 Jacqueline A. Wright1†
Jacqueline A. Wright1† Cassandra Bazile1†Emily S. Clark1
Cassandra Bazile1†Emily S. Clark1 Gianluca Carlesso2
Gianluca Carlesso2 Justin Boucher1Eden Kleiman1Tamer Mahmoud2
Justin Boucher1Eden Kleiman1Tamer Mahmoud2 Lily I. Cheng3
Lily I. Cheng3 Darlah M. López-Rodríguez1
Darlah M. López-Rodríguez1 Anne B. Satterthwaite4Norman H. Altman5Eric L. Greidinger6
Anne B. Satterthwaite4Norman H. Altman5Eric L. Greidinger6 Wasif N. Khan1*
Wasif N. Khan1*- 1Department of Microbiology and Immunology, Miller School of Medicine, University of Miami, Miami, FL, United States
- 2Early Oncology Discovery, Early Oncology R&D, AstraZeneca, Gaithersburg, MD, United States
- 3Oncology Safety/Pathology, Clinical Pharmacology and Safety Sciences, AstraZeneca, Gaithersburg, MD, United States
- 4Department of Immunology, University of Texas Southwestern Medical Center, Dallas, TX, United States
- 5Department of Pathology, Miller School of Medicine, University of Miami, Miami, FL, United States
- 6Department of Medicine, Miller School of Medicine, University of Miami, Miami, FL, United States
While apoptosis plays a role in B-cell self-tolerance, its significance in preventing autoimmunity remains unclear. Here, we report that dysregulated B cell apoptosis leads to delayed onset autoimmune phenotype in mice. Our longitudinal studies revealed that mice with B cell-specific deletion of pro-apoptotic Bim (BBimfl/fl) have an expanded B cell compartment with a notable increase in transitional, antibody secreting and recently described double negative (DN) B cells. They develop greater hypergammaglobulinemia than mice lacking Bim in all cells and accumulate several autoantibodies characteristic of Systemic Lupus Erythematosus (SLE) and related Sjögren’s Syndrome (SS) including anti-nuclear, anti-Ro/SSA and anti-La/SSB at a level comparable to NODH2h4 autoimmune mouse model. Furthermore, lymphocytes infiltrated the tissues including submandibular glands and formed follicle-like structures populated with B cells, plasma cells and T follicular helper cells indicative of ongoing immune reaction. This autoimmunity was ameliorated upon deletion of Bruton’s tyrosine kinase (Btk) gene, which encodes a key B cell signaling protein. These studies suggest that Bim-mediated apoptosis suppresses and B cell tyrosine kinase signaling promotes B cell-mediated autoimmunity.
Introduction
A vast B cell repertoire is generated by a random recombination of exons that are assembled to encode a diverse set of B cell receptors (BCRs) necessary to mount antigen-specific immune responses against a large variety of pathogen-derived antigens as well as potential neoantigens. The process called V(D)J recombination inherently produces a large number of self-reactive B cell clones. Therefore, exquisitely controlled mechanisms of immune tolerance operate during development in the bone marrow (BM) to eliminate autoreactive clones (1, 2). The primary mechanisms of tolerance in the BM include receptor editing, clonal deletion by apoptosis and anergy. Central tolerance eliminates most of the autoreactive B cells immediately after completing and displaying their assembled BCRs. Additional tolerance checkpoints purge the autoreactive B cell clones that variably escape these controls in the BM or are generated in the germinal centers during an immune response (3, 4).
While relatively strong BCR signaling results in the immature B cell negative selection, tonic or low level BCR signaling is continuously required for the survival and maturation of newly formed immature B cells in the BM as well as after their emigration to the spleen as transitional 1 (T1) B cells (5–9). We and others have shown that B cells at the T1 stage remain sensitive to apoptosis to serve as a second tolerance check point allowing deletion of autoreactive B cells in the periphery (7–9). These studies also showed that the window of opportunity for self-tolerance is limited as progression into transitional 2 (T2) B cells allows BCR engagement to promote positive selection (7, 10–13). BCR signaling in T2 cells induces sustained NF-kB activation, upregulation of BAFFR (TNFRSF13c) and more robust synergy between BCR and BAFFR. Excess availability of BAFF and increased BAFFR signaling can sway BCR-engaged autoreactive transitional B cell clones to undergo maturation into follicular (Fo) and marginal zone (MZ) B cells and can promote autoimmunity (6, 7, 14–17). BAFF function becomes particularly important when other B cell coreceptors positively influence autoreactive B cell activation (5, 6, 14, 16, 18). For example, self RNA and DNA reactive B cell clones receive the first antigen specific signal via the BCRs, which endocytose nucleic acids and deliver them to endosomal TLR7 or TLR9. TLRs can then provide the second signal to activate autoreactive B cells (19–23). Availability of BAFF can enhance positive selection of BCR and TLR activated autoreactive B cells and promote their maturation. Thus, TLR and BAFFR can synergize to dysregulate autoreactive BCR signaling towards B cell survival and maturation (18). Recent studies with systemic lupus erythematosus (SLE) patients have identified polymorphisms in genes that dysregulate signaling downstream of BCR, BAFFR and TLRs, supporting synergy between these receptors can promote positive selection of autoreactive B cells leading to autoimmunity (18, 24–26).
Importantly, autoimmune diseases associated with autoAbs are highly common in humans. In fact, both SLE and SS involve B cell hyperactivity that contributes to the development of autoimmune disease manifestation (27). However, whether or not apoptosis-mediated B cell immune tolerance prevents autoimmune disease is an active area of research (16, 28, 29). Clonal deletion of autoreactive B cells by apoptosis can be mediated by cell-extrinsic Fas/FasL – dependent pathway and cell intrinsic mechanisms controlled by pro-apoptotic members of the Bcl-2 family (30, 31). In fact, both pathways may eventually converge on the executioner caspases to induce cell death. Therefore, dysregulated apoptosis is a contributing factor to the escape of autoreactive B cells. However, the physiological role for cell intrinsic pathways, including BH3-only proapoptotic members of the Bcl-2 family, such as Bcl-2-like 11 (Bim) remains elusive (32, 33). Bim is particularly critical in facilitating apoptosis when autoreactive B cells are destined to die (34). Although genetic alterations have been made in apoptosis pathways that variably promote SLE-like disease including Bim-/- (C57BL/6 X 129Ssv) (35), B6.Faslpr/lpr and B6.Bim-/-.Faslpr/lpr (31, 36), the contributions of specific immune cells lacking Bim to autoimmune pathology are not yet fully elucidated. Global deletion of Bim (Bim-/-) results in SLE-like autoimmune disease in mice with splenomegaly and lymphadenopathy accompanied with lymphocyte infiltration and autoantibody production on a mixed C57BL/6x129Sv genetic background (35). Because Bim is also required for negative selection of T cells at multiple checkpoints and in myeloid cells, the contribution of apoptosis regulation in specific cell types to autoimmune pathology in Bim-/- mice remained unknown. Subsequent adaptive transfer experiments demonstrated that Bim-deficient dendritic cells (DCs) can drive autoimmune pathology (37, 38). More recently, conditional gene deletion approach was used to demonstrate that myeloid cell-specific deletion of Bim led to a severe SLE disease whereas they noted CD4 T cell- or B cell-specific Bim gene deletion did not result in significant manifestation of autoimmunity (39). Another excellent study interrogating Bim function in B cells, did not observe autoimmune phenotype and focused on B cell lymphoma genesis (39, 40). However detailed B cell analysis, particularly relating to autoimmunity was not shown in either study (39, 40). Cumulatively, prior studies implicate Bim in suppressing autoimmunity mediated by myeloid and dendritic cells whereas its role in restraining T and B cells in promoting autoimmune disease remains work in progress.
Here we report detailed longitudinal studies in mice with CD19-Cre mediated B cell-specific Bim deletion the revealing a delayed onset SLE/SS-like autoimmune disease in C57BL/6 background. The BBimfl/fl mice had more exaggerated B cell expansion than the Bim null mice, which display a mild autoimmunity in the C57BL/6 background, whereas the increase in T cell numbers was comparable. Autoantibody production included Anti-dsDNA, Anti-SSA, anti-SSB autoantibodies (autoAbs) characteristic of SLE and SS, and their levels were comparable or exceeded autoantibody levels in age-matched NODH2h4 mice. These autoimmune phenotypes were accompanied with lymphocyte infiltration of salivary submandibular glands (SG) which were populated by B cell, Tfh and plasma cells. Thus, Bim-mediated B cell apoptosis suppresses a wide range of autoAbs production and dysregulated apoptosis in B cells promotes T cell activation and participation in autoimmunity. The autoimmunity in BBimfl/fl mice was remediated by deletion of Btk, a key B cell signaling tyrosine kinase, suggesting contribution of altered B cell signaling to autoimmune pathology supporting utility of Btk and tyrosine kinase inhibitors in autoimmunity (41, 42).
Methods
Mice
Adult B57BL/6 (B6) mice were originally obtained from Jackson Laboratory, Bar Harbor, ME and were subsequently bred in house. Bimfl/fl mice were generated by Korsmeyer group (43) and were obtained from S. Zinkel, Vanderbilt University. B lineage specific Bim-deficient mice were generated by intercrossing Bimfl/fl mice with CD19cre mice (44). All BBimfl/fl used were heterozygous for CD19cre. It has been shown that mice expressing CD19cre have no discernable effect on B cell development (44). The original Bim-/- mice in B6.129S1-Bcl2l11tm1.1Ast/J56 had been backcrossed into C57BL/6J (Jackson Laboratory). Spleens from the Bim-deficient mice were kindly provided by Richard T Libby, Flaum Eye Institute, University of Rochester Medical Center, referred here as Bim-/-. The generation of Btk-/- mice have been previously described (45). Mice were treated humanely in accordance with federal, state, and institutional guidelines.
Cell Isolation and In Vitro Culture Conditions
Spleens were removed and mechanically disrupted, generating a single cell suspension. Red blood cells were then lysed using RBC lysis buffer (Biolegend) according to manufacturer’s instructions. Splenic B cells were enriched by negative selection to avoid inadvertent activation, either by autoMACS depletion using anti-CD43 microbeads (Miltenyi Biotec) or using anti-CD43 microbeads B cell enrichment kit (BD Biosciences). B cell purity was determined to be between 92-98% as determined by flow cytometric analysis using antibodies directed against CD19 and IgM (Table 1). For B cell proliferation assays, purified B cells were cultured in RPMI (Hyclone laboratories) supplemented with 10% fetal calf serum, 55nM β-mercaptoethanol, 2nM L-glutamine and 100IU penicillin/streptomycin in a 37°C humidified incubator. In vitro cultures were left nonstimulated or treated with F(ab’)2 goat anti-mouse IgM (10μg/ml; Jackson ImmunoResearch Laboratories), recombinant human BAFF purified from Chinese Hamster ovary cells (46) (100ng/ml), anti-CD40 (2.5μg/ml; BD Bioscience), LPS (2.5μg/ml Sigma- Aldrich), or CpG (1.0μg/ml) at the times indicated. To measure cell death, purified B cells were stimulated with LPS (1μg/ml), CPG (ODN-1826 1μg/ml; InvivoGen), CL097 (1μg/ml; InvivoGen), or Fa(b’)2 goat anti-mouse IgM (1μg/ml) in the absence or in the presence of IL-21 (25ng/ml; RnD System) for 48 hours at 37°C. To assess cytokine production, total splenocytes were stimulated with CPG or CL097 for 24 hours and PMA (50ng/ml) and ionomycin (500ng/ml) were added to the culture for the last 4 hours.

Table 1 Antibodies used in this study.
Flow Cytometry
For phenotypic analysis, single-cell suspensions were prepared from spleens, inguinal lymph nodes, submandibular lymph nodes, tertiary lymphoid structures (TLS) and the blood of WT, BBimfl/fl, Bim−/−, Btk−/− and BBimfl/fl × Btk−/− mice. Cells were stained with fluorescently labelled antibodies in various combinations to identify; B cells and subpopulations including T1, MZ, pMZ, An1, CD21lo and, CD4 and CD8 T cells and their subpopulations including T follicular cells, as defined below. Antibodies, fluorochrome labelling and sources are detailed in Table 1 and indicated in figure legends.
Intracellular cytokine staining was carried out by first staining cell surface markers in PBS with 2% serum after incubation with FcR-block (CD16), washed and stained with antibodies to various cytokines (IFN-γ, IFN-α, IL-6, IL-10 and TNF-α) using the BD Biosciences fix/perm kit. Dead cells and doublets were excluded from the FCM analysis by Live/Dead dye (BD Biosciences) and SSC-W/SCC-H and FCS-W/FSC-H gating protocols. Dead cells and cells undergoing apoptosis were detected by staining with AnnexinV and 7AAD. All flow cytometry data was acquired on a BD LSR II flow cytometer and analyzed using the FlowJo software package (Tree Star).
Definition of Cell Types by Flow Cytometry
Cell types were defined by the following markers: T1 B cells (CD19+, IgMhi, IgDlo, CD21-, CD23-) MB B cells (CD19+, IgMhi, IgDlo, CD21hi, CD23lo) FoB1 cells (CD19+, IgMlo, IgDhi CD21int CD23+), FoB2 cells (CD19+, IgMhi, IgDhi CD21int CD23+) Plasma cells (B220+CD138+) and Anergic B cells (B220+ AA4+ IgD+ IgMlo). T cells (CD19- CD3+ CD5+), CD4+ T cells (CD19- CD3+ CD4+ CD8-), CD8+ T cells (CD19- CD3+ CD4- CD8+), Tfh cells (CD4+ PD1+ CXCR5+), effector memory T cells (CD44+ CD62L-), Central memory T cells (CD44+CD62L+), Naïve T cells (CD44- CD62L-).
3H-Thymidine Incorporation Cell Proliferation Assay
Cell proliferation was measured by 3H-Thymidine (Perkin Elmer) incorporation into replicative strands of DNA. Cells were cultured in U bottom microplates and stimulated with the indicated agonists for the specified times either in the continuous presence of 1 microcurie per well of 3H-Thymidine for 48 hours or 3H-Thymidine was added in the last 16 hours of the 72 hours incubation period at 37°C in humidified. The 3H-Thymidine labeled DNA was captured on fiber filter disks, which were then placed in a liquid scintillation counting vials before counting on a scintillation beta-counter.
Quantitative PCR
RNA was extracted from freshly isolated cells (ex vivo) or after culture in vitro with agonists using the RNeasy Mini Kit (Qiagen) and used to synthesize cDNA. RNA was quantified on a NanoDrop 1000 prior to use in the RT-PCR reactions. Reverse Transcription was carried out using equivalent amounts of RNA, dNTP, M-MLV reverse transcriptase, RNase inhibitor, nuclease free water, Random Hexamer, 10xPCR buffer and MgCl2 (All from Applied Biosystems). Taqman Real time reactions used TaqmanUniversal Master Mix or Taqman Fast Advanced Master Mix (Applied Biosystems) and changes in gene expression were determined by running samples on the Stratagene Max 3000p Detection Systems or Step One Real Time System (Applied Biosystems). Primer/Probe combinations were obtained from applied biosystems; (Mm00477631_m1), Bmf (Mm00506773_m1), Btk (Mm00442712_m1), IFNa1 (Mm03030145_gH), IFNb (Mm00439552_s1), IFNg (Mm00801778_m1), IL-6 (MM00446190), IL12p10 (Mm00434169_m1), Mcl-1 (Mm01257351_g1) MCP-1 (Mm00441242_m1), TNFa (Mm00443258_m1), TLR7 (Mm00446590), and TLR9 (Mm00446193_m1). The relative mRNA fold induction for each gene was calculated relative to 18S ribosomal RNA or GAPDH expression.
Autoantibody Array
Sera from mice of indicated ages were hybridized to an array containing approximately 75 autoantigens as described (47). Briefly, IgM and IgG antibodies were detected with Cy3 and Cy5 coupled secondary antibodies. Data were normalized to total Ig levels and clustered by antigen. Red indicates greater reactivity than the average for each antigen, green indicates lower reactivity than the average for each antigen.
ELISA Detection of Antibody Isotypes
Blood samples were obtained by retro-orbital bleeding of mice using heparin containing microcentrifuge tubes. Samples were centrifuged to pellet the RBC before the serum was drawn off and stored at -80 until analysis. For the determination of Igs (total Ig, IgM, IgG1, IgG2a, IgG2c, and IgG3) in the serum/plasma using SBA clonotyping system according to the manufacturer’s instructions (Southern Biotechnology Associates). Briefly, plates were coated with 5 μg/ml of capture Ab, and serum (diluted 1/1,000) was incubated and bound Igs were revealed by HRP-labeled secondary Abs. Results are plotted as the concentration of each Ig isotype.
Serum autoAbs against ANAs, nRNP, SSA, and SSB were measured using ELISA kits (Alpha Diagnostics, San Antonio, TX, USA). Sera were diluted 20-fold before the assay and the manufacturer protocol was followed. Positive values for autoreactive antibodies were determined by the manufacturers cut off value. Soluble BAFF was measured using a mouse BAFF quantikine ELISA respectively (R&D systems). Serum was diluted 50-fold and the manufacturer protocol was followed and ELISA plates were developed and absorbance (absorbance 450nm) using microplate ELISA reader.
ELISpot for Mouse IgM and IgG
Detection and enumeration of B cells secreting IgM and IgG was determined by ELISpot according to the manufacturer’s instructions. Briefly, antigen was coated onto the ELIspot plate and B cells from the indicated genotypes were incubated on the plate for 16-24 hours before spots were detected. After the incubation time, the plate was washed and biotinylated antigen was added. The plate was washed again prior to the addition diluted Streptavidin-ALP which was incubated for 1 hour. At the completion of the incubation time, the plate was washed and the substrate solution (BCIP/NBT-plus) was added and incubated until distinct spots emerged. The color development was then stopped by washing the plate with tap water and allowed to dry before reading on a ELISpot reader.
Cytokine Bead Array
Serum cytokine levels were determined by cytokine bead array in accordance with the manufacturer’s guidelines (551287 BD Biosciences). Briefly, serum samples were mixed cytokine labeled capture beads. Each capture bead mixture has a distinct fluorescence when acquired by FCM. The intensity of brightness in the PE channel reveals the cytokine concentration. FCAP Array software (BD Biosciences) was used to calculate results.
Immunohistochemistry
Spleens, LN, intestines, liver, kidney, salivary glands and other tissues from WT and BBimfl/fl mice were formalin (10%) fixed immediately upon harvest, paraffin embedded for H&E staining or frozen in for immunofluorescence staining. Sections were stained with anti- mouse CD19 biotinylated (revealed by streptavidin Alexa Fluor 488) and anti-mouse CD3 (revealed by Alexa Fluor 647 conjugated goat anti-mouse Ab) to visualize B and T cells. Slides were analyzed using a Zeiss Axiovert 200M fluorescence microscope and the Axiovision 4.6 data analysis software program. For H&E staining spleen, kidneys, Pancreas, salivary glands, liver, lungs, and intestines were harvested from control and mutant mice. Tissues were reviewed and scored by a board-certified veterinary pathologist. Scores of inflammation and glomerulonephritis were determined and scored as (0, none; 1, mild; 2, moderate; 3, severe; and 4 extensive). Slides were analyzed and imaged using the Olympus VS120 slide scanner.
Statistical Analyses
Data collected were compared by two-tailed Students t test. Values of *p ≤ 0.05 were considered statistically significant.
Results
B Cell-Specific Deletion of Bcl2l11 (BBimfl/fl) Leads to Expansion of B and T Cells
To address the significance of B cell-intrinsic regulation of apoptosis in establishing tolerance in the wild type B cell repertoire, we crossed a CD19-cre mouse to a mouse carrying Loxp flanked Bim gene (Bcl2l11) alleles in C57BL/6 background, previously described by Korsmeyer group (43). The mouse produced by this intercross was confirmed for Bim deletion in the B lineage, termed BBimfl/fl (Supplementary Figure 1A). We found that BBimfl/fl mice developed splenomegaly and lymphadenopathy at a relatively early age with a corresponding increase in splenic weight, size and cellularity (Figures 1A, B and Supplementary Figure 2A). Splenic lymphoid follicles appeared normal in young BBimfl/fl mice (Figure 1C), but with age the lymphoid follicles in the BBimfl/fl mice were enlarged and fused due to enlarged B cell zones and became disorganized in one year and older mice (Supplementary Figure 2D). The overall splenic B cell numbers were increased 2-3 fold in young adult BBimfl/fl mice relative to littermate control mice (Figure 1D), consistent with recent reports of B cell-specific Bim deletion (39, 48). In contrast to the previous reports, the increase in B cell numbers and ratios was maintained into relatively old age (Figure 1D).
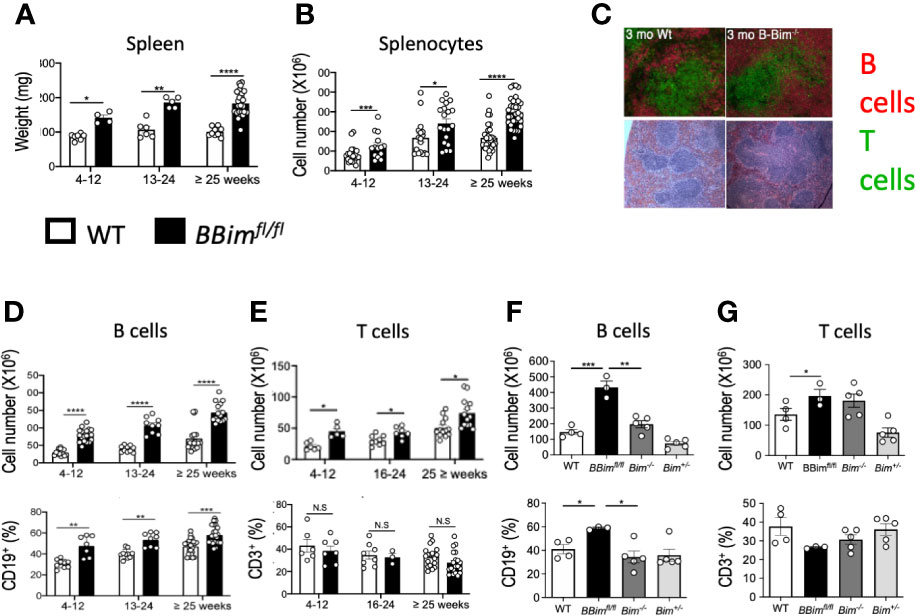
Figure 1 B cell lineage-specific deletion of Bim resulted in expansion of splenic B and T cells. Spleen weights and splenocyte numbers, B cells and T cells were identified by FCM using established cell surface markers as described in materials and methods. (A) Spleen weights at different ages from BBimfl/fl and WT mice. (B) Total splenocyte numbers. (C) Immunofluorescence image showing splenic follicles (top panel) stained for B220+ B cells (red) and CD3+ T cells (green) and H&E images (bottom panel) showing follicular structure and lymphocyte organization (top panel) of spleens from 3 month old mice. (D) Numbers (top panel) and proportions (bottom panel) of splenic (D) B cells and (E) T cells. (F, G) Quantification of splenic B and T cells in BBimfl/fl relative to Bim-/- mice. Numbers (top panel) and proportions (bottom panel) of (F) B cells and (G) T cells. Data is representative of >3 independent experiments. Not significant (N.S) P > 0.05, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001, calculated by Students T- test and Mann-Whitney non-parametric test.
The overall T cells were also variably increased in the BBimfl/fl relative to control mice but to a lesser extent relative to the expansion of B cells (Figure 1E). Like B cells, the increases in T cells did not wane with age (Figure 1E).
Prior studies have reported that the SLE-like phenotype observed in systemically Bim-deficient mice (Bim-/-) in a mixed genetic background (SV129 x C57BL/6) was much milder in pure C57BL/6 background (35, 36, 49). Therefore, we compared overall B and T cell proportions and numbers in the BBimfl/fl mice with Bim-/- mice, both in C57BL/6 background. In agreement with the prior reports, we found that the numbers of B cells were increased (1.5-2-fold) in Bim-/- mice relative to WT. However, the expansion of B cells in the BBimfl/fl mice was greater (3-fold) relative to Bim-/- mice (Figure 1F), whereas T cells were increased comparably (1.5-2-fold) between the two genotypes (Figure 1G). These data suggest a critical B cell-autonomous role for Bim in B cell homeostasis, which is more apparent in the Bim-sufficient milieu in the BBimfl/fl mice. In addition, Bim-sufficient T cell compartment also appears to be influenced by dysregulated B cells directly and/or indirectly including an increase in T cells.
In young BBimfl/fl mice (4-12 wk old) the numbers of all mature FoB1 and FoB2 and T1 B cell subsets were increased 2-4 fold, except B1 B cells (Figures 2A–C and data not shown). In contrast, the proportions of MZ and precursor (pMZ) B cells were significantly decreased in BBimfl/fl mice with age relative to WT mice, which also reflected in a modest decrease in their numbers (Figures 2D, E). In what may be a related finding, the reduction in CD21 expression was greater in BBimfl/fl than WT B cells (Figure 2E, bottom panels). We noted that CD21- and CD23- double negative (DN) B cells were significantly increased in BBimfl/fl spleens and progressively further increased with age relative to WT (Figure 2F). Given increased numbers of T1 B cells escape deletion in the BBimfl/f, we wondered whether anergic (An1/T3) B cells that have a shorter life span and are hypersensitive to apoptosis may live longer and accumulate, consistent with previous findings (34). Indeed. the anergic An1/T3 B cells increased in the BBimfl/fl mice (Figure 2G). The abnormal increases in T1 and An1/T3 B cells may be consequential for autoAb production as plasma B cells were also increased (Figure 2H). An increase in T1 B cells, which serves as a first peripheral tolerance checkpoint suggests that this otherwise apoptosis-sensitive population survives negative selection. Likewise, An1 B cells also acquire resistance to apoptosis and may contribute to break in tolerance in BBimfl/fl mice. The potential for T-B interaction was investigated by evaluating CD40 on B cell and helper T cells. The cell surface CD40 expression is reduced in lupus patient B cells that resemble ABC B cells (50), but there was no difference in the CD40 levels in WT and in BBimfl/fl B cells (Figure 2I). The CD4 and CD8 T cell numbers were increased with age but proportions were not significantly altered (Figures 2J–M). The T cell subset analysis revealed that effector CD4 T cells were increased (Figures 2N, O), but CD8 T cells were not (Figures 2P, Q). These data suggest that dysregulated apoptosis in B cells by loss of Bim promotes accumulation of autoreactive B cell populations and CD21loCD23lo B cells, which may be related to SLE-associated Tbet+ B cells (51), along with an increase in CD4 T effector and Tfh cells (Figures 2R, S) raise the possibility of autoimmune pathogenesis in BBimfl/fl mice. This potential is also supported by the findings that BBimfl/fl mice die earlier than their WT counterparts (Supplementary Figure 2E).
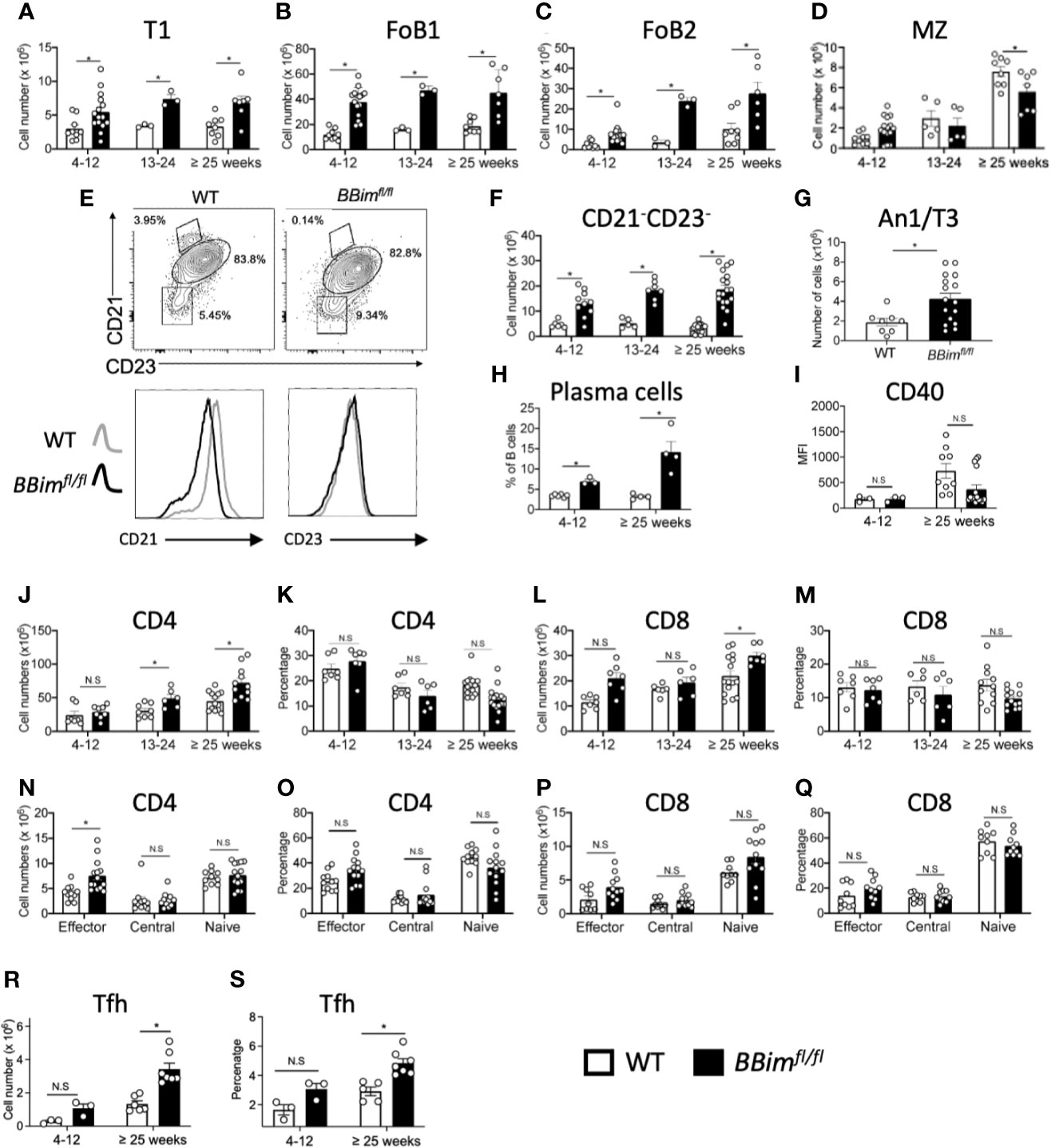
Figure 2 BBimfl/fl mice have expanded effector B and T cell populations in the spleen. Splenic B and T cell subpopulations were identified by FCM using established cell surface markers described in materials and methods. (A–D) Numbers of T1, FoB1, FoB2 and MZ plus pre-MZ B cells subsets in BBimfl/fl and WT mice in the indicated age groups. (E) Representative FCM plots showing gating strategy to quantify CD21 and CD23 double negative (DN) B cells within total B cells (top panels) and histograms displaying CD21 and CD23 expression showing reduced CD21 expression in B cells from BBimfl/fl relative to WT mice (bottom panels). (F) Numbers of CD21- CD23- DN B cells in BBimfl/fl and WT mice. (G) Numbers of anergic (An1/T3) B cells. (H) Numbers of plasma cells. (I) MFI values of CD40 in B cells (J–Q) Quantification of splenic CD4 and CD8 T cell populations on CD3+ gated cells in the ≥ 25 week old BBimfl/fl and WT mice. Representative graphs displaying (J) CD4 T cell numbers and (K) proportions and (L) CD8 T cell numbers and (M) proportions. (N) Cell numbers and (O) proportions of CD4 effector, central memory and naïve CD4 subsets. (P) CD8 T cell numbers and (Q) proportions of CD8 effector, central memory and naïve T cells. (R) Cell numbers and (S) percentages of Tfh cells. Data is representative of >3 independent experiments. Not significant (N.S) P > 0.05, *P ≤ 0.05, calculated by Students T- test and Mann-Whitney non-parametric test.
BBimfl/fl Mice Display Systemic Autoimmunity
Our data suggests that Bim mediated apoptosis is critical for B cell homeostasis and loss of this regulatory function may lead to autoimmune pathogenesis, which is not consistent with previous reports (39, 40, 52). We therefore, aimed to better define the pathophysiological effects of B cell specific loss of Bim, we analyzed various tissues for immune cell infiltration and damage in cohorts of BBimfl/fl and WT mice at different ages. We found that with age (6 months and older), the BBimfl/fl mice displayed lymphocytic infiltration in the liver and the lungs (Supplementary Figure 2B), as previously described for Bim-/- mice (35, 43). In addition, the majority of the BBimfl/fl mice had immune cell infiltration in the SGs, and kidneys (Figures 3A–C and Supplementary Figure 2C). Given these tissues are affected in SLE and SS they were further analyzed by immunohistochemistry. We found that glomeruli in BBimfl/fl mice were disorganized and damaged (Figure 3C upper panels) with evidence of T cell infiltration and to a lesser extent B cell infiltration, (Figure 3C lower panels). Consistently, BBimfl/fl mice also showed an increase in Bun/Crea ratio in the blood (Figure 3D). The glomerular damage was present in 40% of BBimfl/fl mice but not observed in the WT controls (Figures 3E, F).
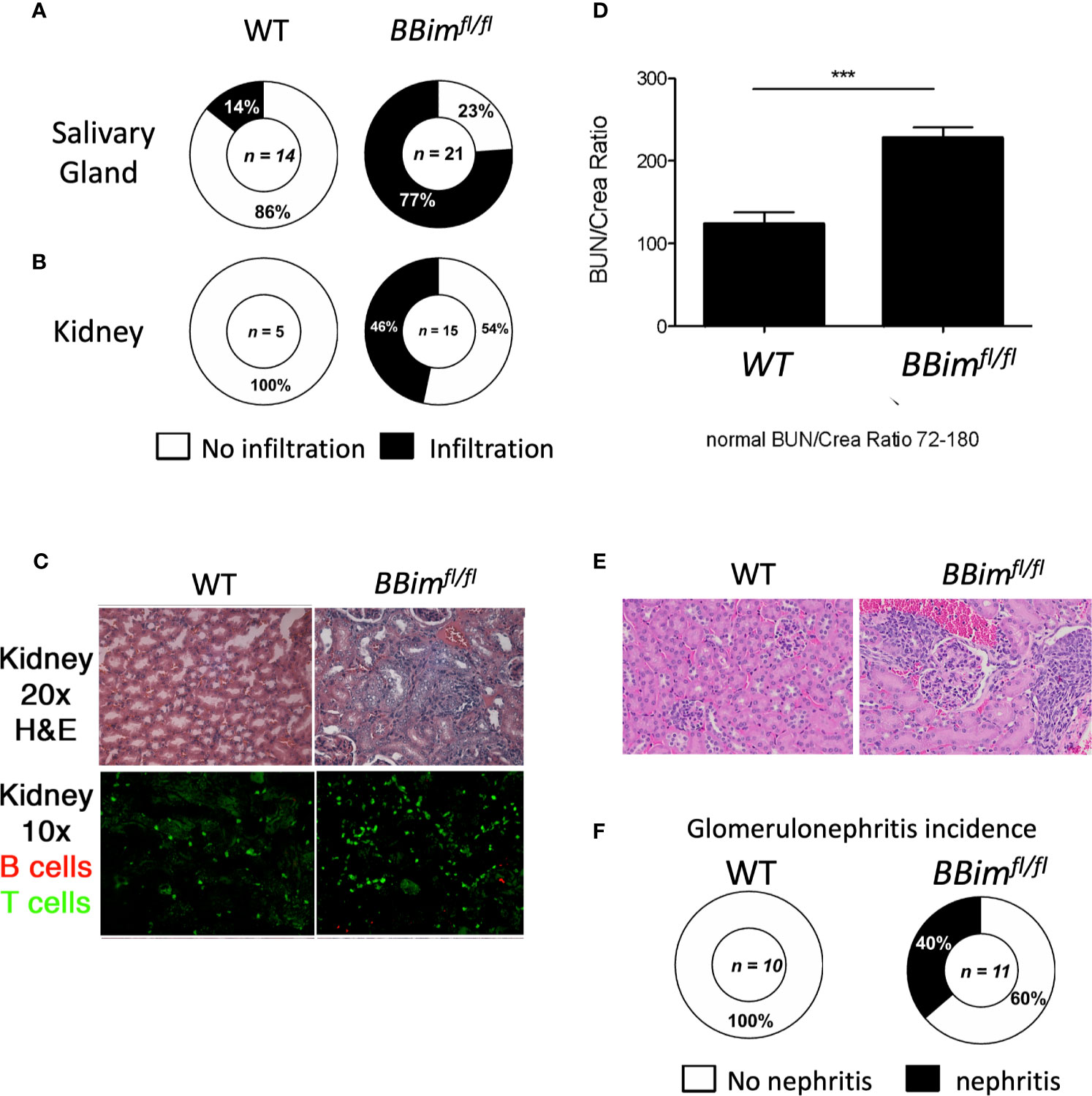
Figure 3 BBimfl/fl mice exhibit multiorgan lymphocyte infiltration and kidney damage. Tissues from ≥ 6 month old WT (n ≥3) and BBimfl/fl (n ≥8) mice were assessed for tissue damage. Representative pie charts show incidence of inflammation in the (A) salivary glands, and (B) kidneys. (C) Representative H&E images showing glomeruli in kidney sections (top panels) and immunofluorescence images of kidney sections (bottom panels) showing infiltrated T cells (green) and B cells (red) (D) Blood urea nitrogen and plasma creatine levels (BUN/crea) in the blood (age ≥ 6 month old) (E) H&E stained images of kidney sections showing enlarged glomeruli (glomerulonephritis) in BBimfl/fl relative to WT mice. (F) Incidence of kidney damage in the BBimfl/fl and WT mice. ***P ≤0.001, calculated by Students T-test.
Enlarged SGs are among the characteristic features of SS, therefore, BBimfl/fl SGs were more closely examined by histology and expanded to immune cell phenotype by FCM. Histological examination of the SGs revealed significant lymphoid hyperplasia, consisting of small lymphocytes (Figure 4A). Furthermore, immune cell infiltrates in the SGs formed follicle-like structures populated with B cells and plasma cells (PCs) (Figures 4B, C). These data suggest chronic inflammation, which may result in the loss of SG acini and impaired SG function. To uncover any T cell contribution to the PC formation FCM analysis of submandibular LNs was performed. The results revealed a two-fold increase in the T follicular helper cells (PD1+CXCR5+Bcl6+) in BBimfl/fl relative to WT mice (Figures 4D, E and Supplementary Figures 3A–D). Tertiary lymphoid structures (TLSs) are often formed at the site of inflammation and are associated with autoimmune inflammation (8, 53). We found that BBimfl/fl mice had TLSs in the proximity of SGs as well as in the abdomen as a network of strings of lymph node-like structures (Supplementary Figure 4A). The TLS in the BBimfl/fl mice were composed of different proportions of B cells, T cells and dendritic cells in different mice. B cell phenotype was also distinct containing different ratios of CD23-CD21+ MZ-like and CD21+CD23+ FoB like B cells (Supplementary Figure 4B). The extent of infiltration and neogenesis of TLS varied between mice. Taken together, these data support the notion that apoptosis-resistant B cells escape self-tolerance mechanisms in BBimfl/fl mice and can mediate SS- and SLE-like autoimmune pathology. Our findings differ with recent studies reporting no obvious autoimmune pathology in mice with B cell-specific Bim deletion, perhaps due to analysis limited to relatively young mice (39, 48).
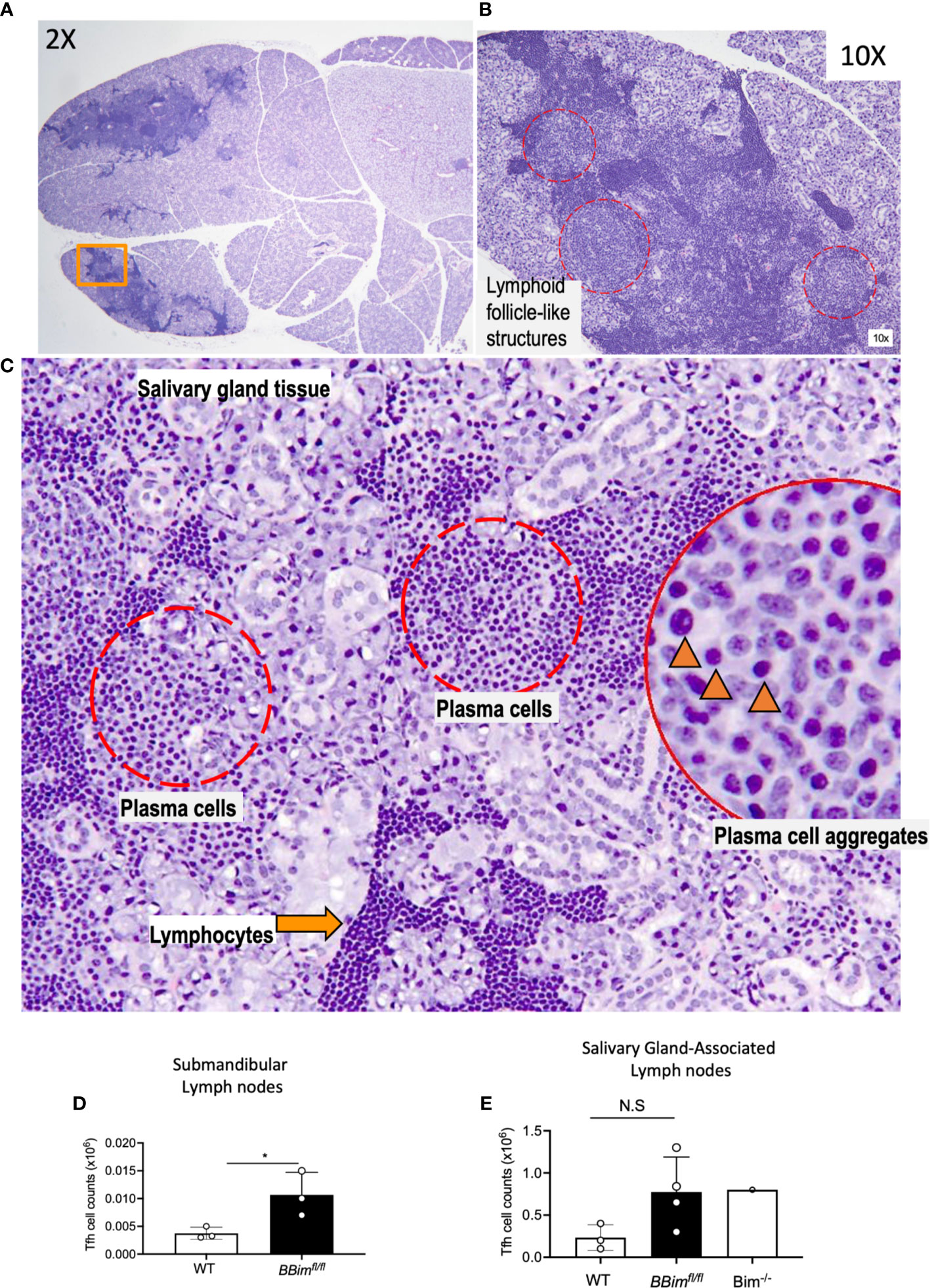
Figure 4 Salivary glands in BBimfl/fl mice display ongoing immune reaction. (A) Salivary gland from an 8.5 month old BBimfl/fl mouse showing aggregates of lymphoid infiltrates (H&E, 2x magnification). (B) Region of salivary gland indicated by an orange rectangle in (A), showing pale areas with of lymphoid follicle-like structures (red dotted circles, 10x). (C) Higher magnification (20x) of one of these areas demonstrates plasma cell aggregates (red dotted circles); plasma cells are identified by ovoid cells with abundant pale basophilic cytoplasm and eccentric nucleus (inset, black arrowheads, 40x), adjacent to lymphocytic infiltrates identified by smaller cells with scant cytoplasm and prominent dark round nucleus (orange arrow). (D) Numbers of Tfh cells in the submandibular lymph nodes and (E) salivary gland associated lymph nodes. Not significant (N.S) P > 0.05, *P ≤ 0.05, calculated by Students T- test.
SLE and Sjögen’s Signature autoAbs in BBimfl/fl Mice
To extend our findings of systemic autoimmunity in BBimfl/fl mice, serum immunoglobulins were measured by ELISA. While both young and old BBimfl/fl mice displayed increased circulating IgG2a/c (Figures 5A, B, right panels). The IgM levels were low in young mice but increased in older mice (Figures 5A, B, left panels), whereas IgG1 increased only in the young mice and IgG3 was not changed (Supplementary Figures 5A, B). Consistently, IgG2a/c antibody secreting cells (ASCs) were also increased in the older BBimfl/fl mice (Figure 5C).
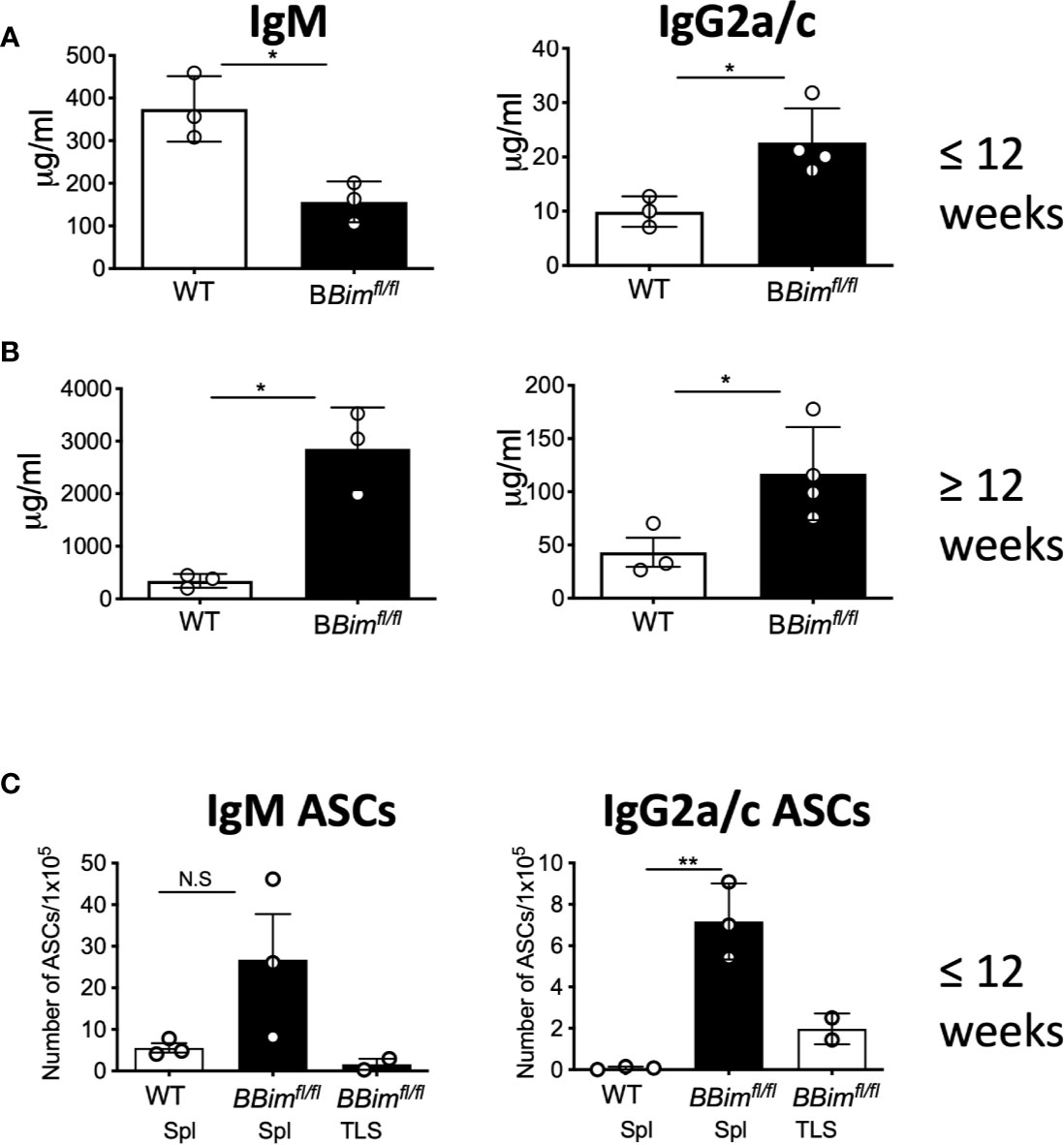
Figure 5 BBimfl/fl mice exhibit hypergammaglobulinemia and increased ASCs. Quantification of serum immunoglobulins and ASCs in WT and BBimfl/flmice. (A) IgM and IgG2a/c levels in serum from mice ≤ 12 weeks old (B) IgM and IgG2a/c levels in mice ≥ 12 weeks old mice. Numbers of (C) IgM and IgG2a/c ASCs in splenocytes in mice ≥ 12 weeks old. Data is representative of >3 independent experiments. Not significant (N.S) P > 0.05, *P ≤ 0.05, **P ≤ 0.01, calculated by Students T-test.
For a comprehensive analysis of autoAb breadth and specificity, we used an autoantigen (autoAg) array representing more than 90 of the most common autoantigens in serum from mice grouped by ages; 2-3, 6-9 months, one-year and over one-year. IgM and/or IgG autoAbs to several known autoantigens particularly those characteristic of SLE and SS were present in the serum from mice of the indicated genotypes and ages (Figures 6A, B). Each serum specimen was taken from a different mouse so each time point would be independent. Many autoAbs showed overexpression in the majority of BBimfl/fl mice, whereas this pattern was not seen with any autoantibodies at any time points for the WT or NODH2h4 mice. Among the earliest autoAbs to emerge were all of the tested SS-associated antibodies (Ro52/SS-A, Ro60/SS-A, La/SS-B, CENP-B), and a variety of lupus-associated autoantibodies targeting DNA complexes, ribonucleoproteins, and connective tissue/structural antigens, collectively including nuclear, cytoplasmic, membrane-bound, and extracellular targets. Over time, many additional lupus-associated autoantibodies emerged. All the SS and SLE autoantibodies showed persistence over time, and showed progression from IgM to also IgG expression (Figures 6A, B).
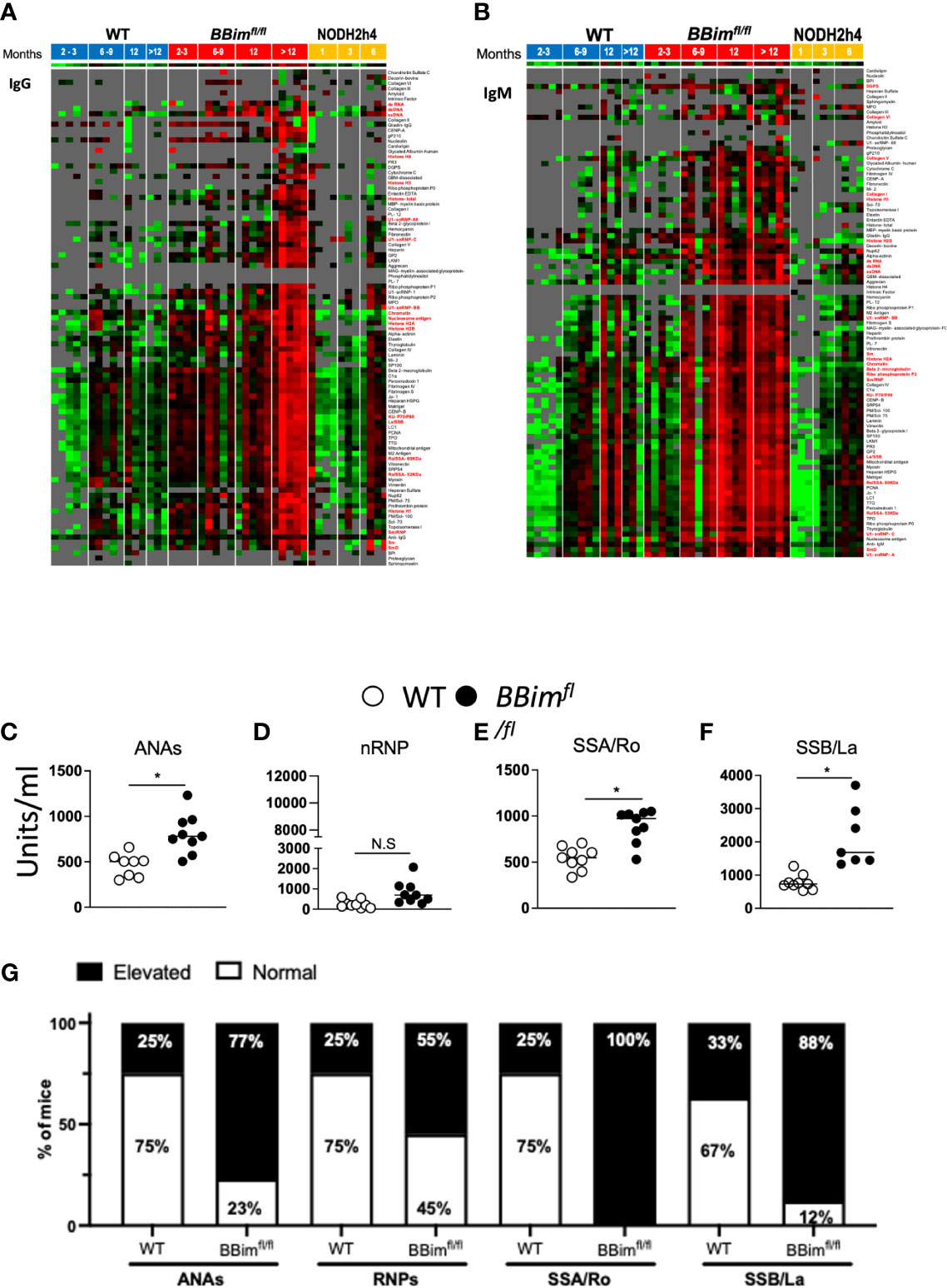
Figure 6 BBimfl/fl mice exhibit elevated titers of SLE/SS signature autoAbs. Serum from mice of indicated age groups were screened for autoAbs by hybridizing to an array containing over 90 autoantigens. Each serum specimen was taken from a different mouse so each time point would be independent. Red boxes indicate greater reactivity than the average for each antigen and green boxes indicate lower reactivity than the average for each antigen. Reactivities close to the mean are displayed in black/gray. Some of the SS and SLE-associated autoAb names are highlighted in red. (A, B) Heat maps with clustering of WT (left), BBimfl/fl (middle) and NODH2h4 (right). Supervised clustering of autoAbs was performed with normalized signal intensities for baseline IgG and IgM autoAbs. (A) IgG and (B) IgM autoAbs showing higher reactivity (red) appeared in serum of BBimfl/fl mice by 6-9 months. At 12 months, additional IgM antibodies emerged and persisted, which were again followed by the subsequent development of IgG antibodies in most instances. In each case tested (with the exception of PR3 antibodies (vasculitis-associated) which remained persistently IgM only), IgM antibodies appeared at time points either concurrent with or prior to the emergence of IgG antibodies, and both IgM and IgG persisted through all subsequent time points. The autoantibodies that emerged included all of the tested SS-associated antibodies (Ro52/SS-A, Ro60/SS-A, La/SS-B, CENP-B), and a variety of lupus-associated autoantibodies (chromatin, ssDNA, alpha-actinin, vitronectin, snRNP, Beta2-GPI, PCNA, nucleosome Ag, Sm-D, Histone H2B, peroxiredoxin 1, ribophosphoprotein P0, myosin, Heparan HSPG, Matrigel, Vitronectin, Heparin, collagen IV). Additional autoantibodies emerging in the same time frame included those targeting some autoimmune hepatitis antigens (LKM1, mitochondrial Ag, LC-1), some myositis-associated antigens (SRP54, Jo-1, Nup6.2), celiac disease –associated targets (DGPS, TTG), as well as the following antigens: C1alpha, the Crohn’s-associated antigen GP2, and the thyroiditis-associated antigen TPO. At 12 months, additional IgM antibodies emerged and persisted, followed by the development of IgG in most instances includingU1-snRNP-BB, U1-RNP-C, laminin, hemocyanin, aggrecan, fibrinogen s, autoimmune hepatitis antigens (M2 antigen, SP100), and a scleroderma/myositis-associated target (PM-Scl75). However, some of IgM autoAbs were not accompanied/followed by IgG autoAbs, exemplified by the myositis –associated antigen PL12 and neuropathy-associated myelin associated glycoprotein-Fc (MAG) and collagen V (in older mice).In addition, aged BBimfl/fl mice developed some additional IgM autoAbs to lupus-associated autoantigens in the majority of tested mice including GBM-associated, and prothrombin protein, which were associated with concurrent emergence of IgG against the same targets. Several autoAbs were upregulated only in aged mice and only as IgG, including additional lupus-associated specificities (U1-snRNP68, U1-snRNP1, Sm, total histone, histone H2A, Histone H1, Heparan Sulfate, Entactin-EDTA, collagen II, fibronectin, elastin, fibrinogen IV, ribophosphoprotein P1, ribophosphoprotein p2), a myositis-associated target (Mi-2), some scleroderma-associated specificities (CENP A, Ku P70-p80, Scl-70, Topoisomerase I, PM-Scl100), an autoimmune hepatitis-associated target (gp210), a thyroiditis-associated antigen (thyroglobulin), and the vasculitis-associated target MPO. (C–F) Serum fromBBimfl/fl and WT mice (age ≥ 24 weeks old) were assessed for autoAbs distinctive for SS and SLE by ELISA. (G) Representative bar graphs show the frequency of elevated (filled bar) and normal (open bar) autoantigens titers in WT and BBimfl/fl mice. Elevated titers were determined by calculating the positive index as stated in the ELISA kit protocol (alpha diagnostic international). Data is representative of >3 independent experiments. Not significant (N.S) P > 0.05, *P ≤ 0.05, calculated by Students T-test and non-parametric Mann-Whitney test.
The presence of autoAbs to autoantigens characteristic of SS and SLE including anti-dsDNA, -sm/RNP, -La/SSB, -Ro/SSA were confirmed by ELISA (Figures 6C–G). Some of older BBimfl/fl mice had a tendency of increased autoreactive ASCs in the LNs associated with the sSGs relative to WT controls, although only anti-SSA IgM was statistically significant (Supplementary Figure 6). Together, autoAb array, ELISA and ELISpot data demonstrate that SLE/SS characteristic autoAbs were elevated much more frequently among BBimfl/fl mice relative to WT controls.
Dysregulation of B cells and autoAb production in SLE-like autoimmune disease are influenced by both innate (TLR) and adaptive (CD40) pathways and Tfh secreted IL-21 which regulates B cell differentiation into plasma cells, memory B cells and CD11chi ABC B cells (50, 54, 55). IL-21 can regulate B cell proliferation, differentiation or apoptosis in a context-dependent manner (56, 57). For example, IL-21 can promote both proliferation and differentiation as well as apoptosis in B cells costimulated with anti-CD40, whereas this combination can rescue BCR and TLR9 induced cell death (23, 55–57). Mechanistically, IL-21 inhibits TLR4 and TLR9 induced proliferation by downregulating anti-apoptosis members of the Bcl-2 family and inducing Bim-dependent apoptosis (23, 55–57). Due to relevance of both adaptive and innate pathways in autoAb production (58, 59), we determined whether B cells in the BBimfl/fl mouse model are protected from IL-21 induced apoptosis under TLR or CD40 costimulatory conditions (Figures 7A–D and Supplementary Figures 7A–C). The BBimfl/fl B cells were more resistant to IL-21-induced apoptosis relative to WT controls in response to stimulation via TLR4 (LPS), TLR9 (CpG),TLR7 (CL097), and anti-CD40 Abs (Figures 7A–D and Supplementary Figure 7A). Although, the anti-CD40 Abs enhanced B cell viability similarly in both WT and BBimfl/fl B cells, IL-21 induced significant apoptosis in WT B cells but not BBimfl/fl B cells (Figure 7D and Supplementary Figure 7A), as previously shown (56). Similar results were obtained with costimulation of peripheral blood B cells with IL-21 plus CL097 and IL-21 plus anti-CD40 (Supplementary Figure 7B, C). The rescue of IL-21 induced apoptosis by CD40 may require use of optimal dose and presentation of CD40L (e.g., membrane bound CD40L) as was recently reported (55). These data suggest that B cells from BBimfl/fl mice are significantly protected from IL-21 induced apoptosis under both innate and adaptive costimulatory conditions and that innate, adaptive or both mechanisms may contribute to autoAb production targeting autoantigens associated SS and SLE in BBimfl/fl mice.
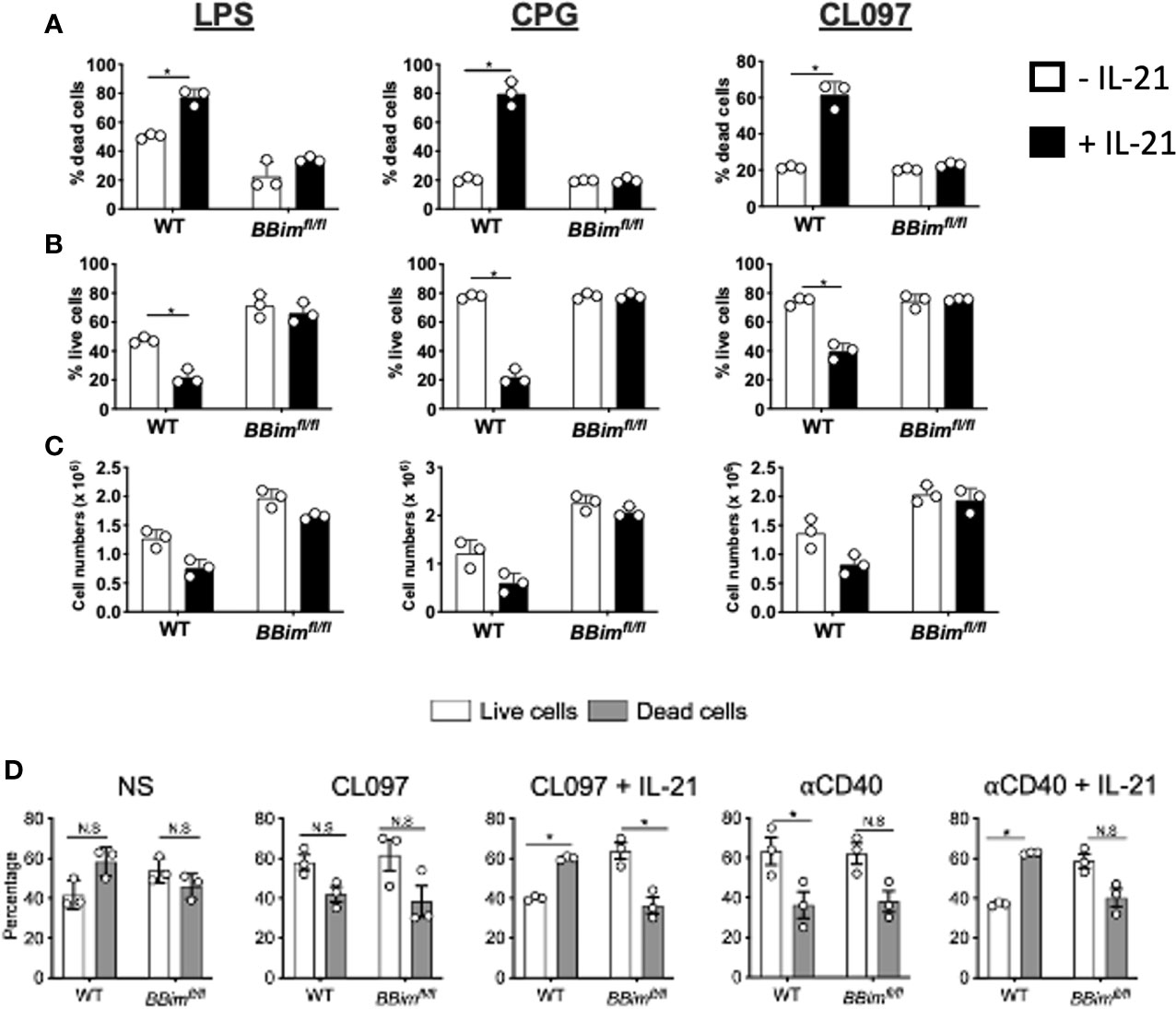
Figure 7 BBimfl/fl B cells are resistant apoptosis costimulated with IL-21 and via TLR4, TLR7, TLR9 or CD40. Purified B cells from WT and BBimfl/fl mice were incubated for 48 hours with the indicated TLR agonists or anti-CD40 in the absence or in the presence of IL-21 (25ng/ml) and then analyzed for (A) dead cells and (B) live cells distinguished using a fixable viability dye (Invitrogen). (C) Numbers of live B cells in the cultures (trypan blue-) (D) Splenocytes from BBimfl/fl and WT mice were treated with TLR7 agonist (CL097) or anti-CD40 Abs in the absence or in the presence of IL-21 (25ng/ml), stained with annexin V and 7AAD and analyzed by FCM. Bar graphs showing percentages of B cells that are live (white; 7AAD- Annexin V-) and dead (gray; 7AAD+/- Annexin V+) after culture for 48 hours. Not significant (N.S) P > 0.05, *P ≤ 0.05, calculated by Students T-test.
BBimfl/fl B Cells Can Proliferate Upon BCR and TLR Stimulation
To understand the underpinnings of autoimmunity in BBimfl/fl mice, B cells were analyzed for spontaneous and induced survival and proliferation. Comparable expression of Ki67 in B cells isolated from WT and BBimfl/fl mice indicated that Bim-deficiency does not impair B cell proliferation in vivo (Figure 8E). To assess BCR induced proliferation in vivo, we immunized mice with TNP-Ficoll, a prototypical T cell-independent type-2 antigen that induces clonal B cell expansion and production of antibody response without T cell help (Figures 8A, B). The data demonstrated that the numbers of IgM and IgG3 ASCs were greater in the BBimfl/fl than in the WT control mice. These results demonstrate that B cells in BBimfl/fl mice are capable of proliferation in vivo and can do so upon engagement of the BCR with antigen.
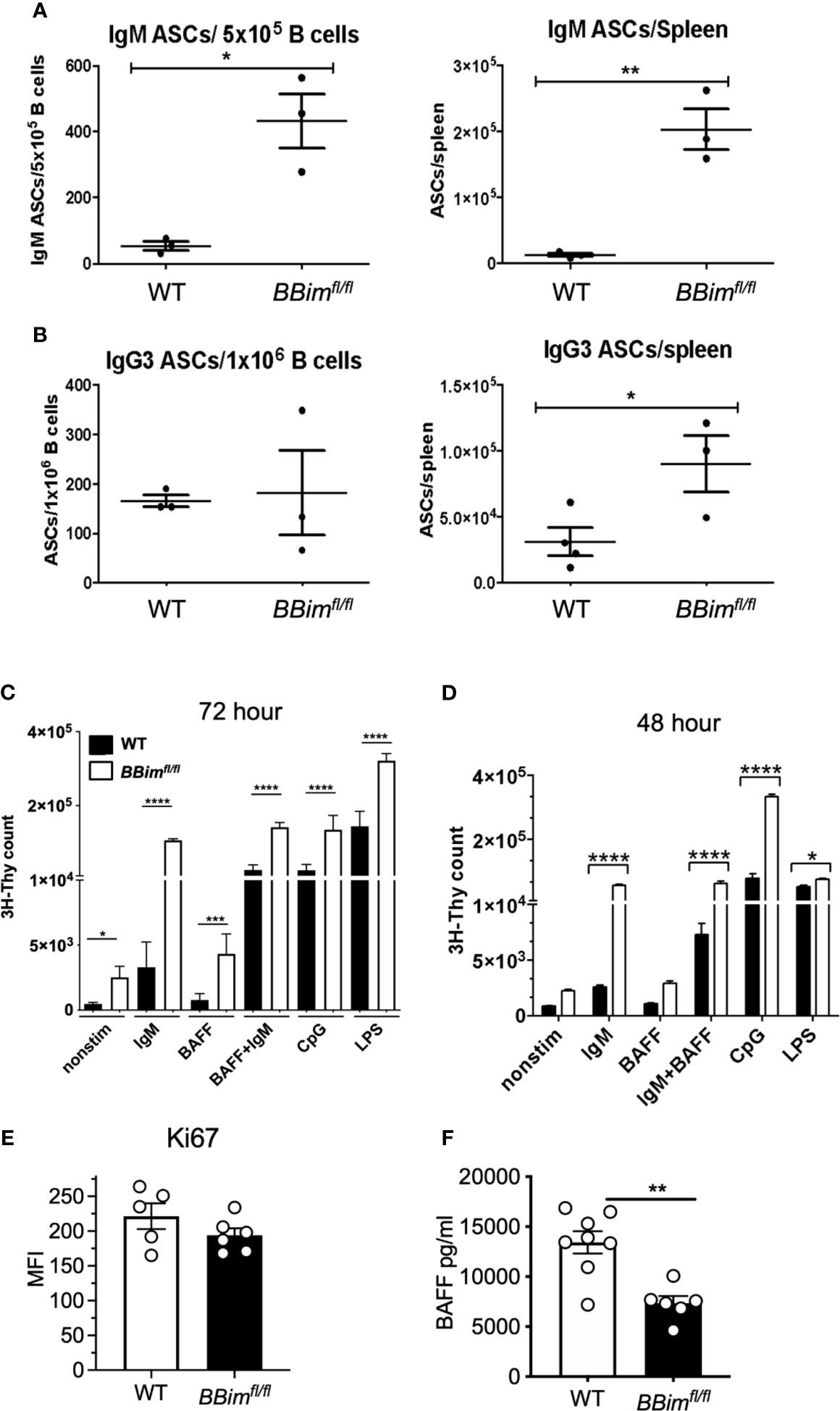
Figure 8 BCR and TLR induced BBimfl/fl B cell proliferation is enhanced by BAFF. B cells from WT and BBimfl/fl mice were analyzed for differences in activation and proliferation. WT and BBimfl/fl mice were immunized with TNP-Ficoll, before measuring B cell and antibody response by ELISpot. Quantification of (A) IgM and (B) IgG3 antibody secreting B cells from BBimfl/fl and WT mice. (C) 3H-Thymidine incorporation assay after treatment with indicated agonist for72 hours. (D) 3H-Thymidine incorporation of B cells treated with the indicated agonist for 48 hours. (E) MFI of Ki67 in B cells from BBimfl/fl and WT mice. (F) Circulating BAFF levels in serum from BBimfl/fl and WT mice. *P ≤ 0.05, **P ≤ 0.01, ****P ≤ 0.0001, calculated by Students T-test.
Because altered homeostasis of the B cell compartment is likely the driving force in the development of autoimmunity in BBimfl/fl mice, we sought to determine mechanisms of B cell expansion. Therefore, the ability of BBimfl/fl B cells to proliferate in vitro was assessed in response to stimulation via TLR4, TLR9 and the BCR with or without combination with BAFF. Purified B cells were cultured in the presence or absence of anti-IgM (Fab2), BAFF, LPS, and CpG individually or in the indicated combinations for 72 hrs or for a shorter duration (48h) and B cell proliferation measured by DNA synthesis by 3H-Thymidine incorporation (Figures 8C, D). The results showed that BBimfl/fl B cells proliferated more than the WT controls and BAFF enhanced BCR stimulatory effect, suggesting that BBimfl/fl B cells can respond to stimulation via key receptors that regulate B cell survival and proliferation. A greater effect of BAFF on BCR induced proliferation of the mutant compared to WT cells could result from in vivo BAFF deprivation as indicated by reduced circulating BAFF in BBimfl/fl mice (Figure 8F). Overall, our data shows that Bim-deficient B cells isolated from BBimfl/fl mice can proliferate in vivo and can be induced to proliferate in vitro. These results are consistent with prior studies showing that Bim-deficient B cells can proliferate normally (43), whereas they differ with another study reporting defective proliferation (60). The reasons for these differences are not clear as both prior studies used Bim-/- mice generated in the Korsmeyer laboratory (43). In fact, our data indicates that B cells isolated from BBimfl/fl mice can proliferate better under certain conditions, possibly due to the differences in the in vivo milieu in mice with B cell-specific deletion compared to the mice lacking Bim in all cells, as indicated by gene expression differences in B cells from the two strains (Supplementary Figure 8). An alternative possibility is that the proportion of BBimfl/fl B cells that undergo cell division is reduced relative to WT, but their numbers are increased due to resistance to BCR and TLR activation induced cell death in cultures (61, 62). These BBimfl/fl B cells undergo DNA synthesis and incorporate 3H-Thymidine independent of the history of cell cycle and number of cell divisions. Consistent with this interpretation, prior studies have shown that while there are substantial numbers of non-dividing cells in Bim-deleted B cell cultures, the number of B cells that had undergone cell division was not significantly different relative to the WT B cells (40). Together, these results suggest that at least a proportion of B cells from BBimfl/fl mice undergo cell division and BAFF may also contribute to B cell expansion in addition to prolonged survival.
To further dissect the underlying reasons for the differences in proliferation of B cells in BBimfl/fl mice and those isolated from Bim-/- mice previously reported, we compared gene expression profiles between the two genotypes. This analysis included genes that may display compensatory expression such as Bim-related genes encoding BH-3 only pro-apoptotic, along with the anti-apoptotic genes of the Bcl-2 family as well as TLRs and the TLR and BCR signal mediator, Btk, which has been implicated in B cell mediated autoimmunity in mice (63). RNA from purified B cells isolated from Bim-/-, Bim-/+, BBimfl/fl and WT mice was subjected to qRT-PCR. We found that Bmf mRNA was increased in BBimfl/fl B cells (3-4 fold) relative to those isolated from Bim-/-, Bim-/+ and WT mice (Supplementary Figure 8A). The expression of Blcl2 or Mcl1 was reduced in Bim-/- B cells relative to WT and B-Bimfl/fl B cells (Supplementary Figures 8C, D). These results suggest differences in gene expression profiles in B cells lacking Bim based on whether they developed in a Bim-deficient versus Bim-sufficient milieu. Increased BH-3 only pro-apoptotic Bmf, which functions in a similar manner to Bim in B cells as deletion of both genes compounds the increase in B cell phenotype (52), suggest some functional compensation of loss of Bim in the BBimfl/fl B cells. Interestingly, Btk mRNA was also modestly increased in the BBimfl/fl B cells relative to Bim-/- B cells (Supplementary Figure 8B). In contrast, TLR7 and TLR9 mRNA were largely comparable among the genotypes (Supplementary Figures 8E, F), suggesting altered TLR expression is not responsible for increased proliferation via TLR7 or TLR9 (Figures 8C, D). In this context, both Btk and BH3 only proteins are involved in regulating intracellular Ca2+ flux that is important for B cell proliferation. Thus, an increase in Bmf and Btk may contribute to restoring BBimfl/fl B cell proliferation relative to B cells from Bim-/- mice. The nature of this compensation and the inductive signals that drive Bmf and Btk expression remain to be elucidated by careful comparative studies.
Deletion of Btk in BBimfl/fl Mice Reduced Symptoms of Autoimmunity
BCR and TLR signaling control B cell selection, growth, activation and differentiation into antibody secreting cells (ASCs). There is a modest increase in Btk gene expression in BBimfl/fl mice (Supplementary Figure 8) and B cells from BBimfl/fl mice express increased levels of certain cytokines (IL-6, IL-10 and IFNa) in response to stimulation via TLR7 and TLR9 (Supplementary Figure 9), both of which utilize Btk for signaling. To test Btk function in the observed autoimmunity, Btk-/- (45) and BBimfl/fl mice were intercrossed (DKO). Deletion of Btk did not alter overall proportion of splenic B cells in the DKO mice (Figure 9A), however, B cell subpopulation distribution was altered and reduced some characteristic features of autoimmune pathology in the BBimfl/fl mice (Figure 9). Specifically the proportion of mature splenic FoB1 cells (IgMloIgDhi) was decreased, but not of mature FoB2 cells (IgMhiIgDhi, Figures 9B, C), the proportion of immature transitional (T1, IgMhiIgDlo/-) B cells was increased (Figure 9D), whereas MZ (Figure 9E) and anergic B cells (An1 or T3, Figure 9F) was decreased in the DKO mice compared to BBimfl/fl or WT mice. These outcomes are consistent with our and others previous findings that loss of Btk selectively reduces FoB1 cells and affects An1 B cell survival (64). These cellular alterations was accompanied with reduced lymphocytic tissue infiltration in the DKO mice (Supplementary Figure 2B, right panels). There was a modest increase in the proportions of IL-6+ and IFNα+ B cells in the BBimfl/fl relative to control B cells (Supplementary Figures 9A, C). However, Btk deletion had no significant effect on the circulating cytokines tested including (Supplementary Figure 10).

Figure 9 Deletion of Btk in BBimfl/fl mice reduced symptoms of autoimmunity. (A) Graph displaying splenic B cell percentages in WT, BBimfl/fl, Btk-/- and Btk-/-BBimfl/fl mice. (B–F) Representative graphs comparing percentages of the listed B cells subpopulations in WT, BBimfl/fl, Btk-/-, and Btk-/- BBimfl/fl mice. (G) Basal IgM and switched IgG2a antibodies in the serum of young (≤ 12 weeks old) and old (≥ 12 weeks old) mice. (H, I) Heat map with clustering of autoAbs in the serum from WT (left), BBimfl/fl (middle) and Btk-/-BBimfl/fl (right) mice of indicated ages (H) IgM and (I) IgG autoAbs. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, calculated by Students T-test.
We also noticed that Btk deletion decreased overall basal serum IgM and IgG2a (Figure 9G). To determine if overall reduction in the immunoglobulin levels in the DKO mice had an effect on the accumulation of autoAbs, we analyzed serum using autoantigen arrays. Results show that both IgM and IgG autoAbs were decreased in the DKO mice (Figures 9H, I). However, IgM autoAbs were reduced in both young and old (Figure 9H), IgG autoAbs were decreased only in the old DKO mice (Figure 9H). Taken together, these results indicate that Btk-dependent signaling likely contributes to aberrant B cell activation and autoimmune pathology in the BBimfl/fl mice. This interpretation is consistent with previous reports that overexpression of Btk transgene led to SLE-like autoimmune disease and expression of SYK, another B cell kinase is increased in B cells from lupus patients (63, 65, 66).
Discussion
Self-reactive B cells arise routinely during BCR diversification and are purged to avoid autoimmunity by receptor editing, anergy and apoptosis. Failure of any of these mechanisms could cause autoimmune disease, however, the precise contribution of each of these tolerance mechanisms in eliminating autoreactive B cells and preventing autoimmunity is unclear. To examine the contribution of B cell apoptosis in preventing autoAb production and autoimmune disease, we rendered B cells tolerance-compromised by B cell-specific deletion of proapoptotic Bcl-2 family member Bim, a known regulator of immune tolerance in B and T cells (34, 37). Our longitudinal analysis of BBimfl/fl mice indicates that apoptosis contributes to the elimination of autoreactive B cells significantly enough that dysregulation of apoptosis leads to the development of autoimmunity. However, the manifestation of autoimmune disease becomes apparent only with age; although autoAbs and tissue damage can be detected at a relatively young age (< 6 months), the pathogenesis is clearly evident after six months. Nonetheless, our data shows that autoAbs against several prototypical autoantigens associated with SLE and SS are present in BBimfl/fl mice at elevated level compared to NODH2h4, a widely used autoimmune mouse model (Figure 6). One possible explanation of the delayed onset of autoimmunity in BBimfl/fl mice is that the events that trigger autoreactive B cell activation such as inflammation, innate and/or T cell mediated signals take a while to accumulate and manifest in older mice. Furthermore, BBimfl/fl B cells are not entirely resistant to apoptosis as they may employ other proapoptotic pathways including Noxa, Puma and Bmf as shown in BAK and BAX double knockout mice (43). These data are consistent with prior findings that autoreactive B cell hyperactivity and autoantibody production in autoimmune diseases involves B cell intrinsic innate TLR signals and adaptive T helper cell signals (via CD40) as well as homeostatic regulation by BAFF (23, 67–69) and that depletion of B cells is beneficial for patients with several autoimmune diseases including SLE, RA, SS and MS (70).
Bim is expressed in all tissues including hematopoietic cells and it is a key physiological facilitator of apoptosis in lymphocytes purging autoreactive B and T cells (37, 71). Prior gene targeting experiments have demonstrated that systemic deletion of the gene encoding Bim leads to a systemic SLE-like autoimmune condition in a mixed 129SV x C57BL/6 genetic background. Subsequent reports indicated that SLE-like autoimmunity in Bim-deficient mice gets much milder when bred to pure C57BL/6 background. One explanation for a milder autoimmune response and pathology in Bim-deficient mice is reduced functionality of immune cells. For example, T cells in Bim-deficient mice are defective in TCR-induced activation and IL-2 production due to impaired calcium signaling (72). Additionally, T cell development is impaired at the DN to DP stages in the thymus altering T cellular composition and repertoire in the Bim-deficient mice (73). Alternatively, global loss of Bim in the whole body reduces release of self-antigens from dead and dying host cells that trigger autoimmune response. The data presented here demonstrates that B cell-specific deletion of Bim in C57BL/6 background can lead to SLE and SS-like autoimmune disease with age. We hypothesize that Bim-sufficient T cells and other immunocytes are more potent in promoting B cell activation in BBimfl/fl mice contributing to progression of autoimmunity. Our data showing expanded B cell compartment is consistent with prior studies using mb-cre or CD23-cre mediated B cell specific Bim deletion (40). The B cell expansion does not appear to be entirely due to extended survival as BBimfl/fl B cells could proliferate in vitro and during an immune response in vivo. The delayed appearance and mild autoimmune disease in the BBimfl/fl mice may explain why autoimmunity escaped detection particularly in younger mice (39, 74).
The immune cell expansion in the BBimfl/fl mice was not limited to B cells; an increase in T cells also contributed to the splenomegaly and lymphadenopathy. In this regard, autoimmune disease observed in mice lacking Bim selectively in the myeloid cell-lineage was accompanied with B cell expansion (39). However, mice with a B lineage specific deletion of Bim, described here provide an opportunity to investigate B cell subset-specific role in the development of autoimmunity. While FoB cells were markedly increased, B1 and MZ B cells which are often associated with the development of autoimmunity (75), were not increased suggesting B cell subtype-specific function of Bim in autoimmune pathogenesis. It is possible that the B1 and MZ B cell subsets are changed phenotypically in BBimfl/fl mice and contribute to autoimmunity in the BBimfl/fl mice. Future experiments will determine the effector functions and localization of B1 and MZ present in BBimfl/fl mice. In addition to mature FoB cells, we observed an increase in immature T1 and anergic T3 (An1) B cells. Like immature B cells in the bone marrow, T1 B cells are targeted for negative selection in the periphery to remove autoreactive B cells (7, 11, 18). Furthermore, T3 anergic B cell population is a rich source of autoreactive B cells (76). An increase in these B cell populations may contribute to break in B cell tolerance in BBimfl/fl mice.
SS has been long thought to be a T cell mediated disease especially in the initiation of the autoimmune process within the submandibular SG, however, there is growing evidence that B cells play multiple pathophysiological roles and may be important in the development of SS (77, 78). We found that BBimfl/fl mice displayed particularly strong SS phenotype with lymphocytic infiltration of SG, consisting of B cells, plasma cells and Tfh cells (Figures 2, 4). The BBimfl/fl mice displayed hypergammaglobulinemia and had elevated autoAbs, including anti-SSB and anti-SSA autoAbs that are characteristic of SS (Figures 5, 6). These data suggest that break in B cell self-tolerance can initiate the autoimmune process that lead to SS-like autoimmunity. However, whether anti-SSB and anti-SSA specific B cell expansion and activation occurs with the help of T cells and/or depends on the second signal via the TLRs and BAFFR remains to be determined. Thus, our data reveal a novel role for B cells in the initiation and progression of SS. It is unclear how B cells initiate autoimmune reaction. One possibility is that this role is associated with BBimfl/fl B cell function as autoantigen presenting cells, as suggested by their ability to produce inflammatory cytokines like myeloid cells to initiate adaptive immune response (39). Further comparative studies may reveal distinct and overlapping functions of apoptosis resistant B and myeloid cells in autoimmunity.
We found that deletion of Btk reduced autoimmune pathology and autoantibody accumulation in BBimfl/fl mice (Figure 9). These data suggest that Btk contributes to the hyperresponsiveness of BBimfl/fl B cells to BCR and TLR signaling and differentiation into antibody producing cells. These data are consistent with appearance of SLE-like autoimmune disease in mice overexpressing Btk (65, 79), whereas pharmacological inhibition of Btk kinase by PCI-32765 decreased the disease symptoms in several autoimmune models (80). It is possible that loss of Btk function in myeloid cells contributed to the reduction in autoimmune pathology in the Btk-/-BBimfl/fl mice. Future myeloid-specific Btk deletion experiments will address this possibility.
We have demonstrated here that loss of Bim in B cells alone is sufficient to cause SLE/SS-like autoimmunity in C57BL/6 background, notwithstanding, delayed onset. We propose a model in which B cell-specific loss of Bim promotes autoimmunity in several ways. First, by allowing the survival of autoreactive T1 B cells that can go through maturation, damage host tissues by promoting activation of the innate and T cells leading to tissue immune cell infiltration, notably of the SGs and secrete autoAbs that possibly form immune complexes leading to kidney damage. Although, the B cells in BBimfl/fl mice primarily display prolonged survival, they may accumulate sufficient activation signals by self-nucleic acids over time and may present RNA/DNA complexed protein autoantigens to activate autoreactive T cells. This T and B cell interaction likely further promotes proliferation of immune cells and the release of inflammatory cytokines which reinforce the innate and humoral immune response to self-antigens as has been previously proposed (20). In support of this we observed an increase in mature FoB and tolerance susceptible T1 and anergic T3 B cells and the ability of BBimfl/fl B cells to undergo cell division in vitro in response to key immune response regulatory receptors, anti-IgM, BAFF-R and TLR, suggesting in vivo priming. Primed B cells in this model would be more responsive to unmethylated CpGs and RNA/protein complexes found in serum or apoptotic bodies from neighboring cells undergoing normal apoptotic processes in the Bim-sufficient mileue in the BBimfl/fl mice, fueling a feedback loop whereby B cell activation regulates the immune response to react against self-antigens. In support of this idea, genetic models have shown that impairment of effector cell apoptosis participates in the breakdown of tolerance through chronic signaling caused by repeated exposures to self-antigen resulting in autoimmunity (81). With the predisposition to autoimmunity, BBimfl/fl mice can serve as a model for interrogation of genetic and environmental factors that trigger B cell mediated autoimmune disease. Amelioration of autoimmune pathology by Btk deletion in our studies is consistent with our and others’ prior studies indicating contribution of altered B cell signaling to autoimmunity and support the therapeutic use of Btk inhibitors in autoimmune diseases (41, 42, 79).
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics Statement
The animal study was reviewed and approved by Office of the Vice Provost for Research Institutional Animal Care & Use Committee Office (IACUC) University of Miami FL 33136.
Author Contributions
JAW, CB, ESC, DL-R, GC, and WNK designed research; JAW, ESC, CB, DL-R, GC, LIC, and TM, performed research; JAW, ESC, CB, DL-R, GC ELG and WNK analyzed data; NHA and LIC performed pathology and JAW, CB, ESC, GC, ELG and WNK wrote the paper. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by National Institutes of Health grant R21AI088511-01 and Intramural Funding Program, Sylvester Comprehensive Cancer Center, University of Miami to (WK).
Conflict of Interest
Authors GC, LC, and TM are employed by AstraZeneca.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank, Dr. Oliver Umland (Diabetes Research Institute, University of Miami) for his expertise and help with flow cytometry and sorting experiments and Sylvester Comprehensive Cancer Center, Flow Cytometry Shared Resource. We thank Ms. Daniela Rocca for technical assistance. We would also like to thank Dr. Richard T Libby Flaum Eye Institute, University of Rochester Medical Center, Rochester, NY for providing spleens from Bim-deficient mice.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.705307/full#supplementary-material
References
1. Pelanda R, Schwers S, Sonoda E, Torres RM, Nemazee D, Rajewsky K. Receptor Editing in a Transgenic Mouse Model: Site, Efficiency, and Role in B Cell Tolerance and Antibody Diversification. Immunity (1997) 7:765–75. doi: 10.1016/S1074-7613(00)80395-7
2. Pelanda R, Torres RM. Central B-Cell Tolerance: Where Selection Begins. Cold Spring Harb Perspect Biol (2012) 4:a007146. doi: 10.1101/cshperspect.a007146
3. Meffre E, Casellas R, Nussenzweig MC. Antibody Regulation of B Cell Development. Nat Immunol (2000) 1:379–85. doi: 10.1038/80816
4. Mayer CT, Nieke JP, Gazumyan A, Cipolla M, Wang Q, Oliveira TY, et al. An Apoptosis-Dependent Checkpoint for Autoimmunity in Memory B and Plasma Cells. Proc Natl Acad Sci USA (2020) 117:24957–63. doi: 10.1073/pnas.2015372117
5. Meyer-Bahlburg A, Rawlings DJ. B Cell Autonomous TLR Signaling and Autoimmunity. Autoimmun Rev (2008) 7:313–6. doi: 10.1016/j.autrev.2007.11.027
6. Stadanlick JE, Kaileh M, Karnell FG, Scholz JL, Miller JP, Quinn WJ 3rd, et al. Tonic B Cell Antigen Receptor Signals Supply an NF-kappaB Substrate for Prosurvival BLyS Signaling. Nat Immunol (2008) 9:1379–87. doi: 10.1038/ni.1666
7. Castro I, Wright JA, Damdinsuren B, Hoek KL, Carlesso G, Shinners NP, et al. B Cell Receptor-Mediated Sustained C-Rel Activation Facilitates Late Transitional B Cell Survival Through Control of B Cell Activating Factor Receptor and NF-κb2. J Immunol (2009) 182:7729–37. doi: 10.4049/jimmunol.0803281
8. Khan WN, Wright JA, Kleiman E, Boucher JC, Castro I, Clark ES. B-Lymphocyte Tolerance and Effector Function in Immunity and Autoimmunity. Immunol Res (2013) 57:335–53. doi: 10.1007/s12026-013-8466-z
9. Meyer-Bahlburg A, Andrews SF, Yu KO, Porcelli SA, Rawlings DJ. Characterization of a Late Transitional B Cell Population Highly Sensitive to BAFF-Mediated Homeostatic Proliferation. J Exp Med (2008) 205:155–68. doi: 10.1084/jem.20071088
10. Petro JB, Castro I, Lowe J, Khan WN. Bruton's Tyrosine Kinase Targets NF-kappaB to the Bcl-X Promoter via a Mechanism Involving Phospholipase C-Gamma2 Following B Cell Antigen Receptor Engagement. FEBS Lett (2002) 532:57–60. doi: 10.1016/S0014-5793(02)03623-2
11. Hoek KL, Antony P, Lowe J, Shinners N, Sarmah B, Wente SR, et al. Transitional B Cell Fate is Associated With Developmental Stage-Specific Regulation of Diacylglycerol and Calcium Signaling Upon B Cell Receptor Engagement. J Immunol (2006) 177:5405–13. doi: 10.4049/jimmunol.177.8.5405
12. Andrews SF, Dai X, Ryu BY, Gulick T, Ramachandran B, Rawlings DJ. Developmentally Regulated Expression of MEF2C Limits the Response to BCR Engagement in Transitional B Cells. Eur J Immunol (2012) 42:1327–36. doi: 10.1002/eji.201142226
13. Kleiman E, Salyakina D, De Heusch M, Hoek KL, Llanes JM, Castro I, et al. Distinct Transcriptomic Features Are Associated With Transitional and Mature B-Cell Populations in the Mouse Spleen. Front Immunol (2015) 6. doi: 10.3389/fimmu.2015.00030
14. Cancro MP. Signalling Crosstalk in B Cells: Managing Worth and Need. Nat Rev Immunol (2009) 9:657–61. doi: 10.1038/nri2621
15. Mackay F, Schneider P, Rennert P, Browning J. BAFF AND APRIL: A Tutorial on B Cell Survival. Annu Rev Immunol (2003) 21:231–64. doi: 10.1146/annurev.immunol.21.120601.141152
16. Mackay F, Silveira PA, Brink R. B Cells and the BAFF/APRIL Axis: Fast-Forward on Autoimmunity and Signaling. Curr Opin Immunol (2007) 19:327–36. doi: 10.1016/j.coi.2007.04.008
17. Mackay F, Woodcock SA, Lawton P, Ambrose C, Baetscher M, Schneider P, et al. Mice Transgenic for BAFF Develop Lymphocytic Disorders Along With Autoimmune Manifestations. J Exp Med (1999) 190:1697–710. doi: 10.1084/jem.190.11.1697
18. Rawlings DJ, Metzler G, Wray-Dutra M, Jackson SW. Altered B Cell Signalling in Autoimmunity. Nat Rev Immunol (2017) 17:421–36. doi: 10.1038/nri.2017.24
19. Fillatreau S, Manfroi B, Dorner T. Toll-Like Receptor Signalling in B Cells During Systemic Lupus Erythematosus. Nat Rev Rheumatol (2021) 17:98–108. doi: 10.1038/s41584-020-00544-4
20. Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-Like Receptor 7 and TLR9 Dictate Autoantibody Specificity and Have Opposing Inflammatory and Regulatory Roles in a Murine Model of Lupus. Immunity (2006) 25:417–28. doi: 10.1016/j.immuni.2006.07.013
21. Green NM, Laws A, Kiefer K, Busconi L, Kim YM, Brinkmann MM, et al. Murine B Cell Response to TLR7 Ligands Depends on an IFN-Beta Feedback Loop. J Immunol (2009) 183:1569–76. doi: 10.4049/jimmunol.0803899
22. Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG Complexes Activate B Cells by Dual Engagement of IgM and Toll-Like Receptors. Nature (2002) 416:603–7. doi: 10.1038/416603a
23. Sindhava VJ, Oropallo MA, Moody K, Naradikian M, Higdon LE, Zhou L, et al. A TLR9-Dependent Checkpoint Governs B Cell Responses to DNA-Containing Antigens. J Clin Invest (2017) 127:1651–63. doi: 10.1172/JCI89931
24. Braley-Mullen H, Sharp GC, Medling B, Tang H. Spontaneous Autoimmune Thyroiditis in NOD.H-2h4 Mice. J Autoimmun (1999) 12:157–65. doi: 10.1006/jaut.1999.0272
25. Saeed M. Lupus Pathobiology Based on Genomics. Immunogenetics (2017) 69:1–12. doi: 10.1007/s00251-016-0961-7
26. Manjarrez-Orduno N, Marasco E, Chung SA, Katz MS, Kiridly JF, Simpfendorfer KR, et al. CSK Regulatory Polymorphism Is Associated With Systemic Lupus Erythematosus and Influences B-Cell Signaling and Activation. Nat Genet (2012) 44:1227–30. doi: 10.1038/ng.2439
27. Theodorou E, Nezos A, Antypa E, Ioakeimidis D, Koutsilieris M, Tektonidou M, et al. B-Cell Activating Factor and Related Genetic Variants in Lupus Related Atherosclerosis. J Autoimmun (2018) 92:87–92. doi: 10.1016/j.jaut.2018.05.002
28. Goodnow CC, Vinuesa CG, Randall KL, Mackay F, Brink R. Control Systems and Decision Making for Antibody Production. Nat Immunol (2010) 11:681–8. doi: 10.1038/ni.1900
29. Tischner D, Woess C, Ottina E, Villunger A. Bcl-2-Regulated Cell Death Signalling in the Prevention of Autoimmunity. Cell Death Dis (2010) 1:e48. doi: 10.1038/cddis.2010.27
30. Hughes P, Bouillet P, Strasser A. Role of Bim and Other Bcl-2 Family Members in Autoimmune and Degenerative Diseases. Curr Dir Autoimmunity KARGER (2005), 74–94. doi: 10.1159/000090773
31. Hutcheson J, Scatizzi JC, Siddiqui AM, Haines GK 3rd, Wu T, Li QZ, et al. Combined Deficiency of Proapoptotic Regulators Bim and Fas Results in the Early Onset of Systemic Autoimmunity. Immunity (2008) 28:206–17. doi: 10.1016/j.immuni.2007.12.015
32. Tsubata T. B-Cell Tolerance and Autoimmunity. F1000Res (2017) 6:391. doi: 10.12688/f1000research.10583.1
33. Huang DC, Strasser A. BH3-Only Proteins-Essential Initiators of Apoptotic Cell Death. Cell (2000) 103:839–42. doi: 10.1016/S0092-8674(00)00187-2
34. Oliver PM, Vass T, Kappler J, Marrack P. Loss of the Proapoptotic Protein, Bim, Breaks B Cell Anergy. J Exp Med (2006) 203:731–41. doi: 10.1084/jem.20051407
35. Bouillet P, Metcalf D, Huang DC, Tarlinton DM, Kay TW, Kontgen F, et al. Proapoptotic Bcl-2 Relative Bim Required for Certain Apoptotic Responses, Leukocyte Homeostasis, and to Preclude Autoimmunity. Science (1999) 286:1735–8. doi: 10.1126/science.286.5445.1735
36. Hughes PD, Belz GT, Fortner KA, Budd RC, Strasser A, Bouillet P. Apoptosis Regulators Fas and Bim Cooperate in Shutdown of Chronic Immune Responses and Prevention of Autoimmunity. Immunity (2008) 28:197–205. doi: 10.1016/j.immuni.2007.12.017
37. Bouillet P, Purton JF, Godfrey DI, Zhang LC, Coultas L, Puthalakath H, et al. BH3-Only Bcl-2 Family Member Bim Is Required for Apoptosis of Autoreactive Thymocytes. Nature (2002) 415:922–6. doi: 10.1038/415922a
38. Chen M, Huang L, Wang J. Deficiency of Bim in Dendritic Cells Contributes to Overactivation of Lymphocytes and Autoimmunity. Blood (2007) 109:4360–7. doi: 10.1182/blood-2006-11-056424
39. Tsai F, Homan PJ, Agrawal H, Misharin AV, Abdala-Valencia H, Haines GK 3rd, et al. Bim Suppresses the Development of SLE by Limiting Myeloid Inflammatory Responses. J Exp Med (2017) 214:3753–73. doi: 10.1084/jem.20170479
40. Liu R, King A, Bouillet P, Tarlinton DM, Strasser A, Heierhorst J. Proapoptotic BIM Impacts B Lymphoid Homeostasis by Limiting the Survival of Mature B Cells in a Cell-Autonomous Manner. Front Immunol (2018) 9:592. doi: 10.3389/fimmu.2018.00592
41. Kendall PL, Moore DJ, Hulbert C, Hoek KL, Khan WN, Thomas JW. Reduced Diabetes in Btk-Deficient Nonobese Diabetic Mice and Restoration of Diabetes With Provision of an Anti-Insulin IgH Chain Transgene. J Immunol (2009) 183:6403–12. doi: 10.4049/jimmunol.0900367
42. Zarrin AA-O, Bao KA-O, Lupardus P, Vucic D. Kinase Inhibition in Autoimmunity and Inflammation. Nat Rev Drug Discov (2021) 20:39–63. doi: 10.1038/s41573-020-0082-8
43. Takeuchi O, Fisher J, Suh H, Harada H, Malynn BA, Korsmeyer SJ. Essential Role of BAX,BAK in B Cell Homeostasis and Prevention of Autoimmune Disease. Proc Natl Acad Sci USA (2005) 102:11272–7. doi: 10.1073/pnas.0504783102
44. Rickert RC, Roes J, Rajewsky K, lymphocyte-specific B. Cre-Mediated Mutagenesis in Mice. Nucleic Acids Res (1997) 25:1317–8. doi: 10.1093/nar/25.6.1317
45. Khan WN, Alt FW, Gerstein RM, Malynn BA, Larsson I, Rathbun G, et al. Defective B Cell Development and Function in Btk-Deficient Mice. Immunity (1995) 3:283–99. doi: 10.1016/1074-7613(95)90114-0
46. Thompson JS, Bixler SA, Qian F, Vora K, Scott ML, Cachero TG, et al. BAFF-R, a Newly Identified TNF Receptor That Specifically Interacts With BAFF. Science (2001) 293:2108–11. doi: 10.1126/science.1061965
47. Li QZ, Zhou J Fau - Wandstrat AE, Wandstrat Ae Fau - Carr-Johnson F, Carr-Johnson F Fau - Branch V, Branch V Fau - Karp DR, Karp Dr Fau - Mohan C, et al. Protein Array Autoantibody Profiles for Insights Into Systemic Lupus Erythematosus and Incomplete Lupus Syndromes. Immunity (2018) 49(4):725–39.e6. doi: 10.1016/j.immuni.2018.08.015
48. Liu L, Allman WR, Coleman AS, Takeda K, Lin TL, Akkoyunlu M. Delayed Onset of Autoreactive Antibody Production and M2-Skewed Macrophages Contribute to Improved Survival of TACI Deficient MRL-Fas/Lpr Mouse. Sci Rep (2018) 8:1308. doi: 10.1038/s41598-018-19827-8
49. Egle A, Harris AW, Bouillet P, Cory S. Bim is a Suppressor of Myc-Induced Mouse B Cell Leukemia. Proc Natl Acad Sci USA (2004) 101:6164–9. doi: 10.1073/pnas.0401471101
50. Hao Y, O'Neill P, Naradikian MS, Scholz JL, Cancro MP. A B-Cell Subset Uniquely Responsive to Innate Stimuli Accumulates in Aged Mice. Blood (2011) 118:1294–304. doi: 10.1182/blood-2011-01-330530
51. Jenks SA, Cashman KS, Zumaquero E, Marigorta UM, Patel AV, Wang X, et al. Distinct Effector B Cells Induced by Unregulated Toll-Like Receptor 7 Contribute to Pathogenic Responses in Systemic Lupus Erythematosus. Front Immunol (2021). doi: 10.3389/fimmu.2021.658048
52. Hübner A, Cavanagh-Kyros J, Rincon M, Flavell RA, Davis RJ. Functional Cooperation of the Proapoptotic Bcl2 Family Proteins Bmf and Bim In Vivo. Mol Cell Biol (2009) 30:98–105. doi: 10.1128/MCB.01155-09
53. Corsiero E, Delvecchio FR, Bombardieri M, Pitzalis C. B Cells in the Formation of Tertiary Lymphoid Organs in Autoimmunity, Transplantation and Tumorigenesis. Curr Opin Immunol (2019) 57:46-52. doi: 10.1016/j.coi.2019.01.004
54. Wei C, Anolik J, Cappione A, Zheng B, Pugh-Bernard A, Brooks J, et al. A New Population of Cells Lacking Expression of CD27 Represents a Notable Component of the B Cell Memory Compartment in Systemic Lupus Erythematosus. J Immunol (2007) 178:6624–33. doi: 10.4049/jimmunol.178.10.6624
55. Wang J, Li T, Zan H, Rivera CE, Yan H, Xu Z. LUBAC Suppresses IL-21-Induced Apoptosis in CD40-Activated Murine B Cells and Promotes Germinal Center B Cell Survival and the T-Dependent Antibody Response. Front Immunol (2021) 12:658048. doi: 10.3389/fimmu.2021.658048
56. Jin H, Carrio R, Yu A, Malek TR. Distinct Activation Signals Determine Whether IL-21 Induces B Cell Costimulation, Growth Arrest, or Bim-Dependent Apoptosis. J Immunol (2004) 173:657–65. doi: 10.4049/jimmunol.173.1.657
57. Mehta DS, Wurster AL, Whitters MJ, Young DA, Collins M, Grusby MJ. IL-21 Induces the Apoptosis of Resting and Activated Primary B Cells. J Immunol (2003) 170:4111–8. doi: 10.4049/jimmunol.170.8.4111
58. Johnson JL, Scholz JL, Marshak-Rothstein A, Cancro MP. Molecular Pattern Recognition in Peripheral B Cell Tolerance: Lessons From Age-Associated B Cells. Curr Opin Immunol (2019) 61:33–8. doi: 10.1016/j.coi.2019.07.008
59. Naradikian MS, Myles A, Beiting DP, Roberts KJ, Dawson L, Herati RS, et al. Cutting Edge: IL-4, IL-21, and IFN-Gamma Interact To Govern T-Bet and CD11c Expression in TLR-Activated B Cells. J Immunol (2016) 197:1023–8. doi: 10.4049/jimmunol.1600522
60. Craxton A, Draves KE, Clark EA. Bim Regulates BCR-Induced Entry of B Cells Into the Cell Cycle. Eur J Immunol (2007) 37:2715–22. doi: 10.1002/eji.200737327
61. Banerjee A, Grumont R, Gugasyan R, White C, Strasser A, Gerondakis S. NF-kappaB1 and c-Rel cooperate to promote the survival of TLR4-activated B cells by neutralizing Bim via distinct mechanisms. Blood (2008) 112(13):5063–73. doi: 10.1182/blood-2007-10-120832
62. Gerondakis S, Grumont Rj Fau - Banerjee A, Banerjee A. Regulating B-Cell Activation and Survival in Response to TLR Signals. Blood (2012) 119(16):3744–56. doi: 10.1182/blood-2011-12-397919
63. Rip J, de Bruijn MJW, Appelman MK, Pal Singh S, Hendriks RW, Corneth OBJ. Toll-Like Receptor Signaling Drives Btk-Mediated Autoimmune Disease. Front Immunol (2019) 10:95. doi: 10.3389/fimmu.2019.00095
64. Nyhoff LE, Clark ES, Barron BL, Bonami RH, Khan WN, Kendall PL. Bruton's Tyrosine Kinase Is Not Essential for B Cell Survival Beyond Early Developmental Stages. J Immunol (2018) 200:2352–61. doi: 10.4049/jimmunol.1701489
65. Kil LP, de Bruijn MJ, van Nimwegen M, Corneth OB, van Hamburg JP, Dingjan GM, et al. Btk Levels Set the Threshold for B-Cell Activation and Negative Selection of Autoreactive B Cells in Mice. Blood (2012) 119:3744–56. doi: 10.1182/blood-2011-12-397919
66. Fleischer SJ, Giesecke C, Mei HE, Lipsky PE, Daridon C, Dorner T. Increased Frequency of a Unique Spleen Tyrosine Kinase Bright Memory B Cell Population in Systemic Lupus Erythematosus. Arthritis Rheumatol (2014) 66:3424–35. doi: 10.1002/art.38854
67. Ettinger R, Kuchen S, Lipsky PE. The Role of IL-21 in Regulating B-Cell Function in Health and Disease. Immunol Rev (2008) 223:60–86. doi: 10.1111/j.1600-065X.2008.00631.x
68. Green NM, Marshak-Rothstein A. Toll-Like Receptor Driven B Cell Activation in the Induction of Systemic Autoimmunity. Semin Immunol (2011) 23:106–12. doi: 10.1016/j.smim.2011.01.016
69. Groom JR, Fletcher CA, Walters SN, Grey ST, Watt SV, Sweet MJ, et al. BAFF and MyD88 Signals Promote a Lupuslike Disease Independent of T Cells. J Exp Med (2007) 204:1959–71. doi: 10.1084/jem.20062567
70. Lee DSW, Rojas OL, Gommerman JL. B Cell Depletion Therapies in Autoimmune Disease: Advances and Mechanistic Insights. Nat Rev Drug Discov (2021) 20:179–99. doi: 10.1038/s41573-020-00092-2
71. Enders A, Bouillet P, Puthalakath H, Xu Y, Tarlinton DM, Strasser A. Loss of the Pro-Apoptotic BH3-Only Bcl-2 Family Member Bim Inhibits BCR Stimulation-Induced Apoptosis and Deletion of Autoreactive B Cells. J Exp Med (2003) 198:1119–26. doi: 10.1084/jem.20030411
72. Ludwinski MW, Sun J, Hilliard B, Gong S, Xue F, Carmody RJ, et al. Critical Roles of Bim in T Cell Activation and T Cell-Mediated Autoimmune Inflammation in Mice. J Clin Invest (2009) 119:1706–13. doi: 10.1172/JCI37619
73. Li YF, Xu S, Huang Y, Ou X, Lam KP. Tyrosine Kinase C-Abl Regulates the Survival of Plasma Cells. Sci Rep (2017) 7:40133. doi: 10.1038/srep40133
74. Ludwig LM, Roach LE, Katz SG, LaBelle JL. Loss of BIM in T Cells Results in BCL-2 Family BH3-Member Compensation But Incomplete Cell Death Sensitivity Normalization. Apoptosis (2020) 25:247–60. doi: 10.1007/s10495-020-01593-6
75. Amezcua Vesely MC, Schwartz M, Bermejo DA, Montes CL, Cautivo KM, Kalergis AM, et al. FcgammaRIIb and BAFF Differentially Regulate Peritoneal B1 Cell Survival. J Immunol (2012) 188:4792–800. doi: 10.4049/jimmunol.1102070
76. Nojima T, Reynolds AE, Kitamura D, Kelsoe G, Kuraoka M. Tracing Self-Reactive B Cells in Normal Mice. J Immunol (2020) 205(1):90–101. doi: 10.4049/jimmunol.1901015
77. Nguyen CQ, Hu MH, Li Y, Stewart C, Peck AB. Salivary Gland Tissue Expression of Interleukin-23 and Interleukin-17 in Sjogren's Syndrome: Findings in Humans and Mice. Arthritis Rheum (2008) 58:734–43. doi: 10.1002/art.23214
78. Cornec D, Devauchelle-Pensec V, Tobon GJ, Pers JO, Jousse-Joulin S, Saraux A. B Cells in Sjogren's Syndrome: From Pathophysiology to Diagnosis and Treatment. J Autoimmun (2012) 39:161–7. doi: 10.1016/j.jaut.2012.05.014
79. Hutcheson J, Vanarsa K, Bashmakov A, Grewal S, Sajitharan D, Chang BY, et al. Modulating Proximal Cell Signaling by Targeting Btk Ameliorates Humoral Autoimmunity and End-Organ Disease in Murine Lupus. Arthritis Res Ther (2012) 14:R243. doi: 10.1186/ar4086
80. Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, et al. The Bruton Tyrosine Kinase Inhibitor PCI-32765 Blocks B-Cell Activation and Is Efficacious in Models of Autoimmune Disease and B-Cell Malignancy. Proc Natl Acad Sci USA (2010) 107:13075–80. doi: 10.1073/pnas.1004594107
Keywords: autoimmunity, apoptosis, Bcl-2-like 11 (Bim), B cell tolerance, systemic lupus - erythematosus, autoantibodies, Sjogren’s syndrome, Bruton’s tyrosine kinase (Bkt)
Citation: Wright JA, Bazile C, Clark ES, Carlesso G, Boucher J, Kleiman E, Mahmoud T, Cheng LI, López-Rodríguez DM, Satterthwaite AB, Altman NH, Greidinger EL and Khan WN (2021) Impaired B Cell Apoptosis Results in Autoimmunity That Is Alleviated by Ablation of Btk. Front. Immunol. 12:705307. doi: 10.3389/fimmu.2021.705307
Received: 05 May 2021; Accepted: 30 July 2021;
Published: 26 August 2021.
Edited by:
Jayanta Chaudhuri, Memorial Sloan Kettering Cancer Center, United StatesReviewed by:
Keishi Fujio, The University of Tokyo, JapanDavid A. Fruman, University of California, Irvine, United States
Copyright © 2021 Wright, Bazile, Clark, Carlesso, Boucher, Kleiman, Mahmoud, Cheng, López-Rodríguez, Satterthwaite, Altman, Greidinger and Khan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wasif N. Khan, d25raGFuQG1pYW1pLmVkdQ==
†These authors share first authorship