 Ioana Sonya Ciulean1*
Ioana Sonya Ciulean1* Joe Fischer1
Joe Fischer1 Andrea Quaiser1
Andrea Quaiser1 Christoph Bach1
Christoph Bach1 Hinrich Abken2
Hinrich Abken2 Uta Sandy Tretbar1
Uta Sandy Tretbar1 Stephan Fricke1
Stephan Fricke1 Ulrike Koehl1,3
Ulrike Koehl1,3 Dominik Schmiedel1,3†
Dominik Schmiedel1,3† Thomas Grunwald1*†
Thomas Grunwald1*†- 1Fraunhofer Institute for Cell Therapy and Immunology (IZI), Leipzig, Germany
- 2Leibniz Institute for Immunotherapy, Division of Genetic Immunotherapy, Regensburg, Germany
- 3Institute for Clinical Immunology, University of Leipzig, Leipzig, Germany
Head and neck squamous cell carcinoma (HNSCC) is a major challenge for current therapies. CAR-T cells have shown promising results in blood cancers, however, their effectiveness against solid tumors remains a hurdle. Recently, CD44v6-directed CAR-T cells demonstrated efficacy in controlling tumor growth in multiple myeloma and solid tumors such as HNSCC, lung and ovarian adenocarcinomas. Apart from CAR-T cells, CAR-NK cells offer a safe and allogenic alternative to autologous CAR-T cell therapy. In this paper, we investigated the capacity of CAR-NK cells redirected against CD44v6 to execute cytotoxicity against HNSCC. Anti-CD44v6 CAR-NK cells were generated from healthy donor peripheral blood-derived NK cells using gamma retroviral vectors (gRVs). The NK cell transduction was optimized by exploring virus envelope proteins derived from the baboon endogenous virus envelope (BaEV), feline leukemia virus (FeLV, termed RD114-TR) and gibbon ape leukemia virus (GaLV), respectively. BaEV pseudotyped gRVs induced the highest transduction rate compared to RD114-TR and GaLV envelopes as measured by EGFP and surface CAR expression of transduced NK cells. CAR-NK cells showed a two- to threefold increase in killing efficacy against various HNSCC cell lines compared to unmodified, cytokine-expanded primary NK cells. Anti-CD44v6 CAR-NK cells were effective in eliminating tumor cell lines with high and low CD44v6 expression levels. Overall, the improved cytotoxicity of CAR-NK cells holds promise for a therapeutic option for the treatment of HNSCC. However, further preclinical trials are necessary to test in vivo efficacy and safety, as well to optimize the treatment regimen of anti-CD44v6 CAR-NK cells against solid tumors.
Introduction
Cancer immunotherapy has witnessed significant success with the emergence of chimeric antigen receptor T-cell (CAR-T) therapy, particularly in the treatment of blood cancers. FDA-approved anti-CD19 CAR-T therapies such as Kymriah®, Yescarta®, and Tecartus® (1–3) demonstrated long-lasting remission effect in patients with various B-cell malignancies (4), paving the way towards CAR therapy development for other malignancies. However, the translation of CAR-T success to solid tumors is still challenging. Though currently clinically manageable (5), life-threatening side effects associated with CAR-T therapies such as cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS) (5–7) led the search for alternative immune cell types. In this context, the use of NK cells for developing CAR-based cellular therapy options offers several advantages over T cells. First, CAR-NK cells are associated with a reduced risk of inducing graft-versus-host disease (GvHD) due to their non-MHC-restricted mechanism of target recognition (8, 9). Therefore, they can be administered as an allogeneic, off-the-shelf cell therapy product generated from healthy donor cells instead of autologous patient-individual immune cells. NK cells can be obtained from various sources including peripheral blood (10), umbilical cord blood (11, 12), induced pluripotent stem cells (13) or cell lines like NK-92 (14). Regarding their off-the-shelf use, it was previously reported that one unit of cord blood could yield approximately 100 doses of CAR-NK cells (11), potentially undercutting the $375,000 per infusion cost of CAR-T cells (15). Moreover, CAR-NK cell products are considered safer for administration because they possess a distinct cytokine profile from the proinflammatory T cell cytokines associated with CRS occurrence (16). Their safe administration was demonstrated in a clinical trial (clinical trials.gov NCT03056339) (17).
Head and neck squamous cell carcinomas (HNSCC) represent a major challenge in cancer treatment. According to GLOBCAN estimates, in 2020 a number of 377.713 new cases and 177.757 new deaths worldwide were attributed to cancers of the lip and oral cavity (18). Current treatment plans for HNSCC involve a multimodal approach, with surgery or radiation therapy being a curative option for early-stage disease (19). However, locally advanced cases carry a higher risk of recurrence and distant metastases, resulting in worse prognosis (20). The introduction of targeted immunotherapeutic options, such as cetuximab (21), an anti-EGFR monoclonal antibody, and later the inclusion of immune checkpoint inhibitors targeting PD-1, like nivolumab and pembrolizumab (22, 23), broadened treatment possibilities for patients with locally advanced HNSCC. However, a need for effective immune cell therapies remains. To date, only a few surface markers have been studied as possible targets for CAR therapy in HNSCC. These include EGFR (24, 25), HER2 (26), PD-L1 (27), MUC1 (28) and CD44v6 (29). CD44v6, an isoform of the CD44 glycoprotein, emerges as a promising target for cellular therapy in HNSCC. CD44v6 is expressed in various malignancies like multiple myeloma (30), acute myeloid leukemia (31), breast (32), gastric (33) and colorectal cancers (34), as well as head and neck cancers (35). CD44v6 is also expressed by 1.5-3.6% healthy immune cells of the myeloid lineage (36), while oral mucosa keratinocytes were found to express the CD44v1 and v2 isoforms of CD44 (37). Preclinical studies demonstrated the potential of CAR-T cell therapy targeting CD44v6 in HNSCC (29), lung and ovary adenocarcinoma (38), as well as myeloid leukemia and multiple myeloma (39). Clinical trials utilizing CD44v6 CAR-T cells have been launched for both blood cancers (clinical trials.gov NCT04097301) and solid tumors (clinical trials.gov NCT04427449), highlighting the therapeutic potential of this target. Recent research has shown the efficacy of CD44v6 CAR-NK cells against triple-negative breast cancer in vitro (40), while in a phase 2 clinical trial, anti-PD-L1 CAR-NK cells in combination with the anti-PD-1-antibody pembrolizumab and the IL-15 superagonist complex N-803 are used to treat recurrent or metastatic gastric or head and neck cancers (clinical trials.gov NCT04847466).
In this in vitro proof of principle study, we explore the use of gamma retroviral vector-generated CAR-NK cells against CD44v6 to augment the innate cytotoxicity of NK cells against HNSCC cell lines. In a side-by-side comparison, we tested the efficacy of three viral envelopes in transducing primary NK cells and checked for the impact on NK cell surface markers.
Materials and methods
Ethics statement
This work was performed with NK cells from anonymous healthy blood donors and was approved by the State Chamber of Physicians of Saxony, Germany, under the ethical vote number EK-BR-79/21-1.
Cell lines and primary cells culture
HEK293-T cells (ACC 635) obtained from DSMZ (Braunschweig, Germany) were grown in DMEM GlutaMAX (Gibco, New York, United States) supplemented with 10% fetal calf serum (FCS; Gibco, New York, United States) and 1% (v/v) penicillin-streptomycin (pen./strep., Gibco, New York, United States). Prof. Reidar Grénman, University of Turku, Finland kindly provided the human head and neck cancer cell lines UT-SCC-14 (tongue squamous cell carcinoma) and UT-SCC-42B (laryngeal squamous cell carcinoma). UT-SCC-14 and UT-SCC-42B cells were grown in DMEM high glucose medium (Sigma-Aldrich, St. Louise, United States) supplemented with 10% (v/v) FCS and 1% (v/v) GlutaMax (Gibco, New York, United States). The tongue squamous cell carcinoma derived SCC-25 cell line (ACC 617) obtained from DSMZ (Braunschweig, Germany) was kept in DMEM/F12 high glucose medium (Gibco, New York, United States) supplemented with 10% (v/v) and 1% (v/v) GlutaMax. The glioblastoma cell line LN-299 purchased from ATCC (CRL-2611) was cultivated in DMEM high glucose medium (Sigma-Aldrich, St. Louise, United States) supplemented with 10% (v/v) FCS. All adherent cancer cell lines were harvested after a 20-30 min incubation with accutase cell detachment solution (Corning, New York, United States). NK-92, a malignant non-Hodgkin’s lymphoma cell line (ATCC CRL-2407) was kept in RPMI 1640 HEPES modified medium (Sigma-Aldrich, St. Louise, United States) supplemented with 20% (v/v) FCS, 1% (v/v) GlutaMax, 1% (v/v) MEM non-essential amino-acids (Sigma-Aldrich, St. Louise, United States) and 100 IU/ml recombinant rhIL-2 Proleukin®S (Clinigen Healthcare B.V, Schiphol, Netherlands). All cell lines were cultivated at 37°C, 5% (v/v) CO2, and 95% (v/v) humidity and were split every 2 to 3 days. Regular testing for Mycoplasma was performed for all cell lines using a LookOut® Mycoplasma PCR Detection Kit (Sigma-Aldrich, St. Louise, United States).
Primary NK cells were purified from healthy donor buffy coats by negative selection using the RosetteSep™ human NK cell enrichment cocktail according to the manufacturer’s instructions (STEMCELL Technologies, Vancouver, Canada). After NK cell isolation, 1 × 106 cells per well were cultivated in a 24-well plate, in 1 ml of NK MACS® medium (Miltenyi Biotec, Bergisch-Gladbach, Germany) supplemented with 5% (v/v) human serum (Sigma-Aldrich, St. Louise, United States), 500 IU/ml of recombinant rhIL-2 Proleukin®S and 140 IU/ml of IL-15 (Miltenyi Biotec, Bergisch-Gladbach, Germany). NK cell purity was determined by flow cytometry post-isolation. All NK cell-isolations had a purity higher than 72% on the day of isolation and higher than 95% on the day of transduction.
Transgene constructs
The anti-CD44v6 CAR construct was cloned in the pBullet (41) gamma retroviral vector by replacing the BW431/26-scFv-Fc-CD28-CD3ζ CAR (42). An enhanced GFP (EGFP) reporter gene flanked by the PacI (5´) and XhoI (3´) restriction sites (Eurofins, Luxembourg, Luxembourg) was first cloned into the original vector construct, downstream of the CAR construct. For the construction of the anti-CD44v6 specific CAR, the anti-CD44v6 single chain variable fragment (scFv) sequence was synthetized in a lentivirus expression plasmid from Invitrogen GeneArt (Thermo Fisher, Waltham, MA, USA). In brief, the anti-CD44v6 scFv with a VL-VH orientation consists of the following encoding sequences: CD8a signaling peptide, sequences of the VL and VH region of the humanized BIWA8 antibody (43) connected by a (G4S)3 linker. The scFv was amplified from the lentivirus expression plasmid via PCR using the following primers (Biomers.net GmbH, Ulm, Germany): BIWA_for (AGCCACCATGGCCTTACCAGTGAC) and BIWA_VL-VH_rev (GCATGGATCCAGGCTGCTCACGGTCACCAGG). PCR products were purified using a Macherey-Nagel™ NucleoSpin™ Gel and PCR Clean-up Kit according to the manufacturer’s instructions (Machery-Nagel GmbH & Co.KG, Düren, Germany). Ligation of the scFv into the pBullet gRV CAR backbone was performed with 50 ng vector and 16 ng of scFv insert at a molar insert to vector ratio of 3:1, using T4 DNA Ligase (New England Biolabs, Ipswich, UK) according to the manufacturer’s instructions and incubated overnight at 16°C. For the bacterial transformation step, 4 to 8 µl of the ligation mixture was added to 50 µl of chemical competent NEB stable E. coli (C3040, New England Biolabs, Ipswich, UK) and incubated on ice for 30 min. This was followed by a 30 s heat shock at 42°C and a cool down step of 2 min on ice. 950 µl of NEB® 10-beta/stable Outgrowth (New England Biolabs, Ipswich, UK) medium was added and the bacteria was incubated at 30°C, 400 rpm for 1 hour (h). Next, the transformed bacteria were plated on LB agar plates containing 100 µg/ml ampicillin. Bacterial colonies were grown overnight at 30°C and clones were picked on the next day and expanded in appropriate LB medium. Plasmids were isolated from bacterial liquid cultures using the E.Z.N.A.® DNA Mini Kit according to the manufacturer’s instructions (Omega Bio-tek Inc., Norcross, USA). DNA sequences were checked by Sanger sequencing (Microsynth AG, Balgach, Switzerland), the clone with the correct sequence was further expanded and plasmid-working stocks were generated using the EndoFree Plasmid Maxi Kit (10) according to the manufacturer’s instructions (Qiagen GmbH, Venlo, Netherlands).
Lentivirus vectors encoding firefly luciferase (F.luc) and puromycin (Puro) were used to generate tumor target cell lines that have a stable expression of the luciferase enzyme. The used pGreenFire_Puro_Luciferase plasmid was generated by exchanging the GFP reporter in the pGreenFire1-CMV Plasmid (TR011PA-1-SBI System Biosciences) backbone with a puromycin resistance gene.
Gamma retroviral vector generation, concentration and titration
Transient transfection of HEK293-T cells using a calcium phosphate transfection kit (Thermo Fisher, Waltham, MA, USA) was used to generate gRV vector particles. The viral packaging system used included a plasmid expressing the Gag and Pol retroviral core proteins (pHIT60) (41) and one of the three envelope plasmids: envelope derived from the baboon endogenous virus envelope (BaEVRless, BaEV for short) (44), feline leukemia virus (FeLV, termed RD114-TR, RD114 for short) (45) and gibbon ape leukemia virus (GaLV) (41), respectively. One day before transfection, 7 × 106 HEK293-T cells were seeded per flask into 10 x T175 flasks. In total, 35 μg plasmid DNA (plasmids expressing transfer gene, Gag/Pol and envelope in a ratio of 5:3:2) was mixed with 1.26 ml 2M calcium chloride and topped with sterile ultra-filtrated water up to 10.5 ml. The 2X HEPES buffered saline was added dropwise to the DNA mix for a final 1:1 dilution and incubated for 10 min at room temperature. The culture medium was exchanged to 35 ml DMEM GlutaMAX supplemented with 1.5% (v/v) FCS and 1% (v/v) pen./strep. solution before the transfection solution was added dropwise over the HEK293-T cells. Eight hours later, the medium was replaced with fresh DMEM GlutaMAX supplemented with 10% (v/v) FCS and 10.000 U/ml penicillin; 10 mg/ml streptomycin; the flasks were further incubated for approximately 72 h. Three days after transfection, the cell culture supernatant was collected, centrifuged down for 5 min, 800 x g to get rid of cell debris and then filtered through a 0.45 μm filter. Part of the viral vector suspension was kept and frozen, while the rest of the suspension was mixed at a 1:3 ratio with the Retro-X concentrator solution (Takara Bio Inc., Kusatsu, Japan) and incubated at 4°C overnight. The following day, the vector suspension was centrifuged for 45 min, at 4°C and 1,500 x g; the supernatant was discarded, and pellets were resuspended in 2-3 ml DMEM GlutaMAX medium supplemented with 10% (v/v) FCS and 1% (v/v) pen./strep. for a 75-90 x concentration effect. Concentrated viral vector stocks were aliquoted and frozen at -80°C. To determine vector titers, frozen vector aliquots were titrated on HEK293-T cells using six serial dilutions of the vector particles. HEK293-T transduction efficacy was checked by flow cytometry after 4 days and was quantified as percentage of the EGFP+ cells.
Anti-CD44v6 CAR-NK generation
Primary NK (pNK) cell transduction was done in 24-well flat-bottom plates for suspension cells using 0.25 x 106 cells/well. Unconcentrated and concentrated viral vectors from the same stocks were used to transduce pNKs on day 6 post isolation and IL-2/IL-15 cytokine-expansion. From the viral vector stocks, unconcentrated vectors (UNC) and three MOIs of 0.5; 1 and 5 concentrated viral vectors were used. Briefly, DMEM GlutaMAX with 10% (v/v) FCS and 1% (v/v) pen./strep. was added in the 24 well plate and topped with virus up to 500 µl per well. Vectofusin-1 (Miltenyi Biotec, Bergisch-Gladbach, Germany) was added at a final concentration of 10 μg/ml, the solution was mixed and incubated for 5-10 min at room temperature. pNKs were finally added to the wells, mixed and centrifuged down at 400 × g, 32°C for 90 min to increase transduction efficacy. Afterwards, the plates were set in the incubator for a 30 min rest. Finally, 500 µl of fresh NK MACS with 5% (v/v) human serum, IL-2 and IL-15 were added and the NK cells were gently resuspended by pipetting. pNK cells were maintained at a concentration of approximately 1 x 106 cells/ml; medium was refreshed every 3-4 days and cells were moved to 12-well flat-bottom plates on day 7 post transduction. Transduction rates were assessed on days 3, 7, 10 and 14 post-transduction using flow cytometry to detect EGFP signal and surface CAR expression through anti-IgG1 staining (Miltenyi Biotec, Bergisch-Gladbach, Germany). Transduction efficacy was considered to be the sum of EGFP+; EGFP+CAR+ and CAR+ cells. NK cells that underwent transduction without viral vectors served as process control (PC pNKs) and cytokine-expanded NK cells (EXP pNKs) were included as negative controls.
Lentiviral vector generation tumor target cell transduction with firefly luciferase
The lentiviral vectors encoding firefly luciferase (F.luc) were produced by transfection of HEK293-T cells using the calcium phosphate transfection kit (Thermo Fisher, Waltham, MA, USA). The packaging system assembled VSVG-pseudotyped lentiviral vectors by using the pMDLg/pRRE (Addgene #12251) plasmid encoding HIV-1 Gag and Pol and the pRSV-Rev (Addgene #12253) plasmid. Briefly, 3 x 106 HEK293-T cells were seeded one day in advance in a T75 flask. In total, 30 μg total plasmid DNA (plasmids expressing transfer gene, VSV-G, Gag/Pol and Rev in a ratio of 16.2 µg: 4.5 µg: 3 µg: 6.3 µg) was mixed with 54 µl 2M calcium chloride and topped with cell culture water up to 500 µl, and 2X HEPES buffered saline was added dropwise to a final 1:1 dilution. Cell culture medium was changed from 15 ml DMEM GlutaMAX supplemented with 10% (v/v) FCS and 1% (v/v) pen./strep. to 15 ml DMEM GlutaMAX supplemented with 1.5% (v/v) FCS and 1% (v/v) pen./strep. before the transfection solution was added dropwise to the medium. Eight hours later, the medium was replaced with fresh DMEM GlutaMAX supplemented with 10% (v/v) FCS and 1% (v/v) pen./strep and the flasks were further incubated until the next day. After 24 h, cell culture medium was refreshed and the medium containing lentiviral vectors was filtered with a 0.45 µm filter and added over pre-seeded tumor target cells (UT-SCC-14, UT-SCC-42B, SCC-25 and LN-299). This procedure was repeated twice with an extended incubation time of 72 h after the third addition of lentiviral vectors. Subsequently, the transduced cells were expanded and selected with ascending puromycin (Gibco, New York, United States) concentration (1-6 µg/ml). Luciferase expression was confirmed using the Bright-Glo™ Luciferase Assay System (Promega GmbH, Walldorf, Germany) in a 1:1 dilution with 2.5x104 cells/well. After a 2 min incubation time, luminescence was detected using a Centro XS3 LB960 Luminometer (Berthold Technologies GmbH & Co.KG, Bad Wildbach, Germany).
Cytotoxicity luminescence assay for testing NK-effector cell functionality
To determine specific killing of the CAR-NK cells, a kinetic cytotoxicity assay was performed based on the activity of living, luciferase expressing tumor cells. Luciferase engineered tumor cells (UT-SCC-14-F.luc; UT-SCC-42B-F.luc, SCC-25-F.luc and LN-299-F.luc cells) were seeded at a concentration of 1 x 104 cells/well into a 96-high binding LUMITRAC™ well plate (Greiner Bio-One GmbH, Frickenhausen, Germany) and incubated for two hours before co-culture started. Effector cells (expanded NK, CD44v6/CD19 CAR-NK, NK92 and CD44v6 CAR-NK92 cells) were added to the plates in cytokine free medium at indicated effector to target ratios (2.5:1; 1:1and 0.1:1) and incubated up to 24 h at 37°C. For the readout, D-Luciferin (Perkin Elmer, Waltham, MA) was added to the wells to a final concentration of 0.15 mg/ml and luminescence was measured using a Centro XS3 LB960 Luminometer (Berthold Technologies GmbH & Co.KG, Bad Wildbach, Germany) after 4, 6, 8 and 24 h of co-culture. Tumor cell viability was calculated as previously described (46): ((Emission − Background)/(Tumor cell alone − Background)) × 100%. The cytotoxic effect was determined by subtracting tumor cell viability: cytotoxicity (%) = 100% - tumor cell viability (%). Each condition was tested in triplicates and the following controls were used: tumor cells alone, medium only (background) and maximum killing (1% Triton X-100).
Flow cytometry
CAR expression on the NK cell surface, CD44v6 and PD-L1 expression on target cells were assessed on a BD FACSCanto II, while NK cell purity and phenotyping were analyzed on a MACSQuant X. Acquired data was evaluated with the FlowJo v10.7 software (Becton Dickinson and Company, Franklin Lakes, NJ, USA). Staining was carried out according to the manufacturer’s instructions using the following antibodies: CD44v6-PE (Becton, Dickson and Company, New Jersey, USA); PD-L1-PE-Vio770; IgG1-APC; CD8-FITC; CD4-PE; CD19-PE-Vio770; CD16-PE-Vio 615; CD14-APC, CD3-VioBlue; CD45-VioGreen; NKG2A-PE; NKp44-Vio Bright B515; NKG2D-APC; NKp46-PE-Vio 615; CD16-VioGreen; CD56-APC-Vio 770; PD-1-PE-Vio770; CD69-PerCP-Vio 700; DNAM-1-Vio Bright R720; TIGIT-PE. Unless otherwise specified, all antibodies were supplied from Miltenyi Biotec, Bergisch Gladbach, Germany.
Statistical analysis
Statistical analysis was performed with the GraphPad Prism 6 software (GraphPad, San Diego, CA, United States). Statistical significance was determined by applying the two-way ANOVA and Turkey’s multiple comparison tests; p values < 0.05 were considered significant.
Results
Engineering of anti-CD44v6 CAR-NK cells
A second-generation CAR targeting CD44v6 was constructed using the coding sequence of the recognition domain from the humanized anti-CD44v6 antibody bivatuzumab, clone BIWA8 (43). The single chain variable fragment (scFv) targeting CD44v6 is connected through an IgG1 hinge region to the CD8a transmembrane, CD28 costimulatory and CD3ζ signaling domains. The enhanced GFP (EGFP) reporter gene was co-expressed (Figure 1A). The entire CAR construct is encoded by the previously described gamma retroviral vector pBullet (41). Different versions of the CAR construct were tested (Supplementary Figure 1A), but the decision to proceed with the CAR-P2A-EGFP construct was made to facilitate monitoring both transfection and transduction efficiency through the EGFP transgene expression (Supplementary Figure 1B). Transduction rates in primary NK were quantified as the total amount of EGFP+, EGFP+CAR+ and CAR+ cells as exemplified in Figure 1B. From the tested pseudotyped vectors, the BaEV pseudotyped gamma retroviral vectors (BaEV-gRV) induced the highest transduction rates in primary NK cells compared to the RD114 (RD114-gRV) and GaLV- pseudotyped gRV (GALV-gRV) (Figure 1C). By day 14 post transduction, BaEV-gRV induced mean transduction rates of 21% at an MOI of 1 for the CD44v6 CAR construct and 17.8% for a CD19 CAR construct. At the same MOI, a mean 7.6% transduction rate was obtained with RD114-gRV, while the GALV-gRV transduction was below the detection limit of 1% (Figure 1C). Using different MOIs of the viral vector stocks yield a titration effect in pNKs, for which the highest mean levels of CAR expression of 39.3% corresponded with the use of concentrated BaEV-gRV at an MOI of 5 (Figure 1C). Concentrated viral vector stocks were generated as outlined in the methods section, with additional information on vector titers available in Supplementary Table 1.
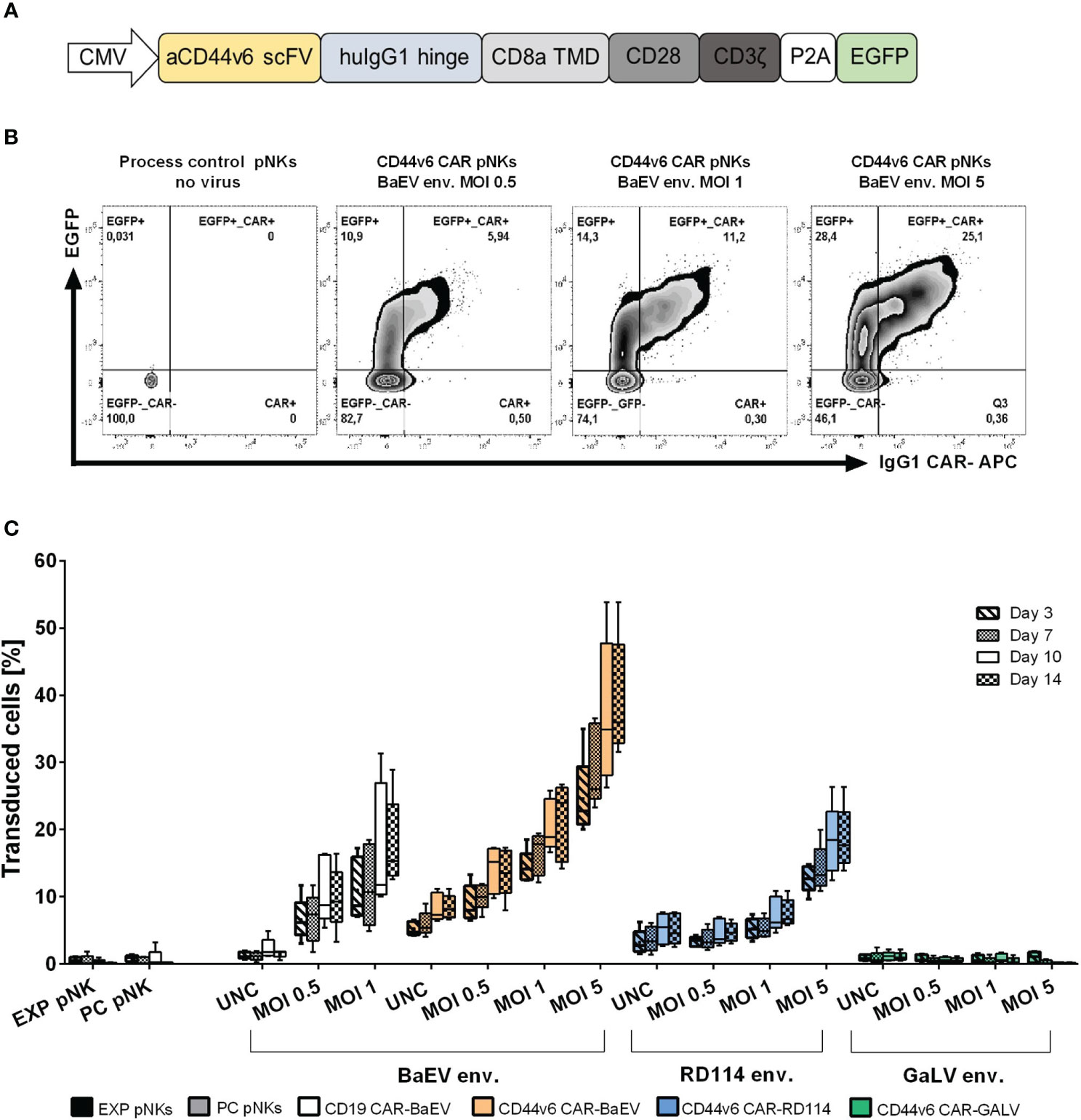
Figure 1 Optimization of the anti-CD44v6 retroviral vector CAR delivery and expression in primary NK cells. (A) Schematic representation of the anti-CD44v6 CAR construct used to generate CAR-NK cells. The anti-CD44v6 scFv (aCD44v6 scFv) derives from the BIWA8 clone of the anti-CD44v6 humanized antibody bivatuzumab. A human IgG1 hinge domain (huIgG1 hinge) links the scFv to a CD8 transmembrane domain (CD8a TMD), CD28 costimulatory domain (CD28) and CD3ζ signaling domain (CD3ζ). The genes for the enhanced GFP (EGFP) reporter and the CAR are connected through a P2A self-cleaving peptide sequence. The constitutive expression of the CAR gene in primary NK is controlled by the cytomegalovirus (CMV) promoter. The expression cassette is cloned into a gamma retroviral vector (gRV). (B) Example of gating strategy and comparison between different MOIs of the gamma retroviral vectors produced in HEK293-T cells and used to transduce primary NK cells. The two-parameter dot plots represent EGFP+, EGFP+CAR+ and CAR+ expression in transduced primary NKs with gRV pseudotyped with BaEV on day 10 post transduction. CAR surface expression was detected by staining with an APC IgG1 monoclonal antibody. (C) NK-cell transduction and percentage of total transduced cells (EGFP+, EGFP+CAR+, CAR+) at days 3, 7, 10 and 14 post transductions with gRV pseudotyped with BaEV, RD114 and GaLV. Unconcentrated viral vectors (UNC) and three MOIs of 0.5, 1 and 5 concentrated viral vectors were used to transduce NK cells. Expanded NK cells (EXP pNKs) and the process control NK cells (PC pNKs) serve as controls. Data of five independent experiments using five different healthy NK cells donors is presented as mean and standard deviation.
Primary NK cell phenotype profile post transduction with gamma retrovirus pseudotyped with BaEV and RD114
In order to see whether transduction with different viral envelopes affects NK cell receptor expression, we checked the level of activation and inhibitory markers on the surface of donor-derived and cytokine-expanded NK cells 10 days post transduction (Figures 2A–H). We had a closer look at the two fractions within the transduced cells subpopulation namely untransduced CD56+ cells (CD56+EGFP-) and transduced CD56+ cell fraction (CD56+EGFP+). Due to the fact that only BaEV-gRV and RD114-gRV induced detectable levels of CD56+EGFP+ cells, surface marker profiles are presented only in these versions of genetically engineered NK cells. While the NKG2D and DNAM-1 activation markers were present on NK cells at similar levels on the isolation day and 10 days post cytokine-stimulation (Figures 2A, E), other activation markers like NKp44, NKp46 and CD69 were upregulated during cytokine-expansion of NK cells (Figures 2B, C, D). The inhibitory marker NKG2A was found at higher levels in NK cells that were cytokine stimulated, with similar expression in all transduced cells (Figure 2F). TIGIT and PD-1 immune checkpoint inhibitor expression was also upregulated on NK cells after cytokine expansion (Figures 2G, H). No difference in the expression of investigated surface markers was detected between the BaEV and RD114 viral envelope proteins. The activation marker NKp46 was upregulated in the CD56+EGFP+ cell fraction as compared to the CD56+EGFP- fraction. In two donors, higher PD-1 expression was found in expanded NK cells, process control NK cells, as well as in the untransduced, CD56+EGFP- cells, compared to CD56+EGFP+ cells (Supplementary Figure 2A).
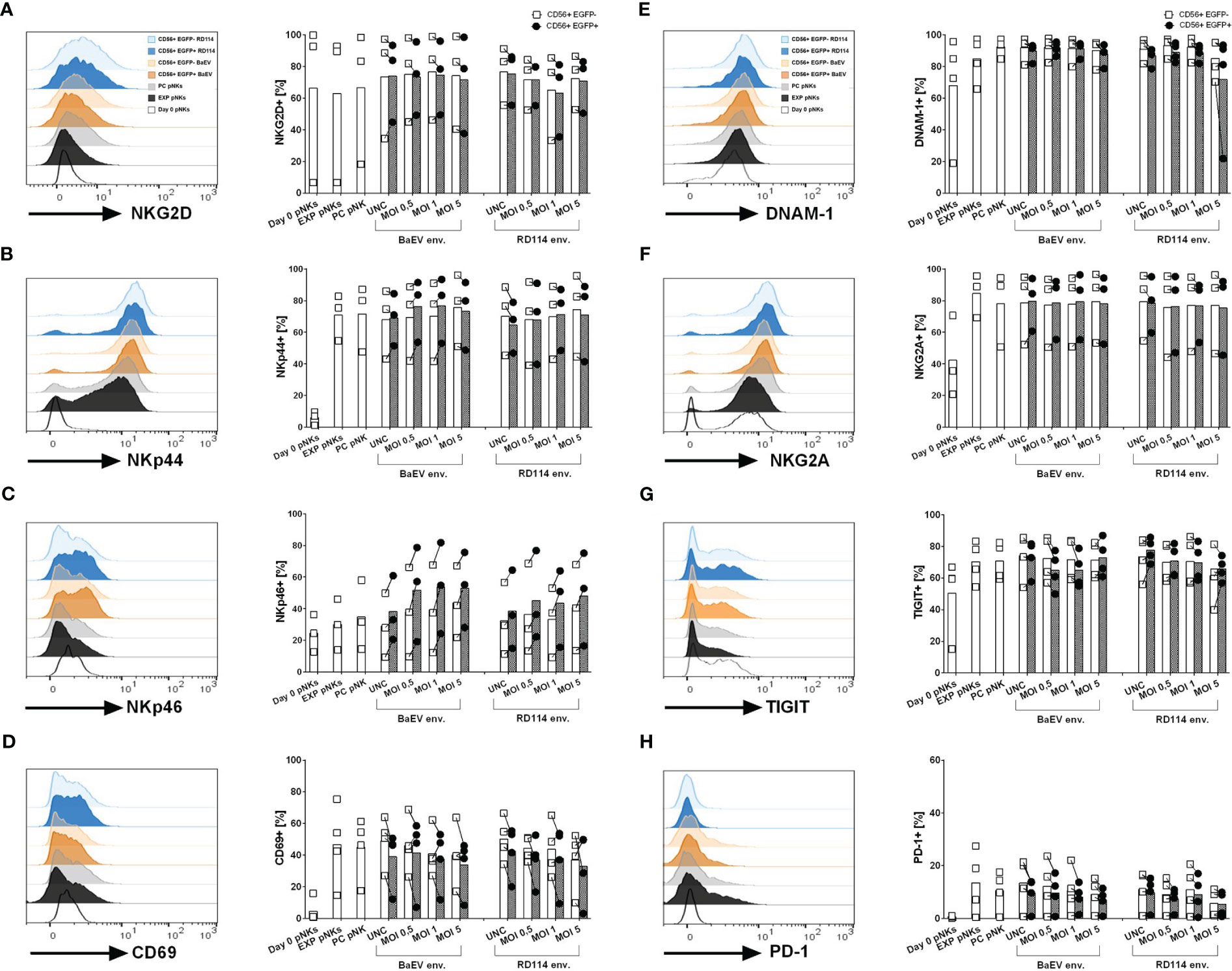
Figure 2 Surface marker expression on healthy donor NK and genetically modified NK cells was analyzed using flow cytometry. The histograms (A–H) illustrate exemplary data showcasing surface marker expression on different cell populations: unstimulated pNK cells on day 0 (Day 0 pNKs – white), expanded NK cells (EXP pNKs - black), no-vector transduced, process control NK cells (PC pNKs - grey) and viral vector transduced NK cells on day 10 post transduction. For the viral vector transduced pNKs, the following populations are highlighted: CD56+EGFP+ subpopulation from pNK cells transduced with BaEV MOI 5 (dark orange), CD56+EGFP- subpopulation from pNK cells transduced with BaEV MOI 5 (light orange), CD56+EGFP+ subpopulation from pNK cells transduced with RD114 MOI 5 (dark blue), and CD56+EGFP- subpopulation from pNK cells transduced with RD114 MOI 5 (light blue). The column dot plots display the mean and individual data of the investigated markers (n=3 NK donors for activation/inhibitory markers, n=4 NK donors for check point inhibitors). Percentages of the transduced pNK cell subpopulation (CD56+EGFP+) and untransduced subpopulation (CD56+EGFP-) expressing the activation markers NKG2D (A), NKp44 (B), NKp46 (C), CD69 (D), inhibitory markers such as DNAM-1 (E), NKG2A (F), as well as the check point inhibitors TIGIT (G) and PD-1 (H).
Anti-CD44v6 CAR-NK cells display redirected killing activity against HNSCC cell lines compared to controls
Functionality of genetically modified NK cells was determined through co-cultures with firefly-luciferase (F.luc) expressing target cells and quantified at different time points by measuring the activity of living target cells that reduced the D-luciferin substrate to oxyluciferin as shown in Figure 3A. Using this setup, killing kinetics was measured in the same well at different time points. For this assay, F.luc positive HNSCC cell lines UT-SCC-14-F.luc, UT-SCC-42B-F.luc and SCC-25-F.luc were analyzed for their CD44v6 surface expression (Figures 3B–D) and NK cells transduced with an MOI of 1 from each of the CAR variants were chosen as effector cells. Transgene expression level was in average between 21% for BaEV-gRV, 7.6% for RD114-gRV and below 1% for GALV-gRV. An anti-CD19 CAR (CD19 CAR-BaEV) as well as expanded NK cells (EXP pNKs) and a no-virus process control pNKs (PC pNKs) were used as controls. All effector cells were tested functionally on day 17 post isolation and cytokine expansion using the luminescence-based cytotoxicity assay. In the case of the UT-SCC-14-F.luc target cells, CD44v6 CAR-NKs transduced with BaEV-gRV induced significantly higher killing than cytokine-expanded NK cells at effector to target ratios of 2.5 to 1 and 1 to 1 even at the early time points of 4, 6 and 8 h of the co-culture (Figure 3B). After 24 h, a significant CAR effect was seen only at the lowest 0.1 to 1 effector to target ratio against the same cell line (Figure 3E). Similar effects were visible in the case of the UT-SCC-42B-F.luc cells, but a significant difference between the CD44v6 CAR-BaEV and the expanded NK cells control was recorded starting with 6 h post co-culture setup (Figures 3C, F). For the SCC-25-F.luc cell line, a higher donor variation in killing efficacy was seen (Figure 3D). Significant difference between the CD44v6 CAR-BaEV and the expanded NK cells control in killing SCC-25-F.luc targets was visible after 6 h of co-culture and remained consistent for 24 h (Figure 3G). Overall, anti-CD44v6 CAR-NK cells outperformed cytokine-expanded primary NKs from the same donor with an approximately two- to threefold increase in killing efficacy. We recorded a different killing kinetics for two donors found to have high PD-1 expression (Supplementary Figure 2A). PD-1/PD-L1 inhibition is an established treatment option for HNSCC patients (47). The tested cell lines were also found to express PD-L1 at different levels (Supplementary Figure 2B). NK cells from donors expressing PD-1 exhibited poorer performance against UT-SCC-42B and SCC-25 HNSCC cell lines during the functionality test (Supplementary Figure 2C). A direct correlation between PD-1 expression and the cytotoxicity of effector NK cells was indicated by the difference between CAR-NK and expanded NK cell cytotoxicity. As PD-1 expression increased, the discrepancy in killing efficacy decreased (Supplementary Figures 2D, E).
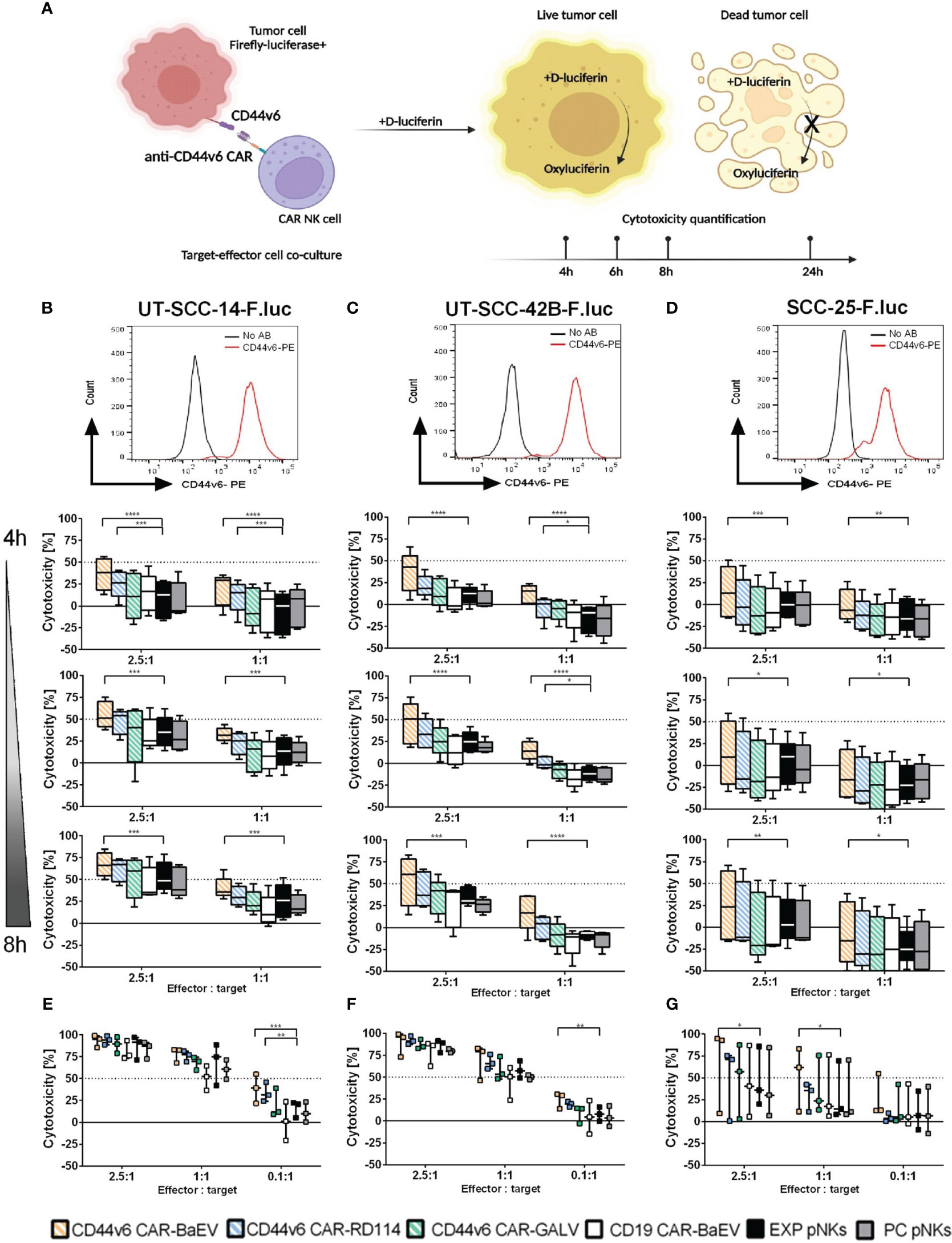
Figure 3 Killing efficacy of anti-CD44v6 CAR-NK cells against different HNSCC cell lines after 4, 6, 8 and 24 h of co-culture. (A) Schematic representation of the kinetic killing assay in which target cells genetically modified to express firefly luciferase (F.luc) are co-cultured with effector NK cells. Killing efficacy is monitored by recording living cells through the use of D-luciferin as substrate. This substrate is reduced to oxyluciferin by living cells. The bioluminescent signal is quantified and normalized to indicate cell killing. (B–D) CD44v6 positive UT-SCC-14-F.luc; UT-SCC-42B-F.luc and SCC-25-F.luc cells are set in co-culture with EXP pNKs; PC pNKs and CD44v6 CAR-NKs at 2.5:1 and 1:1 effector to target ratios. Killing efficacy is quantified after 4, 6 and 8 h of co-culture (n=5 donors). (E–G) Killing efficacy against the same target cell lines was checked after 24 h using 2.5:1, 1:1 and 0.1:1 effector to target ratios (n=3 donors). Data is presented as mean and standard deviation of 5 or 3 independent experiments. Descriptive statistics were calculated using two-way ANOVA and Turkey’s multiple comparison test (* p<0.05; **p<0.01; ***p<0.001; ****p<0.0001).
To determine if the anti-CD44v6 CAR-NK cells also target other types of tumor cells, genetically modified NK cells were set in co-culture with a glioblastoma cell line, LN-299-F.luc. Although LN-299-F.luc cells express CD44v6 to a lesser extent than HNSCC cell lines (Supplementary Figure 3A), specific CAR mediated killing was induced at an effector to target ratio of 1 to 1 after 4 h (Supplementary Figure 3B) which was maintained up to 8 h of co-culture (Supplementary Figures 3C, D).
We also conducted efficacy testing comparing primary NK cell-derived CARs with CAR-NK-92 cells. Anti-CD44v6 CAR-NK-92 cells were generated from the NK-92 cell line using a BaEV-gRV with a co-expressed EGFP reporter. Transduction efficacy in NK-92 cells was still over 93% after long-term culture (Supplementary Figure 4A), while positively transduced primary NK cells at an MOI of 1 had an average CAR expression of 21% (Supplementary Figure 4B). We compared CAR-mediated cytotoxicity using transduced primary NKs and transduced NK-92 cells after 4, 6 and 8 h of co-culture with target HNSCC cell lines (Supplementary Figures 4C, D, E). Killing efficacy of the tested CARs was target cell line depended, with higher killing seen in UT-SCC-14-F.luc and UT-SCC-42B-F.luc targets. For these two target cell lines, significant difference between primary NKs and NK-92 was detected starting at 4 h post co-culture, and remained constant throughout the 8 h of experiment (Supplementary Figures 4C, D). The SCC-25 cell line, was effectively targeted and killed by both CAR-NK cells at similar rates throughout the assay (Supplementary Figure 4E). Despite their high CAR expression, the difference in efficacy of CD44v6 CAR-NK92 compared to the NK-92 control was marginal. An advantage of NK cell redirection with CAR molecules was detected only at later time points, and seemed to be more pronounced against the SCC-25 target cell line (Supplementary Figures 4C, D, E).
Discussion
Effective treatment of HNSCC still remains challenging despite current advancements in immunotherapy. Approved immunotherapies like PD-1 checkpoint inhibitors have limited efficacy, extending overall survival with around 13 months in combination with chemotherapy (48). Despite intense pre-clinical efforts, the success of CAR-T cell therapy has yet to be transferred for solid tumor treatment (49). In this setting, natural killer cells provide a good alternative to the T effector-cell. Through the use of CARs, NK cells are redirected towards specific surface antigens on tumor cells, allowing for antigen-specific targeting and enhanced killing (50). In this study, we genetically modified NK cells to express CARs redirected against the CD44v6 surface protein, a target associated with HNSCC during metastasis and disease progression (51, 52). Various CD44v6 expression levels were previously reported in HNSCC, depending on the choice of antibodies, detection method and scoring systems used. Overall, CD44v6 was found to be higher expressed than other surface markers in HNSCC, underlining its use as a good option for solid tumor targeted therapy (53). Prior research demonstrated the usefulness of CD44v6 as a solid tumor target in both CAR-T and CAR-NK preclinical applications (29, 38, 40), as well as in phase I/II clinical trials (clinical trials.gov NCT04097301 and NCT04427449).
In the presented study, a second-generation anti-CD44v6 CAR was successfully expressed in primary NK cells when using a pseudotyped gRV vector. Safety concerns in regard to insertional oncogenesis limited gRV use in gene therapy applications despite their efficiency in genetic modification of cells (54). However, vector optimization (55) enabled gRV use in approved cell therapies such as the anti-CD19 CAR-T Yescarta® (56). Furthermore, gRV were successfully and safely tested in a clinical trial using genetically modified NK cells (17). Efficient genetic modification of primary NK cells relies on the utilization of an optimal envelope for vector pseudotyping. In this paper we describe the comparative use of three different envelopes for NK cell transduction: BaEV, RD114 and GALV. Alpha retroviral vectors pseudotyped with RD114 were previously reported to induce higher transduction rates in blood-derived primary NK cells compared to gamma retroviral and lentiviral vectors when using CAR constructs (57). Transduction rates of up to 87.4% were also achieved by utilizing RD114-pseudotyped gRV to express a CD19-CAR into cord-blood derived NK cells (11). In contrast to published data, we obtained mean transduction rates of 7.6% in blood derived primary NKs when using RD114-gRV, and rates below detection limit for GALV-gRV modified NK cells. We achieved the highest transduction rates when using BaEV-gRV, which is in line with prior studies by Colamartino et al. (58) and Bari et al. (59). These studies demonstrated a transduction efficacy of over 38% in primary NK cells when using lentiviral vectors pseudotyped with BaEV at MOIs of 5, as opposed to the RD114 and vesicular stomatitis virus G protein (VSV-G) pseudotyped variants.
For the present study, the design of the CAR molecule was based on a T cell specific construct (42), with intracellular signaling domains that, although not NK cell specific, were previously shown to induce efficient and targeted killing for an anti-CD19 CAR-NK cell therapy; and consequently with notable therapeutic outcome in a phase I clinical trial (17, 60).
Examination of NK cell phenotype post transduction revealed an increase in the expression of markers such as NKp44, NKp46, CD69 and NKG2A post cytokine-expansion of NK cells. No correlation with the used viral envelope proteins and NK cell surface marker expression was found. Similar NK cell phenotypes were reported when using either RD114 on alpha retroviral (57), BaEV on lentiviral (58) or RD114 on gamma retroviral vectors (11). We noticed an upregulation of NKp46 expression in the transduced CD56+EGFP+ cell subpopulation, while other studies reported a slightly higher NKp44 positive population in transduced cells as compared to expanded cells (11). The differences in receptor expression observed in expanded and transduced NK cells could be attributed to the source of NK cells (peripheral blood versus cord blood) or to the expansion methods used (cytokine-expansion versus feeder-cell expansion).
The efficacy of NK cells in HNSCC was and is being investigated in various clinical trials, that include the use of off-the-shelf NK products combined with monoclonal antibodies (clinical trials.gov NCT05674526), autologous and ex vivo expanded NK cells (clinical trials.gov NCT00717184) with or without bispecific antibodies (clinical trials.gov NCT05099549) as well as genetically modified NK cells expressing anti-PD-L1 (clinical trials.gov NCT04847466) or anti-NKG2D-ligand CARs (clinical trials.gov NCT03415100). In our study, we demonstrate two- to threefold enhanced cytotoxicity against HNSCC in vitro when using CD44v6-targeted CAR-NK cells, as compared to expanded non-engineered NK cells. Similar results were obtained with anti-CD44v6 CAR-NK cells directed against triple negative breast cancer (40) and anti-CD44v6 CAR-T cells targeting HNSCC (29), or lung and ovarian carcinomas (38). Using anti-CD44v6 CAR-NK cells, we also saw effective killing against the glioblastoma cell line LN-299 expressing low levels of CD44v6. Currently, an anti-HER2-CAR-NK-92 cell-based therapy is under investigation in a phase I/II clinical trial (clinical trials.gov NCT03383978) for glioblastoma. However, additional CAR targets are likely beneficial for the treatment of such a challenging tumor. The CD44v6 splice variant is expressed also by brain tumor stem-like cells (BTSC), implying a promising target for redirected therapy of brain tumors (61). These findings highlight CD44v6 as a promising target for CAR therapy across multiple cancer types.
Inter-donor variability in primary NK cells can significantly influence NK cell phenotype, inherent transduction efficacy and overall function (62). Prior studies revealed the occurrence of functionally different capabilities in eliminating tumor cells between different NK cell subtypes and individuals (63, 64). In accordance to these findings, we have seen donor variability in transduction efficacy and basal functionality of expanded NK cells, as well as donor-to-donor variation in NK cell phenotypes post cytokine-expansion. PD-1 expression on primary NK cells was reported in different types of cancer (65–67) and linked to poor anti-tumor activity (68). We found PD-1 expression in two of our tested donors in cytokine-expanded NK cells and the CD56+EGFP- subpopulation of transduced cells. Both the use of IL-2 and IL-15 as well as a combination of IL-12, IL-15, and IL-18 may induce PD-1 expression on expanding NK cells (67, 69). We noticed that PD-1 expression in the CD56+EGFP- subpopulation of CAR-NK cells can affect their overall cytotoxicity, suggesting that the presence of this immune check point could be a limiting factor for CAR-therapy (as shown in Supplementary Figure 2). Nevertheless, further PD-1 blocking experiments are required to determine the validity of the effect of PD-1 presence on NK cells. In support of this, recent preclinical research has highlighted the advantages of incorporating competitive PD-1 blocking by co-expression alongside the CAR construct (40). Moreover, a phase II clinical trial evaluating anti-PD-L1 CAR-NK cells together with an immune check point inhibitor for the treatment of advanced HNSCC is currently recruiting (clinical trials.gov NCT04847466).
In terms of effector cells, NK-92 cells were reported to have higher cytotoxicity compared to donor-derived NK cells due to increased granzyme content and fewer inhibitory killer Ig-like receptors (KIR) receptors (70). In contrast, we recorded a superior cytotoxic effect of the expanded primary NK and anti-CD44v6 CAR-NK compared to the NK-92 or anti-CD44v6 CAR-NK-92 cell lines, despite a stable, over 95% CAR expression in the last-mentioned effector cell type. Furthermore, there seemed to be a marginal effect of the CAR presence on NK-92 cells. These observations may be explained by the different expansion and activation methods applied to the two cell types. The IL-2/IL-15 cytokine combination used for primary NK cell culture might have induced stronger signaling in these cells through the IL-2/IL-15 receptor complex, potentially enhancing their killing efficacy compared to the IL-2 stimulated NK-92 cells (71). Furthermore, there seemed to be a tumor cell line susceptibility to effector cell killing, with UT-SCC-14-F.luc and UT-SCC-42B-F.luc cell lines being more efficiently killed by primary NK cells as opposed to NK-92 cells, likely as a result of a more favorable match between primary NK cell receptors and tumor target ligands (72).
While our findings show that anti-CD44v6 CAR-NK cell therapy has specific anti-tumor cell line efficacy, further testing is required to optimize the efficacy of this cell-based therapy against HNSCC. A more comprehensive examination using patient derived tumor material or 3D tumor models would underline the potential of this CAR-NK cell therapy under the appropriate microenvironment. Furthermore, in vivo preclinical testing would aid in understanding CAR-NK cell longevity and homing patterns to various body compartments, as well as determining whether it induces off-tumor effects.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by the State Chamber of Physicians of Saxony, Germany. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin because blood donors are anonymous and volunteers.
Author contributions
ISC: Conceptualization, Formal Analysis, Investigation, Methodology, Visualization, Writing – original draft, Writing – review & editing, Project administration. JF: Investigation, Methodology, Writing – review & editing, Visualization. AQ: Visualization, Writing – original draft, Formal Analysis. CB: Investigation, Methodology, Writing – review & editing. HA: Resources, Writing – review & editing. UST: Methodology, Resources, Writing – review & editing. SF: Resources, Writing – review & editing, Conceptualization. UK: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – review & editing. DS: Conceptualization, Formal Analysis, Methodology, Project administration, Supervision, Visualization, Writing – original draft, Writing – review & editing. TG: Conceptualization, Formal Analysis, Methodology, Resources, Supervision, Visualization, Writing – original draft, Writing – review & editing, Project administration, Funding acquisition.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by a Marie Curie network grant of the European Commission (H2020-MSC-ITN-765104-MATURE-NK) to UK and TG. ISC was an early stage research fellow in the project.
Acknowledgments
We would like to thank Prof. Reidar Grénman, University of Turku, Finland and Prof. Achim Aigner, Rudolf-Boehm-Institute for Pharmacology and Toxicology, Leipzig, Germany for kindly providing the human head and neck cancer cell lines UT-SCC-14 and UT-SCC-42B. Also, many thanks to Katharina Ruppel, Fraunhofer Institute for Cell Therapy and Immunology (IZI) for providing the anti-CD19 CAR plasmid. Figure 3 was generated using BioRender.com.
Conflict of interest
SF receives consultant and/or speaker fees from Novartis Pharma GmbH, Janssen-Cilag GmbH, Vertex Pharmaceuticals Germany GmbH, Kite/Gilead Sciences GmbH, MSGO GmbH, Bristol-Myers Squibb GmbH & Co. KGaA. UK receives consultant and/or speaker fees from AstraZeneca, Affimed, Glycostem, GammaDelta, Zelluna, Miltenyi Biotec and Novartis Pharma GmbH, Bristol-Myers Squibb GmbH & Co. KGaA.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1290488/full#supplementary-material
References
1. Mullard A. FDA approves first CAR T therapy. Nat Rev Drug Discov (2017) 16(10):669. doi: 10.1038/nrd.2017.196
2. Dolgin E. FDA okays second CAR-T for Kite. Nat Biotechnol (2020) 38:9. doi: 10.1038/s41587-020-0676-z
3. European Medicines Agency. First two CAR-T cell medicines recommended for approval in the European Union: Development of Kymriah and Yescarta supported through PRIME (2018). Available at: https://www.ema.europa.eu/en/news/first-two-car-t-cell-medicines-recommended-approval-european-union (Accessed 2023 Jun 26).
4. Cappell KM, Sherry RM, Yang JC, Goff SL, Vanasse DA, McIntyre L, et al. Long-term follow-up of anti-CD19 chimeric antigen receptor T-cell therapy. J Clin Oncol (2020) 38:32. doi: 10.1200/JCO.20.01467
5. Hayden PJ, Roddie C, Bader P, Basak GW, Bonig H, Bonini C, et al. Management of adults and children receiving CAR T-cell therapy: 2021 best practice recommendations of the European Society for Blood and Marrow Transplantation (EBMT) and the Joint Accreditation Committee of ISCT and EBMT (JACIE) and the European Haematology Association (EHA). Ann Oncol (2022) 33:3. doi: 10.1016/j.annonc.2021.12.003
6. Azoulay E, Shimabukuro-Vornhagen A, Darmon M, Bergwelt-Baildon Mv. Critical care management of chimeric antigen receptor T cell-related toxicity. Be aware and prepared. Am J Respir Crit Care Med (2019) 200(1):20–3. doi: 10.1164/rccm.201810-1945ED
7. Sterner RC, Sterner RM. Immune effector cell associated neurotoxicity syndrome in chimeric antigen receptor-T cell therapy. Front Immunol (2022) 13:879608. doi: 10.3389/fimmu.2022.879608
8. Rafei H, Daher M, Rezvani K. Chimeric antigen receptor (CAR) natural killer (NK)-cell therapy: leveraging the power of innate immunity. Br J Haematol (2021) 193(2):216–30. doi: 10.1111/bjh.17186
9. Kärre K, Ljunggren HG, Piontek G, Kiessling R. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature (1986) 319(6055):675–8. doi: 10.1038/319675a0
10. Du Z, Ng YY, Zha S, Wang S. piggyBac system to co-express NKG2D CAR and IL-15 to augment the in vivo persistence and anti-AML activity of human peripheral blood NK cells. Mol Ther Methods Clin Dev (2021) 23:582–96. doi: 10.1016/j.omtm.2021.10.014
11. Liu E, Tong Y, Dotti G, Shaim H, Savoldo B, Mukherjee M, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia (2018) 32(2):520–31. doi: 10.1038/leu.2017.226
12. Daher M, Basar R, Gokdemir E, Baran N, Uprety N, Nunez Cortes AK, et al. Targeting a cytokine checkpoint enhances the fitness of armored cord blood CAR-NK cells. Blood (2021) 137(5):624–36. doi: 10.1182/blood.2020007748
13. Tang SY, Zha S, Du Z, Zeng J, Zhu D, Luo Y, et al. Targeted integration of EpCAM-specific CAR in human induced pluripotent stem cells and their differentiation into NK cells. Stem Cell Res Ther (2021) 12(1):580. doi: 10.1186/s13287-021-02648-4
14. Burger MC, Forster M-T, Romanski A, Straßheimer F, Macas J, Zeiner PS, et al. Intracranial injection of NK cells engineered with a HER2-targeted chimeric antigen receptor in patients with recurrent glioblastoma. Neuro Oncol (2023). doi: 10.1093/neuonc/noad087
15. Choi G, Shin G, Bae S. Price and prejudice? The value of chimeric antigen receptor (CAR) T-cell therapy. Int J Environ Res Public Health (2022) 19:19. doi: 10.3390/ijerph191912366
16. Khawar MB, Sun H. CAR-NK cells: from natural basis to design for kill. Front Immunol (2021) 12:707542. doi: 10.3389/fimmu.2021.707542
17. Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med (2020) 382(6):545–53. doi: 10.1056/NEJMoa1910607
18. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2021) 71(3):209–49. doi: 10.3322/caac.21660
19. Pfister DG, Spencer S, Adelstein D, Adkins D, Anzai Y, Brizel DM, et al. Head and neck cancers, version 2.2020, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw (2020) 18(7):873–98. doi: 10.6004/jnccn.2020.0031
20. Braakhuis BJM, Brakenhoff RH, Leemans CR. Treatment choice for locally advanced head and neck cancers on the basis of risk factors: biological risk factors. Ann Oncol (2012) 23 Suppl 10:x173–7. doi: 10.1093/annonc/mds299
21. Hitt R, Irigoyen A, Cortes-Funes H, Grau JJ, García-Sáenz JA, Cruz-Hernandez JJ. Phase II study of the combination of cetuximab and weekly paclitaxel in the first-line treatment of patients with recurrent and/or metastatic squamous cell carcinoma of head and neck. Ann Oncol (2012) 23(4):1016–22. doi: 10.1093/annonc/mdr367
22. Ferris RL, Blumenschein G, Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab vs investigator's choice in recurrent or metastatic squamous cell carcinoma of the head and neck: 2-year long-term survival update of CheckMate 141 with analyses by tumor PD-L1 expression. Oral Oncol (2018) 81:45–51. doi: 10.1016/j.oraloncology.2018.04.008
23. Uppaluri R, Campbell KM, Egloff AM, Zolkind P, Skidmore ZL, Nussenbaum B, et al. Neoadjuvant and adjuvant pembrolizumab in resectable locally advanced, human papillomavirus-unrelated head and neck cancer: A multicenter, phase II trial. Clin Cancer Res (2020) 26(19):5140–52. doi: 10.1158/1078-0432.CCR-20-1695
24. Haist C, Poschinski Z, Bister A, Hoffmann MJ, Grunewald CM, Hamacher A, et al. Engineering a single-chain variable fragment of cetuximab for CAR T-cell therapy against head and neck squamous cell carcinomas. Oral Oncol (2022) 129:105867. doi: 10.1016/j.oraloncology.2022.105867
25. Papa S, Adami A, Metoudi M, Beatson R, George MS, Achkova D, et al. Intratumoral pan-ErbB targeted CAR-T for head and neck squamous cell carcinoma: interim analysis of the T4 immunotherapy study. J Immunother Cancer (2023) 11:6. doi: 10.1136/jitc-2023-007162
26. Rosewell Shaw A, Porter CE, Watanabe N, Tanoue K, Sikora A, Gottschalk S, et al. Adenovirotherapy delivering cytokine and checkpoint inhibitor augments CAR T cells against metastatic head and neck cancer. Mol Ther (2017) 25(11):2440–51. doi: 10.1016/j.ymthe.2017.09.010
27. Robbins Y, Greene S, Friedman J, Clavijo PE, van Waes C, Fabian KP, et al. Tumor control via targeting PD-L1 with chimeric antigen receptor modified NK cells. Elife (2020) 9. doi: 10.7554/eLife.54854
28. Mei Z, Zhang K, Lam AK-Y, Huang J, Qiu F, Qiao B, et al. MUC1 as a target for CAR-T therapy in head and neck squamous cell carinoma. Cancer Med (2020) 9(2):640–52. doi: 10.1002/cam4.2733
29. Haist C, Schulte E, Bartels N, Bister A, Poschinski Z, Ibach TC, et al. CD44v6-targeted CAR T-cells specifically eliminate CD44 isoform 6 expressing head/neck squamous cell carcinoma cells. Oral Oncol (2021) 116:105259. doi: 10.1016/j.oraloncology.2021.105259
30. Liebisch P, Eppinger S, Schöpflin C, Stehle G, Munzert G, Döhner H, et al. CD44v6, a target for novel antibody treatment approaches, is frequently expressed in multiple myeloma and associated with deletion of chromosome arm 13q. Hematologica (2005) 90(4):489–93.
31. Legras S, Günthert U, Stauder R, Curt F, Oliferenko S, Kluin-Nelemans HC, et al. A strong expression of CD44-6v correlates with shorter survival of patients with acute myeloid leukemia. Blood (1998) 91(9):3401–13. doi: 10.1182/blood.V91.9.3401
32. Qiao G-L, Song L-N, Deng Z-F, Chen Y, Ma L-J. Prognostic value of CD44v6 expression in breast cancer: a meta-analysis. Onco Targets Ther (2018) 11:5451–7. doi: 10.2147/OTT.S156101
33. Almeida GM, Pereira C, Park J-H, Lemos C, Campelos S, Gullo I, et al. CD44v6 high membranous expression is a predictive marker of therapy response in gastric cancer patients. Biomedicines (2021) 9:9. doi: 10.3390/biomedicines9091249
34. Wang JL, Su WY, Lin YW, Xiong H, Chen YX, Xu J. CD44v6 overexpression related to metastasis and poor prognosis of colorectal cancer: A meta-analysis. Onco Targets Ther (2017) 8:8. doi: 10.18632/oncotarget.14163
35. Chen J, Zhou J, Lu J, Xiong H, Shi X Gong L. Significance of CD44 expression in head and neck cancer: a systemic review and meta-analysis. Cancer (2014) 14:15. doi: 10.1186/1471-2407-14-15
36. Neu S, Geiselhart A, Sproll M, Hahn D, Kuc¸ S, Niethammer D, et al. Expression of CD44 isoforms by highly enriched CD34-positive cells in cord blood, bone marrow and leukaphereses. Bone Marrow Transplant (1997) 20:7. doi: 10.1038/sj.bmt.1700940
37. Athanassiou-Papaefthymiou M, Shkeir O, Kim D, Divi V, Matossian M, Owen JH, et al. Evaluation of CD44 variants expression in oral, head and neck squamous cell carcinomas using a triple approach and its clinical significance. Int J Immunopathol Pharmacol (2014) 27:3. doi: 10.1177/039463201402700304
38. Porcellini S, Asperti C, Corna S, Cicoria E, Valtolina V, Stornaiuolo A, et al. CAR T cells redirected to CD44v6 control tumor growth in lung and ovary adenocarcinoma bearing mice. Front Immunol (2020) 11:99. doi: 10.3389/fimmu.2020.00099
39. Casucci M, Di Nicolis Robilant B, Falcone L, Camisa B, Norelli M, Genovese P, et al. CD44v6-targeted T cells mediate potent antitumor effects against acute myeloid leukemia and multiple myeloma. Blood (2013) 122(20):3461–72. doi: 10.1182/blood-2013-04-493361
40. Raftery MJ, Franzén AS, Radecke C, Boulifa A, Schönrich G, Stintzing S, et al. Next generation CD44v6-specific CAR-NK cells effective against triple negative breast cancer. Int J Mol Sci (2023) 24:10. doi: 10.3390/ijms24109038
41. Weijtens ME, Willemsen RA, Hart EH, Bolhuis RL. A retroviral vector system ‘STITCH’ in combination with an optimized single chain antibody chimeric receptor gene structure allows efficient gene transduction and expression in human T lymphocytes. Gene Ther (1998) 5:9. doi: 10.1038/sj.gt.3300696
42. Hombach A, Wieczarkowiecz A, Marquardt T, Heuser C, Usai L, Pohl C, et al. Tumor-specific T cell activation by recombinant immunoreceptors: CD3 zeta signaling and CD28 costimulation are simultaneously required for efficient IL-2 secretion and can be integrated into one combined CD28/CD3 zeta signaling receptor molecule. J Immunol (2001) 167(11):6123–31. doi: 10.4049/jimmunol.167.11.6123
43. Adolf G, Ostermann E, Patzelt E, Sproll M, Heider KH, Miglietta J, et al. Antibodies specific for CD44v6. (2002), United States patent. US20030190319A1. USA: Boehringer Ingelheim International GmbH, Boehringer Ingelheim Pharmaceuticals Inc.
44. Girard-Gagnepain A, Amirache F, Costa C, Lévy C, Frecha C, Fusil F, et al. Baboon envelope pseudotyped LVs outperform VSV-G-LVs for gene transfer into early-cytokine-stimulated and resting HSCs. Blood (2014) 124(8):1221–31. doi: 10.1182/blood-2014-02-558163
45. Sandrin V, Boson B, Salmon P, Gay W, Nègre D, Le Grand R, et al. Lentiviral vectors pseudotyped with a modified RD114 envelope glycoprotein show increased stability in sera and augmented transduction of primary lymphocytes and CD34+ cells derived from human and nonhuman primates. Blood (2002) 100(3):823–32. doi: 10.1182/blood-2001-11-0042
46. Helsen CW, Hammill JA, Lau VWC, Mwawasi KA, Afsahi A, Bezverbnaya K, et al. The chimeric TAC receptor co-opts the T cell receptor yielding robust anti-tumor activity without toxicity. Nat Commun (2018) 9(1):3049. doi: 10.1038/s41467-018-05395-y
47. Botticelli A, Cirillo A, Strigari L, Valentini F, Cerbelli B, Scagnoli S, et al. Anti-PD-1 and anti-PD-L1 in head and neck cancer: A network meta-analysis. Front Immunol (2021) 12:705096. doi: 10.3389/fimmu.2021.705096
48. Burtness B, Harrington KJ, Greil R, Soulières D, Tahara M, de Castro G Jr, et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): a randomised, open-label, phase 3 study. Lancet (2019) 394:1915–28. doi: 10.1016/S0140-6736(19)32591-7
49. Marofi F, Motavalli R, Safonov VA, Thangavelu L, Yumashev AV, Alexander M, et al. CAR T cells in solid tumors: challenges and opportunities. Stem Cell Res Ther (2021) 12(1):81. doi: 10.1186/s13287-020-02128-1
50. Ruppel KE, Fricke S, Köhl U, Schmiedel D. Taking lessons from CAR-T cells and going beyond: tailoring design and signaling for CAR-NK cells in cancer therapy. Front Immunol (2022) 13:822298. doi: 10.3389/fimmu.2022.822298
51. Wang Z, Zhao K, Hackert T, Zöller M. CD44/CD44v6 a reliable companion in cancer-initiating cell maintenance and tumor progression. Front Cell Dev Biol (2018) 6:97. doi: 10.3389/fcell.2018.00097
52. Kanke M, Fujii M, Kameyama K, Kanzaki J, Tokumaru Y, Imanishi Y, et al. Role of CD44 variant exon 6 in invasion of head and neck squamous cell carcinoma. Arch Otolaryngol Head Neck Surg (2000) 126:10. doi: 10.1001/archotol.126.10.1217
53. Odenthal J, Rijpkema M, Bos D, Wagena E, Croes H, Grenman R, et al. Targeting CD44v6 for fluorescence-guided surgery in head and neck squamous cell carcinoma. Sci Rep (2018) 8(1):10467. doi: 10.1038/s41598-018-28059-9
54. Deichmann A, Schmidt M. Biosafety considerations using gamma-retroviral vectors in gene therapy. Curr Gene Ther (2013) 13(6):469–77. doi: 10.2174/15665232113136660004
55. Ghani K, Boivin-Welch M, Roy S, Dakiw-Piaceski A, Barbier M, Pope E, et al. Generation of high-titer self-inactivated γ-retroviral vector producer cells. Mol Ther Methods Clin Dev (2019) 14:90–9. doi: 10.1016/j.omtm.2019.05.013
56. Irving M, Lanitis E, Migliorini D, Ivics Z, Guedan S. Choosing the right tool for genetic engineering: clinical lessons from chimeric antigen receptor-T cells. Hum Gene Ther (2021) 32(19-20):1044–58. doi: 10.1089/hum.2021.173
57. Suerth JD, Morgan MA, Kloess S, Heckl D, Neudörfl C, Falk CS, et al. Efficient generation of gene-modified human natural killer cells via alpharetroviral vectors. J Mol Med (Berl) (2016) 94(1):83–93. doi: 10.1007/s00109-015-1327-6
58. Colamartino ABL, Lemieux W, Bifsha P, Nicoletti S, Chakravarti N, Sanz J, et al. Efficient and robust NK-cell transduction with baboon envelope pseudotyped lentivector. Front Immunol (2019) 10:2873. doi: 10.3389/fimmu.2019.02873
59. Bari R, Granzin M, Tsang KS, Roy A, Krueger W, Orentas R, et al. A distinct subset of highly proliferative and lentiviral vector (LV)-transducible NK cells define a readily engineered subset for adoptive cellular therapy. Front Immunol (2019) 10:2001. doi: 10.3389/fimmu.2019.02001
60. Kruschinski A, Moosmann A, Poschke I, Norell H, Chmielewski M, Seliger B, et al. Engineering antigen-specific primary human NK cells against HER-2 positive carcinomas. Proc Natl Acad Sci U.S.A. (2008) 105(45):17481–6. doi: 10.1073/pnas.0804788105
61. Jijiwa M, Demir H, Gupta S, Leung C, Joshi K, Orozco N, et al. CD44v6 regulates growth of brain tumor stem cells partially through the AKT-mediated pathway. PloS One (2011) 6(9):e24217. doi: 10.1371/journal.pone.0024217
62. Laskowski TJ, Biederstädt A, Rezvani K. Natural killer cells in antitumour adoptive cell immunotherapy. Nat Rev Cancer (2022) 22(10):557–75. doi: 10.1038/s41568-022-00491-0
63. Foley B, Ta C, Barnes S, de JE, Nguyen M, Cheung LC, et al. Identifying the optimal donor for natural killer cell adoptive therapy to treat paediatric B- and T-cell acute lymphoblastic leukaemia. Clin Transl Immunol (2020) 9(7):e1151. doi: 10.1002/cti2.1151
64. Schwane V, van Huynh-Tran H, Vollmers S, Yakup VM, Sauter J, Schmidt AH, et al. Distinct signatures in the receptor repertoire discriminate CD56bright and CD56dim natural killer cells. Front Immunol (2020) 11:568927. doi: 10.3389/fimmu.2020.568927
65. Pesce S, Greppi M, Tabellini G, Rampinelli F, Parolini S, Olive D, et al. Identification of a subset of human natural killer cells expressing high levels of programmed death 1: A phenotypic and functional characterization. J Allergy Clin Immunol (2017) 139(1):335–346.e3. doi: 10.1016/j.jaci.2016.04.025
66. MacFarlane AW, Jillab M, Plimack ER, Hudes GR, Uzzo RG, Litwin S, et al. PD-1 expression on peripheral blood cells increases with stage in renal cell carcinoma patients and is rapidly reduced after surgical tumor resection. Cancer Immunol Res (2014) 2(4):320–31. doi: 10.1158/2326-6066.CIR-13-0133
67. Judge SJ, Dunai C, Aguilar EG, Vick SC, Sturgill IR, Khuat LT, et al. Minimal PD-1 expression in mouse and human NK cells under diverse conditions. J Clin Invest (2020) 130(6):3051–68. doi: 10.1172/JCI133353
68. Niu C, Li M, Zhu S, Chen Y, Zhou L, Xu D, et al. PD-1-positive Natural Killer Cells have a weaker antitumor function than that of PD-1-negative Natural Killer Cells in Lung Cancer. Int J Med Sci (2020) 17(13):1964–73. doi: 10.7150/ijms.47701
69. Wagner AK, Kadri N, Tibbitt C, van de Ven K, Bagawath-Singh S, Oliinyk D, et al. PD-1 expression on mouse intratumoral NK cells and its effects on NK cell phenotype. iScience (2022) 25(10):105137. doi: 10.1016/j.isci.2022.105137
70. Klingemann H. The NK-92 cell line-30 years later: its impact on natural killer cell research and treatment of cancer. Cytotherapy (2023) 25(5):451–7. doi: 10.1016/j.jcyt.2022.12.003
71. Mao Y, van Hoef V, Zhang X, Wennerberg E, Lorent J, Witt K, et al. IL-15 activates mTOR and primes stress-activated gene expression leading to prolonged antitumor capacity of NK cells. Blood (2016) 128(11):1475–89. doi: 10.1182/blood-2016-02-698027
Keywords: CAR-NK cells, head and neck cancer, CD44v6, NK cells, solid tumor, immune cell therapy, chimeric antigen receptor
Citation: Ciulean IS, Fischer J, Quaiser A, Bach C, Abken H, Tretbar US, Fricke S, Koehl U, Schmiedel D and Grunwald T (2023) CD44v6 specific CAR-NK cells for targeted immunotherapy of head and neck squamous cell carcinoma. Front. Immunol. 14:1290488. doi: 10.3389/fimmu.2023.1290488
Received: 07 September 2023; Accepted: 26 October 2023;
Published: 10 November 2023.
Edited by:
Payam Behzadi, ShahreQods, IranReviewed by:
Marit Inngjerdingen, University of Oslo, NorwayIan CD Johnston, Miltenyi Biotec, Germany
Copyright © 2023 Ciulean, Fischer, Quaiser, Bach, Abken, Tretbar, Fricke, Koehl, Schmiedel and Grunwald. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ioana Sonya Ciulean, c29ueWEuY2l1bGVhbkBpemkuZnJhdW5ob2Zlci5kZQ==; Thomas Grunwald, dGhvbWFzLmdydW53YWxkQGl6aS5mcmF1bmhvZmVyLmRl
†These authors contributed equally to this work and share last authorship