 Ritu Mahesh Ramamurthy
Ritu Mahesh Ramamurthy Anthony Atala
Anthony Atala Christopher D. Porada
Christopher D. Porada Graҫa Almeida-Porada
Graҫa Almeida-Porada- Fetal Research and Therapy Program, Wake Forest Institute for Regenerative Medicine, Winston-Salem, NC, United States
The FDA has predicted that at least 10-20 gene therapy products will be approved by 2025. The surge in the development of such therapies can be attributed to the advent of safe and effective gene delivery vectors such as adeno-associated virus (AAV). The enormous potential of AAV has been demonstrated by its use in over 100 clinical trials and the FDA’s approval of two AAV-based gene therapy products. Despite its demonstrated success in some clinical settings, AAV-based gene therapy is still plagued by issues related to host immunity, and recent studies have suggested that AAV vectors may actually integrate into the host cell genome, raising concerns over the potential for genotoxicity. To better understand these issues and develop means to overcome them, preclinical model systems that accurately recapitulate human physiology are needed. The objective of this review is to provide a brief overview of AAV gene therapy and its current hurdles, to discuss how 3D organoids, microphysiological systems, and body-on-a-chip platforms could serve as powerful models that could be adopted in the preclinical stage, and to provide some examples of the successful application of these models to answer critical questions regarding AAV biology and toxicity that could not have been answered using current animal models. Finally, technical considerations while adopting these models to study AAV gene therapy are also discussed.
Introduction – gene therapy today
Over the past two decades, gene therapy has evolved from a hypothetical concept to a clinical reality for numerous genetic disorders, fulfilling its promise of a single-treatment cure in some cases (1). As of June 23, there were 7 gene therapy products approved by the FDA, several of which use CAR-T cells (2), and it is predicted that 10-20 products will be approved by 2025 (3).
Gene therapy involves introducing a functional copy of a gene into a cell to compensate for a defect in its own endogenous copy of that gene or to suppress a defective gene that encodes a harmful product. While gene therapy can take many forms to achieve its objective, these forms all fall into one of the following categories: gene addition, gene silencing, and gene editing/gene replacement, and they are comprised of two components - the vector and its encoded payload, which can include the therapeutic transgene, appropriate regulatory elements to confer transgene expression, and, in the case of gene-editing, the necessary machinery to mediate this process, e.g., CRISPR/Cas9, ZFNs, TALENS, prime/base editors. The process of gene delivery itself can be achieved by exposing the desired target cells to the vector ex vivo, and then infusing the cells to act as vehicles to carry the therapeutic gene, or by directly administering the vector in vivo. Success of gene therapy is primarily dependent upon the selection of an appropriate vector that can deliver its payload to the desired location with high efficiency (and ideally specificity, as well). For this to occur, the vector must also have the ability to evade barriers imposed by the host’s immune system.
Among the myriad gene delivery platforms available, vectors based upon viruses remain the most widely used. Millions of years of evolution has honed the innate capacity of viruses to transfer their genetic material into host cells, and this ability can be exploited to achieve delivery of therapeutic genes at far higher efficiency than with any nonviral vector that has been created to-date. Viral vectors used for gene therapy can be classified into integrating and non-integrating vectors. The choice is made depending on the desired expression profile (transient or prolonged), the size of the genetic payload (each virus has an ideal genome size which must be observed for efficient packaging), and the inherent biology of the target cell type/tissue (is the cell quiescent or dividing, is it short- or long-lived, which viral receptors does it express on its surface, etc.). Currently, the viral vectors that have shown the most promise in preclinical studies and in clinical trials are those based upon adeno-associated virus (AAV), lentiviruses (i.e., HIV), and murine retroviruses (e.g., MMLV, MSCV). While lentiviral and murine retroviral vectors are most often used in the context of ex vivo gene delivery, modifying cells in vitro for subsequent infusion, AAV has proven highly successful in an in vivo setting (4).
AAV gene therapy
Clinical landscape
Due to the tremendous potential it has demonstrated in both decades of preclinical studies and in past and ongoing clinical trials, AAV is currently a very popular choice for accomplishing in vivo gene delivery. Clinical data covering over 3000 patients treated over more than 20 years has demonstrated that AAV is a safe and well-tolerated gene delivery vector that can be highly effective (5, 6). Moreover, depending upon the target cell/tissue, AAV can achieve long-term transgene expression after a single infusion, with the longest reported period of > 15 years in primates (7). There are only two FDA-approved AAV gene therapy products on the market today (Luxturna and Zolgensma), but there are ~136 ongoing clinical trials involving AAV products to treat 55 different diseases (ClinicalTrials.gov). It is also expected that 50 trials will be completed in the next 3 years, of which 80% will be phase 2 (5). Currently, inherited disorders affecting the eye, lysosomal storage, and coagulation (hemophilia A and B) account for the majority of clinical trials. Among these, hemophilia A has the most ongoing trials, most of which are in late phases of development and a high likelihood of commercialization. Of the many ongoing clinical gene therapy trials, the vast majority involve gene addition - only 3 gene silencing and 3 gene editing trials are currently ongoing. For more detailed information on the clinical landscape of AAV, the reader is referred to the very thorough recent reviews by Au et al. and Kuzmin et al. (5, 6).
AAV biology
AAV is a non-enveloped virus with an icosahedral capsid of approximately 22 nm in size. Wild type AAV contains a 4.7 kb single-stranded DNA genome flanked by inverted terminal repeats (ITRs). The genome consists of the rep and cap genes, which are required for replication/packaging of the genome and synthesis of viral capsid, respectively. The cap gene gives rise to three viral capsid proteins (VP1-3) and assembly-activating protein (AAP) which, as the name suggests, helps in the assembly of 60 VP subunits at a 1:1:10 ratio of VP1:VP2:VP3. Nine variable regions are present on each subunit, and they determine the tropism and intracellular trafficking of the vector, as well as the serotype (4). These regions of the capsid proteins also serve as the recognition domains for neutralizing antibodies (Nabs) (8, 9). AAV has very high seroprevalence (40-70% of human population harbor neutralizing antibodies, depending upon the AAV serotype) (10–12). This is one of the motivations behind the search for novel naturally occurring variants, and the quest to create new bioengineered variants, that could evade the pre-existing immunity present in the majority of the population and thereby allow the widespread use of AAV-based vectors. Bioengineering approaches are also being undertaken in an effort to increase AAV’s transduction efficiency and to enable transduction of specific cell types in vivo. Such targeted transduction would increase both safety and efficiency, since limiting transduction to only the desired target cell would avoid transduction of undesirable cells/tissues (e.g., germline) and would ensure the entire administered dose of vector was delivered to the target cells, thus maximizing clinical benefit. Such targeting would also have the added safety benefit of being able to achieve therapeutic levels of transduction of the target cells/tissues with a lower dose of vector.
Engineering AAV
When constructing a recombinant AAV vector to be used for gene delivery, the rep and cap genes are removed and replaced with a cassette containing the therapeutic transgene and a promoter to drive its expression, while the ITRs are conserved, since they are essential for vector genome packaging (4). Importantly, removal of the rep gene renders AAV replication-defective, greatly increasing the safety of AAV. Selection of the appropriate capsid and promoter are the first steps to designing an effective AAV-based gene delivery vector. Since the capsid protein determines both the tropism, and thereby which cells can/will be transduced, and the antigenic profile of the vector (and its potential for being eliminated by pre-existing immunity or triggering an immune response) (9), bioengineering strategies have focused primarily on genetically modifying the capsid protein to achieve desired characteristics for the specific disease setting being targeted (4). To-date, hundreds of natural and bioengineered AAV variants have been reported, and tremendous progress has been made towards enhancing tropism for specific cells/tissues and for evading pre-existing anti-capsid immunity (13–18).
The promoter provides the opportunity to restrict expression of the therapeutic transgene to certain tissues, or even to specific cell types. Choosing a tissue-specific promoter avoids off-target expression of the transgene in undesirable cells, e.g., antigen-presenting cells (APCs) that could lead to an immune response, elimination of transduced cells, and loss of therapeutic effect. On the other hand, the use of a promoter that has been engineered to drive high-level constitutive expression, such as CAG, CBA, and CMV, or simply including the appropriate enhancer regions, can enable higher-than-normal expression of the transgene, allowing one to achieve a therapeutic effect from even a small number of transduced cells. Each choice has its own pros and cons. More than 25 trials have used tissue-specific promoters (5), yet these studies have shown that off-target effects still occur, due to “leakiness” of the promoter, allowing its promiscuous expression in non-target cells. Moreover, these studies have demonstrated the use of a tissue-specific promoter also results in a marked reduction in the effective dose of vector that reaches the target tissue, due to transduction of cells that will not contribute to expression of the transgene. While the ability to manufacture larger quantities of AAV has enabled the use of native promoters to drive more physiologically-relevant/appropriate expression of the therapeutic transgene, recent preclinical studies have suggested this approach can lead to severe toxicity due to the high vector copy number needed to achieve sufficient expression (19). As a result, the trend has been to rely on strong, constitutively active promoters, with 45% of trials between 2015 and 2019 employing the CAG, CBA, or CMV promoters discussed above (5). The use of one of these promoters does not guarantee success, however, as the high expression levels of such promoters often triggers their transcriptional silencing by methylation. In addition, supraphysiologic levels of expression of the transgene often come at the expense of reduced expression of endogenous genes that are forced to compete for limiting resources within the cells, leading to cell stress (6).
In addition to altering the capsid and the transgene cassette, the design of the vector backbone also provides an engineering opportunity to improve transduction efficiency. AAV naturally exists in a form with a signle-stranded DNA genome. After entering the target cells, the single-stranded AAV genome, upon reaching the nucleus, relies on the host cell’s machinery to be converted to a double-stranded form that can be transcribed. This conversion is a rate-limiting step in the transduction process. To attempt to overcome this bottleneck, researchers have developed what are known as “self-complementary” AAV vectors, the genome of which is double-stranded by design. As such, the vector genome undergoes transcription immediately upon nuclear entry, thereby increasing transduction efficiency and also decreasing the dose of vector required (20). This increase in transduction efficiency does not, however, come without a cost. By rendering the AAV genome self-complementary, the carrying capacity of resultant AAV vector is halved, severely limiting which transgens can be delivered by these vectors. In addition, some studies employing self-complementary AAV vectors have reported an increase in the innate immune response following transduction (21, 22). At the present time, the majority of the clinical trials are using AAV vectors with a single-stranded genome (6).
Hurdles to be overcome with AAV gene delivery
The simplicity of its genome organization, which facilitates engineering and production, its ability to mediate sustained transgene expression in a variety of quiescent cells, its wide range of tissue tropism and high efficiency of transduction, and its established safety profile all contribute to the extensive application of AAV in gene therapy (23, 24). Despite its many advantageous properties, however, it is also important to acknowledge aspects of AAV that hinder its more widespread clinical use in gene therapy trials and pose potential risks to subjects receiving AAV vectors.
(i) Immunogenicity/seropositivity: Two of the major hurdles to the use of AAV-based vectors in clinical gene therapy are its high degree of immunogenicity and the fact that, depending upon the AAV serotype in question, 40-70% of people in the general population harbor neutralizing antibodies (NAbs), depending upon the AAV serotype (11, 12, 20, 25, 26). At the present time, clinical trials avoid the issue of pre-existing immunity to AAV by simply excluding any patients that harbor NAbs to the serotype of AAV being used as a vector. As a result, the vast majority of patients who could benefit from gene therapy are ineligible to receive this potentially life-saving treatment. Even in patients with no pre-existing immunity, a single injection of AAV can trigger a robust immune response to both the AAV capsid and the transgene product, causing cell-mediated elimination of transduced cells, loss of therapeutic effect, and potentially lead to toxicity in the transduced tissue (27–34). To prevent this deleterious cascade arising from T cell-mediated immunity, many AAV clinical trials pre-emptively administer a course of corticosteroids to the patients (32). However, novel strategies are constantly being developed (14, 16) to overcome these issues. These strategies run the gamut, starting at the design level with mutating critical antigenic sites on the capsid to generate novel AAV variants with no seroprevalence, all the way to strategies that are only incorporated at the time of AAV administration, such as chemical shielding of AAV antigens, elimination of antibody-producing cells, or removal of NAbs through plasmapheresis or using the IgG-degrading enzyme, IdeZ (35). While promising, these approaches are still in early stages of development and will require extensive preclinical testing and validation before clinical implementation can be considered.
(ii) Genomic integration: Wild-type AAV integrates into a specific locus (AAV-S1) on human chromosome 19, through actions of the rep gene (36–38). Unlike wild-type AAV, replication-defective AAV vectors lack the rep gene, and as such the majority of genomes that entered the target cell were assumed to remain episomal (39). However, studies over the past 2 decades have provided evidence that host (both animal and human) cell DNA-modifying enzymes can mediate genomic integration of AAV vectors, albeit at very low frequency (39–41). The extremely low incidence of genomic integration in these studies provided a sense of security with respect to the potential for AAV vectors to cause genotoxicity. The low likelihood of this risk being of concern for human patients receiving AAV vectors was further supported by a number of studies performed in juvenile and adult mice, all of which demonstrated a lack of AAV-induced mutagenesis in large cohorts of animals followed for relatively long times (23, 42–46). Unfortunately, however, the story proved to be very different when therapeutic, and even reporter-encoding, AAV vectors were administered to neonatal (1-2 day-old) mice, with some studies reporting AAV-induced hepatocellular carcinoma (HCC) in 100% of recipients (47–54). Interestingly, the majority of the AAV integrations that were present in the tumors were in the Rian (RNA imprinted and accumulated in nucleus) locus, which is orthologous to the human long non-coding RNA MEG8, increased expression of which has been correlated with poor prognosis in HCC patients (55). Needless to say, these findings raise significant concerns for human safety. Although to-date no AAV-induced genotoxic events have been confirmed in humans, these findings highlight the need for more in-depth studies in human-based systems to better determine the true risk of AAV-mediated genotoxicity in human patients, and the impact that the patient’s age may have on this risk. This need is further underscored by an elegant 10-year follow-up of AAV-mediated FVIII expression in adult dogs, which detected >1700 integration events and evidence of clonal expansion in liver tissue (56) and the recent demonstration that a powerful a new multiplex linear amplification-mediated polymerase chain reaction (M-LAM-PCR) assay was able to identify integration events and persistence of these integrated AAV genomes in both nonhuman primate tissues and human clinical liver biopsies post-AAV gene therapy (57, 58).
(iv) Tropism/Vector Biodistribution: Given these afore-detailed limitations and the lack of data in human-based model systems that accurately recapitulate normal physiology, it is not surprising that AAV clinical GT trials are experiencing immunological and/or inflammatory responses and levels of transgene expression that are often lower than what was expected based on preclinical studies (31, 59–64). Another issue that has hampered the translation of AAV vectors from animal models into human patients is the marked species-species differences that exist in AAV vector tropism (65), which precludes extrapolation of results between species and raises the critical question of how accurately even the best animal models can predict tropism/vector biodistribution, transduction efficiency, and eventual treatment success in humans. AAV’s tropism is governed by the specific molecular interaction between the viral capsid and moieties on the surface of the cell, with the interactions differing by serotype (66). Cellular uptake occurs after initial binding to moieties such as glycans and proteoglycans that are ubiquitously expressed in cells (67). This is followed by a series of secondary protein interactions which facilitate internalization (66–71). While species-species variation in tropism can be explained, in part, by differential expression of these receptors on animal vs. human cells, it has also been observed to be due to differences in capsid sequence which could vary the processing of the virus after its entry into the cell, thereby affecting eventual gene transfer (72). For example, studies have shown serotype-dependent difference in hepatic transduction efficiency may not always be due to the ability of different serotypes to enter hepatocytes but may instead be due to variation in post-entry processing such as translocation to the nucleus and conversion of single-stranded genome to double-stranded (59, 72–75), again emphasizing the importance of performing studies in the specific human tissues and cell types to be targeted to be able to draw meaningful conclusions and accurately predict clinical outcome. Since it is difficult to obtain multiple biopsies from patients in the clinical setting, especially those with hemophilia, which account for a substantial percentage of the ongoing trials, information on tropism and vector biodistribution has not been reported in detail in any published clinical trials to-date (29–32, 76), leaving this critical safety and efficacy issue unanswered.
In an effort to fill this void, Lisowski et al. (77) and others (78, 79) have made use of FRG® mice whose liver was partially repopulated with human hepatocytes to evaluate the clinical utility of natural and genetically-engineered variants/serotypes of AAV These studies led to the identification of two variants that transduced human hepatocytes with high efficiency in vivo, AAV5 and AAV-LK03, both of which are currently being used in liver-directed clinical GT trial for hemophilia A. These data represent an important first step to defining the relative efficiency with which various AAV serotypes transduce human liver. However, because the only humanized tissue was the liver (specifically, only the hepatocytes) (77, 78), it is impossible to derive information on the specificity of these AAV serotypes for hepatocytes, i.e., the true tropism in humans, only the efficiency with which they transduce these cells when there are no other human cells to compete for binding. As such, this model system leaves the critical questions of off-target effects and dilution of effective vector dose from uptake by non-target cells unanswered.
Novel models to combat hurdles of AAV gene therapy
Although animals and humanized animals provide invaluable model systems for preclinical evaluation of gene therapy-based treatments, their failure to accurately recapitulate the myriad nuances of human responses to gene therapy has often led to unexpected outcomes, with respect to both efficacy and safety, during clinical trials. These shortcomings highlight the need to develop novel preclinical models that can more accurately recapitulate human physiology and genetics to replace, or at least supplement, existing models in preclinical studies, providing information of direct human relevance. A similar concern exists in the pharmaceutical world, where even the best animal surrogates often fail to accurately predict efficacy and toxicity of new drugs (80–83). To overcome the inherent problems with extrapolating animal data to humans, the WFIRM-led X.C.E.L. program created an integrated microfluidic human “body-on-a-chip” (hBOAC) platform, in which up to 12 different human 3D organoids retain normal physiology/functionality for weeks to months, that can be used to screen new drugs, define effects of environmental toxins/stressors, and test countermeasures (84–94). This novel system was able to predict toxicities of former FDA-approved drugs that were recalled due to unexpected human toxicities, sometimes only years after market release (84, 85, 94), demonstrating the immense potential of individual 3D human organoids and the integrated hBOAC platform to provide information that is otherwise not attainable with current animal models, and thus fill this critical knowledge gap and enable more direct translation of preclinical findings with new pharmaceuticals into clinical success. In the following sections, we will discuss how we envision this promising platform being adopted for use in gene therapy studies to enable more accurate predictions of the human response to gene therapy with respect to transduction efficiency, tissue and cellular tropism, and immunogenicity, as well as serving as a model in which to study the incidence and genomic location of AAV vector integration events in the context of an all-human system.
3D organoids
Organoids, while fairly new to the fields of biomedical and pharmaceutical research, originated several decades ago in the developmental sciences, born from the need for a model that could allow the study of the complex process of organogenesis (95). Organoids are three-dimensional (3D) structures derived from tissue-specific cells that self-organize and undergo spatially-restricted lineage commitment to recapitulate the structural characteristics, cytoarchitecture, and functional properties of the organ/tissue from which they are derived (96, 97). For example, human intestinal organoids have been shown to carry out key functions of the gastrointestinal tract including establishment and maintenance of an appropriate epithelial barrier, mucus production, absorption, and secretion of biomolecules (98). As detailed in the informative review by Hofer et al. (97), organoids have now been developed, representing all of the major human tissues. A critical scientific advance that greatly facilitated the fabrication of various organoids possible was the advent of induced pluripotent stem cells (iPSCs), which allowed generation of various highly-specialized cell types (99) which would otherwise be inaccessible. The availability of these tissue-specific cells allowed fabrication of more complex organoids that more closely replicate the anatomy and physiology of the native tissue. Moreover, iPSC technology has made possible the generation of patient-specific and disease-specific organoids, opening whole new avenues in research and drug testing. Aside from iPSCs, cells used to fabricate organoids include embryonic stem cells, cell lines, and adult stem/progenitor cells, and tissue-specific primary cells derived from tissue samples. To create organoids, cells are usually combined together in physiologically relevant percentages within a protein-rich medium that resembles the composition of the natural extracellular matrix (ECM) of the specific tissue to be modeled. The organoids can either be formed through self-aggregation to yield spheroids or by immobilizing the cells within a hydrogel matrix. The architectural complexity can be further increased by fabricating in a system that allows stratification, e.g., including a membrane to create an air-liquid interface in the case of lung organoids.
Microphysiological systems (MPS) and organ-on-a-chip
MPS are microfluidic systems fabricated at a biologically relevant scale with the application of fluid flow and mechanical stressors to create a dynamic environment that more accurately recapitulates the physiology and microenvironment of the native tissue. The inclusion of these additional makes it possible to generate biomolecular gradients and to create mechanical cues such as shear stress and contraction/relaxation, allowing them to reproduce more of the physiological subtleties that are unique to each tissue than conventional static cultures. By combining MPS-based models and organoids, one obtains what are referred to as an “organ-on-a-chip”; integrating multiple distinct organ-on-a-chip devices together and enabling tissue-to-tissue communication via shared circulating media yields what has been dubbed a “body-on-a-chip”. These systems have proven to be very powerful preclinical models for testing pharmaceuticals, reproducing such complex integrated multi-organ physiologic responses as metabolism of a drug by the liver, with the resultant metabolites then trafficking to other organs and exerting toxicity (84, 93, 94, 100). Their small size/scale allows for better control of the microenvironment and also requires minimal resources. The immense promise of these organ-on-a-chip and body-on-a-chip platforms as preclinical tools is eloquently stated in the recently published report by Marx et al. (100), in which the authors detail a roadmap to regulatory acceptance of organ-on-a-chip models and highlight the potential for such systems to supplement and, perhaps, ultimately replace animal models for preclinical testing of drug efficacy and toxicity. Organ-on-a-chip systems are also being integrated with artificial intelligence platforms, further increasing the predictive power of these models and preclinical data derived from them. It is envisioned that the organ-on-a-chip platforms will reach the necessary qualification level within the next decade and will dramatically decrease the use of animal testing. Currently, the success rate for drugs in clinical development is a meager 15%, due in large part to the discrepancies seen between results in an animals and those observed when the drug moves to clinical trials. It is anticipated that the adoption of these all-human organ-on-a-chip models in the preclinical drug development pipeline will greatly improve this success rate, enabling safer and more effective drugs to enter the market at a lower cost to patients who need them. Although originally developed for drug screening, in the next sections, we will highlight how we envision the field of AAV gene therapy can also greatly benefit from implementing these organ-on-a-chip platforms.
Technical considerations for adapting organ-on-a-chip and body-on-a-chip models to study AAV gene therapy
The tremendous progress in biofabrication techniques has enabled the development of body-on-a-chip platforms and their use as preclinical models to probe human response to therapies to advance at an exponential rate in recent years (93, 101–104), and we foresee it only being a matter of time before these systems are extensively applied to gene therapy studies, because of their unique ability to provide answers to critical questions that elude even the best current models (Figure 1). Looking specifically at AAV-based gene therapy studies, it is important to identify technical considerations that are relevant to gene delivery. In general, a body-on-chip platform consists of 3 key components: (i) organoids formed with a normal repertoires of cell types in physiologically relevant frequencies; (ii) a universal media that is able to support all cell types present in each of the different organoids; and (iii) microfluidics and microfabrication techniques that enable physiological interaction of multiple organ compartments and create the necessary physical/chemical cues. In addition, it is often desirable to incorporate physical, biochemical, and optical sensing modalities to facilitate the performance of real-time, nondestructive analyses on the various organoids and their shared “circulation” (97). It is also important to consider possible interactions of AAV with any of the materials used to fabricate the device. For example, polydimethylsiloxane (PDMS) is a widely employed polymer with many attractive properties for microfluidic device fabrication, such as biocompatibility, low toxicity, optical transparency, elastomeric properties, gas permeability, ease of fabrication, and low manufacturing costs (105, 106). Unfortunately, it suffers from serious protein adsorption problems due to its hydrophobic nature (106, 107), a property that has the potential to result in binding of proteins of the AAV capsid and thereby interfere with transduction. Therefore, to adapt existing microfluidic “on-a-chip” platforms for studies employing AAV, it will likely be necessary to either opt for an entirely different material or to develop a suitable anti-fouling surface-treatment to avoid vector adsorption to the PDMS. Luckily, such treatments are currently an area of very active investigation (106, 108).
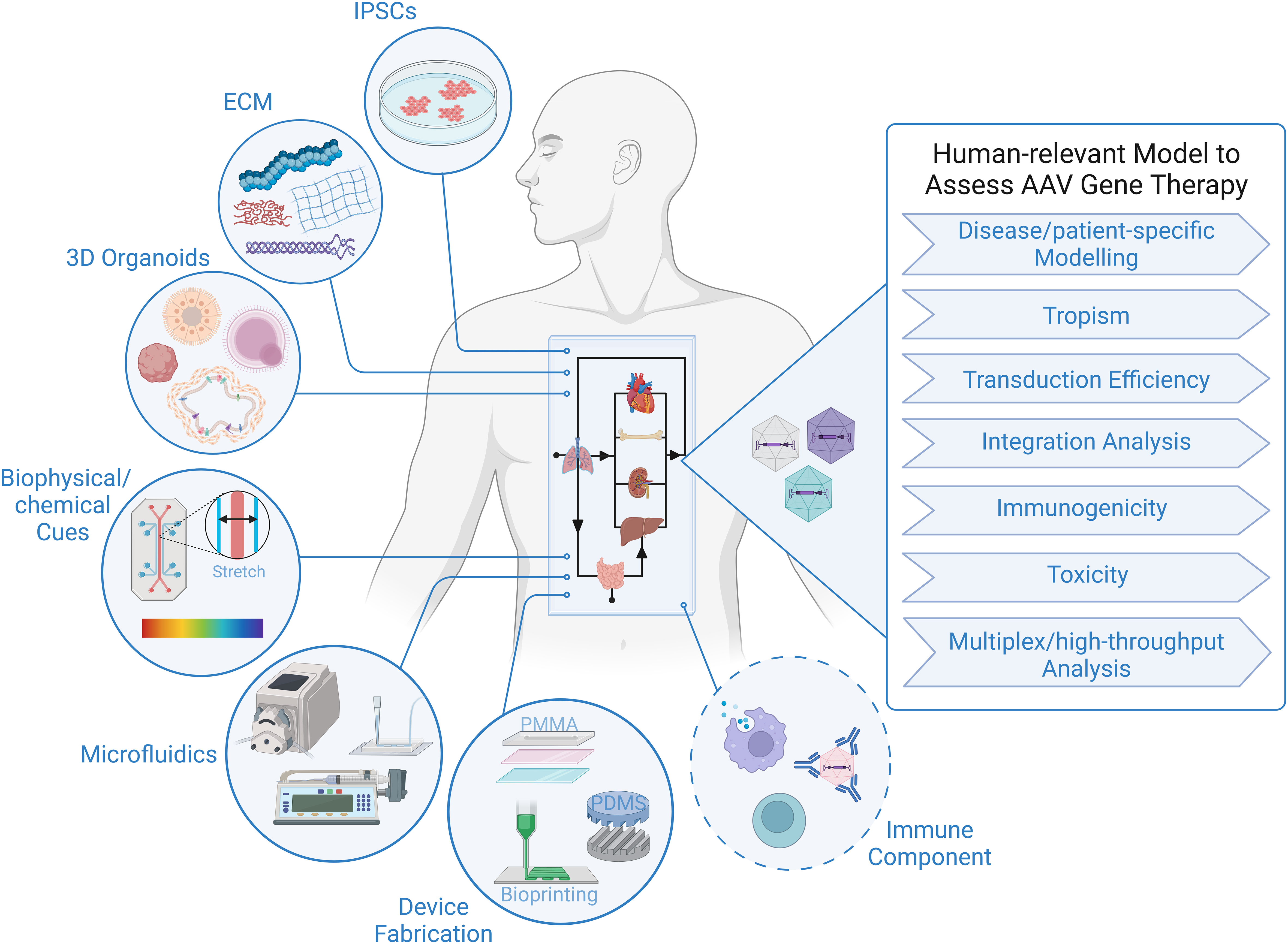
Figure 1 Human-relevant Preclinical Model to Accelerate AAV Gene Therapy Studies. Representation of critical components constituting body-on-a-chip platform that can be used to assess outcome of AAV gene therapy in the context of a human system. Addition of an immune component is essential to increase the predictive value of a model to study viral gene therapy.
As a completely human system, the organ-on-a-chip and body-on-a-chip platforms have the potential to accurately predict transduction efficiency of specific human cell types, tropism/selectivity of transduction when simultaneously presented with multiple human tissues/cells, true cell-specificity of promoters within human tissues/organs, and the incidence and precise genomic location of AAV insertion events within the human genome. There are also certain inherent design aspects of these platforms that are beneficial for gene therapy studies. For example, microfluidic devices function at a very low volume, and therefore, high vector-to-cell ratios/multiplicity of infection (MOI) can easily be achieved without the need for large quantities of viral vector. The small size also allows for high-throughput screening of vectors by connecting an array of devices, which increases the power of analysis with minimal resources (109, 110). The compartmentalization/stratification allows exploration of the impact different routes of gene delivery, e.g., systemic administration vs. direct injection into a specific target tissue, has on the efficiency and specificity of vector delivery to the desired target organ.
Although many sources of cells can be employed to create the organoids in the body-on-a-chip, iPSCs have some advantages with respect to gene therapy, as their use not only allows fabrication of a body-on-chip in which all tissues and all cells possess an identical genetic makeup, but it also provides the opportunity for rapid high-throughput screening of a diverse population (111), and it allows the derivation of patient-specific cells that can be used for disease modelling and developing personalized approaches to gene therapy that are optimized for the specific patient to be treated (103, 112). Due to the inability to biopsy most tissues in human patients who receive AAV-based gene therapy, insertion event data is simply not available for human subjects, with the exception of a single study (57). Human body-on-a-chip platforms provide the unique opportunity to define patterns of AAV genome insertion and answer thew critical question of whether the frequency and pattern of genomic integration vary as a function of the recipient’s age and the tissue that is targeted, as has been observed in mice (58). The use of patient-derived cells to create the body-on-a-chip would also make it possible to assess the specific patient’s risk of insertional mutagenesis. No other system is currently available that can screen for patient-specific genotoxic events and provide invaluable data concerning the underlying molecular mechanisms governing any observed patient-to-patient differences.
Since pre-existing anti-capsid immunity and the development of an immune response to the vector and transgene post-gene delivery are a major hurdle limiting more widespread clinical use of AAV vectors, the inclusion of an immune component to the body-on-a-chip would tremendously increase the utility of this platform and markedly enhance its preclinical value. In order for an AAV vector to successfully achieve long-term transgene expression in vivo, key immune-related roadblocks must be overcome: (i) the presence of pre-existing neutralizing antibodies (NAbs) against the capsid, the presence of which can markedly reduce transduction; (ii) triggering of an adaptive immune response and NAbs by the capsid, which can prevent future re-administration; (iii) triggering of a cytotoxic T lymphocyte (CLT)-mediated response by the capsid, which can lead to the elimination of transduced cells and loss of therapeutic effect; and (iv) the transgene product can induce B cell- and T cell-mediated responses that can lead to generation of antibodies and CTLs against the transgene product (113). While it was initially supposed that innate immunity would be transient and inconsequential for AAV-mediated gene therapy, it is now appreciated that both the innate and adaptive arms of the immune system pose formidable barriers to successful AAV (113). Despite their critical role in the human response to gene therapy and the ultimate success or failure or such treatments, immune components have thus far not been addressed in any detail in organ-on-a-chip or body-on-a-chip systems. Being hematopoietic in nature, all immune cells are derived from hematopoietic stem/progenitor cells (HSPC) that reside within the bone marrow. We and others have created human bone marrow-on-a-chip systems to gain insight into the complex process of normal and malignant hematopoiesis in an all-human system (86, 114–119). Despite being very new, these platforms have already provided an unprecedented view into the differing interactions of healthy and malignant human HSPC with specific cells of the various bone marrow niches, often in real-time, and molecules involved in these processes. However, they have not yet been utilized to study immune ontogeny, likely due, at least in part, to the need for T cell precursors to traffic to and mature within the thymus to achieve full function, and B cell precursors to mature within germinal centers in secondary lymphoid organs such as the lymph nodes, ileal Peyer’s patches, and the spleen. As such, a platform containing bone marrow, thymus, and one of the secondary lymphoid tissues would be needed to study T cell and B cell development and function “on-a-chip” – an accomplishment that is likely going to take quite a bit of work to make a reality. As a first step towards this goal, Seet et al. described (120) a serum-free, artificial thymic organoid (ATO) system that supports the efficient and reproducible in vitro differentiation and positive selection of conventional human T cells from HSPC. Moreover, they showed that ATO-derived T cells exhibited mature naive phenotypes, a diverse T cell receptor (TCR) repertoire and TCR-dependent function, and that ATOs initiated with TCR-engineered HSPC produced T cells with antigen-specific cytotoxicity. While these ATOs provide a robust tool for studying human T cell differentiation, they have yet to be incorporated into an on-a-chip platform, precluding their integration with other hematopoietic tissues. A further major step was very recently made towards achieving at least part of a functioning human immune system on-a-chip by Ingber and colleagues (121), who showed that primary human blood B- and T-lymphocytes autonomously assemble into ectopic lymphoid follicles (LFs) when cultured in a 3D extracellular matrix gel, and that B cells in these germinal center-like LFs exhibit plasma cell differentiation upon activation. Most excitingly, the authors demonstrated that when these human LFs-on-a-chip were inoculated with commercial influenza vaccine, plasma cell formation and production of anti-hemagglutinin IgG was observed, and a repertoire of cytokines similar to vaccinated humans was secreted over clinically relevant timescales.
It is clear that ongoing advances in the organoid field will one day make a human immune system-on-a-chip a reality (122). In the meantime, however, it is important to note that most tissues possess an intrinsic immune surveillance component in the form of tissue-resident antigen presenting cells (APC) such as dendritic cells (DC) and macrophages. One of the first steps in detection of foreign entities by the innate immune system is the recognition of structural motifs called pathogen-associated molecular patterns (PAMPs), by pattern recognition receptors (PRRs) expressed on APC. Thus, while it is not yet possible to incorporate an entire functioning immune system, simply including the appropriate APC in a given organoid/tissue-on-a-chip dramatically improves the predictive power of these models, as activation of any of the innate immunity pathways can readily be detected through differential expression of genes involved in innate immunity. For example, the liver-on-a-chip platform we developed includes Kupffer cells at a physiologically appropriate frequency to enable investigation of immune and inflammatory responses within this system (94).
Current application of 3D organoids and MPS platforms to study AAV gene therapy
While organoids and MPS have already been used extensively in studies focused on drug testing, disease modelling, and personalized medicine (reviewed in (103)), to-date only a fairly limited number of reports have employed organoids to study AAV gene therapy (123–145). Most of the human organoid-based AAV studies thus far have been performed on retinal organoids (123–131). The initial focus on organoids designed to model the human retina can likely be attributed to several factors. The first of these is the eye’s presumed immune-privileged status, which has allowed effective in vivo gene transfer to this organ and at least partial phenotypic correction of several different defects with very minimal to no immune reactivity in both preclinical and clinical studies (146–148), although more recent reports have questioned the true immune-privileged status of the intraocular space and the lack of an immune response following AAV delivery to the eye (149). A second factor driving the relatively large number of studies exploring AAV gene delivery to the retina with human organoids is the availability of well-established in vitro models, many of which are comprised of all the major cell types in the eye and have the ability to build functional synaptic connections and recapitulate photosensitivity (123–131). A small number of studies have also been performed with AAV vectors in human cerebral organoids to determine the optimal capsid for achieving efficient transduction and transgene expression within the human brain, to develop novel means of combatting latent HIV infection within CNS glial cells, and to test the ability of AAV-based treatment to provide phenotypic correction in the setting of genetic diseases that impact the CNS (132–136). Lung and gut/intestinal organoids have also been utilized to successfully identify AAV serotypes that yield optimal transduction in the respective tissue, to create and test chimeric AAV/Bocavirus vectors, to test AAV/RNAi-based treatments for SARS-CoV-2, and to test the ability of AAV vectors to correct the cystic fibrosis phenotype, comparing results obtained in human organoids to those in CF mice (137–140). Organoids of the human kidney have also been employed to test the ability of a novel synthetic AAV vector to achieve efficient gene transfer to the mesenchymal cells of this organ (145). We and others have begun using human liver organoids to define the basic biology, therapeutic efficacy, and potential toxicity of liver-directed AAV gene delivery (141–144) (more detail appears in a subsequent section below). Collectively, these studies have employed human organoids to answer various questions regarding AAV biology and behavior in a human system, and they have provided valuable information regarding the expression of various AAV receptors and the transduction efficiency of different AAV serotypes/engineered variants. They have also begun to define the transduction pattern in organoids comprised of heterogeneous cell populations (vector tropism) and the expression profile and localization of transgene product (thereby testing the true selectivity of various “cell-specific” promoters), and they have explored some of the mechanisms of cell attachment of AAV vectors to different cell types within these human tissues. These studies have also used organoids to validate animal study outcomes in an all-human setting and to test novel approaches to gene delivery/editing to target infectious agents such as HIV and COVID-19 (132, 139). By using iPSCs, either derived from patients or engineered via CRISPR/Cas9 gene editing, to fabricate these organoids, investigators have provided critical proof-of-concept for the value of using organoids to model a specific disease, e.g., retinitis pigmentosa (127), CRX-Leber congenital amaurosis (131), and lysosomal β-galactosidase deficiency (135), and test the therapeutic effect of AAV-mediated gene correction, often in a patient-specific, i.e., personalized medicine, setting.
Surprisingly, despite the rapid transition from static organoids to organ-on-a-chip and body-on-a-chip platforms in drug-based studies, organoid studies investigating AAV gene delivery have thus far been limited to individual 3D organoids in static conditions with the notable exception of the recent report by Achberger et al. (150), in which 3D human iPSC-derived retinal organoids were incorporated into a dynamic environment in a microfluidic device to create a retina-on-a-chip, fabricated with more than seven different essential retinal cells. The microfluidic device enabled not only the incorporation of a dynamic environment, but also a dual chamber setup, one housing the organoids and the other acting as vasculature. These two chambers were separated by a membrane barrier to protect the organoids from shear force. The compartmentalization and vasculature-like perfusion of the system enabled a physiological subretinal-like injection of the AAV particles and a nutrient supply via a choroidal-like vasculature. Seven variants of AAV were characterized for cell tropism and transduction efficiency, with the optical accessibility of this novel platform providing the opportunity for in situ live-cell imaging. This study represents the first time a human iPSC-derived organ-on-a-chip was utilized to test transduction efficacy of AAV gene therapy using a highly translational route of administration, and as the authors convincingly argue, the presesented data demonstrate the immense potential of human organ-on-a-chip (and ultimately body-on-a-chip) models as the next generation of screening platforms for future gene therapeutic studies.
Another key area in AAV biology and AAV-mediated gene transfer for which organoids and tissue-/body-on-a-chip platforms are ideally suited is the identification of existing AAV serotypes or the engineering of novel AAV capsid variants that enable passage of AAV across biological obstacles such as the blood-brain-barrier (BBB) and the pulmonary epithelial barrier (PEB). As many diseases could benefit from efficient delivery of AAV vectors to the brain or the lung, the identification of AAV serotypes/variants that can efficiently cross the BBB or PEB would have a substantial clinical impact. Human brain organoids that accurately model the functionality of the BBB have already been developed and used extensively to study the ability of therapeutics to access the central nervous system (89, 151–155), to better understand alterations to the BBB that occur in various disease settings and during infection and the neuroinflammation that ensues due to BBB disruption (89, 153, 156–162), to study tumor metastasis (163–168), to examine receptor-mediated antibody transcytosis (169–171), and to develop and test novel nanoparticles for their ability to transit the BBB (172, 173). Similarly, lung organoids have also been used to study epithelial barrier function under normal homeostasis and in response to infection, exposure to nanomaterials, environmental toxicants, and small particulates, as well as for screening of new pharmaceuticals (174–185). Despite the fairly widespread use of organoids and tissue-/body-on-a-chip systems to study barrier function and its impact on health, disease, and pharmacological intervention, the use of these systems to explore AAV’s ability to transit these key biological barriers represents a highly promising, but is as-yet unexplored, avenue of research.
To our knowledge, we are the only group to-date to use organoids to begin to study the human immune response to AAV vectors. These studies, which are still in the early stages, utilized human liver organoids comprised of all major cell types present in the human liver in physiologically relevant frequencies: 78% human primary hepatocytes, 2% biliary cells, 5% hepatic stellate cells, and 5% endothelial cells, and included 10% Kupffer cells, enabling the evaluation of innate immune responses. Importantly, these liver organoids recapitulate the function of the native liver – hepatocytes express p450 reductase and secrete serum albumin, are tightly interwoven with stellate cells expressing vimentin, and exhibit microvilli, and bile canaliculus-like structures form within 7 days, demonstrating a rudimentary biliary system. Moreover, these organoids metabolize drugs, respond to inflammatory stimuli by initiating fibrosis and steatosis, and they demonstrate acetaminophen-induced toxicity that is reversed by its clinical countermeasure N-acetyl cysteine (84, 92–94, 186, 187). Based on the accuracy with which it recapitulates in vivo biology and function, we hypothesized that these liver organoids could provide a unique and powerful system in which to define the cellular, molecular, and functional impacts of transduction of the human liver with AAV3b and AAV5 to model ongoing clinical gene therapy trials using these serotypes of AAV to treat hemophilia A (188, 189), and to determine the true tropism of these serotypes in this all-human system. Our results to-date have shown the utility of this human liver organoid platform to determine the cellular tropism, transduction efficiency, and potential of these two clinically employed serotypes of AAV to induce inflammation and innate immune response following liver-directed gene transfer (142). Studies are currently ongoing to transition to a body-on-a-chip platform with a simulated circulatory system to better replicate the systemic in vivo AAV gene delivery being employed in hemophilia A gene therapy clinical trials and to incorporate the bone marrow-on-a-chip we have developed (4) and either the lymphoid follicle (LF)-on-a-chip described by Goyal et al. (121) or the tonsil organoids Wagar et al. (190) used to characterize the varying response donors with differing immune status (vaccination status, age, immunosuppressive state, etc.) exhibited to influenza, which we predict will greatly enhance the amount and quality of the data concerning the immune response to AAV that can be garnered from this platform.
Discussion and future directions to further improve preclinical AAV studies
The objective of this review has been to provide an overview of AAV gene therapy, to underscore its immense therapeutic potential, and to discuss the hurdles that currently limit its more widespread application to the correction of genetic diseases in a larger percentage of patients who would benefit from such intervention (Table 1). We highlighted how the lack of robust preclinical models that can accurately recapitulate human biology and thus predict clinical response is one of the main limitations that needs to be addressed. To progress from this impasse, we discussed how 3D organoids, MPS, and organ/body-on-a-chip platforms can sere as powerful alternatives to current preclinical models and how they have already begun to do so in the realm of pharmaceuticals. To support this tenet, we provided examples of successful application of these techniques to answer questions that animal models have failed to adequately and accurately address. In addition to the advantages stemming from their being an “all-human” model, we also stressed the ability of these on-a-chip systems to enable the creation and exploration of disease-specific and/or patient-specific models for personalized medicine and the further value such options brings to these models. We also provided a brief list of technical considerations to be applied in the context of assessing gene therapy using organ/body-on-a-chip platforms, including the need to incorporate a functional immune component, which would exponentially increase the value of these models. Despite the many unique advantages that organoids and tissue-/body-on-a-chip platforms possess compared to traditional 2D culture systems and animal models, it is important to acknowledge that they are not without limitations. The first of these, as we have discussed, pertains to the need to integrate a more physiologically relevant and functional immune component to better model the in vivo setting and accurately predict the potential for an immune response and/or inflammatory reaction following delivery of a given AAV vector. Second, while the use of microfluidic allows the organoids/tissue chips to communicate with one another and approximates a very primitive circulatory system, to truly model the human body, it will be necessary to create media that accurately models the components of human serum and to devise a means of coating the microfluidic channels with appropriate endothelial cells to recreate the important biological effects the reticuloendothelial system exerts on vectors and other agents that are administered systemically. The third issue that bears mention is the source of cells used to create the organoids/tissue-on-a-chip. While iPS cells are certainly convenient and enable the investigator to generate essentially any cell type that is needed, one must very carefully interpret results obtained with iPS-derived cells, as they often possess a phenotype that is developmentally immature and may not accurately model an adult patient (191–194). In addition, the source of cell used to generate the iPS cells can also exert an effect on the phenotype and function of the subsequently differentiated tissue-specific cells as a result of the irreversible epigenetic “memory” each cell type possesses (195–198). Even when the appropriate cell types are used to create the organoid/tissue-on-a-chip, the most complex physiological aspects and cell-cell communication networks of certain tissues (the brain for example) will likely be almost impossible to recapitulate with complete accuracy in vitro. As these technologies continue to mature, however, it is safe to assume that the fidelity with which these systems mirror their in vivo counterparts will steadily improve. With these caveats in mind, we hope this review has served to highlight the immense potential organ-on-a-chip and body-on-a-chip platforms have to revolutionize preclinical testing, and that the examples we have provided have convinced the reader that these systems represent a powerful translational model that can provide critical information regarding transduction efficiency, tropism, potential immunogenicity, and genotoxicity, thus greatly aid in translating safer and more effective AAV-based gene therapies to the clinic to treat/cure a wide array of genetic disorders.

Table 1 Strategies of studies employing organoids and microphysiological systems to overcome the hurdles of AAV gene therapy.
Author contributions
RR wrote original draft of manuscript. CP, AA, and GA-P edited and added to the draft to yield the final document. All authors contributed to the article and approved the submitted version.
Funding
The authors declare that this study received funding from Pfizer Global Medical Grants System; grant #61589649. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article, or the decision to submit it for publication.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bennett J, Wellman J, Marshall KA, McCague S, Ashtari M, DiStefano-Pappas J, et al. Safety and durability of effect of contralateral-eye administration of AAV2 gene therapy in patients with childhood-onset blindness caused by RPE65 mutations: A follow-on phase 1 trial. Lancet (2016) 388(10045):661–72. doi: 10.1016/S0140-6736(16)30371-3
2. Administration, U.F.a.D. Approved cellular and gene therapy products (2022). Available at: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/approved-cellular-and-gene-therapy-products.
3. Statement from FDA commissioner Scott gottlieb, M.D. and Peter marks, M.D., Ph.D., director of the center for biologics evaluation and research on new policies to advance development of safe and effective cell and gene therapies (2019). Available at: https://www.fda.gov/news-events/press-announcements/statement-fda-commissioner-scott-gottlieb-md-and-peter-marks-md-phd-director-center-biologics.
4. Li C, Samulski RJ. Engineering adeno-associated virus vectors for gene therapy. Nat Rev Genet (2020) 21(4):255–72. doi: 10.1038/s41576-019-0205-4
5. Kuzmin DA, Shutova MV, Johnston NR, Smith OP, Fedorin VV, Kukushkin YS, et al. The clinical landscape for AAV gene therapies. Nat Rev Drug Discovery (2021) 20(3):173–5. doi: 10.1038/d41573-021-00017-7
6. Au HKE, Isalan M, Mielcarek M. Gene therapy advances: A meta-analysis of AAV usage in clinical settings. Front Med (Lausanne) (2021) 8:809118–31. doi: 10.3389/fmed.2021.809118
7. Sehara Y, Fujimoto KI, Ikeguchi K, Katakai Y, Ono F, Takino N, et al. Persistent expression of dopamine-synthesizing enzymes 15 years after gene transfer in a primate model of parkinson's disease. Hum Gene Ther Clin Dev (2017) 28(2):74–9. doi: 10.1089/humc.2017.010
8. Xie Q, Bu W, Bhatia S, Hare J, Somasundaram T, Azzi A, et al. The atomic structure of adeno-associated virus (AAV-2), a vector for human gene therapy. Proc Natl Acad Sci U.S.A. (2002) 99(16):10405–10. doi: 10.1073/pnas.162250899
9. Govindasamy L, Padron E, McKenna R, Muzyczka N, Kaludov N, Chiorini JA, et al. Structurally mapping the diverse phenotype of adeno-associated virus serotype 4. J Virol (2006) 80(23):11556–70. doi: 10.1128/JVI.01536-06
10. Gonçalves MA. Adeno-associated virus: from defective virus to effective vector. Virol J (2005) 2:43–59. doi: 10.1186/1743-422X-2-43
11. Calcedo R, Vandenberghe LH, Gao G, Lin J, Wilson JM. Worldwide epidemiology of neutralizing antibodies to adeno-associated viruses. J Infect Dis (2009) 199(3):381–90. doi: 10.1086/595830
12. Mikals K, Nam HJ, Vliet Van K, Vandenberghe LH, Mays LE, McKenna R, et al. The structure of AAVrh32.33, a novel gene delivery vector. J Struct Biol (2014) 186(2):308–17. doi: 10.1016/j.jsb.2014.03.020
13. Madigan VJ, Asokan A. Engineering AAV receptor footprints for gene therapy. Curr Opin Virol (2016) 18:89–96. doi: 10.1016/j.coviro.2016.05.001
14. Barnes C, Scheideler O, Schaffer D. Engineering the AAV capsid to evade immune responses. Curr Opin Biotechnol (2019) 60:99–103. doi: 10.1016/j.copbio.2019.01.002
15. Li W, Asokan A, Wu Z, Dyke Van T, DiPrimio N, Johnson JS, et al. Engineering and selection of shuffled AAV genomes: A new strategy for producing targeted biological nanoparticles. Mol Ther (2008) 16(7):1252–60. doi: 10.1038/mt.2008.100
16. Bartel M, Schaffer D, Büning H. Enhancing the clinical potential of AAV vectors by capsid engineering to evade pre-existing immunity. Front Microbiol (2011) 2:204–13. doi: 10.3389/fmicb.2011.00204
17. Gao G, Vandenberghe LH, Wilson JM. New recombinant serotypes of AAV vectors. Curr Gene Ther (2005) 5(3):285–97. doi: 10.2174/1566523054065057
18. Büning H, Huber A, Zhang L, Meumann N, Hacker U. Engineering the AAV capsid to optimize vector-host-interactions. Curr Opin Pharmacol (2015) 24:94–104. doi: 10.1016/j.coph.2015.08.002
19. Hinderer C, Katz N, Buza EL, Dyer C, Goode T, Bell P, et al. Severe toxicity in nonhuman primates and piglets following high-dose intravenous administration of an adeno-associated virus vector expressing human SMN. Hum Gene Ther (2018) 29(3):285–98. doi: 10.1089/hum.2018.015
20. Wang D, Tai PWL, Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discovery (2019) 18(5):358–78. doi: 10.1038/s41573-019-0012-9
21. Martino AT, Suzuki M, Markusic DM, Zolotukhin I, Ryals RC, Moghimi B, et al. The genome of self-complementary adeno-associated viral vectors increases toll-like receptor 9-dependent innate immune responses in the liver. Blood (2011) 117(24):6459–68. doi: 10.1182/blood-2010-10-314518
22. Wu T, Töpfer K, Lin SW, Li H, Bian A, Zhou XY, et al. Self-complementary AAVs induce more potent transgene product-specific immune responses compared to a single-stranded genome. Mol Ther (2012) 20(3):572–9. doi: 10.1038/mt.2011.280
23. Colella P, Ronzitti G, Mingozzi F. Emerging issues in AAV-mediated in vivo gene therapy. Mol Ther Methods Clin Dev (2018) 8:87–104. doi: 10.1016/j.omtm.2017.11.007
24. Korneyenkov MA, Zamyatnin AA Jr. Next step in gene delivery: Modern approaches and further perspectives of AAV tropism modification. Pharmaceutics (2021) 13(5):750–65. doi: 10.3390/pharmaceutics13050750
25. Klamroth R, Hayes G, Andreeva T, Gregg K, Suzuki T, Mitha IH, et al. Global seroprevalence of pre-existing immunity against AAV5 and other AAV serotypes in people with hemophilia a. Hum Gene Ther (2022) 33(7-8):432–41. doi: 10.1089/hum.2021.287
26. Louis Jeune V, Joergensen JA, Hajjar RJ, Weber T. Pre-existing anti-adeno-associated virus antibodies as a challenge in AAV gene therapy. Hum Gene Ther Methods (2013) 24(2):59–67. doi: 10.1089/hgtb.2012.243
27. Long BR, Veron P, Kuranda K, Hardet R, Mitchell N, Hayes GM, et al. Early phase clinical immunogenicity of valoctocogene roxaparvovec, an AAV5-mediated gene therapy for hemophilia A. Mol Ther (2021) 29(2):597–610.doi: 10.1016/j.ymthe.2020.12.008
28. Manno CS, Pierce GF, Arruda VR, Glader B, Ragni M, Rasko JJ, et al. Successful transduction of liver in hemophilia by AAV-factor IX and limitations imposed by the host immune response. Nat Med (2006) 12(3):342–7. doi: 10.1038/nm1358
29. Miesbach W, Meijer K, Coppens M, Kampmann P, Klamroth R, Schutgens R, et al. Gene therapy with adeno-associated virus vector 5-human factor IX in adults with hemophilia b. Blood (2018) 131(9):1022–31. doi: 10.1182/blood-2017-09-804419
30. Nathwani AC, Reiss UM, Tuddenham EG, Rosales C, Chowdary P, McIntosh J, et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia b. N Engl J Med (2014) 371(21):1994–2004. doi: 10.1056/NEJMoa1407309
31. Nathwani AC, Tuddenham EG, Rangarajan S, Rosales C, McIntosh J, Linch DC, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia b. N Engl J Med (2011) 365(25):2357–65. doi: 10.1056/NEJMoa1108046
32. Rangarajan S, Walsh L, Lester W, Perry D, Madan B, Laffan M, et al. AAV5-factor VIII gene transfer in severe hemophilia a. N Engl J Med (2017) 377(26):2519–30. doi: 10.1056/NEJMoa1708483
33. Samelson-Jones BJ, Arruda VR. Protein-engineered coagulation factors for hemophilia gene therapy. Mol Ther Methods Clin Dev (2019) 12:184–201. doi: 10.1016/j.omtm.2018.12.007
34. Samelson-Jones BJ, Finn JD, George LA, Camire RM, Arruda VR. Hyperactivity of factor IX padua (R338L) depends on factor VIIIa cofactor activity. JCI Insight (2019) 5:e128683–e128697. doi: 10.1172/jci.insight.128683
35. Elmore ZC, Oh DK, Simon KE, Fanous MM, Asokan A. Rescuing AAV gene transfer from neutralizing antibodies with an IgG-degrading enzyme. JCI Insight (2020) 5(19):e139881–92. doi: 10.1172/jci.insight.139881
36. Kotin RM, Siniscalco M, Samulski RJ, Zhu XD, Hunter L, Laughlin CA, et al. Site-specific integration by adeno-associated virus. Proc Natl Acad Sci U.S.A. (1990) 87(6):2211–5. doi: 10.1073/pnas.87.6.2211
37. Samulski RJ, Zhu X, Xiao X, Brook JD, Housman DE, Epstein N, et al. Targeted integration of adeno-associated virus (AAV) into human chromosome 19. EMBO J (1991) 10(12):3941–50. doi: 10.1002/j.1460-2075.1991.tb04964.x
38. Young SM Jr., McCarty DM, Degtyareva N, Samulski RJ. Roles of adeno-associated virus rep protein and human chromosome 19 in site-specific recombination. J Virol (2000) 74(9):3953–66. doi: 10.1128/JVI.74.9.3953-3966.2000
39. McCarty DM, Young SM Jr., Samulski RJ. Integration of adeno-associated virus (AAV) and recombinant AAV vectors. Annu Rev Genet (2004) 38:819–45. doi: 10.1146/annurev.genet.37.110801.143717
40. Nakai H, Montini E, Fuess S, Storm TA, Grompe M, Kay MA. AAV serotype 2 vectors preferentially integrate into active genes in mice. Nat Genet (2003) 34(3):297–302. doi: 10.1038/ng1179
41. Nowrouzi A, Penaud-Budloo M, Kaeppel C, Appelt U, Le Guiner C, Moullier P, et al. Integration frequency and intermolecular recombination of rAAV vectors in non-human primate skeletal muscle and liver. Mol Ther (2012) 20(6):1177–86. doi: 10.1038/mt.2012.47
42. Bell P, Wang L, Lebherz C, Flieder DB, Bove MS, Wu D, et al. No evidence for tumorigenesis of AAV vectors in a large-scale study in mice. Mol Ther (2005) 12(2):299–306. doi: 10.1016/j.ymthe.2005.03.020
43. Ferla R, Alliegro M, Dell'Anno M, Nusco E, Cullen JM, Smith SN, et al. Low incidence of hepatocellular carcinoma in mice and cats treated with systemic adeno-associated viral vectors. Mol Ther Methods Clin Dev (2021) 20:247–57. doi: 10.1016/j.omtm.2020.11.015
44. Kao CY, Yang SJ, Tao MH, Jeng YM, Yu IS, Lin SW. Incorporation of the factor IX padua mutation into FIX-triple improves clotting activity in vitro and in vivo. Thromb Haemost (2013) 110(2):244–56. doi: 10.1160/TH13-02-0154
45. Li H, Malani N, Hamilton SR, Schlachterman A, Bussadori G, Edmonson SE, et al. Assessing the potential for AAV vector genotoxicity in a murine model. Blood (2011) 117(12):3311–9. doi: 10.1182/blood-2010-08-302729
46. Embury JE, Frost S, Charron CE, Madrigal E, Perera OP, Poirier AE, et al. Hepatitis virus protein X-phenylalanine hydroxylase fusion proteins identified in PKU mice treated with AAV-WPRE vectors. Molecular Therapy (2007) 15(Suppl 1):S131.
47. Bell P, Moscioni AD, McCarter RJ, Wu D, Gao G, Hoang A, et al. Analysis of tumors arising in male B6C3F1 mice with and without AAV vector delivery to liver. Mol Ther (2006) 14(1):34–44. doi: 10.1016/j.ymthe.2006.03.008
48. Chandler RJ, LaFave MC, Varshney GK, Burgess SM, Venditti CP. Genotoxicity in mice following AAV gene delivery: A safety concern for human gene therapy? Mol Ther (2016) 24(2):198–201. doi: 10.1038/mt.2016.17
49. Chandler RJ, LaFave MC, Varshney GK, Trivedi NS, Carrillo-Carrasco N, Senac JS, et al. Vector design influences hepatic genotoxicity after adeno-associated virus gene therapy. J Clin Invest (2015) 125(2):870–80. doi: 10.1172/JCI79213
50. Donsante A, Miller DG, Li Y, Vogler C, Brunt EM, Russell DW, et al. AAV vector integration sites in mouse hepatocellular carcinoma. Science (2007) 317(5837):477. doi: 10.1126/science.1142658
51. Donsante A, Vogler C, Muzyczka N, Crawford JM, Barker J, Flotte T, et al. Observed incidence of tumorigenesis in long-term rodent studies of rAAV vectors. Gene Ther (2001) 8(17):1343–6. doi: 10.1038/sj.gt.3301541
52. Li Y, Miller CA, Shea LK, Jiang X, Guzman MA, Chandler RJ, et al. Enhanced efficacy and increased long-term toxicity of CNS-directed, AAV-based combination therapy for krabbe disease. Mol Ther (2021) 29(2):691–701. doi: 10.1016/j.ymthe.2020.12.031
53. Walia JS, Altaleb N, Bello A, Kruck C, LaFave MC, Varshney GK, et al. Long-term correction of sandhoff disease following intravenous delivery of rAAV9 to mouse neonates. Mol Ther (2015) 23(3):414–22. doi: 10.1038/mt.2014.240
54. Zhong L, Malani N, Li M, Brady T, Xie J, Bell P, et al. Recombinant adeno-associated virus integration sites in murine liver after ornithine transcarbamylase gene correction. Hum Gene Ther (2013) 24(5):520–5. doi: 10.1089/hum.2012.112
55. Lou J, Yan W, Li QY, Zhu AK, Tan BQ, Dong R, et al. LncRNA MEG8 plays an oncogenic role in hepatocellular carcinoma progression through miR-367-3p/14-3-3zeta/TGFbetaR1 axis. Neoplasma (2021) 68(2):273–82. doi: 10.4149/neo_2020_200730N785
56. Nguyen GN, Everett JK, Kafle S, Roche AM, Raymond HE, Leiby J, et al. A long-term study of AAV gene therapy in dogs with hemophilia a identifies clonal expansions of transduced liver cells. Nat Biotechnol (2021) 39(1):47–55. doi: 10.1038/s41587-020-0741-7
57. Gil-Farina I, Fronza R, Kaeppel C, Lopez-Franco E, Ferreira V, D, et al. Recombinant AAV integration is not associated with hepatic genotoxicity in nonhuman primates and patients. Mol Ther (2016) 24(6):1100–5. doi: 10.1038/mt.2016.52
58. Sabatino DE, Bushman CFD, Chandler RJ, Crystal RG, Davidson BL, Dolmetsch R, et al. Evaluating the state of the science for adeno-associated virus (AAV) Integration:An integrated perspective. Mol Ther (2022). doi: 10.1016/j.ymthe.2022.06.004
59. Davidoff AM, Gray JT, Ng CY, Zhang Y, Zhou J, Spence Y, et al. Comparison of the ability of adeno-associated viral vectors pseudotyped with serotype 2, 5, and 8 capsid proteins to mediate efficient transduction of the liver in murine and nonhuman primate models. Mol Ther (2005) 11(6):875–88. doi: 10.1016/j.ymthe.2004.12.022
60. Jiang H, Couto LB, Patarroyo-White S, Liu T, Nagy D, Vargas JA, et al. Effects of transient immunosuppression on adenoassociated, virus-mediated, liver-directed gene transfer in rhesus macaques and implications for human gene therapy. Blood (2006) 108(10):3321–8. doi: 10.1182/blood-2006-04-017913
61. Kay MA, Manno CS, Ragni MV, Larson PJ, Couto LB, McClelland A, et al. Evidence for gene transfer and expression of factor IX in haemophilia b patients treated with an AAV vector. Nat Genet (2000) 24(3):257–61. doi: 10.1038/73464
62. Nathwani AC, Gray JT, McIntosh J, Ng CY, Zhou J, Spence Y, et al. Safe and efficient transduction of the liver after peripheral vein infusion of self-complementary AAV vector results in stable therapeutic expression of human FIX in nonhuman primates. Blood (2007) 109(4):1414–21. doi: 10.1182/blood-2006-03-010181
63. Nathwani AC, Gray JT, Ng CY, Zhou J, Spence Y, Waddington SN, et al. Self-complementary adeno-associated virus vectors containing a novel liver-specific human factor IX expression cassette enable highly efficient transduction of murine and nonhuman primate liver. Blood (2006) 107(7):2653–61. doi: 10.1182/blood-2005-10-4035
64. Pipe SW, Reddy KR, Chowdary P. Gene therapy: Practical aspects of implementation. Haemophilia (2022) 28(Suppl 4):44–52. doi: 10.1111/hae.14545
65. Watakabe A, Reddy KR, Chowdary P. Comparative analyses of adeno-associated viral vector serotypes 1, 2, 5, 8 and 9 in marmoset, mouse and macaque cerebral cortex. Neurosci Res (2015) 93:144–57. doi: 10.1016/j.neures.2014.09.002
66. Srivastava A. In vivo tissue-tropism of adeno-associated viral vectors. Curr Opin Virol (2016) 21:75–80. doi: 10.1016/j.coviro.2016.08.003
67. Pillay S, Zou W, Cheng F, Puschnik AS, Meyer NL, Ganaie SS, et al. Adeno-associated virus (AAV) serotypes have distinctive interactions with domains of the cellular AAV receptor. J Virol (2017) 91(18):e00391–400. doi: 10.1128/JVI.00391-17
68. Kashiwakura Y, Tamayose K, Iwabuchi K, Hirai Y, Shimada T, Matsumoto K, et al. Hepatocyte growth factor receptor is a coreceptor for adeno-associated virus type 2 infection. J Virol (2005) 79(1):609–14. doi: 10.1128/JVI.79.1.609-614.2005
69. Ling C, Lu Y, Kalsi J.K, Jayandharan G.R, Li B, Ma W, et al. Human hepatocyte growth factor receptor is a cellular coreceptor for adeno-associated virus serotype 3. Hum Gene Ther (2010) 21(12):1741–7. doi: 10.1089/hum.2010.075
70. Qing K, Mah C, Hansen J, Zhou S, Dwarki V, Srivastava A. Human fibroblast growth factor receptor 1 is a co-receptor for infection by adeno-associated virus 2. Nat Med (1999) 5(1):71–7. doi: 10.1038/4758
71. Wu Z, Asokan A, Grieger JC, Govindasamy L, Agbandje-McKenna M, Samulski RJ. Single amino acid changes can influence titer, heparin binding, and tissue tropism in different adeno-associated virus serotypes. J Virol (2006) 80(22):11393–7. doi: 10.1128/JVI.01288-06
72. Nonnenmacher M, Weber T. Intracellular transport of recombinant adeno-associated virus vectors. Gene Ther (2012) 19(6):649–58. doi: 10.1038/gt.2012.6
73. Thomas CE, Storm TA, Huang Z, Kay MA. Rapid uncoating of vector genomes is the key to efficient liver transduction with pseudotyped adeno-associated virus vectors. J Virol (2004) 78(6):3110–22. doi: 10.1128/JVI.78.6.3110-3122.2004
74. Pipe S, Leebeek FWG, Ferreira V, Sawyer EK, Pasi J. Clinical considerations for capsid choice in the development of liver-targeted AAV-based gene transfer. Mol Ther Methods Clin Dev (2019) 15:170–8. doi: 10.1016/j.omtm.2019.08.015
75. Vercauteren K, Hoffman BE, Zolotukhin I, Keeler GD, Xiao JW, Basner-Tschakarjan E, et al. Superior in vivo transduction of human hepatocytes using engineered AAV3 capsid. Mol Ther (2016) 24(6):1042–9. doi: 10.1038/mt.2016.61
76. George LA, Sullivan SK, Giermasz A, Rasko JEJ, Samelson-Jones BJ, Ducore J, et al. Hemophilia b gene therapy with a high-Specific-Activity factor IX variant. N Engl J Med (2017) 377(23):2215–27. doi: 10.1056/NEJMoa1708538
77. Lisowski L, Dane AP, Chu K, Zhang Y, Cunningham SC, Wilson EM, et al. Selection and evaluation of clinically relevant AAV variants in a xenograft liver model. Nature (2014) 506(7488):382–6. doi: 10.1038/nature12875
78. Wang L, Bell P, Somanathan S, Wang Q, He Z, Yu H, et al. Comparative study of liver gene transfer with AAV vectors based on natural and engineered AAV capsids. Mol Ther (2015) 23(12):1877–87. doi: 10.1038/mt.2015.179
79. Li S, Ling C, Zhong L, Li M, Su Q, He R, et al. Efficient and targeted transduction of nonhuman primate liver with systemically delivered optimized AAV3B vectors. Mol Ther (2015) 23(12):1867–76. doi: 10.1038/mt.2015.174
80. Bailey J, Balls M. Recent efforts to elucidate the scientific validity of animal-based drug tests by the pharmaceutical industry, pro-testing lobby groups, and animal welfare organisations. BMC Med Ethics (2019) 20(1):16–22. doi: 10.1186/s12910-019-0352-3
81. Bailey J, Thew M, Balls M. Predicting human drug toxicity and safety via animal tests: can any one species predict drug toxicity in any other, and do monkeys help? Altern Lab Anim (2015) 43(6):393–403. doi: 10.1177/026119291504300607
82. Fujiwara R, Yoda E, Tukey RH. Species differences in drug glucuronidation: Humanized UDP-glucuronosyltransferase 1 mice and their application for predicting drug glucuronidation and drug-induced toxicity in humans. Drug Metab Pharmacokinet (2018) 33(1):9–16. doi: 10.1016/j.dmpk.2017.10.002
83. Zhong X, Gutierrez C, Xue T, Hampton C, Vergara MN, Cao LH, et al. Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat Commun (2014) 5:4047–60. doi: 10.1038/ncomms5047
84. Skardal A, Murphy SV, Devarasetty M, Mead I, Kang HW, Seol YJ, et al. Multi-tissue interactions in an integrated three-tissue organ-on-a-chip platform. Sci Rep (2017) 7(1):8837–8852. doi: 10.1038/s41598-017-08879-x
85. Zhang YS, Aleman J, Shin SR, Kilic T, Kim D, Shaegh Mousavi SA, et al. Multisensor-integrated organs-on-chips platform for automated and continual in situ monitoring of organoid behaviors. Proc Natl Acad Sci U.S.A. (2017) 114(12):E2293–E2302. doi: 10.1073/pnas.1612906114
86. Aleman J, George SK, Herberg S, Devarasetty M, Porada CD, Skardal A, et al. Deconstructed microfluidic bone marrow on-A-Chip to study normal and malignant hemopoietic cell-niche interactions. Small (2019) 15(43):e1902971–83. doi: 10.1002/smll.201902971
87. Forsythe SD, Devarasetty M, Shupe T, Bishop C, Atala A, Soker S, et al. Environmental toxin screening using human-derived 3D bioengineered liver and cardiac organoids. Front Public Health (2018) 6:103–12. doi: 10.3389/fpubh.2018.00103
88. Lang R, Stern MM, Smith L, Liu Y, Bharadwaj S, Liu G, et al. Three-dimensional culture of hepatocytes on porcine liver tissue-derived extracellular matrix. Biomaterials (2011) 32(29):7042–52. doi: 10.1016/j.biomaterials.2011.06.005
89. Nzou G, Wicks RT, Wicks EE, Seale SA, Sane CH, Chen A, et al. Human cortex spheroid with a functional blood brain barrier for high-throughput neurotoxicity screening and disease modeling. Sci Rep (2018) 8(1):7413–22. doi: 10.1038/s41598-018-25603-5
90. Pendergraft SS, Sadri-Ardekani H, Atala A, Bishop CE. Three-dimensional testicular organoid: A novel tool for the study of human spermatogenesis and gonadotoxicity in vitro. Biol Reprod (2017) 96(3):720–32. doi: 10.1095/biolreprod.116.143446
91. Sittadjody S, Saul JM, Joo S, Yoo JJ, Atala A, Opara EC. Engineered multilayer ovarian tissue that secretes sex steroids and peptide hormones in response to gonadotropins. Biomaterials (2013) 34(10):2412–20. doi: 10.1016/j.biomaterials.2012.11.059
92. Skardal A, Aleman J, Forsythe S, Rajan S, Murphy S, Devarasetty M, et al. Drug compound screening in single and integrated multi-organoid body-on-a-chip systems. Biofabrication (2020) 12(2):025017. doi: 10.1088/1758-5090/ab6d36
93. Skardal A, Shupe T, Atala A. Organoid-on-a-chip and body-on-a-chip systems for drug screening and disease modeling. Drug Discovery Today (2016) 21(9):1399–411. doi: 10.1016/j.drudis.2016.07.003
94. Rajan SAP, Aleman J, Wan M, Zarandi Pourhabibi N, Nzou G, Murphy S, et al. Probing prodrug metabolism and reciprocal toxicity with an integrated and humanized multi-tissue organ-on-a-chip platform. Acta Biomater (2020) 106:124–35. doi: 10.1016/j.actbio.2020.02.015
95. Clevers H. Modeling development and disease with organoids. Cell (2016) 165(7):1586–97. doi: 10.1016/j.cell.2016.05.082
96. Lancaster MA, Knoblich JA. Organogenesis in a dish: Modeling development and disease using organoid technologies. Science (2014) 345(6194):1247125–1247134. doi: 10.1126/science.1247125
97. Hofer M, Lutolf MP. Engineering organoids. Nat Rev Mater (2021) 6(5):402–20. doi: 10.1038/s41578-021-00279-y
98. Zachos NC, Kovbasnjuk O, Foulke-Abel J, In J, Blutt SE, Jonge HR, et al. Human Enteroids/Colonoids and intestinal organoids functionally recapitulate normal intestinal physiology and pathophysiology. J Biol Chem (2016) 291(8):3759–66. doi: 10.1074/jbc.R114.635995
99. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell (2007) 131(5):861–72. doi: 10.1016/j.cell.2007.11.019
100. Marx U, Akabane T, Andersson TB, Baker E, Beilmann M, Beken S, et al. Biology-inspired microphysiological systems to advance patient benefit and animal welfare in drug development. Altex (2020) 37(3):365–94. doi: 10.14573/altex.2001241
101. Kimura H, Sakai Y, Fujii T. Organ/body-on-a-chip based on microfluidic technology for drug discovery. Drug Metab Pharmacokinet (2018) 33(1):43–8. doi: 10.1016/j.dmpk.2017.11.003
102. Sung JH, Wang YI, Sriram Narasimhan N, Jackson M, Long C, Hickman JJ, et al. Recent advances in body-on-a-Chip systems. Anal Chem (2019) 91(1):330–51. doi: 10.1021/acs.analchem.8b05293
103. Ingber DE. Human organs-on-chips for disease modelling, drug development and personalized medicine. Nat Rev Genet (2022) 23(8):1–25. doi: 10.1038/s41576-022-00466-9
104. Esch MB, King TL, Shuler ML. The role of body-on-a-chip devices in drug and toxicity studies. Annu Rev BioMed Eng (2011) 13:55–72. doi: 10.1146/annurev-bioeng-071910-124629
105. Ng JM, Gitlin I, Stroock AD, Whitesides GM. Components for integrated poly(dimethylsiloxane) microfluidic systems. Electrophoresis (2002) 23(20):3461–73. doi: 10.1002/1522-2683(200210)23:20<3461::AID-ELPS3461>3.0.CO;2-8
106. Zhang H, Chiao M. Anti-fouling coatings of poly(dimethylsiloxane) devices for biological and biomedical applications. J Med Biol Eng (2015) 35(2):143–55. doi: 10.1007/s40846-015-0029-4
107. De Stefano P, Bianchi E, Dubini G. The impact of microfluidics in high-throughput drug-screening applications. Biomicrofluidics (2022) 16(3):031501. doi: 10.1063/5.0087294
108. Gao P, Wang Y, Wang J, Wang F, Ma W, Zhang Z, et al. Rational design of durable anti-fouling coatings with high transparency, hardness, and flexibility. ACS Appl Mater Interfaces (2022) 14(25):29156–66. doi: 10.1021/acsami.2c04279
109. Schuster B, Junkin M, Kashaf SS, Romero-Calvo I, Kirby K, Matthews J, et al. Automated microfluidic platform for dynamic and combinatorial drug screening of tumor organoids. Nat Commun (2020) 11(1):5271–82. doi: 10.1038/s41467-020-19058-4
110. Chi CW, et al. Microfluidic cell chips for high-throughput drug screening. Bioanalysis (2016) 8(9):921–37. doi: 10.4155/bio-2016-0028
111. Williamson A, Singh S, Fernekorn U, Schober A. The future of the patient-specific body-on-a-chip. Lab Chip (2013) 13(18):3471–80. doi: 10.1039/c3lc50237f
112. Moreira A, Müller M, Costa PF, Kohl Y. Advanced in vitro lung models for drug and toxicity screening: The promising role of induced pluripotent stem cells. Adv Biol (Weinh) (2022) 6(2):e2101139–22101186. doi: 10.1002/adbi.202101139
113. Muhuri M, Maeda Y, Ma H, Ram S, Fitzgerald KA, Tai PW, et al. Overcoming innate immune barriers that impede AAV gene therapy vectors. J Clin Invest (2021) 131(1):e143780-e14794. doi: 10.1172/JCI143780
114. Chou DB, Frismantas V, Milton Y, David R, Pop-Damkov P, Ferguson D, et al. On-chip recapitulation of clinical bone marrow toxicities and patient-specific pathophysiology. Nat BioMed Eng (2020) 4(4):394–406. doi: 10.1038/s41551-019-0495-z
115. Marturano-Kruik A, Nava MM, Yeager K, Chramiec A, Hao L, Robinson S, et al. Human bone perivascular niche-on-a-chip for studying metastatic colonization. Proc Natl Acad Sci U.S.A. (2018) 115(6):1256–61. doi: 10.1073/pnas.1714282115
116. Nelson MR, Ghoshal D, Mejias JC, Rubio DF, Keith E, Roy K. A multi-niche microvascularized human bone marrow (hBM) on-a-chip elucidates key roles of the endosteal niche in hBM physiology. Biomaterials (2021) 270:120683–91. doi: 10.1016/j.biomaterials.2021.120683
117. Santos Rosalem G, Torres Gonzales LA, Casas Las EB, Mathias FAS, Ruiz JC, Carvalho MGR. Microfluidics and organ-on-a-chip technologies: A systematic review of the methods used to mimic bone marrow. PloS One (2020) 15(12):e0243840. doi: 10.1371/journal.pone.0243840
118. Souquet B, Opitz M, Vianay B, Brunet S, Thery M. Manufacturing a bone marrow-On-A-Chip using maskless photolithography. Methods Mol Biol (2021) 2308:263–78. doi: 10.1007/978-1-0716-1425-9_20
119. Torisawa YS, Spina CS, Mammoto T, Mammoto A, Weaver JC, Tat T, et al. Bone marrow-on-a-chip replicates hematopoietic niche physiology in vitro. Nat Methods (2014) 11(6):663–9. doi: 10.1038/nmeth.2938
120. Seet CS, He C, Bethune MT, Li S, Chick B, Gschweng EH, et al. Generation of mature T cells from human hematopoietic stem and progenitor cells in artificial thymic organoids. Nat Methods (2017) 14(5):521–30. doi: 10.1038/nmeth.4237
121. Goyal G, Prabhala P, Mahajan G, Bausk B, Gilboa T, Xie L, et al. Ectopic lymphoid follicle formation and human seasonal influenza vaccination responses recapitulated in an organ-on-a-Chip. Adv Sci (Weinh) (2022) 9(14):e2103241. doi: 10.1002/advs.202103241
122. Bar-Ephraim YE, Kretzschmar K, Clevers H. Organoids in immunological research. Nat Rev Immunol (2020) 20(5):279–93. doi: 10.1038/s41577-019-0248-y
123. Khabou H, Garita-Hernandez M, Chaffiol A, Reichman S, Jaillard C, Brazhnikova E, et al. Noninvasive gene delivery to foveal cones for vision restoration. JCI Insight (2018) 3(2):e96029–47. doi: 10.1172/jci.insight.96029
124. Tornabene P, Trapani I, Minopoli R, Centrulo M, Lupo M, de Simone S, et al. Intein-mediated protein trans-splicing expands adeno-associated virus transfer capacity in the retina. Sci Transl Med (2019) 11(492). doi: 10.1126/scitranslmed.aav4523
125. Gonzalez-Cordero A, Goh D, Kruczek K, Naeem A, Fernando M, Holthaus Kleine SM, et al. Assessment of AAV vector tropisms for mouse and human pluripotent stem cell-derived RPE and photoreceptor cells. Hum Gene Ther (2018) 29(10):1124–39. doi: 10.1089/hum.2018.027
126. Quinn PM, Buck TM, Mulder AA, Ohonin C, Alves CH, Vos RM, et al. Human iPSC-derived retinas recapitulate the fetal CRB1 CRB2 complex formation and demonstrate that photoreceptors and müller glia are targets of AAV5. Stem Cell Rep (2019) 12(5):906–19. doi: 10.1016/j.stemcr.2019.03.002
127. Lane A, Jovanovic K, Shortall C, Ottaviani D, Panes AB, Schwarz N, et al. Modeling and rescue of RP2 retinitis pigmentosa using iPSC-derived retinal organoids. Stem Cell Rep (2020) 15(1):67–79. doi: 10.1016/j.stemcr.2020.05.007
128. McClements ME, Steward H, Atkin W, Goode EA, Gándara C, Chichagova V, et al. Tropism of AAV vectors in photoreceptor-like cells of human iPSC-derived retinal organoids. Transl Vis Sci Technol (2022) 11(4):3–14. doi: 10.1167/tvst.11.4.3
129. Völkner M, Pavlou M, Büning H, Michalakis S, Karl MO. Optimized adeno-associated virus vectors for efficient transduction of human retinal organoids. Hum Gene Ther (2021) 32(13-14):694–706. doi: 10.1089/hum.2020.321
130. Garita-Hernandez M, Routet F, Guibbal L, Khabou H, Toualbi L, Riancho L, et al. AAV-mediated gene delivery to 3D retinal organoids derived from human induced pluripotent stem cells. Int J Mol Sci (2020) 21(3). doi: 10.3390/ijms21030994
131. Kruczek K, Qu Z, Gentry J, Fadl BR, Gieser L, Hiriyanna S, et al. Gene therapy of dominant CRX-leber congenital amaurosis using patient stem cell-derived retinal organoids. Stem Cell Rep (2021) 16(2):252–63. doi: 10.1016/j.stemcr.2020.12.018
132. Kunze C, Börner K, Kienle E, Orschmann T, Rusha E, Schneider M, et al. Synthetic AAV/CRISPR vectors for blocking HIV-1 expression in persistently infected astrocytes. Glia (2018) 66(2):413–27. doi: 10.1002/glia.23254
133. Depla JA, Sogorb-Gonzalez M, Mulder L.A, Heine V.M, Konstantinova P, van Deventer SJ, et al. Cerebral organoids: A human model for AAV capsid selection and therapeutic transgene efficacy in the brain. Mol Ther Methods Clin Dev (2020) 18:167–75. doi: 10.1016/j.omtm.2020.05.028
134. Westhaus A, Cabanes-Creus M, Jonker T, Sallard E, Navarro RG, Zhu E, et al. AAV-p40 bioengineering platform for variant selection based on transgene expression. Hum Gene Ther (2022) 33(11-12):664–82. doi: 10.1089/hum.2021.278
135. Latour YL, Yoon R, Thomas SE, Grant C, Li C, Sena-Esteves M, et al. Human GLB1 knockout cerebral organoids: A model system for testing AAV9-mediated GLB1 gene therapy for reducing GM1 ganglioside storage in GM1 gangliosidosis. Mol Genet Metab Rep (2019) 21:100513–20. doi: 10.1016/j.ymgmr.2019.100513
136. Allende ML, Cook EK, Larman BC, Nugent A, Brady JM, Golebiowski D, et al. Cerebral organoids derived from sandhoff disease-induced pluripotent stem cells exhibit impaired neurodifferentiation. J Lipid Res (2018) 59(3):550–63. doi: 10.1194/jlr.M081323
137. Fakhiri J, Schneider MA, Puschhof J, Stanifer M, Schildgen V, Holderbach S, et al. Novel chimeric gene therapy vectors based on adeno-associated virus and four different mammalian bocaviruses. Mol Ther Methods Clin Dev (2019) 12:202–22. doi: 10.1016/j.omtm.2019.01.003
138. Meyer-Berg H, Zhou Yang L, Pilar de Lucas M, Zambrano A, Hyde SC, Gill DR. Identification of AAV serotypes for lung gene therapy in human embryonic stem cell-derived lung organoids. Stem Cell Res Ther (2020) 11(1):448–53. doi: 10.1186/s13287-020-01950-x
139. Becker J, Stanifer ML, Leist SR, Stolp B, Maiakovska O, West A, et al. Ex vivo and in vivo suppression of SARS-CoV-2 with combinatorial AAV/RNAi expression vectors. Mol Ther (2022) 30(5):2005–23. doi: 10.1016/j.ymthe.2022.01.024
140. Vidović D, Carlon MS, da Cunha MF, Dekkers JF, Hollenhorst MI, Bijvelds MJ, et al. rAAV-CFTRΔR rescues the cystic fibrosis phenotype in human intestinal organoids and cystic fibrosis mice. Am J Respir Crit Care Med (2016) 193(3):288–98. doi: 10.1164/rccm.201505-0914OC
141. Dai Z, Song G, Balakrishnan A, Yang T, Yuan Q, Möbus S, et al. Growth differentiation factor 11 attenuates liver fibrosis via expansion of liver progenitor cells. Gut (2020) 69(6):1104–15. doi: 10.1136/gutjnl-2019-318812
142. Ramamurthy RM, Zheng WT, George S, Wan M, Zhou Y, Lu B, et al. Using a human liver tissue equivalent (hLTE) platform to define the functional impact of liver-directed AAV gene therapy. Blood (2021) 138:2938. doi: 10.1182/blood-2021-147917
143. Wei J, Ran G, Wang X, Jiang N, Liang J, Lin X, et al. Gene manipulation in liver ductal organoids by optimized recombinant adeno-associated virus vectors. J Biol Chem (2019) 294(38):14096–104. doi: 10.1074/jbc.RA119.008616
144. Paulk NK, Pekrun K, Zhu E, Nygaard S, Li B, Xu J, et al. Bioengineered AAV capsids with combined high human liver transduction in vivo and unique humoral seroreactivity. Mol Ther (2018) 26(1):289–303. doi: 10.1016/j.ymthe.2017.09.021
145. Ikeda Y, Sun Z, Ru X, Vandenberghe LH, Humphreys BD. Efficient gene transfer to kidney mesenchymal cells using a synthetic adeno-associated viral vector. J Am Soc Nephrol (2018) 29(9):2287–97. doi: 10.1681/ASN.2018040426
146. Botto C, Rucli M, Tekinsoy MD, Pulman J, Sahel JA, Dalkara D. Early and late stage gene therapy interventions for inherited retinal degenerations. Prog Retin Eye Res (2022) 86:100975. doi: 10.1016/j.preteyeres.2021.100975
147. Hu ML, Edwards TL, O'Hare F, Hickey DG, Wang JH, Liu Z, et al. Gene therapy for inherited retinal diseases: Progress and possibilities. Clin Exp Optom (2021) 104(4):444–54. doi: 10.1080/08164622.2021.1880863
148. Schon C, Biel M, Michalakis S. Retinal gene delivery by adeno-associated virus (AAV) vectors: Strategies and applications. Eur J Pharm Biopharm (2015) 95(Pt B):343–52. doi: 10.1016/j.ejpb.2015.01.009
149. Bucher K, Rodriguez-Bocanegra E, Dauletbekov D, Fischer MD. Immune responses to retinal gene therapy using adeno-associated viral vectors - implications for treatment success and safety. Prog Retin Eye Res (2021) 83:100915–39. doi: 10.1016/j.preteyeres.2020.100915
150. Achberger K, Cipriano M, Düchs MJ, Schön C, Michelfelder S, Stierstorfer B, et al. Human stem cell-based retina on chip as new translational model for validation of AAV retinal gene therapy vectors. Stem Cell Rep (2021) 16(9):2242–56. doi: 10.1016/j.stemcr.2021.08.008
151. Bergmann S, Lawler SE, Qu Y, Fadzen CM, Wolfe JM, Regan MS, et al. Blood-brain-barrier organoids for investigating the permeability of CNS therapeutics. Nat Protoc (2018) 13(12):2827–43. doi: 10.1038/s41596-018-0066-x
152. Cakir B, Xiang Y, Tanaka Y, Kural MH, Parent M, Kang YJ, et al. Engineering of human brain organoids with a functional vascular-like system. Nat Methods (2019) 16(11):1169–75. doi: 10.1038/s41592-019-0586-5
153. Mukhopadhyay M. Recreating the blood-CNS barrier in vitro. Nat Methods (2020) 17(9):875. doi: 10.1038/s41592-020-0950-5
154. Park JC, Jang SY, Lee D, Lee J, Kang U, Chang H, et al. A logical network-based drug-screening platform for alzheimer's disease representing pathological features of human brain organoids. Nat Commun (2021) 12(1):280–92. doi: 10.1038/s41467-020-20440-5
155. Tournier N, Goutal S, Mairinger S, Hernandez-Lozano I, Filip T, Sauberer M, et al. Complete inhibition of ABCB1 and ABCG2 at the blood-brain barrier by co-infusion of erlotinib and tariquidar to improve brain delivery of the model ABCB1/ABCG2 substrate [(11)C]erlotinib. J Cereb Blood Flow Metab (2021) 41(7):1634–46. doi: 10.1177/0271678X20965500
156. Adams Y, Jensen AR. 3D organoid assay of the impact of infected erythrocyte adhesion on the blood-brain barrier. Methods Mol Biol (2022) 2470:587–99. doi: 10.1007/978-1-0716-2189-9_44
157. Adams Y, Jensen AR. Cerebral malaria - modelling interactions at the blood-brain barrier in vitro. Dis Model Mech (2022) 15(7). doi: 10.1242/dmm.049410
158. Chen X, Sun G, Tian E, Zhang M, Davtyan H, Beach TG, et al. Modeling sporadic alzheimer's disease in human brain organoids under serum exposure. Adv Sci (Weinh) (2021) 8(18):e2101462–77. doi: 10.1002/advs.202101462
159. Logan S, Arzua T, Canfield SG, Seminary ER, Sison SL, Ebert AD, et al. Studying human neurological disorders using induced pluripotent stem cells: From 2D monolayer to 3D organoid and blood brain barrier models. Compr Physiol (2019) 9(2):565–611. doi: 10.1002/cphy.c180025
160. Nzou G, Wicks RT, VanOstrand NR, Mekky GA, Seale SA, El-Taibany A, et al. Multicellular 3D neurovascular unit model for assessing hypoxia and neuroinflammation induced blood-brain barrier dysfunction. Sci Rep (2020) 10(1):9766–80. doi: 10.1038/s41598-020-66487-8
161. Pellegrini L, Albecka A, Mallery DL, Kellner MJ, Paul D, Carter AP, et al. SARS-CoV-2 infects the brain choroid plexus and disrupts the blood-CSF barrier in human brain organoids. Cell Stem Cell (2020) 27(6):951–961.e5. doi: 10.1016/j.stem.2020.10.001
162. Simoes Da Gama C, Morin-Brureau M. Study of BBB dysregulation in neuropathogenicity using integrative human model of blood-brain barrier. Front Cell Neurosci (2022) 16:863836–55. doi: 10.3389/fncel.2022.863836
163. Erb U, Schwerk C, Schroten H, Karremann M. Review of functional in vitro models of the blood-cerebrospinal fluid barrier in leukaemia research. J Neurosci Methods (2020) 329:108478–86. doi: 10.1016/j.jneumeth.2019.108478
164. Le Joncour V, Karaman S, Laakkonen PM. Predicting in vivo payloads delivery using a blood-brain tumor-barrier in a dish. J Vis Exp (2019) 146:(video). doi: 10.3791/59384
165. Pasqualini C, Kozaki T, Bruschi M, Nguyen THH, Minard-Colin V, Castel D, et al. Modeling the interaction between the microenvironment and tumor cells in brain tumors. Neuron (2020) 108(6):1025–44. doi: 10.1016/j.neuron.2020.09.018
166. Choe MS, Kim JS, Yeo HC, Bae CM, Han HJ, Baek K, et al. A simple metastatic brain cancer model using human embryonic stem cell-derived cerebral organoids. FASEB J (2020) 34(12):16464–75. doi: 10.1096/fj.202000372R
167. Quaranta V, Linkous A. Organoids as a systems platform for SCLC brain metastasis. Front Oncol (2022) 12:881989. doi: 10.3389/fonc.2022.881989
168. Liu W, Song J, Du X, Zhou Y, Li Y, Li R, et al. AKR1B10 (Aldo-keto reductase family 1 B10) promotes brain metastasis of lung cancer cells in a multi-organ microfluidic chip model. Acta Biomater (2019) 91:195–208. doi: 10.1016/j.actbio.2019.04.053
169. Kassianidou E, Simonneau C, Gavrilov A, Villasenor R. High throughput blood-brain barrier organoid generation and assessment of receptor-mediated antibody transcytosis. Bio Protoc (2022) 12(8):e4399–410. doi: 10.21769/BioProtoc.4399
170. Simonneau C, Duschmale M, Gavrilov A, Brandenberg N, Hoehnel S, Ceroni C, et al. Investigating receptor-mediated antibody transcytosis using blood-brain barrier organoid arrays. Fluids Barriers CNS (2021) 18(1):43. doi: 10.1186/s12987-021-00276-x
171. Veszelka S, Meszaros M, Porkolab G, Szecsko A, Kondor N, Ferenc G, et al. A triple combination of targeting ligands increases the penetration of nanoparticles across a blood-brain barrier culture model. Pharmaceutics (2021) 14(1):86–104. doi: 10.3390/pharmaceutics14010086
172. Sokolova V, Mekky G, van der Meer SB, Seeds MC, Atala AJ, Epple M. Transport of ultrasmall gold nanoparticles (2 nm) across the blood-brain barrier in a six-cell brain spheroid model. Sci Rep (2020) 10(1):18033. doi: 10.1038/s41598-020-75125-2
173. Sokolova V, Nzou G, van der Meer SB, Ruks T, Heggen M, Loza K, et al. Ultrasmall gold nanoparticles (2 nm) can penetrate and enter cell nuclei in an in vitro 3D brain spheroid model. Acta Biomater (2020) 111:349–62. doi: 10.1016/j.actbio.2020.04.023
174. Ainslie GR, Davis M, Ewart L, Lieberman LA, Rowlands DJ, Thorley AJ, et al. Microphysiological lung models to evaluate the safety of new pharmaceutical modalities: A biopharmaceutical perspective. Lab Chip (2019) 19(19):3152–61. doi: 10.1039/C9LC00492K
175. Artzy-Schnirman A, Hobi N, Schneider-Daum N, Guenat OT, Lehr CM, Sznitman J. Advanced in vitro lung-on-chip platforms for inhalation assays: From prospect to pipeline. Eur J Pharm Biopharm (2019) 144:11–7. doi: 10.1016/j.ejpb.2019.09.006
176. Huh D, Leslie DC, Matthews BD, Fraser JP, Jurek S, Hamilton GA, et al. A human disease model of drug toxicity-induced pulmonary edema in a lung-on-a-chip microdevice. Sci Transl Med (2012) 4(159):159ra147–55. doi: 10.1126/scitranslmed.3004249
177. Huh D, Matthews BD, Mammoto A, Montoya-Zavala M, Hsin HY, Ingber DE. Reconstituting organ-level lung functions on a chip. Science (2010) 328(5986):1662–8. doi: 10.1126/science.1188302
178. Konar D, Devarasetty M, Yildiz DV, Atala A, Murphy SV. Lung-On-A-Chip technologies for disease modeling and drug development. BioMed Eng Comput Biol (2016) 7(Suppl 1):17–27. doi: 10.4137/BECB.S34252
179. Paolicelli G, Luca A, Jose SS, Antonini M, Teloni I, Fric J, et al. Using lung organoids to investigate epithelial barrier complexity and IL-17 signaling during respiratory infection. Front Immunol (2019) 10:323–8. doi: 10.3389/fimmu.2019.00323
180. Schulze F, Gao X, Virzonis D, Damiati S, Schneider MR, Kodzius R. Air quality effects on human health and approaches for its assessment through microfluidic chips. Genes (Basel) (2017) 8(10):244–69. doi: 10.3390/genes8100244
181. Sengupta A, Roldan N, Kiener M, Froment L, Raggi G, Imler T, et al. A new immortalized human alveolar epithelial cell model to study lung injury and toxicity on a breathing lung-On-Chip system. Front Toxicol (2022) 4:840606–23. doi: 10.3389/ftox.2022.840606
182. Stucki AO, Stucki JD, Hall SR, Felder M, Mermoud Y, Schmid RA, et al. A lung-on-a-chip array with an integrated bio-inspired respiration mechanism. Lab Chip (2015) 15(5):1302–10. doi: 10.1039/C4LC01252F
183. Xu C, Zhang M, Chen W, Jiang L, Chen C, Qin J. Assessment of air pollutant PM2.5 pulmonary exposure using a 3D lung-on-Chip model. ACS Biomater Sci Eng (2020) 6(5):3081–90. doi: 10.1021/acsbiomaterials.0c00221
184. Zamprogno P, Wuthrich S, Achenbach S, Thoma G, Stucki JD, Hobi N, et al. Second-generation lung-on-a-chip with an array of stretchable alveoli made with a biological membrane. Commun Biol (2021) 4(1):168–78. doi: 10.1038/s42003-021-01695-0
185. Zhang M, Xu C, Jiang L, Qin J. A 3D human lung-on-a-chip model for nanotoxicity testing. Toxicol Res (Camb) (2018) 7(6):1048–60. doi: 10.1039/C8TX00156A
186. Bhise NS, Manoharan V, Massa S, Tamayol A, Ghaderi M, Miscuglio M, et al. A liver-on-a-chip platform with bioprinted hepatic spheroids. Biofabrication (2016) 8(1):014101–13. doi: 10.1088/1758-5090/8/1/014101
187. Vyas D, Baptista PM, Brovold M, Moran E, Gaston B, Booth C, et al. Self-assembled liver organoids recapitulate hepatobiliary organogenesis in vitro. Hepatology (2018) 67(2):750–61. doi: 10.1002/hep.29483
188. High KA. Gene therapy for hemophilia: The clot thickens. Hum Gene Ther (2014) 25(11):915–22. doi: 10.1089/hum.2014.2541
189. High KA, et al. A phase I/II trial of investigational spk-8011 in hemophilia a demonstrates durable expression and prevention of bleeds. Blood (2018) 132:487. doi: 10.1182/blood-2018-99-115495
190. Wagar LE, Salahudeen A, Constantz CM, Wendel BS, Lyons MM, Mallajosyula V, et al. Modeling human adaptive immune responses with tonsil organoids. Nat Med (2021) 27(1):125–35. doi: 10.1038/s41591-020-01145-0
191. de Lange WJ, Farrell ET, Kreitzer CR, Jacobs DR, Lang D, Glukhov AV, et al. Human iPSC-engineered cardiac tissue platform faithfully models important cardiac physiology. Am J Physiol Heart Circ Physiol (2021) 320(4):H1670–86. doi: 10.1152/ajpheart.00941.2020
192. Nam SA, Seo E, Kim JW, Kim HW, Kim HL, Kim K, et al. Graft immaturity and safety concerns in transplanted human kidney organoids. Exp Mol Med (2019) 51(11):1–13. doi: 10.1038/s12276-019-0336-x
193. Schwartz RE, Fleming HE, Khetani SR, Bhatia SN. Pluripotent stem cell-derived hepatocyte-like cells. Biotechnol Adv (2014) 32(2):504–13. doi: 10.1016/j.biotechadv.2014.01.003
194. Togo K, Fukusumi H, Shofuda T, Ohnishi H, Yamazaki H, Hayashi MK, et al. Postsynaptic structure formation of human iPS cell-derived neurons takes longer than presynaptic formation during neural differentiation in vitro. Mol Brain (2021) 14(1):149–65. doi: 10.1186/s13041-021-00851-1
195. Efrat S. Epigenetic memory: Lessons from iPS cells derived from human beta cells. Front Endocrinol (Lausanne) (2020) 11:614234–38. doi: 10.3389/fendo.2020.614234
196. Khoo TS, Jamal R, Abdul Ghani NA, Alauddin H, Hussin NH, Abdul Murad NA. Retention of somatic memory associated with cell identity, age and metabolism in induced pluripotent stem (iPS) cells reprogramming. Stem Cell Rev Rep (2020) 16(2):251–61. doi: 10.1007/s12015-020-09956-x
197. Noguchi H, Miyagi-Shiohira C, Nakashima Y. Induced tissue-specific stem cells and epigenetic memory in induced pluripotent stem cells. Int J Mol Sci (2018) 19(4). doi: 10.3390/ijms19040930
Keywords: AAV, gene therapy, AAV immunogenicity, organoids, microphysiological system, organ-on-chips, body-on-a-chip
Citation: Ramamurthy RM, Atala A, Porada CD and Almeida-Porada G (2022) Organoids and microphysiological systems: Promising models for accelerating AAV gene therapy studies. Front. Immunol. 13:1011143. doi: 10.3389/fimmu.2022.1011143
Received: 03 August 2022; Accepted: 01 September 2022;
Published: 26 September 2022.
Edited by:
Li Ou, Genemagic Biosciences, United StatesReviewed by:
Xiaojing Shi, University of Southern California, United StatesKanut Laoharawee, University of Minnesota Twin Cities, United States
Xintao Zhang, University of North Carolina at Chapel Hill, United States
Copyright © 2022 Ramamurthy, Atala, Porada and Almeida-Porada. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Graҫa Almeida-Porada, Z2FsbWVpZGFAd2FrZWhlYWx0aC5lZHU=