagosomal overloading with apoptotic DNA may induce TLR9-independent sensing (95). This was further supported by the observation that lethality in mice deficient for lysosomal DNase-II was rescued by crossing to STING-/- or to cGAS-/-, but not TLR9-/- mice (96, 97). More recently, DNA of dying cardiomyocytes engulfed by macrophages was shown to induce cGAS-STING upon transfer from endosomes to the cytosol (98) and DNA delivered to endosomes of monocytes in type-I diabetes patients was observed to trigger STING-dependent inflammation (99). Interestingly, transfer of ingested tumor DNA in the cytosol of dendritic cells is thought to use the same transfer mechanism of cross-presentation, a specialized trafficking route that grants cytosolic access to exogenous antigens (100). Mechanistically, protein partners such as HMGB1 and LL-37, initially identified as enhancers of immunogenicity in TLR9-mediated responses, possess membrane destabilizing properties and promote the endosomal escape of engulfed self-DNA by mechanical disruption (100–104). Besides the cargo mechanical properties, endosomal membrane stability is regulated by cellular factors, including actin dynamics. For instance, endosomal membrane exchange and integrity during maturation are governed by actin nucleators, Rab GTPases, phosphoinositides, and microtubule motors (105–107). In addition, the process of endosomes sealing via recruitment of ESCRT (108) may be related to actin remodeling (109). Evidence comes also from neuronal diseases where enlarged endosomes correlate to activation of cytosolic sensing by ingested DNA. For instance, the accumulation of B amyloid fibrils within endosomes was recently shown to combine with self-DNA enhancing its immunogenicity and IFN-I stimulatory activity (110). Although not yet identified, it is interesting to speculate how deregulated cGAS-STING may also be involved in lysosomal storage diseases such as Niemann Pick Disease type C and Gaucher disease, both characterized by defective endosomal segregation, abnormal trafficking and pathogenic inflammation (111). Importantly, our recent data provided direct evidence that innate cells deficient for the actin regulator WASp carry enlarged and immature endosomes that leak ingested DNA in the cytosol (see below). Remarkably, cross-presentation is increased in WASp deficient DCs (112), further suggesting that cross-presentation and cross-sensing may use similar mechanisms and regulation to coordinate antigen processing and innate activation. Conversely, innate signaling may actively increase membrane permeability to potentiate cross-sensing, which is made possible via the orchestration of cytoskeletal dynamics. Of note, two recent studies point to the role of cytoskeletal proteins in enhancing endosome to cytosol transfer in cross-presentation (113, 114). In summary, actin dynamics are linked to the integrity of mitochondrial, nuclear, and endosomal membranes and therefore regulate the DNA sensing by ensuring confinement of self-DNA.
Actin-Related Immunodeficiencies and the Link to Self-DNA Sensing
A plethora of emerging genetic diseases characterized by autoinflammation and immune dysregulation have been associated with cytoskeletal defects and recently reviewed in (115). The recent identification, the limited number of patients, and the lack of appropriated cellular and animal model still preclude precise identification of the driving mechanisms of innate activation, yet their clinical characteristics suggest a possible impaired control of self-DNA recognition that deserve further investigation. Here we will focus on the prototype cytoskeletal-related immune disease, Wiskott-Aldrich syndrome (WAS), the first example of a cytoskeletal-related primary immune deficiency with deregulated type-I interferon driven by cGAS-STING activation. The disease arises from mutations in (WASp), the hematopoietic founder of the WASp family of nucleation promoting factors (NPFs). NPFs of the WASp family include several members that share the ability to activate the Arp2/3 complex and to promote the generation of branched acting fibers on existing actin filaments. WASp activity is regulated by Rho GTPases, SH2-containing tyrosine kinases, Src kinases, and phosphatidyl-inositol (4,5)-bisphosphate (116, 117). NPFs expression levels are highly cell-type specific. While WASp is exclusively present in immune cells that have little to no expression of the other NPFs, non-immune cells express varying the degree of the other NPFs but have no WASp (Figure 1). Control of peripheral membrane structures has been attributed to N-WASp, WASp, WAVE, and WHIMP, whereas WASH, JMY, and WHAMM were associated with ER-Golgi and endosome trafficking. Compatibly with its abundance and the poor expression of the other NPFs, WASp was shown to multitask at the cell periphery and intracellularly in immune cells (80, 118). WASp regulates multiple immune cell functions, ranging from BCR and TCR receptor signaling in lymphocytes to migration and phagocytosis in myeloid cells (81). Defects in these processes provide a logical explanation for weak responses to pathogens and increased susceptibility to infections. Somehow paradoxically, the majority of WAS patients also develop a spectrum of autoimmune manifestations with a complex and multifactorial origin, not yet fully elucidated (119). Patients who develop autoantibodies against a wide range of self-antigens, including DNA-associated proteins and mitochondrial antigens, show altered peripheral T-cell tolerance and cell-intrinsic flaws in platelets which promote excessive inflammation and autoreactive B cells activation (120–124). A further emerging key aspect in WAS is the deregulated production of inflammatory cytokines by cells of the innate immune system. Intriguingly, up to 20% of WAS patients still suffer from recurrent inflammatory manifestations even after hematopoietic stem cell transplant (125). In search of a mechanism to link WASp and inflammation, our group uncovered that pDCs in patients and mouse models produce constitutive amounts of type-I IFN and display an enhanced response to stimulation with TLR9 agonists (126). Imaging of individual cells revealed altered trafficking of engulfed multimeric agonists of TLR9, establishing the first direct link between actin dynamics and negative regulation of TLR9 signaling. In addition, WASp null neutrophils are prone to secrete NETs, and their depletion in animal models rescued circulating IFN-I to normal levels, indicating that a combination of cell-intrinsic and cell-extrinsic cues cooperate to exacerbate IFN-I production (127). To further investigate the cellular basis of enhanced IFN-I production we later moved to examine myeloid cells exposed to DNA-immune complexes (DNA-IC), an interferogenic trigger abundant in WAS patients. We found that low doses of DNA-ICs, which are normally tolerated by normal cells, induce IFN-I release in WASp deficient cells. Mechanistically, we found that WASp promotes nucleation of endosomal F-actin and its deficiency causes a delay in endolysosomal maturation, prolonging the transit time of ingested DNA-ICs. Interestingly, stalling in maturation-defective organelles facilitated leakage of DNA-ICs into the cytosol, inducing activation of the STING pathway. Genetic deletion of STING corrected excessive responses of WASp null cells, uncovering cytosolic DNA sensing as a contributor of innate immune dysregulation in WAS (118). Building on this pilot discovery, we hypothesize that WASp may control cGAS-STING mediated IFN-I production by mechanisms beyond endosomal maturation and integrity such as STING trafficking at the ER-Golgi, post-Golgi transfer, and autophagosomal degradation (80, 118) (Figure 2). Moreover, by interacting with phosphoinositides (128), WASp may participate in cGAS anchoring at the plasma membrane, a mechanism that prevents recognition of cytosolic self-DNA in myeloid cells (129). Recently it was observed that the formation of actin cages around damaged mitochondria and mitophagy are reduced in WASp null cells (81), suggesting there may be an exaggerated release of mtDNA in the cytosol of WASp deficient cells that could contribute to overall chronic activation of IFN genes. Finally, WASp is also present inside the nucleus. Even if its nuclear function has been uncoupled from actin polymerization, it is tempting to speculate that WASp may limit gDNA leakage and control the epigenetic state of interferon-stimulated genes, for instance in a mechanosensitive fashion. Along this line, it would be interesting to investigate whether, in innate cells, WASp might function as a linker between extracellular matrix stiffness and self-DNA sensing or inflammatory phenotypes.
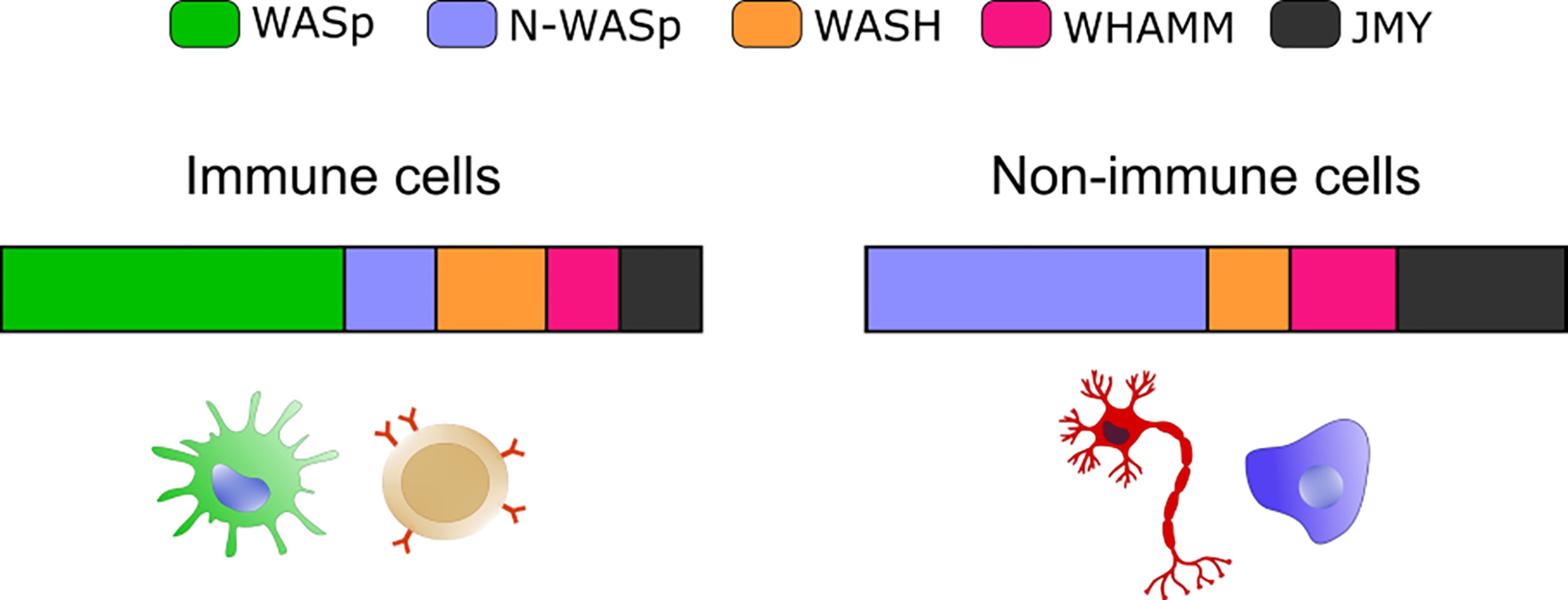
Figure 1 Expression of selected NPFs in immune and non-immune cells. Bar graphs show the relative abundance of the indicated NPF. The “immune cells” plot integrates values from: B cells (bone marrow), Macrophage (bone marrow and spleen) and Classical Monocyte (lung). The “non-immune cells” includes: Epithelial Cell (lung), Fibroblastic Cell (heart), Neuron (brain), Hepatocyte (liver) and Endothelial cell (mammary gland). Plots were obtained by merging expression from different cell types and plotting the resulting average value. Data are from the Tabula Muris website (https://tabula-muris.ds.czbiohub.org/) FACS sorted single cell types RNA-seq.
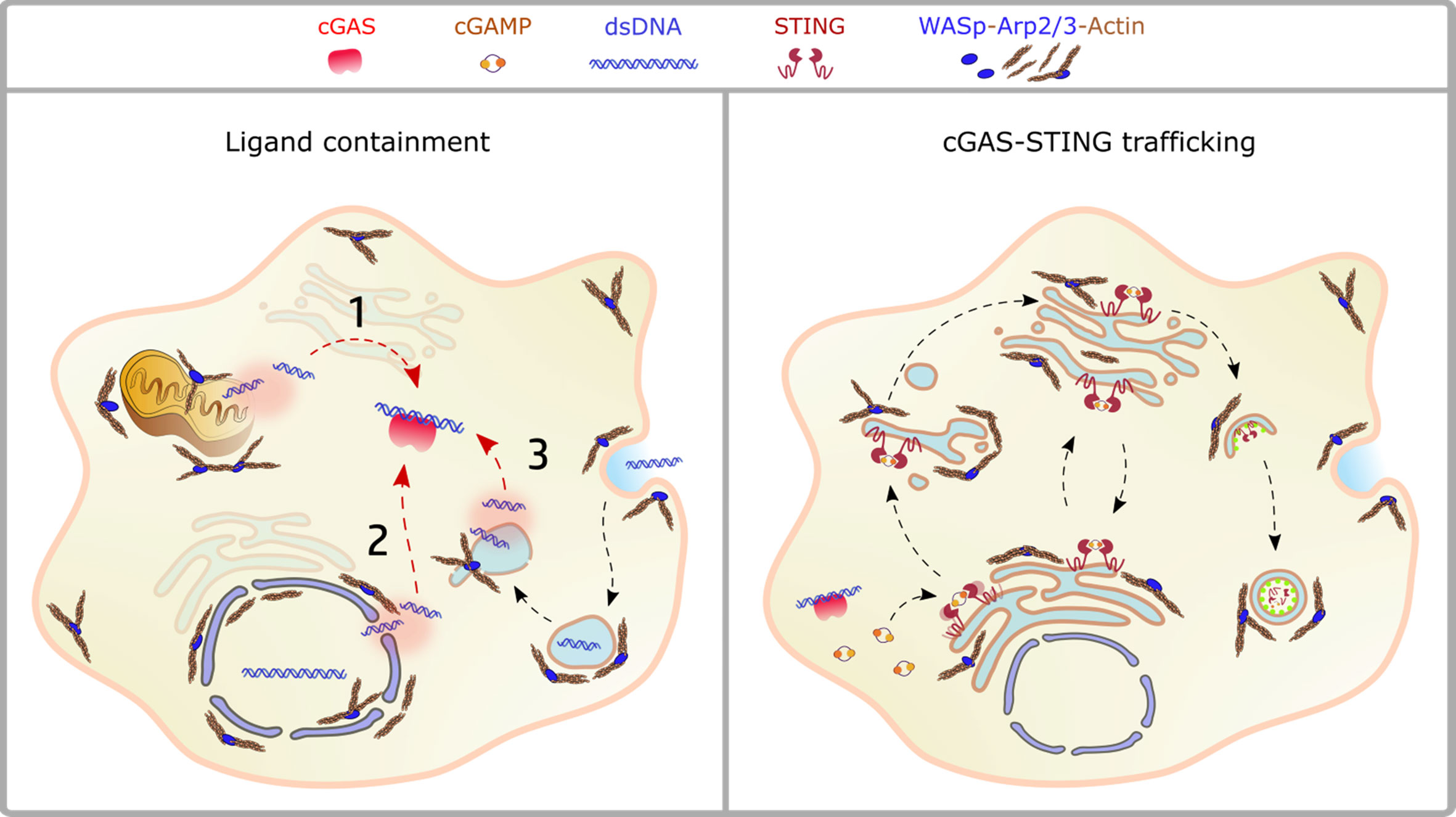
Figure 2 Crosstalk between actin dynamics and cGAS-STING pathway. Left panel: ligand availability into the cytosol may derive from mitochondrial dysfunction (1), nuclear damage (2), endolysosomal leakage (3). The actin cytoskeleton controls both morphology and stability of these compartments, as discussed in the text. Right panel: the scheme depicts STING trafficking steps highlighting the processes controlled by actin dynamics. NPFs of the WASp family regulate vesicles budding from the ER, cargo loading onto COPII vesicles, Golgi stability and post-Golgi trafficking steps, including incorporation into autophagosomes and phagolysosomes.
Concluding Remarks
The interest in the cGAS-STING system of cytosolic DNA detection has boomed in the last 5 years with an impressive number of reports describing the structural characteristic of cGAS, the range of microbial and endogenous DNA that can bind to it, the trafficking of STING, and the regulatory mechanism ensuring self/non-self discrimination. In hand with basic discovery, the role of cGAS-STING has been appreciated far beyond microbial recognition especially in pathological conditions such as cancer and autoimmune diseases. While the general rules for cGAS/STING functioning have been established, little is known about cell-type- and tissue-specific regulation of DNA sensing. Future studies will be important to discriminate the mechanism of cGAS/STING activation in long-lived, poorly dividing cells as opposed to highly proliferative, short-lived and mobile cells such as immune cells. The coordination of DNA detection by the actin cytoskeleton, a fundamental scaffold that supports most cellular functions, such as endocytosis, intracellular trafficking, and migration, is emerging and will be an important area for future research. The initial finding of enhanced cGAS-STING activation in cells deficient for a prototype regulator of actin dynamics anticipates the importance of this layer of regulation and prompts further analysis in this direction.
Author Contributions
RA, GP, and FB wrote the articles. RA and GP prepared Figures. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Italian Telethon to FB (GGP GGP14281).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Crowl JT, Gray EE, Pestal K, Volkman HE, Stetson DB. Intracellular Nucleic Acid Detection in Autoimmunity. Ann Rev Immuno (2017) 35(1):313–36. doi: 10.1146/annurev-immunol-051116-052331
2. Kumar V. a STING to Inflammation and Autoimmunity. J Leukoc Biol (2019) 106:171–85. doi: 10.1002/JLB.4MIR1018-397RR
3. Motwani M, Pesiridis S, Fitzgerald KA. DNA Sensing by the Cgas-STING Pathway in Health and Disease. Nat Rev Genet (2019) 20:657–74. doi: 10.1038/s41576-019-0151-1
4. Pelka K, Shibata T, Miyake K, Latz E. Nucleic Acid-Sensing Tlrs and Autoimmunity: Novel Insights From Structural and Cell Biology. Immunol Rev (2016) 269:60–75. doi: 10.1111/imr.12375
5. Soni C, Reizis B. DNA as a Self-Antigen: Nature and Regulation. Curr Opin Immunol (2018) 55:31–7. doi: 10.1016/j.coi.2018.09.009
6. Banchereau J, Pascual V. Type I Interferon in Systemic Lupus Erythematosus and Other Autoimmune Diseases. Immunity (2006) 25:383–92. doi: 10.1016/j.immuni.2006.08.010
7. Barrat FJ, Elkon KB, Fitzgerald KA. Importance of Nucleic Acid Recognition in Inflammation and Autoimmunity. Annu Rev Med (2016) 67:323–36. doi: 10.1146/annurev-med-052814-023338
8. Soni C, Reizis B. Self-DNA At the Epicenter of SLE: Immunogenic Forms, Regulation, and Effects. Front Immunol (2019) 10:1601. doi: 10.3389/fimmu.2019.01601
9. Khan S, Godfrey V, Zaki MH. Cytosolic Nucleic Acid Sensors in Inflammatory and Autoimmune Disorders. Int Rev Cell Mol Biol (2019) 344:215–53. doi: 10.1016/bs.ircmb.2018.10.002
10. Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, et al. Mutations in the Gene Encoding the 3′-5′ DNA Exonuclease TREX1 Cause Aicardi-Goutières Syndrome At the AGS1 Locus. Nat Genet (2006) 38:917–20. doi: 10.1038/ng1845
11. Napirei M, Karsunky H, Zevnik B, Stephan H, Mannherz HG, Moroy T. Features of Systemic Lupus Erythematosus in Dnase1-Deficient Mice. Nat Genet (2000) 25:177–81. doi: 10.1038/76032
12. Yasutomo K, Horiuchi T, Kagami S, Tsukamoto H, Hashimura C, Urushihara M, et al. Mutation of Dnase1 in People With Systemic Lupus Erythematosus. Nat Genet (2001) 28:313–4. doi: 10.1038/91070
13. Lan YY, Londono D, Bouley R, Rooney MS, Hacohen N. Dnase2a Deficiency Uncovers Lysosomal Clearance of Damaged Nuclear DNA Via Autophagy. Cell Rep (2014) 9:180–92. doi: 10.1016/j.celrep.2014.08.074
14. Rodero MP, Tesser A, Bartok E, Rice GI, Della Mina E, Depp M, et al. Type I Interferon-Mediated Autoinflammation Due to Dnase Ii Deficiency. Nat Commun (2017) 8:2176. doi: 10.1038/s41467-017-01932-3
15. Al-Mayouf SM, Sunker A, Abdwani R, Abrawi SA, Almurshedi F, Alhashmi N, et al. Loss-of-Function Variant in Dnase1l3 Causes a Familial Form of Systemic Lupus Erythematosus. Nat Genet (2011) 43:1186–8. doi: 10.1038/ng.975
16. Sisirak V, Sally B, D’Agati V, Martinez-Ortiz W, Ozcakar ZB, David J, et al. Digestion of Chromatin in Apoptotic Cell Microparticles Prevents Autoimmunity. Cell (2016) 166:88–101. doi: 10.1016/j.cell.2016.05.034
17. Rehwinkel J, Gack MU. Rig-I-Like Receptors: Their Regulation and Roles in RNA Sensing. Nat Rev Immunol (2020) 20:537–51. doi: 10.1038/s41577-020-0288-3
18. Schlee M, Hartmann G. Discriminating Self From Non-Self in Nucleic Acid Sensing. Nat Rev Immunol (2016) 16:566–80. doi: 10.1038/nri.2016.78
19. Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, et al. Toll-Like Receptor Recognizes Bacterial DNA. Nature (2000) 408:740–5. doi: 10.1038/35047123
20. Barton GM, Kagan JC. A Cell Biological View of Toll-Like Receptor Function: Regulation Through Compartmentalization. Nat Rev Immunol (2009) 9:535–42. doi: 10.1038/nri2587
21. Fitzgerald KA, Kagan JC. Toll-like Receptors and the Control of Immunity. Cell (2020) 180:1044–66. doi: 10.1016/j.cell.2020.02.041
22. Lee BL, Barton GM. Trafficking of endosomal Toll-Like Receptors. Trends Cell Biol (2014) 24:360–9.
23. Chockalingam A, Brooks JC, Cameron JL, Blum LK, Leifer CA. TLR9 Traffics Through the Golgi Complex to Localize to Endolysosomes and Respond to Cpg Dna. Immunol Cell Biol (2009) 87:209–17. doi: 10.1038/icb.2008.101
24. Lee BL, Moon JE, Shu JH, Yuan L, Newman ZR, Schekman R, et al. UNC93B1 Mediates Differential Trafficking of Endosomal Tlrs. eLife (2013) 2013(27):e00291. doi: 10.7554/eLife.00291
25. Ewald SE, Lee BL, Lau L, Wickliffe KE, Shi GP, Chapman HA, et al. the Ectodomain of Toll-Like Receptor 9 is Cleaved to Generate a Functional Receptor. Nature (2008) 456:658–62. doi: 10.1038/nature07405
26. Ewald SE, Barton GM. Nucleic Acid Sensing Toll-Like Receptors in Autoimmunity. Curr Opin Immunol (2011) 23:3–9. doi: 10.1016/j.coi.2010.11.006
27. Bauer S, Kirschning CJ, Hacker H, Redecke V, Hausmann S, Akira S, et al. Human TLR9 Confers Responsiveness to Bacterial DNA Via Species-Specific Cpg Motif Recognition. Proc Natl Acad Sci USA (2001) 98:9237–42. doi: 10.1073/pnas.161293498
28. Caielli S, Athale S, Domic B, Murat E, Chandra M, Banchereau R, et al. Oxidized Mitochondrial Nucleoids Released by Neutrophils Drive Type I Interferon Production in Human Lupus. J Exp Med (2016) 213:697–713. doi: 10.1084/jem.20151876
29. Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting Neutrophils are Major Inducers of Type I IFN Production in Pediatric Systemic Lupus Erythematosus. Sci Transl Med (2011) 3:73ra20. doi: 10.1126/scitranslmed.3001201
30. Guiducci C, Ott G, Chan JH, Damon E, Calacsan C, Matray T, et al. Properties Regulating the Nature of the Plasmacytoid Dendritic Cell Response to Toll-Like Receptor 9 Activation. J Exp Med (2006) 203:1999–2008. doi: 10.1084/jem.20060401
31. Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, Gizinski A, Yalavarthi S, Knight JS, et al. Nets are a Source of Citrullinated Autoantigens and Stimulate Inflammatory Responses in Rheumatoid Arthritis. Sci Transl Med (2013) 5:178ra140. doi: 10.1126/scitranslmed.3005580
32. West MA, Wallin RP, Matthews SP, Svensson HG, Zaru R, Ljunggren HG, et al. Enhanced Dendritic Cell Antigen Capture Via Toll-Like Receptor-Induced Actin Remodeling. Science (2004) 305:1153–7. doi: 10.1126/science.1099153
33. Achuthan A, Elsegood C, Masendycz P, Hamilton JA, Scholz GM. Cpg DNA Enhances Macrophage Cell Spreading by Promoting the Src-Family Kinase-Mediated Phosphorylation of Paxillin. Cell Signal (2006) 18:2252–61. doi: 10.1016/j.cellsig.2006.05.007
34. Stovall SH, Yi AK, Meals EA, Talati AJ, Godambe SA, English BK. Role of Vav1- and Src-Related Tyrosine Kinases in Macrophage Activation by Cpg Dna. J Biol Chem (2004) 279:13809–16. doi: 10.1074/jbc.M311434200
35. Sanjuan MA, Rao N, Lai KT, Gu Y, Sun S, Fuchs A, et al. Cpg-Induced Tyrosine Phosphorylation Occurs Via a Tlr9-Independent Mechanism and is Required for Cytokine Secretion. J Cell Biol (2006) 172:1057–68. doi: 10.1083/jcb.200508058
36. Gotoh K, Tanaka Y, Nishikimi A, Nakamura R, Yamada H, Maeda N, et al. Selective Control of Type I IFN Induction by the Rac Activator DOCK2 During TLR-Mediated Plasmacytoid Dendritic Cell Activation. J Exp Med (2010) 207:721–30. doi: 10.1084/jem.20091776
37. Chu T, Ni M, Chen C, Akilesh S, Hamerman JA. Cutting Edge: Bcap Promotes Lupus-Like Disease and TLR-Mediated Type I Ifn Induction in Plasmacytoid Dendritic Cells. J Immunol (2019) 202:2529–34. doi: 10.4049/jimmunol.1801267
38. Gruber E, Heyward C, Cameron J, Leifer C. Toll-Like Receptor Signaling in Macrophages is Regulated by Extracellular Substrate Stiffness and Rho-Associated Coiled-Coil Kinase (ROCK1/2). Int Immunol (2018) 30:267–78. doi: 10.1093/intimm/dxy027
39. Treanor B, Depoil D, Gonzalez-Granja A, Barral P, Weber M, Dushek O, et al. the Membrane Skeleton Controls Diffusion Dynamics and Signaling Through the B Cell Receptor. Immunity (2010) 32:187–99. doi: 10.1016/j.immuni.2009.12.005
40. Freeman SA, Jaumouille V, Choi K, Hsu BE, Wong HS, Abraham L, et al. Toll-Like Receptor Ligands Sensitize B-Cell Receptor Signalling by Reducing Actin-Dependent Spatial Confinement of the Receptor. Nat Commun (2015) 6:6168. doi: 10.1038/ncomms7168
41. Babdor J, Descamps D, Adiko AC, Tohme M, Maschalidi S, Evnouchidou I, et al. Irap(+) Endosomes Restrict Tlr9 Activation and Signaling. Nat Immunol (2017) 18:509–18. doi: 10.1038/ni.3711
42. Cheng H, Li Y, Zuo XB, Tang HY, Tang XF, Gao JP, et al. Identification of a Missense Variant in Lnpep That Confers Psoriasis Risk. J Invest Dermatol (2014) 134:359–65. doi: 10.1038/jid.2013.317
43. Bhattacharya K, Swaminathan K, Peche VS, Clemen CS, Knyphausen P, Lammers M, et al. Novel Coronin7 Interactions With Cdc42 and N-Wasp Regulate Actin Organization and Golgi Morphology. Sci Rep (2016) 6:25411. doi: 10.1038/srep25411
44. Dong R, Saheki Y, Swarup S, Lucast L, Harper JW, De Camilli P. Endosome-ER Contacts Control Actin Nucleation and Retromer Function Through VAP-Dependent Regulation of PI4P. Cell (2016) 166:408–23. doi: 10.1016/j.cell.2016.06.037
45. Gurel PS, Ge P, Grintsevich EE, Shu R, Blanchoin L, Zhou ZH, et al. Inf2-Mediated Severing Through Actin Filament Encirclement and Disruption. Curr Biol (2014) 24:156–64. doi: 10.1016/j.cub.2013.12.018
46. Kage F, Steffen A, Ellinger A, Ranftler C, Gehre C, Brakebusch C, et al. Fmnl2 and -3 Regulate Golgi Architecture and Anterograde Transport Downstream of Cdc42. Sci Rep (2017) 7:9791. doi: 10.1038/s41598-017-09952-1
47. Ablasser A, Hur S. Regulation of cGAS- and RLR-Mediated Immunity to Nucleic Acids. Nat Immunol (2020) 21:17–29. doi: 10.1038/s41590-019-0556-1
48. Hopfner K-PP, Hornung V. Molecular Mechanisms and Cellular Functions of Cgas–STING Signalling. Nat Rev Mol Cell Biol (2020) 21:1–21. doi: 10.1038/s41580-020-0244-x
49. Luteijn RD, Zaver SA, Gowen BG, Wyman SK, Garelis NE, Onia L, et al. SLC19A1 Transports Immunoreactive Cyclic Dinucleotides. Nature (2019) 573:434–8. doi: 10.1038/s41586-019-1553-0
50. Zhou C, Chen X, Planells-Cases R, Chu J, Wang L, Cao L, et al. Transfer of Cgamp Into Bystander Cells Via LRRC8 Volume-Regulated Anion Channels Augments STING-Mediated Interferon Responses and Anti-Viral Immunity. Immunity (2020) 52:767–81. doi: 10.1016/j.immuni.2020.03.016
51. Lahey LJ, Mardjuki RE, Wen X, Hess GT, Ritchie C, Carozza JA, et al. LRRC8A:C/E Heteromeric Channels are Ubiquitous Transporters of Cgamp. Mol Cell (2020) 80:578–591.e575. doi: 10.1016/j.molcel.2020.10.021
52. Gui X, Yang H, Li T, tan X, Shi P, Li M, et al. Autophagy Induction Via STING Trafficking is a Primordial Function of the Cgas Pathway. Nature (2019) 567:262–6. doi: 10.1038/s41586-019-1006-9
53. Wu J, Dobbs N, Yang K, Yan N. Interferon-Independent Activities of Mammalian Sting Mediate Antiviral Response and Tumor Immune Evasion. Immunity (2020) 53:115–26.e115. doi: 10.1016/j.immuni.2020.06.009
54. Gkirtzimanaki K, Kabrani E, Nikoleri D, Polyzos A, Blanas A, Sidiropoulos P, et al. Ifnalpha Impairs Autophagic Degradation of Mtdna Promoting Autoreactivity of Sle Monocytes in a Sting-Dependent Fashion. Cell Rep (2018) 25:921–933 e925. doi: 10.1016/j.celrep.2018.09.001
55. Huang LS, Hong Z, Wu W, Xiong S, Zhong M, Gao X, et al. Mtdna Activates Cgas Signaling and Suppresses the YAP-Mediated Endothelial Cell Proliferation Program to Promote Inflammatory Injury. Immunity (2020) 52:475–486.e475. doi: 10.1016/j.immuni.2020.02.002
56. Kim J, Gupta R, Blanco LP, Yang S, Shteinfer-Kuzmine A, Wang K, et al. VDAC Oligomers Form Mitochondrial Pores to Release Mtdna Fragments and Promote Lupus-Like Disease. Science (2019) 366:1531–6. doi: 10.1126/science.aav4011
57. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil Extracellular Traps Enriched in Oxidized Mitochondrial DNA Are Interferogenic and Contribute to Lupus-Like Disease. Nat Med (2016) 22:146–53. doi: 10.1038/nm.4027
58. Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, et al. Mitochondrial DNA That Escapes From Autophagy Causes Inflammation and Heart Failure. Nature (2012) 485:251–5. doi: 10.1038/nature10992
59. Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, et al. Parkin and PINK1 Mitigate STING-Induced Inflammation. Nature (2018) 561:258–62. doi: 10.1038/s41586-018-0448-9
60. West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, et al. Mitochondrial Dna Stress Primes the Antiviral Innate Immune Response. Nature (2015) 520:553–7. doi: 10.1038/nature14156
61. Gratia M, Rodero MP, Conrad C, Bou Samra E, Maurin M, Rice GI, et al. Bloom Syndrome Protein Restrains Innate Immune Sensing of Micronuclei by Cgas. J Exp Med (2019) 216:1199–213. doi: 10.1084/jem.20181329
62. Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, et al. Cgas Surveillance of Micronuclei Links Genome Instability to Innate Immunity. Nature (2017) 548:461–5. doi: 10.1038/nature23449
63. Guan J, Lu C, Jin Q, Lu H, Chen X, Tian L, et al. Mlh1 Deficiency-Triggered Dna Hyperexcision by Exonuclease 1 Activates the Cgas-Sting Pathway. Cancer Cell (2021) 39:109–21.e105. doi: 10.1016/j.ccell.2020.11.004
64. Balka KR, De Nardo D. Molecular and Spatial Mechanisms Governing STING Signalling. FEBS J (2020). doi: 10.1111/febs.15640
65. Lacruz RS, Feske S. Diseases Caused by Mutations in ORAI1 and STIM1. Ann N Y Acad Sci (2015) 1356:45–79. doi: 10.1111/nyas.12938
66. Srikanth S, Woo JS, Wu B, El-Sherbiny YM, Leung J, Chupradit K, et al. the Ca(2+) Sensor Stim1 Regulates the Type I Interferon Response by Retaining the Signaling Adaptor Sting At the Endoplasmic Reticulum. Nat Immunol (2019) 20:152–62. doi: 10.1038/s41590-018-0287-8
67. Deng Z, Chong Z, Law CS, Mukai K, Ho FO, Martinu T, et al. A Defect in COPI-Mediated Transport of STING Causes Immune Dysregulation in COPA Syndrome. J Exp Med (2020) 217:1–11.e20201045. doi: 10.1084/jem.20201045
68. Lepelley A, Martin-Niclós MJ, Bihan ML, Marsh JA, Uggenti C, Rice GI, et al. Mutations in COPA Lead to Abnormal Trafficking of STING to the Golgi and Interferon Signaling. J Exp Med (2020) 217:1–16.e20200600. doi: 10.1084/jem.20200600
69. Zhang BC, Nandakumar R, Reinert LS, Huang J, Laustsen A, Gao ZL, et al. Steep Mediates Sting Er Exit and Activation of Signaling. Nat Immunol (2020) 21:868–79. doi: 10.1038/s41590-020-0730-5
70. Li Y, James SJ, Wyllie DH, Wynne C, Czibula A, Bukhari A, et al. TMEM203 Is a Binding Partner and Regulator of STING-Mediated Inflammatory Signaling in Macrophages. Proc Natl Acad Sci USA (2019) 116(33):16479–88. doi: 10.1073/pnas.1901090116
71. Jing Y, Dai X, Yang L, Kang D, Jiang P, Li N, et al. STING Couples With PI3K to Regulate Actin Reorganization During BCR Activation. Sci Adv (2020) 6:eaax9455. doi: 10.1126/sciadv.aax9455
72. van Vliet AR, Giordano F, Gerlo S, Segura I, Van Eygen S, Molenberghs G, et al. the Er Stress Sensor Perk Coordinates Er-Plasma Membrane Contact Site Formation Through Interaction With Filamin-a and F-Actin Remodeling. Mol Cell (2017) 65:885–99.e886. doi: 10.1016/j.molcel.2017.01.020
73. Guet D, Mandal K, Pinot M, Hoffmann J, Abidine Y, Sigaut W, et al. Mechanical Role of Actin Dynamics in the Rheology of the Golgi Complex and in Golgi-Associated Trafficking Events. Curr Biol (2014) 24:1700–11. doi: 10.1016/j.cub.2014.06.048
74. Ravichandran Y, Goud B, Manneville JB. The Golgi Apparatus and Cell Polarity: Roles of the Cytoskeleton, the Golgi Matrix, and Golgi Membranes. Curr Opin Cell Biol (2020) 62:104–13. doi: 10.1016/j.ceb.2019.10.003
75. Kruppa AJ, Kendrick-Jones J, Buss F. Myosins, Actin and Autophagy. Traffic (2016) 17:878–90. doi: 10.1111/tra.12410
76. Kast DJ, Zajac AL, Holzbaur EL, Ostap EM, Dominguez R. Whamm Directs the Arp2/3 Complex to the ER for Autophagosome Biogenesis Through an Actin Comet Tail Mechanism. Curr Biol (2015) 25:1791–7. doi: 10.1016/j.cub.2015.05.042
77. Coutts AS, La Thangue NB. Actin Nucleation by WH2 Domains At the Autophagosome. Nat Commun (2015) 6:7888. doi: 10.1038/ncomms8888
78. Mi N, Chen Y, Wang S, Chen M, Zhao M, Yang G, et al. CapZ Regulates Autophagosomal Membrane Shaping by Promoting Actin Assembly Inside the Isolation Membrane. Nat Cell Biol (2015) 17:1112–23. doi: 10.1038/ncb3215
79. Lee JY, Koga H, Kawaguchi Y, Tang W, Wong E, Gao YS, et al. Hdac6 Controls Autophagosome Maturation Essential for Ubiquitin-Selective Quality-Control Autophagy. EMBO J (2010) 29:969–80. doi: 10.1038/emboj.2009.405
80. Lee PP, Lobato-Marquez D, Pramanik N, Sirianni A, Daza-Cajigal V, Rivers E, et al. Wiskott-Aldrich Syndrome Protein Regulates Autophagy and Inflammasome Activity in Innate Immune Cells. Nat Commun (2017) 8:1576. doi: 10.1038/s41467-017-01676-0
81. Rivers E, Rai R, Lotscher J, Hollinshead M, Markelj G, Thaventhiran J, et al. Wiskott Aldrich Syndrome Protein Regulates non-Selective Autophagy and Mitochondrial Homeostasis in Human Myeloid Cells. Elife (2020) 9:e55547. doi: 10.7554/eLife.55547
82. Bartolak-Suki E, Imsirovic J, Nishibori Y, Krishnan R, Suki B. Regulation of Mitochondrial Structure and Dynamics by the Cytoskeleton and Mechanical Factors. Int J Mol Sci (2017) 18(8):1812. doi: 10.3390/ijms18081812
83. Korobova F, Ramabhadran V, Higgs HN. An Actin-Dependent Step in Mitochondrial Fission Mediated by the ER-Associated Formin INF2. Science (2013) 339:464–7. doi: 10.1126/science.1228360
84. Moore AS, Coscia SM, Simpson CL, Ortega FE, Wait EC, Heddleston JM, et al. Actin Cables and Comet Tails Organize Mitochondrial Networks in Mitosis. Nature (2021) 591:659–64. doi: 10.1038/s41586-021-03309-5
85. Hsieh CW, Yang WY. Triggering Mitophagy With Photosensitizers. Methods Mol Biol (2019) 1880:611–9. doi: 10.1007/978-1-4939-8873-0_40
86. Hurst V, Shimada K, Gasser SM. Nuclear Actin and Actin-Binding Proteins in DNA Repair. Trends Cell Biol (2019) 29:462–76. doi: 10.1016/j.tcb.2019.02.010
87. Lamm N, Read MN, Nobis M, Van Ly D, Page SG, Masamsetti VP, et al. Nuclear F-Actin Counteracts Nuclear Deformation and Promotes Fork Repair During Replication Stress. Nat Cell Biol (2020) 22:1460–70. doi: 10.1038/s41556-020-00605-6
88. Schrank BR, Aparicio T, Li Y, Chang W, Chait BT, Gundersen GG, et al. Nuclear Arp2/3 Drives Dna Break Clustering for Homology-Directed Repair. Nature (2018) 559:61–6. doi: 10.1038/s41586-018-0237-5
89. Kidiyoor GR, Li Q, Bastianello G, Bruhn C, Giovannetti I, Mohamood A, et al. Atr is Essential for Preservation of Cell Mechanics and Nuclear Integrity During Interstitial Migration. Nat Commun (2020) 11:4828. doi: 10.1038/s41467-020-18580-9
90. Hartlova A, Erttmann SF, Raffi FA, Schmalz AM, Resch U, Anugula S, et al. Dna Damage Primes the Type I Interferon System Via the Cytosolic Dna Sensor Sting to Promote Anti-Microbial Innate Immunity. Immunity (2015) 42:332–43. doi: 10.1016/j.immuni.2015.01.012
91. Raab M, Gentili M, de Belly H, Thiam HR, Vargas P, Jimenez AJ, et al. Escrt Iii Repairs Nuclear Envelope Ruptures During Cell Migration to Limit Dna Damage and Cell Death. Science (2016) 352:359–62. doi: 10.1126/science.aad7611
92. Vietri M, Schink KO, Campsteijn C, Wegner CS, Schultz SW, Christ L, et al. Spastin and Escrt-Iii Coordinate Mitotic Spindle Disassembly and Nuclear Envelope Sealing. Nature (2015) 522:231–5. doi: 10.1038/nature14408
93. Maciejowski J, Hatch EM. Nuclear Membrane Rupture and its Consequences. Annu Rev Cell Dev Biol (2020) 36:85–114. doi: 10.1146/annurev-cellbio-020520-120627
94. Mu X, Tseng C, Hambright WS, Matre P, Lin CY, Chanda P, et al. Cytoskeleton Stiffness Regulates Cellular Senescence and Innate Immune Response in Hutchinson-Gilford Progeria Syndrome. Aging Cell (2020) 19:e13152. doi: 10.1111/acel.13152
95. Kawane K, Ohtani M, Miwa K, Kizawa T, Kanbara Y, Yoshioka Y, et al. Chronic Polyarthritis Caused by Mammalian Dna That Escapes From Degradation in Macrophages. Nature (2006) 443:998–1002. doi: 10.1038/nature05245
96. Ahn J, Gutman D, Saijo S, Barber GN. STING Manifests Self DNA-Dependent Inflammatory Disease. Proc Natl Acad Sci USA (2012) 109:19386–91. doi: 10.1073/pnas.1215006109
97. Gao D, Li T, Li XD, Chen X, Li QZ, Wight-Carter M, et al. Activation of Cyclic GMP-AMP Synthase by Self-DNA Causes Autoimmune Diseases. Proc Natl Acad Sci USA (2015) 112:E5699–705. doi: 10.1073/pnas.1516465112
98. King KR, Aguirre AD, Ye YX, Sun Y, Roh JD, Ng RP, et al. IRF3 and Type I Interferons Fuel a Fatal Response to Myocardial Infarction. Nat Med (2017) 23:1481–7. doi: 10.1038/nm.4428
99. Zentsova I, Parackova Z, Kayserova J, Palova-Jelinkova L, Vrabcova P, Volfova N, et al. Monocytes Contribute to DNA Sensing Through the TBK1 Signaling Pathway in Type 1 Diabetes Patients. J Autoimmun (2019) 105:102294. doi: 10.1016/j.jaut.2019.06.005
100. Xu MM, Pu Y, Han D, Shi Y, Cao X, Liang H, et al. Dendritic Cells But Not Macrophages Sense Tumor Mitochondrial DNA for Cross-Priming Through Signal Regulatory Protein α Signaling. Immunity (2017) 47:363–373.e365. doi: 10.1016/j.immuni.2017.07.016
101. Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, et al. Neutrophils Activate Plasmacytoid Dendritic Cells by Releasing Self-DNA-Peptide Complexes in Systemic Lupus Erythematosus. Sci Transl Med (2011) 3:73ra19. doi: 10.1126/scitranslmed.3001180
102. Lande R, Gregorio J, Facchinetti V, Chatterjee B, Wang YH, Homey B, et al. Plasmacytoid Dendritic Cells Sense Self-Dna Coupled With Antimicrobial Peptide. Nature (2007) 449:564–9. doi: 10.1038/nature06116
103. Chamilos G, Gregorio J, Meller S, Lande R, Kontoyiannis DP, Modlin RL, et al. Cytosolic Sensing of Extracellular Self-Dna Transported Into Monocytes by the Antimicrobial Peptide Ll37. Blood (2012) 120:3699–707. doi: 10.1182/blood-2012-01-401364
104. Yang H, Biermann MH, Brauner JM, Liu Y, Zhao Y, Herrmann M. New Insights into Neutrophil Extracellular Traps: Mechanisms of Formation and Role in Inflammation. Front Immunol (2016) 7. doi: 10.3389/fimmu.2016.00302
105. Duleh SN, Welch MD. WASH and the Arp2/3 Complex Regulate Endosome Shape and Trafficking. Cytoskeleton (Hoboken) (2010) 67:193–206. doi: 10.1002/cm.20437
106. Wegner CS, Malerod L, Pedersen NM, Progida C, Bakke O, Stenmark H, et al. Ultrastructural Characterization of Giant Endosomes Induced by Gtpase-Deficient Rab5. Histochem Cell Biol (2010) 133:41–55. doi: 10.1007/s00418-009-0643-8
107. Kaur G, Lakkaraju A. Early Endosome Morphology in Health and Disease. Adv Exp Med Biol (2018) 1074:335–43.
108. Skowyra ML, Schlesinger PH, Naismith TV, Hanson PI. Triggered Recruitment of ESCRT Machinery Promotes Endolysosomal Repair. Science (2018) 360:eaar5078. doi: 10.1126/science.aar5078
109. MacDonald E, Brown L, Selvais A, Liu H, Waring T, Newman D, et al. Hrs-Wash Axis Governs Actin-Mediated Endosomal Recycling and Cell Invasion. J Cell Biol (2018) 217:2549–64. doi: 10.1083/jcb.201710051
110. Roy ER, Wang B, Wan YW, Chiu G, Cole A, Yin Z, et al. Type I Interferon Response Drives Neuroinflammation and Synapse Loss in Alzheimer Disease. J Clin Invest (2020) 130:1912–30. doi: 10.1172/JCI133737
111. Vitner EB, Farfel-Becker T, Ferreira NS, Leshkowitz D, Sharma P, Lang KS, et al. Induction of the Type I Interferon Response in Neurological Forms of Gaucher Disease. J Neuroinflamm (2016) 13:104. doi: 10.1186/s12974-016-0570-2
112. Baptista MA, Keszei M, Oliveira M, Sunahara KK, Andersson J, Dahlberg CI, et al. Deletion of Wiskott-Aldrich Syndrome Protein Triggers Rac2 Activity and Increased Cross-Presentation by Dendritic Cells. Nat Commun (2016) 7:12175. doi: 10.1038/ncomms12175
113. Belabed M, Mauvais FX, Maschalidi S, Kurowska M, Goudin N, Huang JD, et al. Kinesin-1 Regulates Antigen Cross-Presentation Through the Scission of Tubulations From Early Endosomes in Dendritic Cells. Nat Commun (2020) 11:1817. doi: 10.1038/s41467-020-15692-0
114. Weimershaus M, Mauvais FX, Saveanu L, Adiko C, Babdor J, Abramova A, et al. Innate Immune Signals Induce Anterograde Endosome Transport Promoting Mhc Class I Cross-Presentation. Cell Rep (2018) 24:3568–81. doi: 10.1016/j.celrep.2018.08.041
115. Papa R, Penco F, Volpi S, Gattorno M. Actin Remodeling Defects Leading to Autoinflammation and Immune Dysregulation. Front Immunol (2020) 11:604206. doi: 10.3389/fimmu.2020.604206
116. Burianek LE, Soderling SH. Under Lock and Key: Spatiotemporal Regulation of WASP Family Proteins Coordinates Separate Dynamic Cellular Processes. Semin Cell Dev Biol (2013) 24:258–66. doi: 10.1016/j.semcdb.2012.12.005
117. Alekhina O, Burstein E, Billadeau DD. Cellular Functions of WASP Family Proteins At a Glance. J Cell Sci (2017) 130:2235–41. doi: 10.1242/jcs.199570
118. Piperno GM, Naseem A, Silvestrelli G, Amadio R, Evelia C-LK, Nalan L, et al. Wiskott-Aldrich Syndrome Protein Restricts Cgas-STING Activation by Dsdna Immune Complexes. JCI Insight (2020) 5(17):e132857. doi: 10.1172/jci.insight.132857
119. Catucci M, Castiello MC, Pala F, Bosticardo M, Villa A. Autoimmunity in Wiskott-Aldrich Syndrome: An Unsolved Enigma. Front Immunol (2012) 3:209. doi: 10.3389/fimmu.2012.00209
120. Marangoni F, Trifari S, Scaramuzza S, Panaroni C, Martino S, Notarangelo LD, et al. Wasp Regulates Suppressor Activity of Human and Murine Cd4(+)Cd25(+)Foxp3(+) Natural Regulatory T Cells. J Exp Med (2007) 204:369–80. doi: 10.1084/jem.20061334
121. Castiello MC, Bosticardo M, Pala F, Catucci M, Chamberlain N, van Zelm MC, et al. Wiskott-Aldrich Syndrome Protein Deficiency Perturbs the Homeostasis of B-Cell Compartment in Humans. J Autoimmun (2014) 50:42–50. doi: 10.1016/j.jaut.2013.10.006
122. Crestani E, Volpi S, Candotti F, Giliani S, Notarangelo LD, Chu J, et al. Broad Spectrum of Autoantibodies in Patients With Wiskott-Aldrich Syndrome and X-Linked Thrombocytopenia. J Allergy Clin Immunol (2015) 136(5):1401–4.e1–3. doi: 10.1016/j.jaci.2015.08.010
123. Sereni L, Castiello MC, Marangoni F, Anselmo A, di Silvestre D, Motta S, et al. Autonomous Role of Wiskott-Aldrich Syndrome Platelet Deficiency in Inducing Autoimmunity and Inflammation. J Allergy Clin Immunol (2018) 142:1272–84. doi: 10.1016/j.jaci.2017.12.1000
124. Candotti F. Clinical Manifestations and Pathophysiological Mechanisms of the Wiskott-Aldrich Syndrome. J Clin Immunol (2018) 38:13–27. doi: 10.1007/s10875-017-0453-z
125. Rivers E, Worth A, Thrasher AJ, Burns SO. How I Manage Patients With Wiskott Aldrich Syndrome. Br J Haematol (2019) 185:647–55. doi: 10.1111/bjh.15831
126. Prete F, Catucci M, Labrada M, Gobessi S, Castiello MC, Bonomi E, et al. Wiskott-Aldrich syndrome protein-mediated actin dynamics control type-I interferon production in plasmacytoid dendritic cells. J Exp Med (2013) 210:355–74. doi: 10.1084/jem.20120363
127. Cervantes-Luevano KE, Caronni N, Castiello MC, Fontana E, Piperno GM, Naseem A, et al. Neutrophils Drive Type I Interferon Production and Autoantibodies in Patients With Wiskott-Aldrich Syndrome. J Allergy Clin Immunol (2018) 142(5):P1605–17.E4. doi: 10.1016/j.jaci.2017.11.063
128. Senju Y, Kalimeri M, Koskela EV, Somerharju P, Zhao H, Vattulainen I, et al. Mechanistic Principles Underlying Regulation of the Actin Cytoskeleton by Phosphoinositides. Proc Natl Acad Sci USA (2017) 114:E8977–86. doi: 10.1073/pnas.1705032114
Keywords: self-DNA, actin cytoskeleton, cGAS-STING pathway, phagocytes, Wiskott-Aldrich syndrome
Citation: Amadio R, Piperno GM and Benvenuti F (2021) Self-DNA Sensing by cGAS-STING and TLR9 in Autoimmunity: Is the Cytoskeleton in Control? Front. Immunol. 12:657344. doi: 10.3389/fimmu.2021.657344
Received: 22 January 2021; Accepted: 06 April 2021;
Published: 18 May 2021.
Edited by:
Samuel Cern Cher Chiang, Cincinnati Children’s Hospital Medical Center, United StatesReviewed by:
Erin Janssen, Boston Children’s Hospital and Harvard Medical School, United StatesLisa S. Westerberg, Karolinska Institutet (KI), Sweden
Kerstin Nundel, University of Massachusetts Medical School, United States
Copyright © 2021 Amadio, Piperno and Benvenuti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Federica Benvenuti, YmVudmVudXRAaWNnZWIub3Jn