 Susan F. Steinberg
Susan F. Steinberg- Department of Molecular Pharmacology and Therapeutics, Columbia University, New York, NY, United States
Conventional models view β1-adrenergic receptors (β1ARs) as full-length proteins that activate signaling pathways that influence contractile function and ventricular remodeling - and are susceptible to agonist-dependent desensitization. This perspective summarizes recent studies from my laboratory showing that post-translational processing of the β1-adrenergic receptor N-terminus results in the accumulation of both full-length and N-terminally truncated forms of the β1AR that differ in their signaling properties. We also implicate oxidative stress and β1AR cleavage by elastase as two novel mechanisms that would (in the setting of cardiac injury or inflammation) lead to altered or decreased β1AR responsiveness.
1 Introduction
β-Adrenergic receptors (βARs) are among the most intensively studied members of the G protein-coupled receptor superfamily primarily because they control physiologic mechanisms that impact on the pathogenesis of cardiovascular disease; they are clinically important targets for drug discovery. Conventional models hold that βARs function to rapidly adjust cardiac output by recruiting a Gs-adenylyl cyclase pathway that leads to the accumulation of cAMP, activation of protein kinase A, and phosphorylation of membrane and sarcomeric proteins involved in excitation-contraction coupling (Figure 1). βAR-driven inotropic and chronotropic responses provide hemodynamic support in the setting of acute heart failure. However, with chronic heart failure, agonist-occupied βARs are stabilized in an active conformation that is phosphorylated by G protein-coupled receptor kinase (GRK); GRK-phosphorylated receptors then recruit β-arrestin, an adapter protein that acts to both sterically interdict βAR-G protein interactions and scaffold binding partners that trigger a second wave of signaling to Gs-independent growth regulatory responses such as extracellular-signal regulated kinase (ERK) and AKT (1, 2). While these pathways generally have been implicated in cardioprotection, studies in cardiomyocytes also link chronic βAR activation the activation of proapoptotic pathways and a spectrum of changes (including cardiomyocyte hypertrophy and apoptosis, interstitial fibrosis, and contractile dysfunction) that contribute to the pathogenesis of heart failure (3, 4).
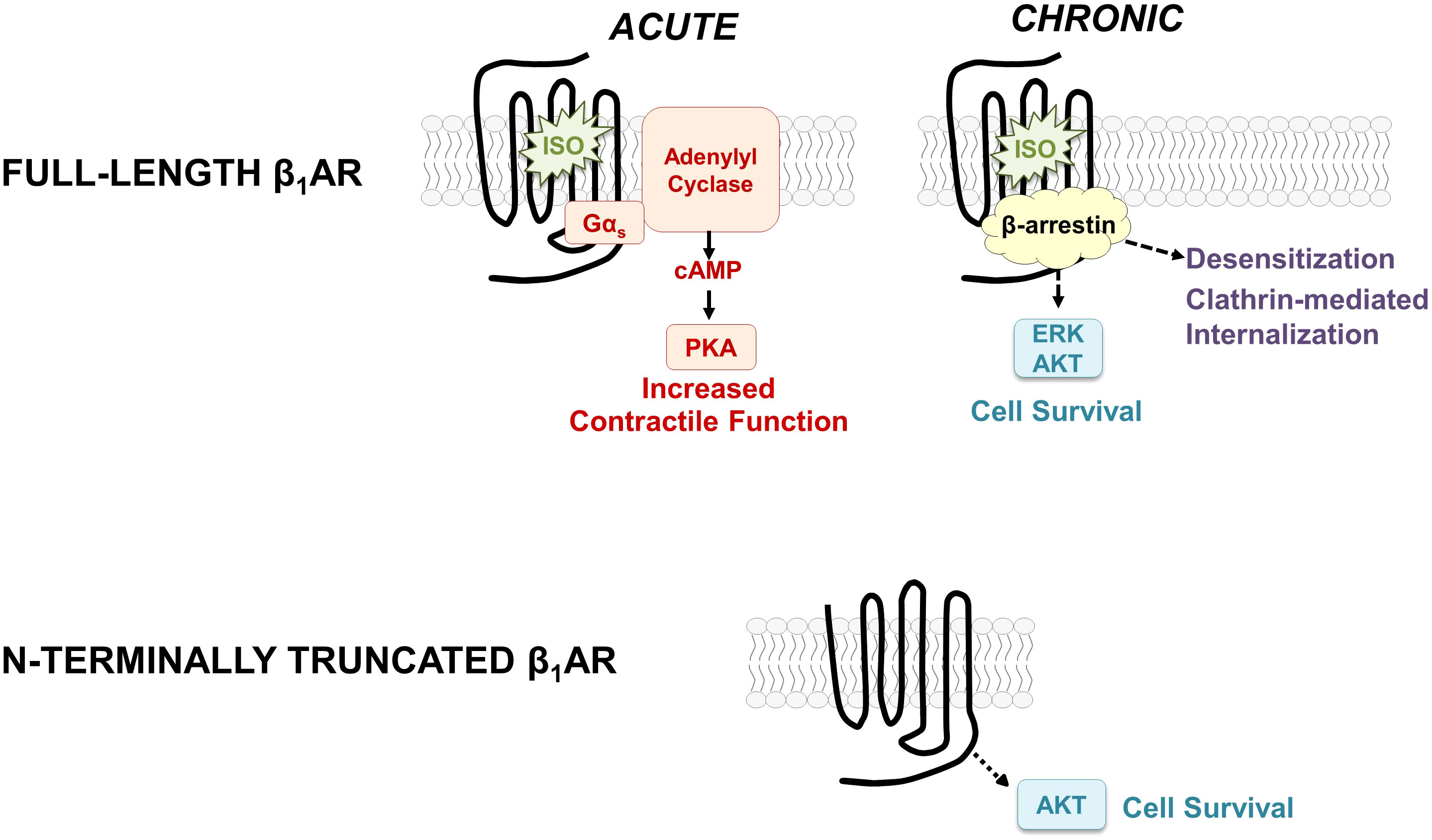
Figure 1 Schematic showing the classical paradigm for signaling by full-length β1ARs (top) and the distinct signaling properties of the N-terminally truncated form of the β1AR (bottom).
It is worth noting that conventional models describing the molecular basis for βAR signaling responses derive from literature heavily biased towards an analysis of the β2AR. While the literature has generally tended to assume that the signaling properties of β1ARs and β2ARs are similar (and that β2ARs can serve as meaningful surrogates for the β1AR subtype), this ignores a substantial body of evidence showing that β1ARs are relatively resistant to agonist-induced GRK-dependent phosphorylation, they engage β-arrestin only weakly (5), and they show little-to-no agonist-induced internalization (6, 7). These differences should not be surprising, since the 54% overall homology between β1AR and β2AR subtypes is largely confined to their transmembrane, ligand-binding pockets; their N- and C-termini are quite distinct (8). This perspective summarizes our recent studies focusing on the β1AR N-terminus, the relatively short/unstructured extracellular portion of the receptor that differs in length, sequence, and post-translational processing from the β2AR N-terminus. Our studies characterize an O-glycan regulated N-terminal cleavage mechanism that is specific for the β1AR (does not apply to the β2AR) that results in the generation of N-terminally truncated β1ARs with signaling properties that differ from that described for full-length β1ARs (9, 10). Our results force a reexamination of current concepts regarding the molecular basis for β1AR-dependent signaling responses, since conventional models have been derived from literature predicated on the assumption that β1ARs signal exclusively as full-length receptors. This perspective focuses on the novel notion β1AR cleavage results in the generation of distinct β1AR species that differ in their signaling properties.
2 Post-translational processing of the β1AR
2.1 β1AR O-glycosylation
β1ARs and β2ARs both contain sites for N-glycosylation, but β1ARs uniquely serve as targets for O-glycosylation and proteolytic cleavage (Figure 2A). In way of background, N-glycosylation results from the en bloc transfer of a preformed complex glycan structure to an asparagine residue in a N-x-S/T consensus motif, with the glycan then further trimmed/modified to yield higher-order glycan structures. In contrast, O-glycosylation is a non-template driven post-translational modification that is initiated by the enzymatic transfer of a single α-GalNAc to an acceptor serine or threonine residue (a reaction catalyzed by a member of the multi-gene family of polypeptide GalNAc-transferases that differ in their tissue distribution and substrate specificities), followed by the step-wise enzymatic transfer of additional sugars (galactose, GlcNac, fucose, etc.) to yield a spectrum of complex higher-order linear and/or branched glycan structures (11). This mechanism allows for a high level of structural diversity/microheterogeneity, even at single sites within a given protein. Evidence that clusters of O-glycans can serve as barriers to prevent protein cleavage by proteases (11) provided the rationale to examine whether O-glycosylation play a role in the maturational processing of β1ARs.
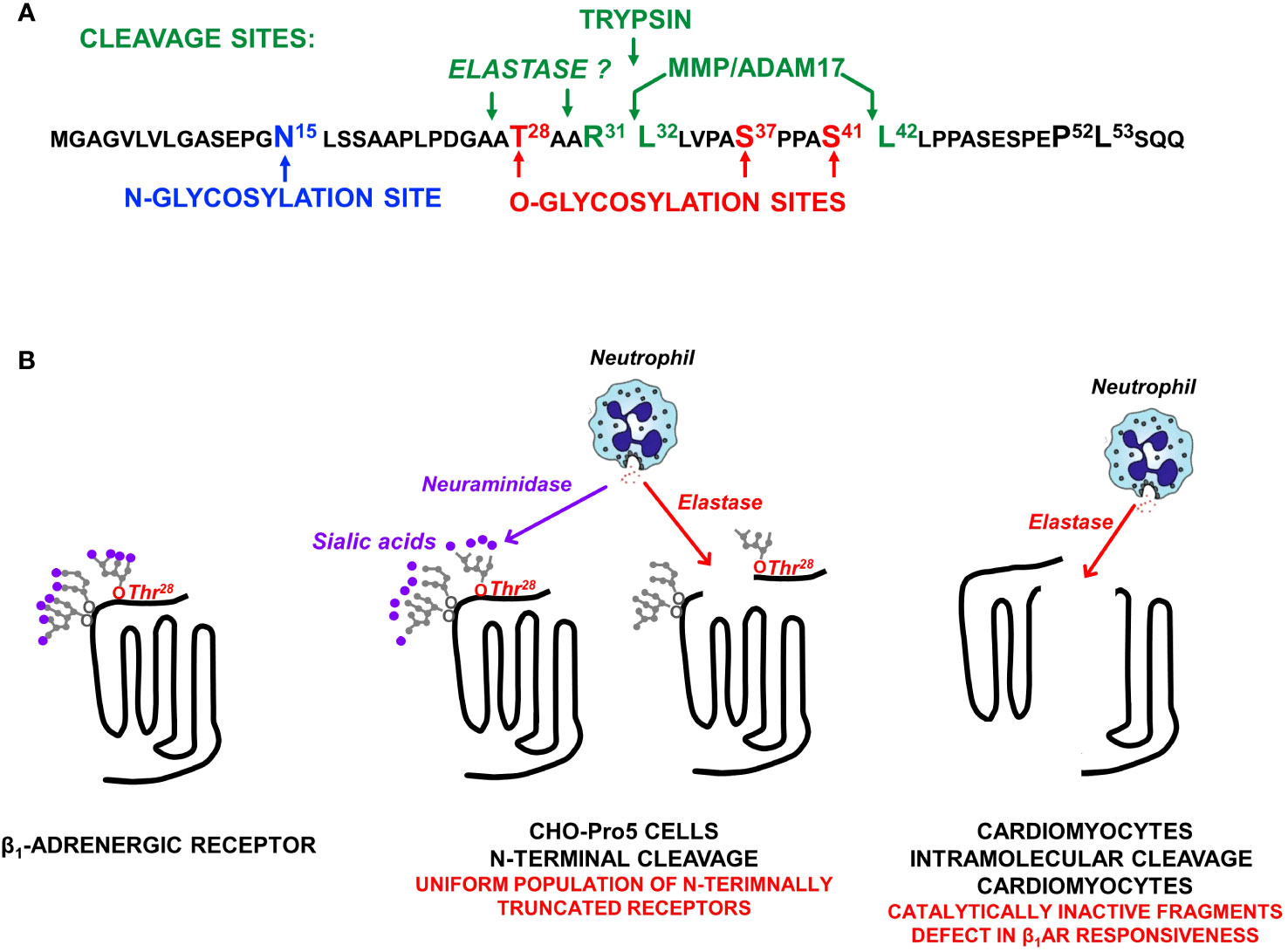
Figure 2 (A) The β1AR N-terminal sequence showing the single N-glycosylation site at N15, O-glycosylation sites at T28, S37, and S41, and the major ADAM17 cleavage site at R31↓L32 as well as a second ADAM17 cleavage site at S41↓L42 (that is exposed only when S41 O-glycosylation is prevented). (B) β1AR N-terminal cleavage by neutrophil elastase occurs only when the O-linked glycan at T28 is not capped by a terminal sialic acid; O-linked glycans at S37 and S41 do not impact on elastase-dependent β1AR N-terminal cleavage. Studies in neonatal rat ventricular cardiomyocytes expose an additional intramolecular cleavage site for elastase (that is not dependent on N-terminal glycan desialylation) that disrupts catecholamine responsiveness.
We used a range of biochemical and mutagenesis approaches [described in detail in previous publications (9, 10)] to map the major β1AR N-terminal O-linked glycosylation sites to Ser37 and Ser41 and show that O-glycan structures are heavily sialylated (a feature that will become important in the section that follows). We showed that O-glycosylation is required for full-length β1AR expression; β1ARs accumulate as N-terminally truncated species under conditions that prevent β1AR O-glycosylation [suggesting that mucin-like O-glycans at the N-terminus act as barriers to prevent protease cleavage at adjacent sites (9)]. We also established the importance of this N-terminal processing mechanism, showing that N-terminal truncation alters the β1AR’s signaling bias between cAMP/PKA vs. ERK pathways and that N-terminally truncated β1ARs acquire a unique function to constitutively activate AKT and protect against doxorubicin-induced cardiomyocyte apoptosis [i.e., the β1AR acquires a cardioprotective phenotype as a result of N-terminal truncation, Figure 1, bottom (9, 10)]. These studies implicate the β1AR N-terminus as a heretofore-unrecognized structural determinant of β1AR responsiveness.
It is worth noting that the O-glycan-regulated N-terminal cleavage mechanism identified in our studies provides the first credible explanation for the molecular heterogeneity observed for native β1AR in various cardiac preparations. Our findings also emphasize that immunblotting or immunohistochemistry studies that rely on antibodies to N-terminal epitope tags to track β1AR expression and/or localization should be interpreted with caution, since these techniques do not capture N-terminally truncated forms of the β1AR.
2.2 β1AR N-terminal cleavage
We used mutagenesis and pharmacologic strategies [described in (9, 10)] to map the major O-glycan-regulated β1AR N-terminal cleavage site to R31↓L32 and show that cleavage at this site is attributable to the cellular actions of a disintegrin and metalloproteinase 17 [ADAM17; Figure 2A (9, 10)]. We also identified a secondary ADAM17-dependent N-terminal cleavage at S41↓L42 that is specifically inhibited by the O-glycan modification at S41 (10).
The observation that the β1AR N-terminus can serve as a target for proteolytic cleavage by ADAM17 raised the obvious question of whether full-length β1ARs on the cell surface are cleaved by other proteases. Since the R31↓L32 cleavage site conforms to a consensus trypsin cleavage motif, we examined whether β1ARs are cleaved by trypsin. We showed that trypsin cleaves full-length β1ARs and that cleavage is specifically at the N-terminal R31↓L32 cleavage site (Figure 2A (12),). This mechanism should interest laboratories that interrogate β1AR signaling in isolated adult cardiomyocytes – cells typically extracted from intact ventricular using trypsin digests (13). The in vivo significance of cardiomyocyte β1AR cleavage by trypsin (a digestive enzyme found in the gastrointestinal tract) is dubious.
Therefore, we turned our attention to other, more pathophysiologically relevant proteases. Preliminary studies performed in CHO-Pro5 cells failed to identify significant levels of β1AR cleavage by various proteases. Since terminal charged sialic acid residues have been implicated as regulators of glycoprotein cleavage (14–16) and O-glycan attachments on the β1AR N-terminus are heavily sialylated (9), we repeated the protease screens in cells treated with neuraminidase (an enzyme that removes terminal sialic acid residues). These studies exposed an action of elastase to cleave full-length (but not N-terminally truncated) β1ARs under conditions that disrupt glycoprotein sialylation [Figure 2B (17)]. We then used a mutagenesis strategy to identify the sialylated O-glycan attachment that prevents elastase cleavage. Preliminary studies effectively ruled out roles for previously identified N- or O-glycosylation sites at N15, S37, or S41. Therefore, we considered a possible role for T28, a residue previously reported to be O-glycosylated in the context of reductionist in vitro assays (18). These studies showed that a sialylated O-glycan at T28 plays only a minor role in the maturational processing of β1ARs to full-length receptors, but it fully protects β1ARs from elastase-dependent cleavage (17). This novel mechanism for β1AR regulation is predicted to have pathophysiologic importance, given that neuraminidase is released along with elastase by activated neutrophils at sites of inflammation or injury (Figure 2).
We then used a mutagenesis strategy in an attempt to map the elastase cleavage site. Our studies excluded possible roles for previously identified MMP/ADAM17-sensitive sites at R31↓L32 or S41↓L42 indicating that elastase cleavage must be at another site on the β1AR N-terminus (17). In this regard, it is intriguing to note that T28 is strategically positioned adjacent to an elastase consensus cleavage motif (i.e., elastase typically cleaves scissile bonds C-terminal to small residues such as Ala, Gly, or Val; Figure 2A).
An elastase-dependent cleavage mechanism restricted to the β1AR N-terminus would generate a uniform population of signaling-competent N-terminally truncated receptors – a molecular form of the β1AR that constitutively couples to the cardioprotective AKT pathway. Hence, this type of proteolytic cleavage mechanism is predicted to afford survival advantage in the setting of heart failure.
2.3 β1AR cleavage at an intramolecular site
The actions of elastase also were examined in the more physiologically relevant cardiomyocyte context. Here, elastase treatment (even under conditions that do not disrupt protein O-glycosylation or sialylation) leads to a decrease in the abundance of the β1AR in association with the accumulation of ~40 kD N-terminal and ~25 kD C-terminal fragments, consistent with an intramolecular cleavage in extracellular loop 2 (Figure 2B (17),). The additional observation that elastase treatment results in a pronounced defect in isoproterenol-dependent (but not basal or forskolin-dependent) cAMP accumulation (17) supports the conclusion that the β1AR fragments that accumulate in elastase-treated cardiomyocytes are signaling-incompetent.
Our studies linking elastase treatment to an intramolecular cleavage that disrupts β1AR responsiveness were performed in neonatal cardiomyocyte cultures. It is important to note that differences in β1AR glycosylation or trafficking patterns between neonatal and adult cardiomyocytes or in cardiomyocytes that have been induced to hypertrophy could in theory lead to differences in β1AR protease-sensitivity. Hence, the functional consequences of β1AR cleavage would depend upon whether cleavage is restricted to the N-terminus (resulting in a uniform population of cardioprotective β1ARs) or whether cleavage is at an intramolecular site that disrupts catecholamine responsiveness. Mechanisms that fine-tune β1AR protease-sensitivity are the subject of ongoing studies.
2.4 β1AR regulation by oxidative stress
There is considerable evidence that chronic heart failure (which is associated with elevated catecholamine levels) leads to a loss of cardiac reserve due to decreased expression and signaling by βARs. This loss of catecholamine responsiveness traditionally has been attributed to homologous desensitization and/or βAR down-regulation. However, the notion that a single mechanism underlies the heart failure-induced defects in signaling by both the β1AR (the predominant βAR subtype and principal driver of catecholamine-driven sympathetic responses in the healthy heart) and the β2AR subtype is difficult to reconcile with clinical studies showing that β1ARs and β2ARs are regulated differently in heart failure; heart failure leads to a selective downregulation of the β1AR subtype that is not accompanied by a commensurate loss of β2ARs. This formulation also is at odds with cell-based studies showing that β2ARs undergo agonist-dependent desensitization/down-regulation, but β1ARs are relatively resistant agonist-induced desensitization/internalization (6, 19). In this regard, it is worth noting that much like the N-termini, β1AR and β1AR C-termini and intracellular loops show little sequence homology; sites on the β2AR that serve as substrates for GRK phosphorylation and/or docking sites for β-arrestin are not conserved in the β1AR subtype. This raises a fundamental question as to the mechanism underlying the defect in β1AR responsiveness in heart failure. Our recent studies address this conundrum by showing that oxidative stress (a stimulus that contributes to the pathogenesis of heart failure and various other cardiomyopathic syndromes) decreases β1AR expression and isoproterenol responsiveness in cardiomyocytes; oxidative stress does not lead to changes in the expression of the β2AR subtype (20). Hence, these studies implicate oxidative stress as a mechanism that selectively decreases β1AR (but not β2AR) expression that would underly the decreased cardiac catecholamine responsiveness that is a hallmark of heart failure.
3 Discussion
Studies in model cell types have traditionally ignored possible differences in the biological controls and cellular actions of β1ARs vs. β2ARs. However, studies in genetic models of receptor overexpression provide compelling evidence that the deleterious effects of chronic sympathetic overdrive that contribute to the pathogenesis of cardiac hypertrophy and heart failure can be attributed to the cardiac actions of the β1AR subtype. These studies show that moderate levels of transgenic β1AR overexpression leads to maladaptive cardiac remodeling and heart failure (21, 22) whereas even high levels of transgenic β2AR overexpression are relatively well tolerated (23–25). Our studies add an additional dimension to the analysis by showing that the β1AR subtype accumulates as both full-length and N-terminally truncated forms and that these distinct molecular forms of the β1AR display importance differences in their coupling to pro- vs. anti-apoptotic signaling pathways. The notion that the β1AR N-terminus functions as a novel molecular determinant of β1AR signaling responses suggests that therapeutic strategies designed to influence β1AR N-terminal cleavage might be exploited for the treatment of heart failure. Our studies also implicate oxidative stress and proteolytic cleavage as two pathophysiologicaly relevant stimuli that act to either disrupt or alter catecholamine-driven β1AR growth and/or injury responses in the setting of heart failure, cardiac inflammation, or myocardial infarction-induced cardiac injury. Collectively, the novel signaling paradigms for cardiomyocyte β1ARs identified in our studies add a new dimension to our understanding of the evolution of heart failure and other cardiomyopathic disorders.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was approved by Columbia University Institutional Animal Care and Use Committee. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
SS: Conceptualization, Funding acquisition, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was funded by NHLBI grant RO1-HL138468.
Conflict of interest
The authors declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Tilley DG, Kim IM, Patel PA, Violin JD, Rockman HA. β-Arrestin mediates β1-adrenergic receptor-epidermal growth factor receptor interaction and downstream signaling. J Biol Chem (2009) 284:20375–86. doi: 10.1074/jbc.M109.005793
2. Kim IM, Tilley DG, Chen J, Salazar NC, Whalen EJ, Violin JD, et al. β-blockers alprenolol and carvedilol stimulate β-arrestin-mediated EGFR transactivation. Proc Natl Acad Sci USA (2008) 105:14555–60. doi: 10.1073/pnas.0804745105
3. Port JD, Bristow MR. Altered β-adrenergic receptor gene regulation and signaling in chronic heart failure. J Mol Cell Cardiol (2001) 33:887–905. doi: 10.1006/jmcc.2001.1358
4. Lohse MJ, Engelhardt S, Eschenhagen T. What is the role of β-adrenergic signaling in heart failure? Circ Res (2003) 93:896–906. doi: 10.1161/01.RES.0000102042.83024.CA
5. Shiina T, Kawasaki A, Nagao T, Kurose H. Interaction with β-arrestin determines the difference in internalization behavor between β1- and β2-adrenergic receptors. J Biol Chem (2000) 275:29082–90. doi: 10.1074/jbc.M909757199
6. Green SA, Liggett SB. A proline-rich region of the third intracellular loop imparts phenotypic β1-versus β2-adrenergic receptor coupling and sequestration. J Biol Chem (1994) 269:26215–9. doi: 10.1016/S0021-9258(18)47181-5
7. Eichel K, Jullie D, von Zastrow M. β-Arrestin drives MAP kinase signalling from clathrin-coated structures after GPCR dissociation. Nat Cell Biol (2016) 18:303–10. doi: 10.1038/ncb3307
8. Frielle T, Collins S, Daniel KW, Caron MG, Lefkowitz RJ, Kobilka BK. Cloning of the cDNA for the human β1-adrenergic receptor. Proc Natl Acad Sci USA (1987) 84:7920–4. doi: 10.1073/pnas.84.22.7920
9. Park M, Reddy GR, Wallukat G, Xiang YK, Steinberg SF. β1-adrenergic receptor O-glycosylation regulates N-terminal cleavage and signaling responses in cardiomyocytes. Sci Rep (2017) 7:7890. doi: 10.1038/s41598-017-06607-z
10. Zhu J, Steinberg SF. β1-adrenergic receptor N-terminal cleavage by ADAM17; the mechanism for redox-dependent downregulation of cardiomyocyte β1-adrenergic receptors. J Mol Cell Cardiol (2021) 154:70–9. doi: 10.1016/j.yjmcc.2021.01.012
11. Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, et al, editors. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press (2009).
12. Zhu J, Steinberg SF. Trypsin cleavage of the β1-adrenergic recepetor. Am J Physiol (2022) 322:H486–91. doi: 10.1152/ajpheart.00005.2022
13. Rybin VO, Pak E, Alcott S, Steinberg SF. Developmental changes in β2-adrenergic receptor signaling in ventricular myocytes. Mol Pharmacol (2003) 63:1338–48. doi: 10.1124/mol.63.6.1338
14. Hane M, Matsuoka S, Ono S, Miyata S, Kitajima K, Sato C. Protective effects of polysialic acid on proteolytic cleavage of FGF2 and ProBDNF/BDNF. Glycobiology (2015) 25:1112–24. doi: 10.1093/glycob/cwv049
15. Sumer-Bayraktar Z, Grant OC, Venkatakrishnan V, Woods RJ, Packer NH, Thaysen-Andersen M. Asn347 Glycosylation of corticosteroid-binding globulin fine-tunes the host immune response by modulating proteolysis by Pseudomonas aeruginosa and neutrophil elastase. J Biol Chem (2016) 291:17727–42. doi: 10.1074/jbc.M116.735258
16. Rutledge EA, Enns CA. Cleavage of the transferrin receptor is influenced by the composition of the O-linked carbohydrate at position 104. J Cell Physiol (1996) 168:284–93. doi: 10.1002/(SICI)1097-4652(199608)168:2<284::AID-JCP7>3.0.CO;2-L
17. Zhu J, Steinberg SF. β1-adrenergic receptor cleavage and regulation by elastase. JACC Basic to Trans Sci (2023) 8:976–88. doi: 10.1016/j.jacbts.2023.02.002
18. Goth CK, Tuhkanen HE, Khan H, Lackman JJ, Wang S, Narimatsu Y, et al. Site-specific O-Glycosylation by polypeptide N-acetylgalactosaminyltransferase 2 (GalNAc-transferase T2) co-regulates β1-adrenergic receptor N-terminal cleavage. J Biol Chem (2017) 292:4714–26. doi: 10.1074/jbc.M116.730614
19. Suzuki T, Nguyen CT, Nantel F, Borin H, Valiqueae M, Frielle T, et al. Distinct regulation of β1- and β2-adrenergic receptors in Chinese hamster fibroblasts. Mol Pharmacol (1992) 41:542–8.
20. Park M, Steinberg SF. Carvedilol prevents redox inactivation of cardiomyocyte β1-adrenergic receptors. JACC; Basic to Trans Sci (2018) 3:521–32. doi: 10.1016/j.jacbts.2018.06.002
21. Engelhardt S, Hein L, Wiesmann F, Lohse MJ. Progressive hypertrophy and heart failure in β1-adrenergic receptor transgenic mice. Proc Natl Acad Sci USA (1999) 96:7059–64. doi: 10.1073/pnas.96.12.7059
22. Bisognano JD, Weinberger HD, Bohlmeyer TJ, Pende A, Raynolds MV, Sastravaha A, et al. Myocardial-directed overexpression of the human β1-adrenergic receptor in transgenic mice. J Mol Cell Cardiol (2000) 32:817–30. doi: 10.1006/jmcc.2000.1123
23. Milano CA, Allen LF, Rockman HA, Dolber PC, McMinn TR, Chien KR, et al. Enhanced myocardial function in transgenic mice overexpressing the β2-adrenergic receptor. Science (1994) 264:582–6. doi: 10.1126/science.8160017
24. Turki J, Lorenz JN, Green SA, Donnelly ET, Jacinto M, Liggett SB. Myocardial signaling defects and impaired cardiac function of a human β2-adrenergic receptor polymorphism expressed in transgenic mice. Proc Natl Acad Sci U.S.A. (1996) 93:10483–8. doi: 10.1073/pnas.93.19.10483
Keywords: β1-adrenergic receptors, elastase, oxidative stress, cardiomyocytes, proteolysis
Citation: Steinberg SF (2023) Redox and proteolytic regulation of cardiomyocyte β1-adrenergic receptors – a novel paradigm for the regulation of catecholamine responsiveness in the heart. Front. Immunol. 14:1306467. doi: 10.3389/fimmu.2023.1306467
Received: 03 October 2023; Accepted: 21 November 2023;
Published: 04 December 2023.
Edited by:
Wei Huang, University of Cincinnati, United StatesReviewed by:
Xingyu He, University of Cincinnati, United StatesCopyright © 2023 Steinberg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Susan F. Steinberg, c2ZzMUBjb2x1bWJpYS5lZHU=