 Rinaldo Rodrigues dos Passos Junior1,2
Rinaldo Rodrigues dos Passos Junior1,2 Gisele Facholi Bomfim3
Gisele Facholi Bomfim3 Fernanda R. Giachini1,2
Fernanda R. Giachini1,2 Rita C. Tostes4
Rita C. Tostes4 Victor Vitorino Lima1*
Victor Vitorino Lima1*- 1Institute of Biological and Health Sciences, Federal University of Mato Grosso, Barra do Garças, Brazil
- 2Institute of Biological Sciences, Federal University of Goias, Goiânia, Brazil
- 3Institute of Health Sciences, Federal University of Mato Grosso, Sinop, Brazil
- 4Department of Pharmacology, Ribeirão Preto Medical School, University of São Paulo, Ribeirão Preto, Brazil
The O-linked β-N-acetylglucosamine modification (O-GlcNAcylation) of proteins dynamically regulates protein function, localization, stability, and interactions. This post-translational modification is intimately linked to cardiovascular disease, including hypertension. An increasing number of studies suggest that components of innate and adaptive immunity, active players in the pathophysiology of hypertension, are targets for O-GlcNAcylation. In this review, we highlight the potential roles of O-GlcNAcylation in the immune system and discuss how those immune targets of O-GlcNAcylation may contribute to arterial hypertension.
Stating the Problem
O-GlcNAcylation on serine (Ser), threonine (Thr) and tyrosine (Tyr) residues of nuclear, cytosolic, and mitochondrial proteins is one of the most abundant post-translational modifications that modulate phosphorylation, stability, and activity of multiple cellular signaling pathways and transcription regulatory cascades (1, 2).
O-GlcNAc is dynamically, reversibly, and rapidly cycled on and off proteins by two specific enzymes: O-GlcNAc transferase (OGT), which catalyzes the addition of the O-GlcNAc moiety; and O-GlcNAcase (OGA), that catalyzes its removal (Figure 1). This modification is responsive to several stimuli, including nutrient availability. An overabundance of nutrients can drastically shift substrates to the hexosamine biosynthetic pathway (HBP), favoring the synthesis of the O-GlcNAc precursor, UDP-GlcNAc, manly via recruitment of the rate-limiting enzyme glutamine:fructose-6-phosphate aminotransferase (GFAT) (2, 3). However, many studies show that protein O-GlcNAcylation excess occurs in response to factors that do not fit neatly with the nutrient availability concept, including oxidative stress (4, 5), renin-angiotensin system (RAS) (6–8) and endothelin-1 (ET-1) (9–12).
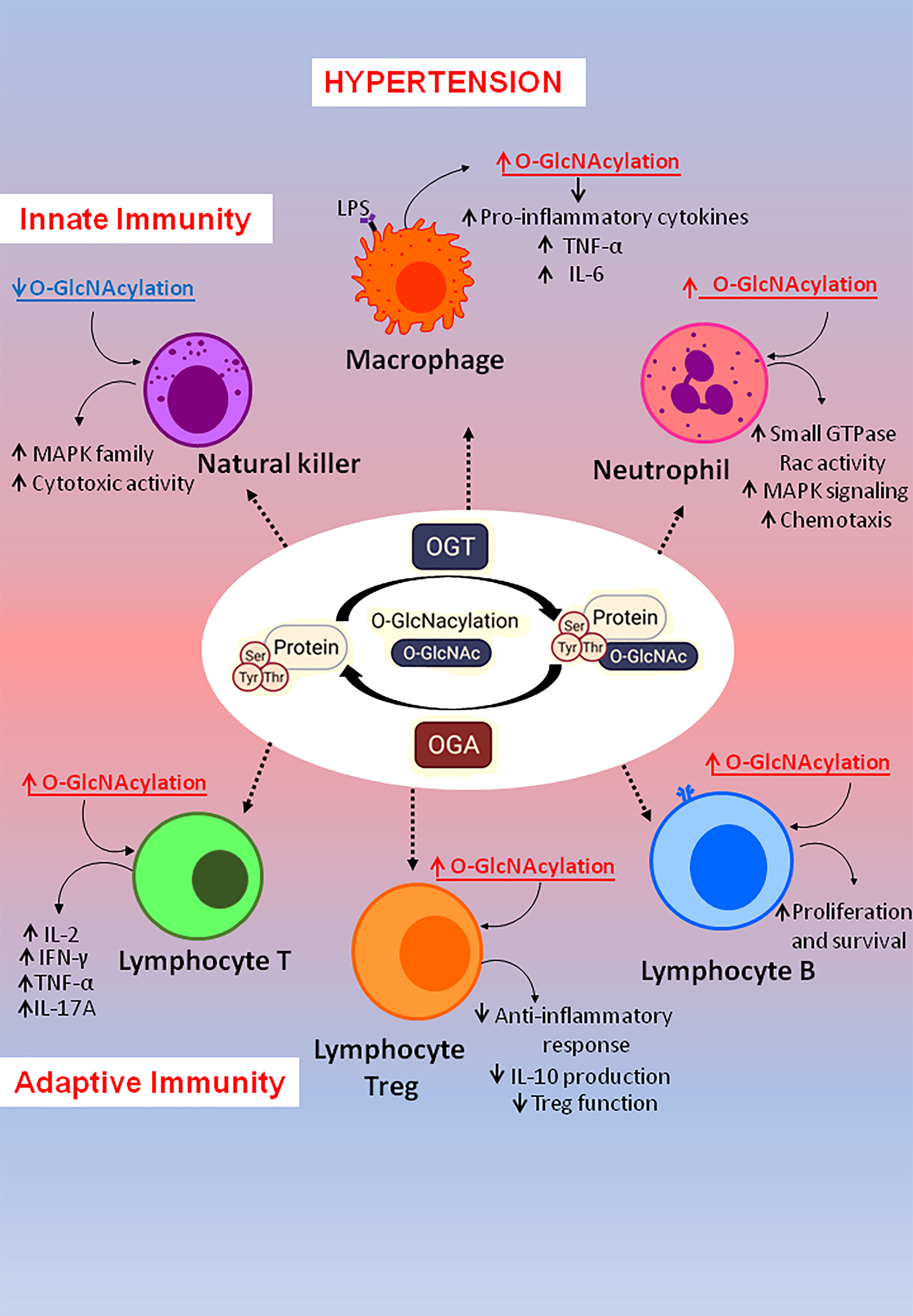
Figure 1 Factors linked to hypertension also related to O-GlcNAcylation levels. Hypertension is a multifactorial disease involving many factors and events, which are also associated with excessive chronic O-GlcNAcylation levels.
O-GlcNAc-modified proteins have been implicated in a diverse array of cellular functions, including signaling, transcription, apoptosis, and inflammation (2, 13). Given its diverse roles, protein O-GlcNAcylation has been associated with a diverse array of pathological conditions, such as arterial hypertension.
Hypertension affects over 1.2 billion individuals worldwide and is a multifactorial disease involving immune cells activation, inflammation, oxidative stress, activation of the sympathetic and renin angiotensin aldosterone (RAAS) systems, and others. This might explain the fact that the etiology of hypertension is known in only 10% of the patients (14). Many therapeutical classes of drugs targeting many different molecules are available to treat hypertension based on the classical view of blood pressure (BP) control by neural, vascular and renal mechanisms. Still, 20% of hypertensive patients do not adequately achieve BP control (15). With this refractory response to the regular anti-hypertensive therapeutic arsenal in mind, scientific advances have been targeting the immune system, as a possible therapeutic target to treat hypertension (16).
The present review will provide a brief overview of the scientific advances related to the role of O-GlcNAcylation on hypertension progression. Then, the focus will be to set on potential mechanisms whereby O-GlcNAcylation of components of the immune system could impact hypertension.
O-GlcNAcylation and Arterial Hypertension
It is well-recognized that hyperglycemia, linked to the diabetic condition, negatively impacts cardiovascular function and, consequently, blood pressure control (17). Yet, little is known regarding how augmented flux of glucose, or other subproducts from the nucleotide and fatty acid metabolism, through the hexosamine biosynthetic pathway (HBP) contributes to the pathophysiology of hypertension.
The HBP is highly sensitive to the major metabolites produced in glucose, amino acid, nucleotide, and fatty acid metabolism. Therefore, an overabundance of nutrients (18), as seen in diabetic or hyperlipidemic subjects, may overload the HBP flux, leading to increased protein O-GlcNAcylation (19). In addition to diabetes, increased O-GlcNAc levels have been reported in different experimental models of hypertension (6, 20, 21), cardiac hypertrophy (22, 23), cardiac dysfunction (22, 24, 25), as well as in response to agonists such as angiotensin II (Ang II) (6) and endothelin-1 (ET-1) (9, 10). The fact that O-GlcNAc impacts biological functions that are not totally linked to altered nutrient availability, raised the suggestion that other mechanisms regulating O-GlcNAcylation may participate in the pathophysiology of hypertension.
Increases in arterial blood pressure can be generated by a variety of events, including modifications in cardiac output, vascular dysfunction, and target organ damage (14, 17). Interestingly, well-known mechanisms whereby blood pressure (BP) and cardiac function may be altered are also targeted by O-GlcNAcylation (Figure 1).
Excessive chronic O-GlcNAcylation has been shown both in humans and animal models of myocardial dysfunction, cardiac remodeling, aortic banding, and hypertension (22, 25–27). Hyperglycemia in Zucker (diabetic fatty) rats leads to high O-GlcNAc levels along with attenuated cardiomyocytes calcium peak and prolonged time to relaxation and, consequently, to impaired cardiac contraction and relaxation in these animals (28).
Chronic increases in O-GlcNAc levels also leads to increased risk of ventricular arrhythmias, which has been linked to increased O-GlcNAcylation of cardiac voltage-gated sodium channels (26).
Cardiac hypertrophy is usually seen in different stages of hypertension (29, 30). In the long-term, cardiac O-GlcNAcylation is also increased in cardiac hypertrophy conditions (22, 31). High levels of O‐GlcNAc are seen in hypertrophic hearts in response to phenylephrine (32, 33). However, hypertrophy was not observed in cardiomyocytes treated with an inhibitor of GFAT (23), a rate-limiting enzyme in the HBP pathway. These results suggest that high levels of O-GlcNAc are associated with cardiac hypertrophy and inhibition of the O-GlcNAc pathway may work as a pharmacological tool to block hypertrophy progression during hypertension.
Considering the evidence linking the O-GlcNAc involvement with ventricular arrhythmias, cardiac hypertrophy and other cardiac dysfunctions, it is reasonable that O-GlcNAc may influence important factors related to hypertension (24). In this sense, experimental models of hypertension, such as spontaneously hypertensive rats (SHR), have significantly higher systolic blood pressure, hypertrophy and increased protein O-GlcNAcylation in the left ventricle (LV). This study showed that increased O-GlcNAc in pressure overload conditions, including chronic hypertension and aortic banding, is associated with increased OGT protein, suggesting that OGT levels might be an important mechanism for cardiac protein O-GlcNAcylation. Of importance, O-GlcNAc levels are 65% higher in LV biopsies from patients with severe aortic stenosis, compared with controls (22).
Furthermore, a correlation between increased O-GlcNAcylation and GFAT expression was reported in SHR (20, 22). Silva-Aguiar et al. showed that adult SHR with established hypertension display increased renal cortical O-GlcNAcylation. They proposed that changes in protein located at the proximal tubule are associated with an increase in O-GlcNAcylation in the renal cortex of adult SHR. Conversely, no changes in O-GlcNAc levels or blood pressure were observed in young SHR, suggesting that increased cortical O-GlcNAcylation could be related to the development of hypertension. In agreement, a GFAT inhibitor reduced global O-GlcNAcylation and also significantly decreased blood pressure in SHRs (20).
ET-1, a potent vasoconstrictor peptide and growth-promoting factor (34), also plays an important role in the physiological control of blood pressure and in the genesis and development of arterial (35) and pulmonary hypertension (36). ET-1 levels are increased in the vasculature of deoxycorticosterone acetate (DOCA)-salt hypertensive rats (Schiffrin, 2005). Moreover, increased vascular GFAT expression and O-GlcNAcylation was correlated with time-dependent increases in blood pressure and vascular dysfunction in DOCA-salt hypertensive rats (21). In agreement, in vitro and in vivo treatment with ET-1 increases vasoconstriction and the vascular content of O-GlcNAc-modified proteins. Interestingly, these effects were not observed when vessels were previously transfected with antibodies against OGT or incubated with an OGT inhibitor (10).
Chronically increased BP occurring similarly to augmented O-GlcNAc tissular levels may be an additional mechanism eliciting end-organ damage (20). For example, chronically increased O-GlcNAcylation was positively correlated with renal damage (12, 37), and patients with nephropathy display increased glomerular and tubular O-GlcNAcylation (38). Moreover, a number of studies also provide evidence for an interplay between protein O-GlcNAcylation and Ang II, a well-known vasoactive that increases blood pressure (29), induces cardiac hypertrophy (39) and kidney damage (40, 41). Ang II increases O-GlcNAcylation in mesangial cells (6) and heart from mice (7). Conversely, Ang 1–7 and Mas-receptor inhibition reduced protein O-GlcNAcylation by repressing GFAT1 activity (8).
Gellai et al. showed that RAAS inhibitors inhibit diabetes-induced O-GlcNAcylation in the kidney, by increasing OGA expression (12). In line with this view, changes of protein O-GlcNAcylation levels resulted in a concomitant alteration in angiotensinogen, OGT, and GFAT transcripts (42). Furthermore, in vivo glucosamine-treatment increased the expression of angiotensinogen in adipose tissue (43). Therefore, O-GlcNAcylation regulates local and systemic RAAS, which may contribute to the progression of hypertension. Finally, high levels of O-GlcNAcylation driven by high glucose or glucosamine treatment leads to impaired vascular endothelial and smooth muscle cells function in human and rat (11, 25, 44–46), a phenotype closely associated with hypertension (29). In this sense, there is direct evidence that elevated levels of protein O-GlcNAcylation in endothelial cells impairs endothelium-dependent relaxation (44, 47–49), demonstrating that O-GlcNAcylation leads to endothelial dysfunction. Furthermore, O-GlcNAcylation of vascular smooth muscle cells augments vascular response to contractile agonists (50, 51) and favors vascular calcification (52–54), resulting in high blood pressure (25).
Possible Mechanisms Whereby the Crosstalk Between O-GlcNAcylation and the Immune System Impacts Arterial Hypertension
Classically, the main function of the immune system is to defend a host against pathogen invasion. However, as extensively reviewed in the literature, overactivation of immune system components contributes to non-infectious disease, like hypertension (16, 55, 56). The immune response is complex and has two interconnected systems: the innate immunity, which mediates early reactions, and adaptive immunity, which is a late and more specific response. Both systems are activated and contribute to high blood pressure and tissue damage in hypertension. The interplay between O-GlcNAcylation and the immune system has gained great interest in the last few years when several studies have been performed, highlighting how this post-translational modification impacts the immune cells. In this chapter we will highlight potential mechanisms whereby O-GlcNAcylation of immune components may contribute to hypertension (Figure 2).
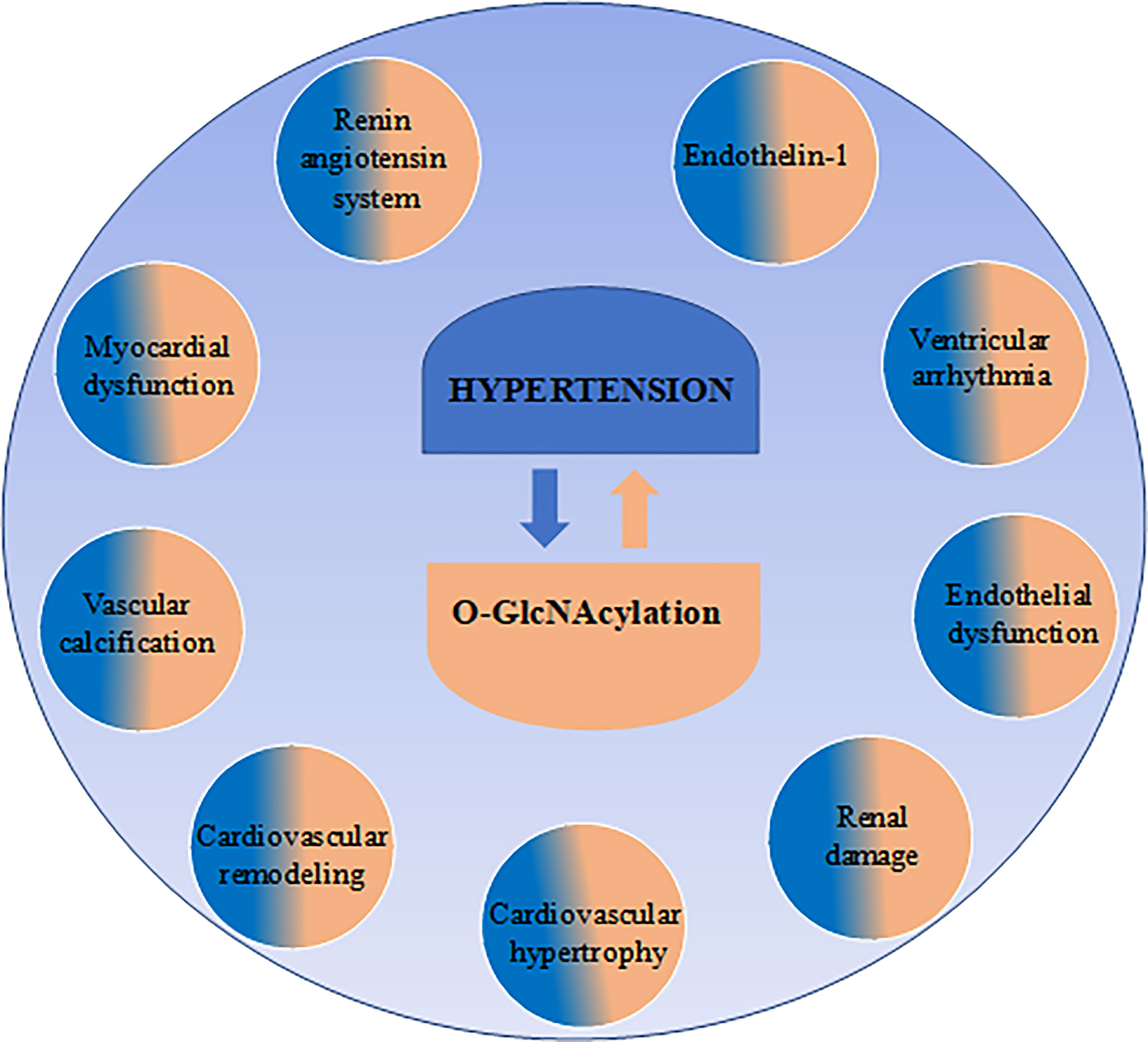
Figure 2 Immune targets of O-GlcNAcylation may contribute to arterial hypertension. O-GlcNAcylation is a dynamic and reversible modification in protein residues by the actions of two specific enzymes: O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA). Regarding innate immunity: O-GlcNAc plays important roles in macrophages and neutrophils by favoring pro-inflammatory cytokine production, neutrophil infiltration and motility, which are associated with the severity of hypertension. Additionally, in natural killer (NK)-cells, reduced O-GlcNAc modification was found to favor NK cell cytotoxicity by enhancing MAPK family activity. Regarding adaptive immunity: O-GlcNAcylation was found to target T cells, T reg cells and B cells. In lymphocytes T, global elevation of O-GlcNAc modification favors IL-2 production and cell proliferation, as well as Th1 pro-inflammatory IL-17A and IFNγ cytokine secretion, which are associated with hypertension. In lymphocyte Treg was found that O-GlcNAc actions impaired anti-inflammatory response through STAT-3 O-GlcNAcylation, affecting STAT3–IL-10 axis and impairing Treg function. Additionally, in B cells, protein O-GlcNAcylation enhances B cell proliferation and survival. Interestingly, B cells were found to be related to increased blood pressure.
Involvement of Innate Immunity in Hypertension: Possible Roles of O-GlcNAcylation
The innate immune response is initiated by the recognition of antigens originated from debris and other molecules from damaged cells, known as DAMPs (damage associated molecular patterns); or alternatively, from antigens originated from pathogens, called PAMPs (pathogen associated molecular patterns) (57). DAMPs and PAMPs are recognized by pattern recognition receptors (PRR), including Toll-like receptors (TLR) family, which are expressed in several cells.
There are 11 subtypes of TLRs, and the participation of TLR2, TLR3, TLR4, TLR7, and TLR9 receptors in hypertension has been described (58, 59). Activation of TLR4 is directly involved on vascular inflammation, vascular dysfunction, and hypertension (60, 61). Vascular TLR4 expression is increased in cardiovascular disease, including several animal models of hypertension (SHR, Ang II infusion, DOCA-Salt), atherosclerosis and others (18, 62). Treatment with a neutralizing anti-TLR4 antibody decreases blood pressure, vascular inflammation, and cardiac remodeling in hypertensive rats (60, 61, 63). TLR4 overexpression aggravates vascular smooth muscle cells proliferation and vascular remodeling in hypertension (64). TLR9 stimulation produces systemic maternal inflammation and vascular dysfunction that lead to hypertension (65). A TLR9 antagonist decreased blood pressure in SHR and TLR9 agonist impairs mesenteric resistance arteries’ function and increases local ROS (66). Mitochondrial DNA is a DAMP that activate TLR9. Echem et al. (63) demonstrated that only male SHR exhibit high levels of plasma mitDNA and the antagonism of TLR9 normalizes mitDNA-induced increased aortic contractions elicited to phenylephrine (67). TLR9 also negatively modulates cardiac vagal tone and baroreflex in mice (68).
After the PRR recognize specific antigens, innate immunity cells are activated and contribute to the inflammatory profile in hypertension. Circulating and endothelial cell adhering monocytes are increased in many hypertensive animal models and hypertensive patients (69–74). Monocytes isolated from hypertensive patients are pre-activated and secrete high levels of IL-1β after Ang II stimulation (75). Of importance, macrophage infiltration is recognized as a classical histological characteristic of end-organ injury in hypertension (70, 76).
As previously mentioned, TLRs are known to initiate innate immune response in several cells, such as macrophages, neutrophils, natural killer (NK), dendritic cells, and mast cells (77). Thus, regulation of TLRs activation is a critical step to ensure adequate immune responses.
Macrophages can switch to a distinct functional phenotype in response to physiological and microenvironmental signals and stimuli. The classically activated M1 macrophages are known to possess a pro-inflammatory phenotype, secreting pro-inflammatory cytokines, aiming to kill pathogens. They may be characterized by TLR-2, TLR-4, CD80, CD86, iNOS, and MHC-II surface phenotypes, secreting pro-inflammatory cytokines and chemokines, promoting cell proliferation and tissue repair (78, 79). Beyond involvement in the innate immune response, there is direct evidence that macrophages per se may affect blood pressure. For example, patients with hypertension display macrophage infiltration into the vascular wall, myocardium and kidneys (80). Moreover, Ang II-induced increased blood pressure is attenuated in macrophages-deficient mice, showing the contribution of these cells to hypertension (81).
When macrophages are activated by LPS/TLR4, M1 macrophage polarization is induced, resulting in reduced HBP activity and attenuated protein O-GlcNAcylation (82). However, increased O-GlcNAcylation in LPS-stimulated macrophages, or after intraperitoneal injection of LPS in mice have also been reported (83). In this case, when macrophages are activated by LPS, OGT is activated, enhancing protein O-GlcNAcylation, which in turn favors cytokine production, and a pro-inflammatory environment.
Among the pro-inflammatory cytokines produced by M1 macrophage and other immune cells, tumor necrosis factor alfa (TNF-α) and interleukin (IL)-6 are crucial to this inflammatory profile. Therefore, NF-κB activation is a key component of the stimulation of innate immunity by PRR recognition of PAMPs/DAMPs, inducing the production of pro-inflammatory cytokines, which plays an essential role in the hypertension (84). Interesting, O-GlcNAcylation was found to modulate this transcription factor (85). NF-κB transcriptional activity is induced by OGT, and OGT colocalizes with promoter regions of NF-κB, enhancing RelA acetylation upon TNF-α stimulus, as observed in human embryonic kidney (HEK 293) cells (86). c-Rel, one of the five NF-κB subunits, is a target for O-GlcNAcylation at serine350, a mandatory post-translational modification required for c-Rel binding to the DNA, allowing transcription, as demonstrated by RAMOS in a B lymphocyte line and in Jurkat cells (immortalized line of human T lymphocytes (87). LPS also favors O-GlcNAcylation of c-Rel in the iNOS promoter in BV2 microglia cells (88). In RAW 264.7 cells, a cell line of mouse macrophages, overexpression of OGT inhibited NF-κB reporter activity upon LPS stimulus, resulting in NF-κB/iNOS transcription suppression (89). Therefore, these data on macrophages indicate that immune dysfunction may be elicited both by augmented and diminished protein O-GlcNAcylation.
Neutrophil lymphocyte ratio is an inflammatory marker, which has strong independent association with the severity of hypertension (90–93). During inflammation, neutrophils are the first cells to arrive at the injured site, ready to orchestrate the repair of tissue damage induced by macrophages. Neutrophils are polymorphonuclear leukocytes that enter the circulation and migrate towards other tissues, are responsible for patrolling the organism and searching for pathogens and other signs of infection (94). To reach sites of injury, neutrophils migrate under the regulation of intracellular signaling in a mechanism called chemotaxis. These signaling pathways release molecules that stimulate neutrophils to migrate towards the injury site (95). One of the most commonly used polymorphonuclear leukocytes stimuli, the chemotactic tripeptide formyl-methionine-leucine-phenylalanine (fMLF), binds to cell surface receptors and induces protein phosphorylation within seconds.
O-GlcNAc-modified proteins are increased in polymorphonuclear leukocytes stimulated with fMLF-stimulus, and glucosamine increases O-GlcNAc and enhances neutrophils motility (96). In addition, pharmacological increases in O-GlcNAc, using glucosamine or PUGNAc (OGA inhibitor), increases the activity of the small GTPase Rac and MAPK signaling in neutrophils (97). These protein kinases are involved in neutrophil chemotaxis and Rac is known to activate MAPK (98). This evidence shows that O-GlcNAcylation is required for neutrophil signaling transduction, including chemotaxis. In fact, augmented O-GlcNAcylation evokes both neutrophil chemotaxis and mobility (96, 97). Importantly, the downregulation of the immune system appears as a key factor in the treatment of hypertension. As a proof of this concept, a selective antagonist of β1-adrenoceptors, nebivolol, efficiently reduced innate immune responses in hypertensive patients, through decreased levels of neutrophils (99). Therefore, the role of O-GlcNAc during neutrophil chemotaxis and mobility may be a predictor of ongoing vascular inflammation in various cardiovascular disorders such as hypertension.
NK cells are a large granular type of cytotoxic lymphocytes that are essential for innate immune response, acting rapidly against a great number of pathogens and constantly interacting with other immune cells, such as macrophages and dendritic cells (100). Compared to normotensive rats, SHRs have a strong increase in the number of NK cells (101). Depletion of NK cells, using an anti-NK antibody, protects against Ang II-induced vascular dysfunction (102). Furthermore, an association between increased proportions of NK-cells and levels of systolic blood pressure has been detected in a large multi-ethnic cohort (103).
NK cells recognize infected cells through activation of receptors such as natural killer group 2D (NKG2D), which require the actions of the transcription factor enhancer of zeste homolog 2 (EZH2) (104, 105). Interestingly, few studies found that EZH2 O-GlcNAcylation is required for EZH2 protein stability and enzymatic function in human breast and colorectal cancer cells (78, 79, 106). Upon activation through its receptors, NK cells exert their cytotoxic activity killing aberrant cells, such as infected and tumorigenic cells, through the release of cytotoxic molecules stored in exocytic organelles (107). The cytotoxic vesicles release depends on MAPK family and extracellular signal-regulated kinases (ERK) activation (108). ERK-2 MAPK-dependent pathway becomes activated and mediates the movement of intracellular granules (109). A study reported that protein O-GlcNAcylation may be involved in the cytotoxic signal transduction of NK cells. There is reduced O-GlcNAc modification during NK cell cytotoxicity and inhibiting NK cytotoxicity by GST-sHLA-G1α chain restores O-GlcNAcylation in NK92 cells (110). Moreover, glucosamine treatment exerts an inhibitory effect on NK-92 cell cytotoxic vesicles release by increasing O-GlcNAc modification of ERK downstream proteins, increasing ERK nuclear localization and altering granules migration (111). The same study also showed that after, glucosamine treatment, the transcription factor FOXO1 presented reduced phosphorylation and increased O-GlcNAcylation. Interestingly, FOXO1 was found to negatively regulate NK cells’ function (112). Altogether, these data elucidate the role of protein O-GlcNAcylation on NK cell function.
The wide range of responses elicited by O-GlcNAcylation in innate immune cells indicates an important, but still incomplete unknown, role for this post-translational modification on inmate responses.
Involvement of Adaptive Immunity in Hypertension: Possible Roles of O-GlcNAcylation
Adaptive immunity, also referred to as acquired or specific immunity, is the second and long-lasting line of the host’s defense against non-self particles or pathogens. The main characteristic of the adaptive immune response is the clonal expansion of lymphocytes, such as T and B lymphocytes. There are two primary subtypes of T cells: cytotoxic T cells (CTLs), and helper T cells (Th). Experimental and clinical studies have shown the importance of adaptive immune system in hypertension (113). The main cells subtypes that contribute to hypertension are Th1, Th17, regulatory T cells (Treg), T CD8 and B cell.
Possible Target: T Cells
The association of T lymphocytes with hypertension has been supported by many studies showing that mice lacking T- and B-lymphocytes exhibit attenuated hypertension in response to Ang II (114–117).
Th1, Th2, Th17 and Treg are subtypes of CD4 T cells. Th1 cells express the transcription factor T-bet and release the cytokine IFN-γ. T-bet deficient mice infused with Ang II is protected against renal injury, but not from high blood pressure (118). Isolated T cells from the spleen of Ang II-infused rats exhibit an imbalance of Th1/Th2 subsets, with increased IFN-γ (Th1 cytokine) and decreased IL-4 (Th2 cytokine) (119). IFN-γ knockout mice are protected from Ang II-induced vascular and kidney dysfunction (102, 120). Although some studies show the contribution of IFN-γ in hypertension, other cell subtypes like LT CD8 and NK-T can also release this cytokine. Therefore, the association between the activation of the immune system and hypertension is clear.
The role of O-GlcNAcylation, as well as OGT, on T cells activation has been investigated before (121–123). One of the earliest studies in this field demonstrated that murine T lymphocytes activation resulted in decreased levels of O-GlcNAc-modified cytosolic proteins with a concomitant increase of this post-translational modification in the nucleus, establishing the role for protein O-GlcNAcylation in the early stages of T-cell activation (123). This was further supported by a study that found that blocking O-GlcNAc cycling disrupts early T cell development in mice (124).
The nuclear factor of activated T cells (NFAT) allows transcriptional induction and release of IL-2, as well as other cytokines such as IL-4, IFNγ, and TNFα, as observed in activated T cells (125). NFAT is a target for O-GlcNAc and silencing OGT through siRNA-mediated knockdown, impairs activation of NFAT and NFκB, reducing IL-2 production, and consequently preventing T cell receptors (TCR)-induced activation (121). Thus, OGT is required for T and B cell activation. These data was further supported by Lund et al. (126), who found that activation of T cells through the TCR resulted in a global elevation of O-GlcNAc levels. Yet, in the absence of O-GlcNAc, IL-2 production and T cell proliferation were compromised (126). Controversially, augmented O-GlcNAc levels in heart tissue and rat cardiomyocyte-derived cell line suppressed NFAT and NF-κB activity through GSK-3β protein O-GlcNAcylation (127). It seems that O-GlcNAcylation antagonizes NFAT effects, since GSK-3β is known to negatively regulate NFAT activity (128). Collectively, these data show that this post-translational modification is crucial for T cell activation, whereas aberrant protein O-GlcNAcylation may be deleterious.
Th17 and γδ T cell release IL-17, which is associated with hypertension. Kim et al. (129) demonstrated that transfer of Th17 cells from adult SHR accelerates the development of hypertension in juvenile SHR (129). Ang II infusion increases IL‐17A production by T cells and IL‐17 protein in the aortic media and the heart (130). Hypertension is not sustained in IL-17A-/- mice infused with Ang II. These mice are also protected against aortic stiffening and cardiac fibrosis (130, 131).
Regarding O-GlcNAcylation evoking immune response through T cell, Ramakrishnan et al. (87) demonstrated that hyperglycemia promoted NF-κB and c-Rel O-GlcNAcylation, promoting autoimmunity through enhancing the release of cytokines by helper Th cells (87). Elevated O-GlcNAc levels, through OGA inhibitor, correlate with increased Th17 and Th1 pro-inflammatory IL-17A and IFNγ cytokines secretion by murine and human CD4+ T cells (132). Additionally, Liu et al. (133) found that the microRNA-15b contributes to multiple sclerosis by negatively regulating Th17 cell differentiation, by targeting the OGT enzyme (133). Moreover, microRNA-15b suppressed retinoic acid-related orphan receptor (ROR)γT activation by diminishing NFκB protein O-GlcNAcylation. These findings provide evidence of the importance of O-GlcNAcylation in CD4+ T cells differentiation, since (ROR)γT is known to play a key role in the differentiation of Th17 cell lineage.
Possible Target: Regulatory T Cells (Treg)
Another specialized subpopulation of CD4+ T cells is the Treg. These cells play a key role in maintaining the homeostasis of the immune system, regulating the balance between pro-inflammatory and anti-inflammatory responses, and preventing autoimmune responses
In SHR, peripheral blood and splenic Treg cells are markedly diminished, whereas Th17 cells are enhanced (134). In fact, Treg have a protective effect in hypertension. A clinical study demonstrated that during hypertension, end organ damage and arterial stiffness in children is associated with decreased population of circulating Treg (135). Adoptive transfer of Treg prevents Ang II–induced hypertension, vascular damage, and vascular immune cell infiltration (114). Treg adoptive transfer also prevents kidney macrophages infiltration, vascular dysfunction and vascular oxidative stress induced by aldosterone (136).
Treg cells express high levels of IL-2 receptor α (IL-2Rα) chain in its surface and the forkhead box protein P3 (FOXP3) transcription factor in the nucleus, which are important for Treg function and cell fate (137, 138). O-GlcNAc-deficient mice due Treg cell-specific deletion of OGT, display reduced FOXP3 expression, impaired Treg function, and aggressive and lethal autoimmunity (139). Furthermore, deficiency in protein O-GlcNAcylation results in attenuation of IL-2/STAT5 activity in Treg cells. IL-2R activity is known to depend on STAT5 to regulate FOXP3 expression and promote Treg development, thus IL-2/STAT5 is required for FOXP3-induced differentiation of Treg (140). The same importance can be given to Th2 cells, once STAT5 activation is also crucial for Th2 differentiation (141).
IL-10 (STAT3/IL-10) is an anti-inflammatory cytokine released by Treg and other immune cells. IL-10 knockout mice infused with Ang II exhibit greater vascular contractions and IL-10 infusion prevents blood pressure increase in Ang II-infused mice (142). O-GlcNAcylation of signal transducer and activator of the transcription (STAT)-3 resulted in defective STAT3 phosphorylation and IL-10 production, affecting STAT3–IL-10 signaling in macrophages, increasing disease severity in colitis models while inhibition of OGT-mediated O-GlcNAcylation protects against intestinal inflammation (143). The STAT3–IL-10 axis is essential for an anti-inflammatory response, since STAT3 is a key transcriptional factor mediating IL-10 production (144, 145). Thus, STAT-3 protein O-GlcNAcylation impairs its activation, affecting STAT3–IL-10 anti-inflammatory responses. This is further supported by the fact that elevated protein O-GlcNAcylation enhances innate immune responses by increasing NF-κB signaling, and by counterbalancing the anti-inflammatory STAT3-IL-10 signaling in macrophages (82).
Possible Target: Cytotoxic T Cells
Once activated through TCR stimulation, T cells differentiate into cytotoxic effector cells (CD8+) and undergo clonal expansion and maturation to become activated CD8+ T cells (146). In addition to CD4 T cells, CD8 cells are also activated and increased in hypertension. Trott et al. (2014) reported that an oligoclonal population of CD8+ cells accumulate in the kidney and contribute to sodium retention and volume expansion, and vascular rarefaction during hypertension development (147). Resistant hypertension patients submitted to renal denervation and with blood pressure control show low values of CD4, CD8 and naïve CD8 T cells, leanding to the suggestion that T cells can be cellular biomarkers that predict the response to renal denervation (148).
CD8 T cells travel through the blood towards tissues looking for cells infected by pathogens, and inducing cell lysis and apoptosis in order to resolve the infection (149). This process involves many post-translational modifications such as phosphorylation and O-GlcNAcylation. Interestingly, protein O-GlcNAcylation is strongly involved in both transcriptional and translational processes that prompt T cells formation and proliferation, in effector-like T cells and memory-like T cells, establishing the role for O-GlcNAc in CD8+ T cells function (150).
Possible Target: B Cells
B cells are bone marrow-derived cells that play crucial functions in adaptive immunity, such as antibody production, antigen presentation, and cytokine production and release (151). In the bone marrow, progenitor B cells undergo pro-B, early pre-B, and late pre-B stages to become immature B cells (152). B cells also contribute to Ang II hypertension. B cells genetically deficient mice infused with Ang II present lower blood pressure and B cell transfer rescues this response. Knockout mice are also protected from Ang II-induced collagen deposition and aortic stiffening (117).
Augmented protein O-GlcNAcylation, by inhibiting the OGA enzyme, enhances B cell activation and apoptosis induced by B cell receptor (BCR). B cell-mediated apoptosis occurs through protein O-GlcNAcylation of lymphocyte-specific protein-1 (Lsp1), resulting in protein kinase C-β1-mediated increased levels of Lsp1 phosphorylation with consequent activation of apoptosis-related signaling (153). OGT-deleted mouse exhibit decreased number of mature B increased apoptosis in these cells and defective activation of the B-cell receptor signaling cascade (154), suggesting that O-GlcNAcylation is required for B cell homeostasis and antibody responses since an cells. Inhibition of O-GlcNAc in pre-B cells reduces growth and proliferation due to a decrease in c-Myc expression upon decreased O-GlcNAc, which is important for normal B cell proliferation and cell cycle progression (155). Collectively, these data highlight an important role for protein O-GlcNAcylation in regulating B cells homeostasis.
Conclusion
Since chronic increases in O-GlcNAcylation levels lead to increased risk of ventricular arrhythmias, myocardial dysfunction, cardiac remodeling, organ damage, aortic banding, and vascular dysfunction, all of them well-known mechanisms whereby blood pressure, it is plays this post-translational modification has been associated with arterial hypertension. On the other hand, recent efforts have been made to determine the role of immune response during high blood pressure and tissue damage in hypertension.
Considering that less than 10% of the patients know the primary cause of their hypertensive disease, one may speculate that the association between O-GlcNAcylation and the activation of the immune system may represent a new clinical approach to the treatment of hypertension. In this sense, current findings in the literature show the many players of innate and adaptive immunity, which are directly involved on vascular inflammation, vascular dysfunction, and hypertension are also targeted by O-GlcNAcylation. As an example, O-GlcNAc-pathway is able to modulate activity, production or pro-inflammatory environment of key component of innate immune response such as, Toll-like receptors, NF-κB pathway, circulating and endothelial cell adhering monocytes, macrophage infiltration and neutrophil lymphocyte ratio, players that are strongly association with the severity of hypertension. Furthermore, it seems clear that O-GlcNAcylation, as well as OGT, play an important role on T and B cell activation, immune cells that are enrolled in the hypertensive disease. Consequently, this post-translational modification modulates IL-2, as well as other cytokines such as IL-4, IFNγ, and TNFα, as observed in activated T cells. Besides, this modification is involved in antibody production, antigen presentation, cytokine production and release through B cell activation. Therefore, the investigation of new potential therapies, specifically aimed to modulate the impact of O-GlcNAc-modified proteins in the innate and adaptive immunity cells to treat or prevent hypertension should be further encouraged.
Author Contributions
VL, RT, and FG designed the review. RP, GB, FG, and VL wrote the manuscript. VL, FG, and RT revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Fundação de Amparo à Pesquisa do Estado de Mato Grosso (FAPEMAT, 003/2021 to VL); by Conselho Nacional de Desenvolvimento Cientı́fico e Tecnológico (CNPq, 141502/2020-7 to RP); the Conselho Nacional de Desenvolvimento Cientı́fico e Tecnológico (CNPq, 306166/2019-4 to FG) and São Paulo State Research Foundation (FAPESP; Grant 2013/08216–2 – Research Center in Inflammatory Diseases).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bourre G, Cantrelle FX, Kamah A, Chambraud B, Landrieu I, Smet-Nocca C. Direct Crosstalk Between O-GlcNAcylation and Phosphorylation of Tau Protein Investigated by NMR Spectroscopy. Front Endocrinol (Lausanne) (2018) 9:595. doi: 10.3389/fendo.2018.00595
2. Chatham JC, Zhang J, Wende AR. Role of O-Linked N-Acetylglucosamine Protein Modification in Cellular (Patho)Physiology. Physiol Rev (2021) 101(2):427–93. doi: 10.1152/physrev.00043.2019
3. Marshall S, Bacote V, Traxinger RR. Discovery of a Metabolic Pathway Mediating Glucose-Induced Desensitization of the Glucose Transport System. Role of Hexosamine Biosynthesis in the Induction of Insulin Resistance. J Biol Chem (1991) 266(8):4706–12. doi: 10.1016/S0021-9258(19)67706-9
4. Lima VV, Spitler K, Choi H, Webb RC, Tostes RC. O-GlcNAcylation and Oxidation of Proteins: Is Signalling in the Cardiovascular System Becoming Sweeter? Clin Sci (Lond) (2012) 123(8):473–86. doi: 10.1042/CS20110638
5. Souza-Silva L, Alves-Lopes R, Silva Miguez J, Dela Justina V, Neves KB, Mestriner FL, et al. Glycosylation With O-Linked Beta-N-Acetylglucosamine Induces Vascular Dysfunction via Production of Superoxide Anion/Reactive Oxygen Species. Can J Physiol Pharmacol (2018) 96(3):232–40. doi: 10.1139/cjpp-2017-0225
6. James LR, Ingram A, Ly H, Thai K, Cai L, Scholey JW. Angiotensin II Activates the GFAT Promoter in Mesangial Cells. Am J Physiol Renal Physiol (2001) 281(1):F151–62. doi: 10.1152/ajprenal.2001.281.1.F151
7. Salma W, Franekova V, Lund T, Hoper A, Ludvigsen S, Lund J, et al. Dietary Calanus Oil Antagonizes Angiotensin II-Induced Hypertension and Tissue Wasting in Diet-Induced Obese Mice. Prostaglandins Leukot Essent Fatty Acids (2016) 108:13–21. doi: 10.1016/j.plefa.2016.03.006
8. Dierschke SK, Toro AL, Barber AJ, Arnold AC, Dennis MD. Angiotensin-(1-7) Attenuates Protein O-GlcNAcylation in the Retina by EPAC/Rap1-Dependent Inhibition of O-GlcNAc Transferase. Invest Ophthalmol Vis Sci (2020) 61(2):24. doi: 10.1167/iovs.61.2.24
9. Lima VV, Giachini FR, Hardy DM, Webb RC, Tostes RC. O-GlcNAcylation: A Novel Pathway Contributing to the Effects of Endothelin in the Vasculature. Am J Physiol Regul Integr Comp Physiol (2011) 300(2):R236–50. doi: 10.1152/ajpregu.00230.2010
10. Lima VV, Giachini FR, Carneiro FS, Carneiro ZN, Saleh MA, Pollock DM, et al. O-GlcNAcylation Contributes to Augmented Vascular Reactivity Induced by Endothelin 1. Hypertension (2010) 55(1):180–8. doi: 10.1161/HYPERTENSIONAHA.109.143818
11. Lima VV, Giachini FR, Carneiro FS, Carvalho MH, Fortes ZB, Webb RC, et al. O-GlcNAcylation Contributes to the Vascular Effects of ET-1 via Activation of the RhoA/Rho-Kinase Pathway. Cardiovasc Res (2011) 89(3):614–22. doi: 10.1093/cvr/cvq338
12. Gellai R, Hodrea J, Lenart L, Hosszu A, Koszegi S, Balogh D, et al. Role of O-Linked N-Acetylglucosamine Modification in Diabetic Nephropathy. Am J Physiol Renal Physiol (2016) 311(6):F1172–81. doi: 10.1152/ajprenal.00545.2015
13. Chatham JC, Young ME, Zhang J. Role of O-Linked N-Acetylglucosamine (O-GlcNAc) Modification of Proteins in Diabetic Cardiovascular Complications. Curr Opin Pharmacol (2021) 57:1–12. doi: 10.1016/j.coph.2020.08.005
14. Rossier BC, Bochud M, Devuyst O. The Hypertension Pandemic: An Evolutionary Perspective. Physiol (Bethesda) (2017) 32(2):112–25. doi: 10.1152/physiol.00026.2016
15. Siddiqui M, Calhoun DA. Refractory Versus Resistant Hypertension. Curr Opin Nephrol Hypertens (2017) 26(1):14–9. doi: 10.1097/MNH.0000000000000286
16. Bomfim GF, Cau SBA, Bruno AS, Fedoce AG, Carneiro FS. Hypertension: A New Treatment for an Old Disease? Targeting the Immune System. Br J Pharmacol (2019) 176(12):2028–48. doi: 10.1111/bph.14436
17. Petrie JR, Guzik TJ, Touyz RM. Diabetes, Hypertension, and Cardiovascular Disease: Clinical Insights and Vascular Mechanisms. Can J Cardiol (2018) 34(5):575–84. doi: 10.1016/j.cjca.2017.12.005
18. Roshan MH, Tambo A, Pace NP. The Role of TLR2, TLR4, and TLR9 in the Pathogenesis of Atherosclerosis. Int J Inflam 2016 (2016) p:1532832. doi: 10.1155/2016/1532832
19. Dierschke SK, Dennis MD. Retinal Protein O-GlcNAcylation and the Ocular Renin Angiotensin System: Signaling Cross-Roads in Diabetic Retinopathy. Curr Diabetes Rev (2021) 18:9–23. doi: 10.2174/1573399817999210111205933
20. Silva-Aguiar RP, Bezerra NCF, Lucena MC, Sirtoli GM, Sudo RT, Zapata-Sudo G, et al. O-GlcNAcylation Reduces Proximal Tubule Protein Reabsorption and Promotes Proteinuria in Spontaneously Hypertensive Rats. J Biol Chem (2018) 293(33):12749–58. doi: 10.1074/jbc.RA118.001746
21. Lima VV, Giachini FR, Choi H, Carneiro FS, Carneiro ZN, Fortes ZB, et al. Impaired Vasodilator Activity in Deoxycorticosterone Acetate-Salt Hypertension is Associated With Increased Protein O-GlcNAcylation. Hypertension (2009) 53(2):166–74. doi: 10.1161/HYPERTENSIONAHA.108.116798
22. Lunde IG, Aronsen JM, Kvaloy H, Qvigstad E, Sjaastad I, Tonnessen T, et al. Cardiac O-GlcNAc Signaling Is Increased in Hypertrophy and Heart Failure. Physiol Genomics (2012) 44(2):162–72. doi: 10.1152/physiolgenomics.00016.2011
23. Mailleux F, Gelinas R, Beauloye C, Horman S, Bertrand L. O-GlcNAcylation, Enemy or Ally During Cardiac Hypertrophy Development? Biochim Biophys Acta (2016) 1862(12):2232–43. doi: 10.1016/j.bbadis.2016.08.012
24. Vaidyanathan K, Durning S, Wells L. Functional O-GlcNAc Modifications: Implications in Molecular Regulation and Pathophysiology. Crit Rev Biochem Mol Biol (2014) 49(2):140–63. doi: 10.3109/10409238.2014.884535
25. Ng YH, Okolo CA, Erickson JR, Baldi JC, Jones PP. Protein O-GlcNAcylation in the Heart. Acta Physiol (Oxf) (2021) 233(1):e13696. doi: 10.1111/apha.13696
26. Yu P, Hu L, Xie J, Chen S, Huang L, Xu Z, et al. O-GlcNAcylation of Cardiac Nav1.5 Contributes to the Development of Arrhythmias in Diabetic Hearts. Int J Cardiol (2018) 260:74–81. doi: 10.1016/j.ijcard.2018.02.099
27. Lima VV, Rigsby CS, Hardy DM, Webb RC, Tostes RC. O-GlcNAcylation: A Novel Post-Translational Mechanism to Alter Vascular Cellular Signaling in Health and Disease: Focus on Hypertension. J Am Soc Hypertens (2009) 3(6):374–87. doi: 10.1016/j.jash.2009.09.004
28. Fulop N, Mason MM, Dutta K, Wang P, Davidoff AJ, Marchase RB, et al. Impact of Type 2 Diabetes and Aging on Cardiomyocyte Function and O-Linked N-Acetylglucosamine Levels in the Heart. Am J Physiol Cell Physiol (2007) 292(4):C1370–8. doi: 10.1152/ajpcell.00422.2006
29. Cicalese SM, da Silva JF, Priviero F, Webb RC, Eguchi S, Tostes RC. Vascular Stress Signaling in Hypertension. Circ Res (2021) 128(7):969–92. doi: 10.1161/CIRCRESAHA.121.318053
30. Yan K, Wang K, Li P. The Role of Post-Translational Modifications in Cardiac Hypertrophy. J Cell Mol Med (2019) 23(6):3795–807. doi: 10.1111/jcmm.14330
31. Gelinas R, Mailleux F, Dontaine J, Bultot L, Demeulder B, Ginion A, et al. AMPK Activation Counteracts Cardiac Hypertrophy by Reducing O-GlcNAcylation. Nat Commun (2018) 9(1):374. doi: 10.1038/s41467-017-02795-4
32. Cannon MV, Sillje HH, Sijbesma JW, Vreeswijk-Baudoin I, Ciapaite J, van der Sluis B, et al. Cardiac LXRalpha Protects Against Pathological Cardiac Hypertrophy and Dysfunction by Enhancing Glucose Uptake and Utilization. EMBO Mol Med (2015) 7(9):1229–43. doi: 10.15252/emmm.201404669
33. Facundo HT, Brainard RE, Watson LJ, Ngoh GA, Hamid T, Prabhu SD, et al. O-GlcNAc Signaling Is Essential for NFAT-Mediated Transcriptional Reprogramming During Cardiomyocyte Hypertrophy. Am J Physiol Heart Circ Physiol (2012) 302(10):H2122–30. doi: 10.1152/ajpheart.00775.2011
34. Chen Y, Su X, Qin Q, Yu Y, Jia M, Zhang H, et al. New Insights Into Phenotypic Switching of VSMCs Induced by Hyperhomocysteinemia: Role of Endothelin-1 Signaling. BioMed Pharmacother (2020) 123:109758. doi: 10.1016/j.biopha.2019.109758
35. Schiffrin EL. Does Endothelin-1 Raise or Lower Blood Pressure in Humans? Nephron (2018) 139(1):47–50. doi: 10.1159/000487346
36. Goto K, Hama H, Kasuya Y. Molecular Pharmacology and Pathophysiological Significance of Endothelin. Jpn J Pharmacol (1996) 72(4):261–90. doi: 10.1254/jjp.72.261
37. Park MJ, Kim DI, Lim SK, Choi JH, Han HJ, Yoon KC, et al. High Glucose-Induced O-GlcNAcylated Carbohydrate Response Element-Binding Protein (ChREBP) Mediates Mesangial Cell Lipogenesis and Fibrosis: The Possible Role in the Development of Diabetic Nephropathy. J Biol Chem (2014) 289(19):13519–30. doi: 10.1074/jbc.M113.530139
38. Degrell P, Cseh J, Mohas M, Molnar GA, Pajor L, Chatham JC, et al. Evidence of O-Linked N-Acetylglucosamine in Diabetic Nephropathy. Life Sci (2009) 84(13-14):389–93. doi: 10.1016/j.lfs.2009.01.007
39. Cao Y, Wang Q, Liu C, Wang W, Lai S, Zou H, et al. Capn4 Aggravates Angiotensin II-Induced Cardiac Hypertrophy by Activating the IGF-AKT Signaling Pathway. J Biochem 171(1):53–61 (2021). doi: 10.1093/jb/mvab100
40. Ayyadevara S, Mercanti F, Wang X, Mackintosh SG, Tackett AJ, Prayaga SV, et al. Age- and Hypertension-Associated Protein Aggregates in Mouse Heart Have Similar Proteomic Profiles. Hypertension (2016) 67(5):1006–13. doi: 10.1161/HYPERTENSIONAHA.115.06849
41. Cheema MU, Poulsen ET, Enghild JJ, Hoorn EJ, Fenton RA, Praetorius J, et al. Aldosterone and Angiotensin II Induce Protein Aggregation in Renal Proximal Tubules. Physiol Rep (2013) 1(4):e00064. doi: 10.1002/phy2.64
42. Tanaka T, Sohmiya K, Kono T, Terasaki F, Horie R, Ohkaru Y, et al. Thiamine Attenuates the Hypertension and Metabolic Abnormalities in CD36-Defective SHR: Uncoupling of Glucose Oxidation From Cellular Entry Accompanied With Enhanced Protein O-GlcNAcylation in CD36 Deficiency. Mol Cell Biochem (2007) 299(1-2):23–35. doi: 10.1007/s11010-005-9032-3
43. Einstein FH, Fishman S, Bauman J, Thompson RF, Huffman DM, Atzmon G, et al. Enhanced Activation of a “Nutrient-Sensing” Pathway With Age Contributes to Insulin Resistance. FASEB J (2008) 22(10):3450–7. doi: 10.1096/fj.08-109041
44. Federici M, Menghini R, Mauriello A, Hribal ML, Ferrelli F, Lauro D, et al. Insulin-Dependent Activation of Endothelial Nitric Oxide Synthase is Impaired by O-Linked Glycosylation Modification of Signaling Proteins in Human Coronary Endothelial Cells. Circulation (2002) 106(4):466–72. doi: 10.1161/01.CIR.0000023043.02648.51
45. Akimoto Y, Kreppel LK, Hirano H, Hart GW. Hyperglycemia and the O-GlcNAc Transferase in Rat Aortic Smooth Muscle Cells: Elevated Expression and Altered Patterns of O-GlcNAcylation. Arch Biochem Biophys (2001) 389(2):166–75. doi: 10.1006/abbi.2001.2331
46. Ganguly R, Sahu S, Chavez RJ, Raman P. Trivalent Chromium Inhibits TSP-1 Expression, Proliferation, and O-GlcNAc Signaling in Vascular Smooth Muscle Cells in Response to High Glucose In Vitro. Am J Physiol Cell Physiol (2015) 308(2):C111–22. doi: 10.1152/ajpcell.00256.2014
47. Du XL, Edelstein D, Dimmeler S, Ju Q, Sui C, Brownlee M. Hyperglycemia Inhibits Endothelial Nitric Oxide Synthase Activity by Posttranslational Modification at the Akt Site. J Clin Invest (2001) 108(9):1341–8. doi: 10.1172/JCI11235
48. Makino A, Dai A, Han Y, Youssef KD, Wang W, Donthamsetty R, et al. O-GlcNAcase Overexpression Reverses Coronary Endothelial Cell Dysfunction in Type 1 Diabetic Mice. Am J Physiol Cell Physiol (2015) 309(9):C593–9. doi: 10.1152/ajpcell.00069.2015
49. Masaki N, Feng B, Breton-Romero R, Inagaki E, Weisbrod RM, Fetterman JL, et al. O-GlcNAcylation Mediates Glucose-Induced Alterations in Endothelial Cell Phenotype in Human Diabetes Mellitus. J Am Heart Assoc (2020) 9(12):e014046. doi: 10.1161/JAHA.119.014046
50. Lima VV, Lobato NS, Filgueira FP, Webb RC, Tostes RC, Giachini FR. Vascular O-GlcNAcylation Augments Reactivity to Constrictor Stimuli by Prolonging Phosphorylated Levels of the Myosin Light Chain. Braz J Med Biol Res (2014) 47(10):826–33. doi: 10.1590/1414-431X20144001
51. Kim DH, Seok YM, Kim IK, Lee IK, Jeong SY, Jeoung NH. Glucosamine Increases Vascular Contraction Through Activation of RhoA/Rho Kinase Pathway in Isolated Rat Aorta. BMB Rep (2011) 44(6):415–20. doi: 10.5483/BMBRep.2011.44.6.415
52. Meigs JB, Larson MG, D'Agostino RB, Levy D, Clouse ME, Nathan DM, et al. Coronary Artery Calcification in Type 2 Diabetes and Insulin Resistance: The Framingham Offspring Study. Diabetes Care (2002) 25(8):1313–9. doi: 10.2337/diacare.25.8.1313
53. Heath JM, Sun Y, Yuan K, Bradley WE, Litovsky S, Dell'Italia LJ, et al. Activation of AKT by O-Linked N-Acetylglucosamine Induces Vascular Calcification in Diabetes Mellitus. Circ Res (2014) 114(7):1094–102. doi: 10.1161/CIRCRESAHA.114.302968
54. Byon CH, Kim SW. Regulatory Effects of O-GlcNAcylation in Vascular Smooth Muscle Cells on Diabetic Vasculopathy. J Lipid Atheroscler (2020) 9(2):243–54. doi: 10.12997/jla.2020.9.2.243
55. Bomfim GF, Rodrigues FL, Carneiro FS. Are the Innate and Adaptive Immune Systems Setting Hypertension on Fire? Pharmacol Res (2017) 117:377–93. doi: 10.1016/j.phrs.2017.01.010
56. Norlander AE, Madhur MS, Harrison DG. The Immunology of Hypertension. J Exp Med (2018) 215(1):21–33. doi: 10.1084/jem.20171773
57. Matzinger P. An Innate Sense of Danger. Ann N Y Acad Sci (2002) 961:341–2. doi: 10.1111/j.1749-6632.2002.tb03118.x
58. Bomfim G, Szasz T, Carvalho M, Webb RJ. The Toll Way to Hypertension: Role of the Innate Immune Response. Endocrinol Metabol Syndrome S (2011) 8:2161–1017. doi: 10.4172/2161-1017.1000117
59. Dela Justina V, Giachini FR, Sullivan JC, Webb RC. Toll-Like Receptors Contribute to Sex Differences in Blood Pressure Regulation. J Cardiovasc Pharmacol (2020) 76(3):255–66. doi: 10.1097/FJC.0000000000000869
60. Bomfim GF, Echem C, Martins CB, Costa TJ, Sartoretto SM, Dos Santos RA, et al. Toll-Like Receptor 4 Inhibition Reduces Vascular Inflammation in Spontaneously Hypertensive Rats. Life Sci (2015) 122:1–7. doi: 10.1016/j.lfs.2014.12.001
61. Bomfim GF, Dos Santos RA, Oliveira MA, Giachini FR, Akamine EH, Bomfim RC, et al. Toll-Like Receptor 4 Contributes to Blood Pressure Regulation and Vascular Contraction in Spontaneously Hypertensive Rats. Clin Sci (Lond) (2012) 122(11):535–43. doi: 10.1042/CS20110523
62. Biancardi VC, Bomfim GF, Reis WL, Al-Gassimi S, Nunes KP. The Interplay Between Angiotensin II, TLR4 and Hypertension. Pharmacol Res (2017) 120:88–96. doi: 10.1016/j.phrs.2017.03.017
63. Echem C, Bomfim GF, Ceravolo GS, Oliveira MA, Santos-Eichler RA, Bechara LR, et al. Anti-Toll Like Receptor 4 (TLR4) Therapy Diminishes Cardiac Remodeling Regardless of Changes in Blood Pressure in Spontaneously Hypertensive Rats (SHR). Int J Cardiol (2015) 187:243–5. doi: 10.1016/j.ijcard.2015.03.190
64. Qi HM, Cao Q, Liu Q. TLR4 Regulates Vascular Smooth Muscle Cell Proliferation in Hypertension via Modulation of the NLRP3 Inflammasome. Am J Transl Res (2021) 13(1):314–25.
65. Goulopoulou S, Matsumoto T, Bomfim GF, Webb RC. Toll-Like Receptor 9 Activation: A Novel Mechanism Linking Placenta-Derived Mitochondrial DNA and Vascular Dysfunction in Pre-Eclampsia. Clin Sci (Lond) (2012) 123(7):429–35. doi: 10.1042/CS20120130
66. McCarthy CG, Wenceslau CF, Goulopoulou S, Ogbi S, Baban B, Sullivan JC, et al. Circulating Mitochondrial DNA and Toll-Like Receptor 9 Are Associated With Vascular Dysfunction in Spontaneously Hypertensive Rats. Cardiovasc Res (2015) 107(1):119–30. doi: 10.1093/cvr/cvv137
67. Echem C, Costa TJD, Oliveira V, Giglio Colli L, Landgraf MA, Rodrigues SF, et al. Mitochondrial DNA: A New Driver for Sex Differences in Spontaneous Hypertension. Pharmacol Res (2019) 144:142–50. doi: 10.1016/j.phrs.2019.04.008
68. Rodrigues FL, Silva LE, Hott SC, Bomfim GF, da Silva CA, Fazan R Jr, et al. Toll-Like Receptor 9 Plays a Key Role in the Autonomic Cardiac and Baroreflex Control of Arterial Pressure. Am J Physiol Regul Integr Comp Physiol (2015) 308(8):R714–23. doi: 10.1152/ajpregu.00150.2014
69. Crowley SD, Song YS, Sprung G, Griffiths R, Sparks M, Yan M, et al. A Role for Angiotensin II Type 1 Receptors on Bone Marrow-Derived Cells in the Pathogenesis of Angiotensin II-Dependent Hypertension. Hypertension (2010) 55(1):99–108. doi: 10.1161/HYPERTENSIONAHA.109.144964
70. Huang L, Wang A, Hao Y, Li W, Liu C, Yang Z, et al. Macrophage Depletion Lowered Blood Pressure and Attenuated Hypertensive Renal Injury and Fibrosis. Front Physiol (2018) 9:473. doi: 10.3389/fphys.2018.00473
71. Clozel M, Kuhn H, Hefti F, Baumgartner HR. Endothelial Dysfunction and Subendothelial Monocyte Macrophages in Hypertension. Effect of Angiotensin Converting Enzyme Inhibition. Hypertension (1991) 18(2):132–41.doi: 10.1161/01.hyp.18.2.132
72. Hartner A, Porst M, Gauer S, Prols F, Veelken R, Hilgers KF. Glomerular Osteopontin Expression and Macrophage Infiltration in Glomerulosclerosis of DOCA-Salt Rats. Am J Kidney Dis (2001) 38(1):153–64. doi: 10.1053/ajkd.2001.25209
73. Asagami T, Reaven GM, Tsao PS. Enhanced Monocyte Adherence to Thoracic Aortae From Rats With Two Forms of Experimental Hypertension. Am J Hypertens (1999) 12(9 Pt 1):890–3. doi: 10.1016/S0895-7061(99)00072-2
74. Wirtz PH, von Kanel R, Frey K, Ehlert U, Fischer JE. Glucocorticoid Sensitivity of Circulating Monocytes in Essential Hypertension. Am J Hypertens (2004) 17(6):489–94. doi: 10.1016/j.amjhyper.2004.01.010
75. Dorffel Y, Latsch C, Stuhlmuller B, Schreiber S, Scholze S, Burmester GR, et al. Preactivated Peripheral Blood Monocytes in Patients With Essential Hypertension. Hypertension (1999) 34(1):113–7. doi: 10.1161/01.HYP.34.1.113
76. Hilgers KF, Hartner A, Porst M, Mai M, Wittmann M, Hugo C, et al. Monocyte Chemoattractant Protein-1 and Macrophage Infiltration in Hypertensive Kidney Injury. Kidney Int (2000) 58(6):2408–19. doi: 10.1046/j.1523-1755.2000.00424.x
77. Hoppstadter J, Dembek A, Linnenberger R, Dahlem C, Barghash A, Fecher-Trost C, et al. Toll-Like Receptor 2 Release by Macrophages: An Anti-Inflammatory Program Induced by Glucocorticoids and Lipopolysaccharide. Front Immunol (2019) 10:1634. doi: 10.3389/fimmu.2019.01634
78. Lo PW, Shie JJ, Chen CH, Wu CY, Hsu TL, Wong CH. O-GlcNAcylation Regulates the Stability and Enzymatic Activity of the Histone Methyltransferase EZH2. Proc Natl Acad Sci USA (2018) 115(28):7302–7. doi: 10.1073/pnas.1801850115
79. Jiang M, Xu B, Li X, Shang Y, Chu Y, Wang W, et al. O-GlcNAcylation Promotes Colorectal Cancer Metastasis via the miR-101-O-GlcNAc/EZH2 Regulatory Feedback Circuit. Oncogene (2019) 38(3):301–16. doi: 10.1038/s41388-018-0435-5
80. Justin Rucker A, Crowley SD. The Role of Macrophages in Hypertension and Its Complications. Pflugers Arch (2017) 469(3-4):419–30. doi: 10.1007/s00424-017-1950-x
81. De Ciuceis C, Amiri F, Brassard P, Endemann DH, Touyz RM, Schiffrin EL. Reduced Vascular Remodeling, Endothelial Dysfunction, and Oxidative Stress in Resistance Arteries of Angiotensin II-Infused Macrophage Colony-Stimulating Factor-Deficient Mice: Evidence for a Role in Inflammation in Angiotensin-Induced Vascular Injury. Arterioscler Thromb Vasc Biol (2005) 25(10):2106–13. doi: 10.1161/01.ATV.0000181743.28028.57
82. Li X, Gong W, Wang H, Li T, Attri KS, Lewis RE, et al. O-GlcNAc Transferase Suppresses Inflammation and Necroptosis by Targeting Receptor-Interacting Serine/Threonine-Protein Kinase 3. Immunity (2019) 50(3):576–590 e6. doi: 10.1016/j.immuni.2019.01.007
83. Ryu IH, Do SI. Denitrosylation of S-Nitrosylated OGT Is Triggered in LPS-Stimulated Innate Immune Response. Biochem Biophys Res Commun (2011) 408(1):52–7. doi: 10.1016/j.bbrc.2011.03.115
84. Rodriguez-Iturbe B, Ferrebuz A, Vanegas V, Quiroz Y, Mezzano S, Vaziri ND. Early and Sustained Inhibition of Nuclear Factor-kappaB Prevents Hypertension in Spontaneously Hypertensive Rats. J Pharmacol Exp Ther (2005) 315(1):51–7. doi: 10.1124/jpet.105.088062
85. Yang WH, Park SY, Nam HW, Kim DH, Kang JG, Kang ES, et al. NFkappaB Activation Is Associated With Its O-GlcNAcylation State Under Hyperglycemic Conditions. Proc Natl Acad Sci USA (2008) 105(45):17345–50. doi: 10.1073/pnas.0806198105
86. Allison DF, Wamsley JJ, Kumar M, Li D, Gray LG, Hart GW, et al. Modification of RelA by O-Linked N-Acetylglucosamine Links Glucose Metabolism to NF-kappaB Acetylation and Transcription. Proc Natl Acad Sci USA (2012) 109(42):16888–93. doi: 10.1073/pnas.1208468109
87. Ramakrishnan P, Clark PM, Mason DE, Peters EC, Hsieh-Wilson LC, Baltimore D. Activation of the Transcriptional Function of the NF-kappaB Protein C-Rel by O-GlcNAc Glycosylation. Sci Signal (2013) 6(290):ra75. doi: 10.1126/scisignal.2004097
88. Hwang SY, Hwang SY, Kim SY, Han IO. O-GlcNAcylation and P50/P105 Binding of C-Rel Are Dynamically Regulated by LPS and Glucosamine in BV2 Microglia Cells. Br J Pharmacol (2013) 169(7):1551–60. doi: 10.1111/bph.12223
89. Hwang SY, Hwang SY, Kim SY, Han IO. O-GlcNAc Transferase Inhibits LPS-Mediated Expression of Inducible Nitric Oxide Synthase Through an Increased Interaction With Msin3a in RAW264.7 Cells. Am J Physiol Cell Physiol (2013) 305(6):C601–8. doi: 10.1152/ajpcell.00042.2013
90. Cimen T, Sunman H, Efe TH, Erat M, Sahan HF, Algul E, et al. The Relationship Between 24-Hour Ambulatory Blood Pressure Load and Neutrophil-to-Lymphocyte Ratio. Rev Port Cardiol (2017) 36(2):97–105. doi: 10.1016/j.repc.2016.07.009
91. Sunbul M, Gerin F, Durmus E, Kivrak T, Sari I, Tigen K, et al. Neutrophil to Lymphocyte and Platelet to Lymphocyte Ratio in Patients With Dipper Versus Non-Dipper Hypertension. Clin Exp Hypertens (2014) 36(4):217–21. doi: 10.3109/10641963.2013.804547
92. Liu X, Zhang Q, Wu H, Du H, Liu L, Shi H, et al. Blood Neutrophil to Lymphocyte Ratio as a Predictor of Hypertension. Am J Hypertens (2015) 28(11):1339–46. doi: 10.1093/ajh/hpv034
93. Dagli N, Dogdu O, Senarslan O, Yucel H, Hakki K, Akpek M, et al. Relation of Neutrophil/Lymphocyte Ratio to Resistant Hypertension. Eur Res J (2016) 2(3):211–8. doi: 10.18621/eurj.2016.5000185856
94. Mayadas TN, Cullere X, Lowell CA. The Multifaceted Functions of Neutrophils. Annu Rev Pathol (2014) 9:181–218. doi: 10.1146/annurev-pathol-020712-164023
95. Nuzzi PA, Lokuta MA, Huttenlocher A. Analysis of Neutrophil Chemotaxis. Methods Mol Biol (2007) 370:23–36. doi: 10.1007/978-1-59745-353-0_3
96. Kneass ZT, Marchase RB. Neutrophils Exhibit Rapid Agonist-Induced Increases in Protein-Associated O-GlcNAc. J Biol Chem (2004) 279(44):45759–65. doi: 10.1074/jbc.M407911200
97. Kneass ZT, Marchase RB. Protein O-GlcNAc Modulates Motility-Associated Signaling Intermediates in Neutrophils. J Biol Chem (2005) 280(15):14579–85. doi: 10.1074/jbc.M414066200
98. Kim D, Haynes CL. The Role of P38 MAPK in Neutrophil Functions: Single Cell Chemotaxis and Surface Marker Expression. Analyst (2013) 138(22):6826–33. doi: 10.1039/c3an01076g
99. Hussain M, Saeed M, Babar MZM, Atif MA, Akhtar L. Nebivolol Attenuates Neutrophil Lymphocyte Ratio: A Marker of Subclinical Inflammation in Hypertensive Patients. Int J Hypertens 2017 (2017) p:7643628. doi: 10.1155/2017/7643628
100. Freud AG, Mundy-Bosse BL, Yu J, Caligiuri MA. The Broad Spectrum of Human Natural Killer Cell Diversity. Immunity (2017) 47(5):820–33. doi: 10.1016/j.immuni.2017.10.008
101. Schreckenberg R, Wolf A, Troidl C, Simsekyilmaz S, Schluter KD. Pro-Inflammatory Vascular Stress in Spontaneously Hypertensive Rats Associated With High Physical Activity Cannot Be Attenuated by Aldosterone Blockade. Front Cardiovasc Med (2021) 8:699283. doi: 10.3389/fcvm.2021.699283
102. Kossmann S, Schwenk M, Hausding M, Karbach SH, Schmidgen MI, Brandt M, et al. Angiotensin II-Induced Vascular Dysfunction Depends on Interferon-Gamma-Driven Immune Cell Recruitment and Mutual Activation of Monocytes and NK-Cells. Arterioscler Thromb Vasc Biol (2013) 33(6):1313–9. doi: 10.1161/ATVBAHA.113.301437
103. Delaney JAC, Olson NC, Sitlani CM, Fohner AE, Huber SA, Landay AL, et al. Natural Killer Cells, Gamma Delta T Cells and Classical Monocytes Are Associated With Systolic Blood Pressure in the Multi-Ethnic Study of Atherosclerosis (MESA). BMC Cardiovasc Disord (2021) 21(1):45. doi: 10.1186/s12872-021-01857-2
104. Wensveen FM, Jelencic V, Polic B. NKG2D: A Master Regulator of Immune Cell Responsiveness. Front Immunol (2018) 9:441. doi: 10.3389/fimmu.2018.00441
105. Yin J, Leavenworth JW, Li Y, Luo Q, Xie H, Liu X, et al. Ezh2 Regulates Differentiation and Function of Natural Killer Cells Through Histone Methyltransferase Activity. Proc Natl Acad Sci USA (2015) 112(52):15988–93. doi: 10.1073/pnas.1521740112
106. Chu CS, Lo PW, Yeh YH, Hsu PH, Peng SH, Teng YC, et al. O-GlcNAcylation Regulates EZH2 Protein Stability and Function. Proc Natl Acad Sci USA (2014) 111(4):1355–60. doi: 10.1073/pnas.1323226111
107. Krzewski K, Coligan JE. Human NK Cell Lytic Granules and Regulation of Their Exocytosis. Front Immunol (2012) 3:335. doi: 10.3389/fimmu.2012.00335
108. Li C, Ge B, Nicotra M, Stern JN, Kopcow HD, Chen X, et al. JNK MAP Kinase Activation Is Required for MTOC and Granule Polarization in NKG2D-Mediated NK Cell Cytotoxicity. Proc Natl Acad Sci USA (2008) 105(8):3017–22. doi: 10.1073/pnas.0712310105
109. Trotta R, Fettucciari K, Azzoni L, Abebe B, Puorro KA, Eisenlohr LC, et al. Differential Role of P38 and C-Jun N-Terminal Kinase 1 Mitogen-Activated Protein Kinases in NK Cell Cytotoxicity. J Immunol (2000) 165(4):1782–9. doi: 10.4049/jimmunol.165.4.1782
110. Yao AY, Tang HY, Wang Y, Feng MF, Zhou RL. Inhibition of the Activating Signals in NK92 Cells by Recombinant GST-sHLA-G1a Chain. Cell Res (2004) 14(2):155–60. doi: 10.1038/sj.cr.7290215
111. Bozic J, Stoka V, Dolenc I. Glucosamine Prevents Polarization of Cytotoxic Granules in NK-92 Cells by Disturbing FOXO1/ERK/paxillin Phosphorylation. PloS One (2018) 13(7):e0200757. doi: 10.1371/journal.pone.0200757
112. Deng Y, Kerdiles Y, Chu J, Yuan S, Wang Y, Chen X, et al. Transcription Factor Foxo1 Is a Negative Regulator of Natural Killer Cell Maturation and Function. Immunity (2015) 42(3):457–70. doi: 10.1016/j.immuni.2015.02.006
113. Mikolajczyk TP, Guzik TJ. Adaptive Immunity in Hypertension. Curr Hypertens Rep (2019) 21(9):68. doi: 10.1007/s11906-019-0971-6
114. Barhoumi T, Kasal DA, Li MW, Shbat L, Laurant P, Neves MF, et al. T Regulatory Lymphocytes Prevent Angiotensin II-Induced Hypertension and Vascular Injury. Hypertension (2011) 57(3):469–76. doi: 10.1161/HYPERTENSIONAHA.110.162941
115. Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, et al. Role of the T Cell in the Genesis of Angiotensin II Induced Hypertension and Vascular Dysfunction. J Exp Med (2007) 204(10):2449–60. doi: 10.1084/jem.20070657
116. Crowley SD, Song YS, Lin EE, Griffiths R, Kim HS, Ruiz P. Lymphocyte Responses Exacerbate Angiotensin II-Dependent Hypertension. Am J Physiol Regul Integr Comp Physiol (2010) 298(4):R1089–97. doi: 10.1152/ajpregu.00373.2009
117. Chan CT, Sobey CG, Lieu M, Ferens D, Kett MM, Diep H, et al. Obligatory Role for B Cells in the Development of Angiotensin II-Dependent Hypertension. Hypertension (2015) 66(5):1023–33. doi: 10.1161/HYPERTENSIONAHA.115.05779
118. Zhang JD, Patel MB, Song YS, Griffiths R, Burchette J, Ruiz P, et al. A Novel Role for Type 1 Angiotensin Receptors on T Lymphocytes to Limit Target Organ Damage in Hypertension. Circ Res (2012) 110(12):1604–17. doi: 10.1161/CIRCRESAHA.111.261768
119. Shao J, Nangaku M, Miyata T, Inagi R, Yamada K, Kurokawa K, et al. Imbalance of T-Cell Subsets in Angiotensin II-Infused Hypertensive Rats With Kidney Injury. Hypertension (2003) 42(1):31–8. doi: 10.1161/01.HYP.0000075082.06183.4E
120. Kamat NV, Thabet SR, Xiao L, Saleh MA, Kirabo A, Madhur MS, et al. Renal Transporter Activation During Angiotensin-II Hypertension is Blunted in Interferon-Gamma-/- and Interleukin-17A-/- Mice. Hypertension (2015) 65(3):569–76. doi: 10.1161/HYPERTENSIONAHA.114.04975
121. Golks A, Tran TT, Goetschy JF, Guerini D. Requirement for O-Linked N-Acetylglucosaminyltransferase in Lymphocytes Activation. EMBO J (2007) 26(20):4368–79. doi: 10.1038/sj.emboj.7601845
122. Swamy M, Pathak S, Grzes KM, Damerow S, Sinclair LV, van Aalten DM, et al. Glucose and Glutamine Fuel Protein O-GlcNAcylation to Control T Cell Self-Renewal and Malignancy. Nat Immunol (2016) 17(6):712–20. doi: 10.1038/ni.3439
123. Kearse KP, Hart GW. Lymphocyte Activation Induces Rapid Changes in Nuclear and Cytoplasmic Glycoproteins. Proc Natl Acad Sci USA (1991) 88(5):1701–5. doi: 10.1073/pnas.88.5.1701
124. Abramowitz LK, et al. Blocked O-GlcNAc Cycling Disrupts Mouse Hematopoeitic Stem Cell Maintenance and Early T Cell Development. Sci Rep (2019) 9(1):12569. doi: 10.1038/s41598-019-48991-8
125. Chow CW, Rincon M, Davis RJ. Requirement for Transcription Factor NFAT in Interleukin-2 Expression. Mol Cell Biol (1999) 19(3):2300–7. doi: 10.1128/MCB.19.3.2300
126. Lund PJ, Elias JE, Davis MM. Global Analysis of O-GlcNAc Glycoproteins in Activated Human T Cells. J Immunol (2016) 197(8):3086–98. doi: 10.4049/jimmunol.1502031
127. Nakagawa T, Furukawa Y, Hayashi T, Nomura A, Yokoe S, Moriwaki K, et al. Augmented O-GlcNAcylation Attenuates Intermittent Hypoxia-Induced Cardiac Remodeling Through the Suppression of NFAT and NF-kappaB Activities in Mice. Hypertens Res (2019) 42(12):1858–71. doi: 10.1038/s41440-019-0311-x
128. Beals CR, et al. Nuclear Export of NF-ATc Enhanced by Glycogen Synthase Kinase-3. Science (1997) 275(5308):1930–4. doi: 10.1126/science.275.5308.1930
129. Kim JY, Lee E, Koo S, Kim CW, Kim I. Transfer of Th17 From Adult Spontaneous Hypertensive Rats Accelerates Development of Hypertension in Juvenile Spontaneous Hypertensive Rats. BioMed Res Int 2021 (2021) p:6633825. doi: 10.1155/2021/6633825
130. Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, et al. Interleukin 17 Promotes Angiotensin II-Induced Hypertension and Vascular Dysfunction. Hypertension (2010) 55(2):500–7. doi: 10.1161/HYPERTENSIONAHA.109.145094
131. Wu J, Thabet SR, Kirabo A, Trott DW, Saleh MA, Xiao L, et al. Inflammation and Mechanical Stretch Promote Aortic Stiffening in Hypertension Through Activation of P38 Mitogen-Activated Protein Kinase. Circ Res (2014) 114(4):616–25. doi: 10.1161/CIRCRESAHA.114.302157
132. Machacek M, Saunders H, Zhang Z, Tan EP, Li J, Li T, et al. Elevated O-GlcNAcylation Enhances Pro-Inflammatory Th17 Function by Altering the Intracellular Lipid Microenvironment. J Biol Chem (2019) 294(22):8973–90. doi: 10.1074/jbc.RA119.008373
133. Liu R, Ma X, Chen L, Yang Y, Zeng Y, Gao J, et al. MicroRNA-15b Suppresses Th17 Differentiation and Is Associated With Pathogenesis of Multiple Sclerosis by Targeting O-GlcNAc Transferase. J Immunol (2017) 198(7):2626–39. doi: 10.4049/jimmunol.1601727
134. Ni X, Li XZ, Fan ZR, Wang A, Zhang L, et al. Increased Expression and Functionality of the Gap Junction in Peripheral Blood Lymphocytes Is Associated With Hypertension-Mediated Inflammation in Spontaneously Hypertensive Rats. Cell Mol Biol Lett (2018) 23:40. doi: 10.1186/s11658-018-0106-0
135. Gackowska L, Michalkiewicz J, Helmin-Basa A, Klosowski M, Niemirska A, Obrycki L, et al. Regulatory T-Cell Subset Distribution in Children With Primary Hypertension Is Associated With Hypertension Severity and Hypertensive Target Organ Damage. J Hypertens (2020) 38(4):692–700. doi: 10.1097/HJH.0000000000002328
136. Kasal DA, Barhoumi T, Li MW, Yamamoto N, Zdanovich E, Rehman A, et al. T Regulatory Lymphocytes Prevent Aldosterone-Induced Vascular Injury. Hypertension (2012) 59(2):324–30. doi: 10.1161/HYPERTENSIONAHA.111.181123
137. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic Self-Tolerance Maintained by Activated T Cells Expressing IL-2 Receptor Alpha-Chains (CD25). Breakdown of a Single Mechanism of Self-Tolerance Causes Various Autoimmune Diseases. J Immunol (1995) 155(3):1151–64.
138. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 Programs the Development and Function of CD4+CD25+ Regulatory T Cells. Nat Immunol (2003) 4(4):330–6. doi: 10.1038/ni904
139. Liu B, Salgado OC, Singh S, Hippen KL, Maynard JC, Burlingame AL, et al. The Lineage Stability and Suppressive Program of Regulatory T Cells Require Protein O-GlcNAcylation. Nat Commun (2019) 10(1):354. doi: 10.1038/s41467-019-08300-3
140. Burchill MA, Yang J, Vogtenhuber C, Blazar BR, Farrar MA. IL-2 Receptor Beta-Dependent STAT5 Activation Is Required for the Development of Foxp3+ Regulatory T Cells. J Immunol (2007) 178(1):280–90. doi: 10.4049/jimmunol.178.1.280
141. Zhu J, Cote-Sierra J, Guo L, Paul WE. Stat5 Activation Plays a Critical Role in Th2 Differentiation. Immunity (2003) 19(5):739–48. doi: 10.1016/S1074-7613(03)00292-9
142. Lima VV, Zemse SM, Chiao CW, Bomfim GF, Tostes RC, Clinton Webb R. Interleukin-10 Limits Increased Blood Pressure and Vascular RhoA/Rho-Kinase Signaling in Angiotensin II-Infused Mice. Life Sci (2016) 145:137–43. doi: 10.1016/j.lfs.2015.12.009
143. Li X, Zhang Z, Li L, Gong W, Lazenby AJ, Swanson BJ, et al. Myeloid-Derived Cullin 3 Promotes STAT3 Phosphorylation by Inhibiting OGT Expression and Protects Against Intestinal Inflammation. J Exp Med (2017) 214(4):1093–109. doi: 10.1084/jem.20161105
144. Hutchins AP, Diez D, Miranda-Saavedra D. The IL-10/STAT3-Mediated Anti-Inflammatory Response: Recent Developments and Future Challenges. Brief Funct Genomics (2013) 12(6):489–98. doi: 10.1093/bfgp/elt028
145. Takeda K, Clausen BE, Kaisho T, Tsujimura T, Terada N, Förster I, et al. Enhanced Th1 Activity and Development of Chronic Enterocolitis in Mice Devoid of Stat3 in Macrophages and Neutrophils. Immunity (1999) 10(1):39–49. doi: 10.1016/S1074-7613(00)80005-9
146. Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two Subsets of Memory T Lymphocytes With Distinct Homing Potentials and Effector Functions. Nature (1999) 401(6754):708–12. doi: 10.1038/44385
147. Trott DW, Thabet SR, Kirabo A, Saleh MA, Itani H, Norlander AE, et al. Oligoclonal CD8+ T Cells Play a Critical Role in the Development of Hypertension. Hypertension (2014) 64(5):1108–15. doi: 10.1161/HYPERTENSIONAHA.114.04147
148. Delgado Silva J, Almeida JS, Rodrigues-Santos P, Santos Rosa M, Goncalves L. Activated Double-Negative T Cells (CD3(+)CD4(-)CD8(-)HLA-DR(+)) Define Response to Renal Denervation for Resistant Hypertension. Clin Immunol (2020) 218:108521. doi: 10.1016/j.clim.2020.108521
149. Ariotti S, Beltman JB, Chodaczek G, Hoekstra ME, van Beek AE, Gomez-Eerland R. Tissue-Resident Memory CD8+ T Cells Continuously Patrol Skin Epithelia to Quickly Recognize Local Antigen. Proc Natl Acad Sci USA (2012) 109(48):19739–44. doi: 10.1073/pnas.1208927109
150. Lopez Aguilar A, Gao Y, Hou X, Lauvau G, Yates JR, Wu P. Profiling of Protein O-GlcNAcylation in Murine CD8(+) Effector- and Memory-Like T Cells. ACS Chem Biol (2017) 12(12):3031–8. doi: 10.1021/acschembio.7b00869
151. Rodriguez-Pinto D. B Cells as Antigen Presenting Cells. Cell Immunol (2005) 238(2):67–75. doi: 10.1016/j.cellimm.2006.02.005
152. Winkler TH, Martensson IL. The Role of the Pre-B Cell Receptor in B Cell Development, Repertoire Selection, and Tolerance. Front Immunol (2018) 9:2423. doi: 10.3389/fimmu.2018.02423
153. Wu JL, Wu HY, Tsai DY, Chiang MF, Chen YJ, Gao S, et al. Temporal Regulation of Lsp1 O-GlcNAcylation and Phosphorylation During Apoptosis of Activated B Cells. Nat Commun (2016) 7:12526. doi: 10.1038/ncomms12526
154. Wu JL, Chiang MF, Hsu PH, Tsai DY, Hung KH, Wang YH, et al. O-GlcNAcylation Is Required for B Cell Homeostasis and Antibody Responses. Nat Commun (2017) 8(1):1854. doi: 10.1038/s41467-017-01677-z
Keywords: O-glcnacylation modification, immune system (IS), hypertension, adaptive immunity (ADIM), innate immunity (basic sciences)
Citation: dos Passos Junior RR, Bomfim GF, Giachini FR, Tostes RC and Lima VV (2022) O-Linked β-N-Acetylglucosamine Modification: Linking Hypertension and the Immune System. Front. Immunol. 13:852115. doi: 10.3389/fimmu.2022.852115
Received: 10 January 2022; Accepted: 21 February 2022;
Published: 17 March 2022.
Edited by:
Parameswaran Ramakrishnan, Case Western Reserve University, United StatesReviewed by:
Patrick E. Fields, University of Kansas Medical Center, United StatesJunfeng Ma, Georgetown University, United States
Copyright © 2022 dos Passos Junior, Bomfim, Giachini, Tostes and Lima. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Victor Vitorino Lima, dnZsaW1hdWZtdEBnbWFpbC5jb20=