 Alexander Grotemeyer
Alexander Grotemeyer Rhonda Leah McFleder1
Rhonda Leah McFleder1 Jingjing Wu
Jingjing Wu Jörg Wischhusen
Jörg Wischhusen Chi Wang Ip
Chi Wang Ip- 1Department of Neurology, University Hospital Würzburg, Würzburg, Germany
- 2Section for Experimental Tumor Immunology, Department of Obstetrics and Gynecology, University Hospital of Würzburg, Würzburg, Germany
Parkinson’s disease (PD) is a progressive and debilitating chronic disease that affects more than six million people worldwide, with rising prevalence. The hallmarks of PD are motor deficits, the spreading of pathological α-synuclein clusters in the central nervous system, and neuroinflammatory processes. PD is treated symptomatically, as no causally-acting drug or procedure has been successfully established for clinical use. Various pathways contributing to dopaminergic neuron loss in PD have been investigated and described to interact with the innate and adaptive immune system. We discuss the possible contribution of interconnected pathways related to the immune response, focusing on the pathophysiology and neurodegeneration of PD. In addition, we provide an overview of clinical trials targeting neuroinflammation in PD.
1 Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disease after Alzheimer’s disease (AD) (1–4). Motor symptoms such as bradykinesia, rigidity, resting tremor, and postural instability (5, 6) are hallmarks of PD and essential for staging. PD patients also exhibit a wide variety of non-motor symptoms, ranging from anosmia, rapid eye movement (REM) sleep disorders, and constipation to severe psychiatric symptoms such as dementia (6). Some of these non-motor symptoms are observed long before motor counterparts occur and are therefore essential to early PD diagnosis (7). From the defined histological description, a staging model arose in which progression of PD symptoms can be matched with intracellular deposits of pathological α-synuclein aggregates (pαSYN) or Lewy pathology (LP) of the corresponding brain areas (8–11). However, LP is also observed in the brains of older patients with no PD symptoms during their lifetime, indicating that LP is not specific as a standalone for PD diagnosis (10, 12). Moreover, it remains unclear why pαSYN occurs, why pαSYN spreads from cell to cell (10, 13–16), and how cell death is mediated by pαSYN (17). Since α-synuclein (αSYN) has been described as a critical mediator of inflammation and immune responses, and is released from inflamed neurons, αSYN may trigger a self-amplifying proinflammatory response (18). Intriguingly, neuroinflammation has emerged as a key aspect in PD (19–22) – but the extent to which neuroinflammation contributes to the development and maintenance of PD is controversial. Hence, knowledge transfer from neuroinflammatory diseases, where the concept of immune system-induced neuronal cell damage is established, could help to elucidate the still-obscure role of the immune system in PD.
Immunomodulatory therapies are established as effective treatments in ‘classical’ inflammatory neurological diseases, such as multiple sclerosis (MS), neuromyelitis optica spectrum disorders (NMOSD), or myasthenia gravis (MG) (23–27). Although the pathophysiology differs regarding the antigen in NMOSD and MG, common therapeutic strategies are implemented (28). In both entities, antibodies are pathophysiologically relevant and inhibition of the complement system has proven beneficial (29, 30). This supports the idea that similar inflammatory processes can be addressed across different neuroinflammatory diseases.
Various concepts have been tested as a causal treatment for PD, including vaccination against αSYN (31, 32). However, none have yet proven successful and traditional treatment of PD is still symptomatic and largely based on dopaminergic medication (33–35). To determine the relevance of neuroinflammation in PD pathogenesis, large-scale clinical observational studies are essential to generate appropriate hypotheses, which can be subsequently tested in animal models. In our review, we discuss the evidence for neuroinflammation in PD and potential future targets. We further illustrate immunological pathways linked to neuroinflammation and dopaminergic neurodegeneration.
2 Pathomechanisms of Neuroinflammation in PD Patients
The pathophysiologic role of the immune system in PD is still enigmatic. Neuroinflammation was proposed by James Parkinson himself in the initial description of PD (1). There is also early literature discussing PD as an ‘autoimmune disease’ (36). Yet interest in the topic was only rekindled in 1988, when microglia activation was described in brain autopsies of PD and AD patients (21). Since then, evidence has accumulated that neuroinflammation is likely to play a critical role in PD (36–38). Significant elevation of inflammatory cytokines in the blood and cerebrospinal fluid (CSF) of PD patients has now been confirmed in a meta-analysis over 25 clinical studies (39). Increased expression of NLRP3 and caspase-1 genes in peripheral blood mononuclear cells (PBMC) and elevated protein levels of NLRP3, caspase-1, and IL-1B in the blood plasma were found to correlate with PD severity (40), further supporting the perception of PD as a chronic systemic inflammatory disease. Evidence suggests that both innate and adaptive immune responses are involved in PD progression (41–44).
2.1 Innate Immune Response
Microglia are permanently in contact with astrocytes, neurons, and endothelial cells, constantly monitoring surrounding tissue for an ongoing infection or trauma via thin processes (45). In response to injury or infection, resting microglia undergo morphological alterations, change their transcriptional activity, and present antigens via the major histocompatibility complexes (MHC)-I and -II (46–48). Unlike MHC-I, which is constitutively expressed on all nucleated cells and platelets (49–52), MHC-II is specifically expressed on antigen-presenting cells such as microglia, astrocytes, monocytes, macrophages, dendritic cells, and granulocytes, and can be induced upon activation.
As brain-resident innate immune cells, microglia are typically the first responders to altered homeostasis within the CNS. Microglia activation increases the amount of nuclear factor ‘kappa-light-chain-enhancer’ of activated B-cells (NFKB) and NLR family pyrin domain-containing 3 (NLRP3), with subsequent upregulation of nicotinamide adenine dinucleotide phosphate (NADPH)-oxidase and cytokines such as interleukin-1 beta (IL-1B) and tumor necrosis factor alpha (TNF) (53, 54). Phagocytic activity and MHC-II expression are also enhanced (55, 56). An increase in activated microglia has been observed in both PD patients and healthy older people, indicating that various triggers might induce a proinflammatory shift with age (21, 57). Compared to age-matched controls, PD patients exhibited significantly elevated levels of proinflammatory cytokines such as IL-1B, IL-6, and TNF in CSF and blood (39, 58). Furthermore, ([11C](R)-PK11195)-based positron emission tomography (PET) has revealed a higher density of activated microglia in the midbrain and putamen of early PD patients (59, 60), which correlated with decreased activity of dopamine transporter ligands ([11C] CFT). Similar results were described in patients suffering from a REM sleep behavior disorder (60, 61). While the presence of chronic inflammatory processes in PD is now widely accepted, the underlying reason for neuroinflammation is still unclear. Due to its function as a damage-associated molecular pattern (DAMP) (53, 62, 63), pαSYN might trigger and maintain a proinflammatory shift of the immune system. In addition, DAMPs may be released from dying or damaged cells. Well-known DAMPs triggering an innate immune response upon interaction with pattern recognition receptors (PRRs) are IL-1α or mitochondrial reactive oxygen species (mROS), which activate NLRP3. Consecutive NLRP3 activation leads to increased IL-1B synthesis as a trigger for further innate immune responses (64). Initial microglia activation in PD (21, 65) may therefore result from PRR-mediated responses to DAMPs.
In addition to DAMPs, pathogen-associated molecular patterns such as viral RNA or the bacterial cell wall component lipopolysaccharide (LPS) may also maintain neuroinflammation. While a correlation between viral infection and PD has not strictly been established, preceding infections might still modulate the risk for developing PD (66). For example, there is evidence for toxin-related PD, which implies that toxins released by infectious agents might also be responsible for some cases of ‘sporadic’ PD. Other toxins such as the pesticide rotenone have neurotoxic and neuroinflammatory effects that may kill dopaminergic cells and thereby cause PD in people exposed to higher concentrations (67).
Possible links between PD and infection have become even more topical with the still-ongoing worldwide COVID-19 pandemic. While there is very little consolidated knowledge regarding possible interactions between the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and PD, there are some interesting observations on pathogenic coronaviruses and neuroinflammation or, more specifically, PD. Antibodies against coronavirus antigens have been found in the CSF of PD patients (68). In addition, intranasal infection of hACE2 transgenic mice with SARS-CoV-1 caused severe CNS infections that spread from the olfactory bulb to the basal ganglia and midbrain within approximately four days (69). Considering that COVID-19 patients often experience anosmia, SARS-CoV-2 is also likely to reach the CNS via the olfactory bulb. Disconcertingly, in vitro assays have shown that the nucleocapsid(N)-protein of SARS-CoV-2 accelerates pαSyn-aggregation by a factor of 10 (70). Thus, it is concerning that COVID-19-related inflammation may trigger or accelerate PD in patients at risk. Yet to date, there are only a few case studies describing the development of PD after SARS-CoV-2 infection (71, 72). Whether infection with SARS-CoV-2 impacts the development of PD remains to be explored.
2.2 Adaptive Immune Response in PD: T Cells Friends and Foes
Neuroinflammation in PD is not confined to the innate immune system; it also involves adaptive immune responses. Several findings indicate that various T cell subpopulations may contribute to PD pathophysiology (73).
Neuroinflammation within the substantia nigra (SN) is well-described in PD. Significant infiltration of both CD4+ and CD8+ T cells into the SN of PD patients has been described (42), particularly elevated levels of CD8+ T cells (74, 75). It is possible that CD8+ T cells have an important role in early PD, even before pαSYN can be detected in the SN, as relevant infiltration into the SN has been observed in very early PD that subsides with disease progression (74). Altered counts of CD4+ T cell populations in the blood of PD patients have also been reported, but remain controversial. Many studies describe a decrease in the overall CD4+ T cell population in PD patients (44, 76–78). However, both elevated (76) and reduced Treg cell counts (79) were found in the blood of PD patients, although the studies used a relatively non-specific marker (CD4+CD25+) to identify Treg cells. Increased numbers of CD4+FoxP3+ Treg have been observed in the blood of PD (aged 46-80 years) and AD (62-82 years) patients and healthy older people (51-87 years) compared to healthy young controls (23-40 years) (80). An increase in Treg cell activity with age was also seen, which was significantly more pronounced in PD and AD patients compared to young controls (80). In contrast, decreased levels of CD4+CD25highCD127low Treg cells (44, 81) and reduced suppressive activity of Treg cells has also been found in PD patients (77). Furthermore, peripherally-reduced Treg cells and increased Th1 cell counts were found to correlate with the severity of motor dysfunction (assessed by Unified Parkinson’s Disease Rating Scale III [UPDRS III]) (44, 81). In addition, decreased Th2 cell numbers and a relative increase in Th1 cells were seen in PD patients (44). Despite these discrepancies, most studies highlight an increased peripheral immune response in PD, with a reduced amount of Treg cells (CD4+CD25highCD127low) (44, 81) (Table 1).

Table 1 Overview of human and animal studies on T cells with special regard to Treg cells.
These population and activity shifts of T cells, along with an increase in HLA-DR positive antigen-presenting microglia in PD patients (82), support the idea that CD4+ T cells might contribute to neurodegeneration in PD. Studies on the role of Th17 cells in PD have generally confirmed this concept. Addition of IL-17 to autologous cocultures between T cells and pluripotent stem cells (iPSC)-derived mid brain neurons (MBN) from PD patients also induced NFKB-dependent cell death. iPSC-derived MBN from PD patients further showed higher expression of IL-17 receptors, together with enhanced susceptibility towards death induction by recombinant IL-17 or autologous Th17 cells (73). Interestingly, Th17 cells are increased in PD patients, which is consistent with the results of previous studies (81, 83, 84). However, one study in PD patients reported a decrease in the Th17 cell count, along with unchanged levels of IL-17 (44). Based on these studies, Th17 cells and IL-17 are likely to favor the progression of PD, but their distinct role remains unresolved (85, 86). Consequently, a comparison of different analytical approaches might be needed to rule out possible methodological issues.
In addition to flow cytometric analyses, gene expression analyses and genome-wide association studies that investigate alterations in immune response pathways are playing an increasing role in PD research. Expression quantitative trait loci (eQTLs) are obtained by analyzing the genome and transcriptome of patients with correlating alleles and the expression level of transcripts. Based on CD4+ T cells and monocytes from a multi-ethnic cohort of 461 healthy individuals, susceptibility alleles for MS, rheumatoid arthritis, type 1 diabetes, and PD were found to be overrepresented in CD4+ T cells, while disease- and trait-associated cis-eQTLs associated with AD and PD were primarily overrepresented in monocytes (87). Further evidence for the relevance of the immune system in PD pathogenesis comes from the association of PD with an immune haplotype, including the DRB5*01 and DRB1*15:01 alleles, and from a non-coding polymorphism in the region that might increase MHC-II expression (88). Finally, a very recent study stratified PD patients by their T cell responsiveness to α-SYN as a proxy for an ongoing inflammatory autoimmune response (89). Gene expression analysis in peripheral memory T cells then revealed a clear PD-specific gene signature with transcriptomic markers for inflammation, oxidative stress, phosphorylation, autophagy of mitochondria, cholesterol metabolism, and chemokine signaling via CX3CR1, CCR5, and CCR1.
2.3 Adaptive Immune Response in PD: B Cells and Humoral Immunity
While T cells are the more prevalent adaptive immune cells in CSF (90), B and T cell immunity are closely intertwined (91). Although immunohistochemical analyses have found B cell-rich tertiary lymphoid structures in most brains from patients with MS, but not in the two PD patients investigated (92), there is ample evidence for the involvement of B cells, antibodies, and humoral immune effector mechanisms in PD (93–95). In a postmortem study of 13 patients with idiopathic PD and three with genetic PD (compared with 12 controls), all PD patients showed IgG (mostly IgG1) but no IgM binding to dopaminergic neurons, while Lewy bodies were strongly immunolabelled with IgG (94). Nearby activated microglia expressed the high-affinity activating IgG receptor FcγRI, which implies a significant role for antibody-dependent cell-mediated cytotoxicity. The inhibitory IgG receptor FcγRII was absent in all cases. Subsequent studies on B cell-related adaptive immune responses in PD support the involvement of B cells and humoral immune effector mechanisms (96–98). Remarkably, some patient sera also contained antibodies capable of neutralizing pαSYN ‘seeding’ (forming of αSYN aggregates) in vitro (99). Elevated levels of serum antibodies against αSYN have also been reported in a comparison of sera from PD patients with AD patients and controls (100). Of note, B cell numbers were found to be reduced in PD patients (96, 101), along with an increase in IgA levels (102) that correlated with non-motor symptoms (96). Moreover, cytokine expression of B cells was altered in PD (96). Cytokine-dependent interactions between B cells and Th1 and Th17 cells (103) imply that B cells may also modulate proinflammatory Th17 cells in PD patients (73). In addition, there are relevant interactions between the humoral immune response and the complement system, as described in a comprehensive review (104).
Given the paucity of studies focusing on the distinct function of B cells in PD or other neurodegenerative diseases (103, 105), a more profound understanding of B cell function might reveal new opportunities for disease modification. Again, the targeted transfer of knowledge from ‘classical’ neuroinflammatory disease would be useful (103).
2.4 Anti-Inflammatory Disease Modification
Since proinflammatory changes have been consistently observed in PD, several studies analyzed whether suppressing inflammation might modify the disease course of PD. A population-based, case–control study found that patients receiving immunosuppressive therapy had a decreased risk of developing PD (106). Interestingly, the greatest risk reduction was conveyed by drugs affecting T cells such as the inosine monophosphate dehydrogenase inhibitors, azathioprine and mycophenolat mofetil (106), while a protective effect was revealed for corticosteroids. However, this finding might be biased by smoking behavior. Smoking is described as a possible protective factor for PD, and smokers frequently need corticosteroids due to pulmonary maladies (106, 107). Moreover, cigarette smoke induces chronic inflammation, which results in immune cell exhaustion and generally attenuates the function of many immune responses (108). Therefore, smoking can be considered an immune-inhibitory activity (109, 110). The potential effectiveness of anti-inflammatory treatment is further exemplified by a lowered risk for developing PD in patients receiving immunosuppressive therapy (111).
Accordingly, patients with inflammatory bowel disease (IBD) receiving an anti-TNF treatment had a 78% reduction in the incidence of PD compared to IBD patients who did not receive this therapy (112). However, the lack of direct comparison between the incidence of PD in treated IBD patients compared with non-IBD patients (112) means that the ability of anti-TNF treatment to reduce PD incidence in non-IBD patients remains unanswered. Other research has indicated that severity of IBD is inversely associated with the risk of developing PD (-15%) (113). However, although patients suffering from severe IBD are more likely to receive anti-inflammatory drugs, immune-modulatory treatment was not directly assessed. Accordingly, further studies are necessary to verify a positive or negative correlation between PD and anti-inflammatory treatment, both for IBD and for non-IBD patients.
One of the most frequently used anti-inflammatory drugs is ibuprofen, a reversible cyclooxygenase-2 (COX-2) inhibitor and non-steroidal anti-inflammatory drug (NSAID) (114). Intriguingly, ibuprofen users showed a lower risk for PD, whereas no protective effect could be demonstrated for acetylsalicylic acid (ASA) (114). In a prospective study of subjects without PD who were assessed at baseline for NSAID intake, 0.2% developed PD during a 6-year follow-up (115). A significant dose-response relationship between ibuprofen intake (tablets/week) and reduced PD risk was found. Again, this effect was not observed for ASA, other NSAIDs (e.g., indomethacin), or acetaminophen. Although all NSAIDs and ASA are known to inhibit COX-2, only ibuprofen was shown to have some impact on PD prevention. Pre-clinical findings from in vitro models indicate that ibuprofen prevents oxidative damage induced by LPS injection and better protects dopaminergic neurons against glutamate neurotoxicity than ASA (116, 117). The neuroprotective effects of ibuprofen have been confirmed in studies that considered confounder variables of PD, such as age, smoking, and caffeine intake (115, 118). However, the putative neuroprotective effect of ibuprofen in PD remains controversial; other research focusing on all NSAIDs rather than on ibuprofen alone found that NSAIDs had no neuroprotective effects (107). In this context, it is noteworthy that ibuprofen is known to reduce inflammation via inhibition of COX-2, but also via modulation of the angiotensin pathway by shifting the ACE/ACE-2 (angiotensin converting enzyme) receptor ratio towards ACE-2 (119, 120) (discussed in detail in Section 4.2).
Drugs such as β-agonists and β-antagonists have been shown to affect the incidence of PD in opposing directions, with β-agonists seeming to be somewhat protective (121, 122). However, further evidence is needed to confirm whether β-agonists might act directly on immune cells and are thus capable of modulating PD-related inflammation (122). It should be noted that β-agonists are often used in patients suffering from chronic obstructive pulmonary disease, a common consequence of frequent smoking. The fact that smoking was reported to lower the incidence of PD in one study represents an epidemiological bias (121).
Given that observational studies have yielded controversial data regarding the neuroprotective effects of different drugs and immunomodulators (123–128), randomized, controlled trials are needed to better decipher which drugs have distinct neuroprotective effects in PD.
2.5 Current Clinical Trials for Anti-Inflammatory Disease Modification in PD
Current clinical trials on anti-inflammatory therapy in PD focus on the treatment of glucose metabolism, improvement of oxidative stress, and regulation of gut microbiota.
2.5.1 Regulation of Glucose Metabolism
Impaired insulin signaling resulting in altered energy balance and impaired cell repair mechanisms has been observed in PD patients (129–131). Glucagon-like peptide-1 (GLP-1) is a growth factor and insulin-stimulating hormone that can activate the same effector molecules as insulin, and its receptor (GLP1R) is expressed in neurons. In addition to the known effects of GLP1R on blood sugar regulation and stimulation of the hypothalamus to regulate appetite, studies have shown that GLP1R agonists have neuroprotective effects in different animal models of PD. One agonist, exendin-4, can enhance cognitive function (132) and attenuate neurodegeneration in 6-hydroxydopamin (6-OHDA) rats (133). In 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated mice, the newer GLP-1 mimetics liraglutide and lixisenatide can improve motor impairment and reduce dopaminergic cell loss in the SN (134, 135). In a human A53T αSYN (hA53T-αSYN) transgenic mouse model, a long-acting GLP1R agonist reduced behavioral deficits and neurodegeneration (136). It is noteworthy that activation of GLP1R can inhibit the production of proinflammatory cytokines in microglia, such as TNF and IL-1B (137, 138). The neuroprotective effect of semaglutide treatment is currently under investigation (139).
2.5.2 Regulation of Gut Microbiota
The gastrointestinal (GI) tract and CNS have complex mutual interactions (140) that are affected by the intestinal flora (141–143). Therefore, a healthy and stable intestinal flora is important for immunity, homeostatic balance of barrier integrity, and metabolism (144). Interestingly, changes in the intestinal flora may alter nerve development and even cause neurodegenerative disorders (145). Conversely, traumatic brain injury may lead to impaired intestinal barrier function (146, 147). GI dysfunction in PD patients, particularly obstipation, is commonly known to be one of the initial symptoms that precedes motor impairment (148). Changes in intestinal microbial flora can alter the barrier function and permeability of the gut and subsequently influence the immune system (140, 149), and have been linked to PD (149). It is noteworthy that pαSYN can be found in the enteric nervous system (150).
A speculative hypothesis is that the methylation status of the SNCA gene might be influenced by the gut microbiome. It is accepted that duplications and triplications of the SNCA gene confer a higher risk of developing PD (151, 152). Decreased methylation of the SNCA gene, which would lead to increased transcription, could likewise affect αSYN expression and influence the risk of PD (153). Postulating a potential role of the gut microbiota as an epigenetic factor for DNA methylation, an ongoing clinical study is investigating the composition of the GI bacteria in the stool of PD patients and healthy controls by extracting total bacterial DNA from the samples (154).
2.5.3 Regulation of Oxidative Stress
Oxidative stress in the brain plays a key role in the development of PD (155, 156), and results from an imbalance between reactive oxygen species (ROS) and the ability to scavenge reactive intermediates (157). ROS, which include superoxide, nitric oxide, hydroxyl radical, hydrogen peroxide, and peroxynitrite, can be generated from various sources including the electron transport chain or activated immune cells. Interestingly, increased oxidative stress seems to be target-specific and, in the case of PD, related to LP and the pαSYN contained in Lewy bodies, where pαSYN nitration (an oxidation marker) was found to be elevated (158–160). Furthermore, the SN of early PD patients has been found to contain reduced amounts of the antioxidant glutathione (GSH), which correlated with PD severity (159).
Increasing the availability of reduced GSH N-acetylcysteine (NAC) can potentially reduce the damage to neurons caused by oxidative stress. In a current clinical trial, NAC is being used to protect PD patients from oxidative damage (161). Aside from NAC, other supplements with antioxidative properties are under investigation. In the CNS, vitamin B3 (niacin) is considered a key mediator of neuronal development and survival (162). Niacin is a cofactor of nicotinamide adenine dinucleotide and NADPH, which are necessary for the scavenging of oxidants and needed for GSH regeneration (157). Niacin can improve the integrity of mitochondria and hence the energy supply of neurons. By enhancing antioxidant defense mechanisms, niacin can protect the normal structure and function of neurons exposed to oxidative stress, thereby delaying the progression of PD (163). Furthermore, niacin binds to the G-protein-coupled hydroxycarboxylic acid receptor 2, which is upregulated in a proinflammatory environment and which may downregulate inflammation (164–166). Since PD patients are known to have low levels of niacin due to mitochondrial malfunctioning, supplementation of niacin could be beneficial (166). A clinical trial is evaluating the effectiveness of niacin or niacinamide supplementation on inflammation and severity of PD symptoms (167).
3 Animal Models and Their Contribution Towards Understanding PD Pathomechanisms
Animal models play an important role in understanding PD pathophysiology, including neuroinflammatory processes. Next to the 6-OHDA model (168), one of the most often used PD models is induced by MPTP delivery (169), which persists extensively in the SN and results in PD symptoms with inhalation and cutaneous contact (169). Immune responses in the various MPTP models used are, however, quite different.
3.1 Innate Immune Responses in Animal Models for PD
Toxins such as MPTP, rotenone, and 6-OHDA act as DAMPs and initiate a strong innate immune response with microglia activation and subsequent neurodegeneration in the SN (170–172). This immune response resembles the microglia activation found in human brain autopsies (21, 173). In the 6-OHDA model, neurodegeneration is accompanied by a gradual repolarization of microglia from an anti-inflammatory M2 to a pro-inflammatory M1 phenotype (174). Cytokine production by M1 cells is intracellularly initiated by NFKB (53, 175, 176), which in turn induces interleukin and procaspase-1 transcription. Together with the inflammasome NLRP3, caspase-1 activates IL-1B. Moreover, other proinflammatory proteins such as iNOS and TNF are also released from M1 cells (53, 175, 176) and contribute to neurodegeneration in PD (177). Consequently, inhibition of NFKB protects MPTP mice from neurodegeneration (178). M2 macrophages, in contrast, contribute to neuroprotection and release neurotrophic factors (176) (Figure 1). Since TLR-4 knock-out reduced the number of MHC-II+ microglial cells and partially protected MPTP-treated mice from dopaminergic degeneration in the SN, MPTP triggers also neuroinflammation via TLR-4 (179). However, the canonical ligand for TLR-4 is LPS, which is commonly found on cell walls of gram-negative bacteria (180) In LPS-driven models, LPS is either systemically administered or focally injected into the SN (181). Since microglia activation after delivery of LPS or another of the aforementioned toxins results in the same downward cascades, all of these models lend themselves to studying innate immune responses (181–183). However, in mice lacking the mitophagy inducers PINK and Parkin, LPS triggers the release of mitochondria-derived vesicles (184, 185). This can prime CD8+ T cell responses against mitochondrial antigens (184, 185). External stressors such as LPS can thus also induce adaptive immune responses, in particular in transgenic models with impaired mitochondrial functions.
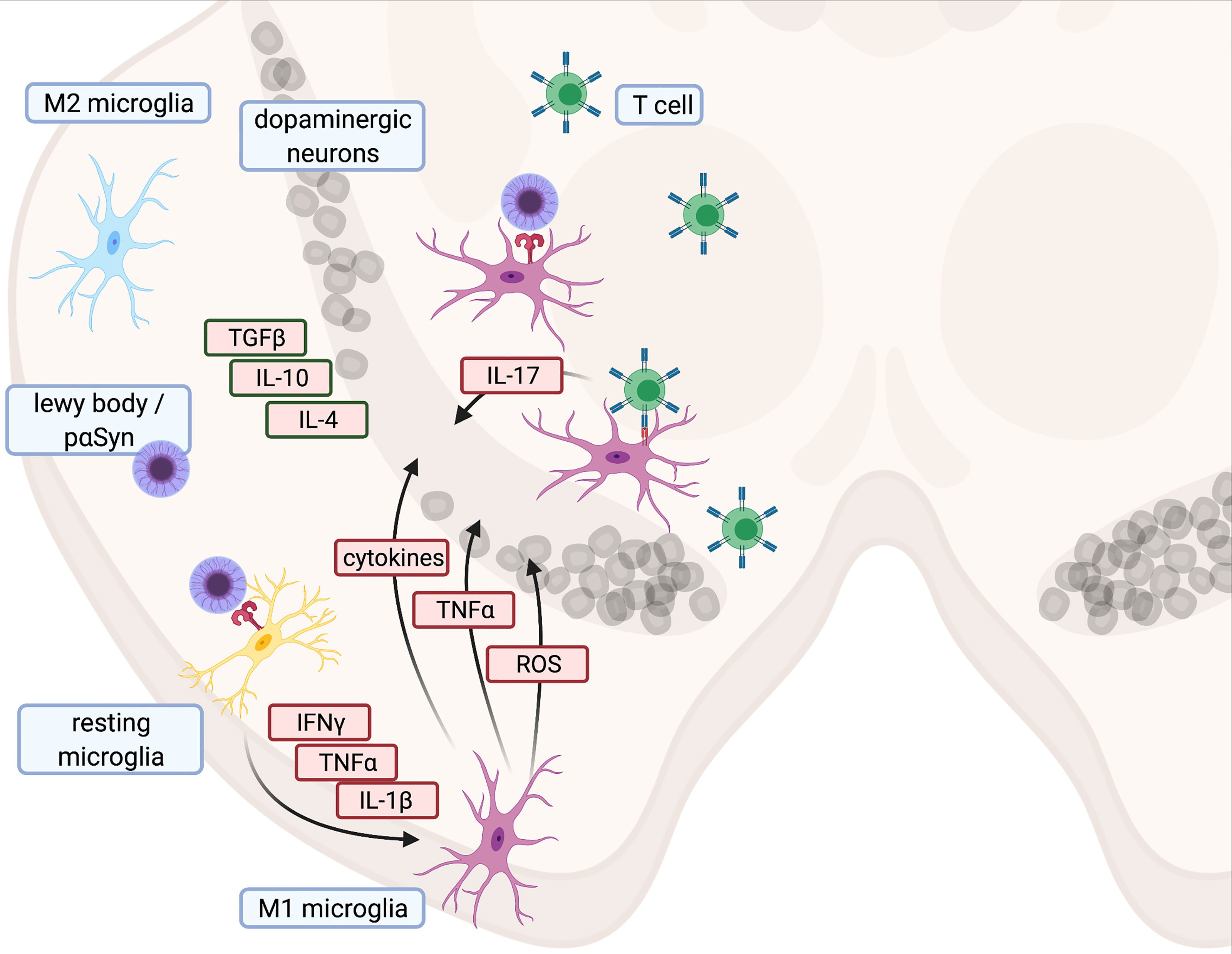
Figure 1 Illustration of proinflammatory cascade and main cytokines contributing to cell death in Parkinson’s disease - Created with BioRender.com.
For MPTP, but not 6-OHDA, a Lewy body-like pathology has been found in non-human primates (186). An even more prominent Lewy body-like histopathology was observed in viral vector-based models overexpressing human mutations of αSYN (e.g., A53T) (187). Furthermore, viral vector-based models demonstrate a similar innate immune response, as seen in toxin-based models. pαSYN, which is believed to enter the cell via TLR-2 to unleash its toxic potential (53, 188, 189), thus apparently acts like a DAMP (190). Interestingly, orthotopic injection of an αSYN-encoding AAV into the SN only induced neurodegeneration in the presence of Fc gamma receptors (191), which implies an antibody-dependent activation of the complement cascade and/or antibody-dependent cellular cytotoxicity that may be mediated by microglia, for example (192). However, unlike astrocytes, microglia do not directly contribute to a proinflammatory environment after exposure to pαSYN (193). Nevertheless, progressive neurodegeneration can be induced by microglia-specific overexpression of pαSYN in the absence of pαSYN in dopaminergic cells (194). The critical role of CD4+ T cells was confirmed by showing that virus-based αSYN-overexpression can only induce microglia activation and neurodegeneration when MHC-II is present (195). Therefore, viral vector-based models require a complex interplay between different types of immune cells, which may also best reflect observations made in human PD.
Since animal research in PD has focused mostly on alterations within the CNS, our understanding of peripheral innate and adaptive immune processes in PD pathology is still limited. Recent studies have provided evidence that increased gut permeability and gut microbiota are likely to contribute to PD development (149, 196). This has resulted in PD becoming more and more accepted as a systemic disease (197). For instance, overexpression of human TNF (hTNF) in a mouse model leads to microglia activation in various regions of the brain (198, 199), while immunomodulation with infliximab could attenuate microglia activation (199). Since infliximab cannot penetrate the blood-brain barrier, this finding indicates that effective treatment of neuroinflammation could be achieved by modulating peripheral immune mechanisms.
3.2 Adaptive Immune System in Animal Models for PD
Several animal models support the hypothesis that the adaptive immune response in PD is mainly T cell driven, whereas the role of B cells remains enigmatic (41, 42, 73, 75, 88, 200). Nevertheless, the mechanisms of T cells vary across different animal models (42, 201).
3.2.1 MPTP Model
Variations in frequency and dosage of MPTP injections in mice result in different PD models with diverse disease kinetics. The subacute MPTP model is generated by administering MPTP once daily over five consecutive days, whereas an acute model is induced by delivering MPTP every two hours for four times in only one day (202). For the acute model, it is important to note that in contrast to humans, the initial immune response decreases significantly within 30 days, and MPTP-injected mice may recover over time (203). Moreover, pαSYN is not found in this model per se. However, if the adaptive immune response in the acute model is maintained by peripherally administering cytokines such as RANTES (Regulated on Activation, Normal T cell Expressed and Secreted) and eotaxin twice a week, the inflammation is sustained and pαSYN accumulates in the SN of the rodents (203). In this model, pαSYN formation is linked to a sustained proinflammatory environment.
CD4+ and CD8+ T cell infiltration in the SN of acute MPTP mice supports the idea that T cells contribute to neurodegeneration in this PD model (42). CD4+ or recombinant activating gene 1 (RAG1)-deficient mice, which lack mature lymphocytes, show attenuated neurodegeneration upon treatment with MPTP. In contrast, neurodegeneration is unrestrained in CD8+ T cell-deficient mice (42). Blockade of RANTES and eotaxin in the MPTP mouse and non-human primate model reduces microglia activation, CD4+ and CD8+ T cell infiltration, and neurodegeneration (204, 205). Continuous T cell trafficking and induction of pαSYN in the SN of MPTP mice is also driven by supplementation of RANTES and/or eotaxin, but not by TNF and IL-1B (203). Furthermore, MPTP-treated mice show altered and dysfunctional CD4+CD25+ Treg cells. Administration of natural Treg cells leads to attenuation of SN degeneration (206). As Th17 cells appear to contribute to nigrostriatal degeneration (206, 207), the beneficial effect of Treg may be due to their ability to inhibit TH17 cells. However, while mechanistic insights from the aforementioned studies are based on the acute MPTP model, CD4+ T cell infiltration occurs earlier in the subacute model (208). Given the differences between the models, the choice of the most appropriate model may determine the outcome (209).
3.2.2 6-OHDA Model
This model is generated by injecting 6-OHDA either directly into the SN, the medial forebrain bundle, or the striatum (168, 210, 211). The initial cytotoxic effect of 6-OHDA is known to be mediated via oxidative stress (212) and impaired mitochondrial function (213). Data on the role of the adaptive immune response in the 6-OHDA model are still scarce. But it is known that 6-OHDA administration leads to infiltration of CD4+ and Treg cells in the CNS (76, 174). In contrast to MPTP, 6-OHDA must only be administered once to induce continuous neuroinflammation and neurodegeneration (174, 201). Apart from neuroinflammation, 6-OHDA-treated rats also show decreased circulating Treg cells, indicating that this model can reflect aspects of human PD (174). However, while CD4-/- or Rag1-/- knock-out mice injected with MPTP were partly protected from severe neurodegeneration (42), 6-OHDA-treated Rag1-/- mice showed even more pronounced neurodegeneration in the SN than immune-competent mice (201) or rats (214). Accordingly, the adaptive immune system is mostly neuroprotective in the 6-OHDA rodent model. Thus, toxin-based models provide contradictory data on the role of the adaptive immune system. Based on the epidemiological data linking PD and adaptive immune responses in humans, these models do not seem to adequately recapitulate the pathomechanisms underlying the human disease.
3.2.3 Viral Vector and Preformed Fibrils Models
Creating a model that shows a large overlap with the human phenotype and pathophysiology is the main objective when generating new animal models. Regarding chronic disease progression, mild-to-moderate nigrostriatal degeneration, Lewy-like pathology, and neuroinflammation, the AAV1/2-A53T-αSYN model resembles human PD much more closely than conventional toxin-induced models (75, 187, 215). Recently, CD4+ and CD8+ cells infiltrating the SN were found in the AAV1/2-A53T-αSYN model (75). Ten weeks after AAV1/2-A53T-αSYN injection, CD4+ and CD8+ cells in the spleen were also activated. Moreover, RAG deficiency protected dopaminergic SN neurons, indicating that CD4+ and CD8+ crucially contribute to neurodegeneration in this model (75). This reflects findings made in PD patients, thereby demonstrating a higher immune-related face validity of the AAV1/2-A53T-αSYN mouse model compared to the toxin-induced MPTP and 6-OHDA models. Comparable results, including gradual dopaminergic cell loss and CD3+ infiltration in the SN, were also achieved by viral vector-based application of human αSYN in the SN of mice (216, 217) or by directly injecting preformed αSYN fibrils (αSYN-PFF) into the striatum of mice (62). The immune response triggered by the presence of pαSYN is thus conserved and comparable between different models. This is in line with the recently revealed role of αSYN as a critical mediator of inflammatory and immune responses (18).
Aside from PD models with direct injection of pαSYN or αSYN-PFF into the brain, there are also models based on peripheral injection of αSYN-PFF. Intriguingly, spread of pαSYN from the periphery to the brain is observed, accompanied by motor deficits (218, 219). However, in a more recent study where strong involvement of the adaptive immune response in the brain was confirmed, further inflammatory stress by LPS was required to achieve spreading of pαSYN into the brain (220). Accordingly, ongoing neuroinflammation contributes to the progression and maintenance of PD (197, 221–225).
3.2.4 Conclusion for the Role of the Immune System in PD Animal Models
These animal models provide substantial support for the hypothesis that the immune system participates in the neurodegeneration observed in PD. However, for investigating adaptive immunity, choosing the right animal model is key. Central and peripheral immune responses must both be considered. Finally, since the overall role of the immune response in PD animal models is still understudied, interactions between pαSYN, microglia, astrocytes, the adaptive immune system, and peripheral inflammation need to be explored.
4 Signaling Cascades Related to PD and Neuroinflammation as Possible Therapeutic Targets
Pathophysiology of PD is manifold and includes several well-investigated pathways, while others may still be unknown. There are several important signaling pathways related to inflammation and neurodegeneration.
4.1 COX-2 and Disease Modification With NSAIDs
ASA is a standard drug for secondary prophylaxis of ischemic stroke and a potent non-competitive inhibitor of COX-2. Since COX-2 is expressed in many cell types, including platelets and microglia, it is involved in many physiological processes. Intriguingly, the protective role of COX-2 inhibitors on neuroprotection remains controversial. An early administration of high ASA doses showed a neuroprotective effect in MPTP mice, while other COX-2 inhibitors including ibuprofen did not (226). In contrast, the protective effect of ASA in PD patients is comparatively low, while ibuprofen is associated with decreased disease risk compared to other NSAIDs (114, 115). Furthermore, some studies have shown that NSAID intake has no effect on PD development, but instead correlates with an increased risk for PD (107, 227). It is important to note that none of these studies compared equivalent dosages. In MPTP mice, however, COX-2 is upregulated and known to be involved in the oxidation of dopamine, forming dopamine quinone (DAQ) (202). Interestingly, COX-2-deficient mice are protected against induction of PD, which was not explained by reduced inflammation but by prevention of DAQ formation (202, 228, 229). DAQ conjugates with GSH to form GSHDAQ. After conversion to 5cysDAQ, GSHDAQ inhibits mitochondrial complex I. In turn, this leads to decreased degradation of ROS and increased DAQ levels. Subsequently, this vicious circle results in microglia and astrocyte activation and a shift towards a proinflammatory state (175), accompanied by exhaustion of the pentose-phosphate pathway (PPP). However, there are multiple pathways that result in upregulation of COX-2 in brain neurons, including increased N-methyl-D-Aspartate (NMDA) receptor-dependent activity (230), cerebral ischemia, seizures, and diseases such as AD or PD (231). Another important function of COX-1 and COX-2 is the further processing of arachidonic acid. COX-1 induction leads to a substantial increase in thromboxane A2 (TXA2) in human endothelial cells, while induction of COX-2 by IL-1B leads to a minor increase in TXA2 but a substantial induction of the proinflammatory proteins prostacyclin (PGI2) and prostaglandin E2 (PGE2) (232).
PGI2 and PGE2 are physiologically involved in body temperature regulation. PGI2 and TXA2 have important roles in the maintenance of vascular homeostasis and can mainly be described as agonist and antagonist (233). Among the different PGE2 receptors (EP1-4), EP2 contributes to neurodegeneration in the presence of pαSYN in PD (234). EP2-deficient microglia from mice showed increased clearance of pαSYN on brain slices from Lewy-body patients. Furthermore, EP2-/- MPTP mice were spared from neurodegeneration in the SN (234). However, this mechanism only seems to apply for neurotoxicity associated with chronic inflammatory processes. In acute pathological processes such as brain ischemia or NMDA-induced toxicity, EP2 activation exerts protein kinase A-dependent protective effects (231). Interestingly, EP2 is also neuroprotective in the 6-OHDA primary cell culture (235). The cause of this dichotomous action of EP2 is not yet understood (231).
Beside possible neuroprotective effects, COX-2 inhibitors increase intestinal permeability that correlates with their potency to inhibit COX-2, thus ultimately leading to local inflammatory side effects (236, 237). Furthermore, since increased intestinal permeability is discussed as an important pathological axis of PD development, prolonged treatment of PD with potent NSAIDs is not advisable, which could explain the controversial results (149, 196).
4.2 Oxidative Stress, PPP, and Renin-Angiotensin System
4.2.1 Oxidative Stress and PPP
Under steady-state conditions, ROS are maintained in a stable range and participate in the regulation of cell growth, differentiation, apoptosis signals, and enzyme activity by modulating the production of ceramide, kinase regulatory proteins, and activation of transcription factor NFKB (238, 239). Oxidative stress is a potent and highly conserved defense mechanism against intruders such as bacteria or fungi (240). By stimulating the production of inflammatory factors, ROS play an essential role in pathogen clearance (238). Free radical scavengers such as GSH act as a counterpart to ROS and protect cells from excessive oxidative stress. Maintaining the reduced state of GSH by oxidation of NADPH is therefore important for tissue homeostasis. Under pathological conditions, such as in PD, the level of ROS outpaces the compensatory mechanisms. ROS are located upstream of many inflammatory signaling pathways, upregulate the expression of other inflammatory factors, interfere with signaling pathways that regulate growth factors, and often alter signal transduction. High levels of ROS can be generated (241–244) by M1 microglia, but also the renin-angiotensin system (RAS) or NADPH-oxidase and its homologues, the so-called NOX enzymes (240). High ROS concentrations may cause apoptosis, structural damage, and cell death (159). ROS can therefore directly exert neurotoxicity and amplify neurotoxic inflammatory responses (157, 159). Understanding the role of pathways such as the PPP, which expurgate excessive amounts of ROS, might be crucial for finding an effective treatment against PD. The PPP connects glucose metabolism with NADPH production and nucleotide precursor synthesis. In this pathway, glucose-6-phosphate (G6P) is metabolized to produce NADPH and ribose-5-phosphate. In nearly all cells, the main task of PPP is to provide ribose-5-phosphate. In neurons, however, its main task is to provide NADPH to eliminate ROS (245). The definite role of PPP in PD remains still uncertain (245), even though there is increasing evidence that PD is associated with disorders of glucose metabolism (246–248).
The rate-limiting enzyme of the oxidative part of the PPP is G6P dehydrogenase (G6PD), which catalyzes the reaction between G6P and NADP+. Through consumption of NADPH, oxidative stress leads to the accumulation of large amounts of NADP+ and subsequently to the activation of G6DP due to the increased concentration of oxidants (249). In MPTP mice, neurodegeneration was shown to correlate with NADPH oxidase upregulation (242). Moreover, G6PD expression was positively associated with microglial activation and dopaminergic neurodegeneration (245), indicating that oxidative stress is closely linked to neurodegeneration.
Another important protein related to oxidative stress is GSH, an important antioxidant in brain neurons. The level of GSH in cultured cells is directly related to the production of NADPH by PPP (250). Increased ROS and decreased GSH levels have been described in many PD models and patients (155, 251–254).
Maintaining or adding NADPH in therapeutic dosages might help to overcome high levels of ROS, keep a stable level of GSH in the brain, and finally result in neuroprotection.
4.2.2 Renin-Angiotensin(-Aldosterone) System
The renin-angiotensin[-aldosterone] system (RA[A]S) is well-known in the context of blood pressure regulation (255). Stimuli such as increased activity of kidney baroreceptors, low plasma sodium levels, and activation of β1-receptors result in release of renin from the kidney into the blood circulation. Renin then cleaves liver-synthesized angiotensinogen into angiotensin I (AngI). Subsequently, AngI is cleaved via ACE to angiotensin II (AngII). Binding of AngII to the G-protein-coupled AngII receptor type 1 (AT1R) is essential for blood circulation. AT1Rs are commonly expressed in the vascular system, the heart, and the brain. Aside from binding to AT1R, AngII also induces release of aldosterone from the adrenal cortex (256). Drugs for the treatment of arterial hypertension therefore interfere with the RA[A]S by inhibiting renin, ACE, AT1R, or the aldosterone receptor (257). Investigations into the role of the RA[A]S in the brain, and more specifically in dopaminergic neurons, have deepened our understanding of neurodegeneration in PD. Different groups have demonstrated in both 6-OHDA and MPTP mice that neurodegeneration can be accelerated or decelerated through AT1R activation or blockade, respectively (243, 255, 258, 259). However, the RAS includes many complex crosslinks with pro- and anti-inflammatory endpoints (Figure 2). The main axis consists of core substrates including angiotensinogen, AngI/II, and renin. AngII mainly binds AT1R, but also the AngII receptor (AT2R), which initiates anti-inflammatory and anti-oxidative cascades (123, 260). This contrasts with AT1R, which initiates and maintains neuroinflammation and oxidative stress (260–262). AngII can be cleaved to AngIII by aminopeptidase-A, and further to AngIV and Ang3-7 by aminopeptidase N or carboxypeptidase P and prolyl-oligopeptidase, respectively. AngIII binds to AT1R and AT2R, whereas AngIV has a low affinity to AT1R and AT2R but a high affinity to the AngIV receptor (Figure 2). AngIV and analogues were shown to have a positive effect on synaptogenesis and improved clinical outcomes after stroke and subarachnoid hemorrhage by increasing cerebral blood flow (263–266).
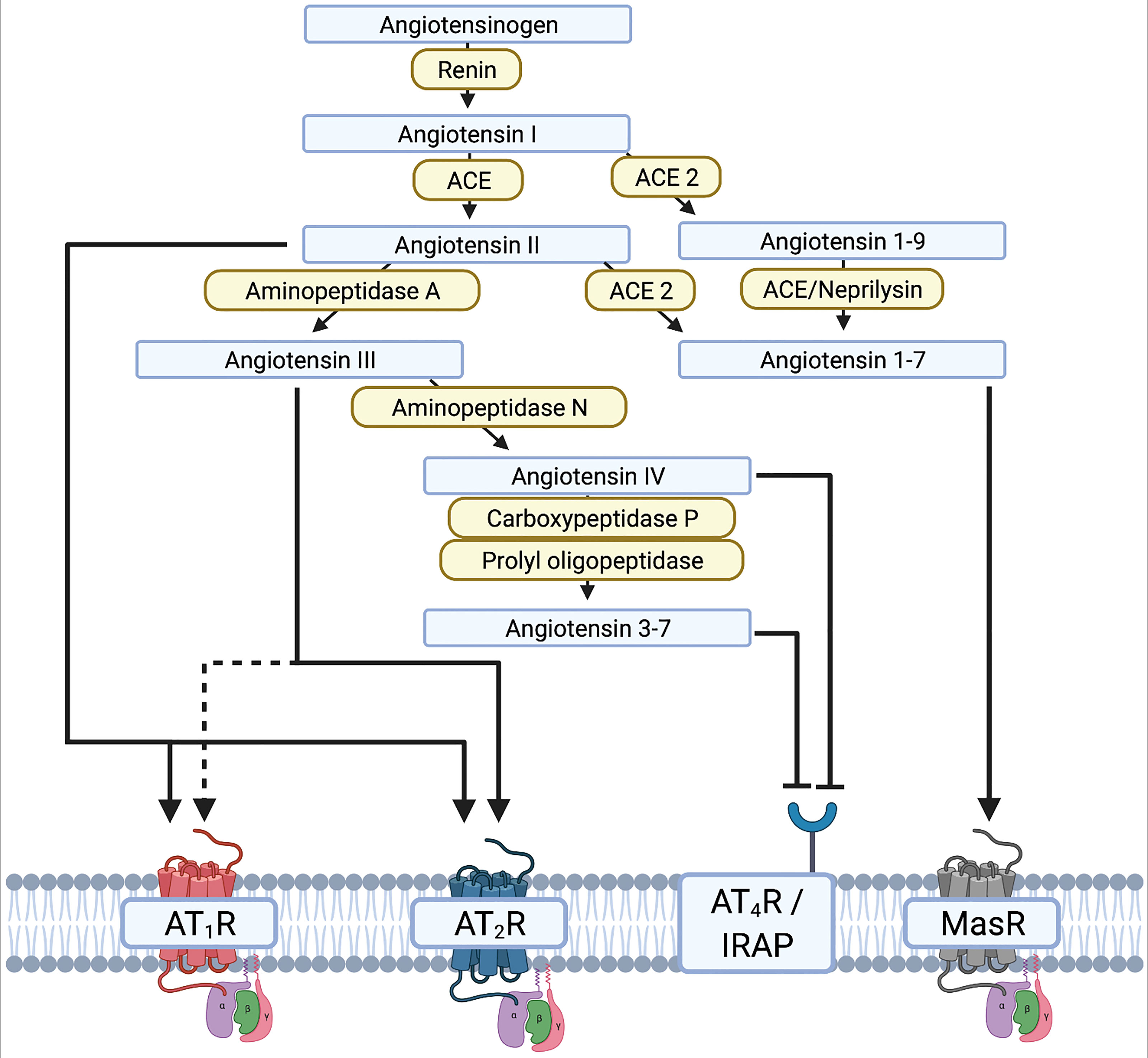
Figure 2 Illustration of the renin-angiotensin system (RAS) and the different interactions of the renin-angiotensin-[aldosterone] system RAAS substrates on receptors. - Created with BioRender.com.
The neurodegenerative effect mediated by AngII and the AngII-AT1R complex are mediated by an increased release of ROS via the NADPH-oxidase complex in microglia and dopaminergic neurons, resulting in a vicious proinflammatory circle (123). The increased release of ROS is mediated by increased nuclear expression of AT1R and NOX in dopaminergic neurons of PD patients, resulting in oxidative DNA damage and cell loss (267). In contrast, the cytoplasmic and membrane expression of AT1R is reduced (267). Under normal conditions, the AT1R-NOX interaction maintains cell homeostasis and physiological levels of dopamine. The pathological shift of AT1R expression from the membrane/cytoplasm to the nucleus reflects the disease process and contributes to secondary neurodegeneration due to increased oxidative stress. Interestingly, this shift can be observed relatively early. An altered ratio of membrane/cytoplasm to nuclear AT1R expression has already been observed in patients with a neuropathological diagnosis of PD who had not yet developed clinical manifestations or significant dopaminergic neurodegeneration (267). Yet to date, a potential re-distribution of other AT receptors in the brain of human PD patients has rarely been explored. However, ACE2 cleaves AngII and AngI to Ang1-9 and Ang1-7, respectively, and thereby shifts the RAS away from the proinflammatory main axis to the more anti-inflammatory side axis. Altogether, the RAS is a complex system that may be amenable to therapeutic modulation. The potential of targeting the RAS in PD and AD has therefore been discussed in considerable detail in dedicated review articles (123, 261).
Of note, unwanted alterations in the subtle balance between the pro- and the anti-inflammatory axis of the RAS occur during COVID-19 infections, where ACE2 is the main entry receptor for the pathogenic SARS-CoV-2 virus (268). Neurological consequences of the strong inflammation observed in severe COVID-19 remain to be studied.
5 Conclusions/Outlook
Even 200 years after the first description of PD, its pathophysiology is still far from being fully understood. Fortunately, research towards a causal treatment of PD has made substantial progress in the last 15 years. Realizing that (systemic) inflammation plays an essential role of PD, at least in its maintenance, might pave the way for new therapeutic pathways. Knowledge transfer from classical inflammatory diseases or diseases that lead to inflammatory processes will likely have a major impact on the therapy of PD, and allow new development and/or a repurposing of drugs that are already available. In addition, research in early diagnostics for PD is important. Ideally, neurodegeneration can be halted by initiating treatment before severe symptoms have developed. Skin biopsy or sleep analysis, along with new serum/liquor parameters, might lead to a sensitive and specific early diagnosis of PD. In turn, this should open up an earlier treatment window for future PD patients. Treating early with well-tolerated neuroprotective/anti-inflammatory agents certainly holds great promise for this devastating disease.
Author Contributions
AG - Conceptualization, Visualization, Writing - original draft, Writing – review & editing. RM - Writing - review & editing. JWu - Writing - original draft. JWi - Writing - review & editing. CWI - Conceptualization, Supervision, Writing - review & editing. All authors contributed to the article and approved the submitted version.
Funding
CWI is supported by the Interdisciplinary Center for Clinical Research (IZKF) at the University of Würzburg (A-303 to CWI, A-421 to CWI and JWi, N-362 to CWI), by the Deutsche Stiftung Neurologie and the ParkinsonFonds Germany and by the University Research Funds of the State of Bavaria. Moreover, CWI is supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) Project-ID 424778381-TRR 295 (A06) and the VERUM Foundation. CWI and JWi are supported by Aeterna Zentaris. AG is supported by Interdisciplinary Center for Clinical Research (IZKF) at the University of Würzburg (Z-2/79) and is a Gerok-Position holder funded by the CRC TRR 295 ‘Retune’. RM is funded by the Alexander von Humboldt-Stiftung.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Parkinson J. An Essay on the Shaking Palsy. Needly and Jones, London: Whittingham and Rowland for Sherwood (1817).
2. Goedert M, Compston A. Parkinson’s Disease — the Story of an Eponym. Nat Rev Neurol (2017) 14(1):57–62. doi: 10.1038/nrneurol.2017.165
3. Goetz CG. The History of Parkinson’s Disease: Early Clinical Descriptions and Neurological Therapies. Csh Perspect Med (2011) 1(1):a008862. doi: 10.1101/cshperspect.a008862
4. Walusinski O. Jean-Martin Charcot and Parkinson’s Disease: Teaching and Teaching Materials. Rev Neurol (2018) 174(7–8):491–505. doi: 10.1016/j.neurol.2017.08.005
5. Hoehn MM, Yahr MD. Parkinsonism: Onset, Progression, and Mortality. Neurology (1967) 17(5):427–7. doi: 10.1212/WNL.17.5.427
6. Armstrong MJ, Okun MS. Diagnosis and Treatment of Parkinson Disease. JAMA (2020) 323(6):548. doi: 10.1001/jama.2019.22360
7. Berg D, Postuma RB, Adler CH, Bloem BR, Chan P, Dubois B, et al. MDS Research Criteria for Prodromal Parkinson’s Disease: MDS Criteria for Prodromal Pd. Movement Disord (2015) 30(12):1600–11. doi: 10.1002/mds.26431
8. Braak H, Tredici KD, Rüb U, de Vos RAI, Steur ENHJ, Braak E. Staging of Brain Pathology Related to Sporadic Parkinson’s Disease. Neurobiol Aging (2003) 24(2):197–211. doi: 10.1016/S0197-4580(02)00065-9
9. Braak H, Braak E. Pathoanatomy of Parkinson’s Disease. J Neurol (2000) 247(Suppl 2):II3–10. doi: 10.1007/PL00007758
10. Surmeier DJ, Obeso JA, Halliday GM. Selective Neuronal Vulnerability in Parkinson Disease. Nat Rev Neurosci (2017) 18(2):101–13. doi: 10.1038/nrn.2016.178
11. Braak H, Ghebremedhin E, Rüb U, Bratzke H, Tredici KD. Stages in the Development of Parkinson’s Disease-Related Pathology. Cell Tissue Res (2004) 318(1):121–34. doi: 10.1007/s00441-004-0956-9
12. Dijkstra AA, Voorn P, Berendse HW, Groenewegen HJ, Bank NB, Rozemuller AJM, et al. Stage-Dependent Nigral Neuronal Loss in Incidental Lewy Body and Parkinson’s Disease. Movement Disord (2014) 29(10):1244–51. doi: 10.1002/mds.25952
13. Cheng J, Lu Q, Song L, Ho MS. α -Synuclein Trafficking in Parkinson’s Disease: Insights From Fly and Mouse Models. Asn Neuro (2018) 10:175909141881258. doi: 10.1177/1759091418812587
14. Kalia LV, Lang AE. Parkinson’s Disease. Lancet (2015) 386(9996):896–912. doi: 10.1016/S0140-6736(14)61393-3
15. Killinger BA, Kordower JH. Spreading of Alpha-Synuclein - Relevant or Epiphenomenon? J Neurochem (2019) 150(5):605–11. doi: 10.1111/jnc.14779
16. Visanji NP, Brooks PL, Hazrati L-N, Lang AE. The Prion Hypothesis in Parkinson’s Disease: Braak to the Future. Acta Neuropathol Commun (2013) 1(1):2. doi: 10.1186/2051-5960-1-2
17. Hijaz BA, Volpicelli-Daley LA. Initiation and Propagation of α-Synuclein Aggregation in the Nervous System. Mol Neurodegener (2020) 15(1):18. doi: 10.1186/s13024-020-00368-6
18. Alam MM, Yang D, Li X-Q, Liu J, Back TC, Trivett A, et al. Alpha Synuclein, the Culprit in Parkinson Disease, Is Required for Normal Immune Function. Cell Rep (2022) 38(2):110090. doi: 10.1016/j.celrep.2021.110090
19. Ransohoff RM. How Neuroinflammation Contributes to Neurodegeneration. Science (2016) 353(6301):777–83. doi: 10.1126/science.aag2590
20. Rogers J, Webster S, Lue L-F, Brachova L, Civin WH, Emmerling M, et al. Inflammation and Alzheimer’s Disease Pathogenesis. Neurobiol Aging (1996) 17(5):681–6. doi: 10.1016/0197-4580(96)00115-7
21. McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive Microglia are Positive for HLA-DR in the Substantia Nigra of Parkinson’s and Alzheimer’s Disease Brains. Neurology (1988) 38(8):1285–5. doi: 10.1212/WNL.38.8.1285
22. McGeer PL, McGeer EG. Targeting Microglia for the Treatment of Alzheimer’s Disease. Expert Opin Ther Tar (2014) 19(4):497–506. doi: 10.1517/14728222.2014.988707
23. Wang S, Breskovska I, Gandhy S, Punga AR, Guptill JT, Kaminski HJ. Advances in Autoimmune Myasthenia Gravis Management. Expert Rev Neurother (2018) 18(7):573–88. doi: 10.1080/14737175.2018.1491310
24. Venkataramaiah S, Kamath S. Management of Myasthenia Gravis. J Neuroanaesth Crit Care (2019) 06(02):153–9. doi: 10.1055/s-0039-1689739
25. Hardy TA, Reddel SW, Barnett MH, Palace J, Lucchinetti CF, Weinshenker BG. Atypical Inflammatory Demyelinating Syndromes of the CNS. Lancet Neurol (2016) 15(9):967–81. doi: 10.1016/S1474-4422(16)30043-6
26. Hartung H-P, Aktas O. Old and New Breakthroughs in Neuromyelitis Optica. Lancet Neurol (2020) 19(4):280–1. doi: 10.1016/S1474-4422(20)30062-4
27. Wesley S, Hafler DA. Chapter 51 - Multiple Sclerosis In: Rose NR, Mackay IR. The Autoimmune Diseases (Sixth Edition). Academic Press. (2020). p. 961–986. doi: 10.1016/B978-0-12-812102-3.00051-8
28. Chamberlain JL, Huda S, Whittam DH, Matiello M, Morgan BP, Jacob A. Role of Complement and Potential of Complement Inhibitors in Myasthenia Gravis and Neuromyelitis Optica Spectrum Disorders: A Brief Review. J Neurol (2021) 268:1643–64. doi: 10.1007/s00415-019-09498-4
29. Toyka K, Brachman D, Pestronk A, Kao I. Myasthenia Gravis: Passive Transfer From Man to Mouse. Science (1975) 190(4212):397–9. doi: 10.1126/science.1179220
30. Bennett JL, Lam C, Kalluri SR, Saikali P, Bautista K, Dupree C, et al. Intrathecal Pathogenic Anti-Aquaporin-4 Antibodies in Early Neuromyelitis Optica. Ann Neurol (2009) 66(5):617–29. doi: 10.1002/ana.21802
31. Braczynski AK, Schulz JB, Bach J-P. Vaccination Strategies in Tauopathies and Synucleinopathies. J Neurochem (2017) 143(5):467–88. doi: 10.1111/jnc.14207
32. Foltynie T, Langston JW. Therapies to Slow, Stop, or Reverse Parkinson’s Disease. J Park Dis (2018) 8(s1):S115–21. doi: 10.3233/JPD-181481
33. Birkmayer W, Hornykiewicz O. [The L-3,4-Dioxyphenylalanine (DOPA)-Effect in Parkinson-Akinesia]. Wien Klin Wochenschr (1961) 73:787–8.
34. Czech H, Zeidman LA. Walther Birkmayer, Co-Describer of L-Dopa, and His Nazi Connections: Victim or Perpetrator? J Hist Neurosci (2014) 23(2):160–91. doi: 10.1080/0964704X.2013.865427
35. Olanow CW. Levodopa is the Best Symptomatic Therapy for PD: Nothing More, Nothing Less. Movement Disord (2019) 34(6):812–5. doi: 10.1002/mds.27690
36. Mogi M, Harada M, Narabayashi H, Inagaki H, Minami M, Nagatsu T. Interleukin (IL)-1β, IL-2, IL-4, IL-6 and Transforming Growth Factor-α Levels are Elevated in Ventricular Cerebrospinal Fluid in Juvenile Parkinsonism and Parkinson’s Disease. Neurosci Lett (1996) 211(1):13–6. doi: 10.1016/0304-3940(96)12706-3
37. Mogi M, Harada M, Kondo T, Riederer P, Inagaki H, Minami M, et al. Interleukin-1β, Interleukin-6, Epidermal Growth Factor and Transforming Growth Factor-α are Elevated in the Brain From Parkinsonian Patients. Neurosci Lett (1994) 180(2):147–50. doi: 10.1016/0304-3940(94)90508-8
38. Mogi M, Harada M, Riederer P, Narabayashi H, Fujita K, Nagatsu T. Tumor Necrosis Factor-Alpha (TNF-Alpha) Increases Both in the Brain and in the Cerebrospinal Fluid From Parkinsonian Patients. Neurosci Lett (1994) 165(1–2):208–10. doi: 10.1016/0304-3940(94)90746-3
39. Qin X-Y, Zhang S-P, Cao C, Loh YP, Cheng Y. Aberrations in Peripheral Inflammatory Cytokine Levels in Parkinson Disease: A Systematic Review and Meta-Analysis. JAMA Neurol (2016) 73(11):1316. doi: 10.1001/jamaneurol.2016.2742
40. Fan Z, Pan Y-T, Zhang Z-Y, Yang H, Yu S-Y, Zheng Y, et al. Systemic Activation of NLRP3 Inflammasome and Plasma α-Synuclein Levels Are Correlated With Motor Severity and Progression in Parkinson’s Disease. J Neuroinflamm (2020) 17(1):11. doi: 10.1186/s12974-019-1670-6
41. Sommer A, Fadler T, Dorfmeister E, Hoffmann A-C, Xiang W, Winner B, et al. Infiltrating T Lymphocytes Reduce Myeloid Phagocytosis Activity in Synucleinopathy Model. J Neuroinflamm (2016) 13(1):174. doi: 10.1186/s12974-016-0632-5
42. Brochard V, Combadière B, Prigent A, Laouar Y, Perrin A, Beray-Berthat V, et al. Infiltration of CD4+ Lymphocytes Into the Brain Contributes to Neurodegeneration in a Mouse Model of Parkinson Disease. J Clin Invest (2009) 119(1):182–92. doi: 10.1172/JCI36470
43. Stone DK, Reynolds AD, Mosley RL, Gendelman HE. Innate and Adaptive Immunity for the Pathobiology of Parkinson’s Disease. Antioxid Redox Sign (2009) 11(9):2151–66. doi: 10.1089/ars.2009.2460
44. Kustrimovic N, Comi C, Magistrelli L, Rasini E, Legnaro M, Bombelli R, et al. Parkinson’s Disease Patients Have a Complex Phenotypic and Functional Th1 Bias: Cross-Sectional Studies of CD4+ Th1/Th2/T17 and Treg in Drug-Naïve and Drug-Treated Patients. J Neuroinflamm. (2018) 15(1):205. doi: 10.1186/s12974-018-1248-8
45. Michell-Robinson MA, Touil H, Healy LM, Owen DR, Durafourt BA, Bar-Or A, et al. Roles of Microglia in Brain Development, Tissue Maintenance and Repair. Brain (2015) 138(5):1138–59. doi: 10.1093/brain/awv066
46. Eyolfson E, Khan A, Mychasiuk R, Lohman AW. Microglia Dynamics in Adolescent Traumatic Brain Injury. J Neuroinflamm (2020) 17(1):326. doi: 10.1186/s12974-020-01994-z
47. Cherry JD, Tripodis Y, Alvarez VE, Huber B, Kiernan PT, Daneshvar DH, et al. Microglial Neuroinflammation Contributes to Tau Accumulation in Chronic Traumatic Encephalopathy. Acta Neuropathol Commun (2016) 4(1):112. doi: 10.1186/s40478-016-0382-8
48. Marques CP, Hu S, Sheng W, Lokensgard JR. Microglial Cells Initiate Vigorous Yet non-Protective Immune Responses During HSV-1 Brain Infection. Virus Res (2006) 121(1):1–10. doi: 10.1016/j.virusres.2006.03.009
49. Chapman LM, Aggrey AA, Field DJ, Srivastava K, Ture S, Yui K, et al. Platelets Present Antigen in the Context of MHC Class I. J Immunol Baltim Md 1950 (2012) 189(2):916–23. doi: 10.4049/jimmunol.1200580
50. Kao K. Plasma and Platelet HLA in Normal Individuals: Quantitation by Competitive Enzyme-Linked Immunoassay. Blood (1987) 70(1):282–6. doi: 10.1182/blood.V70.1.282.282
51. Kao K, Cook D, Scornik J. Quantitative Analysis of Platelet Surface HLA by W6/32 Anti-HLA Monoclonal Antibody. Blood (1986) 68(3):627–32. doi: 10.1182/blood.V68.3.627.627
52. Zufferey A, Schvartz D, Nolli S, Reny J-L, Sanchez J-C, Fontana P. Characterization of the Platelet Granule Proteome: Evidence of the Presence of MHC1 in Alpha-Granules. J Proteomics (2014) 101:130–40. doi: 10.1016/j.jprot.2014.02.008
53. Panicker N, Sarkar S, Harischandra DS, Neal M, Kam T-I, Jin H, et al. Fyn Kinase Regulates Misfolded α-Synuclein Uptake and NLRP3 Inflammasome Activation in Microgliafyn Mediates NLRP3 Inflammasome Activation in PD. J Exp Med (2019) 216(6):1411–30. doi: 10.1084/jem.20182191
54. Ferreira SA, Romero-Ramos M. Microglia Response During Parkinson’s Disease: Alpha-Synuclein Intervention. Front Cell Neurosci (2018) 12:247. doi: 10.3389/fncel.2018.00247
55. Colonna M, Butovsky O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu Rev Immunol (2017) 35(1):441–68. doi: 10.1146/annurev-immunol-051116-052358
56. Prinz M, Priller J. The Role of Peripheral Immune Cells in the CNS in Steady State and Disease. Nat Neurosci (2017) 20(2):136–44. doi: 10.1038/nn.4475
57. Srinivasan K, Friedman BA, Larson JL, Lauffer BE, Goldstein LD, Appling LL, et al. Untangling the Brain’s Neuroinflammatory and Neurodegenerative Transcriptional Responses. Nat Commun (2016) 7(1):11295. doi: 10.1038/ncomms11295
58. Chen X, Hu Y, Cao Z, Liu Q, Cheng Y. Cerebrospinal Fluid Inflammatory Cytokine Aberrations in Alzheimer’s Disease, Parkinson’s Disease and Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis. Front Immunol (2018) 9:2122. doi: 10.3389/fimmu.2018.02122
59. Ouchi Y, Yoshikawa E, Sekine Y, Futatsubashi M, Kanno T, Ogusu T, et al. Microglial Activation and Dopamine Terminal Loss in Early Parkinson’s Disease. Ann Neurol (2005) 57(2):168–75. doi: 10.1002/ana.20338
60. Ouchi Y, Yagi S, Yokokura M, Sakamoto M. Neuroinflammation in the Living Brain of Parkinson’s Disease. Parkinsonism Relat D (2009) 15:S200–4. doi: 10.1016/S1353-8020(09)70814-4
61. Stokholm MG, Iranzo A, Østergaard K, Serradell M, Otto M, Svendsen KB, et al. Assessment of Neuroinflammation in Patients With Idiopathic Rapid-Eye-Movement Sleep Behaviour Disorder: A Case-Control Study. Lancet Neurol (2017) 16(10):789–96. doi: 10.1016/S1474-4422(17)30173-4
62. Earls RH, Menees KB, Chung J, Barber J, Gutekunst C-A, Hazim MG, et al. Intrastriatal Injection of Preformed Alpha-Synuclein Fibrils Alters Central and Peripheral Immune Cell Profiles in Non-Transgenic Mice. J Neuroinflamm (2019) 16(1):250. doi: 10.1186/s12974-019-1636-8
63. Kuhbandner K, Hoffmann A, Alvarado MNG, Seyler L, Bäuerle T, Winkler J, et al. Alpha-Synuclein: A Modulator During Inflammatory CNS Demyelination. J Mol Neurosci (2020) 70:1038–49. doi: 10.1007/s12031-020-01498-8
64. Dinarello CA. Overview of the IL-1 Family in Innate Inflammation and Acquired Immunity. Immunol Rev (2017) 281(1):8–27. doi: 10.1111/imr.12621
65. McGeer PL, McGeer EG. Inflammation and the Degenerative Diseases of Aging. Ann Ny Acad Sci (2004) 1035(1):104–16. doi: 10.1196/annals.1332.007
66. Hirsch EC, Standaert DG. Ten Unsolved Questions About Neuroinflammation in Parkinson’s Disease. Mov Disord Off J Mov Disord Soc (2021) 36(1):16–24. doi: 10.1002/mds.28075
67. Pouchieu C, Piel C, Carles C, Gruber A, Helmer C, Tual S, et al. Pesticide Use in Agriculture and Parkinson’s Disease in the AGRICAN Cohort Study. Int J Epidemiol (2018) 47(1):299–310. doi: 10.1093/ije/dyx225
68. Fazzini E, Fleming J, Fahn S. Cerebrospinal Fluid Antibodies to Coronavirus in Patients With Parkinson’s Disease. Movement Disord (1992) 7(2):153–8. doi: 10.1002/mds.870070210
69. Netland J, Meyerholz DK, Moore S, Cassell M, Perlman S. Severe Acute Respiratory Syndrome Coronavirus Infection Causes Neuronal Death in the Absence of Encephalitis in Mice Transgenic for Human ACE2. J Virol (2008) 82(15):7264–75. doi: 10.1128/JVI.00737-08
70. Semerdzhiev SA, Fakhree MAA, Segers-Nolten I, Blum C, Claessens MMAE. Interactions Between SARS-CoV-2 N-Protein and α-Synuclein Accelerate Amyloid Formation. ACS Chem Neurosci (2022) 13:143–50. doi: 10.1101/2021.04.12.439549
71. Brundin P, Nath A, Beckham JD. Is COVID-19 a Perfect Storm for Parkinson’s Disease? Trends Neurosci (2020) 43(12):931–3. doi: 10.1016/j.tins.2020.10.009
72. Makhoul K, Jankovic J. Parkinson’s Disease After COVID-19. J Neurol Sci (2021) 422:117331. doi: 10.1016/j.jns.2021.117331
73. Sommer A, Maxreiter F, Krach F, Fadler T, Grosch J, Maroni M, et al. Th17 Lymphocytes Induce Neuronal Cell Death in a Human iPSC-Based Model of Parkinson’s Disease. Cell Stem Cell (2018) 23(1):123–31.e6. doi: 10.1016/j.stem.2018.06.015
74. Galiano-Landeira J, Torra A, Vila M, Bové J. CD8 T Cell Nigral Infiltration Precedes Synucleinopathy in Early Stages of Parkinson’s Disease. Brain (2020) 143(12):3717–33. doi: 10.1093/brain/awaa269
75. Karikari AA, McFleder RL, Ribechini E, Blum R, Bruttel V, Knorr S, et al. Neurodegeneration by α-Synuclein-Specific T Cells in AAV-A53T-α-Synuclein Parkinson’s Disease Mice. Brain Behav Immun (2022) 101:194–210. doi: 10.1016/j.bbi.2022.01.007
76. Bas J, Calopa M, Mestre M, Mollevı́ DG, Cutillas B, Ambrosio S, et al. Lymphocyte Populations in Parkinson’s Disease and in Rat Models of Parkinsonism. J Neuroimmunol (2001) 113(1):146–52. doi: 10.1016/S0165-5728(00)00422-7
77. Saunders JAH, Estes KA, Kosloski LM, Allen HE, Dempsey KM, Torres-Russotto DR, et al. CD4+ Regulatory and Effector/Memory T Cell Subsets Profile Motor Dysfunction in Parkinson’s Disease. J Neuroimmune Pharm (2012) 7(4):927–38. doi: 10.1007/s11481-012-9402-z
78. Jiang S, Gao H, Luo Q, Wang P, Yang X. The Correlation of Lymphocyte Subsets, Natural Killer Cell, and Parkinson’s Disease: A Meta-Analysis. Neurol Sci (2017) 38(8):1373–80. doi: 10.1007/s10072-017-2988-4
79. Baba Y, Kuroiwa A, Uitti RJ, Wszolek ZK, Yamada T. Alterations of T-Lymphocyte Populations in Parkinson Disease. Parkinsonism Relat D (2005) 11(8):493–8. doi: 10.1016/j.parkreldis.2005.07.005
80. Rosenkranz D, Weyer S, Tolosa E, Gaenslen A, Berg D, Leyhe T, et al. Higher Frequency of Regulatory T Cells in the Elderly and Increased Suppressive Activity in Neurodegeneration. J Neuroimmunol (2007) 188(1–2):117–27. doi: 10.1016/j.jneuroim.2007.05.011
81. Chen Y, Qi B, Xu W, Ma B, Li L, Chen Q, et al. Clinical Correlation of Peripheral CD4+-Cell Sub-Sets, Their Imbalance and Parkinson’s Disease. Mol Med Rep (2015) 12(4):6105–11. doi: 10.3892/mmr.2015.4136
82. McGeer PL, Itagaki S, Akiyama H, McGeer EG. Rate of Cell Death in Parkinsonism Indicates Active Neuropathological Process. Ann Neurol (1988) 24(4):574–6. doi: 10.1002/ana.410240415
83. Chen S, Liu Y, Niu Y, Xu Y, Zhou Q, Xu X, et al. Increased Abundance of Myeloid-Derived Suppressor Cells and Th17 Cells in Peripheral Blood of Newly-Diagnosed Parkinson’s Disease Patients. Neurosci Lett (2017) 648:21–5. doi: 10.1016/j.neulet.2017.03.045
84. Chen Y, Yu M, Liu X, Qu H, Chen Q, Qian W, et al. Clinical Characteristics and Peripheral T Cell Subsets in Parkinson’s Disease Patients With Constipation. Int J Clin Exp Patho (2015) 8(3):2495–504.
85. Storelli E, Cassina N, Rasini E, Marino F, Cosentino M. Do Th17 Lymphocytes and IL-17 Contribute to Parkinson’s Disease? A Systematic Review of Available Evidence. Front Neurol (2019) 10:13. doi: 10.3389/fneur.2019.00013
86. Shukla AW, Fox SH. Th17 Lymphocyte Spearheads the Immune Attack in Parkinson’s Disease: New Evidence for Neuronal Death. Movement Disord (2018) 33(10):1560–0. doi: 10.1002/mds.27496
87. Raj T, Rothamel K, Mostafavi S, Ye C, Lee MN, Replogle JM, et al. Polarization of the Effects of Autoimmune and Neurodegenerative Risk Alleles in Leukocytes. Sci New York NY (2014) 344(6183):519–23. doi: 10.1126/science.1249547
88. Garretti F, Agalliu D, Arlehamn CSL, Sette A, Sulzer D. Autoimmunity in Parkinson’s Disease: The Role of α-Synuclein-Specific T Cells. Front Immunol (2019) 10:303. doi: 10.3389/fimmu.2019.00303
89. Dhanwani R, Lima-Junior JR, Sethi A, Pham J, Williams G, Frazier A, et al. Transcriptional Analysis of Peripheral Memory T Cells Reveals Parkinson’s Disease-Specific Gene Signatures. NPJ Park Dis (2022) 8(1):30. doi: 10.1038/s41531-022-00282-2
90. de Graaf MT, Smitt PAES, Luitwieler RL, van Velzen C, van den Broek PDM, Kraan J, et al. Central Memory CD4+ T Cells Dominate the Normal Cerebrospinal Fluid. Cytom Part B Clin Cytom (2011) 80B(1):43–50. doi: 10.1002/cyto.b.20542
91. Hong S, Zhang Z, Liu H, Tian M, Zhu X, Zhang Z, et al. B Cells Are the Dominant Antigen-Presenting Cells That Activate Naive CD4+ T Cells Upon Immunization With a Virus-Derived Nanoparticle Antigen. Immunity (2018) 49(4):695–708.e4. doi: 10.1016/j.immuni.2018.08.012
92. Bell L, Lenhart A, Rosenwald A, Monoranu CM, Berberich-Siebelt F. Lymphoid Aggregates in the CNS of Progressive Multiple Sclerosis Patients Lack Regulatory T Cells. Front Immunol (2020) 10:3090. doi: 10.3389/fimmu.2019.03090
93. Rowe DB, Le W, Smith RG, Appel SH. Antibodies From Patients With Parkinson’s Disease React With Protein Modified by Dopamine Oxidation. J Neurosci Res (1998) 53(5):551–8. doi: 10.1002/(SICI)1097-4547(19980901)53:5<551::AID-JNR5>3.0.CO;2-8
94. Orr CF, Rowe DB, Mizuno Y, Mori H, Halliday GM. A Possible Role for Humoral Immunity in the Pathogenesis of Parkinson’s Disease. Brain (2005) 128(11):2665–74. doi: 10.1093/brain/awh625
95. Han M, Nagele E, DeMarshall C, Acharya N, Nagele R. Diagnosis of Parkinson’s Disease Based on Disease-Specific Autoantibody Profiles in Human Sera. PLoS One (2012) 7(2):e32383. doi: 10.1371/journal.pone.0032383
96. Li R, Tropea TF, Baratta LR, Zuroff L, Diaz-Ortiz ME, Zhang B, et al. Abnormal B-Cell and Tfh-Cell Profiles in Patients With Parkinson Disease: A Cross-Sectional Study. Neurol - Neuroimmunol Neuroinflamm (2022) 9(2):e1125. doi: 10.1212/NXI.0000000000001125
97. Sun C, Yu W, Zhao Z, Song C, Liu Y, Jia G, et al. Peripheral Humoral Immune Response Is Associated With the Non-Motor Symptoms of Parkinson’s Disease. Front Neurosci-switz (2019) 13:1057. doi: 10.3389/fnins.2019.01057
98. Wang P, Luo M, Zhou W, Jin X, Xu Z, Yan S, et al. Global Characterization of Peripheral B Cells in Parkinson’s Disease by Single-Cell RNA and BCR Sequencing. Front Immunol (2022) 13:814239. doi: 10.3389/fimmu.2022.814239
99. Li X, Koudstaal W, Fletcher L, Costa M, van Winsen M, Siregar B, et al. Naturally Occurring Antibodies Isolated From PD Patients Inhibit Synuclein Seeding In Vitro and Recognize Lewy Pathology. Acta Neuropathol (2019) 137(5):825–36. doi: 10.1007/s00401-019-01974-5
100. Shalash A, Salama M, Makar M, Roushdy T, Elrassas HH, Mohamed W, et al. Elevated Serum α-Synuclein Autoantibodies in Patients With Parkinson’s Disease Relative to Alzheimer’s Disease and Controls. Front Neurol (2017) 8:720. doi: 10.3389/fneur.2017.00720
101. Ray A, Basu S, Williams CB, Salzman NH, Dittel BN. A Novel IL-10–Independent Regulatory Role for B Cells in Suppressing Autoimmunity by Maintenance of Regulatory T Cells via GITR Ligand. J Immunol (2012) 188(7):3188–98. doi: 10.4049/jimmunol.1103354
102. Hoffman PM, Robbins DS, Oldstone MBA, Gibbs CJ, Gajdusek DC. Humoral Immunity in Guamanians With Amyotrophic Lateral Sclerosis and Parkinsonism-Dementia. Ann Neurol (1981) 10(2):193–6. doi: 10.1002/ana.410100210
103. Sabatino JJ, Pröbstel A-K, Zamvil SS. B Cells in Autoimmune and Neurodegenerative Central Nervous System Diseases. Nat Rev Neurosci (2019) 20(12):728–45. doi: 10.1038/s41583-019-0233-2
104. Orsini F, Blasio DD, Zangari R, Zanier ER, Simoni M-GD. Versatility of the Complement System in Neuroinflammation, Neurodegeneration and Brain Homeostasis. Front Cell Neurosci (2014) 8:380. doi: 10.3389/fncel.2014.00380
105. Ahn JJ, Abu-Rub M, Miller RH. B Cells in Neuroinflammation: New Perspectives and Mechanistic Insights. Cells (2021) 10(7):1605. doi: 10.3390/cells10071605
106. Racette BA, Gross A, Vouri SM, Camacho-Soto A, Willis AW, Nielsen SS. Immunosuppressants and Risk of Parkinson Disease. Ann Clin Transl Neur. (2018) 5(7):870–5. doi: 10.1002/acn3.580
107. Hancock DB, Martin ER, Stajich JM, Jewett R, Stacy MA, Scott BL, et al. Smoking, Caffeine, and Nonsteroidal Anti-Inflammatory Drugs in Families With Parkinson Disease. Arch Neurol-Chicago (2007) 64(4):576. doi: 10.1001/archneur.64.4.576
108. Qiu F, Liang C-L, Liu H, Zeng Y-Q, Hou S, Huang S, et al. Impacts of Cigarette Smoking on Immune Responsiveness: Up and Down or Upside Down? Oncotarget (2016) 8(1):268–84. doi: 10.18632/oncotarget.13613
109. Sopori M. Effects of Cigarette Smoke on the Immune System. Nat Rev Immunol (2002) 2(5):372–7. doi: 10.1038/nri803
110. Ansel H, David M, Yiyun H, Nabeel N, Jim R, Richard C, et al. Imaging Dynamic Neuroimmune Responses to LPS in Tobacco Smokers: A [11C]PBR28 PET Study. J Nucl Med (2019) 60(Suppl 1):488.
111. Fan J, Nielsen SS, Faust IM, Racette BA. Transplant and Risk of Parkinson Disease. Parkinsonism Relat D (2019) 63:149–55. doi: 10.1016/j.parkreldis.2019.02.013
112. Peter I, Dubinsky M, Bressman S, Park A, Lu C, Chen N, et al. Anti-Tumor Necrosis Factor Therapy and Incidence of Parkinson Disease Among Patients With Inflammatory Bowel Disease. JAMA Neurol (2018) 75(8):939. doi: 10.1001/jamaneurol.2018.0605
113. Camacho-Soto A, Gross A, Nielsen SS, Dey N, Racette BA. Inflammatory Bowel Disease and Risk of Parkinson’s Disease in Medicare Beneficiaries. Parkinsonism Relat D (2018) 50:23–8. doi: 10.1016/j.parkreldis.2018.02.008
114. Chen H, Jacobs E, Schwarzschild MA, McCullough ML, Calle EE, Thun MJ, et al. Nonsteroidal Antiinflammatory Drug Use and the Risk for Parkinson’s Disease. Ann Neurol (2005) 58(6):963–7. doi: 10.1002/ana.20682
115. Gao X, Chen H, Schwarzschild MA, Ascherio A. Use of Ibuprofen and Risk of Parkinson Disease(E–Pub Ahead of Print). Neurology (2011) 76(10):863–9. doi: 10.1212/WNL.0b013e31820f2d79
116. Asanuma M, Nishibayashi-Asanuma S, Miyazaki I, Kohno M, Ogawa N. Neuroprotective Effects of non-Steroidal Anti-Inflammatory Drugs by Direct Scavenging of Nitric Oxide Radicals. J Neurochem (2001) 76(6):1895–904. doi: 10.1046/j.1471-4159.2001.00205.x
117. Casper D, Yaparpalvi U, Rempel N, Werner P. Ibuprofen Protects Dopaminergic Neurons Against Glutamate Toxicity In Vitro. Neurosci Lett (2000) 289(3):201–4. doi: 10.1016/S0304-3940(00)01294-5
118. Bower JH, Maraganore DM, Peterson BJ, Ahlskog JE, Rocca WA. Immunologic Diseases, Anti-Inflammatory Drugs, and Parkinson Disease: A Case-Control Study. Neurology (2006) 67(3):494–6. doi: 10.1212/01.wnl.0000227906.99570.cc
119. Fang L, Karakiulakis G, Roth M. Are Patients With Hypertension and Diabetes Mellitus at Increased Risk for COVID-19 Infection? Lancet Respir Med (2020) 8(4):e21. doi: 10.1016/S2213-2600(20)30116-8
120. Qiao W, Wang C, Chen B, Zhang F, Liu Y, Lu Q, et al. Ibuprofen Attenuates Cardiac Fibrosis in Streptozotocin-Induced Diabetic Rats. Cardiology (2015) 131(2):97–106. doi: 10.1159/000375362
121. Hopfner F, Höglinger GU, Kuhlenbäumer G, Pottegård A, Wod M, Christensen K, et al. β-Adrenoreceptors and the Risk of Parkinson’s Disease. Lancet Neurol (2020) 19(3):247–54. doi: 10.1016/S1474-4422(19)30400-4
122. Magistrelli L, Comi C. Beta2-Adrenoceptor Agonists in Parkinson’s Disease and Other Synucleinopathies. J Neuroimmune Pharmacol (2020) 15(1):74–81. doi: 10.1007/s11481-018-09831-0
123. Perez-Lloret S, Otero-Losada M, Toblli JE, Capani F. Renin-Angiotensin System as a Potential Target for New Therapeutic Approaches in Parkinson’s Disease. Expert Opin Inv Drug (2017) 26(10):1163–73. doi: 10.1080/13543784.2017.1371133
124. Jeong S, Jang W, Shin DW. Association of Statin Use With Parkinson’s Disease: Dose–response Relationship. Movement Disord (2019) 34(7):1014–21. doi: 10.1002/mds.27681
125. Samii A, Carleton BC, Etminan M. Statin Use and the Risk of Parkinson Disease: A Nested Case Control Study. J Clin Neurosci Off J Neurosurg Soc Australas (2008) 15(11):1272–3. doi: 10.1016/j.jocn.2008.01.016
126. Poly TN, Islam MM, Walther BA, Yang H-C, Nguyen P-A, Huang C-W, et al. Exploring the Association Between Statin Use and the Risk of Parkinson’s Disease: A Meta-Analysis of Observational Studies. Neuroepidemiology (2017) 49(3–4):142–51. doi: 10.1159/000480401
127. Shi Q, Liu S, Fonseca VA, Thethi TK, Shi L. Effect of Metformin on Neurodegenerative Disease Among Elderly Adult US Veterans With Type 2 Diabetes Mellitus. BMJ Open (2019) 9(7):e024954. doi: 10.1136/bmjopen-2018-024954
128. Kuan Y-C, Huang K-W, Lin C-L, Hu C-J, Kao C-H. Effects of Metformin Exposure on Neurodegenerative Diseases in Elderly Patients With Type 2 Diabetes Mellitus. Prog Neuro-Psychoph (2017) 79(Pt B):77–83. doi: 10.1016/j.pnpbp.2017.06.002
129. Athauda D, Foltynie T. Insulin Resistance and Parkinson’s Disease: A New Target for Disease Modification? Prog Neurobiol (2016) 145–146:98–120. doi: 10.1016/j.pneurobio.2016.10.001
130. Aviles-Olmos I, Limousin P, Lees A, Foltynie T. Parkinson’s Disease, Insulin Resistance and Novel Agents of Neuroprotection. Brain (2013) 136(Pt 2):374–84. doi: 10.1093/brain/aws009
131. Yang L, Wang H, Liu L, Xie A. The Role of Insulin/IGF-1/PI3K/Akt/Gsk3β Signaling in Parkinson’s Disease Dementia. Front Neurosci-switz (2018) 12:73. doi: 10.3389/fnins.2018.00073
132. During MJ, Cao L, Zuzga DS, Francis JS, Fitzsimons HL, Jiao X, et al. Glucagon-Like Peptide-1 Receptor is Involved in Learning and Neuroprotection. Nat Med (2003) 9(9):1173–9. doi: 10.1038/nm919
133. Chen S, Yu S-J, Li Y, Lecca D, Glotfelty E, Kim HK, et al. Post-Treatment With PT302, a Long-Acting Exendin-4 Sustained Release Formulation, Reduces Dopaminergic Neurodegeneration in a 6-Hydroxydopamine Rat Model of Parkinson’s Disease. Sci Rep-uk (2018) 8(1):10722. doi: 10.1038/s41598-018-28449-z
134. Bertilsson G, Patrone C, Zachrisson O, Andersson A, Dannaeus K, Heidrich J, et al. Peptide Hormone Exendin-4 Stimulates Subventricular Zone Neurogenesis in the Adult Rodent Brain and Induces Recovery in an Animal Model of Parkinson’s Disease. J Neurosci Res (2008) 86(2):326–38. doi: 10.1002/jnr.21483
135. Liu W, Li Y, Jalewa J, Saunders-Wood T, Li L, Hölscher C. Neuroprotective Effects of an Oxyntomodulin Analogue in the MPTP Mouse Model of Parkinson’s Disease. Eur J Pharmacol (2015) 765:284–90. doi: 10.1016/j.ejphar.2015.08.038
136. Yun SP, Kam T-I, Panicker N, Kim S, Oh Y, Park J-S, et al. Block of A1 Astrocyte Conversion by Microglia is Neuroprotective in Models of Parkinson’s Disease. Nat Med (2018) 24(7):931–8. doi: 10.1038/s41591-018-0051-5
137. Lee C-H, Jeon SJ, Cho KS, Moon E, Sapkota A, Jun HS, et al. Activation of Glucagon-Like Peptide-1 Receptor Promotes Neuroprotection in Experimental Autoimmune Encephalomyelitis by Reducing Neuroinflammatory Responses. Mol Neurobiol (2018) 55(4):3007–20. doi: 10.1007/s12035-017-0550-2
138. Wu H-Y, Tang X-Q, Mao X-F, Wang Y-X. Autocrine Interleukin-10 Mediates Glucagon-Like Peptide-1 Receptor-Induced Spinal Microglial β-Endorphin Expression. J Neurosci (2017) 37(48):11701–14. doi: 10.1523/JNEUROSCI.1799-17.2017
139. National Library of Medicine (U.S.). GLP1R in Parkinson's Disease. (2018) Identifier NCT03659682. clinicaltrials.gov-NCT03659682.
140. Zhu X, Han Y, Du J, Liu R, Jin K, Yi W. Microbiota-Gut-Brain Axis and the Central Nervous System. Oncotarget (2017) 8(32):53829–38. doi: 10.18632/oncotarget.17754
141. Bienenstock J, Kunze W, Forsythe P. Microbiota and the Gut–Brain Axis. Nutr Rev (2015) 73(suppl 1):28–31. doi: 10.1093/nutrit/nuv019
142. Wang H-X, Wang Y-P. Gut Microbiota-Brain Axis. Chin Med J-peking (2016) 129(19):2373–80. doi: 10.4103/0366-6999.190667
143. Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell (2016) 167(6):1469–80.e12. doi: 10.1016/j.cell.2016.11.018
144. Olszak T, An D, Zeissig S, Vera MP, Richter J, Franke A, et al. Microbial Exposure During Early Life has Persistent Effects on Natural Killer T Cell Function. Sci New York NY (2012) 336(6080):489–93. doi: 10.1126/science.1219328
145. Quigley EMM. Microbiota-Brain-Gut Axis and Neurodegenerative Diseases. Curr Neurol Neurosci (2017) 17(12):94. doi: 10.1007/s11910-017-0802-6
146. Bansal V, Costantini T, Kroll L, Peterson C, Loomis W, Eliceiri B, et al. Traumatic Brain Injury and Intestinal Dysfunction: Uncovering the Neuro-Enteric Axis. J Neurotraum (2009) 26(8):1353–9. doi: 10.1089/neu.2008.0858
147. Katzenberger RJ, Chtarbanova S, Rimkus SA, Fischer JA, Kaur G, Seppala JM, et al. Death Following Traumatic Brain Injury in Drosophila is Associated With Intestinal Barrier Dysfunction. Elife (2015) 4:e04790. doi: 10.7554/eLife.04790
148. Sun M-F, Shen Y-Q. Dysbiosis of Gut Microbiota and Microbial Metabolites in Parkinson’s Disease. Ageing Res Rev (2018) 45:53–61. doi: 10.1016/j.arr.2018.04.004
149. Forsyth CB, Shannon KM, Kordower JH, Voigt RM, Shaikh M, Jaglin JA, et al. Increased Intestinal Permeability Correlates With Sigmoid Mucosa Alpha-Synuclein Staining and Endotoxin Exposure Markers in Early Parkinson’s Disease. PloS One (2011) 6(12):e28032. doi: 10.1371/journal.pone.0028032
150. Braak H, de Vos RAI, Bohl J, Tredici KD. Gastric α-Synuclein Immunoreactive Inclusions in Meissner’s and Auerbach’s Plexuses in Cases Staged for Parkinson’s Disease-Related Brain Pathology. Neurosci Lett (2006) 396(1):67–72. doi: 10.1016/j.neulet.2005.11.012
151. Chartier-Harlin M-C, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, et al. α-Synuclein Locus Duplication as a Cause of Familial Parkinson’s Disease. Lancet (2004) 364(9440):1167–9. doi: 10.1016/S0140-6736(04)17103-1
152. Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, et al. α-Synuclein Locus Triplication Causes Parkinson9s Disease. Science (2003) 302(5646):841–1. doi: 10.1126/science.1090278
153. Jowaed A, Schmitt I, Kaut O, Wüllner U. Methylation Regulates Alpha-Synuclein Expression and Is Decreased in Parkinson’s Disease Patients’ Brains. J Neurosci (2010) 30(18):6355–9. doi: 10.1523/JNEUROSCI.6119-09.2010
154. National Library of Medicine (U.S.). Gut Microbiota and Parkinson's Disease. (2018) Identifier NCT03710668. clinicaltrials.gov-NCT03710668.
155. Coles LD, Tuite PJ, Öz G, Mishra UR, Kartha RV, Sullivan KM, et al. Repeated-Dose Oral N-Acetylcysteine in Parkinson’s Disease: Pharmacokinetics and Effect on Brain Glutathione and Oxidative Stress. J Clin Pharmacol (2018) 58(2):158–67. doi: 10.1002/jcph.1008
156. Smeyne M, Smeyne RJ. Glutathione Metabolism and Parkinson’s Disease. Free Radic Biol Med (2013) 62:13–25. doi: 10.1016/j.freeradbiomed.2013.05.001
157. Dias V, Junn E, Mouradian MM. The Role of Oxidative Stress in Parkinson’s Disease. J Park Dis (2013) 3(4):461–91. doi: 10.3233/JPD-130230
158. Giasson BI, Duda JE, Murray IVJ, Chen Q, Souza JM, Hurtig HI, et al. Oxidative Damage Linked to Neurodegeneration by Selective α-Synuclein Nitration in Synucleinopathy Lesions. Science (2000) 290(5493):985–9. doi: 10.1126/science.290.5493.985
159. Andersen JK. Oxidative Stress in Neurodegeneration: Cause or Consequence? Nat Med (2004) 10(Suppl 7):S18–25. doi: 10.1038/nrn1434
160. Bandookwala M, Sengupta P. 3-Nitrotyrosine: A Versatile Oxidative Stress Biomarker for Major Neurodegenerative Diseases. Int J Neurosci (2020) 130(10):1–16. doi: 10.1080/00207454.2020.1713776
161. clinicaltrials.gov-NCT02445651. Physiological Effects of Nutritional Support in Patients With Parkinson’s Disease (201). Available at: https://clinicaltrials.gov/ct2/show/NCT02445651.
162. Weiss R, Schilling E, Grahnert A, Kölling V, Dorow J, Ceglarek U, et al. Nicotinamide: A Vitamin Able to Shift Macrophage Differentiation Toward Macrophages With Restricted Inflammatory Features. Innate Immun (2015) 21(8):813–26. doi: 10.1177/1753425915602545
163. Jia H, Li X, Gao H, Feng Z, Li X, Zhao L, et al. High Doses of Nicotinamide Prevent Oxidative Mitochondrial Dysfunction in a Cellular Model and Improve Motor Deficit in a Drosophila Model of Parkinson’s Disease. J Neurosci Res (2008) 86(9):2083–90. doi: 10.1002/jnr.21650
164. Wakade C, Chong R, Bradley E, Thomas B, Morgan J. Upregulation of GPR109A in Parkinson’s Disease. PLoS One (2014) 9(10):e109818. doi: 10.1371/journal.pone.0109818
165. Giri B, Belanger K, Seamon M, Bradley E, Purohit S, Chong R, et al. Niacin Ameliorates Neuro-Inflammation in Parkinson’s Disease via GPR109A. Int J Mol Sci (2019) 20(18):4559. doi: 10.3390/ijms20184559
166. Seamon M, Purohit S, Giri B, Baban B, Morgan J, Chong R, et al. Niacin for Parkinson’s Disease. Clin Exp Neuroimmunol (2019) 11(1):47–56. doi: 10.1111/cen3.12553
167. clinicaltrials.gov-NCT03808961. Niacin for Parkinsons Disease (2019). Available at: https://clinicaltrials.gov/ct2/show/NCT03808961.
168. Ungerstedt U. 6-Hydroxy-Dopamine Induced Degeneration of Central Monoamine Neurons. Eur J Pharmacol (1968) 5(1):107–10. doi: 10.1016/0014-2999(68)90164-7
169. Langston JW. MPTP And Parkinson’s Disease. Trends Neurosci (1985) 8:79–83. doi: 10.1016/0166-2236(85)90031-1
170. Parillaud VR, Lornet G, Monnet Y, Privat A-L, Haddad AT, Brochard V, et al. Analysis of Monocyte Infiltration in MPTP Mice Reveals That Microglial CX3CR1 Protects Against Neurotoxic Over-Induction of Monocyte-Attracting CCL2 by Astrocytes. J Neuroinflamm. (2017) 14(1):60. doi: 10.1186/s12974-017-0830-9
171. Lee E, Hwang I, Park S, Hong S, Hwang B, Cho Y, et al. MPTP-Driven NLRP3 Inflammasome Activation in Microglia Plays a Central Role in Dopaminergic Neurodegeneration. Cell Death Differ (2018) 26(2):213–28. doi: 10.1038/s41418-018-0124-5
172. Simola N, Morelli M, Carta AR. The 6-Hydroxydopamine Model of Parkinson’s Disease. Neurotox Res (2007) 11(3–4):151–67. doi: 10.1007/BF03033565
173. McGeer PL, Kawamata T, Walker DG, Akiyama H, Tooyama I, McGeer EG. Microglia in Degenerative Neurological Disease. Glia (1993) 7(1):84–92. doi: 10.1002/glia.440070114
174. Ambrosi G, Kustrimovic N, Siani F, Rasini E, Cerri S, Ghezzi C, et al. Complex Changes in the Innate and Adaptive Immunity Accompany Progressive Degeneration of the Nigrostriatal Pathway Induced by Intrastriatal Injection of 6-Hydroxydopamine in the Rat. Neurotox Res (2017) 32(1):71–81. doi: 10.1007/s12640-017-9712-2
175. Singh SS, Rai SN, Birla H, Zahra W, Rathore AS, Singh SP. NF-κb-Mediated Neuroinflammation in Parkinson’s Disease and Potential Therapeutic Effect of Polyphenols. Neurotox Res (2019) 37(3):491–507. doi: 10.1007/s12640-019-00147-2
176. Tang Y, Le W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol Neurobiol (2015) 53(2):1181–94. doi: 10.1007/s12035-014-9070-5
177. Hunot S, Boissière F, Faucheux B, Brugg B, Mouatt-Prigent A, Agid Y, et al. Nitric Oxide Synthase and Neuronal Vulnerability in Parkinson’s Disease. Neuroscience (1996) 72(2):355–63. doi: 10.1016/0306-4522(95)00578-1
178. Ghosh A, Roy A, Liu X, Kordower JH, Mufson EJ, Hartley DM, et al. Selective Inhibition of NF-κb Activation Prevents Dopaminergic Neuronal Loss in a Mouse Model of Parkinson’s Disease. Proc Natl Acad Sci (2007) 104(47):18754–9. doi: 10.1073/pnas.0704908104
179. Noelker C, Morel L, Lescot T, Osterloh A, Alvarez-Fischer D, Breloer M, et al. Toll Like Receptor 4 Mediates Cell Death in a Mouse MPTP Model of Parkinson Disease. Sci Rep-uk (2013) 3(1):1393. doi: 10.1038/srep01393
180. Qureshi ST, Larivière L, Leveque G, Clermont S, Moore KJ, Gros P, et al. Endotoxin-Tolerant Mice Have Mutations in Toll-Like Receptor 4 (Tlr4). J Exp Med (1999) 189(4):615–25. doi: 10.1084/jem.189.4.615
181. Mao Z, Liu C, Ji S, Yang Q, Ye H, Han H, et al. The NLRP3 Inflammasome is Involved in the Pathogenesis of Parkinson’s Disease in Rats. Neurochem Res (2017) 42(4):1104–15. doi: 10.1007/s11064-017-2185-0
182. Heng Y, Zhang Q-S, Mu Z, Hu J-F, Yuan Y-H, Chen N-H. Ginsenoside Rg1 Attenuates Motor Impairment and Neuroinflammation in the MPTP-Probenecid-Induced Parkinsonism Mouse Model by Targeting α-Synuclein Abnormalities in the Substantia Nigra. Toxicol Lett (2016) 243:7–21. doi: 10.1016/j.toxlet.2015.12.005
183. Liu J-Q, Zhao M, Zhang Z, Cui L-Y, Zhou X, Zhang W, et al. Rg1 Improves LPS-Induced Parkinsonian Symptoms in Mice via Inhibition of NF-κb Signaling and Modulation of M1/M2 Polarization. Acta Pharmacol Sin (2020) 41(4):523–34. doi: 10.1038/s41401-020-0358-x
184. Matheoud D, Sugiura A, Bellemare-Pelletier A, Laplante A, Rondeau C, Chemali M, et al. Parkinson’s Disease-Related Proteins PINK1 and Parkin Repress Mitochondrial Antigen Presentation. Cell (2016) 166(2):314–27. doi: 10.1016/j.cell.2016.05.039
185. Matheoud D, Cannon T, Voisin A, Penttinen A-M, Ramet L, Fahmy AM, et al. Intestinal Infection Triggers Parkinson’s Disease-Like Symptoms in Pink1–/– Mice. Nature (2019) 571(7766):565–9. doi: 10.1038/s41586-019-1405-y
186. Huang B, Wu S, Wang Z, Ge L, Rizak JD, Wu J, et al. Phosphorylated α-Synuclein Accumulations and Lewy Body-Like Pathology Distributed in Parkinson’s Disease-Related Brain Areas of Aged Rhesus Monkeys Treated With MPTP. Neuroscience (2018) 379:302–15. doi: 10.1016/j.neuroscience.2018.03.026
187. Ip CW, Klaus L-C, Karikari AA, Visanji NP, Brotchie JM, Lang AE, et al. AAV1/2-Induced Overexpression of A53T-α-Synuclein in the Substantia Nigra Results in Degeneration of the Nigrostriatal System With Lewy-Like Pathology and Motor Impairment: A New Mouse Model for Parkinson’s Disease. Acta Neuropathol Commun (2017) 5(1):11. doi: 10.1186/s40478-017-0416-x
188. Haque ME, Akther M, Jakaria M, Kim I, Azam S, Choi D. Targeting the Microglial NLRP3 Inflammasome and Its Role in Parkinson’s Disease. Mov Disord (2020) 35(1):20–33. doi: 10.1002/mds.27874
189. Labzin LI, Heneka MT, Latz E. Innate Immunity and Neurodegeneration. Annu Rev Med (2018) 69(1):437–49. doi: 10.1146/annurev-med-050715-104343
190. Béraud D, Twomey M, Bloom B, Mittereder A, Ton V, Neitzke K, et al. α-Synuclein Alters Toll-Like Receptor Expression. Front Neurosci-switz (2011) 5:80. doi: 10.3389/fnins.2011.00080
191. Cao S, Theodore S, Standaert DG. Fcγ Receptors Are Required for NF-κb Signaling, Microglial Activation and Dopaminergic Neurodegeneration in an AAV-Synuclein Mouse Model of Parkinson’s Disease. Mol Neurodegener (2010) 5(1):42. doi: 10.1186/1750-1326-5-42
192. Daëron M. Fc RECEPTOR BIOLOGY. Annu Rev Immunol (1997) 15(1):203–34. doi: 10.1146/annurev.immunol.15.1.203
193. Rostami J, Fotaki G, Sirois J, Mzezewa R, Bergström J, Essand M, et al. Astrocytes Have the Capacity to Act as Antigen-Presenting Cells in the Parkinson’s Disease Brain. J Neuroinflamm. (2020) 17(1):119. doi: 10.1186/s12974-020-01776-7
194. Bido S, Muggeo S, Massimino L, Marzi MJ, Giannelli SG, Melacini E, et al. Microglia-Specific Overexpression of α-Synuclein Leads to Severe Dopaminergic Neurodegeneration by Phagocytic Exhaustion and Oxidative Toxicity. Nat Commun (2021) 12(1):6237. doi: 10.1038/s41467-021-26519-x
195. Harms AS, Cao S, Rowse AL, Thome AD, Li X, Mangieri LR, et al. MHCII Is Required for -Synuclein-Induced Activation of Microglia, CD4 T Cell Proliferation, and Dopaminergic Neurodegeneration. J Neurosci (2013) 33(23):9592–600. doi: 10.1523/JNEUROSCI.5610-12.2013
196. Spielman LJ, Gibson DL, Klegeris A. Unhealthy Gut, Unhealthy Brain: The Role of the Intestinal Microbiota in Neurodegenerative Diseases. Neurochem Int (2018) 120:149–63. doi: 10.1016/j.neuint.2018.08.005
197. Schonhoff AM, Williams GP, Wallen ZD, Standaert DG, Harms AS. Innate and Adaptive Immune Responses in Parkinson’s Disease. Prog Brain Res (2020) 252:169–216. doi: 10.1016/bs.pbr.2019.10.006
198. Süß P, Kalinichenko L, Baum W, Reichel M, Kornhuber J, Loskarn S, et al. Hippocampal Structure and Function are Maintained Despite Severe Innate Peripheral Inflammation. Brain Behav Immun (2015) 49:156–70. doi: 10.1016/j.bbi.2015.05.011
199. Süß P, Hoffmann A, Rothe T, Ouyang Z, Baum W, Staszewski O, et al. Chronic Peripheral Inflammation Causes a Region-Specific Myeloid Response in the Central Nervous System. Cell Rep (2020) 30(12):4082–95.e6. doi: 10.1016/j.celrep.2020.02.109
200. Arlehamn CSL, Dhanwani R, Pham J, Kuan R, Frazier A, Dutra JR, et al. α-Synuclein-Specific T Cell Reactivity Is Associated With Preclinical and Early Parkinson’s Disease. Nat Commun (2020) 11(1):1875. doi: 10.1038/s41467-020-15626-w
201. Ip CW, Beck SK, Volkmann J. Lymphocytes Reduce Nigrostriatal Deficits in the 6-Hydroxydopamine Mouse Model of Parkinson’s Disease. J Neural Transm (2015) 122(12):1633–43. doi: 10.1007/s00702-015-1444-y
202. Teismann P. COX-2 in the Neurodegenerative Process of Parkinson’s Disease. Biofactors (2012) 38(6):395–7. doi: 10.1002/biof.1035
203. Chandra G, Roy A, Rangasamy SB, Pahan K. Induction of Adaptive Immunity Leads to Nigrostriatal Disease Progression in MPTP Mouse Model of Parkinson’s Disease. J Immunol (2017) 198(11):4312–26. doi: 10.4049/jimmunol.1700149
204. Chandra G, Rangasamy SB, Roy A, Kordower JH, Pahan K. Neutralization of RANTES and Eotaxin Prevents the Loss of Dopaminergic Neurons in a Mouse Model of Parkinson Disease. J Biol Chem (2016) 291(29):15267–81. doi: 10.1074/jbc.M116.714824
205. Roy A, Mondal S, Kordower JH, Pahan K. Attenuation of Microglial RANTES by NEMO-Binding Domain Peptide Inhibits the Infiltration of CD8+ T Cells in the Nigra of Hemiparkinsonian Monkey. Neuroscience (2015) 302:36–46. doi: 10.1016/j.neuroscience.2015.03.011
206. Reynolds AD, Stone DK, Hutter JAL, Benner EJ, Mosley RL, Gendelman HE. Regulatory T Cells Attenuate Th17 Cell-Mediated Nigrostriatal Dopaminergic Neurodegeneration in a Model of Parkinson’s Disease. J Immunol (2010) 184(5):2261–71. doi: 10.4049/jimmunol.0901852
207. Liu Z, Huang Y, Cao B-B, Qiu Y-H, Peng Y-P. Th17 Cells Induce Dopaminergic Neuronal Death via LFA-1/ICAM-1 Interaction in a Mouse Model of Parkinson’s Disease. Mol Neurobiol (2017) 54(10):7762–76. doi: 10.1007/s12035-016-0249-9
208. Martin HL, Santoro M, Mustafa S, Riedel G, Forrester JV, Teismann P. Evidence for a Role of Adaptive Immune Response in the Disease Pathogenesis of the MPTP Mouse Model of Parkinson’s Disease. Glia (2015) 64(3):386–95. doi: 10.1002/glia.22935
209. Luchtman DW, Shao D, Song C. Behavior, Neurotransmitters and Inflammation in Three Regimens of the MPTP Mouse Model of Parkinson’s Disease. Physiol Behav (2009) 98(1–2):130–8. doi: 10.1016/j.physbeh.2009.04.021
210. Heuer A, Smith GA, Dunnett SB. Comparison of 6-Hydroxydopamine Lesions of the Substantia Nigra and the Medial Forebrain Bundle on a Lateralised Choice Reaction Time Task in Mice. Eur J Neurosci (2012) 37(2):294–302. doi: 10.1111/ejn.12036
211. Ungerstedt U, Arbuthnott GW. Quantitative Recording of Rotational Behavior in Rats After 6-Hydroxy-Dopamine Lesions of the Nigrostriatal Dopamine System. Brain Res (1970) 24(3):485–93. doi: 10.1016/0006-8993(70)90187-3
212. Zigmond MJ, Hastings TG, Abercrombie ED. Neurochemical Responses to 6-Hydroxydopamine and L-Dopa Therapy: Implications for Parkinson’s Disease. Ann Ny Acad Sci (1992) 648(1 Neurotoxins a):71–86. doi: 10.1111/j.1749-6632.1992.tb24525.x
213. Kupsch A, Schmidt W, Gizatullina Z, Debska-Vielhaber G, Voges J, Striggow F, et al. 6-Hydroxydopamine Impairs Mitochondrial Function in the Rat Model of Parkinson’s Disease: Respirometric, Histological, and Behavioral Analyses. J Neural Transm (2014) 121(10):1245–57. doi: 10.1007/s00702-014-1185-3
214. Wheeler CJ, Seksenyan A, Koronyo Y, Rentsendorj A, Sarayba D, Wu H, et al. T-Lymphocyte Deficiency Exacerbates Behavioral Deficits in the 6-OHDA Unilateral Lesion Rat Model for Parkinson’s Disease. J Neurol Neurophysiol (2014) 05(03):209. doi: 10.4172/2155-9562.1000209
215. Koprich JB, Johnston TH, Reyes MG, Sun X, Brotchie JM. Expression of Human A53T Alpha-Synuclein in the Rat Substantia Nigra Using a Novel AAV1/2 Vector Produces a Rapidly Evolving Pathology With Protein Aggregation, Dystrophic Neurite Architecture and Nigrostriatal Degeneration With Potential to Model the Pathology of Parkinson’s Disease. Mol Neurodegener (2010) 5(1):43. doi: 10.1186/1750-1326-5-43
216. Theodore S, Cao S, McLean PJ, Standaert DG. Targeted Overexpression of Human α-Synuclein Triggers Microglial Activation and an Adaptive Immune Response in a Mouse Model of Parkinson Disease. J Neuropathol Exp Neurol (2008) 67(12):1149–58. doi: 10.1097/NEN.0b013e31818e5e99
217. Williams GP, Schonhoff AM, Jurkuvenaite A, Gallups NJ, Standaert DG, Harms AS. CD4 T Cells Mediate Brain Inflammation and Neurodegeneration in a Mouse Model of Parkinson Disease. Brain (2021) 144(7):2047–59. doi: 10.1093/brain/awab103
218. Harms AS, Delic V, Thome AD, Bryant N, Liu Z, Chandra S, et al. α-Synuclein Fibrils Recruit Peripheral Immune Cells in the Rat Brain Prior to Neurodegeneration. Acta Neuropathol Commun (2017) 5(1):85. doi: 10.1186/s40478-017-0494-9
219. Kim S, Kwon S-H, Kam T-I, Panicker N, Karuppagounder SS, Lee S, et al. Transneuronal Propagation of Pathologic α-Synuclein From the Gut to the Brain Models Parkinson’s Disease. Neuron (2019) 103(4):627–41.e7. doi: 10.1016/j.neuron.2019.05.035
220. Ramos JMP, Iribarren P, Bousset L, Melki R, Baekelandt V, Perren AVd. Peripheral Inflammation Regulates CNS Immune Surveillance Through the Recruitment of Inflammatory Monocytes Upon Systemic α-Synuclein Administration. Front Immunol (2019) 10:80. doi: 10.3389/fimmu.2019.00080
221. Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, et al. Mutations in the DJ-1 Gene Associated With Autosomal Recessive Early-Onset Parkinsonism. Science (2002) 299(5604):256–9. doi: 10.1126/science.1077209
222. Guo M. PINK1/PARKIN And Mitochondrial Dynamics In Neurodegeneration. Free Radical Bio Med (2017) 112:16. doi: 10.1016/j.freeradbiomed.2017.10.369
223. Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, et al. Mutations in the Parkin Gene Cause Autosomal Recessive Juvenile Parkinsonism. Nature (1998) 392(6676):605–8. doi: 10.1038/33416
224. Valente EM, Abou-Sleiman PM, Caputo V, Muqit MMK, Harvey K, Gispert S, et al. Hereditary Early-Onset Parkinson’s Disease Caused by Mutations in PINK1. Science (2004) 304(5674):1158–60. doi: 10.1126/science.1096284
225. Weindel CG, Bell SL, Vail KJ, West KO, Patrick KL, Watson RO. LRRK2 Maintains Mitochondrial Homeostasis and Regulates Innate Immune Responses to Mycobacterium tuberculosis. ELife (2020) 9:e51071. doi: 10.7554/eLife.51071
226. Aubin N, Curet O, Deffois A, Carter C. Aspirin and Salicylate Protect Against MPTP-Induced Dopamine Depletion in Mice. J Neurochem (1998) 71(4):1635–42. doi: 10.1046/j.1471-4159.1998.71041635.x
227. Ren L, Yi J, Yang J, Li P, Cheng X, Mao P. Nonsteroidal Anti-Inflammatory Drugs Use and Risk of Parkinson Disease. Medicine (2018) 97(37):e12172. doi: 10.1097/MD.0000000000012172
228. Teismann P, Tieu K, Choi D-K, Wu D-C, Naini A, Hunot S, et al. Cyclooxygenase-2 is Instrumental in Parkinson’s Disease Neurodegeneration. Proc Natl Acad Sci (2003) 100(9):5473–8. doi: 10.1073/pnas.0837397100
229. Feng Z-H, Wang T-G, Li D-D, Fung P, Wilson BC, Liu B, et al. Cyclooxygenase-2-Deficient Mice are Resistant to 1-Methyl-4-Phenyl1, 2, 3, 6-Tetrahydropyridine-Induced Damage of Dopaminergic Neurons in the Substantia Nigra. Neurosci Lett (2002) 329(3):354–8. doi: 10.1016/S0304-3940(02)00704-8
230. Yamagata K, Andreasson KI, Kaufmann WE, Barnes CA, Worley PF. Expression of a Mitogen-Inducible Cyclooxygenase in Brain Neurons: Regulation by Synaptic Activity and Glucocorticoids. Neuron (1993) 11(2):371–86. doi: 10.1016/0896-6273(93)90192-T
231. Andreasson K. Emerging Roles of PGE2 Receptors in Models of Neurological Disease. Prostag Oth Lipid M (2010) 91(3–4):104–12. doi: 10.1016/j.prostaglandins.2009.04.003
232. Caughey GE, Cleland LG, Penglis PS, Gamble JR, James MJ. Roles of Cyclooxygenase (COX)-1 and COX-2 in Prostanoid Production by Human Endothelial Cells: Selective Up-Regulation of Prostacyclin Synthesis by COX-2. J Immunol (2001) 167(5):2831–8. doi: 10.4049/jimmunol.167.5.2831
233. Moncada S. Prostacyclin and Arterial Wall Biology. Arterioscler Off J Am Hear Assoc Inc (1982) 2(3):193–207. doi: 10.1161/01.ATV.2.3.193
234. Jin J, Shie F-S, Liu J, Wang Y, Davis J, Schantz AM, et al. Prostaglandin E2 Receptor Subtype 2 (EP2) Regulates Microglial Activation and Associated Neurotoxicity Induced by Aggregated α-Synuclein. J Neuroinflamm (2007) 4(1):2. doi: 10.1186/1742-2094-4-2
235. Carrasco E, Werner P, Casper D. Prostaglandin Receptor EP2 Protects Dopaminergic Neurons Against 6-OHDA-Mediated Low Oxidative Stress. Neurosci Lett (2008) 441(1):44–9. doi: 10.1016/j.neulet.2008.05.111
236. Davies NM, Wright MR, Jamali F. Antiinflammatory Drug-Induced Small Intestinal Permeability: The Rat Is a Suitable Model. Pharmaceut Res (1994) 11(11):1652–6. doi: 10.1023/A:1018978308752
237. Bjarnason I, Williams P, Smethurst P, Peters TJ, Levi AJ. Effect of non-Steroidal Anti-Inflammatory Drugs and Prostaglandins on the Permeability of the Human Small Intestine. Gut (1986) 27(11):1292–7. doi: 10.1136/gut.27.11.1292
238. Roy J, Galano J, Durand T, Guennec JL, Lee JC. Physiological Role of Reactive Oxygen Species as Promoters of Natural Defenses. FASEB J (2017) 31(9):3729–45. doi: 10.1096/fj.201700170R
239. Lingappan K. NF-κb in Oxidative Stress. Curr Opin Toxicol (2018) 7:81–6. doi: 10.1016/j.cotox.2017.11.002
240. Groemping Y, Rittinger K. Activation and Assembly of the NADPH Oxidase: A Structural Perspective. Biochem J (2005) 386(3):401–16. doi: 10.1042/BJ20041835
241. Rodriguez-Pallares J, Rey P, Parga JA, Muñoz A, Guerra MJ, Labandeira-Garcia JL. Brain Angiotensin Enhances Dopaminergic Cell Death via Microglial Activation and NADPH-Derived ROS. Neurobiol Dis (2008) 31(1):58–73. doi: 10.1016/j.nbd.2008.03.003
242. Wu D-C, Teismann P, Tieu K, Vila M, Jackson-Lewis V, Ischiropoulos H, et al. NADPH Oxidase Mediates Oxidative Stress in the 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Model of Parkinson’s Disease. Proc Natl Acad Sci (2003) 100(10):6145–50. doi: 10.1073/pnas.0937239100
243. Parga JA, Rodriguez-Perez AI, Garcia-Garrote M, Rodriguez-Pallares J, Labandeira-Garcia JL. Angiotensin II Induces Oxidative Stress and Upregulates Neuroprotective Signaling From the NRF2 and KLF9 Pathway in Dopaminergic Cells. Free Radic Biol Med (2018) 129:394–406. doi: 10.1016/j.freeradbiomed.2018.10.409
244. Belarbi K, Cuvelier E, Destée A, Gressier B, Chartier-Harlin M-C. NADPH Oxidases in Parkinson’s Disease: A Systematic Review. Mol Neurodegener (2017) 12(1):84. doi: 10.1186/s13024-017-0225-5
245. Tu D, Gao Y, Yang R, Guan T, Hong J-S, Gao H-M. The Pentose Phosphate Pathway Regulates Chronic Neuroinflammation and Dopaminergic Neurodegeneration. J Neuroinflamm (2019) 16(1):255. doi: 10.1186/s12974-019-1659-1
246. Firbank MJ, Yarnall AJ, Lawson RA, Duncan GW, Khoo TK, Petrides GS, et al. Cerebral Glucose Metabolism and Cognition in Newly Diagnosed Parkinson’s Disease: ICICLE-PD Study. J Neurol Neurosurg Psychiatry (2016) 88(4):310–6. doi: 10.1136/jnnp-2016-313918
247. Marques A, Dutheil F, Durand E, Rieu I, Mulliez A, Fantini ML, et al. Glucose Dysregulation in Parkinson’s Disease: Too Much Glucose or Not Enough Insulin? Parkinsonism Relat D (2018) 55:122–7. doi: 10.1016/j.parkreldis.2018.05.026
248. Pagano G, Polychronis S, Wilson H, Giordano B, Ferrara N, Niccolini F, et al. Diabetes Mellitus and Parkinson Disease. Neurology (2018) 90(19):e1654–62. doi: 10.1212/WNL.0000000000005475
249. Stanton RC. Glucose-6-Phosphate Dehydrogenase, NADPH, and Cell Survival. IUBMB Life (2012) 64(5):362–9. doi: 10.1002/iub.1017
250. Dunn L, Allen GFG, Mamais A, Ling H, Li A, Duberley KE, et al. Dysregulation of Glucose Metabolism Is an Early Event in Sporadic Parkinson’s Disease. Neurobiol Aging (2014) 35(5):1111–5. doi: 10.1016/j.neurobiolaging.2013.11.001
251. Braak H, Sastre M, Bohl JRE, de Vos RAI, Tredici KD. Parkinson’s Disease: Lesions in Dorsal Horn Layer I, Involvement of Parasympathetic and Sympathetic Pre- and Postganglionic Neurons. Acta Neuropathol (2007) 113(4):421–9. doi: 10.1007/s00401-007-0193-x
252. Chinta SJ, Andersen JK. Reversible Inhibition of Mitochondrial Complex I Activity Following Chronic Dopaminergic Glutathione Depletion In Vitro: Implications for Parkinson’s Disease. Free Radical Bio Med (2006) 41(9):1442–8. doi: 10.1016/j.freeradbiomed.2006.08.002
253. Mischley LK, Lau RC, Shankland EG, Wilbur TK, Padowski JM. Phase IIb Study of Intranasal Glutathione in Parkinson’s Disease. J Park Dis (2017) 7(2):289–99. doi: 10.3233/JPD-161040
254. Sian J, Dexter DT, Lees AJ, Daniel S, Agid Y, Javoy-Agid F, et al. Alterations in Glutathione Levels in Parkinson’s Disease and Other Neurodegenerative Disorders Affecting Basal Ganglia. Ann Neurol (1994) 36(3):348–55. doi: 10.1002/ana.410360305
255. Grammatopoulos TN, Jones SM, Ahmadi FA, Hoover BR, Snell LD, Skoch J, et al. Angiotensin Type 1 Receptor Antagonist Losartan, Reduces MPTP-Induced Degeneration of Dopaminergic Neurons in Substantia Nigra. Mol Neurodegener (2007) 2(1):1. doi: 10.1186/1750-1326-2-1
256. Laghlam D, Jozwiak M, Nguyen LS. Renin-Angiotensin-Aldosterone System and Immunomodulation: A State-Of-the-Art Review. Cells (2021) 10(7):1767. doi: 10.3390/cells10071767
257. te Riet L, van Esch JHM, Roks AJM, van den Meiracker AH, Danser AHJ. Hypertension. Circ Res (2015) 116(6):960–75. doi: 10.1161/CIRCRESAHA.116.303587
258. Rey P, Lopez-Real A, Sanchez-Iglesias S, Muñoz A, Soto-Otero R, Labandeira-Garcia JL. Angiotensin Type-1-Receptor Antagonists Reduce 6-Hydroxydopamine Toxicity for Dopaminergic Neurons. Neurobiol Aging (2007) 28(4):555–67. doi: 10.1016/j.neurobiolaging.2006.02.018
259. Joglar B, Rodriguez-Pallares J, Rodriguez-Perez AI, Rey P, Guerra MJ, Labandeira-Garcia JL. The Inflammatory Response in the MPTP Model of Parkinson’s Disease is Mediated by Brain Angiotensin: Relevance to Progression of the Disease. J Neurochem (2009) 109(2):656–69. doi: 10.1111/j.1471-4159.2009.05999.x
260. Chabrashvili T, Kitiyakara C, Blau J, Karber A, Aslam S, Welch WJ, et al. Effects of ANG II Type 1 and 2 Receptors on Oxidative Stress, Renal NADPH Oxidase, and SOD Expression. Am J Physiology-regulatory Integr Comp Physiol (2003) 285(1):R117–24. doi: 10.1152/ajpregu.00476.2002
261. Wright JW, Kawas LH, Harding JW. A Role for the Brain RAS in Alzheimer’s and Parkinson’s Diseases. Front Endocrinol (2013) 4:158. doi: 10.3389/fendo.2013.00158
262. Muñoz A, Rey P, Guerra MJ, Mendez-Alvarez E, Soto-Otero R, Labandeira-Garcia JL. Reduction of Dopaminergic Degeneration and Oxidative Stress by Inhibition of Angiotensin Converting Enzyme in a MPTP Model of Parkinsonism. Neuropharmacology (2006) 51(1):112–20. doi: 10.1016/j.neuropharm.2006.03.004
263. Dalmay F, Pesteil F, Nisse-Durgeat S, Fournier A, Achard JM. Angiotensin IV Decreases Acute Stroke Mortality in the Gerbil. Am J Hypertens (2001) 14(11):A56. doi: 10.1016/S0895-7061(01)01592-8
264. Kramár EA, Harding JW, Wright JW. Angiotensin II- and IV-Induced Changes in Cerebral Blood Flow. Regul Peptides (1997) 68(2):131–8. doi: 10.1016/S0167-0115(96)02116-7
265. Näveri L, Strömberg C, Saavedra JM. Angiotensin IV Reverses the Acute Cerebral Blood Flow Reduction After Experimental Subarachnoid Hemorrhage in the Rat. J Cereb Blood Flow Metab (1994) 14(6):1096–9. doi: 10.1038/jcbfm.1994.143
266. Benoist CC, Wright JW, Zhu M, Appleyard SM, Wayman GA, Harding JW. Facilitation of Hippocampal Synaptogenesis and Spatial Memory by C-Terminal Truncated Nle 1 -Angiotensin IV Analogs. J Pharmacol Exp Ther (2011) 339(1):35–44. doi: 10.1124/jpet.111.182220
267. Zawada WM, Mrak RE, Biedermann J, Palmer QD, Gentleman SM, Aboud O, et al. Loss of Angiotensin II Receptor Expression in Dopamine Neurons in Parkinson’s Disease Correlates With Pathological Progression and Is Accompanied by Increases in Nox4- and 8-OH Guanosine-Related Nucleic Acid Oxidation and Caspase-3 Activation. Acta Neuropathol Commun (2015) 3(1):9. doi: 10.1186/s40478-015-0189-z
Keywords: Parkinson’s disease, neuroinflammation, T cells, microglia, neurodegeneration, animal models, inflammatory cascades
Citation: Grotemeyer A, McFleder RL, Wu J, Wischhusen J and Ip CW (2022) Neuroinflammation in Parkinson’s Disease – Putative Pathomechanisms and Targets for Disease-Modification. Front. Immunol. 13:878771. doi: 10.3389/fimmu.2022.878771
Received: 18 February 2022; Accepted: 20 April 2022;
Published: 18 May 2022.
Edited by:
Serge Nataf, Université Claude Bernard Lyon 1, FranceReviewed by:
Omid Mirmosayyeb, Isfahan University of Medical Sciences, IranHarry Alexopoulos, National and Kapodistrian University of Athens, Greece
Luke Michael Healy, McGill University, Canada
Copyright © 2022 Grotemeyer, McFleder, Wu, Wischhusen and Ip. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chi Wang Ip, SXBfQ0B1a3cuZGU=