 Xuefeng Li
Xuefeng Li Xiaoli Wei3†
Xiaoli Wei3† Jinduan Lin
Jinduan Lin Li Ou
Li Ou- 1The Sixth Affiliated Hospital of Guangzhou Medical University, Qingyuan People’s Hospital; State Key Laboratory of Respiratory Disease, Sino-French Hoffmann Institute, School of Basic Medical Sciences, Guangzhou Medical University, Guangzhou, China
- 2Shenzhen Luohu People’s Hospital, The Third Affiliated Hospital of Shenzhen University, Shenzhen, China
- 3Guangzhou Dezheng Biotechnology Co., Ltd., Guangzhou, China
- 4Genemagic Biosciences, Philadelphia, PA, United States
- 5Department of Pediatrics, University of Minnesota, Minneapolis, MN, United States
Recombinant adeno-associated virus (AAV) is a promising delivery vehicle for in vivo gene therapy and has been widely used in >200 clinical trials globally. There are already several approved gene therapy products, e.g., Luxturna and Zolgensma, highlighting the remarkable potential of AAV delivery. In the past, AAV has been seen as a relatively non-immunogenic vector associated with low risk of toxicity. However, an increasing number of recent studies indicate that immune responses against AAV and transgene products could be the bottleneck of AAV gene therapy. In clinical studies, pre-existing antibodies against AAV capsids exclude many patients from receiving the treatment as there is high prevalence of antibodies among humans. Moreover, immune response could lead to loss of efficacy over time and severe toxicity, manifested as liver enzyme elevations, kidney injury, and thrombocytopenia, resulting in deaths of non-human primates and patients. Therefore, extensive efforts have been attempted to address these issues, including capsid engineering, plasmapheresis, IgG proteases, CpG depletion, empty capsid decoy, exosome encapsulation, capsid variant switch, induction of regulatory T cells, and immunosuppressants. This review will discuss these methods in detail and highlight important milestones along the way.
1 Introduction
Adeno-associated virus (AAV) is a replication-defective virus that infects vertebrates, including human and non-human primates (1). The genome of AAV is approximately 4.8 kb of single-stranded DNA flanked by inverted terminal repeats (ITRs), which are essential for its life cycle (2). AAV has been associated with no known diseases for long, however, a recent study suggested a potential involvement of AAV in a hepatitis outbreak in Scotland (3). Due to its low pathogenicity and risk of insertional mutagenesis, AAV has been developed as ‘biological nanoparticles’ for in vivo delivery of gene therapy (4). Starting from the late 90s, there have been gene therapy clinical trials using AAV delivery (AAV gene therapy for cystic fibrosis) (5). Until now, there have been over 100 clinical trials using wild-type and engineered AAV vectors (6). The first approved AAV gene therapy drug was Glybera for treating lipoprotein lipase deficiency using AAV1 vector (7), which was approved in Europe in 2012. Then, Luxturna, an AAV2-based gene therapy for retinal diseases (8), was approved in the USA in 2017, and Zolgensma, an AAV9-based gene therapy for spinal muscular atrophy in the USA in 2019 (9). The clinical and commercial success of AAV gene therapy encouraged a rapid clinical development. However, among the ever-increasing clinical development of AAV gene therapy, serious adverse events and toxicity, mostly due to immunity elicited by AAV delivery, were observed. Safety concerns culminated when a clinical trial (NCT03199469) for X-linked myotubular myopathy (XLMTM) reported 4 patient deaths, presumably due to hepatotoxicity (10). Further, incidence of hepatocellular carcinoma (11), elevated liver enzymes (12), MRI abnormalities (13), and dorsal root ganglia (DRG) degeneration (14) were reported from multiple clinical trials. It is of particular interest to highlight patient death cases associated with AAV delivery (summarized in Table 1). Due to the small sample size, it may be premature to draw clear conclusions. Yet, these cases more frequently occurred in CNS and muscular diseases and were associated with a high dose. In light of this, FDA organized a two-day Cellular, Tissue, and Gene Therapies Advisory Committee (CTGAC) meeting focused on addressing AAV toxicity issues in 2021. In addition to toxicity, AAV immunity may be also responsible for loss of efficacy over time due to promoter silencing or T cell-mediated elimination of transgene-expressing cells (22).
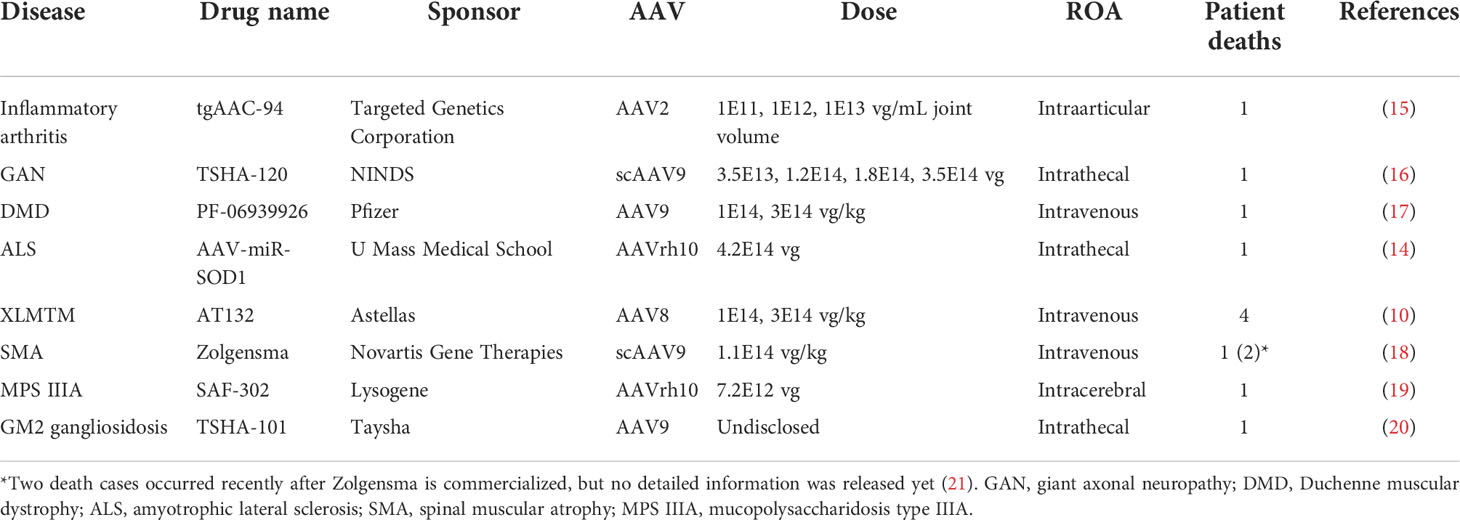
Table 1 Patient death cases that occurred in AAV trials.
2 Immune response against AAV
Immunity against AAV gene therapy is multidimensional, including innate, humoral, and cellular immune responses against capsids, genome sequences, and transgene products. Since the main focus of this review is on the toolkit to overcome AAV immunity, specific mechanisms of AAV immunity are only briefly discussed here as the context (illustrated in Figure 1). Action of innate immunity against AAV gene therapy was mediated through Toll-like receptor 9 (TLR9), which recognizes unmethylated CpG elements in AAV genome (23). The subsequent immunological cascades ultimately results in release of cytokines, inflammation and cytotoxic T cell (CTL) responses (24, 25). There is also evidence indicating the activation of the complement system, a key component of innate immunity, and renal injury in clinical studies (26, 27). Meanwhile, due to natural AAV infection, there is a high prevalence of pre-existing antibodies against AAV in humans (28). Pre-existing antibodies against capsids can significantly inhibit efficient AAV transduction in NHPs (29) and humans (12). Therefore, pre-existing antibodies prevent many patients from being eligible for potentially life-saving treatments. Moreover, humoral immunity against both capsids and transgene products elicited by AAV administration makes repeat dosing challenging (30). In addition, T cell responses may eliminate transgene-expressing cells, resulting in hepatotoxicity and loss of transgene expression in several clinical studies (12, 31–33). Fortunately, the immune response is not always against AAV-mediated gene transfer, evidenced by induction of regulatory T cells (Tregs) that suppress immune response and facilitate sustained transgene expression (34, 35).
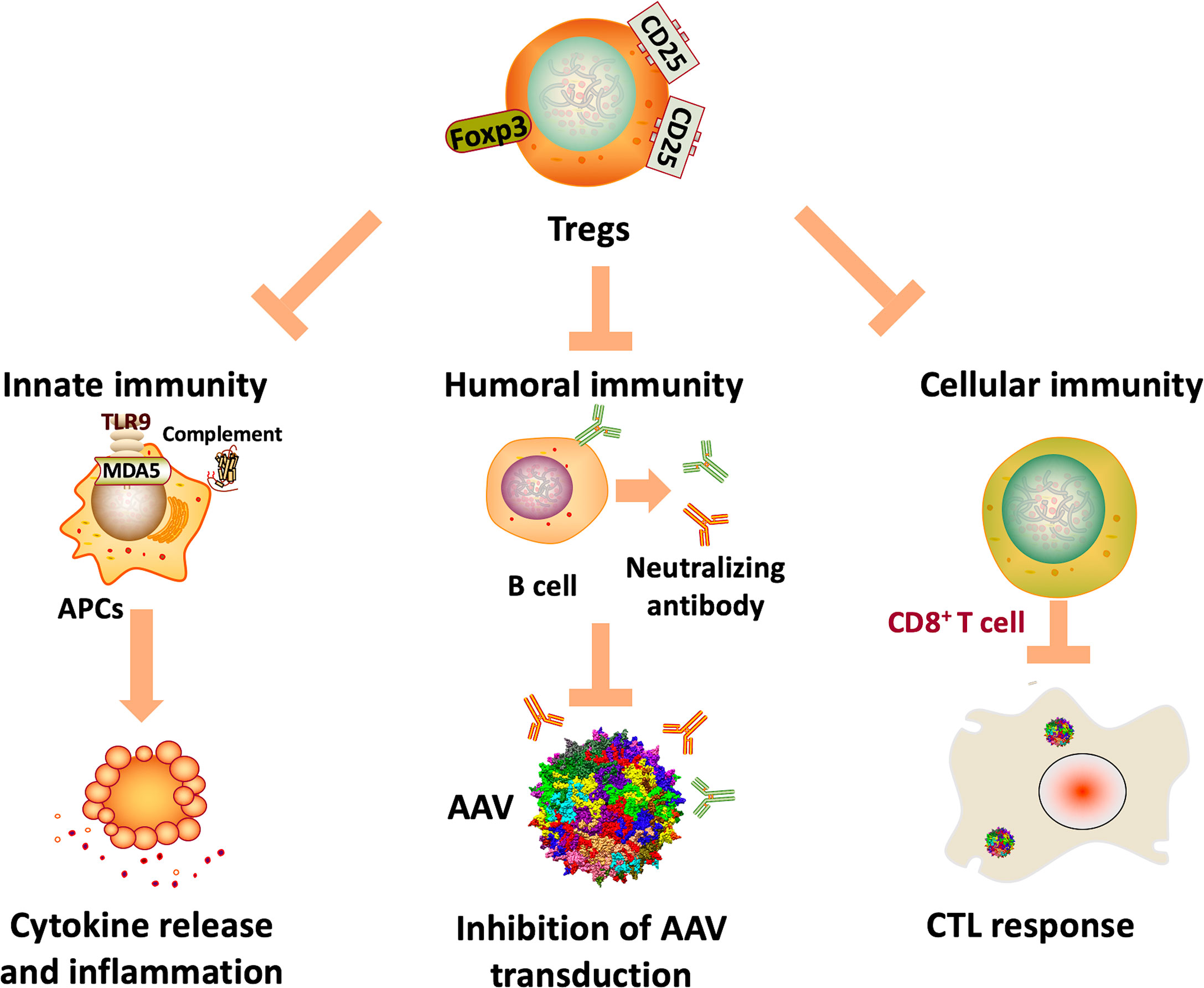
Figure 1 Potential mechanisms of AAV immunity. Innate immunity against AAV genome and transcripts can be activated by TLR9 and MDA5 recognition, resulting in cytokine release, inflammation, and ultimately CTL responses. In addition, the complement system may also be activated, which is associated with thrombotic microangiopathy and adverse events in clinical trials. Also, humoral immunity against capsids can significantly inhibit AAV transduction, limiting the application of AAV gene therapy in many patients. Further, cellular immunity against capsids and transgene products can eliminate transgene-expressing cells, ultimately resulting in loss of efficacy.
3 Toolkit to overcome AAV immunity
There have been extensive efforts to address AAV immunity. In this review, a versatile toolkit for overcoming AAV immunity is discussed in an order of starting from methods that have more clinical applications. Within each section, preclinical data is discussed before clinical data, if any.
3.1 Immunosuppressants
3.1.1 Corticosteroids
Corticosteroids (e.g., methylprednisolone, prednisolone, prednisone) are commonly used immunosuppressants for their global inhibitory effects on innate and adaptive immunity (36). Corticosteroids are most-widely used immunosuppressants for AAV gene therapy clinical studies (Table 2) and also used in approved gene therapy products, Luxturna (37) and Zolgensma (9). However, recent studies suggest that corticosteroids alone may not be sufficient. In the previously mentioned XLMTM trial (NCT03199469), there were 4 patient deaths, presumably resulting from hepatotoxicity (10). These patients receiving high dose IV administration AAV (1 to 3 x1014 vg/kg) were also on a 16-week prednisolone regimen. In another clinical study for ALS, one patient developed meningoradiculitis after intrathecal AAV delivery in spite of methylprednisolone and prednisone usage (14). In contrast, lower T cell responses and NAbs were observed in a second patient who was received a different immunosuppressant regimen that includes rituximab and sirolimus in addition to corticosteroids prior to AAV delivery, indicating the role of immunosuppressant selection and timing. The drawback of corticosteroids are side effects and non-specific immunosuppressing effects. In summary, due to the established safety profile and effects in preventing or alleviating immune responses, corticosteroids are the most frequently used immunosuppressants in AAV clinical studies.
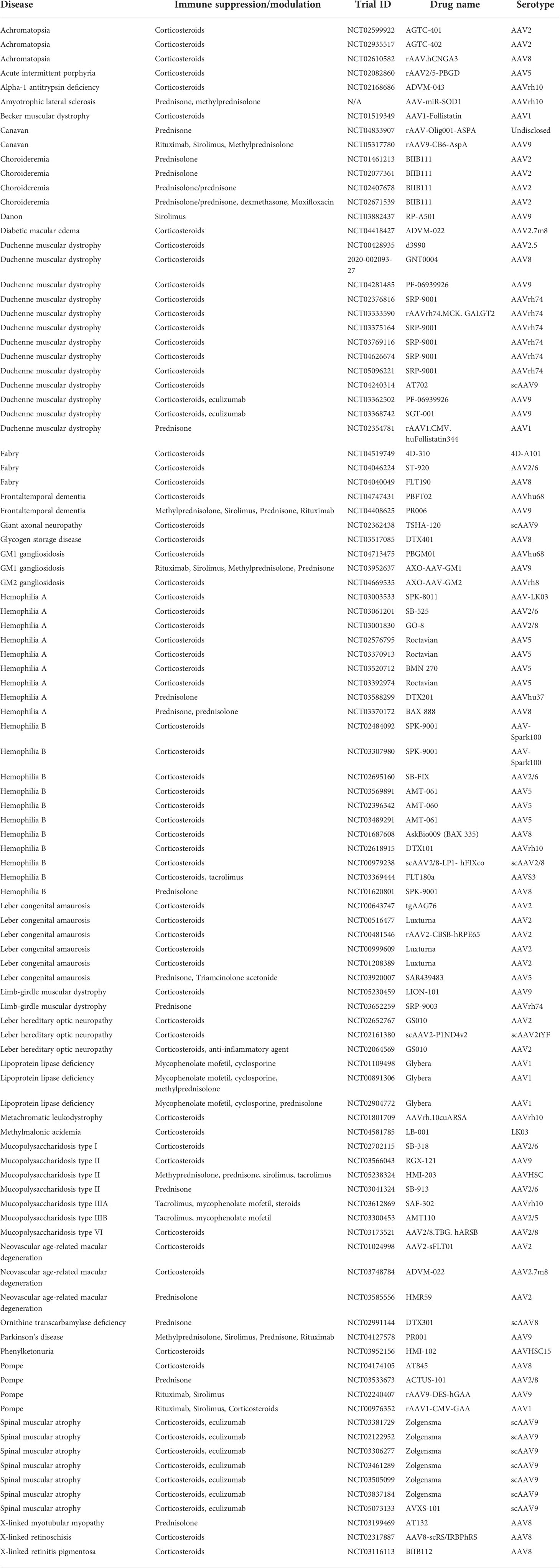
Table 2 AAV clinical studies using immunosuppressants.
3.1.2 Rapamycin
Rapamycin/sirolimus is a macrolide immunosuppressant that inhibits T and B cell activation and induce Tregs through targeting mTOR (38). As shown in one study (39), intraperitoneal administration of rapamycin and prednisolone reduced serum AAV9 NAbs by up to 93% and inhibited activation of B and T cells. Rapamycin/sirolimus was also frequently used in AAV clinical trials for immune suppression (e.g., NCT03882437, NCT02240407). Further, tolerogenic rapamycin nanoparticles, but not rapamycin in free form, have been shown to be able to inhibit both cellular and humoral immune response and induce Tregs in mice (40). A more recent study showed that tolerogenic rapamycin nanoparticles (SVP-rapamycin) prevented anti-capsid humoral and cellular responses, and more importantly, enabled AAV readministration in mice and NHPs (41). SVP-rapamycin also inhibited expansion of antigen-specific B cells. Notably, this effect was antigen-specific as coadministration of SVP-rapamycin (later known as ImmTOR) and AAV8 did not inhibit immunity against AAV5, which is preferable over other non-specific methods (e.g., IgG protease, plasmapheresis. ImmTOR has also been tested in clinical trials. A Phase Ib trial (NCT02648269) is testing whether ImmTOR could induce tolerance of pegadricase, a highly immunogenic enzyme derived from fungus, in hyperuricemia patients. In this study, ImmTOR has been shown to be able to inhibit anti-drug antibody formation in a dose-dependent manner (42). ImmTOR also showed a good safety profile as no treatment emergent serious adverse events (TESAEs) were observed.
3.1.3 Mycophenolate mofetil
Another commonly used immunosuppressant for AAV clinical studies is mycophenolate mofetil (MMF), a compound that inhibits type II inosine monophosphate dehydrogenase and thus suppresses proliferation of T and B cells (43). It was shown that although MMF reduced anti-AAV1 humoral immunity, it also inhibited transduction of ssAAV1 (but not scAAV1) by impairing second strand DNA synthesis in rats (44). This highlights the need to consider using MMF as immunosuppressants for clinical trials using ssAAV delivery. Notably, MMF also induced mild leukopenia and body weight loss. Interestingly, when tested in combination with tacrolimus in NHPs, MMF did not inhibit liver transduction of AAV8 (45). Further studies are needed to understand whether this transduction-inhibitory effects of MMF is species- or serotype-specific.
3.1.4 Complement inhibitors
Complement activation was recently found to be associated with adverse events in AAV clinical trials for DMD (26, 27) and SMA (46). Thus, there has been growing interest in applying complement inhibitors as immunosuppressants. Several FDA-approved complement pathway inhibitors could be potentially applied to inhibit AAV immunity and minimize risk of adverse events. For instance, C1 inhibitor effectively blocked AAV-induced complement activation in both human and mouse serum and whole blood samples. However, the complement blockade did not result in hampering the pro-inflammatory cytokine response towards AAV (47). Also, it had limited effect on anti-AAV antibodies. C5 inhibitor, eculizumab, has also been used in clinical trials to alleviate symptoms of complement activation, resulting in resolved toxicity in most patients (SGT-001, PF-06939926). In addition, Apellis Pharmaceuticals is applying C3 inhibitor, APL-9, to control the complement system during AAV delivery (48).
3.1.5 mAbs
Rituximab is an anti-CD20 mAb that depletes B cells by inducing apoptosis (49). The combined use of rituximab and T helper cell inhibitor ciclosporin depleted antibody against transgene products and B cells in NHPs that had previously received AAV8 administration (50). This immunosuppressant combination also enabled efficient transgene expression after AAV readministration. Rituximab was also used in combination with methotrexate (a cytotoxic drug with antiproliferative effects on both B and T cells) in human rheumatoid arthritis patients (51). However, NAb titer drop was only observed in patients with a titer of <1:1,000. Moreover, the reduction was partial because the NAb titer in most of those patients remained to be above 1:5. Other mAbs have been assessed for suppressing AAV immunity. In one study (52), the combined use of rituximab and rapamycin successfully induced immune tolerance of transgene expression from AAV delivery in hemophilia A mice. Also, only mice receiving both rituximab and rapamycin had no increased inhibitors following rechallenge with intravenous FVIII protein therapy. This strategy is being tested in a clinical trial for Pompe disease (NCT02240407) (53). Preliminary results from this trial showed that the combined use of rituximab and rapamycin prevented humoral immune response and enabled AAV readministration (54). The combination of rituximab, sirolimus, and corticosteroids was also used in a recent clinical trial for GM2-gangliosidosis through CNS-directed AAV delivery (55). No vector-related adverse events was observed. In addition, since CD40/CD40L has been shown be involved in activation of CTL response by AAV (56), blocking this co-stimulatory pathway can be utilized for suppressing immune response against AAV delivery. For instance, dapirolizumab pegol, antibody against CD40 that is being tested in a Phase III trial for systemic lupus erythematosus (NCT04294667), can be used for this purpose. Similarly, CTLA4-IgG (abatacept) can block CD28-mediated immune activation by AAV and thus reduce capsid-specific CTL responses (57). In this study, CTLA4-IgG also inhibited formation of NAbs and enabled a second administration of AAV8 in a murine model of hemophilia B.
3.1.6 Calcineurin inhibitors
Calcineurin inhibitors, including ciclosporin and tacrolimus, inhibit T cell proliferation and activation through targeting IL-2 (58). In one NHP study using intramuscular delivery of AAV8 or AAV9, tacrolimus significantly prolonged transgene expression from less than 8 weeks to beyond 42 weeks (59). However, one study showed that ciclosporin and tacrolimus inhibited Tregs proliferation and activation in vitro (60). Similarly, another study in human patients found that tacrolimus impaired Tregs proliferation, function, and phenotype, while rapamycin had beneficial effects on Tregs (61). In contrast, the combined use of tacrolimus and anti-CD4 antibody reduced antibodies against AAV capsids and transgene products and induced immune tolerance in mice (62). However, these results could not completely rule out the possibility of the inhibitory effects of tacrolimus on Tregs because tacrolimus was used in combination with another immunosuppressant in this study. Ciclosporin and MMF were used in combination in clinical studies of Glybera, which was the first approved gene therapy product in Europe (7, 63). In all patients, anti-AAV antibodies were not affected by immunosuppressants, and T cell response was observed in most patients (7). When the immunosuppression protocol changed the starting date and added methylprednisolone to the combination (63), humoral and cellular responses remained the same as the previous study. Therefore, in light of mixed results, the usage of calcineurin inhibitors as immunosuppressants for AAV gene therapy warrants further studies.
3.1.7 Other pharmacological agents in preclinical development
Ubiquitination of AAV leads to proteasome-mediated degradation, antigen presentation of capsid-derived peptides, and immune activation (64). Thus, there have been efforts to use proteasome inhibitors to reduce AAV immunogenicity. One study showed that an FDA-approved proteasome inhibitor bortezomib reduced antigen presentation in vitro and enhanced transgene expression from AAV2, but not AAV8, in mice (65). Two other studies using AAV8 showed that bortezomib improved transduction in mice (66) and dogs (67). Another proteasome inhibitor, carfilzomib, also enhanced AAV2 transduction in mice, however, to a lesser extent than bortezomib (68). Nevertheless, bortezomib failed to enhance AAV9 transduction in rats (69). The mixed results of bortezomib may result from differences in serotypes, species, AAV dose, and bortezomib dose. In addition, although there were no toxicity findings in these preclinical studies, bortezomib use in humans was associated with peripheral neuropathy, thrombocytopenia, and neutropenia (70). Therefore, further studies are needed for clinical translation of this approach. Agents associated with oxidizing and anti-oxidizing pathways can be also applied as immunosuppressants for AAV gene therapy. Arsenic trioxide (As (2)O (3)), one oxidizing agent and FDA-approved chemotherapeutic drug, has been shown to stabilize AAV by reducing proteasome-mediated degradation (71). As mentioned earlier, AAV degradation resulted in increased antigen presentation and immune activation. Intraperitoneal administration of arsenic trioxide enhanced transduction and transgene expression after intravenous AAV delivery in mice (71). In addition, a synthetic antioxidant MnTBAP inhibited CD4+ T cell mediated immunity in mice after intramuscular AAV delivery in mice (72). MnTBAP functioned through downregulating and inducing reverse internalization of CD4. MnTBAP treatment prevented anti-AAV and anti-transgene cellular immunity, enabling readministration of the same serotype.
Cyclophosphamide, a potent immunosuppressant that inhibits both T and B cell immunity, was used in preclinical AAV studies (73, 74). In a gene therapy study for mucopolysaccharidosis type I (MPS I), after IV AAV6 delivery, transgene expression from a subset of mice reduced to 0 between 3 and 6 weeks post injection (74). When biweekly administration of cyclophosphamide was performed, sustained transgene expression was observed till 120 days. The effects of cyclophosphamide may be towards immunity against transgene products, not capsids, because a parallel study in MPS II mice using the same approach did not observe transgene expression reduction (75). A more recent study using a similar approach but CpG-removed transgene observed no reduction in transgene expression in MPS I mice (76), providing further supporting evidence.
Hydroxychloroquine (HCQ) is an approved anti-malaria drug, and accumulation of hydroxychloroquine inhibits the binding ability of TLR9 to DNA and subsequent immune activation (77). Subretinal AAV administration elicited innate immunity, evidenced by increased expression of IFN-γ, TNF-α, and CXCL10 (78). In this study, HCQ enhanced AAV transduction in NHP retinal pigment epithelial cells and human retina ex vivo. Further, coadministration of AAV with HCQ enhanced mouse retina transduction in vivo, and no evidence of toxicity was observed. It is noteworthy that high-dose systemic administration of HCQ was associated with risk of retinopathies.
3.1.8 Key considerations for immunosuppressant usage
Overall, the timing, duration, and selection of immunosuppressants should be carefully considered. One NHP study showed that the addition of anti-IL-2 receptor antibody (daclizumab) to MMF and sirolimus failed to reduce antibody formation after AAV delivery (31). In contrast, MMF and sirolimus alone successfully reduced antibody formation, which may be explained by the fact that daclizumab had negative impacts on Tregs. A 5-drug immunosuppression regimen, including anti-thymocyte globulin (ATG), tacrolimus, rituximab, MMF, and methylprednisolone, inhibited T cell response after IV AAV5 delivery in NHPs (79). Interestingly, NAb formation occurred upon the removal of immunosuppression. Another NHP study showed that only delayed administration of ATG inhibited anti-drug antibody formation after AAV delivery, while early administration did not (80). FDA recently put a clinical trial for phenylketonuria on hold and lifted the hold after the immunosuppressant protocol incorporates tacrolimus and a shorter course of steroids (81). Other important things when considering immunosuppression regimens include side effects (e.g., infection) and the lack of suitable animal models for investigating and predicting AAV immunity due to the species difference.
3.2 Capsid engineering
3.2.1 Rational design
Capsid engineering for capsids with resistance to antibodies can be performed through rational design (82–88). As shown in previous studies, based on understanding of the epitope responsible for antibody binding, mutations and insertions were introduced to disrupt antibody binding and thus generate novel capsids with reduced antibody affinity (82, 87–89). One caveat is that epitopes on capsids may be adjacent to regions that are important for other functions, e.g., packaging, intracellular transport. Therefore, impacts on normal functions and overall performance of AAV should be assessed during the process of discovering immune-escaping capsids through this approach. A novel capsid CAM130 had reduced NAb affinity against AAV1, but maintained comparable titer, transduction efficiency, and tissue tropism (88). A chimeric capsid, AAV2.5, was generated by inserting 5 amino acids of AAV1 into AAV2 (83). AAV2.5 maintained muscle tropism of AAV1 and receptor binding ability of AAV2 and also had reduced antigen presentation against both AAV1 and AAV2. In this study, AAV2.5 also enabled repeated administration in mice. Further, some wildtype serotypes have lower prevalence of NAbs in humans (28), and thus can be used as the template for capsid engineering. One example is mutagenesis of epitope regions of AAVrh10, which is one of the least seropositive common variants (82). The novel capsid AAVrh10-S671A achieved higher transduction efficiency and was at least 27-fold more resistant to NAbs when tested in mice. In addition, immune-escaping capsids were identified through incubating the rationale-designed AAV library with AAV2 neutralizing antibodies in serum or human immunoglobin (IVIG) (90). The packaging ability, infectivity, and tropism of top candidates were assessed as well. The drawback of this rational design-based approach is its dependence on structural understanding of AAV and identification of antibody-binding epitopes.
In addition, rational design can be also used in capsid engineering for reduced antigen presentations and cellular immunity. After entry into cells, AAV cells can be ubiquitinated for degradation in proteasomes (91), generating increased amount of antigens for immune activation. Since surface-exposed residues on AAV capsids can be ubiquitinated through phosphorylation, mutations of these residues (e.g. tyrosine, lysine, serine) to reduce the likelihood of ubiquitination can generate novel capsids with reduced ubiquitination, antigen presentation, and activated cellular immunity (92–99). One study identified AAV2(Y-F), a novel capsid generated through phenylalanine (F) for tyrosine (Y) substitutions of several surface-expose tyrosine residues, which achieved more sustained transgene expression and reduced CTL responses against transduced hepatocytes than AAV8 in mice (92). A triple Y-to-F mutant of AAV2, termed as AAV2tYF (99), has been applied in four clinical trials for ocular indications (NCT02416622, NCT02599922, NCT03316560, NCT02161380). In addition, mutations of lysine residues of AAV8 led to the identification of AAV-K137R, which had reduced antigen presentation, activation of innate immunity, and NAb formation (97). AAV-K137R also exhibited improved liver transduction in mice.
3.2.2 Directed evolution
Another major category for capsid engineering is directed evolution, a method that mimics the process of natural selection to optimize certain features of DNA or proteins. Methods of AAV directed evolution include error-prone PCR (100–103), random peptide display (104–106), and DNA shuffling (107–110). Liver toxicity has been a major challenge for systemic AAV delivery, evidenced by incidence of liver enzyme elevations. There have been efforts in identifying liver-detargeting capsids for indications that liver is not the primary tissue target (111–113). Through random peptide display, a novel capsid B10 was identified (114). B10 had the superior brain tropism than AAV9 and had significantly reduced liver biodistribution after intravenous administration in mice and marmosets. Similarly, AAVMYO, a novel capsid with improved muscle tropism, was generated through random peptide display (115). AAVMYO was also 9-fold detargeted from liver than AAV9 after intravenous administration in mice. Another way of directed evolution is DNA shuffling, which generates a chimeric capsid containing sequence of multiple wildtype capsid variants. A novel capsid AAV-DJ was generated through DNA shuffling and subsequent high throughput screening in mice (106). AAV-DJ was able to transduce hepatocytes, more efficiently than parental serotypes, in the presence of intravenous human IVIG. A more recent study using the same approach identified a muscle-tropic capsid with improved resistance to NAbs, and the threefold symmetry region of AAV was deemed to be responsible for NAb recognition (116). Another chimeric capsid, SCH9, efficiently transduced neural stem cells and Purkinje cells and mediated higher transgene expression than AAV9 when delivered to mouse brains (117). Moreover, SCH9 showed enhanced resistance to NAbs and comparable packaging efficiency to wildtype capsid variants. In addition, one study screened novel liver-tropic capsids generated by DNA shuffling in a humanized mouse model (118). The library was incubated with human IgG to apply selective pressure for immune-evasive capsids. The top candidate AAV-NP59 was highly specific for human hepatocytes in this xenograft model. AAV-NP59 also elicited reduced anti-AAV IgG than AAV-LK03 (119) and AAV8, but not as good as previously discovered engineer capsid AAV-DJ.
3.3 Methods to overcome capsids-specific antibodies
3.3.1 IgG proteases
It has been previously reported that even low levels of neutralizing antibodies against AAV (1:5–1:10) can completely abrogate transduction with high-titer vectors (12, 46, 120). Also, a substantial subset of humans have been exposed to AAV and have pre-existing NAbs (28). Therefore, NAbs significantly limits the potential of AAV gene therapy due to the exclusion of many patients from treatment. To this end, IgG proteases have attracted attention from gene therapists over the few years (121, 122). Prophylactic administration of IdeZ, an immunoglobulin-degrading enzyme from Streptococcus equi subspecies zooepidemicus, degraded IgG, prevented antibody neutralization, and rescued AAV transduction in passively immunized mice and NHPs with pre-existing NAbs (122). Similarly, IdeS is an endopeptidase able to degrade circulating IgG that is currently being tested in transplant patients (123). IdeS treatment allowed for efficient transduction in NHPs in the presence of pre-existing NAbs, and more importantly, AAV readministration (121). However, IdeS and IdeZ originate from bacteria, and thus it is likely that patients have pre-existing antibodies against IdeS and IdeZ, which may affect the cleavage efficiency. Therefore, one drawback of this approach is that administration of IgG proteases can be performed only once because of antibody formation against these enzymes. There have been efforts to develop methods that enable redosing of IgG proteases through ImmTOR (124). However, these IgG degrading enzymes can only address NAbs, but leave memory T and B cells intact, which can lead to low transduction, reduced durability, inflammation, and adverse events.
3.3.2 Plasmapheresis
One way to reduce the inhibitory effects of pre-existing NAbs is plasmapheresis, a method to remove certain substances from the blood. Plasmapheresis is a widely-used and generally safe procedure in adults and children, even in pathological conditions. In one NHP study, plasmapheresis in sero-positive animals achieved efficient transduction, comparable to sero-negative animals and significantly higher than sero-positive animals without plasmapheresis (125). Also, there was no histopathological evidence of necrosis or lymphocyte infiltration in animals receiving plasmapheresis and AAVrh74 delivery. Plasmapheresis has also been tested in a clinical study, in which 3-5 consecutive plasmapheresis significantly reduced NAb titers in patients (126). The main drawback of plasmapheresis is non-specific removal of all circulating IgG, resulting in hypogammaglobulinemia and thus risk of infection. To this end, previous studies developed a capsid-specific plasmapheresis method using immunoadsorption (127–129). AAV-specific plasmapheresis column was used, enabling near-complete specific removal of anti-AAV IgG and AAV readministration in mice (127), rats (128), and NHPs (129). In addition, when plasmapheresis depletes antibodies in plasma, a quick rebound may come from antibodies in interstitial fluid and de novo production of B cells and antibodies (128). Therefore, AAV delivery should be performed in a short period post plasmapheresis. Another drawback is the need of repeated cycles to deplete IgG.
3.3.3 Empty capsid decoy
Empty capsids are AAV vectors without DNA enclosed and usually side products during AAV packaging. Interestingly, empty capsids were used as decoy for anti-AAV antibodies, and thus allowed for efficient transduction of genome-containing capsids and transgene expression in mice and NHPs (130). Remarkably, even in the presence of high NAb titer (1:3,000), addition of empty capsids achieved ~45% of transgene expression from the control group that did not receive IVIG. Since empty capsids had no or minimal DNA inside, they are not expected to activate TLR9-mediated innate immunity. One concern about this approach is the increase in antigen load and subsequent T cell activation. To this end, mutation was introduced into the receptor binding site of capsid to generate an empty capsid mutant, which could not enter target cells but maintained the ability to absorb antibodies (130). By this means, antigen presentation by transduced cells would be reduced. However, increase in antigen presentation still seems to be inevitable because empty mutant capsid can be uptaken by APCs through receptor-independent pathways, e.g., pinocytosis. One study showed efficient antigen presentation from empty capsids (131). Similar findings were also observed in a murine model of rheumatoid arthritis (132), which showed the addition of empty capsids improved transduction and transgene expression. Another study showed that partially empty capsids containing fragmented genome sequence cogenerated with capsids containing full length genome sequence reduced liver transduction of AAV8 and also caused liver enzyme elevation in mice because they still contain DNA sequences (133). Therefore, it was recommended that decoy capsids should be packaged separately to generate completely empty capsids (133). It was also argued that for certain diseases, e.g., retina, where NAbs and synovial macrophages are absent, empty capsids should be avoided; but for cases like arthritic joints, empty decoys may be considered (134). However, this approach is not widely adopted in clinical studies. This is probably due to the requirement of large quantities of empty capsids (10-fold of full capsids), which creates additional manufacturing and regulatory challenges, and the increased antigen presentation in spite of mutations introduced to inhibit cell entry. Under most circumstances, the presence of empty capsids in clinical grade vectors is undesirable.
3.3.4 Chemical modification of capsids
Another strategy to overcome NAbs is to encapsulate AAV capsids with polymers. One of the first efforts was not particularly successful. One study using polyethylene glycol (PEG) showed a modest improvement in resistance against NAbs at the price of significantly reduced infectivity (135). Another study using glycation on arginine and lysine residues showed that although binding affinity to antibodies decreased by 2 fold, transduction efficiency was reduced by 1,000 fold in vitro, potentially due to the disruption of AAV binding to heparin sulfate (136). Interestingly, as shown in one study, chemical modification led to redirection of tropism from liver to muscle. PEG conjugation activated by tresyl chloride (TMPEG) maintained tissue tropism to liver and muscle and achieved enhanced resistance to NAbs (137). TMPEG conjugation also enabled repeated AAV administration in mice. N-acetylgalactosamine (GalNAc), a ligand targeting hepatocytes, was also conjugated to lysine residues of AAV capsids (138). Significant lower total antibodies and NAbs were observed in mice receiving GalNAc-AAV. A more recent study applied click chemistry to specifically introduce unnatural amino acids (UAA), which can be tethered with various small molecules and polymers, into capsid proteins (139). This study showed that the resultant chemically modified AAV, oligo-AAV, had improved resistance against NAbs compared with previous polymer-conjugated AAVs. However, the packaging yields were significantly lowered. Similarly, another study using the PEGylation at UAA site strategy encountered the issue of decreased titer as well, although antibody recognition was modestly improved (140).
3.3.5 Exo-AAV
Exosome, natural extracellular vesicles secreted from cells, is being developed as non-viral delivery vectors of proteins or nucleic acids (141). As shown in one study (142), exosome-associated AAV (exo-AAV), enabled efficient transduction in the presence of pre-existing NAbs at least at a moderate titer. Moreover, exosome encapsulation improved cellular and nuclear uptake of AAV through a non-canonical pathway, which is independent of known receptors. In this study, exo-AAV also enhanced Tregs expansion, and thus induced immune tolerance. Two other studies showed that exosome-encapsulated AAV8 and AAV9 had comparable or improved infectivity in addition to having 3 to 23-fold higher resistance to NAbs (143, 144). Indeed, exo-AAV may have improved tropism to specific tissues. Exo-AAV9 had an improved ability to cross the BBB and target neurons and astrocytes than AAV9 (145). Exo-AAV had improved ability to cross the inner limiting membrane of the retina after intravitreal injection in mice (146). Exo-AAV also efficiently transduced outer hair cells without detectable toxicity (145). It was also shown that exo-AAV was easier and faster to purify as it requires only simple ultracentrifugation of media. One concern about exo-AAV is the reduced purity and the possibility of containing nucleic acids and proteins as shown in other studies using exosome preparations (147). By overexpressing tetraspanin CD9, an exosome marker, the exosome output from AAV-packaging HEK293 cells increased, resulting in a remarkable increase of AAV1 yields (148). Moreover, these vectors had improved transduction compared with standard exo-AAVs. Finally, a similar approach to exo-AAV is epitope masking using small molecules, e.g., albumin. A study in adenovirus showed that when an albumin-binding domain was inserted to capsid proteins of adenovirus, it can protect vectors from NAb recognition while maintaining infectivity and transduction (149). This strategy may be applied to AAV. While all these encapsulation strategies seem to be promising, they also create a challenge for scalable manufacturing and release tests of clinical-grade vectors.
3.4 Methods to address cellular immunity
3.4.1 Reduce recognition of unmethylated CpG
Unmethylated CpG elements elicit innate immunity and CTL response through TLR9/MyD88-dependent pathway (23). Recombinant AAV vector has abundant hypomethylated CpG motifs due to the viral or bacterial origin of its sequence compared with human DNA, providing the basis for discriminating AAV and human genome sequences by TLR9. Previous animal studies have shown the immune-activating effects of CpG motifs in AAV (23, 150, 151). For instance, CpG-depleted AAVrh32.33 vectors achieved sustained transgene expression and reduced lymphocyte infiltration in mice (152). A more recent study showed CpG deletion from AAV genome reduced CD8+ T cell response in a murine model of hemophilia after intramuscular delivery (152). Further, a recent study reviewed multiple hemophilia B gene therapy trials and found that sustained transgene expression correlated well with low CpG content in AAV vectors, but not other parameters (reviewed in 153). Since CpG removal is usually performed by synonymous codon substitution, one concern is misfolding of transgene products due to species differences in codon usage (154). Moreover, CpG not only exists in the open reading frame (ORF), but also promoters, enhancers, and ITRs. Removing CpG in those regions may have impacts on functionality of AAV vectors (transcription, packaging). To this end, one study developed methods to generate functional CpG-free ITRs (155), but the yields decreased by 3 fold due to reduced genome replication. Another approach is to increase AAV methylation by improving production technologies. By providing sufficient methyl transferase when preparing input DNA vectors (e.g., plasmids, baculovirus) and packaging AAV in host cells (e.g., HEK293, Sf9), CpG methylation of AAV genome could be achieved.
Alternatively, since CpG elements are recognized by TLR9 (23), inhibition of TLR9 recognition can be also used to reduce CpG-activated cellular immunity. TTAGGG repeat commonly found in mammalian telomeres has been shown to be able to inhibit TLR9 signaling and downstream immune activation (156, 157). Incorporating TLR9 inhibitory sequence (TTAGGG motif) in AAV vectors reduced CTL responses and enhanced transgene expression in mice and pigs (158). However, this strategy only delayed but could not prevent CTL response in NHPs, indicating that immune factors other than TLR9 contributed to intra-ocular inflammation in NHPs. Based on this technology, a startup Ally Therapeutics was launched in 2018. Nevertheless, due to ‘disappointing results in animal trials’, the company was shut down in 2021. Still, the approach represents a novel direction that warrants further investigation.
3.4.2 Induction of Tregs
Tregs are a subset of T cells that suppress immune response (159) and have been implicated in contributing to long-term transgene expression in preclinical studies (35, 160, 161) and clinical trials (162–164). A study in mice demonstrated that coadministration of antigen-specific Tregs and AAV successfully reduced both cellular and humoral immune responses against transgene products, and enabled sustained transgene expression (165). Another study showed that adoptive transfer of ex vivo Tregs induced antigen-specific immune suppression and achieved sustained transgene expression after AAV delivery in a murine model of hemophilia B (166). More recently, AAV capsid-specific chimeric antigen receptor Tregs (AAV CAR Tregs) were generated and shown to be able to reduce inflammation, mediate sustained transgene expression, and suppress immune response against both capsids and transgene products after injection into mice (167). In addition to this ex vivo Tregs approach, in vivo induction of Tregs can be applied to suppress AAV immunity through administration of rapamycin (discussed in section 3.1) or liver gene transfer. It has been shown in multiple preclinical (160, 168–171) and clinical studies (34) that liver-targeted AAV delivery induced immune tolerance of transgene products. In summary, induction of Tregs represent a novel approach to provide lasting and targeted immune suppression, which may be preferred over broad-spectrum and transient immune suppression offered by corticosteroids.
3.5 Other methods
3.5.1 Nucleotide sequence to reduce antigen presentation
One viable approach is to limit transgene expression from APCs to reduce antigen presentation and immune responses against AAV gene therapy. This can be done by using tissue-specific promoters or miRNA sequences. Notably, it was recently shown that tissue-specific promoter could not reduce immune responses in a canine study using AAV delivery of the CRISPR system (172). As to miRNA, it has been used to detarget transgene expression from APCs (173, 174). In one study, AAV containing miRNA-142 abrogated CTL and humoral response and enabled sustained transgene expression in mice (173). However, this effect seems to be strain-specific: miRNA-142 attenuated CTL response in WT C57BL/6 mice, but not muscular dystrophy mice (174). Another approach to limit antigen cross presentation is through using a small peptide named ICP47 (175). ICP47, derived from herpes simplex virus, could inhibit MHC class I pathway and antigen cross presentation. Expressing ICP47 from the AAV genome has been shown to reduce CTL responses in mice (176).
3.5.2 Selection of capsid variants or ROAs
Prevalence of AAV2 NAbs in humans was shown to be higher than other capsid variants (28), but cross-reactivity between different capsid variants was relatively low (177). Therefore, using alternative capsid variant with low immunogenicity was shown to be a viable option for achieving efficient transduction in animal studies (178–184). For instance, the first administration of AAV8 in neonatal mice did not prevent a second administration of AAV9 in adults (181). Admittedly, neonatal injection was known to induce immune tolerance (185, 186), making it easier for a second administration. Another study showed that a second administration of AAV6 was not affected by prior administration of AAV2 in adult mice, and vice versa (179). Similarly, increased transgene expression in skeletal muscle was observed after AAV1 administration in AAV2- or AAV5-pretreated mice (180). In addition, AAVrh10-mediated transgene expression was not affected by prior AAV2 or AAV5 administration in mice (182). Capsid variant switch also enabled repeated administration of AAV vectors in rats (183). More importantly, this strategy was shown to be successful in large animal models. In a canine model of hemophilia B, a second administration of AAV8 in AAV2-pretreated dog increased FIX level from <1% to 16% of normal controls and lasted for 2 years (184). Also, no significant liver toxicity and antibody formation was observed in this study. Another approach is to directly use a serotype with low immunogenicity. Since AAV5 is the most structurally divergent of wildtype serotypes, it was hypothesized that AAV5 might be less likely to elicit NAbs (187). One study showed that AAV5 delivery mediated therapeutic level transgene expression in the presence of pre-existing NAbs in NHPs and humans (188). Interestingly, another study found that pre-existing antibody led to significantly reduced transgene expression in NHPs (189).
In addition to capsids, careful selection of route of administration (ROA) can also help overcome AAV immunity. While systemic administration leads to more physical contact between AAV and pre-existing antibodies, local delivery to retina or CNS may be less likely to cause immune responses (190, 191). Notably, one study demonstrated that transgene expression after intrastriatal AAV delivery in rats was inhibited by high circulating NAb titer (1:1,208) (192). Similarly, a mouse study showed that high titer (>1:6,400) circulating NAbs inhibited brain transduction after CNS-directed delivery (190). This study also found that the antibodies in the brain was 0.6% of those in serum, suggesting that a small amount of circulating antibodies cross the blood-brain barrier (BBB). One ramification is that the BBB is compromised in certain neurological diseases (MPS IIIB, Parkinson’s disease) (193, 194), making the inhibitory effects of NAbs more likely. Therefore, this approach should still be considered as long as serum high titer is not extremely high and the BBB is not compromised.
4 Discussion
The potential of AAV-mediated gene therapy is significantly limited by immunogenicity. First of all, pre-existing NAbs exclude many patients (> 20% depending on serotypes) from being eligible for the treatment (177). It has also been shown that anti-AAV NAbs could persist for 15 years (195). When the field moves from rare disease, most of which are pediatric diseases, to more common diseases (e.g., Parkinson’s disease), most of which affect adults and even aged population, it becomes a bigger problem because people have a higher chance of being exposed to AAV as they age. Second, immune responses disallow repeated administration of AAV. Although AAV gene therapy is expected to provide long-term efficacy, there may be a need for repeated administration, especially for pediatric patients whose cells continue to expand. Third, AAV immunity could cause severe toxicity, possibly resulting in patient deaths and clinical holds (10, 26, 27). Over the past few years, more treatment emergent serious adverse events (TESAEs) have been reported as more patients receive AAV delivery (6), generating serious concerns about AAV immunogenicity and toxicity.
As mentioned above, there have been extensive efforts to address different aspects of AAV immunity. Many of these approaches have been applied in clinical studies and achieved their goals, at least, to an extent. One strategy, capsid engineering, is particularly promising as it has the potential to develop next generation vectors that have multi-dimensional improvements over currently available vectors. Current AAV vectors are still limited by the low delivery efficiency to specific tissues, resulting in the need of high dose or invasive administration. Improvement in tropism and transduction efficiency alone will enable the usage of lower dose and thus significantly reduce toxicity from AAV immunity. Moreover, next generation vectors with immune-evading features can help address the issue as well.
In this review, the toolkit to overcome AAV immunity are categorized mainly based on methodology, instead of on targets (capsids, genome, or transgene products). Although less discussed in previous sections, immune responses against transgene products should not be underestimated. CTL responses against transgene products were initially observed in some patients of a DMD trial through intramuscular AAV delivery (196). More recently, CTL responses against transgene products and reduced transgene expression were observed in one alpha-1 antitrypsin patient receiving intramuscular AAV delivery (197). Similar observations occurred in MPS IIIB patients receiving intracranial AAV delivery (198). It is worthwhile to mention that immunity against transgene products may vary on genetic backgrounds (34, 199). For instance, AAV gene therapy in individuals who have residual protein expression may be less likely to cause anti-transgene immunity than in those with no residual endogenous expression. This may be further complicated by the fact that some patients of protein deficiencies may have received protein replacement therapies prior to AAV delivery. In addition, tissue inflammation due to the underlying disease can also increase the risk of transgene immunogenicity (200).
A particular challenge for understanding mechanisms of and developing methods to overcome AAV immunity is the species difference between animal models and patients, especially T cell responses. For instance, unlike in mice, there was expansion of pre-existing capsid-specific CD8+ T cells in humans (31). Similarly, preclinical studies in mice (92), dogs (201), and NHPs (202) could not recapitulate capsid-specific T cell responses in humans. In addition, many animal models were generated through completely disrupting the function of a certain gene, which may not recapitulate human conditions as some patients still have residual protein expression. This fact adds further complication when using preclinical studies in animals to predict AAV immunity in clinical studies. Therefore, there is a critical need to develop more suitable animal models to predict AAV immunity in humans. Other areas that this field should consider to focus on include: 1) mechanistic studies of toxicity events; 2) identification of reliable preclinical models to monitor and predict AAV immunity and toxicity; 3) standardization of methods that measure AAV immunity, e.g., NAb assays, ELISPOT, complement panel; and 4) long-term follow-up of patients who have received AAV delivery and proper archiving and sharing of these data.
Author contributions
XL, XW, JL, and LO attended discussion for the project design. XL and LO drafted the manuscript. XW and JL revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by National Natural Science Foundation of China (81972204), Natural Science Foundation of Guangdong Province (2019A1515011097), Innovation Program of Shenzhen (Grant No. JCYJ20180508165208399), the grant from the State Key Lab of Respiratory Disease, Guangzhou Medical University (SKLRD-Z-202002), and the 111 Project (D18010) from the Ministry of Education of China. The open research funds from the Sixth Affiliated Hospital of Guangzhou Medical University, Qingyuan People's Hospital.
Conflict of interest
Author XW was employed by Guangzhou Dezheng Biotechnology Co., Ltd. Author LO was employed by Genemagic Bio.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Samulski RJ, Muzyczka N. AAV-mediated gene therapy for research and therapeutic purposes. Annu Rev Virol (2014) 1(1):427–51. doi: 10.1146/annurev-virology-031413-085355
2. Grieger JC, Samulski RJ. Adeno-associated virus as a gene therapy vector: vector development, production and clinical applications. Gene Ther Gene Deliv Syst (2005) 99:119–45. doi: 10.1007/10_005
3. Ho A, Orton R, Tayler R, Asamaphan P, Tong L, Smollett K, et al. Adeno-associated virus 2 infection in children with non-AE hepatitis. medRxiv (2022). doi: 10.1101/2022.07.19.22277425
4. Wang D, Tai PW, Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discovery (2019) 18(5):358–78. doi: 10.1038/s41573-019-0012-9
5. Wagner JA, Reynolds T, Moran ML, Moss RB, Wine JJ, Flotte TR, et al. Efficient and persistent gene transfer of AAV-CFTR in maxillary sinus. Lancet (1998) 351(9117):1702–3. doi: 10.1016/S0140-6736(05)77740-0
6. Kuzmin DA, Shutova MV, Johnston NR, Smith OP, Fedorin VV, Kukushkin YS, et al. The clinical landscape for AAV gene therapies. Nat Rev Drug Discovery (2021) 20(3):173–5. doi: 10.1038/d41573-021-00017-7
7. Gaudet D, Méthot J, Déry S, Brisson D, Essiembre C, Tremblay G, et al. Efficacy and long-term safety of alipogene tiparvovec (AAV1-LPLS447X) gene therapy for lipoprotein lipase deficiency: an open-label trial. Gene Ther (2013) 20(4):361–9. doi: 10.1038/gt.2012.43
8. Maguire AM, Simonelli F, Pierce EA, Pugh EN Jr., Mingozzi F, Bennicelli J, et al. Safety and efficacy of gene transfer for leber's congenital amaurosis. N Engl J Med (2008) 358(21):2240–8. doi: 10.1056/NEJMoa0802315
9. Mendell JR, Al-Zaidy S, Shell R, Arnold WD, Rodino-Klapac LR, Prior TW, et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med (2017) 377(18):1713–22. doi: 10.1056/NEJMoa1706198
10. Shieh PB, Bönnemann CG, Müller-Felber W, Blaschek A, Dowling JJ, Kuntz NL, et al. Re:”Moving forward after two deaths in a gene therapy trial of myotubular myopathy” by Wilson and flotte. Hum Gene Ther (2020) 31(15-16):787–7. doi: 10.1089/hum.2020.217
11. de Jong YP, Herzog RW. Liver gene therapy and hepatocellular carcinoma: A complex web. Mol Ther (2021) 29(4):1353–4. doi: 10.1016/j.ymthe.2021.03.009
12. Manno CS, Pierce GF, Arruda VR, Glader B, Ragni M, Rasko JJ, et al. Successful transduction of liver in hemophilia by AAV-factor IX and limitations imposed by the host immune response. Nat Med (2006) 12(3):342–7. doi: 10.1038/nm1358
13. Sondhi D, Kaminsky SM, Hackett NR, Pagovich OE, Rosenberg JB, De BP, et al. Slowing late infantile batten disease by direct brain parenchymal administration of a rh. 10 adeno-associated virus expressing CLN2. Sci Trans Med (2020) 12(572):eabb5413. doi: 10.1126/scitranslmed.abb5413
14. Mueller C, Berry JD, McKenna-Yasek DM, Gernoux G, Owegi MA, Pothier LM, et al. SOD1 suppression with adeno-associated virus and MicroRNA in familial ALS. N Engl J Med (2020) 383(2):151–8. doi: 10.1056/NEJMoa2005056
15. Available at: https://www.fiercebiotech.com/biotech/press-release-fda-probing-death-targeted-genetics-gene-therapy-trial.
16. Bonnemann CG. (2020). AAV Related Immunological Safety and Toxicity: Preliminary Clinical Observations in the GAN and MTM1 Trials. in: Presented at: Virtual Workshop on Systemic Immunogenicity Considerations of AAV-Mediated Gene Therapy, NIH, NCATS.
17. Available at: https://www.fiercebiotech.com/biotech/pfizer-reports-death-patient-duchenne-trial-halts-enrolment.
18. Available at: https://www.fiercepharma.com/marketing/zolgensma-s-european-trial-death-unrelated-to-gene-therapy-as-novartis-touts-new.
19. Philippidis A. “Profoundly saddened” lysogene discloses child's death in phase II/III trial. Hum Gene Ther (2020) 31(21-22):1141–3. doi: 10.1089/hum.2020.29139.bfs
20. Available at: https://www.fiercebiotech.com/biotech/taysha-mulls-tweaks-to-gene-therapy-trial-after-subject-dies.
21. Available at: https://www.fiercepharma.com/pharma/two-deaths-after-novartis-zolgensma-bring-gene-therapys-liver-safety-spotlight-again.
22. Li C, Hirsch M, DiPrimio N, Asokan A, Goudy K, Tisch R, et al. Cytotoxic-t-lymphocyte-mediated elimination of target cells transduced with engineered adeno-associated virus type 2 vector in vivo. J Virol (2009) 83(13):6817–24. doi: 10.1128/JVI.00278-09
23. Zhu J, Huang X, Yang Y. The TLR9-MyD88 pathway is critical for adaptive immune responses to adeno-associated virus gene therapy vectors in mice. J Clin Invest (2009) 119(8):2388–98. doi: 10.1172/JCI37607
24. Martino AT, Suzuki M, Markusic DM, Zolotukhin I, Ryals RC, Moghimi B, et al. The genome of self-complementary adeno-associated viral vectors increases toll-like receptor 9–dependent innate immune responses in the liver. Blood (2011) 117(24):6459–68. doi: 10.1182/blood-2010-10-314518
25. Ashley SN, Somanathan S, Giles AR, Wilson JM. TLR9 signaling mediates adaptive immunity following systemic AAV gene therapy. Cell Immunol (2019) 346:103997. doi: 10.1016/j.cellimm.2019.103997
26. Available at: https://www.fiercebiotech.com/biotech/pfizer-s-dmd-gene-therapy-looks-good-data-refresh-but-safety-concerns-persist.
27. Available at: https://www.fiercebiotech.com/biotech/fda-lifts-clinical-hold-solid-bio-gene-therapy-trial.
28. Boutin S, Monteilhet V, Veron P, Leborgne C, Benveniste O, Montus MF, et al. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum Gene Ther (2010) 21(6):704–12. doi: 10.1089/hum.2009.182
29. Gao GP, Alvira MR, Wang L, Calcedo R, Johnston J, Wilson JM. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc Natl Acad Sci (2002) 99(18):11854–9. doi: 10.1073/pnas.182412299
30. Tang Y, Yan Z, Lin S, Huntemann ED, Feng Z, Park SY, et al. Repeat dosing of AAV2. 5T to ferret lungs elicits an antibody response that diminishes transduction in an age-dependent manner. Mol Ther-Methods Clin Dev (2020) 19:186–200. doi: 10.1016/j.omtm.2020.09.008
31. Mingozzi F, Hasbrouck NC, Basner-Tschakarjan E, Edmonson SA, Hui DJ, Sabatino DE, et al. Modulation of tolerance to the transgene product in a nonhuman primate model of AAV-mediated gene transfer to liver. Blood (2007) 110(7):2334–41. doi: 10.1182/blood-2007-03-080093
32. Nathwani AC, Tuddenham EG, Rangarajan S, Rosales C, McIntosh J, Linch DC, et al. Adenovirus-associated virus vector–mediated gene transfer in hemophilia b. N Engl J Med (2011) 365(25):2357–65. doi: 10.1056/NEJMoa1108046
33. Nathwani AC, Reiss UM, Tuddenham EG, Rosales C, Chowdary P, McIntosh J, et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia b. N Engl J Med (2014) 371(21):1994–2004. doi: 10.1056/NEJMoa1407309
34. Mingozzi F, Liu YL, Dobrzynski E, Kaufhold A, Liu JH, Wang Y, et al. Induction of immune tolerance to coagulation factor IX antigen by in vivo hepatic gene transfer. J Clin Invest (2003) 111(9):1347–56. doi: 10.1172/JCI200316887
35. Cao O, Dobrzynski E, Wang L, Nayak S, Mingle B, Terhorst C, et al. Induction and role of regulatory CD4+ CD25+ T cells in tolerance to the transgene product following hepatic in vivo gene transfer. Blood (2007) 110(4):1132–40. doi: 10.1182/blood-2007-02-073304
36. Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids–new mechanisms for old drugs. N Engl J Med (2005) 353(16):1711–23. doi: 10.1056/NEJMra050541
37. Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (2017) 390(10097):849–60. doi: 10.1016/S0140-6736(17)31868-8
38. Limon JJ, So L, Jellbauer S, Chiu H, Corado J, Sykes SM, et al. mTOR kinase inhibitors promote antibody class switching via mTORC2 inhibition. Proc Natl Acad Sci (2014) 111(47):E5076–85. doi: 10.1073/pnas.1407104111
39. Velazquez VM, Meadows AS, Pineda RJ, Camboni M, McCarty DM, Fu H. Effective depletion of pre-existing anti-AAV antibodies requires broad immune targeting. Mol Ther-Methods Clin Dev (2017) 4:159–68. doi: 10.1016/j.omtm.2017.01.003
40. Maldonado RA, LaMothe RA, Ferrari JD, Zhang AH, Rossi RJ, Kolte PN, et al. Polymeric synthetic nanoparticles for the induction of antigen-specific immunological tolerance. Proc Natl Acad Sci (2015) 112(2):E156–65. doi: 10.1073/pnas.1408686111
41. Meliani A, Boisgerault F, Hardet R, Marmier S, Collaud F, Ronzitti G, et al. Antigen-selective modulation of AAV immunogenicity with tolerogenic rapamycin nanoparticles enables successful vector re-administration. Nat Commun (2018) 9(1):1–13. doi: 10.1038/s41467-018-06621-3
42. Sands E, Kivitz A, DeHaan W, Leung SS, Johnston L, Kishimoto TK. Tolerogenic nanoparticles mitigate the formation of anti-drug antibodies against pegylated uricase in patients with hyperuricemia. Nat Commun (2022) 13(1):1–7. doi: 10.1038/s41467-021-27945-7
43. Lauwerys BR, Wakeland EK. Genetics of lupus nephritis. Lupus (2005) 14(1):2–12. doi: 10.1191/0961203305lu2052oa
44. Montenegro-Miranda PS, Bloemendaal LT, Kunne C, de Waart DR, Bosma PJ. Mycophenolate mofetil impairs transduction of single-stranded adeno-associated viral vectors. Hum Gene Ther (2011) 22(5):605–12. doi: 10.1089/hum.2010.222
45. Jiang H, Couto LB, Patarroyo-White S, Liu T, Nagy D, Vargas JA, et al. Effects of transient immunosuppression on adenoassociated, virus-mediated, liver-directed gene transfer in rhesus macaques and implications for human gene therapy. Blood (2006) 108(10):3321–8. doi: 10.1182/blood-2006-04-017913
46. Day JW, Finkel RS, Mercuri E, Swoboda KJ, Menier M, van Olden R, et al. Adeno-associated virus serotype 9 antibodies in patients screened for treatment with onasemnogene abeparvovec. Mol Ther-Methods Clin Dev (2021) 21:76–82. doi: 10.1016/j.omtm.2021.02.014
47. Ross N, Smith C, Kamal A, Majowicz A, Mingozzi F, Kuranda K. Effects of complement component 1 (C1) inhibition on AAV-based gene transfer efficacy and immunogenicity in mice. Mol Ther (2022) 30:559–60.
48. Available at: https://investors.apellis.com/news-releases/news-release-details/apellis-pharmaceuticals-will-commence-apl-9-program-control.
49. Smith MR. Rituximab (monoclonal anti-CD20 antibody): mechanisms of action and resistance. Oncogene (2003) 22(47):7359–68. doi: 10.1038/sj.onc.1206939
50. Mingozzi F, Chen Y, Murphy SL, Edmonson SC, Tai A, Price SD, et al. Pharmacological modulation of humoral immunity in a nonhuman primate model of AAV gene transfer for hemophilia b. Mol Ther (2012) 20(7):1410–6. doi: 10.1038/mt.2012.84
51. Mingozzi F, Chen Y, Edmonson SC, Zhou S, Thurlings RM, Tak PP, et al. Prevalence and pharmacological modulation of humoral immunity to aav vectors in gene transfer to synovial tissue. Gene Ther (2013) 20(4):417–24. doi: 10.1038/gt.2012.55
52. Biswas M, Palaschak B, Kumar SR, Rana J, Markusic DM. B cell depletion eliminates FVIII memory b cells and enhances AAV8-coF8 immune tolerance induction when combined with rapamycin. Front Immunol (2020) 11:1293. doi: 10.3389/fimmu.2020.01293
53. Corti M, Cleaver B, Clement N, Conlon TJ, Faris KJ, Wang G, et al. Evaluation of readministration of a recombinant adeno-associated virus vector expressing acid alpha-glucosidase in pompe disease: preclinical to clinical planning. Hum Gene Ther Clin Dev (2015) 26(3):185–93. doi: 10.1089/humc.2015.068
54. Byrne BJ, Fuller DD, Smith BK, Clement N, Coleman K, Cleaver B, et al. Pompe disease gene therapy: Neural manifestations require consideration of cns directed therapy. Ann Trans Med (2019) 7(13):290. doi: 10.21037/atm.2019.05.56
55. Flotte TR, Cataltepe O, Puri A, Batista AR, Moser R, McKenna-Yasek D, et al. AAV gene therapy for Tay-Sachs disease. Nat Med (2022) 28(2):251–9. doi: 10.1038/s41591-021-01664-4
56. Zhang YI, Chirmule N, Gao GP, Wilson J. CD40 ligand-dependent activation of cytotoxic T lymphocytes by adeno-associated virus vectors in vivo: role of immature dendritic cells. J Virol (2000) 74(17):8003–10. doi: 10.1128/JVI.74.17.8003-8010.2000
57. Frentsch M, Japp AS, Dingeldey M, Matzmohr N, Thiel A, Scheiflinger F, et al. Blockade of the costimulatory CD28-B7 family signal axis enables repeated application of AAV8 gene vectors. J Thromb Haemostasis (2020) 18(5):1075–80. doi: 10.1111/jth.14757
58. Azzi JR, Sayegh MH, Mallat SG. Calcineurin inhibitors: 40 years later, can’t live without. J Immunol (2013) 191(12):5785–91. doi: 10.4049/jimmunol.1390055
59. Ishii A, Okada H, Hayashita-Kinoh H, Shin JH, Tamaoka A, Okada T, et al. rAAV8 and rAAV9-mediated long-term muscle transduction with tacrolimus (FK506) in non-human primates. Mol Ther-Methods Clin Dev (2020) 18:44–9. doi: 10.1016/j.omtm.2020.05.012
60. Miroux C, Morales O, Ghazal K, Othman SB, De Launoit Y, Pancré V, et al. In vitro effects of cyclosporine a and tacrolimus on regulatory T-cell proliferation and function. Transplantation (2012) 94(2):123–31. doi: 10.1097/TP.0b013e3182590d8f
61. Akimova T, Kamath BM, Goebel JW, Meyers KE, Rand EB, Hawkins A, et al. Differing effects of rapamycin or calcineurin inhibitor on T-regulatory cells in pediatric liver and kidney transplant recipients. Am J Transplant (2012) 12(12):3449–61. doi: 10.1111/j.1600-6143.2012.04269.x
62. McIntosh JH, Cochrane M, Cobbold S, Waldmann H, Nathwani SA, Davidoff AM, et al. Successful attenuation of humoral immunity to viral capsid and transgenic protein following AAV-mediated gene transfer with a non-depleting CD4 antibody and cyclosporine. Gene Ther (2012) 19(1):78–85. doi: 10.1038/gt.2011.64
63. Ferreira V, Petry H, Salmon F. Immune responses to AAV-vectors, the glybera example from bench to bedside. Front Immunol (2014) 582. doi: 10.3389/fimmu.2014.00082
64. Yan Z, Zak R, Luxton GG, Ritchie TC, Bantel-Schaal U, Engelhardt JF. Ubiquitination of both adeno-associated virus type 2 and 5 capsid proteins affects the transduction efficiency of recombinant vectors. J Virol (2002) 76(5):2043–53. doi: 10.1128/jvi.76.5.2043-2053.2002
65. Finn JD, Hui D, Downey HD, Dunn D, Pien GC, Mingozzi F, et al. Proteasome inhibitors decrease AAV2 capsid derived peptide epitope presentation on MHC class I following transduction. Mol Ther (2010) 18(1):135–42. doi: 10.1038/mt.2009.257
66. Nathwani AC, Cochrane M, McIntosh J, Ng CY, Zhou J, Gray JT, et al. Enhancing transduction of the liver by adeno-associated viral vectors. Gene Ther (2009) 16(1):60–9. doi: 10.1038/gt.2008.137
67. Monahan PE, Lothrop CD, Sun J, Hirsch ML, Kafri T, Kantor B, et al. Proteasome inhibitors enhance gene delivery by AAV virus vectors expressing large genomes in hemophilia mouse and dog models: a strategy for broad clinical application. Mol Ther (2010) 18(11):1907–16. doi: 10.1038/mt.2010.170
68. Mitchell AM, Samulski RJ. Mechanistic insights into the enhancement of adeno-associated virus transduction by proteasome inhibitors. J Virol (2013) 87(23):13035–41. doi: 10.1128/JVI.01826-13
69. Chaanine AH, Nonnenmacher M, Kohlbrenner E, Jin D, Kovacic JC, Akar FG, et al. Effect of bortezomib on the efficacy of AAV9. SERCA2a treatment to preserve cardiac function in a rat pressure-overload model of heart failure. Gene Ther (2014) 21(4):379–86. doi: 10.1038/gt.2014.7
70. Merin NM, Kelly KR. Clinical use of proteasome inhibitors in the treatment of multiple myeloma. Pharmaceuticals (2014) 8(1):1–20. doi: 10.3390/ph8010001
71. Mitchell AM, Li C, Samulski RJ. Arsenic trioxide stabilizes accumulations of adeno-associated virus virions at the perinuclear region, increasing transduction in vitro and in vivo. J Virol (2013) 87(8):4571–83. doi: 10.1128/JVI.03443-12
72. Da Rocha S, Bigot J, Onodi F, Cosette J, Corre G, Poupiot J, et al. Temporary reduction of membrane CD4 with the antioxidant MnTBAP is sufficient to prevent immune responses induced by gene transfer. Mol Ther-Methods Clin Dev (2019) 14:285–99. doi: 10.1016/j.omtm.2019.06.011
73. Streck CJ, Dickson PV, Ng CY, Zhou J, Gray JT, Nathwani AC, et al. Adeno-associated virus vector-mediated systemic delivery of IFN-β combined with low-dose cyclophosphamide affects tumor regression in murine neuroblastoma models. Clin Cancer Res (2005) 11(16):6020–9. doi: 10.1158/1078-0432.CCR-05-0502
74. Ou L, DeKelver RC, Rohde M, Tom S, Radeke R, Martin SJS, et al. ZFN-mediated in vivo genome editing corrects murine hurler syndrome. Mol Ther (2019) 27(1):178–87. doi: 10.1016/j.ymthe.2018.10.018
75. Laoharawee K, DeKelver RC, Podetz-Pedersen KM, Rohde M, Sproul S, Nguyen HO, et al. Dose-dependent prevention of metabolic and neurologic disease in murine MPS II by ZFN-mediated in vivo genome editing. Mol Ther (2018) 26(4):1127–36. doi: 10.1016/j.ymthe.2018.03.002
76. Ou L, Przybilla MJ, Ahlat O, Kim S, Overn P, Jarnes J, et al. A highly efficacious PS gene editing system corrects metabolic and neurological complications of mucopolysaccharidosis type I. Mol Ther (2020) 28(6):1442–54. doi: 10.1016/j.ymthe.2020.03.018
77. Kužnik A, Benčina M, Švajger U, Jeras M, Rozman B, Jerala R. Mechanism of endosomal TLR inhibition by antimalarial drugs and imidazoquinolines. J Immunol (2011) 186(8):4794–804. doi: 10.4049/jimmunol.1000702
78. Chandler LC, Barnard AR, Caddy SL, Patrício MI, McClements ME, Fu H, et al. Enhancement of adeno-associated virus-mediated gene therapy using hydroxychloroquine in murine and human tissues. Mol Ther-Methods Clin Dev (2019) 14:77–89. doi: 10.1016/j.omtm.2019.05.012
79. Unzu C, Hervás-Stubbs S, Sampedro A, Mauleón I, Mancheño U, Alfaro C, et al. Transient and intensive pharmacological immunosuppression fails to improve AAV-based liver gene transfer in non-human primates. J Trans Med (2012) 10(1):1–11. doi: 10.1186/1479-5876-10-122
80. Samelson-Jones BJ, Finn JD, Favaro P, Wright JF, Arruda VR. Timing of intensive immunosuppression impacts risk of transgene antibodies after AAV gene therapy in nonhuman primates. Mol Ther-Methods Clin Dev (2020) 17:1129–38. doi: 10.1016/j.omtm.2020.05.001
81. Available at: https://investors.homologymedicines.com/news-releases/news-release-details/homology-medicines-announces-fda-lifted-clinical-hold-phenix.
82. Selot R, Arumugam S, Mary B, Cheemadan S, Jayandharan GR. Optimized AAV rh. 10 vectors that partially evade neutralizing antibodies during hepatic gene transfer. Front Pharmacol (2017) 8:441. doi: 10.3389/fphar.2017.00441
83. Bowles DE, McPhee SW, Li C, Gray SJ, Samulski JJ, Camp AS, et al. Phase 1 gene therapy for duchenne muscular dystrophy using a translational optimized AAV vector. Mol Ther (2012) 20(2):443–55. doi: 10.1038/mt.2011.237
84. Burg M, Rosebrough C, Drouin LM, Bennett A, Mietzsch M, Chipman P, et al. Atomic structure of a rationally engineered gene delivery vector, AAV2. 5. J Struct Biol (2018) 203(3):236–41. doi: 10.1016/j.jsb.2018.05.004
85. Jose A, Mietzsch M, Smith JK, Kurian J, Chipman P, McKenna R, et al. High-resolution structural characterization of a new adeno-associated virus serotype 5 antibody epitope toward engineering antibody-resistant recombinant gene delivery vectors. J Virol (2019) 93(1):e01394–18. doi: 10.1128/JVI.01394-18
86. Li C, Diprimio N, Bowles DE, Hirsch ML, Monahan PE, Asokan A, et al. Single amino acid modification of adeno-associated virus capsid changes transduction and humoral immune profiles. J Virol (2012) 86(15):7752–9. doi: 10.1128/JVI.00675-12
87. Wobus CE, Hügle-Doürr B, Girod A, Petersen G, Hallek M, Kleinschmidt JA. Monoclonal antibodies against the adeno-associated virus type 2 (AAV-2) capsid: epitope mapping and identification of capsid domains involved in AAV-2–cell interaction and neutralization of AAV-2 infection. J Virol (2000) 74(19):9281–93. doi: 10.1128/JVI.74.19.9281-9293.2000
88. Tse LV, Klinc KA, Madigan VJ, Castellanos Rivera RM, Wells LF, Havlik LP, et al. Structure-guided evolution of antigenically distinct adeno-associated virus variants for immune evasion. Proc Natl Acad Sci (2017) 114(24):E4812–21. doi: 10.1073/pnas.1704766114
89. Li C, Narkbunnam N, Samulski RJ, Asokan A, Hu G, Jacobson LJ, et al. Neutralizing antibodies against adeno-associated virus examined prospectively in pediatric patients with hemophilia. Gene Ther (2012) 19(3):288–94. doi: 10.1038/gt.2011.90
90. Maersch S, Huber A, Büning H, Hallek M, Perabo L. Optimization of stealth adeno-associated virus vectors by randomization of immunogenic epitopes. Virology (2010) 397(1):167–75. doi: 10.1016/j.virol.2009.10.021
91. Duan D, Yue Y, Yan Z, Yang J, Engelhardt JF. Endosomal processing limits gene transfer to polarized airway epithelia by adeno-associated virus. J Clin Invest (2000) 105(11):1573–87. doi: 10.1172/JCI8317
92. Martino AT, Basner-Tschakarjan E, Markusic DM, Finn JD, Hinderer C, Zhou S, et al. Engineered AAV vector minimizes in vivo targeting of transduced hepatocytes by capsid-specific CD8+ T cells. Blood (2013) 121(12):2224–33. doi: 10.1182/blood-2012-10-460733
93. Zhong L, Li B, Mah CS, Govindasamy L, Agbandje-McKenna M, Cooper M, et al. Next generation of adeno-associated virus 2 vectors: point mutations in tyrosines lead to high-efficiency transduction at lower doses. Proc Natl Acad Sci (2008) 105(22):7827–32. doi: 10.1073/pnas.0802866105
94. Petrs-Silva H, Dinculescu A, Li Q, Min SH, Chiodo V, Pang JJ, et al. High-efficiency transduction of the mouse retina by tyrosine-mutant AAV serotype vectors. Mol Ther (2009) 17(3):463–71. doi: 10.1038/mt.2008.269
95. Kay CN, Ryals RC, Aslanidi GV, Min SH, Ruan Q, Sun J, et al. Targeting photoreceptors via intravitreal delivery using novel, capsid-mutated AAV vectors. PloS One (2013) 8(4):e62097. doi: 10.1371/journal.pone.0062097
96. Mowat FM, Gornik KR, Dinculescu A, Boye SL, Hauswirth WW, Petersen-Jones SM, et al. Tyrosine capsid-mutant AAV vectors for gene delivery to the canine retina from a subretinal or intravitreal approach. Gene Ther (2014) 21(1):96–105. doi: 10.1038/gt.2013.64
97. Sen D, Gadkari RA, Sudha G, Gabriel N, Kumar YS, Selot R, et al. Targeted modifications in adeno-associated virus serotype 8 capsid improves its hepatic gene transfer efficiency in vivo. Hum Gene Ther Methods (2013) 24(2):104–16. doi: 10.1089/hgtb.2012.195
98. Markusic DM, Herzog RW, Aslanidi GV, Hoffman BE, Li B, Li M, et al. High-efficiency transduction and correction of murine hemophilia b using AAV2 vectors devoid of multiple surface-exposed tyrosines. Mol Ther (2010) 18(12):2048–56. doi: 10.1038/mt.2010.172
99. Ye GJ, Budzynski E, Sonnentag P, Nork TM, Miller PE, Sharma AK, et al. Safety and biodistribution evaluation in cynomolgus macaques of rAAV2tYF-PR1. 7-hCNGB3, a recombinant AAV vector for treatment of achromatopsia. Hum Gene Ther Clin Dev (2016) 27(1):37–48. doi: 10.1089/humc.2015.164
100. Perabo L, Endell J, King S, Lux K, Goldnau D, Hallek M, et al. Combinatorial engineering of a gene therapy vector: directed evolution of adeno-associated virus. J Gene Med: A Cross-disciplinary J Res Sci Gene Transfer Its Clin Appl (2006) 8(2):155–62. doi: 10.1002/jgm.849
101. Maheshri N, Koerber JT, Kaspar BK, Schaffer DV. Directed evolution of adeno-associated virus yields enhanced gene delivery vectors. Nat Biotechnol (2006) 24(2):198–204. doi: 10.1038/nbt1182
102. Huttner NA, Girod A, Perabo L, Edbauer D, Kleinschmidt JA, Büning H, et al. Genetic modifications of the adeno-associated virus type 2 capsid reduce the affinity and the neutralizing effects of human serum antibodies. Gene Ther (2003) 10(26):2139–47. doi: 10.1038/sj.gt.3302123
103. Qian R, Xiao B, Li J, Xiao X. Directed evolution of AAV serotype 5 for increased hepatocyte transduction and retained low humoral seroreactivity. Mol Ther-Methods Clin Dev (2021) 20:122–32. doi: 10.1016/j.omtm.2020.10.010
104. Deverman BE, Pravdo PL, Simpson BP, Kumar SR, Chan KY, Banerjee A, et al. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat Biotechnol (2016) 34(2):204–9. doi: 10.1038/nbt.3440
105. Chan KY, Jang MJ, Yoo BB, Greenbaum A, Ravi N, Wu WL, et al. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat Neurosci (2017) 20(8):1172–9. doi: 10.1038/nn.4593
106. Dalkara D, Byrne LC, Klimczak RR, Visel M, Yin L, Merigan WH, et al. In vivo–directed evolution of a new adeno-associated virus for therapeutic outer retinal gene delivery from the vitreous. Sci Trans Med (2013) 5(189):189ra76–189ra76. doi: 10.1126/scitranslmed.3005708
107. Grimm D, Lee JS, Wang L, Desai T, Akache B, Storm TA, et al. In vitro and in vivo gene therapy vector evolution via multispecies interbreeding and retargeting of adeno-associated viruses. J Virol (2008) 82(12):5887–911. doi: 10.1128/JVI.00254-08
108. Koerber JT, Jang JH, Schaffer DV. DNA Shuffling of adeno-associated virus yields functionally diverse viral progeny. Mol Ther (2008) 16(10):1703–9. doi: 10.1038/mt.2008.167
109. Li W, Asokan A, Wu Z, Van Dyke T, DiPrimio N, Johnson JS, et al. Engineering and selection of shuffled AAV genomes: a new strategy for producing targeted biological nanoparticles. Mol Ther (2008) 16(7):1252–60. doi: 10.1038/mt.2008.100
110. Yang L, Jiang J, Drouin LM, Agbandje-Mckenna M, Chen C, Qiao C, et al. A myocardium tropic adeno-associated virus (AAV) evolved by DNA shuffling and in vivo selection. Proc Natl Acad Sci (2009) 106(10):3946–51. doi: 10.1073/pnas.0813207106
111. Pulicherla N, Shen S, Yadav S, Debbink K, Govindasamy L, Agbandje-McKenna M, et al. Engineering liver-detargeted AAV9 vectors for cardiac and musculoskeletal gene transfer. Mol Ther (2011) 19(6):1070–8. doi: 10.1038/mt.2011.22
112. Rotundo IL, Lancioni A, Savarese M, D'Orsi L, Iacomino M, Nigro G, et al. Use of a lower dosage liver-detargeted AAV vector to prevent hamster muscular dystrophy. Hum Gene Ther (2013) 24(4):424–30. doi: 10.1089/hum.2012.121
113. Wang D, Li S, Gessler DJ, Xie J, Zhong L, Li J, et al. A rationally engineered capsid variant of AAV9 for systemic CNS-directed and peripheral tissue-detargeted gene delivery in neonates. Mol Ther-Methods Clin Dev (2018) 9:234–46. doi: 10.1016/j.omtm.2018.03.004
114. Goertsen D, Flytzanis NC, Goeden N, Chuapoco MR, Cummins A, Chen Y, et al. AAV capsid variants with brain-wide transgene expression and decreased liver targeting after intravenous delivery in mouse and marmoset. Nat Neurosci (2022) 25(1):106–15. doi: 10.1038/s41593-021-00969-4
115. Weinmann J, Weis S, Sippel J, Tulalamba W, Remes A, El Andari J, et al. Identification of a myotropic AAV by massively parallel in vivo evaluation of barcoded capsid variants. Nat Commun (2020) 11(1):1–12. doi: 10.1038/s41467-020-19230-w
116. Li C, Wu S, Albright B, Hirsch M, Li W, Tseng YS, et al. Development of patient-specific AAV vectors after neutralizing antibody selection for enhanced muscle gene transfer. Mol Ther (2016) 24(1):53–65. doi: 10.1038/mt.2015.134
117. Ojala DS, Sun S, Santiago-Ortiz JL, Shapiro MG, Romero PA, Schaffer DV. In vivo selection of a computationally designed SCHEMA AAV library yields a novel variant for infection of adult neural stem cells in the SVZ. Mol Ther (2018) 26(1):304–19. doi: 10.1016/j.ymthe.2017.09.006
118. Paulk NK, Pekrun K, Zhu E, Nygaard S, Li B, Xu J, et al. Bioengineered AAV capsids with combined high human liver transduction in vivo and unique humoral seroreactivity. Mol Ther (2018) 26(1):289–303. doi: 10.1016/j.ymthe.2017.09.021
119. Lisowski L, Dane AP, Chu K, Zhang Y, Cunningham SC, Wilson EM, et al. Selection and evaluation of clinically relevant AAV variants in a xenograft liver model. Nature (2014) 506(7488):382–6. doi: 10.1038/nature12875
120. Scallan CD, Jiang H, Liu T, Patarroyo-White S, Sommer JM, Zhou S, et al. Human immunoglobulin inhibits liver transduction by AAV vectors at low AAV2 neutralizing titers in SCID mice. Blood (2006) 107(5):1810–7. doi: 10.1182/blood-2005-08-3229
121. Leborgne C, Barbon E, Alexander JM, Hanby H, Delignat S, Cohen DM, et al. IgG-cleaving endopeptidase enables in vivo gene therapy in the presence of anti-AAV neutralizing antibodies. Nat Med (2020) 26(7):1096–101. doi: 10.1038/s41591-020-0911-7
122. Elmore ZC, Oh DK, Simon KE, Fanous MM, Asokan A. Rescuing AAV gene transfer from neutralizing antibodies with an IgG-degrading enzyme. JCI Insight (2020) 5(19). doi: 10.1172/jci.insight.139881
123. Jordan SC, Lorant T, Choi J, Kjellman C, Winstedt L, Bengtsson M, et al. IgG endopeptidase in highly sensitized patients undergoing transplantation. N Engl J Med (2017) 377(5):442–53. doi: 10.1056/NEJMoa1612567
124. . Available at: https://ir.selectabio.com/news-releases/news-release-details/selecta-biosciences-and-genovis-enter-exclusive-license.
125. Chicoine LG, Montgomery CL, Bremer WG, Shontz KM, Griffin DA, Heller KN, et al. Plasmapheresis eliminates the negative impact of AAV antibodies on microdystrophin gene expression following vascular delivery. Mol Ther (2014) 22(2):338–47. doi: 10.1038/mt.2013.244
126. Monteilhet V, Saheb S, Boutin S, Leborgne C, Veron P, Montus MF, et al. A 10 patient case report on the impact of plasmapheresis upon neutralizing factors against adeno-associated virus (AAV) types 1, 2, 6, and 8. Mol Ther (2011) 19(11):2084–91. doi: 10.1038/mt.2011.108
127. Bertin B, Veron P, Leborgne C, Deschamps JY, Moullec S, Fromes Y, et al. Capsid-specific removal of circulating antibodies to adeno-associated virus vectors. Sci Rep (2020) 10(1):1–11. doi: 10.1038/s41598-020-57893-z
128. Orlowski A, Katz MG, Gubara SM, Fargnoli AS, Fish KM, Weber T. Successful transduction with AAV vectors after selective depletion of anti-AAV antibodies by immunoadsorption. Mol Ther-Methods Clin Dev (2020) 16:192–203. doi: 10.1016/j.omtm.2020.01.004
129. Salas D, Kwikkers KL, Zabaleta N, Bazo A, Petry H, van Deventer SJ, et al. Immunoadsorption enables successful rAAV5-mediated repeated hepatic gene delivery in nonhuman primates. Blood Adv (2019) 3(17):2632–41. doi: 10.1182/bloodadvances.2019000380
130. Mingozzi F, Anguela XM, Pavani G, Chen Y, Davidson RJ, Hui DJ, et al. Overcoming preexisting humoral immunity to AAV using capsid decoys. Sci Trans Med (2013) 5(194):194ra92–194ra92. doi: 10.1126/scitranslmed.3005795
131. Pei X, Earley LF, He Y, Chen X, Hall NE, Samulski RJ, et al. Efficient capsid antigen presentation from adeno-associated virus empty virions in vivo. Front Immunol (2018) 9:844. doi: 10.3389/fimmu.2018.00844
132. Aalbers CJ, Broekstra N, van Geldorp M, Kramer E, Ramiro S, Tak PP, et al. Empty capsids and macrophage inhibition/depletion increase rAAV transgene expression in joints of both healthy and arthritic mice. Hum Gene Ther (2017) 28(2):168–78. doi: 10.1089/hum.2016.036
133. Gao K, Li M, Zhong L, Su Q, Li J, Li S, et al. Empty virions in AAV8 vector preparations reduce transduction efficiency and may cause total viral particle dose-limiting side effects. Mol Ther-Methods Clin Dev (2014) 1:9. doi: 10.1038/mtm.2013.9
134. Flotte TR. Empty adeno-associated virus capsids: contaminant or natural decoy? Hum Gene Ther (2017) 28(2):147–8. doi: 10.1089/hum.2017.29039.trf
135. Lee GK, Maheshri N, Kaspar B, Schaffer DV. PEG conjugation moderately protects adeno-associated viral vectors against antibody neutralization. Biotechnol Bioeng (2005) 92(1):24–34. doi: 10.1002/bit.20562
136. Horowitz ED, Weinberg MS, Asokan A. Glycated AAV vectors: chemical redirection of viral tissue tropism. Bioconjugate Chem (2011) 22(4):529–32. doi: 10.1021/bc100477g
137. Le HT, Yu QC, Wilson JM, Croyle MA. Utility of PEGylated recombinant adeno-associated viruses for gene transfer. J Controlled Release (2005) 108(1):161–77. doi: 10.1016/j.jconrel.2005.07.019
138. Mével M, Bouzelha M, Leray A, Pacouret S, Guilbaud M, Penaud-Budloo M, et al. Chemical modification of the adeno-associated virus capsid to improve gene delivery. Chem Sci (2020) 11(4):1122–31. doi: 10.1039/C9SC04189C
139. Katrekar D, Moreno AM, Chen G, Worlikar A, Mali P. Oligonucleotide conjugated multi-functional adeno-associated viruses. Sci Rep (2018) 8(1):1–8. doi: 10.1038/s41598-018-21742-x
140. Yao T, Zhou X, Zhang C, Yu X, Tian Z, Zhang L, et al. Site-specific PEGylated adeno-associated viruses with increased serum stability and reduced immunogenicity. Molecules (2017) 22(7):1155. doi: 10.3390/molecules22071155
141. Rezaie J, Ajezi S, Avci ÇB, Karimipour M, Geranmayeh MH, Nourazarian A, et al. Exosomes and their application in biomedical field: difficulties and advantages. Mol Neurobiol (2018) 55(4):3372–93. doi: 10.1007/s12035-017-0582-7
142. Meliani A, Boisgerault F, Fitzpatrick Z, Marmier S, Leborgne C, Collaud F, et al. Enhanced liver gene transfer and evasion of preexisting humoral immunity with exosome-enveloped AAV vectors. Blood Adv (2017) 1(23):2019–31. doi: 10.1182/bloodadvances.2017010181
143. Hudry E, Martin C, Gandhi S, György B, Scheffer DI, Mu D, et al. Exosome-associated AAV vector as a robust and convenient neuroscience tool. Gene Ther (2016) 23(4):380–92. doi: 10.1038/gt.2016.11
144. György B, Fitzpatrick Z, Crommentuijn MH, Mu D, Maguire CA. Naturally enveloped AAV vectors for shielding neutralizing antibodies and robust gene delivery in vivo. Biomaterials (2014) 35(26):7598–609. doi: 10.1016/j.biomaterials.2014.05.032
145. György B, Sage C, Indzhykulian AA, Scheffer DI, Brisson AR, Tan S, et al. Rescue of hearing by gene delivery to inner-ear hair cells using exosome-associated AAV. Mol Ther (2017) 25(2):379–91. doi: 10.1016/j.ymthe.2016.12.010
146. Wassmer SJ, Carvalho LS, György B, Vandenberghe LH, Maguire CA. Exosome-associated AAV2 vector mediates robust gene delivery into the murine retina upon intravitreal injection. Sci Rep (2017) 7(1):1–10. doi: 10.1038/srep45329
147. Pegtel DM, Peferoen L, Amor S. Extracellular vesicles as modulators of cell-to-cell communication in the healthy and diseased brain. Philos Trans R Soc B: Biol Sci (2014) 369(1652):20130516. doi: 10.1098/rstb.2013.0516
148. Schiller LT, Lemus-Diaz N, Ferreira RR, Böker KO, Gruber J. Enhanced production of exosome-associated AAV by overexpression of the tetraspanin CD9. Mol Ther-Methods Clin Dev (2018) 9:278–87. doi: 10.1016/j.omtm.2018.03.008
149. Rojas LA, Condezo GN, Moreno R, Fajardo CA, Arias-Badia M, San Martín C, et al. Albumin-binding adenoviruses circumvent pre-existing neutralizing antibodies upon systemic delivery. J Control Release (2016) 237:78–88. doi: 10.1016/j.jconrel.2016.07.004
150. Xiang Z, Kurupati RK, Li Y, Kuranda K, Zhou X, Mingozzi F, et al. The effect of CpG sequences on capsid-specific CD8+ T cell responses to AAV vector gene transfer. Mol Ther (2020) 28(3):771–83. doi: 10.1016/j.ymthe.2019.11.014
151. Faust SM, Bell P, Cutler BJ, Ashley SN, Zhu Y, Rabinowitz JE, et al. CpG-depleted adeno-associated virus vectors evade immune detection. J Clin Invest (2013) 123(7):2994–3001. doi: 10.1172/JCI68205
152. Bertolini TB, Shirley JL, Zolotukhin I, Li X, Kaisho T, Xiao W, et al. Effect of CpG depletion of vector genome on CD8+ T cell responses in AAV gene therapy. Front Immunol (2021) 12:1618. doi: 10.3389/fimmu.2021.672449
153. Wright JF. Codon modification and PAMPs in clinical AAV vectors: the tortoise or the hare? Mol Ther (2020) 28(3):701–3. doi: 10.1016/j.ymthe.2020.01.026
154. Alexaki A, Hettiarachchi GK, Athey JC, Katneni UK, Simhadri V, Hamasaki-Katagiri N, et al. Effects of codon optimization on coagulation factor IX translation and structure: Implications for protein and gene therapies. Sci Rep (2019) 9(1):1–15. doi: 10.1038/s41598-019-51984-2
155. Pan X, Yue Y, Boftsi M, Wasala LP, Tran NT, Zhang K, et al. Rational engineering of a functional CpG-free ITR for AAV gene therapy. Gene Ther (2022) 29(6):333–45. doi: 10.1038/s41434-021-00296-0
156. Kaminski JJ, Schattgen SA, Tzeng TC, Bode C, Klinman DM, Fitzgerald KA. Synthetic oligodeoxynucleotides containing suppressive TTAGGG motifs inhibit AIM2 inflammasome activation. J Immunol (2013) 191(7):3876–83. doi: 10.4049/jimmunol.1300530
157. Steinhagen F, Zillinger T, Peukert K, Fox M, Thudium M, Barchet W, et al. Suppressive oligodeoxynucleotides containing TTAGGG motifs inhibit cGAS activation in human monocytes. Eur J Immunol (2018) 48(4):605–11. doi: 10.1002/eji.201747338
158. Chan YK, Wang SK, Chu CJ, Copland DA, Letizia AJ, Costa Verdera H, et al. Engineering adeno-associated viral vectors to evade innate immune and inflammatory responses. Sci Trans Med (2021) 13(580):eabd3438. doi: 10.1126/scitranslmed.abd3438
159. Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol (2008) 8(7):523–32. doi: 10.1038/nri2343
160. Markusic DM, Hoffman BE, Perrin GQ, Nayak S, Wang X, LoDuca PA, et al. Effective gene therapy for haemophilic mice with pathogenic factor IX antibodies. EMBO Mol Med (2013) 5(11):1698–709. doi: 10.1002/emmm.201302859
161. Breous E, Somanathan S, Vandenberghe LH, Wilson JM. Hepatic regulatory T cells and kupffer cells are crucial mediators of systemic T cell tolerance to antigens targeting murine liver. Hepatology (2009) 50(2):612–21. doi: 10.1002/hep.23043
162. Brantly ML, Chulay JD, Wang L, Mueller C, Humphries M, Spencer LT, et al. Sustained transgene expression despite T lymphocyte responses in a clinical trial of rAAV1-AAT gene therapy. Proc Natl Acad Sci (2009) 106(38):16363–8. doi: 10.1073/pnas.0904514106
163. Flotte TR, Trapnell BC, Humphries M, Carey B, Calcedo R, Rouhani F, et al. Phase 2 clinical trial of a recombinant adeno-associated viral vector expressing α1-antitrypsin: interim results. Hum Gene Ther (2011) 22(10):1239–47. doi: 10.1089/hum.2011.053
164. Ferreira V, Twisk J, Kwikkers K, Aronica E, Brisson D, Methot J, et al. Immune responses to intramuscular administration of alipogene tiparvovec (AAV1-LPLS447X) in a phase II clinical trial of lipoprotein lipase deficiency gene therapy. Hum Gene Ther (2014) 25(3):180–8. doi: 10.1089/hum.2013.169
165. Gross DA, Leboeuf M, Gjata B, Danos O, Davoust J. CD4+ CD25+ regulatory T cells inhibit immune-mediated transgene rejection. Blood (2003) 102(13):4326–8. doi: 10.1182/blood-2003-05-1454
166. Sarkar D, Biswas M, Liao G, Seay HR, Perrin GQ, Markusic DM, et al. Ex vivo expanded autologous polyclonal regulatory T cells suppress inhibitor formation in hemophilia. Mol Ther-Methods Clin Dev (2014) 1:14030. doi: 10.1038/mtm.2014.30
167. Arjomandnejad M, Sylvia K, Blackwood M, Nixon T, Tang Q, Muhuri M, et al. Modulating immune responses to AAV by expanded polyclonal T-regs and capsid specific chimeric antigen receptor T-regulatory cells. Mol Ther-Methods Clin Dev (2021) 23:490–506. doi: 10.1016/j.omtm.2021.10.010
168. Finn JD, Ozelo MC, Sabatino DE, Franck HW, Merricks EP, Crudele JM, et al. Eradication of neutralizing antibodies to factor VIII in canine hemophilia a after liver gene therapy. Blood (2010) 116(26):5842–8. doi: 10.1182/blood-2010-06-288001
169. Crudele JM, Finn JD, Siner JI, Martin NB, Niemeyer GP, Zhou S, et al. AAV liver expression of FIX-padua prevents and eradicates FIX inhibitor without increasing thrombogenicity in hemophilia b dogs and mice. Blood (2015) 125(10):1553–61. doi: 10.1182/blood-2014-07-588194
170. Zhang P, Sun B, Osada T, Rodriguiz R, Yang XY, Luo X, et al. Immunodominant liver-specific expression suppresses transgene-directed immune responses in murine pompe disease. Hum Gene Ther (2012) 23(5):460–72. doi: 10.1089/hum.2011.063
171. Han SO, Ronzitti G, Arnson B, Leborgne C, Li S, Mingozzi F, et al. Low-dose liver-targeted gene therapy for pompe disease enhances therapeutic efficacy of ERT via immune tolerance induction. Mol Ther-Methods Clin Dev (2017) 4:126–36. doi: 10.1016/j.omtm.2016.12.010
172. Hakim CH, Kumar SR, Pérez-López DO, Wasala NB, Zhang D, Yue Y, et al. Cas9-specific immune responses compromise local and systemic AAV CRISPR therapy in multiple dystrophic canine models. Nat Commun (2021) 12(1):1–12. doi: 10.1038/s41467-021-26830-7
173. Xiao Y, Muhuri M, Li S, Qin W, Xu G, Luo L, et al. Circumventing cellular immunity by miR142-mediated regulation sufficiently supports rAAV-delivered OVA expression without activating humoral immunity. JCI Insight (2019) 4(13). doi: 10.1172/jci.insight.99052
174. Boisgerault F, Gross DA, Ferrand M, Poupiot J, Darocha S, Richard I, et al. Prolonged gene expression in muscle is achieved without active immune tolerance using microrRNA 142.3 p-regulated rAAV gene transfer. Hum Gene Ther (2013) 24(4):393–405. doi: 10.1089/hum.2012.208
175. Ahn K, Gruhler A, Galocha B, Jones TR, Wiertz EJ, Ploegh HL, et al. The ER-luminal domain of the HCMV glycoprotein US6 inhibits peptide translocation by TAP. Immunity (1997) 6(5):613–21. doi: 10.1016/S1074-7613(00)80349-0
176. Shao W, Chen X, Samulski RJ, Hirsch ML, Li C. Inhibition of antigen presentation during AAV gene therapy using virus peptides. Hum Mol Genet (2018) 27(4):601–13. doi: 10.1093/hmg/ddx427
177. Peden CS, Burger C, Muzyczka N, Mandel RJ. Circulating anti-wild-type adeno-associated virus type 2 (AAV2) antibodies inhibit recombinant AAV2 (rAAV2)-mediated, but not rAAV5-mediated, gene transfer in the brain. J Virol (2004) 78(12):6344–59. doi: 10.1128/JVI.78.12.6344-6359.2004
178. Xiao W, Chirmule N, Berta SC, McCullough B, Gao G, Wilson JM. Gene therapy vectors based on adeno-associated virus type 1. J Virol (1999) 73(5):3994–4003. doi: 10.1128/JVI.73.5.3994-4003.1999
179. Halbert CL, Rutledge EA, Allen JM, Russell DW, Miller AD. Repeat transduction in the mouse lung by using adeno-associated virus vectors with different serotypes. J Virol (2000) 74(3):1524–32. doi: 10.1128/JVI.74.3.1524-1532.2000
180. Riviere C, Danos O, Douar AM. Long-term expression and repeated administration of AAV type 1, 2 and 5 vectors in skeletal muscle of immunocompetent adult mice. Gene Ther (2006) 13(17):1300–8. doi: 10.1038/sj.gt.3302766
181. Bočkor L, Bortolussi G, Iaconcig A, Chiaruttini G, Tiribelli C, Giacca M, et al. Repeated AAV-mediated gene transfer by serotype switching enables long-lasting therapeutic levels of hUgt1a1 enzyme in a mouse model of crigler–najjar syndrome type I. Gene Ther (2017) 24(10):649–60. doi: 10.1038/gt.2017.75
182. De BP, Heguy A, Hackett NR, Ferris B, Leopold PL, Lee J, et al. High levels of persistent expression of α1-antitrypsin mediated by the nonhuman primate serotype rh. 10 adeno-associated virus despite preexisting immunity to common human adeno-associated viruses. Mol Ther (2006) 13(1):67–76. doi: 10.1016/j.ymthe.2005.09.003
183. Peden CS, Manfredsson FP, Reimsnider SK, Poirier AE, Burger C, Muzyczka N, et al. Striatal readministration of rAAV vectors reveals an immune response against AAV2 capsids that can be circumvented. Mol Ther (2009) 17(3):524–37. doi: 10.1038/mt.2008.284
184. Wang L, Calcedo R, Nichols TC, Bellinger DA, Dillow A, Verma IM, et al. Sustained correction of disease in naive and AAV2-pretreated hemophilia b dogs: AAV2/8-mediated, liver-directed gene therapy. Blood (2005) 105(8):3079–86. doi: 10.1182/blood-2004-10-3867
185. Hinderer C, Bell P, Louboutin JP, Zhu Y, Yu H, Lin G, et al. Neonatal systemic AAV induces tolerance to CNS gene therapy in MPS I dogs and nonhuman primates. Mol Ther (2015) 23(8):1298–307. doi: 10.1038/mt.2015.99
186. Ou L, Przybilla MJ, Tăbăran AF, Overn P, O’Sullivan MG, Jiang X, et al. A novel gene editing system to treat both Tay–Sachs and sandhoff diseases. Gene Ther (2020) 27(5):226–36. doi: 10.1038/s41434-019-0120-5
187. Martino AT, Markusic DM. Immune response mechanisms against AAV vectors in animal models. Mol Ther-Methods Clin Dev (2020) 17:198–208. doi: 10.1016/j.omtm.2019.12.008
188. Majowicz A, Nijmeijer B, Lampen MH, Spronck L, de Haan M, Petry H, et al. Therapeutic hFIX activity achieved after single AAV5-hFIX treatment in hemophilia b patients and NHPs with pre-existing anti-AAV5 NABs. Mol Ther-Methods Clin Dev (2019) 14:27–36. doi: 10.1016/j.omtm.2019.05.009
189. Long BR, Sandza K, Holcomb J, Crockett L, Hayes GM, Arens J, et al. The impact of pre-existing immunity on the non-clinical pharmacodynamics of AAV5-based gene therapy. Mol Ther-Methods Clin Dev (2019) 13:440–52. doi: 10.1016/j.omtm.2019.03.006
190. Bucher K, Rodríguez-Bocanegra E, Dauletbekov D, Fischer MD. Immune responses to retinal gene therapy using adeno-associated viral vectors–implications for treatment success and safety. Prog Retinal Eye Res (2021) 83:100915. doi: 10.1016/j.preteyeres.2020.100915
191. Treleaven CM, Tamsett TJ, Bu J, Fidler JA, Sardi SP, Hurlbut GD, et al. Gene transfer to the CNS is efficacious in immune-primed mice harboring physiologically relevant titers of anti-AAV antibodies. Mol Ther (2012) 20(9):1713–23. doi: 10.1038/mt.2012.114
192. Sanftner LM, Suzuki BM, Doroudchi MM, Feng L, McClelland A, Forsayeth JR, et al. Striatal delivery of rAAV-hAADC to rats with preexisting immunity to AAV. Mol Ther (2004) 9(3):403–9. doi: 10.1016/j.ymthe.2003.12.005
193. Garbuzova-Davis S, Louis MK, Haller EM, Derasari HM, Rawls AE, Sanberg PR. Blood-brain barrier impairment in an animal model of MPS III b. PloS One (2011) 6(3):e16601. doi: 10.1371/journal.pone.0016601
194. Al-Bachari S, Naish JH, Parker GJ, Emsley HC, Parkes LM. Blood–brain barrier leakage is increased in parkinson’s disease. Front Physiol (2020) 11:593026. doi: 10.3389/fphys.2020.593026
195. George LA, Ragni MV, Rasko JE, Raffini LJ, Samelson-Jones BJ, Ozelo M, et al. Long-term follow-up of the first in human intravascular delivery of AAV for gene transfer: AAV2-hFIX16 for severe hemophilia b. Mol Ther (2020) 28(9):2073–82. doi: 10.1016/j.ymthe.2020.06.001
196. Mendell JR, Campbell K, Rodino-Klapac L, Sahenk Z, Shilling C, Lewis S, et al. Dystrophin immunity in duchenne's muscular dystrophy. New Engl J Med (2010) 363(15):1429–37. doi: 10.1056/NEJMoa1000228
197. Calcedo R, Somanathan S, Qin Q, Betts MR, Rech AJ, Vonderheide RH, et al. Class I-restricted T-cell responses to a polymorphic peptide in a gene therapy clinical trial for α-1-antitrypsin deficiency. Proc Natl Acad Sci (2017) 114(7):1655–9. doi: 10.1073/pnas.1617726114
198. Tardieu M, Zérah M, Gougeon ML, Ausseil J, de Bournonville S, Husson B, et al. Intracerebral gene therapy in children with mucopolysaccharidosis type IIIB syndrome: an uncontrolled phase 1/2 clinical trial. Lancet Neurol (2017) 16(9):712–20. doi: 10.1016/S1474-4422(17)30169-2
199. Cao O, Hoffman BE, Moghimi B, Nayak S, Cooper M, Zhou S, et al. Impact of the underlying mutation and the route of vector administration on immune responses to factor IX in gene therapy for hemophilia b. Mol Ther (2009) 17(10):1733–42. doi: 10.1038/mt.2009.159
200. Ferrand M, Galy A, Boisgerault F. A dystrophic muscle broadens the contribution and activation of immune cells reacting to rAAV gene transfer. Gene Ther (2014) 21(9):828–39. doi: 10.1038/gt.2014.61
201. Sun J, Shao W, Chen X, Merricks EP, Wimsey L, Abajas YL, et al. An observational study from long-term AAV re-administration in two hemophilia dogs. Mol Ther-Methods Clin Dev (2018) 10:257–67. doi: 10.1016/j.omtm.2018.07.011
Keywords: adeno-associated virus (AAV), gene therapy, immunity, toxicity, immunosuppressant, capsid engineering, exosome, CpG
Citation: Li X, Wei X, Lin J and Ou L (2022) A versatile toolkit for overcoming AAV immunity. Front. Immunol. 13:991832. doi: 10.3389/fimmu.2022.991832
Received: 12 July 2022; Accepted: 17 August 2022;
Published: 02 September 2022.
Edited by:
Arun Srivastava, University of Florida, United StatesReviewed by:
Moanaro Biswas, Indiana University, United StatesDongsheng Duan, University of Missouri, United States
Ashley Thomas Martino, St. John’s University, United States
Copyright © 2022 Li, Wei, Lin and Ou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Ou, b3V4eHgwNDVAdW1uLmVkdQ==
†These authors have contributed equally to this work