 Rebeca Kennedy-Batalla1†
Rebeca Kennedy-Batalla1† Daniel Acevedo2,3,4†
Daniel Acevedo2,3,4† Yiyi Luo2,3,4
Yiyi Luo2,3,4 Ana Esteve-Solé2,3,4
Ana Esteve-Solé2,3,4 Alexandru Vlagea3,5
Alexandru Vlagea3,5 Rafael Correa-Rocha1‡
Rafael Correa-Rocha1‡ Ma Elena Seoane-Reula1,6,7*‡
Ma Elena Seoane-Reula1,6,7*‡ Laia Alsina2,3,4,8*‡
Laia Alsina2,3,4,8*‡- 1Laboratory of Immune-Regulation, Gregorio Marañón Health Research Institute (IISGM), Madrid, Spain
- 2Clinical Immunology and Primary Immunodeficiencies Unit, Allergy and Clinical Immunology Department, Hospital Sant Joan de Déu, Barcelona, Spain
- 3Clinical Immunology Unit, Hospital Sant Joan de Déu-Hospital Clínic, Barcelona, Spain
- 4Study Group for Immune Dysfunction Diseases in Children (GEMDIP), Institut de Recerca Sant Joan de Déu, Barcelona, Spain
- 5Immunology Department, Biomedic Diagnostic Center (CDB), Hospital Clínic of Barcelona, Clinical Immunology Unit Hospital Sant Joan de Déu-Hospital Clínic de Barcelona, Barcelona, Spain
- 6Pediatric Immuno-Allergy Unit, Allergy Department, Hospital General Universitario Gregorio Marañón, Madrid, Spain
- 7Primary Immunodeficiencies Unit, Hospital General Universitario Gregorio Marañón, Madrid, Spain
- 8Department of Surgery and Surgical Specializations, Facultat de Medicina i Ciències de la Salut, Universitat de Barcelona, Barcelona, Spain
Regulatory T cells (Treg) are essential for immune balance, preventing overreactive responses and autoimmunity. Although traditionally characterized as CD4+CD25+CD127lowFoxP3hi, recent research has revealed diverse Treg subsets such as Tr1, Tr1-like, and CD8 Treg. Treg dysfunction leads to severe autoimmune diseases and immune-mediated inflammatory disorders. Inborn errors of immunity (IEI) are a group of disorders that affect correct functioning of the immune system. IEI include Tregopathies caused by genetic mutations affecting Treg development or function. In addition, Treg dysfunction is also observed in other IEIs, whose underlying mechanisms are largely unknown, thus requiring further research. This review provides a comprehensive overview and discussion of Treg in IEI focused on: A) advances and controversies in the evaluation of Treg extended subphenotypes and function; B) current knowledge and gaps in Treg disturbances in Tregopathies and other IEI including Treg subpopulation changes, genotype-phenotype correlation, Treg changes with disease activity, and available therapies, and C) the potential of Treg cell-based therapies for IEI with immune dysregulation. The aim is to improve both the diagnostic and the therapeutic approaches to IEI when there is involvement of Treg. We performed a non-systematic targeted literature review with a knowledgeable selection of current, high-quality original and review articles on Treg and IEI available since 2003 (with 58% of the articles within the last 6 years) in the PubMed database.
1 Introduction
Regulatory T cells (Treg) are a subtype of T lymphocytes essential for immune homeostasis. Due to their regulatory function, Treg are able to supress and control immune responses to restore the immune system’s steady state, and they are necessary to tolerate autoantigens. Treg are essential in physiological processes such as pregnancy, host-commensal interactions, tolerance to allergens, and tissue repair, as they help decide between what should be considered threatening or harmless and, hence, allow the immune system to mount a response or suppress it, respectively. Because they are so essential for correct immune function, Treg are also involved in numerous diseases (1, 2). Defective Treg can disturb immune balance, leading to pathologies such as allergies, autoimmune and inflammatory disorders, infections, cancer, and primary immune regulatory disorders (PIRD) (2). This review aims at providing an integrated view of Treg in the context of inborn errors of immunity (IEIs) highlighting what is known and what is not known, the problems in Treg characterisation, and how they affect understanding of underlying pathological mechanisms of IEIs. In addition, it is intended to provide insight into potential Treg cell-based treatments that could be of use in IEI management. We performed a non-systematic targeted literature review with a knowledgeable selection of current, high-quality original and review articles on Treg and IEI available since 2003 (with 58% of the articles within the last 6 years) in the PubMed database.
1.1 History of Treg: from CD4+ Treg to CD8+ Treg
Discovered in 1969 as thymic-derived T cells with suppressive capacity in mice (3–5), Treg have gained great interest in immune regulation and their implications in different diseases. Shortly after their discovery, during the 1970s and 1980s, it was seen that these suppressive T cells were able to slow down autoimmune disease in rodent models (6, 7). During the decade of the 1990s, several discoveries were made. First and foremost, these cells were seen to express high levels of the IL-2 receptor alpha chain (IL-2Rα, CD25) (4, 6, 8) that is now one of the markers used for Treg characterisation. Although discovery of the suppressive cytokines IL-10 and transforming factor-beta (TGF-β) was made in the 1990s, it was not until later that they were associated with Treg function (7). Despite the growing knowledge, human Treg were not discovered until 2001 both in thymus and periphery (4, 6, 7). The identification of the autoimmune scurfy mice and immune dysregulation polyendocrinopathy enteropathy X-linked (IPEX) syndrome in humans caused by loss of function of FOXP3 pointed towards the role of this gene in Treg function (2, 6). A few years later, in 2003, FoxP3 expression was detected in Treg, in mice (6, 7) and later in humans (9, 10). FoxP3 expression was correlated with CD25 levels (7) and in 2006, thanks to the discovery of an inverse correlation between CD127 (IL-7 receptor) and FoxP3, Treg definition by surface markers was defined as follows: CD3+CD4+CD25hiCD127low which is still the most common phenotype used for their characterization by flow cytometry (4, 8, 11). The inducible T-cell costimulator (ICOS) surface molecule was known to be expressed in activated T cells. In 2008, ICOS expression was observed in Treg. According to its expression, Treg could be divided into two subsets depending on the suppressive mechanisms used: ICOS+, which use IL-10 to suppress dendritic cells (DCs) and TGF-β to suppress T cell function, and ICOS-, which only use TGF-β (12, 13). With all the developments in Treg characterization, function, and their disease-related role, in 2013 Abbas et al. proposed, in addition to the phenotypic classification, a unified nomenclature for Treg in order to facilitate their description, dividing them depending on where they completed their development (14). According to the new classification, Treg can be thymus-derived or natural Treg (tTreg or nTreg) if they complete their development in the thymus, or peripherally derived Treg (pTreg) if development occurs in the periphery. Moreover, they can also be induced in vitro and called induced Treg (iTreg) (5–7, 14). tTreg comprise around 80% of circulating Treg and have stable FoxP3 expression (15). pTreg, on the other hand, express ectopic FoxP3 which is not stable and only provides suppressive functions while it is expressed (16).
In mice, FoxP3 seems to be a Treg exclusive marker which makes it possible to characterise Treg as CD25+FoxP3+. On the other hand, human conventional T cells (Tconv) can also gain FoxP3 expression and, currently, there are no distinct markers available to distinguish between true tTreg and pTreg. Classical Treg are commonly described as CD3+CD4+CD25hiCD127lowFoxP3+ (4, 7, 8). However, the best way to identify these cells is by measuring the level of methylation in the Treg-specific demethylated region (TSDR) of the FOXP3 gene. FoxP3 is not the only gene that needs to be demethylated to ensure Treg function. CTLA-4 (cytotoxic T-lymphocyte antigen 4), Eos, GITR (glucocorticoid-induced TNFR-related), and CD25 are also responsible for correct Treg function – all of which can be used as markers to define Treg by flow cytometry (16).
As previously noted, there is still a lack of specific markers to differentiate tTreg and pTreg. Recent investigations have proposed Helios as a possible differentiating factor for tTreg. Helios expression presents a high correlation with tTreg whereas pTreg are mostly Helios–. This has been further confirmed by higher complete demethylation of TSDR of the FOXP3 gene in tTreg compared to not more than 50% in pTreg (17). However, Helios is not yet established as a definitive marker to differentiate tTreg and pTreg as many questions are still unanswered (18).
Although Treg research has always been more focused on CD4 T cell population, CD8 Treg populations were described in mice as early as 1970, when Gershon and Kondo described CD8 T cells from bone marrow responsible for tolerance (19, 20). In 1978, they saw CD8 suppressive capacity on CD4 cells in vitro, further confirmed in 1992 in an allergic encephalomyelitis animal model (21). In 2007, CD8 Treg were seen under different infections and tumour environments in rat models, and it was clear that IFNγ was important for the regulatory function of these cells (22). During this decade, it was also observed that most CD8 T cells with suppressive function were memory cells expressing high CD122 and Ly49 and that they were dependant on Helios expression, in contrast to CD4 Treg that are dependent on FoxP3 (21). These CD122hi Ly49+ CD8+ T cells have been considered the “CD8 Treg” in mice. Whether this subtype exists in humans has been, and probably still is, debatable. Very recently, killer cell immunoglobulin-like receptors (KIR)+ CD8+ T cells were described as the counterpart of Ly49+CD8+ T cells in humans. Because from an evolutionary point of view KIR are the human equivalent of Ly49 in mice, these cells were studied in autoimmune and infectious human diseases such as celiac disease and COVID-19 (23). Because of the novelty of this regulatory subtype, the definition of CD8 Treg is even less established than that for CD4 Treg (24). Other human studies define CD8 Treg depending on their CD28, FoxP3, CD122, or Helios expression (24, 25). The timeline of CD4 and CD8 Treg description in mice and humans is summarized in Figure 1 and the phenotypic markers in humans in Table 1.
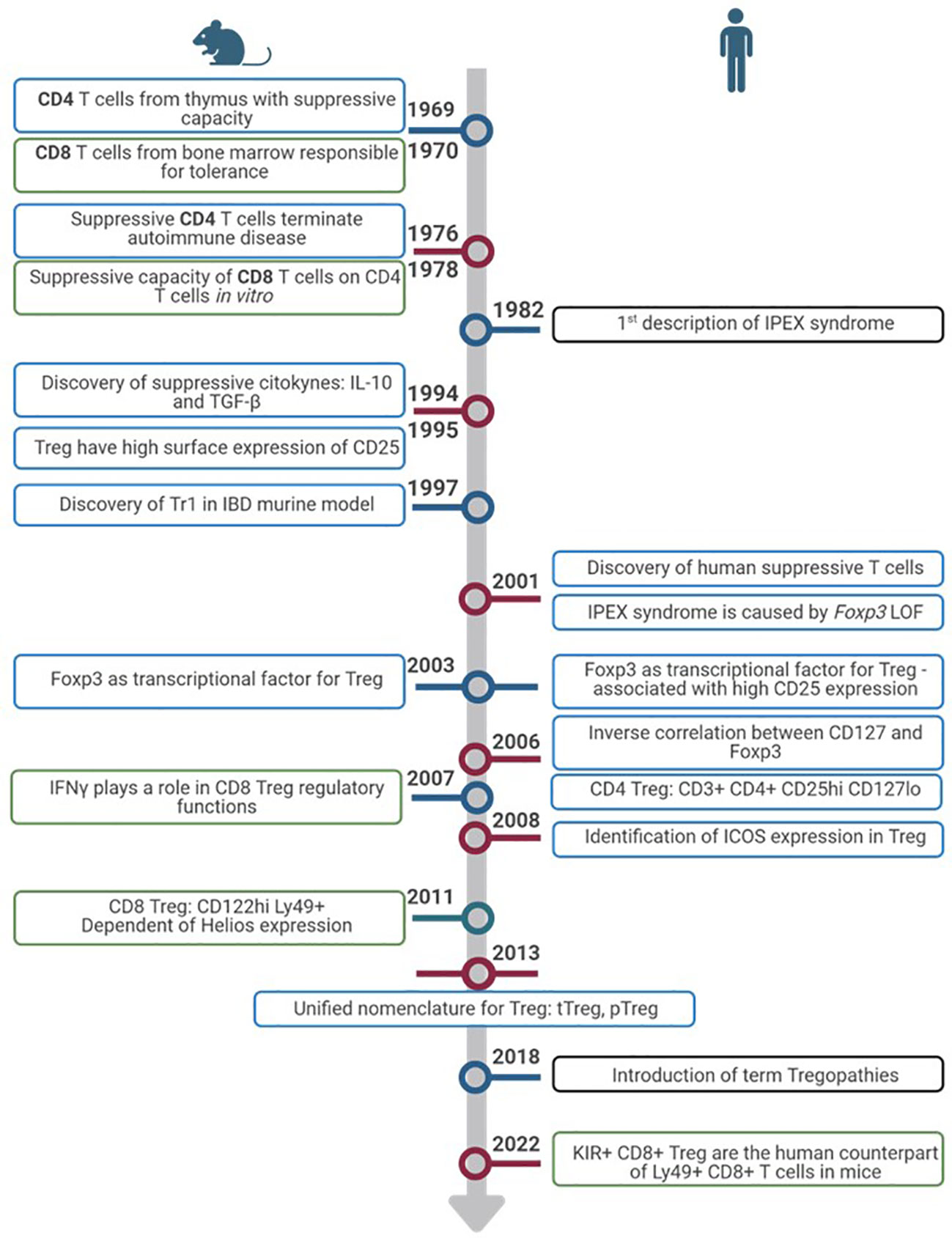
Figure 1 Timeline of Treg discovery. Timeline progression highlights pivotal advances in the exploration of Treg from inception to the present. Discoveries in murine models (left) and humans (right). Colors of the rectangles show advances in CD4+ Treg (blue), CD8+ Treg (green) and IEIs (black).
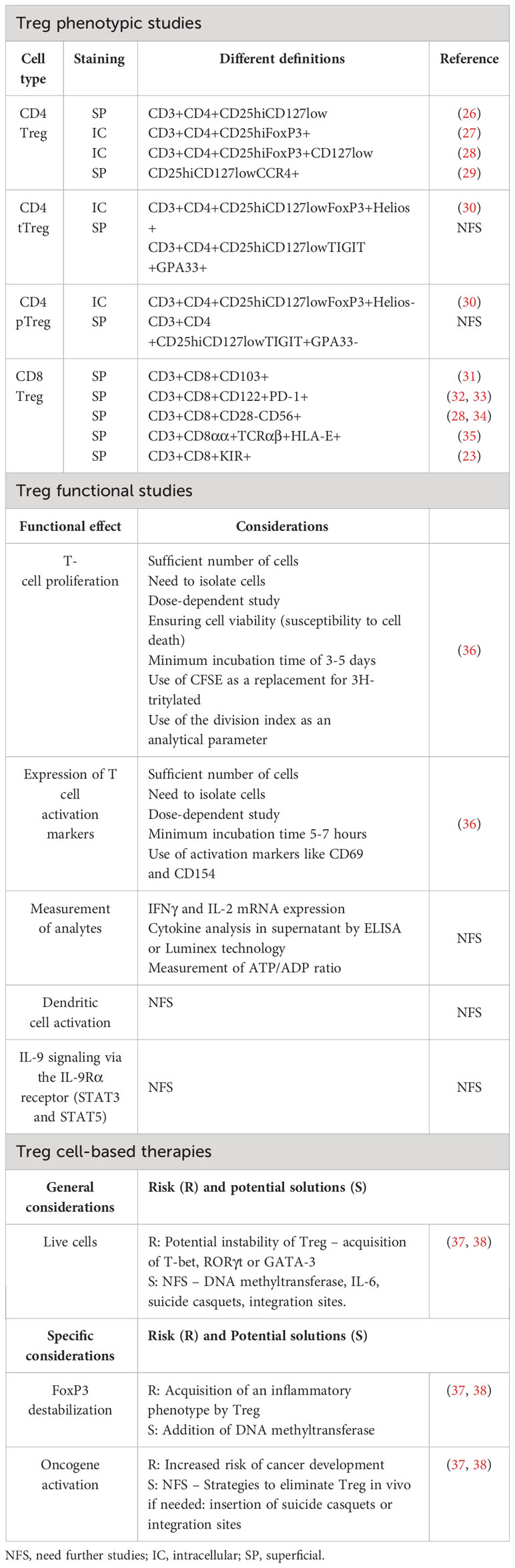
Table 1 Controversies and considerations in the study of Treg and in Treg cell-based therapies in humans.
1.2 Suppressive functions of CD4 and CD8 Treg
FOXP3 is essential for CD4 Treg suppressive function although it is not the only gene necessary. In this context, FoxP3 expression supresses IL-2 production as well as upregulating CD25 and CTLA-4 expression – all involved in Treg mechanisms of action (16). Treg can exert their function directly or indirectly. As direct mechanisms, Tregs secrete suppressive cytokines such as IL-10, TGF-β, and IL-35 that act directly on effector T cells (Teff). They also produce perforin and granzyme-inducing apoptosis in target cells (39). Besides these secreted proteins, Tregs express a number of surface molecules that enable them to perform their inhibitory functions: through CTLA-4 and lymphocyte activation gene 3 (LAG-3) they can inhibit dendritic cells (DCs) (5) and through programmed cell death 1 (PD-1) they directly inhibit B cells (39). Moreover, the high levels of CD25 allow Tregs to highjack IL-2 preventing it from stimulating T and natural killer (NK) cells (2). As indirect mechanisms, Tregs also present ectoenzymes on their membrane, specifically CD39 and CD73, that are responsible for ATP metabolism and control Teff activation. ATP acts as an inflammatory molecule when it is cleaved, AMP acts as an anti-inflammatory signal (5, 39).
Amongst other genes necessary for Treg functionality, there is a group of molecules that enhance Treg inhibitory functions without being selective of Treg. These include ICOS, CTLA-4, PD-1, TIGIT, Helios, and LAG-3.
ICOS increases Treg effectivity and, under certain circumstances, can supress Treg apoptosis, playing an important role in their survival (7). In order to do so, ICOS needs to bind to its ligand, ICOS-L, expressed on antigen-presenting cells (APCs): DCs, B cells, and macrophages. Upon ICOS-L binding, it enhances FoxP3 transcription which, in turn, increases IL-4, TGF-β, and IL-10 secretion. ICOS+ Treg normally co-express co-inhibitory surface molecules important for their immune regulation such as CTLA-4, LAG3, PD-1, and T cell immunoglobulin and ITIM domain (TIGIT). Whether it is a matter of ICOS directly affecting this expression or indirectly contributing to the increased Treg functionality remains unclear (13).
CTLA-4 is a co-inhibitory surface molecule that is constitutively expressed in Treg. It is not a Treg-exclusive marker as it is also present in activated lymphocytes. Binding of CTLA-4 from activated lymphocytes to CD80 and CD86 present on the surface of DCs inhibits them, restoring immune homeostasis once they have completed their task. In this context, CTLA-4 acts as an immune checkpoint in order to prevent an overreactive immune system. However, the role of CTLA-4 in Treg is not completely straight forward. It is suggested that CTLA-4 from Treg binds to CD80/CD86 in DCs and downregulates their expression. Because this signal from DCs is necessary to complete T cell activation during antigen presentation, this downregulation would reduce T cell activation which, in turn, would favour regaining immune homeostasis (40).
PD-1 and its ligand PD-L1 are both highly expressed in Treg surface (7). Binding of PD-L1, on Treg surface, to PD-1, expressed on activated cells, triggers an inhibitory response that negatively affects T cell proliferation and cytotoxicity. This response induces a state of anergy on the activated T cells and can promote the induction of pTreg, by enhancing FoxP3 expression and Treg suppressive activity. Moreover, interaction of PD-1 and DCs’ PD-L1 also prompts pTreg generation (41). In turn, once pTreg are generated, their PD-1 binding to PD-L1 in DCs also generates tolerogenic DCs (7). Hence, PD-1 signalling is needed to directly and indirectly maintain immune homeostasis. PD-1/PD-L1 signalling mechanism is not exclusive to Treg, but, along with CTLA-4 and LAG-3, constitutes one of their cell-to-cell contact suppressive mechanisms (41). Constitutive PD-1 expression is related to T cell exhaustion and is usually used as a CD4+ and CD8+ T cell exhaustion marker (42).
TIGIT is another co-inhibitory receptor that is expressed in Treg but also in activated and memory T cells, NK cells, and some follicular helper T cells (Tfh). It has two ligands, CD155 (or PVR) and CD112 (or nicotine-2), although it binds with greater affinity to CD155. These molecules are expressed in APCs and T cells, among other non-hematopoietic cells (43). In the context of Treg, the effect of TIGIT binding to DCs prompts tolerogenic DCs which express IL-10 and, in turn, suppress T cell activation. Moreover, increased TIGIT expression induces FOXP3 demethylation in Treg. It can also supress T cell priming and block the CD226 activation pathway. Without the presence of TIGIT expression, CD226 acts as an activating receptor when binding to CD155. All of these mechanisms lead to reduced T cell activation (44).
LAG-3 is expressed in Treg, activated T cells (both CD4+ and CD8+), and a subset of NKs. This molecule binds to MHC-II due to its similarity with CD4 co-receptor among other receptors in CD8 T cells and NKs (43). Regarding Treg, LAG-3 is essential for immune homeostasis control by Treg, and is necessary for Treg cell-to-cell mediated suppressive mechanism, together with PD-1 and CTLA-4 (41). Similarly to these molecules, LAG-3 supresses cell proliferation, cytokine secretion, and immune function, playing a role in immune homeostasis (42). Expression of LAG-3 confers CD4+ T cell suppressive activity, whilst its blockade on Treg prevents their suppressive functions (43). LAG-3 constitutive expression is associated with CD4+ and CD8+ T cell exhaustion (42). It is also worth noting that LAG-3 is also expressed on Tr1 (type 1 regulatory T cells) although it is not clear whether it is necessary for their suppressive function (43).
Helios is a member of the IKAROS transcriptional factor family gene, which has been shown to play an important role in Treg function. It is implicated in Treg maintenance as they ensure FoxP3 stable expression under inflammatory conditions, preventing Treg from becoming effector cells (45). Moreover, in Helios-deficient mice, Tregs fail to produce an appropriate cytokine response and may exhibit the expression of cytokines typically associated with other effector CD4 T cells. Development, function, and immune homeostasis in most CD4+ T cells are not affected. However, in the case of Treg, Helios deficiency, although not affecting development, translates into a late onset autoimmune disease, underlying the importance of this transcription factor in the functionality of Treg (46). The schematic representation of Treg mechanisms of action in humans is summarized in Figure 2.
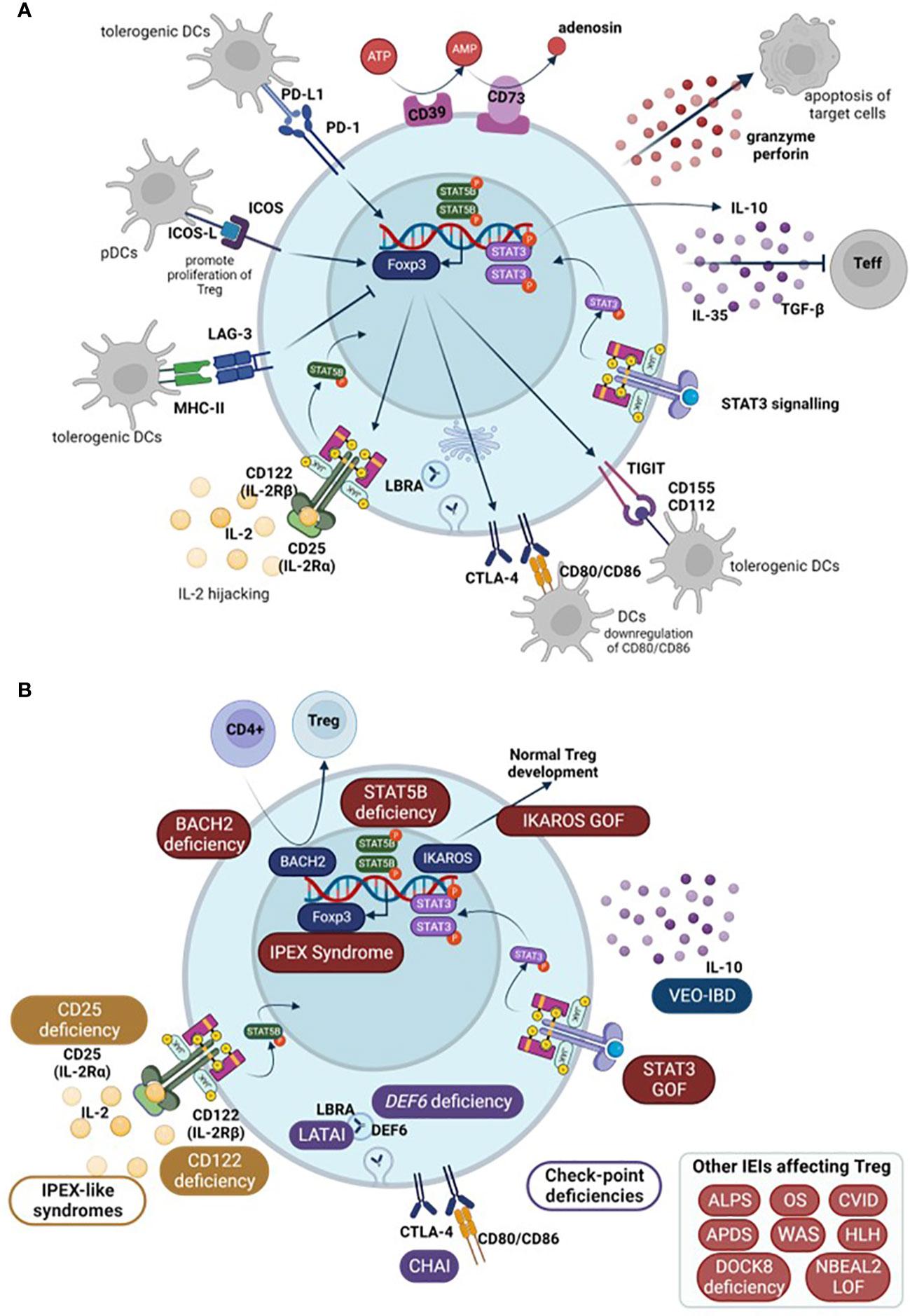
Figure 2 (A) Schematic representation of Treg mechanisms of action. (B) Main Tregopathies and the gene mutation responsible for the disruption of Treg function among other IEIs that affect Treg (in box, down-right). Arrows show normal physiological processes. Teff, effector T cells; DCs, dendritic cells; pDCs, plasmacytoid DCs; IEI, inborn errors of immunity; IPEX, immune dysregulation polyendocrinopathy enteropathy X-linked syndrome; LATAI, LRBA deficiency with autoantibodies, regulatory T (Treg) cell defects, autoimmune infiltration; CHAI, CTLA-4 haploinsufficiency with autoimmune infiltration; ALPS, Autoimmune lymphoproliferative syndrome; OS, Omenn Syndrome; HLH, hemophagocytic lymphohistiocytosis; CVID, Common variable immunodeficiency; VEO-IBD, very early onset inflammatory bowel disease; APDS, Activated PI3K Delta Syndrome; WAS, Wiskott-Aldrich Syndrome; DOCK8, Dedicator of cytokinesis 8 deficiency; NBEAL2, Neurobeachin-like 2.
2 IEI and Treg: the new kid on the block
2.1 From primary immunodeficiencies to inborn errors of immunity to primary immune regulatory disorders
Bruton’s agammaglobulinemia was first discovered seven decades ago. Since then, and thanks to technological advances in recent years such as next-generation sequencing (NGS), whose massive sequencing technology of the human exome and genome has been made affordable worldwide, we have seen a rapid increase in the rate of discovery of new genetic defects in primary immunodeficiency disorders (PIDs). This has allowed experts from the WHO’s International Union of Immunological Societies (IUIS) to collect more than 485 genes causing PIDs by 2022 (47).
PIDs are a group of disorders that affect the proper functioning of the immune system, most of them of genetic origin. Since the immune system protects us against various pathogens, patients with PIDs suffer from a wide range of severe and/or recurrent infections. Although one of the mainstays of immunodeficiency is immunocompromise, which is characteristic of most patients with PIDs, their symptomatology is, nevertheless, increasingly diverse, ranging from autoimmunity, autoinflammation, and severe allergies to neoplastic syndromes (48). The presence of autoimmunity in the context of immunodeficiency suggests a paradox, with two opposite pathologies, where one offers an excess and the other a defect of response, respectively, coexisting in the same clinical entity (49). Due to this increasing phenotype and with the increasing number of genetic defects being discovered, some of them causing gain-of-functions and not only loss-of-functions, the IUIS proposed a paradigm shift in 2019. Since then and to this day, PIDs are known as Inborn Errors of Immunity (IEI), and the classification of these diseases is updated every other year (47, 50).
The latest update of the IUIS in 2022 classifies IEIs into the following 10 groups (47, 51): 1. Combined immunodeficiencies. 2. Combined immunodeficiencies with syndromic characteristics. 3. Predominant antibody deficiencies. 4. Diseases of immune dysregulation. 5. Congenital phagocyte defects. 6. Defects in the intrinsic and innate immunity. 7. Autoinflammatory diseases. 8. Complement deficiencies. 9. Bone marrow failure. 10. Phenocopies of inborn errors of immunity.
Inborn errors of immune dysregulation (group 4) are a heterogeneous group of disorders with clinical variability that affect genes involved in the regulation of the immune system. Those presenting with a phenotype caused by loss of tolerance mechanisms leading to autoimmunity, autoinflammation, lymphoproliferation, and/or severe atopy have come to be recognized as having Primary Immune Regulatory Disorders (PIRD) since 2020 (52, 53). PIRD encompass a diverse set of disorders due to failures in different immune regulatory pathways; hence, this enables a subgrouping into different categories.
2.2 From primary immune regulatory disorders to tregopathies: central role of Treg in the pathophysiology
In 2018, the term tregopathies (54) was first introduced: it referred to a group of IEI with a clinical phenotype later encompassed within PIRD, in which the affected regulatory target is the Treg cell itself. This group initially included mutations in FOXP3, CD25, CTLA-4, LRBA, BACH2, IL10, and gain of function (GOF) of STAT3 (47, 55, 56). Since then, the IUIS expert committee (EC) has added new genes to this category, including mutations in FERMT1, CD122, DEF6, and IKAROS GOF (47, 51) (Figure 2).
Immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (known by the acronym IPEX) was first recognized in 1982 by Powell et al. and was the first disease associated with a defect in Treg. It presents with a wide variety of autoimmune manifestations in early life. IPEX is generated as a consequence of mutations in the transcription factor FoxP3, resulting in a quantitative reduction of protein expression or a loss of protein function (57, 58). However, several studies have shown that the FOXP3 gene is not essential for the establishment of the Treg cell lineage, but rather for its suppressive effector functions, as it controls the expression of different regulatory molecules such as TIGIT, CTLA-4, and the suppression of the IL-2 gene and different effector T-cell cytokines (59–61). Reported mutations causing IPEX are null or nonsense variants and small deletions or insertions in the reading frame (62–64) in the FOXP3 gene that result in a residual expression of the protein, but lacking function. Therefore, Treg cells from these patients are dysfunctional, being unable to inhibit the proliferation and inflammatory functions of effector T cells (Teff) (62, 65, 66). Of the 70 mutations identified, 40% are located in the DNA-binding region (FKH), 23% in the proline-rich terminal region (PRR), 9% in the leucine helix (LZ), 14% in the LZ-FKH loop, and 6% in the non-coding region. In addition, human FOXP3 gene has two main isoforms, a full-length isoform (FOXP3 FL) and an isoform lacking part of exon 2 (FOXP3 ΔE2) (67). A recent study shows that the lack of exon 2 of FOXP3 in mice and patients with IPEX syndrome leads to severe immune disorders (67). Even though Treg with FOXP3 ΔE2 can suppress T cells in culture, in vivo they lose their stability, suggesting that the presence or absence of exon 2 in FOXP3 is critical for normal Treg function (67). Although these FOXP3 mutations are linked to IPEX, there is an increasing number of patients who do not manifest with the classical triad of early-onset intractable diarrhea, type 1 diabetes (T1D), and eczema. Instead, these atypical cases of IPEX manifest with late-onset of symptoms, single-organ involvement, and mild disease phenotypes (68). Thus, a better understanding of the connection between the genotype and phenotype of this disease is needed.
A variety of tests have been developed to assess key molecules, signaling pathways, and cells implicated in IPEX or IPEX-like diseases for diagnostic purposes. Some are clinically available, while others are only available as research tests. On the one hand, Treg levels and FoxP3 expression can be assessed by flow cytometry. For this methodology, it is common to stain cells with CD4/CD25/CD127 to assess Treg cell frequency or numbers (69, 70). In the European Society for Immunodeficiencies (ESID) registry, working definitions for the clinical diagnosis of IEI (71) are used only for patients with no genetic diagnosis. FoxP3 expression by CD4+CD25+ on flow analysis appears as a diagnostic criterion in IPEX and IPEX-like suspected disease. Nevertheless, patients with nonsense mutations have normal FoxP3 frequency but typically decreased median fluorescence intensity (MFI) because of reduced expression levels (55). Thus, the clinical utility of this technique is moderate (55). The suppressive capacity of Treg can also be measured in vitro through a T-cell coculture suppression assay, but this requires cell separation with a significant blood volume. In addition, there are no reference values for this assay yet, so the clinical utility is poor. A clinically useful test is the assessment of IL-2 signaling in vitro through T-cell proliferation in the presence of mitogens or IL-2. Mutations in either IL-2 receptor chains will lead to decreased proliferation.
It is also considered useful to measure IL-10 signaling through studies of STAT3 phosphorylation following IL-10 stimulation. Mutations in the IL-10 receptor lead to a loss of STAT3 phosphorylation (55). IL-10 enhances iTreg differentiation and function probably through the STAT3 and Foxo1 signaling pathway (72). Inhibition of Foxo1 blocks the ability of IL-10 to enhance iTreg differentiation and function, suggesting that the effect of IL-10 is dependent on Foxo1 activation (72). This finding relates to the well-known role of Foxo1 as a contributing factor to Treg function and development (73, 74). In addition, IL-10-induced inhibition of STAT3 phosphorylation also reduces the ability of IL-10 itself to enhance iTreg differentiation, suggesting that STAT3-defective CD4+ T cells respond poorly to IL-10, contributing to immune dysregulation (72).
CD25 deficiency is an immune dysregulation disorder segregating in autosomal recessive form caused by biallelic variants in the IL2RG gene encoding IL-2Rα also known as CD25 protein. CD25 is expressed at high levels by CD4+ CD25+FoxP3+ Treg cells and has a pivotal role in Treg since IL-2 (and CD25) is required for IL-10 production, as mentioned above (75). Patients with CD25 deficiency present with a clinical phenotype overlapping that of IPEX patients, thus being termed as having IPEX-like disease (75). CD25 expression on the T-cell surface is completely abolished in all patients described, enabling diagnosis by immunological phenotyping with flow cytometry. Decreased T-cell proliferative responses to non-specific or antigen-specific stimuli were observed in some patients, although this could be restored with high doses of IL-2 or IL-15 in some of them. In addition, patients tend to have normal or reduced counts of FoxP3+ regulatory T cells, which makes their isolation for functional studies difficult (75). However, recent studies have demonstrated an altered suppressive capacity in CD25-deficient regulatory T cells purified as CD4+TIGIT+CD127- T cells (76).
In humans, CTLA-4 haploinsufficiency causes a dominant immune dysfunction known as CTLA-4 haploinsufficient autoimmune infiltrative disease (CHAI). Patients with CHAI experience autoimmune cytopenia, hypogammaglobulinaemia, infectious and non-infectious lung disease, enteropathy, lymphoproliferation, skin conditions and neurological involvement, and increased infectious susceptibility. In addition, patients with CTLA-4 haploinsufficiency have decreased numbers of circulating B cells and memory B cells, as well as NK and T cells. In some cases, an increase in CD4+ helper T cells is observed. It is important to note that Treg analysis is made challenging by the low CD25 expression observed in many CVID patients (77). Thus, although the expression of Foxp3 and CD25 may be decreased in CTLA-4 and LRBA deficiencies, this may not necessarily indicate a low Treg count (78). CTLA-4 deficient Treg cells also show decreased suppressive capacity, as in patients with IPEX syndrome (79–81).
LRBA deficiency, either homozygous or compound heterozygous, causes CTLA-4 deficient expression on cell surface. It is also known as LRBA disease with autoantibodies, Treg cell defects, autoimmune infiltration, and enteropathy (LATAI disease), and it shares phenotypic and clinical features with CTLA-4 haploinsufficiency. The CTLA-4 protein is constantly recycled between the cell surface and the cell interior. LRBA facilitates the recycling of CTLA-4 from endosomes to the surface of T cells to prevent its degradation in lysosomes. As a result, LRBA-deficient Treg cells show reduced levels of CTLA-4 expression. Similar to CTLA-4 mutated patients, LRBA-deficient T cells are hyperproliferative upon in vitro activation and both Treg cell-mediated suppressor capacity and CD80 transendocytosis are impaired. Also, Treg cell counts are reduced (82, 83).
A very recent study has revealed the interaction of the Neurobeachin-like 2 (NBEAL2) protein with immune cells (84). Loss of function of NBEAL2 leads to grey platelet syndrome (GPS) and some patients develop autoimmune disorders (84). By mass spectrometry it was shown that one of the proteins associated with NBEAL2 is LRBA. Immunoprecipitation further confirmed that NBEAL2 and CTLA-4 interact with each other (84). Interestingly, NBEAL2 deficiency leads to low CTLA-4 expression in Tconv cells, while Treg cells appear to be unaffected (84). Therefore, further studies of this deficiency are needed to elucidate both phenotypic and functional involvement of Treg cells.
BACH2 is a transcription factor that plays a crucial role in B-cell class recombination, somatic hypermutation, T-cell differentiation and function, and alveolar macrophage function. Genetic variations at the BACH2 locus are commonly associated with an increased risk of various autoimmune and inflammatory diseases, such as rheumatoid arthritis, T1D, asthma, multiple sclerosis, vitiligo, Graves’ disease, Crohn’s disease, and celiac disease. In 2017, BACH2 haploinsufficiency was initially described in a patient with lymphocytic colitis, splenomegaly, and progressive humoral deficiency with sinopulmonary infections (85). With further cases reported, the disease is now named BACH2-related immunodeficiency and autoimmunity (BRIDA). Patients have been shown to exhibit decreased levels of Treg in the blood and colon (85), where FoxP3 expression levels decrease, compromising Treg (85). Further functional studies are needed to elucidate the role of Treg in this disease.
The STAT3 signaling cascade plays an important role in the regulation of Treg and Th17 cells. STAT3 gain-of-function (GOF) syndrome is a multi-organ primary immune regulatory disorder characterized by early onset autoimmunity. Early in life, patients present most commonly with lymphoproliferation, autoimmune cytopenias, and growth delay (86). STAT3 induction by IL-6, together with TGF-β, is essential for Th17 cell differentiation. However, IL-6 also inhibits TGF-β-induced upregulation of FoxP3 in naïve CD4+ T cells, favoring polarization towards a potentially pathogenic Th17 phenotype (87). In addition, STAT3 activation increases the expression and secretion of SOCS3 (STAT5 inhibitory protein), a positive regulator of FoxP3 and CD25. STAT3 GOF disease may result from altered upregulation of both FoxP3 and imbalance in Treg/Th17 cell polarization, although it is unclear which factor has a dominant effect. Thus, Treg cell counts are usually reduced in STAT3 GOF (88). Because of the broad role of STAT3 in regulating various signaling, differentiation, growth, and regeneration cascades in different tissues and cell types, it is possible that other cell subsets and pathways also contribute to immune dysfunction in these patients (88).
The disturbances in Treg in Tregopathies are summarized in Table 2. Currently there are overlapping terms to refer to some PIRD disorders: some are based on physiopathology such as Tregopathies (53) or check-point deficiencies (81, 97), while others refer to the clinical phenotype of affected patients: autoimmune lymphoproliferative syndrome (ALPS)-like (92) and IPEX-like (66).
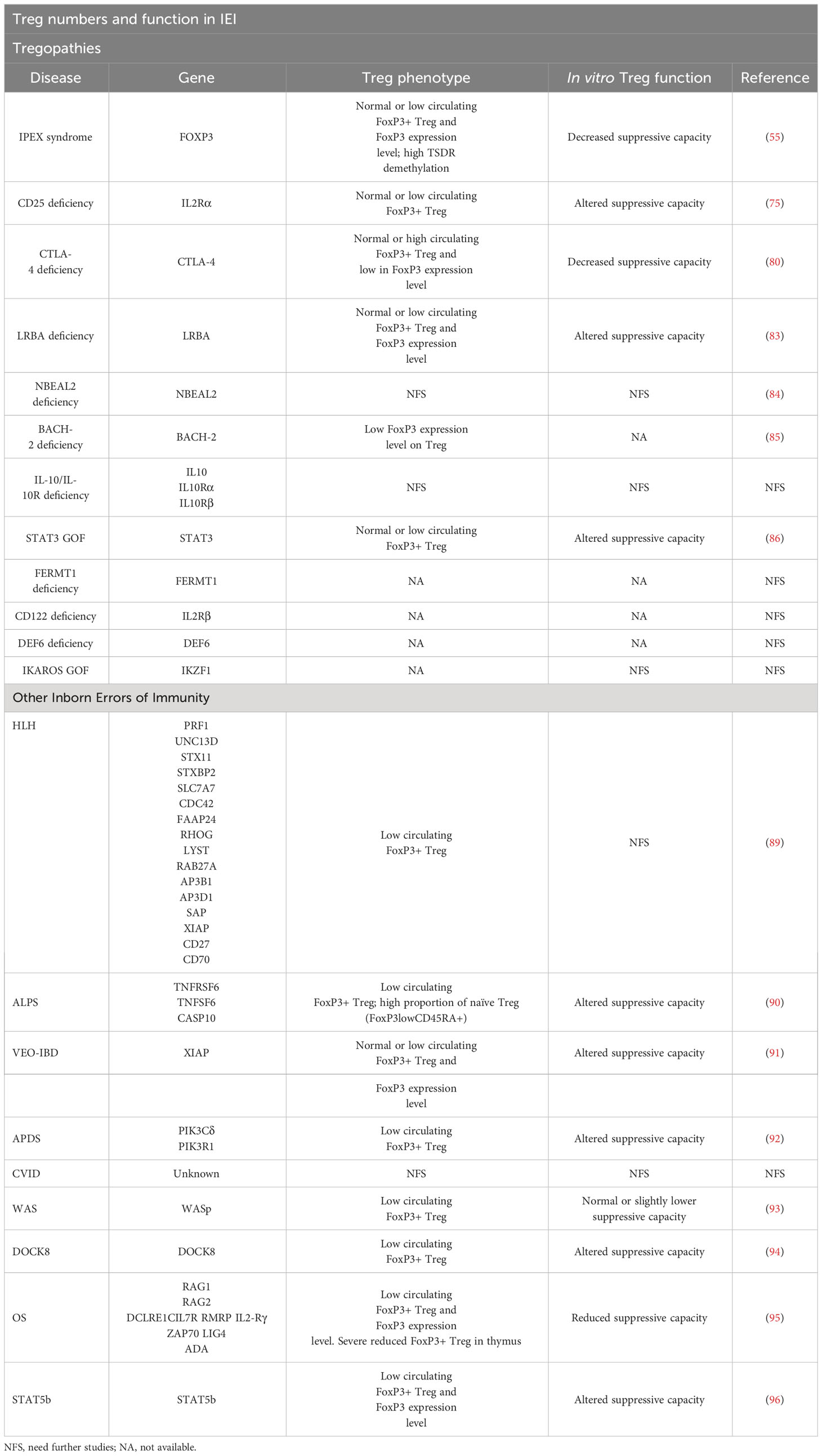
Table 2 Summary of published information on Treg numbers and function in IEI.
2.3 Treg dysfunction in other IEI
The remaining categories of PIRD which are not recognized as Tregopathies include hyperinflammatory disorders, such as hemophagocytic lymphohistocytosis (HLH) with or without intrinsic susceptibility to Epstein-Barr virus, non-malignant lymphoproliferation with autoimmunity (ALPS), inflammatory bowel disease linked to IL-10 signaling (very early onset inflammatory bowel disease, VEO-IBD), and other disorders linked to monogenic autoimmunity but lacking a central role in Treg (51, 53). Although the genetics of these other PIRD do not impact directly on the biology and function of Treg, these cells can be markedly altered in a direct or indirect manner.
HLH is a hyperinflammatory disorder with excessive immune activation resulting from different genetic defects in PRF1, UNC13D, STX11, STXBP2, SLC7A7, CDC42, FAAP24, RHOG, LYST, RAB27A, AP3B1, AP3D1, SAP, XIAP, CD27, or CD70 (51, 98) that impair granule-mediated cytotoxicity. This impedes the cytotoxic effector cells from achieving their goal: to kill infected and transformed cells, with the consequent release of cytokines that promote a state of hyperactivation of the immune response and hyperinflammation. It has been shown that in acute HLH CD8+ T lymphocytes overexpresses CD25, so that by consuming IL-2 from the medium they compete against Treg for this very resource. This results in an indirect Treg cell malfunction and a collapse in Treg cell numbers (89). In the specific case of X-linked inhibitor of apoptosis protein (XIAP), which causes X-linked Lymphoproliferative Disorder (XLP-2) and VEO-IBD, Treg cells are also affected. Suppressor of cytokine signaling 1 (SOCS1) is one of the transcription factors involved in the maintenance of Treg inhibitory function. XIAP binds to SOCS1 promoting its stabilization (99). Mutations in XIAP lead to SOCS1 deficiency and trigger an overactivation of the transcription factors STAT1 and STAT3 with a consequent reduction of the suppressive capacity of Treg, reducing their cell numbers, reducing FoxP3 stability, and even predisposing to IFNγ secretion, which diverts the response from a regulatory function to an inflammatory one (91).
In the case of ALPS, the underlying genetic defect involves the components of the apoptotic Fas-FasL pathway. There are a number of biomarkers linked to ALPS such as elevated levels of CD3+TCRαβ+CD4-CD8- double negative cells, as well as elevated plasma levels of IL-10, soluble FasL (sFasL), and vitamin B12 (92, 100). It has been shown that in ALPS patients the proportion of Treg defined as both CD25hiCD127low and CD25hiFoxP3+Helios+ was lower compared to healthy controls (90). In addition, patients also had a high proportion of naïve Treg (FoxP3lowCD45RA+) and a rare population defined as CD4+CD127lowCD15s+CD45RA+. However, although the proportions of Treg were low, the suppressive capacity on T-cell proliferation was not affected. This study suggests that excessive T cell proliferation in ALPS may not be ascribed to a Treg defect or T cell sensitivity to suppression (90). However, given the paucity of information on Treg in ALPS, further studies are needed.
Furthermore, in recent years and thanks to advances in NGS, novel gene defects outside the Fas-FasL apoptosis pathway with a phenotype and clinical mimicry to ALPS have been identified (ALPS-like). These include the above-mentioned check-point deficiencies (CTLA-4, LRBA and DEF6 deficiencies) but also the Activated PI3K Delta Syndrome (APDS). APDS results from the increased activity of the phosphoinositide-3-kinase δ (PI3Kδ) pathway (101). This enzyme is predominantly expressed by leukocytes and plays a very important role in immune cell function. Animal models indicate that impaired PI3Kδ in mice leads to colitis mostly due to compromised Treg function, suggesting PI3Kδ plays a role in Treg function (102). However, it should be pointed out that the mouse model of PI3Kδ does not recapitulate ALPS in humans. In most of these ALPS-like disorders, total Treg CD4+CD25+FoxP3+ levels are lower compared to healthy controls, as well as lower CTLA-4 expression levels and an expansion of circulating follicular CD4+CXCR5+CD45RA- (cTFH) T cells and polarization towards a Th1 phenotype (CCR6-CXCR3+) (92). Also, in most ALPS-like phenotypes there is a reduction in the T cell compartment, while senescent CD4+ T cells are increased.
Clinical phenotypes of lymphoproliferation and autoimmunity are found in other entities such as common variable immunodeficiency (CVID). In CVID, the involvement of Treg cells is still under study; recent studies have reported decreases in CD4 Treg in patients with CVID (103), while others have found conflicting results (104). For example, some researchers have observed that CD4 Treg (identified by markers such as CD25hiCD127lowFoxP3+) are decreased in CVID patients, especially those with associated autoimmune diseases (105). However, other studies have found no significant changes in CD4 Treg in relation to disease severity or the presence of autoimmunity (104). Some researchers have also explored the function of these cells and found that, despite differences in levels, the suppressive function of CD4 Treg appears to be maintained overall (106). However, in general, there is still a lack of consensus on how CD4 Treg relate to the pathophysiology of CVID and its association with autoimmunity (103, 107, 108). In addition, a recent study analyzed CD8 Treg (CD8+CD25hiCD183+Foxp3+) in patients with CVID, finding significant reductions in CD8 Treg ratios in these patients (109). However, no significant differences in CD8 Treg ratios were observed between patients with CVID with or without autoimmunity (109). Because CVID is an umbrella term for a probable heterogeneous group of diseases with different pathophysiological mechanisms (110), the study of Treg in this disease is challenging. Further research with larger series and more specific approaches are needed to fully understand the role of CD4 and CD8 Tregs in the pathogenesis and clinical manifestations of CVID and its relationship with autoimmunity. The disturbances in Treg in IEI other than Tregopathies are summarized in Table 2.
Wiskott-Aldrich syndrome (WAS) is an X-linked IEI characterized by lymphocyte dysfunction leading to opportunistic viral and bacterial infections, thrombocytopenia, eczema, autoimmune disorders, and cancer (93, 111, 112). More than 200 mutations have been described in the gene encoding the WAS protein (WASp) (93, 111). Mutations resulting in loss of WASp expression correlate with a more severe disease phenotype (93). Its deficiency is mainly associated with defects in T-lymphocytes. In particular, lymphocytes are unable to reorganize the actin cytoskeleton in response to T-cell receptor (TCR) engagement, leading to incomplete cell activation, and decreased cell proliferation and survival. Although WASp has not been shown to play a crucial role in the production of Tregs in the thymus, it is required for Treg expansion and survival in the periphery (93, 111). WASp deficiency leads to a decrease in the percentage of peripheral Treg (although FoxP3 expression is maintained), with a strong impact on activated Treg as evidenced by a decrease in activation markers and migration receptors (93, 112). Despite this, WASp-deficient Treg show normal or slightly lower suppressor capacity against T cells, suggesting that the impact of WASp mainly affects the survival and expansion of peripheral Treg, but not their suppressor function (93). Notably, in a study of WASp-deficient mice, Tregs were defective in their suppressor function in vitro, but in WAS patients this defect in function was less clear (113). In terms of clinical implications, although alterations in Treg may correlate with the high frequency of autoimmunity in WAS patients, more research is needed to improve understanding of this correlation.
Dedicator of cytokinesis 8 (DOCK8) deficiency is a rare IEI characterized by a constellation of symptoms including severe immunodeficiency, elevated IgE levels, allergies, and autoimmune disorders (94, 114). While the precise pathophysiology of immune dysregulation remains only partially understood, it is suggested that DOCK8 regulates the suppressive function of Tregs by promoting STAT5 phosphorylation in response to IL-2 signaling, which is essential for Treg maintenance (94). Furthermore, DOCK8 is involved in CD25 recycling (94). Patients with DOCK8 deficiency have been shown to exhibit reduced levels of Treg with impaired suppressive function (94). Remarkably, in a study using contact hypersensitivity (CHS) models, Treg lacking DOCK8 acquired a pathogenic phenotype expressing both FoxP3 and T-bet, along with IFNγ, suggesting that Tregs polarized towards a potentially inflammatory phenotype rather than maintaining their suppressive function (94, 114).
Omenn syndrome (OS) is an severe recessive autosomal combined immunodeficiency, ascribed to hypomorphic mutations in the activating recombination genes (RAG1 and RAG2), but also in other genes (DCLRE1C, IL7R, RMRP, IL2-Rγ, ZAP70, LIG4, ADA) (115), and impairing, but not abolishing, the recombination process of the V(D)J genes. OS manifests with generalized erythroderma, alopecia, lymphadenopathy, hepatomegaly, and diarrhea, and the immunological phenotype is characterized by a reduced number of circulating B lymphocytes (95, 115). In one study, patients with OS were shown to have a variable number of circulating Treg with low FoxP3 expression and reduced or no T-cell suppressive capacity in vitro, together with a severe reduction of Treg cells in the thymus (95). These results provided the first evidence of a defect in Treg development and function in OS, implying that both central and peripheral tolerance are compromised in this disease.
STAT5b deficiency is a rare autosomal recessive disease involving both marked growth failure and severe immunodeficiency (116). Normal transcription of IL-2Rα, FOXP3, Bcl-2 and growth hormone (GH) genes are controlled by STAT5b signaling (96, 116). Immunophenotypically, STAT5b-deficient patients have decreased Treg numbers, low expression of FoxP3 and CD25, and an impaired ability to suppress T-cell proliferation (96, 116). Patients present with severe eczema, arthritis, autoimmune thyroiditis, and thrombocytopenic purpura, which are thought to be associated with Treg dysfunction (96, 116), demonstrating that STAT5b plays a critical role in Treg maintenance and function. Interestingly, an overshadowed cytokine, IL-9, activates the transcription factors STAT3 and STAT5 whose cellular targets include Treg (117). In an IL-9Rα-deficient mouse model of autoimmune encephalomyelitis, Tregs have a reduced ability to suppress T-cell proliferation (117). In addition, in vivo and in vitro assays have shown that IL-9 promotes Treg suppressive function and survival through STAT3 and STAT5 signaling (117). This suggests that IL-9 has an important role in Treg function and maintenance. Therefore, it could be of interest to include the study of IL-9Rα or IL-9 function in Treg in patients with immune dysregulation immunodeficiencies.
3 Treg cell-based therapies
The diagnosis and management of IEIs pose challenges due to the confluence of overlapping and multisystemic manifestations and their variable underlying genetic etiologies (54). Also, there is a variety of treatment options for the management of immune dysregulation in IEIs depending on the specific type, severity, and pathway involved in the disorder. These include systemic immunosuppressants, enzyme replacement therapy, small molecules, biological therapies (such as monoclonal antibodies), and adoptive cell therapies (ACT) such as hematopoietic stem cell transplantation (HSCT), gene therapy (54, 118, 119) and, in the near future, cell-based therapies. The aim of this review is to ascertain available Treg cell-based therapies more comprehensively and, as such, the other therapies will be briefly summarized.
3.1 Current therapeutic approaches for immune dysregulation in IEI
Systemic immunosuppressants, such as glucocorticoids and calcineurin inhibitors, exhibit non-specific effects and are typically employed in cases where specific diagnosis is still lacking, but there is a requirement to manage inflammation (54, 118). As for targeted therapies, there is a range of pharmaceutical agents with the ability to selectively counteract the aberrant pathways and/or molecules involved in specific diseases, thereby tailoring drug-based therapies to the precise needs of each individual patient. Within these possibilities, monoclonal antibodies are used as a more personalized approach to treat some IEIs. This is the case of alemtuzumab (anti-CD52) (56), emapalumab (anti-IFNγ) (120, 121) and anakinra (IL-1R agonist) (122) used to treat HLH. Other drugs include rituximab (anti-CD20) used for autoimmune and lymphoproliferative disorders (121) and tocilizumab (anti-IL6R) for GOF STAT3 (123, 124). Moreover, a recent clinical trial explored the efficacy and safety of tadekinig alpha (an IL-18 binding protein) in autoinflammatory conditions driven by IL-18 (Arnold) such as NLRC4 GOF and XIAP deficiency (125). Regarding IL-10 related VEO-IBD, IL-1 inhibitors have been suggested based on the assumption that IL-10 LOF mutations can interact with activation through the inflammasome which would lead to increased IL-1 levels (126). Abatacept (CTLA-4 fusion protein) is used in CTLA-4 and LRBA deficiencies (127). Additionally, due to its mechanism of action inducing T cell anergy it has been tested in a mouse model for IPEX with very promising results and superior efficacy to sirolimus and anti-CD4 treatments (128). Teplizumab, a CD3-directed monoclonal antibody, has been approved to treat T1D and is being tested in pediatric patients with stage 2 of this disease (129). Considering the occurrence of T1D in IPEX patients, this therapeutic approach could be highly beneficial in effectively managing these patients (63). Besides monoclonals, other drugs targeting specific affected pathways are being used. For example, sirolimus (an mTor pathway inhibitor) has been used in LRBA biallelic mutations and CTLA-4 haploinsufficiency (56), as well as ALPS and APDS (120) and CVID (130). This is also the case for Jakinibs (direct JAK/STAT inhibitors) like ruxolitinib (JAK1 and JAK2 inhibitor) and tofacitinib (JAK1 and JAK3 inhibitor) that have been used in STAT3 GOF mutations (56, 120) and HLH (131).
HSCT is a curative option for IEIs. Within the context of IEIs, the initial application of HSCT was documented in SCID cases as early as 1968 (120, 132). This procedure carries substantial risks, including graft-versus-host disease (GVHD) and the administration of conditioning agents that result in grave immunosuppression, thereby elevating the risk of severe infections (54). Furthermore, limitations encompass the availability of compatible donors, the age of patients, and the severity of the disease and/or end-organ damage (118). Due to the importance of conditioning regimes for survival, HSCT recommendation guidelines are established by the EBMT/ESID inborn errors working party guidelines (IEWP) (118, 126, 133). PIRD has particular challenges for HSCT. This approach becomes essential in patients that cannot be managed by immunosuppressive treatments (125). In this context, patients show an increased risk of alloreactivity to HSCT indicating a need for further research into the use of targeted treatment as a bridge for HSCT (134). Moreover, while this approach has demonstrated efficacy in resolving clinical symptoms for a subset of patients, the long-term survival rates remain relatively low compared to conventional IEIs. For PIRD, the survival rate at the 5-year mark is approximately 67% (135) whilst conventional IEIs exhibit a 90% (134). Regarding HSCT in PIRD, even when the donor is a close relative, genetic screening becomes necessary due to the disease’s delayed and variable clinical onset or incomplete penetrance, which could also potentially impact other family members (134). Therefore, comprehensive research is imperative to render HSCT in PIRD a safer, more efficient and viable therapeutic approach.
Gene therapy was first used in SCID-ADA as early as 1993, followed by SCID-X1 (132). This strategy entails the modification of HSC and other immune cells involved in the disease process. Subsequent to its inception, significant advances have been made such as using self-inactivating lentiviruses, rendering this approach a promising and safer alternative compared to its earlier iterations (119, 120). Indeed, in 2016, Strimvelis™ was approved in Europe for the treatment for ADA-SCID. It consists of autologous CD34+ cells (HSC) transduced with a gamma-retrovirus that encodes for the human ADA cDNA sequence. There are, however, important limitations for this gene-adding approach. Transduction can limit the success and number of cells available for treatment, and gene manipulation can generate insertional mutagenesis that can turn on oncogenes and lead to clonal expansion or leukemic proliferation. Moreover, preexisting mutations or availability of autologous cells for gene modification could rule out the patient for the procedure (136). A more pinpointed approach is gene editing where stable genetic modification is introduced at the specific targeted site that is mutated and responsible for the IEIs. This process employs engineered nucleases, with three primary types being utilized: Zinc-Finger Nucleases (ZFNs), Transcription activator-like effector nucleases (TALENs), and CRISPR/Cas. These molecular tools enable researchers to precisely edit the genetic material, offering a more targeted and efficient approach to treating genetic-based immune disorders (119). This technique is still in preclinical phases but demonstrates remarkable promise. Regarding gene therapy in PIRD, is has been evaluated in IPEX syndrome and HLH. In IPEX, two primary gene therapy strategies are currently under development. The first consists in forcing ectopic and stable FoxP3 expression on CD4+ T cells, thereby transforming them into FoxP3 engineered Treg-like cells. This approach is already being tested in IPEX patients where a clinical trial (NCT05241444) is evaluating the feasibility and safety profile of these Treg-like cells, called CD4LVFoxP3. The second approach, which is still in preclinical studies, uses CRISPR-Cas to modify hematopoietic stem and progenitor cells (HSPCs) for the development of long-lasting and functional Treg and Teff cells that can normally express FoxP3. This latter approach is capable of restoring FoxP3 levels independent of the specific mutation of the patient and it also enables cell marking for monitoring and purification purposes (137). In HLH, there is also a gene therapy approach under development for perforin deficiency (138). It consists in transducing autologous murine CD8+ T cells with a gammaretroviral vector with corrected perforin gene to generate fully functional CD8+ T cells, which are subsequently transferred back to the mouse. Indeed, this strategy has demonstrated success in vivo in an HLH murine model (139). These promising findings offer hope for the potential application of gene therapy in the treatment of IPEX and HLH in the future.
3.2 Adoptive cell therapy with Treg
With the first human treated with Treg in 2009, ACT using Treg is a rapidly expanding field that offers substantial advantages and holds great promise for medical applications (140). Numerous clinical trials are currently being carried out with Treg cell-based therapies to manage GVHD (39), T1D (141) and transplant rejection (142). As highlighted throughout this review, Tregs exhibit suppressive and immunoregulatory functions that render them ideal for addressing diseases characterized by immune activation. Nevertheless, care must be taken when employing Treg cell-based therapeutic approaches, which are summarized in Table 1. Firstly, Treg isolated from blood is a mix of pTreg and tTreg, and tTregs have been shown to be more stable under lymphopenia conditions in mouse models. Secondly, Tregs possess the ability to modulate their local microenvironments, enabling them to convert their surroundings into a suppressive milieu regardless of their own persistence. This phenomenon is referred to as infectious tolerance. Consequently, infused Treg may not necessarily need to endure indefinitely, but rather, they can persist as long as it takes for the microenvironment to undergo transformation and establish a suppressive environment. Furthermore, it is important to consider that when Tregs for therapy are derived from Teff, they may exhibit different mechanisms of action compared to their natural counterpart (37). Numerous studies have demonstrated that antigen-specific Tregs are more efficient and safer than polyclonal Tregs as they migrate to the specific site where the antigen itself is expressed, reducing the risk of general immunosuppression. However, antigen-specific Tregs are present in very low numbers in blood, and the expansion protocol needed for their use as ACT is very challenging (38). Therefore, polyclonal Treg could be a more feasible approach although it could generate more side effects due to bystander suppression (143).
Taking all these aspects into account, various mechanisms are being explored to generate antigen-specific Treg. One method involves the use of an engineered T cell receptor (TCR), where an antigen-specific TCR is obtained from a Teff and introduced into Treg. These transduced cells gather in target tissues expressing the specific antigen, exerting their function directly or by bystander suppression. Importantly, they are MHC restricted so matching MHC between donor and recipient is needed (143). This technique has demonstrated remarkable potential in mouse models of multiple sclerosis and hemophilia, exhibiting promising results (37). Another approach involves chimeric antigen receptor (CAR) Treg cells, which are designed to recognize specific proteins. This strategy proves particularly useful for conditions involving tissue destruction. Compared to engineered TCR Treg, CAR Tregs require high antigen expression to activate but are MHC independent which makes them a more versatile option for widespread application in patients. In fact, engineered CAR Treg against HLA-A2, commonly mismatched in transplantation, have been proven to protect from GVHD in different mouse models (143). A third approach involved overexpressing FoxP3 in antigen-specific Teff. By transducing FoxP3 into CD4+ Teff, stable and heightened expression of this transcription factor is achieved, effectively converting effector T cells into functional Treg cells (37, 144). Similarly, the most frequently used approach in Treg based therapies comprises the obtaining of Treg cells from peripheral blood and stimulating them with anti CD3/CD28 beads along with high IL-2 concentration to achieve a significant expansion of polyclonal Treg. Although this expansion yields a large number of cells, the expanded Treg cells are less specific and potent than antigen-specific Treg (37). An important limitation in approaches that use peripheral blood as the Treg source is the low number of Treg cells that can be obtained, making it challenging to achieve sufficient cell numbers for therapy. Additionally, the expansion process itself can lead to an exhausted phenotype of the Treg, potentially impacting functionality, stability, and efficacy (39).
An exceptionally innovative recent approach consists in generating effective Treg from pediatric thymus, which is routinely discarded in pediatric cardiac surgeries. Referred to as thyTreg, these cells are derived from fresh thymocytes and are induced to differentiate and expanded under controlled laboratory conditions. Notably, thyTreg show highly advantageous properties as they are immature with an undifferentiated phenotype, have greater suppressor capacity, and display increased stability of FoxP3 (39). Given the promising characteristics, thyTreg are currently being evaluated in a clinical trial to prevent rejection in heart transplant patients (NCT04924491). If this therapy succeeds, thyTreg could be further developed for allogenic use as a potentially promising therapy for some PIRD.
There are different methods for obtaining Treg for ACT, generally involving isolation of specific cells from elected source, expansion, characterization of the resulting cell product, reinfusion, and monitoring of the infused cells (38). Throughout this section, different sources for Treg obtention have been mentioned. These sources are continuously explored and allow the isolation of other immune cell types in addition to Treg. These include peripheral blood and umbilical cord blood, and, more recently, Treg cells directly derived from the thymus (where T cells are developed). Each source has its own advantages and challenges, and ongoing research aims to optimize the selection and utilization of these sources to enhance the efficacy and safety of Treg cell-based therapies (39).
3.3 Adoptive cell therapy with Treg in PIRD
IPEX syndrome, the benchmark for studying Treg function, highlights the potential benefits adoptive Treg cell therapy could have in IEIs. Indeed, the treatment of the equivalent phenotype in mice, known as scurfy, has shown promising results, leading to an increased lifespan of diseased mice (144). CD4+ T cells were isolated from scurfy mice and transduced with a lentiviral vector containing the whole FOXP3 transcript, as well as the transcript encoding a truncated surface-expressed protein (CD271), facilitating their selection and further detection in vivo (145). Resulting CD4LVFoxP3 cells were expanded in vitro, resulting in a pool of CD4+ T cells with FoxP3 expression that showed both suppressive function and sustained stability of FoxP3 (137). Initially achieved in scurfy mice, this accomplishment was subsequently extended to encompass CD4+ T cells sourced from IPEX patients (63). This approach demonstrated the feasibility of appropriately transducing previously affected T cells from IPEX patients, characterized by a FoxP3 mutation, resulting in the conversion into CD4+ FoxP3+ T cells. Moreover, these CD4LVFoxP3 exhibited the same functional traits as naturally occurring Treg, including suppressive functions and expression of CD25hi CD127lo CTLA-4+ ICOS+ Helios+ (137). Indeed, upon infusion of CD4LVFoxP3 cells into scurfy mice, notable immunomodulatory effects were observed, leading to symptom improvement (146).
These findings offer valuable insights into the potential therapeutic applications of Treg cell-based interventions for immune dysregulation disorders such as IPEX syndrome (144). To our knowledge, IPEX syndrome remains the only IEI subject with comprehensive investigation aimed at the development of a Treg cell-based therapeutic approach aimed at curing the disorder.
4 Discussion
4.1 Changes and controversies in the phenotypical definition of Treg
Treg cells have a unique phenotypic molecular signature characterized by high expression of CD25 and FoxP3, both in tTreg or pTreg (14). Although in mice the expression of Helios and low expression of Neuropilin 1 (Nrp-1) are good molecular discriminants between tTreg and pTreg cells (30, 147), in humans this discrimination is problematic and somewhat unclear. In human tTreg, Helios expression is not universal and, similar to FoxP3, its expression has been reported in activated T cells (148, 149). FoxP3 and Helios provide information about the stability and suppressive power of the cell (150) with Helios+ Treg having been reported to be more stable and with a greater suppressive capacity (151). In mice, Nrp-1 expression indicates greater Treg stability; however, in humans, this protein is virtually undetectable in peripheral Treg (152). Because of this, the use of Helios as a differentiating marker between tTreg and pTreg is a matter of ongoing debate. Nevertheless, because of the information it provides, measuring Helios along with FoxP3 should be included as a routine marker in laboratories whenever possible (153).
Characterization of Treg by flow cytometry may not be so simple. Firstly, and as mentioned earlier, there is no specific unique marker for Treg. Secondly, the relatively low expression of FoxP3 requires the use of fluorochrome-labelled monoclonal antibodies of sufficient intensity to detect it. Within Treg, the degree of FoxP3 expression shows a dynamic range from naïve or resting Treg cells, with low expression, to activated Treg with higher expression (154, 155). Therefore, separate identification among Tconv, resting Treg (rTreg), naïve Treg, and activated Treg would be achieved by the relatively high expression of FoxP3 and by additional markers such as CD45RA, thus defining rTreg as CD45RA+FoxP3low activated Treg cells with suppressive activity (aTreg) as CD45RA-FoxP3hi and T cells without inhibitory capacity as CD45RA-FoxP3low/- (155).
Further complicating the matter, intracellular staining requires a fixation process. Although some molecules, such as formaldehyde, largely preserve the native conformation of proteins, they can also generate cross-linking between proteins, polysaccharides, and lipids that may hide the reactive epitopes for fluorochrome-labelled antibodies (150). Transcription factors are not suitable for the isolation of viable cells, and they require more sample handling, time, and resources, reinforcing the clear need to identify unique surface markers for Treg characterization. One possible solution found through omics analysis is a transmembrane protein known as GPA33 that is expressed by a set of Treg with tTreg-like characteristics (156–158). Additionally, TIGIT appears to inhibit the activation of the mammalian mTORC1 target of rapamycin signaling pathway, maintaining the stability and expression of FoxP3 in Treg (159). Also, elevated TIGIT expression in tTreg (CD4+FoxP3+Helios+) has been reported to be associated with cell lineage stability and suppressive capacity (160). In conclusion, a possible alternative to the use of Helios and FoxP3 as markers of lineage, cell stability, and suppressive force would be the replacement of these markers with GPA33 and TIGIT, respectively. However, further studies are needed to clarify their validity for the identification of stable human Treg.
Given all this information, a crucial question is how human Treg should be defined. Concerning CD4+ cells, there are several ways to define them, either as CD25hiCD127low, CD25hiFoxP3+, FoxP3+, CD25hiCD127lowFoxP3+ or CD25hiCD127lowCCR4+ (69, 70). At present, Treg cells are characterized in the literature as any of the above definitions, therefore emphasizing the need for a consensual and unified characterization to study this cell type. Based on current knowledge, the most rigorous way to define them is as CD25hiCD127lowFoxP3+Helios+, which ensures the selection of tTreg with stable FoxP3 expression. However, as discussed above, the use of FoxP3 and Helios is not possible when working with viable cells, and sometimes all four markers cannot be used together due to instrumentation limitations. If this is the case, at least two of them should be used, either as CD25hiCD127low or CD25hiFoxP3+ (69, 70). Using each marker separately is not a valid option, as Tconv cells may overexpress CD25, FoxP3, and Helios, and reduce CD127 expression under stimulation conditions (154, 161, 162). CCR4 is currently recognized as a Treg effector cell marker, and therefore can be used to complete the phenotypic definition of Treg when used alongside the markers already mentioned (29).
The complexity of characterizing Treg increases if CD8+ regulatory cells are to be studied, given their recent role in viral infections and tumorigenesis (22). CD8+ Treg can be divided into different groups, including CD8+CD103+, CD8+CD122+, CD8+CD28-, and CD8αα+HLA-E+TCRαβ+ cells (163). CD8+CD103+ Treg have been implicated in studies of kidney transplantation rejection and intestinal graft-versus-host disease (164). TGFβ is directly related to the level of CD103 (αE7β integrin) expression, and it is thought that its suppressive mechanism is through cell-cell contact. However, other studies show that, under inflammatory conditions, CD8 Treg releases TGF-β conferring a protective effect (31, 165, 166). Although these cells are CD28+ and do not express FoxP3, CTLA-4, GITR, LAG-3, or CD25, a stable human CD8+ regulatory type (hCD8+) can be induced by adding TGF-β and Rapamycin (RAPA) together with αCD3/CD28 and IL-2 in vitro expressing FoxP3, CD103, and PD-1 (167). Nevertheless, further studies are needed to determine the differences among these cell types.
CD8+CD122+ Treg cells have been shown to exert suppressive function in transplantation and autoimmunity settings (32, 168), especially within the memory compartment (CD44hiCD62LhiCCR7+CD127-). However, CD8+CD122+ cells do not express FoxP3, suggesting that they are part of a different group from CD8+FoxP3+ (hCD8 Treg) induced CD8+ cells (169–171). A comparative study showed that a subgroup of CD8+CD122+ Treg cells expressing PD1 (CD8+CD122+PD-1+) versus CD4+FoxP3+ Treg cells are more effective in the context of allograft rejection, with this protein being mainly responsible for the inhibitory effects (33, 170). In spite of this, PD-1 is a marker of T-cell exhaustion and functional impairment, so PD-1 expression on CD8+ Treg requires additional research (172).
Other populations have also been recognized as regulatory cells. These include CD8+CD28- cells which have been associated with different clinical conditions such as pregnancy, infectious diseases, cancer, and organ transplantation. Several studies have shown that their suppressive capacity is dependent on IL-10 or TGF-β (34, 173). Although some research groups have demonstrated FoxP3 expression in this cell type (174, 175), other groups have not confirmed this (176, 177). Similarly, loss of FoxP3 and CD28 in this cell type correlates with the expression of CD56, granzyme A, and perforin exhibiting cytotoxic activity (178). In addition, a unique phenotype of CD8αα+TCRαβ+Qa-1 restricted Treg cells (in mice) and HLA-E in humans, expressing surface molecules typical of NK cells, has been demonstrated (35), as well as the recent discovery of a CD8+KIR+ Treg cell subpopulation, which appears to play an important role in the maintenance of peripheral tolerance in the context of antigen-specific viral infections and its relationship to autoimmunity (179). All these findings are of significant importance for understanding the regulatory mechanism of CD8+ cells, but significant further work is needed to clarify precise phenotypic identification, and the possible relationship with CD4+FoxP3+Helios+ Treg.
Currently, the reliable identification of Treg requires a panel with different surface markers, together with function analyses. Taking into account the major problems discussed here, dedicated research is needed to improve our understanding of Treg cells biology and the phenotypic plasticity of the cells in different biological and pathological contexts. These phenotypic controversies are summarized in Table 1.
4.2 Changes and controversies in the functional studies of Treg
Because phenotypic characterization of human Treg is challenging, measuring the suppressive capacity of Treg in artificial in vitro systems can additionally be used for Treg characterization. Nevertheless, this functional assay may or may not accurately represent their functionality in vivo, and the assays currently used may be misinterpreted. Despite these limitations, if assays are rigorous and carefully controlled, the data obtained provide a reliable indication of the regulatory potential of Treg.
Traditionally, what has been considered the gold standard to date is the measurement of the suppressive capacity of Treg, particularly in relation to inhibition of proliferation of conventional CD4+ and CD8+ T cells. Previously, cell proliferation measurements had certain limitations given the use of radioactive isotopes (3H-tritylated) and the lack of precision in identifying the proliferating cells. Although it is very sensitive and requires a small number of cells, this technique lacks suppression of proliferation and cannot confirm Treg regulatory function (92, 180–182). Consequently, it is useful to measure inflammatory cytokine production in the supernatant in this type of assay because 1) the analysis of cytokines is independent of Treg activity, and 2) Tregs suppress the action of inflammatory cytokines (183). However, a quicker solution is to replace the suppression assay with the measurement of IL-2 or IFNγ mRNA expression, as well as flow cytometric expression of CD69 and CD154 on Tconv cells, as these alternatives can be performed in 5-7h after co-culture with Treg cells (183, 184). Indeed, commercial companies such as Becton Dickinson (BD) already offer kits to determine Treg suppression based on these two surface markers on Tconv cells after culture at the clinical level (185).
Current flow cytometry-optimized assays using fluorescent reactive amines such as CFSE (Carboxyfluorescein succinimidyl ester) are more efficient and accurate. CFSE labelling of Teff allows independent analysis of Treg from effector cells, as well as proliferation-parallel analysis of surface marker expression and cytokine secretion (186). Although to date this technique is widely used, it should be noted that by analyzing cells that have divided at least once, there will be cells with variable numbers of divisions, so this may underestimate the suppressive capacity of Treg. Therefore, a more accurate approach is the use of cytometry analysis software such as FlowJO (TreeStar) as it provides parameters such as proliferation index (average number of divisions), percentage of divided cells (percentage of cells in the initial population that have divided at least once), and division index (average number of divisions a cell has undergone in the initial population). By basing it on cell divisions rather than the total number of cells, the calculation of the division index allows for a more accurate assessment (36).
Although the in vitro suppression assay is a powerful tool, the co-culture system is risky and can lead to misleading data or misinterpretation. Hence, the need for rigorous controls. First, titration of cells in the co-culture is essential. Whilst some studies show that high titers of 1:1 or 1:2 (Treg : Teff) are unlikely to occur under physiological conditions, suppression should be detected at ratios as low as 1 Treg:16 Teff (181, 187). It is essential, however, to have a co-culture control of non-Treg : Teff cells under the same conditions to confirm that the suppression is specifically due to Treg. An additional consideration is that Treg cells have cytotoxic function (Treg CD8+) as one of the mechanisms of suppression; hence, a granzyme/perforin study of the cell culture is desirable (59, 188).
Second, it is also crucial to confirm cell viability. As Tregs are prone to cell death and can be exposed to prolonged purification processes, it is necessary to confirm their viability throughout the suppression assay (189). To this end, good control should include the presence and absence of IL-2 in the assay.
Finally, in order to obtain correct titers of Treg, they need to be purified. Different purification methods exist, but this is a major challenge given the lack of well-defined markers. Therefore, it is not advisable to purify Treg solely based on the use of CD4- and CD25-labelled magnetic beads. Methods to check for Treg cell purity include 1) phenotypic assessment in resting Treg cells at least 10 days after TCR activation to allow decrease of FoxP3 expression on T cells after activation (190) and 2) evaluation of the TSDR region from the FOXP3 promoter. Females inactivate one of their X chromosomes by methylation; since FOXP3 is located on the X chromosome this test may be more feasible in male patients (191, 192), which concstitutes a major limitation in human studies.
Ultimately, since Treg cells exert their effects beyond the classical inhibition of CD4+ T cell proliferation, it would be interesting to also include other Treg-cellular targets, such as antigen-presenting cells (APCs). An interesting proposal would be to measure inhibitory cytokines such as IL-10, IL-35, or TGF-β, although special care must be taken with the available antibody conjugates and with the release kinetics of these cytokines if they are measured with flow cytometry; new strategies to measure cell activation through the ATP/ADP ratio may even be tried. The functional controversies are summarized in Table 1.
4.3 Concerns regarding Treg cell-based therapies
Treg cell-based therapies present certain concerns that need to be carefully considered. As live cells, Tregs possess the inherent risk of becoming unstable, potentially leading to the generation of undesirable inflammatory responses or exhibiting effector cell properties. Notably, destabilization of FoxP3 and the consequent transformation of Treg into an effector phenotype could trigger tissue destruction. Furthermore, the plasticity of Treg cells allow them to express transcriptional factors that characterize other T cell lineages such as T-bet, GATA-3, and RORγt that can activate transcriptional programs that allow them to migrate and counteract the effect of the cell whose transcriptional factor is matching. This plasticity is a double-edge-sword, as in this context, it makes Treg quite reactive and liable to lose suppressive capacity, secreting inflammatory cytokines under certain circumstances (37, 38). Addressing these issues is crucial to ensure the safety and efficacy of these therapies. The addition of molecules like DNA methyltransferase could help reduce the risk of FoxP3 destabilization. Another factor of concern is the potential risk of oncogene activation subsequent to cell transduction, analogous to occurrences observed in other gene therapy methodologies (37). Moreover, some patients have shown increased susceptibility to opportunistic infections due to the immunosuppressive nature of (38).
To address these issues, strategies to eliminate infused Treg cells if they become overly active or unstable are being thoroughly examined. Among the most advanced approaches are the insertion of suicide casquets or integration sites into the Treg cell, allowing for its inhibition or elimination if necessary (37).
These concerns highlight the importance of ongoing research and vigilance in the development and application of Treg cell-based therapies. Addressing these challenges will be crucial to ensure the safety and effectiveness of these innovative treatments for a wide range of medical conditions. The controversies in Treg cell-based therapies are summarized in Table 1.
5 Conclusions
The field of Treg has come a long way recently, with its definition and itsfunction-dysfunction studies correlating with pathology and cell-based therapies. Yet some controversies remain. Therefore, continuous update and integration of recent information is needed to offer a response to clinical needs, which arise in patients with PIRD, but also in other immunodeficiencies, allergy, and neoplastic syndromes, in order to develop future therapies aimed at restoring Treg functionality.
Author contributions
RK-B: Conceptualization, Investigation, Software, Writing – original draft, Writing – review & editing. DA: Conceptualization, Investigation, Software, Writing – original draft, Writing – review & editing. YL: Writing – review & editing. AE-S: Writing – review & editing. AV: Writing – review & editing. RC-R: Funding acquisition, Resources, Supervision, Writing – review & editing. ES-R: Funding acquisition, Project administration, Resources, Supervision, Writing – review & editing. LA: Funding acquisition, Project administration, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the projects PI18/00223, FI19/00208, and PI21/00211 to LA, integrated in the Plan Nacional de I+D+I and cofinanced by the ISCIII -Subdirección General de Evaluación y Formento de la Investigación Sanitaria -and the Fondo Europeo de Desarrollo Regional (FEDER), by Pla Estratègic de Recerca i Innovació en Salut (PERIS), Departament de Salut, Generalitat de Catalunya (SLT006/17/00199 to LA), by a 2017 Leonardo Grant for Researchers and Cultural Creators, BBVA Foundation [IN(17)_BBM_CLI_0357] to LA, by a 2017 Beca de Investigación de la Sociedad Española De Inmunología Clínica Alergología y Asma Pediátrica to LA, by a 2022 Convocatòria de Beques de Recerca IRSJD -Carmen de Torres 2022 (2022AR-IRSJD-CdTorres) and CERCA Programme/Generalitat de Catalunya. It was also supported by grants from “Fundación Familia Alonso” (FFA-2019), from Instituto de Salud Carlos III (ISCIII) co-financed by FEDER funds (ICI20/00063; PI21/00189) granted to RCR. RKB is supported by research funds from IISGM. Project PI21/01325 was granted to ES-R and also supported this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ohkura N, Sakaguchi S. Transcriptional and epigenetic basis of Treg cell development and function: its genetic anomalies or variations in autoimmune diseases. Cell Res (2020) 30:465–74. doi: 10.1038/s41422-020-0324-7
2. Plitas G, Rudensky AY. Regulatory T cells: Differentiation and function. Cancer Immunol Res (2016) 4:721–5. doi: 10.1158/2326-6066.CIR-16-0193
3. Nishizuka Y, Sakakura T. Thymus and reproduction: sex-linked dysgenesia of the gonad after neonatal thymectomy in mice. Science (1969) 166:753–5. doi: 10.1126/science.166.3906.753
4. Dominguez-Villar M, Hafler DA. Regulatory T cells in autoimmune disease. Nat Immunol (2018) 19:665–73. doi: 10.1038/s41590-018-0120-4
5. Romano M, Fanelli G, Albany CJ, Giganti G, Lombardi G. Past, present, and future of regulatory T cell therapy in transplantation and autoimmunity. Front Immunol (2019) 31:43(10). doi: 10.3389/fimmu.2019.00043
6. Sakaguchi S, Mikami N, Wing JB, Tanaka A, Ichiyama K, Ohkura N. Annual review of immunology regulatory T cells and human disease. Annu Rev Immunol (2020) 26(38):541–66. doi: 10.1146/annurev-immunol-042718
7. Shevyrev D, Tereshchenko V. Treg heterogeneity, function, and homeostasis. Front Immunol (2020) 10:3100. doi: 10.3389/fimmu.2019.03100
8. Lucca LE, Dominguez-Villar M. Modulation of regulatory T cell function and stability by co-inhibitory receptors. Nat Rev Immunol (2020) 20:680–93. doi: 10.1038/s41577-020-0296-3
9. Grant CR, Liberal R, Mieli-Vergani G, Vergani D, Longhi MS. Regulatory T-cells in autoimmune diseases: Challenges, controversies and-yet-unanswered questions. Autoimmun Rev (2015) 14:105–16. doi: 10.1016/j.autrev.2014.10.012
10. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. J Immunol (2017) 198:981–5. doi: 10.1126/science.1079490
11. Liu W, Putnam AL, Xu-Yu Z, Szot GL, Lee MR, Zhu S, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med (2006) 203:1701–11. doi: 10.1084/jem.20060772
12. Ito T, Hanabuchi S, Wang YH, Park WR, Arima K, Bover L, et al. Two functional subsets of FOXP3+ Regulatory T cells in human thymus and periphery. Immunity (2008) 28:870–80. doi: 10.1016/j.immuni.2008.03.018
13. Li DY, Xiong XZ. ICOS+ Tregs: A functional subset of tregs in immune diseases. Front Immunol (2020) 28:2104. doi: 10.3389/fimmu.2020.02104
14. Abbas AK, Benoist C, Bluestone JA, Campbell DJ, Ghosh S, Hori S, et al. Regulatory T cells: Recommendations to simplify the nomenclature. Nat Immunol (2013) 14:307–8. doi: 10.1038/ni.2554
15. Klein L, Robey EA, Hsieh CS. Central CD4 + T cell tolerance: deletion versus regulatory T cell differentiation. Nat Rev Immunol (2019) 19:7–18. doi: 10.1038/s41577-018-0083-6
16. Pellerin L, Jenks JA, Bégin P, Bacchetta R, Nadeau KC. Regulatory T cells and their roles in immune dysregulation and allergy. Immunol Res (2014) 58:358–68. doi: 10.1007/s12026-014-8512-5
17. Morina L, Jones ME, Oguz C, Kaplan MJ, Gangaplara A, Fitzhugh CD, et al. Co-expression of Foxp3 and Helios facilitates the identification of human T regulatory cells in health and disease. Front Immunol (2023) 7:1114780(14). doi: 10.3389/fimmu.2023.1114780
18. Cheru N, Hafler DA, Sumida TS. Regulatory T cells in peripheral tissue tolerance and diseases. Front Immunol (2023) 1:1154575(14). doi: 10.3389/fimmu.2023.1154575
19. Vieyra-Lobato MR, Vela-Ojeda J, Montiel-Cervantes L, López-Santiago R, Moreno-Lafont MC. Description of CD8+ regulatory T lymphocytes and their specific intervention in Graft-versus-Host and infectious diseases, autoimmunity, and cancer. J Immunol Res (2018) 5(2018):3758713. doi: 10.1155/2018/3758713
20. Gershon RK, Kondo K. Cell interactions in the induction of tolerance: the role of thymic lymphocytes. Immunology (1970) 18(5):723–37.
21. Mishra S. CD8+ Regulatory T cell – A mystery to be revealed. Front Immunol (2021) 18:708874(12). doi: 10.3389/fimmu.2021.708874
22. Guillonneau C, Hill M, Hubert FX, Chiffoleau E, Hervé C, Li XL, et al. CD40Ig treatment results in allograft acceptance mediated by CD8+CD45RClow T cells, IFN-γ, and indoleamine 2,3-dioxygenase. J Clin Invest (2007) 117:1096–106. doi: 10.1172/JCI28801
23. Li J, Zaslavsky M, Su Y, Guo J, Sikora MJ, van Unen V, et al. KIR+CD8+ T cells suppress pathogenic T cells and ar active in autoimmune diseases and COVID-19. Science (2022) 376(6590):eabi9591. doi: 10.1126/science.abi9591
24. Sakowska J, Glasner P, Dukat-Mazurek A, Rydz A, Zieliński M, Pellowska I, et al. Local T cell infiltrates are predominantly associated with corneal allograft rejection. Transpl Immunol (2023) 79:101852. doi: 10.1016/j.trim.2023.101852
25. Bolivar-Wagers S, Larson JH, Jin S, Blazar BR. Cytolytic CD4+ and CD8+ Regulatory T-cells and implications for developing immunotherapies to combat graft-versus-host disease. Front Immunol (2022) 13:864748. doi: 10.3389/fimmu.2022.864748
26. Yu N, Li X, Song W, Li D, Yu D, Zeng X, et al. CD4(+) CD25(+) CD127 (low/-) T cells: a more specific Treg population in human peripheral blood. Inflammation (2012) 35(6):1773–80. doi: 10.1007/s10753-012-9496-8
27. Chatenoud L. Natural and induced T CD4+CD25+FOXP3+ regulatory T cells. Methods Mol Biol (2011) 677:3–13. doi: 10.1007/978-1-60761-869-0_1
28. Aly MG, Ibrahim EH, Karakizlis H, Weimer R, Opelz G, Morath C, et al. CD4+CD25+CD127-foxp3+ and CD8+CD28- tregs in renal transplant recipients: phenotypic patterns, association with immunosuppressive drugs, and interaction with effector CD8+ T cells and CD19+IL-10+ Bregs. Front Immunol (2021) 12:716559. doi: 10.3389/fimmu.2021.716559
29. Baatar D, Olkhanud P, Sumitomo K, Taub D, Gress R, Biragyn A. Human peripheral blood T regulatory cells (Tregs), functionally primed CCR4+ Tregs and unprimed CCR4– tregs, regulate effector T cells using fasL. J Immunol (2007) 178:4891–900. doi: 10.4049/jimmunol.178.8.4891
30. Thornton AM, Korty PE, Tran DQ, Wohlfert EA, Murray PE, Belkaid Y, et al. Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J Immunol (2010) 184(7):3433–41. doi: 10.4049/jimmunol.0904028
31. Lu L, Yu Y, Li G, Pu L, Zhang F, Zheng S, et al. CD8+CD103+ regulatory T cells in spontaneous tolerance of liver allografts. Int Immunopharmacol (2009) 9:546–8. doi: 10.1016/J.INTIMP.2009.01.021
32. Rifa’i M, Kawamoto Y, Nakashima I, Suzuki H. Essential roles of CD8+CD122+ Regulatory T cells in the maintenance of T cell homeostasis. J Exp Med (2004) 200:1123–34. doi: 10.1084/JEM.20040395
33. Dai Z, Zhang S, Xie Q, Wu S, Su J, Li S, et al. Natural CD8+CD122+ T cells are more potent in suppression of allograft rejection than CD4+CD25+ regulatory T cells. Am J Transplant (2014) 14:39–48. doi: 10.1111/ajt.12515
34. Fenoglio D, Ferrera F, Fravega M, Balestra P, Battaglia F, Proietti M, et al. Advancements on phenotypic and functional characterization of non–antigen-specific CD8+CD28– regulatory T cells. Hum Immunol (2008) 69:745–50. doi: 10.1016/J.HUMIMM.2008.08.282
35. Fanchiang SS, Cojocaru R, Othman M, Khanna R, Brooks MJ, Smith T, et al. Global expression profiling of peripheral Qa-1-restricted CD8αα+TCRαβ+ regulatory T cells reveals innate-like features: Implications for immune-regulatory repertoire. Hum Immunol (2012) 73:214–22. doi: 10.1016/j.humimm.2011.07.306
36. Mcmurchy AN, Levings MK. Suppression assays with human T regulatory cells: A technical guide. Eur J Immunol (2012) 42:27–34. doi: 10.1002/eji.201141651
37. Raffin C, Vo LT, Bluestone JA. Treg cell-based therapies: challenges and perspectives. Nat Rev Immunol (2020) 20:158–72. doi: 10.1038/s41577-019-0232-6
38. Baeten P, Van Zeebroeck L, Kleinewietfeld M, Hellings N, Broux B. Improving the efficacy of regulatory T cell therapy. Clin Rev Allergy Immunol (2022) 62:363–81. doi: 10.1007/s12016-021-08866-1
39. Bernaldo-de-Quirós E, Pion M, Martínez-Bonet M, Correa-Rocha R. A new generation of cell therapies employing regulatory T cells (Treg) to induce immune tolerance in pediatric transplantation. Front Pediatr (2022) 10:862807. doi: 10.3389/fped.2022.862807
40. Liu Y, Zheng P. Preserving the CTLA-4 checkpoint for safer and more effective cancer immunotherapy. Trends Pharmacol Sci (2020) 41:4–12. doi: 10.1016/j.tips.2019.11.003
41. Gianchecchi E, Fierabracci A. Inhibitory receptors and pathways of lymphocytes: The role of PD-1 in Treg development and their involvement in autoimmunity onset and cancer progression. Front Immunol (2018) 9:2374. doi: 10.3389/fimmu.2018.02374
42. Chocarro L, Blanco E, Zuazo M, Arasanz H, Bocanegra A, Fernández-Rubio L, et al. Understanding lag-3 signaling. Int J Mol Sci (2021) 22(10):5282. doi: 10.3390/ijms22105282
43. Anderson AC, Joller N, Kuchroo VK. Lag-3, tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity (2016) 44:989–1004. doi: 10.1016/j.immuni.2016.05.001
44. Lee DJ. The relationship between TIGIT+ regulatory T cells and autoimmune disease. Int Immunopharmacol (2020) 83:106378. doi: 10.1016/j.intimp.2020.106378
45. Polak K, Marchal P, Taroni C, Ebel C, Kirstetter P, Kastner P, et al. CD4+ regulatory T cells lacking Helios and Eos. Biochem Biophys Res Commun (2023) 674:83–9. doi: 10.1016/j.bbrc.2023.06.087
46. Chougnet C, Hildeman D. Helios-controller of Treg stability and function. Transl Cancer Res (2016) 5:S338–41. doi: 10.21037/tcr.2016.07.37
47. Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. Human inborn errors of immunity: 2022 update on the classification from the international union of immunological societies expert committee Vol. 42. New York City, USA: Springer US (2022) p. 1473–507. doi: 10.1007/s10875-022-01289-3
48. Yamashita M, Inoue K, Okano T, Morio T. Inborn errors of immunity—recent advances in research on the pathogenesis. Inflammation Regener (2021) 41(1):9. doi: 10.1186/s41232-021-00159-6
49. Schmidt RE, Grimbacher B, Witte T. Autoimmunity and primary immunodeficiency: Two sides of the same coin? Nat Rev Rheumatol (2018) 14:7–18. doi: 10.1038/nrrheum.2017.198
50. Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human inborn errors of immunity: 2019 update on the classification from the international union of immunological societies expert committee. J Clin Immunol (2020) 40:24–64. doi: 10.1007/s10875-019-00737-x
51. Bousfiha A, Moundir A, Tangye SG, Picard C, Jeddane L, Al-Herz W, et al. The 2022 update of IUIS phenotypical classification for human inborn errors of immunity. J Clin Immunol (2022) 42:1508–20. doi: 10.1007/s10875-022-01352-z
52. Flinn AM, Gennery AR. Primary immune regulatory disorders: Undiagnosed needles in the haystack? Orphanet J Rare Dis (2022) 17:1–9. doi: 10.1186/s13023-022-02249-1
53. Chan AY, Torgerson TR. Primary immune regulatory disorders: A growing universe of immune dysregulation. Curr Opin Allergy Clin Immunol (2020) 20:582–90. doi: 10.1097/ACI.0000000000000689
54. Cepika AM, Sato Y, Liu JMH, Uyeda MJ, Bacchetta R, Roncarolo MG. Tregopathies: Monogenic diseases resulting in regulatory T-cell deficiency. J Allergy Clin Immunol (2018) 142:1679–95. doi: 10.1016/j.jaci.2018.10.026
55. Torgerson TR. IPEX and IPEX-like disorders. In: Stiehm’s immune deficiencies: inborn errors of immunity. Amsterdam, Netherlands: Elsevier (2020). p. 617–32. doi: 10.1016/B978-0-12-816768-7.00026-0
56. Kolukısa B, Barış S. Primary immune regulatory disorders and targeted therapies. Turkish J Hematol (2021) 38:1–14. doi: 10.4274/tjh.galenos.2021.2020.0724
57. Chatila TA, Blaeser F, Ho N, Lederman HM, Voulgaropoulos C, Helms C, et al. JM2, encoding a fork head-related protein, is mutated in X-linked autoimmunity-allergic dysregulation syndrome. J Clin Invest. (2000) 106(12):R75–81. doi: 10.1172/JCI11679
58. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet (2001) 27:20–1. doi: 10.1038/83713
59. Shevach EM. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity (2009) 30:636–45. doi: 10.1016/j.immuni.2009.04.010
60. Zorn E, Nelson EA, Mohseni M, Porcheray F, Kim H, Litsa D, et al. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood (2006) 108:1571–9. doi: 10.1182/blood-2006-02-004747
61. Borsellino G, Kleinewietfeld M, Di Mitri D, Sternjak A, Diamantini A, Giometto R, et al. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: Hydrolysis of extracellular ATP and immune suppression. Blood (2007) 110:1225–32. doi: 10.1182/blood-2006-12-064527
62. Bacchetta R, Barzaghi F, Roncarolo MG. From IPEX syndrome to FOXP3 mutation: A lesson on immune dysregulation. Ann N Y Acad Sci (2016) 1417:5–22. doi: 10.1111/nyas.13011
63. Ben-skowronek I. Ipex syndrome: Genetics and treatment options. Genes (Basel) (2021) 12:1–13. doi: 10.3390/genes12030323
64. Kirchner A, Sanchez IM, Zalan A, Bhat G, Bain M. Identification of a novel variant of FOXP3 resulting in severe immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome highlights potential pitfalls of molecular testing. Pediatr Dermatol (2022) 39:483–5. doi: 10.1111/PDE.14936
65. Ochs HD, Gambineri E, Torgerson TR. IPEX. FOXP3 and regulatory T-cells: A model for autoimmunity. Immunol Res (2007) 38:112–21. doi: 10.1007/s12026-007-0022-2
66. Gambineri E, Mannurita SC, Hagin D, Vignoli M, Anover-Sombke S, DeBoer S, et al. Patients with the phenotype of immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome. Front Immunol (2018) 9:2411/BIBTEX. doi: 10.3389/FIMMU.2018.02411/BIBTEX
67. Du J, Wang Q, Yang S, Chen S, Fu Y, Spath S, et al. FOXP3 exon 2 controls Treg stability and autoimmunity. Sci Immunol (2022) 7(72):eabo5407. doi: 10.1126/sciimmunol.abo5407
68. Consonni F, Ciullini Mannurita S, Gambineri E. Atypical presentations of IPEX: expect the unexpected. Front Pediatr (2021) 9:643094/BIBTEX. doi: 10.3389/FPED.2021.643094/BIBTEX
69. Maecker HT, McCoy JP, Nussenblatt R. Standardizing immunophenotyping for the human immunology project. Nat Rev Immunol (2012) 12:191–200. doi: 10.1038/nri3158
70. Cossarizza A, Chang HD, Radbruch A, Abrignani S, Addo R, Akdis M, et al. Guidelines for the use of flow cytometry and cell sorting in immunological studies (third edition). Eur J Immunol (2021) 51:2708–3145. doi: 10.1002/eji.202170126
71. Brodszki N, Frazer-Abel A, Grumach AS, Kirschfink M, Litzman J, Perez E, et al. European society for immunodeficiencies (ESID) and european reference network on rare primary immunodeficiency, autoinflammatory and autoimmune diseases (ERN RITA) complement guideline: deficiencies, diagnosis, and management. J Clin Immunol (2020) 40:576–91. doi: 10.1007/s10875-020-00754-1
72. Hsu P, Santner-Nanan B, Hu M, Skarratt K, Lee CH, Stormon M, et al. IL-10 potentiates differentiation of human induced regulatory T cells via STAT3 and foxo1. J Immunol (2015) 195:3665–74. doi: 10.4049/jimmunol.1402898
73. Ren H, Chen Y, Zhu Z, Xia J, Liu S, Hu Y, et al. FOXO1 regulates Th17 cell-mediated hepatocellular carcinoma recurrence after hepatic ischemia-reperfusion injury. Cell Death Dis (2023) 14:367. doi: 10.1038/s41419-023-05879-w
74. Graves DT, Milovanova TN. Mucosal immunity and the FOXO1 transcription factors. Front Immunol (2019) 10:2530. doi: 10.3389/fimmu.2019.02530
75. Caudy AA, Reddy ST, Chatila T, Atkinson JP, Verbsky JW. CD25 deficiency causes an immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like syndrome, and defective IL-10 expression from CD4 lymphocytes. J Allergy Clin Immunol (2007) 119:482–7. doi: 10.1016/j.jaci.2006.10.007
76. Roth TL, Puig-Saus C, Yu R, Shifrut E, Carnevale J, Li PJ, et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature (2018) 559:405–9. doi: 10.1038/s41586-018-0326-5
77. Verma N, Burns SO, Walker LSK, Sansom DM. Immune deficiency and autoimmunity in patients with CTLA-4 (CD152) mutations. Clin Exp Immunol (2017) 190:1–7. doi: 10.1111/cei.12997
78. Hou TZ, Verma N, Wanders J, Kennedy A, Soskic B, Janman D, et al. Identifying functional defects in patients with immune dysregulation due to LRBA and CTLA-4 mutations. Blood (2017) 129(11):156–1468. doi: 10.1182/blood-2016-10-745174
79. Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med (2014) 20:1410–6. doi: 10.1038/nm.3746
80. Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science (2014) 345:1623–7. doi: 10.1126/science.1255904
81. Gámez-Díaz L, Grimbacher B. Immune checkpoint deficiencies and autoimmune lymphoproliferative syndromes. BioMed J (2021) 44:400–11. doi: 10.1016/j.bj.2021.04.005
82. Charbonnier LM, Janssen E, Chou J, Ohsumi TK, Keles S, Hsu JT, et al. Regulatory T-cell deficiency and immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like disorder caused by loss-of-function mutations in LRBA. J Allergy Clin Immunol (2015) 135:217–227.e9. doi: 10.1016/j.jaci.2014.10.019
83. Lo B, Zhang K, Lu W, Zheng L, Zhang Q, Kanellopoulou C, et al. Autoimmune disease. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science (2015) 24;349(6246):436–40. doi: 10.1126/science.aaa1663
84. Delage L, Carbone F, Riller Q, Zachayus JL, Kerbellec E, Buzy A, et al. NBEAL2 deficiency in humans leads to low CTLA-4 expression in activated conventional T cells. Nat Commun (2023) 14:3728. doi: 10.1038/s41467-023-39295-7
85. Afzali B, Grönholm J, Vandrovcova J, O’Brien C, Sun HW, Vanderleyden I, et al. BACH2 immunodeficiency illustrates an association between super-enhancers and haploinsufficiency. Nat Immunol (2017) 18:813–23. doi: 10.1038/ni.3753
86. Vogel TP, Leiding JW, Cooper MA, Forbes Satter LR. STAT3 gain-of-function syndrome. Front Pediatr (2023) 10:770077. doi: 10.3389/fped.2022.770077
87. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature (2006) 441:235–8. doi: 10.1038/nature04753
88. Pillemer BBL, Xu H, Oriss TB, Qi Z, Ray A. Deficient SOCS3 expression in CD4+CD25+FoxP3+ regulatory T cells and SOCS3-mediated suppression of Treg function. Eur J Immunol (2007) 37:1622082–2089. doi: 10.1002/EJI.200737193
89. Humblet-Baron S, Franckaert D, Dooley J, Bornschein S, Cauwe B, Schönefeldt S, et al. IL-2 consumption by highly activated CD8 T cells induces regulatory T-cell dysfunction in patients with hemophagocytic lymphohistiocytosis. J Allergy Clin Immunol (2016) 138:200–209.e8. doi: 10.1016/j.jaci.2015.12.1314
90. Mazerolles F, Stolzenberg MC, Pelle O, Picard C, Neven B, Fischer A, et al. Autoimmune lymphoproliferative syndrome-fas patients have an abnormal regulatory T cell (Treg) phenotype but display normal natural Treg-suppressive function on T cell proliferation. Front Immunol (2018) 9:718. doi: 10.3389/fimmu.2018.00718
91. Hsieh WC, Hsu TS, Chang YJ, Lai MZ. IL-6 receptor blockade corrects defects of XIAP-deficient regulatory T cells. Nat Commun (2018) 9(1):463. doi: 10.1038/s41467-018-02862-4
92. López-Nevado M, González-Granado LI, Ruiz-García R, Pleguezuelo D, Cabrera-Marante O, Salmón N, et al. Primary immune regulatory disorders with an autoimmune lymphoproliferative syndrome-like phenotype: immunologic evaluation, early diagnosis and management. Front Immunol (2021) 12:671755. doi: 10.3389/fimmu.2021.671755
93. Humblet-Baron S, Sather B, Anover S, Becker-Herman S, Kasprowicz DJ, Khim S, et al. Wiskott-Aldrich syndrome protein is required for regulatory T cell homeostasis. J Clin Invest (2007) 117:407–18. doi: 10.1172/JCI29539
94. Singh AK, Eken A, Hagin D, Komal K, Bhise G, Shaji A, et al. DOCK8 regulates fitness and function of regulatory T cells through modulation of IL-2 signaling. JCI Insight (2017) 2(19):e94275. doi: 10.1172/jci.insight.94275
95. Cassani B, Poliani PL, Moratto D, Sobacchi C, Marrella V, Imperatori L, et al. Defect of regulatory T cells in patients with Omenn syndrome. J Allergy Clin Immunol (2010) 125:209–16. doi: 10.1016/j.jaci.2009.10.023
96. Cohen AC, Nadeau KC, Tu W, Hwa V, Dionis K, Bezrodnik L, et al. Cutting edge: decreased accumulation and regulatory function of CD4+CD25high T cells in human STAT5b deficiency. J Immunol (2006) 177:2770–4. doi: 10.4049/jimmunol.177.5.2770
97. Lo B, Fritz JM, Su HC, Uzel G, Jordan MB, Lenardo MJ. CHAI and LATAIE: new genetic diseases of CTLA-4 checkpoint insufficiency. Blood (2016) 128:1037. doi: 10.1182/BLOOD-2016-04-712612
98. Canna SW, Marsh RA. Pediatric hemophagocytic lymphohistiocytosis. Blood. (2020) 135(16):1332–43. doi: 10.1182/blood.2019000936
99. Takahashi R, Nishimoto S, Muto G, Sekiya T, Tamiya T, Kimura A, et al. SOCS1 is essential for regulatory T cell functions by preventing loss of Foxp3 expression as well as IFN-γ and IL-17A production. J Exp Med (2011) 208:2055–67. doi: 10.1084/JEM.20110428
100. Oliveira JB, Bleesing JJ, Dianzani U, Fleisher TA, Jaffe ES, Lenardo MJ, et al. Revised diagnostic criteria and classification for the autoimmune lymphoproliferative syndrome (ALPS): Report from the 2009 NIH International Workshop. Blood (2010) 116(14):e35–40. doi: 10.1182/blood-2010-04-280347
101. Michalovich D, Nejentsev S. Activated PI3 kinase delta syndrome: From genetics to therapy. Front Immunol (2018) 9:369. doi: 10.3389/fimmu.2018.00369
102. Luo Y, Xia Y, Wang W, Li Z, Jin Y, Gong Y, et al. Identification of a novel de novo gain-of-function mutation of PIK3CD in a patient with activated phosphoinositide 3-kinase δ syndrome. Clin Immunol (2018) 197:60–7. doi: 10.1016/j.clim.2018.08.007
103. Więsik-Szewczyk E, Rutkowska E, Kwiecień I, Korzeniowska M, Sołdacki D, Jahnz-Różyk K. Patients with common variable immunodeficiency complicated by autoimmune phenomena have lymphopenia and reduced treg, Th17, and NK cells. J Clin Med (2021) 10:3356. doi: 10.3390/jcm10153356
104. Kutukculer N, Azarsiz E, Aksu G, Karaca NE. CD4+CD25+Foxp3+ T regulatory cells, Th1 (CCR5, IL-2, IFN-γ) and Th2 (CCR4, IL-4, Il-13) type chemokine receptors and intracellular cytokines in children with common variable immunodeficiency. Int J Immunopathol Pharmacol (2016) 29:241–51. doi: 10.1177/0394632015617064
105. Arandi N, Mirshafiey A, Jeddi-Tehrani M, Abolhassani H, Sadeghi B, Mirminachi B, et al. Evaluation of CD4+CD25+FOXP3+ regulatory T cells function in patients with common variable immunodeficiency. Cell Immunol (2013) 281:129–33. doi: 10.1016/J.CELLIMM.2013.03.003
106. Cunill V, Clemente A, Lanio N, Barceló C, Andreu V, Pons J, et al. Follicular T cells from smB- common variable immunodeficiency patients are skewed toward a Th1 phenotype. Front Immunol (2017) 8:174. doi: 10.3389/fimmu.2017.00174
107. Gupta S, Demirdag Y, Gupta AA. Members of the regulatory lymphocyte club in common variable immunodeficiency. Front Immunol (2022) 13:864307. doi: 10.3389/fimmu.2022.864307
108. Azizi G, Mirshafiey A, Abolhassani H, Yazdani R, Jafarnezhad-Ansariha F, Shaghaghi M, et al. Circulating helper T-cell subsets and regulatory T cells in patients with common variable immunodeficiency without known monogenic disease. J Investig Allergol Clin Immunol (2018) 28:172–81. doi: 10.18176/jiaci.0231
109. Yesillik S, Agrawal S, Gollapudi SV, Gupta S. Phenotypic analysis of CD4+ Treg, CD8+ Treg, and breg cells in adult common variable immunodeficiency patients. Int Arch Allergy Immunol (2019) 180:150–8. doi: 10.1159/000501457
110. Tessarin G, Baronio M, Lougaris V. Monogenic forms of common variable immunodeficiency and implications on target therapeutic approaches. Curr Opin Allergy Clin Immunol (2023) 23(6):461–6. doi: 10.1097/ACI.0000000000000947
111. Hsu AP. Not too little, not too much: the impact of mutation types in Wiskott-Aldrich syndrome and RAC2 patients. Clin Exp Immunol (2023) 212:137–46. doi: 10.1093/cei/uxad001
112. Lexmond WS, Goettel JA, Lyons JJ, Jacobse J, Deken MM, Lawrence MG, et al. FOXP3+ Tregs require WASP to restrain Th2-mediated food allergy. J Clin Invest (2016) 126:4030–44. doi: 10.1172/JCI85129
113. Adriani M, Aoki J, Horai R, Thornton AM, Konno A, Kirby M, et al. Impaired in vitro regulatory T cell function associated with Wiskott-Aldrich syndrome. Clin Immunol (2007) 124(1):41–8. doi: 10.1016/j.clim.2007.02.001
114. Wilkie H, Janssen E, Leyva-Castillo JM, Geha RS. DOCK8 expression in regulatory T cells maintains their stability and limits contact hypersensitivity. J Invest Dermatol (2021) 141:1503–1511.e3. doi: 10.1016/j.jid.2020.09.027
115. Villa A, Notarangelo LD, Roifman CM. Omenn syndrome: Inflammation in leaky severe combined immunodeficiency. J Allergy Clin Immunol (2008) 122:1082–6. doi: 10.1016/j.jaci.2008.09.037
116. Nadeau K, Hwa V, Rosenfeld RG. STAT5b deficiency: An unsuspected cause of growth failure, immunodeficiency, and severe pulmonary disease. J Pediatr (2011) 158:701–8. doi: 10.1016/j.jpeds.2010.12.042
117. Elyaman W, Bradshaw EM, Uyttenhove C, Dardalhon V, Awasthi A, Imitola J, et al. IL-9 induces differentiation of TH17 cells and enhances function of FoxP3+ natural regulatory T cells. Proc Natl Acad Sci U.S.A. (2009) 106:12885–90. doi: 10.1073/PNAS.0812530106
118. Castiello MC, Ferrari S, Villa A. Correcting inborn errors of immunity: From viral mediated gene addition to gene editing. Semin Immunol (2023) 66:101731. doi: 10.1016/j.smim.2023.101731
119. Liu X, Li G, Liu Y, Zhou F, Huang X, Li K. Advances in CRISPR/Cas gene therapy for inborn errors of immunity. Front Immunol (2023) 14:1111777. doi: 10.3389/fimmu.2023.1111777
120. Slatter M, Lum SH. Personalized hematopoietic stem cell transplantation for inborn errors of immunity. Front Immunol (2023) 14:1162605. doi: 10.3389/fimmu.2023.1162605
121. Bucciol G, Meyts I. Recent advances in primary immunodeficiency: From molecular diagnosis to treatment. F1000Res (2020) 9:F1000. doi: 10.12688/f1000research.21553.1
122. Eloseily EM, Weiser P, Crayne CB, Haines H, Mannion ML, Stoll ML, et al. Benefit of anakinra in treating pediatric secondary hemophagocytic lymphohistiocytosis. Arthritis Rheumatol (2020) 72:326–34. doi: 10.1002/art.41103
123. Forbes LR, Vogel TP, Cooper MA, Castro-Wagner J, Schussler E, Weinacht KG, et al. Jakinibs for the treatment of immune dysregulation in patients with gain-of-function signal transducer and activator of transcription 1 (STAT1) or STAT3 mutations. J Allergy Clin Immunol (2018) 142:1665–9. doi: 10.1016/j.jaci.2018.07.020
124. Wang W, Liu L, Hui X, Wang Y, Ying W, Zhou Q, et al. Efficacy of tocilizumab therapy in a patient with severe pancytopenia associated with a STAT3 gain-of-function mutation. BMC Immunol (2021) 22(1):19. doi: 10.1186/s12865-021-00411-1
125. Arnold DE, Chellapandian D, Leiding JW. The use of biologic modifiers as a bridge to hematopoietic cell transplantation in primary immune regulatory disorders. Front Immunol (2021) 12:692219. doi: 10.3389/fimmu.2021.692219
126. Pinto MV, Neves JF. Precision medicine: The use of tailored therapy in primary immunodeficiencies. Front Immunol (2022) 13:1029560. doi: 10.3389/fimmu.2022.1029560
127. Krausz M, Uhlmann A, Rump IC, Ihorst G, Goldacker S, Sogkas G, et al. The ABACHAI clinical trial protocol: Safety and efficacy of abatacept (s.c.) in patients with CTLA-4 insufficiency or LRBA deficiency: A non controlled phase 2 clinical trial. Contemp Clin Trials Commun (2022) 30:101008. doi: 10.1016/j.conctc.2022.101008
128. Gerbaux M, Roos E, Willemsen M, Staels F, Neumann J, Bücken L, et al. CTLA4-ig effectively controls clinical deterioration and immune condition in a murine model of foxp3 deficiency. J Clin Immunol (2023) 4 3(6):1393–402. doi: 10.1007/s10875-023-01462-2
129. Keam SJ, Deyà-Martínez A, Esteve-Solé A, Vélez-Tirado N, Celis V, Costa J, et al. Teplizumab: first approval Sirolimus as an alternative treatment in patients with granulomatous-lymphocytic lung disease and humoral immunodeficiency with impaired regulatory T cells. Drugs Pediatr Allergy Immunol (2023) 8329:439–45425–32. doi: 10.1111/pai.12890
130. Deyà-Martínez A, Esteve-Solé A, Vélez-Tirado N, Celis V, Costa J, Cols M, et al. Sirolimus as an alternative treatment in patients with granulomatous-lymphocytic lung disease and humoral immunodeficiency with impaired regulatory T cells. Pediatr Allergy Immunol (2018) 29:425–32. doi: 10.1111/pai.12890
131. Keenan C, Nichols KE, Albeituni S. Use of the JAK inhibitor ruxolitinib in the treatment of hemophagocytic lymphohistiocytosis. Front Immunol (2021) 12:614704. doi: 10.3389/fimmu.2021.614704
132. Fischer A. Gene therapy for inborn errors of immunity: past, present and future. Nat Rev Immunol (2023) 23:397–408. doi: 10.1038/s41577-022-00800-6
133. Lankester AC, Albert MH, Booth C, Gennery AR, Güngör T, Hönig M, et al. EBMT/ESID inborn errors working party guidelines for hematopoietic stem cell transplantation for inborn errors of immunity. Bone Marrow Transplant (2021) 56:2052–62. doi: 10.1038/s41409-021-01378-8
134. Margarit-Soler A, Deyà-Martínez À, Canizales JT, Vlagea A, García-García A, Marsal J, et al. Case report: Challenges in immune reconstitution following hematopoietic stem cell transplantation for CTLA-4 insufficiency-like primary immune regulatory disorders. Front Immunol (2022) 13:1070068. doi: 10.3389/fimmu.2022.1070068
135. Chan AY, Leiding JW, Liu X, Logan BR, Burroughs LM, Allenspach EJ, et al. Hematopoietic cell transplantation in patients with primary immune regulatory disorders (PIRD): A primary immune deficiency treatment consortium (PIDTC) survey. Front Immunol (2020) 11:239. doi: 10.3389/fimmu.2020.00239
136. Ferrua F, Aiuti A. Twenty-five years of gene therapy for ADA-SCID: from bubble babies to an approved drug. Hum Gene Ther (2017) 28:972–81. doi: 10.1089/hum.2017.175
137. Borna S, Lee E, Sato Y, Bacchetta R. Towards gene therapy for IPEX syndrome. Eur J Immunol (2022) 52:705–16. doi: 10.1002/eji.202149210
138. Panchal N, Ghosh S, Booth C. T cell gene therapy to treat immunodeficiency. Br J Haematol (2021) 192:433–43. doi: 10.1111/bjh.17070
139. Ghosh S, Carmo M, Calero-Garcia M, Ricciardelli I, Bustamante Ogando JC, Blundell MP, et al. T-cell gene therapy for perforin deficiency corrects cytotoxicity defects and prevents hemophagocytic lymphohistiocytosis manifestations. J Allergy Clin Immunol (2018) 142:904–913.e3. doi: 10.1016/j.jaci.2017.11.050
140. Trzonkowski P, Bieniaszewska M, Juścińska J, Dobyszuk A, Krzystyniak A, Marek N, et al. First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127- T regulatory cells. Clin Immunol (2009) 133:22–6. doi: 10.1016/j.clim.2009.06.001
141. Serr I, Drost F, Schubert B, Daniel C. Antigen-specific treg therapy in type 1 diabetes – challenges and opportunities. Front Immunol (2021) 12:712870. doi: 10.3389/fimmu.2021.712870
142. Bernaldo-de-Quirós E, Cózar B, López-Esteban R, Clemente M, Gil-Jaurena JM, Pardo C, et al. A novel GMP protocol to produce high-quality treg cells from the pediatric thymic tissue to be employed as cellular therapy. Front Immunol (2022) 13:893576. doi: 10.3389/fimmu.2022.893576
143. Steiner R, Pilat N. The potential for Treg-enhancing therapies in transplantation. Clin Exp Immunol (2023) 211:122–37. doi: 10.1093/cei/uxac118
144. Passerini L, Bacchetta R. Forkhead-Box-P3 gene transfer in human CD4+ T conventional cells for the generation of stable and efficient regulatory T cells, suitable for immune modulatory therapy. Front Immunol (2017) 8:1282. doi: 10.3389/fimmu.2017.01282
145. Delville M, Bellier F, Leon J, Klifa R, Lizot S, H´ EìE Vinçon, et al. A combination of cyclophosphamide and interleukin-2 allows CD4 1 T cells converted to Tregs to control scurfy syndrome. Blood (2021) 137(17):2326–36. doi: 10.1182/blood.2020009187
146. Sato Y, Passerini L, Piening BD, Uyeda MJ, Goodwin M, Gregori S, et al. Human-engineered Treg-like cells suppress FOXP3-deficient T cells but preserve adaptive immune responses in vivo. Clin Transl Immunol (2020) 9(11):e1214. doi: 10.1002/cti2.1214
147. Weiss JM, Bilate AM, Gobert M, Ding Y, de Lafaille MAC, Parkhurst CN, et al. Neuropilin 1 is expressed on thymus-derived natural regulatory T cells, but not mucosagenerated induced Foxp3+ T reg cells. J Exp Med (2012) 209:1723–42. doi: 10.1084/jem.20120914
148. Himmel ME, MacDonald KG, Garcia RV, Steiner TS, Levings MK. Helios+ and Helios- cells coexist within the natural FOXP3+ T regulatory cell subset in humans. J Immunol (2013) 190(5):2001–8. doi: 10.4049/jimmunol.1201379
149. Akimova T, Beier UH, Wang L, Levine MH, Hancock WW. Helios expression is a marker of T cell activation and proliferation. PloS One (2011) 6(8):e24226. doi: 10.1371/journal.pone.0024226
150. Cossarizza A, Chang HD, Radbruch A, Acs A, Adam D, Adam-Klages S, et al. Guidelines for the use of flow cytometry and cell sorting in immunological studies (second edition). Eur J Immunol (2019) 49:1457–973. doi: 10.1002/eji.201970107
151. Bin Dhuban K, d’Hennezel E, Nashi E, Bar-Or A, Rieder S, Shevach EM, et al. Coexpression of TIGIT and FCRL3 identifies helios+ Human memory regulatory T cells. J Immunol (2015) 194(8):3687–96. doi: 10.4049/JIMMUNOL.1401803
152. Milpied P, Renand A, Bruneau J, Mendes-Da-Cruz DA, Jacquelin S, Asnafi V, et al. Neuropilin-1 is not a marker of human Foxp3+ Treg. Eur J Immunol (2009) 39:1466–71. doi: 10.1002/eji.200839040
153. Lam AJ, Lin DTS, Gillies JK, Uday P, Pesenacker AM, Kobor MS, et al. Optimized CRISPR-mediated gene knockin reveals FOXP3-independent maintenance of human Treg identity. Cell Rep (2021) 3;36(5):109494. doi: 10.1016/j.celrep.2021.109494
154. Allan SE, Crome SQ, Crellin NK, Passerini L, Steiner TS, Bacchetta R, et al. Activation-induced FOXP3 in human T effector cells does not suppress proliferation or cytokine production. Int Immunol (2007) 19:345–54. doi: 10.1093/INTIMM/DXM014
155. Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the foxP3 transcription factor. Immunity (2009) 30:899–911. doi: 10.1016/j.immuni.2009.03.019
156. Heath JK, White SJ, Johnstone CN, Catimel B, Simpson RJ, Moritz RL, et al. The human A33 antigen is a transmembrane glycoprotein and a novel member of the immunoglobulin superfamily. Proc Natl Acad Sci U.S.A. (1997) 21;94(2):469–74. doi: 10.1073/pnas.94.2.469
157. Opstelten R, de Kivit S, Slot MC, van den Biggelaar M, Iwaszkiewicz-Grześ D, Gliwiński M, et al. GPA33: A marker to identify stable human regulatory T cells. J Immunol (2020) 204:3139–48. doi: 10.4049/jimmunol.1901250
158. Cuadrado E, van den Biggelaar M, de Kivit S, yen CY, Slot M, Doubal I, et al. Proteomic analyses of human regulatory T cells reveal adaptations in signaling pathways that protect cellular identity. Immunity (2018) 48:1046–1059.e6. doi: 10.1016/j.immuni.2018.04.008
159. Sato K, Yamashita-Kanemaru Y, Abe F, Murata R, Nakamura-Shinya Y, Kanemaru K, et al. DNAM-1 regulates Foxp3 expression in regulatory T cells by interfering with TIGIT under inflammatory conditions. Proc Natl Acad Sci USA (2021) 25;118(21):e2021309118. doi: 10.1073/pnas.2021309118
160. Fuhrman CA, Yeh W-I, Seay HR, Saikumar Lakshmi P, Chopra G, Zhang L, et al. Divergent phenotypes of human regulatory T cells expressing the receptors TIGIT and CD226. J Immunol (2015) 195:145–55. doi: 10.4049/jimmunol.1402381
161. Ziegler SF. FOXP3: Not just for regulatory T cells anymore. Eur J Immunol (2007) 37:21–3. doi: 10.1002/eji.200636929
162. Lam AJ, Uday P, Gillies JK, Levings MK. Helios is a marker, not a driver, of human Treg stability. Eur J Immunol (2022) 52:75–84. doi: 10.1002/eji.202149318
163. Crusio WE, Dong H, Radeke H, Rezaei N, Steinlein O, Xiao J. T regulatory cells in human health and diseases, advances in experimental medicine and biology. Amsterdam, Netherlands: Springer (2022). Available at: http://www.springer.com/series/5584.
164. El-Asady R, Yuan R, Liu K, Wang D, Gress RE, Lucas PJ, et al. TGF-β–dependent CD103 expression by CD8+ T cells promotes selective destruction of the host intestinal epithelium during graft-versus-host disease. J Exp Med (2005) 201:1647–57. doi: 10.1084/JEM.20041044
165. Myers L, Croft M, Kwon BS, Mittler RS, Vella AT. Peptide-specific CD8 T regulatory cells use IFN-γ to elaborate TGF-β-based suppression. J Immunol (2005) 174:7625–32. doi: 10.4049/JIMMUNOL.174.12.7625
166. Ho J, Kurtz CC, Naganuma M, Ernst PB, Cominelli F, Rivera-Nieves J. A CD8+/CD103high T cell subset regulates TNF-mediated chronic murine ileitis. J Immunol (2008) 180:2573–80. doi: 10.4049/JIMMUNOL.180.4.2573
167. Sun J, Yang Y, Huo X, Zhu B, Li Z, Jiang X, et al. Efficient therapeutic function and mechanisms of human polyclonal CD8 + CD103 + Foxp3 + Regulatory T cells on collagen-induced arthritis in mice. J Immunol Res (2019) 2019:8575407. doi: 10.1155/2019/8575407
168. Suciu-Foca N, Manavalan JS, Cortesini R. Generation and function of antigen-specific suppressor and regulatory T cells. Transpl Immunol (2003) 11:235–44. doi: 10.1016/S0966-3274(03)00052-2
169. Suzuki H, Shi Z, Okuno Y, Isobe K. Are CD8+CD122+ cells regulatory T cells or memory T cells. Hum Immunol (2008) 69:751–4. doi: 10.1016/J.HUMIMM.2008.08.285
170. Dai H, Wan N, Zhang S, Moore Y, Wan F, Dai Z. Cutting edge: programmed death-1 defines CD8+CD122+ T cells as regulatory versus memory T cells. J Immunol (2010) 185:803–7. doi: 10.4049/JIMMUNOL.1000661
171. Lerret NM, Houlihan JL, Kheradmand T, Pothoven KL, Zhang ZJ, Luo X. Donor-specific CD8+Foxp3+ T cells protect skin allografts and facilitate induction of conventional CD4+Foxp3 + regulatory T cells. Am J Transplant (2012) 12:2335–47. doi: 10.1111/j.1600-6143.2012.04120.x
172. Simon S, Labarriere N. PD-1 expression on tumor-specific T cells: Friend or foe for immunotherapy? Oncoimmunology (2017) 7(1):e1364828. doi: 10.1080/2162402X.2017.1364828
173. Miller WP, Srinivasan S, Panoskaltsis-Mortari A, Singh K, Sen S, Hamby K, et al. GVHD after haploidentical transplantation: a novel, MHC-defined rhesus macaque model identifies CD28– CD8+ T cells as a reservoir of breakthrough T-cell proliferation during costimulation blockade and sirolimus-based immunosuppression. Blood (2010) 116:5403–18. doi: 10.1182/BLOOD-2010-06-289272
174. Frisullo G, Nociti V, Iorio R, Plantone D, Patanella AK, Tonali PA, et al. CD8+Foxp3+ T cells in peripheral blood of relapsing-remitting multiple sclerosis patients. Hum Immunol (2010) 71:437–41. doi: 10.1016/j.humimm.2010.01.024
175. Manavalan JS, Kim-Schulze S, Scotto L, Naiyer AJ, Vlad G, Colombo PC, et al. Alloantigen specific CD8+ CD28- FOXP3+ T suppressor cells induce ILT3+ ILT4+ tolerogenic endothelial cells, inhibiting alloreactivity. Int Immunol (2004) 16:1055–68. doi: 10.1093/intimm/dxh107
176. Korecka-Polak A, Duszota A, Wierzbicki P, Niemczyk M, Bocian K, Kłosowska D, et al. Human peripheral blood CD8+CD28-t cells of renal allograft recipients do not express FOXP3 protein. Transplant Proc (2011) 43(8):2917–21. doi: 10.1016/j.transproceed.2011.08.016
177. Scotto L, Naiyer AJ, Galluzzo S, Rossi P, Manavalan JS, Kim-Schulze S, et al. Overlap between molecular markers expressed by naturally occurring CD4 +CD25 + regulatory T cells and antigen specific CD4 +CD25 + and CD8 +CD28 - T suppressor cells. Hum Immunol (2004) 65:1297–306. doi: 10.1016/j.humimm.2004.09.004
178. Baeten D, Louis S, Braud C, Braudeau C, Ballet C, Moizant F, et al. Phenotypically and functionally distinct CD8+ lymphocyte populations in long-term drug-free tolerance and chronic rejection in human kidney graft recipients. J Am Soc Nephrol (2006) 17(1):294–304. doi: 10.1681/ASN.2005020178
179. Li J, Zaslavsky M, Su Y, Guo J, Sikora MJ, van Unen V, et al. KIR+CD8+ T cells suppress pathogenic T cells and are active in autoimmune diseases and COVID-19. Science (2022) 376(6590):eabi9591. doi: 10.1126/science.abi9591
180. Thornton AM, Shevach EM. CD4+CD25+ Immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J Exp Med (1998) 188:287–96. doi: 10.1084/JEM.188.2.287
181. Crellin NK, Garcia RV, Hadisfar O, Allan SE, Steiner TS, Levings MK. Human CD4+ T cells express TLR5 and its ligand flagellin enhances the suppressive capacity and expression of FOXP3 in CD4+CD25+ T regulatory cells. J Immunol (2005) 175:8051–9. doi: 10.4049/JIMMUNOL.175.12.8051
182. Baecher-Allan C, Wolf E, Hafler DA. Functional analysis of highly defined, FACS-isolated populations of human regulatory CD4+CD25+ T cells. Clin Immunol (2005) 115:10–8. doi: 10.1016/J.CLIM.2005.02.018
183. Allan SE, Alstad AN, Merindol N, Crellin NK, Amendola M, Bacchetta R, et al. Generation of potent and stable human CD4+ T regulatory cells by activation-independent expression of FOXP3. Mol Ther (2008) 16:194–202. doi: 10.1038/sj.mt.6300341
184. Oberle N, Eberhardt N, Falk CS, Krammer PH, Suri-Payer E. Rapid suppression of cytokine transcription in human CD4+CD25– T cells by CD4+Foxp3+ Regulatory T cells: independence of IL-2 consumption, TGF-β, and various inhibitors of TCR signaling. J Immunol (2007) 179:3578–87. doi: 10.4049/JIMMUNOL.179.6.3578
185. BD Life Sciences. Sorting and Downstream Functional Assessment of Regulatory T Cells Isolating live cells with the BD FACSMelody TM cell sorter . Available at: https://www.bdbiosciences.com/content/dam/bdb/marketing-documents/BD-FACSMelody-Sorting-Downstream-Assessment-Regulatory-TCells-DS.pdf.
186. Crome SQ, Clive B, Wang AY, Kang CY, Chow V, Yu J, et al. Inflammatory effects of ex vivo human th17 cells are suppressed by regulatory T cells. J Immunol (2010) 185:3199–208. doi: 10.4049/JIMMUNOL.1000557
188. Sakaguchi S, Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T. Regulatory T cells: how do they suppress immune responses? Int Immunol (2009) 21:1105–11. doi: 10.1093/INTIMM/DXP095
189. Taams LS, Smith J, Rustin MH, Salmon M, Poulter LW, Akbar AN. Human anergic/suppressive CD4 + CD25 + T cells: a highly differentiated and apoptosis-prone population. Eur J Immunol (2001) 31(4):1122–31. doi: 10.1002/1521-4141(200104)31:4<1122::aid-immu1122>3.0.co;2-p
190. Baecher-Allan C, Brown JA, Freeman GJ, Hafler DA. CD4+CD25high regulatory cells in human peripheral blood. J Immunol (2001) 167:1245–53. doi: 10.4049/JIMMUNOL.167.3.1245
191. Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, et al. Epigenetic control of the foxp3 locus in regulatory T cells. PloS Biol (2007) 5(2):e38. doi: 10.1371/JOURNAL.PBIO.0050038
Keywords: Treg, IPEX syndrome, immune tolerance, primary immunodeficiency, primary immune regulatory disorders, cell-based therapies, immune dysregulation, Helios
Citation: Kennedy-Batalla R, Acevedo D, Luo Y, Esteve-Solé A, Vlagea A, Correa-Rocha R, Seoane-Reula ME and Alsina L (2024) Treg in inborn errors of immunity: gaps, knowns and future perspectives. Front. Immunol. 14:1278759. doi: 10.3389/fimmu.2023.1278759
Received: 16 August 2023; Accepted: 13 December 2023;
Published: 08 January 2024.
Edited by:
Veronica De Rosa, National Research Council (CNR), ItalyReviewed by:
Peter Hsu, Children’s Hospital at Westmead, AustraliaEkaterini Simoes Goudouris, Federal University of Rio de Janeiro, Brazil
Copyright © 2024 Kennedy-Batalla, Acevedo, Luo, Esteve-Solé, Vlagea, Correa-Rocha, Seoane-Reula and Alsina. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laia Alsina, bGFpYS5hbHNpbmFAc2pkLmVz; Ma Elena Seoane-Reula, ZWxlbmFzZW9hbmU3MUBnbWFpbC5jb20=
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work and share last authorship