 Yoshiko Matsuda
Yoshiko Matsuda Takeshi Watanabe
Takeshi Watanabe Xiao-Kang Li
Xiao-Kang Li- 1Division of Transplantation Immunology, National Research Institute for Child Health and Development, Tokyo, Japan
- 2Laboratory of Immunology, Institute for Frontier Life and Medical Sciences, Kyoto University, Kyoto, Japan
Both acute and chronic antibody-mediated allograft rejection (AMR), which are directly mediated by B cells, remain difficult to treat. Long-lived plasma cells (LLPCs) in bone marrow (BM) play a crucial role in the production of the antibodies that induce AMR. However, LLPCs survive through a T cell-independent mechanism and resist conventional immunosuppressive therapy. Desensitization therapy is therefore performed, although it is accompanied by severe side effects and the pathological condition may be at an irreversible stage when these antibodies, which induce AMR development, are detected in the serum. In other words, AMR control requires the development of a diagnostic method that predicts its onset before LLPC differentiation and enables therapeutic intervention and the establishment of humoral immune monitoring methods providing more detailed information, including individual differences in the susceptibility to immunosuppressive agents and the pathological conditions. In this study, we reviewed recent studies related to the direct or indirect involvement of immunocompetent cells in the differentiation of naïve-B cells into LLPCs, the limitations of conventional methods, and the possible development of novel control methods in the context of AMR. This information will significantly contribute to the development of clinical applications for AMR and improve the prognosis of patients who undergo organ transplantation.
Introduction
Experiments by using thymectomized mice or chickens conducted during the mid-1960s show that T cell mediated immunity in tissue and organ allografts. Consequently, most immunosuppressive therapies for preventing allograft rejection were targeted on T cells, and the studies of the in vitro effects of immunosuppressive agents against the proliferating T cells significantly contributed to controlling T cell-mediated rejection (TCMR) (1). Although T cells mediate the activation of the humoral immune response to transplanted grafts through activating B cells, efforts to control antibody-mediated allograft rejection (AMR) using conventional immunosuppressive therapy have been still challenging until now (2–16).
Therefore, further understanding of B cell biology related to the differentiation of long-lived plasma cells (LLPCs) and the regulation of the production of antibodies that induce the development of AMR are required to improve disease prognosis.
During AMR development, naïve-B cells recognize donor-specific human leukocyte antigen (HLA) and differentiate into activated B cells. These activated B cells undergo negative selection in germinal centers (GCs), and only cells with high affinity for donor-specific HLA survive, subsequently differentiate into memory B cells (MBCs) or migrate into the bone marrow (BM), and differentiate into LLPCs. LLPCs maintain long-term donor-specific HLA antibodies (DSAs) production (17–19) and conventional immunosuppressive therapy is ineffective for removing these PCs, because their survival is independent on the activities of T cells and expression of CD20 is reduced on these PCs (19, 20). Moreover, the intramedullary environment may prevent these PCs from undergoing apoptosis induced by desensitization therapy (21). Therefore, the development of accurate and rapid techniques to evaluate humoral immune activation targeting transplanted grafts, independent of antibodies, are urgently required to control AMR.
Identifying of the antigen specificity of MBCs circulating in the peripheral blood, as measured using an ELISpot assay or in vitro assay system, will be useful to evaluate the activation of the humoral immune response to donor-specific HLA antigens (22–27). Furthermore, microarray techniques and next-generation sequencing (NGS) may provide detailed information that will contribute to therapeutic control of AMR, including the identification of antibodies that injure transplants and individual differences in patients’ susceptibilities to immunosuppressive agents (28–36).
In addition, the components of humoral immunity involved in immune tolerance as well as in the repair of injured tissues are attracting attention, particularly in the fields of autoimmune diseases, severe infectious diseases, and others (37–42). Here, we discuss the possibility of AMR control by referring to the involvement mechanism of these components in the humoral immunity-associated pathology and the possibilities of these components as well as novel humoral immune monitoring in solving the problems associated with conventional AMR control.
Molecular Pathophysiology in AMR
In the pathways or events leading to the development AMR, molecules such as donor-specific HLA antigens, non-HLA antigens, and self-antigens are recognized by antigen-presenting cells (APCs) that express the major histocompatibility complex (MHC) II on their surface. These latter molecules are presented to follicular helper T cells (Tfhs) through the interaction of MHC II with the T cell receptor (TCR), resulting in the activation of Tfhs (17). The B cell receptor (BCR), which is expressed on the surface of naïve-B cells, is activated through cross-linking to the aforementioned antigens in a T cell-dependent manner, and B cells monoclonally proliferate after stimulation by activated Tfhs in secondary lymphoid tissues. These activated B cells form GCs. After immunoglobulin class switching, they undergo affinity maturation and the cells with low affinity for foreign antigens or high affinity for self-antigens undergo apoptosis as a negative selection mechanism, followed by differentiation into MBCs or LLPCs that bind these antigens with high affinity. MBCs migrate to secondary lymphoid tissue to counter the ensuing invasion by foreign antigens, and LLPCs persist and continue to produce antibodies that induce AMR (17, 18, 43–45) (Figure 1).
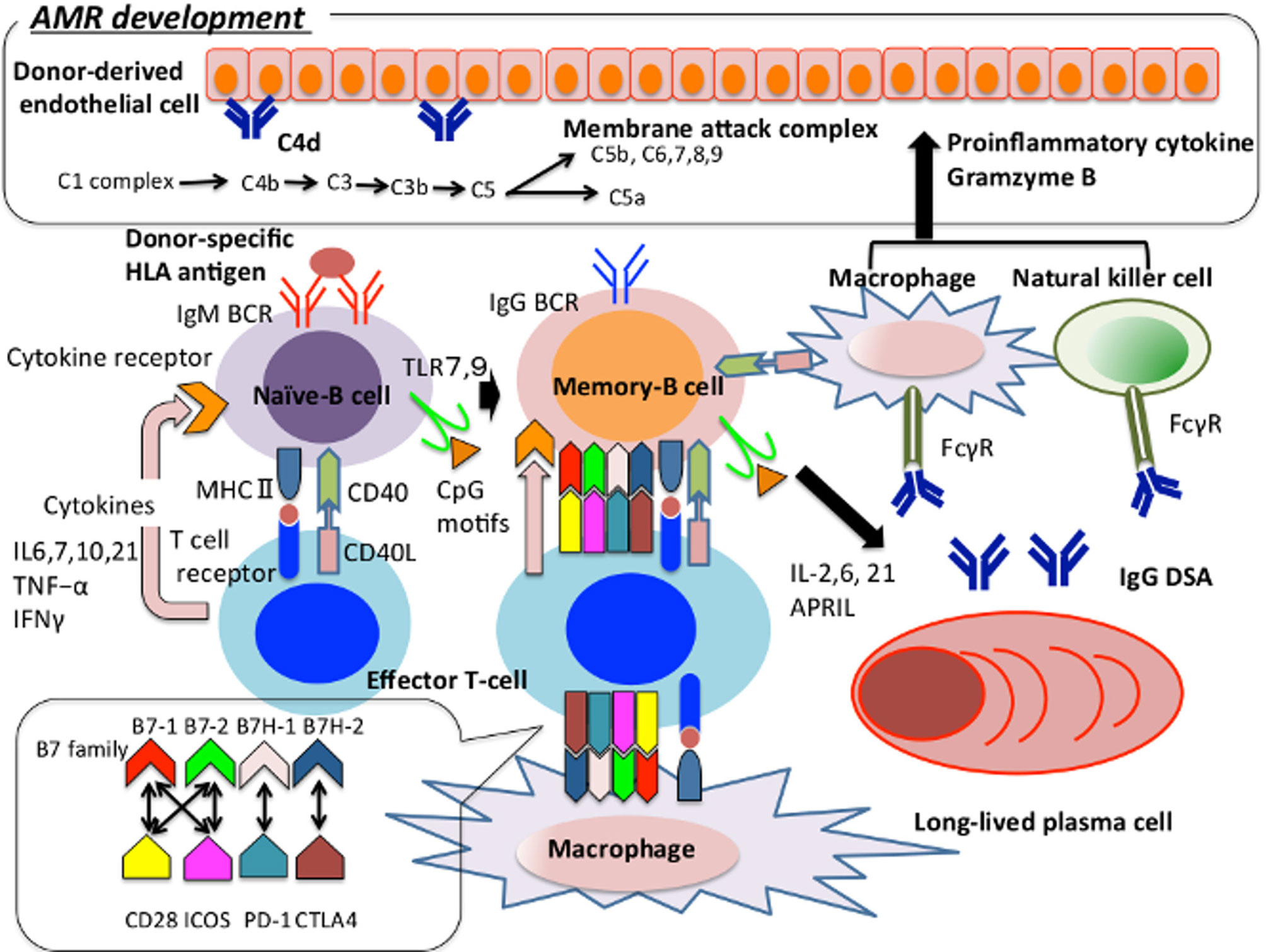
Figure 1 The pathway of naïve-B cell differentiation into plasma cell in the context of AMR development. We show how immunocompetent cells are involved in the onset of AMR, focusing on B cells. Naïve-B cell recognises donor-specific HLA antigen and present the antigen peptides from MHCII to T cell receptor and activate T cells. The activated T cells regulate the growth and survival of B cells through the production of IL4, 6, 7, 10, 21, TNF-α, IFNγ, etc (46). Activated B cell migrates to secondary lymphoid tissue, class-switches to IgG and then undergoes gene conversion and hypersomatic mutation in the germinal center to have a high affinity for donor-specific HLA antigen. These cells migrate into the bone marrow and differentiate into long-lived plasma cells, and keep producing IgG DSAs for a long term. Furthermore, TLR 7/9 is expressed on the surface of naïve-B cells. TLR7 plays an important role in T cell activation and germinal center B-cell development (24). TLR9 recognizes the CpG motif and supports the survival and proliferation of B cells through IL-6 production and T cell activation (47). T cells regulate B cells’ activation and sustain the interaction between follicular helper T cells and germinal center B cells through the association between the B7 family and CD28, ICOS, PD-1, CTLA-4 expressed by B cells, and CD 40 ligand and CD40 expressed by B cells. Regarding macrophages and natural killer cells function as antigen-presenting cells and induce B cells and T cells activation. IgG-type DSA binds to Fcγ on the macrophages and natural killer cells’ surface and activates them. Complement activates adaptive immunity against thymus dependent antigen, or it activates the membrane attack complex and induces inflammation and tissue damage on vascular endothelial cells, thereby supporting the onset of AMR. AMR, Antibody-mediated allograft rejection; APRIL, A proliferation-inducing ligand; BCR, B cell receptor; CD40L, CD40 ligand; CTLA-4, Cytotoxic T-lymphocyte associated antigen 4; DSA, Donor-specific HLA antibody; FcγR, Fc gamma receptor; IFN, Interferon; ICOS, Inducible T-cell co-stimulator; MHC, Major histocompatibility complex; PD-1, Programmed cell death – 1; TNF, Tumor Necrosis Factor; TLR, Tool-like receptor.
Regarding the involvement of T cell in AMR development, Tfhs reside in the peripheral secondary lymphoid tissues and strongly promote the production of antigen-specific antibodies through the support of the GC reactions. The B7 (CD80/86) family of costimulatory molecules B7-1 and B7-2 binds to CD28 and cytotoxic T-lymphocyte associated antigen 4 (CTLA-4) on the surface of T cells binds more strongly to CD80/86 than to CD28 to release control over the generation of Tfhs (2). Human B7 homolog 1 (B7H-1) binds programmed death-ligand 1 (PD-L1) expressed by activated T cells and inhibits T cell activation through the interaction between PD-L1 and programmed cell death1 (PD-1). Human B7 homolog 2 (B7H-2) binds inducible T cell costimulator (ICOS) expressed by CD4/CD8 T cells to increase the secretion of interferon (IFN)-γ and interleukin (IL)-10. ICOS-ligand is expressed on the surface of B cells and supports their migration into B follicles (3, 4). In addition, Thfs support the formation of the GC through the interaction between CD40 and CD40-ligand (CD40-L) (5–9); and the G-protein-coupled receptor sphingosine-1-phosphate receptor 2 (S1PR2) cooperates with CXCR5 to retain Thfs in the GC (10). Members of the signaling lymphocyte activation molecule (SLAM) family act to maintain stable contacts between Tfhs and GC B cells to maintain the differentiation of Thfs, formation of the GC, and proliferation and survival of LLPCs and MBCs (11–16). In contrast, follicular regulatory T cells (Tfrs) inhibit the production of IL-21 by Thfs, and the CTLA-4-mediated signals cooperate with IL-10 and transforming growth factor (TGF)-beta to support the inhibitory effect on humoral immunity (48).
Macrophages (MPs) and natural killer (NK) cells also play an important role in the development of AMR (49). MPs present antigens to Tfhs via MHC class II, serve as a source of co-stimulatory signaling and cytokines required for activation of T cells, and subsequently activate B cells (50). Moreover, IgG DSAs have been revealed to bind to Fc gamma receptor (FcγR) such as FcγRIIIa on the cell membrane of MPs and NK cells, and subsequently, these cells are activated. These activated cells produce proinflammatory cytokines such as IFN-γ, Tumor Necrosis Factor (TNF), and granzyme B, which induce coagulation, inflammation, vascular permeability, and leukocyte trafficking on the vascular endothelium (51).
Regarding the involvement of the complement pathway in AMR development, the humoral immune response to thymus-dependent (TD) antigens requires complementary activation, which is required for the localization of the antigen and C3 ligand to follicular dendritic cells (FDCs). These events maintain the long-term memory function of B cells (52).
As the detailed involvement mechanism in B cell activation and proliferation, the interaction of C3 fragments with CD21 is required for the internalization of antigens by B cell and their presentation. CD21 is a receptor that binds to complement C3d and Epstein–Barr virus and is expressed by mature B cells and FDCs. CD-21 plays a crucial role in B cell activation (53). The internalized antigen is presented to T cells by MHCs II and I, and it then activates T cells (54–57). Therefore, complement activation is required for the uptake of antigen by B cell and inhibiting CD21-mediated signal with polyclonal antibodies significantly inhibits antigen uptake and presentation. Furthermore, B cell uptake of C3d-coated antigen and engagement of the BCR and CD21 by C3d-opsonized antigen is required for the formation and maintenance of the GC and differentiation of MBCs into PCs (57, 58). CD35, an antagonist of CD21, inhibits B cell activation (59), and the CD21/CD19/Target of the Antiproliferative Antibody-1 (Tapa-1) receptor reduces the threshold of survival of follicular B cells and serves as a unique signal that promotes their survival in the GCs, which, in turn, contributes to adaptive immunity (52).
Therefore, complement activation indirectly induces the development of AMR through B cell activation or directly induces inflammation and damage in vascular endothelial cells through formation of the membrane attack complex (60).
Challenges of Conventional AMR Control
Although the pathway described in “MOLECULAR PATHOPHYSIOLOGY IN AMR” is inhibited by immunosuppressive therapy, AMR control remains challenging.
The cause is that signals from immunocompetent cells, including T cells, to B cells are not well regulated, and humoral immune responses to transplanted graft have not been accurately detailed.
Furthermore, B cell subsets include IL-10-producing regulatory B cells (Bregs) (CD19+CD24highCD38high transitional immature B cells), whose reduction is associated with the incidence of posttransplant rejection (61, 62). Regulatory T cells (Tregs) induce tolerance to transplanted grafts through suppression of the humoral immune response. Calcineurin inhibitor (CNI) and mammalian Target of Rapamycin (mTOR) reduce the levels of Bregs, whereas mTOR expands and CNI reduces the levels of Tregs (63). Under pathological conditions, common variable immunodeficiency (CVID) is a pathology characterized by the loss of resistance to bacterial and viral infections due to decreased antibody production. This disease is associated with decreased expression of a proliferation-inducing ligand (APRIL), which supports the growth and survival of MBCs and PCs. An anti-APRIL antibody may serve as a therapeutic agent for SLE (64, 65). In contrast, APRIL supports the growth and survival of naïve-B cells in vitro, and administration of an anti-APRIL antibody may therefore suppress immune tolerance mediated by naïve-B cells (66).
In the other words, some immunosuppressive agents may inhibit the normal functions of these regulatory cells and the identification of the mechanisms through which the populations of these regulatory cells are reduced by immunosuppressive therapy and the differences of drug susceptibilities in subset-specific lymphocyte may help development of more appropriated immunosuppressive agents that maintain the functions of these regulatory cells. For example, IL-2 lengthens the survival and enhances the suppressive effects of Tregs, which increases the survival rates of transplanted grafts, because the decrease in the number of Tregs by CNI is the result of limiting the activity of IL-2 (67).
In addition, desensitization therapies such as plasmapheresis and low-dose intravenous immunoglobulin (IVig), alone or combined with recombinant antithymocyte globulin (rATG), do not significantly influence the number of CD138+ antibody-secreting cells (ASCs), alloantibody production, and the frequency of HLA-producing ASCs in the BM before and after treatment (68). The resistance of ASCs to IVig and rATG may be explained by the inhibition of apoptosis mediated by the microenvironment of the BM and soluble and factors produced from the BM niche (21). Alternatively, the interaction of some receptors expressed on LLPC and their ligands expressed on niche play a critical role in the migration of LLPCs to the BM and their longevity in the BM (20, 69, 70). In the other words, the removal of antibody from the serum using desensitization therapy may not be mediated by the reduction of the numbers and functions of ASCs, but by antibody adsorption to the transplanted graft (68). Thus, further elucidation of the mechanisms of drug resistance in LLPCs and blockade of the signals supporting the migration of LLPCs into the BM and their longevity in the BM may contribute to the development of more effective immunosuppressive therapy.
As a diagnostic method, it is necessary for AMR control to establish a humoral immune monitoring method to evaluate AMR pathology in more detail and predict the development of AMR before antibodies, which induce AMR development, are detected in the serum (71, 72). In addition, it has been reported that the antigen specificity of the progenitor cells of LLPCs can be determined using an in vitro assay system (22–27), but there are no unambiguous criteria for predicting whether antibodies against donor-specific HLA antigens cause humoral immunity-mediated injury in transplanted grafts (73–75). It is therefore necessary to develop a method that evaluates the reactivity of the antibody to transplanted grafts.
Development in AMR Control
Agents Targeting Complement System
The involvement of the complement system in the development of AMR has been reported. In the field of transplantation, the effects of C1-q–positive DSAs on the development of AMR and the incidence of glomerulopathy, as well as the prognosis of transplantation outcomes, have been reported administration of the humanized anti-C5 monoclonal antibody eculizumab, a C5 inhibitor, inhibits the cleavage of C5 to C5a and C5b and the formation of the membrane attack complex C5b-9, and this drug is effective against acute AMR, as indicated by its effective improvement of histopathology in lung transplantation (76–79). In immunologically high-risk cases, the incidence of biopsy-proven AMR following heart or kidney transplantation was significantly lower than that achieved using conventional antibody reduction therapy (80–83).
In addition, C1 esterase inhibitor (C1 INHs) effectively prevents ischemia reperfusion injury (IRI)/delayed graft function (DGF), which has been reported to be involved in the development of AMR through B cell activation, DSA development, and C1q-positive DSA production (84–86). Thus, C1 INHs can be expected to be effective in the maintenance of transplanted graft function by suppressing the aforementioned processes (87, 88). In contrast, no significant inhibitory effects on DSA production and the development of chronic AMR were observed in long-term DSA-positive patients, and no therapeutic effect on AMR was observed in patients with complement-negative DSA (86). Thus, this complement inhibitor should be considered in combination with antibody reduction therapy for AMR control, and it has been reported that transplanted renal graft function and pathological findings are improved by treatment with the standard AMR treatments such as plasmapheresis, IVig, and human plasma-derived C1 INHs (88, 89).
The classic complement pathway inhibitor anti-C1s inhibits C4d deposition, but it does not affect DSA levels and graft function or significantly improve the pathological findings associated with AMR control and clinical prognosis (90, 91). Therefore, it is necessary to elucidate the mechanism by which these complement inhibitors participate in the development of AMR in more detail and improve treatment outcomes in consideration of the administration time and method.
Agents Targeting Costimulatory Signaling
As a clinical potential of agents targeting costimulatory signaling, the fusion protein belatacept comprises the Fc fragment of human IgG1 linked to the extracellular domain of CTLA-4 and inactivates T cells selectively (92).
In immunosuppressed patients who received of administration of belatacept, their survival and function of their grafts are significantly higher after 7 years of kidney transplantation (93). Compared with cyclosporine-based immunosuppression, this therapy is efficacious for maintaining the function of the transplanted grafts, reduces cardiovascular complications, improves the metabolic profile, and mitigates posttransplant lymphoproliferative disease associated with Epstein-Barr virus infection 1–2 years after transplantation (94). In mice model, an example of an immunosuppressive therapy that may effectively treat DSA-specific MBCs is provided using findings collapsing the GC in a mouse model 7 and 14 days after allo-sensitization directly involves B cells, independent of intervention by graft-specific CD4+ Tfhs. Therefore, administration of CTLA- 4 Ig may exert a therapeutic effect on antigen-challenged B cells (95). In contrast, belatacept inhibits the expansion of the Tregs population, which is mediated through the methylation of CpG islands within the Treg-specific demethylation region of the gene encoding FoxP3 (96, 97).
The CTLA-4 Ig fusion protein abatacept, which inhibits CD28-mediated costimulatory signals by binding to CD80/CD86 on the surface of APCs, effectively suppresses T cell activation and cytokine production, which are associated with the development of rheumatoid arthritis (98–100). In the field of transplantation, the function of the transplanted graft persists, and no obvious immunological consequences are observed through the conversion from treatment of a CNI early after transplantation (100).
Action of conventional immunosuppressive therapies and these novel therapies targeting the complement system and co-stimulatory signaling in immune system and their clinical potential for AMR control, their clinical potential for AMR control, are presented in Table 1 and the pathways of differentiation of naïve-B cells into LLPCs and the immunosuppressive agents that influence these processes are shown in Figure 2. In addition, the involvement mechanism and clinical potential of cytokines involved in the differentiation of naïve-B cells into LLPCs in the context of AMR are summarized in Table 2A, and the clinical applicability of these molecules and possible use in AMR control are summarized in Table 2B.

Table 1 Effects of immunosuppressive therapy on the immune system and its clinical role in AMR control.
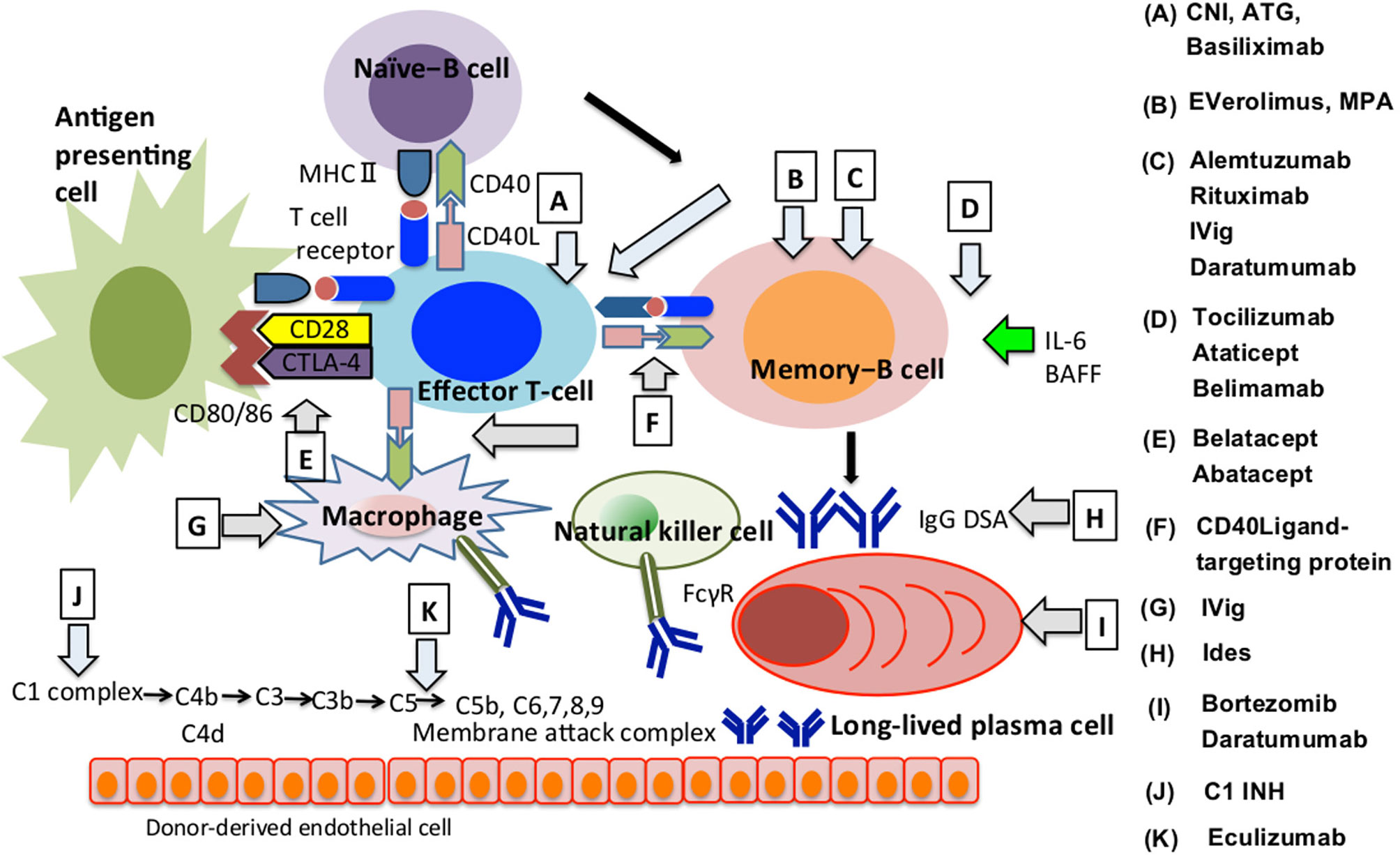
Figure 2 How immunosuppressive agents influence the AMR development. We summarised how immunosuppressants are involved in regulating AMR onset at each B cell differentiation stage, direct or indirect involvement of T cells, macrophages or natural killer cells and the complement system. The alphabet represents immunosuppressants that are effective in the differentiation process indicated by arrows. AMR, Antibody-mediated allograft rejection; ATG, Antithymocyte globulin; BAFF, B Cell Activating Factor; CD40L, CD40 ligand; CTLA-4, Cytotoxic T-lymphocyte associated antigen 4; DSA, Donor-specific HLA antibody; FcγR, Fc gamma receptor; C1 INH, Human plasma-derived C1 esterase inhibitor; CNI, Calcineurin inhibitor; Ides, IgG-degrading enzyme of streptococcus pyogenes; IVig, Intravenous Immunoglobulin; MHC, Major histocompatibility complex; MPA, mycophenolic acid.
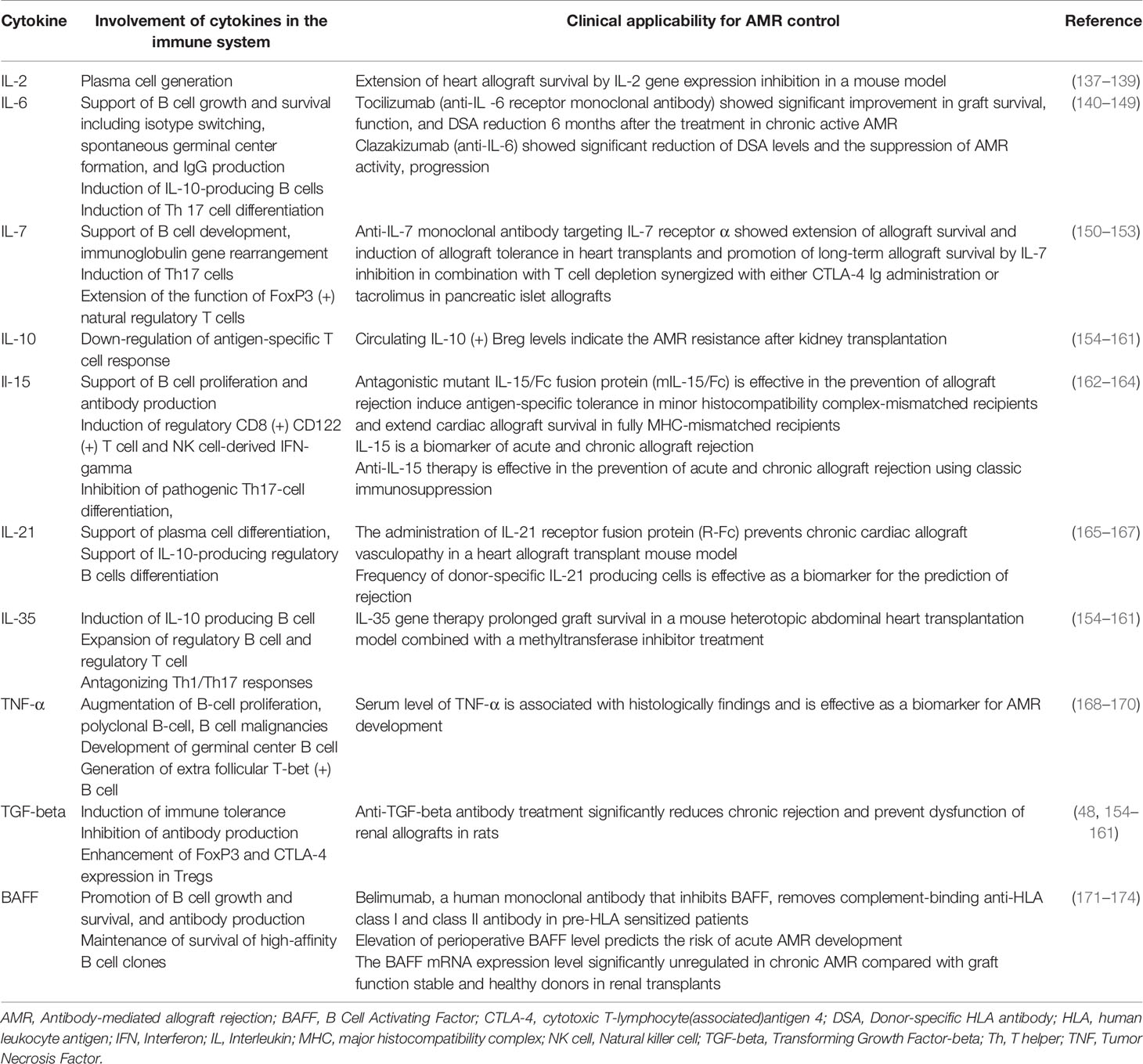
Table 2A Involvement of cytokines in the immune system and clinical applicability for AMR.
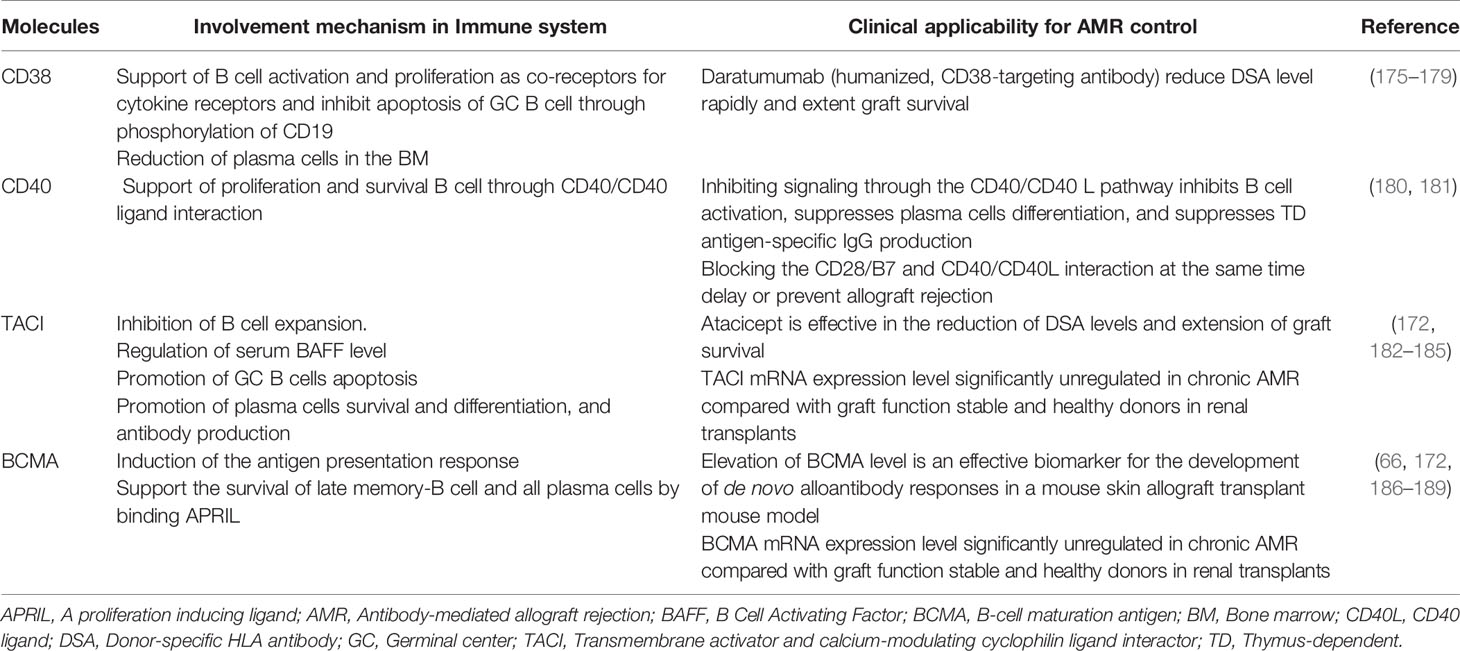
Table 2B Molecules expressed by B cells and clinical applicability for AMR control.
Agents Targeting Molecules Expressed in B Cell
In addition to these listed molecules, CD19-mediated signaling reduces the threshold for BCR-mediated signal and promotion of B cell development (190). Under pathological conditions, inebilizumab (humanised anti-CD19 monoclonal antibody) significantly reduced autoantibodies and relapse rates in patients with neuromyelitis optica (191).
Although antibodies targeting CD20 are used for the treatment of AMR, CD19 is expressed at all B cell differentiation stages, is more widely expressed than CD20 and is expressed in PCs. Therefore, CD19-targeting therapy may be more useful than CD20-targeting therapy in AMR treatment (19, 20) and the safety and tolerability of inebilizumab alone or in combination with VIB4920 (Fc-deficient CD40 Ligand antagonist) are currently being investigated in highly sensitised patients on the waiting list for kidney transplantation at clinicalTrials.gov (study identifier NCT04174677) (192).
CD138, a member of the integral membrane family of heparan sulfate proteoglycans, is highly expressed on PCs (193). CD138 increases heparan sulfate levels in ASCs, and APRIL and IL-6 support the growth and survival of ASCs by binding to heparan sulfate (194).
In other words, CD138 plays an important role in the maintenance of long-term humoral immunity (195).
In the field of transplantation, the relative abundance of CD138-positive cells is closely related to AMR development, the degree of humoral immunity-associated injury progression, and DSA production (196), and a higher frequency of CD138-positive cells and the co-existence of CD20-positive cells are associated with poor allograft function and poor response to treatment after acute rejection including TCMR and AMR (197). In addition, pathological findings indicate that the CD138-positive PCs infiltrates are associated with AMR development (196). Therefore, CD138-targeting therapy may be applicable for AMR control and the maintenance of graft function after further elucidation of the involvement mechanism of CD138-mediated signaling in the activation of humoral immunity to transplanted grafts (43). However, as described in B−cell based therapy, B cells contain a subset that has an immune regulatory function. Therefore, any clinical application for AMR control needs to be fully examined for the susceptibility of these agents for each of the B cell subsets, and the effect it will have upon their function.
B-Cell Based Therapy
In this section, we will discuss B cell subsets with immunoregulatory function because the expansion of regulatory cells that induces immune tolerance that may lead to the reduction of immunosuppressants.
Bregs mediate immune tolerance through mechanisms that involve the production of cytokines such as IL-10, IL-35, and TGF-beta as well as through cell–cell contact (154–161). Although IL-10 has been the focus of attention, IL-10 and IL-35 produced from Breg play important roles as negative regulators of immunity, which are involved in the antigen-presenting function and cytokine secretion of macrophages, dendritic cells, and B cells and activation of inflammatory T cells (156). B-1 cells and marginal zone (MZ) B cells exhibit innate-like immune functions (198). MZ-autoreactive B cells are required to maintain tolerance to self-antigens and the rapid acquisition of immunoregulatory function through the maintenance of the levels of natural IgM antibody and IL-10 (199, 200). Furthermore, ILBs serve as important sources of Bregs that play crucial roles in autoimmunity, inflammation, and infection in the models of autoimmune disease (201, 202).
As other B cell subsets that have been reported to be involved in the induction of immune tolerance, naïve-B cells function as APCs and induce the conversion of CD4+ CD25− T cells into CD25+ Foxp3− Tregs, which express lymphocyte activation gene 3 (LAG3), ICOS, glucocorticoid-induced TNF receptor family-regulated gene (GITR), OX40, PD1, and CTLA-4, to sustain immune tolerance through the production of anti-inflammatory cytokines such as IL-10. These findings indicate that naïve-B cells cooperate with Tregs to induce T cell tolerance, maintain the homeostasis of Tregs, and suppress inflammation by functioning as APCs (203, 204). In addition, IgM+CD138hiTACI+CXCR4+CD1dintTim1int PCs expressing the transcription factor Blimp1 produce IL-10, IL-35 during infections with Salmonella species. Furthermore, CD138+ PCs provide the major source of IL-35 and IL-10 in patients with experimental autoimmune encephalomyelitis (EAE) (156).
In the field of transplantation, IL-10 produced by Bregs is associated with drug resistance in AMR, and higher frequencies of transitional B cells and naïve-B cells and high production of IL-10 are related to the induction of immune tolerance following kidney transplantation. In contrast, the results are opposite in cases of chronic AMR (205), and CD138+ PCs may be involved the induction of immune tolerance and the expansion of these B cells may mitigate the adverse effects of immunosuppressants and thereby improve the prognosis of patients who undergo transplantation.
Development of Diagnostic Methods
Monitoring Humoral Immunity Using Memory-B Cell
The MBC pool has recently attracted more attention than humoral antibodies as a potential diagnostic tool to monitor the humoral immune response to donor-specific HLA antigens, which is evaluated according to the frequency of DSA-specific MBCs circulating in peripheral blood. For example, to determine the antigen specificities of MBCs circulating in the periphery, B cells or peripheral blood mononuclear cells (PBMCs) are induced to differentiate into ASCs with mitogens and cytokines suitable for the proliferation of MBCs and survival in a polyclonal activation-dependent manner. These findings were acquired through analysis of culture supernatants using a solid-phase assay platform with HLA-coated multiplex beads (23–29).
In the field of transplantation, current studies focus on the diagnostic value of IgG antibodies against HLA expressed in donor-derived vascular endothelial cells. On the other hands, there is currently no consensus on the clinical role of IgM antibodies (206).
We examined the clinical potential of DSA-specific IgM-MBCs as early diagnosis and humoral immune monitoring in the context of AMR; DSA-specific IgM-MBCs may achieve higher sensitivity when employed for conventional immunosuppressive therapy compared with IgG-MBCs (207). Therefore, the detection of DSA-specific IgM-MBCs contributes to the inhibition of AMR development by enabling early intervention using less invasive immunosuppression after elucidation of the optimal conditions for inducing DSA-specific IgM-MBC differentiation into IgG-PCs, the process that leads to AMR. In addition, the availability of an in vitro assay system capable of inducing the differentiation of MBCs into ASCs may lead to the introduction of more effective immunosuppressive therapy that recognizes differences in pathology or in patients’ drug sensitivities (208).
However, it is unclear whether in vitro drug susceptibility data can be applied to patients because conventional two-dimensional culture may not reproduce the three-dimensional structure or function such as lymphoid tissue or organs in the living body. Organ-on-a-chip technology is attracting attention because it reproduces organs in functional units while maintaining the in vivo three-dimensional organ structure and physiological function. Thus, pharmacokinetic analysis using this model may be useful for resolving the aforementioned problem (209–212).
Microarray Technique
Attempts are being made to apply microarray technology or NGS to the field of transplantation. Microarrays simultaneously analyze the expression of tens of thousands of genes and obtain information about the transcriptional profiles. Furthermore, pathological changes in gene expression can be obtained, which will provide information to increase the accuracies of classification, diagnosis, and prognosis of diseases (28, 29). Errors frequently occur in analyses of single genes when small data sets are employed. Recently, Chen et al. focused on transcripts that are known to be associated with disease. Microarray evaluation of pathogenesis-based transcript sets, corresponding to events that mainly occur during allograft rejection, is useful for the diagnosis of AMR as well as identification of pathology and prediction of clinical course. For example, biopsy-based microarrays identified 45 genes upregulated in pediatric kidney transplants as well as in pediatric and adult heart transplants undergoing acute rejection. Among them, serum PECAM1 shows the greatest promise as a biomarker (89% sensitivity and 75% specificity) for analyzing renal transplant rejection, suggesting that microarray analysis will be useful for discovering new serum protein biomarkers by mining publicly available data sets (30). Thus, it is possible to identify the gene group associated with graft rejection and pathological condition by enabling comprehensive gene analysis; early diagnosis of the development of rejection is possible and genes related to immune status or drug resistance has also been identified (213).
Next-Generation Sequencing
NGS determines several million base pairs per run and provides the ability to distinguish different isoforms and allelic expression, which is an advantage over microarray analysis and detects somatic mutations with high accuracy and high specificity, which enables identification of candidate genes that cause disease. As an example of the application of NGS to transplantation, comparison of the complementary determining region 3 (CDR3) of the TCR beta chains expressed in AMR (+) and AMR (−), may lead to the prediction of the development of rejection before patients undergo transplantation (31). In addition, this technique can be applied to increase our understanding of the diversity of the variable regions of heavy and light chains of BCRs. Thus, detailed characterization of this repertoire, including reactivity with antigen, becomes possible and may be applied to predict the production of donor-specific antigen-reactive antibodies (32–34) and both specific sequences and the full length of genes including introns and untranslated regions are analyzed by NGS, and thus, HLA alleles can be analyzed in transplantation, ensuring better histocompatibility between donors and recipients (214, 215).
In addition, gene polymorphisms affect the distribution and drug metabolism. For example, Single Nucleotide Polymorphism (SNP) of CYP3A4/3A5, ABCB1 in CNI, CYP3A5 in sirolimus, TPMT in azathioprine, UGT1A9, ABCC2 in Mycophenolic acid (MPA), and MDR1 in tacrolimus affects their pharmacokinetics. Thus, NGS analysis of these SNP will likely provide useful information for developing more effective immunosuppressive therapy, which considers differences in pathology and drug susceptibility (35, 36).
In the other words, these techniques have potential application in developing strategies for controlling AMR and improving the prognosis of transplanted grafts.
Novel Appliable Components in AMR
As mentioned above, we are closely evaluating the possibility of using an IgM antibody in AMR control (207). Therefore, we evaluate its possible use not only as an early diagnosis tool but also as a treatment method. Here, we discuss the involvement, as well as their clinical applicability of humoral immunity-associated components involved in IgM antibody in AMR control whilst referring to reports on the involvement mechanisms in pathological conditions in fields outside of transplants.
Role of IgM Receptors
IgM is the first immunoglobulin produced in response to antigen challenge, and B cell activation is enhanced by stimulatory IgM Fc receptor (FcμR)-mediated signals during the early stage of the immune response. The levels of IgG exceed those of IgM during the late stage of the adaptive immune response, during which the activation of B cells is inhibited by FcγRIIB-mediated inhibitory signals, indicating that stimulatory or inhibitory signals transduced by IgM or IgG may regulate B cell activation and antibody production (216).
FcμR is mainly expressed by B, T, and NK cells in humans and by B cells of mice (216–219). The expression levels of mouse FcμR differ depending on the B cell subset, and FcµR expressed in the trans-Golgi network restricts the transport of IgM-BCR to the B cell surface. FcμR expression is downregulated during GC reactions and at a higher level in plasma blast cells compared with those expressed by PCs and expressed in class-switched B cells (187). In contrast, FcμR-deficient MZBs are significantly mitigated in a mouse knockout model, in which the number of B cell subsets is altered or tonic signaling through the reduction of BCR expression (216, 219, 220). The FcμR-mediated signal contributes further to the maturation of B cell subsets and enhances B cell survival in response to treatment with anti-IgM antibodies (216, 221). These findings indicate that signals transmitted by the FcμR and BCR may cooperate to activate B cells and maintain their survival. In addition, this signal regulates the expression of IgM-BCR in immature B cells, which regulates the signal emitted when antigen activates the BCR through their cross-linking by antigen binding (220). In FcμR KO mice, the levels of natural IgM antibodies in the peripheral blood circulation are increased, and the formation of the GC, MBCs, differentiation of PCs, and production of antigen-specific IgG1 antibodies are reduced. These findings support the conclusion that the FcμR is required for the maintenance of the adaptive immune response and the control of the homeostasis of B-1/B-2 cells (216, 219).
Under pathological conditions, FcμR-mediated signaling controls the production of harmful autoreactive IgG antibodies and is involved in the development of autoimmune and inflammatory diseases, chronic lymphocytic leukemia, and others (37). In a model of severe human multiple sclerosis, the development of EAE is suppressed through the regulation of the functions of dendritic cell and Tregs (222). FcμR-mediated signal transduction increases self-Ag-triggered BCR signaling in immature B cells and contributes to the deletion, anergy, or both of autoreactive immature B cells in the BM, which induces immune tolerance (38).
Soluble IgM is a ligand for CD22 and forms a complex with an antigen. This complex suppresses the CD22-mediated BCR signaling via its binding to CD22 expressed on the B cell surface. CD22, which is expressed on the surface of mature B cells, is an inhibitory receptor. Specifically, phosphorylated CD22 signals through the BCR to downregulate B cell activation that prevents the overactivation of the immune system and development of autoimmune disease (223, 224).
In CD22-deficient mice, BCR ligation promotes the mobilization of intracellular calcium and inhibits BCR signaling. Furthermore, CD22 signaling contributes to the differentiation of B cells and is required for the expansion of B1-b cells after BCR ligation (225, 226). The response to the TD antigen is normal during adaptive immunity; however, Thfs do not support B cells in CD22-deficient mice, because the CD22-CD22-ligand (CD22L) interaction is required for the activation of T cells (224, 226). In CD22-deficient mice, naïve-B cells differentiate into GCB cells but cannot differentiate into MBCs or PCs (221, 222). CD22-mediated inhibition of the BCR signal is associated with B cell tolerance, and CD22-CD22L interactions are required to maintain self-tolerance (227, 228).
Under pathological conditions, CD22 is involved in the control of autoimmune diseases and genetic variants of CD22 are related to the susceptibility of individuals to autoimmune diseases through a defect in B cell tolerance (39, 40). In patients with autoimmune diseases such as rheumatism, T1-diabetes, and SLE, inactivating mutations are frequently found in the CD22-ligand (229–231). Although CD22 is not a major cause of susceptibility to SLE in humans, CD22 deficiency may exert additive or synergistic effects on susceptibility to disease. Moreover, CD22 regulates the B cell response in autoimmune disease through regulation of BCRs and Tool-like receptors (TLRs) (224, 232).
Role of Scavenger Protein
Accumulation of foreign pathogens, apoptotic or necrotic dead cells, and their debris causes chronic inflammation and induces an autoimmune response, which must be eliminated to prevent their onset.
Apoptosis inhibitor of macrophage (AIM, also called CD5L) is a circulating protein that is a member of the scavenger receptor cysteine-rich superfamily. Normally, high levels of AIM bind to IgM pentamers and circulate in the peripheral blood in the inactivated state (233).
In B cells, AIM cooperates with TGF–beta1 to suppress B cell proliferation strongly and persistently and inhibit antibody production. TGF-beta1-mediated increased expression of AIM receptors on the surface of B cells is required for AIM to exert an effect on these cells (234). In addition, it has been reported that IL-10 protects transplanted grafts from recipient immunity and exert anti-inflammatory effects by inhibiting NLRP3 inflammasome activation or MPs with proinflammatory phenotypes by increasing AIM expression. Therefore, regulation of AIM expression may be related to the induction of immune tolerance (35).
Under pathological conditions, IgM dissociates from AIM during the recovery from renal injury through the enhanced clearance of a luminal obstruction during acute renal injury (234–236). Specifically, free AIM bound to debris interacts with kidney injury molecule 1 (KIM-1) that is expressed by injured tubular epithelial cells, resulting in enhanced phagocytic clearance of AIM-bound debris by epithelial cells (234, 237–239). Thus, free AIM is involved in the repair of acute kidney injury, because delayed or deficient removal of dead cells, or both, may cause secondary inflammation and fibrosis in tissues and may impair the repair of tissue damage and the regeneration of such tissues (237). Alternatively, macrophages produce AIM (236), which is involved in the pathophysiology of inflammatory colitis through the maintenance of the survival of macrophages, and the elimination of dead cells and toxic substances in hepatitis by supporting the phagocytic activity of macrophages (239–241). In the field of transplantation, it has been reported that the blood concentration of free AIM increases during acute cellular rejection in cardiac allograft rejection (242).
Role of IgM Antibody
Natural IgMs exert anti-inflammatory effects through clearing pathogens, scavenging toxins, inhibiting the production of inflammatory mediators, neutralizing cytokines, and scavenging complement to directly protect antigens from humoral immune attack (207, 243–248). These antibodies suppress inflammation and injury of target tissues caused by IgG autoantibodies through anti-idiotype activity, competitive inhibition of binding of IgG autoantibody to antigens, and suppression of IgG autoantibody production via signals from the FcµR expressed on the surface of B cells (249). Furthermore, these antibodies significantly inhibit T cell proliferation in lectin-stimulated PBMCs in vitro through the suppression of IL-2 production that enhances the functions of immunocompetent cells, bacterial aggregation, and opsonic activity (37). A polyclonal antibody preparation designated trimodulin, which contains IgM (~23%), IgA (~21%), and IgG (~56%), decreases the levels of TLR2, 4 as well as those of coagulation receptors (CD11b and CD64) in monocytes and inhibits lymphocyte proliferation and regulate of the production of pro- and anti-inflammatory cytokines, including TNF-α and IL-10. Therefore, these antibodies achieve a protective effect to alleviate the inflammatory reaction to target organs through these extents of involvement in immune system (243, 246, 247, 250–252).
In a model of renal ischemia-reperfusion-induced injury, the tissue-protective effects of IgM antibodies that recognize and inhibit the activities of danger-associated molecular patterns reflect human pathology. These mechanisms support the regeneration of damaged hepatocytes in a model of liver ischemia (42). In patients with sepsis, IgM-enriched IVig ameliorates pathology and reduces mortality through the improvement of the peripheral circulation, such that the numbers of circulating B cells and levels of IgM are significantly reduced in nonsurvivors compared with survivors, indicating that IgM is required to achieve successful therapy (253–256).
In an in vitro model of xenotransplantation, compared with IVig, IgM-enriched IVig more strongly inhibits the classical complement pathway and complement-dependent cytotoxicity caused by the deposition of C4 and C3 on the cell surface of pig cells treated with human serum and suppresses the development of hyperacute rejection of a xenotransplanted graft (257).
In allograft transplantation, IgM inhibits complement activity 10-times more than IgG (258). In the case of de novo DSA production after lung transplant recipients; treatment with IgM-enriched IVig is associated with the DSA-clearance effect, which improves the prognosis of transplantation. In early DSA-positive cases, treatment with IgM-enriched IVig and rituximab was superior to that with plasma exchange and rituximab, and the patients were comparable to the DSA-negative group in 1-year survival after transplantation; treatment with IgM-enriched IVig was superior to treatment with plasma exchange and rituximab in DSA clearance (259). In addition, a tissue biopsy confirms no AMR after treatment only by IgM-enriched IVig injection, when AMR develops after heart transplantation (258).
Clinical Potential of Humoral Immunity–Associated Components in AMR
Further elucidation of the involvement of AIM in the humoral immune response to transplanted grafts will be applicable for AMR control. Newly discovered cytokines, antibodies, and receptors involved in antibody production may be expected to be translated to the clinical application in early diagnosis, management, and prognosis prediction. These efforts require the identification of the roles of these components in the underlying mechanisms of AMR.
In the field of transplantation, FcμR-mediated signaling maintains the homeostasis of B1/B2 cells (216, 219) and potentially alleviates the pathology of AMR (38), and elucidation of the involvement of this signaling in AMR development will likely contribute to the development of therapies designed to protect target grafts from humoral immune responses mounted by recipients.
CD22-mediated signaling may be expected to be useful as B cell depletion therapy in AMR control, because IVig is administered as a treatment for AMR, and one of the mechanisms is the induction of mature B cell apoptosis via binding between sialylated IVig and CD22 (71). It is therefore important to evaluate the involvement of CD22-targeting agents in the activation of humoral immunity against transplanted grafts in the same manner as they are applied to the treatment of autoimmune diseases to effectively manage AMR (231, 232).
As a diagnostic method, clinical application of these receptors to new diagnostic methods will be achieved if the functions and structural properties of these receptors can be shown to be involved in humoral immunity to transplanted grafts.
As scavenger protein, it has also been reported that an increase in AIM blood concentrations is associated with the development of acute rejection following heart transplantation, indicating that the blood concentration of free AIM may increase in the early stage of humoral immunity activation in transplanted grafts, and it is expected to be useful as an early diagnostic method for AMR (242). Therefore, the utility of AIM as a diagnostic modality can be ascertained after elucidating its AIM in the development of AMR. In addition, AIM controls inflammation and repairs damaged tissues via phagocytosis of apoptotic cells.
Although IVig has been commonly used for AMR control, the significance of IgM-enriched IVig has been reported in the field of severe infection and organ ischemia (42, 253–258). IgM-enriched IVig may exert more strongly inhibits complement activation as compared with IVig (258, 259). Therefore, identifying the detailed mechanism of IgM involvement in humoral immune activation to transplanted grafts and the significance of IgM-enriched IVig compared with that of IVig will likely lead to further clinical application of IgM-enriched IVig to manage AMR.
Significantly, in addition to the potential induction of immune tolerance of humoral immunity-associated components to transplanted grafts (35, 243, 247, 248, 250–252), AIM and IgM-enriched IVig have a tissue repair and regeneration effect through the clearance of injured tissue (42, 224, 233–235). Therefore, humoral immunity associated injury after the onset of AMR, which was considered irreversible, could be recovered by clinical application of these humoral immunity-associated components.
We summarized clinical potential of these components in the context of AMR control (Figure 3).
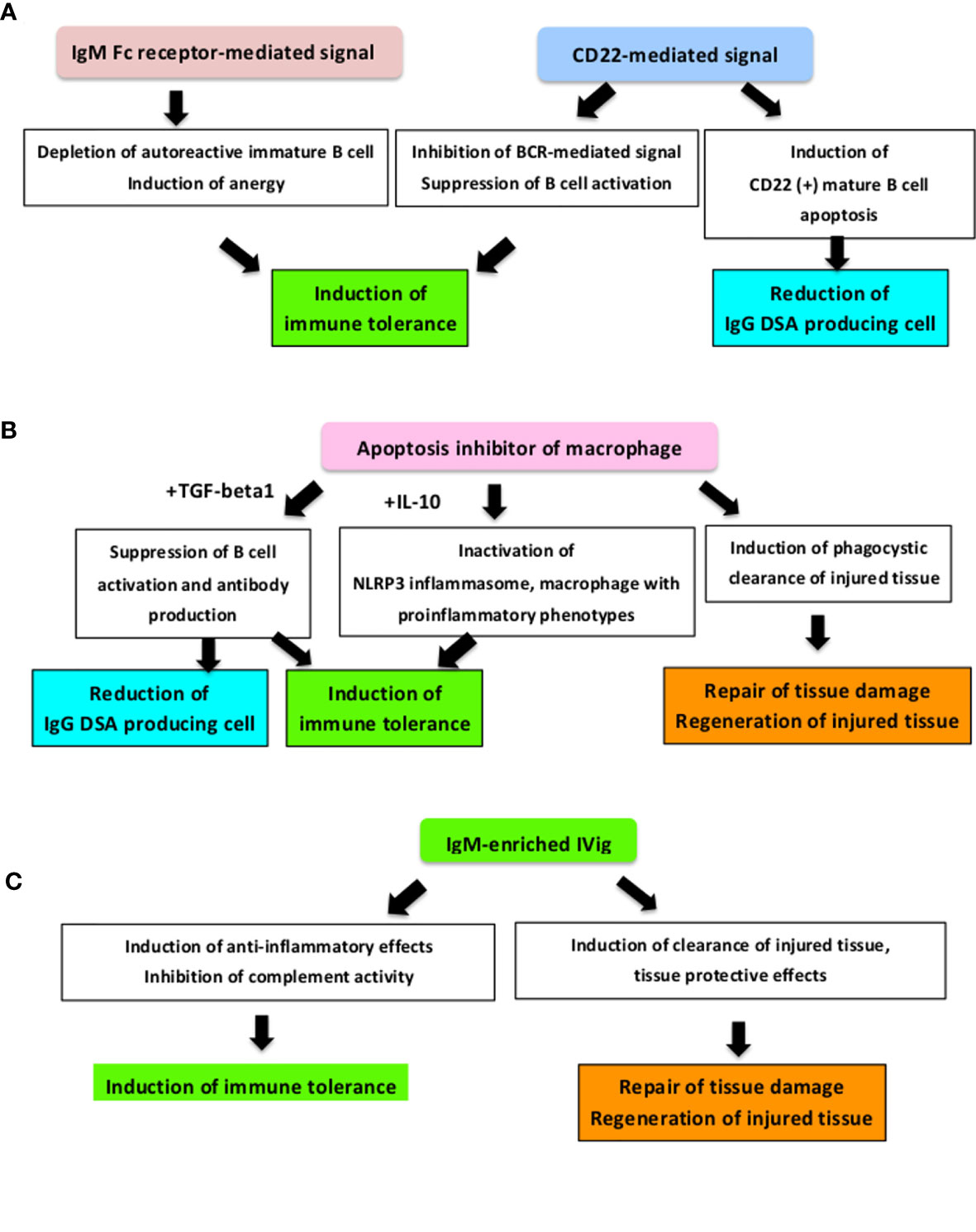
Figure 3 How these novel applicable components improve the prognosis of AMR. We summarised the clinical potential of novel humoral immunity-associated components in AMR control and AMR development mechanisms. (A) As an IgM receptor, the FCu receptor-mediated signal controls harmful autoreactive IgG antibodies production and controls autoimmunity and inflammation through regulating immunoregulatory cells (37, 38, 216, 219, 222). In addition, this signal is associated with immune tolerance induction by promoting deletion and energy of immature B cells (38). CD22-mediated signaling also induces immune tolerance via CD22-CD22L interaction and CD22-mediated inhibition of the BCR-mediated signal (227, 228) and CD22 (+) mature B cell apoptosis (71). (B) As a scavenger protein, it has been reported that free Apoptosis inhibitor of macrophage (AIM) works to suppress inflammation, tissue injury and tissue regeneration through phagocytic activity during acute kidney injury (233–241) and AIM mediates immune tolerance induction by IL-10 and suppresses the immune response by inducing macrophages with immunosuppressive phenotype activation (35) and cooperates with TGF-beta to suppress B cell proliferation and antibody production (233). (C) As a polyclonal IgM antibody, IgM-enriched IVig strongly suppresses cell proliferation, has a inhibitory effect in complement activity (257, 258), and alleviates the humoral immunity-associated pathological condition in severe infection, organ ischemia, and transplants (42, 253–256, 258, 259). BCR, B cell receptor; DSA, Donor-specific HLA antibody; IVig, Intravenous immunoglobulin; TGF, Transforming Growth Factor.
Concluding Remarks
In this study, we discussed the pathways by which naïve-B cells are sensitized to donor-specific HLA antigens and differentiate into LLPCs, which produce DSAs. This study also presented evidence regarding the mechanism by which immunocompetent cells participate in the signal transduction pathways that contribute to AMR and the mechanisms of immunosuppressive therapy designed to suppress the development of AMR (17–19).
Although immunosuppressive therapies that may be useful for suppressing each process during AMR development have been developed, AMR control remains challenging. The main cause of poor AMR control is that positivity for antibodies to donor-specific HLA is used as a reference as one of the diagnostic methods for AMR (17); however, LLPCs, which produce antibodies inducing AMR, may be difficult to remove by applying conventional immunosuppressive therapy and the pathological condition progresses irreversibly when these antibodies are present in serum (20, 71, 72, 260). In addition, the information needed to improve management after transplantation including individual differences in pathological conditions and drug susceptibility has not been discovered.
It has been reported that humoral monitoring methods for better evaluating the humoral immune response to the transplanted graft might contribute to resolving this problem by analyzing the antigenic specificity of MBCs circulating in peripheral blood using in vitro assay systems (22–27) and further possibility of gene-based control method in the context of AMR as following; Microarray techniques can be used to elucidate the molecular mechanisms that induce the migration of LLPCs to BM and promote their longevity (259). Thus, these techniques may permit inhibition of the differentiation of LLPCs. Further; NGS analysis of antibody reactivity to transplanted grafts can be applied to predict the antibodies that cause AMR (28–36). This knowledge, combined with the identification of genetic polymorphisms associated with drug sensitivity, will undoubtedly contribute to the development of optimal management strategies.
In addition to the development of these diagnostic methods, recent studies revealed relevant factors, such as anti-inflammatory effects, reduction of harmful IgG autoantibodies production, tissue regeneration, and their clinical applicability in the fields of autoimmune diseases and inflammatory disease and severe infection disease, and the others (37–42). Therefore, in the field of transplantation, by further clarifying the mechanism of their involvement in AMR, methods for improving the control of AMR including early diagnosis, suppression of AMR development, and alleviation of pathology may be developed.
Although IgG antibody has been the main focus in the field of transplants, we focused on the clinical potential of detecting DSA-specific IgM and IgG-MBC differentiation using in vitro assay systems as an early diagnostic method and biomarkers that enable the inhibition of AMR development with less invasive therapeutic intervention (207). The findings supported the clinical applicability of the detection of DSA-specific IgM-MBC differentiation after elucidating the optimal conditions for inducing the differentiation of DSA-specific IgM-MBC differentiation into IgG-PCs, the detailed mechanism leading to AMR.
Alternatively, IVig has been administered for AMR control mainly, but IgM-enriched IVig has been reported to improve the pathological condition and prognosis in severe infectious diseases and organ ischemia (42, 253–256). Therefore, further elucidation of how signal mediated by IgM-enriched-IVig are involved in the AMR development may contribute to the establishment of AMR control.
Therefore, focusing on the involvement mechanism of humoral immunity-associated components in the pathological conditions regardless of the difference in fields and conventional knowledge and elucidating the mechanism by which these humoral immunity-associated components participate in AMR development will likely be applicable to the development of new diagnostic and therapeutic methods for improving AMR management.
Author Contributions
YM designed and wrote the paper. TW revised the paper. TW and X-KL provided excellent advice. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Bunjes D, Hardt C, Roöllinghoff M, Wagner H. Cyclosporin A Mediates Immu- Nosuppression of Primary Cytotoxic T Cell Responses by Impairing the Release of Interleukin 1 and Interleukin 2. Eur J Immunol (1981) 11(8):657–61. doi: 10.1002/eji.1830110812
2. Wang CJ, Heuts F, Ovcinnikovs V, Wardzinski L, Bowers C, Schmidt EM, et al. CTLA-4 Controls Follicular Helper T-Cell Differentiation by Regulating the Strength of CD28 Engagement. Proc Natl Acad Sci USA (2015) 112:524–9. doi: 10.1073/pnas.1414576112
3. Xu H, Li X, Liu D, Li J, Zhang X, Chen X, et al. Follicular T-Helper Cell Recruitment Governed by Bystander B Cells and ICOS-Driven Motility. Nature (2013) 496:523–7. doi: 10.1038/nature12058
4. Lu D, Ni Z, Liu X, Feng S, Dong X, Shi X, et al. Beyond T Cells: Understanding the Role of PD-1/PD-L1 in Tumor-Associated Macrophages. J Immunol Res (2019) 2019:1919082. doi: 10.1155/2019/1919082
5. Crotty S. A Brief History of T Cell Help to B Cells. Nat Rev Immunol (2015) 15:185–9. doi: 10.1038/nri3803
6. McAdam AJ, Greenwald RJ, Levin MA, Chernova T, Malenkovich N, Ling V, et al. Icos Is Critical for CD40-Mediated Antibody Class Switching. Nature (2001) 409:102–5. doi: 10.1038/35051107
7. Tafuri A, Shahinian A, Bladt F, Yoshinaga SK, Jordana M, Wakeham A, et al. ICOS Is Essential for Effective T-Helper-Cell Responses. Nature (2001) 409:105–9. doi: 10.1038/35051113
8. Dong C, Juedes AE, Temann UA, Shresta S, Allison JP, Ruddle NH, et al. ICOS Co-Stimulatory Receptor Is Essential for T-Cell Activation and Function. Nature (2001) 409:97–101. doi: 10.1038/35051100
9. Dong C, Temann UA, Flavell RA. Cutting Edge: Critical Role of Inducible Costimulator in Germinal Center Reactions. J Immunol (2001) 166:3659–62. doi: 10.4049/jimmunol.166.6.3659
10. Moriyama S, Takahashi N, Green JA, Hori S, Kubo M, Cyster JG, et al. Sphingosine-1-Phosphate Receptor 2 Is Critical for Follicular Helper T Cell Retention in Germinal Centers. J Exp Med (2014) 211:1297–305. doi: 10.1084/jem.20131666
11. Qi H, Cannons JL, Klauschen F, Schwartzberg PL, Germain RN. SAP-Controlled T-B Cell Interactions Underlie Germinal Centre Formation. Nature (2008) 455:764–9. doi: 10.1038/nature07345
12. Cannons JL, Qi H, Lu KT, Dutta M, Gomez-Rodriguez J, Cheng J, et al. Optimal Germinal Center Responses Require a Multistage T Cell:B Cell Adhesion Process Involving Integrins, SLAM-Associated Protein, and CD84. Immunity (2010) 32:253–65. doi: 10.1016/j.immuni.2010.01.010
13. Czar MJ, Kersh EN, Mijares LA, Lanier G, Lewis J, Yap G, et al. Altered Lymphocyte Responses and Cytokine Production in Mice Deficient in the X-Linked Lymphoproliferative Disease Gene SH2D1A/DSHP/SAP. Proc Natl Acad Sci USA (2001) 98:7449–54. doi: 10.1073/pnas.131193098
14. Crotty S, Kersh EN, Cannons J, Schwartzberg PL, Ahmed R. SAP Is Required for Generating Long Term Humoral Immunity. Nature (2003) 421:282–7. doi: 10.1038/nature01318
15. Cannons JL, Yu LJ, Jankovic D, Crotty S, Horai R, Kirby M, et al. SAP Regulates T Cell-Mediated Help for Humoral Immunity by a Mechanism Distinct From Cytokine Regulation. J Exp Med (2006) 203:1551–65. doi: 10.1084/jem.20052097
16. Yusuf I, Kageyama R, Monticelli L, Johnston RJ, Ditoro D, Hansen K, et al. Germinal Center T Follicular Helper Cell IL-4 Production Is Dependent on Signaling Lymphocytic Activation Molecule Receptor (CD150). J Immunol (2010) 185:190–202. doi: 10.4049/jimmunol.0903505
17. Matsuda Y, Sarwal MM. Unraveling the Role of Allo-Antibodies and Transplant Injury. Front Immunol (2016) 7:432. doi: 10.1159/000501460
18. Perera J, Huang H. The Development and Function of Thymic B Cells. Cell Mol Life Sci (2015) 72(14):2657–63. doi: 10.1007/s00018-015-1895-1
19. Mei HE, Wirries I, Frölich D, Brisslert M, Giesecke C, Grün JR, et al. A Unique Population of IgG-Expressing Plasma Cells Lacking CD19 Is Enriched in Human Bone Marrow. Blood (2015) 125:1739–48. doi: 10.1182/blood-2014-02-555169
20. Brynjolfsson SF, Persson Berg L, Olsen Ekerhult T, Rimkute I, Wick MJ, Mårtensson IL, et al. Long-Lived Plasma Cells in Mice and Men. Front Immunol (2018) 16:2673. doi: 10.3389/fimmu.2018.02673
21. Lynch RJ, Silva IA, Chen BJ, Punch JD, Cascalho M, Platt JL. Cryptic B Cell Response to Renal Transplantation. Am J Transpl (2013) 13:1713–23. doi: 10.1111/ajt.12308
22. Jahnmatz M, Kesa G, Netterlid E, Buisman AM, Thorstensson R, Ahlborg N. Optimization of a Human IgG B-Cell ELISpot Assay for the Analysis of Vaccine-Induced B-Cell Responses. J Immunol Methods (2013) 391:50. doi: 10.1016/j.jim.2013.02.009
23. Pinna D, Corti D, Jarrossay D, Sallusto F, Lanzavecchia A. Clonal Dissection of the Human Memory B-Cell Repertoire Following Infection and Vaccination. Eur J Immunol (2009) 39:1260. doi: 10.1002/eji.200839129
24. Lúcia M, Luque S, Crespo E, Melilli E, Cruzado JM, Martorell J. Preformed Circulating HLA-Specific Memory B Cells Predict High Risk of Humoral Rejection in Kidney Transplantation. Kidney Int (2015) 88:874. doi: 10.1038/ki.2015.205
25. Han M, Rogers JA, Lavingia B. Stastny P.Peripheral Blood B Cells Producing Donor-Specific HLA Antibodies In Vitro. Hum Immunol (2009) 70:29. doi: 10.1016/j.humimm.2008.10.013
26. Snanoudj R, Claas FHJ, Heidt S, Legendre C, Chatenoud L, Candon S. Restricted Specificity of Peripheral Alloreactive Memory B Cells in HLA-Sensitized Patients Awaiting a Kidntransplant. Kidney Int (2015) 87:1230. doi: 10.1038/ki.2014.390
27. Heidt S, Roelen DL, de Vaal YJ, Kester MG, Eijsink C. Thomas, sA Novel ELISPOT Assay to Quantify HLA-Specific B Cells in HLA-Immunized Individuals. al Am J Transpl (2012) 12:1469–78. doi: 10.1111/j.1600-6143.2011.03982.x
28. Butte AJ, Sigdel TK, Wadia PP, Miklos DB, Sarwal MM. Protein Microarrays Discover Angiotensinogen and PRKRIP1 as Novel Targets for Autoantibodies in Chronic Renal Disease. Mol Cell Proteomics (2011) 10(3):M110.000497. doi: 10.1074/mcp.M110.000497
29. Yang JY, Sigdel TK, Sarwal MM. Self-Antigens and Rejection: A Proteomic Analysis. Curr Opin Organ Transpl (2016) 21(4):362–7. doi: 10.1097/MOT.0000000000000328
30. Chen R, Sigdel TK, Li L, Kambham N, Dudley JT, Hsieh SC, et al. Differentially Expressed RNA From Public Microarray Data Identifies Serum Protein Biomarkers for Cross-Organ Transplant Rejection and Other Conditions. PloS Comput Biol (2010) 6(9):e1000940. doi: 10.1371/journal.pcbi.1000940
31. Aschauer C, Jelencsics K, Hu K, Heinzel A, Vetter J, Fraunhofer T, et al. Next Generation Sequencing Based Assessment of the Alloreactive T Cell Receptor Repertoire in Kidney Transplant Patients During Rejection: A Prospective Cohort Study. BMC Nephrol (2019) 20(1):346. doi: 10.1186/s12882-019-1541-5
32. Goldstein LD, Chen YJ, Wu J, Chaudhuri S, Hsiao YC, Schneider K. Single-Cell B-Cell Receptor Sequencing Enables Rapid Discovery of Diverse Antigen-Reactive Antibodies. Commun Biol (2019) 2:304. doi: 10.1038/s42003-019-0551-y
33. Kim D, Park D. Deep Sequencing of B Cell Receptor Repertoire. BMB Rep (2019) 52(9):540–7. doi: 10.5483/BMBRep.2019.52.9.192
34. Luque S,M, Bestard O. Refinement of Humoral Immune Monitoring in Kidney Transplantation: The Role of “Hidden” Alloreactive Memory B Cells. Transpl Int (2017) 30(10):955–68. doi: 10.1111/tri.13014
35. Su L, Yin L, Yang J, Sun L. Correlation Between Gene Polymorphism and Blood Concentration of Calcineurin Inhibitors in Renal Transplant Recipients: An Overview of Systematic Reviews. Med (Baltimore) (2019) 98(26):e16113. doi: 10.1097/MD.0000000000016113
36. Anglicheau D, Verstuyft C, Laurent-Puig P, Becquemont L, Schlageter MH, Cassinat B, et al. Association of the Multidrug Resistance-1 Gene Single-Nucleotide Polymorphisms With the Tacrolimus Dose Requirements in Renal Transplant Recipients. J Am Soc Nephrol (2003) 14(7):1889–96. doi: 10.1097/01.asn.0000073901.94759.36
37. Nguyen TT, Baumgarth N. Natural IgM and the Development of B Cell-Mediated Autoimmune Diseases. Crit Rev Immunol (2016) 36:163–77. doi: 10.1615/CritRevImmunol.2016018175
38. Honjo K, Kubagawa Y, Jones DM, Dizon B, Zhu Z, Ohno H, et al. Altered Ig Levels and Antibody Responses in Mice Deficient for the Fc Receptor for IgM (FcmuR). Proc Natl Acad Sci USA (2012) 109:15882–7. doi: 10.1073/pnas.1206567109
39. Angata T. Associations of Genetic Polymorphisms of Siglecs With Human Diseases. Glycobiology (2014) 24:785–93. doi: 10.1093/glycob/cwu043
40. Cariappa A, Takematsu H, Liu H, Diaz S, Haider K, Boboila C, et al. B Cell Antigen Receptor Signal Strength and Peripheral B Cell Development Are Regulated by a 9-O-Acetyl Sialic Acid Esterase. J Exp Med (2009) 206:125–38. doi: 10.1084/jem.20081399
41. Haruta I, Kato Y, Hashimoto E, Minjares C, Kennedy S, Uto H. Association of AIM, a Novel Apoptosis Inhibitory Factor, With Hepatitis via Supporting Macrophage Survival and Enhancing Phagocytotic Function of Macrophages. J Biol Chem (2001) 276:22910–4. doi: 10.1074/jbc.M100324200
42. Krautz C, Maier SL, Brunner M, Langheinrich M, Giamarellos-Bourboulis EJ, Gogos C, et al. Reduced Circulating B Cells and Plasma IgM Levels Are Associated With Decreased Survival in Sepsis—A Meta-Analysis. J Crit Care (2018) 45:71–5. doi: 10.1016/j.jcrc.2018.01.013
43. Woodle ES, Tremblay S, Rossi A, Rojas CC, Alloway R, Roskin K, et al. Plasma Cell Targeting to Prevent Antibody-Mediated Rejection. Am J Transplant (2020) 20 Suppl 4:33–41. doi: 10.1111/ajt.15889
44. Roco JA, Mesin L, Binder SC, Nefzger C, Gonzalez-Figueroa P, Canete PF, et al. Class-Switch Recombination Occurs Infrequently in Germinal Centers. Immunity (2016) 51:337–50. doi: 10.1016/j.immuni.2019.07.001
45. Su H, Zhang CY, Lin JH, Hammes HP, Zhang C. The Role of Long-Lived Plasma Cells in Antibody-Mediated Rejection of Kidney Transplantation: An Update. Kidney Dis (Basel) (2019) 5(4):211–9. doi: 10.1159/000501460.Epub 2019 Aug 20
46. de Goër de Herve MG, Durali D, Dembele B, Giuliani M, Tran TA, Azzarone B, et al. Interferon-Alpha Triggers B Cell Effector 1 (Be1) Commitment. PloS One (2011) 6(4):e19366. doi: 10.1371/journal.pone.0019366
47. Rookhuizen DC, DeFranco AL. Toll-Like Receptor 9 Signaling Acts on Multiple Elements of the Germinal Center to Enhance Antibody Responses. Proc Natl Acad Sci USA (2014) 111:E3224–33. doi: 10.1073/pnas.1323985111
48. Chen W, Bai J, Huang H, Bi L, Kong X, Gao Y, et al. Low Proportion of Follicular Regulatory T Cell in Renal Transplant Patients With Chronic Antibody-Mediated Rejection. Sci Rep (2017) 2. 7(1):1322. doi: 10.1038/s41598-017-01625-3
49. Yazdani S, Callemeyn J, Gazut S, Lerut E, de Loor H, Wevers M. Natural Killer Cell Infiltration Is Discriminative for Antibody-Mediated Rejection and Predicts Outcome After Kidney Transplantation. Kidney Int (2019) 95(1):188–98. doi: 10.1016/j.kint.2018.08.027
50. Guerriero JL. Macrophages: Their Untold Story in T Cell Activation and Function. Int Rev Cell Mol Biol (2019) 342:73–93. doi: 10.1016/bs.ircmb.2018.07.001
51. Charreau B. Cellular and Molecular Crosstalk of Graft Endothelial Cells During AMR: Effector Functions and Mechanisms. Transplantation (preprint). doi: 10.1097/TP.0000000000003741
52. Carroll MC. The Role of Complement in B Cell Activation and Tolerance. Adv Immunol (2000) 74:61–88. doi: 10.1016/s0065-2776(08)60908-6
53. Smith NA, Coleman CB, Gewurz BE, Rochford R. CD21 (Complement Receptor 2) Is the Receptor for Epstein-Barr Virus Entry Into T Cells. J Virol (2020) 94(11):e00428–20. doi: 10.1128/JVI.00428-20
54. Dempsey PW, Allison ME, Akkaraju S, Goodnow CC, Fearon DT. C3d of Complement as a Molecular Adjuvant: Bridging Innate and Acquired Immunity. Science (1996) 271:348–50. doi: 10.1126/science.271.5247.348
55. Lanzavecchia A. Receptor-Mediated Antigen Uptake and Its Effect on Antigen Presentation to Class II-Restricted T Lymphocytes. Annu Rev Immunol (1990) 8:773–93. doi: 10.1146/annurev.iy.08.040190.004013
56. Brimnes MK, Hansen BE, Nielsen LK, Dziegiel MH, Nielsen CH. Uptake and Presentation of Myelin Basic Protein by Normal Human B Cells. PloS One (2014) 9(11):e113388. doi: 10.1371/journal.pone.0113388
57. Heesters BA, Chatterjee P, Kim YA, Gonzalez SF, Kuligowski MP, Kirchhausen T, et al. Endocytosis and Recycling of Immune Complexes by Follicular Dendritic Cells Enhances B Cell Antigen Binding and Activation. Immunity (2013) 38(6):1164–75. doi: 10.1016/j.immuni.2013.02.023
58. Suzuki K, Grigorova I, Phan TG, Kelly LM, Cyster JG. Visualizing B Cell Capture of Cognate Antigen From Follicular Dendritic Cells. J Exp Med (2009) 206(7):1485–93. doi: 10.1084/jem.20090209
59. Jozsi M, Prechl J, Bajtay Z, Erdei A. Complement Receptor Type 1 (CD35) Mediates Inhibitory Signals in Human B Lymphocytes. J Immunol (2002) 168(6):2782–2788.42. doi: 10.1097/01.asn.0000073901.94759.36
60. Barnett AN, Asgari E, Chowdhury P, Sacks SH, Dorling A, Mamode N. The Use of Eculizumab in Renal Transplantation. Clin Transplant (2013) 27(3):E216–29. doi: 10.1111/ctr.12102
61. van de Veen W, Stanic B, Yaman G, Wawrzyniak M, Söllner S, Akdis DG, et al. IgG4 Production Is Confined to Human IL-10-Producing Regulatory B Cells That Suppress Antigen-Specific Immune Responses. J Allergy Clin Immunol (2013) 131(4):1204–12. doi: 10.1016/j.jaci.2013.01.014
62. Tebbe B, Wilde B, Ye Z, Wang J, Wang X, Jian F, et al. Renal Transplant Recipients Treated With Calcineurin-Inhibitors Lack Circulating Immature Transitional CD19+CD24hiCD38hi Regulatory B-Lymphocytes. PloS One (2016) 11(4):e0153170. doi: 10.1371/journal.pone.0153170
63. Latorre I, Esteve-Sole A, Redondo D, Giest S, Argilaguet J, Alvarez S, et al. Calcineurin and mTOR Inhibitors Have Opposing Effects on Regulatory T Cells While Reducing Regulatory B Cell Populations in Kidney Transplant Recipients. Transpl Immunol (2016) 35:1–6. doi: 10.1016/j.trim.2016.01.004
64. Knight AK, Radigan L, Marron T, Langs A, Zhang L, Cunningham-Rundles C. High Serum of BAFF, APRIL, and TACI in Common Variable Immunodeficiency. Clin Immunol (2007) 124(2):182–9. doi: 10.1016/j.clim.2007.04.012
65. Nakayamada S, Tanaka Y. BAFF- and APRIL-Targeted Therapy in Systemic Autoimmune Diseases. Inflammation Regen (2016) 36:6. doi: 10.1186/s41232-016-0015-4
66. Matsuda Y, Haneda M, Kadomatsu K, Kobayashi T. A Proliferation-Inducing Ligand Sustains the Proliferation of Human Naïve (CD27–) B Cells and Mediates Their Differentiation Into Long-Lived Plasma Cells In Vitro via Transmembrane Activator and Calcium Modulator and Cyclophilin Ligand Interactor and B-Cell Mature Antigen. Cell Immunol (2015) 295(2):127–36. doi: 10.1016/j.cellimm.2015.02.011
67. Whitehouse G, Gray E, Mastoridis S, Merritt E, Kodela E, Yang JHM, et al. IL-2 Therapy Restores Regulatory T-Cell Dysfunction Induced by Calcineurin Inhibitors. Proc Natl Acad Sci USA (2017) 114(27):7083–8. doi: 10.1073/pnas.1620835114
68. Perry DK, Pollinger HS, Burns JM, Rea D, Ramos E, Platt JL. Alloantibody-Secreting Cells Demonstrating Resistance to Desensitization With IVIG and rATG. Am J Transpl (2008) 8(1):133–43. doi: 10.1111/j.1600-6143.2007.02039.x
69. Lightman SM, Utley A, Lee KP. Survival of Long-Lived Plasma Cells (LLPC): Piecing Together the Puzzle. Front Immunol (2019) 10:965. doi: 10.3389/fimmu.2019.00965
70. Nguyen DC, Garimalla S, Xiao H, Kyu S, Albizua I, Galipeau J. Factors of the Bone Marrow Microniche That Support Human Plasma Cell Survival and Immunoglobulin Secretion. Nat Commun (2018) 9(1):3698. doi: 10.1038/s41467-018-05853-7
71. Fehr T, Gaspert A. Antibody-Mediated Kidney Allograft Rejection: Therapeutic Options and Their Experimental Rationale. Transpl Int (2012) 25:623–32. doi: 10.1111/j.1432-2277.2012.01453.x
72. Marrari M, Duquesnoy RJ. Detection of Donor-Specific HLA Antibodies Before and After Removal of a Rejected Kidney Transplant. Transpl Immunol (2010) 22.3–4:105–9. doi: 10.1016/j.trim.2009.12.005
73. Cooper JE, Gralla J, Chan L, Wiseman AC. Clinical Significance of Post Kidney Transplant De Novo DSA in Otherwise Stable Grafts. Clin Transpl (2011) 359–64.
74. Knight R, Eagar T, Nguyen D, Graviss E, Kuten S, Patel S. Stable Renal Allograft Function Is a Benign Finding. Transplantation (2018) 102:S39. doi: 10.1097/01.tp.0000542596.39174.b7
75. Lee DR, Kim BC, Kim JP, Kim IG, Jeon MY. C3d-Binding Donor-Specific HLA Antibody Is Associated With a High Risk of Antibody-Mediated Rejection and Graft Loss in Stable Kidney Transplant Recipients: A Single-Center Cohort Study. Transplant Proc (2018) 50(10):3452–9. doi: 10.1016/j.transproceed.2018.06.037
76. Viglietti D, Bouatou Y, Kheav VD, et al. Complement-Binding Anti-HLAantibodies Are Independent Predictors of Response to Treatment in Kidney Recipients With Antibody-Mediated Rejection. Kidney Int (2018) 94(4):773–87. doi: 10.1016/j.kint.2018.03.015
77. Shiina Y, Suzuki H, Kaiho T, Matsumoto H, Hata A, Yamamoto, et al. Complement Inhibition as Potential Novel Therapy for Antibody-Mediated Rejection After Lung Transplantation in Murine Antibody-Mediated Rejection Model. J Heart Lung Transplant 2020 (2020) .39:4. doi: 10.1097/01.asn.0000073901.94759.36
78. Stegall MD, Diwan T, Raghavaiah S, Cornell LD, Burns J, Dean PG, et al. Terminal Complement Inhibition Decreases Antibody-Mediated Rejection in Sensitized Renal Transplant Recipients. Am J Transplant (2011) 11(11):2405–13. doi: 10.1111/j.1600-6143.2011.03757.x
79. Stegall MD, Chedid MF, Cornell LD. The Role of Complement in Anti- Body-Mediated Rejection in Kidney Transplantation. Nat Rev Nephrol (2012) 8(11):670–8. doi: 10.1038/nrneph.2012.212
80. Patel JK, Coutance G, Loupy A, Dilibero D, Hamilton M, Kittleson M, et al. Complement Inhibition for Prevention of Antibody Mediated Rejection in Immunologically High-Risk Heart Allograft Recipients, Terminal Complement Inhibition Decreases Antibody-Mediated Rejection in Sensitized Renal Transplant Recipients. Am J Transplant preprint. doi: 10.1111/ajt.16420
81. Locke JE, M ro CM, Singer AL, Segev DL, Haas M, Hillel AT, et al. The Use of Antibody to Complement Protein C5 for Salvage Treatment of Severe Antibody-Mediated Rejection. Am J Transplant (2009) 9(1):231–5. doi: 10.1111/j.1600-6143.2008.02451
82. Lefaucheur C, Viglietti D, Hidalgo LG, Ratner LE, Bagnasco SM, Batal I, et al. Complement-Activating Anti-HLA Antibodies in Kidney Transplantation: Allograft Gene Expression Profiling and Response to Treatment. J Am Soc Nephrol (2018) 29(2):620–35. doi: 10.1681/ASN.2017050589
83. Böhmig GA, Wahrmann M, Eskandary F, Rostaing L. Novel Approaches to Block Complement. Transplantation (2018) 102(11):1837–43. doi: 10.1097/TP.0000000000002267
84. Cippà PE, Liu J, Sun B, Kumar S, Naesens M, McMahon AP. A Late B Lymphocyte Action in Dysfunctional Tissue Repair Following Kidney Injury and Transplantation. Nat Commun (2019) 10(1):1157. doi: 10.1038/s41467-019-09092-2
85. Huang E, Vo A, Choi J, Ammerman N, Lim K, Sethi S, et al. Three-Year Outcomes of a Randomized, Double-Blind, Placebo-Controlled Study Assessing Safety and Efficacy of C1 Esterase Inhibitor for Prevention of Delayed Graft Function in Deceased Donor Kidney Transplant Recipients. Clin J Am Soc Nephrol (2020) 15(1):109–16. doi: 10.2215/CJN.04840419
86. Viglietti D, Gosset C, Loupy A, Deville L, Verine J, Zeevi A, et al. C1 Inhibitor in Acute Antibody-Mediated Rejection Nonresponsive to Conventional Therapy in Kidney Transplant Recipients: A Pilot Study. Am J Transpl (2016) 16(5):1596–603. doi: 10.1111/ajt.13663
87. Chun N, Fairchild RL, Li Y, Liu J, Zhang M, Baldwin WM, et al. Complement Dependence of Murine Costimulatory Blockade-Resistant Cellular Cardiac Allograft Rejection. Am J Transpl (2017) 17(11):2810–9. doi: 10.1111/ajt.14328
88. Jordan SC, Choi J, Aubert O, Haas M, Loupy A, Huang E, et al. A Phase I/II, Double-Blind, Placebo-Controlled Study Assessing Safety and Efficacy of C1 Esterase Inhibitor for Prevention of Delayed Graft Function in Deceased Donor Kidney Transplant Recipients. Am J Transpl (2018) 18(12):2955–64. doi: 10.1111/ajt.14767
89. Berger M, Lefaucheur C, Jordan SC. Update on C1 Esterase Inhibitor in Human Solid Organ Transplantation. Transplantation (2019) 103(9):1763–75. doi: 10.1097/TP.0000000000002717
90. Eskandary F, Jilma B, Mühlbacher J, Wahrmann M, Regele H, Kozakowski N, et al. Anti-C1s Monoclonal Antibody BIVV009 in Late Antibody-Mediated Kidney Allograft Rejection-Results From a First-in-Patient Phase 1 Trial. Am J Transpl (2018) 18(4):916–26. doi: 10.1111/ajt.14528
91. Freire PC, Muñoz CH, Derhaschnig U, Schoergenhofer C, Firbas C, Parry GC, et al. Specific Inhibition of the Classical Complement Pathway Prevents C3 Deposition Along the Dermal-Epidermal Junction in Bullous Pemphigoid. Heil J Invest Dermatol (2019) 139(12):2417–2424.e2. doi: 10.1016/j.jid.2019.04.025
92. Schwarz C, Mahr B, Muckenhuber M, Wekerle T. Belatacept/CTLA4Ig: An Update and Critical Appraisal of Preclinical and Clinical Results. Expert Rev Clin Immunol (2019) 14(7):583–92. doi: 10.1080/1744666X.2018.1485489
93. Vincenti F, Rostaing L, Grinyo J, Rice K, Steinberg S, Gaite L, et al. Belatacept and Long-Term Outcomes in Kidney Transplantation. N Engl J Med (2016) 374(4):333–43. doi: 10.1056/NEJMoa1506027
94. Jain D, Rajab A, Young JS, Yin D, Nadasdy T, Chong AS, et al. Reversing Donor-Specific Antibody Responses and Antibody-Mediated Rejection With Bortezomib and Belatacept in Mice and Kidney Transplant Recipients. Am J Transplant (2020) 20(10):2675–85. doi: 10.1111/ajt.15881
95. Young JS, Chen J, Miller ML, Vu V, Tian C, Moon JJ, et al. Delayed Cytotoxic T Lymphocyte-Associated Protein 4-Immunoglobulin Treatment Reverses Ongoing Alloantibody Responses and Rescues Allografts From Acute Rejection. Am J Transplant (2016) 16(8):2312–23. doi: 10.1111/ajt.13761
96. Levitsky J, Miller J, Huang X, Chandrasekaran D, Chen L, Mathew JM. Inhibitory Effects of Belatacept on Allospecific Regulatory T-Cell Generation in Humans. Transplantation (2013) 96(8):689–96. doi: 10.1097/TP.0b013e31829f1607
97. Alvarez, Salazar EK, Cortés-Hernández A, Alemán-Muench GR, Alberú J, Rodríguez-Aguilera JR, Recillas-Targa F, et al. Methylation of FOXP3 TSDR Underlies the Impaired Suppressive Function of Tregs From Long-Term Belatacept-Treated Kidney Transplant Patients. Front Immunol (2017) 8:219. doi: 10.3389/fimmu.2017.00219
98. Cagnotto G, Willim M, Nilsson JÅ, Compagno M, Jacobsson LTH, Saevarsdottir S, et al. Abatacept in Rheumatoid Arthritis: Survival on Drug, Clinical Outcomes, and Their Predictors-Data From a Large National Quality Register. Turesson C Arthritis Res Ther (2020) 22(1):15. doi: 10.1186/s13075-020-2100-y
99. Burghuber CK, Manook M, Ezekian B, Gibby AC, Leopardi FV, Song M, et al. Dual Targeting: Combining Costimulation Blockade and Bortezomib to Permit Kidney Transplantation in Sensitized Recipients. Am J Transpl (2019) 19(3):724–36. doi: 10.1111/ajt.15067
100. Badell IR, Karadkhele GM, Vasanth P, Farris AB 3rd, Robertson JM, Larsen CP. Abatacept as Rescue Immunosuppression After Calcineurin Inhibitor Treatment Failure in Renal Transplantation. Am J Transpl (2019) 19(8):2342–9. doi: 10.1111/ajt.15319
101. Edwards SR, Wandless TJ. The Rapamycin-Binding Domain of the Protein Kinase Mammalian Target of Rapamycin Is a Destabilizing Domain. J Biol Chem (2007) 282(18):13395–401. doi: 10.1074/jbc.M700498200.Epub 2007 Mar 9
102. Liefeldt L, Brakemeier S, Glander P, Waiser J, Lachmann N, Schönemann C, et al. Donor-Specific HLA Antibodies in a Cohort Comparing Everolimus With Cyclosporine After Kidney Transplantation. Am J Transpl (2012) 12(5):1192–8. doi: 10.1111/j.1600-6143.2011.03961
103. Hay N, Sonenberg N. Upstream and Downstream of mTOR. Genes Dev (2004) 18(16):1926–45. doi: 10.1101/gad.1212704
104. Grimbert P, Thaunat O. mTOR Inhibitors and Risk of Chronic Antibody-Mediated Rejection After Kidney Transplantation: Where Are We Now? Transpl Int (2017) 30(7):647–57. doi: 10.1111/tri.12975
105. Chapman NM, Zeng H, Nguyen TM, Wang Y, Vogel P, Dhungana Y, et al. mTOR Coordinates Transcriptional Programs and Mitochondrial Metabolism of Activated Treg Subsets to Protect Tissue Homeostasis. Nat Commun (2018) 9(1):2095. doi: 10.1038/s41467-018-04392-5
106. Mele TS, Halloran PF. The Use of Mycophenolate Mofetil in Transplant Recipients. Immunopharmacology (2000) 47(2-3):215–45. doi: 10.1016/s0162-3109(00)00190-9
107. Lederer SR, Friedrich N, Banas B, Welser G, Albert ED, Sitter T. Effects of Mycophenolate Mofetil on Donor-Specific Antibody Formation in Renal Transplantation. Clin Transpl (2005) 19(2):168–74. doi: 10.1111/j.1399-0012.2005.00261.x
108. Ransom JT. Mechanism of Action of Mycophenolate Mofetil. Ther Drug Monit (1995) 17(6):681–4. doi: 10.1097/00007691-199512000-00023
109. Moniodis A, Townsend K, Rabin A, Aloum O, Stempel J, Burkett P, et al. Comparison of Extracorporeal Photopheresis and Alemtuzumab for the Treatment of Chronic Lung Allograft Dysfunction. J Heart Lung Transpl (2018) 37:340–8. doi: 10.1016/j.healun.2017.03.017
110. Xu H, Mehta AK, Gao Q, Lee HJ, Ghali A, Guasch A, et al. B Cell Reconstitution Following Alemtuzumab Induction Under a Belatacept-Based Maintenance Regimen. Am J Transplant (2020) 20(3):653–62. doi: 10.1111/ajt.15639
111. Nückel H, Frey UH, Röth A, Dührsen U, Siffert W. Alemtuzumab Induces Enhanced Apoptosis In Vitro in B-Cells From Patients With Chronic Lymphocytic Leukemia by Antibody-Dependent Cellular Cytotoxicity. Eur J Pharmacol (2005) 514(2-3):217–24. doi: 10.1016/j.ejphar.2005.03.024
112. Ravichandran AK, Schilling JD, Novak E, Pfeifer J, Ewald GA, Joseph SM. Rituximab Is Associated With Improved Survival in Cardiac Allograft Patients With Antibody-Mediated Rejection: A Single Center Review. Clin Transpl (2013) 27(6):961–7. doi: 10.1111/ctr.12277
113. Benazzo A, Schwarz S, Geleff S, Weber D, Murakozy G, Lambers C, et al. Donor-Specific Antibodies and Antibody-Mediated Rejection After Alemtuzumab Induction Therapy: A Retrospective Analysis of a High-Volume Lung Transplant Center. J Heart Lung Transplant (2019) 38(4):S166–167. doi: 10.1016/j.healun.2019.01.399
114. Roberts DM, Jiang SH, Chadban SJ. The Treatment of Acute Antibody-Mediated Rejection in Kidney Transplant Recipients-A Systematic Review. Transplantation (2012) 27.94(8):775–83. doi: 10.1097/TP.0b013e31825d1587
115. Velidedeoglu E, Cavaillé-Coll MW, Bala S, Belen OA, Wang Y, Albrecht R. Summary of 2017 FDA Public Workshop: Antibody-Mediated Rejection in Kidney Transplantation. Transplantation (2018) 102(4):557–68. doi: 10.1097/TP.0000000000002049
116. Moreso F, Crespo M, Ruiz JC, Torres A, Gutierrez-Dalmau A, Osun A, et al. Treatment of Chronic Antibody Mediated Rejection With Intravenous Immunoglobulins and Rituximab: A Multicenter, Prospective, Randomized, Double-Blind Clinical Trial. Am J Transpl (2018) 18(4):927–35. doi: 10.1111/ajt.14520
117. Lefaucheur C, Loupy A, Vernerey D, Duong-Van-Huyen JP, Suberbielle C, Anglicheau D, et al. Antibody-Mediated Vascular Rejection of Kidney Allografts: A Population-Based Study. Lancet (2013) 381(9863):313–9. doi: 10.1016/S0140-6736(12)61265-3
118. Ding Y, Francis J, Gautam A, Pelletier L, Sanchorawala V, Quillen K. Durable Renal Response After Combination of Bortezomib, Corticosteroids, Rituximab, and Plasmapheresis for Late Antibody-Mediated Renal Transplant Rejection. Clin Nephrol (2018) 89(4):252–9. doi: 10.5414/CN109278
119. Tajima T, Hata K, Okajima H, Nishikori M, Yasuchika K, Kusakabe J, et al. Bortezomib Against Refractory Antibody-Mediated Rejection After ABO-Incompatible Living-Donor Liver Transplantation: Dramatic Effect in Acute-Phase? Transplant Direct (2019) 19: 5(10):e491. doi: 10.1097/TXD.0000000000000932
120. Moreno Gonzales MA, Gandhi MJ, Schinstock CA, Moore NA, Smith BH, Braaten NY, et al. 32 Doses of Bortezomib for Desensitization Is Not Well Tolerated and Is Associated With Only Modest Reductions in Anti-HLA Antibody. Transplantation (2017) 101(6):1222–7. doi: 10.1097/TP.0000000000001330
121. Lonze BE, Tatapudi VS, Weldon EP, Min ES, Ali NM, Deterville CL, et al. IdeS (Imlifidase): A Novel Agent That Cleaves Human IgG and Permits Successful Kidney Transplantation Across High-Strength Donor-Specific Antibody. Ann Surg (2018) 268(3):488–96. doi: 10.1097/SLA.0000000000002924
122. Jordan SC, Lorant T, Choi J, Kjellman C, Winstedt L, Bengtsson M, et al. IgG Endopeptidase in Highly Sensitized Patients Undergoing Transplantation. N Engl J Med (2017) 377(5):442–53. doi: 10.1056/NEJMoa1612567
123. Bujnowska A, Michon M, Konopelski P, Hryniewiecka E, Jalbrzykowska A, Perkowska-Ptasinska A, et al. Outcomes of Prolonged Treatment With Intravenous Immunoglobulin Infusions for Acute Antibody-Mediated Rejection in Kidney Transplant Recipients. Transplant Proc (2018) 50(6):1720–5. doi: 10.1016/j.transproceed.2018.02.110
124. de Sousa MV, Gonçalez AC, de Lima Zollner R, Mazzali M. Treatment of Antibody-Mediated Rejection After Kidney Transplantation: Immunological Effects, Clinical Response, and Histological Findings. Ann Transplant (2020) 25:e925488. doi: 10.12659/AOT.925488
125. Liu J, Farmer JD Jr, Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin Is a Common Target of Cyclophilin-Cyclosporin A and FKBP-FK506 Complexes. Cell (1991) 66(4):807–15. doi: 10.1016/0092-8674(91)90124-H
126. Nankivell BJ, Shingde M, Keung KL, Fung CL, Borrows RJ, O’Connell PJ, et al. The Causes, Significance and Consequences of Inflammatory Fibrosis in Kidney Transplantation: The Banff I-IFTA Lesion. Am J Transpl (2018) 18(2):364–76. doi: 10.1111/ajt.14609
127. Sommerer C, Meuer S, Zeier M, Giese T. Calcineurin Inhibitors and NFAT-Regulated Gene Expression. Clin Chim Acta (2012) 413(17-18):1379–86. doi: 10.1016/j.cca.2011.09.041
128. Wiebe C, Rush DN, Nevins TE, Birk PE, Blydt-Hansen T, Gibson IW, et al. Class II Eplet Mismatch Modulates Tacrolimus Trough Levels Required to Prevent Donor-Specific Antibody Development. J Am Soc Nephrol (2017) 28(11):3353–62. doi: 10.1681/ASN.2017030287
129. Kidney Disease: Improving Global Outcomes (KDIGO) Transplant Work Group. KDIGO Clinical Practice Guideline for the Care of Kidney Transplant Recipients. Am J Transpl (2009) 9 Suppl 3:S1–155. doi: 10.1111/j.1600-6143.2009.02834.x
130. Bamoulid J, Staeck O, Crépin T, Halleck F, Saas P, Brakemeier S, et al. Anti-Thymocyte Globulins in Kidney Transplantation: Focus on Current Indications and Long-Term Immunological Side Effects. Nephrol Dial Transpl (2017) 32(10):1601–8. doi: 10.1093/ndt/gfw368
131. Burton SA, Amir N, Asbury A, Lange A, Hardinger KL. Treatment of Antibody-Mediated Rejection in Renal Transplant Patients: A Clinical Practice Survey. Clin Transpl (2015) 29(2):118–23. doi: 10.1111/ctr.12491
132. Chapman TM, Keating GM. Basiliximab: A Review of Its Use as Induction Therapy in Renal Transplantation. Drugs (2003) 63(24):2803–35. doi: 10.2165/00003495-200363240-00009
133. Cornell LD, Schinstock CA, Gandhi MJ, Kremers WK, Stegall MD. Positive Crossmatch Kidney Transplant Recipients Treated With Eculizumab: Outcomes Beyond 1 Year. Am J Transplant (2015) 15(5):1293–302. doi: 10.1111/ajt.13168
134. Schinstock CA, Bentall AJ, Smith BH, Cornell LD, Everly M, Gandhi MJ, et al. Long-Term Outcomes of Eculizumab-Treated Positive Crossmatch Recipients: Allograft Survival, Histologic Findings, and Natural History of the Donor-Specific Antibodies. Am J Transpl (2019) 19(6):1671–83. doi: 10.1111/ajt.15175
135. Ding Y, Francis J, Gautam A, Pelletier L, Sanchorawala V, Quillen K. Durable Renal Response After Combination of Bortezomib, Corticosteroids, Rituximab, and Plasmapheresis for Late Antibody-Mediated Renal Transplant Rejection. Clin Nephrol (2018) 89(4):252–9. doi: 10.5414/CN109278
136. Sakurai H, Shigemori N, Hisada Y, Ishizuka T, Kawashima K, Sugita T. Suppression of NF-Kappa B and AP-1 Activation by Glucocorticoids in Experimental Glomerulonephritis in Rats: Molecular Mechanisms of Anti-Nephritic Action. Biochim Biophys Acta (1997) 1362(2-3):252–62. doi: 10.1016/s0925-4439(97)00068-9
137. Hipp N, Symington H, Pastoret C, Caron G, Monvoisin C, Tarte K, et al. IL-2 Imprints Human Naive B Cell Fate Towards Plasma Cell Through ERK/ELK1-Mediated BACH2 Repression. Nat Commun (2017) 8(1):1443. doi: 10.1038/s41467-017-01475-7
138. Le Gallou S, Caron G, Delaloy C, Rossille D, Tarte K, Fest T. IL-2 Requirement for Human Plasma Cell Generation: Coupling Differentiation and Proliferation by Enhancing MAPK-ERK Signaling. J Immunol (2012) 189(1):161–73. doi: 10.4049/jimmunol.1200301
139. Qu X, Kirken RA, Tian L, Wang M, Bennett CF, Stepkowski SM. Selective Inhibition of IL-2 Gene Expression by IL-2 Antisense Oligonucleotides Blocks Heart Allograft Rejection. Transplantation (2001) 72(5):915–23. doi: 10.1097/00007890-200109150-00029
140. McHugh J. Systemic Lupus Erythematosus: B Cell-Derived IL-6 Promotes Disease. Nat Rev Rheumatol (2017) 13(11):633. doi: 10.1038/nrrheum.2017.163
141. Maeda K, Mehta H, Drevets DA, Coggeshall KM. IL-6 Increases B-Cell IgG Production in a Feed-Forward Proinflammatory Mechanism to Skew Hematopoiesis and Elevate Myeloid Production. Blood (2010) 115(23):4699–706. doi: 10.1182/blood-2009-07-230631
142. Rosser EC, Oleinika K, Tonon S, Doyle R, Bosma A, Carter NA, et al. Regulatory B Cells Are Induced by Gut Microbiota-Driven Interleukin-1beta and Interleukin-6 Production. Nat Med (2014) 20(11):1334–9. doi: 10.1038/nm.3680
143. Mauer J, Chaurasia B, Goldau J, Vogt MC, Ruud J, Nguyen KD, et al. Signaling by IL-6 Promotes Alternative Activation of Macrophages to Limit Endotoxemia and Obesity-Associated Resistance to Insulin. Nat Immunol (2014) 15(5):423–30. doi: 10.1038/ni.2865
144. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal Developmental Pathways for the Generation of Pathogenic Effector TH17 and Regulatory T Cells. Nature (2006) 441(7090):235–8. doi: 10.1038/nature04753
145. Kimura A, Kishimoto T. IL-6: Regulator of Treg/Th17 Balance. Eur J Immunol (2010) 40(7):1830–5. doi: 10.1002/eji.201040391
146. Korn T, Mitsdoerffer M, Croxford AL, Awasthi A, Dardalhon VA, Galileos G, et al. IL-6 Controls Th17 Immunity In Vivo by Inhibiting the Conversion of Conventional T Cells Into Foxp3+ Regulatory T Cells. Proc Natl Acad Sci USA (2008) 105(47):18460–5. doi: 10.1073/pnas.0809850105
147. Schneider A, Long SA, Cerosaletti K, Ni CT, Samuels P, Kita M, et al. In Active Relapsing-Remitting Multiple Sclerosis, Effector T Cell Resistance to Adaptive T(regs) Involves IL-6-Mediated Signaling. Sci Transl Med (2013) 5(170):170ra15. doi: 10.1126/scitranslmed.3004970
148. Doberer K, Duerr M, Halloran PF, Eskandary F, Budde K, Regele H, et al. A Randomized Clinical Trial of Anti-IL-6 Antibody Clazakizumab in Late Antibody-Mediated Kidney Transplant Rejection. J Am Soc Nephrol (2021) 32(3):708–22. doi: 10.1681/ASN.2020071106
149. Choi J, Aubert O, Vo A, Loupy A, Haas M, Puliyanda D, et al. Assessment of Tocilizumab (Anti-Interleukin-6 Receptor Monoclonal) as a Potential Treatment for Chronic Antibody-Mediated Rejection and Transplant Glomerulopathy in HLA-Sensitized Renal Allograft Recipients. Am J Transpl (2017) 17(9):2381–9. doi: 10.1111/ajt.14228
150. Vazquez MI, Catalan-Dibene J, Zlotnik. A. B Cells Responses and Cytokine Production Are Regulated by Their Immune Microenvironment. Cytokine (2015) 74(2):318–26. doi: 10.1016/j.cyto.2015.02.007
151. Corfe SA, Paige CJ. The Many Roles of IL-7 in B Cell Development; Mediator of Survival, Proliferation and Differentiation. Semin Immunol (2012) 24(3):198–208. doi: 10.1016/j.smim.2012.02.001
152. Milne CD, Paige CJ. IL-7: A Key Regulator of B Lymphopoiesis. Semin Immunol (2006) 18(1):20– 30. doi: 10.1016/j.smim.2005.10.003
153. Mai HL, Boeffard F, Longis J, Danger R, Martinet B, Haspot F, et al. IL-7 Receptor Blockade Following T Cell Depletion Promotes Long-Term Allograft Survival. J Clin Invest (2014) 124(4):1723–33. doi: 10.1172/JCI66287
154. Luo Y, Luo F, Zhang K, Wang S, Zhang H, Yang X, et al. Elevated Circulating IL-10 Producing Breg, But Not Regulatory B Cell Levels, Restrain Antibody-Mediated Rejection After Kidney Transplantation. Front Immunol (2021) 11:627496. doi: 10.3389/fimmu.2020.627496
155. Lin X, Wang X, Xiao F, Ma K, Liu L, Wang X, et al. IL-10-Producing Regulatory B Cells Restrain the T Follicular Helper Cell Response in Primary Sjögren’s Syndrome. Cell Mol Immunol (2019) 16(12):921–31. doi: 10.1038/s41423-019-0227-z
156. Shen P, Roch T, Lampropoulou V, O’Connor RA, Stervbo U, Hilgenberg E, et al. IL-35-Producing B Cells Are Critical Regulators of Immunity During Autoimmune and Infectious Diseases. Nature (2014) 507(7492):366–70. doi: 10.1038/nature12979
157. Guo H, Wang W, Zhao N, He X, Zhu L, Jiang X. Inhibiting Cardiac Allograft Rejection With Interleukin-35 Therapy Combined With Decitabine Treatment in Mice. Transpl Immunol (2013) 29(1-4):99–104. doi: 10.1016/j.trim.2013.10.001
158. Choi JK, Egwuagu CE. Interleukin 35 Regulatory B Cells. J Mol Biol (2021) 433(1):166607. doi: 10.1016/j.jmb.2020.07.019
159. Tsuchida Y, Sumitomo S, Ishigaki K, Suzuki A, Kochi Y, Tsuchiya H, et al. Antibody Production by Human B Cells. PloS One (2017) 12(1):e0169646. doi: 10.1371/journal.pone.0169646
160. Guan Q, Li S, Gao S, Chen H, Nguan CY, Du C. Reduction of Chronic Rejection of Renal Allografts by Anti-Transforming Growth Factor-β Antibody Therapy in a Rat Model. Am J Physiol Renal Physiol (2013) 305(2):F199–207. doi: 10.1152/ajprenal.00665.2012
161. Rosser EC. Regulatory B Cells: Origin, Phenotype, and Function. Immunity (2015) 42(4):607–12. doi: 10.1016/j.immuni.2015.04.005
162. Pandiyan P, Yang XP, Saravanamuthu SS, Zheng L, Ishihara S, O’Shea JJ, et al. The Role of IL-15 in Activating STAT5 and Fine-Tuning IL-17A Production in CD4 T Lymphocytes. J Immunol (2012) 189(9):4237–46. doi: 10.4049/jimmunol.1201476
163. Zheng XX, Gao W, Donskoy E, Neuberg M, Ruediger M, Strom TB, et al. An Antagonist Mutant IL-15/Fc Promotes Transplant Tolerance. Transplantation (2006) 81(1):109–16. doi: 10.1097/01.tp.0000188139.11931.98
164. Conti F, Frappier J, Dharancy S, Chereau C, Houssin D, Weill B, et al. Interleukin-15 Production During Liver Allograft Rejection in Humans. Calmus Y Transpl (2003) 76(1):210–6. doi: 10.1097/01.TP.0000067530.95852.67
165. Wang S, Wang J, Kumar V, Karnell JL, Naiman B, Gross PS, et al. IL-21 Drives Expansion and Plasma Cell Differentiation of Autoreactive CD11chiT-Bet+ B Cells in SLE. Nat Commun (2018) 9(1):1758. doi: 10.1038/s41467-018-03750-7
166. Khattar M, Baum CE, Schroder P, Breidenbach JD, Haller ST, Chen W, et al. Interleukin 21 (IL-21) Regulates Chronic Allograft Vasculopathy (CAV) in Murine Heart Allograft Rejection. Stepkowski s. PloS One (2019) 14(11):e0225624. doi: 10.1371/journal.pone.0225624
167. van Besouw NM, Yan L, de Kuiper R, Klepper M, Reijerkerk D, Dieterich M, et al. The Number of Donor-Specific IL-21 Producing Cells Before and After Transplantation Predicts Kidney Graft Rejection. Front Immunol (2019) 10:748. doi: 10.3389/fimmu.2019.00748
168. Popescu M, Cabrera-Martinez B, Winslow GM. TNF-α Contributes to Lymphoid Tissue Disorganization and Germinal Center B Cell Suppression During Intracellular Bacterial Infection. J Immunol (2019) 203(9):2415–24. doi: 10.4049/jimmunol.1900484
169. Tay C, Liu YH, Hosseini H, Kanellakis P, Cao A, Peter K, et al. B-Cell-Specific Depletion of Tumour Necrosis Factor Alpha Inhibits Atherosclerosis Development and Plaque Vulnerability to Rupture by Reducing Cell Death and Inflammation. Cardiovasc Res (2016) 111(4):385–97. doi: 10.1093/cvr/cvw186
170. Sonkar GK, Usha Singh RG. Evaluation of Serum Tumor Necrosis Factor Alpha and Its Correlation With Histology in Chronic Kidney Disease, Stable Renal Transplant and Rejection Cases. Saudi J Kidney Dis Transpl (2009) 20(6):1000–4.
171. Furie R, Rovin BH, Houssiau F, Malvar A, Teng YKO, Contreras G, et al. Two-Year, Randomized, Controlled Trial of Belimumab in Lupus Nephritis. N Engl J Med (2020) 383(12):1117–28. doi: 10.1056/NEJMoa2001180
172. Afzali S, Salehi S, Shahi A, Esmaeili M, Farashi Bonab S, Peykari A, et al. Investigating the Role of BAFF and Its Receptors in Renal Transplant Recipients With Chronic Antibody-Mediated Rejection. J Immunol Res (2021) 2021:6654992. doi: 10.1155/2021/6654992
173. Wang XZ, Wan Z, Xue WJ, Zheng J, Li Y, Ding CG, et al. B-Cell Activating Factor Predicts Acute Rejection Risk in Kidney Transplant Recipients: A 6-Month Follow-Up Study. Front Immunol (2019) 10:1046. doi: 10.3389/fimmu.2019.01046
174. Pongpirul W, Chancharoenthana W, Pongpirul K, Leelahavanichkul A, Kittikowit W, Jutivorakool K, et al. B-Cell Activating Factor, a Predictor of Antibody Mediated Rejection in Kidney Transplantation Recipients. Nephrol (Carlton) (2018) 23(2):169–74. doi: 10.1111/nep.12972
175. Krejcik J, Casneuf T, Nijhof IS, Verbist B, Bald J, Plesner T, et al. Daratumumab Depletes CD38+ Immune Regulatory Cells, Promotes T-Cell Expansion, and Skews T-Cell Repertoire in Multiple Myeloma. Blood (2016) 128(3):384–94. doi: 10.1182/blood-2015-12-687749
176. Casneuf T, Xu XS, Adams HC 3rd, Axel AE, Chiu C, Khan I, et al. Effects of Daratumumab on Natural Killer Cells and Impact on Clinical Outcomes in Relapsed or Refractory Multiple Myeloma. Blood Adv (2017) 1(23):2105–14. doi: 10.1182/bloodadvances.2017006866
177. van de Donk N. Immunomodulatory Effects of CD38-Targeting Antibodies. Immunol Lett (2018) 199:16–22. doi: 10.1016/j.imlet.2018.04.005
178. Kwun J, Matignon M, Manook M, Guendouz S, Audard V, Kheav D, et al. Daratumumab in Sensitized Kidney Transplantation: Potentials and Limitations of Experimental and Clinical Use. J Am Soc Nephrol (2019) 30(7):1206–19. doi: 10.1681/ASN.2018121254
179. Feng X, Zhang LI, Acharya C, An G, Wen K, Qiu L, et al. Targeting CD38 Suppresses Induction and Function of T Regulatory Cells to Mitigate Immunosuppression in Multiple Myeloma. Clin Cancer Res (2017) 23(15):4290–300. doi: 10.1158/1078-0432.CCR-16-3192
180. Daoussis D, Andonopoulos AP, Liossis SN. Targeting CD40L: A Promising Therapeutic Approach. Clin Diagn Lab Immunol (2004) 11(4):635–41. doi: 10.1128/CDLI.11.4.635-641.2004
181. Karnell JL, Albulescu M, Drabic S, Wang L, Moate R, Baca M, et al. Protein Reduces Autoantibodies and Improves Disease Activity in Patients With Autoimmunity. Sci Transl Med (2019) 11(489):eaar6584. doi: 10.1126/scitranslmed.aar6584
182. Kreuzaler M, Rauch M, Salzer U, Birmelin J, Rizzi M, Grimbacher B, et al. Soluble BAFF Levels Inversely Correlate With Peripheral B Cell Numbers and the Expression of BAFF Receptors. J Immunol (2012) 188(1):497–503. doi: 10.4049/jimmunol.1102321
183. Tsuji S, Stein L, Kamada N, Nuñez G, Bram R, Vallance BA, et al. TACI Deficiency Enhances Antibody Avidity and Clearance of an Intestinal Pathogen. J Clin Invest (2014) 124(11):4857–66. doi: 10.1172/JCI74428
184. Ou X, Xu S, Lam KP. Deficiency in TNFRSF13B (TACI) Expands T-Follicular Helper and Germinal Center B Cells via Increased ICOS-Ligand Expression But Impairs Plasma Cell Survival. Proc Natl Acad Sci USA (2012) 109(38):15401–6. doi: 10.1073/pnas.1200386109
185. Salazar-Camarena DC, Ortiz-Lazareno PC, Cruz A, Oregon-Romero E, Machado-Contreras JR, Muñoz-Valle JF, et al. Association of BAFF, APRIL Serum Levels, BAFF-R, TACI and BCMA Expression on Peripheral B-Cell Subsets With Clinical Manifestations in Systemic Lupus Erythematosus. Lupus (2016) 25(6):582–92. doi: 10.1177/0961203315608254
186. Yang M, Hase H, Legarda-Addison D, Varughese L, Seed B, Ting AT. B Cell Maturation Antigen, the Receptor for a Proliferation-Inducing Ligand and B Cell-Activating Factor of the TNF Family, Induces Antigen Presentation in B Cells. J Immunol (2005) 175(5):2814–24. doi: 10.4049/jimmunol.175.5.2814
187. Huang HW, Chen CH, Lin CH, Wong CH, Lin KI. B-Cell Maturation Antigen Is Modified by a Single N-Glycan Chain That Modulates Ligand Binding and Surface Retention. Proc Natl Acad Sci USA (2013) 110(27):10928–33. doi: 10.1073/pnas.1309417110
188. O’Connor BP, Raman VS, Erickson LD, Cook WJ, Weaver LK, Ahonen C, et al. BCMA Is Essential for the Survival of Long-Lived Bone Marrow Plasma Cells. J Exp Med (2004) 199(1):91–8. doi: 10.1084/jem.20031330
189. Jordan SC, Ammerman N, Choi J, Huang E, Peng A, Sethi S, et al. The Role of Novel Therapeutic Approaches for Prevention of Allosensitization and Antibody-Mediated Rejection. Am J Transpl (2020) 20 Suppl 4:42–56. doi: 10.1111/ajt.15913
190. Xu Y, Fairfax K, Light A, Huntington ND, Tarlinton DM. CD19 Differentially Regulates BCR Signalling Through the Recruitment of PI3K. Autoimmunity (2014) 47(7):430–7. doi: 10.3109/08916934.2014.921810
191. Cree BAC, Bennett JL, Kim HJ, Weinshenker BG, Pittock SJ, Wingerchuk DM, et al. Inebilizumab for the Treatment of Neuromyelitis Optica Spectrum Disorder (N-MOmentum): A Double-Blind, Randomised Placebo-Controlled Phase 2/3 Trial. Lancet (2019) 394(10206):1352–63. doi: 10.1016/S0140-6736(19)31817-3
192. Frampton JE. Inebilizumab: First Approval. Drugs (2020) 80(12):1259–64. doi: 10.1007/s40265-020-01370-4
193. Kharabi Masouleh B, Ten Dam GB, Wild MK, Seelige R, van der Vlag J, Rops AL, et al. Role of the Heparan Sulfate Proteoglycan Syndecan-1 (CD138) in Delayed-Type Hypersensitivity. J Immunol (2009) 182(8):4985–93. doi: 10.4049/jimmunol.0800574
194. McCarron MJ, Park PW, Fooksman DR. CD138 Mediates Selection of Mature Plasma Cells by Regulating Their Survival. Blood (2017) 129(20):2749–59. doi: 10.1182/blood-2017-01-761643
195. Khodadadi L, Cheng Q, Radbruch A, Hiepe F. The Maintenance of Memory Plasma Cells. Front Immunol (2019) 10:721. doi: 10.3389/fimmu.2019.00721
196. Carpio VN, Noronha Ide L, Martins HL, Jobim LF, Gil BC, Külzer AS, et al. Expression Patterns of B Cells in Acute Kidney Transplant Rejection. Exp Clin Transpl (2014) 12(5):405–14.
197. Bhat ZY, Bostwick DG, Hossain D, Zeng X. Participation of Functionally Active Plasma Cells in Acute Rejection and Response to Therapy in Renal Allografts. DNA Cell Biol (2014) 33(7):448–54. doi: 10.1089/dna.2014.2371
198. Viau M, Zouali M. B-Lymphocytes, Innate Immunity, and Autoimmunity. Clin Immunol (2005) 114(1):17–26. doi: 10.1016/j.clim.2004.08.019
199. Lenert P, Brummel R, Field EH, Ashman RF. TLR-9 Activation of Marginal Zone B Cells in Lupus Mice Regulates Immunity Through Increased IL-10 Production. J Clin Immunol (2005) 25(1):29–40. doi: 10.1007/s10875-005-0355-6
200. Zhang X. Regulatory Functions of Innate-Like B Cells. Cell Mol Immunol (2013) 10(2):113–21. doi: 10.1038/cmi.2012.63
201. Ran Z, Yue-Bei L, Qiu-Ming Z, Huan Y. Regulatory B Cells and Its Role in Central Nervous System Inflammatory Demyelinating Diseases. Front Immunol (2020) 11:1884. doi: 10.3389/fimmu.2020.01884
202. Tsay GJ, Zouali M. The Interplay Between Innate-Like B Cells and Other Cell Types in Autoimmunity. Front Immunol (2018) 16:1064. doi: 10.3389/fimmu.2018.01064
203. Wang L, Ray A, Jiang X, Wang JY, Basu S, Liu X, et al. T Regulatory Cells and B Cells Cooperate to Form a Regulatory Loop That Maintains Gut Homeostasis and Suppresses Dextran Sulfate Sodium-Induced Colitis. Mucosal Immunol (2015) 8(6):1297–312. doi: 10.1038/mi.2015.20
204. Chien CH, Chiang BL. Regulatory T Cells Induced by B Cells: A Novel Subpopulation of Regulatory T Cells. J BioMed Sci 18; (2017) 24(1):86. doi: 10.1186/s12929-017-0391-3
205. Chesneau M, Pallier A, Braza F, Lacombe G, Le Gallou S, Baron D, et al. Unique B Cell Differentiation Profile in Tolerant Kidney Transplant Patients. Am J Transpl (2014) 14(1):144–55. doi: 10.1111/ajt.12508
206. Lefaucheur C, Viglietti D, Bentlejewski C, van Huyen JP D, Vernerey D, Aubert O, et al. IgG Donor-Specific Anti-Human HLA Antibody Subclasses and Kidney Allograft Antibody-Mediated Injury. J Am Soc Nephrol (2016) 27(1):293–304. doi: 10.1681/ASN.2014111120
207. Imamura R, Matsuda Y, Tsutahara K, Nonomura N, Takahara S. Impact of Immunoglobulin M-Type Donor-Specific Human Leukocyte Antigen-Antibody Levels in Supernatants From Cultured Peripheral Blood Mononuclear Cells as Predictors of Antibody-Mediated Rejection. Pathogens (2020) 9(9):733. doi: 10.3390/pathogens9090733
208. Haneda M, Owaki M, Kuzuya T, Iwasaki K, Miwa Y, Kobayashi T. Comparative Analysis of Drug Action on B-Cell Proliferation and Differentiation for Mycophenolic Acid, Everolimus, and Prednisolone. Transplantation (2014) 97(4):405–12. doi: 10.1097/01.TP.0000441826.70687.f6
209. Kim J, Koo BK, Knoblich JA. Human Organoids: Model Systems for Human Biology and Medicine. Nat Rev Mol Cell Biol (2020) 21(10):571–84. doi: 10.1038/s41580-020-0259-3
210. Broutier L, Mastrogiovanni G, Verstegen MM, Francies HE, Gavarró LM, Bradshaw CR, et al. Human Primary Liver Cancer-Derived Organoid Cultures for Disease Modeling and Drug Screening. Nat Med (2017) 23(12):1424–35. doi: 10.1038/nm.4438
211. Nanki Y, Chiyoda T, Hirasawa A, Ookubo A, Itoh M, Ueno M, et al. Patient-Derived Ovarian Cancer Organoids Capture the Genomic Profiles of Primary Tumours Applicable for Drug Sensitivity and Resistance Testing. Sci Rep (2020) 10(1):12581. doi: 10.1038/s41598-020-69488-9
212. Peelen DM, Hoogduijn MJ, Hesselink DA, Baan CC. Advanced In Vitro Research Models to Study the Role of Endothelial Cells in Solid Organ Transplantation. Front Immunol (2021) 12:607953. doi: 10.3389/fimmu.2021.607953
213. Sivozhelezov V, Braud C, Giacomelli L, Pechkova E, Giral M, Soulillou JP, et al. Immunosuppressive Drug-Free Operational Immune Tolerance in Human Kidney Transplants Recipients. Part II. Non-statistical gene microarray analysis. J Cell Biochem (2008) 103(6):1693–706. doi: 10.1002/jcb.21557
214. Wittig M, Anmarkrud JA, Kässens JC, Koch S, Forster M, Ellinghaus E, et al. Development of a High-Resolution NGS-Based HLA-Typing and Analysis Pipeline. Nucleic Acids Res (2015) 43(11):e70. doi: 10.1093/nar/gkv184
215. Liu C. A Long Road/Read to Rapid High-Resolution HLA Typing: The Nanopore Perspective. Hum Immunol (2020) S0198-8859(20):30189–0. doi: 10.1016/j.humimm.2020.04.009
216. Ouchida R, Mori H, Hase K, Takatsu H, Kurosaki T, Tokuhisa T, et al. Critical Role of the IgM Fc Receptor in IgM Homeostasis, B-Cell Survival, and Humoral Immune Responses. Proc Natl Acad Sci USA (2012) 109:E2699–706. doi: 10.1073/pnas.1210706109
217. Kubagawa H, Oka S, Kubagawa Y, Torii I, Takayama E, Kang DW, et al. Identity of the Elusive IgM Fc Receptor (FcmuR) in Humans. J Exp Med (2009) 206:2779–93. doi: 10.1084/jem.20091107
218. Shima H, Takatsu H, Fukuda S, Ohmae M, Hase K, Kubagawa H, et al. Identification of TOSO/FAIM3 as an Fc Receptor for IgM. Int Immunol (2010) 22:149–56. doi: 10.1093/intimm/dxp121
219. Liu J, Zhu H, Qian J, Xiong E, Zhang L, Wang YQ, et al. Fcmicro Receptor Promotes the Survival and Activation of Marginal Zone B Cells and Protects Mice Against Bacterial Sepsis. Front Immunol (2018) 9:160. doi: 10.3389/fimmu.2018.00160
220. Nguyen TT, Klasener K, Zurn C, Castillo PA, Brust-Mascher I, Imai DM, et al. The IgM Receptor FcmuR Limits Tonic BCR Signaling by Regulating Expression of the IgM BCR. Nat Immunol (2017) 18:321–33. doi: 10.1038/ni.3677
221. Choi SC, Wang H, Tian L, Murakami Y, Shin DM, Borrego F, et al. Mouse IgM Fc Receptor, FCMR, Promotes B Cell Development and Modulates Antigen-Driven Immune Responses. J Immunol (2013) 190:987–96. doi: 10.4049/jimmunol.1202227
222. Brenner D, Brustle A, Lin GH, Lang PA, Duncan GS, Knobbe-Thomsen CB, et al. Toso Controls Encephalitogenic Immune Responses by Dendritic Cells and Regulatory T Cells. Proc Natl Acad Sci USA (2014) 111:1060–5. doi: 10.1073/pnas.1323166111
223. Adachi T, Harumiya S, Takematsu H, Kozutsumi Y, Wabl M, Fujimoto M, et al. CD22 Serves as a Receptor for Soluble IgM. Eur J Immunol (2012) 42(1):241–7. doi: 10.1002/eji.201141899
224. Clark EA, Giltiay NV. CD22: A Regulator of Innate and Adaptive B Cell Responses and Autoimmunity. Front Immunol (2018) 9:2235. doi: 10.3389/fimmu.2018.02235
225. Haas KM, Johnson KL, Phipps JP, Do C. CD22 Promotes B-1b Cell Responses to T Cell-Independent Type 2 Antigens. J Immunol (2018) 200(5):1671–81. doi: 10.4049/jimmunol.1701578
226. Collins BE, Blixt O, DeSieno AR, Bovin N, Marth JD, Paulson JC. Masking of CD22 by Cis Ligands Does Not Prevent Redistribution of CD22 to Sites of Cell Contact. Proc Natl Acad Sci USA (2004) 101:6104–9. doi: 10.1073/pnas.0400851101
227. Macauley MS, Pfrengle F, Rademacher C, Nycholat CM, Gale AJ, von Drygalski A, et al. Antigenic Liposomes Displaying CD22 Ligands Induce Antigen-Specific B Cell Apoptosis. J Clin Invest (2013) 123:3074–83. doi: 10.1172/JCI69187
228. Chappell CP, Clark EA. STALing B Cell Responses With CD22. J Clin Invest (2013) 123:2778–80. doi: 10.1172/JCI69670
229. Pillai S, Cariappa A, Pirnie SP. Esterases and Autoimmunity: The Sialic Acid Acetylesterase Pathway and the Regulation of Peripheral B Cell Tolerance. Trends Immunol (2009) 30:488–93. doi: 10.1016/j.it.2009.07.006
230. Surolia I, Pirnie SP, Chellappa V, Taylor KN, Cariappa A, Moya J, et al. Functionally Defective Germline Variants of Sialic Acid Acetylesterase in Autoimmunity. Nature (2010) 466:243–7. doi: 10.1038/nature09115
231. Chellappa V, Taylor KN, Pedrick K, Donado C, Netravali IA, Haider K, et al. M89V Sialic Acid Acetyl Esterase (SIAE) and All Other Non-Synonymous Common Variants of This Gene Are Catalytically Normal. PloS One (2013) 8:e53453. doi: 10.1371/journal.pone.0053453
232. Dawidowicz K, Dieudé P, Avouac J, Wipff J, Hachulla E, Diot E, et al. Association Study of B-Cell Marker Gene Polymorphisms in European Caucasian Patients With Systemic Sclerosis. Clin Exp Rheumatol (2011) 29:839–42.
233. Kim TH, Yang K, Kim M, Kim HS, Kang JL. Apoptosis Inhibitor of Macrophage (AIM) Contributes to IL-10-Induced Anti-Inflammatory Response Through Inhibition of Inflammasome Activation. Cell Death Dis (2021) 12(1):19. doi: 10.1038/s41419-020-03332-w
234. Sugisawa R, Komatsu G, Hiramoto E, Takeda N, Yamamura KI, Arai S, et al. Independent Modes of Disease Repair by AIM Protein Distinguished in AIM-Felinized Mice. Sci Rep (2018) 8(1):13157. doi: 10.1038/s41598-018-31580-6
235. Yusa S, Ohnishi S, Onodera T, Miyazaki T. AIM, a Murine Apoptosis Inhibitory Factor, Induces Strong and Sustained Growth Inhibition of B Lymphocytes in Combination With TGF-Beta1. Eur J Immunol (1999) 29(4):1086–93. doi: 10.1002/(SICI)1521-4141(199904)29:04<1086:AID-IMMU1086>3.0.CO;2-X
236. Arai S, Miyazaki T. A Scavenging System Against Internal Pathogens Promoted by the Circulating Protein Apoptosis Inhibitor of Macrophage (AIM). Semin Immunopathol (2018) 40(6):567–75. doi: 10.1007/s00281-018-0717-6
237. Sugisawa R, Hiramoto E, Matsuoka S, Iwai S, Takai R, Yamazaki T. AIM on the Susceptibility of Cats to Renal Disease. Sci Rep 2016 (2016) 6:35251. doi: 10.1038/srep35251
238. Arai S, Kitada K, Yamazaki T, Takai R, Zhang X, Tsugawa Y, et al. Macrophage Protein Enhances Intraluminal Debris Clearance and Ameliorates Acute Kidney Injury in Mice. Nat Med (2016) 22(2):183–93. doi: 10.1038/nm.4012
239. Ichimura T, Asseldonk EJ, Humphreys BD, Gunaratnam L, Duffield JS, Bonventre JV. Kidney Injury Molecule-1 Is a Phosphatidylserine Receptor That Confers a Phagocytic Phenotype on Epithelial Cells. J Clin Invest (2008) 118(5):1657–68. doi: 10.1172/JCI34487
240. Yang L, Brooks CR, Xiao S, Sabbisetti V, Yeung MY, Hsiao LL, et al. KIM-1-Mediated Phagocytosis Reduces Acute Injury to the Kidney. J Clin Invest (2015) 125(4):1620–36. doi: 10.1172/JCI75417
241. Ono Y, Kanmura S, Morinaga Y, Oda K, Kawabata K, Arima S, et al. Inhibitor of Macrophages as a Possible Diagnostic Marker in Patients With Crohn’s Disease. BMC Gastroenterol (2017) 17(1):40. doi: 10.1186/s12876-017-0591-z
242. Tarazón E, Corbacho-Alonso N, Barderas MG, Gil-Cayuela C, García-Manzanares M, Feijóo-Bandín S, et al. Plasma CD5L and Non-Invasive Diagnosis of Acute Heart Rejection. J Heart Lung Transplant (2020) 39(3):257–66. doi: 10.1016/j.healun.2019.11.004
243. Rodriguez-Zhurbenko N, Quach TD, Hopkins TJ, Rothstein TL, Hernandez AM. Human B-1 Cells and B-1 Cell Antibodies Change With Advancing Age. Front Immunol (2019) 10:483. doi: 10.3389/fimmu.2019.00483
244. Iyer SS, Cheng G. Role of Interleukin 10 Transcriptional Regulation in Inflammation and Autoimmune Disease. Crit Rev Immunol (2012) 32(1):23–63. doi: 10.1615/critrevimmunol.v32.i1.30
245. Shankar-Hari M, Spencer J, Sewell WA, Rowan KM, Singer M. Bench-To-Bedside Review: Immunoglobulin Therapy for Sepsis - Biological Plausibility From a Critical Care Perspective. Crit Care (2012) 16(2):206. doi: 10.1186/cc10597
246. Matsuda Y, Hiramitsu T, Li XK, Watanabe T. Characteristics of Immunoglobulin M Type Antibodies of Different Origins from the Immunologic and Clinical Viewpoints and Their Application in Controlling Antibody-Mediated Allograft Rejection. Pathogens (2020) 10(1):4. doi: 10.3390/pathogens10010004
247. Morbach HO, Eichhorn EM, Liese JG, Girschick HJ. Reference Values for B Cell Subpopulations From Infancy to Adulthood. Clin Exp Immunol (2010) 162:271–9. doi: 10.1111/j.1365-2249.2010.04206.x
248. Notley CA, Brown MA, Wright GP, Ehrenstein MR. Natural IgM Is Required for Suppression of Inflammatory Arthritis by Apoptotic Cells. J Immunol (2011) 186(8):4967–72. doi: 10.4049/jimmunol.1003021
249. Aschetto R, Clemente N, Pagni A, Esposito T, Longhini F, Mercalli F, et al. A Double Blind Randomized Experimental Study on the Use of IgM-Enriched Polyclonal Immunoglobulins in an Animal Model of Pneumonia Developing Shock. Immunobiology (2017) 222(12):1074–80. doi: 10.1016/j.imbio.2017.09.002
250. Barratt-Due A, Sokolov A, Gustavsen A, Hellerud BC, Egge K, Pischke SE, et al. Polyvalent Immunoglobulin Significantly Attenuated the Formation of IL-1β in Escherichia Coli-Induced Sepsis in Pigs. Immunobiology (2013) 218(5):683–9. doi: 10.1016/j.imbio.2012.08.268
251. Duerr C, Bacher A, de Martin A, Sachet M, Sadeghi K, Baumann S, et al. The Novel Polyclonal Ab Preparation Trimodulin Attenuates Ex Vivo Endotoxin-Induced Immune Reactions in Early Hyperinflammation. Innate Immunity (2019) 25(6):374–88. doi: 10.1177/1753425919853333
252. Marshall K, Jin J, Atkinson C, Alawieh A, Qiao F, Lei B, et al. Natural Immunoglobulin M Initiates an Inflammatory Response Important for Both Hepatic Ischemia Reperfusion Injury and Regeneration in Mice. Hepatology (2018) 67(2):721–35. doi: 10.1002/hep.29512
253. Alejandria MM, Lansang MA, Dans LF, Mantaring JBV. Intravenous Immunoglobulin for Treating Sepsis and Septic Shock. Cochrane Database Syst Rev (2002) 1:CD001090. doi: 10.1002/14651858
254. Toyoda M, Shin BH, Ge S, Mirocha J, Thomas D, Chu M, et al. Impact of Desensitization on Antiviral Immunity in HLA-Sensitized Kidney Transplant Recipients. J Immunol Res (2017) 2017:5672523. doi: 10.1155/2017/5672523
255. Morath C, Opelz G, Zeier M, Süsal CJ. Clinical Relevance of HLAantibody Monitoring After Kidney Transplantation. Immunol Res (2014) 2014:845040. doi: 10.1155/2014/845040
256. Roos A, Rieben R, Faber-Krol MC, Daha MR. IgM-Enriched Human Intravenous Immunoglobulin Strongly Inhibits Complement-Dependent Porcine Cell Cytotoxicity Mediated by Human Xenoreactive Antibodies. Xenotransplantation (2003) 10(6):596–605. doi: 10.1034/j.1399-3089.2003.00063.x
257. Barten Markus J, Buchholz C, Bernhardt A, Rybczynski M, Grahn H, Reichenspurner H. IgM-Enriched Human Intravenous Immunoglobulin for Treatment of Early Antibody Mediated Rejection After Heart Transplantation. Transplantation (2018) 102:S828. doi: 10.1097/01.tp.0000543877.96384.6e
258. Ius F, Sommer W, Kieneke D, Tudorache I, Kühn C, Avsar M. IgM-Enriched Human Intravenous Immunoglobulin-Based Treatment of Patients With Early Donor Specific Anti-HLA Antibodies After Lung Transplantation. Transplantation (2016) 100(12):2682–92. doi: 10.1097/TP.0000000000001027
259. De Vos J, Hose D, Rème T, Tarte K, Moreaux J, Mahtouk K, et al. Microarray-Based Understanding of Normal and Malignant Plasma Cells. Immunol Rev 2006 (2006) 210:86–104. doi: 10.1111/j.0105-2896.2006.00362.x
Keywords: antibody-mediated allograft rejection, naïve-B cell, memory-B cell, germinal center B cell, long-lived plasma cell, B cell biology
Citation: Matsuda Y, Watanabe T and Li X-K (2021) Approaches for Controlling Antibody-Mediated Allograft Rejection Through Targeting B Cells. Front. Immunol. 12:682334. doi: 10.3389/fimmu.2021.682334
Received: 18 March 2021; Accepted: 17 June 2021;
Published: 01 July 2021.
Edited by:
Cheng Yang, Fudan University, ChinaReviewed by:
Chao Hu, Fudan University, ChinaSongjie Cai, Brigham and Women’s Hospital and Harvard Medical School, United States
Copyright © 2021 Matsuda, Watanabe and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yoshiko Matsuda, WW9zaGlrb21hdHN1ZGF5QGdtYWlsLmNvbQ==