 Divya Chopra1†
Divya Chopra1† Rachel A. Arens2†Watcharee Amornpairoj1Michelle A. Lowes3
Rachel A. Arens2†Watcharee Amornpairoj1Michelle A. Lowes3 Marjana Tomic-Canic1
Marjana Tomic-Canic1 Natasa Strbo4Hadar Lev-Tov1*
Natasa Strbo4Hadar Lev-Tov1* Irena Pastar1*
Irena Pastar1*- 1Wound Healing and Regenerative Medicine Research Program, Dr. Phillip Frost Department of Dermatology and Cutaneous Surgery, University of Miami Miller School of Medicine, Miami, FL, United States
- 2College of Medicine, University of Toledo College of Medicine and Life Sciences, Toledo, OH, United States
- 3Laboratory for Investigative Dermatology, The Rockefeller University, New York, NY, United States
- 4Department of Microbiology and Immunology, University of Miami Miller School of Medicine, Miami, FL, United States
Hidradenitis Suppurativa (HS) is a chronic multifactorial inflammatory skin disease with incompletely understood mechanisms of disease pathology. HS is characterized by aberrant activation of the innate immune system, resulting in activation of pathways that aim to protect against pathogenic microorganisms, and also contribute to failure to resolve inflammation. Imbalance in innate immunity is evident in deregulation of host antimicrobial peptides (AMPs) and the complement system associated with the microbiome dysbiosis. The pathology is further complicated by ability of pathogens associated with HS to overcome host immune response. Potential roles of major AMPs, cathelicidin, defensins, dermcidin, S100 proteins, RNAse 7 and complement proteins are discussed. Dysregulated expression pattern of innate immunity components in conjunction with bacterial component of the disease warrants consideration of novel treatment approaches targeting both host immunity and pathogenic microbiome in HS.
Introduction
Hidradenitis suppurativa (HS), also known as acne inversa, is a chronic, highly burdensome, inflammatory cutaneous disease that is associated with systemic manifestations. HS has a global prevalence ranging between 0.00033% to 4.10% (1). The onset of HS usually occurs in young adulthood to middle adulthood (1–3). Patients generally develop one or a few painful nodules or abscesses in intertriginous areas, such as armpits, groin and perianal area, in the early stages. The disease often progresses to form tunnels (also called sinus tracts or fistulas) and scars in more advanced stages. HS is recognized as a multifactorial disease; current evidence points to genetic predisposition, hormonal imbalance, lifestyle factors, and some unique features of the affected skin areas contribute to disease development (1, 2, 4).
Aberrant activation of the innate immune system is another major trait of HS. The intertriginous areas affected by HS are prone to increased mechanical friction causing cutaneous microinjuries in skinfolds which can lead to stimulation of innate immune responses and allow for the invasion of microorganisms to hair follicle and dermis (1, 2, 5). HS lesions commonly cause disfigurement, itch, malodorous pus, and movement restrictions, impacting patients’ quality of life (1–3, 6). The therapeutic options of HS are currently limited, including medical and surgical therapy, and are supported by limited data. Only a single drug has completed the US Food and Drug Administration approval process based on two large randomized controlled trials. Therefore, HS remains challenging to treat due to the complexity of the disease pathology, limited therapies and an enormous impact of the quality of life of patients (2, 5, 7).
Antimicrobial peptides (AMPs) are major component of the cutaneous innate immunity produced by epithelial and immune cells and possess direct bactericidal activity (7). Multiple AMPs are made in the skin, providing the cutaneous barrier with broad spectrum antimicrobial activity making them an essential aspect of the innate immune system. Some AMPs are constitutively expressed to keep the skin microbiota in balance while others are induced by the presence of pathogens or wounding and are also found deregulated in pathologic conditions including HS (8–12). Furthermore, the complement system, an immune effector system involved in the host response to microbes, is also dysregulated in HS (13, 14). Here we review the role of innate immunity in HS with focus on antimicrobial peptides and the complement system and their role in the pathogenesis of the disease. The interplay between microbial dysbiosis and deregulation of components of the innate immune system in HS pathology is discussed, with the focus on understanding the mechanistic contributions of both host response and microbiota to disease pathology.
Microbial dysbiosis in HS
The balance between commensal microbiota contributes to, and is a hallmark of cutaneous homeostasis (15). In contrast, microbial dysbiosis is a well-established feature of HS (16, 17). Compared to other skin loci, intertriginous areas affected in HS have a high density of pilosebaceous-apocrine units, higher temperature and moisture, but lower oxygen availability all contributing to specific microbiome composition and increased risk of dysbiosis (1, 2, 5, 18). Systemic factors such as obesity, diabetes and nicotine use can also contribute to microbial dysbiosis and HS pathogenesis (1, 19, 20), and may correlate with the altered gut microbiome found in HS patients (21, 22). Although it remains to be resolved if dysbiosis is a primary trigger or a consequence of HS, multiple studies provide insights into the microbiome composition at early and advanced stages of the disease. The predominance of commensal coagulase-negative staphylococci (CoNS) and Propionibacterium has been associated with early HS lesions (23, 24). However, in advanced stages of HS, CoNS and additional commensals including Cutibacterium and P. acnes, are significantly diminished, while pathogenic Staphylococcus aureus persists (24–26). Gram-negative anaerobic pathogens Porphyromonas spp, Peptoniphilus spp and Prevotella spp become predominant in HS tunnels and advanced stages of the disease (24, 27, 28). These pathogens are proposed to contribute to biofilm formation found inlesional skin and tunnels (29–31).
Chronic HS lesions and tunnels contain keratin debris and hair fragments that may serve as surfaces for pathogenic anaerobes to anchor and initiate biofilm growth (29). Polymicrobial biofilms containing Porphyromonas and S. aureus have been associated with pathogenesis of periodontal disease (32); however interspecies interactions and their crosstalk with the host innate immunity in HS remain to be elucidated. Biofilms may not only cause an inappropriate host response contributing to perpetual inflammation but are also challenging to treat due to antibiotic and antimicrobial resistance (31, 33). Bacterial biofilms can also evade components of the innate immune system, including complement and AMPs (34–36).
The role of AMPs in HS pathology
AMPs are a key part of the innate immune system of the skin with a critical role in preventing overgrowth of commensal microbiota, preventing invasion by pathogens, and initiating an inflammatory cascade aimed at wound healing (37). AMPs are especially important in the intertriginous regions as the environment is prime for microorganism growth. Without the appropriate expression of AMPs, the intertriginous skin may be overwhelmed by local commensals and is left vulnerable to colonization by pathogens. Importantly, dysregulation of AMPs that is observed in HS can contribute to the chronicity of inflammation, and microbial dysbiosis. Herein, we outline the role of the major AMPs, cathelicidin, defensins, dermcidin, S100 proteins and RNases in cutaneous homeostasis and their associated roles in HS pathology (Table 1).

Table 1 Overview of major AMPs associated with HS pathology.
Cathelicidin role and function in skin and HS
A single cathelicidin gene in humans, cathelicidin antimicrobial peptide (CAMP) encodes for a precursor protein, human cationic antimicrobial protein, hCAP18, which is proteolytically processed by serine proteases into multiple different peptides, including LL-37 (10). Cathelicidins have broad spectrum antimicrobial activity and are constitutively expressed by follicular keratinocytes and mast cells in healthy skin. The production of LL-37 is induced by cutaneous inflammation, often at the hands of pathogens (10, 38, 39). LL-37 is also present in sweat and wound fluid (38, 78). Specifically, LL-37 is induced in acute wounds with a diminishing concentration as the inflammation resolves and wound progresses to healing (79). Several factors determine expression of LL-37. CAMP is under transcriptional control by Vitamin D response element, and CAMP expression is strongly induced by Vitamin D in all skin cell types (10, 40). Vitamin D deficiency has been associated with multiple inflammatory skin diseases including psoriasis, rosacea, atopic dermatitis, and systemic lupus erythematous (80–82), while association with HS has been reported (83) but remains to be thoroughly investigated. The enzymatic processing of hCAP18 in the skin is controlled by serine proteases kallikrein 5 and kallikrein 7 (KLK5, KLK7), which are also induced by Vitamin D and pH changes (41, 84, 85). KLK and its substrates are found in the dark cells in eccrine sweat glands but not sebaceous glands, hair follicles, keratinocytes, or elsewhere in the pilosebaceous unit (86).
Multiple studies have found LL-37 expression to be increased in the skin of HS patients on both mRNA and protein levels (42–44) (Table 1). In addition to its antimicrobial activity, LL-37 has prominent immunomodulatory activity, including chemotaxis of neutrophils, monocytes, mast cells, and T cells, and can influence Th1/Th17 cell maturation (37, 45). Neutrophils drawn to the inflammation further promote the process by releasing additional cathelicidin from granules (37). Chemotactic function of LL37 attracts CD4 T cells and dendritic cells, which further release TNF-α, IL-6, and Il-12. The upregulation of these inflammatory markers results in the Th1/Th17 phenotype, independent of antigen-presenting cells (43), while LL-37 may also act as T-cell autoantigen in psoriasis (87). Additional studies have found the progression of HS severity to be correlated with increased levels of these pro-inflammatory cytokines promoted by LL-37, including IL-17 and TNF-α (46, 88–90), suggesting the potential therapeutic targeting of LL-37. LL-37 also promotes proliferation of keratinocytes by an anti-apoptotic mechanism (91), which may be contributing to epidermal hyperplasia and proliferation of epithelial strands further stimulating formation of tunnels. Bacterial aggregates in these tunnels may form biofilms stringently attaching to epithelial tunnels and amplifying inflammation both locally and systemically (4, 12), though various mechanisms including increase of lipocalin-2 (6, 92, 93). Overall consistent upregulation of LL-37 in HS may allow for the local inflammation to progress to a systemic disease creating a positive feedback loop due to deregulated inflammation and microbial dysbiosis.
Recent findings revealed a contribution of LL-37 to bacterial antibiotic tolerance in S. aureus. LL-37 induced S. aureus tolerance to daptomycin by activating staphylococcal GraRS two-component system, leading to increased peptidoglycan formation, a key component of biofilm formation (94). LL-37 has also been identified to reduce S. aureus susceptibility to vancomycin (95). These findings underscore complexity of host-pathogen interaction in inflammatory conditions associated with AMP upregulation and warrants further investigation in HS considering the prevalence of S. aureus (96, 97).
The role of defensins in cutaneous immunity and deregulation in HS
Defensins are cysteine-rich cationic peptides grouped into α or β; α-defensins are mainly expressed in leukocytes and Paneth cells while β-defensins are found in various epithelial cells, including keratinocytes (47, 48). There are multiple β-defensins; most important are human β-defensin-1 (hBD-1), hBD-2 and hBD-3, all of which are expressed by keratinocytes in response to inflammation or in response to pathogens (49). Unlike cathelicidins, defensins are not proteolytically processed to generate different defensin-peptides, rather they are encoded by multiple defensin genes. hBD-1, -2, and -3 are encoded for by DEFB1, DEFB4 and DEFB103, respectively. DEFB2, like CAMP, is also under transcriptional regulation of vitamin D (50) and DEFB4 is significantly upregulated in HS lesional skin (44, 46, 51, 52).
hBD-1 and hBD-2 are mainly effective against Gram-negative organisms while hBD-3 has broad spectrum activity (49, 53). Fewer studies have focused on hBD-1; however, it has been consistently found to be decreased in HS lesions (Table 1) (54, 55). As hBD-1 is constitutively expressed in healthy skin (56), it forms an important part of the cutaneous innate immune system. Without sufficient hBD-1 patients may be susceptible to commensal overgrowth in early stages of the disease, allowing for microbial dysbiosis as the disease progresses.
hBD-2 and hBD-3 are not constitutively expressed but upregulated at a transcriptional level in response to microorganisms and also increased levels of TNF-α, a hallmark proinflammatory cytokine of HS (Table 1) (13, 49, 53). Increased lesional hBD-2 was strongly correlated with higher levels of IL-20 and IL-22 (54). hBD-2 has well-described chemotactic abilities, as it attracts dendritic cells, memory T cells, and promotes histamine release from mast cells (57, 58), therefore primarily contributing to prolonged inflammation unable to clear bacterial biofilms in HS tunnels. In addition, both mRNA and protein levels of hBD-2 were released by dermal macrophages at a significantly higher level in HS lesions, further amplifying pro-inflammatory signature of HS (51). In contrast to upregulation of hBD-2, conflicting data were reported on hBD-3 indicating either upregulation (42, 46, 54, 55, 59) or lack of regulation compared to non-lesional skin (44, 60). Both hBD-2 and hBD-3 can stimulate keratinocyte proliferation (98). As the levels of these AMPs increase in HS lesions, keratinocytes multiply within the pilosebaceous unit, leading to hyperkeratosis and plugging of the hair follicle which may serve as fuel for microbial dysbiosis and biofilm formation. Overall, hBDs may primarily contribute to HS pathogenesis by promoting keratinocyte proliferation and inflammatory cell chemotaxis.
Dermcidin function in skin and suppression in HS
Like cathelicidin, dermcidin originates from a single gene, dermcidin (DCD), which encodes for an AMP with broad-spectrum activity. DCD is proteolytically processed by cathepsin D (CatD) into DCD-1 and DCD-1L, and others (9, 99, 100). DCD is constitutively and specifically expressed by eccrine sweat glands, especially on the face and hands, but could not be isolated from apocrine sweat glands (9, 64). Unlike most other AMPs, DCD is anionic and has been shown to form large pores in the membranes of Gram-negative bacteria (65, 66). Interestingly, DCD-1 is amphipathic, forming a large complex alongside the bacterial membrane, which is stabilized by zinc ions in sweat, allowing ion channel formation on the pathogens’ membrane (8). DCD is not expressed by keratinocytes in the presence of inflammation like the other AMPs, leaving the current understanding of its function as maintenance of commensal skin microbiota and prevention of pathogens invasion largely unknown (67).
DCD was significantly downregulated in HS lesional skin on mRNA and protein level (Table 1) (60, 101), suggesting that insufficient levels of DCD may allow overgrowth of Gram-negative bacteria in HS tunnels. However, like the other AMPs, DCD-derived peptides can promote epidermal inflammation via TNF-α, IL-8, CXCL 10 and CCL20 (102), limiting the opportunity for therapeutic targeting of DCD in HS. Further investigation of the role of DCD in HS is needed to shed a light on this AMP and its potential contribution to microbial dysbiosis at different stages of disease.
The role of S100 proteins in HS
The S100 protein family, also known as alarmins, consist of at least 21 calcium-binding cytosolic proteins which can be divided into 3 divisions: those with intracellular function only, those with extracellular function only, and those with both. By altering calcium signaling, S100 proteins can serve as intracellular regulators and extracellular signaling molecules in either a paracrine or autocrine manner (103). The S100 protein family has a wide range of function including regulating cell migration, proliferation, differentiation, and apoptosis as well as modulating inflammation and energy metabolism (104, 105), and are made by keratinocytes, dendritic cells, neutrophils, and macrophages (69, 106).
In healthy skin, S100A7, S100A8 and S100A9 are rarely detected at the protein level, while increased levels are well documented in many inflammatory skin conditions such as psoriasis, atopic dermatitis, and mycosis fungoides. Among the S100 protein family, S100A7 (psoriasin), S100A8 (calgranulin A), S100A9 (calgranulin B), S100A12 (calgranulin C) and S100A15 (koebnerisin) have well documented antimicrobial activity (107). S100A7 and S100A15 have strong bactericidal activity against Escherichia coli with weaker preference for Gram-positive bacteria while S100A8/S100A9 and S100A12 have preferential activity against fungi and viruses (70, 107, 108). S100A8 and S100A9 also form a heterodimer known as S100A8/S100A9 or calprotectin which plays an integral role in acute and chronic inflammation (11).
Multiple studies have confirmed increased levels of S100 proteins in HS (Table 1). Increased expression of S100A7 at both the mRNA and protein levels was found in lesional HS skin (46, 51, 52, 60, 71). S100A8, S100A9, S100A12 and S100A15 have also been shown to be increased in lesional skin in HS (13, 46, 52, 54, 60, 109). Overexpression of S100A15 and S100A12 was noted in perilesional skin as well, which suggests a possible role in early pathogenesis of HS by stimulating keratinocyte proliferation in the perifollicular region (13, 109). The increase of S100 proteins may occur in response to elevated inflammatory cytokines such as interleukin (IL)-17 and IL-22, as in psoriasis (110, 111). The overall increased levels of S100 proteins in HS can further elevate level of inflammation due to known chemotactic function of S100A7 and S100A8/S100A9 complex, subsequently increasing expression of proinflammatory cytokines and inducing a positive feedback loop, while microbial dysbiosis persists. The family of S100 proteins exerts its antimicrobial function through sequestration of Mn+ and Zn+ ions required for bacterial metabolism (112); however, excessive sequestration can also negatively affect host response e.g. impact Zn+-dependent DCD function (8) and further diminish antimicrobial response.
Dysregulation of RNAse 7 in HS
RNAse 7 is one of eight enzymes in the RNAse A superfamily. The members of this family have been demonstrated to have a wide range of action including diverse immunomodulatory, angiogenic, and neurotoxic effects (113). A few members of this family also exert antimicrobial action including RNAse 3, 4, 7, 8, and 9 (114). RNAse 7 has a broad-spectrum antimicrobial activity against Gram -positive and -negative bacteria (113). RNAse 7 is constitutively expressed in keratinocytes and exists in high quantities in all layers of the epidermis with a particular concentration in the stratum corneum and in follicular epithelium (77, 115, 116), indicating its role in HS.
Currently, the relationship of RNAse 7 in HS skin has not been clearly defined and was primarily evaluated at mRNA levels (Table 1). RNAse 7 mRNA has been shown to be increased (46),. or decreased (59) in HS lesional skin when compared to heathy skin; not different between lesional and non-lesional HS skin (44), and increased in lesional HS skin compared to perilesional skin (117). There was significantly increased RNAse 7 mRNA in HS lesional skin compared to chronic venous leg ulcers (44). Overall induced expression of RNAse 7 can be attributed to higher levels IL-1β, IL-17, and IFN-γ in HS known to induce expression of this his AMP (77, 113). RNAse 7 can also promote rapid sensing of bacterial and human DNA by plasmacytoid dendritic cells resulting in enhanced production of TNF-α and IFN-α (118) contributing to perpetual cycle of inflammation in HS.
Complement system and its role in disease pathogenesis
The complement system is a part of innate immunity that acts as first line defense against pathogens and altered host cells by augmenting opsonization of pathogens and promoting inflammatory processes. This system is largely composed of plasma proteins synthesized in the liver but also includes inactive precursor proteins on cell membranes (119). Because serum is restricted in areas were complement activation is needed, these proteins can also be made by a variety of cells including epithelial cells, endothelial cells, and immune cells (120, 121).
Activation of the complement system can be achieved by three distinct pathways – classical (CP), lectin (LP), and alternative (AP), each leading to a common terminal (cytolytic) pathway initiated by different recognition molecules (122, 123). All three pathways ultimately form C3 convertases to cleave C3 into C3a and C3b. C3a is an anaphylatoxin with a largely proinflammatory response including induction of oxidative burst in macrophages and neutrophils, histamine production via basophils to induce vasodilation, and increased vascular permeability (124). C3b acts as an opsonin which will target apoptotic cells, pathogens, and immune complexes for phagocytosis. Once levels of C3b have reached a threshold of activation, C3b can bind to a C3 convertase, either C4bC2a or C3bBb, to form C5 convertase which will cleave C5 to produce C5a. C5a in combination with the membrane attack complex (MAC) which consists of a polymer of C5b, C6, C7, C8, and C9 initiates a terminal pathway to bacterial cell lysis (120, 122, 125).
As a very dangerous cytolytic system, complement activation in the CP and LP pathways are tightly regulated in physiological conditions by many plasma proteins to ensure that only infection will stimulate a full response (122). Both inefficient activation and over stimulation of complement have been found to be associated with increased susceptibility to infections, while the imbalance of complement activation contributes to development of chronic inflammation (122, 126).
Complement is essential in maintaining cutaneous health by maintaining healthy skin microbiome and modulating inflammatory responses (127–129). Human keratinocytes produce several complement proteins including C3, C4, and complement factor B (FB) which is induced by IL- 1α, IFN-γ, and TNF-α (121). Keratinocytes also play a significant role in regulating response of complement proteins. Keratinocytes can synthesize soluble complement inhibitors like factor H and factor I, complement receptors CR1, cC1qR, C5aR1, and CR2 and cell-bound complement regulator proteins membrane cofactor protein (MCP/CD46), decay-accelerating factor (DAF), and protectin (CD59) (14). FH and FI production can be stimulated locally by IFN-γ which can then protect the epidermis from damage caused by C3, C4, and FB (130, 131).
Complement activation in HS is thought to be primarily systemic response (132). All three pathways, CP, LP, and AP, are thought to contribute the complement activation in HS (133). Ghias et al. suggested that the initial stimulus was follicular rupture releasing microbes and keratin which would trigger production of C3a, C3b, and C5a (127). This would in turn lead to bacterial opsonization, keratin phagocytosis, neutrophil chemotaxis, mast cell degranulation, and cell lysis through the MAC complex. Additionally, complement activation triggers inflammasomes to produce IL-1ß. IL-1ß further induces a cascade of proinflammatory cytokine release, AMP production, and DC activation (46, 127). A transcriptome study of the HS skin showed C1q, C2, and factor B genes were upregulated, whereas factor H, factor D, and C7 were downregulated. In the serum proteome, C5a was found upregulated, and C4b, C3, C3b, and iC3b were downregulated (133). Levels of C5a and the components of the MAC, C5b-C9, are significantly elevated in HS patients when compared to healthy subjects. When stratified amongst Hurley stages, levels of C5a and C5b-C9 were highest in patients with mild disease than those with more severe disease. However, results from a recent study reported no evidence of elevated plasma levels of C5b-9 in patients with HS despite decreased C3/C3d ratio (134). These results suggest that complement hyperactivation which is likely a consequence of initial dysbiosis of microbiota (Figure 1), plays an important role in early disease development in HS rather than being a sequela of chronic inflammation in more progressed disease (132), although it is also possible that there is increased activity of complement including opsonization due to increased C3b levels but without increased systemic terminal complement pathway (135).
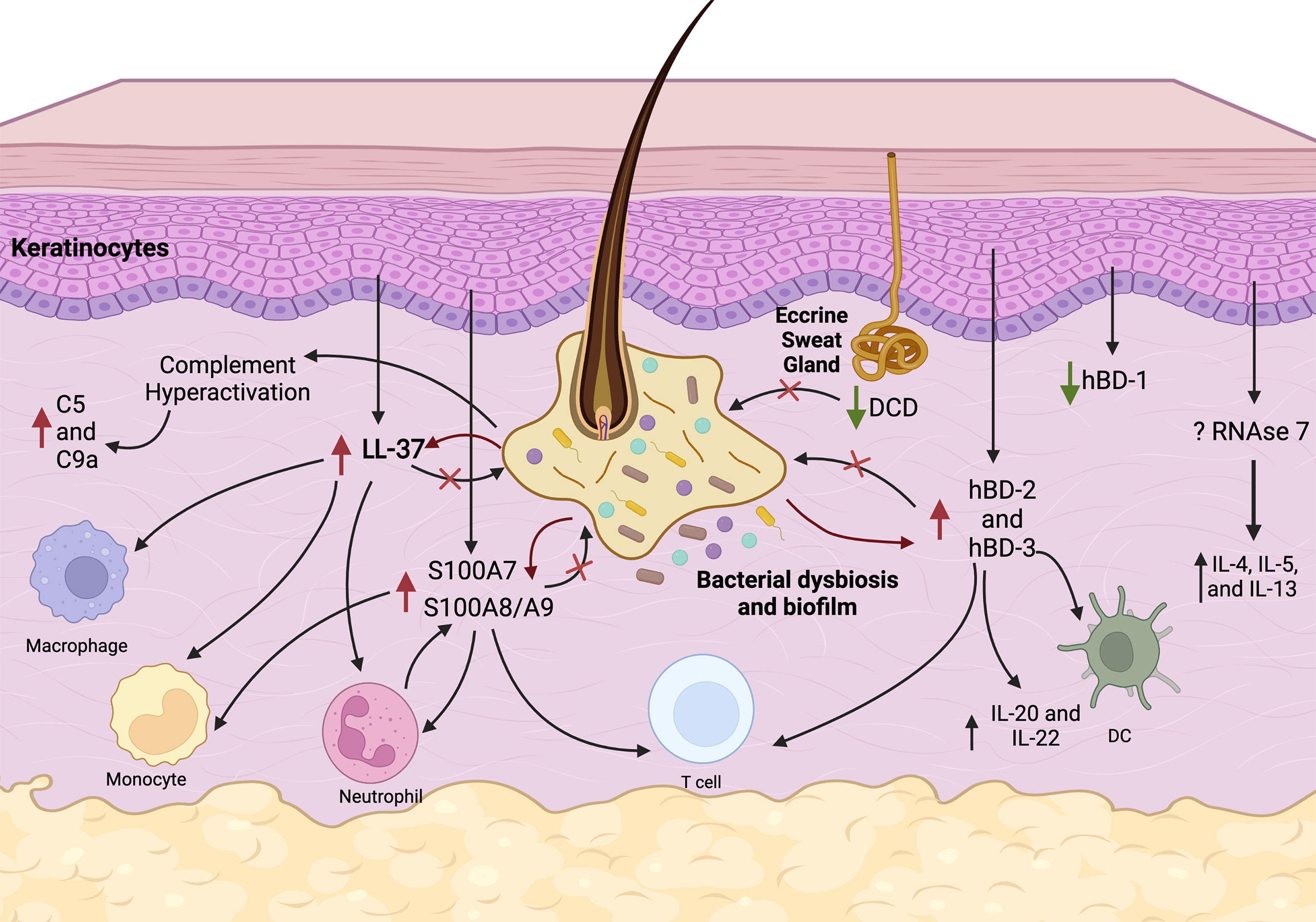
Figure 1 Proposed pathways of AMPs and complement deregulation in HS. Follicular inflammation, microbial dysbiosis and biofilm stimulate keratinocytes to overexpress LL-37 (cathelicidin), S100A7, S100A8/A9, hBD-2, and hBD-3. Immunomodulatory function of these AMPs includes chemotaxis of macrophages, monocytes, neutrophils, T cells, and dendritic cells (DC) and production of cytokines. Complement pathways are hyperactivated in HS causing elevated C5a and C9 levels. Constitutive production of hBD-1 from keratinocytes and DCD production from eccrine sweat glands is inhibited in HS skin which may result in microbial dysbiosis. Overall, these events lead to a persistent vicious cycle of chronic inflammation, which is ineffective in eliminating pathogens and biofilms.
Importantly, C5a is a critical stimulator of TNF-α release. In studies, purified blood mononuclear cells (PBMCs) from HS patients did not produce proinflammatory cytokines upon stimulation with bacterial ligands (136) but did overexpress TNF-α when in the presence of patients’ plasma. These findings support the idea of HS stemming from dysregulated innate immunity in conjunction with microbial dysbiosis. TNF-α in turn stimulates upregulation of C3 (137), FB in the alternative pathway (138), and surface regulatory proteins like CD59 (139). TNF-α overexpression was attenuated after introduction of an anti-C5a monoclonal antibody, suggesting an important relationship between C5a and TNF-α in worsening inflammatory responses in HS (132) and supporting therapeutic targeting of C5Aa (127).
Pathogens have developed complement evasion strategies to escape host innate immunity. Methods of complement evasion are highly conserved across the classes of pathogens including bacteria, virus, and fungi. Common strategies are inactivation via protease secretion, complement disguise using regulatory molecules, and expression of direct inhibitors (140). Streptococcus pyogenes and S. aureus have adapted to develop methods for complement evasion. S. pyogenes utilizes the M protein, a cell membrane-bound polymorphic protein, to defend against complement-mediated phagocytosis by binding regulatory proteins such as FH and C4BP to the bacteria surface. Additionally, MAC-mediated bacterial lysis is inhibited by S. pyogenes due to the thick outer peptidoglycan layer (141). Furthermore, the MAC complex is rendered useless directly by streptococcal inhibitor of complement (SIC) expression (142) and indirectly by expression of vitronectin-binding proteins (143). Similarly, S aureus evades complement through use of regulatory proteins such as FH and inactivation via a staphylococcal complement inhibitor (SCIN). S aureus secretes extracellular protein B (Ecb) and staphylococcal binder of IgG (Sbi) to enhance deposition of FH onto staphylococcal membrane (144, 145). SCIN targets C3BbBb to inhibit formation of C3a, C3b, and C5a and thus preventing bacterial opsonization and decreasing phagocytic cell chemotaxis. S. aureus has also developed mechanisms capable of evading killing by additional host MAC-domain protein, Perforin-2, responsible for elimination of intracellular S. aureus (146–149). Perforin-2 is found suppressed in chronic diabetic wounds associated by microbial dysbiosis and persistent inflammation allowing for intracellular accumulation of S. aureus in epidermal keratinocytes (146). Collectively, these findings warrant further investigation into ability of HS pathogens to escape host innate immunity.
Genetic component of HS
There are multiple genetic factors that correlate with a patient’s susceptibility to develop HS. Approximately 30% of HS patients have a positive family history (2, 5, 150, 151). A recent Dutch nationwide twin cohort study reported a 77% calculated narrow sense heritability in HS which suggests a strong genetic component in pathogenesis of the disease (152).
Mutations in genes encoding γ-secretase, a protease that targets several type-1 transmembrane proteins such as amyloid precursor proteins and the Notch protein family, are considered the most common in HS (153) although HS can be also classified as polygenic condition (151). About 36 mutations in genes encoding γ-secretase, including NCSTN, PSEN1, and PSENEN, were identified in specific patient populations with severe HS (2, 150, 154). These mutations impair γ-secretase activity impairment which may lead to alterations in keratinocytes proliferation and differentiation via impaired Notch signaling. Deregulated Notch signaling in HS may contribute to many of the major pathogenic events in HS. Notch signaling also plays a role in suppressing proinflammatory cytokine release via negative feedback inhibition of TLR4-triggered macrophages usually induced by bacterial lipopolysaccharides (155). Increased proinflammatory cytokine release contributes to activation of Th17-driven inflammation, a major immunologic characteristic of HS (156). Natural killer cells (NKs) whose development is mediated by Notch signaling are diminished in HS (157, 158). Besides Notch signaling, γ-secretase mutations may also induce dysregulation of the complement system. γ-secretase can cleave CD46, a regulatory cofactor of protease Factor I responsible for cleaving C4b and C3b deposited on cells into more benign products to protect host cells (159, 160). Binding of CD46 to C3b dimers induces production of regulatory T cell phenotypes such as T-regulatory cell 1 which then secretes large quantities of IL-10 (161, 162). CD46 expression and function is well documented to be impaired in cutaneous autoimmune disorders such as systemic lupus erythematous and bullous pemphigoid (163). Inhibited γ-secretase may allow for disruption of the protective regulatory function of CD46 and therefore inflammation via augmented complement and T-cell activity (127).
Several other genetic associations have been observed in HS, including NOD2 mutations (164). In addition, RNA expression of NOD2 has been demonstrated to be increased in lesional HS skin as compared to perilesional skin. This response was positively correlated with other AMPs such as hBD2, S100A7, and RNAse 7 (117). However, genomic analysis targeting NOD2 polymorphisms has not yet isolated any of significant correlation to HS (165). Additionally, the presence of over six copies of the DEFB gene cluster, was shown to hold a significantly increased risk of developing more severe HS disease (61). This is hypothesized to be a consequence of the loss of hBD-2’s protective action against bacterial superinfection of the skin, specifically against S. aureus. This same association is not unique for HS as it is also present in psoriasis, a disease that has well-established ties with dysregulation of AMPs including hBD-2 and hBD-3 (61). However, as the most HS patients with a positive family history (>90% in white individuals) do not have γ-secretase or other mutations (2, 5), genetic features contributing to HS development still need to be investigated.
Treatment strategies
Considering the role of bacteria and the immune system in the pathogenesis of HS, the use of antibiotics and biologics with anti-inflammatory effect is a direct extension. Antibiotics, both topical and systemic, are often used as a first line therapy in treating HS. Systemic tetracycline therapy is amongst the most prescribed therapy along with clindamycin, rifampin, and dapsone (166). These classes of antibiotics have well documented anti-inflammatory action that, along with their antibacterial activity, is crucial in treating HS.
Biologic therapies that target specific immune pathways are an emerging class of treatment in HS. Biologics have shown efficacy in several inflammatory conditions such as inflammatory bowel disease (IBD) and psoriasis. Adalimumab, an anti-TNFα monoclonal antibody, that demonstrated efficacy in large placebo controlled trials and is currently the only FDA-approved biologic for HS treatment (167). In addition, several biologics are currently being investigated for efficacy in HS. Anakinra, an IL-1 receptor antagonist, has been demonstrated to decrease disease severity and improve quality of life in two clinical studies (168, 169). Bermekimab is a human IgG1k monoclonal antibody that also targets Il-1 that has been shown to be effective in treating HS patients that have failed anti-TNFα therapy likely due decreased neovascularization and modulation of IL-8 and hBD-2 production (170, 171). Secukinumab, a monoclonal antibody against IL-17, reported achievement of HS clinical response in 67% of patients in a pilot trial (172). A retrospective series and anecdotal data demonstrated that guselkumab, an anti-IL-23 monoclonal antibody, may be efficacious in treating HS (173).
Biologics can be used in conjunction with surgical therapy in patients with severe disease to maximize clinical response (174). Surgical procedures can be focused, such as deroofing procedures or skin-sparing excision with electrosurgical peeling, or more extensive such as wide excisions, and wound management in HS remains a large burden for patients (175). Currently, there is a paucity of HS-specific wound dressing options available. Given the interplay between the immune system and the dysbiosis that is present in HS, combination of therapies that target both of these disease elements simultaneously is likely to have a synergetic effect. However, currently such combination therapy protocols do not exist and quality data to prove the efficacy of this strategy is missing.
Discussion
HS lesional skin is characterized by activation of both innate and adaptive immune response in conjunction with microbial dysbiosis reflected in upregulation of multiple AMPs, complement system proteins, and numerous cytokines (Figure 1). However, it remains to be fully understood whether the dysregulation of immune responses is causative of or consequence of microbiota dysbiosis. In HS, progressive microbial dysbiosis likely drives AMP and complement dysregulation associated with activation of the innate immune response. These features all contribute to excessive inflammation, that is ineffective in eliminating pathogens (Figure 1). The net result is a vicious cycle that results in local tissue destruction, disease progression and potentially, systemic elaboration of cytokines that spreads the disease in genetically, microbiologically, and immunologically susceptible individuals. Future studies should tease out the primary event and the setting required for this vicious progressive cycle to develop, clarify the directionality of this relationship, and identify molecular pathways through which this is mediated. Advancing our understanding of the interplay between microbial dysbiosis and immune dysregulation may provide direction into developing new therapeutics for treating HS.
Initial findings based on aberrant complement activation at the systematic level (132), have resulted in two ongoing clinical trials targeting the C5a–C5aR1 axis (135, 176, 177). However, the variability of results in complement levels amongst studies also suggests benefit in correlating results with patients’ genotype and/or microbiome shifts in response to therapy. In addition, the only US-FDA approved biologic therapeutic adalimumab, an anti-TNF-α monoclonal antibody, has shown limited efficacy (178). Lower efficacy of biologic drugs may in part be a consequence of persistent microbial dysbiosis, particularly bacterial biofilms of anerobic Gram-negative bacteria in HS tunnels at advanced stages of the disease. Like HS, IBD has demonstrated altered expression of AMPs, aberrant inflammatory response, and microbial dysbiosis that has been hypothesized to contribute to resistance to biologic therapy. For example, dysbiosis of bacteria that produce short chain fatty acids (SCFAs) in the gut microbiota of IBD patients has been associated with resistance to anti-TNFα therapy (179). While SCFAs inhibit biofilm formation (180, 181), diminished concentrations of SCFA-producing bacteria in biologic-resistant IBD patients suggests that resistance to therapy has microbial mediated mechanism. The findings in patients with IBD and similar immunologic features of IBD and HS (182) support the importance of prospective studies on microbiome in HS and its changes in patients undergoing biologic therapy. Future research should also aim to investigate combination of therapies targeting the deregulated immune response and microbial dysbiosis should be evaluated.
Based on deregulation of innate immune responses and initial changes in HS microbiome, antibacterial therapy may have important role in limiting progression of early HS. However, considering the rise in antibiotic resistance, antimicrobials may primarily manage disease by stalling inflammation, rather than eliminating pathogens. Additionally, the data presented in this review can reasonably predict the efficacy of a carbapenem antibiotic ertapenem in HS as well as its inability to maintain long term results (183).
Therefore, it is not surprising that the standard of care for moderate to severe disease (i.e. the presence of tunnels) is moving towards a combination of medical and surgical therapy (174). Deroofing is a common surgical procedure in which tunnels are selectively removed. In two large uncontrolled series, the ability of deroofing to prevent recurrence seems to decrease with disease progression. Milder HS was associated with 17% recurrence after deroofing (184), and more severe disease with 41% recurrence (185). The efficacy of deroofing may be related to the elimination of biofilms and reversal of the vicious cycle discussed above. Emerging clinical data suggests that topical anti-biofilm therapy that is administered without removing the diseased skin can eliminate isolated tunnels (186), and new biofilm-eliminating approaches should be considered (187, 188). Future metagenomics studies should examine the antimicrobial resistance in HS lesions and tunnels at advanced stages of the disease and characterize the interaction between the microbiome, biofilms, and innate immunity.
This review has summarized the potential contributions of innate immune system activation and microbial dysbiosis to HS pathogenesis. While studies utilizing single cell omics approaches have already revealed insight into HS pathology and deregulation of host responses (189–191) advancements in spatial transcriptomics and proteomics should be utilized to further understanding of deregulated innate immunity at the host-pathogen interphase. Future longitudinal studies simultaneously evaluating the focal genetic makeup, innate immunity, microbial dysbiosis and bacterial biofilms will provide detailed insights in HS pathogenesis and progression.
Author contributions
IP and HL-T conceived the review and coordinated writing. DC, RA, WA, ML, NS, MT-C, HL-T, and IP were involved in literature search and writing, and had final approval of the submitted and published versions of the manuscript.
Acknowledgments
We are thankful to all current and past members of our research teams for continuous inspiration and support. The figure in this manuscript was created using Biorender.com. This work is in part supported by Hidradenitis Suppurativa Foundation Danby Research Grant (IP and NS).
Conflict of interest
ML has served on the advisory boards for Abbvie, InflaRx, Janssen, and Viela Bio, and consulted for Almirall, BSN medical, Incyte, Janssen, Kymera, Phoenicis, and XBiotech.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Goldburg SR, Strober BE, Payette MJ. Hidradenitis suppurativa: Epidemiology, clinical presentation, and pathogenesis. J Am Acad Dermatol (2020) 82(5):1045–58. doi: 10.1016/j.jaad.2019.08.090
2. Sabat R, Jemec GBE, Matusiak Ł, Kimball AB, Prens E, Wolk K. Hidradenitis suppurativa. Nat Rev Dis Primers (2020) 6(1):18. doi: 10.1038/s41572-020-0149-1
3. Wipperman J, Bragg DA, Litzner B. Hidradenitis suppurativa: Rapid evidence review. Am Fam Phys (2019) 100(9):562–9.
4. Jiang SW, Whitley MJ, Mariottoni P, Jaleel T, MacLeod AS. Hidradenitis suppurativa: Host-microbe and immune pathogenesis underlie important future directions. JID Innov (2021) 1(1):100001. doi: 10.1016/j.xjidi.2021.100001
5. Wolk K, Join-Lambert O, Sabat R. Aetiology and pathogenesis of hidradenitis suppurativa. Br J Dermatol (2020) 183(6):999–1010. doi: 10.1111/bjd.19556
6. Navrazhina K, Frew JW, Gilleaudeau P, Sullivan-Whalen M, Garcet S, Krueger JG. Epithelialized tunnels are a source of inflammation in hidradenitis suppurativa. J Allergy Clin Immunol (2021) 147(6):2213–24. doi: 10.1016/j.jaci.2020.12.651
7. Zouboulis CC, Benhadou F, Byrd AS, Chandran NS, Giamarellos-Bourboulis EJ, Fabbrocini G, et al. What causes hidradenitis suppurativa ?-15 years after. Exp Dermatol (2020) 29(12):1154–70. doi: 10.1111/exd.14214
8. Schittek B. The multiple facets of dermcidin in cell survival and host defense. J Innate Immun (2012) 4(4):349–60. doi: 10.1159/000336844
9. Schittek B, Hipfel R, Sauer B, Bauer J, Kalbacher H, Stevanovic S, et al. Dermcidin: a novel human antibiotic peptide secreted by sweat glands. Nat Immunol (2001) 2(12):1133–7. doi: 10.1038/ni732
10. Gallo RL, Hooper LV. Epithelial antimicrobial defence of the skin and intestine. Nat Rev Immunol (2012) 12(7):503–16. doi: 10.1038/nri3228
11. Yao Y, Frew JW, Thomsen SF, Ring HC. Antimicrobial peptides in hidradenitis suppurativa: a systematic review. Br J Dermatol (2022) 186(2):236–44. doi: 10.1111/bjd.20750
12. Schell SL, Nelson AM. Are antimicrobial peptides a double-edged sword in hidradenitis suppurativa pathophysiology? Br J Dermatol (2022) 186(2):204–5. doi: 10.1111/bjd.20834
13. Navrazhina K, Garcet S, Frew JW, Zheng X, Coats I, Guttman-Yassky E, et al. The inflammatory proteome of hidradenitis suppurativa skin is more expansive than that of psoriasis vulgaris. J Am Acad Dermatol (2022) 86(2):322–30. doi: 10.1016/j.jaad.2021.07.035
14. Panelius J, Meri S. Complement system in dermatological diseases - fire under the skin. Front Med (Lausanne) (2015) 2:3. doi: 10.3389/fmed.2015.00003
15. Byrd AL, Belkaid Y, Segre JA. The human skin microbiome. Nat Rev Microbiol (2018) 16(3):143–55. doi: 10.1038/nrmicro.2017.157
16. Mintoff D, Borg I, Pace NP. The clinical relevance of the microbiome in hidradenitis suppurativa: A systematic review. Vaccines (Basel) (2021) 9(10):1076. doi: 10.3390/vaccines9101076
17. Naik HB, Jo JH, Paul M, Kong HH. Skin microbiota perturbations are distinct and disease severity-dependent in hidradenitis suppurativa. J Invest Dermatol (2020) 140(4):922–5.e3. doi: 10.1016/j.jid.2019.08.445
18. Williams SC, Frew JW, Krueger JG. A systematic review and critical appraisal of metagenomic and culture studies in hidradenitis suppurativa. Exp Dermatol (2021) 30(10):1388–97. doi: 10.1111/exd.14141
19. Thompson KG, Shuster M, Ly BC, Antonescu C, Florea L, Chien AL, et al. Variability in skin microbiota between smokers, former smokers, and nonsmokers. J Am Acad Dermatol (2020) 83(3):942–4. doi: 10.1016/j.jaad.2020.01.042
20. Hutcherson JA, Scott DA, Bagaitkar J. Scratching the surface - tobacco-induced bacterial biofilms. Tob Induc Dis (2015) 13(1):1. doi: 10.1186/s12971-014-0026-3
21. McCarthy S, Barrett M, Kirthi S, Pellanda P, Vlckova K, Tobin AM, et al. Altered skin and gut microbiome in hidradenitis suppurativa. J Invest Dermatol (2022) 142(2):459–68.e15. doi: 10.1016/j.jid.2021.05.036
22. Lam SY, Radjabzadeh D, Eppinga H, Nossent YRA, van der Zee HH, Kraaij R, et al. A microbiome study to explore the gut-skin axis in hidradenitis suppurativa. J Dermatol Sci (2021) 101(3):218–20. doi: 10.1016/j.jdermsci.2020.12.008
23. Lai Y, Di Nardo A, Nakatsuji T, Leichtle A, Yang Y, Cogen AL, et al. Commensal bacteria regulate toll-like receptor 3-dependent inflammation after skin injury. Nat Med (2009) 15(12):1377–82. doi: 10.1038/nm.2062
24. Ring HC, Thorsen J, Saunte DM, Lilje B, Bay L, Riis PT, et al. The follicular skin microbiome in patients with hidradenitis suppurativa and healthy controls. JAMA Dermatol (2017) 153(9):897–905. doi: 10.1001/jamadermatol.2017.0904
25. Guet-Revillet H, Coignard-Biehler H, Jais JP, Quesne G, Frapy E, Poirée S, et al. Bacterial pathogens associated with hidradenitis suppurativa, France. Emerg Infect Dis (2014) 20(12):1990–8. doi: 10.3201/eid2012.140064
26. Riverain-Gillet É, Guet-Revillet H, Jais JP, Ungeheuer MN, Duchatelet S, Delage M, et al. The surface microbiome of clinically unaffected skinfolds in hidradenitis suppurativa: A cross-sectional culture-based and 16S rRNA gene amplicon sequencing study in 60 patients. J Invest Dermatol (2020) 140(9):1847–55.e6. doi: 10.1016/j.jid.2020.02.046
27. Ring HC, Sigsgaard V, Thorsen J, Fuursted K, Fabricius S, Saunte DM, et al. The microbiome of tunnels in hidradenitis suppurativa patients. J Eur Acad Dermatol Venereol (2019) 33(9):1775–80. doi: 10.1111/jdv.15597
28. Guet-Revillet H, Jais JP, Ungeheuer MN, Coignard-Biehler H, Duchatelet S, Delage M, et al. The microbiological landscape of anaerobic infections in hidradenitis suppurativa: A prospective metagenomic study. Clin Infect Dis (2017) 65(2):282–91. doi: 10.1093/cid/cix285
29. Ring HC, Bay L, Nilsson M, Kallenbach K, Miller IM, Saunte DM, et al. Bacterial biofilm in chronic lesions of hidradenitis suppurativa. Br J Dermatol (2017) 176(4):993–1000. doi: 10.1111/bjd.15007
30. Ring HC, Bay L, Kallenbach K, Miller IM, Prens E, Saunte DM, et al. Normal skin microbiota is altered in pre-clinical hidradenitis suppurativa. Acta Derm Venereol (2017) 97(2):208–13. doi: 10.2340/00015555-2503
31. Kathju S, Lasko LA, Stoodley P. Considering hidradenitis suppurativa as a bacterial biofilm disease. FEMS Immunol Med Microbiol (2012) 65(2):385–9. doi: 10.1111/j.1574-695X.2012.00946.x
32. Lima BP, Hu LI, Vreeman GW, Weibel DB, Lux R. The oral bacterium fusobacterium nucleatum binds staphylococcus aureus and alters expression of the staphylococcal accessory regulator sarA. Microb Ecol (2019) 78(2):336–47. doi: 10.1007/s00248-018-1291-0
33. Chen V, Burgess JL, Verpile R, Tomic-Canic M, Pastar I. Novel diagnostic technologies and therapeutic approaches targeting chronic wound biofilms and microbiota. Curr Dermatol Rep (2022) 11(2):60–72. doi: 10.1007/s13671-022-00354-9
34. Schilcher K, Horswill AR. Staphylococcal biofilm development: Structure, regulation, and treatment strategies. Microbiol Mol Biol Rev (2020) 84(3):e00026-19. doi: 10.1128/MMBR.00026-19
35. Joo HS, Otto M. Mechanisms of resistance to antimicrobial peptides in staphylococci. Biochim Biophys Acta (2015) 1848(11 Pt B):3055–61. doi: 10.1016/j.bbamem.2015.02.009
36. Otto M. Bacterial evasion of antimicrobial peptides by biofilm formation. Curr Top Microbiol Immunol (2006) 306:251–8. doi: 10.1007/3-540-29916-5_10
37. Coates M, Blanchard S, MacLeod AS. Innate antimicrobial immunity in the skin: A protective barrier against bacteria, viruses, and fungi. PloS Pathog (2018) 14(12):e1007353. doi: 10.1371/journal.ppat.1007353
38. Frohm M, Agerberth B, Ahangari G, Stâhle-Bäckdahl M, Lidén S, Wigzell H, et al. The expression of the gene coding for the antibacterial peptide LL-37 is induced in human keratinocytes during inflammatory disorders. J Biol Chem (1997) 272(24):15258–63. doi: 10.1074/jbc.272.24.15258
39. Lowry MB, Guo C, Zhang Y, Fantacone ML, Logan IE, Campbell Y, et al. A mouse model for vitamin d-induced human cathelicidin antimicrobial peptide gene expression. J Steroid Biochem Mol Biol (2020) 198:105552. doi: 10.1016/j.jsbmb.2019.105552
40. White JH. Vitamin d as an inducer of cathelicidin antimicrobial peptide expression: past, present and future. J Steroid Biochem Mol Biol (2010) 121(1-2):234–8. doi: 10.1016/j.jsbmb.2010.03.034
41. Morizane S, Yamasaki K, Kabigting FD, Gallo RL. Kallikrein expression and cathelicidin processing are independently controlled in keratinocytes by calcium, vitamin D(3), and retinoic acid. J Invest Dermatol (2010) 130(5):1297–306. doi: 10.1038/jid.2009.435
42. Emelianov VU, Bechara FG, Gläser R, Langan EA, Taungjaruwinai WM, Schröder JM, et al. Immunohistological pointers to a possible role for excessive cathelicidin (LL-37) expression by apocrine sweat glands in the pathogenesis of hidradenitis suppurativa/acne inversa. Br J Dermatol (2012) 166(5):1023–34. doi: 10.1111/j.1365-2133.2011.10765.x
43. Thomi R, Schlapbach C, Yawalkar N, Simon D, Yerly D, Hunger RE. Elevated levels of the antimicrobial peptide LL-37 in hidradenitis suppurativa are associated with a Th1/Th17 immune response. Exp Dermatol (2018) 27(2):172–7. doi: 10.1111/exd.13482
44. Bechara FG, Sand M, Skrygan M, Kreuter A, Altmeyer P, Gambichler T. Acne inversa: evaluating antimicrobial peptides and proteins. Ann Dermatol (2012) 24(4):393–7. doi: 10.5021/ad.2012.24.4.393
45. Lande R, Gregorio J, Facchinetti V, Chatterjee B, Wang YH, Homey B, et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature (2007) 449(7162):564–9. doi: 10.1038/nature06116
46. Hotz C, Boniotto M, Guguin A, Surenaud M, Jean-Louis F, Tisserand P, et al. Intrinsic defect in keratinocyte function leads to inflammation in hidradenitis suppurativa. J Invest Dermatol (2016) 136(9):1768–80. doi: 10.1016/j.jid.2016.04.036
47. Lehrer RI, Lichtenstein AK, Ganz T. Defensins: antimicrobial and cytotoxic peptides of mammalian cells. Annu Rev Immunol (1993) 11:105–28. doi: 10.1146/annurev.iy.11.040193.000541
48. Hancock RE, Lehrer R. Cationic peptides: a new source of antibiotics. Trends Biotechnol (1998) 16(2):82–8. doi: 10.1016/S0167-7799(97)01156-6
49. Harder J, Bartels J, Christophers E, Schroder JM. Isolation and characterization of human beta -defensin-3, a novel human inducible peptide antibiotic. J Biol Chem (2001) 276(8):5707–13. doi: 10.1074/jbc.M008557200
50. Wang TT, Nestel FP, Bourdeau V, Nagai Y, Wang Q, Liao J, et al. Cutting edge: 1,25-dihydroxyvitamin D3 is a direct inducer of antimicrobial peptide gene expression. J Immunol (2004) 173(5):2909–12. doi: 10.4049/jimmunol.173.5.2909
51. Schlapbach C, Yawalkar N, Hunger RE. Human beta-defensin-2 and psoriasin are overexpressed in lesions of acne inversa. J Am Acad Dermatol (2009) 61(1):58–65. doi: 10.1016/j.jaad.2008.12.033
52. Jenei A, Dajnoki Z, Medgyesi B, Gáspár K, Béke G, Kinyó Á, et al. Apocrine gland-rich skin has a non-inflammatory IL-17-Related immune milieu, that turns to inflammatory IL-17-Mediated disease in hidradenitis suppurativa. J Invest Dermatol (2019) 139(4):964–8. doi: 10.1016/j.jid.2018.10.020
53. Harder J, Bartels J, Christophers E, Schröder JM. A peptide antibiotic from human skin. Nature (1997) 387(6636):861. doi: 10.1038/43088
54. Wolk K, Warszawska K, Hoeflich C, Witte E, Schneider-Burrus S, Witte K, et al. Deficiency of IL-22 contributes to a chronic inflammatory disease: pathogenetic mechanisms in acne inversa. J Immunol (2011) 186(2):1228–39. doi: 10.4049/jimmunol.0903907
55. Ardon CB, Wang C, Prens EP, van Straalen KR. Noninvasive assessment of cytokine and antimicrobial peptide levels in hidradenitis suppurativa using transdermal analysis patches. Br J Dermatol (2021) 184(2):343–5. doi: 10.1111/bjd.19462
56. Supp DM, Karpinski AC, Boyce ST. Expression of human beta-defensins HBD-1, HBD-2, and HBD-3 in cultured keratinocytes and skin substitutes. Burns (2004) 30(7):643–8. doi: 10.1016/j.burns.2004.03.012
57. Braff MH, Gallo RL. Antimicrobial peptides: an essential component of the skin defensive barrier. Curr Top Microbiol Immunol (2006) 306:91–110. doi: 10.1007/3-540-29916-5_4
58. Befus AD, Mowat C, Gilchrist M, Hu J, Solomon S, Bateman A. Neutrophil defensins induce histamine secretion from mast cells: mechanisms of action. J Immunol (1999) 163(2):947–53.
59. Hofmann SC, Saborowski V, Lange S, Kern WV, Bruckner-Tuderman L, Rieg S. Expression of innate defense antimicrobial peptides in hidradenitis suppurativa. J Am Acad Dermatol (2012) 66(6):966–74. doi: 10.1016/j.jaad.2011.07.020
60. Coates M, Mariottoni P, Corcoran DL, Kirshner HF, Jaleel T, Brown DA, et al. The skin transcriptome in hidradenitis suppurativa uncovers an antimicrobial and sweat gland gene signature which has distinct overlap with wounded skin. PloS One (2019) 14(5):e0216249. doi: 10.1371/journal.pone.0216249
61. Giamarellos-Bourboulis EJ, Platzer M, Karagiannidis I, Kanni T, Nikolakis G, Ulrich J, et al. High copy numbers of β-defensin cluster on 8p23.1, confer genetic susceptibility, and modulate the physical course of hidradenitis Suppurativa/Acne inversa. J Invest Dermatol (2016) 136(8):1592–8. doi: 10.1016/j.jid.2016.04.021
62. Dréno B, Khammari A, Brocard A, Moyse D, Blouin E, Guillet G, et al. Hidradenitis suppurativa: the role of deficient cutaneous innate immunity. Arch Dermatol (2012) 148(2):182–6. doi: 10.1001/archdermatol.2011.315
63. Mozeika E, Pilmane M, Nürnberg BM, Jemec GB. Tumour necrosis factor-alpha and matrix metalloproteinase-2 are expressed strongly in hidradenitis suppurativa. Acta Derm Venereol (2013) 93(3):301–4. doi: 10.2340/00015555-1492
64. Rieg S, Seeber S, Steffen H, Humeny A, Kalbacher H, Stevanovic S, et al. Generation of multiple stable dermcidin-derived antimicrobial peptides in sweat of different body sites. J Invest Dermatol (2006) 126(2):354–65. doi: 10.1038/sj.jid.5700041
65. Senyürek I, Paulmann M, Sinnberg T, Kalbacher H, Deeg M, Gutsmann T, et al. Dermcidin-derived peptides show a different mode of action than the cathelicidin LL-37 against staphylococcus aureus. Antimicrob Agents Chemother (2009) 53(6):2499–509. doi: 10.1128/AAC.01679-08
66. Steffen H, Rieg S, Wiedemann I, Kalbacher H, Deeg M, Sahl HG, et al. Naturally processed dermcidin-derived peptides do not permeabilize bacterial membranes and kill microorganisms irrespective of their charge. Antimicrob Agents Chemother (2006) 50(8):2608–20. doi: 10.1128/AAC.00181-06
67. Rieg S, Garbe C, Sauer B, Kalbacher H, Schittek B. Dermcidin is constitutively produced by eccrine sweat glands and is not induced in epidermal cells under inflammatory skin conditions. Br J Dermatol (2004) 151(3):534–9. doi: 10.1111/j.1365-2133.2004.06081.x
68. Shanmugam VK, Jones D, McNish S, Bendall ML, Crandall KA. Transcriptome patterns in hidradenitis suppurativa: support for the role of antimicrobial peptides and interferon pathways in disease pathogenesis. Clin Exp Dermatol (2019) 44(8):882–92. doi: 10.1111/ced.13959
69. Lima AL, Karl I, Giner T, Poppe H, Schmidt M, Presser D, et al. Keratinocytes and neutrophils are important sources of proinflammatory molecules in hidradenitis suppurativa. Br J Dermatol (2016) 174(3):514–21. doi: 10.1111/bjd.14214
70. Gläser R, Harder J, Lange H, Bartels J, Christophers E, Schröder JM. Antimicrobial psoriasin (S100A7) protects human skin from escherichia coli infection. Nat Immunol (2005) 6(1):57–64. doi: 10.1038/ni1142
71. Batycka-Baran A, Baran W, Nowicka-Suszko D, Koziol-Gałczyńska M, Bieniek A, Matusiak Ł, et al. Serum concentration and skin expression of S100A7 (Psoriasin) in patients suffering from hidradenitis suppurativa. Dermatology (2021) 237(5):733–9. doi: 10.1159/000510689
72. Eşer E, Engin B, Yüksel P, Kocazeybek BS, Kutlubay Z, Serdaroğlu S, et al. Relationship between fecal calprotectin level and disease activity in patients with hidradenitis suppurativa. Dermatol Ther (2020) 33(2):e13232. doi: 10.1111/dth.13232
73. Wieland CW, Vogl T, Ordelman A, Vloedgraven HG, Verwoolde LH, Rensen JM, et al. Myeloid marker S100A8/A9 and lymphocyte marker, soluble interleukin 2 receptor: biomarkers of hidradenitis suppurativa disease activity? Br J Dermatol (2013) 168(6):1252–8. doi: 10.1111/bjd.12234
74. Lusitani D, Malawista SE, Montgomery RR. Calprotectin, an abundant cytosolic protein from human polymorphonuclear leukocytes, inhibits the growth of borrelia burgdorferi. Infect Immun (2003) 71(8):4711–6. doi: 10.1128/IAI.71.8.4711-4716.2003
75. Büchau AS, Hassan M, Kukova G, Lewerenz V, Kellermann S, Würthner JU, et al. S100A15, an antimicrobial protein of the skin: regulation by e. coli through Toll-like receptor 4 J Invest Dermatol (2007) 127(11):2596–604. doi: 10.1038/sj.jid.5700946
76. Benoit S, Toksoy A, Ahlmann M, Schmidt M, Sunderkötter C, Foell D, et al. Elevated serum levels of calcium-binding S100 proteins A8 and A9 reflect disease activity and abnormal differentiation of keratinocytes in psoriasis. Br J Dermatol (2006) 155(1):62–6. doi: 10.1111/j.1365-2133.2006.07198.x
77. Harder J, Schroder JM. RNase 7, a novel innate immune defense antimicrobial protein of healthy human skin. J Biol Chem (2002) 277(48):46779–84. doi: 10.1074/jbc.M207587200
78. Murakami M, Lopez-Garcia B, Braff M, Dorschner RA, Gallo RL. Postsecretory processing generates multiple cathelicidins for enhanced topical antimicrobial defense. J Immunol (2004) 172(5):3070–7. doi: 10.4049/jimmunol.172.5.3070
79. Heilborn JD, Nilsson MF, Kratz G, Weber G, Sørensen O, Borregaard N, et al. The cathelicidin anti-microbial peptide LL-37 is involved in re-epithelialization of human skin wounds and is lacking in chronic ulcer epithelium. J Invest Dermatol (2003) 120(3):379–89. doi: 10.1046/j.1523-1747.2003.12069.x
80. Yamasaki K, Di Nardo A, Bardan A, Murakami M, Ohtake T, Coda A, et al. Increased serine protease activity and cathelicidin promotes skin inflammation in rosacea. Nat Med (2007) 13(8):975–80. doi: 10.1038/nm1616
81. Hansson L, Bäckman A, Ny A, Edlund M, Ekholm E, Ekstrand Hammarström B, et al. Epidermal overexpression of stratum corneum chymotryptic enzyme in mice: a model for chronic itchy dermatitis. J Invest Dermatol (2002) 118(3):444–9. doi: 10.1046/j.0022-202x.2001.01684.x
82. Karagiannidis I, Nikolakis G, Sabat R, Zouboulis CC. Hidradenitis suppurativa/Acne inversa: an endocrine skin disorder? Rev Endocr Metab Disord (2016) 17(3):335–41. doi: 10.1007/s11154-016-9366-z
83. Kelly G, Sweeney CM, Fitzgerald R, O'Keane MP, Kilbane M, Lally A, et al. Vitamin d status in hidradenitis suppurativa. Br J Dermatol (2014) 170(6):1379–80. doi: 10.1111/bjd.12900
84. Yamasaki K, Schauber J, Coda A, Lin H, Dorschner RA, Schechter NM, et al. Kallikrein-mediated proteolysis regulates the antimicrobial effects of cathelicidins in skin. FASEB J (2006) 20(12):2068–80. doi: 10.1096/fj.06-6075com
85. Jang H, Matsuda A, Jung K, Karasawa K, Matsuda K, Oida K, et al. Skin pH is the master switch of kallikrein 5-mediated skin barrier destruction in a murine atopic dermatitis model. J Invest Dermatol (2016) 136(1):127–35. doi: 10.1038/JID.2015.363
86. Poblete MT, Reynolds NJ, Figueroa CD, Burton JL, Muller-Esterl W, Bhoola KD. Tissue kallikrein and kininogen in human sweat glands and psoriatic skin. Br J Dermatol (1991) 124(3):236–41. doi: 10.1111/j.1365-2133.1991.tb00567.x
87. Lande R, Botti E, Jandus C, Dojcinovic D, Fanelli G, Conrad C, et al. The antimicrobial peptide LL37 is a T-cell autoantigen in psoriasis. Nat Commun (2014) 5:5621. doi: 10.1038/ncomms6621
88. Kelly G, Hughes R, McGarry T, van den Born M, Adamzik K, Fitzgerald R, et al. Dysregulated cytokine expression in lesional and nonlesional skin in hidradenitis suppurativa. Br J Dermatol (2015) 173(6):1431–9. doi: 10.1111/bjd.14075
89. Banerjee A, McNish S, Shanmugam VK. Interferon-gamma (IFN-γ) is elevated in wound exudate from hidradenitis suppurativa. Immunol Invest (2017) 46(2):149–58. doi: 10.1080/08820139.2016.1230867
90. van der Zee HH, de Ruiter L, van den Broecke DG, Dik WA, Laman JD, Prens EP. Elevated levels of tumour necrosis factor (TNF)-α, interleukin (IL)-1β and IL-10 in hidradenitis suppurativa skin: a rationale for targeting TNF-α and IL-1β. Br J Dermatol (2011) 164(6):1292–8. doi: 10.1111/j.1365-2133.2011.10254.x
91. Chamorro CI, Weber G, Grönberg A, Pivarcsi A, Ståhle M. The human antimicrobial peptide LL-37 suppresses apoptosis in keratinocytes. J Invest Dermatol (2009) 129(4):937–44. doi: 10.1038/jid.2008.321
92. Navrazhina K, Garcet S, Gonzalez J, Grand D, Frew JW, Krueger JG. In-depth analysis of the hidradenitis suppurativa serum proteome identifies distinct inflammatory subtypes. J Invest Dermatol (2021) 141(9):2197–207. doi: 10.1016/j.jid.2021.02.742
93. Wolk K, Wenzel J, Tsaousi A, Witte-Händel E, Babel N, Zelenak C, et al. Lipocalin-2 is expressed by activated granulocytes and keratinocytes in affected skin and reflects disease activity in acne inversa/hidradenitis suppurativa. Br J Dermatol (2017) 177(5):1385–93. doi: 10.1111/bjd.15424
94. Ledger EVK, Mesnage S, Edwards AM. Human serum triggers antibiotic tolerance in staphylococcus aureus. Nat Commun (2022) 13(1):2041. doi: 10.1038/s41467-022-29717-3
95. Friberg C, Haaber JK, Vestergaard M, Fait A, Perrot V, Levin BR, et al. Human antimicrobial peptide, LL-37, induces non-inheritable reduced susceptibility to vancomycin in staphylococcus aureus. Sci Rep (2020) 10(1):13121. doi: 10.1038/s41598-020-69962-4
96. Matusiak Ł, Bieniek A, Szepietowski JC. Bacteriology of hidradenitis suppurativa - which antibiotics are the treatment of choice? Acta Derm Venereol (2014) 94(6):699–702. doi: 10.2340/00015555-1841
97. Lapins J, Jarstrand C, Emtestam L. Coagulase-negative staphylococci are the most common bacteria found in cultures from the deep portions of hidradenitis suppurativa lesions, as obtained by carbon dioxide laser surgery. Br J Dermatol (1999) 140(1):90–5. doi: 10.1046/j.1365-2133.1999.02613.x
98. Raziyeva K, Kim Y, Zharkinbekov Z, Kassymbek K, Jimi S, Saparov A. Immunology of acute and chronic wound healing. Biomolecules (2021) 11(5):699–702. doi: 10.3390/biom11050700
99. Rieg S, Steffen H, Seeber S, Humeny A, Kalbacher H, Dietz K, et al. Deficiency of dermcidin-derived antimicrobial peptides in sweat of patients with atopic dermatitis correlates with an impaired innate defense of human skin in vivo. J Immunol (2005) 174(12):8003–10. doi: 10.4049/jimmunol.174.12.8003
100. Baechle D, Flad T, Cansier A, Steffen H, Schittek B, Tolson J, et al. Cathepsin d is present in human eccrine sweat and involved in the postsecretory processing of the antimicrobial peptide DCD-1L. J Biol Chem (2006) 281(9):5406–15. doi: 10.1074/jbc.M504670200
101. Watson SP, Ruggiero M, Abrahams SL, Lapetina EG. Inositol 1,4,5-trisphosphate induces aggregation and release of 5-hydroxytryptamine from saponin-permeabilized human platelets. J Biol Chem (1986) 261(12):5368–72. doi: 10.1016/S0021-9258(19)57224-6
102. Niyonsaba F, Suzuki A, Ushio H, Nagaoka I, Ogawa H, Okumura K. The human antimicrobial peptide dermcidin activates normal human keratinocytes. Br J Dermatol (2009) 160(2):243–9. doi: 10.1111/j.1365-2133.2008.08925.x
103. Donato R, Cannon BR, Sorci G, Riuzzi F, Hsu K, Weber DJ, et al. Functions of S100 proteins. Curr Mol Med (2013) 13(1):24–57. doi: 10.2174/156652413804486214
104. Gonzalez LL, Garrie K, Turner MD. Role of S100 proteins in health and disease. Biochim Biophys Acta Mol Cell Res (2020) 1867(6):118677. doi: 10.1016/j.bbamcr.2020.118677
105. Mischke D, Korge BP, Marenholz I, Volz A, Ziegler A. Genes encoding structural proteins of epidermal cornification and S100 calcium-binding proteins form a gene complex ("epidermal differentiation complex") on human chromosome 1q21. J Invest Dermatol (1996) 106(5):989–92. doi: 10.1111/1523-1747.ep12338501
106. Ma JY, Shao S, Wang G. Antimicrobial peptides: bridging innate and adaptive immunity in the pathogenesis of psoriasis. Chin Med J (Engl) (2020) 133(24):2966–75. doi: 10.1097/CM9.0000000000001240
107. Niyonsaba F, Kiatsurayanon C, Chieosilapatham P, Ogawa H. Friends or foes? host defense (antimicrobial) peptides and proteins in human skin diseases. Exp Dermatol (2017) 26(11):989–98. doi: 10.1111/exd.13314
108. Niyonsaba F, Nagaoka I, Ogawa H. Human defensins and cathelicidins in the skin: beyond direct antimicrobial properties. Crit Rev Immunol (2006) 26(6):545–76. doi: 10.1615/CritRevImmunol.v26.i6.60
109. Batycka-Baran A, Koziol-Galczynska M, Bieniek A, Wolf R, Łaczmański Ł, Szepietowski JC. Expression of koebnerisin (S100A15) and calgranulin a (S100A8) in lesional and perilesional skin in patients suffering from hidradenitis suppurativa. J Eur Acad Dermatol Venereol (2020) 34(8):e402–e4. doi: 10.1111/jdv.16320
110. Guilloteau K, Paris I, Pedretti N, Boniface K, Juchaux F, Huguier V, et al. IL-1{alpha}, and TNF-{alpha} recapitulates some features of psoriasis. J Immunol (2010) 184(9):5263–70. doi: 10.4049/jimmunol.0902464
111. Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med (2006) 203(10):2271–9. doi: 10.1084/jem.20061308
112. Zackular JP, Chazin WJ, Skaar EP. Nutritional immunity: S100 proteins at the host-pathogen interface. J Biol Chem (2015) 290(31):18991–8. doi: 10.1074/jbc.R115.645085
113. Rademacher F, Dreyer S, Kopfnagel V, Gläser R, Werfel T, Harder J. The antimicrobial and immunomodulatory function of RNase 7 in skin. Front Immunol (2019) 10:2553. doi: 10.3389/fimmu.2019.02553
114. Rademacher F, Simanski M, Harder J. RNase 7 in cutaneous defense. Int J Mol Sci (2016) 17(4):560. doi: 10.3390/ijms17040560
115. Wittersheim M, Cordes J, Meyer-Hoffert U, Harder J, Hedderich J, Gläser R. Differential expression and in vivo secretion of the antimicrobial peptides psoriasin (S100A7), RNase 7, human beta-defensin-2 and -3 in healthy human skin. Exp Dermatol (2013) 22(5):364–6. doi: 10.1111/exd.12133
116. Köten B, Simanski M, Gläser R, Podschun R, Schröder JM, Harder J. RNase 7 contributes to the cutaneous defense against enterococcus faecium. PloS One (2009) 4(7):e6424. doi: 10.1371/journal.pone.0006424
117. Gambichler T, Hessam S, Skrygan M, Bakirtzi M, Kasakovski D, Bechara FG. NOD2 signalling in hidradenitis suppurativa. Clin Exp Dermatol (2021) 46(8):1488–94. doi: 10.1111/ced.14773
118. Kopfnagel V, Wagenknecht S, Harder J, Hofmann K, Kleine M, Buch A, et al. RNase 7 strongly promotes TLR9-mediated DNA sensing by human plasmacytoid dendritic cells. J Invest Dermatol (2018) 138(4):872–81. doi: 10.1016/j.jid.2017.09.052
119. Geller A, Yan J. The role of membrane bound complement regulatory proteins in tumor development and cancer immunotherapy. Front Immunol (2019) 10:1074. doi: 10.3389/fimmu.2019.01074
120. Lubbers R, van Essen MF, van Kooten C, Trouw LA. Production of complement components by cells of the immune system. Clin Exp Immunol (2017) 188(2):183–94. doi: 10.1111/cei.12952
121. Giang J, Seelen MAJ, van Doorn MBA, Rissmann R, Prens EP, Damman J. Complement activation in inflammatory skin diseases. Front Immunol (2018) 9:639. doi: 10.3389/fimmu.2018.00639
122. Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I - molecular mechanisms of activation and regulation. Front Immunol (2015) 6:262. doi: 10.3389/fimmu.2015.00262
123. Cortes C, Ohtola JA, Saggu G, Ferreira VP. Local release of properdin in the cellular microenvironment: role in pattern recognition and amplification of the alternative pathway of complement. Front Immunol (2012) 3:412. doi: 10.3389/fimmu.2012.00412
124. Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT. Complement system part II: Role in immunity. Front Immunol (2015) 6:257. doi: 10.3389/fimmu.2015.00257
125. Bubeck D. The making of a macromolecular machine: assembly of the membrane attack complex. Biochemistry (2014) 53(12):1908–15. doi: 10.1021/bi500157z
126. Thurman JM, Yapa R. Complement therapeutics in autoimmune disease. Front Immunol (2019) 10:672. doi: 10.3389/fimmu.2019.00672
127. Ghias MH, Hyde MJ, Tomalin LE, Morgan BP, Alavi A, Lowes MA, et al. Role of the complement pathway in inflammatory skin diseases: A focus on hidradenitis suppurativa. J Invest Dermatol (2020) 140(3):531–6.e1. doi: 10.1016/j.jid.2019.09.009
128. Chehoud C, Rafail S, Tyldsley AS, Seykora JT, Lambris JD, Grice EA. Complement modulates the cutaneous microbiome and inflammatory milieu. Proc Natl Acad Sci U S A (2013) 110(37):15061–6. doi: 10.1073/pnas.1307855110
129. Giacomassi C, Buang N, Ling GS, Crawford G, Cook HT, Scott D, et al. Complement C3 exacerbates imiquimod-induced skin inflammation and psoriasiform dermatitis. J Invest Dermatol (2017) 137(3):760–3. doi: 10.1016/j.jid.2016.11.011
130. Timár KK, Pasch MC, van den Bosch NH, Jarva H, Junnikkala S, Meri S, et al. Human keratinocytes produce the complement inhibitor factor h: synthesis is regulated by interferon-gamma. Mol Immunol (2006) 43(4):317–25. doi: 10.1016/j.molimm.2005.02.009
131. Timár KK, Junnikkala S, Dallos A, Jarva H, Bhuiyan ZA, Meri S, et al. Human keratinocytes produce the complement inhibitor factor I: Synthesis is regulated by interferon-gamma. Mol Immunol (2007) 44(11):2943–9. doi: 10.1016/j.molimm.2007.01.007
132. Kanni T, Zenker O, Habel M, Riedemann N, Giamarellos-Bourboulis EJ. Complement activation in hidradenitis suppurativa: a new pathway of pathogenesis? Br J Dermatol (2018) 179(2):413–9. doi: 10.1111/bjd.16428
133. Hoffman LK, Tomalin LE, Schultz G, Howell MD, Anandasabapathy N, Alavi A, et al. Integrating the skin and blood transcriptomes and serum proteome in hidradenitis suppurativa reveals complement dysregulation and a plasma cell signature. PloS One (2018) 13(9):e0203672. doi: 10.1371/journal.pone.0203672
134. Frew JW. Complement, hidradenitis suppurativa and pathogen-driven positive selection. Br J Dermatol (2019) 180(3):685–6. doi: 10.1111/bjd.17426
135. Prens LM, Ardon CB, van Straalen KR, van der Zee HH, Seelen MAJ, Laman JD, et al. No evident systemic terminal complement pathway activation in hidradenitis suppurativa. J Invest Dermatol (2021) 141(12):2966–9.e1. doi: 10.1016/j.jid.2021.03.037
136. Kanni T, Tzanetakou V, Savva A, Kersten B, Pistiki A, van de Veerdonk FL, et al. Compartmentalized cytokine responses in hidradenitis suppurativa. PloS One (2015) 10(6):e0130522. doi: 10.1371/journal.pone.0130522
137. Page MJ, Bester J, Pretorius E. The inflammatory effects of TNF-α and complement component 3 on coagulation. Sci Rep (2018) 8(1):1812. doi: 10.1038/s41598-018-20220-8
138. Andoh A, Fujiyama Y, Hata K, Araki Y, Takaya H, Shimada M, et al. Counter-regulatory effect of sodium butyrate on tumour necrosis factor-alpha (TNF-alpha)-induced complement C3 and factor b biosynthesis in human intestinal epithelial cells. Clin Exp Immunol (1999) 118(1):23–9. doi: 10.1046/j.1365-2249.1999.01038.x
139. Moutabarrik A, Nakanishi I, Namiki M, Hara T, Matsumoto M, Ishibashi M, et al. Cytokine-mediated regulation of the surface expression of complement regulatory proteins, CD46(MCP), CD55(DAF), and CD59 on human vascular endothelial cells. Lymphokine Cytokine Res (1993) 12(3):167–72.
140. Zipfel PF, Würzner R, Skerka C. Complement evasion of pathogens: common strategies are shared by diverse organisms. Mol Immunol (2007) 44(16):3850–7. doi: 10.1016/j.molimm.2007.06.149
141. Joiner K, Brown E, Hammer C, Warren K, Frank M. Studies on the mechanism of bacterial resistance to complement-mediated killing. III. C5b-9 deposits stably on rough and type 7 s. pneumoniae without causing bacterial killing. J Immunol (1983) 130(2):845–9.
142. Fernie-King BA, Seilly DJ, Willers C, Würzner R, Davies A, Lachmann PJ. Streptococcal inhibitor of complement (SIC) inhibits the membrane attack complex by preventing uptake of C567 onto cell membranes. Immunology (2001) 103(3):390–8. doi: 10.1046/j.1365-2567.2001.01249.x
143. Milis L, Morris CA, Sheehan MC, Charlesworth JA, Pussell BA. Vitronectin-mediated inhibition of complement: evidence for different binding sites for C5b-7 and C9. Clin Exp Immunol (1993) 92(1):114–9. doi: 10.1111/j.1365-2249.1993.tb05956.x
144. Amdahl H, Jongerius I, Meri T, Pasanen T, Hyvärinen S, Haapasalo K, et al. Staphylococcal ecb protein and host complement regulator factor h enhance functions of each other in bacterial immune evasion. J Immunol (2013) 191(4):1775–84. doi: 10.4049/jimmunol.1300638
145. Holmäng S, Hedelin H, Borghede G, Johansson SL. Long-term followup of a bladder carcinoma cohort: questionable value of radical radiotherapy. J Urol (1997) 157(5):1642–6.
146. Pastar I, Sawaya AP, Marjanovic J, Burgess JL, Strbo N, Rivas KE, et al. Intracellular staphylococcus aureus triggers pyroptosis and contributes to inhibition of healing due to perforin-2 suppression. J Clin Invest (2021) 131(24):e133727. doi: 10.1172/JCI133727
147. Strbo N, Pastar I, Romero L, Chen V, Vujanac M, Sawaya AP, et al. Single cell analyses reveal specific distribution of anti-bacterial molecule perforin-2 in human skin and its modulation by wounding and staphylococcus aureus infection. Exp Dermatol (2019) 28(3):225–32. doi: 10.1111/exd.13870
148. Tomic-Canic M, Burgess JL, O'Neill KE, Strbo N, Pastar I. Skin microbiota and its interplay with wound healing. Am J Clin Dermatol (2020) 21(Suppl 1):36–43. doi: 10.1007/s40257-020-00536-w
149. O'Neill K, Pastar I, Tomic-Canic M, Strbo N. Perforins expression by cutaneous gamma delta T cells. Front Immunol (2020) 11:1839. doi: 10.3389/fimmu.2020.01839
150. Ingram JR. The genetics of hidradenitis suppurativa. Dermatol Clin (2016) 34(1):23–8. doi: 10.1016/j.det.2015.07.002
151. Marzano AV, Genovese G, Moltrasio C, Tricarico PM, Gratton R, Piaserico S, et al. Whole-exome sequencing in 10 unrelated patients with syndromic hidradenitis suppurativa: A preliminary step for a genotype-phenotype correlation. Dermatology (2022), 1–10. doi: 10.1159/000521263
152. van Straalen KR, Prens EP, Willemsen G, Boomsma DI, van der Zee HH. Contribution of genetics to the susceptibility to hidradenitis suppurativa in a Large, cross-sectional Dutch twin cohort. JAMA Dermatol (2020) 156(12):1359–62. doi: 10.1001/jamadermatol.2020.3630
153. Li A, Peng Y, Taiclet LM, Tanzi RE. Analysis of hidradenitis suppurativa-linked mutations in four genes and the effects of PSEN1-P242LfsX11 on cytokine and chemokine expression in macrophages. Hum Mol Genet (2019) 28(7):1173–82. doi: 10.1093/hmg/ddy414
154. Frew JW, Vekic DA, Woods J, Cains GD. A systematic review and critical evaluation of reported pathogenic sequence variants in hidradenitis suppurativa. Br J Dermatol (2017) 177(4):987–98. doi: 10.1111/bjd.15441
155. Zhang Q, Wang C, Liu Z, Liu X, Han C, Cao X, et al. Notch signal suppresses toll-like receptor-triggered inflammatory responses in macrophages by inhibiting extracellular signal-regulated kinase 1/2-mediated nuclear factor κB activation. J Biol Chem (2012) 287(9):6208–17. doi: 10.1074/jbc.M111.310375
156. Moran B, Sweeney CM, Hughes R, Malara A, Kirthi S, Tobin AM, et al. Hidradenitis suppurativa is characterized by dysregulation of the Th17:Treg cell axis, which is corrected by anti-TNF therapy. J Invest Dermatol (2017) 137(11):2389–95. doi: 10.1016/j.jid.2017.05.033
157. Felices M, Ankarlo DE, Lenvik TR, Nelson HH, Blazar BR, Verneris MR, et al. Notch signaling at later stages of NK cell development enhances KIR expression and functional maturation. J Immunol (2014) 193(7):3344–54. doi: 10.4049/jimmunol.1400534
158. Giamarellos-Bourboulis EJ, Antonopoulou A, Petropoulou C, Mouktaroudi M, Spyridaki E, Baziaka F, et al. Altered innate and adaptive immune responses in patients with hidradenitis suppurativa. Br J Dermatol (2007) 156(1):51–6. doi: 10.1111/j.1365-2133.2006.07556.x
159. Weyand NJ, Calton CM, Higashi DL, Kanack KJ, So M. Presenilin/gamma-secretase cleaves CD46 in response to neisseria infection. J Immunol (2010) 184(2):694–701. doi: 10.4049/jimmunol.0900522
160. Yamamoto H, Fara AF, Dasgupta P, Kemper C. CD46: the 'multitasker' of complement proteins. Int J Biochem Cell Biol (2013) 45(12):2808–20. doi: 10.1016/j.biocel.2013.09.016
161. Hawkins ED, Oliaro J. CD46 signaling in T cells: linking pathogens with polarity. FEBS Lett (2010) 584(24):4838–44. doi: 10.1016/j.febslet.2010.09.003
162. Kemper C, Chan AC, Green JM, Brett KA, Murphy KM, Atkinson JP. Activation of human CD4+ cells with CD3 and CD46 induces a T-regulatory cell 1 phenotype. Nature (2003) 421(6921):388–92. doi: 10.1038/nature01315
163. Qiao P, Dang E, Cao T, Fang H, Zhang J, Qiao H, et al. Dysregulation of mCD46 and sCD46 contribute to the pathogenesis of bullous pemphigoid. Sci Rep (2017) 7(1):145. doi: 10.1038/s41598-017-00235-3
164. Jfri A, Litvinov IV, Netchiporouk E, O'Brien E. Novel variants of. SAGE. Open Med Case Rep (2020) 8:2050313X20953113. doi: 10.1177/2050313X20953113
165. Nassar D, Hugot JP, Wolkenstein P, Revuz J. Lack of association between CARD15 gene polymorphisms and hidradenitis suppurativa: a pilot study. Dermatology (2007) 215(4):359. doi: 10.1159/000107632
166. Kitts S, Govea R, Maczuga S, Kirby J. Long-term antibiotic use for the treatment of hidradenitis suppurativa consistent with guideline recommendations. Clin Exp Dermatol (2021) 46(3):582–3. doi: 10.1111/ced.14512
167. Kimball AB, Okun MM, Williams DA, Gottlieb AB, Papp KA, Zouboulis CC, et al. Two phase 3 trials of adalimumab for hidradenitis suppurativa. N Engl J Med (2016) 375(5):422–34. doi: 10.1056/NEJMoa1504370
168. Leslie KS, Tripathi SV, Nguyen TV, Pauli M, Rosenblum MD. An open-label study of anakinra for the treatment of moderate to severe hidradenitis suppurativa. J Am Acad Dermatol (2014) 70(2):243–51. doi: 10.1016/j.jaad.2013.09.044
169. Tzanetakou V, Kanni T, Giatrakou S, Katoulis A, Papadavid E, Netea MG, et al. Safety and efficacy of anakinra in severe hidradenitis suppurativa: A randomized clinical trial. JAMA Dermatol (2016) 152(1):52–9. doi: 10.1001/jamadermatol.2015.3903
170. Kanni T, Argyropoulou M, Spyridopoulos T, Pistiki A, Stecher M, Dinarello CA, et al. MABp1 targeting IL-1α for moderate to severe hidradenitis suppurativa not eligible for adalimumab: A randomized study. J Invest Dermatol (2018) 138(4):795–801. doi: 10.1016/j.jid.2017.10.030
171. Gottlieb A, Natsis NE, Kerdel F, Forman S, Gonzalez E, Jimenez G, et al. A phase II open-label study of bermekimab in patients with hidradenitis suppurativa shows resolution of inflammatory lesions and pain. J Invest Dermatol (2020) 140(8):1538–45.e2. doi: 10.1016/j.jid.2019.10.024
172. Prussick L, Rothstein B, Joshipura D, Saraiya A, Turkowski Y, Abdat R, et al. Open-label, investigator-initiated, single-site exploratory trial evaluating secukinumab, an anti-interleukin-17A monoclonal antibody, for patients with moderate-to-severe hidradenitis suppurativa. Br J Dermatol (2019) 181(3):609–11. doi: 10.1111/bjd.17822
173. Kearney N, Byrne N, Kirby B, Hughes R. Successful use of guselkumab in the treatment of severe hidradenitis suppurativa. Clin Exp Dermatol (2020) 45(5):618–9. doi: 10.1111/ced.14199
174. Bechara FG, Podda M, Prens EP, Horváth B, Giamarellos-Bourboulis EJ, Alavi A, et al. Efficacy and safety of adalimumab in conjunction with surgery in moderate to severe hidradenitis suppurativa: The SHARPS randomized clinical trial. JAMA Surg (2021) 156(11):1001–9. doi: 10.1001/jamasurg.2021.3655
175. Moloney S, McGrath BM, Roshan D, Gethin G. The personal impact of daily wound care for hidradenitis suppurativa. Dermatology (2021) 238(4):1–10. doi: 10.1159/000520262
176. GmbH i. studying complement inhibition in patients with moderate to severe hidradenitis suppurativa clinicaltrials (2017). Available at: https://www.clinicaltrials.gov/ct2/show/NCT03001622?id=NCT03001622&draw=2&rank=1.
177. GmbH i. efficacy and safety study of IFX-1 in patients with moderate to severe hidradenitis suppurativa (HS) (SHINE) clinicaltrials (2021). Available at: https://www.clinicaltrials.gov/ct2/show/NCT03487276?id=NCT03487276&draw=2&rank=1.
178. Lowe MM, Naik HB, Clancy S, Pauli M, Smith KM, Bi Y, et al. Immunopathogenesis of hidradenitis suppurativa and response to anti-TNF-α therapy. JCI Insight (2020) 5(19):e139932. doi: 10.1172/jci.insight.139932
179. Yilmaz B, Juillerat P, Øyås O, Ramon C, Bravo FD, Franc Y, et al. Microbial network disturbances in relapsing refractory crohn's disease. Nat Med (2019) 25(2):323–36. doi: 10.1038/s41591-018-0308-z
180. Nakamura K, O'Neill AM, Williams MR, Cau L, Nakatsuji T, Horswill AR, et al. Short chain fatty acids produced by cutibacterium acnes inhibit biofilm formation by staphylococcus epidermidis. Sci Rep (2020) 10(1):21237. doi: 10.1038/s41598-020-77790-9
181. Park T, Im J, Kim AR, Lee D, Jeong S, Yun CH, et al. Short-chain fatty acids inhibit the biofilm formation of streptococcus gordonii through negative regulation of competence-stimulating peptide signaling pathway. J Microbiol (2021) 59(12):1142–9. doi: 10.1007/s12275-021-1576-8
182. Chen WT, Chi CC. Association of hidradenitis suppurativa with inflammatory bowel disease: A systematic review and meta-analysis. JAMA Dermatol (2019) 155(9):1022–7. doi: 10.1001/jamadermatol.2019.0891
183. Braunberger TL, Nartker NT, Nicholson CL, Nahhas AF, Parks-Miller A, Hanna Z, et al. Ertapenem - a potent treatment for clinical and quality of life improvement in patients with hidradenitis suppurativa. Int J Dermatol (2018) 57(9):1088–93. doi: 10.1111/ijd.14036
184. van der Zee HH, Prens EP, Boer J. Deroofing: a tissue-saving surgical technique for the treatment of mild to moderate hidradenitis suppurativa lesions. J Am Acad Dermatol (2010) 63(3):475–80. doi: 10.1016/j.jaad.2009.12.018
185. Ravi S, Miles JA, Steele C, Christiansen MK, Sayed CJ. Patient impressions and outcomes after clinic-based hidradenitis suppurativa surgery. JAMA Dermatol (2022) 158(2):132–41. doi: 10.1001/jamadermatol.2021.4741
186. Weigelt MA, Lev-Tov H. Intralesional anti-biofilm therapy for tunnels in patients with hidradenitis suppurativa. Ital J Dermatol Venerol (2021) 156(5):618–9. doi: 10.23736/S2784-8671.21.06899-1
187. Bettoli V, Manfredini M, Massoli L, Carillo C, Barozzi A, Amendolagine G, et al. Rates of antibiotic resistance/sensitivity in bacterial cultures of hidradenitis suppurativa patients. J Eur Acad Dermatol Venereol (2019) 33(5):930–6. doi: 10.1111/jdv.15332
188. Fischer AH, Haskin A, Okoye GA. Patterns of antimicrobial resistance in lesions of hidradenitis suppurativa. J Am Acad Dermatol (2017) 76(2):309–13.e2. doi: 10.1016/j.jaad.2016.08.001
189. Holgersen EM, Gandhi S, Zhou Y, Kim J, Vaz B, Bogojeski J, et al. Transcriptome-wide off-target effects of steric-blocking oligonucleotides. Nucleic Acid Ther (2021) 31(6):392–403. doi: 10.1089/nat.2020.0921
190. Gudjonsson JE, Tsoi LC, Ma F, Billi AC, van Straalen KR, Vossen ARJV, et al. Contribution of plasma cells and b cells to hidradenitis suppurativa pathogenesis. JCI Insight (2020) 5(19):e139930. doi: 10.1172/jci.insight.139930
Keywords: hidradenitis suppurativa, acne inversa, innate immunity, skin – immunology, antimicrobial peptides (AMPs), complement - immunological terms, microbiome and dysbiosis, biofilm
Citation: Chopra D, Arens RA, Amornpairoj W, Lowes MA, Tomic-Canic M, Strbo N, Lev-Tov H and Pastar I (2022) Innate immunity and microbial dysbiosis in hidradenitis suppurativa – vicious cycle of chronic inflammation. Front. Immunol. 13:960488. doi: 10.3389/fimmu.2022.960488
Received: 03 June 2022; Accepted: 01 July 2022;
Published: 28 July 2022.
Edited by:
Paola Maura Tricarico, Institute for Maternal and Child Health Burlo Garofolo (IRCCS), ItalyReviewed by:
Alexandro Rodriguez-Rojas, University of Veterinary Medicine Vienna, AustriaPaula Borges Mariottoni, Duke University, United States
Copyright © 2022 Chopra, Arens, Amornpairoj, Lowes, Tomic-Canic, Strbo, Lev-Tov and Pastar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Irena Pastar, SXBhc3RhckBtZWQubWlhbWkuZWR1; Hadar Lev-Tov, aGxldnRvdkBtZWQubWlhbWkuZWR1
†These authors have contributed equally to this work