 Xiaoyan Zhan
Xiaoyan Zhan Qiang Li1,2
Qiang Li1,2 Guang Xu
Guang Xu Xiaohe Xiao
Xiaohe Xiao Zhaofang Bai
Zhaofang Bai- 1Department of Hepatology, Fifth Medical Center of Chinese PLA General Hospital, Beijing, China
- 2China Military Institute of Chinese Materia, Fifth Medical Center of Chinese PLA General Hospital, Beijing, China
NLRP3 (NOD-, LRR-, and pyrin domain-containing protein 3) is a cytosolic pattern recognition receptor (PRR) that recognizes multiple pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs). Once activated, NLRP3 initiates the inflammasome assembly together with the adaptor ASC and the effector caspase-1, leading to caspase-1 activation and subsequent cleavage of IL-1β and IL-18. Aberrant NLRP3 inflammasome activation is linked with the pathogenesis of multiple inflammatory diseases, such as cryopyrinassociated periodic syndromes, type 2 diabetes, non-alcoholic steatohepatitis, gout, and neurodegenerative diseases. Thus, NLRP3 is an important therapeutic target, and researchers are putting a lot of effort into developing its inhibitors. The review summarizes the latest advances in the mechanism of NLRP3 inflammasome activation and its pharmacological inhibitors.
1. Introduction
Inflammasomes are a class of complexes made up of cytosolic proteins, mediating inflammatory response to pathogen infection and damage to the host (1). Inflammasome is usually composed of a pattern-recognition receptor (PRR), an adaptor known as ASC, and the effector caspase-1.Once activated by an inflammatory ligand, inflammasomes trigger the auto-processing of caspase-1 into the catalytically active caspase-1, which mediates the cleavage of pro-IL-1β and pro-IL-18 (2, 3) and induces pyroptosis (4).
There are several PRRs that could form inflammasomes, including NLRP1, NLRP3, AIM2, NLRC4, and IFI16 (5–7). NLRP3 inflammasome is the best characterized one. NLRP3-activating mutations have been reported to cause cryopyrinassociated periodic syndromes (CAPS) (8). Moreover, a variety of sterile danger signals in tissues, which induce chronic inflammation also activate NLRP3, so NLRP3 inflammasome is central to the pathogenesis of many chronic inflammatory diseases such as gout, type 2 diabetes, non-alcoholic steatohepatitis (NASH), atherosclerosis, and Alzheimer’s disease (AD) (9–13). Consequently, NLRP3 is regarded as an essential target for pharmacological intervention to treat related inflammatory disorders.
To date, various pharmacological inhibitors of NLRP3 inflammasome have been identified and exhibit therapeutic effects in chronic inflammatory diseases. In this review, we introduce the latest findings in the mechanism of NLRP3 inflammasome activation and the reported inhibitors of NLRP3 inflammasome, which may benefit the treatment of NLRP3 inflammasome-driven diseases.
2. Mechanism of NLRP3 inflammasome activation
NLRP3 is a member of NLR family, containing an aminoterminal PYRIN (PYD) domain, a nucleotide-binding NACHT domain, and a carboxyterminal leucinerich repeat (LRR) domain (Figure 1A). The PYD domain is important for the recruitment of ASC; the NACHT domain contains ATPase activity required for NLRP3 conformational change and oligomerization. The role of LRR domain in NLRP3 activation is controversial, but recent structural studies have shown that the LRR domain is important for the formation of the NLRP3 cage, which is required to disperse the trans-Golgi network at the early stage of NLRP3 inflammasome activation (14). Upon responding to stimuli that cause loss of autoinhibition (15), NLRP3 oligomerizes through interactions between NACHT domains, then recruiting ASC via PYD-PYD interactions, ASC self-associates into helical filaments, which further form a single macromolecular focus-termed ASC speck (16–18). Assembled ASC recruits caspase-1 and enables self-cleavage and activation of caspase-1 (Figure 1B).
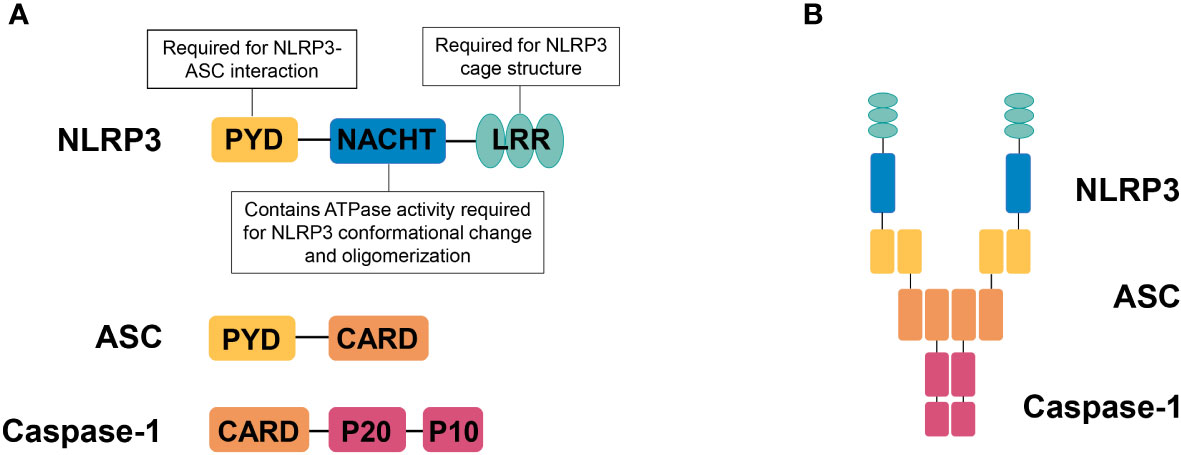
Figure 1 NLRP3 inflammasome. (A) Domain organization of NLRP3, ASC, and caspase-1. NLRP3 contains an aminoterminal PYRIN (PYD) domain, a nucleotidebinding NACHT domain, and a carboxyterminal leucinerich repeat (LRR) domain. The PYD domain is important for recruitment of ASC; the NACHT domain contains ATPase activity required for NLRP3 conformational change and oligomerization. The LRR domain is important for the formation of the NLRP3 cage, which is required to disperse the trans-Golgi network at the early stage of NLRP3 inflammasome activation. (B) NLRP3 inflammasome assembly. Upon activation, NLRP3 recruits ASC via PYD-PYD interactions and ASC recruits caspase-1 via CARD-CARD interactions.
NIMA-related kinase 7 (NEK7) has been identified as a scaffolding protein for NLRP3 activation (19–21). NEK7 is a member of NIMA (“never in mitosis gene a”)–related serine-threonine kinase family and important for mitosis, consisting of a catalytic domain and a short N-terminal domain. Both the NACHT and LRR of NLRP3 are involved in the association with NEK7. NLRP3-NEK7 interaction requires the catalytic domain of NEK7 but not impaired by mutations that abolish the catalytic activity of NEK7, showing that NEK7 binds NLRP3 independently of its kinase activity (19). Meanwhile, NEK7 is dispensable for NLRC4 or AIM2 inflammasome activation, demonstrating it is a specifically essential component of NLRP3 inflammasome (19). The specific structural mechanism of NEK7-licensed NLRP3 inflammasome activation has been revealed by Sharif and colleagues (22). Recently, Le Xiao et al. have reported the cryo-EM structures of the active NLRP3 inflammasome disk; it is proposed that NEK7 breaks the inactive NLRP3 cage to transform NLRP3 into the active NLRP3 inflammasome disk (23).
2.1. Canonical NLRP3 inflammasome activation
Canonical NLRP3 inflammasome activation requires two signals, priming and activation (Figure 2). NLRP3 inflammasome can be primed with various pathogen-associated molecular patterns (PAMPs) to activate NF-κB to induce transcription of NLRP3 and the key proinflammatory cytokines such as pro–IL-1β. Priming signal also mediates multiple post-translational modifications (PTMs) of NLRP3, which licenses NLRP3 for subsequent activation. For example, LPS priming downregulates FBXL2-induced ubiquitination and degradation of NLRP3 and increases its life span. LPS exposure induces level of an E3 ligase FBXO3, which targets FBXL2; the latter recognizes Trp-73 within NLRP3 for interaction and targets Lys-689 (human) for ubiquitin ligation and degradation (24). Moreover, priming triggers phosphorylation of NLRP3 at S194 (mouse) by JNK-1, which facilitates deubiquitylation of NLRP3 and its self-association (25).
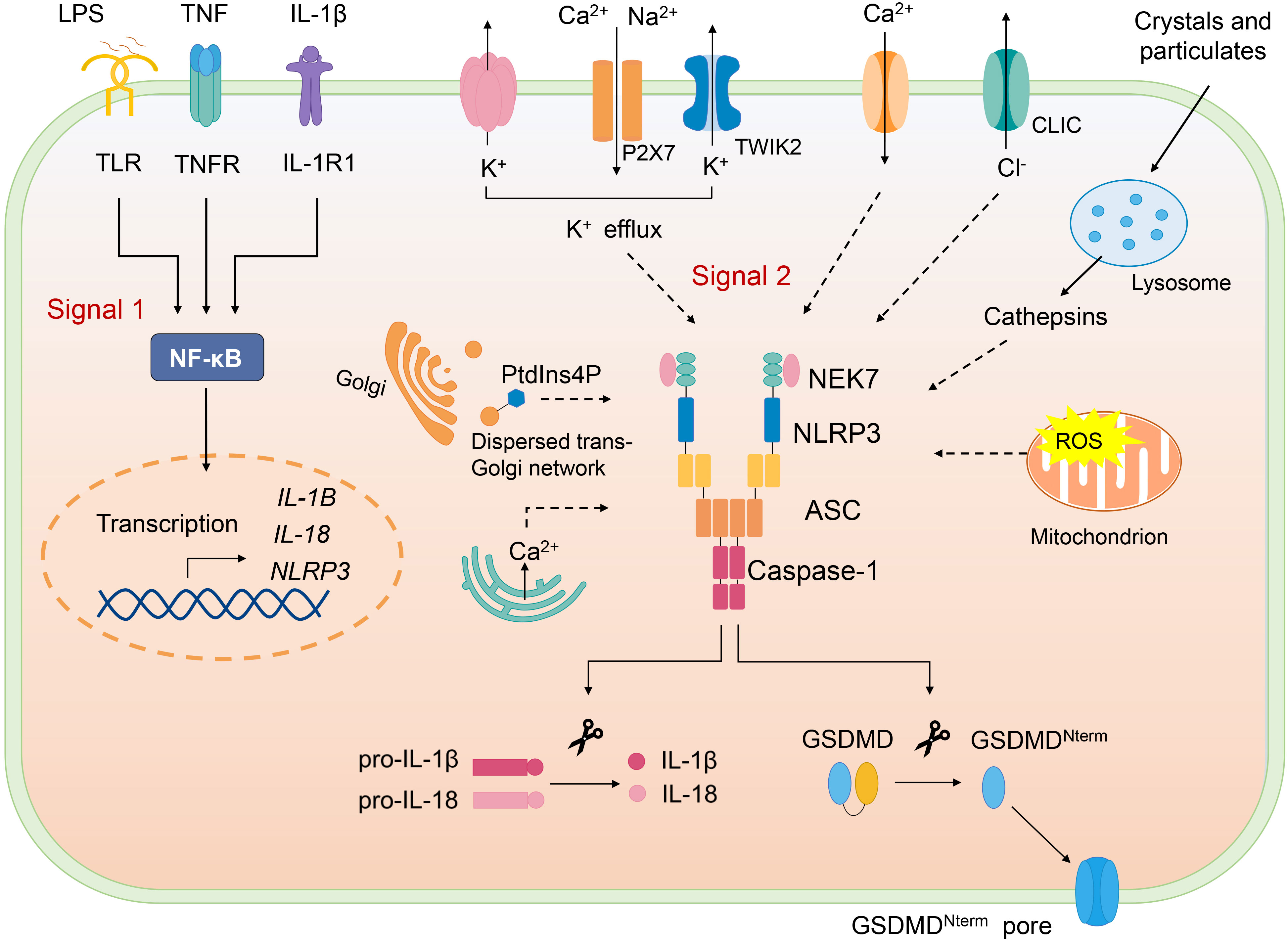
Figure 2 Canonical NLRP3 inflammasome. Canonical NLRP3 inflammasome activation requires two signals: The signal 1 (priming) involves PAMPs or cytokines-induced NF-κB activation and upregulates the gene expression of NLRP3 and the proinflammatory cytokines. Signal 2 is provided by various PAMPs and DAMPs; they activate multiple upstream signaling events, such as K+ efflux, Ca2+ flux, Cl- efflux, mtROS production, release of oxidized mitochondrial DNA, lysosome damage, and Golgi disassembly (dispersed trans-Golgi network). NLRP3 inflammasome assembly triggers the auto-cleavage of caspase-1, which in turn cleaves pro–IL-1β and pro–IL-18. Active caspase-1 also cleaves GSDMD and releases GSDMD-N from the autoinhibition and free GSDMD-N oligomerizes in membranes to form pores and induce pyroptosis.
Following the priming step, NLRP3 is activated by a second signal and initiates its self-oligomerization, activated NLRP3 forms inflammasome together with ASC and pro–caspase-1, causing full activation of NLRP3 inflammasome, which is defined as the “activation step”. Various stimuli, namely, PAMPs and DAMPs can offer the activation signal for NLRP3 inflammasome. To date, several upstream signals, which are interrelated and overlapping, have been proven to trigger assembly of NLRP3 inflammasome.
K+ efflux is considered as an important upstream signal of NLRP3 inflammasome activation. At first, potassium-proton ionophore nigericin and ATP were reported to cause the decrease of intracellular K+, which is indispensable for release of IL-1β, and K+ channel blockers inhibit the processing of IL-1β (26–28). It has recently been reported that P2X7-mediated cations influx promotes K+ efflux through TWIK2 upon ATP stimulation (29). Apart from several NLRP3 activators (such as nigericin) that can permeate the cell membrane to K+, particulate matter can also trigger K+ efflux, highlighting the significant role of K+ efflux in NLRP3 inflammasome activation (30). Nonetheless, NLRP3 activation could also be induced in K+ efflux-independent way. Imiquimod and CL097, the small molecules targeting mitochondria trigger ROS, induce NLRP3 activation independently of K+ efflux (31).
Ca2+ mobilization has been reported to be crucial for NLRP3 activation (32, 33). Upon NLRP3 activation stimulated by various agonists(nigericin, ATP, alum, and monosodium urate [MSU]), Ca2+ is mobilized. Blocking Ca2+ mobilization impairs NLRP3 inflammasome assembly and activation (33). However, how Ca2+ mobilization activates NLRP3 remains not clear. It is suggested that excessive ER release of Ca2+ leads to mitochondrial Ca2+ overload, which, in turn, causes mtROS production, an important trigger of NLRP3 inflammasome activation (34–36). Moreover, inhibitors of Ca2+ mobilization block mtROS production during NLRP3 inflammasome activation by ATP (33). However, a study documents that increased cytosolic Ca2+ induced by Ca2+ ionophore or Ca2+-mobilizing GPCR is not sufficient to activate NLRP3 inflammasome (37).
An increase of extracellular Cl- has been demonstrated to inhibit IL-1β release (38, 39), chloride intracellular channel proteins CLIC1 and CLIC4 deficiency impairs pro-IL-1β transcription and IL-1β secretion (40). Further study shows CLICs act downstream of K+ efflux-mtROS axis, mtROS induces CLICs translocation to plasma membrane and the subsequent chloride efflux, promoting NEK7-NLRP3 interaction, which is essential for inflammasome assembly (41). These researches suggest that chloride efflux is an important for NLRP3 activation. In addition, Cl- efflux has been reported to induce ASC oligomerization but that is inactive in the absence K+ efflux, which is necessary for NLRP3-NEK7 interaction and caspase-1 cleavage (42).
Lysosomal disruption is crucial for crystal-induced NLRP3 inflammasome activation. Multiple crystals could activate NLRP3, including inhaled silica crystals (43), uric acid (44), cholesterol (9), and fibrillar peptide amyloid-β (45). NLRP3 activation by crystals requires phagocytosis of crystals, which results in lysosomal damage and rupture. In a study, sterile lysosomal rupture caused by Leu-Leu-OMe (L-leucyl-L-leucine methyl ester) is sufficient to trigger NLRP3 inflammasome activation, whereas suppression of phagosomal acidification or cathepsin B (using Ca074Me as the inhibitor) blocks NLRP3 activation (43). However, Ca074Me is reported to inhibit multiple cathepsins, and NLRP3 inflammasome activation is mediated by several redundant cathepsins (including cathepsin B) (46).
Mitochondrion dysfunction is also involved in NLRP3 inflammasome activation. Inside the cell, damaged organelles are cleared by autophagy; autophagic proteins deficiency leads to the increased number of dysfunctional mitochondria and mitochondrial DNA (mtDNA) release, thus activating NLRP3 inflammasome (47). Mitochondrion ROS could also trigger NLRP3 activation, blocking key enzymes of the respiratory chain with rotenone or antimycin leads to mtROS production and then activates NLRP3, and inhibition of autophagy/mitophagy also results in accumulation of mtROS and NLRP3 activation (47). AIM2 inflammasome is also responsible for dsDNA recognition; however, oxidized mtDNA preferentially triggers NLRP3 not AIM2 inflammasome (48). CMPK2 is an enzyme that supplies deoxyribonucleotides for mtDNA synthesis, TLR signals via Myd88 and TRIF induce IRF1-dependent transcription of CAMK2, and CAMK2-mediated mtDNA synthesis is required for oxidized mtDNA generation induced by NLRP3 agonists (49). Moreover, mitochondrial lipid cardiolipin was found to interact with NLRP3 directly and induce its activation, suggesting that cardiolipin may serve as a mitochondrial docking site for NLRP3 as well as activating ligand (50). Nevertheless, cardiolipin participates in multiple mitochondrion processes involved in NLRP3 inflammasome activation, such as mitophagy, mitochondrion fission, and fusion (51), so whether cardiolipin directly activates NLRP3 remains further study.
Recently, the role of disassembly of the trans-Golgi network (TGN) in NLRP3 inflammasome activation has been revealed (52). Upon stimulation by NLRP3 agonists, TGN is dissembled into vesicles (dispersed TGN, dTGN), phosphatidylinositol-4-phosphate (PtdIns4P) (negatively charged) on the dTGN forms ion bond with the polybasic region of NLRP3, recruiting NLRP3 to dTGN, where NLRP3 aggregates into puncta, resulting in oligomerization of ASC and subsequent caspase-1 activation. The translocation of NLRP3 to dTGN is crucial for both K+ efflux dependent and independent inflammasome activation (52). During K+ efflux–dependent NLRP3 inflammasome activation, K+ efflux is demonstrated to be required for the recruitment of NLRP3 to TGN, possibly promoting ionic binding via lowering cellular ionic strength, but the subsequent activation does not require K+ efflux. Although stimulated by imiquimod and CL097(K+ efflux-independent stimuli), the recruitment is barely affected by extracellular KCl (52).
NLRP3 inflammasome activation also induces pyroptosis, characterized by pore formation in the plasma membrane, causing membrane rupture and release of cytosolic contents. Gasdermin D (GSDMD) is reported to be the executor of pyroptosis induced by NLRP3 inflammasome agonists (53, 54). GSDMD-N domain possesses the pyroptosis-inducing activity and is inhibited by its C domain (GSDMD-C), upon inflammasome activation, caspase-1 cleaves GSDMD into two parts, the free GSDMD-N oligomerizes in membranes to form pores, thus killing cells from inside (55, 56). In vitro, GSDMD-N is found to kill cell-free bacteria, implying its direct bactericidal effect but further study is needed (56). Moreover, it is demonstrated that GSDMD mediates IL-1β release from living macrophages under the hyperactivation state, GSDMD pores on the membrane facilitate and acts as a conduit for IL-1β secretion (57).
2.2. Non-canonical inflammasome activation
The non-canonical inflammasome refers to the caspase-11–dependent inflammasome (Figure 3) (58). During various bacterial infection, cytoplasmic LPS is recognized by caspase-11, which then mediates non-canonical inflammasome activation (59, 60); thus, caspase-11 plays a critical role in septic shock (59–61). It is demonstrated that mouse caspase-11 (human caspase-4 and caspase-5) directly binds to intracellular LPS followed by its oligomerization and activation (62). Upon activation, Caspase-4/5/11 also cleaves GSDMD and induces pyroptosis (53, 54), the active caspase-11 causes potassium efflux to induce canonical NLRP3 inflammasome activation, leading to IL-1β secretion (63). Oxidized phospholipids (oxPAPC), one class of DAMPs, can bind to caspase-11 and induce inflammasome activation, but oxPAPC only induces caspase-11–dependent IL-1 release, but not pyroptosis (64).
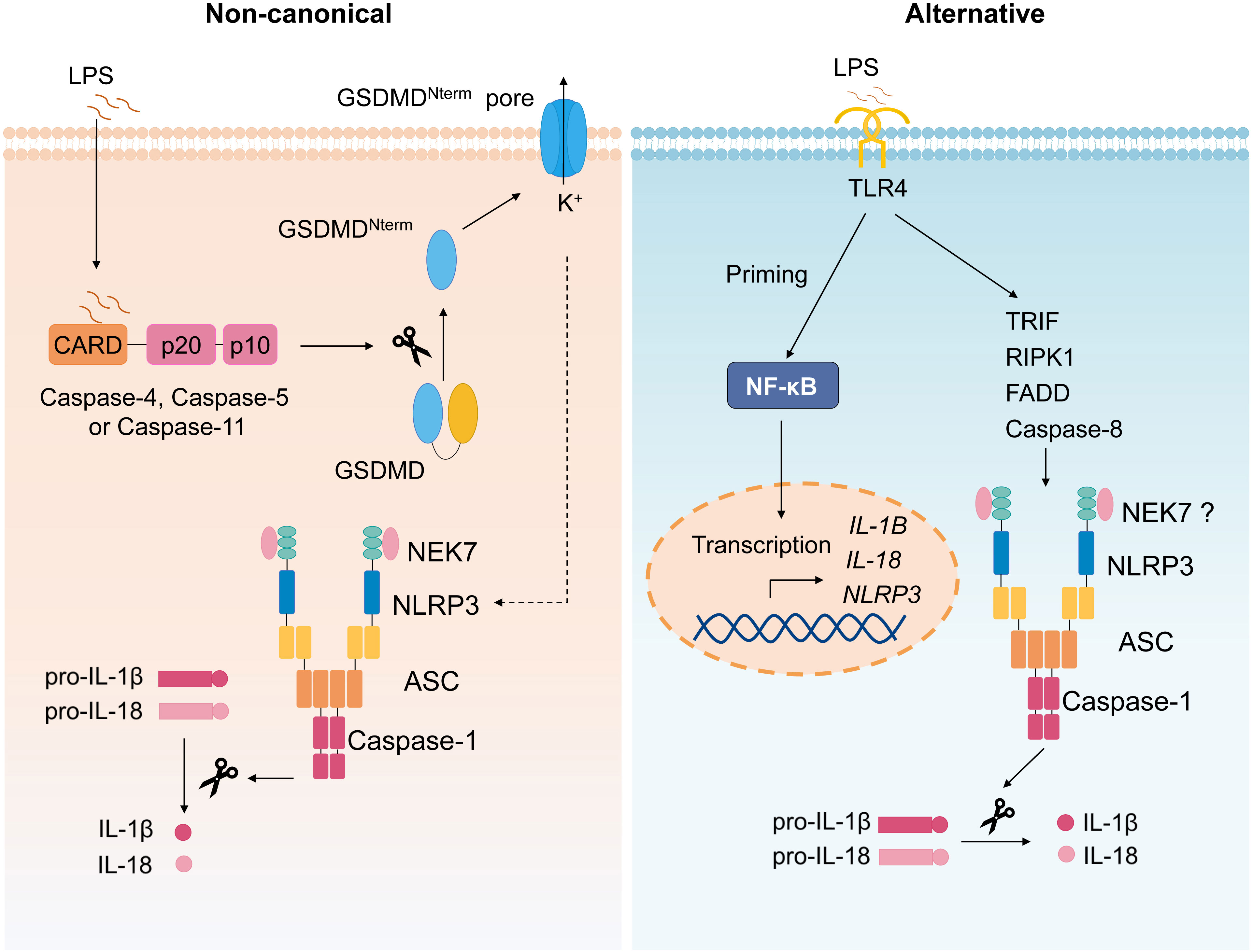
Figure 3 Non-canonical and alternative inflammasome. Non-canonical inflammasome activation is triggered by caspase-4/5/11 activation upon recognition of intracellular LPS, followed by cleavage of GSDMD and subsequent pyroptosis, causing the potassium efflux, which then induces canonical NLRP3 inflammasome. The alternative NLRP3 inflammasome activation refers to the signal 1 only inflammasome response. In monocytes, TLR ligands alone trigger IL-1β secretion via TLR4-TRIF-RIPK1-FADD-CASP8 signaling upstream of NLRP3.
2.3. Alternative inflammasome activation
Alternative NLRP3 inflammasome activation refers to the signal 1 only inflammasome response (Figure 3) (65). Initially, it is reported that TLR ligands alone could stimulate human monocytes to secrete IL-1β (66, 67). Moritz M. Gaidt et al. demonstrated that, in human and porcine monocytes, alternative inflammasome activation was mediated by TLR4-TRIF-RIPK1-FADD-CASP8 signaling upstream of NLRP3. Unlike canonical inflammasome, alternative response does not induce pyroptosome formation or pyroptosis (65). Apart from human and porcine monocytes, murine DCs are also reported to secrete mature IL-1β stimulated by TLRs agonists, which was mediated by NLRP3 inflammasome but independent on the purinergic P2X7 receptor (68).
3. NLRP3 and diseases
Due to the significant role of NLRP3 inflammasome in inflammatory responses, its activation must be tightly controlled. Aberrant NLRP3 inflammasome activation is considered to be responsible for the autoimmune disease CAPS, caused by the gain of function mutant of NLRP3. Many chronic diseases are driven by inflammatory responses in the presence of danger signals formed in pathology situations; these danger signals could stimulate NLRP3 inflammasome to induce secretion of proinflammatory cytokines including IL-1β and IL-18, promoting disease progression.
Gain-of-function mutation of NLRP3 has been documented to trigger CAPS (69–71). The CAPS refers to a class of autoinflammatory disorder with cutaneous, ocular, musculoskeletal, and neurologic disease symptoms combined with chronic inflammation. To date, more than 200 mutations of NLRP3 associated with CAPS have been reported in the INFEVER website (72), almost all the mutations lie in the NACHT domain. In CAPS patients, gain-of-function mutation of NLRP3 leads to the aberrant inflammasome activation and increased IL-1β secretion, resulting in the systemic inflammation and disease systems. Thus, the anti–IL-1β therapy is recommended for CAPS (73). To date, three anti–IL-1β therapies are available including the recombinant form of IL-1RA (Anakinra) (74), the anti-IL-1β monoclonal antibody (Canakinumab) (75), and the dimeric IL-1 receptor fusion protein (Rilonacept) (76).
The main characteristic of atherosclerosis is the plaques in arteries, which is made up of fat, cholesterol, calcium, or other substances in the blood. Recent studies have demonstrated that atherosclerosis is a chronic inflammatory disease; various anti-inflammatory agents are tested in clinical trials to treat atherosclerosis (77, 78). In carotid atherosclerosis plaques, the expression of NLRP3 inflammasome components is elevated, implying the association of NLRP3 with atherosclerosis (79). Furthermore, it has been reported that cholesterol crystal, a hallmark of atherosclerotic lesions (80), activates NLRP3 inflammasome in a process involving lysosomal rupture, and reconstitution with NLRP3-, ASC-, or IL-1α/β–deficient bone marrow in low-density lipoprotein receptor (LDLR)–deficient mice shows reduced early atherosclerosis, suggesting the link between NLRP3 inflammasome and pathogenesis of atherosclerosis (9). Additionally, oxidized LDL (oxLDL) triggers NLRP3 inflammasome activation via CD36-meidated unabated uptake of oxLDL, leading to its transformation into intracellular cholesterol crystals that activate NLRP3. Upon response to oxLDL, CD36 also cooperates with TLR4-TLR6 to activate the priming signal to induce expression of NLRP3 and pro–IL-1β (81). Taken together, DAMPs during AS initiate both priming signal and activation signal to induce NLRP3 inflammasome activation, promoting the pathogenesis of AS.
NALFD (non-alcoholic fatty liver disease) is strongly related with metabolic syndrome; its prevalence is increasing around the world. Non-alcoholic steatohepatitis (NASH) is the progressive subtype of NAFLD, with steatosis, hepatocyte injury (ballooning), and inflammation (82). Although often asymptomatic, NASH can progress to cirrhosis. So far, no medicines have been approved to treat NAFLD or NASH. During the last few years, increasing evidences illustrate the role of NLRP3 in NASH (83–85), elevated hepatic expression of Nlrp3, Asc, and Casp1 associates with liver inflammation in dietary and nutrient deficiency fatty liver diseases (84, 86–88), and NLRP3 activation is critical for progression of NASH evidenced by genetic knockout mice and NLRP3 pharmacological inhibitor (12, 89, 90).
Emerging evidences have shown the essential role of chronic inflammation in type 2 diabetes (T2D) (91). IL-1β is reported to inhibit insulin signaling (92–94); randomized clinical trials shows that blocking IL-1β signaling (using recombinant human IL-1 receptor antagonist) is effective in type 2 diabetes (94). With the discovery that NLRP3 inflammasome mediates caspase-1 activation and IL-1β maturation, its relation with insulin resistance and type 2 diabetes development has been recognized (13, 95–97); NLRP3 deficiency protects mice from obesity-induced insulin resistance (13, 96). Moreover, the level of plasma free fatty acids (FFAs) are elevated in T2D patients and diabetic animals (98); FFAs could activate NLRP3 inflammasome activation through AMPK-ROS signaling axis, possibly elucidating the mechanism by which HFD induces inflammatory response, which promotes insulin resistance (97).
Gout is an inflammatory arthritis associated with precipitation of MSU within the joint (99, 100). IL-1β have been implicated as an important proinflammatory cytokine in gout, inducing the influx of neutrophil into the synovium and joint fluid (101). The link between MSU crystal and IL-1β production remained unknown until Martinon et al. reported that MSU stimulation-activated NLRP3 inflammasome, macrophages deficient in NLRP3, and ASC or caspase-1 were defective in IL-1β secretion in response to MSU, deficiency of ASC, or caspase-1 impaired the neutrophil influx in an animal model of MSU-induced peritonitis (11). Similar results are demonstrated with the causal agent of pseudogout, calcium pyrophosphate dihydrate (CPPD) (11). Therefore, NLRP3 inflammasome is demonstrated to link causal agents of gout and the disease pathogenesis.
Neurotic plaques and neurofibrillary tangles (NFTs) in the brain are hallmarks of (AD), formed by accumulation of amyloid-beta (Aβ) and hyperphosphorylated tau respectively, together leading to neurodegeneration and cognitive decline (102). Aβ activates NLRP3 in a process engaging the lysosomal damage and cathepsin B release (45); cleaved caspase-1 is increased in amyloid-plaque in mice and patients with AD (10, 45). In a model of AD, deficiency of ASC or NLRP3 alleviates amyloid plaque pathology (103), implying that NLRP3 is critical in the progression of amyloid-beta pathology. Moreover, a recent study demonstrates the pivotal role NLRP3 in tau pathology; NLRP3 can be activated by tau monomers and oligomers, then inducing tau hyperphosphorylation and aggregation possibly via regulation of tau kinase (104). α-synuclein, the abnormal aggregation of which is the main pathogenesis of Parkinson’s disease (PD), also activates NLRP3 inflammasome. In patients with PD and PD model mice, abnormal NLRP3 inflammasome activation has been observed; inhibition of NLRP3 inflammasome effectively ameliorated dopaminergic degeneration and pathological α-synuclein accumulation in mice (105). Thus, NLRP3mediated neuroinflammation could be an important driver of PD progression.
Drug-Induced Liver Injury (DILI) is the main reason hindering drug development and for drug withdrawal. There is a growing number of evidences linking NLRP3 inflammasome activation with DILI induced by multiple drugs (Table 1). N-acetyl-p-aminophenol (acetaminophen, APAP) is a commonly used antipyretic and analgesic drug. APAP is safe at normal dose but causes sever liver failure and even death when taken in excess (115). Imaeda and colleagues demonstrated that deficiencies in NLRP3 inflammasome components ameliorated APAP-induced liver injury and death; anti–IL-1β antibody also exhibited protective effect in mice administered with a lethal dose of acetaminophen (106). Additionally, NLRP3 inflammasome activation is implicated in liver injury induced by antituberculosis Drugs (Rifampicin [RIF] and Isoniazid [INH]), NLRP3 inhibitor (INF39 or CP-456773) ameliorated liver injury caused by Isoniazid and Rifampicin (107). Moreover, INH has been reported to induce NLRP3 inflammasome activation in a sirtuin1 (SIRT1)–dependent manner (108). NLRP3 inflammasome activation was also observed in mice treated with Triptolide (TP, a main ingredient of Chinese Traditional Medicine, Tripterygium wilfordii Hook. f), which causes hepatotoxicity. Moreover, caspase-1 inhibitor pretreatment effectively alleviated the TP-induced liver toxicity (110).
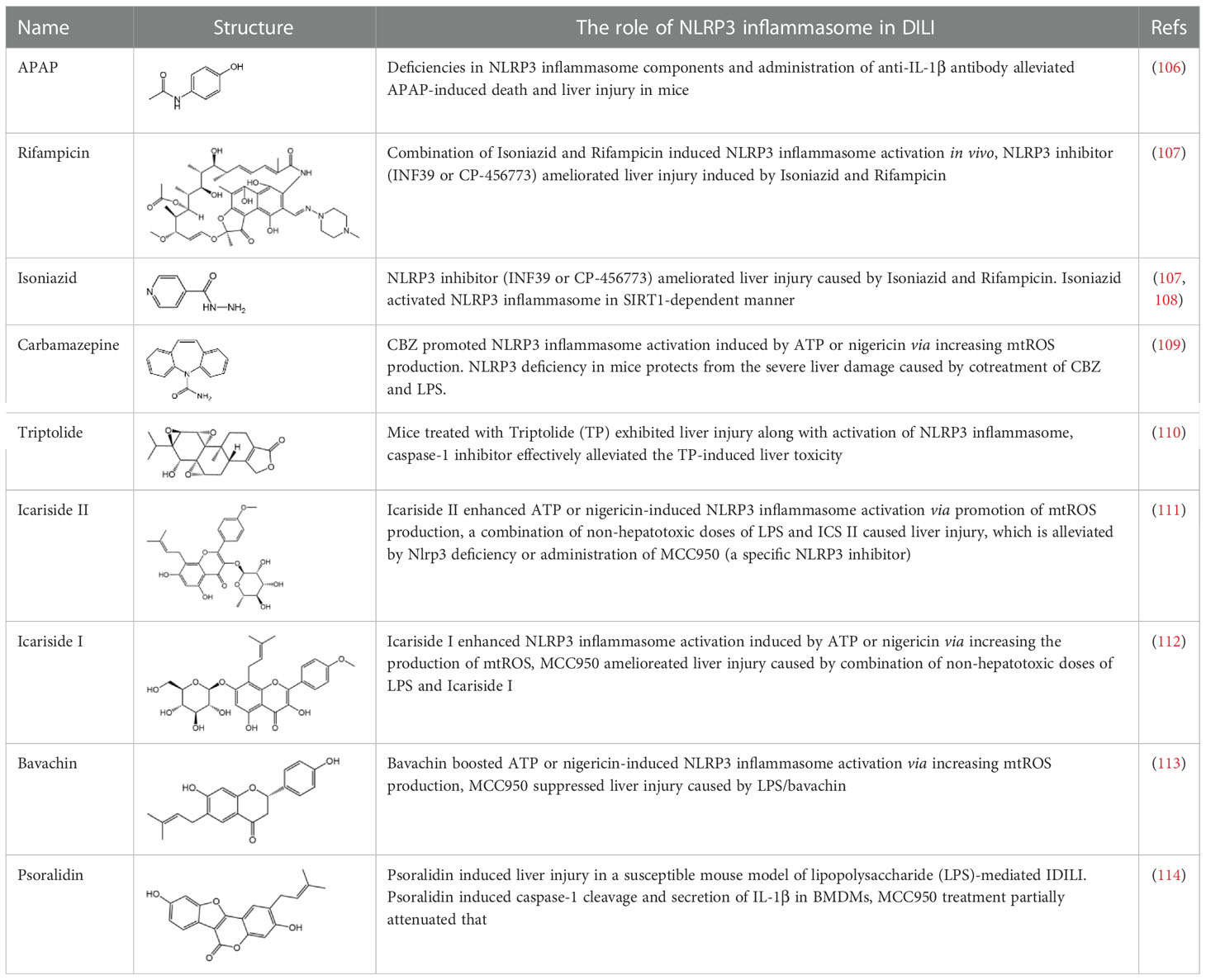
Table 1 The role of NLRP3 inflammasome in Drug-Induced Liver Injury (DILI).
In our studies, we found the widely used antiepileptic agent carbamazepine (CBZ), which causes idiosyncratic liver injury (116) and enhances NLRP3 inflammasome activation through promotion of mtROS production; in vivo data showed that cotreatment of CBZ and LPS caused severe liver damage in WT mice but not in NLRP3−/− mice, suggesting the significance of NLRP3 activation in CBZ-induced liver injury (109). Meanwhile, we have also reported that several components of Epimedii Folium and Psoraleae Fructus, which are traditional Chinese medicine that have been suggested to induce liver injury (117, 118), could enhance NLRP3 activation and caused liver injury in a LPS-mediated susceptibility mouse model of IDILI. These components include icariside II (111), icariside I (112), and bavachin (113). These three components promote NLRP3 activation stimulated by ATP or nigericin via increasing the production of mtROS. Liver injury caused by these compounds could be alleviated by NLRP3 deficiency or MCC950 administration (111–114). Psoralidin is a component of Psoraleae Fructus, induced liver injury in a susceptible mouse model of LPS-mediated IDILI. Psoralidin alone could induce caspase-1 cleavage and IL-1β secretion in BMDMs, but MCC950 only partially attenuated that (114), suggesting that psoralidin activates NLRP3 inflammasome as well as other inflammasomes.
4. Pharmacological inhibitors of NLRP3 inflammasome
The studies revealing the crucial role of NLRP3 in various inflammatory diseases demonstrate that pharmacological intervention aimed at this critical target may hold therapeutic promise. Although IL-1β–targeted therapies have been used in clinic to treat the related inflammatory disease, NLRP3 inflammasome-targeted therapies have a competitive advantage. Recently, more and more inhibitors of NLRP3 inflammasome have been identified (Figure 4 and Table 2).
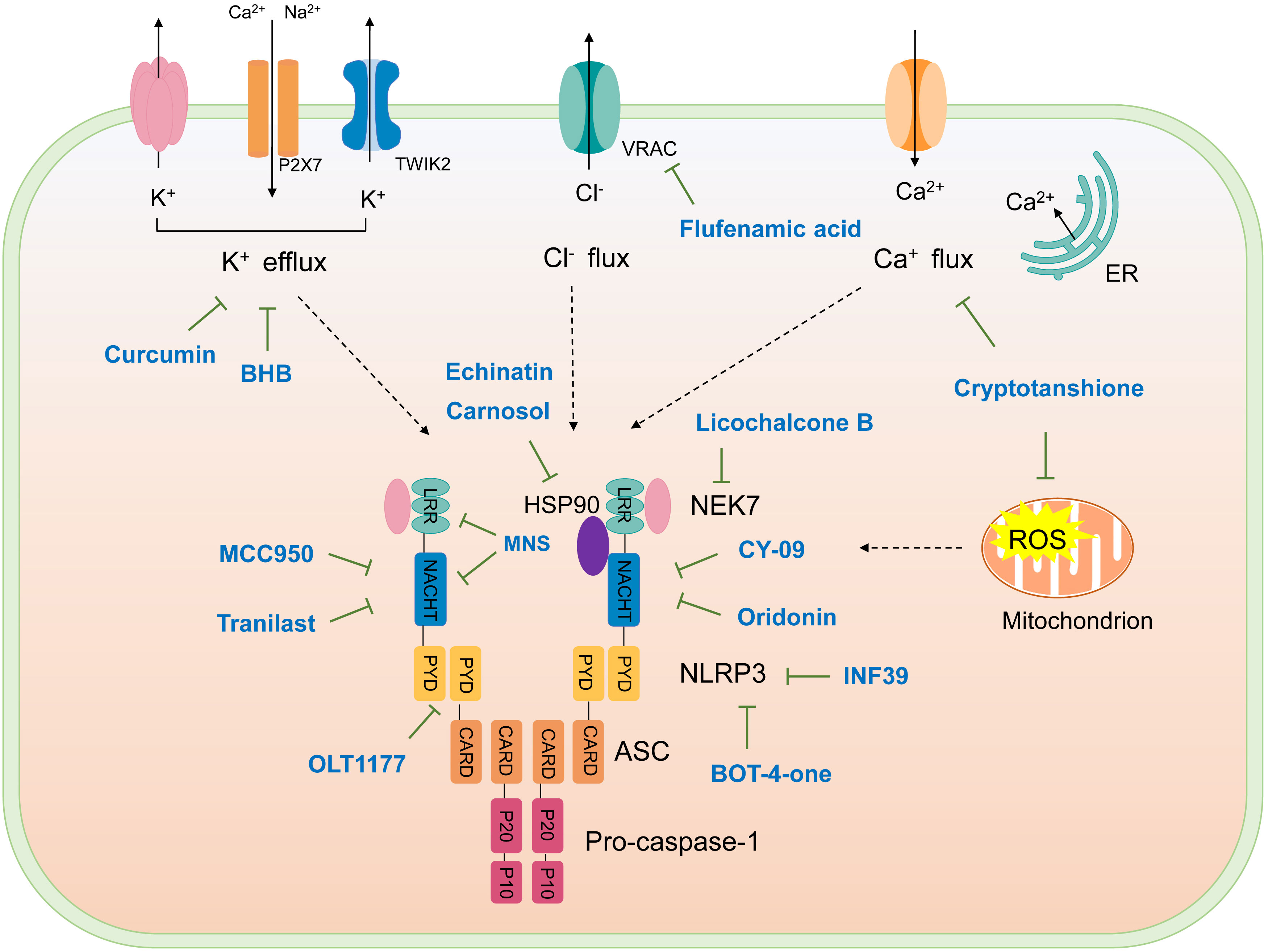
Figure 4 Schematic illustration of targets of NLRP3 inflammasome inhibitors. Upon activation, NLRP3 oligomerizes through interactions between NACHT domains, then recruiting ASC via PYD-PYD interactions, assembled ASC recruits caspase-1, forming NLRP3 inflammasome. NKE7 binds to NLRP3; the interaction is necessary for NLRP3 inflammasome assembly. HSP90 is suggested to bind NLRP3 to promote its activation. K+ efflux, Ca2+ flux, Cl- efflux, and mtROS production are upstream signaling events of NLRP3 activation. The targets of NLRP3 inflammasome inhibitors are shown.
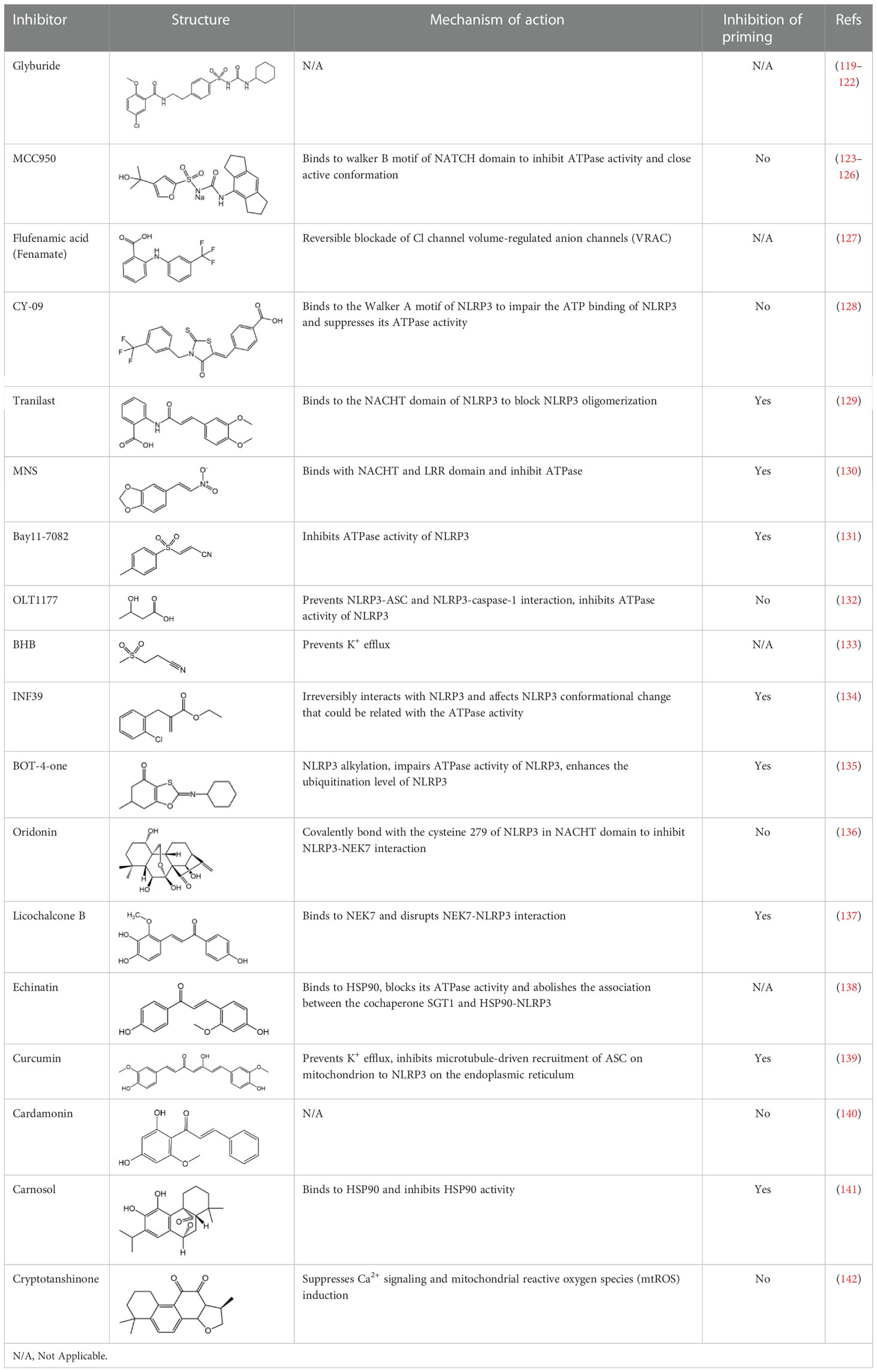
Table 2 Inhibitors of NLRP3 inflammasome.
MCC950, also known as CP-456,773, is the best characterized inhibitor of NLRP3. Initially, MCC950 was reported as a potent IL-1β processing inhibitor via screening of a group of diarylsulfonylureacontaining compounds (124), then Coll and colleagues demonstrate that MCC950 specifically inhibits NLRP3 activation and ASC oligomerization, without affecting K+ efflux, Ca2+ flux or NLRP3–ASC interactions (125). It remains unclear how MCC950 inhibits NLRP3 activation until two recent studies report that MCC950 directly binds to NLRP3 Walker B site within the NACHT domain, inhibiting its ability to hydrolyze ATP and closing the active conformation of NLRP3 (123, 126). MCC950 was even tested to treat rheumatoid arthritis in phase II clinical trials but failed because of the increase of serum liver enzyme levels, which are liver toxicity signals (77).
OLT1177 is a β-sulfonyl nitrile that specifically blocks NLRP3 inflammasome activation. Nanomolar concentrations of OLT1177 suppressed NLRP3 inflammasome activation in vitro but did not impair AIM2 or NLRC4 inflammasome (132). OLT1177 not only prevents NLRP3-ASC and NLRP3-caspase-1 interaction but it also blocks NLRP3 ATPase activity; however, OLT1177 does not affect pro-IL-1β gene expression. In humans receiving OLT1177 at oral doses up to 1000 mg daily for 8 days, no adverse effects or hematological or biochemical changes were observed, showing the safety of OLT117 in human (132). OLT1177 is now tested in clinic to treat acute gout flares and heart failure (78).
Tranilast (TR) is an analog of a tryptophan metabolite used in clinic to treat inflammatory diseases (143). TR also has inhibitory effect on cytokine-induced NF-κB activation (144). TR impaired pro–IL-1β expression induced by LPS in BMDMs, but when BMDMs were stimulated with TR for 30 min after LPS treatment, TR did not affect NLRP3 and pro–IL-1β expression but still inhibited NLRP3 inflammasome activation (129). TR bound to NLRP3 at the NACHT domain and blocked its oligomerization, but it did not affect its ATPase activity. TR exhibited beneficial effects in gouty arthritis, CAPS and type 2 diabetes in mouse models (129).
CY-09 is an analog of CFTR(inh)-172 (C172) without cystic fibrosis transmembrane conductance regulator (CFTR)–inhibitory activity (145), C172 was identified as an inhibitor of NLRP3 from screening a compound library, and as an analog of C172, CY-09 was tested and showed obviously inhibitory effect on NLRP3 activation (128). CY-09 directly binds to NLRP3 at the Walker A motif in NACHT domain, impairing ATP binding of NLRP3 and inhibiting its ATPase activity. Moreover, CY-09 exhibits therapeutic effect on mice model of MSU-induced peritonitis (128).
Several other compounds have been demonstrated to target NLRP3 and inhibit its ATPase activity, such as Bay11-7082, MNS(3,4-methylenedioxy-β-nitrostyrene), IFN39, and BOT-4-one. Bay11-7082 is known as NF-κB inhibitor to block the IKKβ activity; it has been reported to abrogate caspase-1 activation via affecting LPS priming (146, 147). Christine Juliana and co-workers discovered that Bay11-7082 could block NLRP3 activation independently on its effect on NF-κB, through impairing NLRP3 ATPase activity (131). MNS is a reported inhibitor of Syk kinase, but its inhibitory effect on NLRP3 inflammasome is independent on Syk. MNS binds to NLRP3 at the NOD and LRR domain, blocking its ATPase activity possibly through cysteine modification (130). INF39 was identified through the screening of acrylate derivatives designed to inhibit NLRP3, INF39 interrupts NLRP3 activation through direct irreversible binding to NLRP3 and blocking its ATPase activity (134). BOT-4-one is a novel benzoxathiole derivative that has been reported as NF-κB inhibitor through targeting IKKβ (135). BOT-4-one directly targets NLRP3 via alkylation to suppress its ATPase activity, it also enhances the ubiquitination level of NLRP3 (135). However, the safety and protective role of these compounds in NLRP3-driven diseases have not been investigated thoroughly.
Glyburide is a widely used drug approved by FDA to treat type 2 diabetes (148), working by inhibiting ATP-sensitive K+ (KATP) channels in pancreatic β cells (149). It is the first identified compound to inhibit NLRP3 inflammasome activation, but the effect is independent on KATP channels or ATP-binding cassette transporters (119), which was proposed as the glyburide target (121). Glyburide acts upstream of NLRP3 to block IL-1β secretion induced by various stimuli (119), but the direct target needs further study. Several studies report that Glyburide reduces a variety of NLRP3-dependent pathologies such as septic shock, bronchopulmonary dysplasia and Cutaneous leishmaniasis (119, 120, 122).
NSAIDs are characterized by COX inhibitors (150) and approved to treat multiple diseases. Michael J.D. Daniels and colleagues demonstrate Flufenamic acid and mefenamic acid, which belong to the fenamate class of NSAIDs, specifically block NLPR3 inflammasome activation through reversible blockade of Cl- channel volume-regulated anion channels (VRAC), independently of the effect on COX (127). Mefenamic acid also exhibited protective effects in a rodent model of Amyloid beta-induced memory loss and 3× TgAD transgenic mice (a mouse model for AD) (127). The reversible inhibitory effect of fenamate NSAIDs on NLRP3 offers significant clinical benefit, flufenamic acid and mefenamic acid are readily approved by FDA to be used in clinic, they can be quickly repurposed as drugs to treat AD or maybe other NLRP3-driven diseases.
BHB is a type of ketone bodies from the liver of mammals, it serves as alternative energy source during states of energy deficit (151). BHB specifically inhibits NLRP3 inflammasome independently of starvation-regulated mechanisms, but mediated by blockade of potassium efflux and ASC oligomerization (133). The protective role of BHB in NLRP3-driven diseases such as Muckle–Wells syndrome, urate crystal–induced peritonitis, and familial cold autoinflammatory syndrome has been validated in rodent models (133). As a naturally occurring metabolite, BHB has great potential to be developed as a safe drug to treat NLRP3-mediated diseases.
Many Chinese medicinal herbs exhibit obviously anti-inflammatory effects and have been used to treat various inflammatory diseases. Recently, our group and others’ have identified various inhibitors of NLRP3 inflammasome from traditional Chinses medicinal herbs
Oridonin (Ori) is the active component of Rabdosia rubescens, which is a commonly used traditional Chinese medicinal herb to treat inflammatory diseases (152). Ori specifically blocks NLRP3 inflammasome activation, without affecting LPS-induced priming (136). Ori covalently binds to Cys279 of NLRP3 via its carbon-carbon double-bond and block NEK7-NLRP3 interaction, thus blocking subsequent NLRP3 inflammasome assembly (136). Although there is possibility that covalent drugs may exhibit idiosyncratic toxicity or hypersensitivity, they have pharmacological advantages such as enhanced efficacy and prolonged duration of action (153, 154). Thus, Ori may have a significant potential to be used in NLRP3-driven inflammatory diseases.
Licochalcone B is the main component of the traditional Chinese herbal medicine Glycyrrhiza plants (licorice). Our study demonstrates that Licochalcone B exhibits specifically inhibitory effect on NLRP3 inflammasome but does not affect potassium efflux, mtROS production or calcium flux (137). Licochalcone B binds to NEK7 and blocks the interaction between NEK7 and NLRP3. Licochalcone B has been reported to affect NF-κB pathway (155), when administered before LPS priming, it slightly suppresses pro–IL-1β production (137). The protective effect of Licochalcone B has been investigated in mice models of septic shock, NASH, and peritonitis. No changes in biochemical parameters reflecting liver and kidney function were observed in mice treated with 40mg/kg of Licochalcone B for 34 days, suggesting its safety (137).
Echinatin is also the bioactive component of the traditional Chinese herbal medicine Glycyrrhiza plants (licorice). We have identified that Echinatin potently restrains NLRP3 inflammasome activation via affecting the association between SGT1 and HSP90-NLRP3 (138). HSP90 and its client-adaptor SGT1 are essential for NLRP3 activity (156), echinatin directly binds to HSP90 and inhibits its ATPase activity, causing the dissociation of SGT1 from HSP90 and NLRP3, thereby blocking subsequent inflammasome assembly and activation (138). Moreover, HSP90 and SGT1 also play a critical role in NLRC4 activation (156), consistently, echinatin also impairs NLRC4 activation (138) and may also be therapeutic in NLRC4-mediated diseases.
Curcumin is a polyphenolic compound of the rhizomes of turmeric (157), which has been widely used in foods as spice and coloring and traditional medicine. Curcumin plays a beneficial role in various inflammatory diseases including diabetes, rheumatoid arthritis and cardiovascular diseases (158). Curcumin showed specific inhibition for NLRP3, but not for AIM2 or NLRC4 inflammasome (139). Importantly, curcumin blocks potassium efflux and disturbs the mitochondrial transport as well as ASC polymerization, thus preventing NLRP3 inflammasome assembly. Curcumin also ameliorated MSU-induced peritoneal inflammation and HFD-induced insulin resistance through inhibiting NLRP3 inflammasome activation in mice models (139).
Cardamonin is an active ingredient of traditional Chinese medicinal herb Alpinia katsumadai; it has been reported to alleviate inflammatory bowel disease by the inhibition of NLRP3 inflammasome activation (159). One of our recent studies demonstrates that cardamonin specifically inhibits NLRP3 inflammasome activation, it has no effect on the priming stage, but blocks NLRP3-dependent ASC oligomerization (140). In a mouse model, cardamonin obviously ameliorates LPS-induced septic shock and IL-1β production (140).
Carnosol, a natural polyphenol, was first isolated from sage (Salvia carnosa) (160). Carnosol was identified to inhibit NLRP3 inflammasome activation through a high-throughput assay for caspase-1 activity to screen NLRP3 inhibitors (141). Carnosol blocks NLRP3 activation via binding to HSP90 and inhibiting its ATPase activity. The therapeutic effects of carnosol on LPS-induced septic shock and MCD -induced NASH have been verified in mouse models (141). Mice administered with carnosol (120 mg/kg) i.p. for two weeks showed no change in body weight or biochemical parameters of liver and kidney function (141), indicating the potential of carnosol to be a safe candidate for the development of therapeutic drug for NLRP3-driven diseases.
Liu et al. reported cryptotanshinone as a potent inhibitor of NLRP3 activation (142). Cryptotanshinone is a main component of the traditional medicinal herb Salvia miltiorrhiza Bunge, it has been reported to suppress NF-κB activation and reduce inflammation-induced by LPS (161). Cryptotanshinone could block NLRP3 inflammasome activation, the inhibitory effect was independent on its regulation of NF-κB pathway (142) and limited to NLRP3, not affecting AIM2 or NLRC4 inflammasome activation. Moreover, cryptotanshinone impairs calcium flux and the induction of mtROS, it demonstrated significantly preventive property in mice models of septic shock and MCD-induced NASH (142). Further studies about the exact target of cryptotanshinone are awaited.
5. Conclusion and perspectives
NLRP3 inflammasome is central to immune responses triggered by danger signals derived from both pathogens or host. With the intensive studies on its mechanism of activation, much attention has been paid to the relation between aberrant NLRP3 activation and the pathogenesis of various diseases, including those related with metabolic disorder and aging. Currently, the number of individuals affected by the inflammatory conditions is growing rapidly as a result of the improvement of living standards and aging population, there is an increasing demand for NLRP3 inflammasome targeted therapeutics.
The current treatment for NLRP3-mediated diseases is mainly by targeting IL-1β, but IL-1β maturation is also mediated by activation of other types of inflammasomes; blockade of IL-1β may affect the host defense response. Therapeutics targeting NLRP3 inflammasome is more specific, with better efficacy and safety. At present, many NLRP3 inhibitors have been reported; among them, MCC950 is the best studied with high specificity. Many derivatives based on MCC950 are under development, and several derivatives of MCC950 have also entered clinical phase II. In addition, another NLRP3 inhibitor OLT1177 is now tested in clinic for the treatment of acute gout flares and heart failure. With the in-depth understanding of the high-resolution structure and activation mechanism of NLRP3, it will be more conducive to the development of NLRP3 targeted drugs.
Meanwhile, various inhibitors of NLRP3 inflammasome from traditional Chinese medicinal herbs have been reported, many traditional Chinese medicinal herbs have been used in the treatment of inflammatory diseases and show good safety, so it may hold great promise for screening safe and effective inhibitors for NLRP3 inflammasome from traditional Chinese medicinal herbs, which may be developed as candidate therapeutic drugs for treating NLRP3-mediated diseases.
Author contributions
XZ wrote the manuscript, XZ and QL drew the figure and collected the tables. GX contributed to language modification and content adjustment. ZB and XX supervised and revised the manuscript. All authors approved the submitted version.
Funding
This review was supported by National Natural Science Foundation of China (Grant No. 81630100, 81930110, 81874368, 82003984, 81721002), Beijing Natural Science Foundation (Grant No. 7214296), National Science & Technology Major Project “Key New Drug Creation and Manufacturing Program” (Grant No. 2017ZX09301022), Innovation Team and Talents Cultivation Program of National Administration of Traditional Chinese Medicine (Grant No. ZYYCXTD-C-202005).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Franchi L, Munoz-Planillo R, Nunez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol (2012) 13(4):325–32. doi: 10.1038/ni.2231
2. Wang L, Manji GA, Grenier JM, Al-Garawi A, Merriam S, Lora JM, et al. Pypaf7, a novel pyrin-containing Apaf1-like protein that regulates activation of nf-kappa b and caspase-1-Dependent cytokine processing. J Biol Chem (2002) 277(33):29874–80. doi: 10.1074/jbc.M203915200
3. Martinon F, Burns K, Tschopp J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proil-beta. Mol Cell (2002) 10(2):417–26. doi: 10.1016/s1097-2765(02)00599-3
4. Fink SL, Cookson BT. Caspase-1-Dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol (2006) 8(11):1812–25. doi: 10.1111/j.1462-5822.2006.00751.x
5. Martinon F, Mayor A, Tschopp J. The inflammasomes: Guardians of the body. Annu Rev Immunol (2009) 27:229–65. doi: 10.1146/annurev.immunol.021908.132715
6. Davis BK, Wen H, Ting JP. The inflammasome nlrs in immunity, inflammation, and associated diseases. Annu Rev Immunol (2011) 29:707–35. doi: 10.1146/annurev-immunol-031210-101405
7. Jo EK, Kim JK, Shin DM, Sasakawa C. Molecular mechanisms regulating Nlrp3 inflammasome activation. Cell Mol Immunol (2016) 13(2):148–59. doi: 10.1038/cmi.2015.95
8. Broderick L, De Nardo D, Franklin BS, Hoffman HM, Latz E. The inflammasomes and autoinflammatory syndromes. Annu Rev Pathol (2015) 10:395–424. doi: 10.1146/annurev-pathol-012414-040431
9. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. Nlrp3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature (2010) 464(7293):1357–61. doi: 10.1038/nature08938
10. Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, et al. Nlrp3 is activated in alzheimer's disease and contributes to pathology in App/Ps1 mice. Nature (2013) 493(7434):674–8. doi: 10.1038/nature11729
11. Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the Nalp3 inflammasome. Nature (2006) 440(7081):237–41. doi: 10.1038/nature04516
12. Mridha AR, Wree A, Robertson AAB, Yeh MM, Johnson CD, Van Rooyen DM, et al. Nlrp3 inflammasome blockade reduces liver inflammation and fibrosis in experimental Nash in mice. J Hepatol (2017) 66(5):1037–46. doi: 10.1016/j.jhep.2017.01.022
13. Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al. The Nlrp3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med (2011) 17(2):179–88. doi: 10.1038/nm.2279
14. Andreeva L, David L, Rawson S, Shen C, Pasricha T, Pelegrin P, et al. Nlrp3 cages revealed by full-length mouse Nlrp3 structure control pathway activation. Cell (2021) 184(26):6299–312.e22. doi: 10.1016/j.cell.2021.11.011
15. Liston A, Masters SL. Homeostasis-altering molecular processes as mechanisms of inflammasome activation. Nat Rev Immunol (2017) 17(3):208–14. doi: 10.1038/nri.2016.151
16. Cai X, Chen J, Xu H, Liu S, Jiang QX, Halfmann R, et al. Prion-like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell (2014) 156(6):1207–22. doi: 10.1016/j.cell.2014.01.063
17. Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, et al. Unified polymerization mechanism for the assembly of asc-dependent inflammasomes. Cell (2014) 156(6):1193–206. doi: 10.1016/j.cell.2014.02.008
18. Schmidt FI, Lu A, Chen JW, Ruan J, Tang C, Wu H, et al. A single domain antibody fragment that recognizes the adaptor asc defines the role of asc domains in inflammasome assembly. J Exp Med (2016) 213(5):771–90. doi: 10.1084/jem.20151790
19. He Y, Zeng MY, Yang D, Motro B, Nunez G. Nek7 is an essential mediator of Nlrp3 activation downstream of potassium efflux. Nature (2016) 530(7590):354–7. doi: 10.1038/nature16959
20. Schmid-Burgk JL, Chauhan D, Schmidt T, Ebert TS, Reinhardt J, Endl E, et al. A genome-wide crispr (Clustered regularly interspaced short palindromic repeats) screen identifies Nek7 as an essential component of Nlrp3 inflammasome activation. J Biol Chem (2016) 291(1):103–9. doi: 10.1074/jbc.C115.700492
21. Shi H, Wang Y, Li X, Zhan X, Tang M, Fina M, et al. Nlrp3 activation and mitosis are mutually exclusive events coordinated by Nek7, a new inflammasome component. Nat Immunol (2016) 17(3):250–8. doi: 10.1038/ni.3333
22. Sharif H, Wang L, Wang WL, Magupalli VG, Andreeva L, Qiao Q, et al. Structural mechanism for Nek7-licensed activation of Nlrp3 inflammasome. Nature (2019) 570(7761):338–43. doi: 10.1038/s41586-019-1295-z
23. Xiao L, Magupalli VG, Wu H. Cryo-em structures of the active Nlrp3 inflammasome disk. Nature (2022). doi: 10.1038/s41586-022-05570-8
24. Han S, Lear TB, Jerome JA, Rajbhandari S, Snavely CA, Gulick DL, et al. Lipopolysaccharide primes the Nalp3 inflammasome by inhibiting its ubiquitination and degradation mediated by the Scffbxl2 E3 ligase. J Biol Chem (2015) 290(29):18124–33. doi: 10.1074/jbc.M115.645549
25. Song N, Liu ZS, Xue W, Bai ZF, Wang QY, Dai J, et al. Nlrp3 phosphorylation is an essential priming event for inflammasome activation. Mol Cell (2017) 68(1):185–97.e6. doi: 10.1016/j.molcel.2017.08.017
26. Perregaux D, Gabel CA. Interleukin-1 beta maturation and release in response to atp and nigericin. evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J Biol Chem (1994) 269(21):15195–203.
27. Walev I, Reske K, Palmer M, Valeva A, Bhakdi S. Potassium-inhibited processing of il-1 beta in human monocytes. EMBO J (1995) 14(8):1607–14. doi: 10.1002/j.1460-2075.1995.tb07149.x.
28. Surprenant A, Rassendren F, Kawashima E, North RA, Buell G. The cytolytic P2z receptor for extracellular atp identified as a P2x receptor (P2x7). Science (1996) 272(5262):735–8. doi: 10.1126/science.272.5262.735
29. Di A, Xiong S, Ye Z, Malireddi RKS, Kometani S, Zhong M, et al. The Twik2 potassium efflux channel in macrophages mediates Nlrp3 inflammasome-induced inflammation. Immunity (2018) 49(1):56–65.e4. doi: 10.1016/j.immuni.2018.04.032
30. Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM, Nunez G. K(+) efflux is the common trigger of Nlrp3 inflammasome activation by bacterial toxins and particulate matter. Immunity (2013) 38(6):1142–53. doi: 10.1016/j.immuni.2013.05.016
31. Gross CJ, Mishra R, Schneider KS, Medard G, Wettmarshausen J, Dittlein DC, et al. K(+) efflux-independent Nlrp3 inflammasome activation by small molecules targeting mitochondria. Immunity (2016) 45(4):761–73. doi: 10.1016/j.immuni.2016.08.010
32. Lee GS, Subramanian N, Kim AI, Aksentijevich I, Goldbach-Mansky R, Sacks DB, et al. The calcium-sensing receptor regulates the Nlrp3 inflammasome through Ca2+ and camp. Nature (2012) 492(7427):123–7. doi: 10.1038/nature11588
33. Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer AM, et al. Critical role for calcium mobilization in activation of the Nlrp3 inflammasome. Proc Natl Acad Sci U.S.A. (2012) 109(28):11282–7. doi: 10.1073/pnas.1117765109
34. Camello-Almaraz C, Gomez-Pinilla PJ, Pozo MJ, Camello PJ. Mitochondrial reactive oxygen species and Ca2+ signaling. Am J Physiol Cell Physiol (2006) 291(5):C1082–8. doi: 10.1152/ajpcell.00217.2006
35. Csordas G, Hajnoczky G. Sr/Er-mitochondrial local communication: Calcium and ros. Biochim Biophys Acta (2009) 1787(11):1352–62. doi: 10.1016/j.bbabio.2009.06.004
36. Lemasters JJ, Theruvath TP, Zhong Z, Nieminen AL. Mitochondrial calcium and the permeability transition in cell death. Biochim Biophys Acta (2009) 1787(11):1395–401. doi: 10.1016/j.bbabio.2009.06.009
37. Katsnelson MA, Rucker LG, Russo HM, Dubyak GR. K+ efflux agonists induce Nlrp3 inflammasome activation independently of Ca2+ signaling. J Immunol (2015) 194(8):3937–52. doi: 10.4049/jimmunol.1402658
38. Perregaux DG, Laliberte RE, Gabel CA. Human monocyte interleukin-1beta posttranslational processing. evidence of a volume-regulated response. J Biol Chem (1996) 271(47):29830–8. doi: 10.1074/jbc.271.47.29830
39. Verhoef PA, Kertesy SB, Lundberg K, Kahlenberg JM, Dubyak GR. Inhibitory effects of chloride on the activation of caspase-1, il-1beta secretion, and cytolysis by the P2x7 receptor. J Immunol (2005) 175(11):7623–34. doi: 10.4049/jimmunol.175.11.7623
40. Domingo-Fernandez R, Coll RC, Kearney J, Breit S, O'Neill LAJ. The intracellular chloride channel proteins Clic1 and Clic4 induce il-1beta transcription and activate the Nlrp3 inflammasome. J Biol Chem (2017) 292(29):12077–87. doi: 10.1074/jbc.M117.797126
41. Tang T, Lang X, Xu C, Wang X, Gong T, Yang Y, et al. Clics-dependent chloride efflux is an essential and proximal upstream event for Nlrp3 inflammasome activation. Nat Commun (2017) 8(1):202. doi: 10.1038/s41467-017-00227-x
42. Green JP, Yu S, Martin-Sanchez F, Pelegrin P, Lopez-Castejon G, Lawrence CB, et al. Chloride regulates dynamic Nlrp3-dependent asc oligomerization and inflammasome priming. Proc Natl Acad Sci U.S.A. (2018) 115(40):E9371–E80. doi: 10.1073/pnas.1812744115
43. Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, et al. Silica crystals and aluminum salts activate the Nalp3 inflammasome through phagosomal destabilization. Nat Immunol (2008) 9(8):847–56. doi: 10.1038/ni.1631
44. Riteau N, Baron L, Villeret B, Guillou N, Savigny F, Ryffel B, et al. Atp release and purinergic signaling: A common pathway for particle-mediated inflammasome activation. Cell Death Dis (2012) 3:e403. doi: 10.1038/cddis.2012.144
45. Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, et al. The Nalp3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol (2008) 9(8):857–65. doi: 10.1038/ni.1636
46. Orlowski GM, Colbert JD, Sharma S, Bogyo M, Robertson SA, Rock KL. Multiple cathepsins promote pro-Il-1beta synthesis and Nlrp3-mediated il-1beta activation. J Immunol (2015) 195(4):1685–97. doi: 10.4049/jimmunol.1500509
47. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the Nalp3 inflammasome. Nat Immunol (2011) 12(3):222–30. doi: 10.1038/ni.1980
48. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized mitochondrial DNA activates the Nlrp3 inflammasome during apoptosis. Immunity (2012) 36(3):401–14. doi: 10.1016/j.immuni.2012.01.009
49. Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin XJ, et al. New mitochondrial DNA synthesis enables Nlrp3 inflammasome activation. Nature (2018) 560(7717):198–203. doi: 10.1038/s41586-018-0372-z
50. Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity (2013) 39(2):311–23. doi: 10.1016/j.immuni.2013.08.001
51. Dudek J. Role of cardiolipin in mitochondrial signaling pathways. Front Cell Dev Biol (2017) 5:90. doi: 10.3389/fcell.2017.00090
52. Chen J, Chen ZJ. Ptdins4p on dispersed trans-golgi network mediates Nlrp3 inflammasome activation. Nature (2018) 564(7734):71–6. doi: 10.1038/s41586-018-0761-3
53. He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin d is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res (2015) 25(12):1285–98. doi: 10.1038/cr.2015.139
54. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of gsdmd by inflammatory caspases determines pyroptotic cell death. Nature (2015) 526(7575):660–5. doi: 10.1038/nature15514
55. Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature (2016) 535(7610):111–6. doi: 10.1038/nature18590
56. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin d causes pyroptosis by forming membrane pores. Nature (2016) 535(7610):153–8. doi: 10.1038/nature18629
57. Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC. The pore-forming protein gasdermin d regulates interleukin-1 secretion from living macrophages. Immunity (2018) 48(1):35–44.e6. doi: 10.1016/j.immuni.2017.11.013
58. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature (2011) 479(7371):117–21. doi: 10.1038/nature10558
59. Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic lps activates caspase-11: Implications in Tlr4-independent endotoxic shock. Science (2013) 341(6151):1250–3. doi: 10.1126/science.1240988
60. Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, et al. Noncanonical inflammasome activation by intracellular lps independent of Tlr4. Science (2013) 341(6151):1246–9. doi: 10.1126/science.1240248
61. Wang S, Miura M, Jung YK, Zhu H, Li E, Yuan J. Murine caspase-11, an ice-interacting protease, is essential for the activation of ice. Cell (1998) 92(4):501–9. doi: 10.1016/s0092-8674(00)80943-5
62. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular lps. Nature (2014) 514(7521):187–92. doi: 10.1038/nature13683
63. Ruhl S, Broz P. Caspase-11 activates a canonical Nlrp3 inflammasome by promoting k(+) efflux. Eur J Immunol (2015) 45(10):2927–36. doi: 10.1002/eji.201545772
64. Zanoni I, Tan Y, Di Gioia M, Broggi A, Ruan J, Shi J, et al. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science (2016) 352(6290):1232–6. doi: 10.1126/science.aaf3036
65. Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid-Burgk JL, Rapino F, et al. Human monocytes engage an alternative inflammasome pathway. Immunity (2016) 44(4):833–46. doi: 10.1016/j.immuni.2016.01.012
66. Netea MG, Nold-Petry CA, Nold MF, Joosten LA, Opitz B, van der Meer JH, et al. Differential requirement for the activation of the inflammasome for processing and release of il-1beta in monocytes and macrophages. Blood (2009) 113(10):2324–35. doi: 10.1182/blood-2008-03-146720
67. Piccini A, Carta S, Tassi S, Lasiglie D, Fossati G, Rubartelli A. Atp is released by monocytes stimulated with pathogen-sensing receptor ligands and induces il-1beta and il-18 secretion in an autocrine way. Proc Natl Acad Sci U.S.A. (2008) 105(23):8067–72. doi: 10.1073/pnas.0709684105
68. He Y, Franchi L, Nunez G. Tlr agonists stimulate Nlrp3-dependent il-1beta production independently of the purinergic P2x7 receptor in dendritic cells and in vivo. J Immunol (2013) 190(1):334–9. doi: 10.4049/jimmunol.1202737
69. Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and muckle-wells syndrome. Nat Genet (2001) 29(3):301–5. doi: 10.1038/ng756
70. Aganna E, Martinon F, Hawkins PN, Ross JB, Swan DC, Booth DR, et al. Association of mutations in the Nalp3/Cias1/Pypaf1 gene with a broad phenotype including recurrent fever, cold sensitivity, sensorineural deafness, and aa amyloidosis. Arthritis Rheum (2002) 46(9):2445–52. doi: 10.1002/art.10509
71. Aksentijevich I, Nowak M, Mallah M, Chae JJ, Watford WT, Hofmann SR, et al. De novo Cias1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (Nomid): A new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum (2002) 46(12):3340–8. doi: 10.1002/art.10688
72. Sarrauste de Menthiere C, Terriere S, Pugnere D, Ruiz M, Demaille J, Touitou I. Infevers: The registry for fmf and hereditary inflammatory disorders mutations. Nucleic Acids Res (2003) 31(1):282–5. doi: 10.1093/nar/gkg031
73. ter Haar NM, Oswald M, Jeyaratnam J, Anton J, Barron KS, Brogan PA, et al. Recommendations for the management of autoinflammatory diseases. Ann Rheum Dis (2015) 74(9):1636–44. doi: 10.1136/annrheumdis-2015-207546
74. Hawkins PN, Lachmann HJ, McDermott MF. Interleukin-1-Receptor antagonist in the muckle-wells syndrome. N Engl J Med (2003) 348(25):2583–4. doi: 10.1056/NEJM200306193482523
75. Lachmann HJ, Kone-Paut I, Kuemmerle-Deschner JB, Leslie KS, Hachulla E, Quartier P, et al. Use of canakinumab in the cryopyrin-associated periodic syndrome. N Engl J Med (2009) 360(23):2416–25. doi: 10.1056/NEJMoa0810787
76. Hoffman HM, Throne ML, Amar NJ, Sebai M, Kivitz AJ, Kavanaugh A, et al. Efficacy and safety of rilonacept (Interleukin-1 trap) in patients with cryopyrin-associated periodic syndromes: Results from two sequential placebo-controlled studies. Arthritis Rheum (2008) 58(8):2443–52. doi: 10.1002/art.23687
77. Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E. Targeting the Nlrp3 inflammasome in inflammatory diseases. Nat Rev Drug Discovery (2018) 17(9):688. doi: 10.1038/nrd.2018.149
78. Wang L, Hauenstein AV. The Nlrp3 inflammasome: Mechanism of action, role in disease and therapies. Mol Aspects Med (2020) 76:100889. doi: 10.1016/j.mam.2020.100889
79. Shi X, Xie WL, Kong WW, Chen D, Qu P. Expression of the Nlrp3 inflammasome in carotid atherosclerosis. J Stroke Cerebrovasc Dis (2015) 24(11):2455–66. doi: 10.1016/j.jstrokecerebrovasdis.2015.03.024
80. Small DM. George Lyman Duff memorial lecture. progression and regression of atherosclerotic lesions. insights from lipid physical biochemistry. Arteriosclerosis (1988) 8(2):103–29. doi: 10.1161/01.atv.8.2.103
81. Sheedy FJ, Grebe A, Rayner KJ, Kalantari P, Ramkhelawon B, Carpenter SB, et al. Cd36 coordinates Nlrp3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat Immunol (2013) 14(8):812–20. doi: 10.1038/ni.2639
82. Sheka AC, Adeyi O, Thompson J, Hameed B, Crawford PA, Ikramuddin S. Nonalcoholic steatohepatitis: A review. JAMA (2020) 323(12):1175–83. doi: 10.1001/jama.2020.2298
83. Dixon LJ, Flask CA, Papouchado BG, Feldstein AE, Nagy LE. Caspase-1 as a central regulator of high fat diet-induced non-alcoholic steatohepatitis. PLoS One (2013) 8(2):e56100. doi: 10.1371/journal.pone.0056100
84. Csak T, Pillai A, Ganz M, Lippai D, Petrasek J, Park JK, et al. Both bone marrow-derived and non-bone marrow-derived cells contribute to Aim2 and Nlrp3 inflammasome activation in a Myd88-dependent manner in dietary steatohepatitis. Liver Int (2014) 34(9):1402–13. doi: 10.1111/liv.12537
85. Wree A, Eguchi A, McGeough MD, Pena CA, Johnson CD, Canbay A, et al. Nlrp3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology (2014) 59(3):898–910. doi: 10.1002/hep.26592
86. Ganz M, Csak T, Szabo G. High fat diet feeding results in gender specific steatohepatitis and inflammasome activation. World J Gastroenterol (2014) 20(26):8525–34. doi: 10.3748/wjg.v20.i26.8525
87. Szabo G, Petrasek J. Inflammasome activation and function in liver disease. Nat Rev Gastroenterol Hepatol (2015) 12(7):387–400. doi: 10.1038/nrgastro.2015.94
88. Csak T, Ganz M, Pespisa J, Kodys K, Dolganiuc A, Szabo G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology (2011) 54(1):133–44. doi: 10.1002/hep.24341
89. Kamari Y, Shaish A, Vax E, Shemesh S, Kandel-Kfir M, Arbel Y, et al. Lack of interleukin-1alpha or interleukin-1beta inhibits transformation of steatosis to steatohepatitis and liver fibrosis in hypercholesterolemic mice. J Hepatol (2011) 55(5):1086–94. doi: 10.1016/j.jhep.2011.01.048
90. Wree A, McGeough MD, Pena CA, Schlattjan M, Li H, Inzaugarat ME, et al. Nlrp3 inflammasome activation is required for fibrosis development in nafld. J Mol Med (Berl) (2014) 92(10):1069–82. doi: 10.1007/s00109-014-1170-1
91. Hotamisligil GS. Inflammation, metaflammation and immunometabolic disorders. Nature (2017) 542(7640):177–85. doi: 10.1038/nature21363
92. Spranger J, Kroke A, Mohlig M, Hoffmann K, Bergmann MM, Ristow M, et al. Inflammatory cytokines and the risk to develop type 2 diabetes: Results of the prospective population-based European prospective investigation into cancer and nutrition (Epic)-potsdam study. Diabetes (2003) 52(3):812–7. doi: 10.2337/diabetes.52.3.812
93. Jager J, Gremeaux T, Cormont M, Le Marchand-Brustel Y, Tanti JF. Interleukin-1beta-Induced insulin resistance in adipocytes through down-regulation of insulin receptor substrate-1 expression. Endocrinology (2007) 148(1):241–51. doi: 10.1210/en.2006-0692
94. Larsen CM, Faulenbach M, Vaag A, Volund A, Ehses JA, Seifert B, et al. Interleukin-1-Receptor antagonist in type 2 diabetes mellitus. N Engl J Med (2007) 356(15):1517–26. doi: 10.1056/NEJMoa065213
95. Stienstra R, Joosten LA, Koenen T, van Tits B, van Diepen JA, van den Berg SA, et al. The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab (2010) 12(6):593–605. doi: 10.1016/j.cmet.2010.11.011
96. Stienstra R, van Diepen JA, Tack CJ, Zaki MH, van de Veerdonk FL, Perera D, et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc Natl Acad Sci U.S.A. (2011) 108(37):15324–9. doi: 10.1073/pnas.1100255108
97. Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, et al. Fatty acid-induced Nlrp3-asc inflammasome activation interferes with insulin signaling. Nat Immunol (2011) 12(5):408–15. doi: 10.1038/ni.2022
98. Boden G. Interaction between free fatty acids and glucose metabolism. Curr Opin Clin Nutr Metab Care (2002) 5(5):545–9. doi: 10.1097/00075197-200209000-00014
99. Busso N, So A. Mechanisms of inflammation in gout. Arthritis Res Ther (2010) 12(2):206. doi: 10.1186/ar2952
100. Kingsbury SR, Conaghan PG, McDermott MF. The role of the Nlrp3 inflammasome in gout. J Inflammation Res (2011) 4:39–49. doi: 10.2147/JIR.S11330
101. Landis RC, Haskard DO. Pathogenesis of crystal-induced inflammation. Curr Rheumatol Rep (2001) 3(1):36–41. doi: 10.1007/s11926-001-0049-7
102. Ising C, Heneka MT. Functional and structural damage of neurons by innate immune mechanisms during neurodegeneration. Cell Death Dis (2018) 9(2):120. doi: 10.1038/s41419-017-0153-x
103. Venegas C, Kumar S, Franklin BS, Dierkes T, Brinkschulte R, Tejera D, et al. Microglia-derived asc specks cross-seed amyloid-beta in alzheimer's disease. Nature (2017) 552(7685):355–61. doi: 10.1038/nature25158
104. Ising C, Venegas C, Zhang S, Scheiblich H, Schmidt SV, Vieira-Saecker A, et al. Nlrp3 inflammasome activation drives tau pathology. Nature (2019) 575(7784):669–73. doi: 10.1038/s41586-019-1769-z
105. Gordon R, Albornoz EA, Christie DC, Langley MR, Kumar V, Mantovani S, et al. Inflammasome inhibition prevents alpha-synuclein pathology and dopaminergic neurodegeneration in mice. Sci Transl Med (2018) 10(465):eaah4066. doi: 10.1126/scitranslmed.aah4066
106. Imaeda AB, Watanabe A, Sohail MA, Mahmood S, Mohamadnejad M, Sutterwala FS, et al. Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J Clin Invest (2009) 119(2):305–14. doi: 10.1172/JCI35958
107. Su Q, Kuang W, Hao W, Liang J, Wu L, Tang C, et al. Antituberculosis drugs (Rifampicin and isoniazid) induce liver injury by regulating Nlrp3 inflammasomes. Mediators Inflammation (2021) 2021:8086253. doi: 10.1155/2021/8086253
108. Zhang Y, Qu X, Gao H, Zhai J, Tao L, Sun J, et al. Quercetin attenuates Nlrp3 inflammasome activation and apoptosis to protect inh-induced liver injury Via regulating Sirt1 pathway. Int Immunopharmacol (2020) 85:106634. doi: 10.1016/j.intimp.2020.106634
109. Wang Z, Xu G, Zhan X, Liu Y, Gao Y, Chen N, et al. Carbamazepine promotes specific stimuli-induced Nlrp3 inflammasome activation and causes idiosyncratic liver injury in mice. Arch Toxicol (2019) 93(12):3585–99. doi: 10.1007/s00204-019-02606-3
110. Yuan Z, Hasnat M, Liang P, Yuan Z, Zhang H, Sun L, et al. The role of inflammasome activation in triptolide-induced acute liver toxicity. Int Immunopharmacol (2019) 75:105754. doi: 10.1016/j.intimp.2019.105754
111. Wang Z, Xu G, Wang H, Zhan X, Gao Y, Chen N, et al. Icariside II, a main compound in epimedii folium, induces idiosyncratic hepatotoxicity by enhancing Nlrp3 inflammasome activation. Acta Pharm Sin B (2020) 10(9):1619–33. doi: 10.1016/j.apsb.2020.03.006
112. Gao Y, Xu G, Ma L, Shi W, Wang Z, Zhan X, et al. Icariside I specifically facilitates atp or nigericin-induced Nlrp3 inflammasome activation and causes idiosyncratic hepatotoxicity. Cell Commun Signal (2021) 19(1):13. doi: 10.1186/s12964-020-00647-1
113. Qin N, Xu G, Wang Y, Zhan X, Gao Y, Wang Z, et al. Bavachin enhances Nlrp3 inflammasome activation induced by atp or nigericin and causes idiosyncratic hepatotoxicity. Front Med (2021) 15(4):594–607. doi: 10.1007/s11684-020-0809-2
114. Wang Y, Xu G, Wang Z, Li R, Zhan X, Liu H, et al. Psoralidin, a major component of psoraleae fructus, induces inflammasome activation and idiosyncratic liver injury. Int Immunopharmacol (2021) 92:107352. doi: 10.1016/j.intimp.2020.107352
115. Wallace CI, Dargan PI, Jones AL. Paracetamol overdose: An evidence based flowchart to guide management. Emerg Med J (2002) 19(3):202–5. doi: 10.1136/emj.19.3.202
116. Bjornsson E, Olsson R. Outcome and prognostic markers in severe drug-induced liver disease. Hepatology (2005) 42(2):481–9. doi: 10.1002/hep.20800
117. Wang JB, Cui HR, Bai ZF, Xiao XH. [Precision medicine-oriented safety assessment strategy for traditional Chinese medicines: Disease-Syndrome-Based toxicology]. Yao Xue Xue Bao (2016) 51(11):1681–8.
118. Melchart D, Hager S, Albrecht S, Dai J, Weidenhammer W, Teschke R. Herbal traditional Chinese medicine and suspected liver injury: A prospective study. World J Hepatol (2017) 9(29):1141–57. doi: 10.4254/wjh.v9.i29.1141
119. Lamkanfi M, Mueller JL, Vitari AC, Misaghi S, Fedorova A, Deshayes K, et al. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J Cell Biol (2009) 187(1):61–70. doi: 10.1083/jcb.200903124
120. Liao J, Kapadia VS, Brown LS, Cheong N, Longoria C, Mija D, et al. The Nlrp3 inflammasome is critically involved in the development of bronchopulmonary dysplasia. Nat Commun (2015) 6:8977. doi: 10.1038/ncomms9977
121. Hamon Y, Luciani MF, Becq F, Verrier B, Rubartelli A, Chimini G. Interleukin-1beta secretion is impaired by inhibitors of the atp binding cassette transporter, Abc1. Blood (1997) 90(8):2911–5.
122. Carvalho AM, Novais FO, Paixao CS, de Oliveira CI, Machado PRL, Carvalho LP, et al. Glyburide, a Nlrp3 inhibitor, decreases inflammatory response and is a candidate to reduce pathology in leishmania braziliensis infection. J Invest Dermatol (2020) 140(1):246–9.e2. doi: 10.1016/j.jid.2019.05.025
123. Coll RC, Hill JR, Day CJ, Zamoshnikova A, Boucher D, Massey NL, et al. Mcc950 directly targets the Nlrp3 atp-hydrolysis motif for inflammasome inhibition. Nat Chem Biol (2019) 15(6):556–9. doi: 10.1038/s41589-019-0277-7
124. Perregaux DG, McNiff P, Laliberte R, Hawryluk N, Peurano H, Stam E, et al. Identification and characterization of a novel class of interleukin-1 post-translational processing inhibitors. J Pharmacol Exp Ther (2001) 299(1):187–97.
125. Coll RC, Robertson AA, Chae JJ, Higgins SC, Munoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the Nlrp3 inflammasome for the treatment of inflammatory diseases. Nat Med (2015) 21(3):248–55. doi: 10.1038/nm.3806
126. Tapia-Abellan A, Angosto-Bazarra D, Martinez-Banaclocha H, de Torre-Minguela C, Ceron-Carrasco JP, Perez-Sanchez H, et al. Mcc950 closes the active conformation of Nlrp3 to an inactive state. Nat Chem Biol (2019) 15(6):560–4. doi: 10.1038/s41589-019-0278-6
127. Daniels MJ, Rivers-Auty J, Schilling T, Spencer NG, Watremez W, Fasolino V, et al. Fenamate nsaids inhibit the Nlrp3 inflammasome and protect against alzheimer's disease in rodent models. Nat Commun (2016) 7:12504. doi: 10.1038/ncomms12504
128. Jiang H, He H, Chen Y, Huang W, Cheng J, Ye J, et al. Identification of a selective and direct Nlrp3 inhibitor to treat inflammatory disorders. J Exp Med (2017) 214(11):3219–38. doi: 10.1084/jem.20171419
129. Huang Y, Jiang H, Chen Y, Wang X, Yang Y, Tao J, et al. Tranilast directly targets Nlrp3 to treat inflammasome-driven diseases. EMBO Mol Med (2018) 10(4):e8689. doi: 10.15252/emmm.201708689
130. He Y, Varadarajan S, Munoz-Planillo R, Burberry A, Nakamura Y, Nunez G. 3,4-Methylenedioxy-Beta-Nitrostyrene inhibits Nlrp3 inflammasome activation by blocking assembly of the inflammasome. J Biol Chem (2014) 289(2):1142–50. doi: 10.1074/jbc.M113.515080
131. Juliana C, Fernandes-Alnemri T, Wu J, Datta P, Solorzano L, Yu JW, et al. Anti-inflammatory compounds parthenolide and bay 11-7082 are direct inhibitors of the inflammasome. J Biol Chem (2010) 285(13):9792–802. doi: 10.1074/jbc.M109.082305
132. Marchetti C, Swartzwelter B, Gamboni F, Neff CP, Richter K, Azam T, et al. Olt1177, a beta-sulfonyl nitrile compound, safe in humans, inhibits the Nlrp3 inflammasome and reverses the metabolic cost of inflammation. Proc Natl Acad Sci U.S.A. (2018) 115(7):E1530–E9. doi: 10.1073/pnas.1716095115
133. Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D, et al. The ketone metabolite beta-hydroxybutyrate blocks Nlrp3 inflammasome-mediated inflammatory disease. Nat Med (2015) 21(3):263–9. doi: 10.1038/nm.3804
134. Cocco M, Pellegrini C, Martinez-Banaclocha H, Giorgis M, Marini E, Costale A, et al. Development of an acrylate derivative targeting the Nlrp3 inflammasome for the treatment of inflammatory bowel disease. J Med Chem (2017) 60(9):3656–71. doi: 10.1021/acs.jmedchem.6b01624
135. Lee HG, Cho NC, Jeong AJ, Li YC, Rhie SJ, Choi JS, et al. Immunomodulatory activities of the benzoxathiole derivative bot-4-One ameliorate pathogenic skin inflammation in mice. J Invest Dermatol (2016) 136(1):107–16. doi: 10.1038/JID.2015.384
136. He H, Jiang H, Chen Y, Ye J, Wang A, Wang C, et al. Oridonin is a covalent Nlrp3 inhibitor with strong anti-inflammasome activity. Nat Commun (2018) 9(1):2550. doi: 10.1038/s41467-018-04947-6
137. Li Q, Feng H, Wang H, Wang Y, Mou W, Xu G, et al. Licochalcone b specifically inhibits the Nlrp3 inflammasome by disrupting Nek7-Nlrp3 interaction. EMBO Rep (2022) 23(2):e53499. doi: 10.15252/embr.202153499
138. Xu G, Fu S, Zhan X, Wang Z, Zhang P, Shi W, et al. Echinatin effectively protects against Nlrp3 inflammasome-driven diseases by targeting Hsp90. JCI Insight (2021) 6(2):e134601. doi: 10.1172/jci.insight.134601
139. Yin H, Guo Q, Li X, Tang T, Li C, Wang H, et al. Curcumin suppresses il-1beta secretion and prevents inflammation through inhibition of the Nlrp3 inflammasome. J Immunol (2018) 200(8):2835–46. doi: 10.4049/jimmunol.1701495
140. Wang Z, Xu G, Gao Y, Zhan X, Qin N, Fu S, et al. Cardamonin from a medicinal herb protects against lps-induced septic shock by suppressing Nlrp3 inflammasome. Acta Pharm Sin B (2019) 9(4):734–44. doi: 10.1016/j.apsb.2019.02.003
141. Shi W, Xu G, Zhan X, Gao Y, Wang Z, Fu S, et al. Carnosol inhibits inflammasome activation by directly targeting Hsp90 to treat inflammasome-mediated diseases. Cell Death Dis (2020) 11(4):252. doi: 10.1038/s41419-020-2460-x
142. Liu H, Zhan X, Xu G, Wang Z, Li R, Wang Y, et al. Cryptotanshinone specifically suppresses Nlrp3 inflammasome activation and protects against inflammasome-mediated diseases. Pharmacol Res (2021) 164:105384. doi: 10.1016/j.phrs.2020.105384
143. Darakhshan S, Pour AB. Tranilast: A review of its therapeutic applications. Pharmacol Res (2015) 91:15–28. doi: 10.1016/j.phrs.2014.10.009
144. Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, et al. Gpr120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell (2010) 142(5):687–98. doi: 10.1016/j.cell.2010.07.041
145. Sonawane ND, Verkman AS. Thiazolidinone cftr inhibitors with improved water solubility identified by structure-activity analysis. Bioorg Med Chem (2008) 16(17):8187–95. doi: 10.1016/j.bmc.2008.07.044
146. Kahlenberg JM, Lundberg KC, Kertesy SB, Qu Y, Dubyak GR. Potentiation of caspase-1 activation by the P2x7 receptor is dependent on tlr signals and requires nf-Kappab-Driven protein synthesis. J Immunol (2005) 175(11):7611–22. doi: 10.4049/jimmunol.175.11.7611
147. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. Cutting edge: Nf-kappab activating pattern recognition and cytokine receptors license Nlrp3 inflammasome activation by regulating Nlrp3 expression. J Immunol (2009) 183(2):787–91. doi: 10.4049/jimmunol.0901363
148. Riddle MC. Editorial: Sulfonylureas differ in effects on ischemic preconditioning–is it time to retire glyburide? J Clin Endocrinol Metab (2003) 88(2):528–30. doi: 10.1210/jc.2002-021971
149. Ashcroft FM. Atp-sensitive potassium channelopathies: Focus on insulin secretion. J Clin Invest (2005) 115(8):2047–58. doi: 10.1172/JCI25495
150. Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol (1971) 231(25):232–5. doi: 10.1038/newbio231232a0
151. Newman JC, Verdin E. Ketone bodies as signaling metabolites. Trends Endocrinol Metab (2014) 25(1):42–52. doi: 10.1016/j.tem.2013.09.002
152. Ma Z, Hu C, Zhang Y. [Therapeutic effect of rabdosia rubescens aqueous extract on chronic pharyngitis and its safety]. Zhong Nan Da Xue Xue Bao Yi Xue Ban (2011) 36(2):170–3. doi: 10.3969/j.issn.1672-7347.2011.02.014
153. Singh J, Petter RC, Baillie TA, Whitty A. The resurgence of covalent drugs. Nat Rev Drug Discovery (2011) 10(4):307–17. doi: 10.1038/nrd3410
154. Bauer RA. Covalent inhibitors in drug discovery: From accidental discoveries to avoided liabilities and designed therapies. Drug Discovery Today (2015) 20(9):1061–73. doi: 10.1016/j.drudis.2015.05.005
155. Furusawa J, Funakoshi-Tago M, Mashino T, Tago K, Inoue H, Sonoda Y, et al. Glycyrrhiza inflata-derived chalcones, licochalcone a, licochalcone b and licochalcone d, inhibit phosphorylation of nf-kappab P65 in lps signaling pathway. Int Immunopharmacol (2009) 9(4):499–507. doi: 10.1016/j.intimp.2009.01.031
156. Mayor A, Martinon F, De Smedt T, Petrilli V, Tschopp J. A crucial function of Sgt1 and Hsp90 in inflammasome activity links mammalian and plant innate immune responses. Nat Immunol (2007) 8(5):497–503. doi: 10.1038/ni1459
157. Prasad S, Gupta SC, Tyagi AK, Aggarwal BB. Curcumin, a component of golden spice: From bedside to bench and back. Biotechnol Adv (2014) 32(6):1053–64. doi: 10.1016/j.biotechadv.2014.04.004
158. Shehzad A, Rehman G, Lee YS. Curcumin in inflammatory diseases. Biofactors (2013) 39(1):69–77. doi: 10.1002/biof.1066
159. Wang K, Lv Q, Miao YM, Qiao SM, Dai Y, Wei ZF. Cardamonin, a natural flavone, alleviates inflammatory bowel disease by the inhibition of Nlrp3 inflammasome activation Via an Ahr/Nrf2/Nqo1 pathway. Biochem Pharmacol (2018) 155:494–509. doi: 10.1016/j.bcp.2018.07.039
160. Johnson JJ. Carnosol: A promising anti-cancer and anti-inflammatory agent. Cancer Lett (2011) 305(1):1–7. doi: 10.1016/j.canlet.2011.02.005
161. Park EJ, Zhao YZ, Kim YC, Sohn DH. Pf2401-sf, standardized fraction of salvia miltiorrhiza and its constituents, tanshinone I, tanshinone iia, and cryptotanshinone, protect primary cultured rat hepatocytes from bile acid-induced apoptosis by inhibiting jnk phosphorylation. Food Chem Toxicol (2007) 45(10):1891–8. doi: 10.1016/j.fct.2007.04.005
Keywords: pattern recognition receptor, NLRP3, inflammasome, inflammatory diseases, pharmacological inhibitors
Citation: Zhan X, Li Q, Xu G, Xiao X and Bai Z (2023) The mechanism of NLRP3 inflammasome activation and its pharmacological inhibitors. Front. Immunol. 13:1109938. doi: 10.3389/fimmu.2022.1109938
Received: 28 November 2022; Accepted: 29 December 2022;
Published: 18 January 2023.
Edited by:
Qian Yin, Florida State University, United StatesReviewed by:
Jianbin Ruan, UCONN Health, United StatesLe Xiao, Boston Children’s Hospital and Harvard Medical School, United States
Copyright © 2023 Zhan, Li, Xu, Xiao and Bai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaohe Xiao, cGhhcm1hY3lfMzAyQDEyNi5jb20=; Zhaofang Bai, YmFpemYyMDA4QGhvdG1haWwuY29t