 Johan Svenson
Johan Svenson Natalia Molchanova
Natalia Molchanova Christina I. Schroeder
Christina I. Schroeder- 1Cawthron Institute, Nelson, New Zealand
- 2The Molecular Foundry, Lawrence Berkeley National Laboratory, Berkeley, CA, United States
- 3Center for Cancer Research, National Cancer Institute, National Institutes of Health, Frederick, MD, United States
The search for efficient antimicrobial therapies that can alleviate suffering caused by infections from resistant bacteria is more urgent than ever before. Infections caused by multi-resistant pathogens represent a significant and increasing burden to healthcare and society and researcher are investigating new classes of bioactive compounds to slow down this development. Antimicrobial peptides from the innate immune system represent one promising class that offers a potential solution to the antibiotic resistance problem due to their mode of action on the microbial membranes. However, challenges associated with pharmacokinetics, bioavailability and off-target toxicity are slowing down the advancement and use of innate defensive peptides. Improving the therapeutic properties of these peptides is a strategy for reducing the clinical limitations and synthetic mimics of antimicrobial peptides are emerging as a promising class of molecules for a variety of antimicrobial applications. These compounds can be made significantly shorter while maintaining, or even improving antimicrobial properties, and several downsized synthetic mimics are now in clinical development for a range of infectious diseases. A variety of strategies can be employed to prepare these small compounds and this review describes the different compounds developed to date by adhering to a minimum pharmacophore based on an amphiphilic balance between cationic charge and hydrophobicity. These compounds can be made as small as dipeptides, circumventing the need for large compounds with elaborate three-dimensional structures to generate simplified and potent antimicrobial mimics for a range of medical applications. This review highlight key and recent development in the field of small antimicrobial peptide mimics as a promising class of antimicrobials, illustrating just how small you can go.
Introduction
Therapeutic peptides have been used by humans for more than a century, since the first use of insulin in the 1920s and there are now nearly 100 approved peptide drugs on the market used for the treatment of a range of medical conditions (1). In humans, peptides are often produced and used endogenously to respond to altered physiological conditions and we have effective means to regulate their plasma concentration and half-life via different biochemical pathways (2, 3). While these biological systems allow the body to manage homeostasis efficiently, the natural susceptibility of innate immune peptides to enzymatic degradation represents a hurdle which combined with poor oral bioavailability hampers the introduction and developments of new peptide drugs (2, 4). To combat these challenges, modified peptides may be generated incorporating unnatural elements and truncated sequences to tune the ADME-properties (5, 6). However, for some biological targets, successful treatment depends on native peptides with elaborate structures. These complex native structures are essential for optimized target receptor binding and leads to increased production costs (Figure 1) (1, 14, 15).
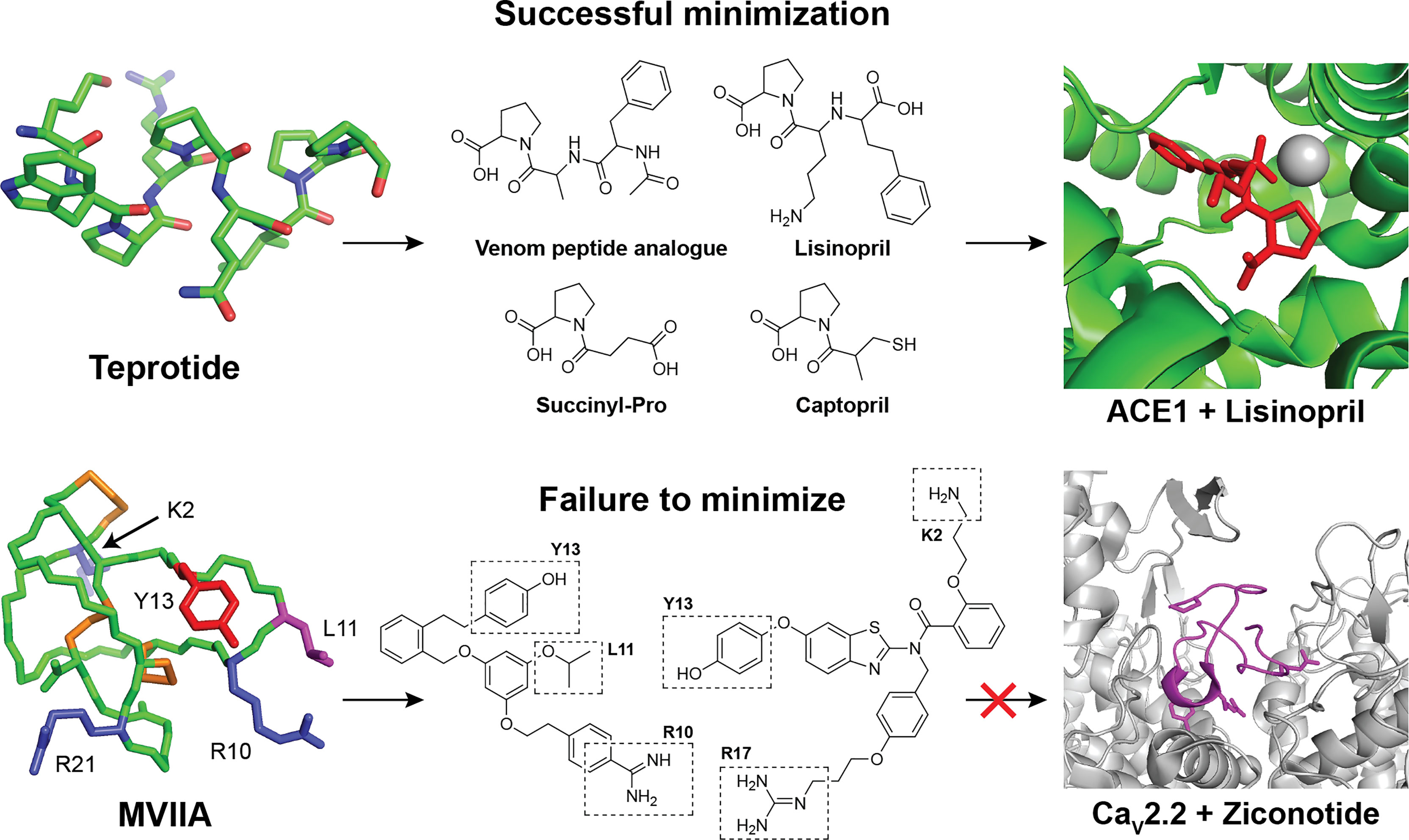
Figure 1 Examples of successful and unsuccessful attempts to minimize natural peptide leads during drug development. Top row: The natural teprotide peptide from the snake Bothrops jararaca is a potent inhibitor of the angiotensin-1-converting enzyme (ACE-1) (7). The native nonapeptide can be effectively truncated and modified to yield a range of simplified and optimized analogs with optimal binding to ACE-1[PDB: 1J36 (8)] (9). Bottom row: The highly complex and bioactive MVIIA peptide [PDB: 1TTK (10)] from the marine snail Conus magus have therapeutic value in the treatment of neuropathic pain by inhibiting N-type (Cav2.2) voltage-gated calcium channels [PDB: 7VFW (11)] (12). Despite extensive analog development and minimization efforts, no improved leads have been reported and the final marketed drug Ziconotide is identical to the native venom peptide (13).
Peptide-based drugs are used to target both intra- and extracellular targets, ion channels, GPCRs, and a variety of enzymes. Currently, the majority of new peptides entering the market are developed as treatments for the major therapeutic challenges including cardiovascular disorders, cancer, and metabolic diseases (1, 2, 16). Microbially produced peptides such as gramicidin, colistin, daptomycin and the lipoglycopeptide vancomycin are further examples of peptides that can be used to combat infectious diseases (17). Despite the success of these microbially derived peptide antibiotics, the advancement of the many endogenously produced defensive antimicrobial peptides (AMPs) in higher organisms have yet to deliver an impact on the drug market (17, 18).
AMPs represent a key component of the innate defence system involved in the initial rapid response to pathogenic intruders (3, 19, 20). AMPs are generally amphiphilic and many rapidly target microbes at the membrane level causing membrane disruption via modes of action less likely to cause resistance (18, 19, 21, 22). Faced with the plethora of naturally producing organisms, the composition and structure of native AMPs differs across species but falls within three groups based on their secondary structure. AMPs are generally short, and commonly adopts either an α-helical or a β-sheet structure with additional examples of extended and random-coil structured AMPs as well (23, 24). Driven by an urgent need to develop effective antimicrobials with novel modes of action due to the emergence of antimicrobial resistance (25), AMPs have been extensively studied and probed for their potential as the next generation antibiotics (18). Well-studied examples include the defensins, LL-37, lactoferricin, indolicidin and magainin (18, 26, 27). With numerous AMPs in clinical trials, it is expected that we will see this class of direct killing and/or immunomodulating compounds available for clinical infection management in the future (18, 24, 26).
The majority of the innate AMPs display highly defined secondary structures and the peptidic nature of these endogenous compounds is hampering the therapeutic development while the dependence of native sequences also results in high production costs (17). AMPs are further faced with obstacles associated with challenging pharmacokinetics, off-target toxicity, low metabolic stability, short half-life, and poor oral bioavailability (4, 28). As such, synthetic innate AMPs and their derivatives have not yet lived up to their heralded potential as the next generation antibiotics (18, 28–30).
Regardless of origin and structural class, the vast majority of AMPs carries a net positive charge (+2 to +11), balanced by a significant number of hydrophobic residues enabling an amphiphilic bioactive structure (31). Recent studies have illustrated that isolating these two functional components allows for the generation of much shorter AMPs and synthetic mimics thereof not adhering to the well-defined secondary structures found in nature (6). The observation that the key requirement for antibacterial activity is sufficient cationic charge balanced by hydrophobic elements has recently allowed the design of diverse AMP mimics as small as dipeptides with maintained and even improved bioactivity over their native counterparts (32–34). These smaller, and more “drug-like” AMP mimics come with several advantages, such as proteolytic resistance (35), potential for oral bioavailability (36, 37) and significantly lower production costs compared to the larger native AMPs (27, 28, 38, 39). The current report describes the state of the art of transferring the AMP pharmacophore onto alternative molecular scaffolds to yield simpler, yet more efficient antimicrobial leads (40). In addition, it answers the questions “How small can you go?” and “Does size matter?” in the search for leads that may offer alternatives to the native AMPs for combating resistant infections in the future.
Making AMPs Shorter
One of the main developmental barriers of the numerous natural AMPs towards their clinical use is the need for long native sequences and a reliance on a maintained helicity for a high bioactivity (28, 41). This dependence is costly and it presents synthetic challenges on large scale as these native sequences also require folding into their native bioactive secondary structure after initial synthesis (42). Furthermore, their size represent an obstacle for oral administration, targeted delivery (28) and they are prone to proteolytic degradation. This is due to their high content of charged and hydrophobic residues, which dictates the process of binding to the active site of many proteolytic enzymes (35, 43).
The most obvious approach to overcome the challenges associated with size is to attempt to make the peptides smaller and this can be achieved by identifying key structural features and trimming away excessive structures (1). In the field of AMPs however this has only been successfully attempted for a limited number of peptides and clinical leads. For notable AMP examples, such as LL-37, human β-defensins, indolicidin (omiganan) and magainin (pexiganan) only minor variation to the native sequence, if any, have been incorporated (24). In contrast, research on the iron binding lactoferrin protein and its antimicrobial cyclic degradation product lactoferricin, have highlighted that most of the antimicrobial activity can be maintained in linear N-terminal fragments (Figure 2) (49). This discovery has led to several shorter linear lactoferrin-derived lead peptides in clinical development such as hlf1-11, PXL01 and PXL150 (24, 26, 50, 51).
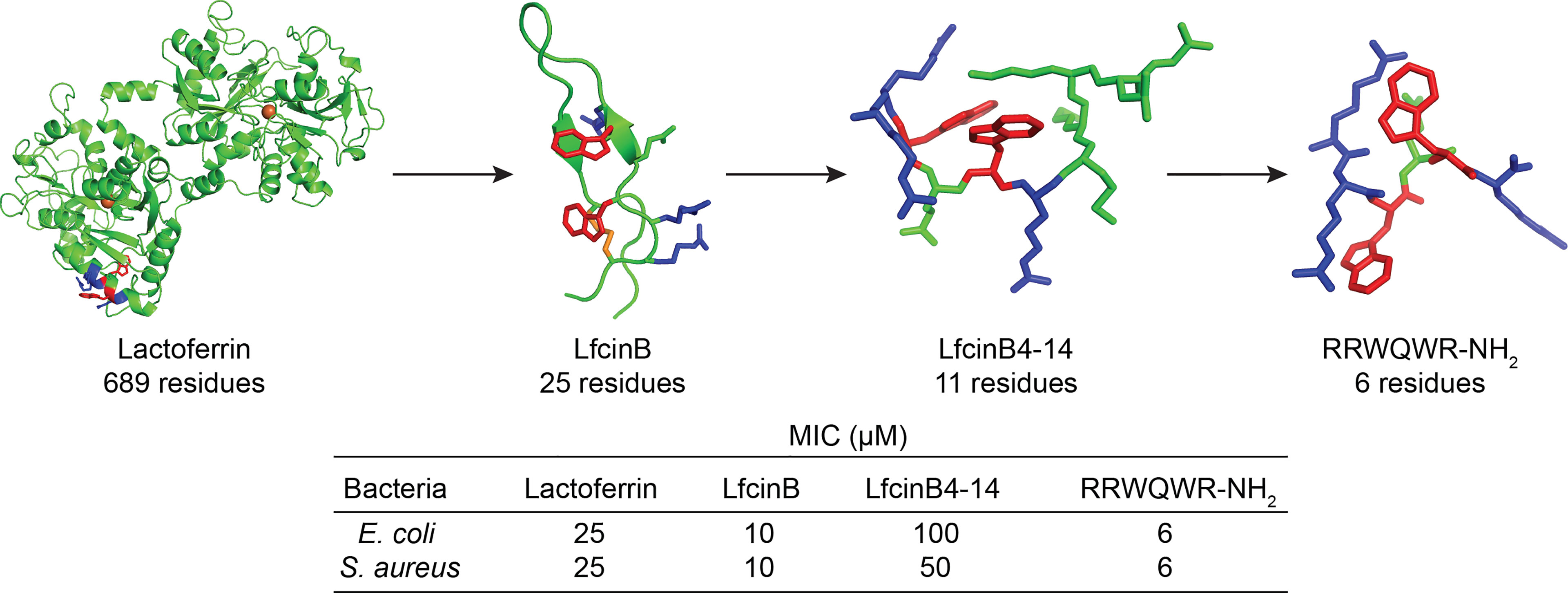
Figure 2 Successful truncation of the native lactoferrin protein, 689 residues, [PDB: 1BLF (44)] via LfcinB, 25 residues [PDB: 1LFC (45)], LfcinB4-14, 11 residues [PDB: 1Y5C (46)], to the antibacterial RRWQWR-NH2 motif (created and minimized in Pymol). Arg – red, Trp – blue, iron – orange ball, and disulfide bond – orange stick). The N-terminal cyclic lactoferricin peptide is formed upon digestion and linearization and additional truncation of lactoferricin yields shorter sequences with maintained antimicrobial activity. MIC-values taken from Svenson et al. (47) and Tomita et al. (48).
The helical N-terminal 15 residue fragment of bovine lactoferricin FKCRRWQWRMKKLGA was studied in detail by Svendsen and co-workers (52, 53). In a series of truncation and alanine scan experiments they showed that this sequence could be significantly improved by amino acid replacement and incorporation of unnatural hydrophobic amino acids as replacement of crucial tryptophan residues (33). By combining these findings with the early identification of an antibacterial RRWQWR motif inside lactoferricin by Tomita and co-workers (48), they embarked upon attempting to further minimize the hexapeptide to generate more “drug-like” compounds (54). By focussing on only using arginine and tryptophan and synthetic amino acids they showed across a series of key papers that a high antimicrobial activity could be obtained once a “sufficient” number of cationic residues and hydrophobic elements were assembled (32, 54, 55). The order of assembly appeared to only play a minor role (32) and this was recently verified in an elegant and extensive study by Dodson and co-workers who assembled and evaluated all (252 out of 254) possible RW peptides up to seven amino acids in length (56). As visually presented in Figure 3, weak activity against Gram-positive bacteria is seen for some tetrapeptides while almost all hexa- and heptapeptides display a high activity, seemingly independent of sequence, provided sufficient charge and hydrophobicity is provided (Figure 3).
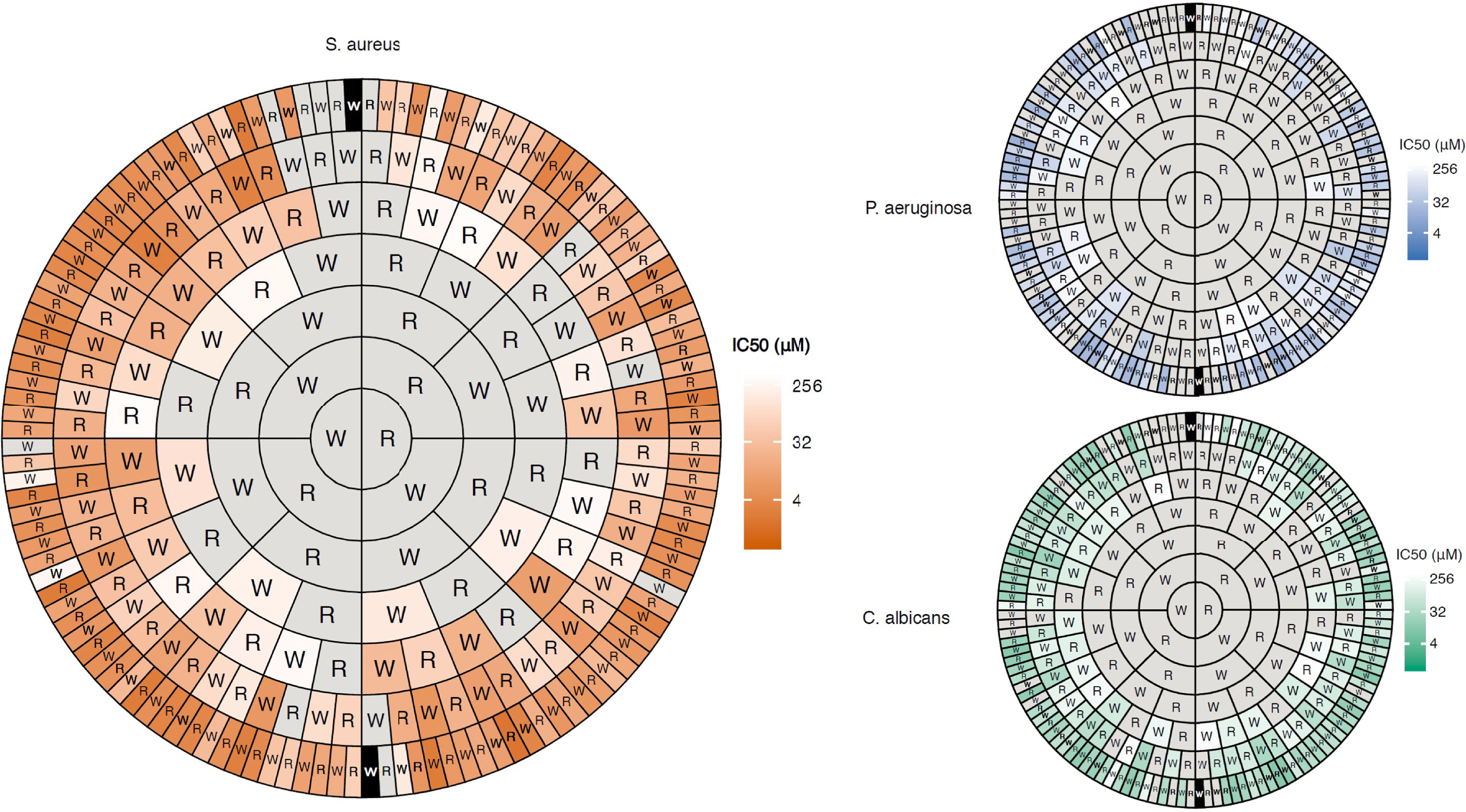
Figure 3 Concentric ring chart representation (Harris-Clark diagram) of the antimicrobial activity of all possible peptides comprised of W and R up to 7 residues long recently developed by Dobson and co-workers (56). The diagram highlights how the antimicrobial activity of the peptides increases with peptide length and that how most peptides are active as heptapeptides if sufficient R and W residues are combined in a non-sequence dependent manner. The data also shows how the compounds display a higher activity (shorter peptides required) against Gram-positive bacteria compared to Gram-negative ones and fungi due to differences in cell wall and membrane composition. Figure reproduced from Clark et al. (56) Commun. Biol. 2021 Vol. 4 Issue 1 Pages 1-14 with permission.
The 2 + 2 Pharmacophore
To further try to identify the minimal antimicrobial motif of these short AMPs, C-terminal modifications were performed which allowed an additional hydrophobic element to be incorporated (32). This was highly successful and by using C-terminal benzyl esters, peptides such as WRW-OBzl and RW-OBzl (Minimal inhibitory concentrations (MIC) against S. aureus 5 and 25 μg/mL, respectively) were generated with the C-terminal group acting as a second hydrophobic residue (32). Through these studies it became clear that the minimum antibacterial motif was at least two positive charges balanced by two units of hydrophobicity (32). Subsequently incorporated bulky unnatural amino acids could be used to increase the bioactivity further towards low micromolar bioactivity against a range of pathogenic bacteria and fungi (57). Despite the tripeptidic nature of these compounds, they strongly associate with plasma proteins (58–60) and ex-vivo metabolism studies using organ extracts highlighted significant enzymatic susceptibility (47). This rapid degradation was ascribed high affinity interactions with the fringing S1 and S1’ binding pockets due to the combination of cationic and hydrophobic residues and illustrated how the natural functionalities needed for bioactivity also generated good substrates for serine proteases and pepsin (35, 61). Reducing the snug fit to enzymatic binding pockets by incorporating “superbulky” hydrophobic residues or cationic analogs that are either longer or shorter than arginine effectively evades enzymatic degradation (36). Following this development, several highly active and stable tripeptides have been generated with LTX-109 (now AMC-109) being the most advanced in clinical development (Figure 4) (6, 63, 64). AMC-109 represent a fast-acting, broad-spectrum, antimicrobial AMP drug candidate and is currently undergoing development by Amicoat (62).
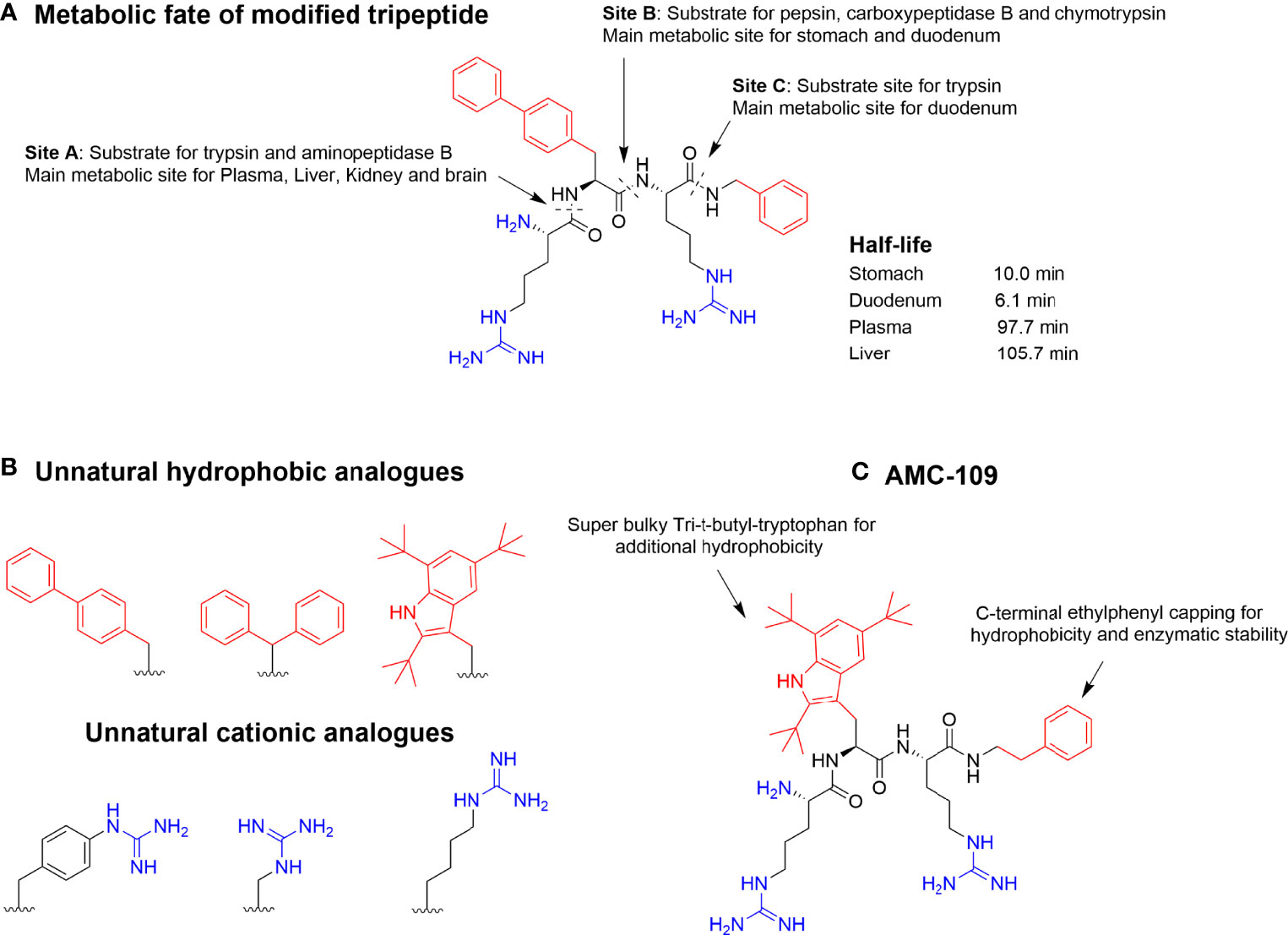
Figure 4 (A) Metabolic susceptibility of a short, modified, and highly active antimicrobial tripeptide in selected key metabolic compartments despite incorporation of bulky unnatural amino acids (47). (B) Incorporation of optimized unnatural cationic and hydrophobic amino acids can be used to steer the metabolic stability together with different C-capping strategies. (C) Optimized lead AMC-109 (previously LTX-109) currently developed by Amicoat AS (Norway) for the eradication of microbial biofilms (62).
Attempts to further refine the 2 + 2 pharmacophore have shown that it is more complex than initially thought and both the basicity of the cationic residues (34) as well as the shape and volume of the hydrophobic moieties are critical for the bioactivity of the final peptides (65). Several studies illustrated that guanidine groups can promote increased hydrogen bonding interactions with the phospholipids of the target microbial membrane, in comparison with amines and quaternary ammonium groups (6, 66). Dissection of the hydrophobic contribution into a minimal hydrophobic volume (65) and its placement (39) instead of a number of hydrophobic residues also generates a more accurate understanding of the essential structural requirements for high antimicrobial activity (65). While the initial “2+2” pharmacophore remains quite accurate for linear peptides, parallel and independent developments have made it apparent that other potent small AMPs and mimics thereof can be prepared using alternative scaffolds to generate potent leads (40). These tailormade synthetic mimics may offer advantages over natural sequences and they have been praised as important classes of compounds for eliminating both multidrug-resistant and pandrug-resistant bacterial isolates (18). The following section will summarize the development of the classes of small (<1000 Da) amphiphilic AMP mimics (Table 1) inspired by the natural cationic AMP pharmacophore which has yielded several leads in late stage clinical development (40). The focus lies on small antimicrobial mimics and our review will not cover immunomodulation (40), larger oligomeric assemblies or polymers which have been reviewed elegantly elsewhere (28, 30, 80–82).
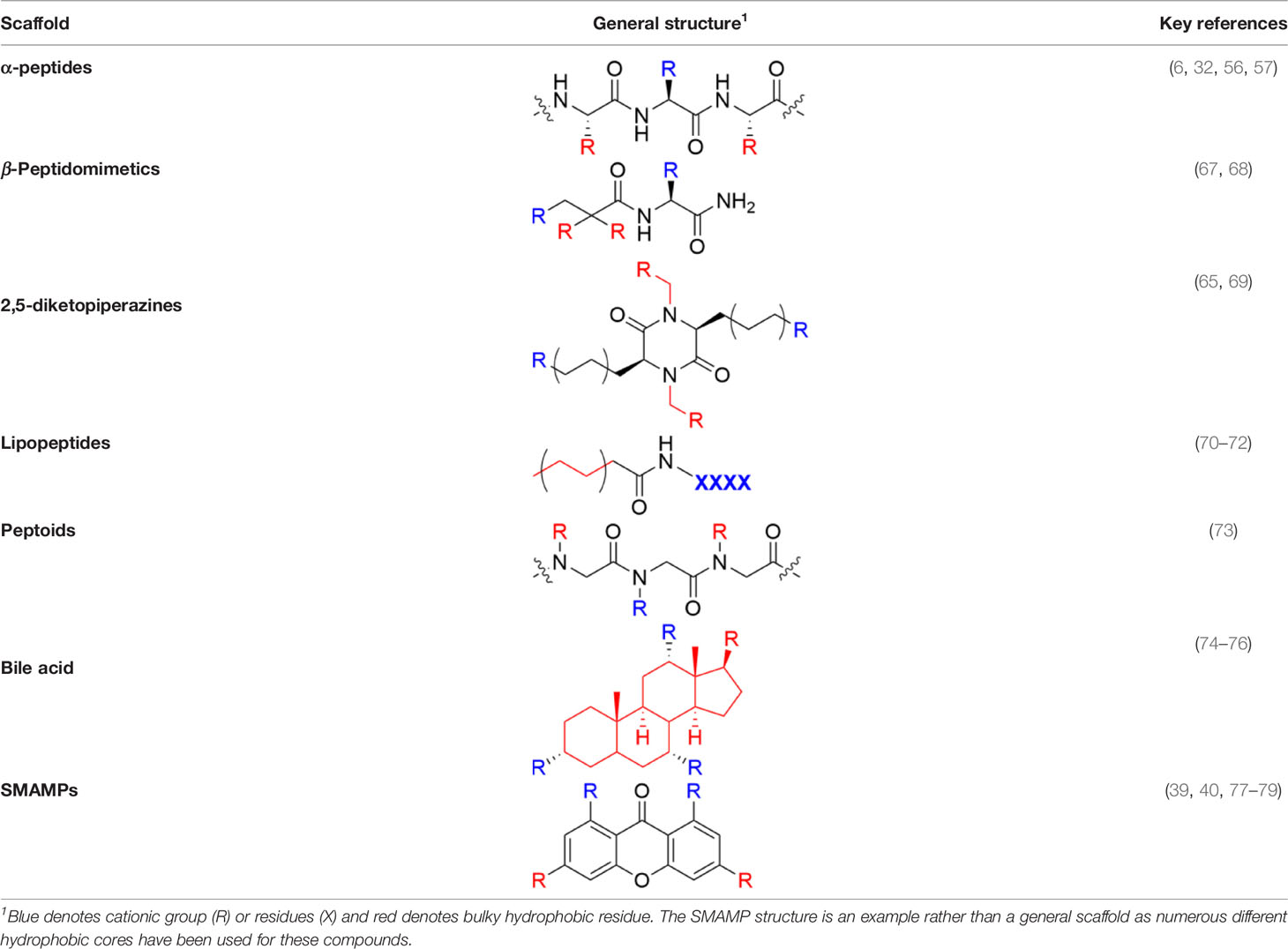
Table 1 Major scaffold types used to generate small bioactive AMP mimics adhering to the 2 + 2 pharmacophore.
β-Peptidomimetics and β2,2-Amino Acid Derivatives
Recent developmental work by the Strøm lab, initially involved in the key fundamental 2 + 2 pharmacophore discovery, focussed on generating simpler AMPs mimics. By transferring the crucial substituents onto an achiral lipophilic 3-amino-2,2-disubstituted propionic acid scaffold they produced a library of β-peptidomimetics (67). Two identical hydrophobic residues were introduced and a substantial antibacterial effect was observed for the bulkier residues, in particular for substituents incorporating one or two t-butyl substituents in analogy to the AMC-109 drug lead (63). A strong link between overall hydrophobicity and antibacterial effect was observed and the compounds were shown to display both low haemolytic activity and resilience towards chymotryptic degradation over 48 h (67). The cationic requirement was initially provided by an arginine residue at the N-terminus but studies on only the β2,2-amino acid scaffold established that two essential cationic group could be included as N and C-terminal substituents, generating smaller compounds (68). These β2,2-amino acid derivatives could be made with significant design freedom with active compounds fulfilling both the 2 + 2 pharmacophore and the Lipinski rules for orally bioavailable drugs (68, 83). The theoretical oral uptake was calculated and also studied experimentally for selected compounds using a phospholipid vesicle-based barrier designed to mimic the intestinal epithelia (68, 84). The permeability studies suggest a moderate oral absorption in humans, which is in alignment with similar experimental studies on modified tripeptides and also highlights how the theoretical uptake models are not necessarily the optimal method for assessing permeation for this class of compounds (37). The effect of a range of both hydrophobic and cationic elements and halogenation have been evaluated in this scaffold and the reported structural observations are similar to those observed for the linear tripeptides (6) with guanidine groups performing better as cationic groups and an overall pattern of dependency on “sufficient hydrophobic bulk” for optimal activity (Figure 5) (86). The compounds have been extensively evaluated as antimicrobials and demonstrated to be highly active against many multi-resistant clinical isolates and also intracellular pathogens such as Chlamydia pneumoniae (86, 87).
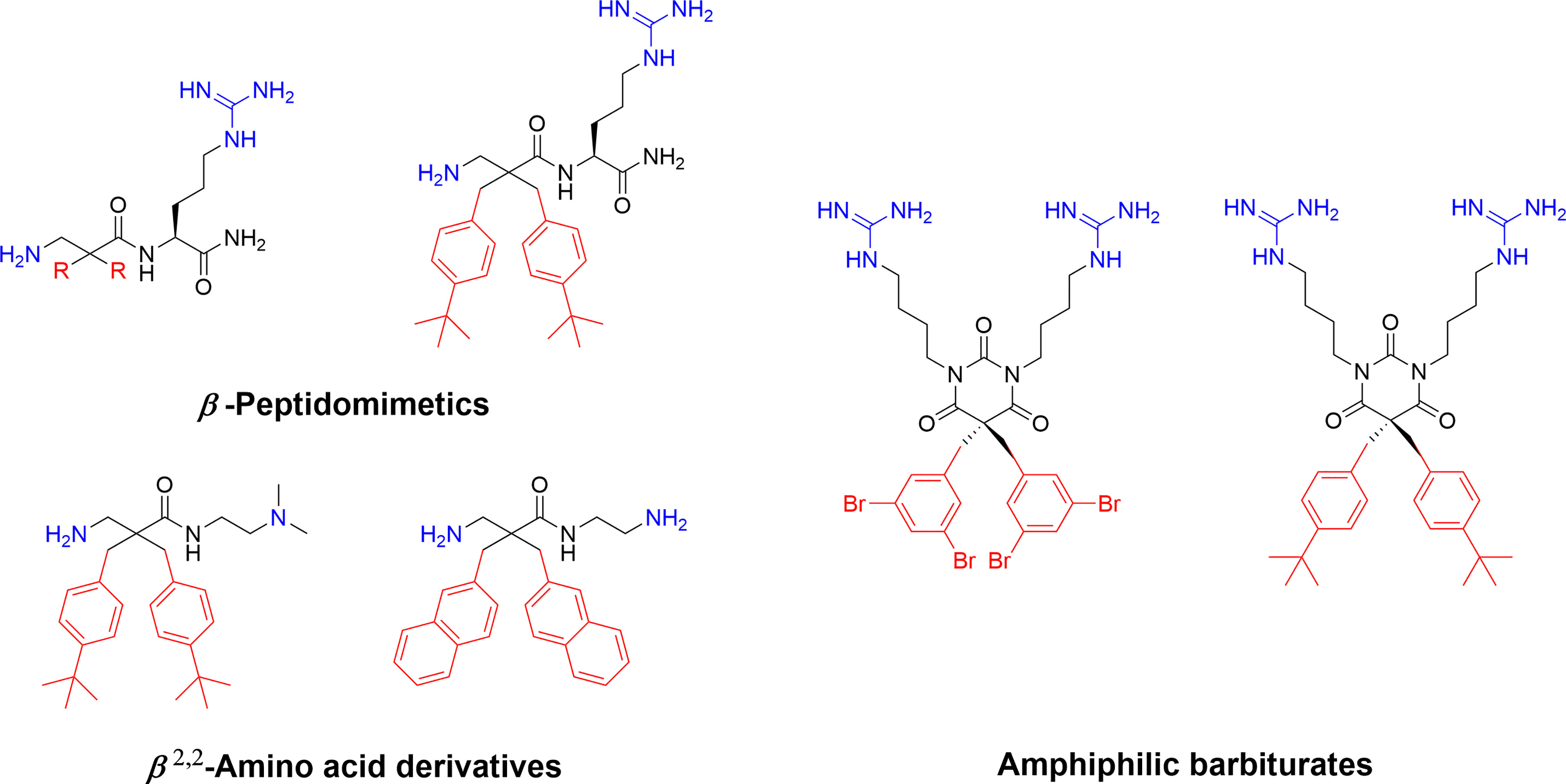
Figure 5 Examples of highly active antimicrobial β-peptidomimetics, β2,2-amino acid derivatives and amphiphilic tetrasubstituted barbiturates develop by Strøm and co-workers (67, 68, 85). The peptidomimetics illustrate how the AMP pharmacophore effectively can be transferred to smaller scaffolds.
The natural marine antimicrobial eusynstyelamides have been isolated from both the marine Arctic bryozoan Tegella cf. spitzbergensis (88) and the Australian ascidian Eusynstyela latericius (89). The eusynstyelamides are composed of a central five-membered dihydroxybutyrolactam ring naturally substituted with two 6-bromo-indoles and two cationic sidechains, containing both guanidine and amine groups (88). As such they are naturally adhering to the AMP pharmacophore and a recent study by Strøm and co-workers describes how the dihydroxybutyrolactam core can be replaced with a simpler achiral barbiturate ring which allows for rapid generation of synthetic analogs (Figure 5) (85). These tetrasubstituted barbiturates were shown to be highly antimicrobial with good cellular selectivity over human erythrocytes and active towards resistant clinical isolates. Introduction of “superbulky” hydrophobic sidechains generated more active analogs compared to the natural compounds. A lead compound was evaluated in vivo in a murine peritonitis model and a single intraperitoneal injection (1.4 mg/kg) resulted in a 98% reduction of the bacterial load of Klebsiella pneumoniae (85).
2,5-Diketopiperazines
2,5 diketopiperazines (DKPs) are cyclic dipeptides and are regarded as privileged structures with the ability to bind to a range of natural receptors (90). 2,5-DKPs present a near ideal scaffold for the development of novel antimicrobial compounds, as they are chemically accessible, highly stable, and amenable to extensive synthetic derivatisation at up to six positions to develop versatile bioactive compounds (69, 91, 92). In addition to freedom in terms of amino acid stereochemistry and choice of sidechains, up to four additional substituents can be further incorporated via different alkylation strategies (90, 91). Taking advantage of the readily available scaffold Labrière et al. recently reported the preparation of a series of 2,5-DKPs adhering to the design principles of the aforementioned linear tripeptides developed by Strøm and co-workers (65). Focussing mainly on diastereomeric mixtures, contribution from both charge and bulk were probed and the results showed that the evaluated DKPs exhibited similar or superior antimicrobial activity in comparison to structurally related linear peptides incorporating unnatural amino acids (65). An enantiopure analog was prepared and shown to display improved activity against all tested bacteria compared to the diastereomeric mixture. Additional work and improvement of the synthetic methodology (69) allowed Grant et al. to generate an enantiopure library of 2,5- DKPs to further investigate the role of stereochemistry on the effect of these compounds (93). The stereoisomers of cyclo(N-Bip-Arg-N-Bip-Arg) were prepared and a clear dependence on the ability to form stable amphiphilic structure for a high activity was verified through spectroscopic and modelling experiments (Figure 6) (93).
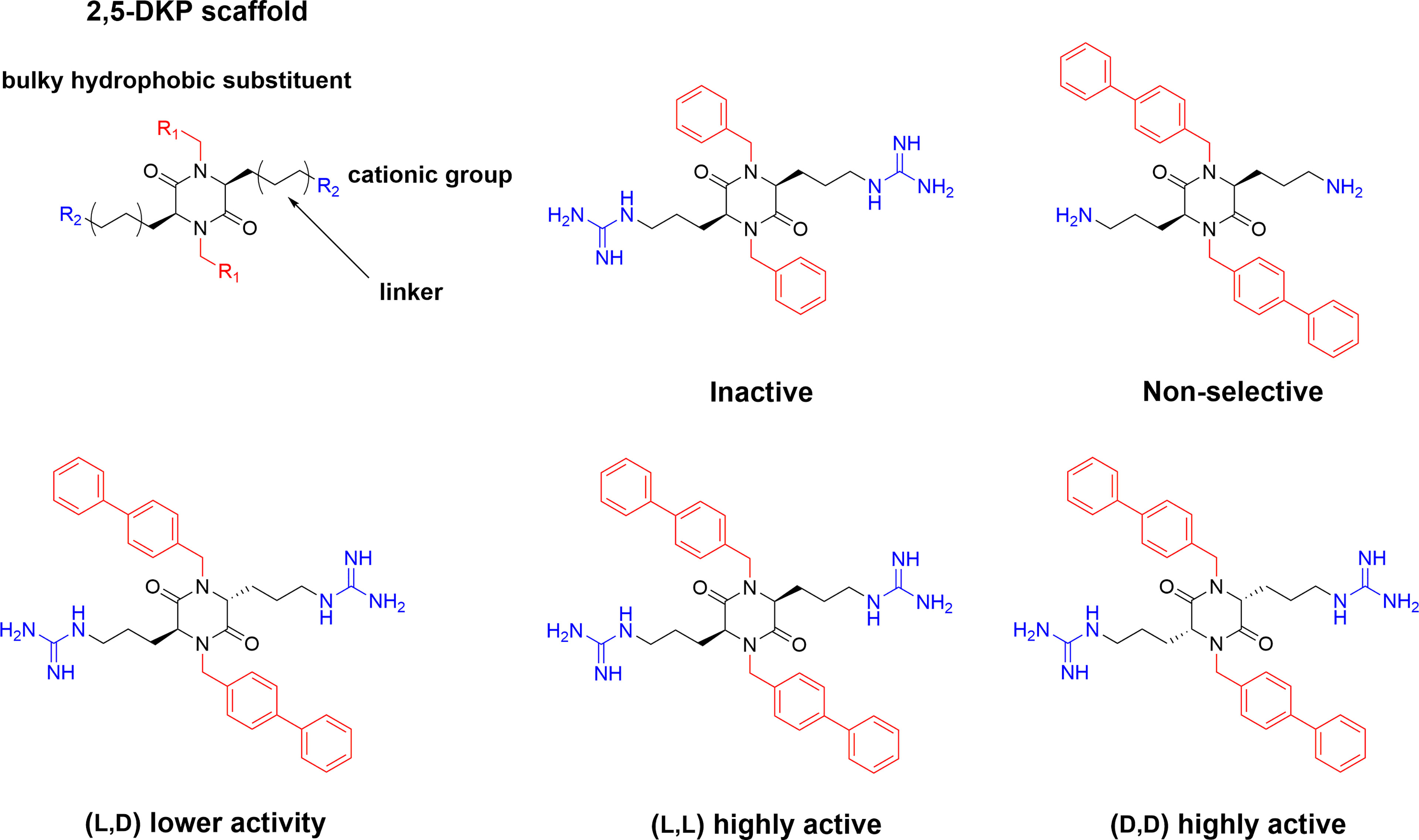
Figure 6 Design of the antimicrobial 2,5-DKP scaffold where the functionalities required for potent bioactivity can readily incorporated. Recent studies report a similar pharmacophore as that described for linear peptides for the shorter cyclic dipeptides (65). Bottom row illustrate stereoisomers of cyclo(N-Bip-Arg-N-Bip-Arg) which display difference in bioactivity due to their ranging ability to form stable amphiphilic solution structures (93).
Ultrashort Lipopeptides
Incorporation of larger hydrophobic residues and unnatural sidechains has been shown to be highly beneficial for antimicrobial activity (6, 57). An alternative way to incorporate this hydrophobic element is though N-terminal modification with fatty acids to generate so called lipopeptides (LiPs) (70). Natural antimicrobial LiPs exist and are nonribosomally produced by some bacteria and fungi (70, 94). This class of antibiotics has demonstrated high activity against multidrug-resistant microorganisms with daptomycin and caspofungin serving as examples of approved antimicrobial LiPs in clinical use (95–97). Natural LiPs are generally smaller than AMPs with short peptide chains composed of either six or seven amino acids coupled to an N-terminal C8-C18 fatty acid chain (70). As such they do not adhere to the antimicrobial pharmacophore but simplified de novo designed ultrashort lipopeptides (USLiPs) have been prepared (71, 98, 99). Shai and co-workers pioneered this area (72) by developing a series of highly potent USLiPs composed of four L and D amino acids with the general KXXK motif (71). The potency and cellular selectivity could be controlled via the alkyl chain length and peptide sequence (71, 100). Since their initial development shorter sequences and both di- and trimeric versions have been prepared (101–103) and they performed successfully in vivo in a mouse model of Candida albicans infection (102) (Figure 7). Effective USLiPs can be prepared with two cationic residues and they act synergistically together with beta-lactams and vancomycin in vitro (104). Beneficial potentiation effects have also been reported for dilipidic tetrabasic lipopeptides in a series of recent report by the Schweizer group (105, 106). These compounds can effectively potentiate the effect of Novobiocin and Rifampicin against Gram-negative bacteria and the effect can be modified by N-methylation of the cationic residues (107). Employing alternative scaffold backbones such as α-AApeptides and α/γ-AA hybrid peptides have also been shown to yield efficient lipidated analogs (108, 109). The history and development of natural and synthetic lipopetides have been reviewed (70, 110) and their industrial potential and application has been recently described (111, 112).
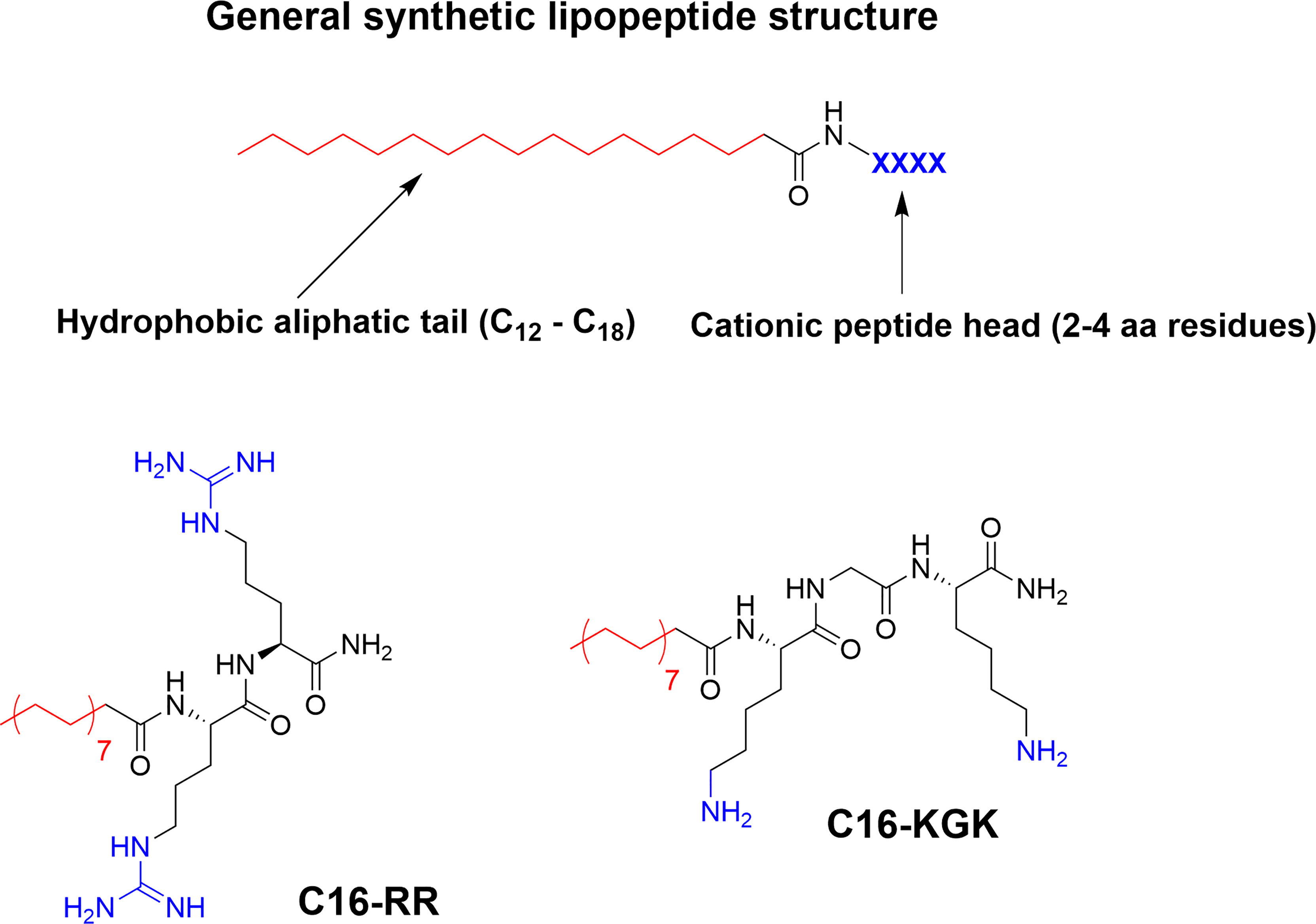
Figure 7 General structure of a USLiP and bioactive examples with a cationic di- to tripeptide providing the cationic charge to sufficiently balance the N-terminal fatty acid tail to generate potent antimicrobial amphiphiles (100, 102).
Peptoids
The natural α-peptides can be efficiently mimicked by using oligo-N-substituted glycines with the sidechains attached to the backbone nitrogens instead of to the α-carbon (73). These peptoids are relatively easy to prepare synthetically and they offer an achiral backbone that is stable to proteolytic degradation. Despite a lack of amide hydrogens for internal hydrogen bonding formation, peptoids can be designed in an amphiphilic manner to adhere to the structural requirements reported for short linear antimicrobial peptides with the same chemical toolbox available to optimize their properties (113–115). Short peptoids have been shown to adopt various self-assembled structures (116, 117) and several potent larger antimicrobial helical peptoids and peptide/peptoid hybrids have been reported with efficacy towards drug-resistant bacteria (73, 114, 118, 119). In analogy to the α-peptides, peptoids can also be used as analogues to linear peptides to generate potent lipopeptoids by attachment of linear alkyl chains (116, 120–122) as shown in Figure 8. Furthermore, the incorporation of halogen atoms into peptoid structures have yielded active leads (115) against a range of ESKAPE pathogens (116). The development of anti-infective peptoids was recently reviewed in detail by Bicker and Cobb (73).
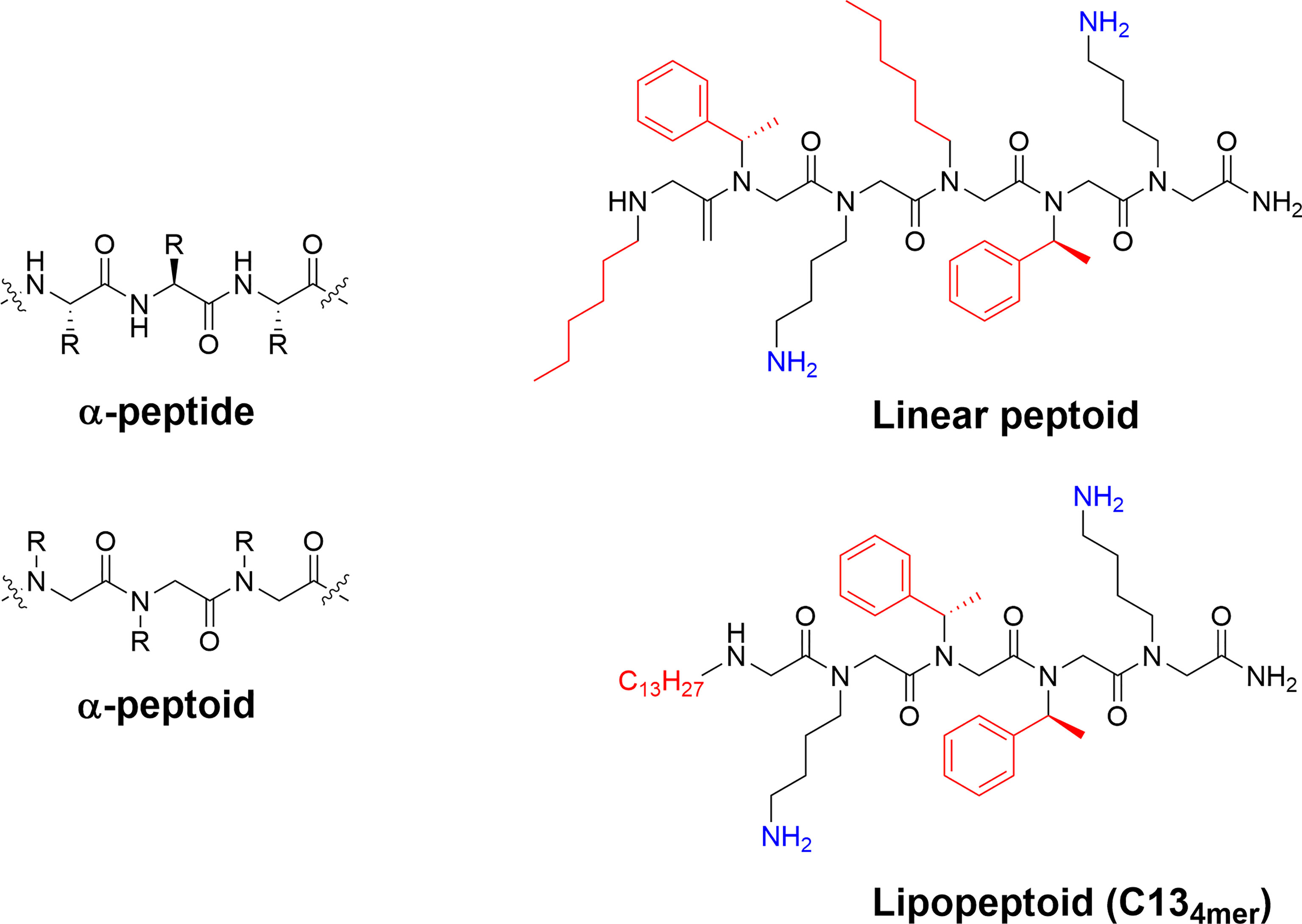
Figure 8 Structure and examples of antimicrobial peptoids. The difference between the backbone of α-peptides and α-peptoids allows for differences in structure and stability of the final compounds. The peptoid scaffold can be used to generate highly active antimicrobial compounds with the linear H-(Nhe-Nspe-NLys)2-NH2 and the lipopeptoid C134mer being active against Mycobacterium tuberculosis and ESKAPE pathogens (73, 116, 120).
Longer α-AApeptides (123) and γ-AApeptides (124) have been used to prepare mimics of AMPs and it is expected that these scaffold can be used also for generating these shorter mimics in analogy to the peptoids (73).
Bile Acid Derivatives (Ceragenins)
The naturally amphiphilic cholic acid is produced endogenously to solubilize lipids as part of the digestion process and several studies have described ways to utilize the scaffold to generate compounds designed for membrane interactions (74, 75). By further fine-tuning the amphiphilic nature of the cholic acid scaffold, Savage and co-workers developed potent nonpeptidic mimics of cationic AMPs, initially known as cationic steroid antibiotics (76), but later renamed “ceragenins” (74). The ceragenins are designed to mimic the facial cationic morphology of natural AMPs and the three free hydroxyls and the carboxy group offer means to develop a range of different ceragenins with optimal activity and cellular selectivity by incorporation of both basic and bulky substituents (75) (Figure 9). The ceragenins CSA-13 and CSA-131 were equipped with an alkyl chain to increases their ability to interact with lipid A and they were particularly active against both bacteria and fungi (74, 125, 126). The CSA-13 ceragenin was initially developed towards clinical use by Ceragenix Pharmaceuticals and there is currently a significant focus on developing surface coatings and nanoparticles incorporating leaching and immobilized ceragenins (127) for prevention of biofilm formation using the second generation CSA-131 lead (128). The Cerashield™ Endotracheal Tube (the CeraShield ETT) is currently being developed by N8 Medical for complications experienced by mechanically-ventilated patients and it has been evaluated in human clinical trials (NCT03716713). This development is parallel with the development of AMC-109 for surface protection by Amicoat AS (62). A plethora of different substituents has been investigated on the bile acid scaffold, such as amino acids (129) and bile-based antimicrobials were recently reviewed by Lin et al. (75).
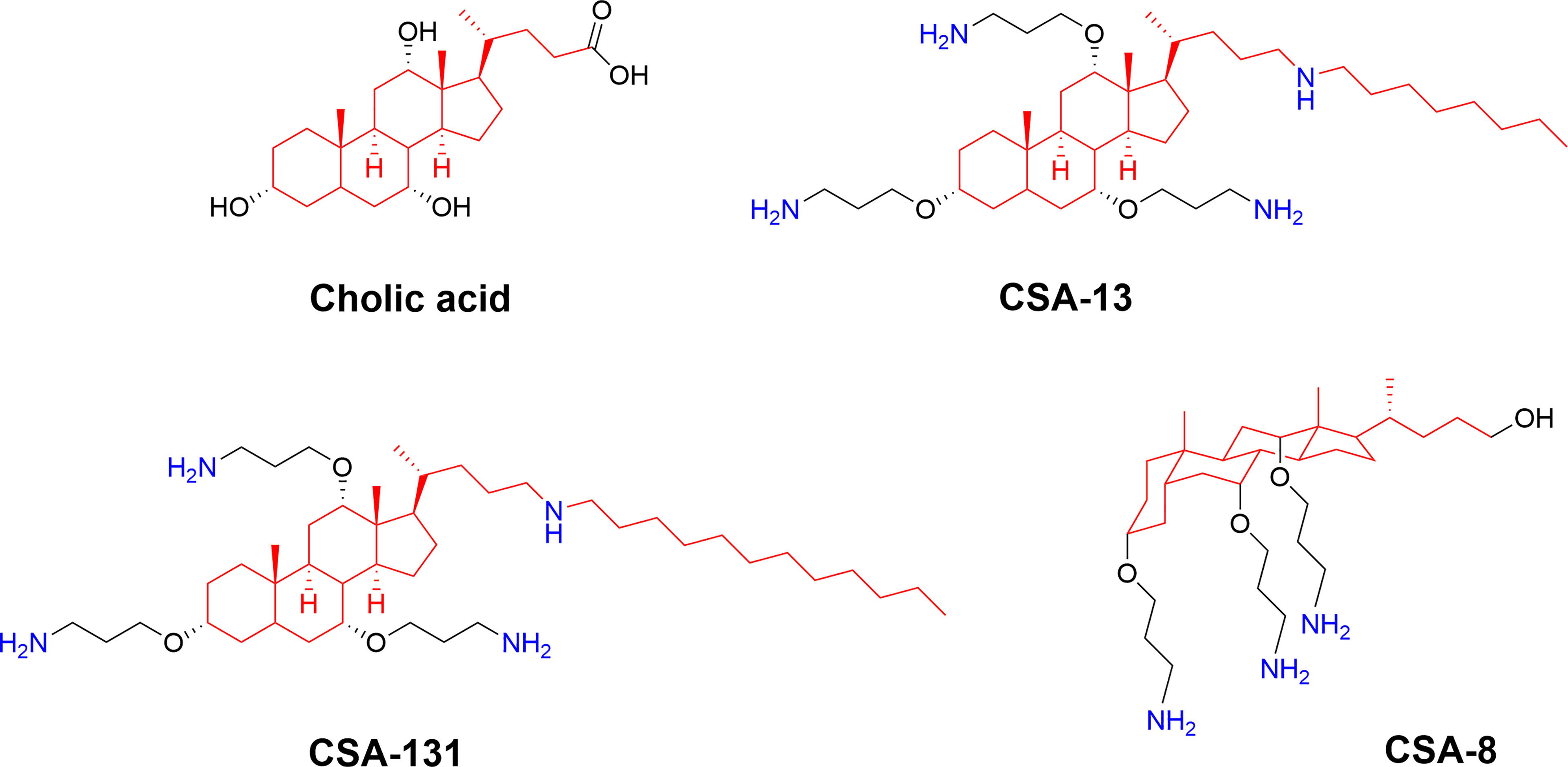
Figure 9 Structures of natural cholic acid and examples of the ceragenins CSA-13, and CSA-131 incorporating amine and alkyl modification. The perspective drawing of CSA-8 illustrates the facial amphiphilicity of the final compounds which is key for bioactivity (74).
Synthetic Mimics of Antimicrobial Peptides (SMAMPs/smHDPs)
The realisation that it is possible to move away from the oligomeric “amino acid scaffold” of α, β-peptides and peptoids for the generation of potent antimicrobial compounds has spurred considerable research into abiotic scaffolds fulfilling the same structural requirements. The development of these diverse compounds was initially led by Tew and Scott and their work has yielded several different effective synthetic mimics of antimicrobial peptides (SMAMPs) and larger polymeric assemblies (40, 130). The diverse SMAMP scaffolds have been designed not to duplicate the natural peptide structures but in an attempt to generate smaller compounds with better pharmacokinetic and tissue distribution properties that can be prepared more cheaply on large scale (40). Whilst producing small compounds displaying both cationic and hydrophobic functionalities can be seen as chemically straightforward, the challenge lies in maintaining both cellular selectivity and therapeutic index. The tools at hand are often a choice between amines, guanidine groups and quaternary ammonium groups balanced with appropriate hydrophobic elements. Much of the research has been focussed on antibiotic-like substances but efforts to include them into different functional polymers have also been made by Lienkamp and co-workers (131, 132). Selected successful SMAMP scaffolds include aryl SMAMPs (133) meta-phenylene ethynylene (77), arylamides (134, 135), diphenyl dibenzopyrole (136), biguanidyl biarylurea (137), and porphyrin (138) (Figure 10). A particular promising arylamide SMAMP based on a central pyrimidine (PMX207) was shown to be promising leads against chloroquine resistant Plasmodium falciparum (137) and further work on the same scaffold (139) produced PMX30063 which is now under clinical development as Brilacidin by Innovation Pharmaceuticals Inc. (formerly Cellceutix) (40). Brilacidin has demonstrated potent bactericidal activity against drug-resistant and drug-susceptible strains of multiple Gram-negative and Gram-positive pathogens with a membrane depolarization mode of action similar to daptomycin (140) and via immunomodulation (140, 141). Several initial clinical trials on brilacidin for the treatment of acute bacterial skin and skin structure infections (ABSSSI) have been conducted and the lead displayed low toxicity and was well tolerated (40). Briliacidin has also been recently reported to be highly active against SARS-CoV-2 in cell culture (141, 142) which illustrates the potential for expanded work on this class of AMP mimics, which has been reviewed by Scott & Tew in 2017 (40).
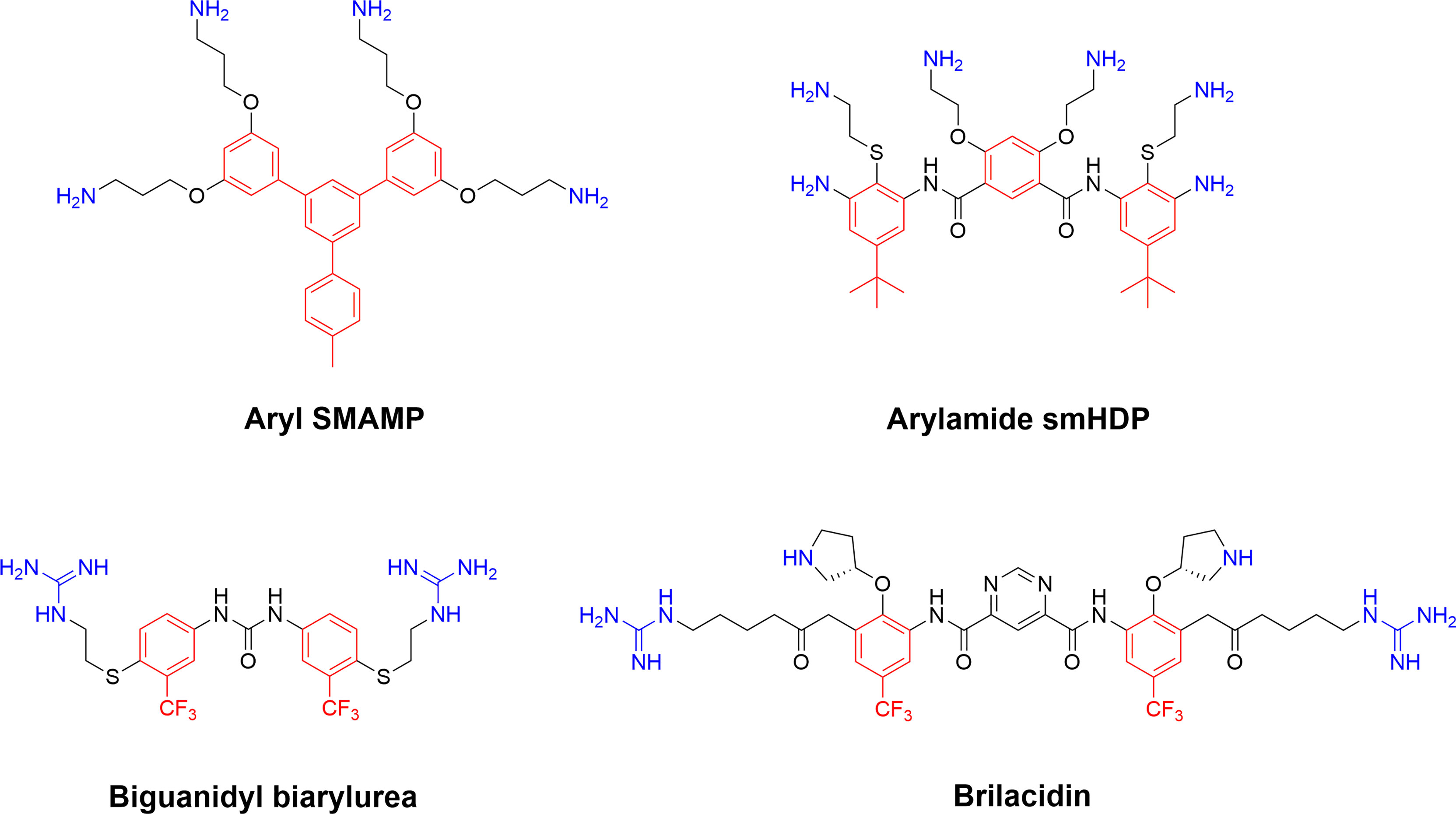
Figure 10 Representative early SMAMPs illustrating the chemical diversity available to generate cationic synthetic amphiphilic compounds adhering to the AMP pharmacophore (40, 133). Briliacidin is currently being developed by Innovation Pharmaceuticals for a range of conditions such as ulcerative proctitis, oral mucositis, ABSSSI and as inhibitors for SARS-CoV-2.
The group of Cai has also recently prepared an extensive series of different SMAMPs (or mimics of host-defence peptides, HDPs) with a focus on synthetic simplicity (78). A series of disubstituted hydantoins were prepared by combining the structure of the hydantoin based antibiotic nitrofurantoin with the structural element needed for bacterial membrane activity (143). The lead compound of the series displayed low micromolar MIC-values against resistant bacteria and exhibited potent in vivo efficacy for the treatment of lungs infected with MRSA in a rat model (143). Further work included small dimeric cyclic guanidine derivatives active against both multidrug-resistant Gram-negative and Gram-positive bacteria. The activity in vitro was subsequently verified in vivo using a MRSA-infected thigh burden mouse model (144). Additional rationally designed SAMP scaffolds evaluated include lysine N-alkylamides (145) reduced amide-based compounds (146) and a range of others based on aromatic linkers (147–149) which have produced effective antimicrobial compounds (Figure 11).
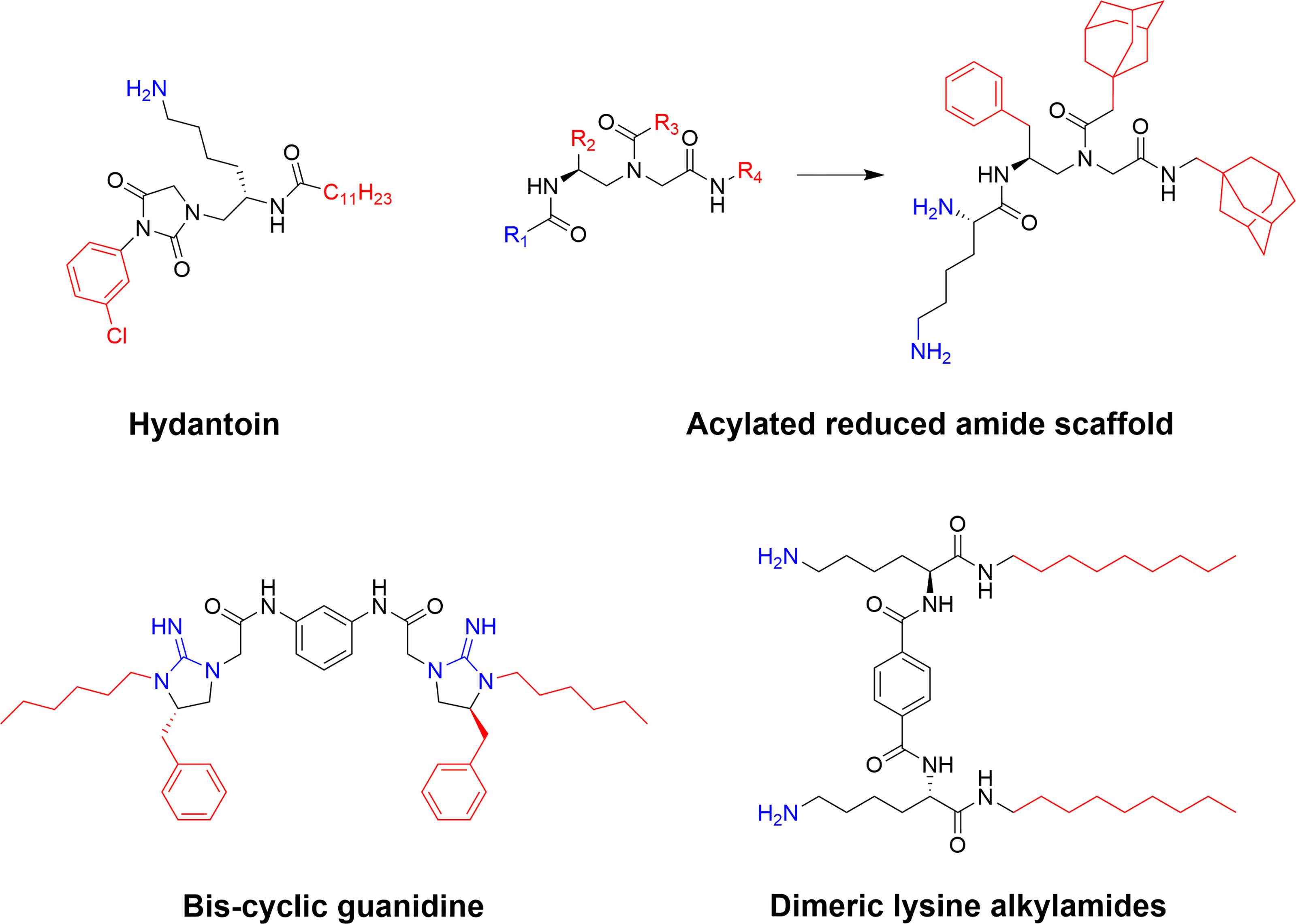
Figure 11 Examples of potent SAMP scaffolds and lead compounds developed by Cai and co-workers (78). Several of these compounds, such as the hydatoin and the cyclic guanidine derivative, display good antimicrobial activity both in vitro and in vivo (143, 144).
Additional SMAMP-type compounds include the recently reported substituted α-hydrazido amino acids developed by Amabili and co-workers (150). The mono-charged α-hydrazido amino acids can be regarded as mimics of β-amino acids and they were designed with a range of both N- and C-terminal lipophilic groups to yield potent amphiphilic compounds (150). The versatile bicyclic norbornane scaffold has also been used for the generation of SMAMPs (151). Pfeffer and co-workers developed norbornane bisether diguanidines that were shown to display submicromolar inhibitory activity against both MRSA and vancomycin-intermediate S. aureus strains (152, 153). The cellular selectivity of the norbornane compounds could be controlled by choice of hydrophobic substituent (153). Building on their work on antibacterial biphenyl compounds (154), Kumar and co-workers also reported biofilm disrupting guanidine functionalized anthranilamides (155) with good selectivity for bacterial cells over mammalian MRC-5 cells in vitro (155). Both Yang et al. (156) and Chen et al. (157) have developed synthetically simple indole based antimicrobials. By using different lipophilic n-acyl side chains at position 1 and a positively charged unusual azepanyl moiety at position 3 a large number of analogs were prepared and shown to display activity against Mycobacterium bovis and M. tuberculosis with both metabolic stability and cellular selectivity (156). Taking a similar approach and starting with ethyl 3-indoleacetate as a cheap starting material, Chen and co-workers recently designed and synthesized membrane-targeting indole-based antimicrobial peptidomimetics. The hydrophobic groups included isoprenyl, geranyl, heptenyl groups and the indole scaffold, while the cationic groups were composed of a range of amino acids or aliphatic amines and guanidines (157). Several active compounds were prepared, and a lead compound displayed high potency against Gram-positive bacteria in a murine model of bacterial keratitis. It was also shown to be more efficient than vancomycin. In addition, the lead compound was shown to not be affected by physiological concentrations of monovalent, divalent, or trivalent cations (Figure 12) (157).
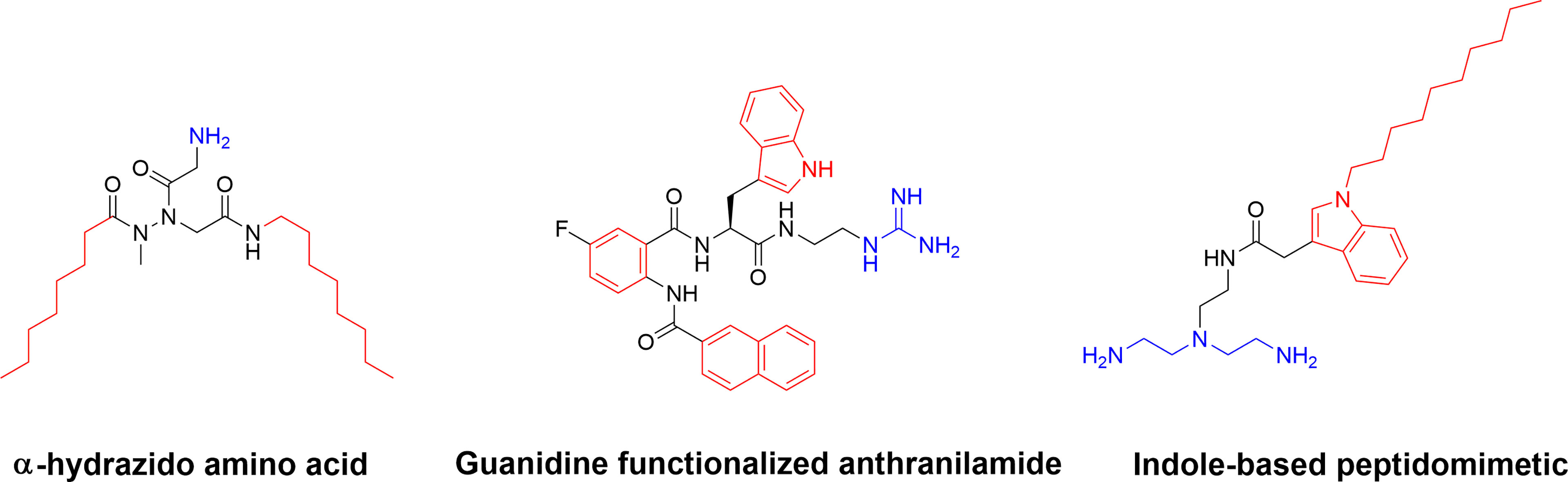
Figure 12 Examples of small and synthetically simple SAMPs illustrating that it is possible to also achieve good antibacterial activity with as single positive charge in some scaffolds.
The phenolic chalcones is a diverse family of bioactive natural products with ranging bioactivities that has been used as scaffolds to yield improved synthetic analogs (158, 159). The scaffolds are readily available for semisynthetic modifications and Lin et al. recently used the isoprenyl chalcone derivative sofalcone as a scaffold to generate potent SMAMPs (160). Sofalcone, prepared from soforadine isolated from the root of the plant Sophora subprostrata, was functionalized with a range of cationic residues and the strongest activity was observed for sofalcone coupled to an RR-dipeptide (160). Previous related work on natural polyphenolic compounds from the group also include the generation of symmetrically substituted xanthone-based amphiphiles (79, 161). The substituted α-mangostin xanthone was used as a hydrophobic core to yield several potent symmetrical SMAMPs which were also effective in vivo in a mouse model of corneal infection by either Staphylococcus aureus or MRSA (79). Additional work by Lin et al. also include the design and semisynthesis of antimicrobial amphiphilic flavones (162) and coumarin derivatives (163). These examples illustrate how natural product scaffolds represent versatile cores for the design of efficient SMAMPs (Figure 13). De novo design of flavonoid-based AMP mimics was recently reviewed (164).
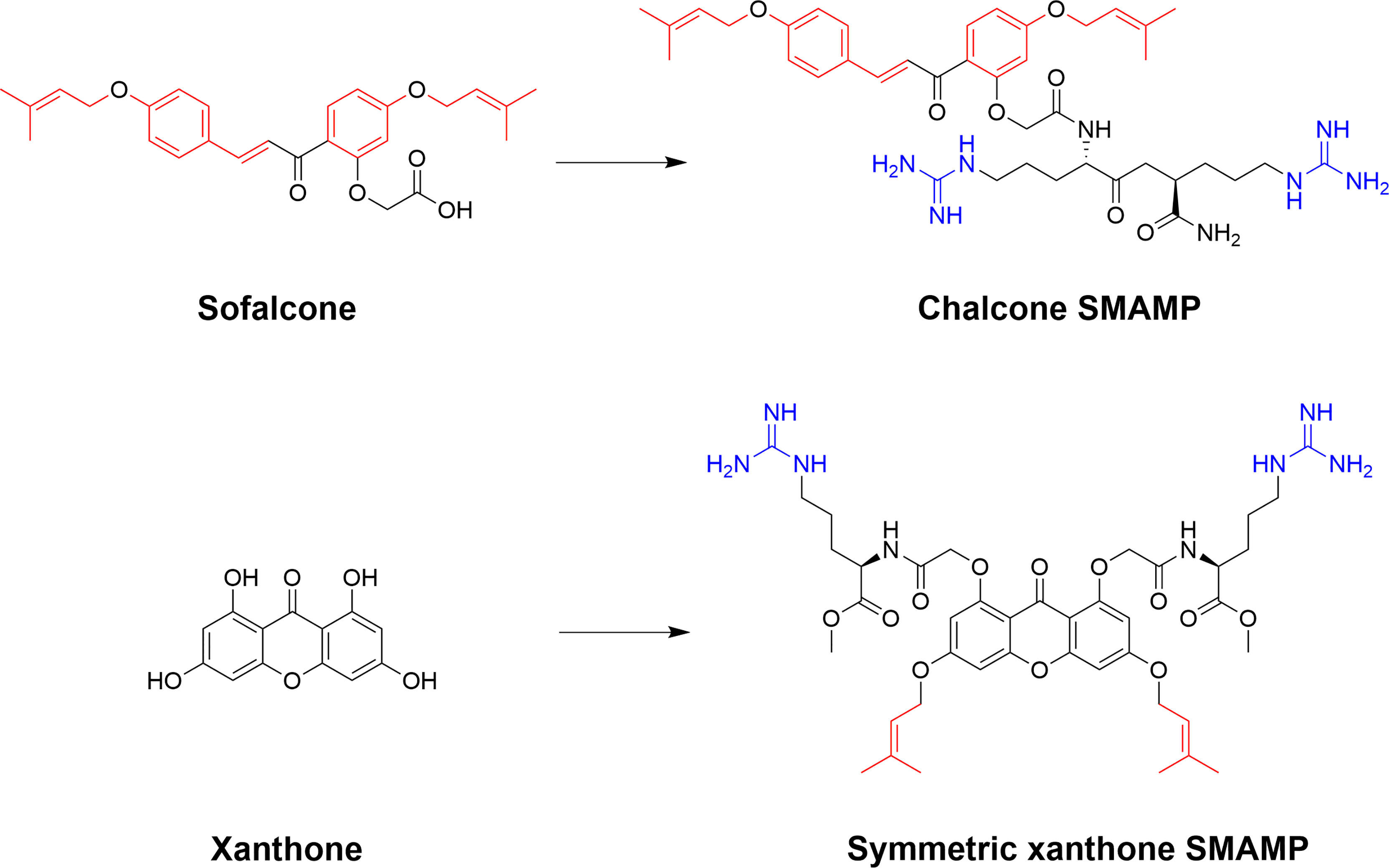
Figure 13 Examples of ways to functionalize natural scaffolds from plants into potent SMAPs. Both chalcones and xanthones offer ample opportunity for synthetic modification to introduce both cationic and hydrophobic residues to achieve the essential balance for high antimicrobial activity and low mammalian cytotoxicity (160, 161).
Structure-Activity Relationship
The mimics included in this review were selected based on their adherence to the 2 + 2 pharmacophore and an optimized balance between the number of cationic charges and the hydrophobic elements. With careful design, the compounds can be tuned to display similar activity against both Gram-positive and Gram-negative bacteria (6, 85). Several compounds have likely been designed without specifically taking this pharmacophore into account but have nevertheless provided compounds adhering to the proposed design principle of Strøm and co-workers (6, 32).
The design of these mimics generally falls into one of three different approaches: 1. the oligomeric approach using amino acids or analogous monomers to assemble a small chain, 2. the scaffold approach in which a natural scaffold is substituted to yield highly active compounds (e.g. ceragenins) or 3. de novo design of purely synthetic compounds (SMAMPs). Our current report illustrates that each approach can be successfully employed to yield very effective antimicrobial compounds suited for clinical development and it is clear that the researcher is not limited to the native AMPs to generate new promising antibiotic leads.
The bioactivity of the majority of native AMPs hinges on a stable amphiphilic structure to allow for optimized membrane interaction (31, 82, 165). AMPs in general comprise up to 50 amino acids and these polypeptides are thus of sufficient size to allow folding into different bioactive motifs (19, 24, 31). The amphiphilicity of an AMP is a reflection of the relative abundance of hydrophilic and hydrophobic residues or domains within the AMP. It is therefore a good descriptor of the balance between the cationic and hydrophobic residues, not just within primary sequence level, but also in terms of the 3-dimensional structure of the AMPs (165). The exact mode of action is expected to depend on the sequence/structure and target species and are often not reported in detail beyond illustrating membrane insertion or disruption (93, 166). Selected mimics have been shown to be active also against marine microorganisms which further suggest multiple modes of action given the spread in cell surface and membrane composition (6, 69, 167).
While our review has illustrated that several synthetic and natural scaffold can be employed to generate improved minimized mimics of AMPs, the majority of the scaffolds are too small to adopt the secondary structures of the native counterparts. Despite the limitations in the ability to form intramolecular bonds and structures stable in solution, increasing evidence suggest that significant gains in activity can be obtained by careful molecular design permitting, or locking, the compounds in facial amphiphilic structures also on this scale (39). Such structures can be obtained by careful sequence optimization or by tuning the stereochemistry. This has been exemplified for numerous of the scaffolds previously described including the tripeptides (166), SMAMPs (133), DKPs and α-hydrazido acids (150). Most studies on these types of small AMP mimics claim that the lead compounds are “amphiphilic” but only a few provide any structural or quantifiable physicochemical data in support, which is also likely dependent, to some extent, on the challenges associated with obtaining crystals.
Tew and co-workers studied the role of amphiphilicity for a series of SMAMPs by incorporating a polar amide bond between the hydrophobic residues. The integy moment (IW) was used to quantify the amphiphilicity of the SMAMPs and confirmed its necessity for the design of optimal SMAMPs (133). This disruption of the amphiphilicity was also reflected in the hydrophobicity of the SMAMPs (133). Only a few other selected published studies have probed the amphiphilicity experimentally. For oligomeric compounds, stereochemistry is a property that is readily tuneable to observe differences in 3D-structures. For this purpose, Isaksson and co-workers prepared all the stereoisomers of LTX-109 and studied the effect on antimicrobial effect (166). It was shown that all L- and all D- isomers retained a high antimicrobial activity while mixed isomers performed poorer in the microbial assays. To develop an understanding of the underlying mechanism the isomers were assessed using nuclear magnetic resonance (NMR) spectroscopy and molecular dynamics (MD) simulations in aqueous solution and in phospholipid bilayers. It was shown that the bioactive compounds were able to adopt a stable amphiphilic structure which was disrupted upon changes in the stereochemistry. This was not only apparent from the differences in antimicrobial activity but also in the solubility and retention time of the compounds. The bioactive amphiphilic compounds were significantly more hydrophobic, and this provided additional support for stable bioactive solution conformations (166). A similar study was recently reported by Grant and co-workers, and they observed similar phenomena for the tetrasubstituted 2,5-DKP scaffold (93). In their study the difference in elution time for the mixed isomers was also strongly reflected in the antimicrobial activity and NMR and MD experiments supported the formation of an optimized amphiphilic bioactive structure (Figure 14) (93).
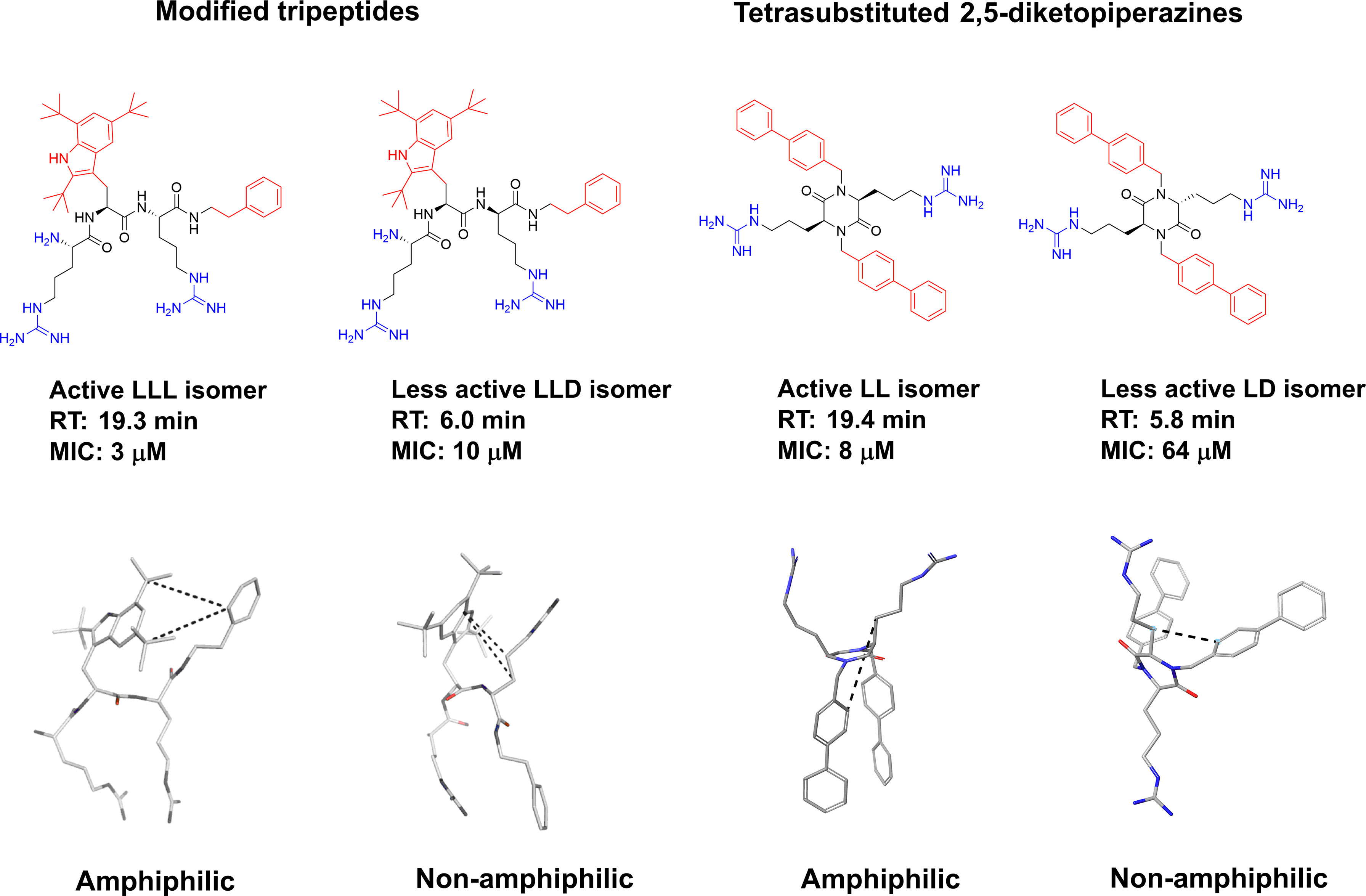
Figure 14 Graphical illustration of the role of stereochemistry on structure and antimicrobial activity of short, modified tripeptidic AMPs and tetrasubstituted 2,5-DKP. Making mixed analogs disrupted the amphiphilicity and creates more hydrophilic and less activity compounds (93, 166). Bottom left structures of LTX-109 are reprinted with permission from Isaksson et al (166). J Med Chem 2011 Vol. 54 Issue 16 Pages 5786-95. Copyright 2011 American Chemical Society.
A plethora of natural and unnatural cationic and hydrophobic building blocks have been employed to generate bioactive compounds and a move from the traditional amino acid-like scaffold provides the chemist with additional freedom to toggle these substituents further. It is clear that several compounds with good efficiency can be obtained slightly outside the 2 + 2 pharmacophore with examples such as the hydantoins and α-hydrazido amino acids that are active despite only displaying a single cationic residue (143, 150). In general, a higher number of charges appear to be needed e.g. a single lysine attached to palmitic acid (16 carbon atoms) does not show any activity (70). The nature of the cationic group is important (168), and many studies generally report increased activity for AMPs displaying guanidine groups over primary amine, histidine and ammonium groups (6, 34, 169, 170). The choice of cationic groups can also have effects of the toxicity of the compounds with several short compounds reporting higher toxicity for compounds incorporating amine substituents (79, 155, 171). It has been established that the exact number of hydrophobic residues is not as crucial and it is rather dependent on the total hydrophobic volume and its optimized molecular distribution (6, 65). Adding more hydrophobic residues usually increases the antimicrobial activity but also at a cost of lowered cellular selectivity (63, 64, 82). Adding too much hydrophobicity renders the compounds inactive, either due to aggregation or poor solubility and has been seen in several studies (69, 149, 172). The optimal balance between charge and hydrophobicity differs depending on scaffold studied and it has not been quantified much further than the 2 + 2 pharmacophore for the compounds covered in this review.
Toxicity and Challenges
From a design perspective, an optimized combination between charge and hydrophobicity is needed. This allows the compounds to obtain the desired amphiphilicity in order to be antimicrobial and membrane active but also display sufficient selectivity and membrane distinction as several AMPs and their shorter mimics are toxic (82). Many of these described compounds have been designed to offer cheap and simple alternatives to AMPs to circumvent some of the limitations faced by natural peptide sequences for combating multidrug-resistant and pandrug-resistant bacterial isolates. Being mimics of cationic AMPs also potentially mean challenges associated with inactivation by high salt concentrations and divalent cations (173), even though studies describing a high salt tolerance and maintained function exist (157, 174). While impressive in vitro and in vivo activity profiles have been reported over the recent years, and several AMP mimics being in clinical development, their clinical safety is less understood. Numerous peptides are approved as drugs (1) and the peptide drug pipeline has been established for many areas. For AMPs, and these synthetic analogs in particular, there are several gaps in the clinical understanding particularly in the area of systemic toxicity (18).
Conclusion
AMPs represent a broad class of diverse natural compounds with great promise for development of novel antibiotics with a lower likelihood of inducing microbial resistance. The clinical progression of AMP leads towards actual real-life use is nevertheless currently hampered (17) with only a small number of compounds on the market targeting a limited number of conditions, due to poor uptake and toxicity (17, 175). The AMPs in use are often small, constrained and contain modifications and noncanonical residues providing them with improved pharmacokinetics properties (30). This observation illustrates key natural methods to produce leads with a higher likelihood of clinical success by synthetic modifications, cyclisation and truncation (176, 177). In the current review we highlight how several of these means have been employed to address the challenges associated with clinical use of native AMPs by developing simplified synthetic mimics of AMPs. It becomes clear, when going through the literature, that ample room for making active and selective low molecular weight AMP mimics room exists, as illustrated by the many highly active mimics reported here. A careful design is nevertheless needed to generate compounds with a sufficient safety profile. These compounds can be cheaply produced in large amounts and with three diverse leads in clinical trials it is expected that these types of small synthetic mimics will play an important role in the future management of microbial infections. Recent studies on synergistic effects in combination with traditional antibiotics, further highlight the imminent role these compounds may have (103, 106, 178, 179). Collectively, it is shown that a high antimicrobial activity can be obtained using diverse low molecular weight compounds and it is clear that shape matters more than size for optimal activity.
Author Contributions
Conceptualization: JS, NM, and CS; Investigation: JS, NM, and CS; Preparation of original draft: JS; Review and editing: JS, NM, and CS; Visualization: CS; Project administration: JS; Funding: JS and CS. All authors have read and agreed to the submitted version of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
JS is grateful for financial support from the Cawthron institute and graphical assistance from Dr. J. Arabashi and Dr. Sam Clark. Dr. P. Savage Dr. M. Strøm are also acknowledged for discussions about the ceragenin and β-peptide development. Work at the Molecular Foundry was supported by the Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. This work was supported [in part] by the Intramural Research Program of the National Cancer Institute, Center for Cancer Research (Grant ZIA BC 012003). The authors further acknowledge Dr. D. Anning for linguistic support.
References
1. Muttenthaler M, King GF, Adams DJ, Alewood PF. Trends in Peptide Drug Discovery. Nat Rev Drug Discov (2021) 20:309–25. doi: 10.1038/s41573-020-00135-8
2. Lau JL, Dunn MK. Therapeutic Peptides: Historical Perspectives, Current Development Trends, and Future Directions. Bioorg Med Chem (2018) 26:2700–7. doi: 10.1016/j.bmc.2017.06.052
3. Guani-Guerra E, Santos-Mendoza T, Lugo-Reyes SO, Teran LM. Antimicrobial Peptides: General Overview and Clinical Implications in Human Health and Disease. Clin Immunol (2010) 135:1–11. doi: 10.1016/j.clim.2009.12.004
4. Di L. Strategic Approaches to Optimizing Peptide ADME Properties. AAPS J (2015) 17:134–43. doi: 10.1208/s12248-014-9687-3
5. Regulska K, Stanisz B, Regulski M, Murias M. How to Design a Potent, Specific, and Stable Angiotensin-Converting Enzyme Inhibitor. Drug Discov Today (2014) 19:1731–43. doi: 10.1016/j.drudis.2014.06.026
6. Svendsen JSM, Grant TM, Rennison D, Brimble MA, Svenson J. Very Short and Stable Lactoferricin-Derived Antimicrobial Peptides: Design Principles and Potential Uses. Acc Chem Res (2019) 52:749–59. doi: 10.1021/acs.accounts.8b00624
7. Acharya KR, Sturrock ED, Riordan JF, Ehlers MR. Ace Revisited: A New Target for Structure-Based Drug Design. Nat Rev Drug Discov (2003) 2:891–902. doi: 10.1038/nrd1227
8. Kim HM, Shin DR, Yoo OJ, Lee H, Lee JO. Crystal Structure of Drosophila Angiotensin I-Converting Enzyme Bound to Captopril and Lisinopril. FEBS Lett (2003) 538:65–70. doi: 10.1016/s0014-5793(03)00128-5
9. Ondetti MA, Williams NJ, Sabo EF, Pluscec J, Weaver ER, Kocy O. Angiotensin-Converting Enzyme Inhibitors From the Venom of Bothrops Jararaca. Isolation, Elucidation of Structure, and Synthesis. Biochemistry (1971) 10:4033–9. doi: 10.1021/bi00798a004
10. Adams DJ, Smith AB, Schroeder CI, Yasuda T, Lewis RJ. Omega-Conotoxin CVID Inhibits a Pharmacologically Distinct Voltage-Sensitive Calcium Channel Associated With Transmitter Release From Preganglionic Nerve Terminals. J Biol Chem (2003) 278:4057–62. doi: 10.1074/jbc.M209969200
11. Dong Y, Gao Y, Xu S, Wang Y, Yu Z, Li Y, et al. Closed-State Inactivation and Pore-Blocker Modulation Mechanisms of Human Cav2.2. Cell Rep (2021) 37:109931. doi: 10.1016/j.celrep.2021.109931
12. Mould J, Yasuda T, Schroeder CI, Beedle AM, Doering CJ, Zamponi GW, et al. The Alpha2delta Auxiliary Subunit Reduces Affinity of Omega-Conotoxins for Recombinant N-Type (Cav2.2) Calcium Channels. J Biol Chemi (2004) 279:34705–14. doi: 10.1074/jbc.M310848200
13. Schmidtko A, Lotsch J, Freynhagen R, Geisslinger G. Ziconotide for Treatment of Severe Chronic Pain. Lancet (2010) 375:1569–77. doi: 10.1016/S0140-6736(10)60354-6
14. Tyndall JD, Pfeiffer B, Abbenante G, Fairlie DP. Over One Hundred Peptide-Activated G Protein-Coupled Receptors Recognize Ligands With Turn Structure. Chem Rev (2005) 105:793–826. doi: 10.1021/cr040689g
15. Gruber C W, Muttenthaler M, Freissmuth M. Ligand-Based Peptide Design and Combinatorial Peptide Libraries to Target G Protein-Coupled Receptors. Curr Pharm Des (2010) 16:3071–88. doi: 10.2174/138161210793292474
16. King GF. Venoms as a Platform for Human Drugs: Translating Toxins Into Therapeutics. Expert Opin Biol Ther (2011) 11:1469–84. doi: 10.1517/14712598.2011.621940
17. Chen CH, Lu TK. Development and Challenges of Antimicrobial Peptides for Therapeutic Applications. Antibiotics (2020) 9:24. doi: 10.3390/antibiotics9010024
18. Magana M, Pushpanathan M, Santos AL, Leanse L, Fernandez M, Ioannidis A, et al. The Value of Antimicrobial Peptides in the Age of Resistance. Lancet Infect Dis (2020) 20:e216–30. doi: 10.1016/S1473-3099(20)30327-3
19. Zasloff M. Antimicrobial Peptides of Multicellular Organisms. Nature (2002) 415:389–95. doi: 10.1038/415389a
20. Mookherjee N, Hancock R. Cationic Host Defence Peptides: Innate Immune Regulatory Peptides as a Novel Approach for Treating Infections. Cell Mol Life Sci (2007) 64:922–33. doi: 10.1007/s00018-007-6475-6
21. Nguyen LT, Haney EF, Vogel HJ. The Expanding Scope of Antimicrobial Peptide Structures and Their Modes of Action. Trends Biotechnol (2011) 29:464–72. doi: 10.1016/j.tibtech.2011.05.001
22. Jenssen H, Hamill P, Hancock RE. Peptide Antimicrobial Agents. Clin Microbiol Rev (2006) 19:491–511. doi: 10.1128/CMR.00056-05
23. Takahashi D, Shukla SK, Prakash O, Zhang G. Structural Determinants of Host Defense Peptides for Antimicrobial Activity and Target Cell Selectivity. Biochimie (2010) 92:1236–41. doi: 10.1016/j.biochi.2010.02.023
24. Mahlapuu M, Hakansson J, Ringstad L, Björn C. Antimicrobial Peptides: An Emerging Category of Therapeutic Agents. Front Cell Infect Microbiol (2016) 6:194. doi: 10.3389/fcimb.2016.00194
25. Tacconelli E, Carrara E, Savoldi A, Harbarth S, Mendelson M, Monnet DL, et al. Discovery, Research, and Development of New Antibiotics: The WHO Priority List of Antibiotic-Resistant Bacteria and Tuberculosis. Lancet Infect Dis (2018) 18:318–27. doi: 10.1016/S1473-3099(17)30753-3
26. Mahlapuu M, Björn C, Ekblom J. Antimicrobial Peptides as Therapeutic Agents: Opportunities and Challenges. Crit Rev Biotechnol (2020) 40:978–92. doi: 10.1080/07388551.2020.1796576
27. Panjla A, Kaul G, Chopra S, Titz A, Verma S. Short Peptides and Their Mimetics as Potent Antibacterial Agents and Antibiotic Adjuvants. ACS Chem Biol (2021) 16:2731–45. doi: 10.1021/acschembio.1c00626
28. Sarkar T, Chetia M, Chatterjee S. Antimicrobial Peptides and Proteins: From Nature’s Reservoir to the Laboratory and Beyond. Front Chem (2021) 9:691532. doi: 10.3389/fchem.2021.691532
29. Kosikowska P, Lesner A. Antimicrobial Peptides (AMPs) as Drug Candidates: A Patent Review (2003–2015). Expert Opin Ther Pat (2016) 26:689–702. doi: 10.1080/13543776.2016.1176149
30. Gan BH, Gaynord J, Rowe SM, Deingruber T, Spring DR. The Multifaceted Nature of Antimicrobial Peptides: Current Synthetic Chemistry Approaches and Future Directions. Chem Soc Rev (2021) 50:7820–80. doi: 10.1039/d0cs00729c
31. Yeaman MR, Yount NY. Mechanisms of Antimicrobial Peptide Action and Resistance. Pharmacol Rev (2003) 55:27–55. doi: 10.1124/pr.55.1.2
32. Strøm MB, Haug BE, Skar ML, Stensen W, Stiberg T, Svendsen JS. The Pharmacophore of Short Cationic Antibacterial Peptides. J Med Chem (2003) 46:1567–70. doi: 10.1021/jm0340039
33. Haug BE, Skar ML, Svendsen JS. Bulky Aromatic Amino Acids Increase the Antibacterial Activity of 15-Residue Bovine Lactoferricin Derivatives. J Pept Sci (2001) 7:425–32. doi: 10.1002/psc.338
34. Svenson J, Karstad R, Flaten GE, Brandsdal BO, Brandl M, Svendsen JS. Altered Activity and Physicochemical Properties of Short Cationic Antimicrobial Peptides by Incorporation of Arginine Analogues. Mol Pharm (2009) 6:996–1005. doi: 10.1021/mp900057k
35. Svenson J, Stensen W, Brandsdal BO, Haug BE, Monrad J, Svendsen JS. Antimicrobial Peptides With Stability Toward Tryptic Degradation. Biochemistry (2008) 47:3777–88. doi: 10.1021/bi7019904
36. Karstad R, Isaksen G, Wynendaele E, Guttormsen Y, De Spiegeleer B, Brandsdal BO, et al. Targeting the S1 and S3 Subsite of Trypsin With Unnatural Cationic Amino Acids Generates Antimicrobial Peptides With Potential for Oral Administration. J Med Chem (2012) 55:6294–305. doi: 10.1021/jm3002058
37. Flaten GE, Kottra G, Stensen W, Isaksen G, Karstad R, Svendsen JS, et al. In Vitro Characterization of Human Peptide Transporter Hpept1 Interactions and Passive Permeation Studies of Short Cationic Antimicrobial Peptides. J Med Chem (2011) 54:2422–32. doi: 10.1021/jm1015704
38. Lin L, Chi J, Yan Y, Luo R, Feng X, Zheng Y, et al. Membrane-Disruptive Peptides/Peptidomimetics-Based Therapeutics: Promising Systems to Combat Bacteria and Cancer in the Drug-Resistant Era. Acta Pharm Sin B (2021) 11:2609–44. doi: 10.1016/j.apsb.2021.07.014
39. Scott RW, DeGrado WF, Tew GN. De Novo Designed Synthetic Mimics of Antimicrobial Peptides. Curr Opin Biotechnol (2008) 19:620–7. doi: 10.1016/j.copbio.2008.10.013
40. Scott R W, Tew G N. Mimics of Host Defense Proteins; Strategies for Translation to Therapeutic Applications. Curr Top Med Chem (2017) 17:576–89. doi: 10.2174/1568026616666160713130452
41. Findlay B, Zhanel GG, Schweizer F. Cationic Amphiphiles, a New Generation of Antimicrobials Inspired by the Natural Antimicrobial Peptide Scaffold. Antimicrob Agents Chemother (2010) 54:4049–58. doi: 10.1128/AAC.00530-10
42. Isidro-Llobet A, Kenworthy MN, Mukherjee S, Kopach ME, Wegner K, Gallou F, et al. Sustainability Challenges in Peptide Synthesis and Purification: From R&D to Production. J Org Chem (2019) 84:4615–28. doi: 10.1021/acs.joc.8b03001
43. Karstad R, Isaksen G, Brandsdal B-O, Svendsen JS, Svenson J. Unnatural Amino Acid Side Chains as S1, S1′, and S2′ Probes Yield Cationic Antimicrobial Peptides With Stability Toward Chymotryptic Degradation. J Med Chem (2010) 53:5558–66. doi: 10.1021/jm1006337
44. Moore SA, Anderson BF, Groom CR, Haridas M, Baker EN. Three-Dimensional Structure of Diferric Bovine Lactoferrin at 2.8 Å Resolution. J Mol Biol (1997) 274:222–36. doi: 10.1006/jmbi.1997.1386
45. Hwang PM, Zhou N, Shan X, Arrowsmith CH, Vogel HJ. Three-Dimensional Solution Structure of Lactoferricin B, an Antimicrobial Peptide Derived From Bovine Lactoferrin. Biochemistry (1998) 37:4288–98. doi: 10.1021/bi972323m
46. Nguyen LT, Schibli DJ, Vogel HJ. Structural Studies and Model Membrane Interactions of Two Peptides Derived From Bovine Lactoferricin. J Pept Sci (2005) 11:379–89. doi: 10.1002/psc.629
47. Svenson J, Vergote V, Karstad R, Burvenich C, Svendsen JS, De Spiegeleer B. Metabolic Fate of Lactoferricin-Based Antimicrobial Peptides: Effect of Truncation and Incorporation of Amino Acid Analogs on the In Vitro Metabolic Stability. J Pharm Exp Ther (2010) 332:1032–9. doi: 10.1124/jpet.109.162826
48. Tomita M, Takase M, Bellamy W, Shimamura S. A review: the active peptide of lactoferrin. Acta Paediatr Jpn (1994) 36:585–91. doi: 10.1111/j.1442-200x.1994.tb03250.x
49. Haug BE, Strøm MB, Svendsen JS. The Medicinal Chemistry of Short Lactoferricin-Based Antibacterial Peptides. Curr Med Chem (2007) 14:1–18. doi: 10.2174/092986707779313435
50. Myhrman E, Hakansson J, Lindgren K, Bjorn C, Sjostrand V, Mahlapuu M. The Novel Antimicrobial Peptide PXL150 in the Local Treatment of Skin and Soft Tissue Infections. Appl Microbiol Biotechnol (2013) 97:3085–96. doi: 10.1007/s00253-012-4439-8
51. Koo HB, Seo J. Antimicrobial Peptides Under Clinical Investigation. Pept Sci (2019) 111:e24122. doi: 10.1002/pep2.24122
52. Strøm MB, Haug BE, Rekdal O, Skar ML, Stensen W, Svendsen JS. Important Structural Features of 15-Residue Lactoferricin Derivatives and Methods for Improvement of Antimicrobial Activity. Biochem Cell Biol (2002) 80:65–74. doi: 10.1139/o01-236
53. Strøm MB, Rekdal O, Svendsen JS. Antibacterial Activity of 15-Residue Lactoferricin Derivatives. J Pept Res (2000) 56:265–74. doi: 10.1034/j.1399-3011.2000.00770.x
54. Strøm MB, Rekdal O, Svendsen JS. Antimicrobial Activity of Short Arginine- and Tryptophan-Rich Peptides. J Pept Sci (2002) 8:431–7. doi: 10.1002/psc.398
55. Haug BE, Svendsen JS. The Role of Tryptophan in the Antibacterial Activity of a 15-Residue Bovine Lactoferricin Peptide. J Pept Sci (2001) 7:190–6. doi: 10.1002/psc.318
56. Clark S, Jowitt TA, Harris LK, Knight CG, Dobson CB. The Lexicon of Antimicrobial Peptides: A Complete Set of Arginine and Tryptophan Sequences. Commun Biol (2021) 4:605. doi: 10.1038/s42003-021-02137-7
57. Haug BE, Stensen W, Stiberg T, Svendsen JS. Bulky Nonproteinogenic Amino Acids Permit the Design of Very Small and Effective Cationic Antibacterial Peptides. J Med Chem (2004) 47:4159–62. doi: 10.1021/jm049582b
58. Svenson J, Brandsdal BO, Stensen W, Svendsen JS. Albumin Binding of Short Cationic Antimicrobial Micropeptides and its Influence on the In Vitro Bactericidal Effect. J Med Chem (2007) 50:3334–9. doi: 10.1021/jm0703542
59. Sivertsen A, Isaksson J, Leiros H-KS, Svenson J, Svendsen J-S, Brandsdal BO. Synthetic Cationic Antimicrobial Peptides Bind With Their Hydrophobic Parts to Drug Site II of Human Serum Albumin. BMC Struct Biol (2014) 14:1–14. doi: 10.1186/1472-6807-14-4
60. Sivertsen A, Brandsdal BO, Svendsen JS, Andersen JH, Svenson J. Short Cationic Antimicrobial Peptides Bind to Human Alpha-1 Acid Glycoprotein With No Implications for the In Vitro Bioactivity. J Mol Recognit (2013) 26:461–9. doi: 10.1002/jmr.2288
61. Hedstrom L. Serine Protease Mechanism and Specificity. Chem Rev (2002) 102:4501–24. doi: 10.1021/cr000033x
62. Hakansson J, Cavanagh JP, Stensen W, Mortensen B, Svendsen JS, Svenson J. In Vitro and In Vivo Antibacterial Properties of Peptide AMC-109 Impregnated Wound Dressings and Gels. J Antibiot (2021) 74:337–45. doi: 10.1038/s41429-021-00406-5
63. Haug BE, Stensen W, Kalaaji M, Rekdal O, Svendsen JS. Synthetic Antimicrobial Peptidomimetics With Therapeutic Potential. J Med Chem (2008) 51:4306–14. doi: 10.1021/jm701600a
64. Stensen W, Turner R, Brown M, Kondori N, Svendsen JS, Svenson J. Short Cationic Antimicrobial Peptides Display Superior Antifungal Activities Toward Candidiasis and Onychomycosis in Comparison With Terbinafine and Amorolfine. Mol Pharm (2016) 13:3595–600. doi: 10.1021/acs.molpharmaceut.6b00654
65. Labriere C, Kondori N, Caous JS, Boomgaren M, Sandholm K, Ekdahl KN, et al. Development and Evaluation of Cationic Amphiphilic Antimicrobial 2,5-Diketopiperazines. J Pept Sci (2018) 24:e3090. doi: 10.1002/psc.3090
66. Meloni BP, Mastaglia FL, Knuckey NW. Cationic Arginine-Rich Peptides (CARPs): A Novel Class of Neuroprotective Agents With a Multimodal Mechanism of Action. Front Neurol (2020) 11:108. doi: 10.3389/fneur.2020.00108
67. Hansen T, Alst T, Havelkova M, Strøm MB. Antimicrobial Activity of Small Beta-Peptidomimetics Based on the Pharmacophore Model of Short Cationic Antimicrobial Peptides. J Med Chem (2010) 53:595–606. doi: 10.1021/jm901052r
68. Hansen T, Ausbacher D, Flaten GE, Havelkova M, Strøm MB. Synthesis of Cationic Antimicrobial β2, 2-Amino Acid Derivatives With Potential for Oral Administration. J Med Chem (2011) 54:858–68. doi: 10.1021/jm101327d
69. Grant TM, Rennison D, Cervin G, Pavia H, Hellio C, Foulon V, et al. Towards Eco-Friendly Marine Antifouling Biocides–Nature Inspired Tetrasubstituted 2, 5-Diketopiperazines. Sci Total Environ (2022) 812:152487. doi: 10.1016/j.scitotenv.2021.152487
70. Mangoni ML, Shai Y. Short Native Antimicrobial Peptides and Engineered Ultrashort Lipopeptides: Similarities and Differences in Cell Specificities and Modes of Action. Cell Mol Life Sci (2011) 68:2267–80. doi: 10.1007/s00018-011-0718-2
71. Makovitzki A, Avrahami D, Shai Y. Ultrashort Antibacterial and Antifungal Lipopeptides. Proc Nat Acad Sci (2006) 103:15997–6002. doi: 10.1073/pnas.0606129103
72. Shai Y, Makovitzky A, Avrahami D. Host Defense Peptides and Lipopeptides: Modes of Action and Potential Candidates for the Treatment of Bacterial and Fungal Infections. Curr Protein Pept Sci (2006) 7:479–86. doi: 10.2174/138920306779025620
73. Bicker KL, Cobb SL. Recent Advances in the Development of Anti-Infective Peptoids. Chem Commun (2020) 56:11158–68. doi: 10.1039/d0cc04704j
74. Lai XZ, Feng Y, Pollard J, Chin JN, Rybak MJ, Bucki R, et al. Ceragenins: Cholic Acid-Based Mimics of Antimicrobial Peptides. Acc Chem Res (2008) 41:1233–40. doi: 10.1021/ar700270t
75. Lin C, Wang Y, Le M, Chen KF, Jia YG. Recent Progress in Bile Acid-Based Antimicrobials. Bioconjug Chem (2021) 32:395–410. doi: 10.1021/acs.bioconjchem.0c00642
76. Savage PB. Design, Synthesis and Characterization of Cationic Peptide and Steroid Antibiotics. Eur J Org Chem (2002) 2002:759–68. doi: 10.1021/ol048845t
77. Tew GN, Clements D, Tang H, Arnt L, Scott RW. Antimicrobial Activity of an Abiotic Host Defense Peptide Mimic. Biochim Biophys Acta (2006) 1758:1387–92. doi: 10.1016/j.bbamem.2006.03.001
78. Zhou M, Zheng M, Cai J. Small Molecules With Membrane-Active Antibacterial Activity. ACS Appl Mater Interfaces (2020) 12:21292–9. doi: 10.1021/acsami.9b20161
79. Koh JJ, Lin S, Aung TT, Lim F, Zou H, Bai Y, et al. Amino Acid Modified Xanthone Derivatives: Novel, Highly Promising Membrane-Active Antimicrobials for Multidrug-Resistant Gram-Positive Bacterial Infections. J Med Chem (2015) 58:739–52. doi: 10.1021/jm501285x
80. Kuroda K, Caputo GA. Antimicrobial Polymers as Synthetic Mimics of Host-Defense Peptides. Wiley Interdiscip Reviews: Nanomed Nanobiotechnol (2013) 5:49–66. doi: 10.1002/wnan.1199
81. Lienkamp K, Madkour AE, Tew GN. Antibacterial Peptidomimetics: Polymeric Synthetic Mimics of Antimicrobial Peptides. In: Polymer Composites–Polyolefin Fractionation–Polymeric Peptidomimetics–Collagens. Berlin: Springer (2010). p. 141–72.
82. Jiang Y, Chen Y, Song Z, Tan Z, Cheng J. Recent Advances in Design of Antimicrobial Peptides and Polypeptides Toward Clinical Translation. Adv Drug Delivery Rev (2021) 170:261–80. doi: 10.1016/j.addr.2020.12.016
83. Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv Drug Deliver Rev (1997) 23:3–25. doi: 10.1016/S0169-409X(96)00423-1
84. Flaten GE, Dhanikula AB, Luthman K, Brandl M. Drug Permeability Across a Phospholipid Vesicle Based Barrier: A Novel Approach for Studying Passive Diffusion. Eur J Pharm Sci (2006) 27:80–90. doi: 10.1016/j.ejps.2005.08.007
85. Paulsen MH, Engqvist M, Ausbacher D, Anderssen T, Langer MK, Haug T, et al. Amphipathic Barbiturates as Mimics of Antimicrobial Peptides and the Marine Natural Products Eusynstyelamides With Activity Against Multi-Resistant Clinical Isolates. J Med Chem (2021) 64:11395–417. doi: 10.1021/acs.jmedchem.1c00734
86. Paulsen MH, Ausbacher D, Bayer A, Engqvist M, Hansen T, Haug T, et al. Antimicrobial Activity of Amphipathic α, α-Disubstituted β-Amino Amide Derivatives Against ESBL–CARBA Producing Multi-Resistant Bacteria; Effect of Halogenation, Lipophilicity and Cationic Character. Eur J Med Chem (2019) 183:111671. doi: 10.1016/j.ejmech.2019.111671
87. Hanski L, Ausbacher D, Tiirola TM, Strøm MB, Vuorela PM. Amphipathic Beta2,2-Amino Acid Derivatives Suppress Infectivity and Disrupt the Intracellular Replication Cycle of Chlamydia Pneumoniae. PloS One (2016) 11:e0157306. doi: 10.1371/journal.pone.0157306
88. Tadesse M, Tabudravu JN, Jaspars M, Strøm MB, Hansen E, Andersen JH, et al. The Antibacterial Ent-Eusynstyelamide B and Eusynstyelamides D, E, and F From the Arctic Bryozoan Tegella Cf. Spitzbergensis. J Nat Prod (2011) 74:837–41. doi: 10.1021/np100499c
89. Tapiolas DM, Bowden BF, Abou-Mansour E, Willis RH, Doyle JR, Muirhead AN, et al. Eusynstyelamides A, B, and C, nNOS Inhibitors, From the Ascidian Eusynstyela Latericius. J Nat Prod (2009) 72:1115–20. doi: 10.1021/np900099j
90. Borthwick AD. 2,5-Diketopiperazines: Synthesis, Reactions, Medicinal Chemistry, and Bioactive Natural Products. Chem Rev (2012) 112:3641–716. doi: 10.1021/cr200398y
91. Tullberg M, Grotli M, Luthman K. Synthesis of Functionalized, Unsymmetrical 1,3,4,6-Tetrasubstituted 2,5-Diketopiperazines. J Org Chem (2007) 72:195–9. doi: 10.1021/jo0619635
92. Saavedra CJ, Cuevas F, Romero-Estudillo I, Boto A. Synthesis of Diketopiperazine Scaffolds With Tailored N-And α-Chains by Selective Modification of Customizable Units. Adv Synth Catal (2020) 362:3158–69. doi: 10.1002/adsc.202000470
93. Grant TM, Rennison D, Krause AL, Mros S, Ferguson SA, Cook GM, et al. Stereochemical Effects on the Antimicrobial Properties of Tetrasubstituted 2,5-Diketopiperazines. ACS Med Chem Lett (2022) 13:632–40. doi: 10.1021/acsmedchemlett.1c00683
94. Fiechter A. Biosurfactants: Moving Towards Industrial Application. Trends Biotechnol (1992) 10:208–17. doi: 10.1016/0167-7799(92)90215-h
95. Tedesco KL, Rybak MJ. Daptomycin. Pharmacotherapy (2004) 24:41–57. doi: 10.1592/phco.24.1.41.34802
97. Baltz RH, Miao V, Wrigley SK. Natural Products to Drugs: Daptomycin and Related Lipopeptide Antibiotics. Nat Prod Rep (2005) 22:717–41. doi: 10.1039/b416648p
98. Juhaniewicz-Debinska J, Lasek R, Tymecka D, Burdach K, Bartosik D, Sek S. Physicochemical and Biological Characterization of Novel Membrane-Active Cationic Lipopeptides With Antimicrobial Properties. Langmuir (2020) 36:12900–10. doi: 10.1021/acs.langmuir.0c02135
99. Zhong C, Zhang F, Zhu N, Zhu Y, Yao J, Gou S, et al. Ultra-Short Lipopeptides Against Gram-Positive Bacteria While Alleviating Antimicrobial Resistance. Eur J Med Chem (2021) 212:113138. doi: 10.1016/j.ejmech.2020.113138
100. Neubauer D, Jaskiewicz M, Bauer M, Golacki K, Kamysz W. Ultrashort Cationic Lipopeptides-Effect of N-Terminal Amino Acid and Fatty Acid Type on Antimicrobial Activity and Hemolysis. Molecules (2020) 25:257. doi: 10.3390/molecules25020257
101. Arnusch CJ, Albada HB, van Vaardegem M, Liskamp RM, Sahl HG, Shadkchan Y, et al. Trivalent Ultrashort Lipopeptides are Potent pH Dependent Antifungal Agents. J Med Chem (2012) 55:1296–302. doi: 10.1021/jm2014474
102. Makovitzki A, Baram J, Shai Y. Antimicrobial Lipopolypeptides Composed of Palmitoyl Di- and Tricationic Peptides: In Vitro and In Vivo Activities, Self-Assembly to Nanostructures, and a Plausible Mode of Action. Biochemistry (2008) 47:10630–6. doi: 10.1021/bi8011675
103. Cirioni O, Kamysz E, Ghiselli R, Kamysz W, Silvestri C, Orlando F, et al. Lipopeptide Laur-CKK-NH2 Dimer Preserves Daptomycin Susceptibility and Enhances its Activity Against Enterococcus Faecalis. J Antimicrob Chemother (2011) 66:859–62. doi: 10.1093/jac/dkr001
104. Kamysz W, Silvestri C, Cirioni O, Giacometti A, Licci A, Della Vittoria A, et al. In Vitro Activities of the Lipopeptides Palmitoyl (Pal)-Lys-Lys-NH2 and Pal-Lys-Lys Alone and in Combination With Antimicrobial Agents Against Multiresistant Gram-Positive Cocci. Antimicrob Agents Chemother (2007) 51:354–8. doi: 10.1128/AAC.00344-06
105. Ramirez D, Berry L, Domalaon R, Brizuela M, Schweizer F. Dilipid Ultrashort Tetrabasic Peptidomimetics Potentiate Novobiocin and Rifampicin Against Multidrug-Resistant Gram-Negative Bacteria. ACS Infect Dis (2020) 6:1413–26. doi: 10.1021/acsinfecdis.0c00017
106. Ramirez D, Berry L, Domalaon R, Li Y, Arthur G, Kumar A, et al. Dioctanoyl Ultrashort Tetrabasic β-Peptides Sensitize Multidrug-Resistant Gram-Negative Bacteria to Novobiocin and Rifampicin. Front Microbiol (2021) 12:803309. doi: 10.3389/fmicb.2021.803309
107. Schweizer L, Ramirez D, Schweizer F. Effects of Lysine N-Ζ-Methylation in Ultrashort Tetrabasic Lipopeptides (UTBLPs) on the Potentiation of Rifampicin, Novobiocin, and Niclosamide in Gram-Negative Bacteria. Antibiotics (2022) 11(3):335. doi: 10.3390/antibiotics11030335
108. Hu Y, Amin MN, Padhee S, Wang RE, Qiao Q, Bai G, et al. Lipidated Peptidomimetics With Improved Antimicrobial Activity. ACS Med Chem Lett (2012) 3:683–6. doi: 10.1021/ml3001215
109. Li Y, Smith C, Wu H, Teng P, Shi Y, Padhee S, et al. Short Antimicrobial Lipo-Alpha/Gamma-AA Hybrid Peptides. Chembiochem (2014) 15:2275–80. doi: 10.1002/cbic.201402264
110. Biniarz P, Lukaszewicz M, Janek T. Screening Concepts, Characterization and Structural Analysis of Microbial-Derived Bioactive Lipopeptides: A Review. Crit Rev Biotechnol (2017) 37:393–410. doi: 10.3109/07388551.2016.1163324
111. Mnif I, Ghribi D. Review Lipopeptides Biosurfactants: Mean Classes and New Insights for Industrial, Biomedical, and Environmental Applications. Biopolymers (2015) 104:129–47. doi: 10.1002/bip.22630
112. Carolin CF, Kumar PS, Ngueagni PT. A Review on New Aspects of Lipopeptide Biosurfactant: Types, Production, Properties and its Application in the Bioremediation Process. J Hazard Mater (2021) 407:124827. doi: 10.1016/j.jhazmat.2020.124827
113. Molchanova N, Hansen PR, Damborg P, Nielsen HM, Franzyk H. Lysine-Based Alpha-Peptide/beta-Peptoid Peptidomimetics: Influence of Hydrophobicity, Fluorination, and Distribution of Cationic Charge on Antimicrobial Activity and Cytotoxicity. ChemMedChem (2017) 12:312–8. doi: 10.1002/cmdc.201600553
114. Chongsiriwatana NP, Patch JA, Czyzewski AM, Dohm MT, Ivankin A, Gidalevitz D, et al. Peptoids That Mimic the Structure, Function, and Mechanism of Helical Antimicrobial Peptides. Proc Nat Acad Sci (2008) 105:2794–9. doi: 10.1073/pnas.0708254105
115. Molchanova N, Nielsen JE, Sorensen KB, Prabhala BK, Hansen PR, Lund R, et al. Halogenation as a Tool to Tune Antimicrobial Activity of Peptoids. Sci Rep (2020) 10:14805. doi: 10.1038/s41598-020-71771-8
116. Nielsen JE, Alford MA, Yung DBY, Molchanova N, Fortkort JA, Lin JS, et al. Self-Assembly of Antimicrobial Peptoids Impacts Their Biological Effects on ESKAPE Bacterial Pathogens. ACS Infect Dis (2022) 8:533–45. doi: 10.1021/acsinfecdis.1c00536
117. Castelletto V, Seitsonen J, Tewari KM, Hasan A, Edkins RM, Ruokolainen J, et al. Self-Assembly of Minimal Peptoid Sequences. ACS Macro Lett (2020) 9:494–9. doi: 10.1021/acsmacrolett.9b01010
118. Molchanova N, Wang H, Hansen PR, Hoiby N, Nielsen HM, Franzyk H. Antimicrobial Activity of Alpha-Peptide/beta-Peptoid Lysine-Based Peptidomimetics Against Colistin-Resistant Pseudomonas Aeruginosa Isolated From Cystic Fibrosis Patients. Front Microbiol (2019) 10:275. doi: 10.3389/fmicb.2019.00275
119. Khara JS, Mojsoska B, Mukherjee D, Langford PR, Robertson BD, Jenssen H, et al. Ultra-Short Antimicrobial Peptoids Show Propensity for Membrane Activity Against Multi-Drug Resistant Mycobacterium Tuberculosis. Front Microbiol (2020) 11:417. doi: 10.3389/fmicb.2020.00417
120. Kapoor R, Eimerman PR, Hardy JW, Cirillo JD, Contag CH, Barron AE. Efficacy of Antimicrobial Peptoids Against Mycobacterium Tuberculosis. Antimicrob Agents Chemother (2011) 55:3058–62. doi: 10.1128/AAC.01667-10
121. Hasan A, Saxena V, Castelletto V, Zimbitas G, Seitsonen J, Ruokolainen J, et al. Chain-End Modifications and Sequence Arrangements of Antimicrobial Peptoids for Mediating Activity and Nano-Assembly. Front Chem (2020) 8:416. doi: 10.3389/fchem.2020.00416
122. Green RM, Bicker KL. Evaluation of Peptoid Mimics of Short, Lipophilic Peptide Antimicrobials. Int J Antimicrob Agents (2020) 56:106048. doi: 10.1016/j.ijantimicag.2020.106048
123. Padhee S, Hu Y, Niu Y, Bai G, Wu H, Costanza F, et al. Non-Hemolytic Alpha-AApeptides as Antimicrobial Peptidomimetics. Chem Commun (2011) 47:9729–31. doi: 10.1039/c1cc13684d
124. Niu Y, Padhee S, Wu H, Bai G, Harrington L, Burda WN, et al. Identification of Gamma-AApeptides With Potent and Broad-Spectrum Antimicrobial Activity. Chem Commun (2011) 47:12197–9. doi: 10.1039/c1cc14476f
125. Leszczyńska K, Namiot A, Cruz K, Byfield F, Won E, Mendez G, et al. Potential of Ceragenin CSA-13 and its Mixture With Pluronic F-127 as Treatment of Topical Bacterial Infections. J Appl Microbiol (2011) 110:229–38. doi: 10.1111/j.1365-2672.2010.04874.x
126. Vila-Farres X, Callarisa AE, Gu X, Savage PB, Giralt E, Vila J. CSA-131, a Ceragenin Active Against Colistin-Resistant Acinetobacter Baumannii and Pseudomonas Aeruginosa Clinical Isolates. Int J Antimicrob Agents (2015) 46:568–71. doi: 10.1016/j.ijantimicag.2015.08.003
127. Niemirowicz K, Durnas B, Tokajuk G, Piktel E, Michalak G, Gu X, et al. Formulation and Candidacidal Activity of Magnetic Nanoparticles Coated With Cathelicidin LL-37 and Ceragenin CSA-13. Sci Rep (2017) 7:4610. doi: 10.1038/s41598-017-04653-1
128. Hashemi MM, Rovig J, Bateman J, Holden BS, Modelzelewski T, Gueorguieva I, et al. Preclinical Testing of a Broad-Spectrum Antimicrobial Endotracheal Tube Coated With an Innate Immune Synthetic Mimic. J Antimicrob Chemother (2018) 73:143–50. doi: 10.1093/jac/dkx347
129. Wu J, Yu TT, Kuppusamy R, Hassan MM, Alghalayini A, Cranfield CG, et al. Cholic Acid-Based Antimicrobial Peptide Mimics as Antibacterial Agents. Int J Mol Sci (2022) 23(9):4623. doi: 10.3390/ijms23094623
130. Sgolastra F, Deronde BM, Sarapas JM, Som A, Tew GN. Designing Mimics of Membrane Active Proteins. Acc Chem Res (2013) 46:2977–87. doi: 10.1021/ar400066v
131. Lienkamp K, Tew GN. Synthetic Mimics of Antimicrobial Peptides–a Versatile Ring-Opening Metathesis Polymerization Based Platform for the Synthesis of Selective Antibacterial and Cell-Penetrating Polymers. Chemistry (2009) 15:11784–800. doi: 10.1002/chem.200900049
132. Boschert D, Schneider-Chaabane A, Himmelsbach A, Eickenscheidt A, Lienkamp K. Synthesis and Bioactivity of Polymer-Based Synthetic Mimics of Antimicrobial Peptides (SMAMPs) Made From Asymmetrically Disubstituted Itaconates. Chemisty (2018) 24:8217–27. doi: 10.1002/chem.201800907
133. Thaker HD, Cankaya A, Scott RW, Tew GN. Role of Amphiphilicity in the Design of Synthetic Mimics of Antimicrobial Peptides With Gram-Negative Activity. ACS Med Chem Lett (2013) 4:481–5. doi: 10.1021/ml300307b
134. Mensa B, Kim YH, Choi S, Scott R, Caputo GA, DeGrado WF. Antibacterial Mechanism of Action of Arylamide Foldamers. Antimicrob Agents Chemother (2011) 55:5043–53. doi: 10.1128/AAC.05009-11
135. Gabriel GJ, Tew GN. Conformationally Rigid Proteomimetics: A Case Study in Designing Antimicrobial Aryl Oligomers. Org Biomol Chemi (2008) 6:417–23. doi: 10.1039/b714490n
136. Ryan LK, Freeman KB, Masso-Silva JA, Falkovsky K, Aloyouny A, Markowitz K, et al. Activity of Potent and Selective Host Defense Peptide Mimetics in Mouse Models of Oral Candidiasis. Antimicrob Agents Chemother (2014) 58:3820–7. doi: 10.1128/AAC.02649-13
137. Love MS, Millholland MG, Mishra S, Kulkarni S, Freeman KB, Pan W, et al. Platelet Factor 4 Activity Against P. Falciparum and its Translation to Nonpeptidic Mimics as Antimalarials. Cell Host Microbe (2012) 12:815–23. doi: 10.1016/j.chom.2012.10.017
138. Ooi N, Miller K, Hobbs J, Rhys-Williams W, Love W, Chopra I, et alXF-73, A Novel Antistaphylococcal Membrane-Active Agent With Rapid Bactericidal Activity. J Antimicrob Chemother (2009) 64(4):735–40. doi: 10.1093/jac/dkp299
139. Tang H, Doerksen RJ, Jones TV, Klein ML, Tew GN. Biomimetic Facially Amphiphilic Antibacterial Oligomers With Conformationally Stiff Backbones. Chem Biol (2006) 13:427–35. doi: 10.1016/j.chembiol.2006.02.007
140. Mensa B, Howell GL, Scott R, DeGrado WF. Comparative Mechanistic Studies of Brilacidin, Daptomycin, and the Antimicrobial Peptide LL16. Antimicrob Agents Chemother (2014) 58:5136–45. doi: 10.1128/AAC.02955-14
141. Hu Y, Jo H, DeGrado WF, Wang J. Brilacidin, a COVID-19 Drug Candidate, Demonstrates Broad-Spectrum Antiviral Activity Against Human Coronaviruses OC43, 229E, and NL63 Through Targeting Both the Virus and the Host Cell. J Med Virol (2022) 94:2188–200. doi: 10.1002/jmv.27616
142. Bakovic A, Risner K, Bhalla N, Alem F, Chang TL, Weston WK, et al. Brilacidin Demonstrates Inhibition of SARS-CoV-2 in Cell Culture. Viruses (2021) 13:271. doi: 10.3390/v13020271
143. Su M, Xia D, Teng P, Nimmagadda A, Zhang C, Odom T, et al. Membrane-Active Hydantoin Derivatives as Antibiotic Agents. J Med Chem (2017) 60:8456–65. doi: 10.1021/acs.jmedchem.7b00847
144. Teng P, Nimmagadda A, Su M, Hong Y, Shen N, Li C, et al. Novel Bis-Cyclic Guanidines as Potent Membrane-Active Antibacterial Agents With Therapeutic Potential. Chem Commun (2017) 53:11948–51. doi: 10.1039/c7cc07285f
145. Niu Y, Wang M, Cao Y, Nimmagadda A, Hu J, Wu Y, et al. Rational Design of Dimeric Lysine N-Alkylamides as Potent and Broad-Spectrum Antibacterial Agents. J Med Chem (2018) 61:2865–74. doi: 10.1021/acs.jmedchem.7b01704
146. Teng P, Huo D, Nimmagadda A, Wu J, She F, Su M, et al. Small Antimicrobial Agents Based on Acylated Reduced Amide Scaffold. J Med Chem (2016) 59:7877–87. doi: 10.1021/acs.jmedchem.6b00640
147. Chu W, Yang Y, Qin S, Cai J, Bai M, Kong H, et al. Low-Toxicity Amphiphilic Molecules Linked by an Aromatic Nucleus Show Broad-Spectrum Antibacterial Activity and Low Drug Resistance. Chem Commun (2019) 55:4307–10. doi: 10.1039/c9cc00857h
148. Chu W, Yang Y, Cai J, Kong H, Bai M, Fu X, et al. Synthesis and Bioactivities of New Membrane-Active Agents With Aromatic Linker: High Selectivity and Broad-Spectrum Antibacterial Activity. ACS Infect Dis (2019) 5:1535–45. doi: 10.1021/acsinfecdis.9b00078
149. Wang M, Gao R, Sang P, Odom T, Zheng M, Shi Y, et al. Dimeric Gamma-AApeptides With Potent and Selective Antibacterial Activity. Front Chem (2020) 8:441. doi: 10.3389/fchem.2020.00441
150. Amabili P, Biavasco F, Brenciani A, Citterio B, Corbisiero D, Ferrazzano L, et al. Simple Amphiphilic Alpha-Hydrazido Acids: Rational Design, Synthesis, and In Vitro Bioactivity Profile of a Novel Class of Potential Antimicrobial Compounds. Eur J Med Chem (2020) 189:112072. doi: 10.1016/j.ejmech.2020.112072
151. Henderson LC, Li J, Nation RL, Velkov T, Pfeffer FM. Developing an Anion Host for Lipid A Binding and Antibacterial Activity. ChemComm (2010) 46:3197–9. doi: 10.1039/B925135A
152. Hickey SM, Ashton TD, Khosa SK, Robson RN, White JM, Li J, et al. Synthesis and Evaluation of Cationic Norbornanes as Peptidomimetic Antibacterial Agents. Org Biomol Chem (2015) 13:6225–41. doi: 10.1039/C5OB00621J
153. Hickey SM, Ashton TD, Boer G, Bader CA, Thomas M, Elliott AG, et al. Norbornane-Based Cationic Antimicrobial Peptidomimetics Targeting the Bacterial Membrane. Eur J Med Chem (2018) 160:9–22. doi: 10.1016/j.ejmech.2018.09.072
154. Kuppusamy R, Yasir M, Berry T, Cranfield CG, Nizalapur S, Yee E, et al. Design and Synthesis of Short Amphiphilic Cationic Peptidomimetics Based on Biphenyl Backbone as Antibacterial Agents. Eur J Med Chem (2018) 143:1702–22. doi: 10.1016/j.ejmech.2017.10.066
155. Kuppusamy R, Yasir M, Yee E, Willcox M, Black DS, Kumar N. Guanidine Functionalized Anthranilamides as Effective Antibacterials With Biofilm Disruption Activity. Org Biomol Chem (2018) 16:5871–88. doi: 10.1039/c8ob01699b
156. Yang T, Moreira W, Nyantakyi SA, Chen H, Aziz DB, Go ML, et al. Amphiphilic Indole Derivatives as Antimycobacterial Agents: Structure-Activity Relationships and Membrane Targeting Properties. J Med Chem (2017) 60:2745–63. doi: 10.1021/acs.jmedchem.6b01530
157. Chen Y, Li H, Liu J, Zhong R, Li H, Fang S, et al. Synthesis and Biological Evaluation of Indole-Based Peptidomimetics as Antibacterial Agents Against Gram-Positive Bacteria. Eur J Med Chem (2021) 226:113813. doi: 10.1016/j.ejmech.2021.113813
158. Jasim HA, Nahar L, Jasim MA, Moore SA, Ritchie KJ, Sarker SD. Chalcones: Synthetic Chemistry Follows Where Nature Leads. Biomolecules (2021) 11:1203. doi: 10.3390/biom11081203
159. Almeida JR, Moreira J, Pereira D, Pereira S, Antunes J, Palmeira A, et al. Potential of Synthetic Chalcone Derivatives to Prevent Marine Biofouling. Sci Total Environ (2018) 643:98–106. doi: 10.1016/j.scitotenv.2018.06.169
160. Lin S, Chen Y, Li H, Liu J, Liu S. Design, Synthesis, and Evaluation of Amphiphilic Sofalcone Derivatives as Potent Gram-Positive Antibacterial Agents. Eur J Med Chem (2020) 202:112596. doi: 10.1016/j.ejmech.2020.112596
161. Lin S, Koh JJ, Aung TT, Lim F, Li J, Zou H, et al. Symmetrically Substituted Xanthone Amphiphiles Combat Gram-Positive Bacterial Resistance With Enhanced Membrane Selectivity. J Med Chem (2017) 60:1362–78. doi: 10.1021/acs.jmedchem.6b01403
162. Lin S, Koh JJ, Aung TT, Sin WLW, Lim F, Wang L, et al. Semisynthetic Flavone-Derived Antimicrobials With Therapeutic Potential Against Methicillin-Resistant Staphylococcus Aureus (MRSA). J Med Chem (2017) 60:6152–65. doi: 10.1021/acs.jmedchem.7b00380
163. Zhong R, Li H, Li H, Fang S, Liu J, Chen Y, et al. Development of Amphiphilic Coumarin Derivatives as Membrane-Active Antimicrobial Agents With Potent In Vivo Efficacy Against Gram-Positive Pathogenic Bacteria. ACS Infect Dis (2021) 7:2864–75. doi: 10.1021/acsinfecdis.1c00246
164. Lin S, Wade JD, Liu S. De Novo Design of Flavonoid-Based Mimetics of Cationic Antimicrobial Peptides: Discovery, Development, and Applications. Acc Chem Res (2021) 54:104–19. doi: 10.1021/acs.accounts.0c00550
165. Kumar P, Kizhakkedathu JN, Straus SK. Antimicrobial Peptides: Diversity, Mechanism of Action and Strategies to Improve the Activity and Biocompatibility In Vivo. Biomolecules (2018) 8:4. doi: 10.3390/biom8010004
166. Isaksson J, Brandsdal BO, Engqvist M, Flaten GE, Svendsen JS, Stensen W. A Synthetic Antimicrobial Peptidomimetic (LTX 109): Stereochemical Impact on Membrane Disruption. J Med Chem (2011) 54:5786–95. doi: 10.1021/jm200450h
167. Trepos R, Cervin G, Pile C, Pavia H, Hellio C, Svenson J. Evaluation of Cationic Micropeptides Derived From the Innate Immune System as Inhibitors of Marine Biofouling. Biofouling (2015) 31:393–403. doi: 10.1080/08927014.2015.1048238
168. Dathe M, Nikolenko H, Meyer J, Beyermann M, Bienert M. Optimization of the Antimicrobial Activity of Magainin Peptides by Modification of Charge. FEBS Lett (2001) 501:146–50. doi: 10.1016/s0014-5793(01)02648-5
169. Arias M, Piga KB, Hyndman ME, Vogel HJ. Improving the Activity of Trp-Rich Antimicrobial Peptides by Arg/Lys Substitutions and Changing the Length of Cationic Residues. Biomolecules (2018) 8:19. doi: 10.3390/biom8020019
170. Haney EF, Barbosa SC, Baquir B, Hancock REW. Influence of Non-Natural Cationic Amino Acids on the Biological Activity Profile of Innate Defense Regulator Peptides. J Med Chem (2019) 62:10294–304. doi: 10.1021/acs.jmedchem.9b01344
171. Kohn EM, Shirley DJ, Arotsky L, Picciano AM, Ridgway Z, Urban MW, et al. Role of Cationic Side Chains in the Antimicrobial Activity of C18G. Molecules (2018) 23:329. doi: 10.3390/molecules23020329
172. Greco I, Molchanova N, Holmedal E, Jenssen H, Hummel BD, Watts JL, et al. Correlation Between Hemolytic Activity, Cytotoxicity and Systemic In Vivo Toxicity of Synthetic Antimicrobial Peptides. Sci Rep (2020) 10:1–13. doi: 10.1038/s41598-020-69995-9
173. Huan Y, Kong Q, Mou H, Yi H. Antimicrobial Peptides: Classification, Design, Application and Research Progress in Multiple Fields. Front Microbiol (2020) 11:582779. doi: 10.3389/fmicb.2020.582779
174. Chu H-L, Yu H-Y, Yip B-S, Chih Y-H, Liang C-W, Cheng H-T, et al. Boosting Salt Resistance of Short Antimicrobial Peptides. Antimicrob Agents Chemother (2013) 57:4050–2. doi: 10.1128/AAC.00252-13
175. Vlieghe P, Lisowski V, Martinez J, Khrestchatisky M. Synthetic Therapeutic Peptides: Science and Market. Drug Discovery Today (2010) 15:40–56. doi: 10.1016/j.drudis.2009.10.009
176. Rezende SB, Oshiro KGN, Junior NGO, Franco OL, Cardoso MH. Advances on Chemically Modified Antimicrobial Peptides for Generating Peptide Antibiotics. Chem Commun (2021) 57:11578–90. doi: 10.1039/d1cc03793e
177. Roberts KD, Zhu Y, Azad MAK, Han ML, Wang J, Wang L, et al. A Synthetic Lipopeptide Targeting Top-Priority Multidrug-Resistant Gram-Negative Pathogens. Nat Commun (2022) 13:1625. doi: 10.1038/s41467-022-29234-3
178. Laulund AS, Schwartz FA, Christophersen L, Høiby N, Svendsen JSM, Stensen W, et al. Lactoferricin-Inspired Peptide AMC-109 Augments the Effect of Ciprofloxacin Against Pseudomonas Aeruginosa Biofilm in Chronic Murine Wounds. J Glob Antimicrob Resist (2022) 29:185–93. doi: 10.1016/j.jgar.2021.12.015
Keywords: antimicrobial peptides, antibiotic, synthetic mimic, amphiphilic, clinical development, peptidomimetics, resistant bacteria
Citation: Svenson J, Molchanova N and Schroeder CI (2022) Antimicrobial Peptide Mimics for Clinical Use: Does Size Matter? Front. Immunol. 13:915368. doi: 10.3389/fimmu.2022.915368
Received: 07 April 2022; Accepted: 29 April 2022;
Published: 26 May 2022.
Edited by:
Ganna Petruk, Lund University, SwedenReviewed by:
Eugenio Notomista, University of Naples Federico II, ItalyFrank Schweizer, University of Manitoba, Canada
Copyright © 2022 Svenson, Molchanova and Schroeder. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Johan Svenson, am9oYW4uc3ZlbnNvbkBjYXd0aHJvbi5vcmcubno=
†ORCID: Johan Svenson, orcid.org/0000-0002-4729-9359
Christina I. Schroeder, orcid.org/0000-0002-6737-6374
Natalia Molchanova, orcid.org/0000-0001-7643-4508