 Xiaoqin Zheng1
Xiaoqin Zheng1 Gang Tan
Gang Tan- 1Department of Ophthalmology, The First Affiliated Hospital, Hengyang Medical School, University of South China, Hengyang, Hunan, China
- 2Department of Orthopedics, The Second Xiangya Hospital, Central South University, Changsha, Hunan, China
In the working-age population worldwide, diabetic retinopathy (DR), a prevalent complication of diabetes, is the main cause of vision impairment. Chronic low-grade inflammation plays an essential role in DR development. Recently, concerning the pathogenesis of DR, the Nod-Like Receptor Family Pyrin Domain Containing 3 (NLRP3) inflammasome in retinal cells has been determined as a causal factor. In the diabetic eye, the NLRP3 inflammasome is activated by several pathways (such as ROS and ATP). The activation of NPRP3 leads to the secretion of inflammatory cytokines interleukin-1β (IL-1β) and interleukin-18 (IL-18), and leads to pyroptosis, a rapid inflammatory form of lytic programmed cell death (PCD). Cells that undergo pyroptosis swell and rapture, releasing more inflammatory factors and accelerating DR progression. This review focuses on the mechanisms that activate NLRP3 inflammasome and pyroptosis leading to DR. The present research highlighted some inhibitors of NLRP3/pyroptosis pathways and novel therapeutic measures concerning DR treatment.
1 Introduction
Diabetic retinopathy (DR), a prevalent diabetes mellitus-related complication, is developed by complex pathophysiological mechanisms triggered by hyperglycemia. The early stages of DR are known as non-proliferative diabetic retinopathy (NPDR) and are characterized by increased vascular permeability, retinal hemorrhage and edema, and the formation of microaneurysms (1). NPDR progresses to a more severe stage of the disease, called proliferative diabetic retinopathy (PDR), which is characterized by the formation of pathological retinal neovascularization and eventually lead to retinal detachment and severely compromise vision (2). In the past, DR has been considered a vascular lesion, but in recent years it has been found that retinal nerve cell dysfunction has been observed prior to retinal vasculopathy, so DR is now considered as a neurovascular lesion (3). There is growing evidence that inflammation is a key player in DR, as high glucose-induced production of advanced glycosylated substances, oxidative stress and vascular endothelial growth factor (VEGF) all contribute to the inflammatory response (4), and chronic low-grade inflammation is detected in all stages of DR (5, 6). One of the most studied is the Nod-Like Receptor Family Pyrin Domain Containing 3 (NLRP3) inflammasome (5, 7), the activation of which not only induces the release of inflammatory cytokines interleukin-18 (IL-18) and interleukin-1β (IL-1β), but also pyroptosis, which releases large amounts of inflammatory cytokines, inducing inflammatory cell death in various retinal cells and accelerating the progression of DR (8–11). Retinal cell death is an essential feature of DR (12). However, most previous studies have focused on apoptosis, necrosis, and autophagy. Therefore, in this review, we focus on the activation mechanism of NLRP3 inflammasome/pyroptosis and its significance in DR, and some certain inhibitors of NLRP3 and pyroptosis were examined concerning their possible therapeutic effect on DR.
2 NLRP3: a star target in the field of inflammation
The NLRP3 inflammasome is a cytoplasmic immune factor that responds to cellular stress signals and constitutes a sensor (NLRP3), an adapter protein (ASC), and an effector (caspase-1). The ASC protein has a PYD as well as a caspase recruitment domain (CARD). In contrast the NLRP3 is a tripartite protein that consists of a central NACHT domain, a carboxy-terminal leucine-rich repeat (LRR) domain, and amino-terminal pyrin domain (PYD) (13). NLRP3 inflammasomes are typically activated by PAMP (such as microbial toxins, viral RNA, and surface components of bacteria) and DAMP (including ATP, uric acid crystals, beta-amyloid peptides, and aluminum adjuvants) (14, 15). NLRP3 interacts with the homotypic NACHT structural domain upon stimuli to undergo self-oligomerization with the oligomerized NLRP3 causing the bound ASC, attracted through homotypic PYD-PYD interactions, to aggregate into ASC specks (macromolecular focal points) (16–18). Afterward, the assembled ASC recruit caspase-1 via homotypic CARD-CARD interactions to generate the NLRP3-ASC-caspase-1 protein complex, called NLRP3 inflammasome (19). The activated NLRP3 inflammasome triggers self-cleavage and activates pro-caspase-1, causing the release of the pro-inflammatory cytokines IL-1β/18. Caspase-1 also cleaves gasdermin D (GSDMD) into N-terminus GSDMD (N-GSDMD), which causes the formation of pores in the cell membrane and triggers pyroptosis, releasing inflammatory factors IL-1β/18 to further promote the inflammatory response (20). Numerous ocular tissues and cells express NLRP3 inflammasome, which is elevated in various ocular disorders (21, 22).
2.1 Priming the NLRP3 inflammasome
At rest, the expression of NLRP3 and IL-1β in macrophages is minimal and cannot be used to assemble or activate NLRP3 inflammasomes, so their activation must be highly regulated. With few exceptions (23, 24), two steps constitute the NLRP3 inflammasome activation (Figure 1): priming (Signal 1) and NLRP3 inflammasome assembly (Signal 2). The priming process has two known functions. The first is to elevate the transcriptional levels of NLRP3 and pro-1L-1β/IL-18. This transcriptional upregulation is triggered by granulocyte-macrophage colony-stimulating factor (GM-CSF) receptors, nucleotide-binding oligomeric structural domain protein 2 (NOD2), TNF receptors TNFR1 and TNFR2, and toll-like receptors (TLRs), which could recognize PAMPs or DAMPs and activate the nuclear factor-κB (NF-κB) (25–27). The second function of priming is to induce post-translational modifications (PTM) of NLRP3, including phosphorylation, ubiquitination, and SUMOylation (28–30), which stabilizes NLRP3 in a signal-competent but auto-suppressed inactive state (14, 23).
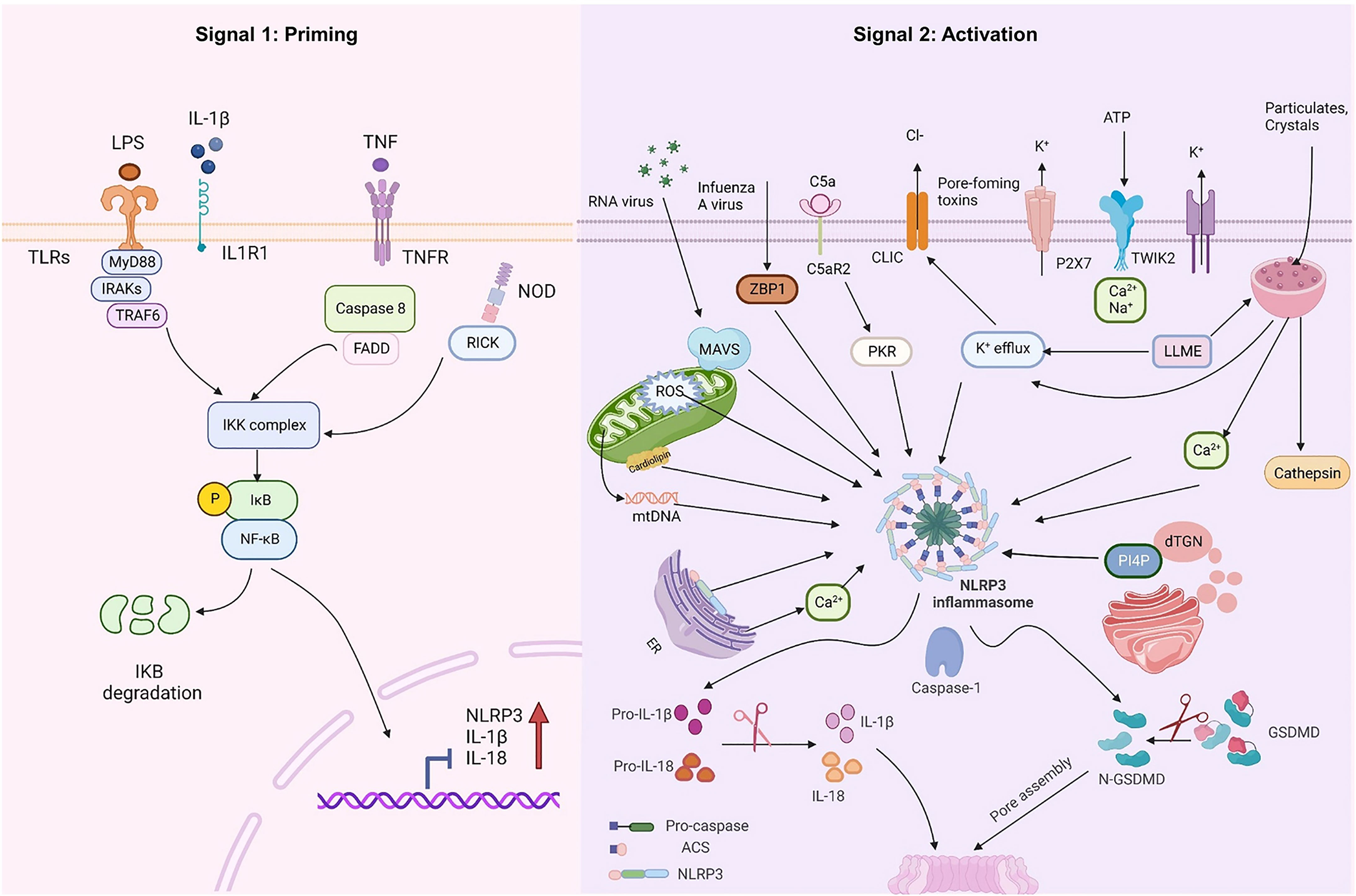
Figure 1 NLRP3 inflammasome priming and activation. The signal 1 (priming, left) is induced by toll-like receptors (TLRs), nucleotide-binding oligomeric structural domain protein (NOD) and tumor necrosis factor receptors (TNFR), which could recognize pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) and upregulate the transcriptional levels of NOD-like receptor thermal protein domain associated protein 3 (NLRP3), proinflammatory cytokines interleukin 1β (IL-1β) and interleukin 18 (IL-18) via the myd88-NF-κB pathway. Signal 2 (activation, right) is induced by various PAMPs or DAMPs, such as particulates, crystals, adenosine triphosphate (ATP), K+ and Cl- efflux, the disruption of lysosomal, the dysfunction of mitochondrial and the production of mitochondrial reactive oxygen species (mtROS) and mitochondrial DNA (mtDNA), NLRP3 is also activated via RNA viruses via mitochondrial antiviral signaling protein (MAVS). Formation of the NLRP3 inflammasome activates caspase 1, which subsequently cleaves pro-IL-1β/IL-18 to IL-1β/18. In addition, GSDMD is also cleaved by caspase-1 and inserts into the membrane, causing pores and pyroptosis. MyD88 myeloiddifferentiationfactor88, IRAKs interleukin-1 receptor-associated kinases, TRAF6 TNF Receptor Associated Factor 6, FADD Fas-associated with death domain protein, IKK inhibitor of Kappa B Kinase, IKB inhibitor kappa B. Created with BioRender.com.
2.2 Signals for activation of the NLRP3 inflammasomes
After the inflammasome receives the priming signal, the next step involves the recognition of agonists and assembling and activating the NLRP3 inflammasome. While most pattern recognition receptors (PRRs) can only be activated in response to one or a few structurally similar PAMPs or DAMPs, the activation of NLRP3 can be induced under multiple unrelated stimuli, including viral, bacterial, and fungal infections, as well as exposure to environmental irritants and endogenous DAMP-mediated sterile inflammation. These factors are unified in terms of causing cellular stress, which is sensed by NLRP3. Various molecular and cellular processes have been suggested as upstream signals for inflammasome assembly and activation, that include ion fluxes (e.g., Ca2+ mobilization, K+ and Cl- efflux), lysosomal disruption, mitochondrial dysfunction, reactive oxygen species (ROS), and mitochondrial DNA (mtDNA) release, metabolic changes and dispersed trans-Golgi (31–36). Recent research has indicated that the NLRP3 inflammasome can also be activated via various mechanisms including the complement system, protein kinase R (PKR) (37, 38), purine receptor (39), necroptotic signaling, and Z-DNA-binding protein 1 (ZBP1) (40–42). Numerous diabetes-linked metabolic factors act as the secondary signals for NLRP3 activation in DR. These signals include adenosine triphosphate (ATP), cholesterol levels, and cellular structural instability, such as lysosomal rupture, dysfunction of the mitochondrial, and molecular or ionic perturbations including K efflux, ROS, and Ca2+ signaling (43). Despite multiple upstream activation events, several pathways are interrelated and overlapped, with ambivalence between the data. Therefore, a consistent model is still unavailable for the activation of NLRP3.
i) Ion flux pathway
K efflux is considered a general upstream event in the activation of the NLRP3 inflammasome. It was observed during the activation of most NLRP3 inflammasomes, except for peptidoglycan, imiquimod, and the related molecule CL097, which can be independent of K efflux (44, 45). However, different activators induced K efflux by diverse pathways. For example, ATP causes K efflux by opening cation channels. Moreover, IL-1β maturation is promoted through K efflux by the purinergic receptor family’s ion channel known as ATP gating of the P2X7 receptor (P2X7R) (46). A recent study has revealed that P2X7 does not function as a cation channel for K efflux even though P2X family receptors are membrane nonselective ion channels for Ca2+, Na+ and K+ (47). P2X7 stimulates the influx of Ca2+ and Na+ after ATP activation and co-ordinates with the K+ efflux-mediated channel tandem pore domain in weak inward rectifying K channel 2 (TWIK2) (48). Subsequently, it induces the binding of downstream NIMA-related kinases (NEK7) to NLRP3, triggering the NLRP3 inflammasome activation (48). A Streptomyces hygroscopicus-derived antibiotic, nigericin, serves as a K+/H+ antiport ionophore that controls the membrane exchange of K+ for H+ across most membranes (49). Moreover, the complement membrane attack complexes or pore-forming microbial toxins induced damage to the cell membrane integrity can also directly result in K efflux (50, 51). Besides ATP and pore-forming toxins, some particles such as cholesterol crystals, calcium pyrophosphate crystals, and silica also trigger K efflux, essential for activating the NLRP3 inflammasome (31).
For calcium signaling, releasing endoplasmic reticulum (ER)-linked intracellular Ca2+ stores or opening plasma membrane channels to allow Ca2+ fluxes into the cytoplasm facilitates the NLRP3 complex formation. Moreover, calcium flux and K efflux are usually coordinated when activating the NLRP3 inflammasome. For instance, ATP induces weak Ca2+ inward flow and coordinates K+ outward flow via its receptor P2X7 (48). Subsequently, the release of ER-linked Ca2+ is promoted by K efflux (32, 52). The activation of NLRP3 promoted by monosodium urate crystals, alum, nigericin, and membrane attack complexes is dependent on K+ efflux and Ca2+ flux (3, 53). Additionally, aside from the K+ and Ca2+, the Cl- channels are also implicated in activating the NLRP3 inflammasome. The most convincing data is that a class of non-steroidal anti-inflammatory drugs (NSAIDs) prevents Cl- migration via inhibiting NLRP3 activation by blocking the volume-regulated anion channel (VRAC) on the plasma membrane (54). Similarly, the translocation of Cl- intracellular channel proteins CLIC1 and CLIC4 to the plasma membrane occurs where they mediate Cl- efflux and participate in NLRP3 inflammasome activation (55, 56). In summary, activating the NLRP3 inflammasome is a complex process resulting from a combination of cellular and molecular effects, with many influencing factors. Changes in the concentration of either ion may affect the activation process, and more research is required to understand precisely how ion movement coordinates with NLRP3 activation.
ii) Mitochondrial dysfunction
Mitochondria is emerging as the focal organelle for the activation of the NLRP3 inflammasome, acting as a docking site for its assembly, danger signal release, mtROS production, etc. (57–59). In the resting state, NLRP3 is localized in the ER, and ASC is dispersed in the cytoplasm. During particle stimulation or activation of the inflammasome by the ion carrier Nigerian bacteriocin, acetylated α-microtubule promotes microtubule-dependent mitochondrial translocation to the ER, which can contribute to inflammasome activation in conjunction with ASC on mitochondria and NLRP3 on the ER (35, 60). At least three proteins are thought to act as linkages between NLRP3 and mitochondria: mitochondrial antiviral signaling protein (MAVS), cardiolipin, and mitofusin 2. An inner membrane phospholipid of the mitochondria, the cardiolipin is exposed to the outer membrane in response to mitochondrial stress and functions as a binding site for the molecules linked with autophagy and apoptosis (61). Additionally, cardiolipin independently binds to caspase-1 and NLRP3, and these associations could activate the inflammasome (57, 59). The second protein, MAVS, is articulatory in the RNA-sensing pathway that is essential in triggering the NLRP3 inflammasome during RNA virus infection and following synthetic RNA polyinosinic-polycytidylic acid stimulation (62, 63). It recruits NLRP3, directing its position towards mitochondria for inflammasome activation (64). Finally, mitofusin 2, found at the ER and MAM contact sites, forms a complex with MAVS during RNA virus infection and contributes to the localization of NLRP3 in mitochondria (65).
Besides, ROS is released in mitochondria continuously because of the by-product of oxidative phosphorylation. Even though mtROS level increases during cellular oxidative stress, the damaged or dysfunctioning mitochondria removal can be achieved through mitochondrial autophagy, thus attenuating mtROS production. Inhibiting mitochondrial autophagy or excessive mitochondrial damage increases activation of the NLRP3 inflammasome (35), indicating the participation of autophagy in the regulation of NLRP3. Imiquimod and related compound CL097 target mitochondria to produce mtROS, activating the NLRP3 inflammasome independent of K efflux (44). The activation of the NLRP3 inflammasome signaling in macrophages by phosphatidylcholine oxidation under cellular stress and injury occurs via mtROS downstream of intracellular Ca2+ signaling (66). Besides the mtROS release and dysfunctioning of mitochondrial, mtDNA could also serve as DAMPs and activate NLRP3 (67–69). Collectively, mtROS and Ca2+ could open mitochondrial permeability transition (MPT) pores during oxidative stress (70). The cytoplasmic release of mtDNA depends on the MPT pore and mtROS (67). Upon stimulation by various NLRP3 inflammasome activators, the cytoplasmic release of mtDNA occurs rapidly and is subsequently oxidized. The oxidized mtDNA can then immunoprecipitate with NLRP3 inflammasomes, thereby activating them. In contrast, non-oxidized mtDNA preferentially stimulates the AIM2 inflammasome (69).
iii) Lysosomal disruption
Lysosomal damage is an important factor in activating the NLRP3 inflammasome, which is closely associated to cellular phagocytosis of granules. Endogenous particles (including cholesterol, Monosodium Urate (MSU) crystals, or amyloid β aggregates and deoxygenated sphingolipid-based lipid crystals) or exogenous particulate matter (e.g., alum, silica, and asbestos) are phagocytosed by the lysosomes and accumulated in the lysosomal lumen, leading to greater acidification and swelling of the lysosome and loss of integrity of the lysosomal membrane and release of cathepsin, thus leading to the activation of NLRP3 inflammasome (71). However, the activation of NLRP3 is not activated by a certain cathepsin, as this pathway is only activated in the absence of multiple cathepsins (72). A soluble enzyme regulator, the Leu-Leu-O-methyl ester (LLME), causes the activation of NLRP3 inflammasome through induced lysosomal rupture and increased K efflux (31, 71). It has also been shown that the Ca2+-CaMKII-TAK1-JNK pathway promotes the oligomerization of ASC and regulates the activation of NLRP3 inflammasome during lysosome rupture (73). These studies highlight a synergistic effect of lysosomes and ion flow channels in activating the NLRP3 inflammasome.
iv) Trans-Golgi disassembly
The Golgi apparatus and its lipid mediators play a crucial role in activating the NLRP3 inflammasome. NLRP3 stimuli enhance the trans-Golgi network disassembly into vesicles, known as dispersed trans-Golgi network (dTGN), in cell reconstruction systems. The subsequent recruitment and aggregation of NLRP3 are mediated by the phosphatidylinositol-4-phosphate (PI4P) on the dTGN, which is necessary for downstream oligomerization of ASC and activation of caspase-1 (74). Even though the K efflux-independent stimuli (imiquimod, target mitochondria) and K efflux-dependent stimuli (Nigerian bacteriocin) both form dTGN and recruit NLRP3, K efflux is just required for the latter function. This observation indicates the distinct convergence of the K efflux- and mitochondria-dependent activation of NLRP3 on Golgi disassembly (74). Research indicated that NLRP3 inflammasome activation depends on sterol regulatory element binding protein 2 (SREBP2) and SREBP cleavage-activating protein (SCAP). SCAP-SREBP2 forms a ternary complex with NLRP3, which translocates into the mitochondria adjacent Golgi apparatus for optimal inflammasome assembly (75). Therefore, NLRP3 activators can trigger various molecular and cellular events, such as mitochondrial dysfunction, ion flux, and lysosomal leakage. Nonetheless, the function of these processes in NLRP3 activation remains largely unclear.
3 Pyroptosis
Inflammasome activation triggers the release and maturation of IL-1β/18 and induces an inflammatory programmed cell death (PCD) termed pyroptosis. Pyroptosis is triggered by pathogenic invasion and has been determined to be a CASP activation-dependent process (76–78), hallmarked by the swelling of cells, formation of pores in the plasma membrane, rupture of membrane, and the secretion of pro-inflammatory cytoplasmic contents into the extracellular space, thereby activating a strong inflammatory response (79, 80). Pyroptosis is primarily observed in bone marrow-derived phagocytes, such as neutrophils, macrophages, and dendritic cells. Additionally, keratin-forming cells, endothelium, epithelium, CD4+ T lymphocytes, and neurons can also undergo pyroptosis (77). Cells that undergo pyroptosis in the ocular structure include astrocytes, endothelial cells, Müller cells, microglia, retinal ganglion cells (RGCs), pericytes, retinal pigment epithelial cells (RPEs), and corneal epithelial cells (CECs) (81–83). Research has recently depicted GSDMD as the executor regulating pyroptosis (84), with an N-terminal cell death domain (GSDMDN-term), a short central junction region, and a C-terminal self-inhibitory structural domain. Activated CASP cleaves the GSDMD at specific protein sites and produces GSDMD-N-terminal and GSDMD-C-terminal. After binding to membrane phosphatidylinositol, phosphatidylserine, and cardiolipin, the N-terminal of GSDMD oligomerizes and inserts itself into the plasma membrane, resulting in the formation of pores, which leads to the release of inflammatory molecules and ultimately triggers pyroptosis (85–88).
3.1 Morphological and biological characteristics of pyroptosis
While pyroptosis is similar to apoptosis and necroptosis, it has several distinguishing features in contrast with other types of PCD. During pyroptosis, cells undergo chromatin condensation and DNA breakage, while the nucleus remains intact, the cell swells, the plasma membrane ruptures, and inflammatory cytokines are released (Table 1) (89). In contrast, apoptosis caused by CASP activation results in membrane blebbing and nuclear fragmentation, while the plasma membrane is left intact and does not cause an inflammatory response in vivo (89). Additionally, no chromatin condensation or loss of plasma membrane integrity occurs in ferroptosis but causes mitochondrial condensation, reduction or loss of mitochondria cristae, and increased membrane density (90). On the other hand, chromatin condensation does not occur in cells undergoing necroptosis; however, their nuclear membranes are ruptured (91). Like GSDMD in pyroptosis, MLKL oligomers act as executive proteins to mediate cell death during necroptosis (92). In contrast, unlike pyroptosis, necroptosis is independent of CASP but requires RIPK3-regulated phosphorylation of MLKL. The phosphorylated MLKL generates a pore complex at the plasma membrane, leading to the secretion of DAMP, cell swelling, and membrane rupturing (93). MLKL forms selective channels in the plasma membrane that induce an influx of ions, increasing intracellular osmotic pressure, inward water flow, and severe cell swelling. However, pyroptosis depends on CASP to cleave the GSDMD directly and form pores. The channels are not selective, and the intracellular and extracellular ionic osmotic pressure gradient disappears. However, the intracellular colloidal osmotic pressure is higher, and water flows inward, leading to increased cell size and subsequent cell lysis, but the swelling is less severe than in necroptosis (85, 89, 94, 95). NLRP3 is a major contributor to the pyroptosis but not to other cell death modes (apoptosis, necroptosis, ferroptosis and autophagy, etc.), but signals (K+ and mtDNA, etc.) associated with the release of other cell death can activate NLRP3, thereby inducing inflammation or pyroptosis (96, 97). In fact, various cell death modes can crosstalk each other through NLRP3.
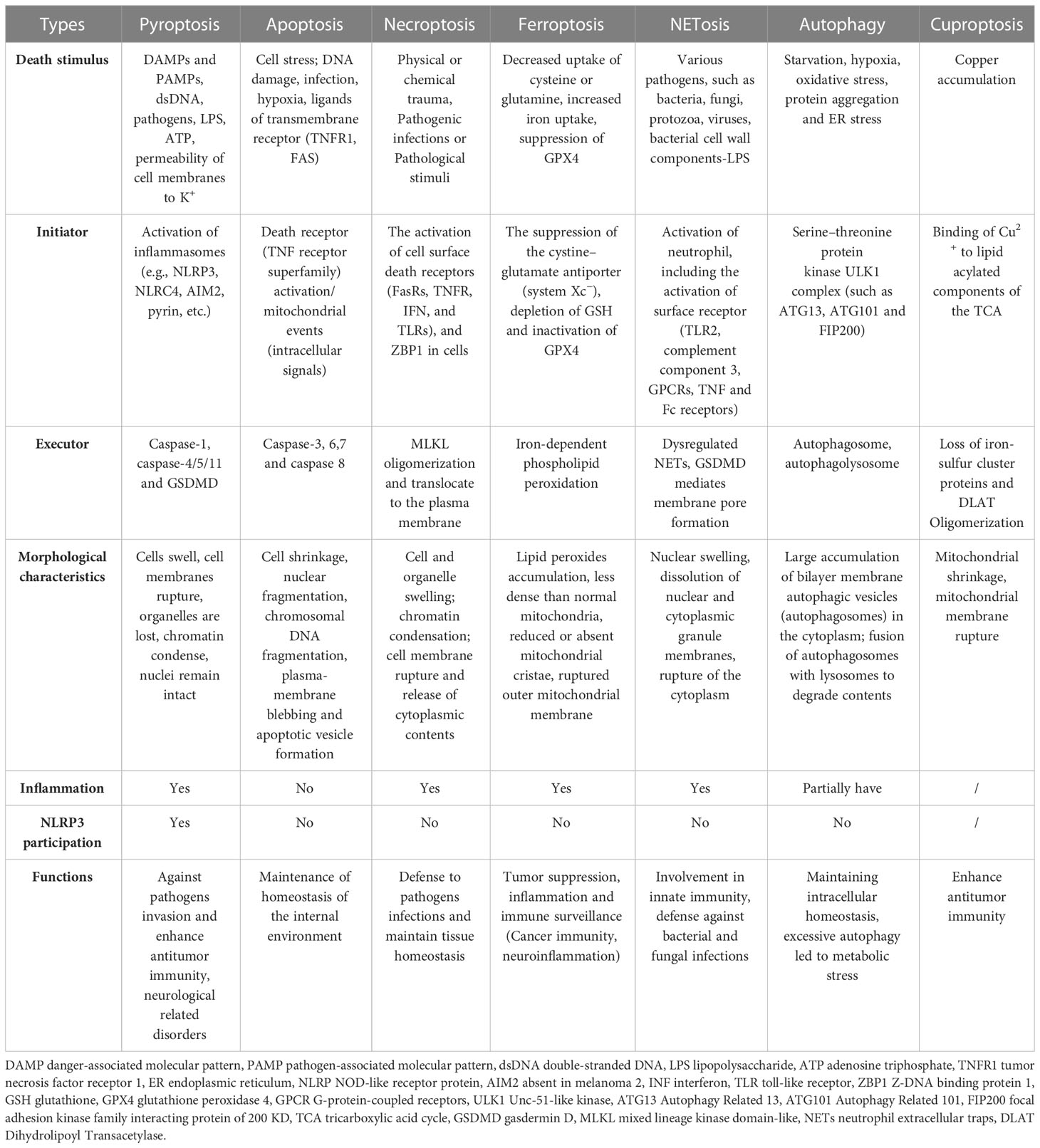
Table 1 The comparison of different modes of cell death.
3.2 Activation mechanisms for pyroptosis
Gasdermin-mediated pyroptosis includes both inflammasome-independent and-dependent pathways (Figure 2). Typically, pyroptosis dependent on inflammasome includes the CASP4/5/11- and CASP1-dependent pathways (non-canonical and canonical, respectively). Recent research has indicated new inflammasome-independent pathways, including the pathways mediated by CASP-3/8 and Granzyme A (GZMA) secreted by cytotoxic lymphocytes, which can sever GSDMB to release the GSDMD-N-terminal fragment, causing cell perforation and inducing other GSDM-mediated pyroptosis (98–100). The canonical pathway is mediated by inflammasome assembly, GSDMD cleavage, and secretion of IL-1β/18. The assembly of inflammasomes begins with PRRs (termed inflammasome sensors), such as NLR, AIM2, and pyrin, which recognize pathogen-associated molecular patterns and risk-associated molecular patterns (PAMP and DAMP). PRRs bind to ASC, a caspase-1 precursor, forming a multi-protein complex and activating caspase-1. The caspase-1, in turn, cleaves GSDMD to create the active domain (GSDMD-N terminal) containing peptide, which causes the cell membrane to become perforated and ultimately ruptures, releasing the contents, leading to an inflammatory reaction (88, 101). Moreover, activated caspase-1 cleaves pro-IL-1β/18, forming extracellularly released activated IL-1β/18, leading to aggregated inflammatory cells and an amplified inflammatory response (20, 84, 86, 102, 103). In the non-canonical pathway, caspase-4/5/11 recognizes intracellular lipopolysaccharide (LPS) directly (104) and cleaves GSDMD, triggering pyroptosis (105). However, caspase-4/5/11 cannot cleave pro-IL-1β/18 but can mediate their maturation and secretion via the NLRP3/caspase-1 pathway in partial cells, indicating the significance of caspase-1 in the production of mature IL-1β and IL-18 (106–108). In addition, cleavage of GSDMD by caspase-4/5/11 could trigger intracellular K efflux (20), causing the activation of NLRP3 inflammasome and accelerating pyroptosis (109–111). Further, in response to LPS stimulation, activated CASP11 cleaves Pannexin-1, induces ATP efflux, and binds to P2X7R, triggering NLRP3-linked pyroptosis (102, 112).
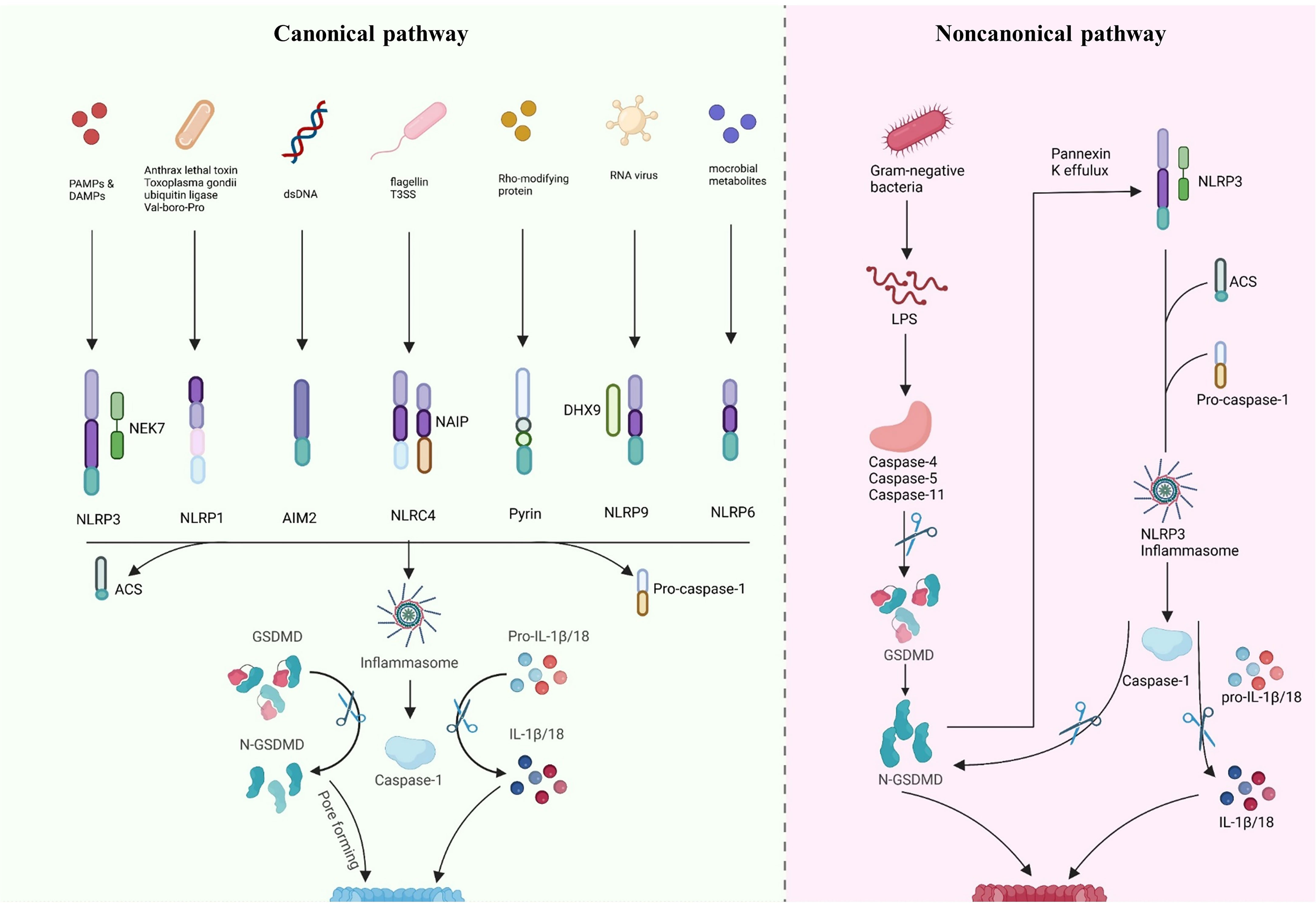
Figure 2 Inflammasome-dependent pathway of pyroptosis. Canonical pathway: inflammasome sensors are activated by different signals, such as dsDNA, flagellin T3SS, Rho-modifying and RNA virus. Activated inflammasome sensors subsequently oligomerization with pro-caspase1 and ASC. Activated caspase1 cleaves pro-IL-1β/18 into mature IL-1β/18. Caspase1 also cleaves GSDMD to release the N-terminal domain (GSDMD-N), which then inserts into the membrane, causes pores and induces pyroptosis. Noncanonical pathway: LPS directly activates caspase-4/5/11, causing GSDMD cleavage and triggering intracellular K efflux, K efflux further activates the NLRP3 pathway and accelerates pyroptosis. Created with BioRender.com.
4 NLRP3 and pyroptosis in diabetic retinopathy
In the working-age population, the most prevalent cause of preventable blindness is diabetic retinopathy, a common and distinct microvascular complication of diabetes (113). The retina is a complex system, usually made up of ten layers (from the inside out): RPE, optic rod and cone layer, and outer membranes consisting of limiting membrane (OLM), nuclear layer (ONL), plexiform layer (OPL), as well as inner membranes such as nuclear layer (INL), plexiform layer (IPL), cell layer (GCL), fiber layer (NFL) and limiting membrane (ILM) (114). In terms of histology, blood cells (endothelial and pericytes), glial cells (astrocytes, Müller cells, and microglia), and retinal neurons form an essential structure called the retinal neurovascular unit (NVU) (115). A growing body of research suggests that the abnormal interactions between inflammation, oxidative stress, mitochondrial dysfunction, advanced glycation end-products (AGEs) and cell death leads to retinal vascular abnormalities, blood-retinal barrier (BRB) disruption and neurological dysfunction (116–118). Recent studies have shown that chronic inflammation plays a crucial role in the pathogenesis of DR, where the NLRP3 inflammasome is of particular significance. Research depicts that peripheral blood mononuclear cells of individuals with DR exhibit greater expression levels of protein and gene of caspase-1, ASC and NLRP3 in contrast with normal individuals. Moreover, increased expression levels of the NLRP3, caspase-1, and the pro-inflammatory factors IL-1β/18 are also evident in the vitreous humor (119). Similarly, patients with proliferative DR show an elevation of ASC and NLRP3 in their fibrovascular membranes (7), while inhibiting NLRP3 inflammasome slows DR progression (120). The activation of NLRP3 can induce an inflammatory response and act as a bridge between neovascularization and the inflammation (121). Most importantly, NLRP3 activation can induce pyroptosis, leading to inflammatory cell death in retinal cells. Cell death is a typical characteristic of DR, which has a major role in the onset and advancement of DR (122). However, a large number of studies in the past have focused on cell death such as apoptosis, autophagy and ferroptosis. Recently, it has been shown that pyroptosis can be observed in neurons, RMEC, Müllers, microglia and RPEs.
4.1 The role of NLRP3 inflammasome in diabetic retinopathy
When the retina is exposed to glucose stimulation, abnormalities in various metabolic pathways lead to cellular oxidative stress and inflammation, and as signal regulators, RGCs may be the first to be damaged, as it has been shown that NLRP3 activation and electroretinogram defects precede the onset of microvascular lesions (123, 124). With RGCs damage, many abnormal signals are released to stimulate microglia and Müller cells to activate and proliferate (125). At the same time the metabolic abnormalities caused by high glucose also stimulate glial cells to activate NLRP3 inflammasomes, releasing anti-inflammatory cytokines to protect the retina (126, 127). However, continued glucose stimulation leads to excessive production of pro-inflammatory cytokines by glial cells and damage to retinal neurovascular units. As glial cell damage and phagocytosis decline, leading to the accumulation of metabolic wastes such as ATP, ROS, mtDNA or toxic chemicals such as serum uric acid (UA) (128), NLRP3 in the retinal vascular endothelium and pericytes is activated and large amounts of the inflammatory cytokines IL-1β and IL-18 are released (129–131). At this point retinal vascular cells begin to deteriorate, the pericytes are shed and leukocytes and macrophages are induced to aggregate and adhere tightly to vascular endothelial cells with the upregulation of adhesion molecules (132, 133), leading to further destruction of the retinal BRB, which leads to vascular leakage, edema and hypoxia. The hypoxic microenvironment disrupts the balance between angiogenic (e.g., VEGF) and anti-angiogenic (e.g., PEDF) regulators, promoting neovascularization (121, 134). Activation of NLRP3 induces the releases of IL-1β and IL-18. IL-1β binding to IL-1R increases retinal vascular permeability, exacerbating hypoxia, IL-18 is involved in pro-angiogenesis with VEGF (5). Most importantly, NLRP3 acts as a major contributor to pyroptosis. Numerous studies have shown that almost all retinal cells can undergo pyroptosis and the cells swell and rupture, releasing inflammatory cytokines and triggering an inflammatory storm. With the progress of DR, excessive activation of NLRP3 induces inflammation and various retinal cell death, triggering an inflammatory storm that leads to structural and functional collapse of the NVU and ultimately impairs vision (Figure 3).
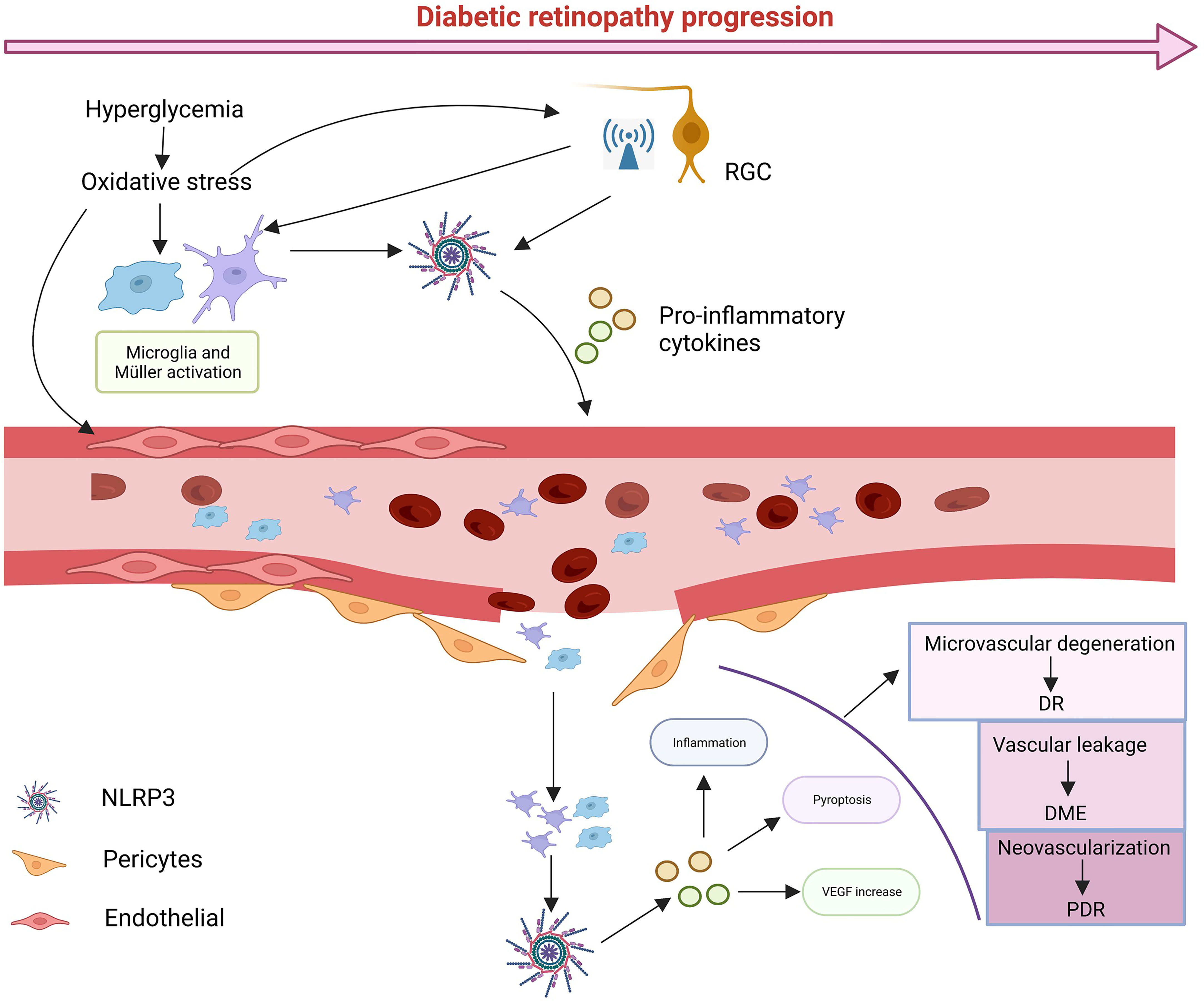
Figure 3 The possible cascade response. In diabetic conditions, glucose-induced metabolic abnormalities and oxidative stress activate NLRP3 in RGCs, leading to high levels of abnormal signaling, while microglia and Müller cells are activated. Sustained glucose further activates NLRP3, which leads to damage to pericytes and vascular endothelial cells, leading blood leakage and creating an ischemic and hypoxic environment, in turn, damaging vascular units. With time, various types of cell death can occur, and ultimately leads to damage to vision. Created with BioRender.com.
4.2 Mechanism of NLRP3/pyroptosis activation in different cells in diabetic retinopathy
Retinal pericytes (RPs) and vascular endothelial cells are essential elements of the retinal microvascular system and the internal of BRB (Figure 4). The loss of RPs can trigger microaneurysm formation, blood leakage, edema and ischemia, and induce proliferative neovascularization of the retina and subsequent loss of endothelial cells, the death of RPs and endothelial cell is a fatal blow to the NVU and accelerates the progression of DR (135, 136). Several models of DR have depicted the activation of NLRP3/caspase-1 and the release of IL-1β in retinal microvascular endothelial cells (RMECs) and retinal endothelial cells (RECs) in vitro and in vivo experiments (137–139). Connexin 43 is a cell-cell communication channel (gap junction) forming protein (140, 141), which is expressed in HRMECs, Müller cells, microglia, RPE, and astrocytes in the retina (142). Research demonstrates that Connexin 43 is upregulated in mouse DR models (142). Hemichannels open under hypoxic-ischemic conditions to form membrane pores, resulting in elevated extracellular ATP (143). A non-selective cation channel is formed when increased extracellular ATP links to a NLRP3 inflammasome activator, the P2X7R, a receptor which detects metabolic stimuli and oxidative stress. This results in initiating pyroptosis in human RMEC due to the subsequent K efflux and Ca2+ influx activating the NLRP3 inflammasome (144–146). Another study demonstrated that P2X7 was expressed at considerably elevated levels in RMEC in the presence of high glucose and LPS and that the persistent presence of high extracellular ATP could induce the P2X7 macropore opening. Activated P2X7 activates NLRP3 inflammasomes via K efflux, ROS, and glutamine efflux, while LPS activates NLRP3 inflammasomes via the caspase-1 and 11 (canonical and non-canonical, respectively) pathways to induce pyroptosis (147). Moreover, Yang et al. observed that RMEC exposed to advanced glycation end product modified bovine serum albumin (AGE-BSA) showed features of pyroptosis such as cell swelling and nucleus fragmentation, and the western blot results showed that expression of NLRP3, GSDMD and caspase-1 were upregulated. These phenomena were inhibited by H3 relaxin and MCC950, suggesting that NLRP3 is activated and induces pyroptosis in RMEC under a variety of pathological conditions (148). ROS, a known DAMP, triggers NLRP3 activation in DR (149–151). In endothelial cell, ROS induce pyroptosis by activating the NLRP3 through the initiation and activation process. The initiation phase refers to upregulation the expression of NLRP3, IL-1β/18 and caspase-1 expression by ROS. The activation phase refers to the promotion of NLRP3 inflammasome assembly and activation by ROS via TXNIP (152). Chen et al. found that minocycline, a tetracycline antibiotic, significantly downregulated ROS production and deceased TXNIP expression, inhibiting NLRP3 activation and decreasing the secretion of IL-1β/18 (138, 153). In a human RMEC model incubated with high glucose, miR-590-3p was downregulated and promoted pyroptosis by activating the NOX4/ROS/TXNIP/NLRP3 pathway and targeting NLRP1. In addition, increased levels of IL-1β further exacerbated cell death by inducing miR-590-3p downregulation through positive feedback (139), suggesting an involvement of the microRNAs in the onset of NLRP3-mediated pyroptosis.
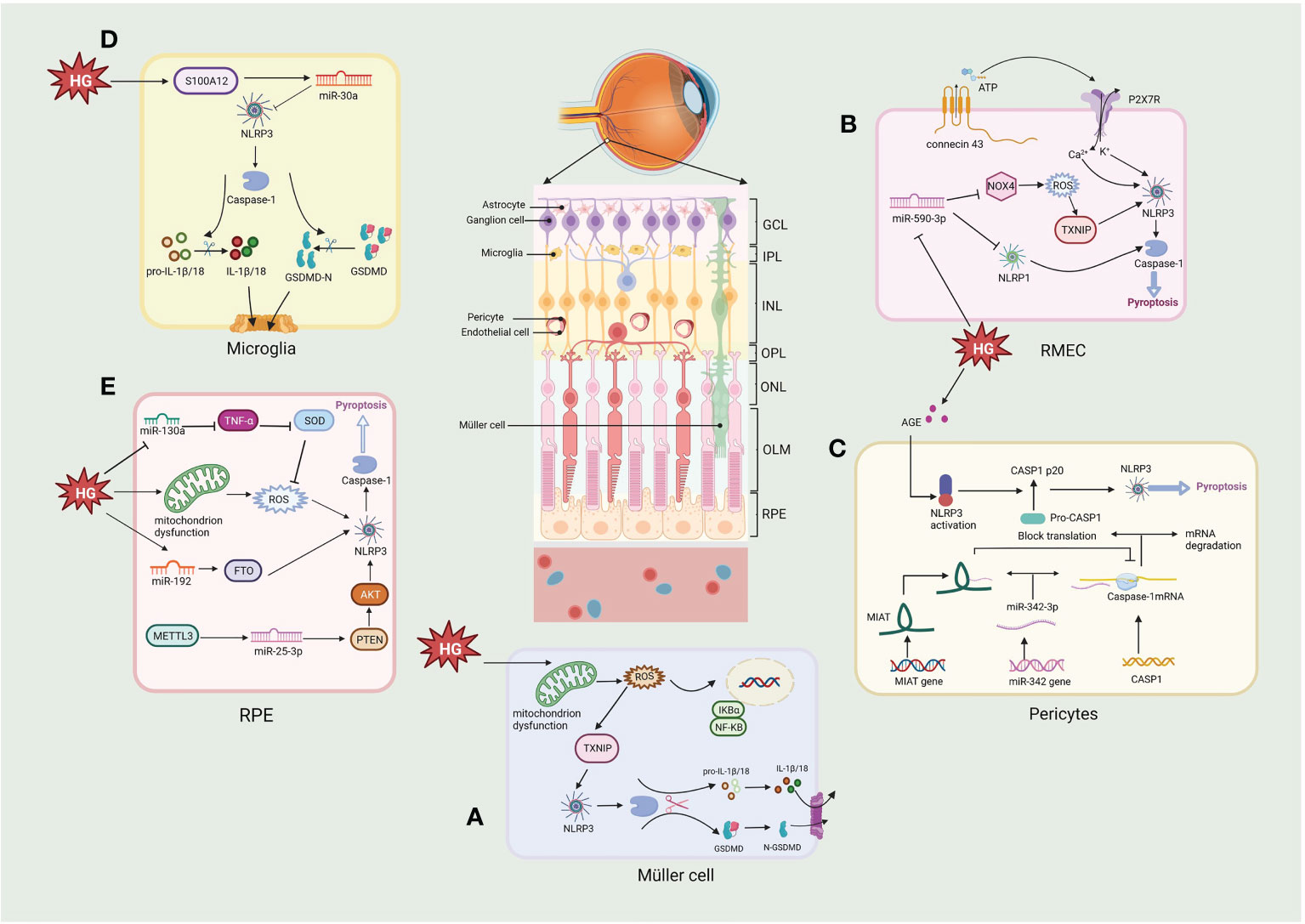
Figure 4 Molecular signaling pathway of NLRP3/pyroptosis in diabetic retinopathy. (A) Müller cell: hyperglycemia activates the NLRP3/pyroptosis via the ROS/TXNIP pathway. (B) Retinal microvascular endothelial cell (RMEC): high glucose induces an increase in extracellular ATP through connexin43 and increased binding to P2X7R, which then causes K+ efflux and Ca2+ influx by activating the NLRP3 inflammasome. High glucose also activates pyroptosis via the miR-590-3p/NOX4/ROS/TXNIP/NLRP3 pathway, with miR-590-3p also targeting NLRP1. (C) Pericytes: increasing lncRNA MIAT competes with CASP1 mRNA for binding to miR-342-3p, thereby inhibiting the CASP1 translation and pericytes pyroptosis. (D) Microglia: hyperglycemia produces S100 protein (S100A12), which induces the activation of NLRP3 via a miR-30a-dependent mechanism. (E) Retinal pigment epithelium: hyperglycemia activates the NLRP3/pyroptosis through triggering the generation of mtROS, also via miR-130a/TNF-α/SOD1/ROS, miR-192/FTO/NLRP3 or METTL3/miR-25-3p/PTEN/Akt/NLRP3 signaling pathway. Created with BioRender.com.
Retinal pericytes (RPs) modulate the production of tight junction proteins and support the vessel wall structurally (154). Gan et al. showed that glucose induced inflammation, pore formation and pyroptosis in RPs with increasing glucose treatment time and dose. The NLRP3 inhibitor glyburide or the caspase-1 inhibitor YVAD could reverse these phenomena, suggesting that glucose induced pyroptosis via NLRP3-caspase1-GSDMD in RPs (130). Another study using AGE-BSA to mimic the DR environment showed increased caspase-1, GSDMD-N, IL-1β/18, and LDH expression in RPs (155). These studies demonstrate that elevated glucose can induce inflammation and pyroptosis of RPs. Furthermore, lncRNA MIAT and CASP1 levels considerably elevated in AGE-BSA-treated RPs, whereas the expression of miR-342-3p was reduced. This observation indicates that CASPA1-dependent pyroptosis of RPs may be promoted due to the overexpressed MIAT which competes with CASP1 for conjugation to miR-342-3p, thus disrupting the inhibitory impact of the miRNA on CASP1 (155). Consequently, the MIAT/miR-342-3p/CASP1 pathway may provide a unique perspective on pericyte loss, contributing to developing novel therapeutic approaches for DR.
The retina’s structural support and nutrient metabolism are associated with Müller cells, the most pre-dominant and extensively dispersed macroglia in the retina (156). Müller cell death promotes loss of BRB integrity, increased vascular permeability and loss of protective effects on neuronal and vascular cells. Loss of Müller cells in diabetes is also associated with aneurysm formation, a clinical feature of DR (156). Whether Müller cells die in DR has been debated, this is because it secretes neurotrophic factors that protect them from hyperglycemia, at least in the beginning of DR. However, recent studies have shown that Müller cells actually begin to die gradually as the course of DR progress and Müller cell death is rapidly accelerated when protective growth factors are diminished (157). Studies have shown that Müller cells died in the DR displayed hypertrophy, a characteristic similar to pyroptosis. However, due to the lack of understanding concerning pyroptosis at the time, it was difficult to distinguish the mode of cell death (158, 159). Further research showed that caspase-1 activation and production of IL-1β were observed in rat retinal Müller cells cultured in a high-glucose environment (160). Moreover, elevated caspase-1, ASC, NLRP3, and IL-1β were also detected in 30mM glucose-treated mouse retinal Müller cells (161). Oxidative stress and the NLRP3 inflammasome are bridged through TXNIP. The expression of TXNIP in Müller cells is upgraded through genomic modifications during chronic hyperglycemia (138, 161, 162). Increased mitochondrial activity in DR produces high ROS as cells attempt to process excess glucose (67, 163). Subsequently, ROS generates oxidized disulfide bonds and releases TXNIP (164, 165); free TXNIP assists NLRP3 assembly with pro-caspase-1 and ASC and activates the NLRP3 inflammasome, thereby activating the pyroptosis (138). In another study it was possible to observe that HG significantly induced the expression of proteins of GSDMD, NLRP3 and caspase-1, after treatment with N-acetylcysteine (NAC), the expression of GSDMD, NLRP3 and caspase-1 were significantly reduced in Müller cells, indicating HG induced pyroptosis in Müller cells via NLRP3/pyroptosis (166). These studies indicate that Müller cells undergo pyroptosis in DR.
In the eye, microglia play an important role in immunity, that surveil the peripapillary environment and process the removal of metabolic wastes from the retina (167). Recent studies have shown that hyperglycemia produces S100 protein (S100A12), which induces microglia activation and inflammatory responses by modulating NLRP3 activity. It also stimulates the miR-30a-dependent secretion of IL-1β/18 from microglia (168). High glucose induces a shift in retinal microglia to the M1 phenotype and promotes the release of pro-inflammatory cytokines (e.g., IL-1β), and subsequently Huang et al. found that microglia showed a significant decrease in cell viability under high glucose (25, 50 or 100 mM) conditions and that lactate dehydrogenase (LDH) release and caspase-1 activity increased with increasing glucose concentration. In addition, protein expression of IL-1β, caspase-1, NLRP3 and cleaved GSDMD was increased. However, pretreatment with either the NLRP3 inhibitor MCC950 or the caspase-1 inhibitor Z-YVAD-FMK significantly inhibited pyroptosis under high glucose (25 mM) conditions, and the results adequately demonstrate that glucose induces pyroptosis via the NLRP3/caspase1-GSDMD pathway in retinal microglia, releasing more inflammatory factors, exacerbating the inflammatory response in the retina and induce NVU damage in the DR (169). In addition, ischemia and reperfusion (I/R) damage underlies many retinal diseases, such as glaucoma, DR and central retinal artery occlusion (170). retinal I/R have been shown to promote pyroptosis of retinal microglia, associated with increased expression of lncRNA-H19. Increased lncRNA-H19 significantly promotes NLRP3/6 inflammasome imbalance, leading to cytokine overproduction and microglia pyroptosis, while lncRNA H19 knockdown effectively inhibits these effects (171). Another report indicated that NLRC5 directly binds to the NLRP3/NLRC4 inflammasome and synergistically drives microglia pyroptosis (172). The above researches suggest that inflammation and pyroptosis of retinal microglia activated by hyperglycemia might play an essential role in the progression of DR.
RPEs form the external BRB and regulate the structure and functioning of the retinal, RPEs act as a cellular barrier separating the neuronal retina and the fenestrated choriocapillaris, and disruption of the RPE barrier plays a pathogenic role in the development of DR (4). A high glucose environment results in pyroptosis of RPE cells through a series of events, starting with the plasma membrane passage of glucose, which then generates mitochondrial ROS, leading to NLRP3 inflammasome activation, cleavage of CASP1, and IL-1β/18 release (173). It was shown that pyroptosis-linked proteins such as Caspase-1, GSDMD, NLRP3, and IL-1β/18 were upregulated in a high glucose environment. In contrast, methyltransferase-like protein 3 (METTL3) overexpression regulated the miR-25-3p/PTEN/Akt/NLRP3 pathway through a DGCR8-dependent approach to reduce the hyperglycemic-induced pyroptosis in RPEs (8). Gu et al. demonstrated that glucose induced the upregulation of NLRP3, GSDMD, IL-1β/18 and caspase-1 in a time- and dose-dependent manner in RPEs, which was later inhibited by the expression of miR-192. The FTO alpha-ketoglutarate-dependent dioxygenase (FTO), a downstream target of miR-192, increases NLRP3 expression by promoting NLRP3 demethylation, while miR-192 overexpression inhibits high glucose-induced RPEs pyroptosis by negatively modulating the FTO/NLRP3 signaling pathway (174). In another study, high glucose (50 mM) increased the expression levels of caspase-1, GSDMD, NLRP3, IL-18/1β in ARPE-19 cells and induced ROS production in a time-dependent manner. In previous studies it was shown that ROS could activate NLRP3, which in turn activate the pyroptosis. Further results verified that the ROS scavenger NAC and the GSDMD inhibitor necrosulfonamide (NSA) reversed the effect of high glucose on pyroptosis in ARPE-19 cells. Mechanistically glucose significantly reduced miR-130a, which activated NLRP3-mediated pyroptosis via the TNF-α/SOD1/ROS axis (175). The redox pathway of Trx is essential for maintaining normal RPE function in the DR. Under hyperglycemia, TXNIP expression is upregulated, leading to cellular oxidative stress, lysosomal dysfunction, and mitochondrial damage. Therefore, Thangal et al. treated RPEs with Auranofin (a TrxR inhibitor), resulting in cellular mitochondrial dysfunction and oxidative stress, along with enhanced activity of the pro-inflammatory caspase-1 in RPEs. These effects of Auranofin on RPEs could be inhibited by the antioxidant NAC, while neither ferrostain-1 no necroptosis-1 (inhibitors of ferroptosis and necroptosis, respectively) did not inhibit this death. In contrast, MCC950 or Ac-YVAD-cmk (Caspase-1 inhibitors) significantly reduced LDH release, suggesting that high glucose-induced oxidative stress promotes cell death via NLRP3-caspase1-pyroptosis rather than other cell death modes, providing sufficient evidence that NLRP3 mediates pyroptotic cell death in DR (176). Overall, there is increasing focus concerning the involvement of RPE-related pyroptosis in the DR.
RGCs are the output neurons that integrate data, while retinal neurons are the core cells that transfer optical signals and create vision (177). Several studies have confirmed neuronal changes, even before clinical vascular changes in DR (178, 179). The immunohistochemical analysis of a diabetic rat model depicted that caspase-1, ASC, and NLRP3 were localized primarily in the inner and outer nuclear layers and the ganglion cell layer. Furthermore, the number of cells expressing the abovementioned factors was considerably elevated in diabetic rats and intravitreal injection of drugs that inhibit NLRP3 inflammasome and IL-1β/18 expression (180). High glucose could lead to hypoxia and an imbalance in the immune response of retinal tissues. Under hypoxic conditions, continuous production and degradation of hypoxia-inducible factor-1 (HIF-1) were detected, activating the IL-6 and IL-8 genes by acting as transcription factors. In addition, pericyte loss may lead to cell-free capillary formation, which is linked to vascular occlusion and further leads to retinal non-perfusion and ischemic-hypoxic damage. Ischemia and hypoxia could further upregulate HIF-1 expression. Pyroptosis contributes to retinal ischemia injury and contributes the death of RGCs through the signaling pathway caspase-8-HIF-1α-NLRP12/NLRP3/NLRP4 in acute glaucoma (181). Caspase-mediated pyroptosis can also be observed in RGCs in various retinal diseases (182). For example, in some optic nerve compression damage models, NLRP3 expression is enhanced in retinal microglia, promoting IL-1β and caspase-1, while knockdown of NLRP3 slows RGC reduction after partial optic nerve compression damage (183). Future studies are essential to detect whether all retinal cells undergo pyroptosis in DR.
In summary, elevated intracellular glucose levels in diabetic patients trigger oxidative stress, leading to intracellular ROS and ATP production, and sustained glucose stimulation leads to mitochondrial dysfunction, which induces additional mtROS leakage and mtDNA release, all of which together activate NLRP3. Activation of NLRP3 can be detected at all stages of DR, suggesting that NLRP3 is involved in the development of DR. Firstly activated NLRP3 inflammasome can provide a platform for the maturation of IL-1β/18, thereby inducing inflammation. Most importantly, the NLRP3 is also involved in pyroptosis, inducing inflammatory cell death. Excessive cell death and inflammation leads to BRB rupture, blood infiltration, edema and retinal detachment, ultimately damaging vision. However, almost all cells are capable of pyroptosis in DR, it is still unclear which cells undergo pyroptosis first or how these cells interact during and after pyroptosis further studies are needed. In addition, NLRP3 may also be involved in other modes of cell death, we just focus on the role of pyroptosis, so the contribution of NLRP3/pyroptosis in DR needs to be further evaluated to provide new directions for early diagnosis or treatment of DR.
5 The activation of NLRP3/pyroptosis in other ophthalmic diseases
NLRP3-mediated pyroptosis is not only present in DR but is also involved in other ophthalmic diseases, for example, NLRP3 inflammasome activation caused by elevated tear osmolarity is the initial signal of corneal inflammation associated with dry eye, and pyroptosis is a prominent result of NLRP3 activation. Zhang et al. observed that hyperosmolarity induced pyroptosis in corneal epithelial cells, and that Calcitriol effectively alleviated damage by inhibiting the NLRP3-ACS- CASP1-GSDMD pathway (184). Chen et al. found that microglia undergo pyroptosis releasing inflammatory cytokines to induce RGC death was associated with glaucomatous vision loss, genetic deletion of the pyroptosis effector GSDMD significantly ameliorated RGCs death and retinal tissue damage in acute glaucoma (181). Particulate matter (PM2.5) induced increased oxidative stress and subsequent NLRP3 inflammasome-mediated pyroptosis was observed in trabecular meshwork cells, leading to ocular hypertension and glaucoma, and NAC ameliorated these symptoms (185). In another study, it was similarly observed that PM2.5 induced pyroptosis in corneal epithelial cells via the ROS/NLRP3/pyroptosis pathway. Activation of NLRP3/pyroptosis was also observed in Candida albicans keratitis, and knockdown of NLRP3 significantly alleviated the pyroptosis and corneal inflammatory response, making it an attractive target for the treatment of fungal keratitis (186). Sun et al. found that MCC950 alleviated Aβ-induced pyroptosis in age-related macular (187). In conclusion, NLRP3, the most studied inflammasome, which induced the pyroptosis in many ocular diseases, therefore targeting the NLRP3/pyroptosis pathway may be a new target for the treatment of many ophthalmic diseases.
6 Therapy targeting NLRP3 and pyroptosis in diabetic retinopathy
Diabetic retinopathy is considered to be a chronic low-grade inflammatory disorder, while the function of NLRP3 inflammasome and pyroptosis activation in the pathogenesis of DR is well established. Therefore, small-molecule inhibitors targeting the inflammasome and pyroptosis in DR might improve clinical outcomes. Fortunately, several inhibitors of NLRP3 and pyroptosis have been identified, including direct inhibitors of NLRP3, caspase-1, and GSDMD, as well as indirect inhibitors that target inflammatory components or associated signaling events. However, animal or human DR model testing of some inhibitors of pyroptosis has not been executed. At the same time, some of the inhibitors tested are potentially risky because their precise target of inhibition is not fully understood. In this review, several pyroptosis inhibitors that have been examined have been summarized (Table 2). For instance, to prevent pore formation and the discharge of inflammatory mediators, the GSDMD inhibitors NSA and disulfiram covalently bind to Cys191 on the GSDMD. This prevents the pyroptosis of retinal cells (175, 189). MCC950 is a highly specific inhibitor of NLRP3, Zhang et al. explored the anti-inflammatory effects of MCC950 treatment on HRECs after HG stimulation and the potential mechanisms to investigate the role of inflammasome-mediated cell death in DR. They demonstrated that MCC950 treatment significantly attenuated the initiation of the NLRP3 inflammasome in HRECs, which in turn reduced the mRNA expression levels of NLRP3, caspase-1 and proIL-1β in HRECs. More importantly, many studies demonstrated that MCC950 inhibits NLRP3 while significantly suppressing GSDMD expression in RMECs, microglia and Müller cells, thereby inhibiting retinal cell pyroptosis, reducing the rate of retinal vascular leakage and delaying the progression of DR in mice, The above studies provide strong evidence that MCC950 is a very promising drug for DR treatment. Mechanistically, MCC950 greatly protects retinal cells from HG-stimulated dysfunction by inhibiting the binding of NEK7 to the NLRP3 inflammasome (120, 191).
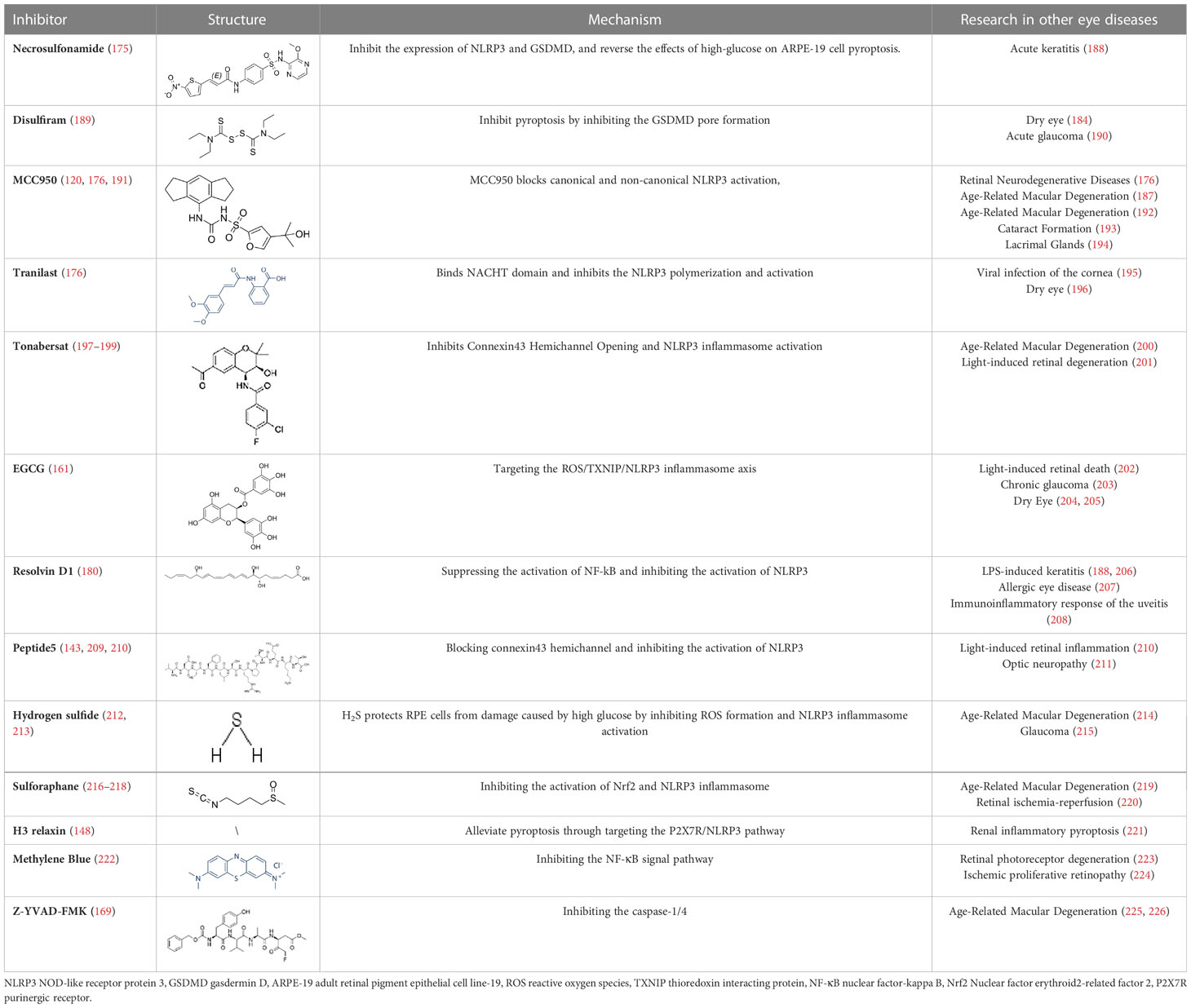
Table 2 Inhibitors target NLRP3/pyroptosis.
Other indirect inhibitors of the inflammasome, such as connexin 43 hemichannel, mediate RPE pyroptosis in DR via the ATP/NLRP3 inflammasome pathway, which can be blocked by peptide5 (142) and tonabersat. Mugisho et al. demonstrated that Peptide5 significantly reduced the incidence of DR-induced beading and vessel dilation, decreased the severity of vitreous and retinal hyper-reflective foci, and reduced subretinal fluid accumulation. In addition, Peptide5 resulted in reduced upregulation of connexin43 and GFAP compared to controls, inhibited the upregulation of the inflammatory markers IL-1β/18. Louie et al. further demonstrated a significant reduction in the expression of NLRP3, pro-inflammatory cytokines IL-1/18 and VEGF in Müller cells via the hemichannel inhibitor tonabersat. The above studies provide evidence that Peptide5, as well as other Connexin43 hemichannel blockades, can inhibit the inflammatory response and cell death by targeting the upstream signaling of NLRP3 (199). In addition, in DR pathogenesis, high glucose stimulates activation of NLRP3 inflammasome mediated by P2X7R, which could be significantly inhibited by H3 Relaxin. H3 Relaxin is a bioactive peptide with an insulin-like structure, which has been reported to be effective in diabetic cardiomyopathy. In STZ-treated retinas, disorganized membrane discs were observed, mitochondria were degraded, the number of synapses and synaptic vesicles in the inner and outer plexiform layer was reduced, and the ganglion cell layer showed swollen ganglion nuclei and dilated endoplasmic reticulum. After administration of high doses of H3 Relaxin, the above phenomena were significantly improved. In addition, the levels of NLRP3, ASC, caspase-1, IL-18/1β and GSDMD were significantly increased in RMECs after STZ treatment. After administration of H3 Relaxin, the expression of all pyroptosis-related proteins decreased. The above results suggest that HG triggered the activation of P2X7R and subsequently NLRP3 inflammasome in RMECs, while H3 Relaxin significantly reduced HG-induced expression and activation of NLRP3 inflammasome by inhibiting P2X7R, which in turn inhibited pyroptosis (148). Epigallocatechin gallate (EGCG), a major bioactive tea compound, could suppress the ROS/TXNIP/NLRP3 inflammasome pathway in Müllers via inhibition of streptozotocin (STZ)-induced DR in a mouse model (161). Garcinia cambogia also prevents the TXNIP/NLRP3 activation via reducing the levels of ASC, NLRP3, cleaved-IL-1β, cleaved-caspase-1, and TXNIP (227). Palbinone has a significant effect on attenuating the inflammatory response of the blood vessels and reducing vascular permeability in the retinal of DR by inhibiting NLRP3 activity, and more importantly, RT-PCR and immunofluorescence staining suggest that Palbinone may be a promising pharmacological agent to inhibit pyroptosis-mediated cell death for the treatment of DR (228). Similarly, rhodopsin has been shown to exert antioxidant effects in a mouse model of DR, activating the Nrf2 pathway and inhibiting NLRP3, caspase1, ASC, and cleaved IL-1β levels in Müller cells (218). Hydrogen sulfide could also protect high glucose-induced RPEs death and inflammatory damage from oxidative stress by suppressing the production of ROS and the activation of NLRP3 inflammasome (229). Resolvin D1 treatment in DR rats effectively reduces NLRP3-mediated inflammatory factor secretion in the retinal tissue by inhibiting the NF-kB pathway (180). As the pyroptosis and inflammasome in several ocular diseases are intensively studied and the number of affected individuals concerning inflammatory disorders is increasing, future clinical translation will be facilitated by specific and direct inhibitors of pyroptosis, with a focus on the use of precision medicine in inflammatory diseases.
7 Conclusion
Previous studies have found inflammatory responses at all stages of DR and NLRP3 as a causative factor in its pathogenesis. In DR, NLRP3 inflammasome can recognize multiple diabetic metabolic factors as endogenous danger signals, in turn activating CASP1. These diabetic metabolic factors include ATP and cytoarchitectural instability, such as rupturing of the lysosome, dysfunction of the mitochondria, and molecular or ionic perturbations, such as K efflux, ROS, and Ca2+ signaling. Activated CASP1 can then cleave GSDMD to C-GSDMD and N-GSDMD; N-GSDMD causes perforation of the cell membrane to form non-selective pores, causing the cells to swell and resulting in pyroptosis. On the other hand, CASP1 cleaves pro-IL-1β/18 to mature IL-1β/18, which are released through GSDMD-induced pores, promoting pyroptosis. However, current research on NLRP3 activation and pyroptosis in DR is still only the tip of the iceberg, and many questions remain unanswered at the molecular mechanism of pyroptosis. For instance, most evidence on pyroptosis is limited to activating the NLRP3 inflammasome, CASP, and other proteins rather than GSDMD activation in Müller cells, pericytes, endothelial cells, and RPE cells. Although evidence indicates the occurrence of high glucose-induced retinal pericyte pyroptosis through the NLRP3-caspase-1-GSDMD pathway, more direct evidence is needed to support the role of GSDMD in retinal cells. Furthermore, although it appears that a variety of cells undergo pyroptosis in DR, it remains unclear which cells undergo pyroptosis first, or whether the interactions between cells undergoing pyroptosis need to be further investigated. Each GSDM family member’s function in various disorders may vary, here, we focused on the role of GSDMD in DR. More research is needed to explore whether other members of the GSDM family are involved in the development of DR. Moreover, in disease models we usually study a single mode of death in isolation, whereas in fact under different pathological conditions the pattern of cell death is dynamic and it is likely that multiple modes of death co-exist. Therefore, it is necessary to look at the mode of cell death as a whole and dynamically observe their role in DR, which may be more relevant for clinical translation and application.
Several promising compounds are currently available that effectively inhibit the onset of pyroptosis, offering promising therapeutic directions for the management and treatment of ocular diseases, including DR. Although we have summarized the therapeutic potential of pyroptosis-related inhibitors in DR, most of the evidence is from cellular or animal studies, and have not yet been validated in clinical trials; therefore, further studies are needed to understand their clinical efficacy. On the other hand, traditional drug delivery by repeated vitreous cavity injections (230, 231) may cause increased intraocular pressure, cataracts, and other complications for the patient (232, 233). Therefore, in addition to investigating effective targeted therapeutic agents, finding better drug delivery methods is necessary to improve the outcome of patients with DR. Recently, gene therapy (183, 234–236) and nanomedicines for pyroptosis have received great attention (237). The findings of this research suggest that utilizing nanomedicine in patients with DR to target the lesion, with the potential to accumulate over time and gradually release, could hold great promise for clinical application and offer new avenues for enhancing clinical outcomes.
Author contributions
Conception and design of study, XZ, JW and GT. Drafting of article, XZ, JW and GT. Revision of draft, XZ and GT. All authors contributed to the article and approved the submitted version.
Funding
This work was financially supported by the National Natural Science Foundation of China (No.81100648, 82171031); Hunan Natural Science Foundation (No.2021JJ30620); The Science and Technology Project of Hunan Health Committee (No.20201966).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Wang W, Lo ACY. Diabetic retinopathy: pathophysiology and treatments. Int J Mol Sci (2018) 19(6):1816. doi: 10.3390/ijms19061816
2. Simó R, Carrasco E, García-Ramírez M, Hernández C. Angiogenic and antiangiogenic factors in proliferative diabetic retinopathy. Curr Diabetes Rev (2006) 2(1):71–98. doi: 10.2174/157339906775473671
3. Oshitari T. The pathogenesis and therapeutic approaches of diabetic neuropathy in the retina. Int J Mol Sci (2021) 22(16):9050. doi: 10.3390/ijms22169050
4. Feng L, Liang L, Zhang S, Yang J, Yue Y, Zhang X. Hmgb1 downregulation in retinal pigment epithelial cells protects against diabetic retinopathy through the autophagy-lysosome pathway. Autophagy (2022) 18(2):320–39. doi: 10.1080/15548627.2021.1926655
5. Raman KS, Matsubara JA. Dysregulation of the Nlrp3 inflammasome in diabetic retinopathy and potential therapeutic targets. Ocul Immunol Inflammation (2022) 30(2):470–8. doi: 10.1080/09273948.2020.1811350
6. Wang X, Antony V, Wang Y, Wu G, Liang G. Pattern recognition receptor-mediated inflammation in diabetic vascular complications. Med Res Rev (2020) 40(6):2466–84. doi: 10.1002/med.21711
7. Chen H, Zhang X, Liao N, Mi L, Peng Y, Liu B, et al. Enhanced expression of Nlrp3 inflammasome-related inflammation in diabetic retinopathy. Invest Ophthalmol Vis Sci (2018) 59(2):978–85. doi: 10.1167/iovs.17-22816
8. Zha X, Xi X, Fan X, Ma M, Zhang Y, Yang Y. Overexpression of Mettl3 attenuates high-glucose induced rpe cell pyroptosis by regulating mir-25-3p/Pten/Akt signaling cascade through Dgcr8. Aging (Albany NY) (2020) 12(9):8137–50. doi: 10.18632/aging.103130
9. Huang C, Qi P, Cui H, Lu Q, Gao X. Circfat1 regulates retinal pigment epithelial cell pyroptosis and autophagy Via mediating M6a reader protein Ythdf2 expression in diabetic retinopathy. Exp Eye Res (2022) 222:109152. doi: 10.1016/j.exer.2022.109152
10. Li W, Yang S, Chen G, He S. Mir-200c-3p regulates pyroptosis by targeting Slc30a7 in diabetic retinopathy. Hum Exp Toxicol (2022) 41:9603271221099589. doi: 10.1177/09603271221099589
11. Wang J-J, Chen Z-L, Wang D-D, Wu K-F, Huang W-B, Zhang L-Q. Linc00174 deteriorates the pathogenesis of diabetic retinopathy Via mir-26a-5p/Pten/Akt signalling cascade-mediated pyroptosis. Biochem Biophys Res Commun (2022) 630:92–100. doi: 10.1016/j.bbrc.2022.09.016
12. Chepchumba E, Yego DJF. Modes of retinal cell death in diabetic retinopathy. J Clin Exp Ophthalmol (2013) 04(05):298. doi: 10.4172/2155-9570.1000298
13. Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E. Targeting the Nlrp3 inflammasome in inflammatory diseases. Nat Rev Drug Discovery (2018) 17(8):588–606. doi: 10.1038/nrd.2018.97
14. Xue Y, Enosi Tuipulotu D, Tan WH, Kay C, Man SM. Emerging activators and regulators of inflammasomes and pyroptosis. Trends Immunol (2019) 40(11):1035–52. doi: 10.1016/j.it.2019.09.005
15. He Y, Hara H, Nunez G. Mechanism and regulation of Nlrp3 inflammasome activation. Trends Biochem Sci (2016) 41(12):1012–21. doi: 10.1016/j.tibs.2016.09.002
16. Cai X, Chen J, Xu H, Liu S, Jiang QX, Halfmann R, et al. Prion-like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell (2014) 156(6):1207–22. doi: 10.1016/j.cell.2014.01.063
17. Lu A, Magupalli Venkat G, Ruan J, Yin Q, Atianand Maninjay K, Vos MR, et al. Unified polymerization mechanism for the assembly of asc-dependent inflammasomes. Cell (2014) 156(6):1193–206. doi: 10.1016/j.cell.2014.02.008
18. Schmidt FI, Lu A, Chen JW, Ruan J, Tang C, Wu H, et al. A single domain antibody fragment that recognizes the adaptor asc defines the role of asc domains in inflammasome assembly. J Exp Med (2016) 213(5):771–90. doi: 10.1084/jem.20151790
19. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell (2002) 10:417–26. doi: 10.1016/s1097-2765(02)00599-3
20. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of gsdmd by inflammatory caspases determines pyroptotic cell death. Nature (2015) 526(7575):660–5. doi: 10.1038/nature15514
21. Lim RR, Wieser ME, Ganga RR, Barathi VA, Lakshminarayanan R, Mohan RR, et al. Nod-like receptors in the eye: uncovering its role in diabetic retinopathy. Int J Mol Sci (2020) 21(3):899. doi: 10.3390/ijms21030899
22. Ildefonso CJ, Biswal MR, Ahmed CM, Lewin AS. The Nlrp3 inflammasome and its role in age-related macular degeneration. Adv Exp Med Biol (2016) 854:59–65. doi: 10.1007/978-3-319-17121-0_9
23. Swanson KV, Deng M, Ting JP. The Nlrp3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol (2019) 19(8):477–89. doi: 10.1038/s41577-019-0165-0
24. He Y, Franchi L, Nunez G. Tlr agonists stimulate Nlrp3-dependent il-1beta production independently of the purinergic P2x7 receptor in dendritic cells and in vivo. J Immunol (2013) 190(1):334–9. doi: 10.4049/jimmunol.1202737
25. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. Cutting edge: nf-kappab activating pattern recognition and cytokine receptors license Nlrp3 inflammasome activation by regulating Nlrp3 expression. J Immunol (2009) 183(2):787–91. doi: 10.4049/jimmunol.0901363
26. Franchi L, Eigenbrod T, Nunez G. Cutting edge: tnf-alpha mediates sensitization to atp and silica Via the Nlrp3 inflammasome in the absence of microbial stimulation. J Immunol (2009) 183(2):792–6. doi: 10.4049/jimmunol.0900173
27. Xing Y, Yao X, Li H, Xue G, Guo Q, Yang G, et al. Cutting edge: Traf6 mediates Tlr/Il-1r signaling-induced nontranscriptional priming of the Nlrp3 inflammasome. J Immunol (2017) 199(5):1561–6. doi: 10.4049/jimmunol.1700175
28. Py Bénédicte F, Kim M-S, Vakifahmetoglu-Norberg H, Yuan J. Deubiquitination of Nlrp3 by Brcc3 critically regulates inflammasome activity. Mol Cell (2013) 49(2):331–8. doi: 10.1016/j.molcel.2012.11.009
29. Song N, Liu Z-S, Xue W, Bai Z-F, Wang Q-Y, Dai J, et al. Nlrp3 phosphorylation is an essential priming event for inflammasome activation. Mol Cell (2017) 68(1):185–97.e6. doi: 10.1016/j.molcel.2017.08.017
30. Barry R, John SW, Liccardi G, Tenev T, Jaco I, Chen CH, et al. Sumo-mediated regulation of Nlrp3 modulates inflammasome activity. Nat Commun (2018) 9(1):3001. doi: 10.1038/s41467-018-05321-2
31. Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM, Nunez G. K(+) efflux is the common trigger of Nlrp3 inflammasome activation by bacterial toxins and particulate matter. Immunity (2013) 38(6):1142–53. doi: 10.1016/j.immuni.2013.05.016
32. Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer AM, et al. Critical role for calcium mobilization in activation of the Nlrp3 inflammasome. Proc Natl Acad Sci (2012) 109(28):11282–7. doi: 10.1073/pnas.1117765109
33. Tang T, Lang X, Xu C, Wang X, Gong T, Yang Y, et al. Clics-dependent chloride efflux is an essential and proximal upstream event for Nlrp3 inflammasome activation. Nat Commun (2017) 8(1):202. doi: 10.1038/s41467-017-00227-x
34. Schorn C, Frey B, Lauber K, Janko C, Strysio M, Keppeler H, et al. Sodium overload and water influx activate the Nalp3 inflammasome. J Biol Chem (2011) 286(1):35–41. doi: 10.1074/jbc.M110.139048
35. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in Nlrp3 inflammasome activation. Nature (2011) 469(7329):221–5. doi: 10.1038/nature09663
36. Seok JK, Kang HC, Cho YY, Lee HS, Lee JY. Regulation of the Nlrp3 inflammasome by post-translational modifications and small molecules. Front Immunol (2020) 11:618231. doi: 10.3389/fimmu.2020.618231
37. Laudisi F, Spreafico R, Evrard M, Hughes TR, Mandriani B, Kandasamy M, et al. Cutting edge: the Nlrp3 inflammasome links complement-mediated inflammation and il-1beta release. J Immunol (2013) 191(3):1006–10. doi: 10.4049/jimmunol.1300489
38. Yu S, Wang D, Huang L, Zhang Y, Luo R, Adah D, et al. The complement receptor C5ar2 promotes protein kinase r expression and contributes to Nlrp3 inflammasome activation and Hmgb1 release from macrophages. J Biol Chem (2019) 294(21):8384–94. doi: 10.1074/jbc.RA118.006508
39. Gratal P, Lamuedra A, Medina JP, Bermejo-Álvarez I, Largo R, Herrero-Beaumont G, et al. Purinergic system signaling in metainflammation-associated osteoarthritis. Front Med (2020) 7:506. doi: 10.3389/fmed.2020.00506
40. Vince JE, Silke J. The intersection of cell death and inflammasome activation. Cell Mol Life Sci (2016) 73(11-12):2349–67. doi: 10.1007/s00018-016-2205-2
41. Conos SA, Chen KW, De Nardo D, Hara H, Whitehead L, Nunez G, et al. Active mlkl triggers the Nlrp3 inflammasome in a cell-intrinsic manner. Proc Natl Acad Sci U.S.A. (2017) 114(6):E961–E9. doi: 10.1073/pnas.1613305114
42. Zheng M, Kanneganti TD. The regulation of the Zbp1-Nlrp3 inflammasome and its implications in pyroptosis, apoptosis, and necroptosis (Panoptosis). Immunol Rev (2020) 297(1):26–38. doi: 10.1111/imr.12909
43. Menini S, Iacobini C, Vitale M, Pugliese G. The inflammasome in chronic complications of diabetes and related metabolic disorders. Cells (2020) 9(8):1812. doi: 10.3390/cells9081812
44. Gross CJ, Mishra R, Schneider KS, Medard G, Wettmarshausen J, Dittlein DC, et al. K(+) efflux-independent Nlrp3 inflammasome activation by small molecules targeting mitochondria. Immunity (2016) 45(4):761–73. doi: 10.1016/j.immuni.2016.08.010
45. Wolf AJ, Reyes CN, Liang W, Becker C, Shimada K, Wheeler ML, et al. Hexokinase is an innate immune receptor for the detection of bacterial peptidoglycan. Cell (2016) 166(3):624–36. doi: 10.1016/j.cell.2016.05.076
46. Surprenant A, Rassendren F, Kawashima E, North RA, Buell G. The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science (1996) 272:735–8. doi: 10.1126/science.272.5262.735
47. Samways DS, Li Z, Egan TM. Principles and properties of ion flow in P2x receptors. Front Cell Neurosci (2014) 8:6. doi: 10.3389/fncel.2014.00006
48. Di A, Xiong S, Ye Z, Malireddi RKS, Kometani S, Zhong M, et al. The Twik2 potassium efflux channel in macrophages mediates Nlrp3 inflammasome-induced inflammation. Immunity (2018) 49(1):56–65.e4. doi: 10.1016/j.immuni.2018.04.032
49. Gong T, Yang Y, Jin T, Jiang W, Zhou R. Orchestration of Nlrp3 inflammasome activation by ion fluxes. Trends Immunol (2018) 39(5):393–406. doi: 10.1016/j.it.2018.01.009
50. Suresh R, Chandrasekaran P, Sutterwala FS, Mosser DM. Complement-mediated ‘bystander’ damage initiates host NLRP3 inflammasome activation. J Cell Sci (2016) 129:1928–39. doi: 10.1242/jcs.179291
51. Hamon MA, Cossart P. K+ efflux is required for histone H3 dephosphorylation by listeria monocytogenes listeriolysin O and other pore-forming toxins. Infect Immun (2011) 79(7):2839–46. doi: 10.1128/IAI.01243-10
52. Yaron JR, Gangaraju S, Rao MY, Kong X, Zhang L, Su F, et al. K+ regulates Ca2+ to drive inflammasome signaling: dynamic visualization of ion flux in live cells. Cell Death Dis (2015) 6(10):e1954–e. doi: 10.1038/cddis.2015.277
53. Triantafilou K, Hughes TR, Triantafilou M, Morgan BP. The complement membrane attack complex triggers intracellular Ca2+ fluxes leading to Nlrp3 inflammasome activation. J Cell Sci (2013) 126(Pt 13):2903–13. doi: 10.1242/jcs.124388
54. Daniels MJ, Rivers-Auty J, Schilling T, Spencer NG, Watremez W, Fasolino V, et al. Fenamate nsaids inhibit the Nlrp3 inflammasome and protect against alzheimer’s disease in rodent models. Nat Commun (2016) 7:12504. doi: 10.1038/ncomms12504
55. Domingo-Fernandez R, Coll RC, Kearney J, Breit S, O’Neill LAJ. The intracellular chloride channel proteins Clic1 and Clic4 induce il-1beta transcription and activate the Nlrp3 inflammasome. J Biol Chem (2017) 292(29):12077–87. doi: 10.1074/jbc.M117.797126
56. Green JP, Yu S, Martin-Sanchez F, Pelegrin P, Lopez-Castejon G, Lawrence CB, et al. Chloride regulates dynamic Nlrp3-dependent asc oligomerization and inflammasome priming. Proc Natl Acad Sci U.S.A. (2018) 115(40):E9371–E80. doi: 10.1073/pnas.1812744115
57. Elliott EI, Miller AN, Banoth B, Iyer SS, Stotland A, Weiss JP, et al. Cutting edge: mitochondrial assembly of the Nlrp3 inflammasome complex is initiated at priming. J Immunol (2018) 200(9):3047–52. doi: 10.4049/jimmunol.1701723
58. Liu Q, Zhang D, Hu D, Zhou X, Zhou Y. The role of mitochondria in Nlrp3 inflammasome activation. Mol Immunol (2018) 103:115–24. doi: 10.1016/j.molimm.2018.09.010
59. Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity (2013) 39(2):311–23. doi: 10.1016/j.immuni.2013.08.001
60. Yu JW, Lee MS. Mitochondria and the Nlrp3 inflammasome: physiological and pathological relevance. Arch Pharm Res (2016) 39(11):1503–18. doi: 10.1007/s12272-016-0827-4
61. Dudek J. Role of cardiolipin in mitochondrial signaling pathways. Front Cell Dev Biol (2017) 5:90. doi: 10.3389/fcell.2017.00090
62. Franchi L, Eigenbrod T, Munoz-Planillo R, Ozkurede U, Kim YG, Arindam C, et al. Cytosolic double-stranded rna activates the Nlrp3 inflammasome Via mavs-induced membrane permeabilization and k+ efflux. J Immunol (2014) 193(8):4214–22. doi: 10.4049/jimmunol.1400582
63. Park S, Juliana C, Hong S, Datta P, Hwang I, Fernandes-Alnemri T, et al. The mitochondrial antiviral protein mavs associates with Nlrp3 and regulates its inflammasome activity. J Immunol (2013) 191(8):4358–66. doi: 10.4049/jimmunol.1301170
64. Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN. The adaptor mavs promotes Nlrp3 mitochondrial localization and inflammasome activation. Cell (2013) 153(2):348–61. doi: 10.1016/j.cell.2013.02.054
65. Ichinohe T, Yamazaki T, Koshiba T, Yanagi Y. Mitochondrial protein mitofusin 2 is required for Nlrp3 inflammasome activation after rna virus infection. Proc Natl Acad Sci U.S.A. (2013) 110(44):17963–8. doi: 10.1073/pnas.1312571110
66. Yeon SH, Yang G, Lee HE, Lee JY. Oxidized phosphatidylcholine induces the activation of Nlrp3 inflammasome in macrophages. J Leukoc Biol (2017) 101(1):205–15. doi: 10.1189/jlb.3VMA1215-579RR
67. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the Nalp3 inflammasome. Nat Immunol (2011) 12(3):222–30. doi: 10.1038/ni.1980
68. Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin XJ, et al. New mitochondrial DNA synthesis enables Nlrp3 inflammasome activation. Nature (2018) 560(7717):198–203. doi: 10.1038/s41586-018-0372-z
69. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized mitochondrial DNA activates the Nlrp3 inflammasome during apoptosis. Immunity (2012) 36(3):401–14. doi: 10.1016/j.immuni.2012.01.009
70. Lemasters JJ, Theruvath Tp Fau - Zhong Z, Zhong Z Fau - Nieminen A-L, Nieminen AL. Mitochondrial calcium and the permeability transition in cell death. Biochim Biophys Acta (2009) 1787(11):1395–401. doi: 10.1016/j.bbabio.2009.06.009
71. Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, et al. Silica crystals and aluminum salts activate the Nalp3 inflammasome through phagosomal destabilization. Nat Immunol (2008) 9(8):847–56. doi: 10.1038/ni.1631
72. Orlowski GM, Colbert JD, Sharma S, Bogyo M, Robertson SA, Rock KL. Correction: multiple cathepsins promote pro-Il-1beta synthesis and Nlrp3-mediated il-1beta activation. J Immunol (2016) 196(1):503. doi: 10.4049/jimmunol.1502363
73. Okada M, Matsuzawa A, Yoshimura A, Ichijo H. The lysosome rupture-activated Tak1-jnk pathway regulates Nlrp3 inflammasome activation. J Biol Chem (2014) 289(47):32926–36. doi: 10.1074/jbc.M114.579961
74. Chen J, Chen ZJ. Ptdins4p on dispersed trans-golgi network mediates Nlrp3 inflammasome activation. Nature (2018) 564(7734):71–6. doi: 10.1038/s41586-018-0761-3
75. Guo C, Chi Z, Jiang D, Xu T, Yu W, Wang Z, et al. Cholesterol homeostatic regulator scap-Srebp2 integrates Nlrp3 inflammasome activation and cholesterol biosynthetic signaling in macrophages. Immunity (2018) 49(5):842–56 e7. doi: 10.1016/j.immuni.2018.08.021
76. Mayahara N. [Dental resin for restoration with unsaturated polyester resin used as base material. effect of catalyst, accelerator and subaccelerator on working time, setting time and peak temperature]. Shika Zairyo Kikai (1989) 8(3):364–74.
77. Vande Walle L, Lamkanfi M. Pyroptosis. Curr Biol (2016) 26(13):R568–R72. doi: 10.1016/j.cub.2016.02.019
78. Man SM, Karki R, Kanneganti T-D. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev (2017) 277(1):61–75. doi: 10.1111/imr.12534
79. Fink SL, Cookson BT. Pyroptosis and host cell death responses during salmonella infection. Cell Microbiol (2007) 9(11):2562–70. doi: 10.1111/j.1462-5822.2007.01036.x
80. Fink SL, Bergsbaken T, Cookson BT. Anthrax lethal toxin and salmonella elicit the common cell death pathway of caspase-1-Dependent pyroptosis Via distinct mechanisms. Proc Natl Acad Sci U States America (2008) 105(11):4312–7. doi: 10.1073/pnas.0707370105
81. Yang M, So K-F, Lam WC, Lo ACY. Novel programmed cell death as therapeutic targets in age-related macular degeneration? Int J Mol Sci (2020) 21(19):7279. doi: 10.3390/ijms21197279
82. Zhao W, Yang H, Lyu L, Zhang J, Xu Q, Jiang N, et al. Gsdmd, an executor of pyroptosis, is involved in il-1β secretion in aspergillus fumigatus keratitis. Exp Eye Res (2021) 202:108375. doi: 10.1016/j.exer.2020.108375
83. Meng C, Gu C, He S, Su T, Lhamo T, Draga D, et al. Pyroptosis in the retinal neurovascular unit: new insights into diabetic retinopathy. Front In Immunol (2021) 12:763092. doi: 10.3389/fimmu.2021.763092
84. He W-T, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin d is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res (2015) 25(12):1285–98. doi: 10.1038/cr.2015.139
85. Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature (2016) 535(7610):111–6. doi: 10.1038/nature18590
86. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin d causes pyroptosis by forming membrane pores. Nature (2016) 535(7610):153–8. doi: 10.1038/nature18629
87. Aglietti RA, Dueber EC. Recent insights into the molecular mechanisms underlying pyroptosis and gasdermin family functions. Trends In Immunol (2017) 38(4):261–71. doi: 10.1016/j.it.2017.01.003
88. Sborgi L, Rühl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, et al. Gsdmd membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J (2016) 35(16):1766–78. doi: 10.15252/embj.201694696
89. Fang Y, Tian S, Pan Y, Li W, Wang Q, Tang Y, et al. Pyroptosis: a new frontier in cancer. BioMed Pharmacother (2020) 121:109595. doi: 10.1016/j.biopha.2019.109595
90. Miyake S, Murai S, Kakuta S, Uchiyama Y, Nakano H. Identification of the hallmarks of necroptosis and ferroptosis by transmission electron microscopy. Biochem Biophys Res Commun (2020) 527(3):839–44. doi: 10.1016/j.bbrc.2020.04.127
91. Chen KW, Demarco B, Broz P. Beyond inflammasomes: emerging function of gasdermins during apoptosis and netosis. EMBO J (2020) 39(2):e103397. doi: 10.15252/embj.2019103397
92. Sun L, Wang X. A new kind of cell suicide: mechanisms and functions of programmed necrosis. Trends In Biochem Sci (2014) 39(12):587–93. doi: 10.1016/j.tibs.2014.10.003
93. Sun Y, Abbondante S, Karmakar M, de Jesus Carrion S, Che C, Hise AG, et al. Neutrophil caspase-11 is required for cleavage of caspase-1 and secretion of il-1β in infection. J Immunol (Baltimore Md: 1950) (2018) 201(9):2767–75. doi: 10.4049/jimmunol.1701195
94. Xia B, Fang S, Chen X, Hu H, Chen P, Wang H, et al. Mlkl forms cation channels. Cell Res (2016) 26(5):517–28. doi: 10.1038/cr.2016.26
95. Kovacs SB, Miao EA. Gasdermins: effectors of pyroptosis. Trends Cell Biol (2017) 27(9):673–84. doi: 10.1016/j.tcb.2017.05.005
96. Huang Y, Xu W, Zhou R. Nlrp3 inflammasome activation and cell death. Cell Mol Immunol (2021) 18(9):2114–27. doi: 10.1038/s41423-021-00740-6
97. Gaidt MM, Hornung V. The Nlrp3 inflammasome renders cell death pro-inflammatory. J Mol Biol (2018) 430(2):133–41. doi: 10.1016/j.jmb.2017.11.013
98. Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of Dfna5 by caspase-3 during apoptosis mediates progression to secondary Necrotic/Pyroptotic cell death. Nat Commun (2017) 8:14128. doi: 10.1038/ncomms14128
99. Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature (2017) 547(7661):99–103. doi: 10.1038/nature22393
100. Zhou Z, He H, Wang K, Shi X, Wang Y, Su Y, et al. Granzyme a from cytotoxic lymphocytes cleaves gsdmb to trigger pyroptosis in target cells. Sci (New York NY) (2020) 368(6494):eaaz7548. doi: 10.1126/science.aaz7548
101. Sollberger G, Strittmatter GE, Garstkiewicz M, Sand J, Beer H-D. Caspase-1: the inflammasome and beyond. Innate Immun (2014) 20(2):115–25. doi: 10.1177/1753425913484374
102. Yu P, Zhang X, Liu N, Tang L, Peng C, Chen X. Pyroptosis: mechanisms and diseases. Signal Transduct Target Ther (2021) 6(1):128. doi: 10.1038/s41392-021-00507-5
103. Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, et al. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature (1992) 356(6372):768–74. doi: 10.1038/356768a0
104. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular lps. Nature (2014) 514(7521):187–92. doi: 10.1038/nature13683
105. Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, et al. Gsdmd P30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci U States America (2016) 113(28):7858–63. doi: 10.1073/pnas.1607769113
106. Shi J, Gao W, Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends In Biochem Sci (2017) 42(4):245–54. doi: 10.1016/j.tibs.2016.10.004
107. Bedoui S, Herold MJ, Strasser A. Emerging connectivity of programmed cell death pathways and its physiological implications. Nat Rev Mol Cell Biol (2020) 21(11):678–95. doi: 10.1038/s41580-020-0270-8
108. Zhaolin Z, Guohua L, Shiyuan W, Zuo W. Role of pyroptosis in cardiovascular disease. Cell Prolif (2019) 52(2):e12563. doi: 10.1111/cpr.12563
109. Baker PJ, Boucher D, Bierschenk D, Tebartz C, Whitney PG, D’Silva DB, et al. Nlrp3 inflammasome activation downstream of cytoplasmic lps recognition by both caspase-4 and caspase-5. Eur J Immunol (2015) 45(10):2918–26. doi: 10.1002/eji.201545655
110. Rühl S, Broz P. Caspase-11 activates a canonical Nlrp3 inflammasome by promoting k(+) efflux. Eur J Immunol (2015) 45(10):2927–36. doi: 10.1002/eji.201545772
111. Schmid-Burgk JL, Gaidt MM, Schmidt T, Ebert TS, Bartok E, Hornung V. Caspase-4 mediates non-canonical activation of the Nlrp3 inflammasome in human myeloid cells. Eur J Immunol (2015) 45(10):2911–7. doi: 10.1002/eji.201545523
112. Yang D, He Y, Muñoz-Planillo R, Liu Q, Núñez G. Caspase-11 requires the pannexin-1 channel and the purinergic P2x7 pore to mediate pyroptosis and endotoxic shock. Immunity (2015) 43(5):923–32. doi: 10.1016/j.immuni.2015.10.009
113. Cheung N, Mitchell P, Wong TY. Diabetic retinopathy. Lancet (2010) 376(9735):124–36. doi: 10.1016/S0140-6736(09)62124-3
114. Wong TY, Cheung CMG, Larsen M, Sharma S, Simó R. Diabetic retinopathy. Nat Rev Dis Primers (2016) 2:16012. doi: 10.1038/nrdp.2016.12
115. Hammes H-P. Diabetic retinopathy: hyperglycaemia, oxidative stress and beyond. Diabetologia (2018) 61(1):29–38. doi: 10.1007/s00125-017-4435-8
116. Rübsam A, Parikh S, Fort PE. Role of inflammation in diabetic retinopathy. Int J Mol Sci (2018) 19(4):942. doi: 10.3390/ijms19040942
117. Antonetti DA, Silva PS, Stitt AW. Current understanding of the molecular and cellular pathology of diabetic retinopathy. Nat Rev Endocrinol (2021) 17(4):195–206. doi: 10.1038/s41574-020-00451-4
118. Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes (2005) 54(6):1615–25. doi: 10.2337/diabetes.54.6.1615
119. Loukovaara S, Piippo N, Kinnunen K, Hytti M, Kaarniranta K, Kauppinen A. Nlrp3 inflammasome activation is associated with proliferative diabetic retinopathy. Acta Ophthalmol (2017) 95(8):803–8. doi: 10.1111/aos.13427
120. Ge K, Wang Y, Li P, Li M, Zhang W, Dan H, et al. Down-expression of the Nlrp3 inflammasome delays the progression of diabetic retinopathy. Microvasc Res (2022) 139:104265. doi: 10.1016/j.mvr.2021.104265
121. Chaurasia SS, Lim RR, Parikh BH, Wey YS, Tun BB, Wong TY, et al. The Nlrp3 inflammasome may contribute to pathologic neovascularization in the advanced stages of diabetic retinopathy. Sci Rep (2018) 8(1):2847. doi: 10.1038/s41598-018-21198-z
122. Al Mamun A, Mimi AA, Zaeem M, Wu Y, Monalisa I, Akter A, et al. Role of pyroptosis in diabetic retinopathy and its therapeutic implications. Eur J Pharmacol (2021) 904:174166. doi: 10.1016/j.ejphar.2021.174166
123. Simó R, Stitt AW, Gardner TW. Neurodegeneration in diabetic retinopathy: does it really matter? Diabetologia (2018) 61(9):1902–12. doi: 10.1007/s00125-018-4692-1
124. Ng DS, Chiang PP, Tan G, Cheung CG, Cheng C-Y, Cheung CY, et al. Retinal ganglion cell neuronal damage in diabetes and diabetic retinopathy. Clin Exp Ophthalmol (2016) 44(4):243–50. doi: 10.1111/ceo.12724
125. Vecino E, Rodriguez FD, Ruzafa N, Pereiro X, Sharma SC. Glia-neuron interactions in the mammalian retina. Prog Retin Eye Res (2016) 51:1–40. doi: 10.1016/j.preteyeres.2015.06.003
126. Tang L, Xu G-T, Zhang J-F. Inflammation in diabetic retinopathy: possible roles in pathogenesis and potential implications for therapy. Neural Regener Res (2023) 18(5):976–82. doi: 10.4103/1673-5374.355743
127. Li W, Liu X, Tu Y, Ding D, Yi Q, Sun X, et al. Dysfunctional Nurr1 promotes high glucose-induced müller cell activation by up-regulating the nf-κb/Nlrp3 inflammasome axis. Neuropeptides (2020) 82:102057. doi: 10.1016/j.npep.2020.102057
128. Thounaojam MC, Montemari A, Powell FL, Malla P, Gutsaeva DR, Bachettoni A, et al. Monosodium urate contributes to retinal inflammation and progression of diabetic retinopathy. Diabetes (2019) 68(5):1014–25. doi: 10.2337/db18-0912
129. Singh LP, Devi TS, Yumnamcha T. The role of txnip in mitophagy dysregulation and inflammasome activation in diabetic retinopathy: a new perspective. JOJ Ophthalmol (2017) 4(4):10. doi: 10.19080/jojo.2017.04.555643
130. Gan J, Huang M, Lan G, Liu L, Xu F. High glucose induces the loss of retinal pericytes partly Via Nlrp3-Caspase-1-Gsdmd-Mediated pyroptosis. BioMed Res Int (2020) 2020:4510628. doi: 10.1155/2020/4510628
131. Platania CBM, Giurdanella G, Di Paola L, Leggio GM, Drago F, Salomone S, et al. P2x7 receptor antagonism: implications in diabetic retinopathy. Biochem Pharmacol (2017) 138:130–9. doi: 10.1016/j.bcp.2017.05.001
132. Cogan DG, Toussaint D, Kuwabara T. Retinal vascular patterns. Iv Diabetic Retinopathy Arch Ophthalmol (1961) 66:366–78. doi: 10.1001/archopht.1961.00960010368014
133. Barouch FC, Miyamoto K, Allport JR, Fujita K, Bursell SE, Aiello LP, et al. Integrin-mediated neutrophil adhesion and retinal leukostasis in diabetes. Invest Ophthalmol Visual Sci (2000) 41(5):1153–8.
134. Simons M, Gordon E, Claesson-Welsh L. Mechanisms and regulation of endothelial vegf receptor signalling. Nat Rev Mol Cell Biol (2016) 17(10):611–25. doi: 10.1038/nrm.2016.87
135. Eshaq RS, Aldalati AMZ, Alexander JS, Harris NR. Diabetic retinopathy: breaking the barrier. Pathophysiology (2017) 24(4):229–41. doi: 10.1016/j.pathophys.2017.07.001
136. Park DY, Lee J, Kim J, Kim K, Hong S, Han S, et al. Plastic roles of pericytes in the blood-retinal barrier. Nat Commun (2017) 8:15296. doi: 10.1038/ncomms15296
137. Jiang Y, Liu L, Curtiss E, Steinle JJ. Epac1 blocks Nlrp3 inflammasome to reduce il-1 in retinal endothelial cells and mouse retinal vasculature. Mediators Inflammation (2017) 2017:2860956. doi: 10.1155/2017/2860956
138. Chen W, Zhao M, Zhao S, Lu Q, Ni L, Zou C, et al. Activation of the Txnip/Nlrp3 inflammasome pathway contributes to inflammation in diabetic retinopathy: a novel inhibitory effect of minocycline. Inflammation Res: Off J Eur Histamine Res Soc [et Al] (2017) 66(2):157–66. doi: 10.1007/s00011-016-1002-6
139. Gu C, Draga D, Zhou C, Su T, Zou C, Gu Q, et al. Mir-590-3p inhibits pyroptosis in diabetic retinopathy by targeting Nlrp1 and inactivating the Nox4 signaling pathway. Invest Ophthalmol Visual Sci (2019) 60(13):4215–23. doi: 10.1167/iovs.19-27825
140. Qiu C, Coutinho P, Frank S, Franke S, L-y L, Martin P, et al. Targeting Connexin43 expression accelerates the rate of wound repair. Curr Biol (2003) 13(19):1697–703. doi: 10.1016/j.cub.2003.09.007
141. Mori R, Power KT, Wang CM, Martin P, Becker DL. Acute downregulation of Connexin43 at wound sites leads to a reduced inflammatory response, enhanced keratinocyte proliferation and wound fibroblast migration. J Cell Sci (2006) 119(Pt 24):5193–203. doi: 10.1242/jcs.03320
142. Mugisho OO, Green CR, Squirrell DM, Bould S, Danesh-Meyer HV, Zhang J, et al. Connexin43 hemichannel block protects against the development of diabetic retinopathy signs in a mouse model of the disease. J Mol Med (Berl) (2019) 97(2):215–29. doi: 10.1007/s00109-018-1727-5
143. Mugisho OO, Green CR, Kho DT, Zhang J, Graham ES, Acosta ML, et al. The inflammasome pathway is amplified and perpetuated in an autocrine manner through Connexin43 hemichannel mediated atp release. Biochim Biophys Acta Gen Subj (2018) 1862(3):385–93. doi: 10.1016/j.bbagen.2017.11.015
144. Mugisho OO, Green CR, Zhang J, Acosta ML, Rupenthal ID. Connexin43 hemichannels: a potential drug target for the treatment of diabetic retinopathy. Drug Discovery Today (2019) 24(8):1627–36. doi: 10.1016/j.drudis.2019.01.011
145. Danesh-Meyer HV, Kerr NM, Zhang J, Eady EK, O’Carroll SJ, Nicholson LFB, et al. Connexin43 mimetic peptide reduces vascular leak and retinal ganglion cell death following retinal ischaemia. Brain (2012) 135(Pt 2):506–20. doi: 10.1093/brain/awr338
146. Danesh-Meyer HV, Zhang J, Acosta ML, Rupenthal ID, Green CR. Connexin43 in retinal injury and disease. Prog Retin Eye Res (2016) 51:41–68. doi: 10.1016/j.preteyeres.2015.09.004
147. Kong H, Zhao H, Chen T, Song Y, Cui Y. Targeted P2x7/Nlrp3 signaling pathway against inflammation, apoptosis, and pyroptosis of retinal endothelial cells in diabetic retinopathy. Cell Death Dis (2022) 13(4):336. doi: 10.1038/s41419-022-04786-w
148. Yang K, Liu J, Zhang X, Ren Z, Gao L, Wang Y, et al. H3 relaxin alleviates migration, apoptosis and pyroptosis through P2x7r-mediated nucleotide binding oligomerization domain-like receptor protein 3 inflammasome activation in retinopathy induced by hyperglycemia. Front Pharmacol (2020) 11:603689. doi: 10.3389/fphar.2020.603689
149. Shi H, Zhang Z, Wang X, Li R, Hou W, Bi W, et al. Inhibition of autophagy induces il-1β release from arpe-19 cells Via ros mediated Nlrp3 inflammasome activation under high glucose stress. Biochem Biophys Res Commun (2015) 463(4):1071–6. doi: 10.1016/j.bbrc.2015.06.060
150. Wang S, Ji L-Y, Li L, Li J-M. Oxidative stress, autophagy and pyroptosis in the neovascularization of Oxygen−Induced retinopathy in mice. Mol Med Rep (2019) 19(2):927–34. doi: 10.3892/mmr.2018.9759
151. Chen Y, Wang L, Pitzer AL, Li X, Li P-L, Zhang Y. Contribution of redox-dependent activation of endothelial Nlrp3 inflammasomes to hyperglycemia-induced endothelial dysfunction. J Mol Med (Berl) (2016) 94(12):1335–47. doi: 10.1007/s00109-016-1481-5
152. Zheng D, Liu J, Piao H, Zhu Z, Wei R, Liu K. Ros-triggered endothelial cell death mechanisms: focus on pyroptosis, parthanatos, and ferroptosis. Front In Immunol (2022) 13:1039241. doi: 10.3389/fimmu.2022.1039241
153. Vincent JA, Mohr S. Inhibition of caspase-1/Interleukin-1beta signaling prevents degeneration of retinal capillaries in diabetes and galactosemia. Diabetes (2007) 56(1):224–30. doi: 10.2337/db06-0427
154. Lechner J, O’Leary OE, Stitt AW. The pathology associated with diabetic retinopathy. Vision Res (2017) 139:7–14. doi: 10.1016/j.visres.2017.04.003
155. Yu X, Ma X, Lin W, Xu Q, Zhou H, Kuang H. Long noncoding rna miat regulates primary human retinal pericyte pyroptosis by modulating mir-342-3p targeting of Casp1 in diabetic retinopathy. Exp Eye Res (2021) 202:108300. doi: 10.1016/j.exer.2020.108300
156. Coughlin BA, Feenstra DJ, Mohr S. Müller cells and diabetic retinopathy. Vision Res (2017) 139:93–100. doi: 10.1016/j.visres.2017.03.013
157. Fu S, Dong S, Zhu M, Sherry DM, Wang C, You Z, et al. Müller glia are a major cellular source of survival signals for retinal neurons in diabetes. Diabetes (2015) 64(10):3554–63. doi: 10.2337/db15-0180
158. Hori S, Mukai N. Ultrastructural lesions of retinal pericapillary müller cells in streptozotocin-induced diabetic rats. Albrecht Von Graefes Arch Klin Exp Ophthalmol (1980) 213(1):1–9. doi: 10.1007/BF02391205
159. Mizutani M, Gerhardinger C, Lorenzi M. Müller cell changes in human diabetic retinopathy. Diabetes (1998) 47(3):445–9. doi: 10.2337/diabetes.47.3.445
160. Trueblood KE, Mohr S, Dubyak GR. Purinergic regulation of high-Glucose-Induced caspase-1 activation in the rat retinal müller cell line rmc-1. Am J Physiol Cell Physiol (2011) 301(5):C1213–C23. doi: 10.1152/ajpcell.00265.2011
161. Du J, Wang Y, Tu Y, Guo Y, Sun X, Xu X, et al. A prodrug of epigallocatechin-3-Gallate alleviates high glucose-induced pro-angiogenic factor production by inhibiting the Ros/Txnip/Nlrp3 inflammasome axis in retinal müller cells. Exp Eye Res (2020) 196:108065. doi: 10.1016/j.exer.2020.108065
162. Devi TS, Lee I, Hüttemann M, Kumar A, Nantwi KD, Singh LP. Txnip links innate host defense mechanisms to oxidative stress and inflammation in retinal Muller glia under chronic hyperglycemia: implications for diabetic retinopathy. Exp Diabetes Res (2012) 2012:438238. doi: 10.1155/2012/438238
163. Calderon GD, Juarez OH, Hernandez GE, Punzo SM, de la Cruz ZD. Oxidative stress and diabetic retinopathy: development and treatment. Eye (Lond) (2017) 31(8):1122–30. doi: 10.1038/eye.2017.64
164. Hwang J, Suh H-W, Jeon YH, Hwang E, Nguyen LT, Yeom J, et al. The structural basis for the negative regulation of thioredoxin by thioredoxin-interacting protein. Nat Commun (2014) 5:2958. doi: 10.1038/ncomms3958
165. Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol (2010) 11(2):136–40. doi: 10.1038/ni.1831
166. Ma M, Zhao S, Li C, Tang M, Sun T, Zheng Z. Transient receptor potential channel 6 knockdown prevents high glucose-induced müller cell pyroptosis. Exp Eye Res (2023) 227:109381. doi: 10.1016/j.exer.2023.109381
167. Rathnasamy G, Foulds WS, Ling E-A, Kaur C. Retinal microglia - a key player in healthy and diseased retina. Prog Neurobiol (2019) 173:18–40. doi: 10.1016/j.pneurobio.2018.05.006
168. Dong N, Wang Y. Mir-30a regulates S100a12-induced retinal microglial activation and inflammation by targeting Nlrp3. Curr Eye Res (2019) 44(11):1236–43. doi: 10.1080/02713683.2019.1632350
169. Huang L, You J, Yao Y, Xie M. High glucose induces pyroptosis of retinal microglia through Nlpr3 inflammasome signaling. Arq Bras Oftalmol (2021) 84(1):67–73. doi: 10.5935/0004-2749.20210010
170. Osborne NN, Casson RJ, Wood JPM, Chidlow G, Graham M, Melena J. Retinal ischemia: mechanisms of damage and potential therapeutic strategies. Prog Retin Eye Res (2004) 23(1):91–147. doi: 10.1016/j.preteyeres.2003.12.001
171. Wan P, Su W, Zhang Y, Li Z, Deng C, Li J, et al. Lncrna H19 initiates microglial pyroptosis and neuronal death in retinal Ischemia/Reperfusion injury. Cell Death Differ (2020) 27(1):176–91. doi: 10.1038/s41418-019-0351-4
172. Deng Y, Fu Y, Sheng L, Hu Y, Su L, Luo J, et al. The regulatory nod-like receptor Nlrc5 promotes ganglion cell death in ischemic retinopathy by inducing microglial pyroptosis. Front In Cell Dev Biol (2021) 9:669696. doi: 10.3389/fcell.2021.669696
173. Li H, Li R, Wang L, Liao D, Zhang W, Wang J. Proanthocyanidins attenuate the high glucose-induced damage of retinal pigment epithelial cells by attenuating oxidative stress and inhibiting activation of the Nlrp3 inflammasome. J Biochem Mol Toxicol (2021) 35(9):e22845. doi: 10.1002/jbt.22845
174. Gu C, Zhang H, Li Q, Zhao S, Gao Y. Mir-192 attenuates high glucose-induced pyroptosis in retinal pigment epithelial cells Via inflammasome modulation. Bioengineered (2022) 13(4):10362–72. doi: 10.1080/21655979.2022.2044734
175. Xi X, Yang Y, Ma J, Chen Q, Zeng Y, Li J, et al. Mir-130a alleviated high-glucose induced retinal pigment epithelium (Rpe) death by modulating tnf-A/Sod1/Ros cascade mediated pyroptosis. BioMed Pharmacother (2020) 125:109924. doi: 10.1016/j.biopha.2020.109924
176. Yumnamcha T, Devi TS, Singh LP. Auranofin mediates mitochondrial dysregulation and inflammatory cell death in human retinal pigment epithelial cells: implications of retinal neurodegenerative diseases. Front Neurosci (2019) 13:1065. doi: 10.3389/fnins.2019.01065
177. Völgyi B. Molecular biology of retinal ganglion cells. Cells (2020) 9(11):2483. doi: 10.3390/cells9112483
178. Srinivasan S, Dehghani C, Pritchard N, Edwards K, Russell AW, Malik RA, et al. Corneal and retinal neuronal degeneration in early stages of diabetic retinopathy. Invest Ophthalmol Visual Sci (2017) 58(14):6365–73. doi: 10.1167/iovs.17-22736
179. Jonsson KB, Frydkjaer-Olsen U, Grauslund J. Vascular changes and neurodegeneration in the early stages of diabetic retinopathy: which comes first? Ophthalmic Res (2016) 56(1):1–9. doi: 10.1159/000444498
180. Yin Y, Chen F, Wang W, Wang H, Zhang X. Resolvin D1 inhibits inflammatory response in stz-induced diabetic retinopathy rats: possible involvement of Nlrp3 inflammasome and nf-κb signaling pathway. Mol Vis (2017) 23:242–50.
181. Chen H, Deng Y, Gan X, Li Y, Huang W, Lu L, et al. Nlrp12 collaborates with Nlrp3 and Nlrc4 to promote pyroptosis inducing ganglion cell death of acute glaucoma. Mol Neurodegener (2020) 15(1):26. doi: 10.1186/s13024-020-00372-w
182. Thomas CN, Berry M, Logan A, Blanch RJ, Ahmed Z. Caspases in retinal ganglion cell death and axon regeneration. Cell Death Discovery (2017) 3:17032. doi: 10.1038/cddiscovery.2017.32
183. Puyang Z, Feng L, Chen H, Liang P, Troy JB, Liu X. Retinal ganglion cell loss is delayed following optic nerve crush in Nlrp3 knockout mice. Sci Rep (2016) 6:20998. doi: 10.1038/srep20998
184. Zhang J, Dai Y, Yang Y, Xu J. Calcitriol alleviates hyperosmotic stress-induced corneal epithelial cell damage Via inhibiting the Nlrp3-Asc-Caspase-1-Gsdmd pyroptosis pathway in dry eye disease. J Inflammation Res (2021) 14:2955–62. doi: 10.2147/JIR.S310116
185. Li L, Xing C, Zhou J, Niu L, Luo B, Song M, et al. Airborne particulate matter (Pm2.5) triggers ocular hypertension and glaucoma through pyroptosis. Part Fibre Toxicol (2021) 18(1):10. doi: 10.1186/s12989-021-00403-4
186. Lian H, Fang X, Li Q, Liu S, Wei Q, Hua X, et al. Nlrp3 inflammasome-mediated pyroptosis pathway contributes to the pathogenesis of candida albicans keratitis. Front In Med (2022) 9:845129. doi: 10.3389/fmed.2022.845129
187. Sun H-J, Jin X-M, Xu J, Xiao Q. Baicalin alleviates age-related macular degeneration Via mir-223/Nlrp3-Regulated pyroptosis. Pharmacology (2020) 105(1-2):28–38. doi: 10.1159/000502614
188. Wang P, Zhu C, Liu M, Yuan Y, Ke B. The inhibiting effect of aspirin triggered-resolvin D1 in non-canonical pyroptosis in rats with acute keratitis. Exp Eye Res (2022) 218:108938. doi: 10.1016/j.exer.2022.108938
189. Deguchi S, Ogata F, Yamaguchi M, Minami M, Otake H, Kanai K, et al. In Situ Gel incorporating disulfiram nanoparticles rescues the retinal dysfunction Via atp collapse in otsuka long-Evans tokushima fatty rats. Cells (2020) 9(10):2171. doi: 10.3390/cells9102171
190. Zhou X, Rong R, Liang G, Li H, You M, Zeng Z, et al. Simultaneously deplete reactive oxygen species and inhibit pyroptosis by Dopamine/Thioketal-containing polymers delivering disulfiram in combination with Cu(Ii) for acute glaucoma. Nano Today (2022) 47:101668. doi: 10.1016/j.nantod.2022.101668
191. Zhang Y, Lv X, Hu Z, Ye X, Zheng X, Ding Y, et al. Protection of Mcc950 against high-Glucose-Induced human retinal endothelial cell dysfunction. Cell Death Dis (2017) 8(7):e2941. doi: 10.1038/cddis.2017.308
192. Wooff Y, Fernando N, Wong JHC, Dietrich C, Aggio-Bruce R, Chu-Tan JA, et al. Caspase-1-Dependent inflammasomes mediate photoreceptor cell death in photo-oxidative damage-induced retinal degeneration. Sci Rep (2020) 10(1):2263. doi: 10.1038/s41598-020-58849-z
193. Lian L, Le Z, Wang Z, Chen Y-A, Jiao X, Qi H, et al. Sirt1 inhibits high glucose-induced Txnip/Nlrp3 inflammasome activation and cataract formation. Invest Ophthalmol Visual Sci (2023) 64(3):16. doi: 10.1167/iovs.64.3.16
194. Cao X, Di G, Bai Y, Zhang K, Wang Y, Zhao H, et al. Aquaporin5 deficiency aggravates Ros/Nlrp3 inflammasome-mediated pyroptosis in the lacrimal glands. Invest Ophthalmol Visual Sci (2023) 64(1):4. doi: 10.1167/iovs.64.1.4
195. Liu Y, Kan M, Li A, Hou L, Jia H, Xin Y, et al. Inhibitory effects of tranilast on cytokine, chemokine, adhesion molecule, and matrix metalloproteinase expression in human corneal fibroblasts exposed to Poly(I:C). Curr Eye Res (2016) 41(11):1400–7. doi: 10.3109/02713683.2015.1127389
196. Ogawa Y, Dogru M, Uchino M, Tatematsu Y, Kamoi M, Yamamoto Y, et al. Topical tranilast for treatment of the early stage of mild dry eye associated with chronic gvhd. Bone Marrow Transplant (2010) 45(3):565–9. doi: 10.1038/bmt.2009.173
197. Lyon HA-OX, Shome A, Rupenthal IA-O, Green CA-O, Mugisho OO. Tonabersat inhibits Connexin43 hemichannel opening and inflammasome activation in an in vitro retinal epithelial cell model of diabetic retinopathy. Int J Mol Sci (2020) 30:22, 298. doi: 10.3390/ijms22010298
198. Mat Nor MN, Rupenthal ID, Green CR, Acosta MA-OX. Connexin hemichannel block using orally delivered tonabersat improves outcomes in animal models of retinal disease. Neurotherapeutics (2020) 17:371–87. doi: 10.1007/s13311-019-00786-5
199. Louie HH, Shome A, Kuo CY, Rupenthal ID, Green CR, Mugisho OO. Connexin43 hemichannel block inhibits Nlrp3 inflammasome activation in a human retinal explant model of diabetic retinopathy. Exp Eye Res (2021) 202:108384. doi: 10.1016/j.exer.2020.108384
200. Kim Y, Griffin JM, Nor MNM, Zhang J, Freestone PS, Danesh-Meyer HV, et al. Tonabersat prevents inflammatory damage in the central nervous system by blocking Connexin43 hemichannels. Neurotherapeutics (2017) 14(4):1148–65. doi: 10.1007/s13311-017-0536-9
201. Mat Nor MN, Rupenthal ID, Green CR, Acosta ML. Differential action of connexin hemichannel and pannexin channel therapeutics for potential treatment of retinal diseases. Int J Mol Sci (2021) 22(4):1755. doi: 10.3390/ijms22041755
202. Zhang B, Rusciano D, Osborne NN. Orally administered epigallocatechin gallate attenuates retinal neuronal death in vivo and light-induced apoptosis in vitro. Brain Res (2008) 1198:141–52. doi: 10.1016/j.brainres.2007.12.015
203. Shen C, Chen L, Jiang L, Lai TYY. Neuroprotective effect of epigallocatechin-3-Gallate in a mouse model of chronic glaucoma. Neurosci Lett (2015) 600:132–6. doi: 10.1016/j.neulet.2015.06.002
204. Tseng C-L, Hung Y-J, Chen Z-Y, Fang H-W, Chen K-H. Synergistic effect of artificial tears containing epigallocatechin gallate and hyaluronic acid for the treatment of rabbits with dry eye syndrome. PloS One (2016) 11(6):e0157982. doi: 10.1371/journal.pone.0157982
205. Huang H-Y, Wang M-C, Chen Z-Y, Chiu W-Y, Chen K-H, Lin IC, et al. Gelatin-epigallocatechin gallate nanoparticles with hyaluronic acid decoration as eye drops can treat rabbit dry-eye syndrome effectively Via inflammatory relief. Int J Nanomed (2018) 13:7251–73. doi: 10.2147/IJN.S173198
206. Petrillo F, Trotta MC, Bucolo C, Hermenean A, Petrillo A, Maisto R, et al. Resolvin D1 attenuates the inflammatory process in mouse model of lps-induced keratitis. J Cell Mol Med (2020) 24(21):12298–307. doi: 10.1111/jcmm.15633
207. Saban DR, Hodges RR, Mathew R, Reyes NJ, Yu C, Kaye R, et al. Resolvin D1 treatment on goblet cell mucin and immune responses in the chronic allergic eye disease (Aed) model. Mucosal Immunol (2019) 12(1):145–53. doi: 10.1038/s41385-018-0089-1
208. Settimio R, Clara DF, Franca F, Francesca S, Michele DA. Resolvin D1 reduces the immunoinflammatory response of the rat eye following uveitis. Mediators Inflammation (2012) 2012:318621. doi: 10.1155/2012/318621
209. Mugisho OO, Green CR, Squirrell DM, Bould S, Danesh-Meyer HV, Zhang J, et al. Connexin43 hemichannel block protects against the development of diabetic retinopathy signs in a mouse model of the disease. FASEB J (2023) 37:e22705. doi: 10.1096/fj.202200778R
210. Guo CX, Mat Nor MN, Danesh-Meyer HV, Vessey KA, Fletcher EL, O’Carroll SJ, et al. Connexin43 mimetic peptide improves retinal function and reduces inflammation in a light-damaged albino rat model. Invest Ophthalmol Visual Sci (2016) 57(10):3961–73. doi: 10.1167/iovs.15-16643
211. Chen Y-S, Green CR, Teague R, Perrett J, Danesh-Meyer HV, Toth I, et al. Intravitreal injection of lipoamino acid-modified Connexin43 mimetic peptide enhances neuroprotection after retinal ischemia. Drug Delivery Transl Res (2015) 5(5):480–8. doi: 10.1007/s13346-015-0249-8
212. Wang PA-O, Chen F, Wang W, Zhang XA-OX. Hydrogen sulfide attenuates high glucose-induced human retinal pigment epithelial cell inflammation by inhibiting ros formation and Nlrp3 inflammasome activation. Mediators Inflamm (2019) 24:8908960. doi: 10.1155/2019/8908960
213. Zhao H, Liu H, Yang Y, Wang HA-O. The role of H(2)S regulating Nlrp3 inflammasome in diabetes. Int J Mol Sci (2022) 23:4818. doi: 10.3390/ijms23094818
214. George AK, Singh M, Homme RP, Majumder A, Sandhu HS, Tyagi SC. A hypothesis for treating inflammation and oxidative stress with hydrogen sulfide during age-related macular degeneration. Int J Ophthalmol (2018) 11(5):881–7. doi: 10.18240/ijo.2018.05.26
215. Huang S, Huang P, Liu X, Lin Z, Wang J, Xu S, et al. Relevant variations and neuroprotecive effect of hydrogen sulfide in a rat glaucoma model. Neuroscience (2017) 341:27–41. doi: 10.1016/j.neuroscience.2016.11.019
216. Ren X, Li C, Liu J, Zhang C, Fu Y, Wang N, et al. Thioredoxin plays a key role in retinal neuropathy prior to endothelial damage in diabetic mice. Oncotarget (2017) 8:61350–64. doi: 10.18632/oncotarget
217. Maeda S, Matsui T, Ojima A, Takeuchi M, Yamagishi S. Sulforaphane inhibits advanced glycation end product-induced pericyte damage by reducing expression of receptor for advanced glycation end products. Nutr Res (2014) 34:807–13. doi: 10.1016/j.nutres.2014.08.010
218. Li S, Yang H, Chen X. Protective effects of sulforaphane on diabetic retinopathy: activation of the Nrf2 pathway and inhibition of Nlrp3 inflammasome formation. Exp Anim (2019) 68(2):221–31. doi: 10.1538/expanim.18-0146
219. Song H, Wang Y-H, Zhou H-Y, Cui K-M. Sulforaphane alleviates lps-induced inflammatory injury in arpe-19 cells by repressing the Pwrn2/Nf-kb pathway. Immunopharmacol Immunotoxicol (2022) 44(6):868–76. doi: 10.1080/08923973.2022.2090954
220. Pan H, He M, Liu R, Brecha NC, Yu ACH, Pu M. Sulforaphane protects rodent retinas against ischemia-reperfusion injury through the activation of the Nrf2/Ho-1 antioxidant pathway. PloS One (2014) 9(12):e114186. doi: 10.1371/journal.pone.0114186
221. Liu JA-O, Yang K, Jin Y, Liu Y, Chen Y, Zhang X, et al. H3 relaxin protects against calcium oxalate crystal-induced renal inflammatory pyroptosis. Cell Prolif (2020) 53:e12902. doi: 10.1111/cpr.12902
222. Hao J, Zhang H, Yu J, Chen X, Yang L. Methylene blue attenuates diabetic retinopathy by inhibiting Nlrp3 inflammasome activation in stz-induced diabetic rats. Ocul Immunol Inflammation (2019) 27:836–43. doi: 10.1080/09273948.2018.1450516
223. Mekala NK, Kurdys J, Depuydt MM, Vazquez EJ, Rosca MG. Apoptosis inducing factor deficiency causes retinal photoreceptor degeneration. the protective role of the redox compound methylene blue. Redox Biol (2019) 20:107–17. doi: 10.1016/j.redox.2018.09.023
224. Rey-Funes M, Larrayoz IM, Fernández JC, Contartese DS, Rolón F, Inserra PIF, et al. Methylene blue prevents retinal damage in an experimental model of ischemic proliferative retinopathy. Am J Physiol Regul Integr Comp Physiol (2016) 310(11):R1011–R9. doi: 10.1152/ajpregu.00266.2015
225. Tseng WA, Thein T, Kinnunen K, Lashkari K, Gregory MS, D’Amore PA, et al. Nlrp3 inflammasome activation in retinal pigment epithelial cells by lysosomal destabilization: implications for age-related macular degeneration. Invest Ophthalmol Visual Sci (2013) 54(1):110–20. doi: 10.1167/iovs.12-10655
226. Gao J, Cui JZ, Wang A, Chen HHR, Fong A, Matsubara JA. The reduction of xiap is associated with inflammasome activation in rpe: implications for amd pathogenesis. J Neuroinflamm (2019) 16(1):171. doi: 10.1186/s12974-019-1558-5
227. Chen J, Li L, Zhou Y, Zhang J, Chen L. Gambogic acid ameliorates high glucose- and palmitic acid-induced inflammatory response in arpe-19 cells Via activating Nrf2 signaling pathway: ex vivo. Cell Stress Chaperones (2021) 26(2):367–75. doi: 10.1007/s12192-020-01182-1
228. Shi Q, Wang J, Cheng Y, Dong X, Zhang M, Pei C. Palbinone alleviates diabetic retinopathy in stz-induced rats by inhibiting Nlrp3 inflammatory activity. J Biochem Mol Toxicol (2020) 34(7):e22489. doi: 10.1002/jbt.22489
229. Wang P, Chen F, Wang W, Zhang X-D. Hydrogen sulfide attenuates high glucose-induced human retinal pigment epithelial cell inflammation by inhibiting ros formation and Nlrp3 inflammasome activation. Mediators Inflammation (2019) 2019:8908960. doi: 10.1155/2019/8908960
230. Maberley D. Photodynamic therapy and intravitreal triamcinolone for neovascular age-related macular degeneration: a randomized clinical trial. Ophthalmology (2009) 116(11):2149–57.e1. doi: 10.1016/j.ophtha.2009.04.032
231. Jonas JB, Kreissig I, Degenring R. Intravitreal triamcinolone acetonide for treatment of intraocular proliferative, exudative, and neovascular diseases. Prog Retin Eye Res (2005) 24(5):587–611. doi: 10.1016/j.ophtha.2009.04.032
232. Thorne JE, Sugar EA, Holbrook JT, Burke AE, Altaweel MM, Vitale AT, et al. Periocular triamcinolone vs. intravitreal triamcinolone vs. intravitreal dexamethasone implant for the treatment of uveitic macular edema: the periocular vs. intravitreal corticosteroids for uveitic macular edema (Point) trial. Ophthalmology (2019) 126(2):283–95. doi: 10.1016/j.ophtha.2018.08.021
233. Gillies MC, Lim LL, Campain A, Quin GJ, Salem W, Li J, et al. A randomized clinical trial of intravitreal bevacizumab versus intravitreal dexamethasone for diabetic macular edema: the bevordex study. Ophthalmology (2014) 121(12):2473–81. doi: 10.1016/j.ophtha.2014.07.002
234. Wan L, Bai X, Zhou Q, Chen C, Wang H, Liu T, et al. The advanced glycation end-products (Ages)/Ros/Nlrp3 inflammasome axis contributes to delayed diabetic corneal wound healing and nerve regeneration. Int J Biol Sci (2022) 18(2):809–25. doi: 10.7150/ijbs.63219
235. Xu C, Lu Z, Luo Y, Liu Y, Cao Z, Shen S, et al. Targeting of Nlrp3 inflammasome with gene editing for the amelioration of inflammatory diseases. Nat Commun (2018) 9(1):4092. doi: 10.1038/s41467-018-06522-5
236. Wu M, Yang Z, Zhang C, Shi Y, Han W, Song S, et al. Inhibition of Nlrp3 inflammasome ameliorates podocyte damage by suppressing lipid accumulation in diabetic nephropathy. Metabolism (2021) 118:154748. doi: 10.1016/j.metabol.2021.154748
Keywords: diabetic retinopathy, inflammation, NLRP3, pyroptosis, treatments
Citation: Zheng X, Wan J and Tan G (2023) The mechanisms of NLRP3 inflammasome/pyroptosis activation and their role in diabetic retinopathy. Front. Immunol. 14:1151185. doi: 10.3389/fimmu.2023.1151185
Received: 25 January 2023; Accepted: 11 April 2023;
Published: 25 April 2023.
Edited by:
Alessandra Mortellaro, San Raffaele Telethon Institute for Gene Therapy (SR-Tiget), ItalyReviewed by:
Zongming Song, Henan Provincial Third People’s Hospital, ChinaUjjaldeep Jaggi, Cedars Sinai Medical Center, United States
Copyright © 2023 Zheng, Wan and Tan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gang Tan, dGFuZ2FuZzk5QGhvdG1haWwuY29t