 Marloes A. H. M. Michels1*
Marloes A. H. M. Michels1* Elena B. Volokhina1,2
Elena B. Volokhina1,2 Nicole C. A. J. van de Kar1
Nicole C. A. J. van de Kar1 Lambertus P.W. J. van den Heuvel1,3,4,5 on behalf of the COMBAT consortium
Lambertus P.W. J. van den Heuvel1,3,4,5 on behalf of the COMBAT consortium- 1Department of Pediatric Nephrology, Amalia Children’s Hospital, Radboud University Medical Center, Nijmegen, Netherlands
- 2Innatoss Laboratories, Oss, Netherlands
- 3Department of Laboratory Medicine, Radboud University Medical Center, Nijmegen, Netherlands
- 4Department of Pediatrics/Pediatric Nephrology, University Hospitals Leuven, Leuven, Belgium
- 5Department of Development and Regeneration, University Hospitals Leuven, Leuven, Belgium
Nephritic factors (NeFs) are autoantibodies promoting the activity of the central enzymes of the complement cascade, an important first line of defense of our innate immune system. NeFs stabilize the complement convertase complexes and prevent their natural and regulator-mediated decay. They are mostly associated with rare complement-mediated kidney disorders, in particular with C3 glomerulopathy and related diseases. Although these autoantibodies were already described more than 50 years ago, measuring NeFs for diagnostic purposes remains difficult, and this also complicates our understanding of their clinical associations. In this review, we address the multifactorial challenges of NeF diagnostics. We describe the diseases NeFs are associated with, the heterogenic mechanisms of action of different NeF types, the different methods available in laboratories used for their detection, and efforts for standardization. Finally, we discuss the importance of proper NeF diagnostics for understanding the clinical impact of these autoantibodies in disease pathophysiology and for considering future complement-directed therapy.
1. Introduction
With the enhanced understanding of the role of the complement system (Figure 1) in the pathogenesis of many diseases (1), the characterization and diagnosis of patients with complement-mediated disorders has gained a novel dimension. In-depth characterization of the complement dysregulation has become an important part of standard patient laboratory tests. More importantly, the acknowledgment of the role of complement has provided us with novel strategies to target complement-mediated diseases. After the success story of the complement inhibitor eculizumab in the treatment of atypical hemolytic uremic syndrome (2–4), great efforts have been made to achieve similar successes for the treatment of other diseases. Numerous new complement-directed therapeutics are currently in the drug development pipeline (5, 6). To keep up with the advances in drug development, it is crucial to have accurate and reliable assays available to characterize the complement activation status and complement-dysregulating factors in each patient.
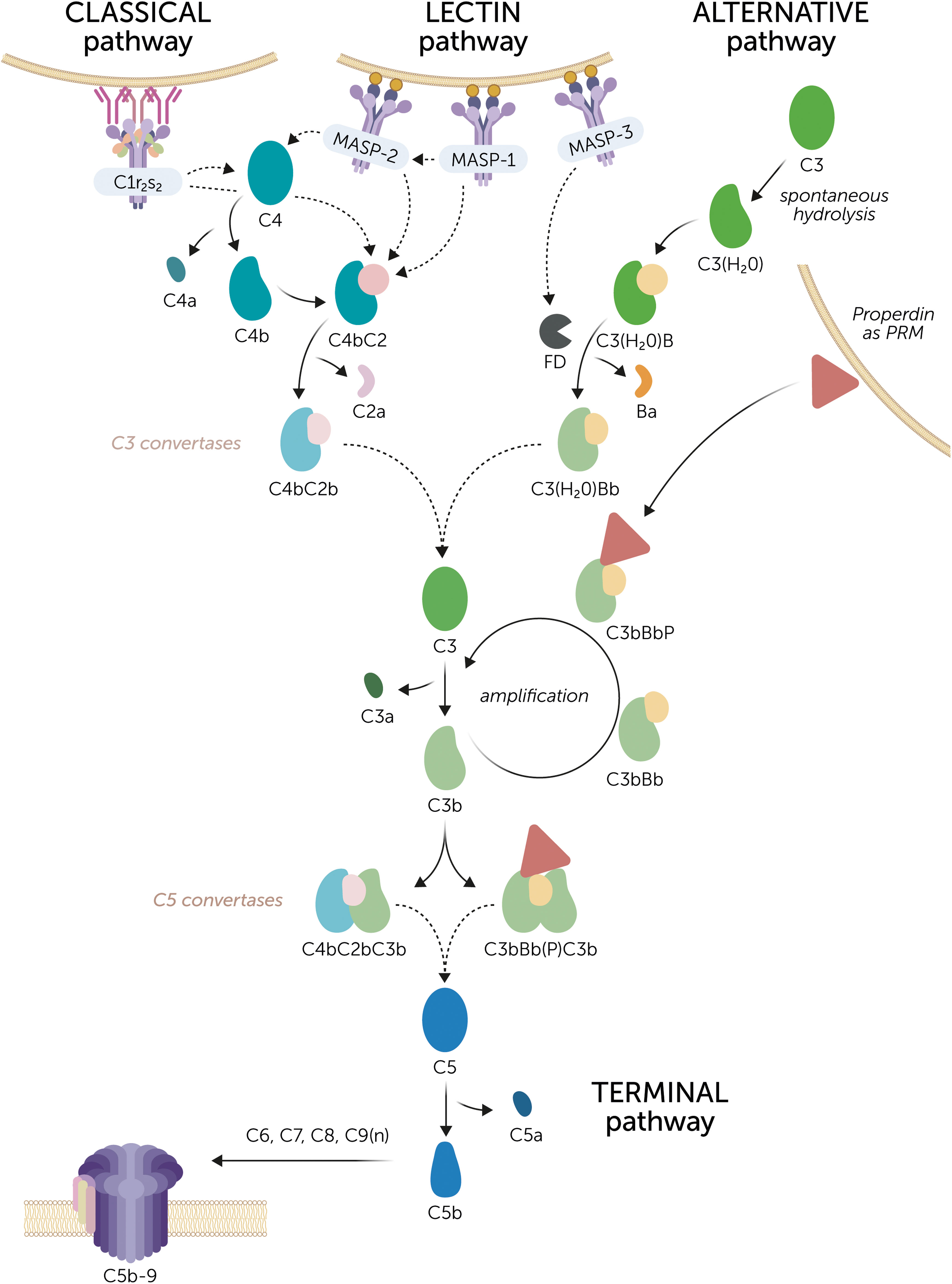
Figure 1 The complement system. Three routes can initiate complement activation. The classical pathway is activated by C1q coupled to the C1r2s2 proteases, together known as the C1 complex that recognizes, for example, antigen-antibody complexes. The lectin pathway is initiated by mannose-binding lectin (MBL), ficolins, or lectins, which recognize specific carbohydrate patterns on foreign surfaces. These pattern recognition molecules (PRMs) are coupled to the MBL-associated serine proteases (MASPs) MASP-1 or MASP-2. MASP-1 is autoactivated and is required to activate MASP-2. Activation of the classical and lectin pathway results in the cleavage of C4 and C2 by the C1r2s2 proteases and MASPs to form the C3 convertase C4bC2b. The alternative pathway is constantly active at a low rate due to the spontaneous hydrolysis of C3. Factor D (FD), which is activated by MASP-3, cleaves factor B to enable the formation of the initial C3 convertase C3(H2O)Bb. The C3 convertases convert C3 into C3a and C3b. Subsequently, the complement activation can be amplified via the alternative pathway. C3b can form new C3 convertase complexes that are stabilized by properdin (C3bBbP). Of note, properdin can also act as a PRM on certain surfaces to initiate alternative pathway activation. Upon continued C3 cleavage and C3b formation, C5 convertase complexes are formed, denoted as C4bC2bC3b and C3bBb(P)C3b, although their exact composition is unknown. C5 convertases cleave C5 into C5a and C5b, after which C5b recruits C6, C7, C8, and multiple copies of C9 to form the C5b-9 complex. This is the terminal complement complex that forms a pore and disrupts the membrane of the target cell. Dashed arrows indicate cleaving interactions.
One of the most challenging assays in the field of complement diagnostics is the detection of nephritic factors (NeFs). NeFs are stabilizing autoantibodies directed against the central enzymes of the complement system: the convertase complexes. By prolonging their activity and preventing their normal regulation, NeFs can contribute to an overactive complement system. Especially in the kidney disease C3 glomerulopathy (C3G), NeFs targeting the complement alternative pathway (AP) C3 convertase, i.e. C3NeFs, are considered the main driver of disease in many patients. However, robust and reliable standardized assays for C3NeF are lacking and this is hampering C3NeF research. According to the External Quality Assessment (EQA) rounds, organized by INSTAND e.V., the average success rate for C3NeF detection in the past five years (2016-2020) was 48% among the participating laboratories (7). This indicates that only half of all laboratories were able to correctly identify the presence or absence of C3NeFs in the reference samples sent around. Similar challenges apply for NeFs stabilizing the convertases of the complement classical pathway (CP), i.e. C4NeFs. Thus, important hurdles are still to be overcome in this field.
In this review, we provide an overview of the challenges in NeF testing and discuss the implications this has on our understanding of their role in disease. The two main factors complicating NeF analysis are the intrinsic heterogeneity of NeFs regarding their function and the large variety of methodologies used by different laboratories. Differences in NeF findings therefore hamper the research to their association with disease and/or disease activity. This underlines the need of standardization to aid research and diagnostics of NeFs and the diseases they are involved in.
2. C3 nephritic factor and C4 nephritic factor
In 1969, the term C3NeF was ascribed to a factor present in the serum of a hypocomplementemic membranoproliferative glomerulonephritis (MPGN) patient that specifically and very efficiently cleaved C3 when the serum was mixed with normal human serum (8). Later studies found that this C3NeF activity was mediated by stabilization of the AP convertase (9) and that C3NeFs had a heterogeneous immunoglobulin nature, mainly IgG (10–13). Therefore, nowadays, the term C3NeF is used to describe autoantibodies directed to neoepitopes of the AP C3 convertase that prolong the enzyme’s half-life, thereby resulting in enhanced cleavage of C3 (Figure 2).
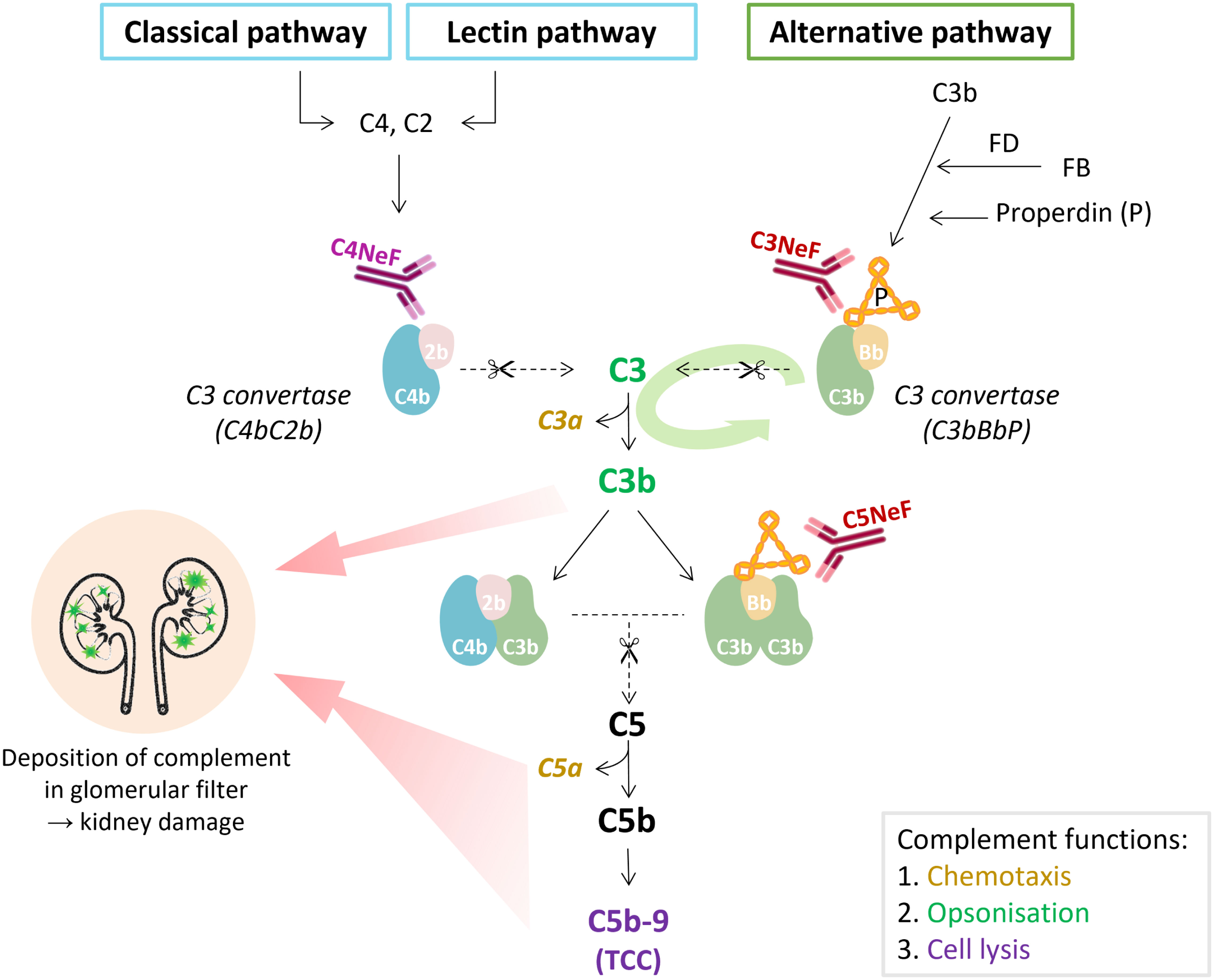
Figure 2 The place of nephritic factors in the complement system. C3 nephritic factors (C3NeF) stabilize the alternative pathway convertase complexes, whereas C4 nephritic factors (C4NeF) stabilize the convertases shared by the classical and lectin pathways. The term C5 nephritic factor (C5NeF) has been proposed for NeFs stabilizing alternative pathway C5 convertases. The green oval arrow illustrates the amplification loop of the alternative pathway. The colors of the complement components correspond to their function as indicated in the gray box. Overactivation of the complement system, mediated by NeFs, may result in the deposition of complement (breakdown) proteins in the glomerulus, inflammatory responses, and kidney injury. In C3 glomerulopathy, C3 degradation products are most abundant in the glomeruli, though terminal pathway proteins are also found. TCC, terminal complement complex.
Analogous to C3NeFs, in 1980 autoantibodies directed against the convertases shared by the CP and lectin pathway (LP) were reported (14, 15). These autoantibodies later became known as C4NeFs due to their capacity to stabilize the C4bC2b convertase complex (Figure 2). In many aspects, C4NeFs are functionally similar to C3NeFs, except for their specificity for convertases in another complement pathway. C4NeFs are IgGs recognizing neoepitopes in the CP/LP convertases to prolong their half-life and increase their ability to cleave C3.
3. Diseases associated with nephritic factors
C3NeFs are best described in the rare but severe kidney disease C3G. C3G is characterized by AP dysregulation leading to the deposition of C3 breakdown fragments in the glomeruli (16) (Figure 2). To establish the C3G diagnosis a kidney biopsy is required. The defining feature is a dominant staining for C3 visualized by immunofluorescence microscopy, i.e., a staining intensity of at least two orders of magnitude greater than staining for any other immune reactant (e.g., IgG, IgM, IgA, and C1q) (16–18). C3G can be subdivided into dense deposit disease (DDD) and C3 glomerulonephritis (C3GN) based on the glomerular complement deposition pattern visualized by electron microscopy. C3GN also encompasses CFHR5 nephropathy, which is a genetically driven familial form of C3G. The majority of the C3G patients show clear signs of AP activation in their blood. This includes increased markers of complement activation, e.g. C3 breakdown products and soluble (s)C5b-9, and low serum C3 levels, indicative of complement consumption due to excessive activation (19–22). C3NeFs are found in approximately 50% of the C3G cases, but the C3NeF prevalence among cohorts varies within a range of 26-75% (Table 1) (20–36). The large differences in these findings may not only be due to the different compositions of the patient cohorts, e.g. age of the patients and number of patients with DDD or C3GN, but also due to the heterogeneity of C3NeFs (see section 4) and the ability of the different assays used to detect all C3NeFs (see section 5).
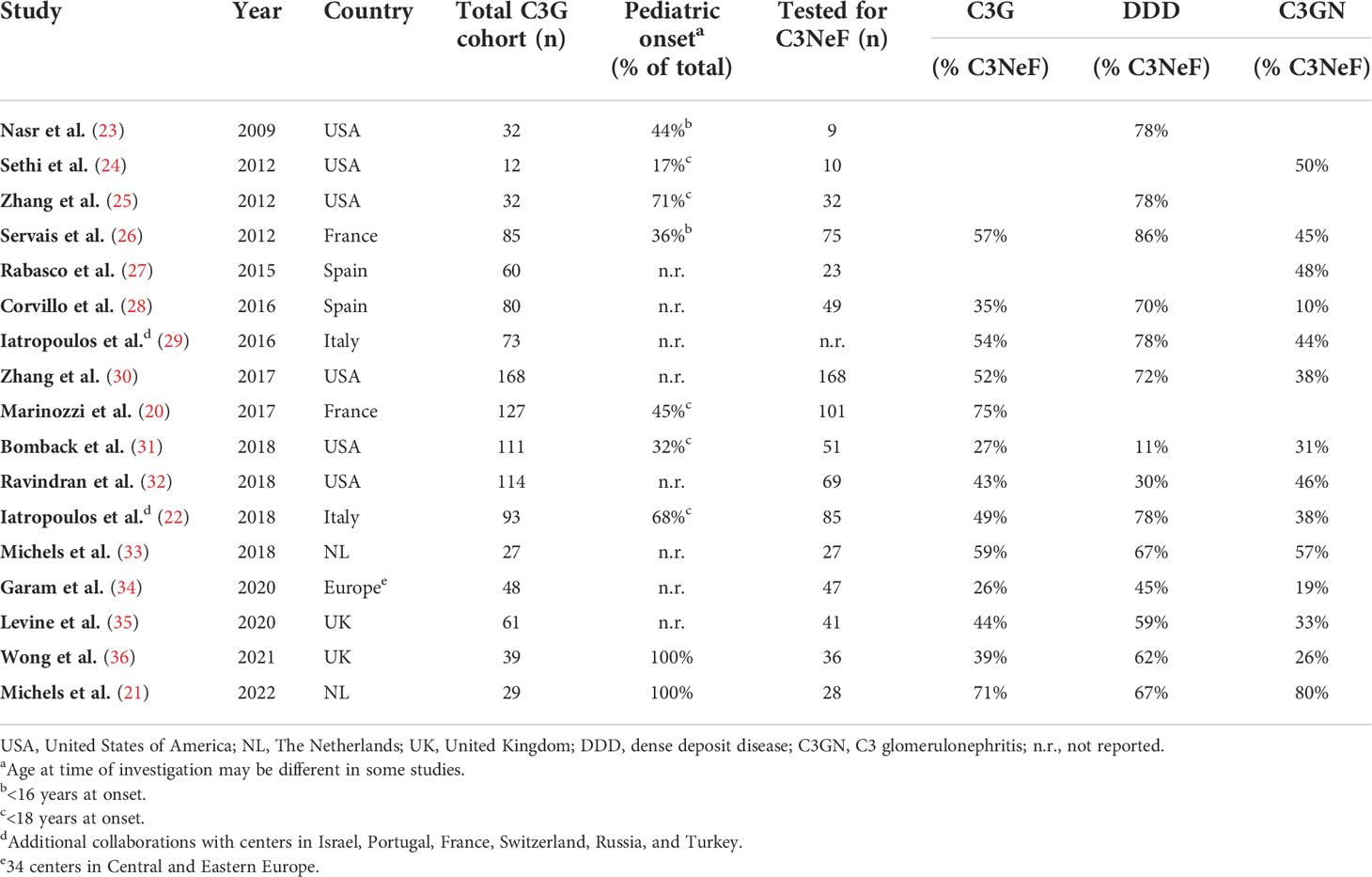
Table 1 Prevalence of C3 nephritic factor (C3NeF) in C3 glomerulopathy (C3G) cohorts.
C3NeFs are also found in a substantial number of patients with idiopathic (primary) immune complex-mediated MPGN (IC-MPGN). The prevalence in patient cohorts is reported between 23% and 54% (22, 26, 29, 34–36). The pattern of glomerular injury in IC-MPGN resembles the injury in C3G, but IC-MPGN is distinguished from C3G by the substantial presence of immunoglobulin deposits, which can cause CP activation in the glomerulus. However, the finding that genetic and acquired abnormalities of the AP, including C3NeFs, are found in both diseases suggests they may share a disease spectrum (37). This is also supported by cases in which the IC-MPGN pattern evolves to a C3G pattern in subsequent biopsies or vice versa.
Next to these glomerular kidney diseases, C3NeFs are common in patients with acquired partial lipodystrophy (APL). APL is an extremely rare disorder characterized by fat loss in the upper half of the body, usually occurring during childhood or adolescence (38). Most patients have C3 hypocomplementemia and in approximately 70-80% of the cases C3NeFs are found, especially in those with low serum C3 levels (38–40). Supposedly as a result of the C3NeF presence, approximately 20% of the patients develop C3G (38, 41, 42).
Lastly, C3NeFs have been described, although much less frequently, in patients with meningococcal infections and disease (43–45), in patients with systemic lupus erythematosus (SLE), who often also showed presence of APL and/or C3G (46–48), and in post-infectious glomerulonephritis (49–51).
C4NeFs have been reported in several diseases as well. The first reports from 1980 identified C4NeFs in a patient with acute post-infectious glomerulonephritis (15) and in patients with SLE (14). Later studies have described C4NeFs in more patients with SLE (52, 53), in patients with MPGN (52, 54, 55), in patients with Sjögren syndrome (52), and in a patient with sepsis caused by an N. meningitidis infection (56). More recently, the focus of C4NeF research has been on C3G and IC-MPGN. C4NeFs have been reported in two C3G cohorts with an incidence of 3% (30) and 8% (57) and in mixed C3G/IC-MPGN cohorts with an incidence of 6% (58) and 14% (59). Interestingly, in a substantial proportion of these patients both C4NeFs and C3NeFs are found (30, 52, 54, 55, 58, 59).
4. Nephritic factor heterogeneity
C3NeFs form a heterogeneous group of immunoglobulins that likely bind to different epitopes of the AP convertase. First, they differ in the extent to which they prolong the half-life of the C3 convertase (20, 25, 60), ranging from tens of minutes to even hours. Second, C3NeFs confer the convertase with varying resistance against the various complement regulatory proteins. C3NeFs may interfere with Factor H (FH) (60–65), complement receptor 1 (CR1) (60, 66), and decay-accelerating factor (DAF) (60, 67), although the efficiency (for each regulator) differs between NeFs of different patients. Few C3NeFs were found that did not or hardly interfere with any of these regulators of extrinsic decay (60). Third, C3NeFs differ in their dependence on the convertase-stabilizing protein properdin. Some C3NeFs are only active in the presence of properdin, i.e. properdin-dependent NeFs, whereas other C3NeFs also, or only (20), act on convertases without properdin, i.e. properdin-independent NeFs (20, 25, 60, 62, 68–70). Finally, it has been shown that NeFs can differ in their ability to affect C5 convertase activity and C5 conversion, sometimes referred to as C5NeF activity (20, 60–62, 71) (Figure 2). It is this marked heterogeneity that has complicated our understanding of C3NeFs since their discovery until today.
Although less studied, C4NeFs also form a heterogeneous group. Convertase stabilization through interference with complement regulator-mediated extrinsic decay has been described for C4 binding protein (C4BP) (30, 56, 72, 73), CR1 (30, 56, 74), and DAF (57, 67). Some studies have shown the specific ability of C4NeFs to also stabilize the C5 convertase complex C4bC2bC3b (56, 73), suggesting also C4NeFs may recognize different epitopes in different patients. However, it is important to note that research on the stabilization of CP/LP and especially AP C5 convertases by NeFs is hampered by the unknown exact structure of the C5 convertase complex and the transient nature between convertase complexes that cleave C3 and C5.
5. Nephritic factor detection methods
A major bottleneck in NeF research, related to the heterogeneous nature of NeFs, is the difficulty and diversity of detection methods, which are often only available in specialized laboratories. Many different assays are being used for NeF identification and tests differ in sensitivity and specificity. C4NeF testing is available in even fewer laboratories than C3NeF testing, so here we focus on the methods described for C3NeF detection, which often have a parallel version for detecting C4NeFs. NeF assays can generally be divided into three groups (Figure 3): [1] binding assays that detect the binding of the NeF to the convertase complex, [2] functional assays that measure the C3 activation products, and [3] functional assays that detect the stabilization of the convertase. C3NeFs can be variably positive in these different assays, hence some laboratories use a combination of them for C3NeF detection (25, 60).
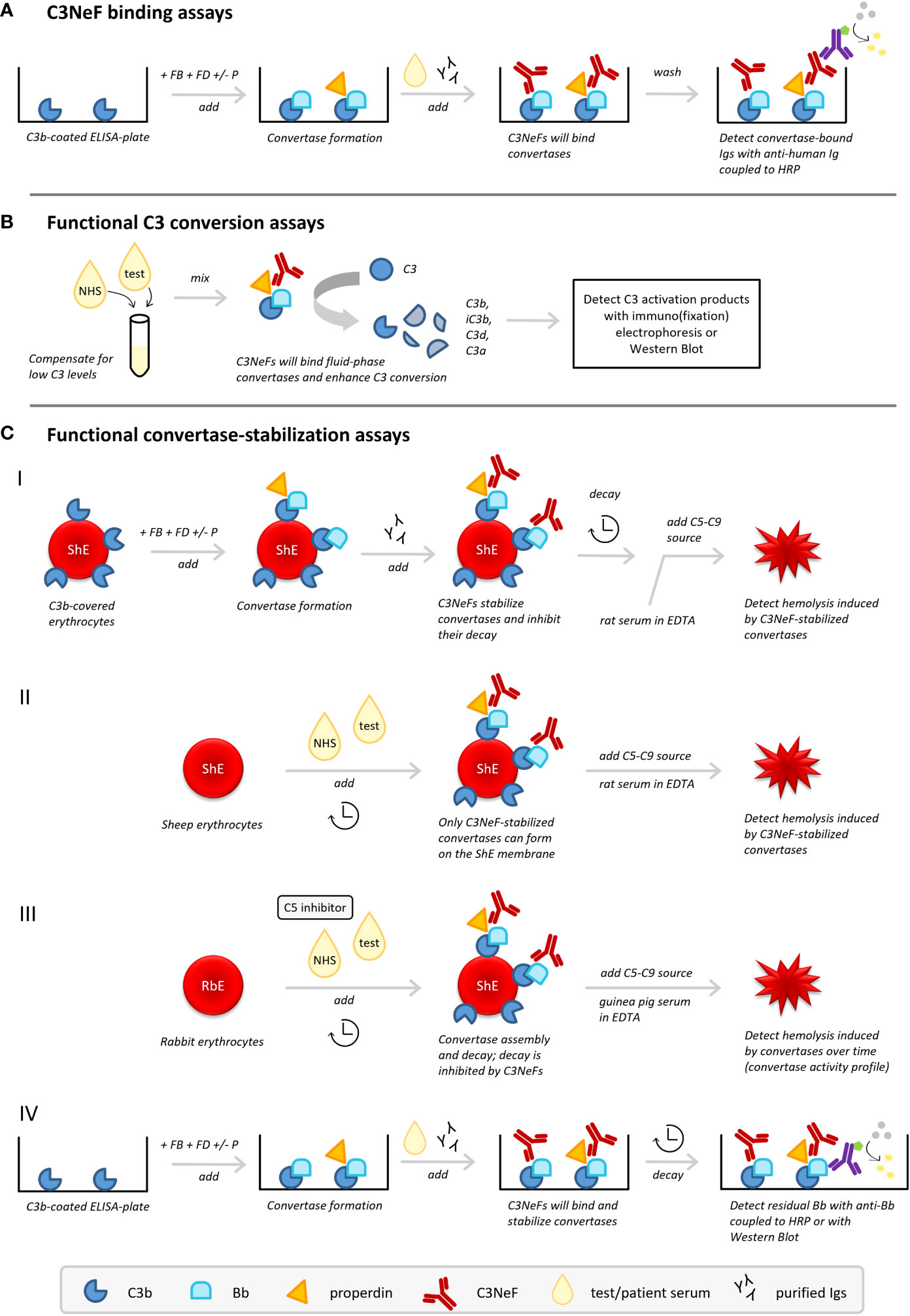
Figure 3 C3 nephritic factor (C3NeF) detection methods. Three types of methods to detect C3NeFs can be distinguished: (A) assays detecting NeF binding to the convertase, (B) functional assays measuring C3 conversion influenced by NeFs, and (C) functional assays measuring convertase stabilization by NeFs. ELISA, enzyme-linked immunosorbent assay; FB, Factor B; FD, Factor D; HRP, horseradish peroxidase; Igs, immunoglobulins; NHS, normal human serum; P, properdin; RbE, rabbit erythrocytes; ShE, sheep erythrocytes.
5.1. C3NeF binding assays
C3NeF binding assays (Figure 3A) are usually enzyme-linked immunosorbent assay (ELISA)-based methods aimed to detect the interaction of the C3NeF antibody with its target, i.e. plate-bound AP convertases formed from purified complement components (25, 60, 61, 75). Typically, plates are coated with C3b; then Factor B (FB) and Factor D (FD), in presence or absence of properdin, are added to allow formation of AP convertase complexes. After washing, or simultaneous with the addition of purified AP components, the test sample is added, i.e. purified patient immunoglobulins or patient sera. If C3NeFs are present, their binding can be detected via horseradish peroxidase-conjugated anti-human immunoglobulin antibodies supporting substrate conversion. Importantly, a C3b-only well should be used as a blank control in these methods.
In general, binding assays are easy to implement in laboratories and directly identify the immunoglobulin nature of the factor interacting with the enzyme complex. However, these assays are not functional assays, so they may detect a binding interaction that might not influence enzyme function. Besides this chance on false-positive results, the assay may also give false-negative results, as the convertases formed on microtiter plates can adopt artificial conformations that may not be recognized by all NeFs (Table 2).
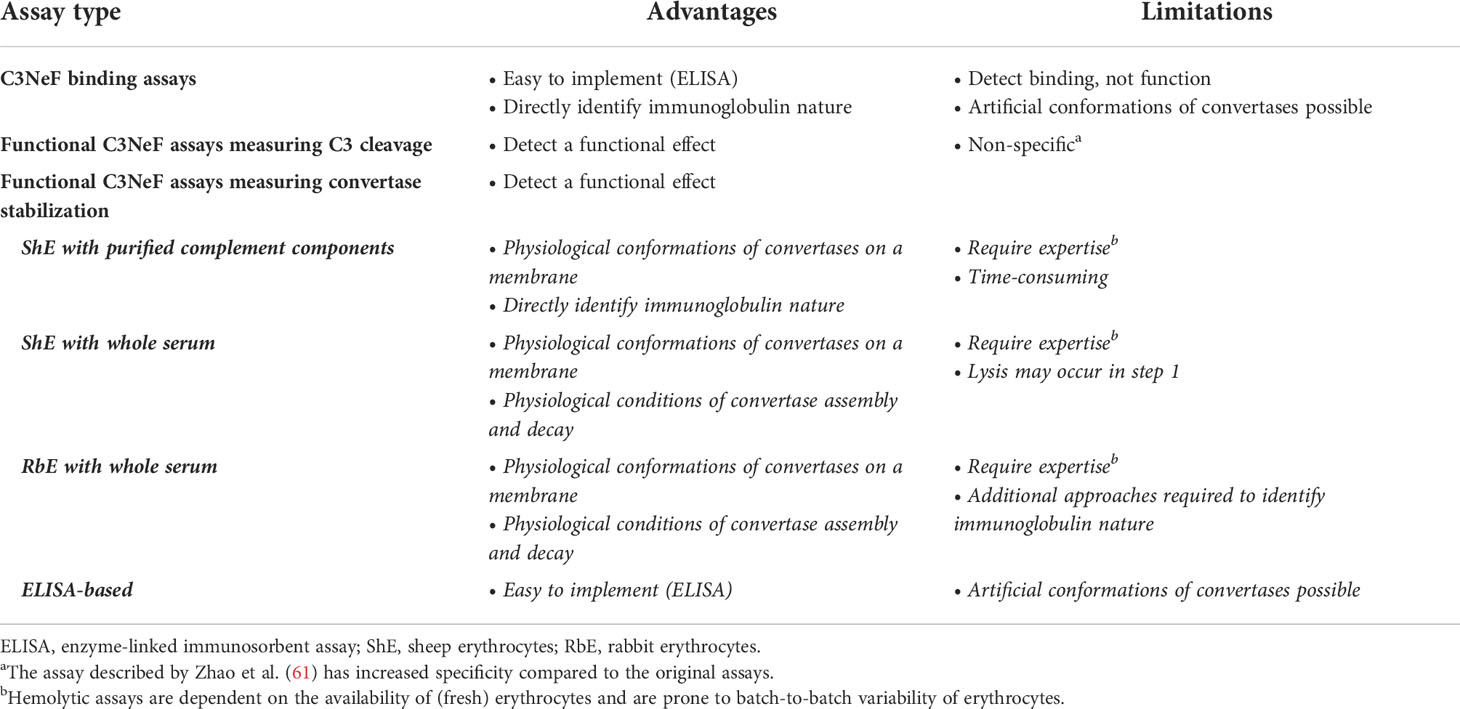
Table 2 Advantages and limitations of the different nephritic factor (NeF) detection assays.
5.2. Functional C3NeF assays measuring C3 activation products
Functional assays measuring the activation products of C3 (Figure 3B) were the first assays described for measuring C3NeF activity (8, 76). These tests measure the release of C3 breakdown products after mixing the test sample with control serum by immunoelectrophoresis or western blotting (25, 60). Many patients with C3G and other diseases related to C3NeF have low serum levels of C3 as a result of consumption. Hence, mixing with control serum to supply uncleaved C3 is essential to allow formation of fluid phase AP convertases to be stabilized by the NeFs from the test sample. C3NeF positivity will then result in increased C3 conversion.
The advantage of these assays is that they are detecting a functional enzyme effect. However, increased C3 breakdown may also be caused by other factors, so a positive outcome does not have to be specific for presence of C3NeFs (Table 2).
Zhao et al. recently described a variant on this assay. Fluid phase C3 convertases were assembled with purified components, which were then mixed with patient immunoglobulins and additional C3 (61). C3 conversion was measured by the release of C3a detected by ELISA.
5.3. Functional C3NeF assays measuring convertase stabilization
Functional assays measuring the convertase-stabilizing function of C3NeFs were developed later, mostly in the form of hemolytic assays (9) (Figure 3C). These assays have supplemented or replaced many of the previous assays and are frequently used today. Although there are variations in the exact procedures, AP hemolytic C3NeF assays generally follow the same principle: to measure the activity of C3NeF-stabilized convertases on the erythrocyte membrane via C5b-9-induced hemolysis. Hemolysis provides the easy readout of released hemoglobin that can be detected spectrophotometrically.
5.3.1. Sheep erythrocyte hemolytic assays with convertases formed from purified components
In the first variant, sheep erythrocytes are used as the platform for formation of AP convertases with purified complement components (Figure 3C.I). Since sheep erythrocytes are rich in sialic acids and therefore naturally protected against human complement attack by recruiting the major AP regulator FH (77, 78), specific approaches need to be applied to cover the sheep erythrocytes with initial C3b. C3b can be deposited on the sensitized, i.e. anti-sheep antibody-treated, sheep erythrocyte membrane via CP convertase intermediates (79, 80) or on unsensitized sheep erythrocytes by using FB-partially inactivated/FH-depleted serum (25). Subsequently, AP convertases are built from purified components (FB and FD, with or without properdin) followed by the addition of patient immunoglobulin fractions as a potential source of C3NeFs. Importantly, convertases are then left to decay for a set time period. After this incubation, the residual activity of convertases is examined by the addition of rat serum diluted in ethylenediaminetetraacetic acid (EDTA) as a heterologous source of C5-C9. This allows stabilized convertases to cleave C5 and induce C5b-9-mediated hemolysis. Rat serum, as well as guinea pig serum, is compatible with human complement and more potent in generating C5b-9; the EDTA prevents de novo convertase formation from these sera. If no C3NeFs are present in the test sample, the convertases are broken down after a certain decay time and no hemolysis is observed. However, if hemolysis is observed, it indicates that convertases have been stabilized by C3NeFs present in the test sample, allowing them to induce C5b-9 formation (20, 25, 81).
5.3.2. Sheep erythrocyte hemolytic assays with convertases formed from whole serum
Another widely applied hemolytic C3NeF assay combines unsensitized sheep erythrocytes with a mix of control serum and test serum (82–84) (Figure 3C.II). This method exploits the ability of C3NeFs to form and stabilize convertase complexes on the surface of the naturally non-activating sheep erythrocyte by overcoming the negative regulation; an action that cannot be fulfilled by serum in the absence of C3NeFs (83). A limited time frame is used to restrict the complement activation by the test sample to convertase assembly and to prevent lysis. Hemolysis is only induced in a standardized second step by addition of rat serum in EDTA-buffer. If no hemolysis is seen, no C3NeFs were present and no convertases could be formed on the sheep erythrocyte membrane. Although this time-dependent separation of convertase assembly and hemolysis will apply for most samples, it should be noted that some patient samples are able to cause (unwanted) hemolysis in the convertase formation step of those assays (85) (Table 2).
5.3.3. Rabbit erythrocyte hemolytic assays with convertases formed from whole serum
Recently, an adapted variant of this assay using rabbit erythrocytes (86, 87) was optimized for C3NeF diagnostics (33) (Figure 3C.III). Rabbit erythrocytes have a low sialic acid content and are therefore potent AP activators. When mixed with control serum and test serum, AP convertases will spontaneously assemble on their membrane. Importantly, a C5 inhibitor, e.g. eculizumab, is added here to strictly prevent hemolysis during this first step and to separate convertase assembly from hemolysis readout. After washing of the erythrocytes, guinea pig serum in EDTA is added to allow preformed convertases to cleave C5 and induce C5b-9-mediated hemolysis. In contrast to previous assays, the full convertase activity profile with convertase assembly and decay is visualized by using different incubation periods for the test serum incubation. Compared to control serum showing a profile with convertase assembly and clear convertase breakdown, serum containing C3NeFs will show a prolonged convertase activity profile with persistently high levels of lysis over time, indicative of stabilized convertases.
Of note, both this assay and the assay described in section 5.3.2 do not directly identify C3NeFs, since the immunoglobulin nature of the convertase-stabilizing factor in serum is not confirmed. Mixing control serum with patient immunoglobulin fractions as an extra test overcomes this limitation and will specify if the convertase-stabilizing factor is an autoantibody (33). On the other hand, assessing the kinetics of convertase assembly and decay in complete serum offers the advantage of the physiological environment. All complement regulators and other serum factors that may influence convertase activity are present, so this maximizes the chance of detecting functionally relevant pathogenic factors (Table 2).
5.3.4. ELISA-based convertase stabilization assays
The principles of convertase stabilization by NeFs in the hemolytic assays above, especially the one described in section 5.3.1, have also been translated to assays analyzing the kinetics of convertase dissociation on a microtiter plate (Figure 3C.IV). AP convertases are built from purified components on a C3b-coated microtiter plate after which serum or patient immunoglobulin fractions are added. Instead of hemolysis, residual plate-bound Bb fragments detected after a certain time of decay are used as a measure of intact convertases stabilized by C3NeFs (60, 61). Recently, also a variant of this ELISA-based assay was described that combined convertase formation and decay on microtiter plates with residual Bb analysis on western blot (62).
Of note, these assays do not assess the C3/C5 cleaving capacities of the C3NeF-stabilized convertases. Furthermore, in hemolytic assays, convertases are formed on a membrane surface in their natural orientation via the reactive thioester of C3, whereas convertases formed on microtiter plates may adopt artificial configurations. Nonetheless, the availability of erythrocytes and batch-to-batch variability are a major challenge of hemolytic assays, making these assays often only available in specialized labs. In contrast, ELISA techniques are much more easy to implement (Table 2).
6. Efforts for assay standardization
As stated before, C3NeF detection in general is problematic, with the EQA rounds from 2016-2020 reporting success rates for identifying the reference samples between 31% (2020) and 75% (2016) (7). Thus, if results of a sample are compared between laboratories, or even between different tests within one laboratory, variable outcomes are obtained. At the moment, a golden standard is lacking and there is a high need for standardization. The same holds true for C4NeF assays.
First, it is important to note that properdin-dependent C3NeFs are missed in assays that study the stability of C3bBb convertases formed from purified components without properdin. In line with this, previous reports describing panels with multiple C3NeF assays have shown that additional NeFs could be identified when properdin was incorporated in the test settings (20, 25, 60, 62). Marinozzi et al. also identified a group of C3NeFs that was only detected in absence of properdin (20), and this is supported by our own unpublished observations. However, it remains to be investigated what the physiological relevance of these NeFs is, as in physiological conditions properdin is always present in the circulation.
Recently, a CFB construct based on the gain-of-function variant p.Lys323Glu was proposed as a standardized positive control for C3NeF assays (88). This genetic variation is associated with atypical hemolytic uremic syndrome and has previously been shown to result in a stabilized convertase complex that is more resistant to negative regulation (89). The construct caused prolonged convertase activity reminiscent of that of patients with C3NeF activity (33, 88). Therefore, this might be a good alternative for patient serum or patient immunoglobulins, of which material is limited. For C4NeF assays, a similar approach could be followed, as recently also C2 gain-of-function variants resulting in stabilized CP convertase complexes were identified (90).
Furthermore, surfaces other than erythrocytes have been described for convertase analysis, namely magnetic beads and liposomes (91, 92). These platforms may be more suitable for standardization due to lower batch-to-batch variability. However, these setups have not yet been validated for C3NeF or C4NeF detection. Besides, magnetic beads are a less physiological model than erythrocytes. The optimal platform for the detection of functionally relevant NeFs should still be determined. Thus, future research may not only focus on improved standardization but also on further assay optimization with different (cellular) targets for convertase assembly.
7. Clinical implications
Assay standardization may also lead to a better understanding of the role of C3NeFs and C4NeFs in disease. For a long time, it has been difficult to relate C3NeF presence with complement (activity) markers and clinical parameters, such as disease activity and outcome. Nonetheless, increased interest in this field of research has led to major progresses during the last decade. Here, we will discuss some of the main findings of the clinical associations of NeFs, with a focus on C3G/IC-MPGN.
7.1. Clinical associations of C3NeFs
By stabilizing AP convertases, C3NeFs increase the potential of C3 conversion, which may lead to consumption of C3. Indeed, previous studies have associated the lowered C3 levels in patients with C3G with the presence of C3NeFs (20, 26, 29, 93). Recently, two studies on pediatric C3G cohorts showed that although C3 levels at presentation did not differ between patients with or without C3NeFs, the last measured C3 levels during follow-up were significantly lower in the patients that had tested positive for C3NeF (21, 36). This indicates the presence of C3NeF was associated with a higher likelihood of continued C3 consumption. In line with this, a previous report showed permanently low C3 during follow-up in 67% (12/18) of the C3NeF-positive children with C3G (94).
In a subset of patients with C3G, the complement system is dysregulated up to the level of the terminal pathway as seen by elevated sC5b-9 levels. This implies that there is also enhanced activation of the C5 convertases in those patients, and it was already hypothesized in 1986 that different NeF types may lie at the basis of this difference (71). Several studies have related properdin-dependent C3NeFs to (stronger) terminal pathway activation and to the C3GN phenotype (20, 62, 68, 69). On itself, C3GN is also associated with more pronounced terminal pathway activation (e.g. higher sC5b-9 and lower C5 and properdin) compared to the DDD subtype (19, 21). In 2017, the term C5 nephritic factor (C5NeF) was coined for NeFs that stabilize the AP C5 convertase. This was based on the finding that these NeFs stabilized convertases formed from purified proteins with properdin (C3bBbP) and were correlated with increased sC5b-9 levels (20). Some patients with C5NeFs were also C3NeF positive, i.e. they also tested positive when the stability of convertases formed without properdin (C3bBb) was assayed. In this regard, single C5NeF positive patients as described by this study correspond to the patients with properdin-dependent C3NeFs, and C3NeF/C5NeF double-positive patients correspond to patients with properdin-independent C3NeFs. Importantly, the exact composition and conformation of AP C5 convertases (and CP/LP C5 convertases) remain unknown. Furthermore, some patients with presumed C5NeFs (isolated or in combination with C3NeFs) do not show C5 convertase dysregulation, i.e. increased sC5b-9 levels, and conversely, some patients without C5NeFs do have increased sC5b-9 levels (20, 62). Moreover, complement markers are not always available. Therefore, the application of C5NeF terminology may be confusing, as it is unclear whether the C5NeF terminology is directly interchangeable with the properdin-dependency of NeFs and whether it may be used regardless of C5 convertase dysregulation. This is why it may be preferable to classify the NeF types based on their dependence on properdin for recognizing the convertase and to refrain from implications on the specific stabilization of the C5 convertase complex.
Even though C3NeFs are the most commonly found AP aberrations in C3G, not much is known about their behavior over time in the course of the disease and their relation to clinical outcome. While some patients have been shown to maintain their C3NeF activity for more than 10 years (58), C3NeF findings may also fluctuate over time, with some patients eventually losing their C3NeFs (26, 33, 93, 94). The longitudinal follow-up of C3NeFs is often not part of standard patient investigations. Moreover, many studies did not find correlations between C3NeF activity and clinical parameters (93, 94), although some studies have shown that patients with C3NeFs were more likely to have a better outcome (20, 29).
7.2. Clinical associations of C4NeFs
Lately, C4NeF research has also mostly focused on C3G and IC-MPGN. One of the most important questions here is how and to what extent a CP-dysregulating factor can contribute to AP-driven disease. In many patients with C3G, the disease is preceded by an infectious trigger, which exposes C4NeF epitopes. C3G shows many clinical similarities with post-infectious glomerulonephritis, such as low serum C3 levels, but in contrast to C3G, the C3 consumption and disease generally resolve within weeks or months (95, 96). However, some (atypical) cases evolve to C3G, indicating that an initial CP-mediated response may evolve to a primarily AP-driven pathology, likely via the AP amplification loop. Possibly, C4NeFs elicit such a similar mechanism in C3G and IC-MPGN.
The majority of C3G/IC-MPGN patients with C4NeFs are characterized by strong complement activation up to the level of the terminal pathway, especially patients positive for both C4NeFs and C3NeFs (30, 54, 58, 59). This double positivity for both C4NeF and C3NeF occurs in many cases and makes it difficult to determine the individual contribution of both NeFs to the disease. Furthermore, C4NeFs may persist over the disease course and during partial remission up to 70 months (58).
Studies on the relationship between C4NeF activity and clinical markers are scarce and often lack power due to the low number of positive patients. Besides, the scarce literature shows conflicting results. In a cohort with 100 hypocomplementemic MPGN patients described in 1994, nephrotic syndrome and poor prognosis occurred more frequently in C3NeF/C4NeF double-positive patients compared to single C3NeF or single C4NeF-positive patients (54). A recent study with 119 IC-MPGN/C3G patients showed that C4NeF-positive and C3NeF/C4NeF double-positive patients less frequently had a lowered eGFR at presentation and that, during the follow-up, none of the 17 patients reached kidney failure requiring kidney replacement therapy (in contrast to 17/92 C4NeF-negative patients) (59). In a study of our group, we did not observe an abnormal clinical presentation or disease course compared to C3G/IC-MPGN patients without C4NeFs (58).
8. Conclusions and future directions
With the increase in complement therapeutics, laboratory analysis of the complement system has become increasingly important (97). For example, it can be key in monitoring complement-directed therapy, as has been shown for the C5 inhibitor eculizumab in atypical hemolytic uremic syndrome (98, 99). It is well-known that accurate NeF analysis is challenging. This is due to the heterogenic nature of NeF autoantibodies, including differences in properdin-dependency, and due to inconsistent outcomes of samples tested in different assays between laboratories or even between different assays within one laboratory. As outlined in this review, each assay has its own advantages and disadvantages. There is a high need for comparison of NeF findings between groups. The considerable inconsistency between cohort findings now complicates the interpretation of the results of the NeF associations with complement biomarkers and/or disease parameters. Assay standardization, in combination with longitudinal analysis of NeFs and all relevant complement and clinical parameters in patients, may help to reveal different pathophysiological mechanisms of complement dysregulation in different patient groups. It may even provide more insight in disease outcome. Iatropoulos et al. showed that disease outcome was better predicted when patients were divided in groups based on cluster analysis, including not only pathology findings but also complement (e.g. NeF presence) and clinical findings, than based on the traditional subdivision into C3GN, DDD, and IC-MPGN (22). Moreover, our group showed in vitro proof that properdin inhibition may be a potential novel therapeutic approach for patients with properdin-dependent NeFs, likely regardless of disease classification (70). Thus, accurate and reliable NeF diagnostics are essential to better understand disease, to characterize patient (sub)groups and to move forward towards more specific complement-directed therapy in different patient groups.
Author contributions
MM wrote the manuscript. EV, NK, and LH critically reviewed the manuscript. All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by the Dutch Kidney Foundation (13OCA27 COMBAT Consortium and 20OK021 PERSPECTIV).
Acknowledgments
This study was performed on behalf of the COMBAT Consortium. This is an interuniversity collaboration in the Netherlands that is formed to study basic mechanisms, assay development, and therapeutic translation of complement-mediated renal diseases. Principal investigators are (in alphabetical order): S. Berger (Department of Internal Medicine-Nephrology, University Medical Center Groningen, Groningen, Netherlands), J. van den Born (Department of Internal Medicine-Nephrology, University Medical Center Groningen, Groningen, Netherlands), P. Gros (Department of Chemistry, Utrecht University, Utrecht, Netherlands), L. van den Heuvel (Department of Pediatric Nephrology, Radboud University Medical Center, Nijmegen, Netherlands), N. van de Kar (Department of Pediatric Nephrology, Radboud University Medical Center, Nijmegen, Netherlands), C. van Kooten (Department of Internal Medicine-Nephrology, Leiden University Medical Center, Leiden, Netherlands), M. Seelen (Department of Internal Medicine-Nephrology, University Medical Center Groningen, Groningen, Netherlands), A. de Vries (Department of Internal Medicine-Nephrology, Leiden University Medical Center, Leiden, Netherlands). NK and BH and are members of the European Reference Network for Rare Kidney Diseases (ERKNet)-Project No 739532.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Pouw RB, Ricklin D. Tipping the balance: Intricate roles of the complement system in disease and therapy. Semin Immunopathol (2021) 43(6):757–71. doi: 10.1007/s00281-021-00892-7
2. Legendre CM, Licht C, Muus P, Greenbaum LA, Babu S, Bedrosian C, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Eng J Med (2013) 368(23):2169–81. doi: 10.1056/NEJMoa1208981
3. Menne J, Delmas Y, Fakhouri F, Licht C, Lommelé Å, Minetti EE, et al. Outcomes in patients with atypical hemolytic uremic syndrome treated with eculizumab in a long-term observational study. BMC Nephrol (2019) 20(1):125. doi: 10.1186/s12882-019-1314-1
4. Dmytrijuk A, Robie-Suh K, Cohen MH, Rieves D, Weiss K, Pazdur R. FDA Report: Eculizumab (Soliris) for the treatment of patients with paroxysmal nocturnal hemoglobinuria. Oncologist (2008) 13(9):993–1000. doi: 10.1634/theoncologist.2008-0086
5. Mastellos DC, Ricklin D, Lambris JD. Clinical promise of next-generation complement therapeutics. Nat Rev Drug Discov (2019) 18(9):707–29. doi: 10.1038/s41573-019-0031-6
6. Zelek WM, Xie L, Morgan BP, Harris CL. Compendium of current complement therapeutics. Mol Immunol (2019) 114:341–52. doi: 10.1016/j.molimm.2019.07.030
7. Frazer-Abel A, Kirschfink M, Prohászka Z. Expanding horizons in complement analysis and quality control. Front Immunol (2021) 12:697313. doi: 10.3389/fimmu.2021.697313
8. Spitzer RE, Vallota EH, Forristal J, Sudora E, Stitzel A, Davis NC, et al. Serum C’3 lytic system in patients with glomerulonephritis. Science (1969) 164(3878):436–7. doi: 10.1126/science.164.3878.436
9. Daha MR, Fearon DT, Austen KF. C3 nephritic factor (C3NeF): Stabilization of fluid phase and cell-bound alternative pathway convertase. J Immunol (1976) 116(1):1–7.
10. Daha MR, Austen KF, Fearon DT. Heterogeneity, polypeptide chain composition and antigenic reactivity of C3 nephritic factor. J Immunol (1978) 120(4):1389–94.
11. Daha MR, van Es LA. Further evidence for the antibody nature of C3 nephritic factor (C3NeF). J Immunol (1979) 123(2):755–8.
12. Davis AE 3rd, Ziegler JB, Gelfand EW, Rosen FS, Alper CA. Heterogeneity of nephritic factor and its identification as an immunoglobulin. Proc Natl Acad Sci USA (1977) 74(9):3980–3. doi: 10.1073/pnas.74.9.3980
13. Scott DM, Amos N, Sissons JG, Lachmann PJ, Peters DK. The immunogloblin nature of nephritic factor (NeF). Clin Exp Immunol (1978) 32(1):12–24.
14. Daha MR, Hazevoet HM, Vanes LA, Cats A. Stabilization of the classical pathway C3 convertase C42, by a factor F-42, isolated from serum of patients with systemic lupus erythematosus. Immunology (1980) 40(3):417–24.
15. Halbwachs L, Leveillé M, Lesavre P, Wattel S, Leibowitch J. Nephritic factor of the classical pathway of complement: Immunoglobulin G autoantibody directed against the classical pathway C3 convertase enzyme. J Clin Invest (1980) 65(6):1249–56. doi: 10.1172/jci109787
16. Pickering MC, D’Agati VD, Nester CM, Smith RJ, Haas M, Appel GB, et al. C3 glomerulopathy: Consensus report. Kidney Int (2013) 84(6):1079–89. doi: 10.1038/ki.2013.377
17. Goodship TH, Cook HT, Fakhouri F, Fervenza FC, Frémeaux-Bacchi V, Kavanagh D, et al. Atypical hemolytic uremic syndrome and C3 glomerulopathy: Conclusions from a “Kidney disease: Improving global outcomes” (KDIGO) controversies conference. Kidney Int (2017) 91(3):539–51. doi: 10.1016/j.kint.2016.10.005
18. Hou J, Markowitz GS, Bomback AS, Appel GB, Herlitz LC, Barry Stokes M, et al. Toward a working definition of C3 glomerulopathy by immunofluorescence. Kidney Int (2014) 85(2):450–6. doi: 10.1038/ki.2013.340
19. Zhang Y, Nester CM, Martin B, Skjoedt MO, Meyer NC, Shao D, et al. Defining the complement biomarker profile of C3 glomerulopathy. Clin J Am Soc Nephrol (2014) 9(11):1876–82. doi: 10.2215/cjn.01820214
20. Marinozzi MC, Chauvet S, Le Quintrec M, Mignotet M, Petitprez F, Legendre C, et al. C5 nephritic factors drive the biological phenotype of C3 glomerulopathies. Kidney Int (2017) 92(5):1232–41. doi: 10.1016/j.kint.2017.04.017
21. Michels MAHM, Wijnsma KL, Kurvers RAJ, Westra D, Schreuder MF, van Wijk JAE, et al. Long-term follow-up including extensive complement analysis of a pediatric C3 glomerulopathy cohort. Pediatr Nephrol (2022) 37(3):601–12. doi: 10.1007/s00467-021-05221-6
22. Iatropoulos P, Daina E, Curreri M, Piras R, Valoti E, Mele C, et al. Cluster analysis identifies distinct pathogenetic patterns in C3 glomerulopathies/immune complex-mediated membranoproliferative GN. J Am Soc Nephrol (2018) 29(1):283–94. doi: 10.1681/asn.2017030258
23. Nasr SH, Valeri AM, Appel GB, Sherwinter J, Stokes MB, Said SM, et al. Dense deposit disease: Clinicopathologic study of 32 pediatric and adult patients. Clin J Am Soc Nephrol (2009) 4(1):22–32. doi: 10.2215/cjn.03480708
24. Sethi S, Fervenza FC, Zhang Y, Zand L, Vrana JA, Nasr SH, et al. C3 glomerulonephritis: Clinicopathological findings, complement abnormalities, glomerular proteomic profile, treatment, and follow-up. Kidney Int (2012) 82(4):465–73. doi: 10.1038/ki.2012.212
25. Zhang Y, Meyer NC, Wang K, Nishimura C, Frees K, Jones M, et al. Causes of alternative pathway dysregulation in dense deposit disease. Clin J Am Soc Nephrol (2012) 7(2):265–74. doi: 10.2215/cjn.07900811
26. Servais A, Noel LH, Roumenina LT, Le Quintrec M, Ngo S, Dragon-Durey MA, et al. Acquired and genetic complement abnormalities play a critical role in dense deposit disease and other C3 glomerulopathies. Kidney Int (2012) 82(4):454–64. doi: 10.1038/ki.2012.63
27. Rabasco C, Cavero T, Roman E, Rojas-Rivera J, Olea T, Espinosa M, et al. Effectiveness of mycophenolate mofetil in C3 glomerulonephritis. Kidney Int (2015) 88(5):1153–60. doi: 10.1038/ki.2015.227
28. Corvillo F, Bravo Garcia-Morato M, Nozal P, Garrido S, Tortajada A, Rodriguez de Cordoba S, et al. Serum properdin consumption as a biomarker of C5 convertase dysregulation in C3 glomerulopathy. Clin Exp Immunol (2016) 184(1):118–25. doi: 10.1111/cei.12754
29. Iatropoulos P, Noris M, Mele C, Piras R, Valoti E, Bresin E, et al. Complement gene variants determine the risk of immunoglobulin-associated MPGN and C3 glomerulopathy and predict long-term renal outcome. Mol Immunol (2016) 71:131–42. doi: 10.1016/j.molimm.2016.01.010
30. Zhang Y, Meyer NC, Fervenza FC, Lau W, Keenan A, Cara-Fuentes G, et al. C4 nephritic factors in C3 glomerulopathy: A case series. Am J Kidney Dis (2017) 70(6):834–43. doi: 10.1053/j.ajkd.2017.07.004
31. Bomback AS, Santoriello D, Avasare RS, Regunathan-Shenk R, Canetta PA, Ahn W, et al. C3 glomerulonephritis and dense deposit disease share a similar disease course in a large United States cohort of patients with C3 glomerulopathy. Kidney Int (2018) 93(4):977–85. doi: 10.1016/j.kint.2017.10.022
32. Ravindran A, Fervenza FC, Smith RJH, De Vriese AS, Sethi S. C3 glomerulopathy: Ten years’ experience at Mayo clinic. Mayo Clin Proc (2018) 93(8):991–1008. doi: 10.1016/j.mayocp.2018.05.019
33. Michels MAHM, van de Kar NCAJ, Okrój M, Blom AM, van Kraaij SAW, Volokhina EB, et al. Overactivity of alternative pathway convertases in patients with complement-mediated renal diseases. Front Immunol (2018) 9:612. doi: 10.3389/fimmu.2018.00612
34. Garam N, Prohászka Z, Szilágyi Á, Aigner C, Schmidt A, Gaggl M, et al. Validation of distinct pathogenic patterns in a cohort of membranoproliferative glomerulonephritis patients by cluster analysis. Clin Kidney J (2020) 13(2):225–34. doi: 10.1093/ckj/sfz073
35. Levine AP, Chan MMY, Sadeghi-Alavijeh O, Wong EKS, Cook HT, Ashford S, et al. Large-scale whole-genome sequencing reveals the genetic architecture of primary membranoproliferative GN and C3 glomerulopathy. J Am Soc Nephrol (2020) 31(2):365–73. doi: 10.1681/asn.2019040433
36. Wong EKS, Marchbank KJ, Lomax-Browne H, Pappworth IY, Denton H, Cooke K, et al. C3 glomerulopathy and related disorders in children: Etiology-phenotype correlation and outcomes. Clin J Am Soc Nephrol (2021) 16(11):1639–51. doi: 10.2215/cjn.00320121
37. Noris M, Daina E, Remuzzi G. Membranoproliferative glomerulonephritis: No longer the same disease and may need very different treatment. Nephrol Dial Transplant (2021) Epub ahead of print. doi: 10.1093/ndt/gfab281
38. Misra A, Peethambaram A, Garg A. Clinical features and metabolic and autoimmune derangements in acquired partial lipodystrophy: Report of 35 cases and review of the literature. Med (Baltimore) (2004) 83(1):18–34. doi: 10.1097/01.md.0000111061.69212.59
39. Corvillo F, Ceccarini G, Nozal P, Magno S, Pelosini C, Garrido S, et al. Immunological features of patients affected by Barraquer-Simons syndrome. Orphanet J Rare Dis (2020) 15(1):9. doi: 10.1186/s13023-019-1292-1
40. Corvillo F, Okrój M, Nozal P, Melgosa M, Sánchez-Corral P, López-Trascasa M. Nephritic factors: An overview of classification, diagnostic tools and clinical associations. Front Immunol (2019) 10:886. doi: 10.3389/fimmu.2019.00886
41. Corvillo F, López-Trascasa M. Acquired partial lipodystrophy and C3 glomerulopathy: Dysregulation of the complement system as a common pathogenic mechanism. Nefrol (Engl Ed) (2018) 38(3):258–66. doi: 10.1016/j.nefro.2017.10.002
42. Garg A. Acquired and inherited lipodystrophies. N Eng J Med (2004) 350(12):1220–34. doi: 10.1056/NEJMra025261
43. Teisner B, Elling P, Svehag SE, Poulsen L, Lamm LU, Sjöholm A. C3 nephritic factor in a patient with recurrent Neisseria meningitidis infections. Acta Pathol Microbiol Immunol Scand C (1984) 92(6):341–9. doi: 10.1111/j.1699-0463.1984.tb00098.x
44. Fijen CA, Kuijper EJ, te Bulte MT, Daha MR, Dankert J. Assessment of complement deficiency in patients with meningococcal disease in the Netherlands. Clin Infect Dis (1999) 28(1):98–105. doi: 10.1086/515075
45. Thompson RA, Yap PL, Brettle RB, Dunmow RE, Chapel H. Meningococcal meningitis associated with persistent hypocomplementaemia due to circulating C3 nephritic factor. Clin Exp Immunol (1983) 52(1):153–6.
46. Walport MJ, Davies KA, Botto M, Naughton MA, Isenberg DA, Biasi D, et al. C3 nephritic factor and SLE: Report of four cases and review of the literature. QJM (1994) 87(10):609–15.
47. Cronin CC, Higgins TJ, Molloy M. Lupus, C3 nephritic factor and partial lipodystrophy. QJM (1995) 88(4):298–9.
48. Hristova MH, Stoyanova VS. Autoantibodies against complement components in systemic lupus erythematosus - role in the pathogenesis and clinical manifestations. Lupus (2017) 26(14):1550–5. doi: 10.1177/0961203317709347
49. Davis CA, McAdams AJ, Wyatt RJ, Forristal J, McEnery PT, West CD. Idiopathic rapidly progressive glomerulonephritis with C3 nephritic factor and hypocomplementemia. J Pediatr (1979) 94(4):559–63. doi: 10.1016/s0022-3476(79)80010-4
50. Frémeaux-Bacchi V, Weiss L, Demouchy C, May A, Palomera S, Kazatchkine MD. Hypocomplementaemia of poststreptococcal acute glomerulonephritis is associated with C3 nephritic factor (C3NeF) IgG autoantibody activity. Nephrol Dial Transplant (1994) 9(12):1747–50.
51. Chauvet S, Berthaud R, Devriese M, Mignotet M, Vieira Martins P, Robe-Rybkine T, et al. Anti-factor B antibodies and acute postinfectious GN in children. J Am Soc Nephrol (2020) 31(4):829–40. doi: 10.1681/asn.2019080851
52. Arakawa M, Seino J, Kinoshita Y, Fukuda K, Saito T, Furuyama T. Nephritic factor (NeF) of alternate pathway (NeFA) and of classical pathway (NeFc). Tohoku J Exp Med (1982) 138(4):401–9. doi: 10.1620/tjem.138.401
53. Daha MR, Hazevoet HM, van Es LA. Heterogeneity, polypeptide chain composition and antigenic reactivity of autoantibodies (F-42) that are directed against the classical pathway C3 convertase of complement and isolated from sera of patients with systemic lupus erythematosus. Clin Exp Immunol (1984) 56(3):614–20.
54. Ohi H, Yasugi T. Occurrence of C3 nephritic factor and C4 nephritic factor in membranoproliferative glomerulonephritis (MPGN). Clin Exp Immunol (1994) 95(2):316–21.
55. Tanuma Y, Ohi H, Watanabe S, Seki M, Hatano M. C3 nephritic factor and C4 nephritic factor in the serum of two patients with hypocomplementaemic membranoproliferative glomerulonephritis. Clin Exp Immunol (1989) 76(1):82–5.
56. Miller EC, Chase NM, Densen P, Hintermeyer MK, Casper JT, Atkinson JP. Autoantibody stabilization of the classical pathway C3 convertase leading to C3 deficiency and neisserial sepsis: C4 nephritic factor revisited. Clin Immunol (2012) 145(3):241–50. doi: 10.1016/j.clim.2012.09.007
57. Blom AM, Corvillo F, Magda M, Stasilojc G, Nozal P, Perez-Valdivia MA, et al. Testing the activity of complement convertases in serum/plasma for diagnosis of C4NeF-mediated C3 glomerulonephritis. J Clin Immunol (2016) 36(5):517–27. doi: 10.1007/s10875-016-0290-5
58. Michels MAHM, van de Kar NCAJ, van Kraaij SAW, Sarlea SA, Gracchi V, Engels F, et al. Different aspects of classical pathway overactivation in patients with C3 glomerulopathy and immune complex-mediated membranoproliferative glomerulonephritis. Front Immunol (2021) 12:715704. doi: 10.3389/fimmu.2021.715704
59. Garam N, Prohaszka Z, Szilagyi A, Aigner C, Schmidt A, Gaggl M, et al. C4 nephritic factor in patients with immune-complex-mediated membranoproliferative glomerulonephritis and C3-glomerulopathy. Orphanet J Rare Dis (2019) 14(1):247. doi: 10.1186/s13023-019-1237-8
60. Paixao-Cavalcante D, Lopez-Trascasa M, Skattum L, Giclas PC, Goodship TH, de Cordoba SR, et al. Sensitive and specific assays for C3 nephritic factors clarify mechanisms underlying complement dysregulation. Kidney Int (2012) 82(10):1084–92. doi: 10.1038/ki.2012.250
61. Zhao F, Afonso S, Lindner S, Hartmann A, Löschmann I, Nilsson B, et al. C3-glomerulopathy autoantibodies mediate distinct effects on complement C3- and C5-convertases. Front Immunol (2019) 10:1030. doi: 10.3389/fimmu.2019.01030
62. Donadelli R, Pulieri P, Piras R, Iatropoulos P, Valoti E, Benigni A, et al. Unraveling the molecular mechanisms underlying complement dysregulation by nephritic factors in C3G and IC-MPGN. Front Immunol (2018) 9:2329. doi: 10.3389/fimmu.2018.02329
63. Jelezarova E, Schlumberger M, Sadallah S, Späth PJ, Schifferli JA, Lutz HU. A C3 convertase assay for nephritic factor functional activity. J Immunol Methods (2001) 251(1-2):45–52. doi: 10.1016/s0022-1759(01)00295-2
64. Ohi H, Watanabe S, Fujita T, Yasugi T. Significance of C3 nephritic factor (C3NeF) in non-hypocomplementaemic serum with membranoproliferative glomerulonephritis (MPGN). Clin Exp Immunol (1992) 89(3):479–84. doi: 10.1111/j.1365-2249.1992.tb06984.x
65. Weiler JM, Daha MR, Austen KF, Fearon DT. Control of the amplification convertase of complement by the plasma protein beta1H. Proc Natl Acad Sci U.S.A. (1976) 73(9):3268–72. doi: 10.1073/pnas.73.9.3268
66. Daha MR, Kok DJ, Van Es LA. Regulation of the C3 nephritic factor stabilized C3/C5 convertase of complement by purified human erythrocyte C3b receptor. Clin Exp Immunol (1982) 50(1):209–14.
67. Ito S, Tamura N, Fujita T. Effect of decay-accelerating factor on the assembly of the classical and alternative pathway C3 convertases in the presence of C4 or C3 nephritic factor. Immunology (1989) 68(4):449–52.
68. Clardy CW, Forristal J, Strife CF, West CD. A properdin dependent nephritic factor slowly activating C3, C5, and C9 in membranoproliferative glomerulonephritis, types I and III. Clin Immunol Immunopathol (1989) 50(3):333–47. doi: 10.1016/0090-1229(89)90141-4
69. Tanuma Y, Ohi H, Hatano M. Two types of C3 nephritic factor: Properdin-dependent C3NeF and properdin-independent C3NeF. Clin Immunol Immunopathol (1990) 56(2):226–38. doi: 10.1016/0090-1229(89)90141-4
70. Michels MAHM, van de Kar NCAJ, van den Bos RM, van der Velden TJAM, van Kraaij SAW, Sarlea SA, et al. Novel assays to distinguish between properdin-dependent and properdin-independent C3 nephritic factors provide insight into properdin-inhibiting therapy. Front Immunol (2019) 10:1350. doi: 10.3389/fimmu.2019.01350
71. Mollnes TE, Ng YC, Peters DK, Lea T, Tschopp J, Harboe M. Effect of nephritic factor on C3 and on the terminal pathway of complement in vivo and in vitro. Clin Exp Immunol (1986) 65(1):73–9.
72. Daha MR, van Es LA. Relative resistance of the F-42-stabilized classical pathway C3 convertase to inactivation by C4-binding protein. J Immunol (1980) 125(5):2051–4.
73. Gigli I, Sorvillo J, Mecarelli-Halbwachs L, Leibowitch J. Mechanism of action of the C4 nephritic factor. Deregulation of the classical pathway of C3 convertase. J Exp Med (1981) 154(1):1–12. doi: 10.1084/jem.154.1.1
74. Fischer E, Kazatchkine MD, Mecarelli-Halbwachs L. Protection of the classical and alternative complement pathway C3 convertases, stabilized by nephritic factors, from decay by the human C3b receptor. Eur J Immunol (1984) 14(12):1111–4. doi: 10.1002/eji.1830141209
75. Skattum L. Analysis of C3 nephritic factors by ELISA. Methods Mol Biol (2019) 1901:177–82. doi: 10.1007/978-1-4939-8949-2_13
76. Peters DK, Martin A, Weinstein A, Cameron JS, Barratt TM, Ogg CS, et al. Complement studies in membrano-proliferative glomerulonephritis. Clin Exp Immunol (1972) 11(3):311–20.
77. Kazatchkine MD, Fearon DT, Austen KF. Human alternative complement pathway: Membrane-associated sialic acid regulates the competition between B and beta1 H for cell-bound C3b. J Immunol (1979) 122(1):75–81.
78. Ferreira VP, Pangburn MK, Cortés C. Complement control protein Factor H: The good, the bad, and the inadequate. Mol Immunol (2010) 47(13):2187–97. doi: 10.1016/j.molimm.2010.05.007
79. Chabannes M, Bordereau P, Martins PV, Dragon-Durey MA. Sheep erythrocyte preparation for hemolytic tests exploring complement functional activities. Methods Mol Biol (2021) 2227:61–7. doi: 10.1007/978-1-0716-1016-9_6
80. Ohi H, Watanabe S, Fujita T, Seki M, Hatano M. Detection of C3bBb-stabilizing activity (C3 nephritic factor) in the serum from patients with membranoproliferative glomerulonephritis. J Immunol Methods (1990) 131(1):71–6. doi: 10.1016/0022-1759(90)90234-m
81. Chabannes M, Frémeaux-Bacchi V, Chauvet S. Detection of C3 nephritic factor by hemolytic assay. Methods Mol Biol (2021) 2227:147–58. doi: 10.1007/978-1-0716-1016-9_15
82. West CD. A hemolytic method for the measurement of nephritic factor. J Immunol Methods (2008) 335(1-2):1–7. doi: 10.1016/j.jim.2007.12.001
83. Rother U. A new screening test for C3 nephritis factor based on a stable cell bound convertase on sheep erythrocytes. J Immunol Methods (1982) 51(1):101–7. doi: 10.1016/0022-1759(82)90386-6
84. López-Trascasa M, Marín MA, Fontán G. C3 nephritic factor determination. A comparison between two methods. J Immunol Methods (1987) 98(1):77–82. doi: 10.1016/0022-1759(87)90438-8
85. Skattum L, Mårtensson U, Sjöholm AG. Hypocomplementaemia caused by C3 nephritic factors (C3 NeF): Clinical findings and the coincidence of C3 NeF type II with anti-C1q autoantibodies. J Intern Med (1997) 242(6):455–64. doi: 10.1111/j.1365-2796.1997.tb00018.x
86. Blom AM, Volokhina EB, Fransson V, Strömberg P, Berghard L, Viktorelius M, et al. A novel method for direct measurement of complement convertases activity in human serum. Clin Exp Immunol (2014) 178(1):142–53. doi: 10.1111/cei.12388
87. Okroj M, Holmquist E, King BC, Blom AM. Functional analyses of complement convertases using C3 and C5-depleted sera. PLoS One (2012) 7(10):e47245. doi: 10.1371/journal.pone.0047245
88. Urban A, Borowska A, Felberg A, van den Heuvel L, Stasilojc G, Volokhina E, et al. Gain of function mutant of complement factor b K323E mimics pathogenic C3NeF autoantibodies in convertase assays. Autoimmunity (2018) 51(1):18–24. doi: 10.1080/08916934.2017.1423286
89. Goicoechea de Jorge E, Harris CL, Esparza-Gordillo J, Carreras L, Arranz EA, Garrido CA, et al. Gain-of-function mutations in complement factor B are associated with atypical hemolytic uremic syndrome. Proc Natl Acad Sci USA (2007) 104(1):240–5. doi: 10.1073/pnas.0603420103
90. Urban A, Volokhina E, Felberg A, Stasiłojć G, Blom AM, Jongerius I, et al. Gain-of-function mutation in complement C2 protein identified in a patient with aHUS. J Allergy Clin Immunol (2020) 146(4):916–9.e11. doi: 10.1016/j.jaci.2020.02.014
91. Yorulmaz Avsar S, Jackman JA, Kim MC, Yoon BK, Hunziker W, Cho NJ. Immobilization strategies for functional complement convertase assembly at lipid membrane interfaces. Langmuir (2017) 33(29):7332–42. doi: 10.1021/acs.langmuir.7b01465
92. Zwarthoff SA, Berends ETM, Mol S, Ruyken M, Aerts PC, Józsi M, et al. Functional characterization of alternative and classical pathway C3/C5 convertase activity and inhibition using purified models. Front Immunol (2018) 9:1691. doi: 10.1034/j.1399-3038.2001.012003166.x.
93. Schwertz R, Rother U, Anders D, Gretz N, Scharer K, Kirschfink M. Complement analysis in children with idiopathic membranoproliferative glomerulonephritis: A long-term follow-up. Pediatr Allergy Immunol (2001) 12(3):166–72.
94. Nicolas C, Vuiblet V, Baudouin V, Macher MA, Vrillon I, Biebuyck-Gouge N, et al. C3 nephritic factor associated with C3 glomerulopathy in children. Pediatr Nephrol (2014) 29(1):85–94. doi: 10.1007/s00467-013-2605-6
95. Al-Ghaithi B, Chanchlani R, Riedl M, Thorner P, Licht C. C3 glomerulopathy and post-infectious glomerulonephritis define a disease spectrum. Pediatr Nephrol (2016) 31(11):2079–86. doi: 10.1007/s00467-015-3311-3
96. Sethi S, Fervenza FC, Zhang Y, Zand L, Meyer NC, Borsa N, et al. Atypical postinfectious glomerulonephritis is associated with abnormalities in the alternative pathway of complement. Kidney Int (2013) 83(2):293–9. doi: 10.1038/ki.2012.384
97. Prohászka Z, Kirschfink M, Frazer-Abel A. Complement analysis in the era of targeted therapeutics. Mol Immunol (2018) 102:84–8. doi: 10.1016/j.molimm.2018.06.001
98. Volokhina E, Wijnsma K, van der Molen R, Roeleveld N, van der Velden T, Goertz J, et al. Eculizumab dosing regimen in atypical HUS: Possibilities for individualized treatment. Clin Pharmacol Ther (2017) 102(4):671–8. doi: 10.1002/cpt.686
Keywords: nephritic factor, complement system, complement dysregulation, complement alternative pathway, assay standardization, C3 glomerulopathy, immune complex-mediated glomerulonephritis
Citation: Michels MAHM, Volokhina EB, van de Kar NCAJ and van den Heuvel LPWJ (2022) Challenges in diagnostic testing of nephritic factors. Front. Immunol. 13:1036136. doi: 10.3389/fimmu.2022.1036136
Received: 04 September 2022; Accepted: 12 October 2022;
Published: 14 November 2022.
Edited by:
Lillemor Skattum, Lund University, SwedenReviewed by:
Yuzhou Zhang, The University of Iowa, United StatesJiaxuan Chen, Affiliated Hospital of Guangdong Medical University, China
Copyright © 2022 Michels, Volokhina, van de Kar and van den Heuvel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marloes A. H. M. Michels, bWFybG9lcy5taWNoZWxzQHJhZGJvdWR1bWMubmw=