 Seema Patel
Seema Patel Heidi R. Tucker2
Heidi R. Tucker2 Himanshu Gogoi
Himanshu Gogoi Samira Mansouri
Samira Mansouri Lei Jin
Lei Jin- 1Division of Pulmonary, Critical Care and Sleep Medicine, Department of Medicine, University of Florida, Gainesville, FL, United States
- 2Department of Immunology and Microbial Disease, Albany Medical College, Albany, NY, United States
The cyclic GMP–AMP synthase–stimulator of interferon genes (cGAS–STING) pathway senses DNA and induces type I interferon (IFN) production. Whether and how the STING pathway crosstalk to other innate immune pathways during pathogen infection, however, remains unclear. Here, we showed that STING was needed for Streptococcus pneumoniae-induced late, not early, stage of lung IFNγ production. Using knockout mice, IFNγ reporter mice, intracellular cytokine staining, and adoptive cell transfer, we showed that cGAS–STING-dependent lung IFNγ production was independent of type I IFNs. Furthermore, STING expression in monocyte/monocyte-derived cells governed IFNγ production in the lung via the production of IL-12p70. Surprisingly, DNA stimulation alone could not induce IL-12p70 or IFNγ in Ly6Chi monocyte. The production of IFNγ required the activation by both DNA and heat-killed S. pneumococcus. Accordingly, MyD88−/− monocyte did not generate IL-12p70 or IFNγ. In summary, the cGAS–STING pathway synergizes with the MyD88 pathway in monocyte to promote late-stage lung IFNγ production during pulmonary pneumococcal infection.
Introduction
During pathogen infections, multiple innate immune signaling pathways are activated. Stimulator of interferon genes (STING) is essential for cytosolic DNA-induced type I interferon (IFN) production but largely dispensable for Toll-like receptors (TLRs) activations (1–3). Currently, it is not clear if and how the STING pathway crosstalks with another innate immune pathway during infections.
Streptococcus pneumoniae is an extracellular bacterial pathogen that causes pneumonia, sinusitis, otitis media, septicemia, and meningitis (4, 5). A recent study found that the STING-mediated cytosolic DNA sensing pathway is activated during pulmonary S. pneumoniae infection (6). However, pneumococcal infection-induced proinflammatory cytokines, including tumor necrosis factor α (TNFα), interleukin (IL)-6, and IL-1β, are largely intact in the STING−/− mice, and the bacterial burden in the lung, spleen, and blood were comparable between STING−/− and wild-type (WT) mice (6). Thus, STING seems to be dispensable for the initial innate immunity to S. pneumoniae including the control of bacterial burden.
IFNγ promotes M1-macrophage development that not only phagocyte and kill the bacteria but also contribute to tissue injury. Streptococcus pneumoniae infection induces lung IFNγ. In patients with S. pneumoniae sepsis, plasma IFNγ was elevated and correlated with increased mortality (7). IFNγ−/− mice are more resistant than the WT mice in developing pneumococcal meningitis (8). For pneumococcal pneumonia, IFNγ−/− mice or pretreatment of mice with anti-IFNγ neutralizing Ab had either no effect on mortality (9, 10) or lead to decreased survival (11, 12). Thus, the role of lung IFNγ production during pulmonary pneumococcal infection remains controversial.
In this report, we found that there were two waves of lung IFNγ productions by different immune cells during pulmonary pneumococcal infection. STING is required for the late, not early, stage of lung IFNγ production. Notably, the production of IFNγ required the activation of both STING and MyD88 pathways indicating previous unknown crosstalk between STING and TLRs pathways during infection.
Materials and Methods
Mice
Mice 8- to 16 weeks old, both males and females, were used for all experiments. STING−/− mice (tmem173<tm1Camb>) have been described previously (13). The STINGflox/flox/TMEM173flox/flox mouse has been described previously (14). The following strains were obtained from The Jackson Laboratory: CCR2−/−, cyclic GMP–AMP synthase (cGAS)−/−, IL-12p70−/−, IFNRA1−/−, MyD88−/−, TLR2−/−, and IFNγ YFP-reporter.
All mice are on a C57BL/6 background. Mice were housed and bred in the Animal Research Facility at the University of Florida. All experiments with mice were performed by the regulations and approval of the Institutional Animal Care and Use Committee from the University of Florida (protocol number 201909362).
Streptococcus pneumoniae Infection
Streptococcus pneumoniae D39 (serotype 2) were grown in tryptic soy broth (TSB) at 37°C to an optical density (OD) of 0.45–0.50 at 600 nm (∼108 CFU/ml). Mice were intranasally administered ∼8–10 × 106 CFU in 50 µl of 1× Ultrapure phosphate-buffered saline (PBS). CFUs were confirmed by colony counting of log10 serial dilutions of bacteria cultured overnight on a TSB with a 10% sheep blood agar plate.
BacLight Green Stained Streptococcus pneumoniae Infection
Streptococcus pneumoniae D39 (serotype 2) were grown in TSB at 37°C to an OD of 0.35–0.4 at 600 nm; BacLight green (1 µg/ml) was added and incubated at 37°C to an OD of 0.45–0.5 at 600 nm (∼108 CFU/ml). Mice were intranasally administered with ∼8–10 × 106 CFU.
Detection of the Lung Cytokine Production
Mice were intranasally infected with S. pneumoniae D39. Mice were sacrificed by CO2 asphyxiation at the indicated time points. The lungs were subsequently perfused with cold PBS, washed in PBS once, and stored in a 1.0-ml tissue protein extraction reagent (T-PER) containing protease inhibitors (Roche, Indianapolis, IN). The lungs were homogenized using a Bertin Technology Minilys tissue homogenizer. Lung homogenates were spun at 14,000×g for 15 min at 4°C. The supernatant was collected and analyzed for cytokine production using ELISA.
Cytokine ELISAs
Cytokine concentrations were measured using ELISA kits from eBiosciences according to the manufacturer’s instructions. The ELISA kits used were IL-1β, IL-6, IL-12/p70, TNFα, monocyte chemoattractant protein-1 (MCP-1), and IFNγ. The IFNβ ELISA kit was from PBI Interferon Source, Piscataway, NJ.
Flow Cytometry Analysis
Mice were intranasally infected with S. pneumoniae D39. Mice were sacrificed by CO2 asphyxiation at the indicated time points. The lungs were subsequently perfused with cold PBS. Excised lungs were cut into small pieces and digested in Roswell Park Memorial Institute (RPMI) containing 200 μg/ml DNase I (Roche) and 25 μg/ml Liberase TM (Roche) at 37°C for 2 h. Red blood cells were then lysed using ACK lysis buffer (Gibco), and a single-cell suspension was prepared and analyzed by BD LSR Fortessa flow cytometry.
The following Abs from Biolegend were used in the flow cytometry: Ly6C (HK1.4), CD11b (M1/70), Ly6G (1A8), CD11c (N418), NK-1.1 (PK136), MHC II (M5/114.15.2), CD103 (2E7), CD3 (145.2C11), CD64 (X54-5/7.1), Siglec F (S17007L), IFNγ (XMG1.2), and IL-12p70 (27537).
Intracellular Staining
The intracellular cytokine staining was performed using the Cytofix/Cytoperm™ kit from BD Biosciences. Briefly, mice were intranasally administered with either S. pneumoniae D39 ∼8–10 × 106 CFU in 50 µl of 1× Ultrapure PBS or PBS alone. The lungs were perfused and harvested at 24 and 48 h postinfection and washed in PBS followed by 2 h of digestion in RPMI containing 200 µg/ml DNAse I (Roche), 25 µg/ml Librase TM (Roche), and Golgi-plug 1 µg/µl (BD Bioscience). Digested lungs were processed to prepare the single lung cell suspension in RPMI containing Golgi-plug 1 µg/µl. The cells were fixed in Cytofix/perm buffer (BD Biosciences) in the dark for 20 min at room temperature (RT). Fixed cells were washed and kept in Perm/Wash buffer at 4°C. The Golgi-plug was present during every step before fixation. Cells were stained with cytokine-specific staining antibodies in perm buffer in the dark for 30 min at RT. Cells were washed, and the single-cell suspension was prepared to be analyzed by BD LSRFortessa flow cytometry.
Ex Vivo Monocyte Culture and Activation
Ly6Chi monocytes were purified from bone marrow cells using EasySep Mouse Monocyte Isolation kit (STEMCELL Technologies). Purified monocytes were cultured in RPMI (Invitrogen) with 10% FBS, 2 mM L-glutamine, 1 mM sodium pyruvate, 10 mM HEPES buffer, 1% non-essential amino acids, 50 μM 2-mercaptoethanol, and1% Pen/Strep. Cells were stimulated with 5 × 106 CFU/ml heat-killed S. pneumoniae (HKSP) (InvivoGen), 2 µg/ml apoptotic DNA transfected with lipofectamine 3000 (15), or both for 17 h at 37°C. To generate apoptotic DNA, we isolated splenocytes from WT mice and cultured those in complete RPMI for 3 days at 37°C without changing the media; cells were used to isolate genomic DNA using a Qiagen kit. The supernatant of stimulated Ly6Chi monocytes was analyzed for cytokine production.
Statistical Analysis
All data were expressed as means ± SEM. Statistical significance was evaluated using Prism 9.1 software to perform one-way ANOVA Tukey’s multiple comparison test.
Results
Streptococcus pneumoniae-Induced Innate Immune Responses Are Largely Intact in STING−/− Mice
We investigated the role of STING in host defense against pulmonary S. pneumoniae infection. Streptococcus pneumoniae-induced lung inflammatory cytokines IL-6, IL-1β, TNFα, and chemokines KC, MCP-1 were not altered in the STING−/− mice (Figures S1A–E). Interestingly, there was no detectable S. pneumoniae-induced lung IFNβ protein (Figure S1F). Lung bacterial burden and total proteins in the BAL fluid, an indication of lung damage, were also not significantly different between STING−/− and WT mice (Figures S1G, H). Pneumococcal infection recruits neutrophils into the lung that are critical for the host defense (16). There was no difference in the total numbers of recruited neutrophils (CD11bhiLy6G+) between WT and STING−/− mice (Figures S1I, J). Thus, STING is largely dispensable for the innate immune responses to pulmonary pneumococcal infection, which is consistent with a recent report (6).
STING Is Required for S. pneumoniae Induced Lung IL-12p70 by Monocyte/Monocyte-Derived Cells
Interestingly, S. pneumoniae-induced lung IL-12p70 was lost in STING−/− mice (Figure 1A). Streptococcus pneumoniae secretes cyclic di-AMP that is a STING ligand. STING can also be activated via cGAS that senses cytosolic DNA (17–19). We found that cGAS−/− mice failed to make lung IL-12p70, suggesting that S. pneumoniae-induced lung IL-12p70 was likely induced by DNA, not cyclic di-AMP (Figure 1B). As a control, S. pneumoniae induced lung TNFα was unaltered in the cGAS−/− mice (Figure S1K).

Figure 1 cGAS–STING mediate S. pneumoniae-induced lung IL-12p70 production by monocyte/monocyte-derived cells. (A, B) STING−/−, cGAS−/−, and WT littermates mice were given PBS or infected (i.n.) with S. pneumoniae (D39 strain, ~5 × 106 CFU). IL-12p70 (24, 48 hpi) were measured by ELISA (n = 3-4 mice per group). Data are representative of two independent experiments. (C) Flow cytometry analysis of IL-12p70 in lung immune cells from PBS or S. p (~5 × 106 CFU) infected C57BL/6J mice at 24 hpi (n = 3 mice per group). Data are representative of three independent experiments. (D) Total cell numbers of IL-12p70+ lung immune cells from (C) were enumerated. (E, F) CCR2−/− and WT littermates mice were infected (i.n.) with S. pneumoniae as in (A) Total cell numbers of lung Ly6Chi monocyte (E) and neutrophils (F) were enumerated (n = 3–4 mice per group). Data are representative of three independent experiments. (G) CCR2−/− and WT littermate mice were infected with S. pneunoniae as in panel (A) IL-12p70 in lung homogenates (24 hpi) were measured by ELISA (n = 3 mice per group). Data are representative of two independent experiments. Graphs represent the mean with error bars indicating SEM. p-values determined by one-way ANOVA Tukey’s multiple comparison test. Significance is represented by asterisk, where *p < 0.05, **p < 0.001, ***p < 0.0001.
We wanted to determine the cellular source of IL-12p70 during S. pneumonia infection by intracellular cytokine stain. Monocyte, monocyte-derived DCs (moDCs), and monocyte-derived macrophage (moMACs) produced IL-12p70 in the lung during pneumococcal infection, while lung conventional DCs (cDC), T cells, or NK cells did not produce IL-12p70 (Figures 1C, D). CCR2 binds to MCP-1 mediating monocyte migration (20). We found that CCR2−/− mice failed to recruit Ly6Chi monocyte (CD11b+Ly6Chi) into the lung during pneumococcal pulmonary infection (Figure 1E). In comparison, the CCR2−/− mice had unaltered lung neutrophils infiltration during the pneumococcal infection (Figure 1F). We then examined IL-12p70 production in CCR2−/− mice. CCR2−/− mice failed to make lung IL-12p70 upon S. pneumoniae infection (Figure 1G). Together, the data suggested that cGAS–STING promoted monocyte production of lung IL-12p70 during S. pneumoniae infection.
Two Waves of Lung IFNγ Production During S. pneumoniae Infection
IL-12p70 drives IFNγ production (21, 22). Streptococcus pneumoniae infection induces strong IFNγ production in the lungs (9–12). We found that IL-12p70−/− mice failed to make IFNγ upon S. pneumoniae infection at 48 hpi, but not at 24 hpi (Figure 2A). We reasoned that there were at least two waves of lung IFNγ production during S. pneumoniae infection, and only the second wave of lung IFNγ was dependent on IL-12p70. CCR2−/− mice lack lung IL-12p70 production. Similar to the IL-12p70−/− mice, CCR2−/− mice were defective in the late, but not early S. pneumoniae-induced lung IFNγ production (Figure 2B).
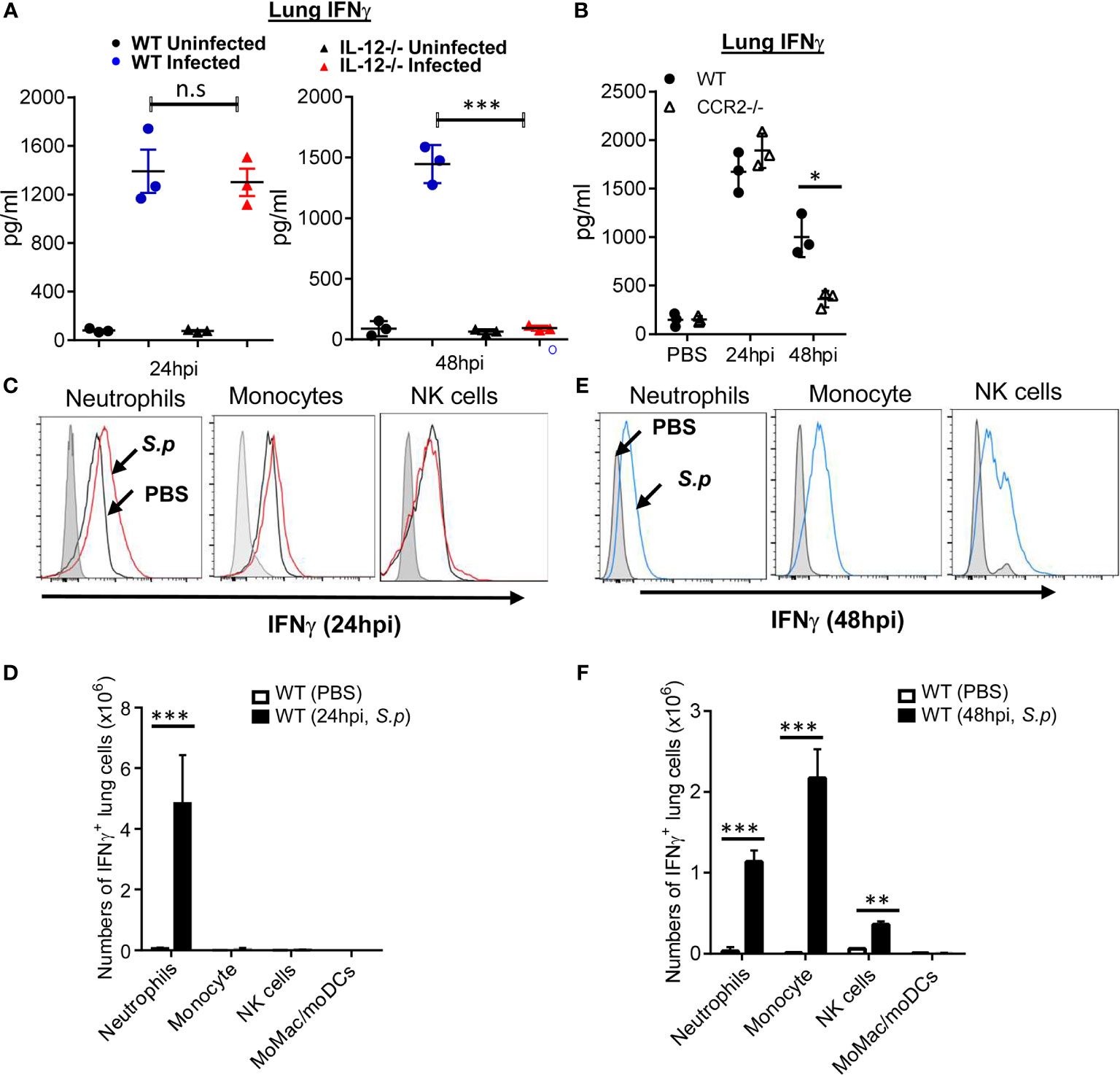
Figure 2 Monocyte and IL-12p70 promote S. pneumoniae-induced late-stage lung IFNγ production. (A) IL-12p70−/− and WT littermates mice were infected (i.n.) with S. pneumoniae (D39 strain, ~5 × 106 CFU). IFNγ in lung homogenates were measured at 24 and 48 hpi by ELISA (n = 3 mice per group). Data are representative of two independent experiments. (B) CCR2−/− and WT littermates mice were infected (i.n.) with S. pneumoniae as in (A). IFNγ in lung homogenates were measured at 24 and 48 hpi by ELISA (n = 3–4 mice per group). Data are representative of three independent experiments. (C, E) Flow cytometry analysis of YFP expression (IFNγ) in lung immune cells from PBS or S. p (~5 × 106 CFU) infected IFNγ reporter mice at 24 hpi (C) and 48 hpi (E) (n = 3–4 mice per group). Data are representative of three independent experiments. (D, F) Total cell numbers of IFNγ+ lung immune cells in (C, E) were enumerated. Graphs represent the mean with error bars indicating SEM. p-values determined by one-way ANOVA Tukey’s multiple comparison test. Significance is represented by asterisk, where *p < 0.05, **p < 0.001, ***p < 0.0001, n.s., not significant.
We hypothesized that different immune cells were responsible for lung IFNγ production during the early and late stages. We used IFNγ-YFP reporter mice to detect lung IFNγ-producing cells. Lung immune cells were analyzed by flow cytometry (Figures S2A–E). We found that, at 24 hpi, neutrophils were the predominant lung IFNγ−producing cells (Figures 2C, D), which is consistent with a recent report (23). However, by 48 hpi, Ly6Chi monocytes and natural killer (NK) cells also produced lung IFNγ-producing cells (Figures 2E, F). Neither CD3+ T cells, macrophages (CD11b+ CD64+ CD11c+/− Ly6Clow) nor dendritic cells (SiglecF− CD11chi MHC IIhi) produced IFNγ at 48 hpi (Figure S2F). Thus, neutrophils produce lung IFNγ at 24 hpi, while monocyte, NK cells, and neutrophils generate lung IFNγ at 48 hpi.
STING Is Required for S. pneumoniae-Induced Type I IFN-Independent, Late-Stage Lung IFNγ Production
STING−/− mice failed to make IL-12p70 during S. pneumoniae infection (Figure 1A). Similar to the CCR2−/− and IL-12p70−/− mice, we observed a significant reduction in lung IFNγ production in the STING−/− mice at 48 hpi, but not at 24 hpi (Figure 3A). cGAS−/− mice also failed to make lung IFNγ at 48 hpi (Figure 3B), suggesting that cytosolic sensing of DNA, not S. pneumoniae cyclic di-AMP, promoted lung IFNγ. Lastly, IFNAR1−/− mice had unaltered IFNγ production in the lung upon S. pneumoniae infection (Figure 3C). Thus, cGAS–STING–IL12p70–IFNγ signaling is likely type I IFN independent.
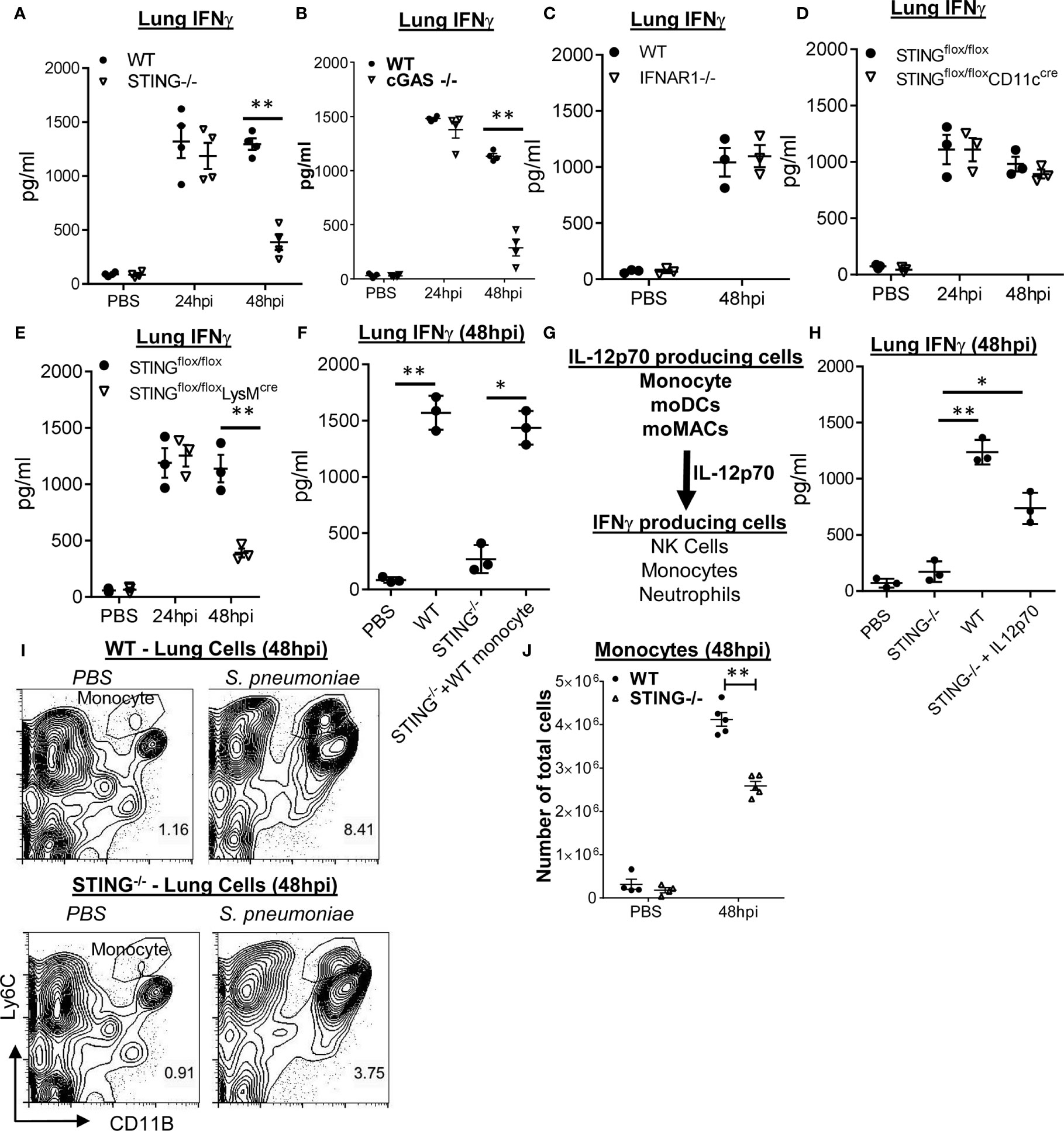
Figure 3 Monocyte expression of STING mediates S. pneumoniae induced late-stage lung IFNγ production. (A, B) STING−/−, cGAS−/−, and WT littermates were given PBS or infected (i.n.) with S. pneumoniae (D39 strain, ~5 × 106 CFU). IFNγ in lung homogenates (24 and 48hpi) were measured by ELISA (n = 3–4 mice per group). Data are representative of two independent experiments. (C) IFNAR1−/− and WT littermate mice were infected with S. pneunoniae as in (A). IFNγ in lung homogenates (48 hpi) were measured by ELISA (n = 3 mice per group). Data are representative of two independent experiments. (D, E) STINGfl/flCD11CCre, STINGfl/flLysMCre, and STINGfl/fl littermates mice were infected (i.n.) with S. pneumoniae as in (A). IFNγ in lung homogenates (24, 48 hpi) were measured by ELISA (n = 3 mice per group). Data are representative of two independent experiments. (F) STING−/− and WT littermates were infected with S. pneumoniae as in panel (A) At 16 hpi, 1 million bone marrow WT Ly6Chi monocytes were adoptively transferred (i.n.) into STING−/− mice (STING−/− + WT monocyte). IFNγ in lung homogenates was measured at 48 hpi by ELISA (n = 3 mice per group). Data were representative of two independent experiments. (G) A diagram of lung IFNγ production by monocyte-derived IL-12p70. (H) STING−/− and WT littermates were infected with S. pneumoniae as in (A). At the 16 hpi, recombinant IL-12p70 (1 µg) was administered (i.n.) into STING−/− mice. IFNγ in lung homogenates was measured at 48 hpi by ELISA (n = 3 mice per group). Data are representative of two independent experiments. (I, J) STING−/− and WT littermates mice were infected (i.n.) with S. pneumoniae as in (A). Ly6Chi monocytes (I) were identified in total lung cells at 48 hpi by flow cytometry. Total cell numbers of lung Ly6Chi monocytes (J) were enumerated (n = 3–4 mice per group). Data were representative of three independent experiments. Graphs represent the mean with error bars indicating SEM. p-values determined by one-way ANOVA Tukey’s multiple comparison test. Significance is represented by asterisk, where *p < 0.05, **p < 0.001.
LysMcreSTINGfl/fl Mice Lack S. pneumoniae Induced Late-Stage Lung IFNγ Production
Monocyte/monocyte-derived cells are critical for lung IL-12p70 and the late-stage IFNγ production (Figures 1 and 2). We hypothesized that STING expression in monocyte drove lung IFNγ production. We examined S. pneumonia-induced lung IFNγ production in CD11ccreSTINGfl/fl and LysMcreSTINGfl/fl mice. CD11ccreSTINGfl/fl mice delete STING gene in alveolar macrophage and dendritic cells (DCs), while LysMcreSTINGfl/fl mice delete STING gene in alveolar macrophage, interstitial macrophage, monocyte/monocyte-derived cells, and neutrophils (24). Notably, neutrophils do not express STING, while NK cells and Ly6Chi monocytes have strong STING expression (13, 14).
We found that LysMcreSTINGfl/fl, not the CD11ccre-STINGfl/fl mice, were defective in the late-stage lung IFNγ production by S. pneumoniae infection (Figures 3D, E), suggesting that STING expressing in interstitial macrophage, monocyte/monocyte-derived cells, not DCs or alveolar macrophage, was likely responsible for S.pneumoniae-induced late-stage lung IFNγ production.
STING Expression in Ly6Chi Monocyte Promotes S. pneumoniae-Induced Lung IFNγ at 48 hpi
To further establish that STING expression in monocyte/monocyte-derived cells is critical for lung IFNγ production, we adoptively transferred (i.n) WT bone marrow Ly6Chi monocyte into STING−/− mice at 16 hpi and determined lung IFNγ production at 48 hpi. We found that STING−/− mice receiving WT Ly6Chi monocyte produced lung IFNγ at 48 hpi (Figure 3F). We concluded that STING expression in monocyte/monocyte-derived cells promotes the late-stage lung IFNγ production during S. pneumoniae infection.
Besides monocyte, neutrophils and NK cells produce lung IFNγ at 48 hpi (Figure 2F). We proposed that STING expression in monocyte/monocyte-derived cells produces IL-12p70 that drove late-stage lung IFNγ production by NK cells and neutrophils during pneumococcal infection (Figure 3G). Indeed, intranasal administration of recombinant IL-12p70 at 16 hpi restored IFNγ production in STING−/− mice (Figure 3H).
We also examined lung monocyte infiltration in STING−/− mice during S. pneumoniae infection. We observed a mild decrease in lung Ly6Chi monocytes in STING−/− mice at 48 hpi (Figures 3I, J). Furthermore, STING−/− had similar S. pneumoniae-induced MCP-1 production as the WT mice (S1D). We, thus, preferred the hypothesis that STING expression in monocyte senses DNA and drives IL-12p70 production to promote late-stage lung IFNγ production.
Activation of the STING Pathway by DNA Is not Sufficient to Induce IL-12p70 and IFNγ in Ly6Chi Monocyte
cGAS–STING pathway senses cytosolic DNA from invading pathogens or self-DNA by damaged host cells. We examined if monocyte/monocyte-derived cells were directly infected by S. pneumoniae at 48 hpi, thus may contain cytosolic pathogen DNA. We infected (i.n.) mice with BacLight Green-stained S. pneumoniae. At 48 hpi, we examined BacLight Green+ cells in the lung. Neutrophils were heavily infected with S. pneumoniae (Figure S3A). Ly6Chi monocyte, NK cells, or moMAC, however, contained few labeled bacteria (Figures S3B–D), indicating that the S. pneumoniae may not directly release pathogen DNA into the cytosol of these cells.
During live infection, besides live bacteria and bacteria components, there were dead host cells in the lung that could release their DNA and activate the cGAS–STING pathway. We activated monocyte with mouse genomic DNA isolated from apoptotic mouse splenocytes (Figure 4A). We isolated Ly6Chi monocytes from the bone marrow and stimulated them with mouse genomic apoptotic DNA. Surprisingly, we found that the apoptotic DNA alone did not generate IL-12p70 or IFNγ in the isolated Ly6Chi monocyte (Figures 4B, C). We also used cGAMP to activate the Ly6Chi monocyte. Again, cGAMP did not induce IL-12p70 (Figure 4B). As control, both DNA and cGAMP activated Ly6Chi monocytes to produce TNFα and IFNβ (Figures S4A, B). Thus, DNA sensing alone cannot activate Ly6Chi monocyte to produce IL-12p70 or IFNγ.
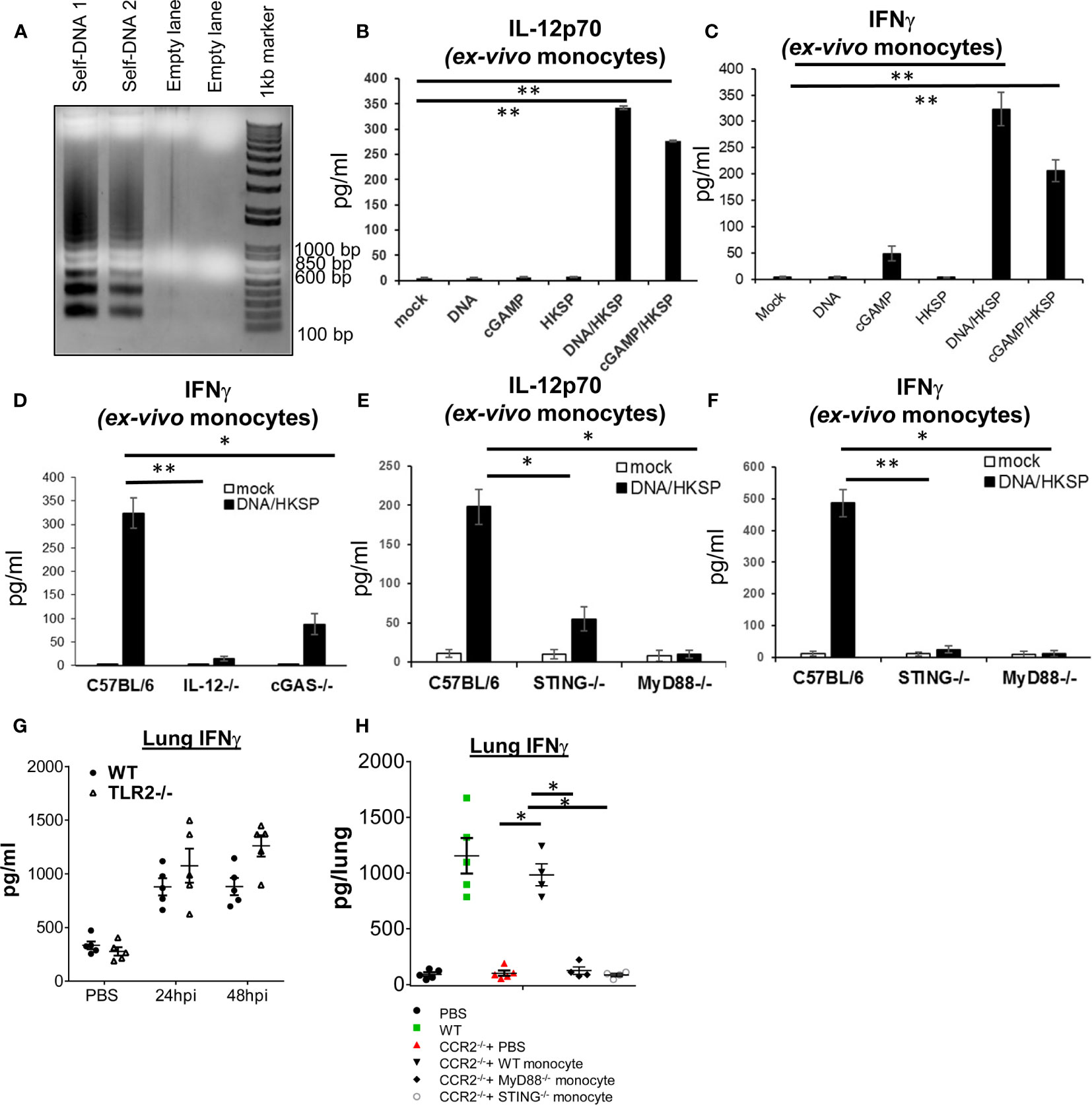
Figure 4 Ly6Chi monocyte production of IL-12p70 and IFNγ require the activation of both cGAS–STING and MyD88 pathways. (A) Splenocytes isolated from a C57BL/6J mouse were cultured ex vivo for 4 days. Genomic DNA was extracted and run on an agarose gel (Self DNA 1 and Self DNA2). (B, C) Ly6Chi monocyte isolated from C57BL/6J mice were activated with self-DNA (1.5 µg/ml), HKSP (5 × 106 CFU/ml), 2′3′-cGAMP (4 µg/ml) or DNA + HKSP, HKSP + 2′3′-cGAMP for 17 h IL-12p70 (B) and IFNγ (C) were measured in the culture supernatant by ELISA. Data are representative of three independent experiments. (D–F) Ly6Chi monocyte isolated from indicated mice were activated with self-DNA (1.5 µg/ml) plus HKSP (5 × 106 CFU/ml) for 17 h as in (B). IFNγ and IL-12p70 were measured in the culture supernatant by ELISA. Data are representative of three independent experiments. (G) TLR2−/− and their WT littermates were infected (i.n.) with PBS or S. p (D39 strain, ~5 × 106 CFU). IFNγ in lung homogenates was measured by ELISA at 24 and 48 hpi (n = 4–5 mice/group). Data are representative of two independent experiments. (H) CCR2−/− and WT littermates were infected with S. pneumoniae (D39 strain, ~8 × 106 CFU). At the 16 hpi, 1 million bone marrow WT, MyD88−/−, or STING−/− Ly6Chi monocytes were adoptively transfer (i.n.) into CCR2−/− mice. IFNγ in lung homogenates were measured at 48 hpi by ELISA (n = 4–5 mice/group). Data are representative of two independent experiments. Graphs represent the mean with error bars indicating SEM. p-values determined by one-way ANOVA Tukey’s multiple comparison test. Significance is represented by asterisk, where *p < 0.05, **p < 0.001.
Heat-Killed Streptococcus pneumoniae and Apoptotic DNA Together Induce IL-12p70 and IFNγ in Ly6Chi Monocyte
During live S. pneumoniae infection, monocyte and monocyte-derived cells likely encounter both PAMP, e.g., TLR agonists, and DAMP, e.g., host DNA released from dead cells. We hypothesized that the cGAS–STING pathway may synergize with the TLR pathway to induce IL-12p70 and IFNγ in Ly6Chi monocyte. We activated monocyte with HKSP plus apoptotic DNA. Indeed, HKSP/DNA stimulation induced IL-12p70 and IFNγ in monocyte (Figures 4B, C). Similarly, HKSP/cGAMP induced IL-12p70 and IFNγ (Figures 4B, C). HKSP alone did not induce IL-12p70 or IFNγ in the monocyte (Figures 4B, C). We concluded that the induction of IL-12p70-IFNγ in monocyte required the synergistic activation of DNA and TLRs signaling.
MyD88 Is Required for HKSP/DNA Induced IL-12p70 and IFNγ Production in Ly6Chi Monocyte
As expected, HKSP/DNA did not stimulate IFNγ production in Ly6Chi monocyte from IL-12p70−/− mice (Figure 4D). cGAS−/− and STING−/− monocyte were also defective in IFNγ production by HKSP/DNA (Figures 4D–F) confirming that the cGAS–STING pathway was required. As control, HKSP activated TNFα in IL-12p70−/− and cGAS−/− monocyte (Figure S4C). HKSP activated the TLR2–MyD88 pathway (25). We found that Ly6Chi monocyte from MyD88−/− mice failed to make IL-12p70 or IFNγ (Figures 4E, F), suggesting that monocyte production of IL-12p70 by HKSP/DNA requires the MyD88 pathway. As a control, MyD88−/− monocyte made IFNβ and TNFα in response to HKSP/DNA (Figures S4D, E).
To ask if TLR2 is required for lung IFNγ production, we infected TLR2−/− mice with S. pneumoniae. Unlike the STING−/− or cGAS−/− mice, the lung IFNγ production was similar in WT and TLR2−/− mice (Figure 4G). We suspected that additional TLRs pathways, such as TLR9 (26), might synergize with the cGAS–STING pathway for lung IFNγ production during pathogen infection, compensating for the loss of TLR2.
To establish that MyD88 are required for S. pneumoniae induced lung IFNγ, we adoptively transferred (i.n.) WT, STING−/−, or MyD88−/− monocytes to CCR2−/− mice and examined the IFNγ production in the lung by S. pneumonia. Unlike the WT monocyte, neither STING−/− nor MyD88−/− monocyte restored lung IFNγ production in the CCR2−/− mice (Figure 4H). Thus, both MyD88 and STING expression in the monocyte are required for lung IFNγ production in vivo.
Discussion
In this report, we showed that STING synergizes with MyD88 to induce IL-12p70 and IFNγ in the lung. STING is particularly required for the late-stage (48 hpi) lung IFNγ production during S. pneumoniae infection. This unique requirement likely reflects the need for IL-12p70 production since the initial lung IFNγ production by S. pneumoniae is IL-12p70 independent (23).
Previously, Temizoz et al. showed that cGAMP, in combination with CpG ODN, stimulated IFNγ production in PBMCs (26). Furthermore, cGAMP and CpG ODN together, acting as an antigen-free anticancer agent, reduced tumor size significantly compared to cGAMP alone in the EG-7 and B16-F10 mouse tumor models (26). They further showed that IL-12p70 was required for the synergistic induction of IFNγ in PBMCs (26). Different from ours, Temizoz et al. showed that type I IFN was needed for the IFNγ production (26). Type I IFN is required for IL-18 production in moMACs (27). IL-18, also known as IFNγ-inducing factor, can induce IFNγ production (21, 22, 28). In our experimental setting, lung production of IFNγ does not require type I IFN. Nevertheless, it is likely that that type I IFN, via the production of IL-18, together with IL-12p70, could further augment IFNγ production.
It has long been known that DCs production of IL-12p70 requires at least two stimuli (29–33). This dual requirement is likely a safeguard to avoid the possible detrimental effects of uncontrolled IL-12p70-medicated Th1 responses. Napolitani et al. showed that in both human and mouse DCs, TLR3 and TLR4 potently synergized with TLR7, TLR8, and TLR9 to induce IL-12p70 and IL-23, leading to enhanced and sustained Th1 responses (29). Here, we found that STING-mediated cytosolic DNA sensing pathway synergize with TLR2 pathway in monocyte for IL-12p70 and IFNγ production. Nevertheless, it is likely that STING pathway can synergize with other PRRs for IL-12p70 and IFNγ production because TLR2−/− mice did not have defect in lung IFNγ production during S. pneumoniae infection. How STING and TLRs synergistically induce IL-12p70 is unclear. TLRs activation takes place on the plasma membrane or endosome, while STING activation happens at the ER–Golgi interface. It is tempting to speculate that a spatiotemporal activation of STING and TLRs may aid in IL-12p70 production.
The discovery of two waves of lung IFNγ production during S. pneumoniae infection may clarify the role of IFNγ in pneumococcal infection. Using IFNγ−/− mice or anti-IFNγ neutralizing Ab, previous studies were inconclusive (9–12). We speculated that the two waves of lung IFNγ may play opposite roles in host defense against pneumococcal infection. The neutrophils-mediated, IL-12p70-independent early lung IFNγ may be beneficial by generating M1 macrophages to neutralize bacteria. The late-stage lung IFNγ in pneumococcal infection, however, may be detrimental because in patients with S. pneumoniae sepsis, IFNγ was elevated and correlated with increased mortality (7). We speculated that persistent IFNγ production may promote sustained inflammation that may enhance tissue damage and mortality.
Ly6Chi monocyte has emerged as a key player in pathogen-induced IFNγ production in the mucosal surface (34, 35). Ly6Chi monocytes are rapidly recruited to sites of infection and differentiate into macrophages and dendritic cells. Two recent studies found that CCR2−/− mice, which lack infiltrating Ly6Chi monocyte, produced significantly less IFNγ in the lung during pulmonary Legionella pneumophila infection (34, 35). Similar to our finding, they found that IL-12p70 is required for IFNγ production and identified infiltrating monocyte as the major source of IL-12p70 (34, 35). Another study found that during vaginal HSV-2 infection, Ly6Chi monocytes produce IL-18, which activates NK cells to produce IFNγ (36). Thus, a new paradigm emerges that during mucosal pathogen infection, infiltrating Ly6Chi monocyte produces IL-12p70 or IL-18 that instructs NK cells or T cells to produce IFNγ.
In summary, the activation of the STING pathway in monocyte/monocyte-derived cells can synergize with the MyD88 pathway to drive IFNγ production during pneumococcal infection that may influence the development of adaptive immunity.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
All experiments with mice were performed by the regulations and approval of the Institutional Animal Care and Use Committee from the University of Florida (protocol number 201909362).
Author Contributions
HRT, SP, and LJ conceived the research. LJ designed the experiments, wrote the manuscript, and supervised the research. SP, HT, HG, SM, and LJ performed experiments and analyzed the data. SP drafted the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by NIH grants AI110606, AI125999, AI132865, and HL152163 (to LJ). SM was supported through The American Association of Immunologists Careers in Immunology Fellowship Program.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Yang Jun and Dr. Bai Guangchun from Albany Medical College for the initial helps with the S. pneumonia culture. We thank Dr. Roy Curtiss III for the help with S. pneumonia infection. We thank the Center for Immunology and Transplantation at the University of Florida for the assistance with flow cytometry. Lastly, we would like to thank members of Jin Lab for helpful discussion and technical support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.699702/full#supplementary-material
The Supplementary Material contains four figures and one table and can be found online.
References
1. Ishikawa H, Barber GN. STING is an Endoplasmic Reticulum Adaptor That Facilitates Innate Immune Signalling. Nature (2008) 455(7213):674–8. doi: 10.1038/nature07317
2. Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, et al. The Adaptor Protein MITA Links Virus-Sensing Receptors to IRF3 Transcription Factor Activation. Immunity (2008) 29(4):538–50. doi: 10.1016/j.immuni.2008.09.003
3. Patel S, Jin L. TMEM173 Variants and Potential Importance to Human Biology and Disease. Genes Immun (2018) 20(1):82–9. doi: 10.1038/s41435-018-0029-9
4. Henriques-Normark B, Tuomanen EI. The Pneumococcus: Epidemiology, Microbiology, and Pathogenesis. Cold Spring Harb Perspect Med (2013) 3(7):1–15. doi: 10.1101/cshperspect.a010215
5. Periselneris J, José RJ, Brown JS. Pulmonary Immune Response to Streptococcus Pneumoniae. Shortness Breath (2014) 3(4):147–58. doi: 10.11138/sob/2014.3.4.147
6. Ruiz-Moreno JS, Hamann L, Jin L, Sander LE, Puzianowska-Kuznicka M, Cambier J, et al. The cGAS/STING Pathway Detects Streptococcus Pneumoniae But Appears Dispensable for Antipneumococcal Defense in Mice and Humans. Infect Immun (2018) 86(3):e00849–17. doi: 10.1128/IAI.00849-17
7. Bjerre A, Brusletto B, Hoiby EA, Kierulf P, Brandtzaeg P. Plasma Interferon-Gamma and Interleukin-10 Concentrations in Systemic Meningococcal Disease Compared With Severe Systemic Gram-Positive Septic Shock. Crit Care Med (2004) 32(2):433–8. doi: 10.1097/01.CCM.0000104950.52577.97
8. Mitchell AJ, Yau B, McQuillan JA, Ball HJ, Too LK, Abtin A, et al. Inflammasome-Dependent IFN-Gamma Drives Pathogenesis in Streptococcus Pneumoniae Meningitis. J Immunol (2012) 189(10):4970–80. doi: 10.4049/jimmunol.1201687
9. Rijneveld AW, Lauw FN, Schultz MJ, Florquin S, Te Velde AA, Speelman P, et al. The Role of Interferon-Gamma in Murine Pneumococcal Pneumonia. J Infect Dis (2002) 185(1):91–7. doi: 10.1086/338122
10. Sun K, Salmon SL, Lotz SA, Metzger DW. Interleukin-12 Promotes Gamma Interferon-Dependent Neutrophil Recruitment in the Lung and Improves Protection Against Respiratory Streptococcus Pneumoniae Infection. Infect Immun (2007) 75(3):1196–202. doi: 10.1128/IAI.01403-06
11. Weber SE, Tian H, Pirofski LA. CD8+ Cells Enhance Resistance to Pulmonary Serotype 3 Streptococcus Pneumoniae Infection in Mice. J Immunol (2011) 186(1):432–42. doi: 10.4049/jimmunol.1001963
12. Yamamoto N, Kawakami K, Kinjo Y, Miyagi K, Kinjo T, Uezu K, et al. Essential Role for the P40 Subunit of Interleukin-12 in Neutrophil-Mediated Early Host Defense Against Pulmonary Infection With Streptococcus Pneumoniae: Involvement of Interferon-Gamma. Microbes Infect (2004) 6(14):1241–9. doi: 10.1016/j.micinf.2004.08.007
13. Jin L, Getahun A, Knowles HM, Mogan J, Akerlund LJ, Packard TA, et al. STING/MPYS Mediates Host Defense Against Listeria Monocytogenes Infection by Regulating Ly6C(hi) Monocyte Migration. J Immunol (2013) 190(6):2835–43. doi: 10.4049/jimmunol.1201788
14. Blaauboer SM, Mansouri S, Tucker HR, Wang HL, Gabrielle VD, Jin L. The Mucosal Adjuvant Cyclic Di-GMP Enhances Antigen Uptake and Selectively Activates Pinocytosis-Efficient Cells In Vivo. Elife (2015) 4:e06670. doi: 10.7554/eLife.06670
15. Patel S, Blaauboer SM, Tucker HR, Mansouri S, Ruiz-Moreno JS, Hamann L, et al. The Common R71H-G230A-R293Q Human TMEM173 Is a Null Allele. J Immunol (2017) 198(2):776–87. doi: 10.4049/jimmunol.1601585
16. Koppe U, Suttorp N, Opitz B. Recognition of Streptococcus Pneumoniae by the Innate Immune System. Cell Microbiol (2012) 14(4):460–6. doi: 10.1111/j.1462-5822.2011.01746.x
17. Barber GN. STING: Infection, Inflammation and Cancer. Nat Rev Immunol (2015) 15(12):760–70. doi: 10.1038/nri3921
18. Wu J, Sun L, Chen X, Du F, Shi H, Chen C, et al. Cyclic GMP-AMP is an Endogenous Second Messenger in Innate Immune Signaling by Cytosolic DNA. Science (2013) 339(6121):826–30. doi: 10.1126/science.1229963
19. Zhang X, Shi H, Wu J, Sun L, Chen C, Chen ZJ. Cyclic GMP-AMP Containing Mixed Phosphodiester Linkages Is an Endogenous High-Affinity Ligand for STING. Mol Cell (2013) 51(2):226–35. doi: 10.1016/j.molcel.2013.05.022
20. Murray PJ. Immune Regulation by Monocytes. Semin Immunol (2018) 35:12–8. doi: 10.1016/j.smim.2017.12.005
21. Freudenberg MA, Merlin T, Kalis C, Chvatchko Y, Stübig H, Galanos C. Cutting Edge: A Murine, IL-12-Independent Pathway of IFN-Gamma Induction by Gram-Negative Bacteria Based on STAT4 Activation by Type I IFN and IL-18 Signaling. J Immunol (2002) 169(4):1665–8. doi: 10.4049/jimmunol.169.4.1665
22. Barbulescu K, Becker C, Schlaak JF, Schmitt E, Meyer zum Buschenfelde KH, Neurath MF. IL-12 and IL-18 Differentially Regulate the Transcriptional Activity of the Human IFN-Gamma Promoter in Primary CD4+ T Lymphocytes. J Immunol (1998) 160(8):3642–7.
23. Gomez JC, Yamada M, Martin JR, Dang H, Brickey WJ, Bergmeier W, et al. Mechanisms of Interferon-Gamma Production by Neutrophils and its Function During Streptococcus Pneumoniae Pneumonia. Am J Respir Cell Mol Biol (2015) 52(3):349–64. doi: 10.1165/rcmb.2013-0316OC
24. Mansouri S, Patel S, Katikaneni DS, Blaauboer SM, Wang W, Schattgen S, et al. Immature Lung TNFR2(-) Conventional DC 2 Subpopulation Activates moDCs to Promote Cyclic Di-GMP Mucosal Adjuvant Responses In Vivo. Mucosal Immunol (2019) 12(1):277–89. doi: 10.1038/s41385-018-0098-0
25. Yoshimura A, Lien E, Ingalls RR, Tuomanen E, Dziarski R, Golenbock D. Cutting Edge: Recognition of Gram-Positive Bacterial Cell Wall Components by the Innate Immune System Occurs via Toll-Like Receptor 2. J Immunol (1999) 163(1):1–5.
26. Temizoz B, Kuroda E, Ohata K, Jounai N, Ozasa K, Kobiyama K, et al. TLR9 and STING Agonists Synergistically Induce Innate and Adaptive Type-II IFN. Eur J Immunol (2015) 45(4):1159–69. doi: 10.1002/eji.201445132
27. Zhu Q, Kanneganti TD. Cutting Edge: Distinct Regulatory Mechanisms Control Proinflammatory Cytokines IL-18 and IL-1beta. J Immunol (2017) 198(11):4210–5. doi: 10.4049/jimmunol.1700352
28. Nakahira M, Ahn HJ, Park WR, Gao P, Tomura M, Park CS, et al. Synergy of IL-12 and IL-18 for IFN-Gamma Gene Expression: IL-12-Induced STAT4 Contributes to IFN-Gamma Promoter Activation by Up-Regulating the Binding Activity of IL-18-Induced Activator Protein 1. J Immunol (2002) 168(3):1146–53. doi: 10.4049/jimmunol.168.3.1146
29. Napolitani G, Rinaldi A, Bertoni F, Sallusto F, Lanzavecchia A. Selected Toll-Like Receptor Agonist Combinations Synergistically Trigger a T Helper Type 1-Polarizing Program in Dendritic Cells. Nat Immunol (2005) 6(8):769–76. doi: 10.1038/ni1223
30. Krummen M, Balkow S, Shen L, Heinz S, Loquai C, Probst HC, et al. Release of IL-12 by Dendritic Cells Activated by TLR Ligation Is Dependent on MyD88 Signaling, Whereas TRIF Signaling is Indispensable for TLR Synergy. J Leukoc Biol (2010) 88(1):189–99. doi: 10.1189/jlb.0408228
31. Snijders A, Kalinski P, Hilkens CM, Kapsenberg ML. High-Level IL-12 Production by Human Dendritic Cells Requires Two Signals. Int Immunol (1998) 10(11):1593–8. doi: 10.1093/intimm/10.11.1593
32. Tada H, Aiba S, Shibata K, Ohteki T, Takada H. Synergistic Effect of Nod1 and Nod2 Agonists With Toll-Like Receptor Agonists on Human Dendritic Cells to Generate Interleukin-12 and T Helper Type 1 Cells. Infect Immun (2005) 73(12):7967–76. doi: 10.1128/IAI.73.12.7967-7976.2005
33. Theiner G, Rossner S, Dalpke A, Bode K, Berger T, Gessner A, et al. TLR9 Cooperates With TLR4 to Increase IL-12 Release by Murine Dendritic Cells. Mol Immunol (2008) 45(1):244–52. doi: 10.1016/j.molimm.2007.02.021
34. Brown AS, Yang C, Fung KY, Bachem A, Bourges D, Bedoui S, et al. Cooperation Between Monocyte-Derived Cells and Lymphoid Cells in the Acute Response to a Bacterial Lung Pathogen. PloS Pathog (2016) 12(6):e1005691. doi: 10.1371/journal.ppat.1005691
35. Casson CN, Doerner JL, Copenhaver AM, Ramirez J, Holmgren AM, Boyer MA, et al. Neutrophils and Ly6Chi Monocytes Collaborate in Generating an Optimal Cytokine Response That Protects Against Pulmonary Legionella Pneumophila Infection. PloS Pathog (2017) 13(4):e1006309. doi: 10.1371/journal.ppat.1006309
Keywords: STING, IFNγ, monocyte, MyD88, Streptococcus pneumoniae (pneumococcus)
Citation: Patel S, Tucker HR, Gogoi H, Mansouri S and Jin L (2021) cGAS–STING and MyD88 Pathways Synergize in Ly6Chi Monocyte to Promote Streptococcus pneumoniae-Induced Late-Stage Lung IFNγ Production. Front. Immunol. 12:699702. doi: 10.3389/fimmu.2021.699702
Received: 24 April 2021; Accepted: 05 August 2021;
Published: 26 August 2021.
Edited by:
Martin Rottenberg, Karolinska Institutet (KI), SwedenReviewed by:
Jianjun Wu, University of Texas Southwestern Medical Center, United StatesJim Vadolas, Hudson Institute of Medical Research, Australia
Copyright © 2021 Patel, Tucker, Gogoi, Mansouri and Jin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lei Jin, bGVpLmppbkBtZWRpY2luZS51ZmwuZWR1
†Present address: Seema Patel, Translational Immunology at BioNTech, Cambridge, MA, United States