 Heba M. Bintalib1,2,3*
Heba M. Bintalib1,2,3* Annick van de Ven4
Annick van de Ven4 Joseph Jacob1,5
Joseph Jacob1,5 Jesper Rømhild Davidsen6,7
Jesper Rømhild Davidsen6,7 Børre Fevang8,9
Børre Fevang8,9 Leif G. Hanitsch10,11Marion Malphettes12
Leif G. Hanitsch10,11Marion Malphettes12 Joris van Montfrans13
Joris van Montfrans13 Paul J. Maglione14
Paul J. Maglione14 Cinzia Milito15
Cinzia Milito15 John Routes16
John Routes16 Klaus Warnatz17,18†
Klaus Warnatz17,18† John R. Hurst1†
John R. Hurst1†- 1University College London (UCL) Respiratory, University College London, London, United Kingdom
- 2Department of Respiratory Care, King Saud bin Abdulaziz University for Health Sciences, Jeddah, Saudi Arabia
- 3King Abdullah International Medical Research Centre, Jeddah, Saudi Arabia
- 4Departments of Internal Medicine & Allergology, Rheumatology & Clinical Immunology, University Medical Center Groningen, Groningen, Netherlands
- 5Satsuma Lab, Centre for Medical Image Computing, University College London (UCL), London, United Kingdom
- 6South Danish Center for Interstitial Lung Diseases (SCILS), Department of Respiratory Medicine, Odense University Hospital, Odense, Denmark
- 7Odense Respiratory Research Unit (ODIN), Department of Clinical Research, University of Southern Denmark, Odense, Denmark
- 8Centre for Rare Disorders, Division of Paediatric and Adolescent Health, Oslo University Hospital, Oslo, Norway
- 9Section of Clinical Immunology and Infectious Diseases, Division of Surgery, Inflammatory Medicine and Transplantation, Oslo University Hospital, Oslo, Norway
- 10Institute of Medical Immunology, Charité - Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin and Humboldt Universität zu Berlin, Augustenburger Platz 1 and Berlin Institute of Health, Berlin, Germany
- 11Berlin Institute of Health at Charité - Universitätsmedizin Berlin, BIH Center for Regenerative Therapies (BCRT), Charitéplatz 1, Berlin, Germany
- 12Department of Clinic Immunopathology, Hôpital Saint-Louis, Paris, France
- 13Department of Pediatric Immunology and Infectious Diseases, Wilhelmina Childrens Hospital, University Medical Center Utrecht (UMC), Utrecht, Netherlands
- 14Section of Pulmonary, Allergy, Sleep, and Critical Care Medicine, Chobanian & Avedisian School of Medicine, Boston University, Boston, MA, United States
- 15Department of Molecular Medicine, Sapienza University of Rome, Rome, Italy
- 16Division of Allergy, Asthma and Immunology, Department of Pediatrics, Medicine, Microbiology and Immunology, Medical College Wisconsin, Milwaukee, WI, United States
- 17Department of Rheumatology and Clinical Immunology, Medical Center - University of Freiburg, Faculty of Medicine, University of Freiburg, Freiburg, Germany
- 18Center for Chronic Immunodeficiency (CCI), Medical Center - University of Freiburg, Faculty of Medicine, University of Freiburg, Freiburg, Germany
Introduction: Common variable immunodeficiency related interstitial lung disease (CVID-ILD, also referred to as GLILD) is generally considered a manifestation of systemic immune dysregulation occurring in up to 20% of people with CVID. There is a lack of evidence-based guidelines for the diagnosis and management of CVID-ILD.
Aim: To systematically review use of diagnostic tests for assessing patients with CVID for possible ILD, and to evaluate their utility and risks.
Methods: EMBASE, MEDLINE, PubMed and Cochrane databases were searched. Papers reporting information on the diagnosis of ILD in patients with CVID were included.
Results: 58 studies were included. Radiology was the investigation modality most commonly used. HRCT was the most reported test, as abnormal radiology often first raised suspicion of CVID-ILD. Lung biopsy was used in 42 (72%) of studies, and surgical lung biopsy had more conclusive results compared to trans-bronchial biopsy (TBB). Analysis of broncho-alveolar lavage was reported in 24 (41%) studies, primarily to exclude infection. Pulmonary function tests, most commonly gas transfer, were widely used. However, results varied from normal to severely impaired, typically with a restrictive pattern and reduced gas transfer.
Conclusion: Consensus diagnostic criteria are urgently required to support accurate assessment and monitoring in CVID-ILD. ESID and the ERS e-GLILDnet CRC have initiated a diagnostic and management guideline through international collaboration.
Systematic review registration: https://www.crd.york.ac.uk/prospero/, identifier CRD42022276337.
Introduction
Common variable immunodeficiency disorders (CVID) are the most prevalent primary symptomatic immunodeficiencies (PID), characterised by hypogammaglobulinemia and impaired immune responses to infections and vaccinations (1, 2). The two major clinical manifestations of CVID are recurrent, mainly bacterial infections and complications secondary to dysregulation of the immune system. Infections can be largely prevented through appropriate use of intravenous or subcutaneous immunoglobulin replacement therapy (IgRT) (3, 4). However, non-infectious complications such as interstitial lung disease, cytopenias, gastrointestinal and hepatic disease, and lymphoproliferative disease are difficult to manage and have become the major causes of morbidity and mortality (4).
Ten to 20% of people with CVID develop CVID-associated interstitial lung disease (CVID-ILD), histologically characterised by granulomatous inflammation and/or lymphocytic infiltrates (5). The condition has also been termed granulomatous and lymphocytic interstitial lung disease (GLILD). CVID-ILD appears alongside other non-infectious complications that increase morbidity and mortality in this group of patients, and thus is considered a manifestation of systemic lymphoproliferation and immune dysregulation (5, 6). There is no single clinical finding or investigation that facilitates the diagnosis of CVID-ILD due to heterogeneity of the disease. CVID-ILDs share clinical and histological characteristics with other conditions, and there is currently no single consensus on the diagnostic criteria for CVID-ILD. The understanding of pathogenesis is limited, and significant gaps in knowledge about diagnosis and management remain (5, 7). No evidence-based guideline for diagnosis or treatment is currently available, and management generally relies on clinicians’ expert opinions (7, 8).
The aim of this systematic review is to provide a comprehensive overview on diagnostic tests employed by clinicians when assessing adult and paediatric patients with CVID for possible CVID-ILD, reporting the utility and risks of these tests, and highlighting tests informing on disease activity or progression.
Method
We searched Ovid-EMBASE, MEDLINE, CINAHL PLUS and PubMed to identify all relevant published articles using the following key words: common variable immunodeficiency, late onset hypogammaglobulinemia, interstitial lung disease, lymphocytic interstitial pneumonitis, granulomatous lymphocytic interstitial lung disease, diagnosis, sign, symptom, clinical feature, characteristic, and manifestation.
Our inclusion criteria were: (1) type of study: we included prospective and retrospective cohort studies, case control studies, case reports, case series and non-randomised controlled trials. (2) population: individuals who fulfilled clinical criteria for common variable immunodeficiency, with or without genetic underlying diagnosis and confirmed or suspected ILD. (3) studies that reported information on diagnostic testing for ILD in patients with CVID. (4) outcomes: utility and, where reported, risks of diagnostic tests. (5) studies were in English. We excluded abstracts, theses, book chapters, review articles, and opinion articles, but searched the reference lists of reviews for primary sources. The original search was done on June 15, 2022, and was updated to December 2nd, 2022. The protocol was registered on PROSPERO (registration: CRD42022276337).
Studies retrieved using the search strategy were entered into Rayyan software (https://www.rayyan.ai/). All titles and abstracts were assessed by two reviewers against the inclusion criteria. Conflicts were settled by a third reviewer. Reviewers read the entire paper if the title and abstract didn’t provide enough information. The references were examined for additional sources. The primary data collected included study design, characteristics of study participants (where reported), description of the diagnostic method, test characteristics, and an evaluation of diagnostic utility. The results were collated for narrative synthesis. Only qualitative data were synthesised.
Qualitative assessment of study methodology
The assessment of study quality was completed by one author. To evaluate bias among observational studies, we used the Newcastle-Ottawa Scale (NOS) and a modified NOS which assesses studies based on three broad perspectives: the selection of the study groups; the comparability of the groups; and the ascertainment of either the exposure or outcome of interest (for cohort, case-control, or cross sectional studies respectively) (9). The Joanna Briggs Institute (JBI) Critical Appraisal Tools was used for case reports and case series (10). This addresses the risk of bias and internal validity and comprises 10 questions about confounding, selection (bias), information bias, and clear reporting.
Results
In total 58 studies describing a total of 796 patients were included (Figure 1). The average age at diagnosis of CVID-ILD in 422 adult and 28 paediatric patients was 40 years and 11 years, respectively. Not all papers were primarily aiming to evaluate diagnostic tests, but all papers that met our inclusion criteria were included. Thirty (52%) studies were performed in Europe, 24 (41%) were in the United States, and the remaining 4 (7%) were in Japan, Australia and Argentina. Among the 58 articles, 40 referred to the condition as GLILD, 16 studies used CVID related ILD and 2 studies described the condition as granulomatous CVID. We will use CVID-ILD in this review.

Figure 1 PRISMA flow diagram of study selection process. From: (11). For more information, visit: http://www.prisma-statement.org/.
The designs of the studies we included, and population characteristics of people included in these studies are reported in Table 1. Results summarising the frequency of the use of the diagnostic tests and the prevalence of abnormalities detected are reported in Table 2. Since most of the included studies involved an observational design, we considered the overall quality of evidence to be low. Supplementary Tables 1–5 provide a summary of the quality assessment.
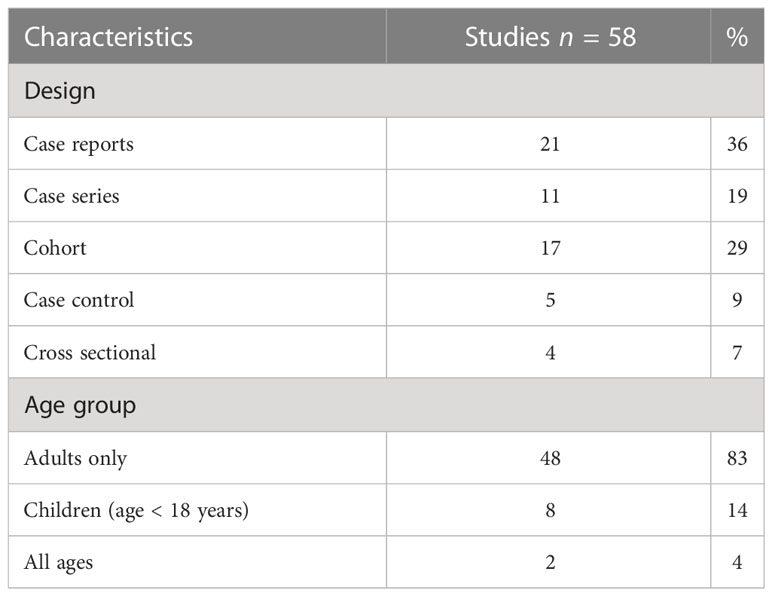
Table 1 Study design and population characteristics.
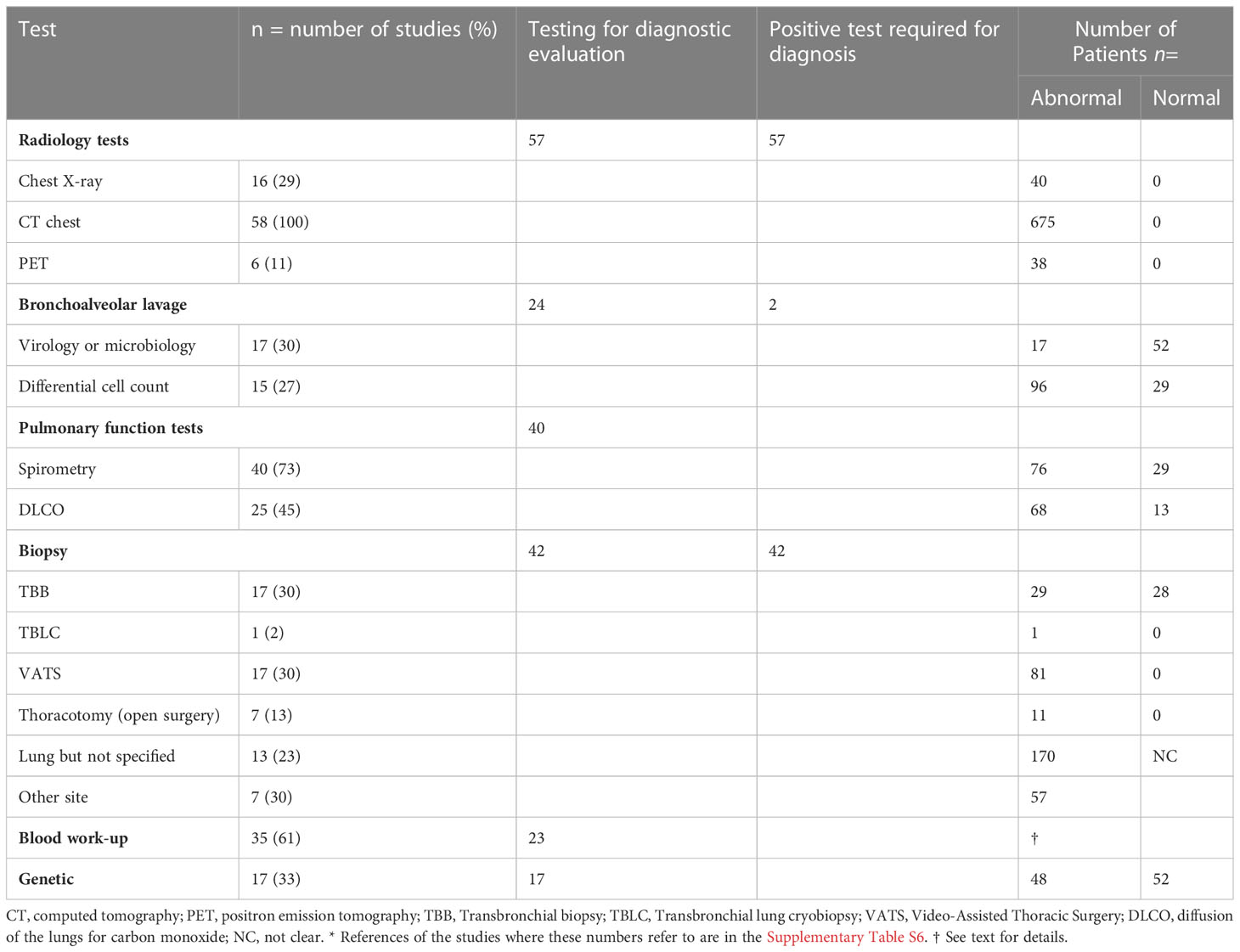
Table 2 Diagnostic tests in the evaluation of suspected CVID-ILD.*
Radiology
Abnormal lung imaging is considered a prerequisite for the diagnosis of CVID-ILD. Radiology studies were therefore the most frequently used tool for the assessment of potential lung involvement. Chest radiographs (CXR) were reported in 16 articles (5, 12–26). Typical pulmonary findings were bilateral patchy and nodular opacities with lower lung field predominance. In all of these studies (high-resolution/thin-section) Computed Tomography (CT) was subsequently performed because plain radiographs were not considered diagnostic.
Thirty-one case reports and case series reported the use of CT in the diagnostic work up (12–22, 26–45). Thirteen observational studies relied primarily on CT as their only criteria for CVID-ILD (2, 23, 27, 46–55), while 13 studies required either histological confirmation (5, 24, 25, 56–63) or an impairment in pulmonary function (64, 65) in addition to the detection of relevant CT abnormalities. As CT imaging was the basis for the diagnosis of CVID-ILD, CT imaging was abnormal in all patients. Studies which relied on CT-features alone to make the diagnosis of CVID-ILD defined typical pulmonary findings as: the presence of micronodules (which were predominantly peri-bronchovascular and more frequently found in the lower lobes), ground glass opacities, consolidation and interlobular septal thickening (Figure 2). In addition, thoracoabdominal lymphadenopathy and splenomegaly were characteristic extrapulmonary features. One recent paper by Smits et al. was added to the review despite being published after our search was completed, as it provides additional insight into the diagnostic criteria used for CVID-ILD (55). The authors recruited patients from the STILPAD study in which appropriate radiographic signs of CVID-ILD were sufficient for diagnosis. According to the authors’ classification, a ‘possible’ diagnosis was made if patients presented radiographic signs of CVID-ILD only, whereas a ‘probable’ diagnosis required either a probability score >50% and radiographic signs of CVID-ILD or histological confirmation of CVID-ILD (55).
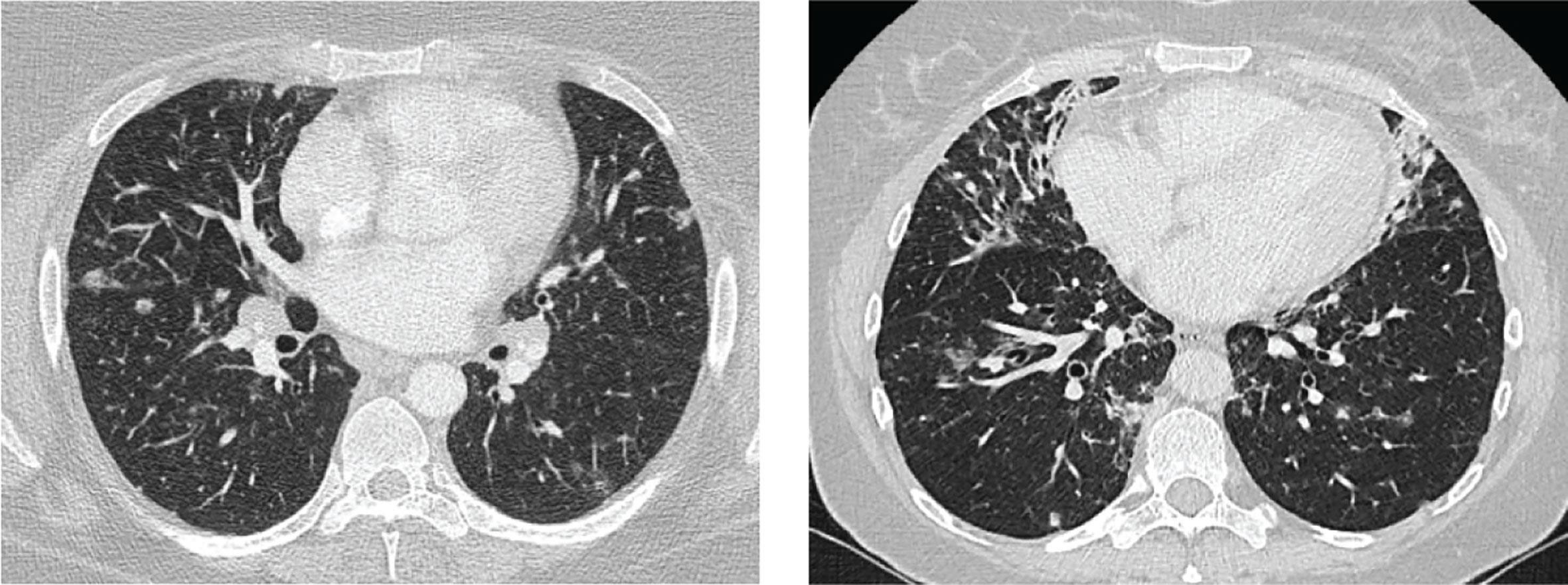
Figure 2 Images of two patients. Left: diffuse nodules and lymphadenopathy. Right: combination of diffuse nodules, reticulation and ground-glass opacities. Apart from CVID-ILD features, there are also signs of airway disease. From reference (51) with permission.
Seven studies reported using positron emission tomography-computed tomography (PET-CT) for assessing possible CVID-ILD (20, 30, 34, 35, 37, 39), assessing disease activity (50), and/or monitoring the response to treatment (30). In a retrospective cohort of 32 patients with CVID-ILD, Fraz et al. found that patients with progressive disease based on lung function tests had significantly higher mean standardized uptake values (SUV) in their lungs at baseline. This suggests a potential role of PET-CT in detecting pulmonary inflammation as part of active or uncontrolled overall disease (50).
The use of Magnetic Resonance Imaging (MRI) was not commonly reported as a diagnostic test for CVID-ILD. However, a few studies suggest that MRI scanning can be used an alternative to CT scanning to detect lung alterations and reduce radiation exposure in people with primary immune deficiencies (66–68).
Pulmonary function tests
Pulmonary function tests (PFT) as assessment tools were reported in 40 studies (5, 13, 15–18, 20–24, 27, 28, 30–36, 38–45, 49, 52–54, 56, 57, 59, 60, 62–64, 69), and included spirometry, measurement of diffusion capacity and assessment of static lung volumes (i.e., total lung capacity (TLC), and residual volume (RV)). Two studies reported PFT abnormalities as potential diagnostic criteria for CVID-ILD in addition to CT (64, 65). In 105 CVID-ILD patients with reported results, 53 (50%) patients had a restrictive lung pattern, while 20 (19%), 29 (28%) and 3 (3%) had obstructive, normal and mixed results, respectively. Gas transfer was low in 68 (57%) patients.
Bronchoalveolar lavage
Bronchoalveolar lavage (BAL) was generally performed to exclude infections, including bacteria, Mycobacteria, fungi and respiratory viruses. Seventeen studies reported BAL culture to exclude infection (12, 13, 16–19, 21, 22, 26, 31, 32, 35, 37, 41, 42, 44, 65) but only four reported polymerase chain reaction (PCR) to exclude cytomegalovirus, Epstein-Barr virus (EBV), HIV and Mycoplasma pneumonia (13, 35, 37, 41). The most common respiratory pathogens reported, where a pathogen was detected, were Staphylococcus aureus, Haemophilus influenzae, Streptococcus pneumoniae, rhinovirus, and cytomegalovirus.
Flow-cytometry analysis including differential cell count was reported in 15 studies verifying significant lymphocytosis in 96/125 (78%) of patients (15–17, 22, 24, 26, 35, 42, 44, 57–59, 64, 65, 69). In addition, where lymphocyte phenotyping was performed this showed a larger proportion of B cells, predominantly CD21low B cells (57, 65). Friedmann et al. reported that patients with CVID-ILD had fewer regulatory T cells, but more T follicular helper (TFH)-like memory cells skewed towards Th1 cells, as well as a greater proportion of B cells (particularly the inflammatory CD21low B cell subtype) in BAL compared to sarcoidosis (65). There are conflicting reports regarding CD4/CD8 ratios in BAL, which have been described as reduced, elevated, and normal (24, 58, 59, 65).
Biopsy
The diagnosis of CVID-ILD was confirmed by biopsy in 31 case reports and series (12–22, 26–45) and was used as an obligatory inclusion criterium for CVID-ILD patients in eleven studies (5, 24, 25, 54, 56–60, 62, 63). Transbronchial biopsy (TBB) was described in 17 studies involving 57 patients, 28 of whom had definitive results, while the remaining patients underwent a supplemental biopsy modality to confirm the diagnosis (16–18, 22, 30, 31, 34, 38, 42, 44, 50, 58, 59, 62–64, 69). The most common diagnostic findings were non-necrotizing granulomatous and lymphocytic inflammation. The use of video-assisted thoracoscopic surgery (VATS) was reported in 17 studies (15, 16, 21, 26, 33, 34, 36, 39–41, 45, 47, 57, 60, 62, 63, 69). The results from 81 patients demonstrated the characteristic histological findings of CVID-ILD. Ten studies reported the use of open biopsy in 11 patients where all had conclusive results (12–14, 18, 28, 29, 42, 60, 63). The most common findings on surgical biopsy were non-necrotising granulomatous inflammation, lymphoid interstitial pneumonitis (LIP), and/or lymphoid hyperplasia, while organising pneumonia (OP), interstitial fibrosis and follicular bronchiolitis were less common. Only one article reported the use of transbronchial cryobiopsy (TBCB), in one patient with no conclusive results (44). One case report used transbronchial fine-needle aspirate (FNA) of pulmonary nodules to exclude malignancy and lymphoma (20). Extrapulmonary biopsy was accepted to substantiate the diagnosis of CVID-ILD in seven studies (24, 28, 32, 37, 42, 54, 57). Lymph node biopsy was the most frequently reported, followed by liver, spleen and skin. The most common finding was non-necrotizing granulomata.
Only one study reported the risk of biopsy-related complications, in this case related to the VATS procedure, where the patient developed pleural empyema (26). Biopsy samples were often also tested for fungi, mycobacteria, pneumonia, EBV, and CMV using culture, special stains and molecular biology.
Blood biomarkers and genetic testing
The blood work-up differed markedly between studies. As a result, drawing conclusions was challenging because no one blood biomarker is has been shown to aid the diagnosis of CVID-ILD. Fraz et al. recently reported that CVID-ILD patients have elevated serum markers of T cell activation and exhaustion reflected by elevated level of TNF, IFN-γ, sCD25, and sTIM-3; increased concentrations of pulmonary epithelium injury biomarkers including CC16, SP-D and MMP-7; and increased levels of ECM remodelling markers compared to patients with other non-infectious complications. Other potential biomarkers have been used to developed diagnostic prediction models and to help avoid biopsy (as discussed further below). Furthermore, different blood biomarkers have been reported to be associated with CVID-ILD progression, and these include increased level of B cell-activating factor (BAFF), IgM in serum, the soluble form of the interleukin-2 receptor (sIL-2R) and neopterin (17, 48, 55, 70). These data are summarised in Table 3. Smits et al. have reported that neopterin levels, in addition to IgM level and sIL-2R, may have the potential to serve as biomarkers for disease activity (55).
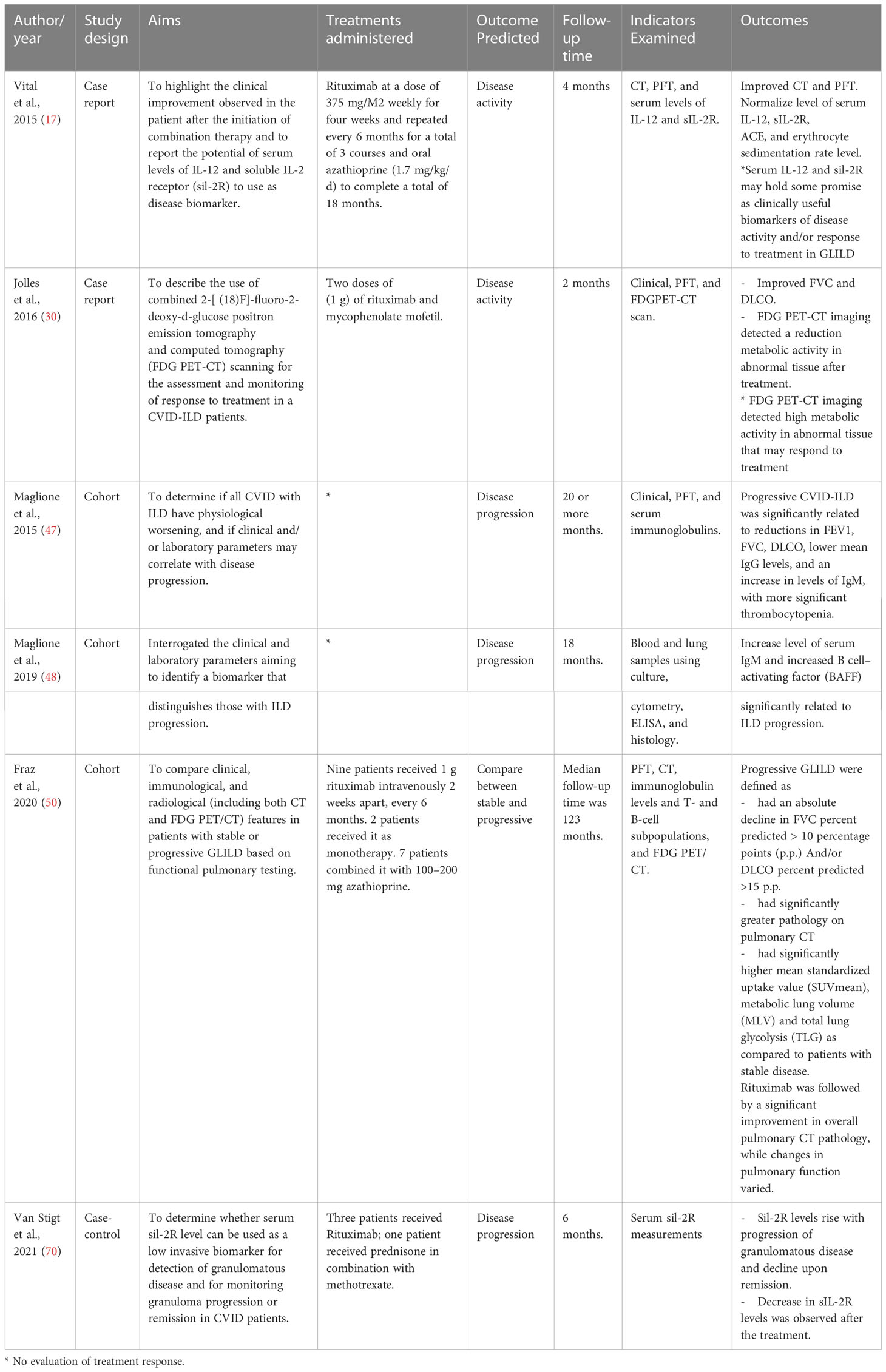
Table 3 Studies that evaluated biomarkers of CVID-ILD disease activity and progression.
Genetic evaluations were reported in seventeen studies (23, 25, 28, 32, 39, 41, 43–45, 48, 50, 56, 57, 62–64, 69). 48 of 100 reported patients had cytotoxic T lymphocyte antigen 4 (CTLA-4) haploinsufficiency, or transmembrane activator and calcium-modulating cyclophilin ligand interactor (TACI) (TNFRSF13B) or signal transducer and activator of transcription 3 (STAT-3) mutations.
Diagnostic prediction models
Four studies developed prediction models for biopsy-positive CVID-ILD based on clinical, laboratory and/or lung physiological parameters to assist predicting the presence of CVID-ILD (54, 56, 57, 59). These are reported in Table 4. All studies reported splenomegaly as a predictor for CVID-ILD, with odds ratios between 8.47 and 23.9 (54, 56, 57, 59). Cinetto et al. proposed a CVID-ILD predictive model based on splenomegaly, CD21lo B cells percentage, autoimmune cytopenia and DLCO percent predicted with Area Under the Curve (AUC) of 0.98 (57). The recent predictive model proposed by Cabanero et al. was based on splenomegaly, lymphadenopathy, low CD8 cell in BAL, and high Baumann’s CVID-ILD composite score, with an AUC of 0.985 (56). Such studies need external validation.
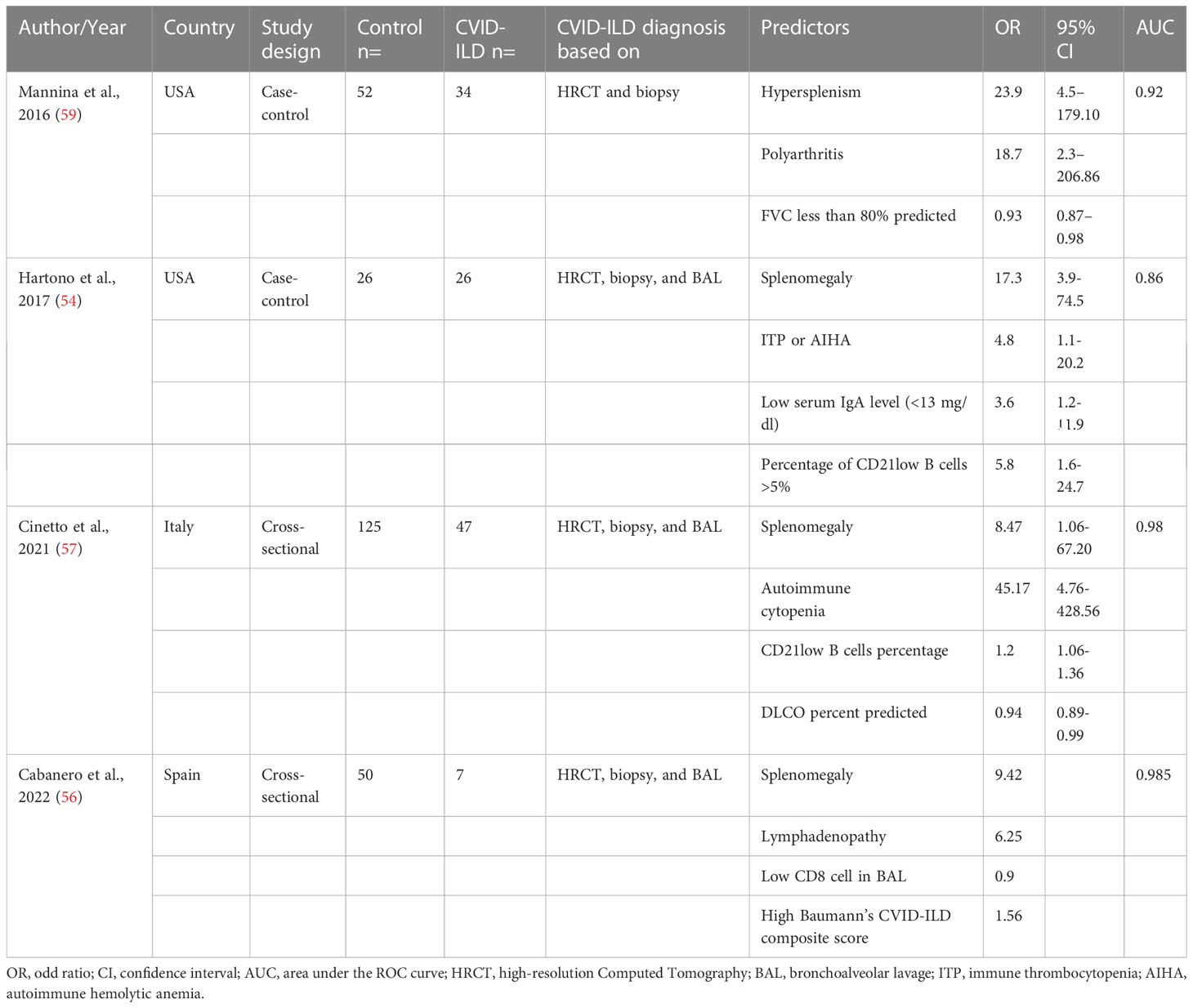
Table 4 Prediction models to screen patients with CVID-ILD.
Discussion
Managing clinically relevant complications in a rare disease is a significant challenge for clinicians, especially in the absence of evidence-based guidelines. The diagnosis and managing of CVID-ILD therefore usually depends on the decisions and experience of individual clinical teams. In this systematic review we reviewed diagnostic methods and criteria for CVID-ILD, and for informing prognosis in CVID-ILD. The key findings are (i) in general, there was diagnostic consistency across studies, (ii) HRCT was the most frequently reported test to detect CVID-ILD, (iii) lung biopsy is required to definitively confirm the diagnosis but some teams make a clinical diagnosis, (iv) BAL was routinely performed to exclude infection, and (v) non-biopsy prediction models for CVID-ILD had good discriminative accuracy but require external validation. A more consistent diagnostic approach would facilitate research collaboration and comparisons across studies (8).
The term GLILD was introduced by Bates et al. to describe a group of CVID patients with histological findings of LIP, lymphoid hyperplasia, follicular bronchiolitis, and/or granulomatous disease (5). However, the term has been interpreted differently across the literature. Some authors consider pulmonary fibrosis and organising pneumonia (OP) as additional features, while others consider the diagnosis to require histologically proven pulmonary granuloma. Thus, there is discussion to reconsider terminology (46, 71–73). We found that three-quarters of the included papers refer to these (histological) manifestations as GLILD, but prefer the term CVID-ILD.
The diagnosis of CVID-ILD has been clearly described in case reports and series. In contrast, the inclusion criteria in observational studies completed for other reasons were often vaguely described, which made it challenging to interpret the results. We found general consistency in the diagnostic approach between studies. However, not all tests were always performed in all subjects, notably biopsy.
Patients with CVID-ILD often have other lymphoproliferative and autoimmune manifestations. Splenomegaly, lymphoproliferative disorders, cytopenias such as thrombocytopenia and autoimmune haemolytic anaemia (AIHA) are the most common extrapulmonary manifestations in these patients. The presence of these features could increase the suspicion of CVID-ILD and were used along with other clinical and laboratory features to develop prediction models (as described above). The purpose of these models was to support the diagnosis of CVID-ILD and/or the risk of future CVID-ILD; however, they need to be validated.
Radiology was the investigation most commonly used during the process of diagnosis. HRCT was the most frequently reported test, in all the included articles, as abnormal results usually first raise suspicion of ILD in CVID. Since plain radiographic studies have low sensitivity to provide sufficient diagnostic information, the diagnosis was generally based on abnormalities revealed on CT scan. Few studies employed CT scoring methods to evaluate lung involvement and progression, which can be complex and therefore time-consuming. Meerburg and colleagues evaluated the Baumann and Hartmann scoring methods in a cohort of 138 people with CVID-ILD (51). They reported Hartmann’s scoring to be more reproducible than Baumann’s and suggested use of radiological scoring to measure outcomes in future studies. Despite widespread use of CT, the potential risk of radiosensitivity in CVID should be considered (74, 75) given that scans may need to be repeated over time. MRI can be utilized to evaluate the lungs in patients with CVID as it is comparable to HRCT in its ability to identify bronchial and parenchymal abnormalities (67, 68). Additionally, MRI does not pose the risk of ionizing radiation, which may be a concern for some patients. However, it is important to note that the spatial resolution of MRI may be lower than that of HRCT, which may limit its sensitivity for detecting some lung abnormalities particularly small nodules.
Lung biopsy was the second most common reported test required for diagnosis, and surgical lung biopsy (SLB) had more conclusive results compared to TBB alongside histological diagnosis of other ILDs (76). Histological findings may be diverse between patients, and indeed diverse between different areas of the lung in individual patients. This variability has implications for the amount of tissue collected during sampling. As the volume of sampling increases, the probability of discovering additional features, if not all, of what is referred to as CVID-ILD increases. Verbsky et al., in their longitudinal retrospective analysis evaluating treatments response, reported 34/39 patients had VATS with sampling of at least two areas of the lung in which all patients exhibited at least three of the four main histological abnormalities considered characteristic of CVID-ILD. This could explain why almost half of the patients who underwent TBB had inconclusive results as the small amount of tissue attributed obtained can result in sampling error and thus may not represent the complete histopathological pattern. In addition, the timing of samples with regard to natural history of the disease (or previous administration of medicines) could contribute to the heterogeneity of histological findings (71). When taking complications into account, other diagnostic histological approaches such as TBCB should be also considered for CVID-ILD (76, 77). Currently, histology is regarded necessary for definitive confirmation because it is still unclear how accurately clinic, laboratory, CT and potentially BAL parameters can exclude alternative diagnoses. Consequently, it has been suggested that a classification of probable vs biopsy-proven CVID-ILD is introduced as used with clinical, radiological and histological classification of other ILD subtypes (55, 78, 79). Three of four non-biopsy prediction models for CVID-ILD had good discriminative accuracy in their development studies (56, 57, 59).
Analysis of BAL was often conducted, primarily to exclude infection, although the BAL differential cell count has been described as an adjunct to positive diagnosis of CVID-ILD. 78% of patients had an increased proportion of lymphocytes which was described as the most prominent feature of BAL with expansion of both T-cells and B-cells, predominantly CD21low B cells, which has been utilized as a predictive parameter in two studies mentioned above. This is in contrast with sarcoidosis where there is no increase in B cells, however a diagnosis of sarcoid instead of CVID-ILD can be more readily clarified by simple measurement of serum immunoglobulins (65).
Our results demonstrate that PFTs including gas transfer are widely used during the diagnostic process. However, results vary from normal to severely impaired, in the latter case typically with a restrictive pattern and reduced gas transfer. PFTs abnormalities can often be found but are not sufficiently sensitive to diagnose CVID-ILD. Gas transfer abnormalities are the most common findings. Future studies need to evaluate how valuable PFTs including DLCO are in determining the need for treatment and to assess changes at follow up. Paediatric articles reported less use of PFTs due to challenges conducting the tests in very young children.
This is the first systematic review to evaluate diagnostic approaches in CVID-ILD. There were some limitations of this study. First, we recognise the heterogeneity of the definition and terminology of ILD in CVID, and methodologies used between studies. Second, we could not summarise risks and benefits of the different diagnostic procedures as these were often not reported. Third, the quality of the evidence is generally low, being based on case reports and case series. Finally, we limited our search to include only English articles. A strength of this review is that we collated all evidence in regard to the clinical approach to diagnosis of CVID-ILD by including case reports and series in our evaluation.
Patients with CVID who experience respiratory symptoms, have abnormal imaging findings, or demonstrate decreased lung function should be evaluated for ILD. The risk of CVID-ILD may increase in patients with other autoimmune conditions. Thus, multidisciplinary discussion is crucial in the diagnosis and management of CVID-ILD, as it facilitates a comprehensive and tailored approach to care that can lead to better outcomes and improved quality of life for patients. In addition, consensus diagnostic criteria are urgently required to support accurate assessment and monitoring in CVID-ILD. The European Society for Immunodeficiencies (ESID) has initiated production of a diagnostic and management guideline through international collaboration. The guideline will promote collaboration and disease management, and reduce unwarranted variation in care.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Author contributions
JH selected the review’s subject and directed the research and writing processes. HB, JH, and KW created the search strategy. HB and JH review papers for inclusion and created the tables. JH and KW gave advice during the synthesis of the results. HB wrote the initial draft. AV, JJ, JD, BF, LH, MM, JM, PJM, CM, JR, KW, and JH evaluated and commented on the draft papers. All authors participated to and approved the final draft of the article.
Funding
There was no specific funding for this study. HB is supported by King Saud bin Abdulaziz University for Health Sciences, Jeddah, Saudi Arabia. This research was funded in whole or in part by the Wellcome Trust [209553/Z/17/Z]. For the purpose of open access, the author has applied a CC-BY public copyright licence to any author accepted manuscript version arising from this submission.
Acknowledgments
The authors would like to acknowledge the assistance of Tope Oyelade, PhD, Institute for Liver and Digestive Health, Division of Medicine, University College London.
Conflict of interest
AV reports fees for educational activities from Takeda outside the submitted work.JJ reports fees from Boehringer Ingelheim, Roche, NHSX, Takeda and GlaxoSmithKline unrelated to the submitted work. JJ was supported by Wellcome Trust Clinical Research Career Development Fellowship 209553/Z/17/Z and the NIHR Biomedical Research Centre at University College London.JD reports fees for advisory board meetings, teaching and educational activities, and congress participation from Boehringer Ingelheim outside the submitted work.PM has received grant support from the National Institutes of Health, AAAAI Foundation, Immune Deficiency Foundation, Takeda, Horizon Pharma, and Boston University and has received consulting fees from Medscape and Pharming.KW reports honoraria for advisory board meetings, teaching and educational activities from TAKEDA, LFB biomedicaments, CSL Behring, Grifols, and Bristol-Myers Squibb outside the submitted work. In addition, KW has received a research grant by Bristol-Myers Squibb for the investigation of Abatacept for interstitial lung disease in CVID.JH has received support to attend meetings, personal payment and payment to his employer from companies that make medicines to treat respiratory disease and immunoglobulin products.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1190235/full#supplementary-material
References
1. Bonilla FA, Barlan I, Chapel H, Costa-Carvalho BT, Cunningham-Rundles C, de la Morena MT, et al. International consensus document (ICON): common variable immunodeficiency disorders. J Allergy Clin Immunol Pract (2016) 4(1):38–59. doi: 10.1016/j.jaip.2015.07.025
2. Fraz MSA, Michelsen AE, Moe N, Aaløkken TM, Macpherson ME, Nordøy I, et al. Raised serum markers of T cell activation and exhaustion in granulomatous-lymphocytic interstitial lung disease in common variable immunodeficiency. J Clin Immunol (2022) 42:1–11. doi: 10.1007/s10875-022-01318-1
3. Orange JS, Grossman WJ, Navickis RJ, Wilkes MM. Impact of trough IgG on pneumonia incidence in primary immunodeficiency: a meta-analysis of clinical studies. Clin Immunol (2010) 137(1):21–30. doi: 10.1016/j.clim.2010.06.012
4. Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood (2012) 119(7):1650–7. doi: 10.1182/blood-2011-09-377945
5. Bates CA, Ellison MC, Lynch DA, Cool CD, Brown KK, Routes JM. Granulomatous-lymphocytic lung disease shortens survival in common variable immunodeficiency. J Allergy Clin Immunol (2004) 114(2):415–21. doi: 10.1016/j.jaci.2004.05.057
6. Schussler E, Beasley MB, Maglione PJ. Lung disease in primary antibody deficiencies. J Allergy Clin Immunol Pract (2016) 4(6):1039–52. doi: 10.1016/j.jaip.2016.08.005
7. Hurst JR, Abbas SH, Bintalib HM, Alfaro TM, Baumann U, Burns SO, et al. Granulomatous–lymphocytic interstitial lung disease: an international research prioritisation. ERJ Open Res (2021) 7(4):00467–2021. doi: 10.1183/23120541.00467-2021
8. Hurst JR, Verma N, Lowe D, Baxendale HE, Jolles S, Kelleher P, et al. British Lung Foundation/United kingdom primary immunodeficiency network consensus statement on the definition, diagnosis, and management of granulomatous-lymphocytic interstitial lung disease in common variable immunodeficiency disorders. J Allergy Clin Immunol Pract (2017) 5(4):938–45. doi: 10.1016/j.jaip.2017.01.021
9. Peterson J, Welch V, Losos M, Tugwell P. The Newcastle-Ottawa scale (NOS) for assessing the quality of nonrandomised studies in meta-analyses. Ottawa: Ottawa Hosp Res Institute (2011) 2(1):1–12.
10. Munn Z, Barker TH, Moola S, Tufanaru C, Stern C, McArthur A, et al. Methodological quality of case series studies: an introduction to the JBI critical appraisal tool. JBI Evid Synth (2020) 18(10):2127–33. doi: 10.11124/JBISRIR-D-19-00099
11. Page MJ, McKenzie JE, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ (2021) 372:n71. doi: 10.1136/bmj.n71
12. Sacco O, Fregonese B, Picco P, Faraci M, Facchetti P, Pistoia V, et al. Common variable immunodeficiency presenting in a girl as lung infiltrates and mediastinal adenopathies leading to severe 'superior vena caval' syndrome. Eur Respir J (1996) 9:1958–61. doi: 10.1183/09031936.96.09091958
13. Davies CW, Juniper MC, Gray W, Gleeson FV, Chapel HM, Davies RJ. Lymphoid interstitial pneumonitis associated with common variable hypogammaglobulinaemia treated with cyclosporin a. Thorax (2000) 55(1):88–90. doi: 10.1136/thorax.55.1.88
14. Thatayatikom A, Thatayatikom S, White AJ. Infliximab treatment for severe granulomatous disease in common variable immunodeficiency: a case report and review of the literature. Ann Allergy Asthma Immunol (2005) 95(3):293–300. doi: 10.1016/S1081-1206(10)61228-8
15. Matsubara M, Koizumi T, Wakamatsu T, Fujimoto K, Kubo K, Honda T. Lymphoid interstitial pneumonia associated with common variable immunoglobulin deficiency. Intern Med (2008) 47(8):763–7. doi: 10.2169/internalmedicine.47.0742
16. Boujaoude Z, Arya R, Rafferty W, Dammert P. Organising pneumonia in common variable immunodeficiency. BMJ Case Rep (2013) 2013. doi: 10.1136/bcr-2013-008905
17. Vitale J, Convers KD, Goretzke S, Guzman M, Noyes B, Parkar N, et al. Serum IL-12 and soluble IL-2 receptor levels as possible biomarkers of granulomatous and lymphocytic interstitial lung disease in common variable immunodeficiency: a case report. J Allergy Clin Immunology: In Practice (2015) 3:273–6. doi: 10.1016/j.jaip.2014.09.019
18. Tashtoush B, Memarpour R, Ramirez J, Bejarano P, Mehta J. Granulomatous-lymphocytic interstitial lung disease as the first manifestation of common variable immunodeficiency. Clin Respir J (2016) 12:337–43. doi: 10.1111/crj.12511
19. Tessarin G, Bondioni MP, Rossi S, Palumbo L, Soresina A, Badolato R, et al. Rituximab as a single agent for granulomatous lymphocytic interstitial lung disease in common variable immune deficiency. J Investig Allergol Clin Immunol (2019) 29(6):470–1. doi: 10.18176/jiaci.0450
20. Cowen JE, Stevenson J, Paravasthu M, Darroch J, Jacob A, Tueger S, et al. Common variable immunodeficiency with granulomatous-lymphocytic interstitial lung disease and preceding neurological involvement: a case-report. BMC Pulmonary Med (2020) 20:205. doi: 10.1186/s12890-020-01231-6
21. Pattanaik D, Ritter S, Fahhoum J. Common variable immunodeficiency (CVID) with granulomatous interstitial lung disease (GLILD) and SARS COVID-19 infection: case report and review of literature. Allergy Asthma Clin Immunol (2021) 17:98. doi: 10.1186/s13223-021-00600-y
22. Perlman DM, Sudheendra MT, Racilla E, Allen TL, Joshi A, Bhargava M. Granulomatous-lymphocytic interstitial lung disease mimicking sarcoidosis. Sarcoidosis Vasculitis Diffuse Lung Diseases (2021) 38(3):e2021025. doi: 10.36141/svdld.v38i3.11114
23. van de Ven AA, de Jong PA, Hoytema van Konijnenburg DP, Kessels OA, Boes M, Sanders EA, et al. Airway and interstitial lung disease are distinct entities in paediatric common variable immunodeficiency. Clin Exp Immunol (2011) 165(2):235–42. doi: 10.1111/j.1365-2249.2011.04425.x
24. Bouvry D, Mouthon L, Brillet PY, Kambouchner M, Ducroix JP, Cottin V, et al. Granulomatosis-associated common variable immunodeficiency disorder: a case-control study versus sarcoidosis. Eur Respir J (2013) 41(1):115–22. doi: 10.1183/09031936.00189011
25. Lopes JP, Ho HE, Cunningham-Rundles C. Interstitial lung disease in common variable immunodeficiency. Front Immunol (2021) 12:605945. doi: 10.3389/fimmu.2021.605945
26. Hasegawa M, Sakai F, Okabayashi A, Sato A, Yokohori N, Katsura H, et al. Intravenous immunoglobulin monotherapy for granulomatous lymphocytic interstitial lung disease in common variable immunodeficiency. Internal Med (2017) 56:2899–902. doi: 10.2169/internalmedicine.7757-16
27. Torigian DA, LaRosa DF, Levinson AI, Litzky LA, Miller WT Jr. Granulomatous-lymphocytic interstitial lung disease associated with common variable immunodeficiency: CT findings. J Thorac Imaging (2008) 23(3):162–9. doi: 10.1097/RTI.0b013e318166d32f
28. Franxman TJ, Howe LE, Baker JR Jr. Infliximab for treatment of granulomatous disease in patients with common variable immunodeficiency. J Clin Immunol (2014) 34(7):820–7. doi: 10.1007/s10875-014-0079-3
29. Rao N, Mackinnon AC, Routes JM. Granulomatous and lymphocytic interstitial lung disease: a spectrum of pulmonary histopathologic lesions in common variable immunodeficiency–histologic and immunohistochemical analyses of 16 cases. Hum Pathol (2015) 46(9):1306–14. doi: 10.1016/j.humpath.2015.05.011
30. Jolles S, Carne E, Brouns M, El-Shanawany T, Williams P, Marshall C, et al. FDG PET-CT imaging of therapeutic response in granulomatous lymphocytic interstitial lung disease (GLILD) in common variable immunodeficiency (CVID). Clin Exp Immunol (2016) 187:138–45. doi: 10.1111/cei.12856
31. Pathria M, Urbine D, Zumberg MS, Guarderas J. Management of granulomatous lymphocytic interstitial lung disease in a patient with common variable immune deficiency. BMJ Case Rep (2016) 2016. doi: 10.1136/bcr-2016-215624
32. Bucciol G, Petrone A, Putti MC. Efficacy of mycophenolate on lung disease and autoimmunity in children with immunodeficiency. Pediatr Pulmonol (2017) 52(10):E73–E6. doi: 10.1002/ppul.23757
33. Limsuwat C, Daroca PJ, Lasky JA. A 56-Year-Old-Man with common variable immunodeficiency and worsening dyspnea. Chest (2017) 154:e27–30. doi: 10.1016/j.pcl.2016.08.007
34. Routes JM, Verbsky JW. Immunodeficiency presenting as an undiagnosed disease. Pediatr Clinics (2017) 64(1):27–37. doi: 10.1016/j.pcl.2016.08.007
35. Zdziarski P, Gamian A, Dworacki G. A case report of lymphoid intestitial pneumonia in common variable immunodeficiency: oligoclonal expansion of effector lymphocytes with preferential cytomegalovirus-specific immune response and lymphoproliferative disease promotion. Med (Baltimore) (2017) 96(23):e7031. doi: 10.1097/MD.0000000000007031
36. Arraya M, Castro Y, Navarro J, Sarmiento E, Fernández-Cruz E, Carbone J. Rituximab for granulomatous lymphocytic interstitial lung disease in a patient with common variable immunodeficiency. is single therapy enough. Int J Clin Reumatol (2018) 13(1):38–42.
37. Deyà-Martínez A, Esteve-Solé A, Vélez-Tirado N, Celis V, Costa J, Cols M, et al. Sirolimus as an alternative treatment in patients with granulomatous-lymphocytic lung disease and humoral immunodeficiency with impaired regulatory T cells. Pediatr Allergy Immunol (2018) 29(4):425–32. doi: 10.1111/pai.12890
38. Shah JL, Amin SB, Verma N, Mohammed TL. Granulomatous-lymphocytic interstitial lung disease in a patient with common variable immunodeficiency. Curr Problems Diagn Radiology (2018) 47:282–4. doi: 10.1067/j.cpradiol.2017.04.007
39. Sood AK, Funkhouser W, ly B, Weston B, Wu EY. Granulomatous-lymphocytic interstitial lung disease in 22q11.2 deletion syndrome: a case report and literature review. Curr Allergy Asthma Rep (2018) 18:14. doi: 10.1007/s11882-018-0768-7
40. Ng J, Wright K, Alvarez M, Hunninghake GM, Wesemann DR. Rituximab monotherapy for common variable immune deficiency-associated granulomatous-lymphocytic interstitial lung disease. Chest (2019) 155:e117–e21. doi: 10.1016/j.chest.2019.01.034
41. Tillman R, Guillerman RP, Trojan T, Silva-Carmona M, Chinn IK. Treatment-responsive granulomatous-lymphocytic interstitial lung disease in a pediatric case of common variable immunodeficiency. Front Pediatr (2019) 7. doi: 10.3389/fped.2019.00105
42. Beaton TJ, Gillis D, Morwood K, Bint M. Granulomatous lymphocytic interstitial lung disease: limiting immunosuppressive therapy-a single-centre experience. Respirology Case Rep (2020) 8. doi: 10.1002/rcr2.565
43. Pac M, Bielecka T, Grzela K, Komarnicka J, Langfort R, Koltan S, et al. Interstitial lung disease in children with selected primary immunodeficiency disorders-a multicenter observational study. Front Immunol (2020) 11:1950. doi: 10.3389/fimmu.2020.01950
44. Ruiz-Alcaraz S, Gaya Garcia-Manso I, Marco-De La Calle FM, Garcia-Mullor MDM, Lopez-Brull H, Garcia-Sevila R. Granulomatous lymphocytic interstitial lung disease: description of a series of 9 cases. Med Clin (Barc) (2021) 156(7):344–8. doi: 10.1016/j.medcli.2020.11.033
45. Strunz PP, Frohlich M, Gernert M, Schwaneck EC, Nagler LK, Kroiss A, et al. Rituximab for the treatment of common variable immunodeficiency (CVID) with pulmonary and central nervous system involvement. Open Rheumatol J (2021) 15:9–15. doi: 10.2174/1874312902115010009
46. Maglione PJ, Overbey JR, Radigan L, Bagiella E, Cunningham-Rundles C. Pulmonary radiologic findings in common variable immunodeficiency: clinical and immunological correlations. Ann Allergy Asthma Immunol (2014) 113:452–9. doi: 10.1016/j.anai.2014.04.024
47. Maglione PJ, Overbey JR, Cunningham-Rundles C. Progression of common variable immunodeficiency interstitial lung disease accompanies distinct pulmonary and laboratory findings. J Allergy Clin Immunology: In Practice (2015) 3:941–50. doi: 10.1016/j.jaip.2015.07.004
48. Maglione PJ, Gyimesi G, Cols M, Radigan L, Ko HM, Weinberger T, et al. BAFF-driven b cell hyperplasia underlies lung disease in common variable immunodeficiency. JCI Insight (2019) 4(5):07. doi: 10.1172/jci.insight.122728
49. Lopez AL, Paolini MV, Fernandez Romero DS. Lung disease in patients with common variable immunodeficiency. Allergol Immunopathol (Madr) (2020) 48(6):720–8. doi: 10.1016/j.aller.2020.04.001
50. Fraz MSA, Moe N, Revheim ME, Stavrinou ML, Durheim MT, Nordoy I, et al. Granulomatous-lymphocytic interstitial lung disease in common variable immunodeficiency-features of CT and 18F-FDG positron emission Tomography/CT in clinically progressive disease. Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.617985
51. Meerburg JJ, Hartmann IJC, Goldacker S, Baumann U, Uhlmann A, Andrinopoulou ER, et al. Analysis of granulomatous lymphocytic interstitial lung disease using two scoring systems for computed tomography scans-a retrospective cohort study. Front Immunol (2020) 11:589148. doi: 10.3389/fimmu.2020.589148
52. von Spee-Mayer C, Echternach C, Agarwal P, Gutenberger S, Soetedjo V, Goldacker S, et al. Abatacept use is associated with steroid dose reduction and improvement in fatigue and CD4-dysregulation in CVID patients with interstitial lung disease. J Allergy Clin Immunology-in Pract (2021) 9(2):760–+. doi: 10.1016/j.jaip.2020.10.028
53. Maarschalk-Ellerbroek LJ, de Jong PA, van Montfrans JM, Lammers JWJ, Bloem AC, Hoepelman AIM, et al. CT screening for pulmonary pathology in common variable immunodeficiency disorders and the correlation with clinical and immunological parameters. J Clin Immunol (2014) 21:642–54. doi: 10.1007/s10875-014-0068-6
54. Hartono S, Motosue MS, Khan S, Rodriguez V, Iyer VN, Divekar R, et al. Predictors of granulomatous lymphocytic interstitial lung disease in common variable immunodeficiency. Ann Allergy Asthma Immunol (2017) 118:614–20. doi: 10.1016/j.anai.2017.01.004
55. Smits B, Goldacker S, Seneviratne S, Malphettes M, Longhurst H, Mohamed OE, et al. The efficacy and safety of systemic corticosteroids as first line treatment for granulomatous lymphocytic interstitial lung disease. J Allergy Clin Immunol (2022). doi: 10.1016/j.jaci.2022.12.813
56. Cabanero-Navalon MD, Garcia-Bustos V, Forero-Naranjo LF, Baettig-Arriagada EJ, Nunez-Beltran M, Canada-Martinez AJ, et al. Integrating clinics, laboratory, and imaging for the diagnosis of common variable immunodeficiency-related granulomatous-lymphocytic interstitial lung disease. Front Immunol (2022) 13. doi: 10.3389/fimmu.2022.813491
57. Cinetto F, Scarpa R, Carrabba M, Firinu D, Lougaris V, Buso H, et al. Granulomatous lymphocytic interstitial lung disease (GLILD) in common variable immunodeficiency (CVID): a multicenter retrospective study of patients from Italian PID referral centers. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.627423
58. Kollert F, Venhoff N, Goldacker S, Wehr C, Lutzen N, Voll RE, et al. Bronchoalveolar lavage cytology resembles sarcoidosis in a subgroup of granulomatous CVID. Eur Respir J (2014) 43(3):922–4. doi: 10.1183/09031936.00025513
59. Mannina A, Chung JH, Swigris JJ, Solomon JJ, Huie TJ, Yunt ZX, et al. Clinical predictors of a diagnosis of common variable immunodeficiency-related granulomatous-lymphocytic interstitial lung disease. Ann Am Thorac Soc (2016) 13(7):1042–9. doi: 10.1513/AnnalsATS.201511-728OC
60. Szczawinska-Poplonyk A, Jonczyk-Potoczna K, Mikos M, Ossowska L, Langfort R. Granulomatous lymphocytic interstitial lung disease in a spectrum of pediatric primary immunodeficiencies. Pediatr Dev Pathology (2021) 24:504–12. doi: 10.1177/10935266211022528
61. Bintalib HM, Lowe DM, Mancuso G, Gkrepi G, Seneviratne SL, Burns SO, et al. Corticosteroid-induced remission and mycophenolate maintenance therapy in granulomatous lymphocytic interstitial lung disease: long-term, longitudinal change in lung function in a single-centre cohort. ERJ Open Res (2022) 8(4). doi: 10.1183/23120541.00024-2022
62. Chase NM, Verbsky JW, Hintermeyer MK, Waukau JK, Tomita-Mitchell A, Casper JT, et al. Use of combination chemotherapy for treatment of granulomatous and lymphocytic interstitial lung disease (GLILD) in patients with common variable immunodeficiency (CVID). J Clin Immunol (2013) 33:30–9. doi: 10.1007/s10875-012-9755-3
63. Verbsky JW, Hintermeyer MK, Simpson PM, Feng M, Barbeau J, Rao N, et al. Rituximab and antimetabolite treatment of granulomatous and lymphocytic interstitial lung disease in common variable immunodeficiency. J Allergy Clin Immunol (2020) 147:704–12.e17. doi: 10.1016/j.jaci.2020.07.021
64. Hanitsch LG, Wittke K, Stittrich AB, Volk HD, Scheibenbogen C. Interstitial lung disease frequently precedes CVID diagnosis. J Clin Immunol (2019) 39(8):849–51. doi: 10.1007/s10875-019-00708-2
65. Friedmann D, Unger S, Keller B, Rakhmanov M, Goldacker S, Zissel G, et al. Bronchoalveolar lavage fluid reflects a TH1-CD21low b-cell interaction in CVID-related interstitial lung disease. Front Immunol (2021) 11. doi: 10.3389/fimmu.2020.616832
66. Serra G, Milito C, Mitrevski M, Granata G, Martini H, Pesce AM, et al. Lung MRI as a possible alternative to CT scan for patients with primary immune deficiencies and increased radiosensitivity. Chest (2011) 140(6):1581–9. doi: 10.1378/chest.10-3147
67. Milito C, Pulvirenti F, Serra G, Valente M, Pesce AM, Granata G, et al. Lung magnetic resonance imaging with diffusion weighted imaging provides regional structural as well as functional information without radiation exposure in primary antibody deficiencies. J Clin Immunol (2015) 35(5):491–500. doi: 10.1007/s10875-015-0172-2
68. Arslan S, Poyraz N, Ucar R, Yesildag M, Yesildag A, Caliskaner AZ. Magnetic resonance imaging may be a valuable radiation-free technique for lung pathologies in patients with primary immunodeficiency. J Clin Immunol (2016) 36(1):66–72. doi: 10.1007/s10875-015-0227-4
69. Nishimura M, Miyata J, Tanigaki T, Nomura S, Serizawa Y, Igarashi S, et al. Successful treatment of granulomatous-lymphocytic interstitial lung disease in a patient with CTLA-4 deficiency. Intern Med (2022) 62:871–5. doi: 10.2169/internalmedicine.0076-22
70. van Stigt AC, Dalm VASH, Nagtzaam NMA, van Rijswijk DA, Barendregt BH, van Hagen PM, et al. Soluble interleukin-2 receptor is a promising serum biomarker for granulomatous disease in common variable immune deficiency. J Clin Immunol (2021) 41(3):694–7. doi: 10.1007/s10875-020-00947-8
71. Dhalla F, Lochlainn DJM, Chapel H, Patel SY. Histology of interstitial lung disease in common variable immune deficiency. Front Immunol (2020) 11:605187. doi: 10.3389/fimmu.2020.605187
72. Larsen BT, Smith ML, Tazelaar HD, Yi ES, Ryu JH, Churg A. GLILD revisited: pulmonary pathology of common variable and selective IgA immunodeficiency. Am J Surg Pathol (2020) 44(8):1073–81. doi: 10.1097/PAS.0000000000001479
73. Patel S, Anzilotti C, Lucas M, Moore N, Chapel H. Interstitial lung disease in patients with common variable immunodeficiency disorders: several different pathologies? Clin Exp Immunol (2019) 198(2):212–23. doi: 10.1111/cei.13343
74. Aghamohammadi A, Allahverdi A, Abolhassani H, Moazzami K, Alizadeh H, Gharagozlou M, et al. Comparison of pulmonary diseases in common variable immunodeficiency and X-linked agammaglobulinaemia. Respirology (2008) 15(2):289–95. doi: 10.1111/j.1440-1843.2009.01679.x
75. Mahmoodi M, Abolhassani H, Mozdarani H, Rezaei N, Azizi G, Yazdani R, et al. In vitro Chromosomal radiosensitivity in patients with common variable immunodeficiency. Cent Eur J Immunol (2018) 43(2):155–61. doi: 10.5114/ceji.2018.77385
76. Davidsen JR, Skov IR, Louw IG, Laursen CB. Implementation of transbronchial lung cryobiopsy in a tertiary referral center for interstitial lung diseases: a cohort study on diagnostic yield, complications, and learning curves. BMC Pulm Med (2021) 21(1):67. doi: 10.1186/s12890-021-01438-1
77. Korevaar DA, Colella S, Fally M, Camuset J, Colby TV, Hagmeyer L, et al. European Respiratory society guidelines on transbronchial lung cryobiopsy in the diagnosis of interstitial lung diseases. Eur Respir J (2022) 60(5). doi: 10.1183/13993003.00425-2022
78. Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, et al. Diagnosis of idiopathic pulmonary fibrosis. an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med (2018) 198(5):e44–68. doi: 10.1164/rccm.201807-1255ST
Keywords: CVID, interstitial lung disease, GLILD, diagnosis, systematic review
Citation: Bintalib HM, van de Ven A, Jacob J, Davidsen JR, Fevang B, Hanitsch LG, Malphettes M, van Montfrans J, Maglione PJ, Milito C, Routes J, Warnatz K and Hurst JR (2023) Diagnostic testing for interstitial lung disease in common variable immunodeficiency: a systematic review. Front. Immunol. 14:1190235. doi: 10.3389/fimmu.2023.1190235
Received: 20 March 2023; Accepted: 17 April 2023;
Published: 08 May 2023.
Edited by:
Eyal Grunebaum, University of Toronto, CanadaReviewed by:
Neslihan Edeer Karaca, Ege University Faculty of Medicine, TürkiyeGuy Gorochov, Sorbonne Universités, France
Luis Ignacio Gonzalez-Granado, University Hospital October 12, Spain
Copyright © 2023 Bintalib, van de Ven, Jacob, Davidsen, Fevang, Hanitsch, Malphettes, van Montfrans, Maglione, Milito, Routes, Warnatz and Hurst. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Heba M. Bintalib, aGViYS5iaW50YWxpYi4yMEB1Y2wuYWMudWs=
†These authors have contributed equally to this work