 Amy M. FitzPatrick
Amy M. FitzPatrick- Independent Researcher, Los Angeles, CA, United States
Sex bias in autoimmune disease (AID) prevalence is known, but the role of estrogen in disease progression is more complex. Estrogen can even be protective in some AIDs; but in systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), and systemic sclerosis (SSc), estrogen, its metabolites, and its receptors have been demonstrated to play critical, localized inflammatory roles. Estrogen is instrumental to the fibrosis seen in RA, SLE, SSc and other disease states, including breast cancer and uterine leiomyomas. Fibrotic diseases tend to share a common pattern in which lymphocyte–monocyte interactions generate cytokines which stimulate the deposition of fibrogenic connective tissue. RA, SLE, SSc and thyroid eye disease (TED) have very similar inflammatory and fibrotic patterns—from pathways to tissue type. The thorough investigations that demonstrated estrogen’s role in the pathology of RA, SLE, and SSc could, and possibly should, be carried out in TED. One might even expect to find an even greater role for estrogen, and sex steroid homeostasis in TED, given that TED is typically sequalae to Graves’ disease (GD), or Hashimoto’s disease (HD), and these are endocrine disorders that can create considerable sex steroid hormone dysregulation. This paper highlights the pathophysiology similarities in 4 AIDs, examines the evidence of sex steroid mediated pathology across 3 AIDs and offers a case study and speculation on how this may be germane to TED.
Introduction
Though a fair portion of the inflammatory cascade in thyroid eye disease (TED) is now well characterized, some key aspects of the disease remain enigmatic. For example, why do orbital fibroblasts in patients with TED express insulin-like growth factor-1 receptor (IGF-1R) at higher levels than people without TED? Why does only a subset of patients with GD or HD manifest TED? Some think it is possible that all GD patients do get TED, but it remains subclinical or very mild in most patients. The question would still remain—what keeps those TED cases subclinical? Furthermore, what, precisely, is the effect of euthyroid status on TED? Anti-thyroid medications, used to achieve euthyroid status, such as methimazole affect T4/T3 synthesis but their impact on the immune system remains to be defined. Therefore, it seems unlikely that the drug itself protects against TED. In fact, someone can achieve euthyroid status with methimazole but have no reduction in levels of thyroid autoantibody. Autoantibody levels are also not a direct predictor of TED. It is possible for a patient to have Graves’ disease with a high TSI (thyroid stimulating immunoglobulin) and not to have TED (1, 2). What prevents TSI from antagonizing the orbital TSHRs in these cases? As with any autoimmune disease, there may be multiple answers to that question. But the possible interaction of the endocrine system with the immune system is not yet represented in the TED literature. This is curious because that interaction has been examined in a number of other AIDs – and since Graves’ and Hashimoto’s diseases disrupt endocrine homeostasis, the endocrine-immune interaction in these diseases could be ripe for pathology.
TED Pathophysiology
Orbital fibroblasts in patients with TED express both thyrotropin receptor (TSHR) and IGF-1R at higher levels than found in fibroblasts of healthy controls. In the case of Graves’ disease, thyroid autoantibodies bind to TSHR, the main autoantigen in TED (3). TSHRs then complex to IGF-1Rs, via β-arrestin, beginning the immune activation (2, 4). Lymphocytes migrate to the orbit. B cells are activated via their recognition of TSHR, and via the CD40:CD40L molecular bridge between B cell and T cell surfaces. B cell proliferation and differentiation ensues. Cytokine releasing T cells are activated, producing adhesion molecules, and facilitating further traffic into the orbit. Activated orbital fibroblasts proliferate, secrete hyaluronan and pro-inflammatory cytokines including IL-1β, IL-1α, IL-6, IL-8, macrophage chemoattractant protein-1 (MCP-1) and transforming growth factor-β (TGF- β). This cascade perpetuates orbital inflammation and facilitates the fibroblast differentiation into adipocytes and myofibroblasts, expanding adipose tissue and fluid accumulation between the muscle fibers. Ultimately, this process increases the volume of the orbital tissues, remodels the orbit, and leads to the characteristic proptosis and congestive features of TED (2, 3, 5).
Typical of fibrosis, tissue proliferation in TED is mediated via the phosphoinositide-3 kinase/AKT/mammalian target of rapamycin (PI3K/AKT/mTOR), adenylyl cyclase/cyclic adenosine monophosphate (cAMP) and MAPK/ERK pathways (Figure 1) (3, 6, 7).
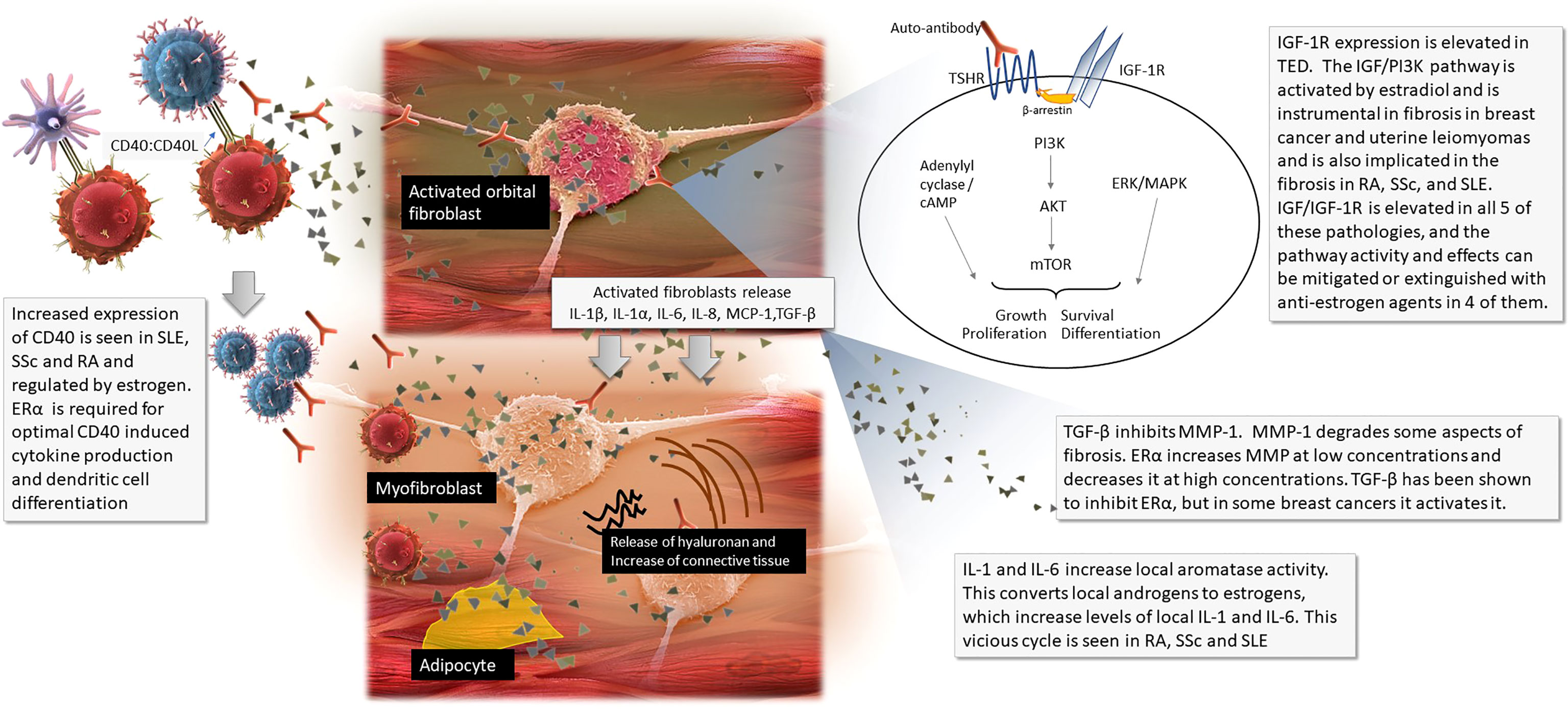
Figure 1 The pathophysiology of thyroid eye disease with callouts of where sex steroids influence other diseases.
Clinical manifestations of this pathophysiology include proptosis, double vision, edema around the orbit and in the eyelids, festoons, eyelid retraction, corneal lacerations, pain and increase in buccal fat. The initial inflammatory and progressive phase typically stops after two years and is followed by a stable fibrotic stage (1, 8). Most cases of TED are mild or self-limiting, but up to ~30% require treatment, usually immunosuppressive (9). Multiple surgeries may also be required and in severe cases, TED can be sight threatening (10). A targeted monoclonal antibody antagonist of IGF-1R is now approved by the FDA and has demonstrated efficacy in the treatment of moderate to severe TED (8). The reduction in the patient’s quality of life has been measured as being on par with cancer (11).
The onset of TED can vary but tends to occur within 18 months of a diagnosis with uncontrolled Graves’ disease. It can also occur in patients with Hashimoto’s disease, and rarely even in people with no thyroidopathy (12).
Epidemiology of TED
The incidence of TED is 16 per 100,000 females and 2.9 per 100,000 males and is found across ethnicities. Like most autoimmunity, the preponderance is higher in women, but as seen in SSc, in severe cases, the ratio reverses to 1:4 females to males (13, 14). The pooled prevalence of TED that occurs in Graves’ disease is 40% (15).
Similarities in Pathophysiology of Three Model Autoimmune Diseases
Fibroblasts overactivation and aberrant differentiation and proliferation are central to TED pathophysiology. Under physiologic conditions, fibroblasts are sentinels that maintain tissues homeostasis and are therefore flexible and responsive to stimuli and their environment. Reductively speaking, fibrotic disease is essentially aberrant or nonterminating wound healing (6, 16).
RA, SLE and SSc have several similarities to TED pathogenesis. They occur in fibroblast-type tissues and can result in a type of fibrosis. IGF is thought to mediate this fibrosis (17–20). These three AIDs also have other similarities to TED, such as overexpression of CD40 causing issues with the CD40:CD40L molecular bridge; the involve several of the same pro-inflammatory cytokines including IL-1β, IL-1α, IL-6; and the PI3K/AKT/mTOR proliferative pathways. The unusual, characteristic two-year burnout seen in TED is also seen in SSc. Estrogen has been shown to play a pivotal role in the each of these inflammatory and fibrotic pathologies in RA, SSc and SLE. A summary of these similarities can be seen in Table 1 and Figure 1 (3, 21–26). Though there are differences among them, given these similarities and the fact that fibrosis and inflammation can have similar pathology across many disease types and are both known to be modulated by estrogen, it seems worth investigating if estrogen, and sex steroid homeostasis, is also instrumental in TED (7, 27).

Table 1 Similar pathogenic pathways in 4 autoimmune diseases.
Sex Steroid Hormones and the Immune System
Most cells of the innate and adaptive immune system express estrogen receptors, and the endocrine system exerts manifold regulation on the immune cells. This interdependency has been investigated for decades and has yielded many interesting observations about the endocrine system as much as the immune. For example, progesterone receptors are not seen in resting states of monocytes but do appear in pregnancy. You cannot always mimic estradiol effects in vitro which are seen in vivo, so there may be temporal aspects that vary. Additionally, hormone receptors are found at different concentrations on different types of immune cells, illustrating flexibility in regulation (28–31).
Estrogen Can Be Anti-Inflammatory or Inflammatory
Estrogen’s impact on the immune system depends on the estrogen receptor type, tissue type, cellular conditions, concentrations of estrogen, concentrations of it relative to other hormones, estrogen metabolites, and, at least in the case of autoimmunity, it may also depend on polymorphisms of estrogen receptor types (25, 32–35).
Estrogen Receptors
Estrogen receptor alpha (ERα) and beta (ERβ) are both nuclear receptors that once bound with their ligand estradiol (E2) translocate to the nucleus where they can regulate gene expression or serve as cofactors with other transcription factors, such as NF-κB (36, 37). They have strong and equal affinity to the E2 and also have affinity for downstream estrogen metabolites—which can also be either anti-inflammatory or inflammatory depending on the type and ratio to each other (27, 32, 38).
ERα and ERβ (from genes ESR1 and ESR2, respectively) are ubiquitous and are expressed on most immune (innate and adaptive) cells in addition to a large assortment of other cells and tissues (38, 39). ERα and ERβ can be co-expressed in a cell, and each receptor regulates different genes. They are also found preferentially and in different concentrations in different tissue types (28, 31, 36, 38–40). For example, CD4+ T cells, which are helper cells express greater amounts of ERα than Erβ, and a greater amount of both ERs than peripheral blood monocytes or CD8+ T cells, which are cytotoxic. B cells express more Erβ, than ERα (41). ERs regulation of different genes and expression at different levels may explain cellular and tissue-specific response to estrogen.
In general, estrogen activity mediated via ERα is proliferative and anti-apoptotic and has therefore been implicated in many proliferative diseases, such as prostate cancer, breast cancer, and autoimmune disorders (23, 31, 34). ERα has recently been demonstrated promote T cell activation and proliferation in autoimmune disease, and to be instrumental to effector functions and dendritic cell development (42–44). The knockout of ERα in SLE murine models resulted in significantly attenuated disease and prolonged survival (45). Other ERα knock out models have demonstrated the receptor’s involvement in physiological lymphoid organ development. Erlandsson et al., showed ERα was mandatory in male mice for development of full-size thymus and spleen, this was not the case in females, who displayed thymic atrophy mediated by ERβ. Of note to TED, these authors noted that a potential background for their findings may have been the down-regulated activity in the growth hormone/IGF-1 axis in males lacking ERα (46). Sánchez-Maldonado, et al., found that 3 ESR1 haplotypes had a reduced risk of erosive arthritis, and that both ESR1 and ESR2 influence the disease (47). Finally, in a colitis mouse model, ERα deletion in T cells reduced their pathogenic potential via increased expression of Foxp3 (42). When the PI3Kδ pathway is activated in lymphocytes, it inhibits FOXO, when it’s not, FOXO is disinhitbited (48). These are just some examples of the manifold ways ERs participate in immune regulation and its pathology, the literature is replete with other examples, as ERs are ubiquitous.
ERβ has been found frequently to be anti-proliferative, and counters ERα’s anti-apoptotic effects (31, 49). In some tissues, ERβ has been shown to prevent or protect against fibrosis (23, 50, 51). ERβ-selective ligands are being investigated as a therapeutic target in Crohn’s disease and rheumatoid arthritis (49, 52).
A third group of more recently discovered and much faster-acting, membrane-bound G-coupled estrogen receptors are also being investigated for their roles in regulating growth factors in cancer and in the immune system (53–55). G protein-coupled estrogen receptor 1 (GPER1) is a Gs-coupled receptor expressed in innate and adaptive immune cells and can facilitate estrogenic activity independently of the other ERs or with their coordination (55, 56).
Estrogen Metabolites
Estradiol and estrone (E1, a weaker estrogen found in all women, but in higher quantities in post-menopausal women) both are converted into hydroxylated metabolites that can also bind with strong or weak affinity to ERs. E1 and E2 are hydroxylated at the C2, C4 and C16 positions and these 6 metabolites are named thusly:

These catechol estrogens can then be methylated into methyoxyestrogens. In general, the 2OH’s and 4OH-E1 are anti-inflammatory at high concentrations, and the 16αOH’s and 4OH-E2 are inflammatory. 16αOH’s have lower affinity tor ERs than estradiol, but they bond covalently and fail to downregulate the receptor, facilitating proliferation in disease such as cancer and autoimmunity (Figure 2) (33, 35, 57–63).
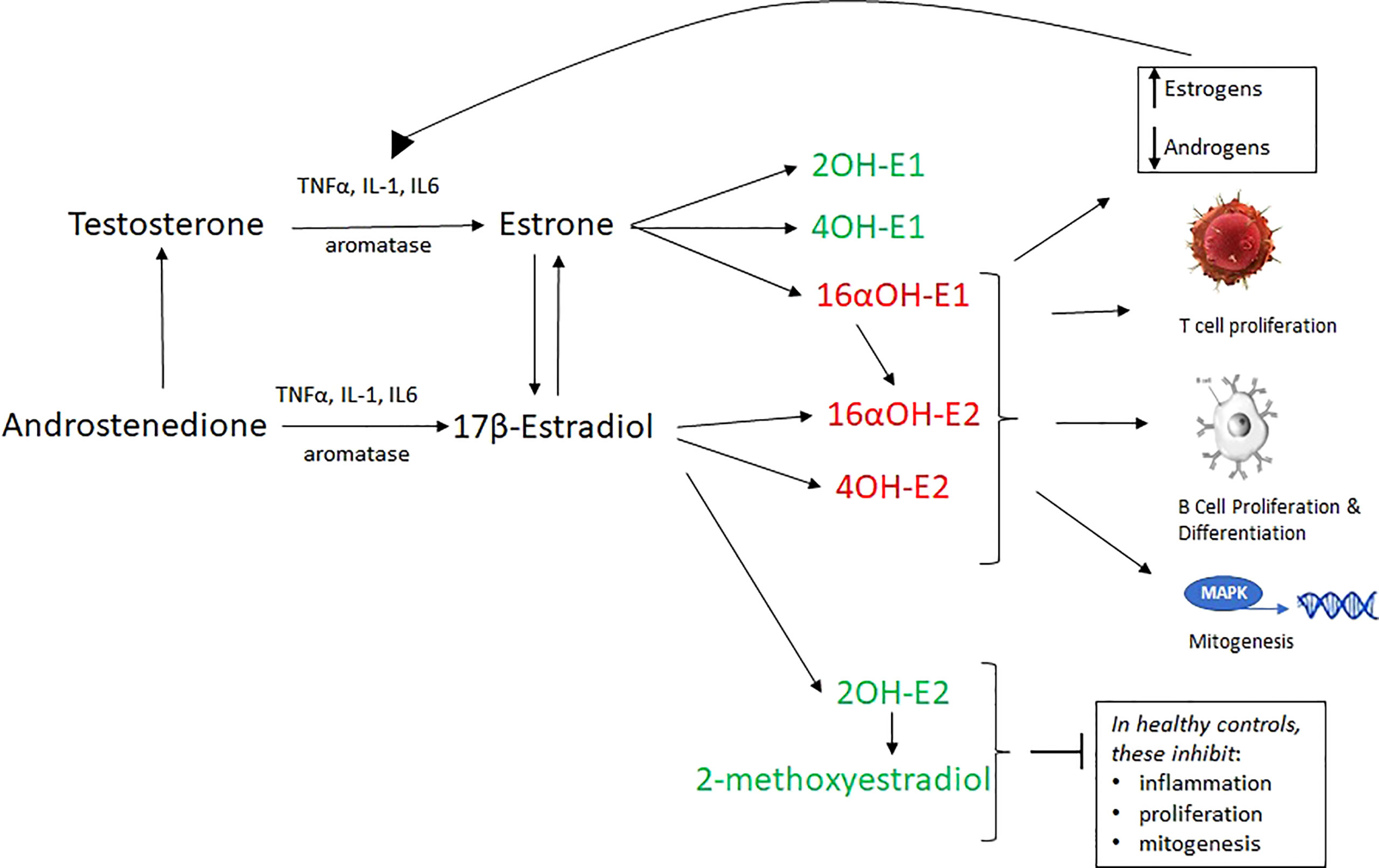
Figure 2 17β-estradiol and estrone, which are derived by the aromatization of testosterone and androstenedione, are converted into 6 hydroxylated metabolites (via cytochrome P450 enzymes) which are inflammatory (red) or non-inflammatory (green). 16α -hydroxylated forms of estrogens covalently bind ERα and allow its proliferative capabilities to flourish. They are important proinflammatory and proproliferative signals. Furthermore, unlike other estrogens, they do not inhibit TNFα. 16αOH-E1 is not only inflammatory, but promotes unscheduled DNA synthesis, mitogenesis, and is a breast cancer tumor initiator. 2-hydroxylated metabolites weakly bind ERα, inhibit the proliferative action of that receptor, and are generally anti-inflammatory, pro-apoptotic. The loss of 2-hydroxylated estrogens (as in RA and SLE) is a proinflammatory signal. Estrogen metabolism in some autoimmune disease favors inflammatory catechol estrogen end products. A vicious cycle wherein IL-6 and other cytokines convert local androgens via aromatase to create more E2, which in turn creates more IL-1 and IL-6. In total, this aberrant estrogen metabolism and increased overall estrogen relative to androgens causes increased T cell proliferation, B cell proliferation, mitogenesis, inflammation and reduced apoptosis.
Sex Steroid Mediated Immune Homeostasis
Progesterone and Testosterone Are Anti-Inflammatory. Low Levels of Either Facilitate Estrogen’s Inflammatory Effects
Progesterone has several anti-inflammatory mechanisms. Like estrogen, it has both membrane and nucelar receptors, but these have not yet been found uniformly across immune cell types as ERs have (64). Progesterone has some capacity to suppress CD4+ T cell proliferation, Th1/Th17 differentiation and effector functions while also being able to enhance Treg differentiation (65–68). Miyaura et al. demonstrated that pregnancy-level progesterone can inhibit differentiation of human Th1 cells, driving them toward an IL-10-secreting phenotype (IL-10 is generally thought to be anti-inflammatory) (65). Additionally, progesterone suppresses IL-6, and reduces IL-6 receptors and suppresses the mTOR pathway which is a proliferative pathway downstream of PI3K used across a wide array of cell types, including lymphocytes, and is instrumental in the fibrosis seen in TED (3, 67, 69–72). Progesterone has also been shown to reduce inflammatory cytokines such as TNFα (67, 73). This partially explains why symptoms in many autoimmune disorders are reduced during the luteal phase of the menstrual cycle (which is dominated by progesterone) and all but eliminated in pregnancy levels of progesterone. Further, these women often see a sharp rebound return of their autoimmune disorder after giving birth, when progesterone plummets.
Testosterone provides a similar inflammatory corrective to estrogen. Like the other steroid hormones, testosterone has influences across immune cell types, cytokines and inflammatory mediators such as C-reactive protein (74). Nasser et al, demonstrated that testosterone therapy was effective compared to placebo in treatment of Crohn’s disease, as measured by Crohn’s Disease Activity Index. The therapy reduced C-reactive protein dramatically and brought leukocytes within normal limits (75). Another example is a murine model that mimics the histology of nonbacterial prostatitis. E2 administration causes dose dependent inflammation in the prostate in castrated mice. E2 exerts this effect on the prostate by stimulating transcript levels of IL-1β, IL-6, macrophage inflammatory protein-2, and iNOS and is accompanied by fibromuscular proliferation, which consisted of fibroblasts, smooth muscle cells, and collagen. The effects increased with time (32, 76).
This homeostatic milieu may be vital to TED, as explained below is the fact that poorly controlled GD or HD facilitates high levels of estrogen and low levels of progesterone and testosterone.
Estrogen and Estrogen Metabolites in RA, SSc and SLE
Lack of sex steroid homeostasis has been illustrated to impact pathology. Local levels of proinflammatory estrogens relative to androgens are significantly elevated in both male and female patients with RA, SLE and SSc compared to controls (77–79). The cause of this is a vicious cycle wherein local inflammatory cytokines such as TNF, IL-1 and IL-6 induce aromatase activity, converting steroid pre-hormones (DHEA, testosterone and androstenedione) into estrogen. Excess estrogen then stimulates IL-1 and IL-6 production (80–82). The local aromatization decreases plasma androgen levels in patients with RA, SLE, and SSc, compared to controls without disease (77–79). In fact, the lowest levels of serum androgen are seen in women with most active SLE disease (78).
This is further complicated by the estrogen metabolism in these diseases which yields a preponderance of the proinflammatory metabolites 16α-hydroxyestrone and 4-hydroxyestradiol (Figure 2) (21, 34, 80, 83). Both of these metabolites have been demonstrated to be mitogenic, angiogenic, and inflammatory in breast cancer, autoimmune disease, pulmonary hypertension, and several other diseases under investigation (57–59, 61, 83–85).
Inflammation and proliferation in these 3 AIDs can be reduced or extinguished in vitro using anti-estrogen agents such as tamoxifen or with aromatase inhibitors like anastrozole (33, 42, 80). Of note, progesterone has also been shown to dampen the growth stimulatory effects of estrogen on RA synoviocytes (26, 67).
The inflammatory effects of 16α-hydroxyestrone (16α-OH) are mitigated by another estrogen metabolite, 2OH-estrogen. The urinary 16α-OH/2OH-estrogen ratio is a marker of inflammation used in breast cancer research, and recently in autoimmune disease. Urinary levels of the anti-mitogenic 2OH estrogens are 10 times lower in RA and SLE patients than in healthy controls, making the 16α-OH/2OH estrogen ratio 20 times higher in RA and SLE patients than in healthy controls. This likely contributes to maintenance of the proliferative state in these diseases (33, 83) (Figure 2). Why these individuals metabolize estrogen toward an inflammatory profile is unknown, but autoimmune and breast cancer research implicates polymorphic genes for the cytochrome P450 enzymes involved in estrogen metabolism, as well as polymorphisms in ERs (47, 61, 86, 87).
In pre-clinical models, 2-methoxyestradiol (2-ME), has been shown to suppress fibrosis in SSc and to induce apoptosis in diverse tumor types and inhibit experimental autoimmune encephalomyelitis (59, 88, 89). Both 2OH-E2 and 2-ME limit estrogen-driven growth and inflammation in autoimmune disease (21, 33, 85).
Khalkhali-Ellis et al., have also demonstrated the dynamic effects of estrogen and progesterone on fibroblast-like synoviocytes in RA, and showed with ER transfected fibroblast-like synoviocytes, cartilage invasion. This was partially mediated by matrix metalloproteinases, which have complex roles in fibrosis, where they can attenuate or augment the fibrotic pathway (26, 90).
Estradiol (E2) Modifies Key Inflammatory Mediators and Cell Metabolism in Autoimmune Disease
E2 Dependent Increase in CD40L Expression Hyperstimulates SLE T Cells, Contributing to the Pathogenesis of SLE
Rider et al., examined T cells in women with SLE vs healthy women. The molecular CD40:40L bridge formed between B and T cells is physiological, but also a point of possible pathology. Increased expression of CD40L can result in spontaneous autoantibody production in autoreactive B cells (91). They bathed SLE T cells and healthy T cells in E2 and had a 3rd group of SLE T cells not bathed in E2 as a second control. They saw a significant increase in CD40L expression (and CD40L mRNA) in the estradiol bathed SLE T cells compared to non-estradiol SLE T cells. These increases could be blocked by an estrogen receptor antagonist. The healthy T cells did not show CD40L expression change with E2 (24). They further demonstrated that E2 is acting via ERα, and in SLE T cells activates calcineurin and CD40L, but not in healthy T cells (25). Perhaps a possible explanation is found in the receptors. SLE cells have been shown to express aberrant polymorphic ERα receptors (92).
Further, In SLE, a SERM (selective estrogen receptor modulator) called raloxifene reduced estrogen induced CD40 overexpression on follicular B cells, making them less responsive to T cell co-stimulation (93).
Though it has been previously noted that increased CD40 expression is unique to orbital fibroblasts, significantly increased CD40 expression is seen in RA, SSc and SLE fibroblasts and fibroblast-like synoviocytes (22, 24). In fact, it is also seen autoimmune pulmonary fibrosis, suggesting this is not an uncommon fibrotic phenomenon. However, it cannot be overstated that estrogen mediated immune system influence is tissue and disease specific. E2 actually blocks the induction of CD40 and CD40L expression on cardiovascular endothelial cells and prevents neutrophil adhesion via the ERα pathway and prevents cardiac fibrosis via the ERβ pathway (50, 51). There is a vast literature on estrogen’s ability to have both anti-inflammatory and inflammatory effects (27).
Looking more directly at the fibrosis in SLE, Corradeti et al. demonstrated metabolism changes conferred due to altered transcription of key genes, as mediated by ERα. Among key results, their findings demonstrate that ERα-mediated metabolism variations exacerbate immune-induced renal damage resulting in increased inflammation and fibrosis. ERα knockout (from both immune and kidney cells) murine models suffered less kidney disease. They also sequenced human patient’s DNA and found similar expression pattern changes (94).
A final inflammatory example is that estrogen induces fibrotic phenotype in SSc, a connective tissue autoimmune disease of unknown origin characterized by fibrosis of the skin, lungs, kidney, and other organs. Focusing on skin fibrosis in SSc, Aida-Yasuoka et al. confirmed the expression ERα and ERβ in primary dermal fibroblasts from twin pairs, six who had SSc, five of whom were healthy. They demonstrate that E2 is an inducer of fibronectin expression, and acting via ERα, exerts profibrotic effects. Further, E2 treatment increased further E2 sensitivity by increasing ERα’s. They were able to block these profibrotic effects with an ER antagonist, fulvestrant. Furthermore, ERβ levels were much higher in the healthy twins than in the fibroblasts of the patients with SSc. Aida-Yasuoka et al. suggest ERβ could play a protective role in SSc as has been seen in cardiac fibrosis. Unsurprisingly, the intracellular pathways in the proliferation of fibrosis implicated (via inhibition of each) were P13K and p38 MAPK (23).
Rationale for Examining the Sex Steroids in TED
When Cutolo and Straub, et al. began examining the exact role of estrogen in the pathophysiology of rheumatoid arthritis, it was speculative. It is clear across autoimmunity that there is a sex bias, but estrogen’s role in direct pathophysiology had not been thoroughly examined. They uncovered a complex and nuanced role for estrogen wherein dose, metabolite, receptor and tissue type made the difference between joint destruction and healthy joint (35, 62). Given the similarity in pathophysiology between RA and SLE, they examined the role of estrogen in this second autoimmune disease – finding it exerted the same influence (34). Then, given the similar fibrogenic pathology, investigators examined estrogen in SSc, discovering, yet again, a salient role (80). I suggest that given the similar fibrogenic and inflammatory cascades, we extend these investigations one disease further – to TED.
Since this would be an entirely new line of inquiry, direct evidence for the role of sex steroids in TED is scant, but the profound sex steroid disruption seen in uncontrolled GD and HD begs the question.
Autoimmune Thyroid Disease Disrupts Sex Steroid Hormone Homeostasis Toward an Inflammatory Milieu
As outlined above, patients with RA, SSc and SLE have slightly higher levels of serum estrogen to androgens, caused by the local levels of increased aromatization (34, 77). E2 levels in women with hyperthyroidism are estimated to be 2-3-fold higher than normal controls, while progesterone levels are often lower than normal (95, 96). Metabolic clearance rate of E2 is decreased in both genders. Testosterone and androstenedione (androgens) are increased, but so is the conversion ratio of androstenedione to estrone, as well as of testosterone to E2 in hyperthyroid women. Bioavailable testosterone levels are subnormal in hyperthyroid males, while free E2 are elevated—making the free testosterone/free E2 ratio lower in hyperthyroid males compared with normal individuals. Progesterone can be reduced in women in either hyper- or hypothyroidism (97–100) (Table 2). These ratios engender an inflammatory milieu similar to those seen in RA, SLE and SSc, and indeed more profoundly disrupted. Could TED’s localized inflammatory cascade also have steroid hormones participation, as in these three model diseases?

Table 2 Synopsis of hormonal changes in male and female thyrotoxicosis and hypothyroidism.
While not strong enough to count as independent evidence, there are some TED statistics that could be explained by sex steroid involvement. For example, risk for TED is equal in pre-pubescent children – but shifts to 4:1 female to male odds after puberty (101).
Though less frequent in men, TED in men tends to be more severe (102). This is similar to SSc, (79) and sex steroid hormones participation this could be manifold:
1. Men have more testosterone which can be converted to estrogen via the IL-6 induced aromatase activity (34, 77, 79);
2. Men lack sufficient progesterone to oppose estrogen, and what little progesterone they have can be inhibited by IGF (103);
3. The baseline testosterone/estrogen imbalances coupled with the increased aromatization, may increase men’s E2 levels to significant levels, as happens in SSc. In SSc, male patients with scleroderma have considerably higher levels of E2 (average, 30.6 pg/mL) than both healthy men (average, 12.9 pg/mL) and postmenopausal women with the disease (24.2 pg/mL). Furthermore, the higher the E2 levels in these men, the higher the mortality rate. It appears that a large portion of testosterone in these patients undergoes aromatization. Of note, post-menopausal women with SSc also have higher E2 levels than healthy post-menopausal women (79).
There are glaringly few case reports in the literature that have examined this possibility. But one is notable. Øgard et al. observed a case where TED manifested in a 56-year-old postmenopausal female, who had no previous history of thyroid disease. She had been a heavy smoker (20/day), so her orbital tissues were primed with oxidative stress. She was given hormone replacement therapy to prevent osteoporosis and within a month she developed TED, with characteristic diplopia and lid retraction. She was then taken off the HRT and treated with corticosteroids. 6 years later, she was placed back on hormone replacement therapy, and again, developed TED. This time the patient refused steroids based on previous adverse events and the TED remitted after 10 months. They also mention a previous, similar case known to them, but did not publish the details (104).
Another case demonstrates a key piece of information for further inquires, that ERα is expressed in orbital fibroblasts. Cury et. al., obtained orbital fibroblasts during orbital decompression surgery from a female patient with inactive moderately severe-to-severe TED. They confirmed ERα expression with rtPCR. They treated the cells with dexamethasone (DEX) and the ERα expression was affected in a dose dependent manner. In the cells treated with 10 nM and 100 nM DEX, ERα gene expression was, respectively, 85% higher and 74% lower, than in the control group (105). Their in vitro data are not conclusive but suggest treatment of TED with glucocorticoids may be partially mediated through ERαs, which is not the first time that has been examined (105–107). Glucocorticoid treatment works only in some patients with TED and often cause a rebound effect, worsening the TED. It’s a leap, but perhaps there is an estrogen marker to be found here about ideal responders?
A third, unpublished case was the impetus for this entire line of inquiry and literature review. I observed a unique case of moderate-to-severe TED which had two starts. This patient was diagnosed with Graves’s disease in April of 2017, a sequala to sepsis. June 15, 2017, she was diagnosed with mild thyroid eye disease. She had photophobia and dry eyes. These were entirely resolved with a week of steroid eye drops; she had no further symptoms. Her thyroid status was difficult to control even with methimazole and propranolol. In November of 2017, she came down with a staphylococcus infection (she still had cytopenias from the sepsis) and her TSI rose considerably (Table 3). Her surgeon noted that the moderate/severe TED likely began then. So did other symptoms. Coincident to this timing was menstrual abnormalities. She had one very long cycle, then concurrent with the appearance of TED, there was a reduction of her menstrual cycle length, which shortened from a regular 27-day cycle to a regular 13-day cycle, which usually indicates lack of progesterone. Every 10th cycle day (CD), her TED signs and symptoms became noticeably worse, and then noticeably better at the start of her menses 4 days later (CD1). Self-described extreme premenstrual dysphoria also began at his same time, which was not in her previous history. After 6 short cycles, she then had three long cycles, and then menses arrested shortly after turning 45, young for her family by 6 years.
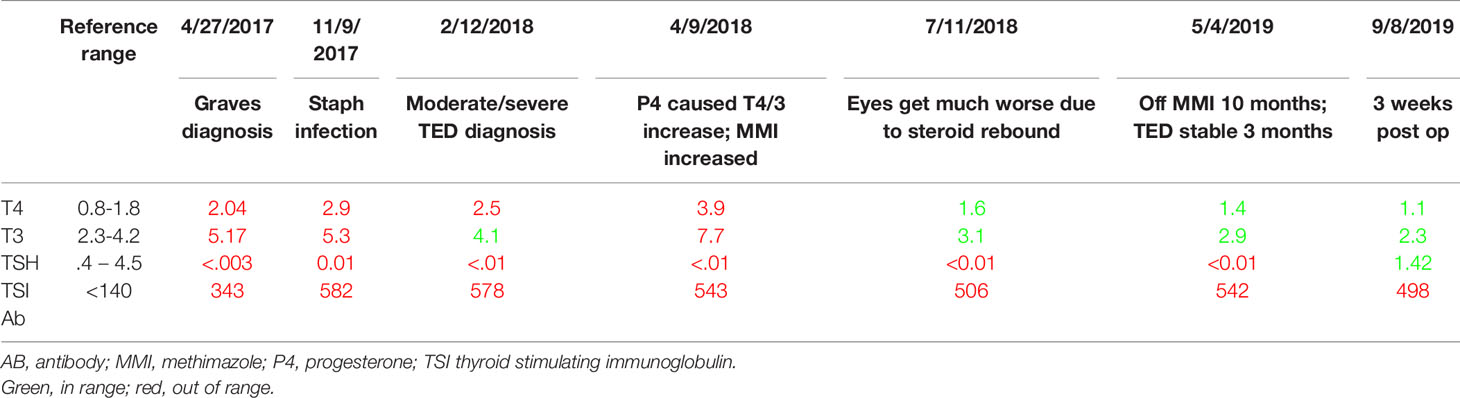
Table 3 Patient thyroid hormones.
Her TED signs and symptoms ebbed and flared in synchrony with the estrogen valleys and peaks of her menstrual cycle, every cycle getting a bit worse. It was not a linear progression. The worst inflammation, edema, dryness, pain, and lid retraction always corresponded to the highest estrogen peak. The highest estrogen levels in a menstrual cycle are in the days just preceding ovulation. Luteinizing hormone tests are a proxy for ovulation timing, and she self-administered these on cycle day (CD) 10 to ensure the effect. These tests administered again on CD 3 of the following cycle to ensure luteinizing hormone was not high the whole cycle.
When her cycles lengthened, she would get a second flare corresponding to the 2nd estrogen peak. She did have some relief and reduction in swelling with high dose progesterone prescribed by her gynecologist, but that had to be discontinued as it increased her T4 considerably (Table 3). In May 2018 she was given a 10-week course of high dose IV methylprednisolone, which temporarily reduced proptosis 1-2 mm, but had a rebound effect and the eye disease became much worse by August. Once the proptosis stabilized it was mainly the lid retraction that ebbed and flowed with her cycle. By the Spring of 2019, her thyroid had been sufficiently damaged by the TSI and by summer she was hypothyroid and entering menopause. She had bilateral orbital decompression and blepharoplasty in August 2019. The lid retraction persisted, and she had a second and final surgery November 2020.
Her estrogen levels throughout were at the upper limit of normal or higher than normal. Her progesterone and testosterone were low, though with as much progesterone as she was taking, it’s impossible to get an accurate read out (Table 4).
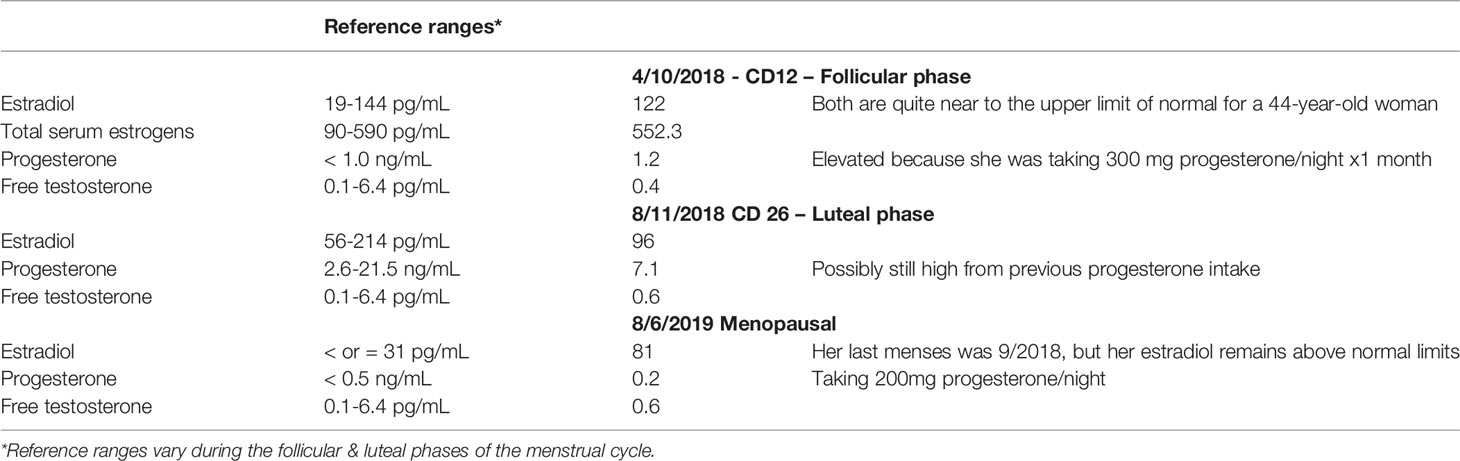
Table 4 Patient steroid hormones.
Regrettably, as I am not an oculoplastic surgeon, I do not have CAS scores for her disease, but we have the below visual diary (Figures 3A, B). All photos were taken in the morning, upon waking, except where noted. It is important to show the whole face, as TED also affects the buccal fat and gives the whole face a boxy appearance.
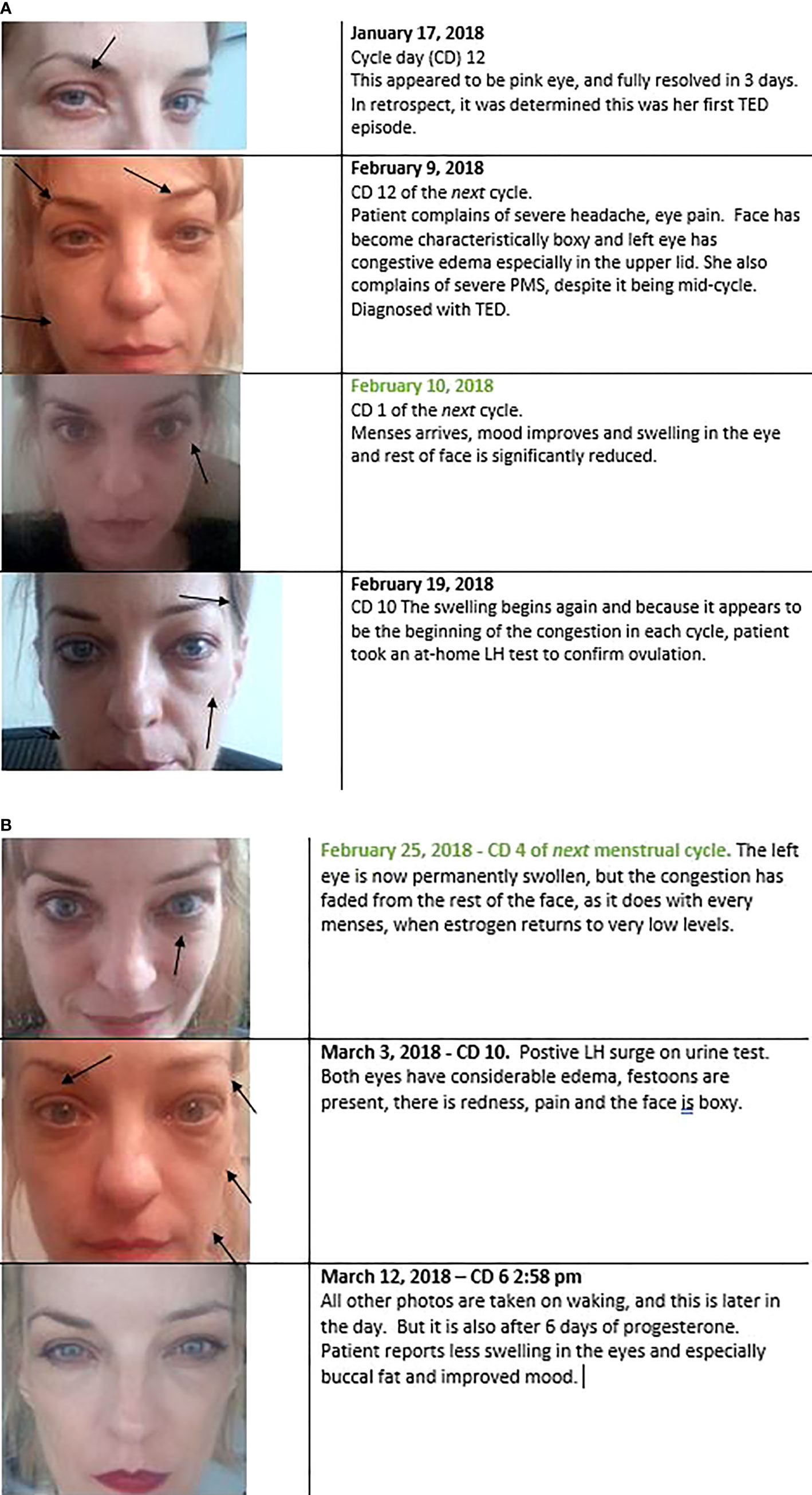
Figure 3 (A, B). Every preovulation or premenstrual period, during concomitant estrogen peaks, patient’s TED signs and symptoms would become dramatically worse. Typically, this started on cycle day 10. Her cycles were short when the TED started (13 days total, indicating a possible lack of progesterone). When menses came and estrogen levels dropped to near zero (Figure 5), periorbital congestion and edema also dropped dramatically. This is evident in the one-day change photographed in the 2nd and 3rd panels. This was not edema-specific, it also occurred in the eyelid retraction, even after bilateral orbital decompression (not shown here). The lids retracted 1-2mm above the iris from cycle days 9 – 13, and then lowered to the level of the iris from cycle days 1 – 4.
The author is well aware that this is far from definitive evidence. The paucity of direct evidence of estrogen’s role in TED does not mean there is no connection – merely that this has not been an area of investigation. This is curious because of all these autoimmune diseases; TED is itself borne out of endocrine disorders. Why has participation of the sex steroid hormones not been thoroughly evaluated in TED when they play clear roles in autoimmune diseases that do not have this underlying hormone dysregulation?
Finally, there is one other area that estrogen research into TED could prove particularly intriguing: the relationship of estrogen and growth hormones. While the below is also not direct evidence, it is further rationale for speculation and investigation.
IGF-1R Has Significant Cross Talk With ERs and E2
IGF-1R is instrumental to TED pathophysiology. For unknown reasons, orbital fibroblasts in patients with TED express higher than normal numbers of IGF-1Rs (1). There is currently a debate in the literature about the presence of an IGF-1R autoantibody in Graves’s disease, but all agree that TSHR once bound by an autoantibody, affixes to IGF-1R via β-arrestin and IGF-1R is necessary for the ensuing inflammatory cascade (4, 108).
Steroid hormones, luteinizing hormone and growth factors regulate expression of IGF-1R (109, 110). E2 and estrogen receptors, specifically, have been shown to modulate IGF-1R in a variety of tissue types and diseases in both animal and human models (111, 112). ERα knock out murine models lower serum levels of IGF-1 compared with their intact ERα+ littermates (113). In breast cancer, in model autoimmune diseases, as in TED, the binding of IGF-1 to IGF-1R triggers the PI3K/AKT/mTOR signaling pathway (17–20, 114, 115). In breast cancer, then a downstream molecule of the AKT/mTOR pathway phosphorylates ERα which binds to promoter of target genes, and upregulates IGF-1, IGF-1R (115, 116) (Figure 4). Estrogen also stimulates cytosolic ER to interact with adaptor proteins, such as Shc or the p85 subunit of PI3K. This complex then translocates to the membrane where it associates with IGF-1R leading to activation of downstream signaling cascades like MAPK (117, 118).
Figure 4 Estrogen and ERα exert genomic and non-genomic effects on IGF-1R, IGF, and other growth factors. Given the increase in E2 in GD and HD, is there a concomitant increase in ERα and IGF-1R, via the mechanisms posited here? If so, it is plausible that estrogen is a disease driver in TED. (Adapted with open access creative commons permission from 115).
ERα rapidly activates IGF-1R in non-carcinogenic cells, as well. IGF-1R and ERα colocalize in in many cell lines (119). Kahlert et al. demonstrated that E2 via ERα, is capable of IGF-1R phosphorylation and that these effects could be blocked with estrogen antagonists. They further showed that constitutively active MAPKK induced an interaction of ERα with the IGF-1R even in the absence of ligand. Their in-depth analysis of these interactions concludes that “taken together, these data demonstrate that ligand bound estrogen receptor α is required for rapid activation of the IGF-1R signaling cascade” (120).
Oesterreich et al. showed that ERα is a pivotal regulator of IGF mitogenesis. They used MCF-7 breast cancer cells which had lost their ERα and showed reduced expression of IGF-signaling and failure to proliferate when stimulated with E2 or IGF-1. Transection-facilitated re-expression of ERα restored the IGF-1-responsives and proliferation (121).
Chan et al., in a murine breast cancer model found that E2 significantly increased basal phosphorylation of IGF-1R, IRS-1, IRS-2, and Akt-1, but that basal phospho p44/42 MAPK was significantly reduced. These effects were extinguished with the anti-estrogen fulvestrant. Furthermore, high doses of fulvestrant increased decreased IGF-1R and its autophosphorylation to approximately 30% of the controls. IRS-1, IRS-2, and c-Raf-1 levels were similarly reduced.
The PI3K pathway is instrumental to cellular metabolism, affording the subsequent development, survival, and proliferation of the cell. For example, PI3Kδ regulates B cell ability to develop from transitional to naïve to plasmablast to antibody secreting plasma cell, and even to undergo class switch recombination (48, 122). Estrogen, via its nuclear and membrane receptors, and IGF-1 complex, and the PI3K pathway can affect gene transcription that affords the cells the metabolic requirements for development (123–126).
Another example of can be found in uterine fibroids (leiomyomas) (112, 127). IGF-1 expression is most abundant in leiomyomas during the proliferative, estrogen dominated phase of the menstrual cycle. Yu et al. have shown that estrogen upregulates the gene encoding IGF-1 through ERα in leiomyoma tissue and cells during this time (112). Finally, estrogen also has direct and rapid effect on growth hormone, which also exerts effects on IGF-1 (128, 129).
Growth Hormone, IGF-1 and Estrogen
Finally, estrogen can regulate IGF via growth hormone (GH). Estrogen has direct and rapid effect on GH (128, 129). At periovulatory levels in healthy women, E2 can increase GH 2-3-fold (129, 130). In that same periovulatory period in healthy women, serum IGF-1 levels (though not its binding proteins) are significantly increased. That measurement is of physiological levels of estrogen, not at the ~2-3x higher, pathological levels seen in GD (95, 100). Consequently, at periovulatory levels in GD of HD, the GH/IGF-1 increase during periovulatory days may be considerable (Figure 5).
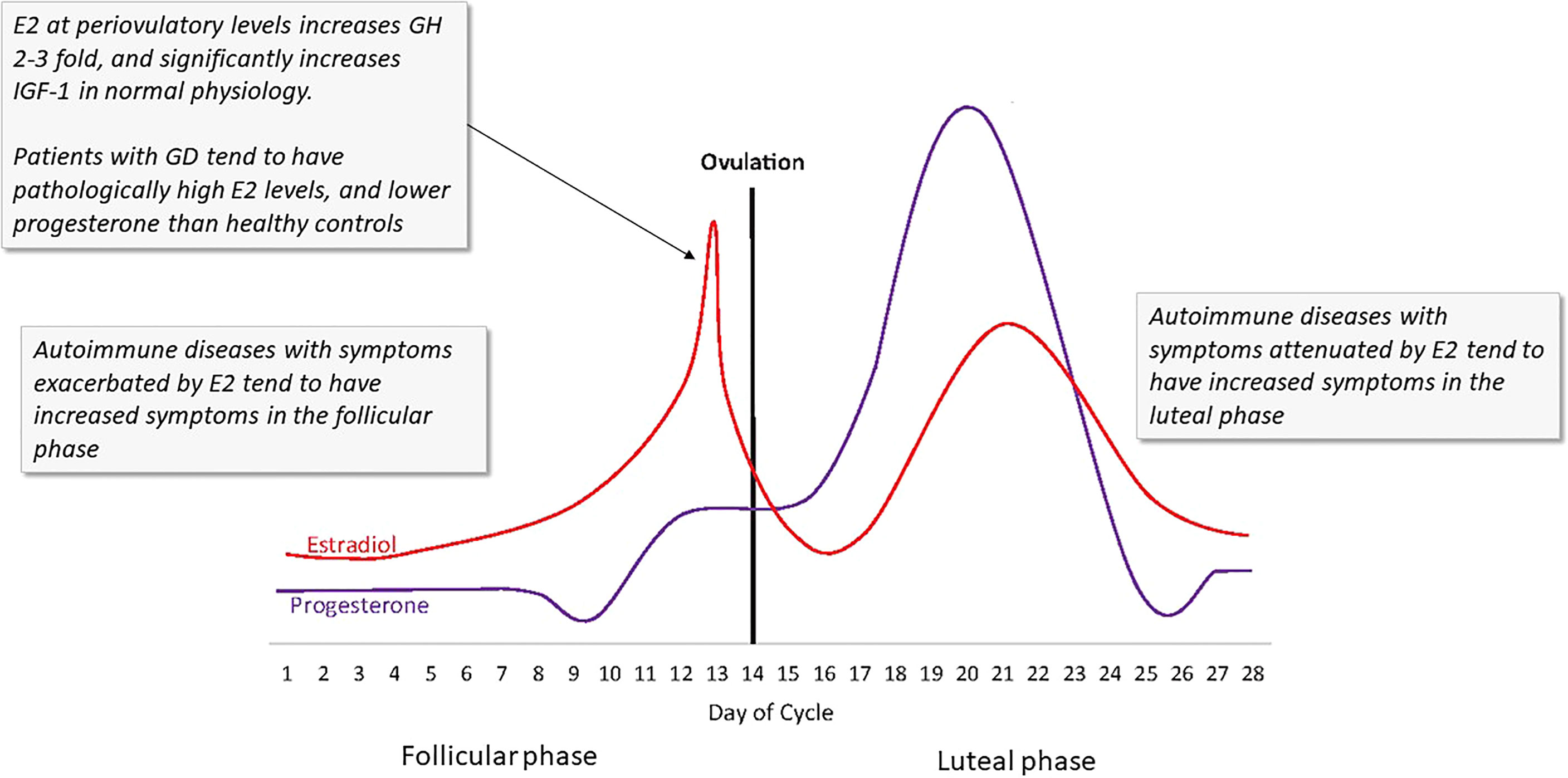
Figure 5 The ebb and flow of steroid hormones in a typical menstrual cycle. Several autoimmune disease symptoms are exacerbated by the rises in estrogen seen in both the first (follicular) and second (luteal) phases of the cycle. In GD and HD, these effects could be more profound due to the lack of steroid hormone homeostasis. Estrogen at periovulatory and premenstrual levels significantly increases IGF-1R, IGF, and GH. (Image copied with permission from 131).
Stromal Metabolism and Estrogen
As noted, fibroblasts, which fall into the larger class of stromal cells, are key to the homeostasis of the tissues they reside on and can function as sentinels. Their own metabolism, and the metabolism of their microenvironment can be influenced by E2. In a series of breast cancer murine model experiments, Péqueux et al, demonstrated that via the ERα microenvironment of stromal cells, E2 can facilitate tumor growth by triggering stromal cells, increasing angiogenesis, improving vessel morphology and architecture (facilitating durable metastasis) and reduced tumor hypoxia. They conclude because of these changes in the vessels, stromal ERα is a necessary component of tumor growth, as it improves oxygen and nutrient delivery, preventing hypoxia and necrosis (132). Specific fibroblast growth factors which mediate angiogenesis and mitogenesis in the stromal cells of endometriosis disease models have also been found to be dependent on aberrant production of E2 and these effects can be extinguished with an aromatase inhibitor (133). Given E2’s direct effects on stromal cells and given its regulation of myriad growth factors, and the consequence of these factors on stromal calls ability to sustain aberrant growth, it would seem likely that in a stromal tissue disease dependent on growth factors, such as TED, that the steroid hormones play a role.
Discussion
There are two large questions in TED that remain enigmatic. 1) Why does euthyroid status matter? What is protective about having thyroid hormone levels within normal limits because that status does not mean the person has low autoantibodies. But those autoantibodies remain in contact with the thyroid, not the orbit more often if they person is euthyroid. The persistence of either hypo- or hyperthyroidism correlates with increased severity of TED (134). What happens when someone remains hyper- or hypothyroid? Other hormones also become dysregulated. Sex steroid hormones, which become meaningfully dysregulated, all influence inflammation, fibrosis, and growth factors. It is my contention that sex steroid homeostasis has the potential to contribute meaningfully to TED, and this should be investigated (Figure 6). If there is merit to this hypothesis, it could explain the importance of euthyroid status on development and severity of TED and might even yield a biomarker. It would also shed light on why TED has been reported to be worse in post-menopausal women and in men.
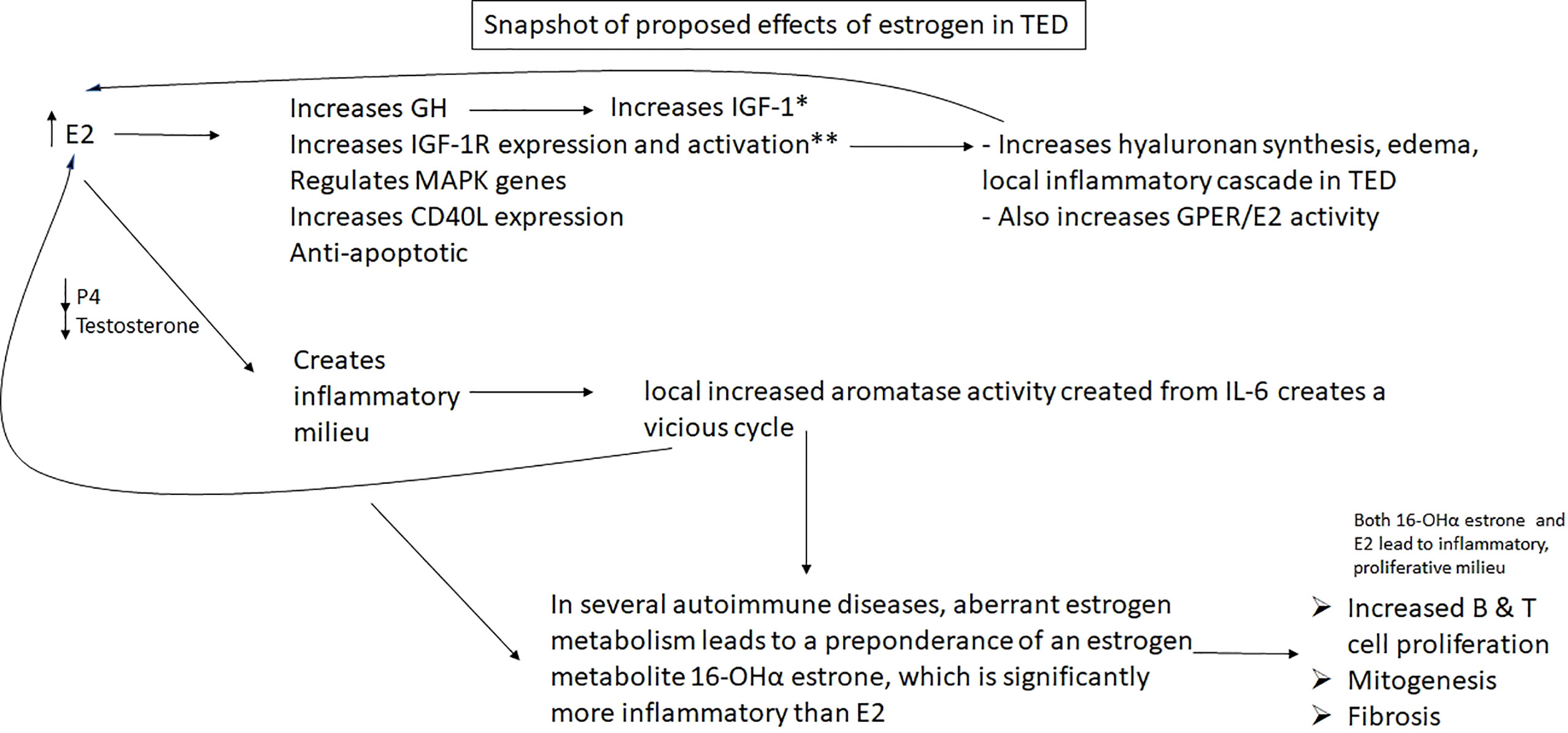
Figure 6 HYPOTHESIS: Excess estradiol and reduced progesterone & testosterone seen in male and female patients with Graves’ and Hashimoto’s disease may increase growth factors and IGF-1 receptors, helping to facilitate the well characterized TED inflammatory pathway elicited by IGF-1R; it also may result in a vicious cycle with local IL-6, which will create more E2 by aromatizing local androgens, which will stimulate the creation of more IL-6, in addition to other downstream inflammatory effects. E2, estradiol; ERα, estrogen receptor α; IGF-1R, insulin-like growth factor 1 receptor; GH, growth hormone; P4, progesterone; *E2 has varying direct effects on IGF-1 depending on age, gender, concentration; **Luteinizing hormone, which is also increased in women in Graves’ disease, meaningfully increases IGF-1R as well.
Question 2 is, why is IGF-1R is overexpressed in fibroblasts of patients with TED? Sex steroid homeostasis is disrupted, skewed toward high estrogen in patients with uncontrolled GD or HD, which are the preponderance of people who get TED. Estrogen regulates IGF-1R and growth hormones. The impetus to investigate if estrogen has contributed to this overexpression or pathology with the growth hormone and its receptor seems axiomatic (Figures 4, 5).
Though the above are speculative, there are corollaries that could serve as models. The type of inflammation and fibrosis seen in TED are not unique. Pathological IGF-1R/IGF-1 mediated tissue proliferation as seen in TED is also not unique. It is seen in breast cancer and uterine leiomyomas. Estrogen is pivotal to all of these pathologies; it certainly could be in TED. As with any investigation in science, these queries into the sex steroid hormones in TED may not yield a theory, or it may bloom in unexpected directions. That happened with rheumatoid arthritis, when unexpected subtypes were revealed based on estrogen pathology – and this shed light on the variance of responses to RA treatments (27).
The extensive and careful investigations on the role of estrogen, estrogen receptors and estrogen metabolites in autoimmunity conducted in RA, SSc and SLE bear repeating in the orbital fibroblasts of patients with thyroid eye disease, as do examinations of estrogen’s effects on IGF-1R. If transitive properties do exist, it will not only shed light on TED, but open a door to further considering IGF-1R’s possible role in these other 3 AIDs – a surface already scratched by Drs. Douglas and Smith in 2010 (135). There are many lines of inquiry possible here, a few initial ones are:
• Orbital fibroblasts (OF) express ERα (105). Does bathing them in E2 modify IGF/IGF1R expression?
○ If yes, can this be extinguished or attenuated with fulvestrant? With progesterone?
▪ Does increased IGF-1R increase ERα activity, engendering a vicious cycle?
○ Does E2 bathing increase expression of its own receptors? Often in the endocrine system, the reverse would happen, but there are examples of ligand saturation increasing its own receptors in the literature.
• Do OFs express GPER and/or ERβ?
○ Does the former participate in any IGF modulation?
○ Are the latter in too low a concentration?
• Are any three of the estrogen receptor types functionally polymorphic compared to healthy controls?
• Do patients with TED have a preponderance of inflammatory estrogen metabolites?
○ This is a simple urine test, and could be examined in healthy controls, patients with GD or HD without TED, and in patients with TED
▪ If yes, do OFs bathed in 16αOH‐estrone, cause any IGF/IGF-1R changes? Does that metabolite bind covalently in these cases?
▪ Do these patients have variants in their cytochrome P450 gene aromatase enzymes (genetic test)
▪ If any of these metabolite studies yield data, could that become a biomarker?
• The unpublished patient case discussed above has high 4OH-estradiol, which is inflammatory, and low 2OH-estradiol; her sister who has Graves’ disease but not TED is getting her estrogen metabolism evaluated
TED occurs almost exclusively in patients with endocrine hormone disorder. Investigating the endocrine system in these patients is an as yet unexplored arena. Based on the above speculations, it could be worthy of exploration.
This review is far from an exhaustive survey on the sex steroid hormones’ influence on the immune system. Interested readers are directed to references for exhaustive findings of this complex interaction (27, 32, 34, 49, 87, 136).
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics Statement
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
All research was funded and conducted solely by the author.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Douglas RS, Gupta S. The Pathophysiology of Thyroid Eye Disease: Implications for Immunotherapy. Curr Opin Ophthalmol (2011) 22(5):385–90. doi: 10.1097/ICU.0b013e3283499446
2. Naik VM, Naik MN, Goldberg R, Smith TJ, Douglas RS. Immunopathogenesis of Thyroid Eye Disease: Emerging Paradigms. Surv Ophthalmol (2010) 55(3):215–26. doi: 10.1016/j.survophthal.2009.06.009
3. Khong JJ, McNab AA, Ebeling PR, Craig JE, Selva D. Pathogenesis of Thyroid Eye Disease: Review and Update on Molecular Mechanisms. Br J Ophthalmol (2016) 100:142–50. doi: 10.1136/bjophthalmol-2015-307399
4. Krieger CC, Boutin A, Jang D, Morgan SJ, Banga JP, Kahaly GJ, et al. Arrestin-Beta-1 Physically Scaffolds TSH and IGF1 Receptors to Enable Crosstalk. Endocrinology (2019) 160(6):1468–79. doi: 10.1210/en.2019-00055
5. Huang Y, Fang S, Li D, Zhou H, Li B, Fan X. The Involvement of T Cell Pathogenesis in Thyroid-Associated Ophthalmopathy. Eye (2019) 33:176–82. doi: 10.1038/s41433-018-0279-9
6. Woeller CF, Roztocil E, Hammond C, Feldon SE. TSHR Signaling Stimulates Proliferation Through PI3K/Akt and Induction of miR-146a and miR-155 in Thyroid Eye Disease Orbital Fibroblasts. Invest Ophthalmol Visual Sci (2019) 60(13):4336–45. doi: 10.1167/iovs.19-27865
7. Wynn T. Fibrotic Disease and the TH1/TH2 Paradigm. Nat Rev Immunol (2004) 4:583–94. doi: 10.1038/nri1412
8. Smith TJ, Kahaly GJ, Ezra DG Fleming JC, Dailey RA, Tang RA, et al. Teprotumumab for Thyroid-Associated Ophthalmopathy. N Engl J Med (2017) 376:1748–61. doi: 10.1056/NEJMoa1614949
9. Bartalena L, Tanda ML. Clinical Practice. Graves’ Ophthalmopathy. N Engl J Med (2009) 360:994–1001. doi: 10.1056/NEJMcp0806317
10. Bartalena L, Pinchera A, Marcocci C. Management of Graves’ Ophthalmopathy: Reality and Perspectives. Endocr Rev (2000) 21:168–99. doi: 10.1210/edrv.21.2.0393
11. Wiersinga WM, Prummel MF, Terwee CB. Effects of Graves’ Ophthalmopathy on Quality of Life. J Endocrinol Invest (2004) 27:259–64. doi: 10.1007/BF03345275
12. Wiersinga WM, Smit T, van der Gaag R, Koornneef L. Temporal Relationship Between Onset of Graves’ Ophthalmopathy and Onset of Thyroidal Graves’ Disease. J Endocrinol Invest (1988) 11:615–9. doi: 10.1007/BF03350193
13. Perros P, Crombie AL, Matthews JN, Kendall-Taylor P. Age and Gender Influence the Severity of Thyroid-Associated Ophthalmopathy: A Study of 101 Patients Attending a Combined Thyroid-Eye Clinic. Clin Endocrinol (Oxf) (1993) 38:367–72. doi: 10.1111/j.1365-2265.1993.tb00516.x
14. Lazarus JH. Epidemiology of Graves' Orbitopathy (GO) and Relationship With Thyroid Disease. Best Pract Res Clin Endocrinol Metab (2012) 26:273–9. doi: 10.1016/j.beem.2011.10.005
15. Chin YH, Ng CH, Lee MH, Koh JWH, Kiew J, Yang SP, et al. Prevalence of Thyroid Eye Disease in Graves’ Disease: A Meta-Analysis and Systematic Review. Clin Endocrinol (Oxf) (2020) 93:363–374. doi: 10.1111/cen.14296
16. Baglole CJ, Ray DM, Bernstein SH, Feldon SE, Smith TJ, Sime PJ, et al. More Than Structural Cells, Fibroblasts Create and Orchestrate the Tumor Microenvironment. Immunol Invest (2006) 35:297–325. doi: 10.1080/08820130600754960
17. Hamaguchi Y, Fujimoto M, Matsushita T, Hasegawa M, Takehara K, Sato S. Elevated Serum Insulin-Like Growth Factor (IGF-1) and IGF Binding Protein-3 Levels in Patients With Systemic Sclerosis: Possible Role in Development of Fibrosis. J Rheumatol (2008) 35(12):2363–71. doi: 10.3899/jrheum.080340
18. Erlandsson MC, Töyrä Silfverswärd S, Nadali M, Turkkila M, Svensson MND, Jonsson IM, et al. IGF-1R Signalling Contributes to IL-6 Production and T Cell Dependent Inflammation in Rheumatoid Arthritis. Biochim Biophys Acta Mol Basis Dis (2017) 1863(9):2158–70. doi: 10.1016/j.bbadis.2017.06.002
19. Mohammed JA, Mok AY, Parbtani A, Matsell DG. Increased Expression of Insulin-Like Growth Factors in Progressive Glomerulonephritis of the MRL/lpr Mouse. Lupus (2003) 12(8):584–90. doi: 10.1191/0961203303lu422oa
20. Garrett SM, Hsu E, Thomas JM, Pilewski JM, Feghali-Bostwick C. Insulin-Like Growth Factor (IGF)-II- Mediated Fibrosis in Pathogenic Lung Conditions. PloS One (2019) 14(11):e0225422. doi: 10.1371/journal.pone.0225422
21. Cutolo M, Villaggio B, Seriolo B, Montagna P, Capellino S, Straub RH, et al. Synovial Fluid Estrogens in Rheumatoid Arthritis. Autoimmun Rev (2004) 3(3):193–8. doi: 10.1016/j.autrev.2003.08.003
22. Fukasawa C, Kawaguchi Y, Harigai M, Sugiura T, Takagi K, Kawamoto M, et al. Increased CD40 Expression in Skin Fibroblasts From Patients With Systemic Sclerosis (SSc): Role of CD40-CD154 in the Phenotype of SSc Fibroblasts. Eur J Immunol (2003) 33(10):2792–800. doi: 10.1002/eji.200324088
23. Aida-Yasuoka K, Peoples C, Yasuoka H, Hershberger P, Thiel K, Cauley JA, et al. Estradiol Promotes the Development of a Fibrotic Phenotype and is Increased in the Serum of Patients With Systemic Sclerosis. Arthritis Res Ther (2013) 15(1):R10. doi: 10.1186/ar4140
24. Rider V, Jones S, Evans M, Bassiri H, Afsar Z, Abdou NI. Estrogen Increases CD40 Ligand Expression in T Cells From Women With Systemic Lupus Erythematosus. J Rheumatol (2001) 28(12):2644–9.
25. Rider V, Li X, Peterson G, Dawson J, Kimler BF, Abdou NI. Differential Expression of Estrogen Receptors in Women With Systemic Lupus Erythematosus. J Rheumatol (2006) 33(6):1093–101.
26. Khalkhali-Ellis Z, Seftor EA, Nieva DR, Handa RJ, Price RH Jr., Kirschmann DA, et al. Estrogen and Progesterone Regulation of Human Fibroblast-Like Synoviocyte Function In Vitro: Implications in Rheumatoid Arthritis. J Rheumatol (2000) 27(7):1622–31.
27. Straub R. The Origin of Chronic Inflammatory Systemic Diseases and Their Sequelae 1st Edition. Amsterdam: Elsevier Academic Press (2015).
28. Stossi F, Barnett DH, Frasor J, Komm B, Lyttle CR, Katzenellenbogen BS. Transcriptional Profiling of Estrogen-Regulated Gene Expression via Estrogen Receptor (ER) α or Erβ in Human Osteosarcoma Cells: Distinct and Common Target Genes for These Receptors. Endocrinology (2004) 145(7):3473–86. doi: 10.1210/en.2003-1682
29. Kovats S. Estrogen Receptors Regulate Innate Immune Cells and Signaling Pathways Cell Immunol (2015) 294(2):63–9. doi: 10.1016/j.cellimm.2015.01.018.
30. Bouman A, Heineman MJ, Faas MM. Sex Hormones and the Immune Response in Humans. Hum Reprod Update (2005) 11:411–23. doi: 10.1093/humupd/dmi008
31. Paterni I, Granchi C, Katzenellenbogen JA, Minutoloa F. Estrogen Receptors Alpha (Erα) and Beta (Erβ): Subtype-Selective Ligands and Clinical Potential. Steroids (2014) 0:13–29. doi: 10.1016/j.steroids.2014.06.012
32. Straub RH. The Complex Role of Estrogens in Inflammation. Endocr Rev (2007) 28(5):521–74. doi: 10.1210/er.2007-0001
33. Capellino S, Montagna P, Villaggio B, Soldano S, Straub RH, Cutolo M. Hydroxylated Estrogen Metabolites Influence the Proliferation of Cultured Human Monocytes: Possible Role in Synovial Tissue Hyperplasia. Clin Exp Rheumatol (2008) 26:903–9.
34. Cutolo M, Sullia A, Straub RH. Estrogen Metabolism and Autoimmunity. Autoimmun Rev (2012) 11(6–7):A460–4. doi: 10.1016/j.autrev.2011.11.014
35. Cutolo M, Brizzolara R, Atzeni F, Capellino S, Straub RH, Puttini PC. The Immunomodulatory Effects of Estrogens: Clinical Relevance in Immune-Mediated Rheumatic Diseases. Ann N Y Acad Sci (2010) 1193:36–42. doi: 10.1111/j.1749-6632.2009.05383.x
36. Tamrazi A, Carlson KE, Daniels JR, Hurth KM, Katzenellenbogen JA. Estrogen Receptor Dimerization: Ligand Binding Regulates Dimer Affinity and Dimer Dissociation Rate. Mol Endocrinol (2002) 16(12):2706–19. doi: 10.1210/me.2002-0250
37. Biswas DK, Singh S, Shi Q, Pardee AB, Iglehart JD. Crossroads of Estrogen Receptor and NF-kappaB Signaling. Sci STKE (2005) 2005(288):pe27. doi: 10.1126/stke.2882005pe27
38. Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, et al. Estrogen Receptors: How do They Signal and What are Their Targets. Physiol Rev (2007) 87:905–31. doi: 10.1152/physrev.00026.2006
39. Couse JF, Lindzey J, Grandien K, Gustafsson JA, Korach KS. Tissue Distribution and Quantitative Analysis of Estrogen Receptor-Alpha (ERalpha) and Estrogen Receptor-Beta (ERbeta) Messenger Ribonucleic Acid in the Wild-Type and ERalphaknockout Mouse. Endocrinology (1997) 138(11):4613–21. doi: 10.1210/endo.138.11.5496
40. Leitman DC, Paruthiyil S, Vivar OI, Saunier EF, Herber CB, Cohen I, et al. Regulation of Specific Target Genes and Biological Responses by Estrogen Receptor Subtype Agonists. Curr Opin Pharmacol (2010) 10(6):629–36. doi: 10.1016/j.coph.2010.09.009
41. Phiel KL, Henderson RA, Adelman SJ, Elloso MM. Differential Estrogen Receptor Gene Expression in Human Peripheral Blood Mononuclear Cell Populations. Immunol Lett (2005) 97(1):107–13. doi: 10.1016/j.imlet.2004.10.007
42. Mohammad I, Starskaia I, Nagy T, Guo J, Yatkin E, Väänänen K, et al. Estrogen Receptor α Contributes to T Cell-Mediated Autoimmune Inflammation by Promoting T Cell Activation and Proliferation. Sci Signal (2018) 11(526). doi: 10.1126/scisignal.aap9415
43. Douin-Echinard V, Laffont S, Seillet C, Delpy L, Krust A, Chambon P, et al. Estrogen Receptor α, But Not β, Is Required for Optimal Dendritic Cell Differentiation and CD40-Induced Cytokine Production. J Immunol (2008) 180(6):3661–9. doi: 10.4049/jimmunol.180.6.3661
44. Bengtsson AK, Ryan EJ, Giordano D, Magaletti DM. Clark EA.17-Estradiol (E2) Modulates Cytokine and Chemokine Expression in Human Monocyte-Derived Dendritic Cells. Blood (2014) 104:1404–10. doi: 10.1182/blood-2003-10-3380
45. Cunningham M, Gilkeson G. Estrogen Receptors in Immunity and Autoimmunity. Clin Rev Allergy Immunol (2011) 40(1):66–73. doi: 10.1007/s12016-010-8203-5
46. Erlandsson MC, Ohlsson C, Gustafsson JA, Carlsten H. Role of Oestrogen Receptors Alpha and Beta in Immune Organ Development and in Oestrogen-Mediated Effects on Thymus. Immunology (2001) 103(1):17–2. doi: 10.1046/j.1365-2567.2001.01212.x
47. Sánchez-Maldonado JM, Cáliz R, Canet L, Bakker O, den Broeder AA, Martínez-Bueno M, et al. Steroid Hormone-Related Polymorphisms Associate With the Development of Bone Erosions in Rheumatoid Arthritis and Help to Predict Disease Progression: Results From the REPAIR Consortium. Sci Rep (2019) 9:14812. doi: 10.1038/s41598-019-51255-0
48. Lucas CL, Kuehn HS, Zhao F, Niemela JE, Deenick EK, Palendira U, et al. Dominant-Activating Germline Mutations in the Gene Encoding the PI(3)K Catalytic Subunit P110δ Result in T Cell Senescence and Human Immunodeficiency. Nat Immunol (2014) 15(1):88–97. doi: 10.1038/ni.2771
49. Deroo BJ, Korach KS. Estrogen Receptors and Human Disease. J Clin Invest (2006) 116(3):561–70. doi: 10.1172/JCI27987
50. Pedram A, Razandi M, O’Mahony F, Lubahn D, Levin ER. Estrogen Receptor-β Prevents Cardiac Fibrosis. Mol Endocrinol (2010) 24(11):2152–65. doi: 10.1210/me.2010-0154
51. Geraldes P, Gagnon S, Hadjadj S, Merhi Y, Sirois MG, Cloutier I, et al. Estradiol Blocks the Induction of CD40 and CD40L Expression on Endothelial Cells and Prevents Neutrophil Adhesion: An ERalpha-Mediated Pathway. Cardiovasc Res (2006) 71(3):566–73. doi: 10.1016/j.cardiores.2006.05.015
52. Harris HA, Albert LM, Leathurby Y, Malamas MS, Mewshaw RE, Miller CP, et al. Evaluation of an Estrogen Receptor-β Agonist in Animal Models of Human Disease. Endocrinology (2003) 144(10):4241–9. doi: 10.1210/en.2003-0550
53. Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A Transmembrane Intracellular Estrogen Receptor Mediates Rapid Cell Signaling. Science (2005) 307:1625–30. doi: 10.1126/science.1106943
54. Thomas P, Pang Y, Filardo EJ, Dong J. Identity of an Estrogen Membrane Receptor Coupled to a G Protein in Human Breast Cancer Cells. Endocrinology (2005) 146:624–32. doi: 10.1210/en.2004-1064
55. Filardo EJ, Thomas P. Minireview: G Protein-Coupled Estrogen Receptor-1, GPER-1: Its Mechanism of Action and Role in Female Reproductive Cancer, Renal and Vascular Physiology Endocrinology (2012) 153(7):2953–62. doi: 10.1210/en.2012-1061.
56. Notas G, Kampa M, Castanas E. G Protein-Coupled Estrogen Receptor in Immune Cells and Its Role in Immune-Related Diseases. Front Endocrinol (2020) 11. doi: 10.3389/fendo.2020.579420
57. Liehr JG, Ricci MJ. 4-Hydroxylation of Estrogens as Marker of Human Mammary Tumors. Proc Natl Acad Sci USA (1996) 93:3294–6. doi: 10.1073/pnas.93.8.3294
58. Osborne MP, Bradlow HL, Wong GY, Telang NT. Upregulation of Estradiol C16 Alpha-Hydroxylation in Human Breast Tissue: A Potential Biomarker of Breast Cancer Risk. J Natl Cancer Inst (1993) 85:1917–20. doi: 10.1093/jnci/85.23.1917
59. Mooberry SL. New Insights Into 2-Methoxyestradiol, a Promising Antiangiogenic and Antitumor Agent. Curr Opin Oncol (2003) 15(6):425–30. doi: 10.1097/00001622-200311000-00004
60. Lustig RH, Mobbs CV, Pfaff DW, Fishman J. Temporal Actions of 16 Alpha-Hydroxyestrone in the Rat: Comparisons of Lordosis Dynamics With Other Estrogen Metabolites and Between Sexes. J Steroid Biochem (1989) 33:417–21. doi: 10.1016/0022-4731(89)90332-4
61. Samavat H, Kurzer MS. Estrogen Metabolism and Breast Cancer. Cancer Lett (2015) 356(200):231–43. doi: 10.1016/j.canlet.2014.04.018
62. Cutolo M, Capellino S, Montagna P, Villaggio B, Sulli A, Seriolo B, et al. New Roles for Estrogen in Arthritis. Clin Exp Rheumatol (2003) 21:687–90.
63. Lahita RG. The Connective Tissue Diseases and the Overall Influence of Gender. Int J Fertil Menopausal Stud (1996) 41(2):156–65.
64. Baker ME. Origin and Diversification of Steroids: Co-Evolution of Enzymes and Nuclear Receptors. Mol Cell Endocrinol (2011) 334:14–20. doi: 10.1016/j.mce.2010.07.013
65. Miyaura H, Iwata M. Direct and Indirect Inhibition of Th1 Development by Progesterone and Glucocorticoids. J Immunol (2002) 168:1087–94. doi: 10.4049/jimmunol.168.3.1087
66. Mao G, Wang J, Kang Y, Tai P, Wen J, Zou Q, et al. Progesterone Increases Systemic and Local Uterine Proportions of CD4+CD25+ Treg Cells During Midterm Pregnancy in Mice. Endocrinology (2010) 151(11):5477–88. doi: 10.1210/en.2010-0426
67. Hughes GH. Progesterone and Autoimmune Disease. Autoimmun Rev (2012) 11(6-7):A502–14. doi: 10.1016/j.autrev.2011.12
68. Dressing G, Goldberg JE, Charles NJ, Schwertfeger KL, Lange CA. Membrane Progesterone Receptor Expression in Mammalian Tissues; a Review of Regulation and Physiological Implications. Steroids (2011) 76(1-2):11–7. doi: 10.1016/j.steroids.2010.09.006
69. Lapp CA, Thomas ME, Lewis JB. Modulation by Progesterone of Interleukin-6 Production by Gingival Fibroblasts. J Periodontol (1995) 66(4):279–84. doi: 10.1902/jop.1995.66.4.279
70. Goddard LM, Ton AN, Org T, Mikkola HK, Iruela-Arispe ML. Selective Suppression of Endothelial Cytokine Production by Progesterone Receptor. Vascul Pharmacol (2013) 59(1-2):36–43. doi: 10.1016/j.vph.2013.06.001
71. Lee JH, Ulrich B, Cho J, Park J, Kim C. Progesterone Promotes Differentiation of Human Cord Blood Fetal T Cells Into T Regulatory Cells But Suppresses Their Differentiation Into Th17 Cells. J Immunol (2011) 187(4):1778–87. doi: 10.4049/jimmunol.1003919
72. Lee JH, Lydon JP, Kim CH. Progesterone Suppresses the mTOR Pathway and Promotes Generation of Induced Regulatory T Cells With Increased Stability. Eur J Immunol (2012) 42(10):2683–96. doi: 10.1002/eji.201142317
73. Hughes GC, Thomas S, Li C, Kaja M, Clark EA. Progesterone Regulates IFN-Alpha Production by Plasmacytoid Dendritic Cells. J Immunol (2008) 180:2029–33. doi: 10.4049/jimmunol.180.4.2029
74. Bianchi VE. The Anti-Inflammatory Effects of Testosterone. J Endocr Soc (2018) 3(1):91–107. doi: 10.1210/js.2018-00186
75. Nasser M, Haider A, Saad F, Kurtz W, Doros G, Fijak M, et al. Testosterone Therapy in Men With Crohn’s Disease Improves the Clinical Course of the Disease: Data From Long-Term Observational Registry Study. Horm Mol Biol Clin Investig (2015) 22(3):111–7. doi: 10.1515/hmbci-2015-0014
76. Robinette CL. Sex-Hormone-Induced Inflammation and Fibromuscular Proliferation in the Rat Lateral Prostate. Prostate (1988) 12:271–86. doi: 10.1002/pros.2990120310
77. Castagnetta LA, Carrubba G, Granata OM, Stefano R, Miele M, Schmidt M, et al. Increased Estrogen Formation and Estrogen to Androgen Ratio in the Synovial Fluid of Patients With Rheumatoid Arthritis. J Rheumatol (2003) 30(12):2597–605.
78. Lahita RG, Bradlow HL, Ginzler E, Pang S, New M. Low Plasma Androgens in Women With Systemic Lupus Erythematosus. Arthritis Rheumatol (1987) 30(3):241–8. doi: 10.1002/art.1780300301
79. Frost DB, Wolf B, Peoples C, Fike J, Silver K, Laffoon M, et al. Estradiol Levels are Elevated in Older Men With Diffuse Cutaneous SSc and are Associated With Decreased Survival. Arthritis Res Ther (2019) 21:85. doi: 10.1186/s13075-019-1870-6
80. Frost DB, Feghali-Bostwick CA. A Positive Feedback Loop Between Estrogen and IL-6 Leads to Fibrosis in Human Skin. Arthritis Rheumatol (2017) 69(suppl 10).
81. Kawasaki T, Ushiyama T, Inoue K, Hukuda S. Effects of Estrogen on Interleukin-6 Production in Rheumatoid Fibroblast-Like Synoviocytes. Clin Exp Rheumatol (2000) 18:743–5.
82. Itoh Y, Hayashi H, Miyazawa K, Kojima S, Akahoshi T, Onozaki K. 17β-Estradiol Induces IL-1α Gene Expression in Rheumatoid Fibroblast-Like Synovial Cells Through Estrogen Receptor α (Erα) and Augmentation of Transcriptional Activity of Sp1 by Dissociating Histone Deacetylase 2 From Erα. J Immunol (2007) 178(5):3059–66. doi: 10.4049/jimmunol.178.5.3059
83. Weidler C, Härle P, Schedel J, Schmidt M, Schölmerich J, Straub RH. Patients With Rheumatoid Arthritis and Systemic Lupus Erythematosus Have Increased Renal Excretion of Mitogenic Estrogens in Relation to Endogenous Antiestrogens. J Rheumatol (2004) 31(3):489–94.
84. Sampson JN, Falk RT, Schairer C, Moore SC, Fuhrman BJ, Dallal CM. Association of Estrogen Metabolism With Breast Cancer Risk in Different Cohorts of Postmenopausal Women. Cancer Res (2017) 77(4):918–25. doi: 10.1158/0008-5472.CAN-16-1717
85. Tofovic SP, Jackson EK. Estrogens in Men: Another Layer of Complexity of Estradiol Metabolism in Pulmonary Hypertension. Am J Respir Crit Care Med (2016) 193(10):1087–90. doi: 10.1164/rccm.201512-2541ED
86. Stark K, Straub RH, Rovenský J, Blažičková S, Eiselt G, Schmidt M. CYB5A Polymorphism Increases Androgens and Reduces Risk of Rheumatoid Arthritis in Women. Arthritis Res Ther (2015) 17:56. doi: 10.1186/s13075-015-0574-9
87. Cutolo M, Straub RH. Sex Steroids and Autoimmune Rheumatic Diseases: State of the Art. Nat Rev Rheumatol (2020) 16:628–44. doi: 10.1038/s41584-020-0503-4
88. Zhu L, Song Y, Li M. 2-Methoxyestradiol Inhibits Bleomycin-Induced Systemic Sclerosis Through Suppression of Fibroblast Activation. J Dermatol Sci (2015) 77(1):63–70. doi: 10.1016/j.jdermsci.2014.10.007
89. Duncan GS, Brenner D, Tusche MW, Brüstle A, Knobbe CB, Elia AJ, et al. 2-Methoxyestradiol Inhibits Experimental Autoimmune Encephalomyelitis Through Suppression of Immune Cell Activation. PNAS (2012) 109(51):21034–9. doi: 10.1073/pnas.1215558110
90. Giannandrea M, Parks WC. Diverse Functions of Matrix Metalloproteinases During Fibrosis. Dis Model Mech (2014) 7(2):193–203. doi: 10.1242/dmm.012062
91. Grammar AC, Slota R, Fischer R, Gur H, Girschick H, Yarboro C, et al. Abnormal Germinal Center Reactions in Systemic Lupus Erythematosus Demonstrated by Blockade of CD154-CD40 Interactions. J Clin Invest (2003) 112(10):1506–20. doi: 10.1172/JCI19301
92. Kassi E, Moutsatsou P. Estrogen Receptor Signaling and Its Relationship to Cytokines in Systemic Lupus Erythematosus. J BioMed Biotechnol (2010) 2010:317452. doi: 10.1155/2010/317452
93. Zhang Y, Saha S, Rosenfeld G, Gonzalez J, Pepeljugoski KP, Peeva E. Raloxifene Modulates Estrogen-mediated B Cell Autoreactivity in NZB/W F1 Mice. The J Rheumatol (2010) 37(8):1646–57. doi: 10.3899/jrheum.090911.
94. Corradetti C, Jog NR, Cesaroni M, Madaio M, Caricchio R. Estrogen Receptor α Signaling Exacerbates Immune-Mediated Nephropathies Through Alteration of Metabolic Activity. J Immunol (2018) 200(2):512–22. doi: 10.4049/jimmunol.1700770
95. Akande EO, Hockaday TD. Plasma Oestrogen and Luteinizing Hormone Concentrations in Thyrotoxic Menstrual Disturbance. Proc R Soc Med (1972) 65(9):789–90.
96. Krassas GE. Thyroid Disease and Female Reproduction. Fertil Steril (2000) 74(6):1063–70. doi: 10.1016/S0015-0282(00)01589-2
97. Abalovich M, Levalle O, Hermes R, et al. Hypothalamic-Pituitary-Testicular Axis and Seminal Parameters in Hyperthyroid Males. Thyroid (1999) 9(9):857–63. doi: 10.1089/thy.1999.9.857
98. Chopra IJ. Gonadal Steroids and Gonadotropins in Hyperthyroidism. Med Clin North Am (1975) 59(5):1109–21. doi: 10.1016/s0025-7125(16)31961-7
99. Southren AL, Olivo J, Gordon GG, VIttek J, Brener J, Rafil F. The Conversion of Androgens to Estrogens in Hyperthyroidism. J Clin Endocrinol Metab (1974) 38(2):207–14. doi: 10.1210/jcem-38-2-207
100. Krassas GE, Poppe K, Glinoer D. Thyroid Function and Human Reproductive Health. Endocrine Rev (2010) 31(5):702–55. doi: 10.1210/er.2009-0041
101. Khong JJ, Finch S, De Silva C, Rylander S, Craig JE, Selva D, et al. Risk Factors for Graves' Orbitopathy; the Australian Thyroid-Associated Orbitopathy Research (ATOR) Study. J Clin Endocrinol Metab (2016) 101(7):2711–20. doi: 10.1210/jc.2015-4294
102. Stan MN, Bahn RS.. Risk factors for development or deterioration of Graves' Ophthalmopathy Thyroid (2010) 20(7):777–83. doi: 10.1089/thy.2010.1634.
103. Cui X, Zhang O, Deng W, Oesterreich S, Lu Y, Mills GB, et al. Progesterone Receptor Expression in Breast Cancer Cells via the Phosphatidylinositol 3-Kinase/Akt/Mammalian Target of Rapamycin Pathway: Progesterone Receptor as a Potential Indicator of Growth Factor Activity in Breast Cancer. Mol Endocrinol (2003) 17(4):575–88. doi: 10.1210/me.2002-0318
104. Øgard CG, Øgard C, Almdal TP. Thyroid-Associated Orbitopathy Developed During Hormone Replacement Therapy. Acta Opthalmol (2001) 79(4):426–7. doi: 10.1034/j.1600-0420.2001.079004426.x
105. Cury SS, Oilveira M, Síbio MT, Clara S, de Azevedo Mel Luvizotto R, Conde S, et al. Gene Expression of Estrogen Receptor-Alpha in Orbital Fibroblasts in Graves’ Ophthalmopathy. Arch Endocrinol Metab (2015) 59(3):273–6. doi: 10.1590/2359-3997000000050
106. Cuzzocrea S, Bruscoli S, Crisafulli C, Mazzon E, Agostini M, Muià C, et al. Estrogen Receptor Antagonist Fulvestrant (ICI 182,780) Inhibits the Anti-Inflammatory Effect of Glucocorticoids. Mol Pharmacol (2007) 71(1):132–44. doi: 10.1124/mol.106.029629
107. Cvoro A, Yuan C, Paruthiyil S, Miller OH, Yamamoto KR, Leitman DC. Cross Talk Between Glucocorticoid and Estrogen Receptors Occurs at a Subset of Proinflammatory Genes. J Immunol (2011) 186(7):4354–60. doi: 10.4049/jimmunol.1002205
108. Smith TJ, Janssen JAMJL. Response to Krieger Et Al. Re: "TSHR/IGF-1r Cross-Talk, Not IGF-1r Stimulating Antibodies, Mediates Graves' Ophthalmopathy Pathogenesis" (Thyroid 2017;27:746-747). Thyroid (2017) 27(11):1458–9. doi: 10.1089/thy.2017.0281
109. Dupont J, Le Roith D. Insulin-Like Growth Factor 1 and Oestradiol Promote Cell Proliferation of MCF-7 Breast Cancer Cells: New Insights Into Their Synergistic Effects Mol Pathol (2001) 54(3):149–54. doi: 10.1136/mp.54.3.149.
110. Le Romancer M, Poulard C, Cohen P, Sentis S, Renoir JM, Corbo L. Cracking the Estrogen Receptor's Posttranslational Code in Breast Tumors Endocrine Reviews (2011) 32(5):597–622. doi: 10.1210/er.2010-0016
111. LeRoith D, Helman L. The New Kid on the Block(ade) of the IGF-1 Receptor Cancer Cell (2014) 5(3) 201–2. doi: 10.1016/S1535-6108(04)00054-6
112. Yu L, Moore AB, Castro L, Gao X, Huynh HL, Klippel M, et al. Estrogen Regulates MAPK-Related Genes Through Genomic and Nongenomic Interactions Between IGF-I Receptor Tyrosine Kinase and Estrogen Receptor-Alpha Signaling Pathways in Human Uterine Leiomyoma Cells. J Signal Transduct (2012) 2012:204236. doi: 10.1155/2012/204236
113. Vidal O, Lindberg M, Sv L, Lubahn DB, Ritzen EM, Gustafsson J, et al. Disproportional Body Growth in Female Estrogen Receptor-Alpha-Inactivated Mice. Biochem Biophys Res Commun (1999) 265:569–71. doi: 10.1006/bbrc.1999.1711
114. Smith TJ, Janssen JAMJL. Insulin-like Growth Factor-I Receptor and Thyroid-Associated Ophthalmopathy Endocr Rev (2019) 40(1):236–67. doi: 10.1210/er.2018-00066.
115. Christopoulos PF, Msaouel P, Koutsilieris M. The Role of the Insulin-Like Growth Factor-1 System in Breast Cancer. Mol Cancer (2015) 14:43. doi: 10.1186/s12943-015-0291-7
116. Choucair A, Pham TH, Omarjee S, Jacquemetton J, Kassem L, Trédan O, et al. The Arginine Methyltransferase PRMT1 Regulates IGF-1 Signaling in Breast Cancer. Oncogene (2019) 38(21):4015–27. doi: 10.1038/s41388-019-0694-9
117. Song RX, Barnes CJ, Zhang Z, Bao Y, Kumar R, Santen RJ. The Role of Shc and Insulin-Like Growth Factor 1 Receptor in Mediating the Translocation of Estrogen Receptor α to the Plasma Membrane. Proc Natl Acad Sci (2004) 101(7):2076–81. doi: 10.1073/pnas.0308334100
118. Erlandsson J, Le Roith D. Insulin-Like Growth Factor 1 and Oestradiol Promote Cell Proliferation of MCF-7 Breast Cancer Cells: New Insights Into Their Synergistic Effects. J Clin Pathol: Mol Pathol (2001) 54:149–54.
119. Driggers PH, Segars JH. Estrogen Action and Cytoplasmic Signaling Pathways. Part II: The Role of Growth Factors and Phosphorylation in Estrogen Signaling. Trends Endocrinol Metab (2002) 13(10):422–7. doi: 10.1016/s1043-2760(02)00634-3
120. Kahlert S, Nuedling S, van Eickels M. Estrogen Receptor α Rapidly Activates the IGF-1 Receptor Pathway. J Biol Chem (2000) 275:18447–53. doi: 10.1074/jbc.M910345199
121. Oesterreich S, Zhang P, Guler RL, Sun X, Curran EM, Welshons WV, et al. Re-Expression of Estrogen Receptor Alpha in Estrogen Receptor Alpha-Negative MCF-7 Cells Restores Both Estrogen and Insulin-Like Growth Factor-Mediated Signaling and Growth. Cancer Res (2001) 61:5771–7.
122. Okkenhaug K, Vanhaesebroeck B. PI3K in Lymphocyte Development, Differentiation and Activation. Nat Rev Immunol (2003) 3(4):317–30. doi: 10.1038/nri1056
123. Imbert-Fernandez Y, Clem BF, O'Neal J, Kerr DA, Spaulding R, Lanceta L, et al. Estradiol Stimulates Glucose Metabolism via 6-Phosphofructo-2-Kinase (PFKFB3). J Biol Chem (2014) 289(13):9440–8. doi: 10.1074/jbc.M113.529990
124. Trefely S, Khoo PS, Krycer JR, Chaudhuri R, Fazakerley DJ, Parker B, et al. Kinome Screen Identifies PFKFB3 and Glucose Metabolism as Important Regulators of the Insulin/Insulin-Like Growth Factor (IGF)-1 Signaling Pathway. J Biol Chem (2015) 290:25834–46. doi: 10.1074/jbc.M115.658815
125. Trenti A, Tedesco S, Boscaro C, Ferri N, Cignarella A, Trevisi L, et al. The Glycolytic Enzyme PFKFB3 Is Involved in Estrogen-Mediated Angiogenesis via GPER1 J Pharmacol Exp Ther (2017) 361:398–407. doi: 10.1124/jpet.116.238212
126. Biello F, Platini F, D'Avanzo F, Cattrini C, Mennitto A, Genestroni S, et al. Insulin/IGF Axis in Breast Cancer: Clinical Evidence and Translational Insights. Biomolecules (2021) 11(1):125. doi: 10.3390/biom11010125
127. Klotz DM, Hewitt SC, Ciana P, Raviscioni M, Lindzey JK, Foley J, et al. Requirement of Estrogen Receptor-α in Insulin-Like Growth Factor-1 (IGF-1)-Induced Uterine Responses and in Vivo Evidence for IGF-1/Estrogen Receptor Cross-Talk. J Biol Chem (2002) 277(10):8531–7. doi: 10.1074/jbc.M109592200
128. Moll GW Jr., Rosenfield RL, Fang VS. Administration of Low-Dose Estrogen Rapidly and Directly Stimulates Growth Hormone Production. Am J Dis Child (1986) 140(2):124–7. doi: 10.1001/archpedi.1986.02140160042027
129. Leung KC, Johannsson G, Leong GM, Ho KKY. Estrogen Regulation of Growth Hormone Action. Endocrine Rev (2004) 25(5):693–721. doi: 10.1210/er.2003-0035
130. Ovesen P, Vahl N, Fisker S, Veldhuis JD, Christiansen JS, Lunde Jørgensen JO. Increased Pulsatile, But Not Basal, Growth Hormone Secretion Rates and Plasma Insulin-Like Growth Factor I Levels During the Periovulatory Interval in Normal Women. J Clin Endocrinol Metab (1998) 83(5):1662–7. doi: 10.1210/jcem.83.5.4761
131. Herzberg SD, Motu’apuaka ML, Lambert W, Fu R, Brady J, Guise JM. The Effect of Menstrual Cycle and Contraceptives on ACL Injuries and Laxity: A Systematic Review and Meta-analysis Orthop J Sports Med (2017) 5(7):2325967117718781. doi: 10.1177/2325967117718781.
132. Péqueux C, Raymond-Letron I, Blacher S, Boudou F, Adlanmerini M, Fouque MJ. Stromal Estrogen Receptor-α Promotes Tumor Growth by Normalizing an Increased Angiogenesis. Cancer Res (2012) 72(12):3010–9. doi: 10.1158/0008-5472.CAN-11-3768
133. Wing LY, Chuang PC, Wu MH, Chen HM, Tsai SJ. Expression and Mitogenic Effect of Fibroblast Growth Factor-9 in Human Endometriotic Implant Is Regulated by Aberrant Production of Estrogen. J Clin Endocrinol Metab (2003) 88(11):5547–54. doi: 10.1210/jc.2003-030597
134. Bartalena L, Marcocci C, Pinchera A. Graves' Ophthalmopathy: A Preventable Disease? Eur J Endocrinol (2002) 146(4):457–61.
135. Smith TJ. Insulin-Like Growth Factor-I Regulation of Immune Function: A Potential Therapeutic Target in Autoimmune Diseases? Pharmacol Rev (2010) 62(2):199–236. doi: 10.1124/pr.109.002469
Keywords: estrogen, thyroid eye disease, TED, Graves’ orbitopathy, estrogen receptor, steroid homeostasis, Graves’ disease, sex steroids
Citation: FitzPatrick AM (2022) Is Estrogen a Missing Culprit in Thyroid Eye Disease? Sex Steroid Hormone Homeostasis Is Key to Other Fibrogenic Autoimmune Diseases – Why Not This One? Front. Immunol. 13:898138. doi: 10.3389/fimmu.2022.898138
Received: 17 March 2022; Accepted: 16 May 2022;
Published: 17 June 2022.
Edited by:
Ruben Dario Motrich, Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), ArgentinaReviewed by:
Adriana Karina Chavez-Rueda, Mexican Social Security Institute (IMSS), MexicoJuan Pablo Mackern-Oberti, Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Argentina
Copyright © 2022 FitzPatrick. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amy M. FitzPatrick, YW1mNDFAY2FhLmNvbHVtYmlhLmVkdQ==