 David E. Spaner1,2,3,4,5*
David E. Spaner1,2,3,4,5*- 1Biology Platform, Sunnybrook Research Institute, Toronto, ON, Canada
- 2Department of Immunology, University of Toronto, Toronto, ON, Canada
- 3Department of Medical Biophysics, University of Toronto, Toronto, ON, Canada
- 4Department of Medical Oncology, Sunnybrook Odette Cancer Center, Toronto, ON, Canada
- 5Department of Medicine, University of Toronto, Toronto, ON, Canada
In the past decade, aberrant O-GlcNAcylation has emerged as a new hallmark of cancer. O-GlcNAcylation is a post-translational modification that results when the amino-sugar β-D-N-acetylglucosamine (GlcNAc) is made in the hexosamine biosynthesis pathway (HBP) and covalently attached to serine and threonine residues in intracellular proteins by the glycosyltransferase O-GlcNAc transferase (OGT). O-GlcNAc moieties reflect the metabolic state of a cell and are removed by O-GlcNAcase (OGA). O-GlcNAcylation affects signaling pathways and protein expression by cross-talk with kinases and proteasomes and changes gene expression by altering protein interactions, localization, and complex formation. The HBP and O-GlcNAcylation are also recognized to mediate survival of cells in harsh conditions. Consequently, O-GlcNAcylation can affect many of the cellular processes that are relevant for cancer and is generally thought to promote tumor growth, disease progression, and immune escape. However, recent studies suggest a more nuanced view with O-GlcNAcylation acting as a tumor promoter or suppressor depending on the stage of disease or the genetic abnormalities, proliferative status, and state of the p53 axis in the cancer cell. Clinically relevant HBP and OGA inhibitors are already available and OGT inhibitors are in development to modulate O-GlcNAcylation as a potentially novel cancer treatment. Here recent studies that implicate O-GlcNAcylation in oncogenic properties of blood cancers are reviewed, focusing on chronic lymphocytic leukemia and effects on signal transduction and stress resistance in the cancer microenvironment. Therapeutic strategies for targeting the HBP and O-GlcNAcylation are also discussed.
Introduction
Upon entry into a cell, glucose is phosphorylated to fructose-6-phosphate before continuing down the glycolysis pathway. About 2-5% of fructose-6-phosphate is normally diverted into the hexosamine biosynthetic pathway (HBP) (Figure 1), a minor metabolic pathway increasingly recognized to have an important role in cancer biology (1). The rate-limiting enzyme of the pathway is glutamine fructose-6-phosphate amidotransferase (GFAT), which has two isoforms (GFAT1 and GFAT2 encoded by GFPT1 and GFPT2, respectively) and generates glucosamine (GlcN)-6-phosphate from fructose-6-phosphate and glutamine. Acetyl-CoA is added by GlcN-6-phosphate acetyl transferase (GNAT) to make N-acetyl glucosamine (GlcNAc)-6-phosphate, which is rearranged to GlcNAc-1-phosphate by GlcNAc-phosphoglucomutase (AGM). GlcNAc-1-phosphate Pryophosphorylase (AGX) adds uridine-diphosphate (UDP) to ultimately form the nucleotide-sugar uridine diphosphate N-acetylglucosamine (UDP-GlcNAc). UDP-GlcNAc is involved in glycosylation of cell-surface lipids and proteins (2) but also employed by O-linked GlcNAc transferase (OGT) to O-GlcNAcylate serine and threonine residues on intracellular proteins. These modifications can be removed by the deglycosylating enzyme, O-GlcNAcase (OGA) encoded by OGA, formerly known as MGEA5 (2, 3) (Figure 1).
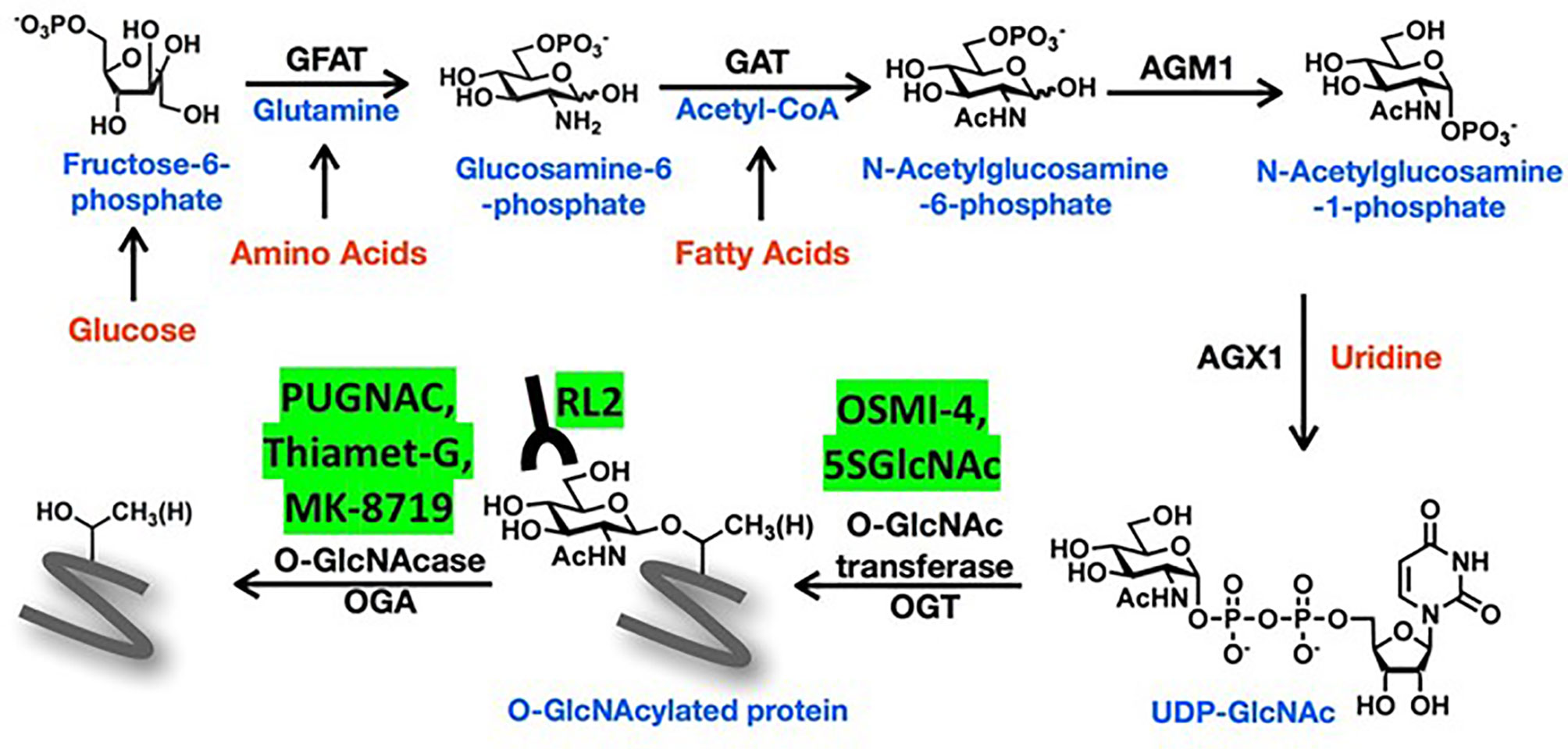
Figure 1 Schema of the hexosamine biosynthetic pathway (HBP). The amino sugar UDP-GlcNAc is generated in the HBP by combining fructose-6-phosphate, glutamine, nucleotides, and acetyl-CoA. More detailed description of the sequential enzyme reactions in the pathway is provided in the text. O-GlcNAc transferase (OGT) uses UDP-GlcNAc to transfer O-GlcNAc moieties onto target proteins that can be recognized by RL2 antibodies and removed by O-GlcNAse (OGA). OGT and OGA inhibitors mentioned in the text are shown in the yellow boxes.
The biological activity of thousands of different proteins can be altered by O-GlcNAcylation (4). Serine and threonine targets of OGT may be phosphorylation sites that are blocked by O-GlcNAc residues to disturb signaling pathways (1). Changes in protein structure by O-GlcNAcylation can also promote phosphorylation and alter cellular localization as well as protein-protein interactions and formation of supramolecular complexes including transcriptional regulators that produce changes in gene and protein expression. Cross-talk between O-GlcNAcylation and ubiquitination can modulate protein expression by either preventing or promoting proteasomal degradation (5).
Antibodies like RL-2 and CTD110.6 (6) are used to detect O-GlcNAc on proteins since antibodies to O-GlcNAcylated forms of specific proteins are generally unavailable (7). Strategies to manipulate O-GlcNAcylated proteins include the use of different concentrations of glucose and glutamine (8) or GlcN and uridine in tissue culture media (9). Along with genetic modification, OGA can be blocked by PUGNAC or Thiamet-G (TMG), and OGT by small molecules like OSMI-4 (10) and metabolic inhibitors like 2-deoxy-2-N-hexanamide-5-thio-D-glucopyranoside (5SGlcNHex) (11, 12) (Figure 1). The glutamine analogue 6-diazo-5-oxo-L-norleucine (DON) is often used in the literature as a GFAT inhibitor but it has multiple substrates that may confuse biological interpretation of the data (13).
Aberrant O-GlcNAcylation occurs in a variety of human diseases including diabetes and dementia (1, 2). O-GlcNAcylation is induced by stressful conditions and thought to act as a cellular survival mechanism (14, 15). Changes in O-GlcNAcylation from altered metabolism or stress can also dysregulate cell signaling networks (1, 14). Since cancer is the result of oncogenic events that cause replicative, proteotoxic, nutrient, and/or oxidative stresses (16, 17) and is driven by dysregulated signaling pathways (18), it is not surprising that high levels of O-GlcNAcylated proteins also characterize and are involved in the basic hallmarks of cancer (19, 20).
O-GlcNAcylation has been studied mainly in solid tumors where it is linked to enhanced glycolysis and aggressive clinical behavior (21). Its effects on transcription factors in cancer have also been reviewed recently (22–24). The purpose of this review is to discuss recent information regarding O-GlcNAcylation in signaling and survival (25) of cancer cells in the context of chronic lymphocytic leukemia, the first blood cancer noted to exhibit aberrant O-GlcNAcylation (2). Studies about O-GlcNAcylation in other blood cancers are also reviewed.
Chronic Lymphocytic Leukemia
CLL is the most common adult leukemia with an incidence of 4.9/100,000 new cases per year in the US (26). It is a cancer of CD19+ B cells that co-express the T cell marker CD5. Males are twice as likely as females to develop CLL and at risk for more aggressive disease (27, 28).
Like any cancer, CLL passes through stages of initiation, promotion, and progression (29, 30). It is initiated by genetic lesions that transform hematopoietic stem cells (31), immature B cells with immunoglobulin heavy chain variable (IGHV) genes in the germline configuration, or more mature antigen-experienced B cells that have undergone somatic hypermutation. Patients with unmutated IGHV disease are considered to have a more aggressive form of CLL (32).
Promoters cause transformed cells to proliferate until they reach sufficient numbers to become clinically evident (33). Hypercholesterolemia may promote tumor growth by altering membrane lipid content and affecting signaling modules in CLL cells (34–36). Signals that drive CLL cells to proliferate are delivered in proliferation centers (PCs) found in lymphoid organs (37) (Figure 2). Proliferative signals are transmitted through the B cell receptor (BCR), Toll-like receptors (TLRs), tumor necrosis factor (TNF) receptors like TNFR1 and TNFR2 (38), non-canonical TNF receptors like CD40, cytokines like type 1 and type 2 interferon (IFN), IL2, IL4, IL15, and IL21 (39–41), and NOTCH family members, particularly NOTCH 1 and 2 (42). Consequently, important signaling pathways in CLL include the NFκB, MEK/ERK, PI3K/AKT/mTOR, janus-kinase (JAK), and NOTCH pathways that can all be affected by O-GlcNAcylation (Figure 2).
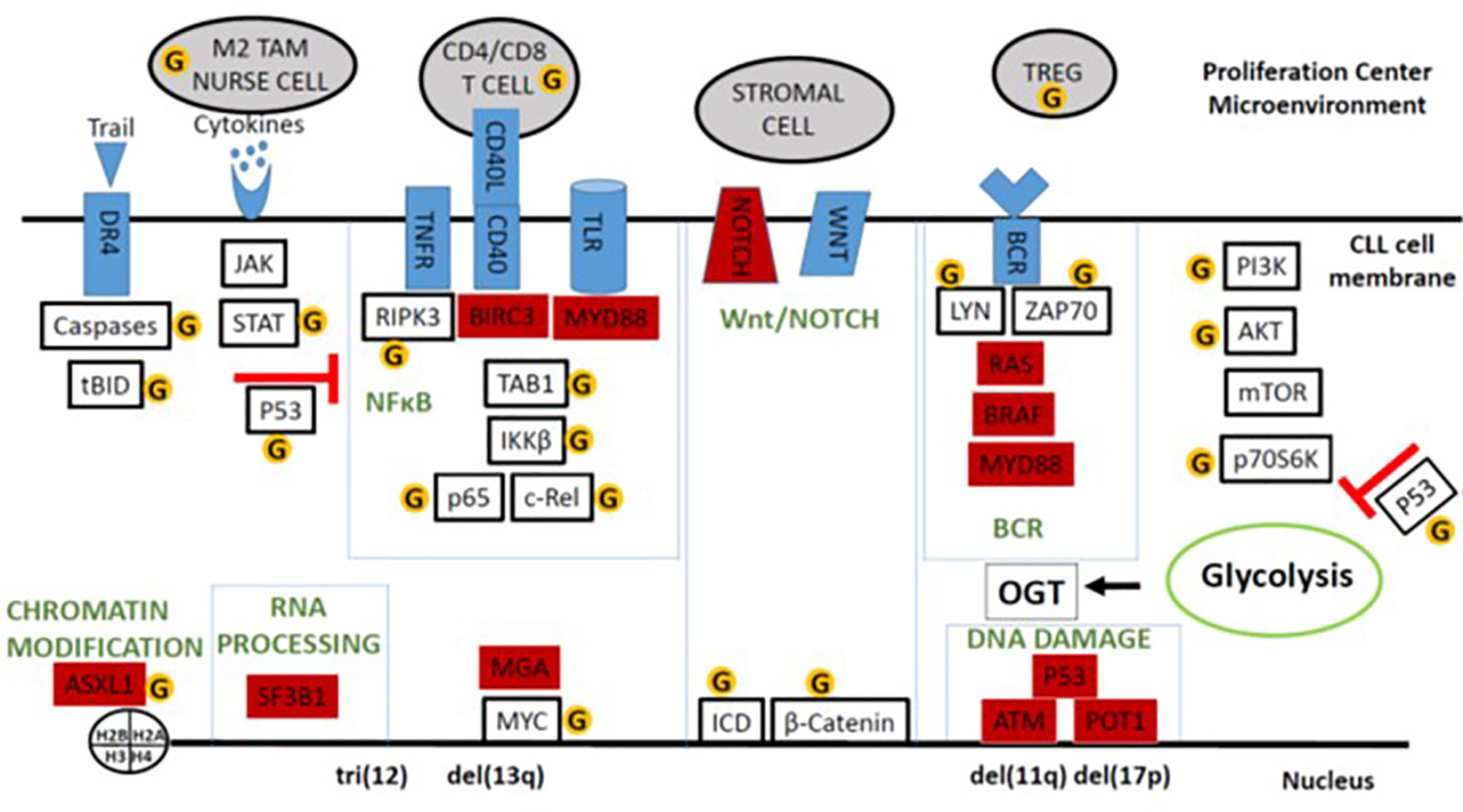
Figure 2 Schema of O-GlcNAcylated oncogenic processes in the CLL microenvironment. Major signaling and system modules that drive growth and progression of CLL cells are organized in the boxes along with important auxiliary pathways like cytokine- and AKT-signaling. Genes frequently mutated in CLL are colored in red. Proteins and cells discussed in the text that can be affected by O-GlcNAcylation are indicated by the yellow circled G’s. The figure emphasizes that O-GlcNAcylation increases the tumor-suppressing activity of wild-type p53 but enhances many of the signaling processes that support tumor growth in the presence of mutant p53 and an impaired p53 axis.
Resting CLL cells traffic from blood to PCs in response to chemokines and their receptors such as CXCR4 (43). CXCR4hiCD5lo cells in blood are about to re-enter PCs while CXCR4loCD5hi CLL cells are recent emigrants that retain some of the transcriptional program in that microenvironment (44). T cells, macrophages, stroma, as well as other leukemia cells produce the cytokines, TLR-ligands, and antigens in PCs that stimulate CLL cells (37). Macrophages in the CLL microenvironment are called “nurse” cells with properties of anti-inflammatory M2 macrophages (45, 46). They support leukemic growth and survival by making factors like the TNF-family member BAFF (45) and through direct protein-protein interactions (47). Nurse cells also impair effective anti-tumor immunity and prevent clearance of CLL cells by secreting immunosuppressive factors such as IL10 (46). Interactions of CLL cells and myeloid cells in the microenvironment cause expansion of regulatory T cells (Tregs) (48, 49) that contribute to immunosuppression and facilitate leukemic growth (Figure 2).
Heterogeneity of CLL
Despite a common CD5+CD19+ phenotype, CLL is marked by a heterogeneous clinical course ranging from a benign disease to one that can be fatal within a few years of diagnosis (32). Cytogenetic abnormalities in CLL cells distinguish 5 major sub-types. Trisomy 12 is found in 10-20% of patients while 40-50%, 10-12%, and 10-15% have deletions at 13q14, 11q22-q23, and 17p13, respectively, and 20-30% of patients display no apparent abnormalities with standard probes (32). Prior to the advent of novel therapies such as kinase and Bcl-2 inhibitors (49, 50), disease severity was associated with these abnormalities with del(17p) > del(11q) > trisomy 12 > normal cytogenetic analysis > isolated del(13q). Other factors associated with aggressive disease include an unmutated IGHV gene suggesting origin in an earlier stage of B cell development, advanced clinical stage, short lymphocyte doubling time in vivo, and high ZAP70 and β2-microglobulin levels (32). Genomic studies have identified over 40 driver mutations in genes such as NOTCH1, SF3B1, ATM, p53, BIRC3, POT1, BRAF, MGA and MYD88 that emphasize the diversity of CLL and affect prognosis (Figure 2) (16).
Dysregulated signaling is central to the biology of CLL. Enhanced responses of leukemia cells to BCR- or TLR-agonists in vitro are associated with aggressive clinical behavior and thought to reflect the events in PCs in vivo (51, 52). Cytokine signaling is also corrupted in clinically aggressive CLL cells that harbor mutations of ATM or TP53. Type 1 IFN inhibits growth of indolent CLL cells and activates the canonical signaling pathway, characterized by prolonged phosphorylation of STAT1 and brief phosphorylation of STAT3. In aggressive CLL cells, IFN causes prolonged STAT3 phosphorylation associated with immunosuppressive factor production and tumor growth in vitro (53).
Many driver mutations associated with more aggressive disease do not activate CLL cells directly but serve to amplify responses to proliferative signals in the CLL microenvironment (16) (Figure 2). NOTCH mutations characteristically prolong the life of the intracellular domain (ICD) that mediates transcription, producing enhanced NOTCH-signaling responses in more aggressive cells (54). Inactivating mutations of BIRC3 produce exaggerated non-canonical NFκB responses (55). Activating mutations in members of the RAS-BRAF-MAPK-ERK pathway lead to enhanced signaling through the BCR and other growth factor receptors (56). SF3B1 mutations promote mis-splicing of MAP3K7 (TAK1), resulting in hyperactivation of NFκB by dysregulating TNFα and TLR-signaling (57). Activating MYD88 mutations enhance signaling through TLRs but are often associated with more indolent disease (58) (Figure 2).
Aberrant activity of AKT and c-MYC are associated with more aggressive forms of CLL, including transformation into a drug-resistant large cell lymphoma called Richter’s transformation (RT) (59). Leukemia cells of patients with high-risk disease and RT express high amounts of activated phosphorylated AKT and a phenotype resembling RT resulted from constitutive activation of AKT in the Eµ-TCL1 CLL mouse model (59). The MYC repressor MGA is recurrently mutated in aggressive forms of CLL (60) and c-MYC activation is a feature of RT (61).
The state of the p53 axis helps clarify the classification of the different molecular sub-types into aggressive or indolent forms (62). P53 is the major tumor-suppressor in most cancers, controlling a myriad of pathways that repair DNA, inhibit glycolysis and proliferation, and promote cell death (63). CLL cells with deletions of chromosome 17 encoding TP53 are associated with an aggressive disease that is resistant to cytotoxic chemotherapy (32). Other subtypes exhibit a similar disease phenotype where the underlying molecular lesions impair the p53 axis independent of mutational damage to TP53. For example, MDM2 negatively regulates p53 by marking it for rapid clearance by the proteasome (62). MDM2 is located on chromosome 12 so that CLL cells with trisomy 12 have higher MDM2 levels causing decreased p53 levels and activity. ATM phosphorylates p53 through CHK2, preventing it from binding MDM2 and being degraded. CLL cells with deficient ATM activity would then have diminished levels and activity of p53 (63). POT1 is a component of the Shelterin complex that protects telomeres by activating p53. Inactivating POT1 mutations diminish the p53 axis and allow chromosomal instability of leukemia cells (64). In contrast, the p53 axis is generally intact in CLL cells with del(13q) and a more indolent course that is responsive to cytotoxic chemotherapy (53, 65). These observations suggest the disparate clinical behavior of CLL subtypes may reflect the underlying status of the p53 pathway (Figure 2).
O-GlcNAcylation IN CLL
Aberrant O-GlcNAcylation may play a role in CLL at a number of stages in disease progression. The sex-based incidence and severity of CLL could in part relate to the location of the OGT gene on the X-chromosome (28, 66). Dietary habits that produce hypercholesterolemia may be linked to the development of CLL in part by increasing O-GlcNAcylation in contributory cell types (67).
Like other cancers, CLL cells are characterized by high levels of O-GlcNAcylated proteins (2). Interestingly, miR-15a on chromosome 13 targets OGT (68) and is deleted in a majority of CLL cells (69). CLL cells express only GFAT1 in contrast to circulating peripheral blood mononuclear cells (PBMCs) that express both isoforms. Total levels of O-GlcNAcylated proteins are significantly higher in CLL cells than PBMCs but the O-GlcNAcome is quite variable, as indicated by the number and density of bands on an immunoblot developed with RL2 antibodies (2). All CLL cells have high levels of O-GlcNAcylated proteins compared to normal lymphocytes but leukemia cells with a lower RL2 index, based arbitrarily on the intensity of all RL2-staining bands on a gel normalized to β-actin, were associated with more aggressive disease indicated by greater hematopoietic impairment, high doubling times, and genetic lesions such as del(11q)and del(17p) (2). This observation is somewhat paradoxical as higher O-GlcNAc levels are often associated with more aggressive clinical behavior in solid tumors (70). However, similar to these findings in CLL, OGT and O-GlcNAcylated protein levels were significantly lower in ovarian cancers resistant to chemotherapy compared to chemosensitive cancers (71).
Reasons for the inverse correlation between total O-GlcNAcylated protein levels and clinical course in CLL are unclear. While high OGT mRNA expression is associated with an adverse outcome (Figure 3A, left panel), the ratio of OGT to OGA proteins is lower in CLL cells with unmutated IGHV genes (U) compared to cells with mutated genes (M) (Figure 3B). Relative changes in glycosylation and deglycosylation could produce lower O-GlcNAcylated protein levels in more aggressive CLL cells and higher levels in more indolent cells (Figure 3B). Differential O-GlcNAcylated protein expression may also reflect proteomic differences (74). Aggressive cells have different numbers and species of proteins (74) that are targets for O-GlcNAcylation compared to indolent cells (75–77). For example, higher levels of ZAP70, a signaling molecule that is O-GlcNAcylated in activated human T cells (4), are found in aggressive CLL cells (32, 51). Similarly, O-GlcNAcylated forms of OGT, p53, c-MYC, and AKT along with UDP-GlcNAc, OGT, and OGA exhibited inter-patient variability but sample sizes were too small to be able to correlate with clinical behavior (2).
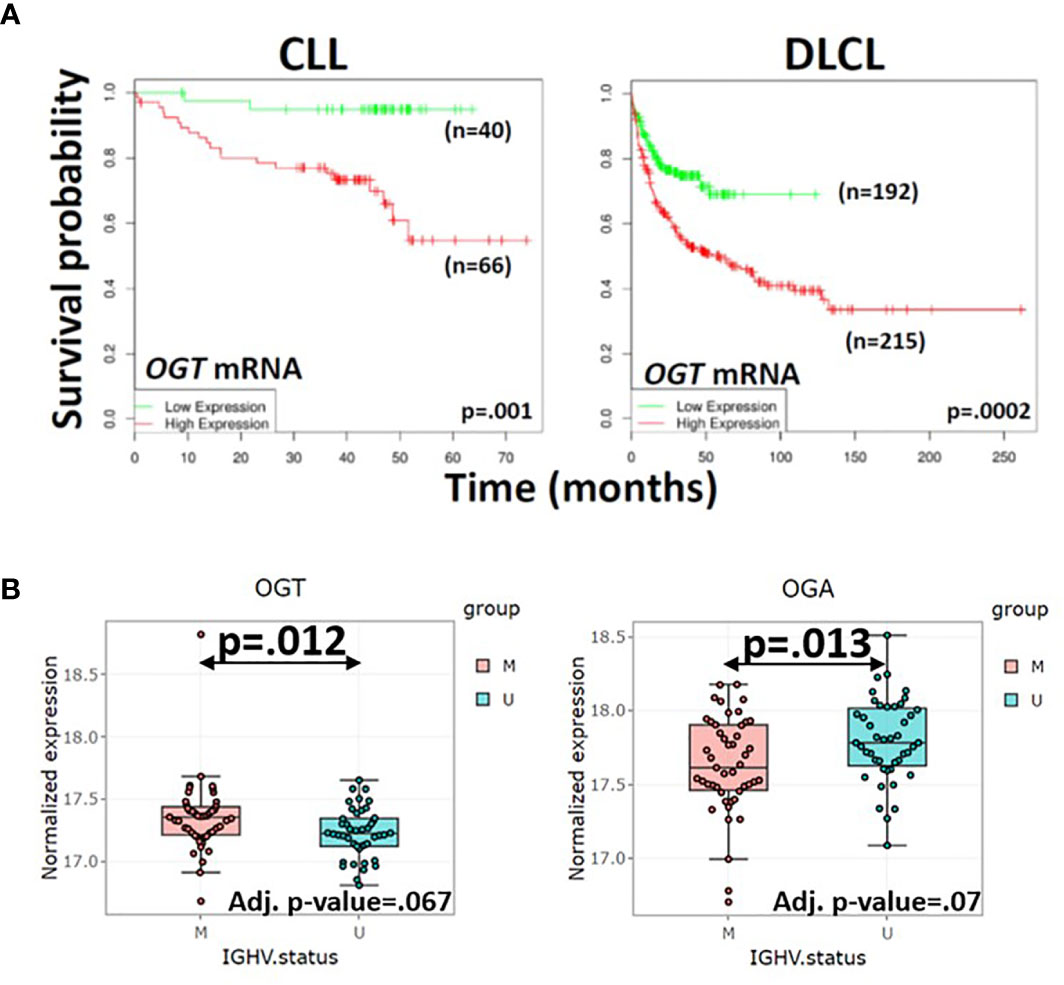
Figure 3 Correlation of OGT mRNA and protein levels in primary blood cancer cells with clinical course. (A) Survival of patients with high (n=66) or low (n=40) expression of OGT in CLL cells (left panel) and survival of diffuse large cell lymphoma patients with rituximab, cyclophosphamide, vincristine, adriamycin, and prednisone (R-CHOP) with high (n=215) or low (n=192) OGT expression in lymph node biopsies (right panel) were respectively compared by in silico analysis of the Herold (72) and Lenz (73) databases using the DRUGSURV bioinformatics analysis tool (http://www.bioprofiling.de/GEO/DRUGSURV). P-values for differences between the curves are 0.001 (left) and 0.0002 (right). (B) Expression of OGT (left) and OGA (right) proteins in 44 patients with aggressive CLL cells more likely to have an impaired p53 axis as indicated by unmutated IGHV genes (U) and 47 patients with more indolent CLL cells and mutated IGHV genes (M) were obtained with the R Shiny app (http://mozi.embl.de/public/proteomExplorer) provided for exploration of the CLL proteome (74). P-values and adjusted P-values are shown in the graphs and suggest more aggressive CLL cells have a lower OGT/OGA ratio, possibly consistent with relatively lower global O-GlcNAcylated protein levels.
The O-GlcNAcome in PCs has not been studied but is likely to differ from the blood compartment due to activating signals in that microenvironment. O-GlcNAcylated proteins, OGT, and OGA were found only in the IgD- fraction of normal tonsils, suggesting they are associated with a state of activation of human B cells in microenvironments that resemble PCs (2). Cellular activity including evidence of glycolysis also increased significantly in circulating CLL cells from patients administered the JAK inhibitor ruxolitinib (78, 79). This finding suggests cytokine-signaling through JAKs may negatively regulate O-GlcNAcylated protein levels but the nature of the cytokines and mechanism are currently unclear.
Effects of O-GlcNAcylation on Oncogenic Signaling Pathways in CLL
P53
The state of the p53 axis helps classify CLL cells (62) and may explain how O-GlcNAcylation can be associated with both aggressive and indolent disease. O-GlcNAcylation at Ser 149 stabilizes wild-type p53 by blocking ubiquitin-dependent proteolysis, facilitating nuclear localization, and transcription of p53 target genes (80, 81). Increased O-GlcNAcylation may then activate an intact p53 pathway to prevent cancer progression, compatible with an indolent clinical course.
In contrast, mutant p53 proteins appear to be unaffected by O-GlcNAcylation (81). Loss of p53 function can lead to upregulated glycolysis and enhanced O-GlcNAcylation by mass action. Impairment of upstream pathways causing disruption of the p53 axis may also result in increased glucose uptake, glycolysis, and HBP activity. For example, mutational inactivation of ATM contributes to cancer progression through a metabolic mechanism rather than its conventional role in DNA repair (82, 83). In the absence of p53, O-GlcNAcylation of other proteins as a result of increased HBP and OGT activity may increase their ability to promote tumor growth, potentially explaining how O-GlcNAcylation can also be associated with more aggressive clinical behavior of CLL cells.
AKT
Similar to p53, the PI3K/AKT pathway can also be affected in diametrically opposed ways by O-GlcNAcylation (21, 84). AKT is activated partially by PDK1-mediated phosphorylation at Thr308 and fully by mTORcomplex2 (mTORC2)-mediated phosphorylation at Ser473 (2). In a number of cancers, including of B cell origin (85), increased O-GlcNAcylation and OGT expression promote PI3K/AKT pathway activity and inhibition of OGT decreases AKT phosphorylation and downstream activity. Activators of the AKT pathway like DDX5 or TCL1 are stabilized by O-GlcNAcylation (86) or induced by O-GlcNAcylated transcription factors (87, 88).
In contrast, use of GlcN to increase O-GlcNAcylated protein levels decreased AKT activity in circulating CLL cells, evidenced by diminished size and lowered phosphorylation at Thr308 (2). Similar results are seen in other cell-types such as adipocytes (89). Why O-GlcNAcylation increases AKT-activity in some conditions and inhibits it in others is not clear but may reflect glucose uptake by the cells. Glucose transporters are decreased when O-GlcNAcylation has inhibitory effects on AKT but upregulated in situations where O-GlcNAcylation promotes AKT activity (84). The proliferative state of the cell may account for different behaviors of glucose transporters. Proliferating cells such as cancer cell models or CLL cells in PC microenvironments are highly glycolytic whereas adipocytes and circulating CLL cells are non-proliferative. The latter use mainly fatty oxidation as a metabolic strategy that is turned off when they are induced to proliferate (90).
NFκB
The theme of differential control of tumorigenesis by O-GlcNAcylation depending on the state of the p53 axis and metabolic state of the leukemia cell appears to also apply to NFκB (37). Several key molecules in the BCR-, TLR- and NFκB signaling pathways that drive the growth of CLL cells are targets for O-GlcNAcylation including LYN, IKKβ, p65, c-REL, and TAB1 (91–95). In the absence of p53, increased O-GlcNAcylation promotes phosphorylation of p65 on Ser536, which is critical for nuclear translocation of NFκB and expression of important genes in CLL growth and migration such as CXCR4 (96, 97). O-GlcNAcylation of IKKβ on Ser733 from enhanced glucose metabolism prevents inactivating phosphorylation at that site and sustains TNFα-induced NFκB activation in transformed human fibroblasts (98).
In non-proliferating macrophages and hepatocytes, O-GlcNAcylation of upstream signaling inputs prevents NFκB activation (99, 100). Polymerization of RIPK3 is required for TLR4 to activate NFκB and make inflammatory molecules like TNFα or assemble necrosomes to kill via necroptosis. O-GlcNAcylation prevents RIPK3 from polymerizing and inhibits TLR4-signaling in murine macrophages (99, 100). CLL cells do not form necrosome complexes but it is unclear if O-GlcNAcylation of RIPK3 is responsible (101). Use of GlcN to increase O-GlcNAcylated protein levels in non-proliferative CLL cells was found to impair TLR7-signaling responses that could be enhanced by an OGT inhibitor but the relevant O-GlcNAcylated proteins were not identified (2).
RAS, WNT, NOTCH, and MYC
The RAS/RAF/MEK/ERK, WNT/β-catenin/c-MYC, and NOTCH/c-MYC pathways are also important in CLL (16, 32, 56) and may be affected by O-GlcNAcylation. Activated RAS upregulates rate limiting enzymes of the HBP pathway, particularly GFAT2 (102, 103), and O-GlcNAcylated protein levels in cancer cells (104, 105). Mitogen-activated protein kinase kinase 2 (MEK2) can be O-GlcNAcylated at Thr13, which enhances phosphorylation at Thr394 and subsequent down-stream activation of ERK1/2 (106).
O-GlcNAcylation stabilizes β-catenin and may enhance cancer cell migration by regulating β-catenin levels (107, 108). Similarly, OGT modulates NOTCH-signaling by O-GlcNAcylating and protecting ICD from proteasomal degradation, leading to increased nuclear translocation and ICD-mediated responses (109, 110) (Figure 2). The WNT/β-catenin and NOTCH pathways increase transcription of c-myc, which mediates aggressive clinical behavior (61). O-GlcNAcylation at Thr58 is thought to stabilize MYC because phosphorylation at this site marks it for degradation. Inhibition of proteasomal destruction by O-GlcNAcylation would lead to stronger MYC activity and tumor progression (111, 112). Variable levels of O-GlcNAcylated c-MYC were seen in primary CLL cells but more samples must be studied to draw correlations with clinical course (2).
STAT Proteins
Members of the mammalian STAT family that includes STAT1-4, STAT5a/b, and STAT6 (113) are important in CLL biology. IL10 activates STAT3 and simultaneously helps control the growth of CLL cells while contributing to the characteristic profound state of immunosuppression (114, 115). Common gamma chain binding cytokines such as IL2, IL4, and IL15 support the growth of CLL cells and are associated with STAT5A/B and STAT6 activation (116). Type 1 and type 2 IFNs activate STAT1-3 and may help control the growth of CLL cells, particularly in the presence of the bruton’s tyrosine kinase-inhibitor ibrutinib (53, 117).
Treatment of mouse mammary epithelial cells with cytokines leads to O-GlcNAcylation of STAT1, STAT3, STAT5, and STAT6 (113). The role of O-GlcNAcylation of STAT6 has not yet been explored but O-GlcNAcylation of STAT5A on Thr92 promotes activating tyrosine phosphorylation in cancer cells (118). O-GlcNAcylation stabilizes phosphorylated STAT1 to increase the duration and strength of type 2 IFN-signaling responses in mesenchymal stem cells (119). O-GlcNAcylation of STAT3 on Thr717/719 inhibits activating STAT3 tyrosine phosphorylation, preventing IL10 production by macrophages (120) and shifting neural stem cell differentiation from neurons to astrocytes (121). Inhibition of O-GlcNAcylation enhanced tyrosine-phosphorylation and STAT3 activity in these systems (120, 121).
These results suggest O-GlcNAcylation promotes STAT1 and inhibits STAT3 activity but may be context and cell-type dependent. In CLL cells, type 1 IFN strongly phosphorylates STAT1 and also phosphorylates STAT3 for various times dependent on the leukemia subtype (53). However, lowered O-GlcNAcylated protein levels were associated with inhibition of STAT3 phosphorylation that was reversed by restoring O-GlcNAcylation with glucosamine (9).
Effects of O-GlcNAcylation on CLL Metabolism
Metabolic reprogramming is another hallmark of cancer cells (19). In contrast to resting cells that generally derive their energy from oxidative phosphorylation in mitochondria, cancer cells often exhibit the “Warburg effect” involving aerobic glycolysis that uses large amounts of glucose and glutamine for fuel and to make nucleosides, amino acids, and fatty acids for proliferation (122). Circulating CLL cells express high levels of pyruvate dehydrogenase kinase 4 (PDK4) that prevent glucose from being metabolized in the mitochondrial tricarboxylic acid cycle (37). They consequently depend on fatty acid oxidation (90) that may be linked to their immunosuppressive phenotype (123). PDK4 and fatty acid oxidation are turned off by the proliferative signals encountered in PCs (37, 90) but the increase in glycolysis is generally not high enough to be seen with 2-deoxy-2-[18F] fluoroglucose/positron emission tomography (FDG/PET) (124). In contrast, development of Richter’s transformation is associated with high glucose utilization detectable by FDG-PET (124).
Metabolism of cancer cells is reprogrammed by oncogenic signaling pathways and reflected by altered flux through the HBP and changes in O-GlcNAcylation (1). Aberrant O-GlcNAcylation can also directly influence the metabolic program of a cancer cell. Sustained high O-GlcNAcylated protein levels generally promote glycolysis and the Warburg effect (125) in a number of ways including O-GlcNAcylation of glycolytic enzymes. For example, the hexokinase HK1 forms glucose-6-phosphate from glucose, the initial step in most glucose-dependent metabolic pathways, and its activity is increased by O-GlcNAcylation (126).
Pyruvate kinase (PK) tetramers catalyze formation of ATP and pyruvate from ADP and phosphoenolpyruvate in the final step of glycolysis. O-GlcNAcylation of the PK isozyme PKM2 promotes less active dimerization and causes upstream glycolytic intermediates to accumulate and be routed into biosynthetic pathways such as the pentose phosphate shunt (PPP) to make ribose sugars for nucleoside formation (127). PK activity also requires binding by fructose-1,6-bisphosphate produced by phosphofructokinase 1 (PFK1). O-GlcNAcylation on Ser529 inhibits PFK1 and also directs glycolytic intermediates into the PPP (126). This O-GlcNAc modification of PFK1 may be unique to cancer cells as it was not seen in dividing normal T cells (126).
In addition to regulating glycolytic enzymes directly, O-GlcNAcylation modulates transcription factors and kinases to promote distinct metabolic programs. As described above, O-GlcNAcylation sustains the activity of c-MYC, a major inducer of glycolysis and glutaminolysis genes (128) as well as NFκB, which increases the rate of aerobic glycolysis in part by increasing transcription of glucose transporters, particularly in the absence of p53 (129).
Hypoxia-inducible factor (HIF1) regulates the expression of genes that contribute to aerobic glycolysis such as PFK1 and HK, glucose transporters such as GLUT1, and lactate dehydrogenase (LDH) (125) while carbohydrate responsive element binding protein (ChREBP) regulates PK isoforms and lipogenic enzymes such as acetyl-CoA carboxylase (ACC) (130, 131). HIF1 is composed of α and β subunits. Hydroxylation by prolyl hydroxylase domain protein 2 (PHD2) using O2 and α-ketoglutarate (α-KG) as substrates marks the α subunit for proteasomal degradation. Akin to hypoxia, O-GlcNAcylation prevents HIF1α destruction and upregulates HIF1 expression by inhibiting production of α-KG (125, 132). O-GlcNAcylation directly protects ChREBP from the proteasome to increase its level and transcriptional activity (130).
AKT/mTOR and AMPK are two other major regulators of the metabolic program of a cancer cell. The AKT/mTOR pathway promotes glucose uptake and utilization along with lipid and protein synthesis that are associated with the Warburg effect (122). In contrast, AMPK is activated by a low energy charge reflecting a high AMP/ATP ratio and phosphorylates substrates such as ACC1 and ACC2 to decrease lipogenesis along with TSC1/2 and Raptor to suppress mTORC1 activity and protein synthesis (133, 134).
O-GlcNAcylation of Akt has context-dependent effects and can sometimes promote or inhibit Akt activity (2, 85) but OGT and O-GlcNAcylation appear to clearly negatively regulate AMPK in solid tumor cells (125). O-GlcNAcylation of AMPK subunits inhibits kinase activity (135) and increases mTOR activity and protein synthesis (125). Inhibition of OGT by shRNA or small molecules to lower O-GlcNAcylated protein levels activated AMPK and decreased HIF1 activity along with growth and proliferation of breast cancer cells (125).
It is unclear how aberrant O-GlcNAcylation affects the metabolic program of CLL cells as they traffic from the circulation to lymph nodes and ultimately evolve a Richter’s transformation. Total O-GlcNAcylated protein levels appear to increase in lymph nodes (2) and likely increase even more as a result of increased Akt activity in a Richter’s transformation (59). Perhaps the relative decrease in O-GlcNAcylated protein levels in the circulation activates AMPK and increases fatty acid oxidation similar to the result of lowering O-GlcNAcylation in breast cancer cells (125).
O-GlcNAcylation and T Cells
Increased T cell numbers are central to the development and evolution of CLL. CD4+ T cell subsets exhibit dysregulation and the ratio of CD4+ to CD8+ T cells becomes increasingly skewed with more aggressive disease (136). Th1 and Th2 cells characterized by IFN-γ and IL-4 production respectively are increased in the blood of CLL patients compared to healthy controls and Th1 cells are even higher in progressive disease (137). Numbers of Th17 cells that make IL-17 are lower than Th1 and Th2 cells but still higher than in normal controls (138) while Tregs that make IL-10 and TGF-β are increased in the blood and lymph nodes of CLL patients (139) and correlate directly with more aggressive disease (140). A decrease in Th17 cells is associated with Treg expansion and disease progression while high Th17 cell numbers correlate with improved survival (141). CD8+ cells are also increased in the blood of CLL patients and correlate inversely with a more benign course (142).
CD4+ T cells are thought to promote the growth of CLL cells. They show evidence of activation in vivo and IFN-γ, IL-4, and IL-17 all increase survival of CLL cells in vitro (117, 143, 144). CD4+ but not CD8+ T cells are required to engraft human CLL cells in immunodeficient mice (145) and cytotoxic drugs like fludarabine may work in part by depleting CD4+ T cells (146).
In contrast, CD8+ T cells are thought to protect against tumor progression. They also show evidence of in vivo activation and depletion of CD8+ T cells hastened development of CLL in a transgenic mouse model (147). However, CD8+ T cells become exhausted, evidenced by decreased cytokine production and cytotoxic ability along with increased expression of checkpoint molecules such as PD1, and ultimately fail to control CLL (147, 148).
The metabolic program employed by a T cell is intimately connected with its phenotype and function (149) and involves many of the factors discussed above. The AKT/mTOR pathway coordinates glycolysis, lipid synthesis, and oxidative phosphorylation in part through HIF1α and c-MYC to promote Th1 and Th2 cell differentiation (150). Similarly, AKT/mTOR positively regulates the functional differentiation of Th17 cells that exhibit aerobic glycolysis and glutamine oxidation (151). In contrast, Tregs and memory CD8+ T cells rely more on AMPK and fatty acid oxidation (123, 152). T cell exhaustion is accompanied by metabolic dysregulation consisting of inhibited AKT/mTOR signaling, suppressed glycolysis, limited spare respiratory capacity, and dysregulated mitochondrial function causing oxidative stress (153).
Given its relationship with cellular metabolism, aberrant O-GlcNAcylation may also be associated with T cell defects in CLL. Activation of T cells is associated with O-GlcNAcylation of over 1000 proteins (4, 154) including c-MYC and NFAT (2, 155). When O-GlcNAcylation is blocked, T cells fail to increase c-MYC, produce cytokines, proliferate, or undergo proper thymic development in mice (156). Functional cytotoxic CD8+ effector T cells require strong HBP activity and contain higher amounts of UDP-GlcNAc than activated CD4+ T cells that are both higher than naïve T cells in mice (156).
O-GlcNAcylation is also involved in the differentiation of T cell subsets. Th2 differentiation requires activation of the mTOR complex mTORC2 (157), which leads to upregulation of GFAT1 in part by regulating the expression of XBP1s (15, 158). STAT5 activity also promotes Th2 differentiation (159) and is enhanced by O-GlcNAcylation (118).
Upregulation of O-GlcNAcylated protein levels by OGA inhibition in CD4+ T cells promoted Th17 function evidenced by increased IL-17 production. Fatty acid and cholesterol ligands for retinoic acid receptor-related orphan receptor gamma (RORγt), the lineage-defining transcription factor of Th17 cells, were increased by enhanced activity of O-GlcNAcylated ACC1, the rate-limiting enzyme in lipogenesis (160). Upregulation of NFκB activity by O-GlcNAcylation (94, 95) may also increase transcription of RORγt (161). Down-regulation of miR-15b, which negatively regulates OGT gene expression, is associated with increased Th17 cells in multiple sclerosis (162).
O-GlcNAcylation is also required for lineage stability and function of Tregs. Genetic deletion of OGT in Treg cells destabilized the lineage-defining transcription factor FoxP3, inhibited IL-2-mediated STAT5 activation that regulates functional capabilities of Tregs, and produced a severe inflammatory phenotype in mice (163).
How O-GlcNAcylation selectively promotes the differentiation of myriad Th subsets is not clear. The magnitude of total O-GlcNAcylation may confer some specificity similar to the switch from glycolysis to fatty acid oxidation that occurs when O-GlcNAcylated protein levels are lowered in breast cancer cells (125) or when CLL cells exit a lymph node (90). For example, Th17 cells and Tregs are linked in that they both require TGF-β to develop from precursors (164). Addition of the pro-inflammatory cytokine IL-6 diverts T cell differentiation from the Treg pathway toward the Th17 pathway (165). Perhaps increased glucose uptake imposed by IL-6 (166) leads to higher O-GlcNAcylated protein levels and a switch to the Th17 phenotype.
Limited information is available about the functional role of O-GlcNAcylation in CD8+ T cells and T cell exhaustion. Down-regulation of glycolysis in exhausted T cells (153) might lead to decreased HBP flux and lower O-GlcNAcylated protein levels. Highly glycolytic tumor cells can “steal” glucose from T cells and potentially lower O-GlcNAcylated protein expression together with mTOR activity, glycolysis, and cytokine production (167) but this mechanism seems unlikely to occur in CLL in the absence of Richter’s transformation (124). CLL cells make factors including exosomes (168) that cause CD8+ T cells to lose glucose transporter expression and mitochondrial mass and undergo oxidative stress consistent with features of exhaustion (148). Evaluation of the public database GSE8835 (169) suggests OGT, OGA and GFPT1 expression trend downward in healthy CD4+ T cells and are significantly down-regulated in CD8+ T cells when they are cultured with CLL cells (170).
Tumor Associated Macrophages (TAMs)
In addition to CLL cells and T cells (Figure 2), O-GlcNAcylation affects other cell-types in the tumor microenvironment such as TAMs that are an important source of oncogenic and immunosuppressive factors. Inflammatory cytokine production by TLR-activated macrophages is limited by O-GlcNAcylation (99, 171). TAMs have properties of M2 macrophages (45, 46) and OGT mediates M2 polarization in human macrophages (172). Increased UDP-GlcNAc has been associated with M2 polarization of murine macrophages (173) but another study found OGT and O-GlcNAcylation did not affect the M2 pathway although M1 polarization was suppressed by inhibiting S6K1 through O-GlcNAcylation of Ser489 (174).
O-GlcNAcylation in Other Hematological Malignancies
Aberrant O-GlcNAcylation appears to be a feature of other hematological malignancies, including myelodysplastic syndromes (MDS), acute myeloid leukemia (AML), acute lymphoblastic leukemia (ALL), mantle cell lymphoma (MCL), diffuse large cell lymphoma (DLCL), and multiple myeloma (MM). A lack of data, some of which appears contradictory, currently limits the understanding of the role of O-GlcNAcylation in blood cancers. Interestingly, cell-lines from hematological malignancies express higher levels of OGT compared to all other solid tumor cell lines (22). Moreover, high OGT expression in primary cells is associated with an adverse clinical outcome in DLCL and MM as well as CLL (Figure 3A), although a complete picture of the role of O-GlcNAcylation requires information on OGT and OGA protein expression along with a catalogue of the cancer specific O-GlcNAcome. While this association suggests OGT is a tumor promoter and novel therapeutic target, there is also evidence in AML and MDS that O-GlcNAcylation helps slow disease progression.
AML
Primary AML cells have increased OGT and GFAT1 expression compared to normal PBMCs (175). Inhibition of HBP activity with the purported GFAT inhibitor DON (13) resulted in growth arrest, differentiation, and death of OCI-AML3 and HL60 AML cell lines in vitro and decreased growth of HL-60 cells in immunodeficient mice without major toxicity (175). Resistance to chemotherapy was also associated with increased O-GlcNAcylated protein levels and the OGT-inhibitor OSMI-1 enhanced killing by doxorubicin in HL-60 cells and primary AML cells from patients with recurrent or resistant disease (176). Similarly, resveratrol inhibited growth of erythroleukemia cells in immunocompetent mice associated with down-regulation of O-GlcNAcylated proteins (9). Taken together, these results suggest OGT and O-GlcNAcylation may promote the growth of AML cells and constitute therapeutic targets in this cancer.
In contrast, OGT was found to stabilize ASXL1 by O-GlcNAcylation on Ser199 (177). ASXL1 is a tumor suppressor in hematologic malignancies that activates gene expression by methylating histones and forming the H3K4me3 mark associated with active transcription. Impaired activity of mutated ASXL1 is associated with failure of differentiation and development of MDS and AML. Knockdown of OGT prevented differentiation of HL-60 cells in response to ATRA while the OGA inhibitor PUGNAC increased O-GlcNAcylation and promoted differentiation as indicated by expression of the myeloid marker CD11b. Pretreatment with PUGNAC also prevented engraftment of leukemic cell-lines expressing a mutant form of ASXL1 in immunodeficient mice but not leukemic cells expressing another oncogene (177). Consistent with these findings, differentiation of leukemic blasts by cannabinoids produced clinical responses associated with increased expression of OGT in vitro and in vivo (178). Cannabinoid-mediated differentiation of Jurkat and MOLMM14 cell-lines used to model acute leukemia in vitro was blocked by gene-silencing of OGT and enhanced by increasing O-GlcNAcylation with the OGA inhibitor TMG (178).
The findings that inhibition of O-GlcNAcylation can both induce (175) and prevent (177, 178) differentiation of AML cells suggest OGT- and O-GlcNAc moieties may promote MDS and AML in some conditions and suppress it in others. Reasons for these discordant results are unclear. It may be that O-GlcNAcylation changes microenvironmental conditions induced by different chemotherapeutic drugs or has different outcomes depending on the stage of disease. For example, OGT and O-GlcNAcylation may have tumor suppressor activity early in the course of MDS and AML when ASXL1 is intact (179) but acquire tumor promoting activity when ASXL1 is inactivated by mutation. O-GlcNAcylation may also have different effects on specific AML subtypes and a detailed characterization of the genetic makeup of the AML cell or perhaps consideration of the state of the p53 axis as in CLL (62) may be required to know if inhibition or enhancement of O-GlcNAcylation is the appropriate treatment strategy.
ALL
Less information is available about ALL. OGT and O-GlcNAcylated proteins are increased while OGA is decreased in CD19+ cells from B-ALL patients compared to healthy donors (85). O-GlcNAcylated protein levels correlated directly with LDH in blood, perhaps reflecting enhanced glycolysis in more aggressive leukemia cells due to high levels of PI3K activity, phospho-AKT, and c-MYC (85). Inhibition of OGT lowered glycolysis and the PI3K/AKT/MYC axis in NALM-6 cells used to model ALL (85).
Lymphoma
OGT transcripts and protein levels are higher in DLCL cell lines and OGT mRNA is higher in primary DLCL cells than normal B cells (127, 180). Nuclear O-GlcNAcylated proteins are also higher in DLCL cell lines and primary cells (180). High OGT expression is associated with poor responses to chemotherapy (180) (Figure 3) and expression of OGA was also inversely associated with survival of DLCL patients (181). OGT was shown to O-GlcNAcylate pyruvate kinase M2 (PKM2), forcing formation of the dimer that shunts glucose into the pentose phosphate pathway to increase proliferation and growth of cancer cells (127). Inhibition of HBP pathway and O-GlcNAcylation with azaserine, another purported competitive GFAT inhibitor, down-regulated NFκB activity and increased killing of DLCL cell-lines. Cells with higher expression of OGT were more sensitive to azaserine (180).
As with AML, other forms of lymphoma gave opposite results. MCL cell-lines and primary cells were protected from bortezomib by alloxan used as an OGT inhibitor. Alloxan is not specific for OGT but the authors also showed the MCL cells were sensitized by several OGA inhibitors including PUGNAC, TMG, and ketoconazole. O-GlcNAcylation prevented degradation of tBID and increased its interactions with BCL2 family members and BAK to cause mitochondrial permeabilization and apoptosis (181).
MM
O-GlcNAcylated proteins may be especially important in myeloma given the role of XBP1 in both the pathogenesis of this blood cancer (182) and the hexosamine pathway (15). OGT levels are high in hematological cancer cell-lines and highest in myeloma models (22). Moreover, analysis of the GEO dataset GSE24080 suggests high OGT mRNA expression in primary myeloma cells is associated with an adverse outcome (183).
Public databases suggest O-GlcNAcylation is actually reduced in myeloma as OGA was significantly higher in 133 primary myeloma samples compared to normal plasma cells from 5 normal donors with similar OGT expression (184, 185). Myeloma microenvironments contain high levels of calcium released by osteolysis. Calcium-signaling in myeloma cell lines was found to decrease total O-GlcNAcylated protein levels associated with up-regulation of the integrins ITGB7 and ITGA4, enhanced motility, and more aggressive clinical behavior. Increasing O-GlcNAcylation by inhibiting calcium-signaling or genetic ablation of OGA resulted in proteasomal degradation of the integrins, decreased motility and decreased growth of RPMI8226 myeloma cells in immunodeficient mice (184). These findings recall results in CLL where higher O-GlcNAcylated protein levels are associated with a more indolent clinical course (2) and suggest disease progression might be slowed by strategies to increase O-GlcNAcylated protein levels in MM cells. In contrast, resistance of RPMI8226 cells to bortezomib, a drug used commonly to treat myeloma, was associated with increased O-GlcNAcylated protein levels and reversed by inhibiting OGT (186).
Overall, O-GlcNAcylation appears to have different effects in cancer cells depending on underlying genetic events and metabolic environments. Therapeutic manipulation of O-GlcNAcylation holds promise in blood cancers but requires knowing when O-GlcNAcylation has tumor promoting or tumor suppressing effects.
O-GlcNAcylation as a Tumor Promoter
Dysregulation of signaling pathways by O-GlcNAcylation orchestrates programs that promote growth and survival of cancer cells. Enhanced AKT-signaling by O-GlcNAcylation (85) inhibits apoptosis by phosphorylating the pro-apoptotic protein Bad and preventing caspase-3 activation (187). Enhanced NFκB activity (92, 94, 96, 188) prevents apoptosis by upregulating MCL-1 (189). O-GlcNAcylation can inhibit cleavage of apoptotic caspases, necroptosis (99, 100, 190), and TRAIL-mediated death by preventing oligomerization of DR5 and transmission of death-receptor signaling (191). Promotion of cancer cell proliferation and inhibition of apoptosis by O-GlcNAcylation is further supported by OGT-mediated O-GlcNAcylation and stabilization of the polycomb group transcription repressor Bmi-1, which inhibits transcription of p53, PTEN, and CDKN1A/CDKN2A genes and prevents their tumor suppressive activities (192).
The link between endoplasmic reticulum (ER) stress and the HBP (14, 15) suggests O-GlcNAcylation may enable cancer cells to survive in harsh microenvironmental environments. Limited nutrients and cytotoxic agents in the microenvironment impose stresses on cancer cells (25). O-GlcNAcylation affects metabolic changes that allow cancer cells to thrive in harsh conditions. For example, O-GlcNAcylation at Ser172 of the glycolytic regulator 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB3) is needed for tumor proliferation in hypoxic conditions. This modification promotes nuclear localization of PKFB3, which then prevents build-up of hypoxia-induced-P27 to allow growth in these conditions (193). O-GlcNAcylation of glucose-6-phosphate dehydrogenase (G6PD) promotes the pentose phosphate pathway that produces antioxidants and ribose sugars for nucletotide synthesis to support tumor growth in hypoxia (194). O-GlcNAcylation of fumarase (FH) prevents FH-catalyzed fumarate from inhibiting KDM2A demethylase activity that would otherwise facilitate expression of genes that mediate cell-cycle arrest in low-glucose (195).
Upregulation of O-GlcNAcylated proteins may also overcome oxidative and ER stresses imposed in the cancer microenvironment by radiation and cytotoxic drugs. Oxidative stress causes DNA damage that can be repaired by the HBP and OGT through O-GlcNAcylation of H2AX, H2AS40, and H2B histones, the Polycomb Related Complex 2 (PRK2) HMT catalytic subunit Ezh2, and the scaffold protein MDC1 to prevent radiation-induced senescence and cell-death (196, 197). Blocking OGT increases sensitivity to oxidative stress and DNA damage, leading to apoptosis or senescence and preventing tumor progression (196, 198).
ER stress in CLL cells is partly related to signaling through the BCR (199, 200) and may increase HBP activity through a IRE1/XBP1s/GFAT axis that can protect cells from death (15). Inhibition of this O-GlcNAcylation-mediated regulatory loop helps sensitize cancer cells to stress (201). Taken together, these observations are consistent with a view that O-GlcNAcylation may sustain cancer cells and that blocking OGT and the hexosamine pathway offers a novel approach to treatment for many cancers including CLL.
O-GlcNAcylation as a Tumor Suppressor
Situations also exist in which O-GlcNAcylation has anti-tumor activity. As described above, O-GlcNAcylation potentiates the tumor suppressive activity of wild-type p53 (80, 81) and down-regulates AKT and NFκB activity in non-proliferating CLL cells (2) (Figure 2). Stabilization of MYC by O-GlcNAcylation promotes cell growth under nutrient-rich conditions but causes activation-induced death in nutrient- and growth factor-poor conditions that can exist in a tumor microenvironment (202). Low OGT activity promotes c-MYC degradation to maintain survival in low glucose of cancer cells that were killed when O-GlcNAcylation was increased by the OGA inhibitor PUGNAC (203). Blocking OGT would then be expected to protect cancer cells in these conditions.
Nuclear factor erythroid 2 like 2 (NRF2) encoded by NFE2L2 is a master regulator of antioxidants that protect cancer cells from oxidative stress (25). KEAP1 is the primary negative regulator of NRF2 and OGT-mediated O-GlcNAcylation at Ser104 is required for efficient ubiquitination and degradation of NRF2. Glucose deprivation lowers O-GlcNAcylation and stabilizes NRF2, allowing cancer cells to survive in harsh conditions. The effects of low glucose on both O-GlcNAcylated proteins and NRF2 levels can be overcome by adding GlcNAc or GlcN to increase UDP-GlcNAc or by inhibiting OGA (204).
Autophagy is another mechanism employed by cancer cells to survive in nutritionally poor conditions. Autophagy is promoted by the formation of a SNARE complex containing the protein SNAP-29 that fuses autophagosomes with endosomes and lysomes. O-GlcNAcylation of SNAP-29 prevents assembly of the complex and negatively regulates autophagy (205, 206) in solid tumors. It is unclear if this mechanism applies to CLL cells or other hematological cancers.
O-GlcNAcylation as a Therapeutic Target
Blocking O-GlcNAcylation
Strategies to block OGT activity include direct inhibitors of OGT (10) and HBP enzymes, particularly GFAT (13). XBP1 inhibitors may also block O-GlcNAcylation (207) given XBP1 is a transcriptional regulator of GFAT (15). Flavonoid compounds like resveratrol can induce rapid proteasomal clearance of O-GlcNAcylated proteins from CLL cells (9).
Specific targeting of glycosyltransferases is difficult and no OGT inhibitors are currently available for clinical use. However, new inhibitors like OSMI-4 have low nanomolar potency and less off-target activity than previous ones (2, 10), suggesting they may form the basis for future phase 1 testing. 5SGlcNAc (11, 12) potently decreases O-GlcNAcylated proteins and UDP-GlcNAc in vitro by generating UDP-5SGlcNAc, a competitive OGT inhibitor with Ki=8 μM (11, 12). Poor aqueous solubility limits the use of such metabolic inhibitors but new analogs have greater activity in vivo and may ultimately be incorporated into leukemia treatment strategies (12). The glutamine antagonist DON inhibits GFAT non-specifically and has anti-cancer activity in vivo at the expense of toxicity that may be minimized with lower concentrations in novel dosing strategies (13).
Inhibition of OGT and O-GlcNAcylation prevent progression of a number of solid tumors (22, 102). Genetic ablation of OGT in pre-B cells downregulates c-MYC and prevents B cell development in mice (91) suggesting OGT may be a novel therapeutic target for B cell cancers such as ALL. Observations that resveratrol decreased O-GlcNAcylated protein levels and slowed tumor growth in CLL patients provide more support for O-GlcNAcylation inhibitors in leukemia (9). However, the anti-leukemic effects were transient and overcome by factors such as IFN that stimulated HBP activity (9). 5SGlcNAc analogues also had only transient effects in vivo (11, 12). Direct inhibition of OGT causes increased transcription of a reservoir pool of non-spliced OGT mRNA to restore O-GlcNAcylation (10). Accordingly, OGT inhibitors may be most effective when used with other therapeutic strategies including combination with GFAT inhibitors.
Consistent with this idea, inhibition of OGT enhances killing of solid tumor cells by conventional chemotherapeutic agents such as anthracyclines (176, 208). OGT promotes NRF1-mediated up-regulation of proteasome subunits (209), which may account in part for the intrinsic resistance of CLL cells to the proteasome inhibitor bortezomib and suggesting it may be overcome by inhibiting OGT (210). OGT inhibitors improve killing of cancer cells by PI3K inhibitors, suggesting OGT mediates resistance to PI3K inhibitors and possibly identifying a strategy to improve outcomes with kinase inhibitors in CLL patients (211, 212).
Enhancing O-GlcNAcylation
In contrast to OGT, OGA inhibitors are available for clinical use. MK-8719 has orphan drug status in the USA for the neurological condition progressive supranuclear palsy (PSP) but has not yet been explored in leukemia (213).
Cancer cells may be sensitized to chemotherapy in some situations by increasing O-GlcNAcylated proteins and protected by inhibiting O-GlcNAcylation. The status of the p53 axis may perhaps indicate these situations as wildtype p53 activity is potentiated by O-GlcNAcylation (80, 81). The OGA inhibitor TMG sensitizes human leukemia cells to the microtubule inhibitor paclitaxel (214) and mantle cell lymphoma cells to bortezomib (181). HBP- and OGT-inhibitors promote resistance of ovarian cancer cells to platinum-based chemotherapy by inducing autophagy (205, 206) and secretion of exosomes that decrease intracellular drug levels (215). O-GlcNAcylation of the death receptor DR4 on Ser424 sensitizes cancer cells to TRAIL-mediated apoptosis and necrosis. TRAIL-induced apoptosis is then increased by the OGA inhibitor TMG but prevented by the OGT inhibitor ST04589 (216).
Glucocorticoids (GCs) remain important drugs for blood cancers including CLL (217) and OGT has been shown to bind the glucocorticoid receptor (GR) and mediate transrepression of NFκB by O-GlcNAcylating RNA polymerase II (218). Inhibition of OGT prevents GC-mediated apoptosis (218).
Taken together, these observations suggest more detailed knowledge of its distinct role in the biological processes that underlie cancer hallmarks is still required to effectively manipulate O-GlcNAcylation as a treatment strategy (19). Global inhibition or enhancement of O-GlcNAcylation may not be the best treatment strategy as OGT is the only enzyme in humans capable of transferring GlcNAc moieties to proteins and inhibiting OGT may cause major toxicities. Consistent with this, mice injected with high doses of 5SGlcNHex became moribund although lower amounts were better tolerated (12). Given the role of O-GlcNAcylation in T cell biology (4, 156, 160), direct OGT inhibition may also cause significant immunosuppression. O-GlcNAcylated and de-glycosylated proteins can both mediate survival of cancer cells in different conditions and cancer cells with intact p53 functioning that may respond to increasing O-GlcNAcylation (81) could possibly be accelerated by HBP and OGT inhibition. If OGT-activity promotes the development and progression of cancers with impaired p53 axes but decreased OGT-activity helps them live in harsh microenvironments, a useful strategy in such cases might involve brief treatments with OGT inhibitors followed by infusion of GlcN or OGA inhibitors to block survival processes induced by prior OGT-inhibition. Another option might be to try to identify cancer cell specific vulnerabilities that are lethal in the presence of a low dose of an OGT inhibitor. For example, the GFAT2 inhibitor cycloserine was found to exhibit synthetic lethality with an OGT inhibitor in prostate cancer cells (219).
Summary and Future Work
Tremendous progress in the understanding of O-GlcNAcylation and its role in cancer has been made in the last decade. O-GlcNAcylation has emerged as a new cancer “hallmark” with an established role in a number of central oncogenic processes. Targeting O-GlcNAcylation offers a novel approach to improve results of current therapies and the lives of patients with hematological and other cancers.
Despite this progress, much remains to be done. More information is needed about how O-GlcNAcylation affects different blood cancers. It seems clear that O-GlcNAcylation can suppress or promote cancer development depending on the stage of disease, type of cancer, and microenvironmental conditions. Development of better mouse models might help clarify the effect of O-GlcNAc during oncogenesis. For example, the role of OGT as a tumor promoter or suppressor in CLL could be addressed in more detail by B cell lineage specific deletion of OGT in TCL1-transgenic mice, considered an excellent model of aggressive IGHV unmutated CLL (59, 200).
Cellular processes affected by aberrant O-GlcNAcylation in different cancers require additional investigation. OGT and O-GlcNAcylation are regulated by cell type-specific mechanisms so that results in one cancer do not necessarily generalize to others. Simply cataloguing proteins that are O-GlcNAcylated in different cancers and in different microenvironments would be helpful to implicate affected processes. Advances in chemoenzymatic labeling strategies for identifying O-GlcNAc modifications by mass-spectrometry (7, 190) make this possible and the results should be correlated with underlying genetic aberrations of the cancer cells.
Also needed is more detailed understanding of how O-GlcNAcylation affects important oncogenic signaling processes. Precise genetic subtyping of individual leukemia cells is required as different types have different levels of O-GlcNAcylation and protein targets that may have disparate effects on signaling responses. These studies should also be done in conditions that more accurately reflect conditions in a cancer microenvironment, including hypoxia, low-glucose, and three-dimensions (220). The role of O-GlcNAcylation in the T cell compartment of CLL (Figure 2) and its relationship to failure to clear leukemia cells also requires further study.
Clinically relevant OGT inhibitors may soon be available (10). As discussed above, results of globally increasing or decreasing O-GlcNAc levels may be limited but short-term blockade of OGT to minimize toxicity may synergize with many cytotoxic drugs and kinase inhibitors. Appropriate modulation of O-GlcNAcylation in T cells may lead to improved immunotherapies (221). The apparent importance of O-GlcNAcylation in hematologic malignancies suggests they may be useful models for evaluating OGT modulators in clinical trials.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
This work was supported by CIHR grant #374817 and the Leukemia and Lymphoma Society of Canada (LLSC).
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Slawson C, Hart GW. O-GlcNAc Signalling: Implications for Cancer Cell Biology. Nat Rev Cancer (2011) 11(9):678–84. doi: 10.1038/nrc3114
2. Shi Y, Tomic J, Wen F, Shaha S, Bahlo A, Harrison R, et al. Aberrant O-GlcNAcylation Characterizes Chronic Lymphocytic Leukemia. Leukemia (2010) 24(9):1588–98. doi: 10.1038/leu.2010.152
3. Zachara NE. Critical Observations That Shaped Our Understanding of the Function(s) of Intracellular Glycosylation (O-GlcNAc). FEBS Lett (2018) 592(23):3950–75. doi: 10.1002/1873-3468.13286
4. Lund PJ, Elias JE, Davis MM. Global Analysis of O-GlcNAc Glycoproteins in Activated Human T Cells. J Immunol (2016) 197(8):3086–98. doi: 10.4049/jimmunol.1502031
5. Ruan HB, Nie Y, Yang X. Regulation of Protein Degradation by O-GlcNAcylation: Crosstalk With Ubiquitination. Mol Cell Proteomics (2013) 12(12):3489–97. doi: 10.1074/mcp.R113.029751
6. Tashima Y, Stanley P. Antibodies That Detect O-Linked β-D-N-Acetylglucosamine on the Extracellular Domain of Cell Surface Glycoproteins. J Biol Chem (2014) 289(16):11132–42. doi: 10.1074/jbc.M113.492512
7. Alteen MG, Tan HY, Vocadlo DJ. Monitoring and Modulating O-GlcNAcylation: Assays and Inhibitors of O-GlcNAc Processing Enzymes. Curr Opin Struct Biol (2021) 68:157–65. doi: 10.1016/j.sbi.2020.12.008
8. Hwang JS, Kwon MY, Kim KH, Lee Y, Lyoo IK, Kim JE, et al. Lipopolysaccharide (LPS)-Stimulated iNOS Induction Is Increased by Glucosamine Under Normal Glucose Conditions But Is Inhibited by Glucosamine Under High Glucose Conditions in Macrophage Cells. J Biol Chem (2017) 292(5):1724–36. doi: 10.1074/jbc.M116.737940
9. Tomic J, McCaw L, Li Y, Hough MR, Ben-David Y, Moffat J, et al. Resveratrol has Anti-Leukemic Activity Associated With Decreased O-GlcNAcylated Proteins. Exp Hematol (2013) 41(8):675–86. doi: 10.1016/j.exphem.2013.04.004
10. Martin SES, Tan ZW, Itkonen HM, Duveau DY, Paulo JA, Janetzko J, et al. Structure-Based Evolution of Low Nanomolar O-GlcNAc Transferase Inhibitors. J Am Chem Soc (2018) 140(42):13542–5. doi: 10.1021/jacs.8b07328
11. Gloster TM, Zandberg WF, Heinonen JE, Shen DL, Deng L, Vocadlo DJ. Hijacking a Biosynthetic Pathway Yields a Glycosyltransferase Inhibitor Within Cells. Nat Chem Biol (2011) 7(3):174–81. doi: 10.1038/nchembio.520
12. Liu TW, Zandberg WF, Gloster TM, Deng L, Murray KD, Shan X, et al. Metabolic Inhibitors of O-GlcNAc Transferase That Act In Vivo Implicate Decreased O-GlcNAc Levels in Leptin-Mediated Nutrient Sensing. Angew Chem Int Ed Engl (2018) 57(26):7644–8. doi: 10.1002/anie.201803254
13. Lemberg KM, Vornov JJ, Rais R, Slusher BS. We're Not "DON" Yet: Optimal Dosing and Prodrug Delivery of 6-Diazo-5-Oxo-L-Norleucine. Mol Cancer Ther (2018) 17(9):1824–32. doi: 10.1158/1535-7163.MCT-17-1148
14. Martinez MR, Dias TB, Natov PS, Zachara NE. Stress-Induced O-GlcNAcylation: An Adaptive Process of Injured Cells. Biochem Soc Trans (2017) 45(1):237–49. doi: 10.1042/BST20160153
15. Wang ZV, Deng Y, Gao N, Pedrozo Z, Li DL, Morales CR, et al. Spliced X-Box Binding Protein 1 Couples the Unfolded Protein Response to Hexosamine Biosynthetic Pathway. Cell (2014) 156(6):1179–92. doi: 10.1016/j.cell.2014.01.014
16. Landau DA, Tausch E, Taylor-Weiner AN, Stewart C, Reiter JG, Bahlo J, et al. Mutations Driving CLL and Their Evolution in Progression and Relapse. Nature (2015) 526(7574):525–30. doi: 10.1038/nature15395
17. Seelige R, Searles S, Bui JD. Innate Sensing of Cancer's Non-Immunologic Hallmarks. Curr Opin Immunol (2018) 50:1–8. doi: 10.1016/j.coi.2017.09.005
18. Luo J, Solimini NL, Elledge SJ. Principles of Cancer Therapy: Oncogene and Non-Oncogene Addiction. Cell (2009) 136(5):823–37. doi: 10.1016/j.cell.2009.02.024
19. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
20. Fardini Y, Dehennaut V, Lefebvre T, Issad T. O-GlcNAcylation: A New Cancer Hallmark? Front Endocrinol (Lausanne) (2013) 4:99. doi: 10.3389/fendo.2013.00099
21. Very N, Vercoutter-Edouart AS, Lefebvre T, Hardivillé S, El Yazidi-Belkoura I. Cross-Dysregulation of O-GlcNAcylation and PI3K/AKT/mTOR Axis in Human Chronic Diseases. Front Endocrinol (Lausanne) (2018) 9:602. doi: 10.3389/fendo.2018.00602
22. Itkonen HM, Loda M, Mills IG. O-GlcNAc Transferase - An Auxiliary Factor or a Full-Blown Oncogene? Mol Cancer Res (2021) 19(4):555–64. doi: 10.1158/1541-7786.MCR-20-0926
23. Hanover JA, Chen W, Bond MR. O-GlcNAc in Cancer: An Oncometabolism-Fueled Vicious Cycle. J Bioenerg Biomembr (2018) 50(3):155–73. doi: 10.1007/s10863-018-9751-2
24. Parker MP, Peterson KR, Slawson C. O-GlcNAcylation and O-GlcNAc Cycling Regulate Gene Transcription: Emerging Roles in Cancer. Cancers (Basel) (2021) 13(7):1666. doi: 10.3390/cancers13071666
25. Li YJ, Sun L, Shi Y, Wang G, Wang X, Dunn SE, et al. PPAR-Delta Promotes Survival of Chronic Lymphocytic Leukemia Cells in Energetically Unfavorable Conditions. Leukemia (2017) 31(9):1905–14. doi: 10.1038/leu.2016.395
26. Available at: https://seer.cancer.gov/statfacts/html/clyl.html.
27. Nabhan C, Aschebrook-Kilfoy B, Chiu BC, Smith SM, Shanafelt TD, Evens AM, et al. The Impact of Race, Ethnicity, Age and Sex on Clinical Outcome in Chronic Lymphocytic Leukemia: A Comprehensive Surveillance, Epidemiology, and End Results Analysis in the Modern Era. Leuk Lymphoma (2014) 55(12):2778–84. doi: 10.3109/10428194.2014.898758
28. Catovsky D, Wade R, Else M. The Clinical Significance of Patients' Sex in Chronic Lymphocytic Leukemia. Haematologica (2014) 99(6):1088–94. doi: 10.3324/haematol.2013.101378
29. Pitot HC. The Natural History of Neoplastic Development: The Relation of Experimental Models to Human Cancer. Cancer (1982) 49(6):1206–11. doi: 10.1002/1097-0142(19820315)49:6<1206::aid-cncr2820490623>3.0.co;2-7
30. Sutton LA, Rosenquist R. Deciphering the Molecular Landscape in Chronic Lymphocytic Leukemia: Time Frame of Disease Evolution. Haematologica (2015) 100(1):7–16. doi: 10.3324/haematol.2014.115923
31. Kikushige Y, Ishikawa F, Miyamoto T, Shima T, Urata S, Yoshimoto G, et al. Self-Renewing Hematopoietic Stem Cell Is the Primary Target in Pathogenesis of Human Chronic Lymphocytic Leukemia. Cancer Cell (2011) 20(2):246–59. doi: 10.1016/j.ccr.2011.06.029
32. Fabbri G, Dalla-Favera R. The Molecular Pathogenesis of Chronic Lymphocytic Leukaemia. Nat Rev Cancer (2016) 16(3):145–62. doi: 10.1038/nrc.2016.8
33. Landgren O, Albitar M, Ma W, Abbasi F, Hayes RB, Ghia P, et al. B-Cell Clones as Early Markers for Chronic Lymphocytic Leukemia. N Engl J Med (2009) 360(7):659–67. doi: 10.1056/NEJMoa0806122
34. Chow S, Buckstein R, Spaner DE. A Link Between Hypercholesterolemia and Chronic Lymphocytic Leukemia. Leuk Lymphoma (2016) 57(4):797–802. doi: 10.3109/10428194.2015.1088651
35. Mozessohn L, Earle C, Spaner D, Cheng SY, Kumar M, Buckstein R. The Association of Dyslipidemia With Chronic Lymphocytic Leukemia: A Population-Based Study. J Natl Cancer Inst (2016) 109(3):1–9. doi: 10.1093/jnci/djw226
36. McCaw L, Shi Y, Wang G, Li YJ, Spaner DE. Low Density Lipoproteins Amplify Cytokine-Signaling in Chronic Lymphocytic Leukemia Cells. EBioMedicine (2017) 15:24–35. doi: 10.1016/j.ebiom.2016.11.033
37. Herishanu Y, Pérez-Galán P, Liu D, Biancotto A, Pittaluga S, Vire B, et al. The Lymph Node Microenvironment Promotes B-Cell Receptor Signaling, NF-kappaB Activation, and Tumor Proliferation in Chronic Lymphocytic Leukemia. Blood (2011) 117(2):563–74. doi: 10.1182/blood-2010-05-284984
38. Waage A, Espevik T. TNF Receptors in Chronic Lymphocytic Leukemia. Leuk Lymphoma (1994) 13(1-2):41–6. doi: 10.3109/10428199409051650
39. Haselager MV, Kater AP, Eldering E. Proliferative Signals in Chronic Lymphocytic Leukemia; What Are We Missing? Front Oncol (2020) 10:592205. doi: 10.3389/fonc.2020.592205
40. Dadashian EL, McAuley EM, Liu D, Shaffer AL 3rd, Young RM, Iyer JR, et al. TLR Signaling Is Activated in Lymph Node-Resident CLL Cells and Is Only Partially Inhibited by Ibrutinib. Cancer Res (2019) 79(2):360–71. doi: 10.1158/0008-5472.CAN-18-0781
41. Pascutti MF, Jak M, Tromp JM, Derks IA, Remmerswaal EB, Thijssen R, et al. IL-21 and CD40L Signals From Autologous T Cells can Induce Antigen-Independent Proliferation of CLL Cells. Blood (2013) 122(17):3010–9. doi: 10.1182/blood-2012-11-467670
42. Ryan RJH, Petrovic J, Rausch DM, Zhou Y, Lareau CA, Kluk MJ, et al. A B Cell Regulome Links Notch to Downstream Oncogenic Pathways in Small B Cell Lymphomas. Cell Rep (2017) 21(3):784–97. doi: 10.1016/j.celrep.2017.09.066
43. Chen SS, Chang BY, Chang S, Tong T, Ham S, Sherry B, et al. BTK Inhibition Results in Impaired CXCR4 Chemokine Receptor Surface Expression, Signaling and Function in Chronic Lymphocytic Leukemia. Leukemia (2016) 30(4):833–43. doi: 10.1038/leu.2015.316
44. Calissano C, Damle RN, Marsilio S, Yan XJ, Yancopoulos S, Hayes G, et al. Intraclonal Complexity in Chronic Lymphocytic Leukemia: Fractions Enriched in Recently Born/Divided and Older/Quiescent Cells. Mol Med (2011) 17(11-12):1374–82. doi: 10.2119/molmed.2011.00360
45. Burger JA, Tsukada N, Burger M, Zvaifler NJ, Dell'Aquila M, Kipps TJ. Blood-Derived Nurse-Like Cells Protect Chronic Lymphocytic Leukemia B Cells From Spontaneous Apoptosis Through Stromal Cell-Derived Factor-1. Blood (2000) 96(8):2655–63. doi: 10.1182/blood.V96.8.2655
46. Murray PJ. Macrophage Polarization. Annu Rev Physiol (2017) 79:541–66. doi: 10.1146/annurev-physiol-022516-034339
47. Boissard F, Tosolini M, Ligat L, Quillet-Mary A, Lopez F, Fournié JJ, et al. Nurse-Like Cells Promote CLL Survival Through LFA-3/CD2 Interactions. Oncotarget (2016) 8(32):52225–36. doi: 10.18632/oncotarget.13660
48. Jitschin R, Braun M, Büttner M, Dettmer-Wilde K, Bricks J, Berger J, et al. CLL-Cells Induce IDOhi CD14+HLA-DRlo Myeloid-Derived Suppressor Cells That Inhibit T-Cell Responses and Promote Tregs. Blood (2014) 124(5):750–60. doi: 10.1182/blood-2013-12-546416
49. Solman IG, Blum LK, Burger JA, Kipps TJ, Dean JP, James DF, et al. Impact of Long-Term Ibrutinib Treatment on Circulating Immune Cells in Previously Untreated Chronic Lymphocytic Leukemia. Leuk Res (2021) 102:106520. doi: 10.1016/j.leukres.2021.106520
50. Scheffold A, Stilgenbauer S. Revolution of Chronic Lymphocytic Leukemia Therapy: The Chemo-Free Treatment Paradigm. Curr Oncol Rep (2020) 22(2):16. doi: 10.1007/s11912-020-0881-4
51. Chiorazzi N, Rai KR, Ferrarini M. Chronic Lymphocytic Leukemia. N Engl J Med (2005) 352(8):804–15. doi: 10.1056/NEJMra041720
52. Longo PG, Laurenti L, Gobessi S, Petlickovski A, Pelosi M, Chiusolo P, et al. The Akt Signaling Pathway Determines the Different Proliferative Capacity of Chronic Lymphocytic Leukemia B-Cells From Patients With Progressive and Stable Disease. Leukemia (2007) 21(1):110–20. doi: 10.1038/sj.leu.2404417
53. Tomic J, Lichty B, Spaner DE. Aberrant Interferon-Signaling Is Associated With Aggressive Chronic Lymphocytic Leukemia. Blood (2011) 117(9):2668–80. doi: 10.1182/blood-2010-05-285999
54. Fabbri G, Holmes AB, Viganotti M, Scuoppo C, Belver L, Herranz D, et al. Common Nonmutational NOTCH1 Activation in Chronic Lymphocytic Leukemia. Proc Natl Acad Sci USA (2017) 114(14):E2911–9. doi: 10.1073/pnas.1702564114
55. Diop F, Moia R, Favini C, Spaccarotella E, De Paoli L, Bruscaggin A, et al. Biological and Clinical Implications of BIRC3 Mutations in Chronic Lymphocytic Leukemia. Haematologica (2020) 105(2):448–56. doi: 10.3324/haematol.2019.219550
56. Giménez N, Martínez-Trillos A, Montraveta A, Lopez-Guerra M, Rosich L, Nadeu F, et al. Mutations in the RAS-BRAF-MAPK-ERK Pathway Define a Specific Subgroup of Patients With Adverse Clinical Features and Provide New Therapeutic Options in Chronic Lymphocytic Leukemia. Haematologica (2019) 104(3):576–86. doi: 10.3324/haematol.2018.196931
57. Lee SC, North K, Kim E, Jang E, Obeng E, Lu SX, et al. Synthetic Lethal and Convergent Biological Effects of Cancer-Associated Spliceosomal Gene Mutations. Cancer Cell (2018) 34(2):225–41.8. doi: 10.1016/j.ccell.2018.07.003
58. Martínez-Trillos A, Pinyol M, Navarro A, Aymerich M, Jares P, Juan M, et al. Mutations in TLR/MYD88 Pathway Identify a Subset of Young Chronic Lymphocytic Leukemia Patients With Favorable Outcome. Blood (2014) 123(24):3790–6. doi: 10.1182/blood-2013-12-543306
59. Kohlhaas V, Blakemore SJ, Al-Maarri M, Nickel N, Pal M, Roth A, et al. Active Akt Signaling Triggers CLL Toward Richter Transformation via Overactivation of Notch1. Blood (2021) 137(5):646–60. doi: 10.1182/blood.2020005734
60. De Paoli L, Cerri M, Monti S, Rasi S, Spina V, Bruscaggin A, et al. MGA, a Suppressor of MYC, Is Recurrently Inactivated in High Risk Chronic Lymphocytic Leukemia. Leuk Lymphoma (2013) 54(5):1087–90. doi: 10.3109/10428194.2012.723706
61. Filip D, Mraz M. The Role of MYC in the Transformation and Aggressiveness of 'Indolent' B-Cell Malignancies. Leuk Lymphoma (2020) 61(3):510–24. doi: 10.1080/10428194.2019.1675877
62. Lodé L, Cymbalista F, Soussi T. Genetic Profiling of CLL: A 'TP53 Addict' Perspective. Cell Death Dis (2016) 7(1):e2042. doi: 10.1038/cddis.2015.415
63. Kastenhuber ER, Lowe SW. Putting P53 in Context. Cell (2017) 170(6):1062–78. doi: 10.1016/j.cell.2017.08.028
64. Calvete O, Martinez P, Garcia-Pavia P, Benitez-Buelga C, Paumard-Hernández B, Fernandez V, et al. A Mutation in the POT1 Gene Is Responsible for Cardiac Angiosarcoma in TP53-Negative Li-Fraumeni-Like Families. Nat Commun (2015) 6:8383. doi: 10.1038/ncomms9383
65. Pettitt AR, Sherrington PD, Stewart G, Cawley JC, Taylor AM, Stankovic T. P53 Dysfunction in B-Cell Chronic Lymphocytic Leukemia: Inactivation of ATM as an Alternative to TP53 Mutation. Blood (2001) 98(3):814–22. doi: 10.1182/blood.v98.3.814
66. Olivier-Van Stichelen S, Abramowitz LK, Hanover JA. X Marks the Spot: Does It Matter That O-GlcNAc Transferase Is an X-Linked Gene? Biochem Biophys Res Commun (2014) 453(2):201–7. doi: 10.1016/j.bbrc.2014.06.068
67. Lima VV, Giachini FR, Matsumoto T, Li W, Bressan AF, Chawla D, et al. High-Fat Diet Increases O-GlcNAc Levels in Cerebral Arteries: A Link to Vascular Dysfunction Associated With Hyperlipidaemia/Obesity? Clin Sci (Lond) (2016) 130(11):871–80. doi: 10.1042/CS20150777
68. You Z, Peng D, Cao Y, Zhu Y, Yin J, Zhang G, et al. P53 Suppresses the Progression of Hepatocellular Carcinoma via miR-15a by Decreasing OGT Expression and EZH2 Stabilization. J Cell Mol Med (2021) 25(19):9168–82. doi: 10.1111/jcmm.16792
69. Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, et al. Frequent Deletions and Down-Regulation of Micro- RNA Genes Mir15 and Mir16 at 13q14 in Chronic Lymphocytic Leukemia. Proc Natl Acad Sci USA (2002) 99(24):15524–9. doi: 10.1073/pnas.242606799
70. Ferrer CM, Sodi VL, Reginato MJ. O-GlcNAcylation in Cancer Biology: Linking Metabolism and Signaling. J Mol Biol (2016) 428(16):3282–94. doi: 10.1016/j.jmb.2016.05.028
71. Zhou F, Yang X, Zhao H, Liu Y, Feng Y, An R, et al. Down-Regulation of OGT Promotes Cisplatin Resistance by Inducing Autophagy in Ovarian Cancer. Theranostics (2018) 8(19):5200–12. doi: 10.7150/thno.27806
72. Herold T, Jurinovic V, Metzeler KH, Boulesteix AL, Bergmann M, Seiler T, et al. An Eight-Gene Expression Signature for the Prediction of Survival and Time to Treatment in Chronic Lymphocytic Leukemia. Leukemia (2011) 25(10):1639–45. doi: 10.1038/leu.2011.125
73. Lenz G, Wright G, Dave SS, Xiao W, Powell J, Zhao H, et al. Lymphoma/Leukemia Molecular Profiling Project. Stromal Gene Signatures in Large-B-Cell Lymphomas. N Engl J Med (2008) 359(22):2313–23. doi: 10.1056/NEJMoa0802885
74. Meier-Abt F, Lu J, Cannizzaro E, Pohly MF, Kummer S, Pfammatter S, et al. The Protein Landscape of Chronic Lymphocytic Leukemia (CLL). Blood (2021), blood.2020009741. doi: 10.1182/blood.2020009741
75. Meier-Abt F, Lu J, Cannizzaro E, Pohly MF, Kummer S, Pfammatter S, et al. The Protein Landscape of Chronic Lymphocytic Leukemia (CLL). Blood (2021) 29:blood.2020009741. doi: 10.1182/blood.2020009741
76. Huang PY, Mactier S, Armacki N, Giles Best O, Belov L, Kaufman KL, et al. Protein Profiles Distinguish Stable and Progressive Chronic Lymphocytic Leukemia. Leuk Lymphoma (2016) 57(5):1033–43. doi: 10.3109/10428194.2015.1094692
77. Johnston HE, Carter MJ, Cox KL, Dunscombe M, Manousopoulou A, Townsend PA, et al. Integrated Cellular and Plasma Proteomics of Contrasting B-Cell Cancers Reveals Common, Unique and Systemic Signatures. Mol Cell Proteomics (2017) 16(3):386–406. doi: 10.1074/mcp.M116.063511
78. Spaner DE, Wang G, McCaw L, Li Y, Disperati P, Cussen MA, et al. Activity of the Janus Kinase Inhibitor Ruxolitinib in Chronic Lymphocytic Leukemia: Results of a Phase II Trial. Haematologica (2016) 101(5):e192–5. doi: 10.3324/haematol.2015.135418
79. Spaner D, Luo Y, Wang G, Gallagher J, Tsui H, Shi Y. Janus-Kinases Restrain Chronic Lymphocytic Leukemia Cells in Patients on Ibrutinib: Results of a Phase II Trial. Cancer Med (2021). doi: 10.1002/cam4.4378
80. Yang WH, Kim JE, Nam HW, Ju JW, Kim HS, Kim YS, et al. Modification of P53 With O-Linked N-Acetylglucosamine Regulates P53 Activity and Stability. Nat Cell Biol (2006) 8(10):1074–83. doi: 10.1038/ncb1470
81. de Queiroz RM, Madan R, Chien J, Dias WB, Slawson C. Changes in O-Linked N-Acetylglucosamine (O-GlcNAc) Homeostasis Activate the P53 Pathway in Ovarian Cancer Cells. J Biol Chem (2016) 291(36):18897–914. doi: 10.1074/jbc.M116.734533
82. Xu L, Ma E, Zeng T, Zhao R, Tao Y, Chen X, et al. ATM Deficiency Promotes Progression of CRPC by Enhancing Warburg Effect. Endocr Relat Cancer (2019) 26(1):59–71. doi: 10.1530/ERC-18-0196
83. Ambrose M, Gatti RA. Pathogenesis of Ataxia-Telangiectasia: The Next Generation of ATM Functions. Blood (2013) 121(20):4036–45. doi: 10.1182/blood-2012-09-456897
84. Rehman S, Obaid A, Naz A, Ali A, Kanwal S, Ahmad J. Model-Based in Silico Analysis of the PI3K/Akt Pathway: The Elucidation of Cross-Talk Between Diabetes and Breast Cancer. PeerJ (2018) 6:e5917. doi: 10.7717/peerj.5917
85. Zhang B, Zhou P, Li X, Shi Q, Li D, Ju X. Bitterness in Sugar: O-GlcNAcylation Aggravates Pre-B Acute Lymphocytic Leukemia Through Glycolysis via the PI3K/Akt/c-Myc Pathway. Am J Cancer Res (2017) 7(6):1337–49.
86. Wu N, Jiang M, Han Y, Liu H, Chu Y, Liu H, et al. O-GlcNAcylation Promotes Colorectal Cancer Progression by Regulating Protein Stability and Potential Catcinogenic Function of DDX5. J Cell Mol Med (2019) 23(2):1354–62. doi: 10.1111/jcmm.14038
87. Jang H, Kim TW, Yoon S, Choi SY, Kang TW, Kim SY, et al. O-GlcNAc Regulates Pluripotency and Reprogramming by Directly Acting on Core Components of the Pluripotency Network. Cell Stem Cell (2012) 11(1):62–74. doi: 10.1016/j.stem.2012.03.001
88. Pekarsky Y, Palamarchuk A, Maximov V, Efanov A, Nazaryan N, Santanam U, et al. Tcl1 Functions as a Transcriptional Regulator and Is Directly Involved in the Pathogenesis of CLL. Proc Natl Acad Sci USA (2008) 105(50):19643–8. doi: 10.1073/pnas.0810965105
89. Vosseller K, Wells L, Lane MD, Hart GW. Elevated Nucleocytoplasmic Glycosylation by O-GlcNAc Results in Insulin Resistance Associated With Defects in Akt Activation in 3T3-L1 Adipocytes. Proc Natl Acad Sci USA (2002) 99(8):5313–8. doi: 10.1073/pnas.072072399
90. Spaner DE, Lee E, Shi Y, Wen F, Li Y, Tung S, et al. PPAR-Alpha Is a Therapeutic Target for Chronic Lymphocytic Leukemia. Leukemia (2013) 27(5):1090–9. doi: 10.1038/leu.2012.329
91. Wu JL, Chiang MF, Hsu PH, Tsai DY, Hung KH, Wang YH, et al. O-GlcNAcylation Is Required for B Cell Homeostasis and Antibody Responses. Nat Commun (2017) 8(1):1854. doi: 10.1038/s41467-017-01677-z
92. Allison DF, Wamsley JJ, Kumar M, Li D, Gray LG, Hart GW, et al. Modification of RelA by O-Linked N-Acetylglucosamine Links Glucose Metabolism to NF-κb Acetylation and Transcription. Proc Natl Acad Sci USA (2012) 109(42):16888–93. doi: 10.1073/pnas.1208468109
93. Pathak S, Borodkin VS, Albarbarawi O, Campbell DG, Ibrahim A, van Aalten DM. O-GlcNAcylation of TAB1 Modulates TAK1-Mediated Cytokine Release. EMBO J (2012) 31(6):1394–404. doi: 10.1038/emboj.2012.8
94. Ramakrishnan P, Clark PM, Mason DE, Peters EC, Hsieh-Wilson LC, Baltimore D. Activation of the Transcriptional Function of the NF-κb Protein C-Rel by O-GlcNAc Glycosylation. Sci Signal (2013) 6(290):ra75. doi: 10.1126/scisignal.2004097
95. Yang WH, Park SY, Nam HW, Kim DH, Kang JG, Kang ES, et al. NFkappaB Activation Is Associated With Its O-GlcNAcylation State Under Hyperglycemic Conditions. Proc Natl Acad Sci USA (2008) 105(45):17345–50. doi: 10.1073/pnas.0806198105
96. Ma Z, Vocadlo DJ, Vosseller K. Hyper-O-GlcNAcylation Is Anti-Apoptotic and Maintains Constitutive NF-κb Activity in Pancreatic Cancer Cells. J Biol Chem (2013) 288(21):15121–30. doi: 10.1074/jbc.M113.470047
97. Ali A, Kim SH, Kim MJ, Choi MY, Kang SS, Cho GJ, et al. O-GlcNAcylation of NF-κb Promotes Lung Metastasis of Cervical Cancer Cells via Upregulation of CXCR4 Expression. Mol Cells (2017) 40(7):476–84. doi: 10.14348/molcells.2017.2309
98. Kawauchi K, Araki K, Tobiume K, Tanaka N. Loss of P53 Enhances Catalytic Activity of IKKbeta Through O-Linked Beta-N-Acetyl Glucosamine Modification. Proc Natl Acad Sci USA (2009) 106(9):3431–6. doi: 10.1073/pnas.0813210106
99. Li X, Gong W, Wang H, Li T, Attri KS, Lewis RE, et al. O-GlcNAc Transferase Suppresses Inflammation and Necroptosis by Targeting Receptor-Interacting Serine/Threonine-Protein Kinase 3. Immunity (2019) 50(3):576–590.e6. doi: 10.1016/j.immuni.2019.01.007
100. Zhang B, Li MD, Yin R, Liu Y, Yang Y, Mitchell-Richards KA, et al. O-GlcNAc Transferase Suppresses Necroptosis and Liver Fibrosis. JCI Insight (2019) 4(21):e127709. doi: 10.1172/jci.insight.127709
101. Maas C, Tromp JM, van Laar J, Thijssen R, Elias JA, Malara A, et al. CLL Cells Are Resistant to Smac Mimetics Because of an Inability to Form a Ripoptosome Complex. Cell Death Dis (2013) 4(8):e782. doi: 10.1038/cddis.2013.305
102. Kim J, Lee HM, Cai F, Ko B, Yang C, Lieu EL, et al. The Hexosamine Biosynthesis Pathway Is a Targetable Liability in KRAS/LKB1 Mutant Lung Cancer. Nat Metab (2020) 2(12):1401–12. doi: 10.1038/s42255-020-00316-0
103. Taparra K, Wang H, Malek R, Lafargue A, Barbhuiya MA, Wang X, et al. O-GlcNAcylation Is Required for Mutant KRAS-Induced Lung Tumorigenesis. J Clin Invest (2018) 128(11):4924–37. doi: 10.1172/JCI94844
104. Zhang X, Ma L, Qi J, Shan H, Yu W, Gu Y. MAPK/ERK Signaling Pathway-Induced Hyper-O-GlcNAcylation Enhances Cancer Malignancy. Mol Cell Biochem (2015) 410(1-2):101–10. doi: 10.1007/s11010-015-2542-8
105. Jiang M, Qiu Z, Zhang S, Fan X, Cai X, Xu B, et al. Elevated O-GlcNAcylation Promotes Gastric Cancer Cells Proliferation by Modulating Cell Cycle Related Proteins and ERK 1/2 Signaling. Oncotarget (2016) 7(38):61390–402. doi: 10.18632/oncotarget.11359
106. Xu Y, Sheng X, Zhao T, Zhang L, Ruan Y, Lu H. O-GlcNAcylation of MEK2 Promotes the Proliferation and Migration of Breast Cancer Cells. Glycobiology (2021) 31(5):571–81. doi: 10.1093/glycob/cwaa103
107. Olivier-Van Stichelen S, Dehennaut V, Buzy A, Zachayus JL, Guinez C, Mir AM, et al. O-GlcNAcylation Stabilizes β-Catenin Through Direct Competition With Phosphorylation at Threonine 41. FASEB J (2014) 28(8):3325–38. doi: 10.1096/fj.13-243535
108. Harosh-Davidovich SB, Khalaila I. O-GlcNAcylation Affects β-Catenin and E-Cadherin Expression, Cell Motility and Tumorigenicity of Colorectal Cancer. Exp Cell Res (2018) 364(1):42–9. doi: 10.1016/j.yexcr.2018.01.024
109. Chen J, Dong X, Cheng X, Zhu Q, Zhang J, Li Q, et al. Ogt Controls Neural Stem/Progenitor Cell Pool and Adult Neurogenesis Through Modulating Notch Signaling. Cell Rep (2021) 34(13):108905. doi: 10.1016/j.celrep.2021.108905
110. Jeon JH, Suh HN, Kim MO, Ryu JM, Han HJ. Glucosamine-Induced OGT Activation Mediates Glucose Production Through Cleaved Notch1 and FoxO1, Which Coordinately Contributed to the Regulation of Maintenance of Self-Renewal in Mouse Embryonic Stem Cells. Stem Cells Dev (2014) 23(17):2067–79. doi: 10.1089/scd.2013.0583
111. Itkonen HM, Minner S, Guldvik IJ, Sandmann MJ, Tsourlakis MC, Berge V, et al. O-GlcNAc Transferase Integrates Metabolic Pathways to Regulate the Stability of C-MYC in Human Prostate Cancer Cells. Cancer Res (2013) 73(16):5277–87. doi: 10.1158/0008-5472.CAN-13-0549
112. Chou TY, Hart GW, Dang CV. C-Myc Is Glycosylated at Threonine 58, a Known Phosphorylation Site and a Mutational Hot Spot in Lymphomas. J Biol Chem (1995) 270(32):18961–5. doi: 10.1074/jbc.270.32.18961
113. Gewinner C, Hart G, Zachara N, Cole R, Beisenherz-Huss C, Groner B. The Coactivator of Transcription CREB-Binding Protein Interacts Preferentially With the Glycosylated Form of Stat5. J Biol Chem (2004) 279(5):3563–72. doi: 10.1074/jbc.M306449200
114. Forconi F, Moss P. Perturbation of the Normal Immune System in Patients With CLL. Blood (2015) 126(5):573–81. doi: 10.1182/blood-2015-03-567388
115. Liang X, Moseman EA, Farrar MA, Bachanova V, Weisdorf DJ, Blazar BR, et al. Toll-Like Receptor 9 Signaling by CpG-B Oligodeoxynucleotides Induces an Apoptotic Pathway in Human Chronic Lymphocytic Leukemia B Cells. Blood (2010) 115(24):5041–52. doi: 10.1182/blood-2009-03-213363
116. Schleiss C, Ilias W, Tahar O, Güler Y, Miguet L, Mayeur-Rousse C, et al. BCR-Associated Factors Driving Chronic Lymphocytic Leukemia Cells Proliferation Ex Vivo. Sci Rep (2019) 9(1):701. doi: 10.1038/s41598-018-36853-8
117. Xia M, Luo TY, Shi Y, Wang G, Tsui H, Harari D, et al. Effect of Ibrutinib on the IFN Response of Chronic Lymphocytic Leukemia Cells. J Immunol (2020) 205(10):2629–39. doi: 10.4049/jimmunol.2000478
118. Freund P, Kerenyi MA, Hager M, Wagner T, Wingelhofer B, Pham HTT, et al. O-GlcNAcylation of STAT5 Controls Tyrosine Phosphorylation and Oncogenic Transcription in STAT5-Dependent Malignancies. Leukemia (2017) 31(10):2132–42. doi: 10.1038/leu.2017.4
119. Jitschin R, Böttcher M, Saul D, Lukassen S, Bruns H, Loschinski R, et al. Inflammation-Induced Glycolytic Switch Controls Suppressivity of Mesenchymal Stem Cells via STAT1 Glycosylation. Leukemia (2019) 33(7):1783–96. doi: 10.1038/s41375-018-0376-6
120. Li X, Zhang Z, Li L, Gong W, Lazenby AJ, Swanson BJ, et al. Myeloid-Derived Cullin 3 Promotes STAT3 Phosphorylation by Inhibiting OGT Expression and Protects Against Intestinal Inflammation. J Exp Med (2017) 214(4):1093–109. doi: 10.1084/jem.20161105
121. White CW 3rd, Fan X, Maynard JC, Wheatley EG, Bieri G, Couthouis J, et al. Age-Related Loss of Neural Stem Cell O-GlcNAc Promotes a Glial Fate Switch Through STAT3 Activation. Proc Natl Acad Sci USA (2020) 117(36):22214–24. doi: 10.1073/pnas.2007439117
122. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science (2009) 324(5930):1029–33. doi: 10.1126/science.1160809
123. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting Edge: Distinct Glycolytic and Lipid Oxidative Metabolic Programs Are Essential for Effector and Regulatory CD4+ T Cell Subsets. J Immunol (2011) 186(6):3299–303. doi: 10.4049/jimmunol.1003613
124. Molica S. FDG/PET in CLL Today. Blood (2014) 123(18):2749–50. doi: 10.1182/blood-2014-03-563700
125. Ferrer CM, Lynch TP, Sodi VL, Falcone JN, Schwab LP, Peacock DL, et al. O-GlcNAcylation Regulates Cancer Metabolism and Survival Stress Signaling via Regulation of the HIF-1 Pathway. Mol Cell (2014) 54(5):820–31. doi: 10.1016/j.molcel.2014.04.026
126. Yi W, Clark PM, Mason DE, Keenan MC, Hill C, Goddard WA 3rd, et al. Phosphofructokinase 1 Glycosylation Regulates Cell Growth and Metabolism. Science (2012) 337(6097):975–80. doi: 10.1126/science.1222278
127. Singh JP, Qian K, Lee JS, Zhou J, Han X, Zhang B, et al. O-GlcNAcase Targets Pyruvate Kinase M2 to Regulate Tumor Growth. Oncogene (2020) 39(3):560–73. doi: 10.1038/s41388-019-0975-3
128. Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, et al. Myc Regulates a Transcriptional Program That Stimulates Mitochondrial Glutaminolysis and Leads to Glutamine Addiction. Proc Natl Acad Sci USA (2008) 105(48):18782–7. doi: 10.1073/pnas.0810199105
129. Kawauchi K, Araki K, Tobiume K, Tanaka N. P53 Regulates Glucose Metabolism Through an IKK-NF-KappaB Pathway and Inhibits Cell Transformation. Nat Cell Biol (2008) 10(5):611–8. doi: 10.1038/ncb1724
130. Sakiyama H, Fujiwara N, Noguchi T, Eguchi H, Yoshihara D, Uyeda K, et al. The Role of O-Linked GlcNAc Modification on the Glucose Response of ChREBP. Biochem Biophys Res Commun (2010) 402(4):784–9. doi: 10.1016/j.bbrc.2010.10.113
131. Yamashita H, Takenoshita M, Sakurai M, Bruick RK, Henzel WJ, Shillinglaw W, et al. A Glucose-Responsive Transcription Factor That Regulates Carbohydrate Metabolism in the Liver. Proc Natl Acad Sci USA (2001) 98(16):9116–21. doi: 10.1073/pnas.161284298
132. Semenza GL. HIF-1: Upstream and Downstream of Cancer Metabolism. Curr Opin Genet Dev (2010) 20(1):51–6. doi: 10.1016/j.gde.2009.10.009
133. Fullerton MD, Galic S, Marcinko K, Sikkema S, Pulinilkunnil T, Chen ZP, et al. Single Phosphorylation Sites in Acc1 and Acc2 Regulate Lipid Homeostasis and the Insulin-Sensitizing Effects of Metformin. Nat Med (2013) 19(12):1649–54. doi: 10.1038/nm.3372
134. Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK Phosphorylation of Raptor Mediates a Metabolic Checkpoint. Mol Cell (2008) 30(2):214–26. doi: 10.1016/j.molcel.2008.03.003
135. Bullen JW, Balsbaugh JL, Chanda D, Shabanowitz J, Hunt DF, Neumann D, et al. Cross-Talk Between Two Essential Nutrient-Sensitive Enzymes: O-GlcNAc Transferase (OGT) and AMP-Activated Protein Kinase (AMPK). J Biol Chem (2014) 289(15):10592–606. doi: 10.1074/jbc.M113.523068
136. Elston L, Fegan C, Hills R, Hashimdeen SS, Walsby E, Henley P, et al. Increased Frequency of CD4+ PD-1+ HLA-DR+ T Cells Is Associated With Disease Progression in CLL. Br J Haematol (2020) 188(6):872–80. doi: 10.1111/bjh.16260
137. Palma M, Gentilcore G, Heimersson K, Mozaffari F, Näsman-Glaser B, Young E, et al. T Cells in Chronic Lymphocytic Leukemia Display Dysregulated Expression of Immune Checkpoints and Activation Markers. Haematologica (2017) 102(3):562–72. doi: 10.3324/haematol.2016.151100
138. Hus I, Bojarska-Junak A, Chocholska S, Tomczak W, Woś J, Dmoszyńska A, et al. Th17/IL-17A Might Play a Protective Role in Chronic Lymphocytic Leukemia Immunity. PloS One (2013) 8(11):e78091. doi: 10.1371/journal.pone.0078091
139. de Weerdt I, Hofland T, de Boer R, Dobber JA, Dubois J, van Nieuwenhuize D, et al. Distinct Immune Composition in Lymph Node and Peripheral Blood of CLL Patients Is Reshaped During Venetoclax Treatment. Blood Adv (2019) 3(17):2642–52. doi: 10.1182/bloodadvances.2019000360
140. Weiss L, Melchardt T, Egle A, Grabmer C, Greil R, Tinhofer I. Regulatory T Cells Predict the Time to Initial Treatment in Early Stage Chronic Lymphocytic Leukemia. Cancer (2011) 117(10):2163–9. doi: 10.1002/cncr.25752
141. Jadidi-Niaragh F, Ghalamfarsa G, Memarian A, Asgarian-Omran H, Razavi SM, Sarrafnejad A, et al. Downregulation of IL-17-Producing T Cells Is Associated With Regulatory T Cell Expansion and Disease Progression in Chronic Lymphocytic Leukemia. Tumour Biol (2013) 34(2):929–40. doi: 10.1007/s13277-012-0628-4
142. Gonzalez-Rodriguez AP, Contesti J, Huergo-Zapico L, Lopez-Soto A, Fernández-Guizán A, Acebes-Huerta A, et al. Prognostic Significance of CD8 and CD4 T Cells in Chronic Lymphocytic Leukemia. Leuk Lymphoma (2010) 51(10):1829–36. doi: 10.3109/10428194.2010.503820
143. Aguilar-Hernandez MM, Blunt MD, Dobson R, Yeomans A, Thirdborough S, Larrayoz M, et al. IL-4 Enhances Expression and Function of Surface IgM in CLL Cells. Blood (2016) 127(24):3015–25. doi: 10.1182/blood-2015-11-682906
144. Zhu F, McCaw L, Spaner DE, Gorczynski RM. Targeting the IL-17/IL-6 Axis can Alter Growth of Chronic Lymphocytic Leukemia In Vivo/In Vitro. Leuk Res (2018) 66:28–38. doi: 10.1016/j.leukres.2018.01.006
145. Bagnara D, Kaufman MS, Calissano C, Marsilio S, Patten PE, Simone R, et al. A Novel Adoptive Transfer Model of Chronic Lymphocytic Leukemia Suggests a Key Role for T Lymphocytes in the Disease. Blood (2011) 117(20):5463–72. doi: 10.1182/blood-2010-12-324210
146. Gauthier M, Durrieu F, Martin E, Peres M, Vergez F, Filleron T, et al. Prognostic Role of CD4 T-Cell Depletion After Frontline Fludarabine, Cyclophosphamide and Rituximab in Chronic Lymphocytic Leukaemia. BMC Cancer (2019) 19(1):809. doi: 10.1186/s12885-019-5971-z
147. Hanna BS, Roessner PM, Yazdanparast H, Colomer D, Campo E, Kugler S, et al. Control of Chronic Lymphocytic Leukemia Development by Clonally-Expanded CD8+ T-Cells That Undergo Functional Exhaustion in Secondary Lymphoid Tissues. Leukemia (2019) 33(3):625–37. doi: 10.1038/s41375-018-0250-6
148. van Bruggen JAC, Martens AWJ, Fraietta JA, Hofland T, Tonino SH, Eldering E, et al. Chronic Lymphocytic Leukemia Cells Impair Mitochondrial Fitness in CD8+ T Cells and Impede CAR T-Cell Efficacy. Blood (2019) 134(1):44–58. doi: 10.1182/blood.2018885863
149. Buck MD, Sowell RT, Kaech SM, Pearce EL. Metabolic Instruction of Immunity. Cell (2017) 169(4):570–86. doi: 10.1016/j.cell.2017.04.004
150. Yang K, Shrestha S, Zeng H, Karmaus PW, Neale G, Vogel P, et al. T Cell Exit From Quiescence and Differentiation Into Th2 Cells Depend on Raptor-Mtorc1-Mediated Metabolic Reprogramming. Immunity (2013) 39(6):1043–56. doi: 10.1016/j.immuni.2013.09.015
151. Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, et al. Metabolic Programming and PDHK1 Control CD4+ T Cell Subsets and Inflammation. J Clin Invest (2015) 125(1):194–207. doi: 10.1172/JCI76012
152. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, et al. Enhancing CD8 T-Cell Memory by Modulating Fatty Acid Metabolism. Nature (2009) 460(7251):103–7. doi: 10.1038/nature08097
153. Bengsch B, Johnson AL, Kurachi M, Odorizzi PM, Pauken KE, Attanasio J, et al. Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8(+) T Cell Exhaustion. Immunity (2016) 45(2):358–73. doi: 10.1016/j.immuni.2016.07.008
154. Woo CM, Lund PJ, Huang AC, Davis MM, Bertozzi CR, Pitteri SJ. Mapping and Quantification of Over 2000 O-Linked Glycopeptides in Activated Human T Cells With Isotope-Targeted Glycoproteomics (Isotag). Mol Cell Proteomics (2018) 17(4):764–75. doi: 10.1074/mcp.RA117.000261
155. Golks A, Tran TT, Goetschy JF, Guerini D. Requirement for O-Linked N-Acetylglucosaminyltransferase in Lymphocytes Activation. EMBO J (2007) 26(20):4368–79. doi: 10.1038/sj.emboj.7601845
156. Swamy M, Pathak S, Grzes KM, Damerow S, Sinclair LV, van Aalten DM, et al. Glucose and Glutamine Fuel Protein O-GlcNAcylation to Control T Cell Self-Renewal and Malignancy. Nat Immunol (2016) 17(6):712–20. doi: 10.1038/ni.3439
157. Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, et al. The Kinase mTOR Regulates the Differentiation of Helper T Cells Through the Selective Activation of Signaling by Mtorc1 and Mtorc2. Nat Immunol (2011) 12(4):295–303. doi: 10.1038/ni.2005
158. Moloughney JG, Kim PK, Vega-Cotto NM, Wu CC, Zhang S, Adlam M, et al. Mtorc2 Responds to Glutamine Catabolite Levels to Modulate the Hexosamine Biosynthesis Enzyme Gfat1. Mol Cell (2016) 63(5):811–26. doi: 10.1016/j.molcel.2016.07.015
159. Zhu J, Cote-Sierra J, Guo L, Paul WE. Stat5 Activation Plays a Critical Role in Th2 Differentiation. Immunity (2003) 19(5):739–48. doi: 10.1016/s1074-7613(03)00292-9
160. Machacek M, Saunders H, Zhang Z, Tan EP, Li J, Li T, et al. Elevated O-GlcNAcylation Enhances Pro-Inflammatory Th17 Function by Altering the Intracellular Lipid Microenvironment. J Biol Chem (2019) 294(22):8973–90. doi: 10.1074/jbc.RA119.008373
161. Ruan Q, Kameswaran V, Zhang Y, Zheng S, Sun J, Wang J, et al. The Th17 Immune Response Is Controlled by the Rel-Rorγ-Rorγ T Transcriptional Axis. J Exp Med (2011) 208(11):2321–33. doi: 10.1084/jem.20110462
162. Liu R, Ma X, Chen L, Yang Y, Zeng Y, Gao J, et al. MicroRNA-15b Suppresses Th17 Differentiation and Is Associated With Pathogenesis of Multiple Sclerosis by Targeting O-GlcNAc Transferase. J Immunol (2017) 198(7):2626–39. doi: 10.4049/jimmunol.1601727
163. Liu B, Salgado OC, Singh S, Hippen KL, Maynard JC, Burlingame AL, et al. The Lineage Stability and Suppressive Program of Regulatory T Cells Require Protein O-GlcNAcylation. Nat Commun (2019) 10(1):354. doi: 10.1038/s41467-019-08300-3
164. Weaver CT, Hatton RD. Interplay Between the TH17 and TReg Cell Lineages: A Co-Evolutionary Perspective. Nat Rev Immunol (2009) 9(12):883–9. doi: 10.1038/nri2660
165. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the Context of an Inflammatory Cytokine Milieu Supports De Novo Differentiation of IL-17-Producing T Cells. Immunity (2006) 24(2):179–89. doi: 10.1016/j.immuni.2006.01.001
166. Kumari N, Das A, Bhatt AN. Interleukin-6 Confers Radio-Resistance by Inducing Akt-Mediated Glycolysis and Reducing Mitochondrial Damage in Cells. J Biochem (2020) 167(3):303–14. doi: 10.1093/jb/mvz091
167. Chang CH, Qiu J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell (2015) 162(6):1229–41. doi: 10.1016/j.cell.2015.08.016
168. Yuan Y, Wang L, Ge D, Tan L, Cao B, Fan H, et al. Exosomal O-GlcNAc Transferase From Esophageal Carcinoma Stem Cell Promotes Cancer Immunosuppression Through Up-Regulation of PD-1 in CD8+ T Cells. Cancer Lett (2021) 500:98–106. doi: 10.1016/j.canlet.2020.12.012
169. Available at: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE8835.
170. Görgün G, Holderried TA, Zahrieh D, Neuberg D, Gribben JG. Chronic Lymphocytic Leukemia Cells Induce Changes in Gene Expression of CD4 and CD8 T Cells. J Clin Invest (2005) 115(7):1797–805. doi: 10.1172/JCI24176
171. Al-Mukh H, Baudoin L, Bouaboud A, Sanchez-Salgado JL, Maraqa N, Khair M, et al. Lipopolysaccharide Induces GFAT2 Expression to Promote O-Linked β-N-Acetylglucosaminylation and Attenuate Inflammation in Macrophages. J Immunol (2020) 205(9):2499–510. doi: 10.4049/jimmunol.2000345
172. Surdziel E, Clay I, Nigsch F, Thiemeyer A, Allard C, Hoffman G, et al. Multidimensional Pooled shRNA Screens in Human THP-1 Cells Identify Candidate Modulators of Macrophage Polarization. PloS One (2017) 12(8):e0183679. doi: 10.1371/journal.pone.0183679
173. Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, et al. Network Integration of Parallel Metabolic and Transcriptional Data Reveals Metabolic Modules That Regulate Macrophage Polarization. Immunity (2015) 42(3):419–30. doi: 10.1016/j.immuni.2015.02.005
174. Yang Y, Li X, Luan HH, Zhang B, Zhang K, Nam JH, et al. OGT Suppresses S6K1-Mediated Macrophage Inflammation and Metabolic Disturbance. Proc Natl Acad Sci USA (2020) 117(28):16616–25. doi: 10.1073/pnas.1916121117
175. Asthana A, Ramakrishnan P, Vicioso Y, Zhang K, Parameswaran R. Hexosamine Biosynthetic Pathway Inhibition Leads to AML Cell Differentiation and Cell Death. Mol Cancer Ther (2018) 17(10):2226–37. doi: 10.1158/1535-7163.MCT-18-0426
176. Liu Y, Cao Y, Pan X, Shi M, Wu Q, Huang T, et al. O-GlcNAc Elevation Through Activation of the Hexosamine Biosynthetic Pathway Enhances Cancer Cell Chemoresistance. Cell Death Dis (2018) 9(5):485. doi: 10.1038/s41419-018-0522-0
177. Inoue D, Fujino T, Sheridan P, Zhang YZ, Nagase R, Horikawa S, et al. A Novel ASXL1-OGT Axis Plays Roles in H3K4 Methylation and Tumor Suppression in Myeloid Malignancies. Leukemia (2018) 32(6):1327–37. doi: 10.1038/s41375-018-0083-3
178. Kampa-Schittenhelm KM, Haverkamp T, Bonin M, Tsintari V, Bühring HJ, Haeusser L, et al. Epigenetic Activation of O-Linked β-N-Acetylglucosamine Transferase Overrides the Differentiation Blockage in Acute Leukemia. EBioMedicine (2020) 54:102678. doi: 10.1016/j.ebiom.2020.102678
179. Chen TC, Hou HA, Chou WC, Tang JL, Kuo YY, Chen CY, et al. Dynamics of ASXL1 Mutation and Other Associated Genetic Alterations During Disease Progression in Patients With Primary Myelodysplastic Syndrome. Blood Cancer J (2014) 4(1):e177. doi: 10.1038/bcj.2013.74
180. Pham LV, Bryant JL, Mendez R, Chen J, Tamayo AT, Xu-Monette ZY, et al. Targeting the Hexosamine Biosynthetic Pathway and O-Linked N-Acetylglucosamine Cycling for Therapeutic and Imaging Capabilities in Diffuse Large B-Cell Lymphoma. Oncotarget (2016) 7(49):80599–611. doi: 10.18632/oncotarget.12413
181. Luanpitpong S, Chanthra N, Janan M, Poohadsuan J, Samart P, U-Pratya Y, et al. Inhibition of O-GlcNAcase Sensitizes Apoptosis and Reverses Bortezomib Resistance in Mantle Cell Lymphoma Through Modification of Truncated Bid. Mol Cancer Ther (2018) 17(2):484–96. doi: 10.1158/1535-7163.MCT-17-0390
182. Carrasco DR, Sukhdeo K, Protopopova M, Sinha R, Enos M, Carrasco DE, et al. The Differentiation and Stress Response Factor XBP-1 Drives Multiple Myeloma Pathogenesis. Cancer Cell (2007) 11(4):349–60. doi: 10.1016/j.ccr.2007.02.015
183. Antonov AV, Krestyaninova M, Knight RA, Rodchenkov I, Melino G, Barlev NA. PPISURV: A Novel Bioinformatics Tool for Uncovering the Hidden Role of Specific Genes in Cancer Survival Outcome. Oncogene (2014) 33(13):1621–8. doi: 10.1038/onc.2013.119
184. Samart P, Luanpitpong S, Rojanasakul Y, Issaragrisil S. O-GlcNAcylation Homeostasis Controlled by Calcium Influx Channels Regulates Multiple Myeloma Dissemination. J Exp Clin Cancer Res (2021) 40(1):100. doi: 10.1186/s13046-021-01876-z
185. Agnelli L, Mosca L, Fabris S, Lionetti M, Andronache A, Kwee I, et al. A SNP Microarray and FISH-Based Procedure to Detect Allelic Imbalances in Multiple Myeloma: An Integrated Genomics Approach Reveals a Wide Gene Dosage Effect. Genes Chromosomes Cancer (2009) 48(7):603–14. doi: 10.1002/gcc.20668
186. Hashimoto E, Okuno S, Hirayama S, Arata Y, Goto T, Kosako H, et al. Enhanced O-GlcNAcylation Mediates Cytoprotection Under Proteasome Impairment by Promoting Proteasome Turnover in Cancer Cells. iScience (2020) 23(7):101299. doi: 10.1016/j.isci.2020.101299
187. Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, et al. Akt Phosphorylation of BAD Couples Survival Signals to the Cell-Intrinsic Death Machinery. Cell (1997) 91(2):231–41. doi: 10.1016/s0092-8674(00)80405-5
188. Ma Z, Chalkley RJ, Vosseller K. Hyper-O-GlcNAcylation Activates Nuclear Factor κ-Light-Chain-Enhancer of Activated B Cells (NF-κb) Signaling Through Interplay With Phosphorylation and Acetylation. J Biol Chem (2017) 292(22):9150–63. doi: 10.1074/jbc.M116.766568
189. Cosimo E, McCaig AM, Carter-Brzezinski LJ, Wheadon H, Leach MT, Le Ster K, et al. Inhibition of NF-κb-Mediated Signaling by the Cyclin-Dependent Kinase Inhibitor CR8 Overcomes Prosurvival Stimuli to Induce Apoptosis in Chronic Lymphocytic Leukemia Cells. Clin Cancer Res (2013) 19(9):2393–405. doi: 10.1158/1078-0432.CCR-12-2170
190. Chuh KN, Batt AR, Zaro BW, Darabedian N, Marotta NP, Brennan CK, et al. The New Chemical Reporter 6-Alkynyl-6-Deoxy-GlcNAc Reveals O-GlcNAc Modification of the Apoptotic Caspases That Can Block the Cleavage/Activation of Caspase-8. J Am Chem Soc (2017) 139(23):7872–85. doi: 10.1021/jacs.7b02213
191. Yang SZ, Xu F, Yuan K, Sun Y, Zhou T, Zhao X, et al. Regulation of Pancreatic Cancer TRAIL Resistance by Protein O-GlcNAcylation. Lab Invest (2020) 100(5):777–85. doi: 10.1038/s41374-019-0365-z
192. Li Y, Wang L, Liu J, Zhang P, An M, Han C, et al. O-GlcNAcylation Modulates Bmi-1 Protein Stability and Potential Oncogenic Function in Prostate Cancer. Oncogene (2017) 36(45):6293–305. doi: 10.1038/onc.2017.223
193. Lei Y, Chen T, Li Y, Shang M, Zhang Y, Jin Y, et al. O-GlcNAcylation of PFKFB3 Is Required for Tumor Cell Proliferation Under Hypoxia. Oncogenesis (2020) 9(2):21. doi: 10.1038/s41389-020-0208-1
194. Rao X, Duan X, Mao W, Li X, Li Z, Li Q, et al. O-GlcNAcylation of G6PD Promotes the Pentose Phosphate Pathway and Tumor Growth. Nat Commun (2015) 6:8468. doi: 10.1038/ncomms9468
195. Wang T, Yu Q, Li J, Hu B, Zhao Q, Ma C, et al. O-GlcNAcylation of Fumarase Maintains Tumour Growth Under Glucose Deficiency. Nat Cell Biol (2017) 19(7):833–43. doi: 10.1038/ncb3562
196. Efimova EV, Appelbe OK, Ricco N, Lee SS, Liu Y, Wolfgeher DJ, et al. O-GlcNAcylation Enhances Double-Strand Break Repair, Promotes Cancer Cell Proliferation, and Prevents Therapy-Induced Senescence in Irradiated Tumors. Mol Cancer Res (2019) 17(6):1338–50. doi: 10.1158/1541-7786.MCR-18-1025
197. Na HJ, Akan I, Abramowitz LK, Hanover JA. Nutrient-Driven O-GlcNAcylation Controls DNA Damage Repair Signaling and Stem/Progenitor Cell Homeostasis. Cell Rep (2020) 31(6):107632. doi: 10.1016/j.celrep.2020.107632
198. Zafir A, Readnower R, Long BW, McCracken J, Aird A, Alvarez A, et al. Protein O-GlcNAcylation Is a Novel Cytoprotective Signal in Cardiac Stem Cells. Stem Cells (2013) 31(4):765–75. doi: 10.1002/stem.1325
199. Krysov S, Steele AJ, Coelho V, Linley A, Sanchez Hidalgo M, Carter M, et al. Stimulation of Surface IgM of Chronic Lymphocytic Leukemia Cells Induces an Unfolded Protein Response Dependent on BTK and SYK. Blood (2014) 124(20):3101–9. doi: 10.1182/blood-2014-04-567198
200. Kriss CL, Pinilla-Ibarz JA, Mailloux AW, Powers JJ, Tang CH, Kang CW, et al. Overexpression of TCL1 Activates the Endoplasmic Reticulum Stress Response: A Novel Mechanism of Leukemic Progression in Mice. Blood (2012) 120(5):1027–38. doi: 10.1182/blood-2011-11-394346
201. Walter LA, Lin YH, Halbrook CJ, Chuh KN, He L, Pedowitz NJ, et al. Inhibiting the Hexosamine Biosynthetic Pathway Lowers O-GlcNAcylation Levels and Sensitizes Cancer to Environmental Stress. Biochemistry (2020) 59(34):3169–79. doi: 10.1021/acs.biochem.9b00560
202. Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H, Brooks M, et al. Induction of Apoptosis in Fibroblasts by C-Myc Protein. Cell (1992) 69(1):119–28. doi: 10.1016/0092-8674(92)90123-t
203. Burén S, Gomes AL, Teijeiro A, Fawal MA, Yilmaz M, Tummala KS, et al. Regulation of OGT by URI in Response to Glucose Confers C-MYC-Dependent Survival Mechanisms. Cancer Cell (2016) 30(2):290–307. doi: 10.1016/j.ccell.2016.06.023
204. Chen PH, Smith TJ, Wu J, Siesser PF, Bisnett BJ, Khan F, et al. Glycosylation of KEAP1 Links Nutrient Sensing to Redox Stress Signaling. EMBO J (2017) 36(15):2233–50. doi: 10.15252/embj.201696113
205. Guo B, Liang Q, Li L, Hu Z, Wu F, Zhang P, et al. O-GlcNAc-Modification of SNAP-29 Regulates Autophagosome Maturation. Nat Cell Biol (2014) 16(12):1215–26. doi: 10.1038/ncb3066
206. Zhou F, Yang X, Zhao H, Liu Y, Feng Y, An R, et al. Down-Regulation of OGT Promotes Cisplatin Resistance by Inducing Autophagy in Ovarian Cancer. Theranostics (2018) 8(19):5200–12. doi: 10.7150/thno.27806
207. Shao A, Xu Q, Spalek WT, Cain CF, Kang CW, Tang CA, et al. Development of Tumor-Targeting IRE-1 Inhibitors for B-Cell Cancer Therapy. Mol Cancer Ther (2020) 19(12):2432–44. doi: 10.1158/1535-7163.MCT-20-0127
208. Lee SJ, Kwon OS. O-GlcNAc Transferase Inhibitor Synergistically Enhances Doxorubicin-Induced Apoptosis in HepG2 Cells. Cancers (Basel) (2020) 12(11):3154. doi: 10.3390/cancers12113154
209. Sekine H, Okazaki K, Kato K, Alam MM, Shima H, Katsuoka F, et al. O-GlcNAcylation Signal Mediates Proteasome Inhibitor Resistance in Cancer Cells by Stabilizing Nrf1. Mol Cell Biol (2018) 38(17):e00252–18. doi: 10.1128/MCB.00252-18
210. Wickremasinghe RG. Why Is CLL Refractory to Bortezomib? Blood (2008) 112(9):3540–1. doi: 10.1182/blood-2008-07-171231
211. Kwei KA, Baker JB, Pelham RJ. Modulators of Sensitivity and Resistance to Inhibition of PI3K Identified in a Pharmacogenomic Screen of the NCI-60 Human Tumor Cell Line Collection. PloS One (2012) 7(9):e46518. doi: 10.1371/journal.pone.0046518
212. Guarente V, Sportoletti P. Lessons, Challenges and Future Therapeutic Opportunities for PI3K Inhibition in CLL. Cancers (Basel) (2021) 13(6):1280. doi: 10.3390/cancers13061280
213. Wang X, Li W, Marcus J, Pearson M, Song L, Smith K, et al. MK-8719, a Novel and Selective O-GlcNAcase Inhibitor That Reduces the Formation of Pathological Tau and Ameliorates Neurodegeneration in a Mouse Model of Tauopathy. J Pharmacol Exp Ther (2020) 374(2):252–63. doi: 10.1124/jpet.120.266122
214. Ding N, Ping L, Shi Y, Feng L, Zheng X, Song Y, et al. Thiamet-G-Mediated Inhibition of O-GlcNAcase Sensitizes Human Leukemia Cells to Microtubule-Stabilizing Agent Paclitaxel. Biochem Biophys Res Commun (2014) 453(3):392–7. doi: 10.1016/j.bbrc.2014.09.097
215. Qian L, Yang X, Li S, Zhao H, Gao Y, Zhao S, et al. Reduced O-GlcNAcylation of SNAP-23 Promotes Cisplatin Resistance by Inducing Exosome Secretion in Ovarian Cancer. Cell Death Discov (2021) 7(1):112. doi: 10.1038/s41420-021-00489-x
216. Lee H, Oh Y, Jeon YJ, Lee SY, Kim H, Lee HJ, et al. DR4-Ser424 O-GlcNAcylation Promotes Sensitization of TRAIL-Tolerant Persisters and TRAIL-Resistant Cancer Cells to Death. Cancer Res (2019) 79(11):2839–52. doi: 10.1158/0008-5472.CAN-18-1991
217. Tung S, Shi Y, Wong K, Zhu F, Gorczynski R, Laister RC, et al. Pparα and Fatty Acid Oxidation Mediate Glucocorticoid Resistance in Chronic Lymphocytic Leukemia. Blood (2013) 122(6):969–80. doi: 10.1182/blood-2013-03-489468
218. Li MD, Ruan HB, Singh JP, Zhao L, Zhao T, Azarhoush S, et al. O-GlcNAc Transferase Is Involved in Glucocorticoid Receptor-Mediated Transrepression. J Biol Chem (2012) 287(16):12904–12. doi: 10.1074/jbc.M111.303792
219. Itkonen HM, Gorad SS, Duveau DY, Martin SE, Barkovskaya A, Bathen TF, et al. Inhibition of O-GlcNAc Transferase Activity Reprograms Prostate Cancer Cell Metabolism. Oncotarget (2016) 7(11):12464–76. doi: 10.18632/oncotarget.7039
220. Scielzo C, Ghia P. Modeling the Leukemia Microenviroment In Vitro. Front Oncol (2020) 10:607608. doi: 10.3389/fonc.2020.607608
Keywords: O-GlcNAc transferase (OGT), O-GlcNAcase (OGase), cancer, metabolism, chronic lymphocytic leukemia, signal transduction, cytokines, O-linked β-D-N-acetylglucosamine (O-GlcNAc)
Citation: Spaner DE (2021) O-GlcNAcylation in Chronic Lymphocytic Leukemia and Other Blood Cancers. Front. Immunol. 12:772304. doi: 10.3389/fimmu.2021.772304
Received: 14 September 2021; Accepted: 02 November 2021;
Published: 18 November 2021.
Edited by:
Parameswaran Ramakrishnan, Case Western Reserve University, United StatesReviewed by:
Chad Slawson, University of Kansas Medical Center Research Institute, United StatesReshmi Parameswaran, Case Western Reserve University, United States
Copyright © 2021 Spaner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David E. Spaner, ZGF2aWQuc3BhbmVyQHN1bm55YnJvb2suY2E=