 Silvere D. Zaongo
Silvere D. Zaongo Yanqiu Liu
Yanqiu Liu Vijay Harypursat
Vijay Harypursat Fangzhou Song
Fangzhou Song Huan Xia
Huan Xia Ping Ma
Ping Ma Yaokai Chen
Yaokai Chen- 1Division of Infectious Diseases, Chongqing Public Health Medical Center, Chongqing, China
- 2Basic Medicine College, Chongqing Medical University, Chongqing, China
- 3Department of Infectious Diseases, Tianjin Second People’s Hospital, Tianjin, China
- 4School of Medicine, Nankai University, Tianjin, China
Antiretroviral therapy (ART), which is a life-long therapeutic option, remains the only currently effective clinical method to treat HIV-1 infection. However, ART may be toxic to vital organs including the liver, brain, heart, and kidneys, and may result in systemic complications. In this context, to consider HIV-1 restriction factors from the innate immune system to explore novel HIV therapeutics is likely to be a promising investigative strategy. In light of this, P-selectin glycoprotein ligand 1 (PSGL-1) has recently become the object of close scrutiny as a recognized cell adhesion molecule, and has become a major focus of academic study, as researchers believe that PSGL-1 may represent a novel area of interest in the research inquiry into the field of immune checkpoint inhibition. In this article, we review PSGL-1’s structure and functions during infection and/or inflammation. We also outline a comprehensive review of its role and potential therapeutic utility during HIV-1 infection as published in contemporary academic literature.
Introduction
Despite several decades of dedicated research, and large financial and human resource investments, no single effective treatment has been developed to eliminate HIV infection (1). To date, the best approach to treat HIV infection remains the administration of antiretroviral therapy (ART), which remains a life-long commitment to the diligent daily ingestion of multiple antiviral drugs (2). However, in the long term, ART can be toxic to vital organs such as the liver, brain, heart, and the kidney (3–10), resulting in systemic physical complications. As such, several alternative therapeutic strategies are being investigated by researchers. It is well recognized that HIV-1 infection triggers pathogen-recognition receptors (PRRs) in infected cells, leading to recognition of viral pathogen-associated molecular patterns. PRRs subsequently initiate a signaling cascade working at restricting the replication and spread of the virus, using innate intracellular antiviral defenses. Such an intrinsic response from the infected cell is, thereafter, disseminated via the action of cytokines and chemokines, which activate and attract innate immune cells to the site of infection. These effector cells can thereby assist in the control of viremia, and regulate immune responses to HIV-1 (11). Unfortunately, in spite of having a well-developed innate immune system composed of restriction factors capable of interrupting the viral replication cycle at particular points, HIV-1 does successfully infect CD4 T-cells and macrophages (11–15). Nevertheless, the restriction factors (which are host proteins) still warrant thorough investigation for their potential therapeutic application. These restriction factors possess a few characteristics in common (16), which can be summarized as: (i) they are usually induced by interferons (IFNs), (ii) they are usually developed from lentiviruses, mounting high selection pressure, leading to a rapid evolution of their coding genes, and resulting in significant production of their amino acid sequences (17), and (iii) in HIV-1 infection, the virus can restrict their actions in order to survive in host cells. Identification of such restriction factors from innate immunity that are involved in HIV-1 modulation and/or control represents a promising approach for the discovery of novel therapeutics to treat HIV infection.
Tripartite motif-containing protein 5-α (TRIM5-α) (18), APOBEC3 (19), tetherin (20), SAM domain and HD domain-containing protein 1 (SAMHD1) (21, 22), and serine incorporator 3 (SERINC3)/SERINC5 (23, 24) are examples of a few restriction factors identified thus far. Recently, P-selectin glycoprotein ligand 1 (PSGL-1) has been identified as another restriction factor, and is generating considerable interest. Indeed, Ying Liu and colleagues revealed, in a proteomic profiling study comparing infected and normal CD4 T-cells, that PSGL-1 is downregulated during HIV-1 infection (25). PSGL-1 sparked the interest of these scientists as it (i) showed a very high likelihood of positive selection to HIV-1, (ii) was specifically expressed in HIV-1 target cells such as lymphocytes and myeloid cells, (iii) was proven to be downregulated in the membrane fraction of the HIV-1-infected CEM-T4 cell line (26), and (iv) was proven to be associated with HIV-1 Gag at HIV-1 assembly sites (27). Following further investigation, they demonstrated that interferon-γ anti-HIV activity is mediated by PSGL-1, and is, on the other hand, antagonized by the HIV-1 viral particle unit (Vpu) (25). Of late, studies of this molecule have shown that PSGL-1 has a novel role in the area of immune checkpoint inhibition (28). PSGL-1 was, for a long time, studied as an adhesion molecule (29, 30), and is now progressively recognized as an important regulator of many facets of immune responses by myeloid cells and T-cells in settings of homeostasis and inflammation (31, 32). Furthermore, the role and potential therapeutic implications of PSGL-1 during HIV-1 infection has become an area of intense focus in academic research. A more thorough understanding of PSGL-1 and its potential functions and functional roles, as described in particular contexts, is warranted.
Herein, we review critical information related to PSGL-1, such as its structure and roles during infections and/or inflammation. We conclude this article with a review of the most recent knowledge related to the role of PSGL-1 during HIV-1 infection.
Definition and Structure
P-selectin glycoprotein ligand 1 (PSGL-1) is coded by a human gene called Selectin P ligand, also known as SELPLG. The protein is also referred to CD162. PSGL-1 is a disulfide-bonded homodimeric type I transmembrane mucin-like glycoprotein, of 120 kilodaltons (KDa) in molecular weight (33–36) that is expressed on most peripheral T-cells and on some B-cells, neutrophils, monocytes, and platelets (37). Upon further post translational modifications (described below), PSGL-1 acquires the capacity to bind P-selectin, which is one of the selectin family of cell adhesion molecules, including E-selectin (endothelial) and L-selectin (leukocytes). The N-terminus of the extracellular domain is necessary to bind P- (with highest affinity), E-, and L-selectin (38–40) through varying levels of affinity (41–45). P-selectin is expressed on platelets and activated endothelial cells (46).
PSGL-1 requires sialyl Lewis x (sLex)-capped O-glycans to bind selectins. This glycosylation is dependent upon glycosyltransferases (including fucosyltransferase-VII, FucT-VII); sialyl 3-galactosyltansferases, ST3Gal-IV and ST3Gal-VI; and β1,4-galactosyltransferase, β1,4GalT-I) (47, 48) sequentially adding carbohydrate moieties to form sLex, which is the dominant sialylated fucosylated O-linked glycan. sLex thereafter mediates selectin binding, with specific enzyme requirements for each of the selectins (47, 49) (Table 1). The enzymes required for PSGL-1 and selectin binding are constitutively expressed by myeloid cells (61), T-cell progenitors (62), and hematopoietic stem cells (HSCs) (38, 63). Although PSGL-1 is highly expressed on resting T cells, selectin binding ability is only conferred during effector T-cell proliferation and differentiation (64, 65). Sulfation of tyrosine residues at the N-terminus of PSGL-1 is necessary for the binding of P- and L-selectin. PSGL-1’s cytoplasmic domain interacts with ezrin/radixin/moesin (ERM) proteins, attaching PSGL-1 to the actin cytoskeleton. PSGL-1 and ERM-family proteins are found in the uropods of polarized leukocytes. The activation of the Src-family kinases Fgr, Hck, and Lyn occurs when PSGL-1 is engaged by P-selectin or E-selectin (66, 67). Interestingly, PSGL-1 must be localized to leukocyte microvilli in order to bind its ligands optimally (68). Structurally, the cytoplasmic and trans-membrane domains of PSGL-1 are both strongly conserved (49), suggesting important functions. PSGL-1’s cytoplasmic domain, which consists of 72 and 69 amino acids for humans and mice, respectively (69), is critical for interactions with scaffolding and signaling molecules (70). Despite variations in PSGL-1’s extracellular domain between organisms, its overall structure and function tend to be regulated in a similar way (71). Regarding PSGL-1 as a therapeutic target, it is critical to understand the various functions that PSGL-1 plays depending on the specific infective and/or inflammatory context involved.

Table 1 Selectins expression and the enzymes required for PSGL-1 binding.
Recognized PSGL-1 Functions During Infections and/or Inflammation
Past in vitro experiments have revealed that PSGL-1 regulates neutrophil rolling and tethering on activated endothelium through P-selectin binding. Later studies using Selplg-/- murine cells (72), confirmed the role of PSGL-1 in modulating the motility of macrophages/monocytes, plasma B-cells, dendritic cells, and T-cells through selectin engagement (47, 49). Thus, PSGL-1 is able to (i) facilitate neutrophil migration into inflamed peritoneum (73), (ii) recruit CD8+ T-cells into the inflamed colon (74), or (iii) recruit CD4+ T-cells into responding lymph nodes (75), intestinal lamina propria (76), and the inflamed retina (77). In short, PSGL-1 plays a critical role in the recruitment of immune cells into sites of inflammation (Figure 1). However, PSGL-1 mutation with the absence of an intracellular tail in cells may result, for instance, in significantly reduced rolling efficiency (70). Moreover, PSGL-1 is involved in neutrophil and monocyte localization. For instance, PSGL-1 mediates the adhesion of these cells to P-selectin, which is expressed on platelets that become attached to inflamed endothelium. Such interactions can enable targeted extravasation into tissues by eliciting chemokine secretion by platelets, as well as inflammatory mediator production by neutrophils (78). Also, neutrophils, via PSGL-1 expressed on their cell membrane, can bind to platelets via P-selectin to achieve extravasation (79). PSGL-1 on other immune cells can interact with P-selectin expressed on endothelial cells to promote extravasation, especially if the inflammation results from the activation by thrombin or histamine (46). PSGL-1 has been implicated in the establishment of cellular complexes that function in pathogen clearance has been suggested, as platelets also utilize PSGL-1 to adhere to vasculature by selectin binding (80). Illustration of this is the host response to Salmonella typhimurium where platelet PSGL-1/P-selectin interaction leads to neutrophil recruitment (81). With regards to the role of PSGL-1 in immune responses, microbes have the capacity to develop mechanisms to neutralize the effects of PSGL-1 by targeting selectin-binding by PSGL-1. Of note, it has been demonstrated that the sialic acid-binding toxin, staphylococcal superantigen-like 5 (SSL5) (secreted by Staphylococcus aureus), has the ability to bind to sLex expressed on PSGL-1 on neutrophils. Consequently, neutrophils can neither be activated nor recruited via PSGL-1/P-selectin binding (82, 83). Due to several endogenous mechanisms, PSGL-1’s role in immune cell recruitment can either be enhanced or inhibited. For example, interactions between PSGL-1 and (i) Sialic acid-binding immunoglobulin-type lectin 5 (Siglec 5) (84) or ADAM8 (proteolytic enzyme) (85) on one hand, and (ii) ADAM28 (86) on the other, were shown to inhibit/block leukocyte rolling on P- and E- selectin, but enhanced binding to P-selectin, respectively.
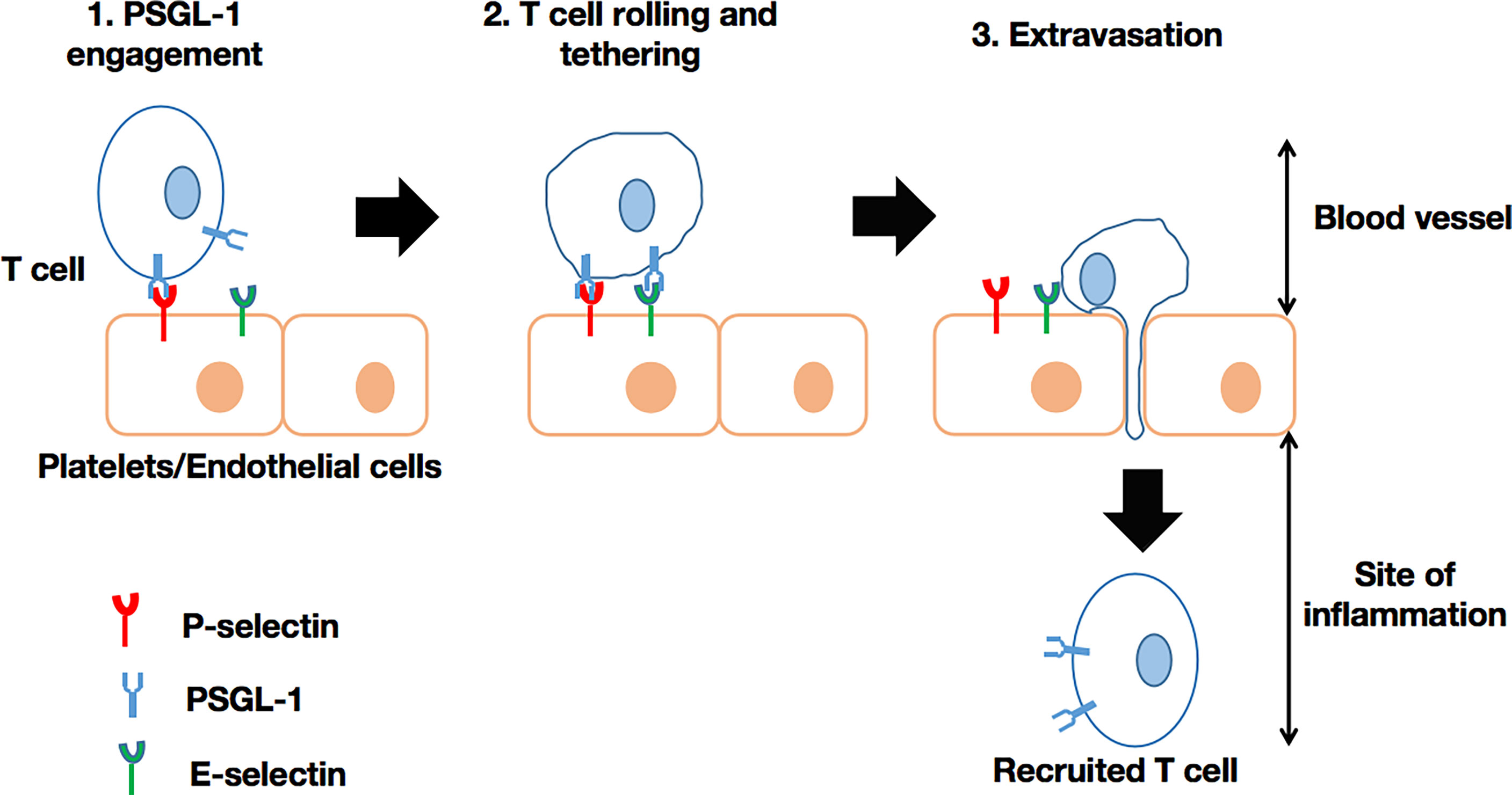
Figure 1 T-cell recruitment via PSGL-1/selectin binding. In the blood vessel, the circulating T-cell is engaged by selectin on platelets (P-selectin) or endothelial cells (P-selectin or E-selectin). Subsequently, the T-cell is able to initiate rolling and the extravasation cascade, leading to its recruitment into the site of inflammation, where it can intervene as an effector cell. This process can also take place in the recruitment of other leukocytes.
PSGL-1, in addition to its role in cell migration, was demonstrated to be an inhibitor of human HSCs proliferation subsequent to ligation by P-selectin or anti-PSGL-1 antibody in vitro (87). On dendritic cells (DCs), PSGL-1 engagement by P-selectin or anti-PSGL-1 antibody was associated with immune inhibition, and an enhanced capacity to induce regulatory T-cells in vitro. Such engagement stimulates the production of c-Fos, indoleamine 2,3 dioxygenase (IDO), interleukin 10 (IL-10), and transforming growth factor (TGF)-β, all recognized for their roles as regulator of cell function (88). PSGL-1 ligation can also stimulate cytokine release by macrophages, DCs and T-cells leading to beneficial responses to pathogens (88–92). Further functions have been attributed to PSGL-1 in microbial infections. Notably, it assists in the control of Streptococcus pneumoniae by supporting neutrophil phagocytosis through binding to the capsular polysaccharide and cell wall autolysin, LytA (93). On the other hand, Enterovirus 71 has been shown to utilize PSGL-1 selectin-binding domain as a primary receptor to infect leukocytes, which aids viral replication (94). Furthermore, PSGL-1 possesses broad-spectrum antiviral activity (against e.g., HIV-1, murine leukemia virus, and influenza A virus) (95), by blocking viral infections through steric hindrance of particle attachment to target cells. A recent study shows that the expression of PSGL-1 in virus-producing cells impairs the incorporation of the SARS-CoV and SARS-CoV-2 spike (S) glycoproteins into pseudovirions, and blocks virus attachment and subsequent infection of target cells (96). Thus, via PSGL-1, bacteria or viruses may promote or inhibit the immune response. Table 2 summarizes the roles of PSGL-1 as reported during some bacterial and viral infections.

Table 2 Summary of the roles of PSGL-1 in bacterial and viral infections.
Finally, a recent publication extensively reviewed the role of PSGL-1 in the development of neoplastic disease. The authors reported that blocking the CD4+ T-cell’s PSGL-1 pathway could possibly be utilized as a novel cancer therapeutic approach to treat tumors (98). Indeed, PSGL-1 activation in the tumor microenvironment can promote CD4+ T-cell exhaustion pathways, which promotes tumor development. Therefore, ligands including chemokines CCL19 and CCL21 (62, 99, 100), Siglec-5 (84, 101), versican, and V-domain immunoglobulin suppressor of T cell activation (VISTA) (102) could possibly bind PSGL-1 on T-cells, and could contribute to inhibitory signaling pathways that dampen T-cell receptor (TCR) signals to induce T-cell exhaustion in tumors. However, there is currently no evidence that blocking PSGL-1 on CD4 T-cells alone would be sufficient to promote tumor growth control without also relieving exhaustion of CD8 T-cells. Further investigation will be necessary to understand the biology of PSGL-1 on T cells as well as other immune cells in tumors.
PSGL-1 and HIV-1 Infection
Within these last two years, studies on PSGL-1 and its putative roles during HIV-1 infection have increased dramatically. Investigations by HIV researchers into PSGL-1 have been stimulated after a study by Liu et.al., was published (25), showing that PSGL-1, which can be induced by interferon-γ in activated CD4+ T-cells, inhibits HIV-1 reverse transcription, and effectively blocks viral infectivity. The same research team then demonstrated that HIV-1 Vpu downregulates PSGL-1 expression in infected CD4+ T-cells after binding to it and inducing its ubiquitination and degradation through the protein ubiquitin ligase SCFβ-TrCP2. To further illustrate the downregulation of PSGL-1 during HIV-1 infection, it is worth considering earlier results of Liang and colleagues (103). They compared PSGL-1 expression on monocytes from healthy individuals (n=26) and treatment-naive participants with primary HIV-1 infection (PHI, n=38) or chronic HIV-1 infection (CHI, n=20). They found that PSGL-1 expression was significantly decreased in the PHI group (P<0.001). PSGL-1 expression on monocytes was partially restored in the CHI group, but did not return to the levels of healthy controls. These results suggest a negative association between PSGL-1 expression and HIV-1 viral load profiles. Indeed, the viral load profile (log10 copies/mL) was almost 4 times higher in the PHI cohort (4.21 ± 0.95) compared to the CHI cohort (1.30 ± 1.68). However, broader, and more in-depth investigations are required to be certain of this possible correlation and its likely determinants, such as age, infection duration, sex, and HIV-1 clades. In contrast, Connor et al., have found that compared with healthy patients, monocytes derived from HIV-infected individuals exhibit greater levels of PSGL-1. In their study, all HIV-infected participants (HIV positive for a longer period of time than in Liang et al.’s study) were under ART (longer than in Liang et al.’s study) suggesting the positive contribution of treatment to PSGL-1 complete restoration and expression (average mean ~1 for HIV- versus ~1.5 for HIV+, p=0.0204). They also observed that c-Myc regulates PSGL-1 expression in monocytes during HIV-1 infection, and that glutamate and sCD40L can promote PSGL-1 expression (104). During their study, Liang et.al., did not analyze the effects of HIV-1 or of its associated particles (Vpu, Vpr and Nef) to explain PSGL-1 disparities observed in HIV-1 positive participants. Five years later, Liu et.al., demonstrated for the first time that overexpression of Vpu but not Nef, Vif, Vpr, nor p55 Gag, significantly decreased PSGL-1 levels in 293T cells, Jurkat cells, and CD4+ T-cells from two donors (25). Although their findings were novel, they looked at intracellular PSGL-1 levels through mRNA profiling only. Just months after the findings of Liu et.al., Fu et.al., provided further details. After confirming that only Vpu negatively regulates intracellular PSGL-1, they also discovered that Vpu and Nef negatively regulates extracellular PSGL-1 levels (95). To date, it is well known that HIV-1 can efficiently suppress PSGL-1’s ability to block viral infectivity by repressing its expression. Fu’s team offered additional information on this process, which was first stated by Liu et al. They confirmed that PSGL-1 in virus-producer cells was not blocking the release of novel viral particles, but did inactivate virion infectivity. In other words, PSGL-1 in the virus-producer cells reduces levels of HIV-1 Env on virions. Consequently, PSGL-1 can inhibit virion attachment to cells in an envelope glycoprotein-independent manner. In fact, without HIV-1 Env structures required for cell attachment, such as gp120 and gp41 (105), novel particles are rendered ineffective at binding to target cells.
When the HIV virion has found target cells (CD4 T-cells), the virus surface glycoprotein (gp120) binds to a cell receptor or co-receptors such as chemokine CC receptor 5 (CCR5) or chemokine CXC receptor 4 (CXCR4), also responsible for the HIV entry into lymphocytes and macrophages. The binding of gp120 results in the exposure of gp41, bringing the HIV virion much closer to the target cell. Subsequently, there is fusion of the viral envelope with the host cell membrane, which is essential for the entry to the inner matrix core of the virus (105). Within this inner core are two strands of viral RNA held together by two small proteins (P6 and P7), three of the viral enzymes (integrase, protease, and reverse transcriptase), and accessory proteins such as Nef, Vpr, and Vif. The viral RNA then undergoes conversion into viral DNA through a process mediated by viral reverse transcriptase before its integration into the host cell genome. In the absence of gp120 and gp41, the aforementioned process does not take place. Importantly, PSGL-1 is incorporated into progeny virions which explains the loss of infectivity, as HIV-1 Env is disorganized by this presence (Figure 2). It is worth noting that the PSGL-1 extracellular domain is fundamental to this process. Fu and colleagues reported that the deletion of much of the intracellular C-terminal domain had no impact on PSGL-1’s ability to restrict HIV-1. On the contrary, the truncation of the extracellular N-terminal domain effectively eliminated the restrictive phenotype. This observation was revealed one month earlier via an e-publication by a Japanese team of researchers (106), who also demonstrated that co-clustering of Gag with PSGL-1 [previously shown to be dependent on the MA domain, especially the HBR (27)] and subsequent release of progeny virions, promote extracellular PSGL-1 down-regulation in infected cells. Very recently (August 2020), Liu et.al (97)., revealed a series of findings showing that PSGL-1 binds to cellular actin filaments (F-actin) to restrict actin dynamics, which consequently inhibit HIV-1 DNA synthesis and HIV-1 reverse transcription (Figure 3). Of note, PSGL-1’s inhibition of HIV-1 reverse transcription is dependent on the cytoplasmic domain, which by itself is sufficient for the inhibition. Importantly, IFN-γ mediates PSGL-1 anti-HIV-1 activity (25), but also increases F-actin intensity/distribution. However, the intensity of F-actin was largely eliminated when PSGL-1 was knocked down by siRNA electroporation. F-actin intensity is inversely related to HIV-1 infection, implying that PSGL-1’s anti-HIV-1 activity is linked to its regulation of F-actin intensity. PSGL-1 cytoplasmic domain binds F-actin and inhibits its depolymerization. Indeed, a highly conserved threonine (T393) was identified as a key residue for the binding of PSGL-1 to actin. Subsequently, PSGL-1’s inhibition of reverse transcription and ability to increase F-actin intensity were greatly reduced by a T393 mutation (97). This represents PSGL-1’s early inhibition role in HIV-1 infection, and is a plausible explanation for this process as it is known that an actin cytoskeleton is required for HIV reverse transcription (107).
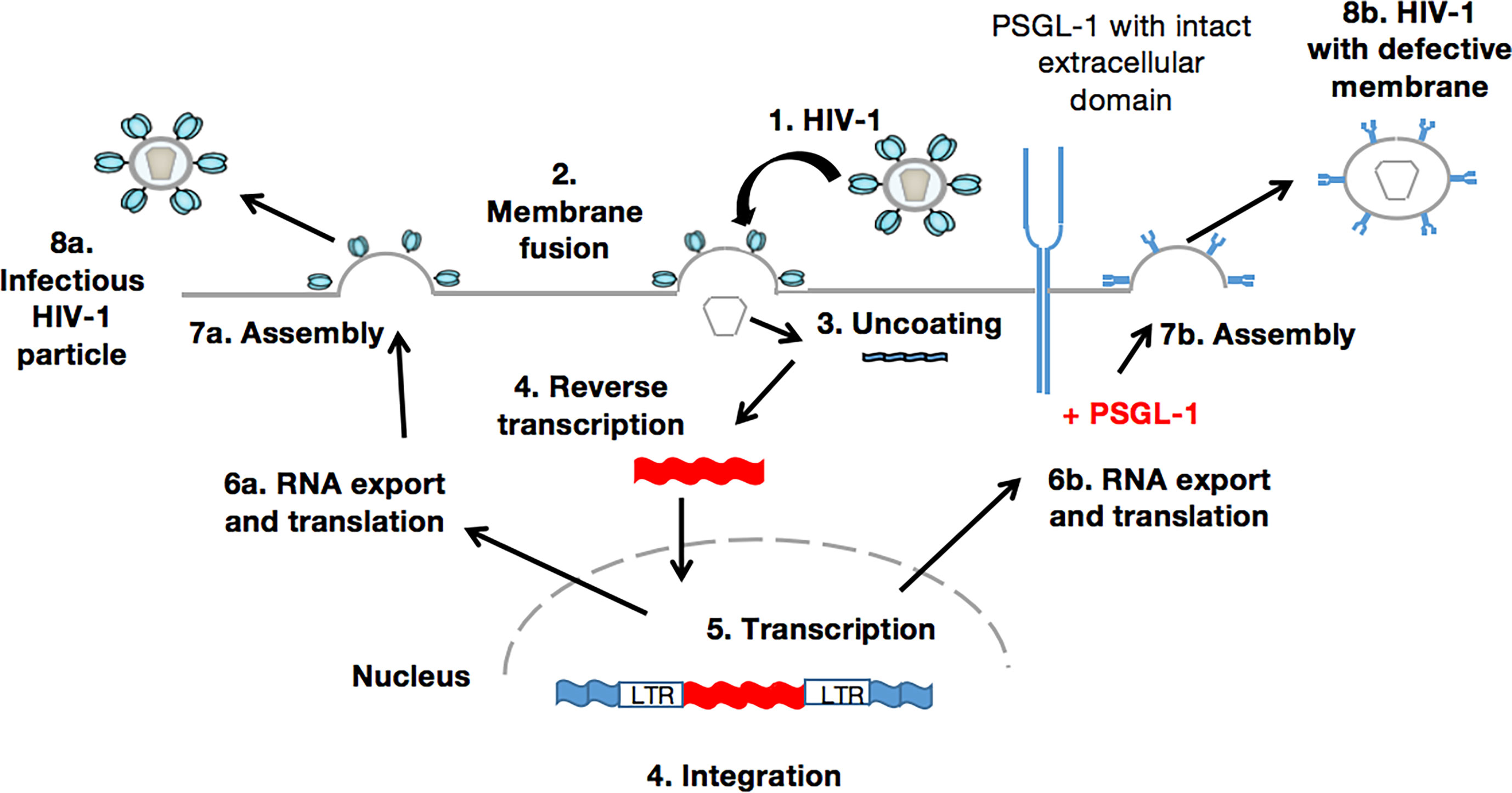
Figure 2 PSGL-1 mediates the production of membrane defective HIV-1 particles. The infected target cell (1-5) produces viral RNAs and proteins that are normally assembled (6a-8a). These new viral particles can infect new cells in which the HIV-1 replication cycle would therefore take place. Conversely, in the presence of PSGL-1 (6b-8b), novel viral particles possess a defective membrane within which PSGL-1 is incorporated. Such a phenotype leads to the production of membrane defective virions that, in the absence of crucial elements such as gp120 and gp41, are unable to attach to or infect new target cells.
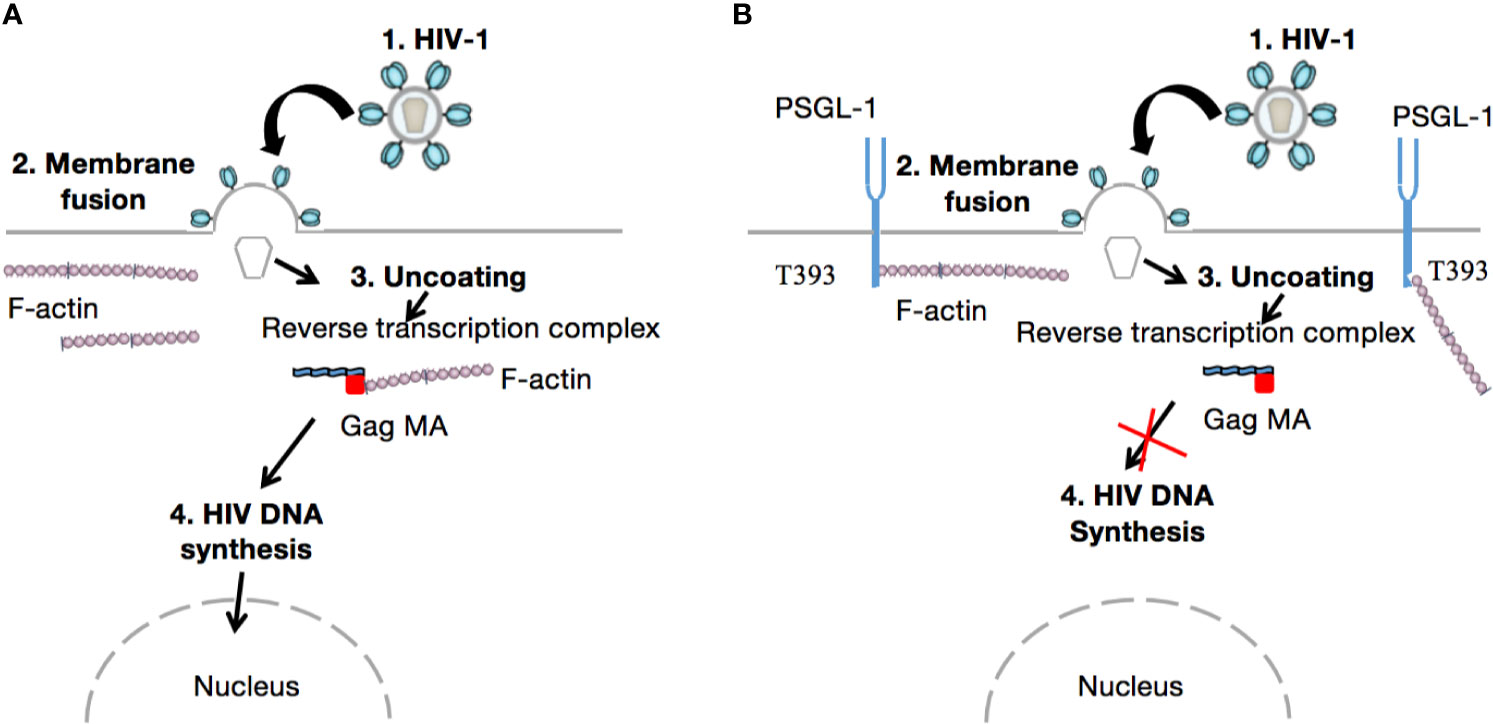
Figure 3 Inhibition of HIV-1 reverse transcription. After attachment of the viral particle to the target cell and incorporation of the inner core into the cell cytoplasm, viral RNA is released (Uncoating). Thereafter, reverse transcription takes place in the cytoskeletal compartment. The viral reverse transcription complex interacts with F-actin via its gag MA domain to achieve cDNA synthesis (A). However, in the presence of PSGL-1, which binds F-actin via its T393 conserved cytoplasmic domain, actin depolymerization or interaction with gag MA within the viral reverse transcription complex cannot occur (B). Consequently, HIV-1 cannot recruit intact actin microfilaments to complete reverse transcription; as reported by Bukrinskaya A et al., in 1998 (107).
With regards to the aforementioned roles that PSGL-1 plays during HIV infection, it is important to inform on its potential applications. Firstly, PSGL-1 can inhibit HIV-1 DNA synthesis and virion infectivity. Compared with HIV DNA synthesis, infectivity inhibition appears to be more potent. Indeed, significant inhibition of virion infectivity can be achieved with a low level of PSGL-1 overexpression (25). Liu and colleagues reported that Vpu has the ability to repress PSGL-1 expression; however we believe that further studies aiming at understanding the interactions between Vpu and PSGL-1 may help develop new anti-HIV drugs. In 2009, Ohba et al. demonstrated that follicular dendritic cells (FDCs) can efficiently activate HIV-1 replication in latently infected monocytes/macrophages. They found that interaction of PSGL-1 with P-selectin is involved in this process, and they further suggested a linkage between PSGL-1 signaling and HIV-1 replication through the activation of nuclear factor k-light chain-enhancer of activated B cells (NF-kB). Indeed, when PSGL-1 on infected monocytes/macrophages interacts with P-selectin on FDCs, a juxtacrine signaling mechanism [with spleen tyrosine kinase (Syk) acting as a downstream effector of PSGL-1 during HIV-1 replication] is activated, and therefore facilitates HIV-1 replication (108). However, this observation is only applicable to monocytes/macrophages as Ohba et al., (in their unpublished data) have found that the PSGL-1/Syk pathway in CD4 T-cells was not prominent. Although this finding seems to discount PSGL-1’s repressive role (reported in contemporary literature, as detailed above) during HIV infection, one may speculate on the benefits of its ability to trigger latently infected cells. We believe that PSGL-1 could be an alternative to latency reversal agents (LRAs) in the “shock and kill” strategy. Such an approach could provide additional tools to finding a cure to HIV infection, especially since it is known that reservoir cells are one of the major impediments to HIV treatment. Secondly, PSGL-1 is an HIV restriction factor, and as such, its expression is triggered by interferon-γ, an immunomodulatory cytokine whose role in innate immunity in HIV infection has only begun to be elucidated (109, 110). Additionally, IL-12 (using Th1 cells from mice) (98), soluble CD40 ligand (sCD40L, using monocytes from HIV negative donor) (104), and glutamate (using monocytes from HIV negative donor) (104) have been proven to be capable of inducing PSGL-1 expression ex vivo. Thus, interferon-γ or IL-12 administration in combination with strategies such as ART, immunotherapy, and gene therapy may provide substantial improvement in the immune responses of HIV-infected individuals. By neutralizing HIV’s repressive role on PSGL-1 expression, immune cells can more readily be recruited to participate in controlling and ameliorating inflammation and/or infection. Importantly, it is now known that during HIV infection there is a fundamental imbalance in the gut microbiome, with a depletion of key immune cells involved in regulating gut microorganisms. CD4+ T-lymphocytes in the gut-associated lymphoid tissues (GALT) (111), Paneth cells (112), macrophages (113), epithelial cells (114), and Th17 cells (115) are all aberrantly represented during HIV infection. Promoting PSGL-1 expression could help in recruiting new cells of the preceding types into the gut associated tissues and re-establishing immune homeostasis, which depends on the overall integrity of the gut microbiome (116), especially in immunological nonresponders. And thirdly, c-Myc regulates PSGL-1 expression in monocytes during HIV-1 infection (104) and this is important, particularly for the treatment of complications associated with HIV, including cardiovascular diseases (CVDs) and neurocognitive disease. It has been reported that sCD40L is implicated in the development of disorders such as inflammatory bowel disease and atherosclerosis (117, 118). Glutamate dysregulation has been demonstrated in a variety of neurocognitive disorders (119). Since sCD40L and glutamate have been proven to be effective at inducing PSGL-1 expression on monocytes (104), an approach targeting PSGL-1 using these factors during HIV infection could be a viable alternative approach to the treatment of CVDs and neurocognitive disease.
Conclusion
Knowledge gaps remain regarding PSGL-1 expression (both intra- and extracellular) and its functions during HIV-1 infection. For example, the impact of HIV-1 clades on PSGL-1 activities requires clarification. Considering that F-actin intensity in infected cells is inversely related to HIV-1 infection, as reported (97), we may therefore suppose that PSGL-1 activity depends upon HIV-1 strain. In other words, PSGL-1 repression is stronger as the strain becomes more virulent. Yet a question remains viz., what are the most repressive viral strains/clades? This is an area that warrants further study. Further questions, such as whether PSGL-1 is an immunological determinant in elite controllers (120–122), long-term non-progressors (123, 124), and immunological non-responders (125, 126), require robust and accurate answers. Finally, since PSGL-1 is expressed by a variety of myeloid cells and T-cells (CD4 and CD8), it is crucial to investigate any potential associations between T-cells and PSGL-1 during HIV infection. Perhaps, a simple index such as the humble CD4+/CD8+ ratio, which is essential for tracking HIV-1 progression (127), could possibly be recruited to track PSGL-1 expression. PSGL-1 is certainly a potential novel therapeutic target for HIV-1, but is also a potential therapeutic target for the treatment of other viral infections as well, when considering its broad-spectrum activities. Further targeted investigation is therefore required to achieve this goal.
Although several studies suggest the benefits of PSGL-1 expression on immune cells during infection/inflammation, some studies present a dissenting picture. For instance, Tinoco et al., have reported that PSGL-1 acts as a negative regulator of T-cell function, which may be exploited by a chronic viral infection to inhibit effector T-cells. They observed that PSGL-1 ligation on exhausted CD8 T-cells (1) inhibits TCR expression, and IL-2 signaling, and (2) upregulates the programmed cell death protein 1 (PD-1) (128). The consequence is their diminished survival with TCR stimulation. Additionally, promoting PSGL-1 expression by DCs, for instance, could limit their immunostimulatory capacity. Indeed, it is known that cDC1 dendritic cells are required to effectively activate both CD4+ and CD8+ T-cells in cellular responses to tumors (129). However, PSGL-1’s role in T-cell activation when expressed on DCs is unknown. Nevertheless, studies have shown that P-selectin engagement with DCs can induce a tolerogenic phenotype that can suppress T-cells (88). These data suggest that PSGL-1, as a potential therapeutic option for HIV, should be explored with caution.
Author Contributions
SZ wrote the manuscript and conceived the figures. VH and YC revised, co-wrote, and provided significant inputs. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by the Medical Research Project of Chongqing Municipal Science and Technology Bureau and Health Commission (grant number 2020GDRC004) and the Key Medical Research Project of Chongqing Municipal Science and Technology Bureau and Health Commission (grant number 2019ZDXM012).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ADAM8, A Disintegrin and metalloproteinase domain-containing protein 8; ADAM28, A Disintegrin and metalloproteinase domain-containing protein 28; ART, Antiretroviral therapy; β1, 4GalT-I, β1, 4-galactosyltransferase; C2GlcNAcT-I, Core 2 β1, 6-glucosaminyltransferase; CCL19, Chemokine (C-C motif) ligand 19; CCL21, Chemokine (C-C motif) ligand 21; CD4, Cluster of differentiation 4; CD8, Cluster of differentiation 8; DCs, Dendritic cells; ERM, Ezrin/radixin/moesin; F-actin, Actin filaments; FucT-IV, Fucosyltransferase-IV; FucT-VII, Fucosyltransferase-VII; Gag, Group-specific antigen; HBR, Highly basic region; HIV-1, Human immunodeficiency virus type 1; HSCs, Hematopoietic stem cells; IDO, Indoleamine 2, 3 dioxygenase; IL-10, Interleukin 10; INFs, Interferons; KDa, Kilodalton; MA, Matrix protein; Nef, Negative factor; PRRs, Pathogen-recognition receptors; PSGL-1, P-selectin glycoprotein ligand 1; SARS-CoV, Severe Acute respiratory syndrome coronavirus; SARS-CoV-2, Severe Acute respiratory syndrome coronavirus 2; Siglec 5, Sialic acid-binding immunoglobulin-type lectin 5; sLex, Sialyl Lewis x; SSL5, Staphylococcal superantigen-like 5; ST3Gal, Sialyl 3-galactosyltransferase; Syk, Spleen tyrosine kinase; TCR, T-cell receptor; TGF, Transforms growth factor; T393, Threonine 393; TPST1, 2, Tyrosine protein sulfotransferases 1, 2; Vif, Viral infectivity factor; VISTA, V-domain immunoglobulin suppressor of T cell activation; Vpu, Viral particle unit; Vpr, Viral protein r.
References
1. Zaongo SD, Xia H, Ma P. HIV Gene Therapy Strategies and Safety: What Do We Know From the Recent Publications? AIDS Rev (2020) 23(3):195–202. doi: 10.24875/AIDSRev.20000008
2. Pace M, Frater J. A Cure for HIV: Is It in Sight? Expert Rev Anti Infect Ther (2014) 12(7):783–91. doi: 10.1586/14787210.2014.910112
3. De Clercq E. Tenofovir Alafenamide (TAF) as the Successor of Tenofovir Disoproxil Fumarate (TDF). Biochem Pharmacol (2016) 119:1–7. doi: 10.1016/j.bcp.2016.04.015
4. Venter WDF, Fabian J, Feldman C. An Overview of Tenofovir and Renal Disease for the HIV-Treating Clinician. South Afr J HIV Med (2018) 19(1):817. doi: 10.4102/sajhivmed.v19i1.817
5. Samuels R, Bayerri CR, Sayer JA, Price DA, Payne BAI. Tenofovir Disoproxil Fumarate-Associated Renal Tubular Dysfunction: Noninvasive Assessment of Mitochondrial Injury. Aids (2017) 31(9):1297–301. doi: 10.1097/qad.0000000000001466
6. Taramasso L, Lorenzini P, Di Biagio A, Lichtner M, Marchetti G, Rossotti R, et al. Incidence and Risk Factors for Liver Enzyme Elevation Among Naive HIV-1-Infected Patients Receiving ART in the ICONA Cohort. J Antimicrob Chemother (2019) 74(11):3295–304. doi: 10.1093/jac/dkz353
7. Debes JD, Bohjanen PR, Boonstra A. Mechanisms of Accelerated Liver Fibrosis Progression During HIV Infection. J Clin Transl Hepatol (2016) 4(4):328–35. doi: 10.14218/jcth.2016.00034
8. Treisman GJ, Soudry O. Neuropsychiatric Effects of HIV Antiviral Medications. Drug Saf (2016) 39(10):945–57. doi: 10.1007/s40264-016-0440-y
9. Vo TT, Varghese Gupta S. Role of Cytochrome P450 2B6 Pharmacogenomics in Determining Efavirenz-Mediated Central Nervous System Toxicity, Treatment Outcomes, and Dosage Adjustments in Patients With Human Immunodeficiency Virus Infection. Pharmacotherapy (2016) 36(12):1245–54. doi: 10.1002/phar.1852
10. Garg H, Joshi A, Mukherjee D. Cardiovascular Complications of HIV Infection and Treatment. Cardiovasc Hematol Agents Med Chem (2013) 11(1):58–66. doi: 10.2174/1871525711311010010
11. Altfeld M, Gale M Jr. Innate Immunity Against HIV-1 Infection. Nat Immunol (2015) 16(6):554–62. doi: 10.1038/ni.3157
12. Malim MH, Bieniasz PD. HIV Restriction Factors and Mechanisms of Evasion. Cold Spring Harb Perspect Med (2012) 2(5):a006940. doi: 10.1101/cshperspect.a006940
13. Yan N, Chen ZJ. Intrinsic Antiviral Immunity. Nat Immunol (2012) 13(3):214–22. doi: 10.1038/ni.2229
14. Towers GJ, Noursadeghi M. Interactions Between HIV-1 and the Cell-Autonomous Innate Immune System. Cell Host Microbe (2014) 16(1):10–8. doi: 10.1016/j.chom.2014.06.009
15. Simon V, Bloch N, Landau NR. Intrinsic Host Restrictions to HIV-1 and Mechanisms of Viral Escape. Nat Immunol (2015) 16(6):546–53. doi: 10.1038/ni.3156
16. Harris RS, Hultquist JF, Evans DT. The Restriction Factors of Human Immunodeficiency Virus. J Biol Chem (2012) 287(49):40875–83. doi: 10.1074/jbc.R112.416925
17. Daugherty MD, Malik HS. Rules of Engagement: Molecular Insights From Host-Virus Arms Races. Annu Rev Genet (2012) 46:677–700. doi: 10.1146/annurev-genet-110711-155522
18. Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J. The Cytoplasmic Body Component TRIM5alpha Restricts HIV-1 Infection in Old World Monkeys. Nature (2004) 427(6977):848–53. doi: 10.1038/nature02343
19. Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a Human Gene That Inhibits HIV-1 Infection and is Suppressed by the Viral Vif Protein. Nature (2002) 418(6898):646–50. doi: 10.1038/nature00939
20. Neil SJ, Zang T, Bieniasz PD. Tetherin Inhibits Retrovirus Release and Is Antagonized by HIV-1 Vpu. Nature (2008) 451(7177):425–30. doi: 10.1038/nature06553
21. Hrecka K, Hao C, Gierszewska M, Swanson SK, Kesik-Brodacka M, Srivastava S, et al. Vpx Relieves Inhibition of HIV-1 Infection of Macrophages Mediated by the SAMHD1 Protein. Nature (2011) 474(7353):658–61. doi: 10.1038/nature10195
22. Laguette N, Sobhian B, Casartelli N, Ringeard M, Chable-Bessia C, Ségéral E, et al. SAMHD1 Is the Dendritic- and Myeloid-Cell-Specific HIV-1 Restriction Factor Counteracted by Vpx. Nature (2011) 474(7353):654–7. doi: 10.1038/nature10117
23. Rosa A, Chande A, Ziglio S, De Sanctis V, Bertorelli R, Goh SL, et al. HIV-1 Nef Promotes Infection by Excluding SERINC5 From Virion Incorporation. Nature (2015) 526(7572):212–7. doi: 10.1038/nature15399
24. Usami Y, Wu Y, Göttlinger HG. SERINC3 and SERINC5 Restrict HIV-1 Infectivity and Are Counteracted by Nef. Nature (2015) 526(7572):218–23. doi: 10.1038/nature15400
25. Liu Y, Fu Y, Wang Q, Li M, Zhou Z, Dabbagh D, et al. Proteomic Profiling of HIV-1 Infection of Human CD4(+) T Cells Identifies PSGL-1 as an HIV Restriction Factor. Nat Microbiol (2019) 4(5):813–25. doi: 10.1038/s41564-019-0372-2
26. Matheson NJ, Sumner J, Wals K, Rapiteanu R, Weekes MP, Vigan R, et al. Cell Surface Proteomic Map of HIV Infection Reveals Antagonism of Amino Acid Metabolism by Vpu and Nef. Cell Host Microbe (2015) 18(4):409–23. doi: 10.1016/j.chom.2015.09.003
27. Grover JR, Veatch SL, Ono A. Basic Motifs Target PSGL-1, CD43, and CD44 to Plasma Membrane Sites Where HIV-1 Assembles. J Virol (2015) 89(1):454–67. doi: 10.1128/jvi.02178-14
28. Tinoco R, Otero DC, Takahashi AA, Bradley LM. PSGL-1: A New Player in the Immune Checkpoint Landscape. Trends Immunol (2017) 38(5):323–35. doi: 10.1016/j.it.2017.02.002
29. Nourshargh S, Alon R. Leukocyte Migration Into Inflamed Tissues. Immunity (2014) 41(5):694–707. doi: 10.1016/j.immuni.2014.10.008
30. Friedl P, Weigelin B. Interstitial Leukocyte Migration and Immune Function. Nat Immunol (2008) 9(9):960–9. doi: 10.1038/ni.f.212
31. Carlow DA, Gossens K, Naus S, Veerman KM, Seo W, Ziltener HJ. PSGL-1 Function in Immunity and Steady State Homeostasis. Immunol Rev (2009) 230(1):75–96. doi: 10.1111/j.1600-065X.2009.00797.x
32. Almulki L, Noda K, Amini R, Schering A, Garland RC, Nakao S, et al. Surprising Up-Regulation of P-Selectin Glycoprotein Ligand-1 (PSGL-1) in Endotoxin-Induced Uveitis. FASEB J (2009) 23(3):929–39. doi: 10.1096/fj.08-118760
33. McEver RP, Cummings RD. Role of PSGL-1 Binding to Selectins in Leukocyte Recruitment. J Clin Invest (1997) 100(11 Suppl):S97–103. doi: 10.1172/JCI119556
34. Moore KL, Stults NL, Diaz S, Smith DF, Cummings RD, Varki A, et al. Identification of a Specific Glycoprotein Ligand for P-Selectin (CD62) on Myeloid Cells. J Cell Biol (1992) 118(2):445–56. doi: 10.1083/jcb.118.2.445
35. Sako D, Chang XJ, Barone KM, Vachino G, White HM, Shaw G, et al. Expression Cloning of a Functional Glycoprotein Ligand for P-Selectin. Cell (1993) 75(6):1179–86. doi: 10.1016/0092-8674(93)90327-m
36. Snapp KR, Craig R, Herron M, Nelson RD, Stoolman LM, Kansas GS. Dimerization of P-Selectin Glycoprotein Ligand-1 (PSGL-1) Required for Optimal Recognition of P-Selectin. J Cell Biol (1998) 142(1):263–70. doi: 10.1083/jcb.142.1.263
37. Tanaka T. Leukocyte Adhesion Molecules. In: Ratcliffe MJH, editor. Encyclopedia of Immunobiology. Oxford: Academic Press Cambridge, Massachusetts (2016). p. 505–11.
38. Spertini O, Cordey AS, Monai N, Giuffrè L, Schapira M. P-Selectin Glycoprotein Ligand 1 Is a Ligand for L-Selectin on Neutrophils, Monocytes, and CD34+ Hematopoietic Progenitor Cells. J Cell Biol (1996) 135(2):523–31. doi: 10.1083/jcb.135.2.523
39. Goetz DJ, Greif DM, Ding H, Camphausen RT, Howes S, Comess KM, et al. Isolated P-Selectin Glycoprotein Ligand-1 Dynamic Adhesion to P- and E-Selectin. J Cell Biol (1997) 137(2):509–19. doi: 10.1083/jcb.137.2.509
40. Xia L, Ramachandran V, McDaniel JM, Nguyen KN, Cummings RD, Mcever RP. N-Terminal Residues in Murine P-Selectin Glycoprotein Ligand-1 Required for Binding to Murine P-Selectin. Blood (2003) 101(2):552–9. doi: 10.1182/blood-2001-11-0036
41. Asa D, Raycroft L, Ma L, Aeed PA, Kaytes PS, Elhammer AP, et al. The P-Selectin Glycoprotein Ligand Functions as a Common Human Leukocyte Ligand for P- and E-Selectins. J Biol Chem (1995) 270(19):11662–70. doi: 10.1074/jbc.270.19.11662
42. Guyer DA, Moore KL, Lynam EB, Schammel CM, Rogelj S, McEver RP, et al. P-Selectin Glycoprotein Ligand-1 (PSGL-1) Is a Ligand for L-Selectin in Neutrophil Aggregation. Blood (1996) 88(7):2415–21. doi: 10.1182/blood.V88.7.2415.bloodjournal8872415
43. Martinez M, Joffraud M, Giraud S, Baïsse B, Bernimoulin MP, Schapira M, et al. Regulation of PSGL-1 Interactions With L-Selectin, P-Selectin, and E-Selectin: Role of Human Fucosyltransferase-IV and -VII. J Biol Chem (2005) 280(7):5378–90. doi: 10.1074/jbc.M410899200
44. Sako D, Comess KM, Barone KM, Camphausen RT, Cumming DA, Shaw GD. A Sulfated Peptide Segment at the Amino Terminus of PSGL-1 Is Critical for P-Selectin Binding. Cell (1995) 83(2):323–31. doi: 10.1016/0092-8674(95)90173-6
45. Tu L, Chen A, Delahunty MD, Moore KL, Watson SR, McEver RP, et al. L-Selectin Binds to P-Selectin Glycoprotein Ligand-1 on Leukocytes: Interactions Between the Lectin, Epidermal Growth Factor, and Consensus Repeat Domains of the Selectins Determine Ligand Binding Specificity. J Immunol (1996) 157(9):3995–4004.
46. Silva M, Videira PA, Sackstein R. E-Selectin Ligands in the Human Mononuclear Phagocyte System: Implications for Infection, Inflammation, and Immunotherapy. Front Immunol (2017) 8:1878. doi: 10.3389/fimmu.2017.01878
47. Ley K, Kansas GS. Selectins in T-Cell Recruitment to Non-Lymphoid Tissues and Sites of Inflammation. Nat Rev Immunol (2004) 4(5):325–35. doi: 10.1038/nri1351
48. Yang WH, Nussbaum C, Grewal PK, Marth JD, Sperandio M. Coordinated Roles of ST3Gal-VI and ST3Gal-IV Sialyltransferases in the Synthesis of Selectin Ligands. Blood (2012) 120(5):1015–26. doi: 10.1182/blood-2012-04-424366
49. Zarbock A, Müller H, Kuwano Y, Ley K. PSGL-1-Dependent Myeloid Leukocyte Activation. J Leukoc Biol (2009) 86(5):1119–24. doi: 10.1189/jlb.0209117
50. Kivisäkk P, Mahad DJ, Callahan MK, Trebst C, Tucky B, Wei T, et al. Human Cerebrospinal Fluid Central Memory CD4+ T Cells: Evidence for Trafficking Through Choroid Plexus and Meninges via P-Selectin. Proc Natl Acad Sci USA (2003) 100(14):8389–94. doi: 10.1073/pnas.1433000100
51. Eppihimer MJ, Russell J, Anderson DC, Wolitzky BA, Granger DN. Endothelial Cell Adhesion Molecule Expression in Gene-Targeted Mice. Am J Physiol (1997) 273(4):H1903–8. doi: 10.1152/ajpheart.1997.273.4.H1903
52. McEver RP, Martin MN. A Monoclonal Antibody to a Membrane Glycoprotein Binds Only to Activated Platelets. J Biol Chem (1984) 259(15):9799–804. doi: 10.1016/S0021-9258(17)42770-0
53. Falati S, Liu Q, Gross P, Merrill-Skoloff G, Chou J, Vandendries E, et al. Accumulation of Tissue Factor Into Developing Thrombi In Vivo Is Dependent Upon Microparticle P-Selectin Glycoprotein Ligand 1 and Platelet P-Selectin. J Exp Med (2003) 197(11):1585–98. doi: 10.1084/jem.20021868
54. Tchernychev B, Furie B, Furie BC. Peritoneal Macrophages Express Both P-Selectin and PSGL-1. J Cell Biol (2003) 163(5):1145–55. doi: 10.1083/jcb.200310079
55. Kansas GS. Selectins and Their Ligands: Current Concepts and Controversies. Blood (1996) 88(9):3259–87. doi: 10.1182/blood.V88.9.3259.bloodjournal8893259
56. Ley K. The Role of Selectins in Inflammation and Disease. Trends Mol Med (2003) 9(6):263–8. doi: 10.1016/s1471-4914(03)00071-6
57. Keelan ET, Licence ST, Peters AM, Binns RM, Haskard DO. Characterization of E-Selectin Expression In Vivo With Use of a Radiolabeled Monoclonal Antibody. Am J Physiol (1994) 266(1 Pt 2):H278–90. doi: 10.1152/ajpheart.1994.266.1.H279
58. Sallusto F, Lenig D, Förster R, Lipp M, Lanzavecchia A. Two Subsets of Memory T Lymphocytes With Distinct Homing Potentials and Effector Functions. Nature (1999) 401(6754):708–12. doi: 10.1038/44385
59. Patel AA, Zhang Y, Fullerton JN, Boelen L, Rongvaux A, Maini AA, et al. The Fate and Lifespan of Human Monocyte Subsets in Steady State and Systemic Inflammation. J Exp Med (2017) 214(7):1913–23. doi: 10.1084/jem.20170355
60. Allport JR, Ding HT, Ager A, Steeber DA, Tedder TF, Luscinskas FW. L-Selectin Shedding Does Not Regulate Human Neutrophil Attachment, Rolling, or Transmigration Across Human Vascular Endothelium In Vitro. J Immunol (1997) 158(9):4365–72.
61. Kieffer JD, Fuhlbrigge RC, Armerding D, Robert C, Ferenczi K, Camphausen RT, et al. Neutrophils, Monocytes, and Dendritic Cells Express the Same Specialized Form of PSGL-1 as Do Skin-Homing Memory T Cells: Cutaneous Lymphocyte Antigen. Biochem Biophys Res Commun (2001) 285(3):577–87. doi: 10.1006/bbrc.2001.5230
62. Rossi FM, Corbel SY, Merzaban JS, Carlow DA, Gossens K, Duenas J, et al. Recruitment of Adult Thymic Progenitors Is Regulated by P-Selectin and Its Ligand PSGL-1. Nat Immunol (2005) 6(6):626–34. doi: 10.1038/ni1203
63. Katayama Y, Hidalgo A, Furie BC, Vestweber D, Furie B, Frenette PS. PSGL-1 Participates in E-Selectin-Mediated Progenitor Homing to Bone Marrow: Evidence for Cooperation Between E-Selectin Ligands and Alpha4 Integrin. Blood (2003) 102(6):2060–7. doi: 10.1182/blood-2003-04-1212
64. Wagers AJ, Kansas GS. Potent Induction of Alpha(1,3)-Fucosyltransferase VII in Activated CD4+ T Cells by TGF-Beta 1 Through a P38 Mitogen-Activated Protein Kinase-Dependent Pathway. J Immunol (2000) 165(9):5011–6. doi: 10.4049/jimmunol.165.9.5011
65. Carlow DA, Williams MJ, Ziltener HJ. Inducing P-Selectin Ligand Formation in CD8 T Cells: IL-2 and IL-12 Are Active In Vitro But Not Required In Vivo. J Immunol (2005) 174(7):3959–66. doi: 10.4049/jimmunol.174.7.3959
66. Yago T, Shao B, Miner JJ, Yao L, Klopocki AG, Maeda K, et al. E-Selectin Engages PSGL-1 and CD44 Through a Common Signaling Pathway to Induce Integrin Alphalbeta2-Mediated Slow Leukocyte Rolling. Blood (2010) 116(3):485–94. doi: 10.1182/blood-2009-12-259556
67. Zarbock A, Abram CL, Hundt M, Altman A, Lowell CA, Ley K. PSGL-1 Engagement by E-Selectin Signals Through Src Kinase Fgr and ITAM Adapters DAP12 and FcR Gamma to Induce Slow Leukocyte Rolling. J Exp Med (2008) 205(10):2339–47. doi: 10.1084/jem.20072660
68. Moore KL, Patel KD, Bruehl RE, Li F, Johnson DA, Lichenstein HS, et al. P-Selectin Glycoprotein Ligand-1 Mediates Rolling of Human Neutrophils on P-Selectin. J Cell Biol (1995) 128(4):661–71. doi: 10.1083/jcb.128.4.661
69. Timmerman I, Daniel AE, Kroon J, van Buul JD. Leukocytes Crossing the Endothelium: A Matter of Communication. Int Rev Cell Mol Biol (2016) 322:281–329. doi: 10.1016/bs.ircmb.2015.10.005
70. Snapp KR, Heitzig CE, Kansas GS. Attachment of the PSGL-1 Cytoplasmic Domain to the Actin Cytoskeleton is Essential for Leukocyte Rolling on P-Selectin. Blood (2002) 99(12):4494–502. doi: 10.1182/blood.v99.12.4494
71. Baïsse B, Galisson F, Giraud S, Schapira M, Spertini O. Evolutionary Conservation of P-Selectin Glycoprotein Ligand-1 Primary Structure and Function. BMC Evol Biol (2007) 7:166. doi: 10.1186/1471-2148-7-166
72. Nuñez-Andrade N, Lamana A, Sancho D, Gisbert JP, Gonzalez-Amaro R, Sanchez-Madrid F, et al. P-Selectin Glycoprotein Ligand-1 Modulates Immune Inflammatory Responses in the Enteric Lamina Propria. J Pathol (2011) 224(2):212–21. doi: 10.1002/path.2850
73. Borges E, Eytner R, Moll T, Steegmaier M, Campbell MA, Ley K, et al. The P-Selectin Glycoprotein Ligand-1 Is Important for Recruitment of Neutrophils Into Inflamed Mouse Peritoneum. Blood (1997) 90(5):1934–42. doi: 10.1182/blood.V90.5.1934
74. Asaduzzaman M, Mihaescu A, Wang Y, Sato T. Thorlacius H. P-Selectin and P-Selectin Glycoprotein Ligand 1 Mediate Rolling of Activated CD8+ T Cells in Inflamed Colonic Venules. J Investig Med (2009) 57(7):765–8. doi: 10.2310/JIM.0b013e3181b918fb
75. Martín-Fontecha A, Baumjohann D, Guarda G, Reboldi A, Hons M, Lanzavecchia A, et al. CD40L+ CD4+ Memory T Cells Migrate in a CD62P-Dependent Fashion Into Reactive Lymph Nodes and License Dendritic Cells for T Cell Priming. J Exp Med (2008) 205(11):2561–74. doi: 10.1084/jem.20081212
76. Haddad W, Cooper CJ, Zhang Z, Brown JB, Zhu Y, Issekutz A, et al. P-Selectin and P-Selectin Glycoprotein Ligand 1 are Major Determinants for Th1 Cell Recruitment to Nonlymphoid Effector Sites in the Intestinal Lamina Propria. J Exp Med (2003) 198(3):369–77. doi: 10.1084/jem.20020691
77. Xu H, Manivannan A, Jiang HR, Liversidge J, Sharp PF, Forrester JV, et al. Recruitment of IFN-Gamma-Producing (Th1-Like) Cells Into the Inflamed Retina In Vivo Is Preferentially Regulated by P-Selectin Glycoprotein Ligand 1:P/E-Selectin Interactions. J Immunol (2004) 172(5):3215–24. doi: 10.4049/jimmunol.172.5.3215
78. Zuchtriegel G, Uhl B, Puhr-Westerheide D, Pörnbacher M, Lauber K, Krombach F, et al. Platelets Guide Leukocytes to Their Sites of Extravasation. PloS Biol (2016) 14(5):e1002459. doi: 10.1371/journal.pbio.1002459
79. Lam FW, Burns AR, Smith CW, Rumbaut RE. Platelets Enhance Neutrophil Transendothelial Migration via P-Selectin Glycoprotein Ligand-1. Am J Physiol-Heart Circulatory Physiol (2011) 300(2):H468–H75. doi: 10.1152/ajpheart.00491.2010
80. Frenette PS, Denis CV, Weiss L, Jurk K, Subbarao S, Kehrel B, et al. P-Selectin Glycoprotein Ligand 1 (PSGL-1) Is Expressed on Platelets and can Mediate Platelet-Endothelial Interactions In Vivo. J Exp Med (2000) 191(8):1413–22. doi: 10.1084/jem.191.8.1413
81. Kum WW, Lee S, Grassl GA, Bidshahri R, Hsu K, Ziltener HJ, et al. Lack of Functional P-Selectin Ligand Exacerbates Salmonella Serovar Typhimurium Infection. J Immunol (2009) 182(10):6550–61. doi: 10.4049/jimmunol.0802536
82. Bestebroer J, van Kessel KP, Azouagh H, Walenkamp AM, Boer IG, Romijn RA, et al. Staphylococcal SSL5 Inhibits Leukocyte Activation by Chemokines and Anaphylatoxins. Blood (2009) 113(2):328–37. doi: 10.1182/blood-2008-04-153882
83. Walenkamp AM, Bestebroer J, Boer IG, Kruizinga R, Verheul HM, van Strijp JA, et al. Staphylococcal SSL5 Binding to Human Leukemia Cells Inhibits Cell Adhesion to Endothelial Cells and Platelets. Cell Oncol (2010) 32(1-2):1–10. doi: 10.3233/clo-2009-0486
84. Pepin M, Mezouar S, Pegon J, Muczynski V, Adam F, Bianchini EP, et al. Soluble Siglec-5 Associates to PSGL-1 and Displays Anti-Inflammatory Activity. Sci Rep (2016) 6:37953. doi: 10.1038/srep37953
85. Domínguez-Luis M, Lamana A, Vazquez J, García-Navas R, Mollinedo F, Sánchez-Madrid F, et al. The Metalloprotease ADAM8 is Associated With and Regulates the Function of the Adhesion Receptor PSGL-1 Through ERM Proteins. Eur J Immunol (2011) 41(12):3436–42. doi: 10.1002/eji.201141764
86. Shimoda M, Hashimoto G, Mochizuki S, Ikeda E, Nagai N, Ishida S, et al. Binding of ADAM28 to P-Selectin Glycoprotein Ligand-1 Enhances P-Selectin-Mediated Leukocyte Adhesion to Endothelial Cells. J Biol Chem (2007) 282(35):25864–74. doi: 10.1074/jbc.M702414200
87. Lévesque JP, Zannettino AC, Pudney M, Niutta S, Haylock DN, Snapp KR, et al. PSGL-1-Mediated Adhesion of Human Hematopoietic Progenitors to P-Selectin Results in Suppression of Hematopoiesis. Immunity (1999) 11(3):369–78. doi: 10.1016/s1074-7613(00)80112-0
88. Urzainqui A, Martínez del Hoyo G, Lamana A, de la Fuente H, Barreiro O, Olazabal IM, et al. Functional Role of P-Selectin Glycoprotein Ligand 1/P-Selectin Interaction in the Generation of Tolerogenic Dendritic Cells. J Immunol (2007) 179(11):7457–65. doi: 10.4049/jimmunol.179.11.7457
89. Hidari KI, Weyrich AS, Zimmerman GA, McEver RP. Engagement of P-Selectin Glycoprotein Ligand-1 Enhances Tyrosine Phosphorylation and Activates Mitogen-Activated Protein Kinases in Human Neutrophils. J Biol Chem (1997) 272(45):28750–6. doi: 10.1074/jbc.272.45.28750
90. Ba XQ, Chen CX, Xu T, Cui LL, Gao YG, Zeng XL. Engagement of PSGL-1 Upregulates CSF-1 Transcription via a Mechanism That May Involve Syk. Cell Immunol (2005) 237(1):1–6. doi: 10.1016/j.cellimm.2005.09.004
91. Celi A, Pellegrini G, Lorenzet R, De Blasi A, Ready N, Furie BC, et al. P-Selectin Induces the Expression of Tissue Factor on Monocytes. Proc Natl Acad Sci USA (1994) 91(19):8767–71. doi: 10.1073/pnas.91.19.8767
92. Damle NK, Klussman K, Dietsch MT, Mohagheghpour N, Aruffo A. GMP-140 (P-Selectin/CD62) Binds to Chronically Stimulated But Not Resting CD4+ T Lymphocytes and Regulates Their Production of Proinflammatory Cytokines. Eur J Immunol (1992) 22(7):1789–93. doi: 10.1002/eji.1830220718
93. Ramos-Sevillano E, Urzainqui A, de Andrés B, González-Tajuelo R, Domenech M, González-Camacho F, et al. PSGL-1 on Leukocytes Is a Critical Component of the Host Immune Response Against Invasive Pneumococcal Disease. PloS Pathog (2016) 12(3):e1005500. doi: 10.1371/journal.ppat.1005500
94. Nishimura Y, Shimojima M, Tano Y, Miyamura T, Wakita T, Shimizu H. Human P-Selectin Glycoprotein Ligand-1 Is a Functional Receptor for Enterovirus 71. Nat Med (2009) 15(7):794–7. doi: 10.1038/nm.1961
95. Fu Y, He S, Waheed AA, Dabbagh D, Zhou Z, Trinité B, et al. PSGL-1 Restricts HIV-1 Infectivity by Blocking Virus Particle Attachment to Target Cells. Proc Natl Acad Sci USA (2020) 117(17):9537–45. doi: 10.1073/pnas.1916054117
96. He S, Waheed AA, Hetrick B, Dabbagh D, Akhrymuk IV, Kehn-Hall K, et al. PSGL-1 Inhibits the Incorporation of SARS-CoV and SARS-CoV-2 Spike Glycoproteins Into Pseudovirions and Impairs Pseudovirus Attachment and Infectivity. Viruses (2020) 13(1):3–6. doi: 10.3390/v13010046
97. Liu Y, Song Y, Zhang S, Diao M, Huang S, Li S, et al. PSGL-1 Inhibits HIV-1 Infection by Restricting Actin Dynamics and Sequestering HIV Envelope Proteins. Cell Discovery (2020) 6:53. doi: 10.1038/s41421-020-0184-9
98. DeRogatis JM, Viramontes KM, Neubert EN, Tinoco R. PSGL-1 Immune Checkpoint Inhibition for CD4(+) T Cell Cancer Immunotherapy. Front Immunol (2021) 12:636238. doi: 10.3389/fimmu.2021.636238
99. Veerman KM, Williams MJ, Uchimura K, Singer MS, Merzaban JS, Naus S, et al. Interaction of the Selectin Ligand PSGL-1 With Chemokines CCL21 and CCL19 Facilitates Efficient Homing of T Cells to Secondary Lymphoid Organs. Nat Immunol (2007) 8(5):532–9. doi: 10.1038/ni1456
100. Veldkamp CT, Kiermaier E, Gabel-Eissens SJ, Gillitzer ML, Lippner DR, DiSilvio FA, et al. Solution Structure of CCL19 and Identification of Overlapping CCR7 and PSGL-1 Binding Sites. Biochemistry (2015) 54(27):4163–6. doi: 10.1021/acs.biochem.5b00560
101. Ali SR, Fong JJ, Carlin AF, Busch TD, Linden R, Angata T, et al. Siglec-5 and Siglec-14 are Polymorphic Paired Receptors That Modulate Neutrophil and Amnion Signaling Responses to Group B Streptococcus. J Exp Med (2014) 211(6):1231–42. doi: 10.1084/jem.20131853
102. Johnston RJ, Su LJ, Pinckney J, Critton D, Boyer E, Krishnakumar A, et al. VISTA Is an Acidic pH-Selective Ligand for PSGL-1. Nature (2019) 574(7779):565–70. doi: 10.1038/s41586-019-1674-5
103. Liang H, Duan Z, Li D, Li D, Wang Z, Ren L, et al. Higher Levels of Circulating Monocyte-Platelet Aggregates are Correlated With Viremia and Increased sCD163 Levels in HIV-1 Infection. Cell Mol Immunol (2015) 12(4):435–43. doi: 10.1038/cmi.2014.66
104. Connor R, Jones LD, Qiu X, Thakar J, Maggirwar SB. Frontline Science: C-Myc Regulates P-Selectin Glycoprotein Ligand-1 Expression in Monocytes During HIV-1 Infection. J Leukoc Biol (2017) 102(4):953–64. doi: 10.1189/jlb.6HI0217-043R
105. Wilen CB, Tilton JC, Doms RW. HIV: Cell Binding and Entry. Cold Spring Harb Perspect Med (2012) 2(8):1–3. doi: 10.1101/cshperspect.a006866
106. Murakami T, Carmona N, Ono A. Virion-Incorporated PSGL-1 and CD43 Inhibit Both Cell-Free Infection and Transinfection of HIV-1 by Preventing Virus-Cell Binding. Proc Natl Acad Sci USA (2020) 117(14):8055–63. doi: 10.1073/pnas.1916055117
107. Bukrinskaya A, Brichacek B, Mann A, Stevenson M. Establishment of a Functional Human Immunodeficiency Virus Type 1 (HIV-1) Reverse Transcription Complex Involves the Cytoskeleton. J Exp Med (1998) 188(11):2113–25. doi: 10.1084/jem.188.11.2113
108. Ohba K, Ryo A, Dewan MZ, Nishi M, Naito T, Qi X, et al. Follicular Dendritic Cells Activate HIV-1 Replication in Monocytes/Macrophages Through a Juxtacrine Mechanism Mediated by P-Selectin Glycoprotein Ligand 1. J Immunol (2009) 183(1):524–32. doi: 10.4049/jimmunol.0900371
109. Hotter D, Kirchhoff F. Interferons and Beyond: Induction of Antiretroviral Restriction Factors. J Leukoc Biol (2018) 103(3):465–77. doi: 10.1002/jlb.3mr0717-307r
110. Rihn SJ, Foster TL, Busnadiego I, Aziz MA, Hughes J, Neil SJD, et al. The Envelope Gene of Transmitted HIV-1 Resists a Late Interferon Gamma-Induced Block. J Virol (2017) 91(7):e02254-16. doi: 10.1128/jvi.02254-16
111. Brenchley JM, Schacker TW, Ruff LE, Price DA, Taylor JH, Beilman GJ, et al. CD4+ T Cell Depletion During All Stages of HIV Disease Occurs Predominantly in the Gastrointestinal Tract. J Exp Med (2004) 200(6):749–59. doi: 10.1084/jem.20040874
112. Zaragoza MM, Sankaran-Walters S, Canfield DR, Hung JK, Martinez E, Ouellette AJ, et al. Persistence of Gut Mucosal Innate Immune Defenses by Enteric α-Defensin Expression in the Simian Immunodeficiency Virus Model of AIDS. J Immunol (2011) 186(3):1589–97. doi: 10.4049/jimmunol.1002021
113. Allers K, Fehr M, Conrad K, Epple HJ, Schürmann D, Geelhaar-Karsch A, et al. Macrophages Accumulate in the Gut Mucosa of Untreated HIV-Infected Patients. J Infect Dis (2014) 209(5):739–48. doi: 10.1093/infdis/jit547
114. Somsouk M, Estes JD, Deleage C, Dunham RM, Albright R, Inadomi JM, et al. Gut Epithelial Barrier and Systemic Inflammation During Chronic HIV Infection. Aids (2015) 29(1):43–51. doi: 10.1097/qad.0000000000000511
115. Favre D, Lederer S, Kanwar B, Ma ZM, Proll S, Kasakow Z, et al. Critical Loss of the Balance Between Th17 and T Regulatory Cell Populations in Pathogenic SIV Infection. PloS Pathog (2009) 5(2):e1000295. doi: 10.1371/journal.ppat.1000295
116. Pickard JM, Zeng MY, Caruso R, Núñez G. Gut Microbiota: Role in Pathogen Colonization, Immune Responses, and Inflammatory Disease. Immunol Rev (2017) 279(1):70–89. doi: 10.1111/imr.12567
117. Danese S, Sans M, Fiocchi C. The CD40/CD40L Costimulatory Pathway in Inflammatory Bowel Disease. Gut (2004) 53(7):1035–43. doi: 10.1136/gut.2003.026278
118. Phipps RP. Atherosclerosis: The Emerging Role of Inflammation and the CD40-CD40 Ligand System. Proc Natl Acad Sci USA (2000) 97(13):6930–2. doi: 10.1073/pnas.97.13.6930
119. Javitt DC. Glutamate as a Therapeutic Target in Psychiatric Disorders. Mol Psychiatry (2004) 9(11):984–97:79. doi: 10.1038/sj.mp.4001551
120. Deeks SG, Walker BD. Human Immunodeficiency Virus Controllers: Mechanisms of Durable Virus Control in the Absence of Antiretroviral Therapy. Immunity (2007) 27(3):406–16. doi: 10.1016/j.immuni.2007.08.010
121. Lambotte O, Boufassa F, Madec Y, Nguyen A, Goujard C, Meyer L, et al. HIV Controllers: A Homogeneous Group of HIV-1-Infected Patients With Spontaneous Control of Viral Replication. Clin Infect Dis (2005) 41(7):1053–6. doi: 10.1086/433188
122. Gebara NY, El Kamari V, Rizk N. HIV-1 Elite Controllers: An Immunovirological Review and Clinical Perspectives. J Virus Erad (2019) 5(3):163–6. doi: 10.1016/S2055-6640(20)30046-7
123. Cao Y, Qin L, Zhang L, Safrit J, Ho DD. Virologic and Immunologic Characterization of Long-Term Survivors of Human Immunodeficiency Virus Type 1 Infection. N Engl J Med (1995) 332(4):201–8. doi: 10.1056/nejm199501263320401
124. Pantaleo G, Menzo S, Vaccarezza M, Graziosi C, Cohen OJ, Demarest JF, et al. Studies in Subjects With Long-Term Nonprogressive Human Immunodeficiency Virus Infection. N Engl J Med (1995) 332(4):209–16. doi: 10.1056/nejm199501263320402
125. Moore RD, Keruly JC. CD4+ Cell Count 6 Years After Commencement of Highly Active Antiretroviral Therapy in Persons With Sustained Virologic Suppression. Clin Infect Dis (2007) 44(3):441–6. doi: 10.1086/510746
126. Kelley CF, Kitchen CM, Hunt PW, Rodriguez B, Hecht FM, Kitahata M, et al. Incomplete Peripheral CD4+ Cell Count Restoration in HIV-Infected Patients Receiving Long-Term Antiretroviral Treatment. Clin Infect Dis (2009) 48(6):787–94. doi: 10.1086/597093
127. McBride JA, Striker R. Imbalance in the Game of T Cells: What can the CD4/CD8 T-Cell Ratio Tell Us About HIV and Health? PloS Pathog (2017) 13(11):e1006624. doi: 10.1371/journal.ppat.1006624
128. Tinoco R, Carrette F, Barraza ML, Otero DC, Magaña J, Bosenberg MW, et al. PSGL-1 Is an Immune Checkpoint Regulator That Promotes T Cell Exhaustion. Immunity (2016) 44(5):1190–203. doi: 10.1016/j.immuni.2016.04.015
Keywords: P-selectin glycoprotein ligand 1, HIV, therapeutic target, infection, inflammation
Citation: Zaongo SD, Liu Y, Harypursat V, Song F, Xia H, Ma P and Chen Y (2021) P-Selectin Glycoprotein Ligand 1: A Potential HIV-1 Therapeutic Target. Front. Immunol. 12:710121. doi: 10.3389/fimmu.2021.710121
Received: 15 May 2021; Accepted: 28 July 2021;
Published: 09 August 2021.
Edited by:
Liwu Li, Virginia Tech, United StatesReviewed by:
Kalyani Pyaram, Kansas State University, United StatesLinda M. Bradley, Sanford Burnham Institute for Medical Research, United States
Valentin P. Yakubenko, East Tennessee State University, United States
Copyright © 2021 Zaongo, Liu, Harypursat, Song, Xia, Ma and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yaokai Chen, eWFva2FpY2hlbkBob3RtYWlsLmNvbQ==; Ping Ma, bWFwaW5ndGlhbmppbkAxNjMuY29t