 Jun Yin1
Jun Yin1 Norin Chaudhry
Norin Chaudhry Yi Huang
Yi Huang- 1The University of Pittsburgh Medical Center (UPMC) Hillman Cancer Center, School of Medicine, University of Pittsburgh, Pittsburgh, PA, United States
- 2Department of Internal Medicine, Division of Hematology, Oncology, and Blood and Marrow Transplantation, Carver College of Medicine, University of Iowa, Iowa City, IA, United States
- 3Fred Hutchinson Cancer Center, University of Washington, Seattle, WA, United States
- 4Holden Comprehensive Cancer Center, University of Iowa, Iowa City, IA, United States
Breast cancer (BC) is the most common non-skin cancer and the second leading cause of cancer death in American women. The initiation and progression of BC can proceed through the accumulation of genetic and epigenetic changes that allow transformed cells to escape the normal cell cycle checkpoint control. Unlike nucleotide mutations, epigenetic changes such as DNA methylation, histone posttranslational modifications (PTMs), nucleosome remodeling and non-coding RNAs are generally reversible and therefore potentially responsive to pharmacological intervention. Epigenetic dysregulations are critical mechanisms for impaired antitumor immunity, evasion of immune surveillance, and resistance to immunotherapy. Compared to highly immunogenic tumor types, such as melanoma or lung cancer, breast cancer has been viewed as an immunologically quiescent tumor which displays a relatively low population of tumor-infiltrating lymphocytes (TIL), low tumor mutational burden (TMB) and modest response rates to immune checkpoint inhibitors (ICI). Emerging evidence suggests that agents targeting aberrant epigenetic modifiers may augment host antitumor immunity in BC via several interrelated mechanisms such as enhancing tumor antigen presentation, activation of cytotoxic T cells, inhibition of immunosuppressive cells, boosting response to ICI, and induction of immunogenic cell death (ICD). These discoveries have established a highly promising basis for using combinatorial approaches of epigenetic drugs with immunotherapy as an innovative paradigm to improve outcomes of BC patients. In this review, we summarize the current understanding of how epigenetic processes regulate immune cell function and antitumor immunogenicity in the context of the breast tumor microenvironment. Moreover, we discuss the therapeutic potential and latest clinical trials of the combination of immune checkpoint blockers with epigenetic agents in breast cancer.
1 Introduction
Breast cancer (BC) is still a major threat and challenge for women’s health worldwide. Like many other types of cancer, breast cancer progression involves multiple steps of uncontrolled cell proliferation and aberrant apoptosis through gene expression changes including gain-of-function of oncogenes or loss-of-function of tumor suppressor genes (TSGs). The loss of TSGs can result from specific DNA mutations, deletion, or frame shifts. Another general mechanism by which normally expressed genes can be changed is so called “epigenetic modifications” (1–3). Three primarily interconnected epigenetic mechanisms have been identified: DNA methylation, post-translational modifications (PTM) of histones, and non-coding RNAs (ncRNA) (4, 5). Several epigenetic-based drugs (DNMT, HDAC and EZH2 inhibitors) have been approved by the FDA for clinical treatment of hematological and solid malignancies (Table 1). In breast cancer, epigenetic alterations drive breast tumor proliferation, invasion and metastasis through genome-wide loss and local gains of DNA methylation within promoters of TSGs, disorders in histone modifications, and abnormal expression of noncoding RNAs (6–9). There is a considerable interest in the potential of epigenetic dysregulation as biomarkers and therapeutic targets in BC. The association between epigenetic aging and clinical outcomes has been recently evaluated in longer term BC survivors to guide cancer care for older women (10). Epigenetic malfunctions can potentially be restored to their normal phenotypes through epigenetic-based therapies (Figure 1). A number of promising epigenetic modifying drugs are under clinical investigation at various stages of breast cancer treatment as a single agent or administered in combination with other therapeutic drugs (11, 12) (Table 2).
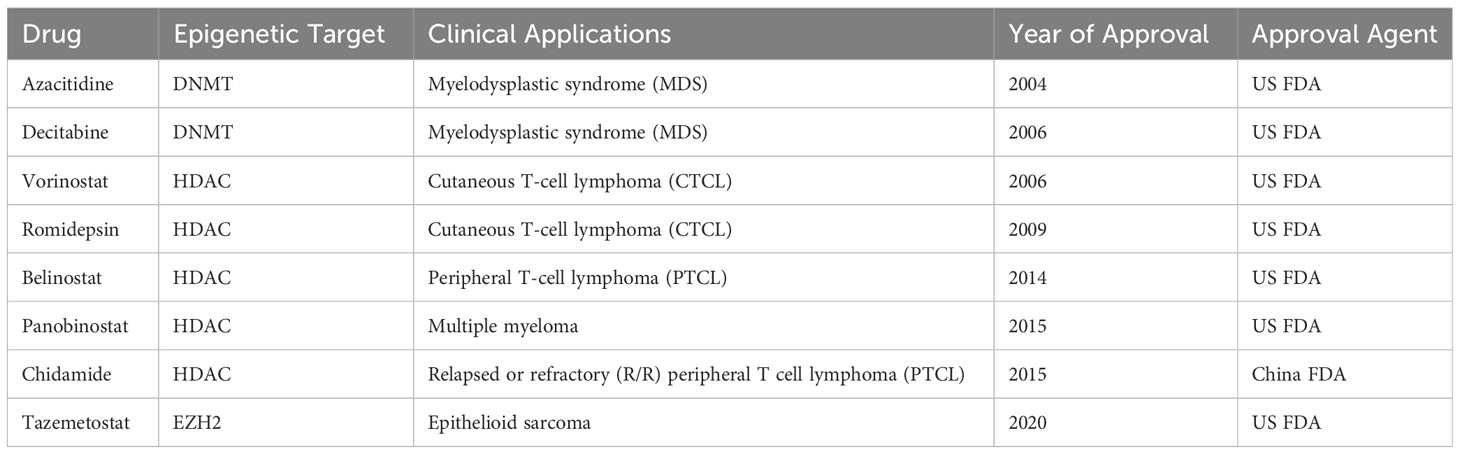
Table 1 Epigenetic drugs approved by the FDA for treatment of cancer.
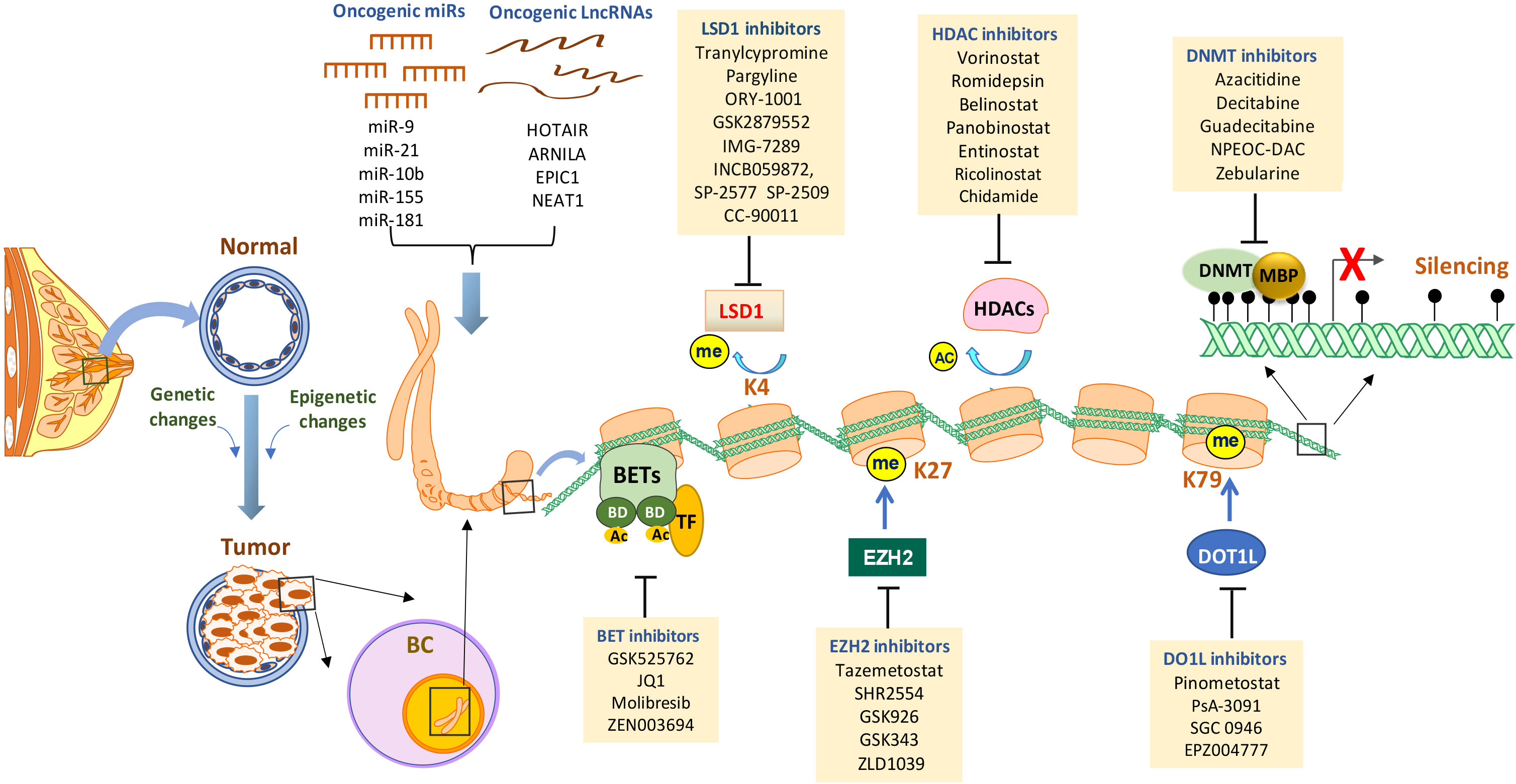
Figure 1 Targeting epigenetic alterations in breast cancer. Epigenetic alterations are critical drivers of breast tumor initiation and progression. The primary epigenetic changes in breast cancer include promoter DNA hypermethylation (black lollipops - methylated CpG dinucleotides), histone posttranslational modifications (enhanced histone deacetylation, aberrant histone methylation, BET amplification, etc), dysregulated expression and activity of miRNAs and lncRNAs. At a gene promoter, DNA methylation can be induced along with abnormal histone modifications and/or other epigenetic alterations that may result in the abnormal loss of genes with important functions in curbing breast tumor. Established or investigational drugs that can inhibit the activity of specific epigenetic modifiers are shown in the boxes. BC, breast cancer; DNMT, DNA methyltransferase; MBD, methyl-binding domain protein; HDAC, histone deacetylase; LSD1, lysine-specific demethylase 1; EZH2, enhancer of zeste homolog 2; DOT1L, DOT1-like histone lysine methyltransferase; BET, bromodomain and extra-terminal; miR, micro-RNA; lncRNA, long non-coding RNA; TF, transcription factors; Ac, acetylation; Me, methylation.
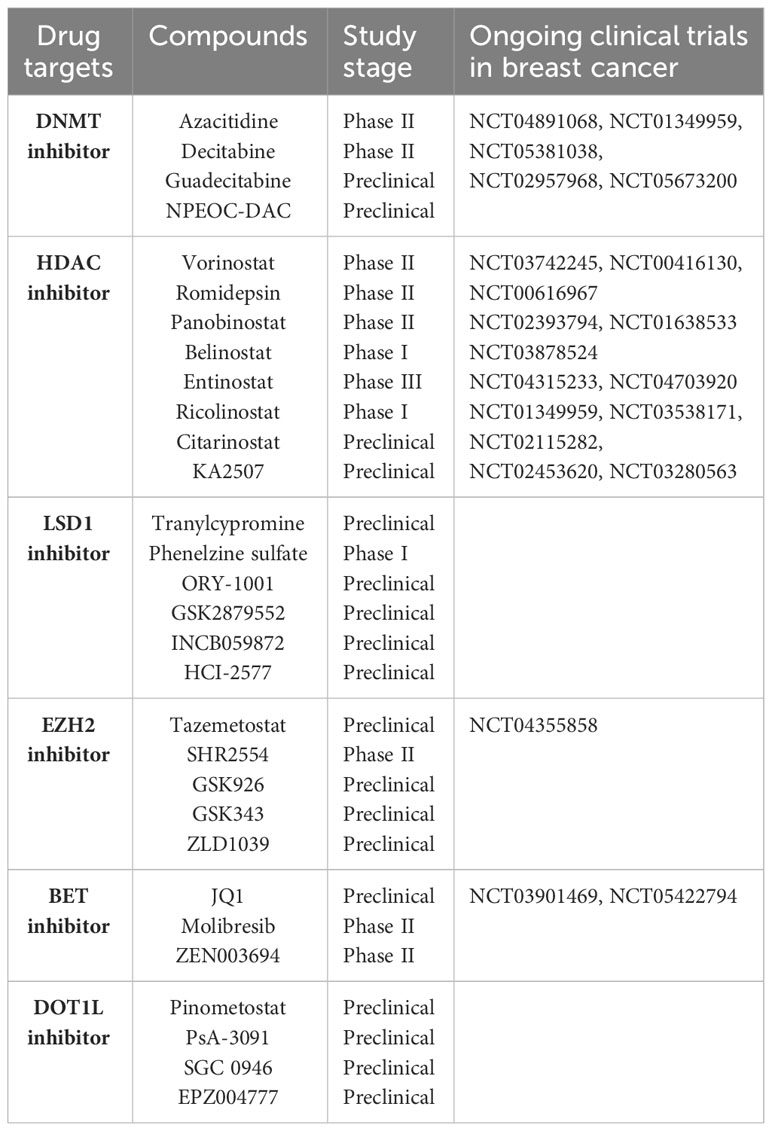
Table 2 Current stages of leading epigenetic drugs for breast cancer therapy.
Immunotherapy has opened a new era in cancer treatment with the FDA approval of several immune checkpoint inhibitors (ICI) which block checkpoint proteins from binding with their partner proteins (e.g. PD-1, PD-L1, CTLA-4, LAG-3) in tumors (13, 14). The immune system plays a critical role in surveillance against the initiation and development of breast cancer by recognizing tumor associated antigen (TAA) and eliciting immunogenic cell death (ICD) (15). Although breast cancer has been traditionally viewed to be immunologically silent, treatment with ICIs has been shown to improve clinical outcomes in some patients with metastatic BC, especially triple negative breast cancer (TNBC) (16, 17). In 2021, Pembrolizumab (anti-PD-1 mAb) in combination with chemotherapy was approved by FDA as a neoadjuvant/adjuvant treatment for high-risk, early-stage TNBCs (18). However, only a small fraction of BC patients can benefit from immunotherapy, and the overall response rates of BC to immunotherapy were only 10-20% (19, 20). Therefore, there is an urgent necessity for the development of more effective clinical strategies for BC patients whose tumors are less immunogenic and refractory to immunotherapy.
Emerging evidence has highlighted key roles of epigenetic alterations in impaired anti-tumor immunity, immune escape, and immunotherapy resistance in breast tumors. Epigenetic regulation influences all aspects of the interaction between BC cells and immune system, thus providing a rational basis for efforts to convert a breast tumor from an immune suppressive (cold) to an immune permissive (hot) state through joint epigenetic therapy. The combination of epigenetically targeted drugs and ICIs is being investigated in clinical trials in a variety of cancer types including BC. In this review, we highlight the evidence of the new roles of epigenetic crosstalk between breast tumors and the immune environment. We also discuss the latest findings on epigenetic biomarkers for immunotherapy, emphasizing the current strategies to improve immunotherapy through reprogramming the breast cancer epigenome. Finally, we summarize the ongoing clinical trials that are evaluating the combination of immune checkpoint blockade with epigenetic agents against breast cancer.
2 Epigenetic biomarkers and targets in breast cancer
2.1 DNA methylation
DNA methylation is catalyzed by a group of enzymes termed DNA methyltransferases (DNMTs) which covalently modify the C-5 position of cytosine residues in CpG islands, using S-adenosylmethionine as a methyl donor. DNMTs are divided into maintenance (DNMT1) and de novo methyltransferases (DNMT3a, DNMT3b) (21, 22). Many cancer-related genes have been shown to be hypermethylated in BC cells. These genes have a wide range of cellular functions that are involved in regulation of hormone activity (e.g. ERα, PR), cell cycle and apoptosis (e.g. RARb, p16, CCND2, SFRPs, RASSF1a, TMS1, APC), DNA damage repair (e.g. BRCA1, 14-3-3σ, HIC1, MLH1), invasion and metastasis (e.g. E-cadherin, TIMP-3) (3, 6, 7, 23, 24). A recent AURORA US Metastasis Project analyzed patients with metastatic BC by RNA sequencing, exome and whole-genome sequencing and global DNA methylation microarrays and revealed that DNA methylation downregulates ER-mediated cell-cell adhesion genes in metastases (25). Thus, DNA methylation represents a novel opportunity to identify biomarkers for early screening and diagnosis, prognosis, outcome prediction, and treatment monitoring of BC (26, 27).
Blockade of DNMT activity is the most effective approach to inhibit DNA methylation. Two DNMT-inhibiting cytosine nucleoside analogues, 5-aza-cytidine (azacitidine) and 5-aza-2’-deoxycytidine (decitabine), received FDA approval for the treatment of hematologic cancers. These two drugs induce antineoplastic activity by inhibition of DNMT activity at low dose and direct cytotoxicity at high dose through incorporation into newly synthesized DNA strands as nucleoside analogs to disrupt normal DNA synthesis. Recently, some new DNMT inhibitors (DNMTi) have been identified, such as guadecitabine (SGI-110), zebularine, and NPEOC-DAC, which exhibit increased resistance to degradation from deamination by cytidine deaminase and prolong plasma half-life (28, 29). Preclinical studies have demonstrated the effectiveness of DNMTi in restoring aberrantly silenced genes and reprograming the epigenome that may block BC cell proliferation and/or sensitize tumors to other therapeutic interventions (30–32). However, early clinical trials showed that DNMTi has limited efficacy as monotherapy in clinical studies in BC, in part due to their toxicity and off-target effects. Current clinical trials are exploring novel strategies through combining DNMTi with other therapies aiming to maximize therapeutic efficacy and minimize side effects. The combination of DNMTi and chemotherapy has been intensively studied in BC preclinical studies and showed enhanced susceptibility compared to chemotherapy alone (33–35). The safety and efficacy of combinations of DNMTi and standard chemotherapeutics are currently being assessed in multiple breast cancer clinical trials.
2.2 Histone acetylation and deacetylation
Histone acetyltransferases (HATs) and histone deacetylases (HDACs) are the enzymes responsible for catalyzing the addition or removal of acetyl groups respectively. Aberrant overexpression of HDACs correlates with aggressive clinicopathological features and poor prognosis of BC (36, 37). The development of HDAC inhibitors (HDACi) has produced encouraging results in the clinic, particularly in the field of hematological malignancy. HDACi are powerful epigenetic modulators that may exert antineoplastic effects by re-expressing silenced TSGs, inducing cancer cell cycle arrest and apoptosis, reducing angiogenesis, and modulating immune response (7, 38). In ER-negative BC cells, overexpression of HDACs have been shown to suppress transcriptional activity of key hormonal receptors, ERα and PR, and treatment with HDACi led to functional reactivation of these silenced receptors either alone or in combination with DNMTi (30, 39, 40). Inhibition of SIRT1, a member of class III HDAC, has been shown to restore multiple epigenetically suppressed TSGs, such as SFRP1, SFRP2, E-cadherin and CRBP1 in BC (41). Thus far, four pan-HDACi, vorinostat (Zolinza), romidepsin (Istodax), panobinostat (Farydak), and belinostat (Beleodaq), have been approved by the FDA for the treatment of hematological malignancies. However, clinical studies found that use of HDACi in solid tumors, including BC, showed limited single-agent activity (42, 43). An early window trial observed limited effects of orally administered vorinostat on chromatin marks and methylated genes in newly diagnosed early-stage BC patients although the treatment led to significant reduction in expression of proliferation-related genes (Ki-67, STK15, Cyclin B1) (42). Another early phase trial of vorinostat in metastatic BC patients did not meet the RECIST response criteria for adequate single-agent activity (43). Several clinical trials are evaluating the efficacy of combining HDACi with other standard cancer therapies (e.g. aromatase inhibitor, chemotherapy, PARP inhibitors, CDK4/6 inhibitors, anti-HER2 therapy) in breast cancer. One recent phase III trial reported that HDAC inhibitors exemestane and entinostat did not improve survival in patients with aromatase inhibitor-resistant advanced BC although pharmacodynamic analysis confirmed target inhibition of HDACi (44). It remains to be determined if there is a role for HDACi in a biomarker-selected BC patient population based on results from ongoing correlative clinical trials. These clinical data suggest that therapeutic potential of current HDACi, although effective in hematological malignancies, is frequently affected by off-target effect, low specificity, unfavorable pharmacokinetics with a relatively short half-life and fast clearance in solid tumor context. These challenges highlight the need for development of the next-generation HDACi with optimized activity and selectivity profiles.
2.3 Histone methylation and demethylation
Histone methylation occurs predominantly on lysine (K) and arginine (R) residues on the H3 and H4 tails which is dynamically regulated by specific lysine methyltransferases (KMT) and demethylases (KDM). There is mounting and compelling evidence that dysregulation of KMTs or KDMs leads to altered histone methylation patterns, which may contribute to BC pathogenesis (6, 7, 45). Enhancer of Zeste Homolog 2 (EZH2), a subunit of Polycomb Repressive Complex 2 (PRC2), catalyzes the addition of methyl groups to histone H3 at lysine 27 (H3K27me), which is typically associated with repression of genes expression (46). EZH2 is often elevated in breast carcinoma and excessive EZH2 expression facilitates invasive tumor growth and aggressive clinical behavior (47, 48). A number of EZH2 inhibitors have been evaluated for their anti-cancer properties, and many clinical trials are under way to examine the antitumor efficacy of EZH2i alone or in combination with other anticancer drugs. In 2020, tazemetostat (Tazverik), a first-in-class small molecule EZH2 inhibitor, was granted accelerated approval by the FDA for patients with advanced or metastatic epithelioid sarcoma who are not eligible for curative surgery. Being the first approval of an epigenetic drug for a solid tumor, tazemetostat represents a new milestone in the cancer epigenetic field.
The histone methyltransferase responsible for H3K79 methylation is an enzyme named DOT1L (disruptor of telomeric silencing 1 like) that is a non-SET domain-containing methyltransferase involved in gene transcription, heterochromatin formation, and DNA repair (49). Oktyabri et al. reported that DOT1L and its target gene, BCAT1 (branched-chain amino acid transaminase) were both up-regulated in BC cells which promotes sphere formation and cell migration in BC cells (50). A recent study showed that inhibition of DOT1L silences ERα gene and blocks the proliferation of antiestrogen-resistant BC cells, indicating that DOT1L is an attractive epigenetic target for treating endocrine therapy-resistant ER+ BC (51). Inhibition of DOT1L and H3K79 methylation has been shown to selectively block proliferation, self-renewal, and metastatic properties in BC cells (52, 53). Byun et al. found that DOT1L is associated with high metastatic potential mediated by the EMT process in TNBCs, and treatment with a novel DOT1L inhibitor, psammaplin A analog (PsA-3091), induced E-cadherin and hindered TNBC progression both in vitro and in vivo (53). These studies suggest that the use of DOT1L inhibitors is emerging as a new therapeutic approach for aggressive BC.
Histone lysine-specific demethylase 1 (LSD1, AOF2, or KDM1A) is the first discovered FAD-dependent histone demethylase that is typically associated with a transcriptional repressor complex such as CoREST, HDAC1/2, BHC80, etc (54–56). The canonical function of LSD1 is the demethylation of lysine 4 of histone 3 (H3K4me), leading to chromatin inaccessibility and gene transcription silencing (54, 55, 57, 58). LSD1 expression is markedly elevated during breast tumor progression from pre-invasive ductal carcinomas in situ (DCIS) to invasive ductal carcinoma (IDC) (59, 60). Analysis of TCGA data indicates a markedly elevated LSD1 expression in the TNBC/basal-like subtype in comparison to other BC groups (61). LSD1 overexpression is significantly associated with worse clinical outcomes in TNBC patients (60, 62). LSD1 has become an important validated epigenetic target for cancer therapy, and numerous small molecule inhibitors have been identified. Several leading LSD1 inhibitors, such as tranylcypromine, ORY-1001, IMG-7289, GSK2879552, INCB059872, CC90011 and SP-2577 (Seclidemstat), have advanced into clinical trials for neoplastic and other diseases.
LSD2 (KDM1B or AOF1) has been identified as a second member of the FAD-dependent histone demethylase family (63). LSD1 and LSD2 share significant similarities in the AO (amine oxidase) catalytic domain and FAD-dependent demethylation activity, but these two enzymes also display different properties such as patterns of chromatin complexes, transcriptional repression mechanisms, and genomic loci (64). We have shown that LSD2 is overexpressed in TNBC cells, which contributes to tumor proliferation and confers CSC-like characteristics (65). We also found that LSD2 overexpression induced global DNA methylation in TNBC. Inhibition of LSD2 reexpressed methylated TSGs and enhanced DNMTi-mediated apoptosis (66). Since LSD2 is part of chromatin-remodeling complexes distinct from those involving LSD1, further study is needed to elucidate the various roles of LSD2 in cell-context-specific settings.
Since the discovery of LSD1, many new KDMs have been identified and characterized. Most of these newly identified HDMs are members of the Jumonji family, which catalyze demethylation of histone lysine residues through a hydroxylation reaction using iron Fe2+ and α-ketoglutarate as cofactors (67–69). Xie et al. have demonstrated that KDM6A (UTX), a key demethylase of H3K27 mark, promotes hormone-responsive breast carcinogenesis through feed-forward transcriptional regulation with ER (70). KDM5B (PLU-1), a JmjC demethylase targeting H3K4me3, is implicated in the proliferative capacity of BC cells through direct transcriptional repression of TSGs including BRCA1 (71). Overexpression of KDM4A was found in about 60% of BC tissue, and several KDM4A inhibitors have been investigated as anticancer agents in BC cells (72). Benedetti et al. reported that dual-KDM inhibitor (MC3324) targeting LSD1 and UTX induces growth arrest and apoptosis in hormone-responsive BC that is associated with a robust increase in H3K4me2 and H3K27me3 (73). These findings suggest that inhibition of FAD-dependent LSD1 or JmjC-based KDMs may represent a novel approach for BC therapy. Compared with the progress of LSD1 inhibitors, the development of agents targeting Jumonji KDMs is still in preclinical stage. The big challenge is that over 30 Jumonji KDMs have been identified, many with overlapping structures and functions. Thus, targeting a single KDM may have limited effect. A better understanding of the chromatin-context specificity of KDMs would help develop more effective and selective KDM inhibitors.
2.4 Bromodomain and extra-terminal family
The bromodomain and extraterminal domain (BET) family is comprised of four conserved mammalian members BRD2, BRD3, BRD4, and BRDT, which contain two similar tandem N-terminal bromodomains (BD1 and BD2). BET proteins are epigenetic readers that regulate gene transcription via binding to acetylated lysine residues on histone proteins and other nuclear factors (74, 75). BET proteins are frequently upregulated in BC cells leading to the activation of genes involved in tumor proliferation and metastasis (76–78). An increasing number of selective and pan-BET inhibitors (BETi) have been developed such as JQ1, I-BET151, I-BET762, OTX-015, ABBV-075, TEN-010, CPI-203, CPI-0610, etc. Li et al. showed that use of hypoxia-cleavable, RGD peptide-modified poly(D,L-lactide-co-glycolide) (PLGA) nanoparticle significantly improved delivery of JQ1 and therapeutic efficacy in suppressing primary breast tumors and bone metastasis (79). Multiple Phase I/II clinical trials have tested the safety and efficacy of BETi in solid tumors, including BC. While BET inhibition has shown promising preclinical anticancer activity in BC, some early clinical studies have reported dose limiting toxicity of BETi, including thrombocytopenia and gastrointestinal disorders, indicating a need for the development of well tolerated BETi and/or optimization of dosing schemes and combinations (76).
2.5 miRNA and lncRNA
MicroRNAs (miRNA) are endogenous non-coding single-stranded RNAs, 21-23-nucleotide in length, known for their pivotal roles in post-transcriptional modification and RNA silencing (80). A growing body of work has suggested that miRNAs play important roles in BC initiation and development. Overexpression of some oncogenic miRNAs (oncomiR), such as miR-9, miR-21, miR-29a, miR-155, miR-181, and miR-10b, etc, is correlated with advanced clinical stage, lymph node metastasis, and poor clinical outcome of BC (81–85). On the other hand, some miRNAs (let-7 family, miR-7, miR-145, miR-200 family, miR-205, miR-335, miR-30a, etc.) function as tumor suppressors to prevent breast cancer development through downregulating oncoproteins coding gene expression (86–89). miRNA-based therapies in BC aim to either inhibit oncomiRs via strategies such as antisense and oligonucleotides or induce tumor-suppressing miRNAs through epigenetic interventions (90, 91). In addition, chemically synthesized miRNA mimics have been used as novel classes of therapeutic agents to restore aberrantly reduced tumor-suppressing miRNAs to their normal levels (92).
Long non-coding RNAs (lncRNA) have been arbitrarily defined as non-protein-coding transcripts longer than 200 nucleotides that lack defined protein coding potential. Abnormal expression and activity of oncogenic lncRNAs, such as HOTAIR, ARNILA, EPIC1, NEAT1, and Lnc015192, accelerates BC proliferation and progression (93–98). HOTAIR is transcriptionally induced by TGFβ in BC cells and is required for EMT and stemness acquisition (99). By multivariate analysis of clinical samples from BC patients receiving neoadjuvant chemotherapy, Xu et al. showed that expression of an oncogenic lncRNA, EPIC1, was associated with ER negativity, HER-2 positivity, higher Ki67 expression, and higher histologic grade (100). These findings suggest that lncRNAs have the potential to serve as prognostic biomarkers and novel therapeutic targets for BC. Currently, several ongoing clinical trials are investigating the role of circulating lncRNAs as potential biomarkers for predicting the risk of recurrence in breast cancer.
3 Impact of epigenetics on cancer-immunity cycle and immune environment in breast cancer
The normal mammary gland ductal layer contains different subsets of immune cells including lymphocytes (B cells and T cells), neutrophils, eosinophils, basophils, mast cells, monocytes, macrophages, dendritic cells, natural killer (NK) cells (101, 102). These immune cells are recruited to distinctive sites during the various stages of mammary gland development where they provide pathogen-specific, long-lasting protection to the breast epithelial layer and help eliminate transformed cells (103). Epigenetic modulations fine-tune gene expression patterns to maintain appropriate differentiation and function of immune cells in a homeostatic environment (104, 105). Growing evidence suggests that epigenetic defects in BC impair immune cell function and antitumor immunity, leading to a more immunosuppressive environment and tumor escape from immune surveillance (106, 107). In this section, we discuss the functional interaction between breast tumor cells and immune cells and how breast tumor hijacks epigenetic mechanisms to escape immune restriction and elimination (Figure 2).
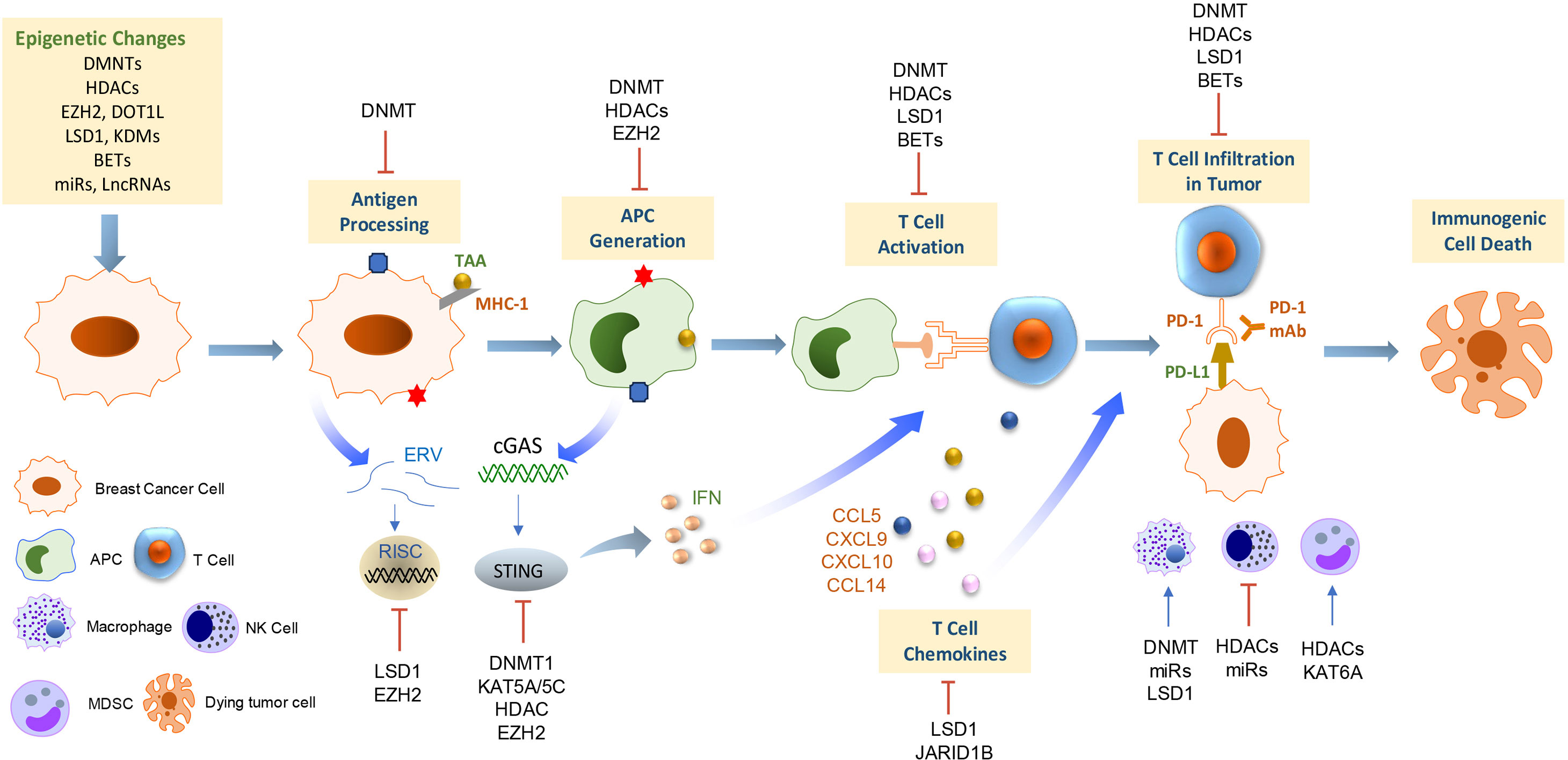
Figure 2 Epigenetic regulation of antitumor immunity and response to immunotherapy in breast cancer. This scheme summarizes the current understanding of the mechanisms underlying the epigenetic regulations on immune cell functions and antitumor immunogenicity in the context of the breast tumor microenvironment. APC, antigen-presenting cells; MDSC, myeloid-derived suppressor cell; NK cells, natural killer cells; TAA, tumor-associated antigen; ERV, endogenous retroviruses; cGAS, cyclic GMP–AMP synthase; STING, stimulator of interferon genes; IFN, interferon.
In 2013, Chen and Mellman described the cancer-immunity cycle model of a series of self-sustaining stepwise events by which adaptive immune responses lead to effective elimination of tumor cells (108). Epigenetic regulations are pivotal factors for many processes in the cancer-immunity cycle (109). A variety of tumor-associated antigens (TAA) that are frequently expressed in BC cells can elicit strong antitumor immune responses. For example, cancer-testis antigens (CTAs) are a group of TAAs whose expression is restricted to malignant cells as well as some germline cells, thereby representing highly promising therapeutic targets for cancer immunotherapy because of their high tumor-specificity pattern and immunogenic nature (110, 111). VCX2, a member of the VCX/Y cancer/testis antigen family, is frequently repressed by promoter CpG methylation. Treatment with the DNMTi, guadecitabine, led to reactivation of VCX2 in several BC cell lines and a patient-derived xenograft (PDX) (112). Antigen presentation by major histocompatibility complex (MHC) is a critical response for adaptive antitumor immunity. MHC class I (MHC-I) molecules present peptide fragments derived from intracellular proteins and transport them to the cell surface (113). MHC-I loss or downregulation has been described as a major escape mechanism for tumors to evade immune surveillance and immunotherapy (114, 115). MHC-I genes are often methylated in human breast cancers, and treatment with guadecitabine can upregulate MHC-I expression in response to interferon γ that potentiates CD8+ T cell activity (116). In a high-throughput library screen of 141 epigenetic compounds, GSK-LSD1 (LSD1i), CUDC-101(inhibitor of HDAC, EGFR, and HER2) and BML-210 (HDACi) displayed significant antitumor effects and up-regulated MHC-I-mediated antigen presentation in orthotopic mammary gland tumors in mice (117). Recent research has explored the function and roles of endogenous retroviruses (ERV) as an additional source of tumor antigens (118). Deblois et al. showed that EZH2 suppressed ERV expression through H3K27me3 deposition, which prevented activation of the viral mimicry response and eluded the immune surveillance in chemotherapy-resistant BC (119). Induction of ERVs by LSD1 inhibition resulted in double-stranded RNA (dsRNA) stress and activation of type 1 IFN, leading to enhanced anti-tumor T cell immunity against growth of BC cells (120). These pieces of evidence suggest that epigenetic drugs can be leveraged to enhance antitumor immunity by increasing TAA tumor antigen expression and presentation.
cGAS (cyclic GMP-AMP synthase) and STING (signaling stimulator of interferon genes) serve as cytosolic DNA sensors in both innate and adaptive immune responses (121, 122). The cytosolic DNA sensing in tumor cells by the cGAS-STING pathway triggers a signaling cascade to induce IFN production and T cell priming that arouses an antitumor immune response (123). Therefore, STING has been regarded as a master regulator of antitumor immunity (124). However, STING is frequently downregulated or silenced in BC and immune cells (125). Amplification and overexpression of MYC proto-oncogene in TNBC prevent the cGAS-STING-dependent innate immunity through binding to DNMT1 promoter and upregulating DNMT1 transcription (126). A recent study revealed that the expression of STING is epigenetically suppressed by the H3K4 demethylases, KDM5B and KDM5C. The use of KDM5 inhibitor or a combination with STING agonists in BC cells triggered a robust induction of STING expression and interferon in a cytosolic DNA-dependent manner (127). Moreover, combined use of EZH2 and HDACi induced IFI16-mediated STING activation and overcame resistance of HER2+ breast tumor to trastuzumab (128). These findings highlight the important roles of epigenetic reprogramming in modulating the cGAS-STING signaling cascade in BC.
After priming in lymph nodes, chemokine-mediated recruitment of CD8+ cytotoxic T lymphocytes (CTLs) into tumor bed is believed to be the most potent effector in the antitumor immune response. CTLs primarily eliminate cancer cells through granule exocytosis (perforin and granzymes) and death ligands, which trigger the inherent apoptotic response. Robust levels of tumor-infiltrating lymphocytes (TILs) are positively associated with favorable clinical outcomes and improved response to chemotherapy and immunotherapy in BC (129, 130). Increased expression of the C-X-C motif chemokine ligands 9 and 10 (CXCL9 and CXCL10), C-C motif chemokine ligand 5 (CCL5), and IFNγ is associated with enhanced presentation of CTLs in the TME. Our study has shown that LSD1 overexpression is negatively correlated with the level of CD8+ T cell attracting chemokines (CCL5, CXCL9, CXCL10) in TNBC (61). Re-expression of these chemokines by LSD1 depletion is associated with increased H3K4me2 levels at proximal promoter regions of chemokine genes. LSD1 inhibitor enhances CD8+ T cell migration which was blocked by concurrent treatment with siRNA or inhibitor of chemokine receptors on T cells, indicating a critical role of LSD1 in governing CD8+ lymphocyte trafficking to the tumor cluster (61). Another study showed that targeting LSD1 induces infiltration of IFNγ/TNFα-expressing CD8+ T cells in mice bearing 4T1 ICI-resistant tumors, which is further augmented by combined immunotherapy (131). These findings suggest the potential of targeting LSD1 in boosting antitumor immunity and overcoming resistance to immunotherapies (132). Li et al. found that JmjC demethylase JARID1B binds to LSD1/NuRD and suppresses angiogenesis and metastasis in BC cells by repressing CCL14, a chemokine-promoting the activation of immune cells (133). Chemokine (C-C motif) ligand 7 (CCL7) is a chemotactic factor and potent attractant of monocytes which plays an important role in regulating antitumor immunity and response to ICI therapy (134, 135). H3K4me3-targeting histone demethylase, Fbxl10, has been found to be recruited to CCL7 promoter and knockdown of Fbxl10 led to inverse regulation of CCL7 (136). Deng et al. showed that hypomethylation of CG sites (cg05224770 and cg07388018) was associated with upregulation of Chemokine Receptor 7 (CCR7) in BC (137). These studies suggest that targeting an aberrant chemokine network in combination with other immunotherapies may augment antitumor immunity and produce clinical benefits in patients with breast cancer.
4 Epigenetic modulation of immunogenic cell death in breast cancer
Immune checkpoint molecules (ICMs) expressed by innate immune cells exert inhibitory effects on adaptive immune responses. ICM family includes programmed cell death protein 1 (PD-1), cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), lymphocyte-activation gene 3 (LAG-3), T cell immunoglobulin and mucin-domain containing-3 (TIM-3), and others (138). While ICMs are crucial regulators for self-tolerance and may prevent the immune system from attacking cells indiscriminately, tumor cells frequently evolve to evade immune surveillance by stimulating immune checkpoint targets and passing an “off” signal to the T cells (139). Gene expression analysis using the METABRIC microarray dataset found that the expression of ICMs is upregulated in many breast tumors, more significantly in basal-like and HER2-enriched subtypes (140). Sasidharan et al. showed that DNA hypomethylation and decreased expression of repressive histone marks H3K27me3 and H3K9me3 contribute to the upregulation of PD-1, CTLA-4, TIM-3, and LAG-3 in breast tumor tissues (141). Overexpression of PD-L1 (CD274), the ligand of PD-1, has been proven to promote immune evasion and tumor growth through enhancing T cell apoptosis in many types of cancer (142). DNA demethylation by ten-eleven translocation (TET) methylcytosine dioxygenases could lead to overexpression of IMCs in BC patients’ blood and tumor tissues (143). Darvin et al. reported that overexpression of PD-L1 in breast cancer-like stem cells was partially independent of promoter CpG methylation and more likely due to posttranslational histone modifications such as lysine tri-methylation and acetylation (144). Although PD-L1 has been correlated with poor prognosis in BC, several clinical trials have reported the positive association of PD-L1 expression with higher response rates to anti-PD-1/PD-L1 antibody therapy (145, 146). Based on these findings, some epigenetic agents could be used to improve antitumor immunity by suppressing the expression of ICMs, while others could be used in combination with ICIs to enhance tumor response to immunotherapy by upregulation of ICM expression.
Myeloid-derived suppressor cells (MDSCs), which commonly express Siglec-3/CD33 and lack HLA-DR and lineage markers, are a group of immature myeloid cells that potently suppress T cell activity and thus contribute to the immune escape of tumors (147). Results from several studies have suggested that histone modifications exert a range of effects on the immunosuppressive function and expansion of MDSCs in BC. The HDACi vorinostat reduces MDSC accumulation in the spleen, blood, and tumor bed but increases the proportion of IFN-γ- or CD8+ T cells in BALB/C mice bearing 4T1 tumors (148). Acetylation of SMAD3 by histone lysine acetyltransferase 6A (KAT6A) promotes metastasis of TNBC through the recruitment of MDSCs. Inhibition of KAT6A in combination with anti-PD-L1 therapy in TNBC-bearing mice reduced MDSC recruitment, upregulated cytokine expression (IL-6, IL-22, and TNFα), and markedly attenuated metastasis of TNBC tumors (149).
Natural Killer (NK) cells are potent effectors of the innate immune system which are best known for killing infected and cancer cells that lack MCH restriction or prior sensitization (150). Chan et al. demonstrated that DNMTi altered gene expression of inhibitory receptors of natural killer (NK) cells. Combining DNMTi with antibodies targeting NK cell inhibitory receptors, TIGIT or KLRG1, effectively reduced the metastatic potential of BC cells (151). Simultaneous implantation of mesenchymal stem cells (MSCs) overexpressing Sirt1 suppressed 4T1 breast tumor growth in mice via chemokine (CXCL10 and IFN-γ)-dependent NK cells recruitment (152). NKG2D is one of the major activating receptors of NK cells that binds to several ligands NKG2DLs. One study showed that silencing of NKG2DL inhibited the miR-17-92 cluster, especially miR-20a, promoting NK cell-mediated cytotoxicity against BC cells and inhibiting immune escape. Treatment with HDACi inhibits members of the miR-17-92 cluster leading to the induction of NKG2DL expression in multiple BC cell lines, suggesting that targeting specific miRNAs with epigenetic modifying drugs may represent a novel approach for augmenting NK cell-mediated antitumor immunity in BC (153).
Tumor-associated macrophages (TAMs) are pivotal in tumor development and anti-cancer therapy. TAMs display a high degree of cellular plasticity and exert context-dependent anti-tumor (M1-like) or pro-tumor (M2-like) functions and polarization states (154). Epigenetic mechanisms have emerged as key controllers of macrophage activation and polarization. For instance, Li et al. reported that TAMs increase DNMT1 expression in breast cancer cells via the IL-6-pSTAT3-ZEB1-DNMT1 axis and DNMT1 is required for TAM-mediated BC metastasis (155). A recent study suggested that miR-182 promotes macrophage alternative activation and drives breast tumor development. Targeting miR-182 inhibitors through delivering TAM-targeting exosomes into macrophages can effectively suppress M2 polarization and block BC progression (156). Another study showed that miR-200c promotes TNBC progression by upregulation of plasminogen activator inhibitor-2 (PAI-2) and M2 phenotype macrophage polarization (157). Hey et al. used 4T1 mouse model and single-cell gene expression data to identify a TAM-specific signature which is associated with altered cytokine production and immune factors in TNBC (158). Moreover, LSD1-CoREST complex has been found to have a role in switching macrophage polarization programs and LSD1 inhibition can prime macrophages toward an anti-tumor M1-like phenotype in TNBC (159). An improved understanding of the complex interactions between TAMs and epigenetic changes is vital to identifying novel TAM-based epigenetic therapies.
5 Combination of epigenetic drugs with immunotherapy in breast cancer: preclinical studies and clinical trials
Recent progress in immunotherapy represents one of the most encouraging advances in the cancer therapeutic field. However, the clinical results suggest that the general response rate for breast cancer patients treated with immune checkpoint inhibitors is around 10-20% (17, 160, 161). While TNBC is more likely to respond to immunotherapy, the overall response rate is still low. Therefore, developing rational combination therapies is a critical approach to improve the efficacy of immunotherapy in BC. The use of epigenetic drugs to prime for response to immunotherapy might lead to new strategies to boost anticancer immune responses and efficacy of immunotherapy.
5.1 Targeting DNA hypermethylation to improve immunotherapy
One preclinical study showed that combined treatment with anti-PD-1 and anti-CTLA-4 antibodies failed to block the growth and metastasis of 4T1 tumors, whereas cotreatment with DNMTi Azacitidine, anti-PD-1 and anti-CTLA-4 antibodies significantly improved treatment outcomes with more than 80% reduction in tumor burden (162). Another study demonstrated that 5-aza-2’-deoxycytidine significantly enhanced the tumor cell-killing effects of MAGE-As co-antigen peptide-specific CTLs and anti-CTLA-4 monoclonal antibody in BC cells (163). Recently, several studies using a combination of DNA methylation inhibitors and immunotherapy have advanced into clinical investigations for breast cancer patients. As shown in Table 3, one completed phase II trial has evaluated the antitumor activity of DNA hypomethylating agent azacitidine (CC-486) in combination with anti-PD-1 antibody (durvalumab) in metastatic ER-positive/HER2-negative BC and other types of cancer (NCT02811497). The published data indicated that the combination was considered safe at the dosages delivered. Although the study did not observe robust pharmacodynamic or clinical activity, lessons learned from this biomarker-rich study will inform continued drug development efforts using these agents (164). Clinical trials conducted in China have assessed the feasibility and safety of anti-PD-1 antibody (camrelizumab) alone or in combination with decitabine and/or chemotherapy in a variety of refractory malignancies including BC (NCT02961101). A completed study in patients with classic Hodgkin lymphoma (cHL) showed that DNMTi plus anti-PD-1 therapy is associated with high response rates and long-term benefits in patients with relapsed/refractory cHL who didn’t respond to PD-1 antibody therapy (165). Several other ongoing studies are investigating the efficacy of decitabine with a chemotherapy/pembrolizumab regimen for patients with locally advanced HER2-negative BC and TNBC (NCT02957968, NCT05673200). The primary goals of these trials are to determine if DNMTi could enhance the antitumor efficacy of immunotherapy and chemotherapy.
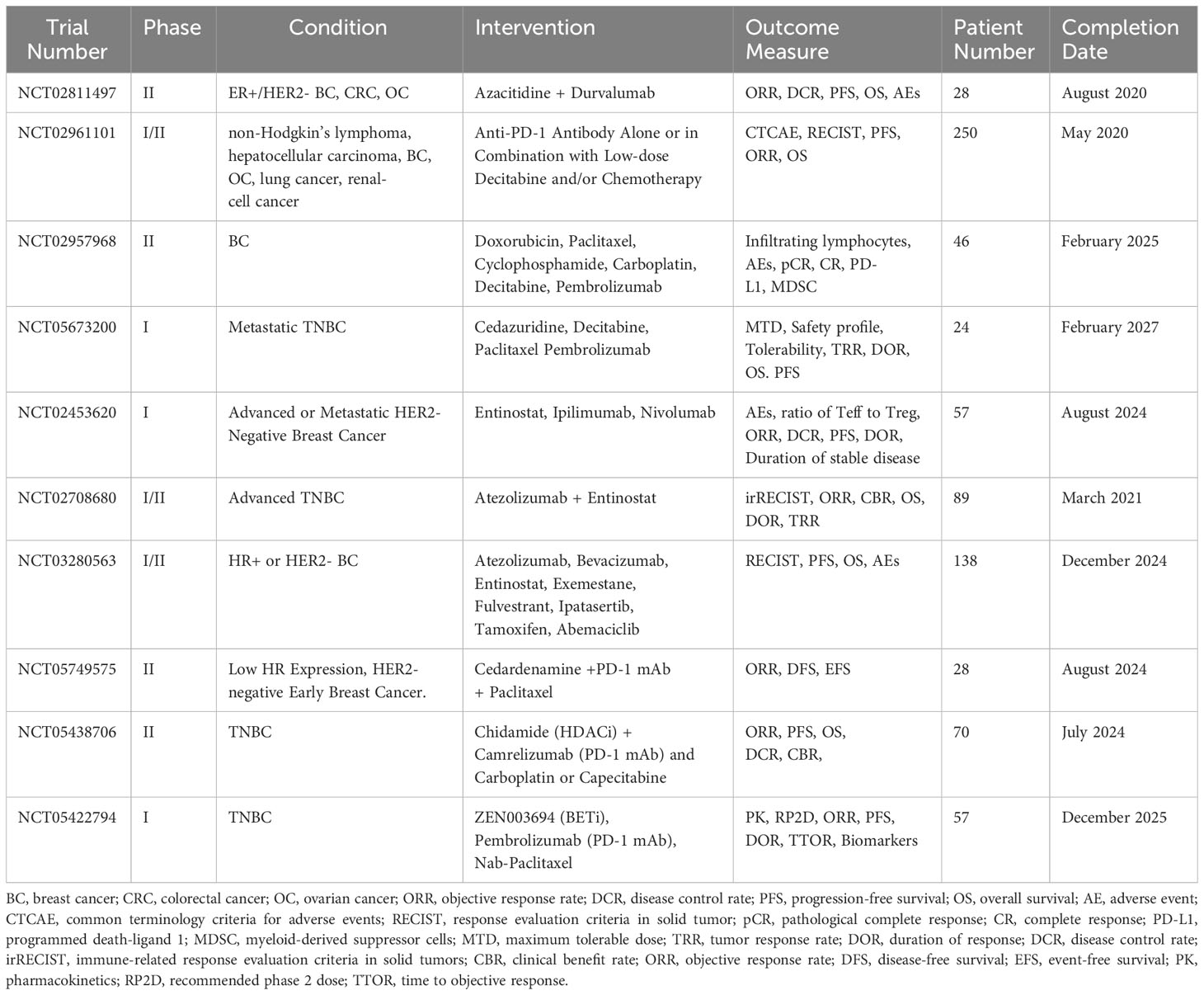
Table 3 Some completed and ongoing clinical trials of epigenetic drugs and immunotherapy in breast cancer (clinicaltrials.gov).
5.2 Combination of HDAC inhibitor with immune checkpoint blockade in breast cancer
A preclinical study demonstrated that HDACi improved in vivo response to PD-1/CTLA-4 blockades in TNBC by up-regulating PD-L1/HLA-DR and down-regulating CD4+ Foxp3+ regulatory T cells (Treg) (166). Another study reported that the combination of HDACi with ICIs altered the infiltration and function of innate immune cells, allowing for a more robust adaptive immune response through suppression of MDSCs and immune-resistant breast tumors (167). Kim et al. demonstrated that the combination of entinostat with CTLA-4 and PD-1 antibodies blocked the growth and metastases of 4T1 breast tumors in mice via suppression of MDSCs (162). The combined use of HDACi as a potential booster for immune checkpoint blockade has been assessed in multiple BC clinical trials (Table 3). One phase I study evaluated the side effects and optimal dose of entinostat and nivolumab (anti-PD-1 mAb) when given together with ipilimumab (anti-CTLA-4) in treating patients with unresectable or metastatic HER2-negative BC (NCT02453620). The preliminary evidence of both clinical efficacy and immune modulation shows that combining entinostat with nivolumab and ipilimumab was safe and tolerable with expected rates of immune-related adverse events (168). This drug combination is being evaluated further in an expansion cohort of HER2-postive BC patients. However, another phase II, randomized, placebo-controlled, double-blinded, multicenter study of atezolizumab (anti-PD-L1 mAb) with entinostat in patients with advanced TNBC did not find prolonged median PFS compared with entinostat alone and the combination resulted in greater toxicity (NCT02708680). One ongoing study is evaluating the efficacy of several immunotherapy-based combination treatments, including entinostat in participants with inoperable locally advanced or metastatic BC who have progressed during or following treatment with a cyclin-dependent kinase (CDK) 4/6 inhibitor (palbociclib, ribociclib, or abemaciclib) (NCT03280563). Other ongoing clinical trials are testing if Chidamide, an HDACi developed in China, could sensitize breast tumors to the antitumor effects of PD-1 antibody and chemotherapeutic agents such as paclitaxel, carboplatin, or capecitabine in relapsed/metastatic HER2-negative or TNBC (NCT05749575, NCT05438706).
5.3 Emerging new epigenetic targets for breast cancer immunotherapy
We have demonstrated that inhibition of LSD1 by RNAi or small molecule inhibitors reactivates T-cell attracting chemokines which may in turn stimulate CD8+ T cell infiltration and sensitize poorly immunogenic TNBC tumors to PD-1 blockade (61). This combination approach also augmented the ratio of CD8+/CD4+ T cells in lymph tissues adjacent to tumor sites at mouse mammary glands, which is considered an important marker of immunological defense against tumor cell dissemination (61). These findings support the promise of LSD1 inhibition as an effective approach to overcoming resistance to ICIs in BC treatment. One ongoing Phase I/II study sponsored by the University of Washington is evaluating the effect of an orally active LSD1 inhibitor, bomedemstat (IMG-7289), and immunotherapy in patients with newly diagnosed extensive stage small cell lung cancer (Es-SCLC) (NCT05191797). It is anticipated that the outcome of this clinical study will provide beneficial information for trials using LSD1 inhibitors in other types of cancer.
BET inhibitors (BETi) have emerged as a novel class of epigenetic drugs that acts as an anti-cancer agent by blocking VEGF production and down-regulating MYC expression in different types of cancer, including BC (169). A number of inhibitors targeting BET have been reported, and many preclinical studies and clinical trials are under way to examine the anti-cancer efficacy of BETi alone or in combination with other therapeutic agents. A recent study has indicated that BRD4 inhibition suppressed PD-L1 expression and changed the proportions of T lymphocyte subsets in mice bearing TNBC tumors (170). Lai et al. developed a mathematical model to show that the BETi and CTLA-4 inhibitor are positively correlated in tumor inhibition and sustain cytotoxic T cell function in BC (171). Furthermore, BET inhibition suppresses the PD-1/PD-L1 axis, improves tumor cell-specific T cell cytotoxic function, and overcomes tumor-mediated T cell exhaustion in TNBC (172). NCI is sponsoring a phase Ib trial using a BET inhibitor, ZEN003694, in combination with pembrolizumab (PD-1 mAb) and nab-paclitaxel for patients with locally advanced or metastatic TNBC (NCT05422794). The outcomes of these trials will provide useful information to address whether targeting dysregulated histone marks would enhance the efficacy of ICIs for breast cancer patients.
6 Conclusions and future prospects
Over the past decades, the rapid development in understanding of epigenetic alterations in breast cancer has facilitated the development of many new diagnostic and treatment tools for this disease. The great potential for epigenetic therapy lies in the knowledge that epigenetic changes can be reversed, allowing restoration of function of aberrantly affected genes in tumor cells. Many drugs targeting dysregulated epigenetic regulators have entered clinical use in the treatment of hematological cancers. The FDA has approved EZH2 inhibitor Tazemetostat for epithelioid sarcoma, the first approval of an epigenetic drug for solid tumor. Increasing and compelling evidence suggests that epigenetic therapy has the potential to convert an immune-repressive (cold) breast tumor into an immune-permissive (hot) one. Recent studies have identified many new roles for epigenetic alterations in promoting immune escape that may represent an opportunity to identify novel therapies for immunotherapy-refractory breast cancer. Given the fact that epigenetic alterations exert effect on both breast tumor cells and immune cells, it is hoped that the combination of epigenetic drugs with immunotherapies can consolidate the antitumor immune response, reprogram the immune suppressive microenvironment, and improve therapeutic efficacy. However, many challenges remain to be addressed and solved in the future. The ultimate goal is to mitigate the off-target effects of epi-drugs and explore more effective site-specific drug delivery approaches. A better understanding of the underlying mechanisms of epigenetic regulation in BC is key for the development of more specific epigenetic-based therapy. In recent years, the progress in epigenome-wide studies has led to the discovery of many previously unknown epigenetic modifiers, especially the histone modifying enzymes. These newly identified epigenetic targets have potential to serve as novel diagnostic and prognostic biomarkers for breast cancer. However, the similarity among enzyme families may elicit cross-reactivity of substrates and inhibitors and impair the specificity of epigenetic drugs. A better insight into the chromatin-context specificity of histone modifying enzymes would facilitate the development of more effective and selective inhibitors. As breast cancer is a heterogeneous disease with multiple subtypes and distinct biological features, we need to further elucidate the diversity of epigenetic regulation in the tumor immune environment. Finally, the epigenetic landscape in immunotherapy-refractory breast tumor populations should be further defined and characterized to identify unique biomarkers in order to optimize the combination of immunotherapy and epigenetic-based therapy with the long-term goal to expand the responder population and improve personalized precision medicine.
Author contributions
JY: Conceptualization, Writing – original draft. TG: Writing – review & editing. NC: Writing – review & editing. ND: Writing – review & editing, Funding acquisition. YH: Funding acquisition, Writing – review & editing, Conceptualization, Data curation, Resources, Software, Supervision, Visualization, Writing – original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by NIH/NCI grant CA260357 (YH) and the Breast Cancer Research Foundation (ND).
Acknowledgments
The authors acknowledge the support from Department of Internal Medicine, Division of Hematology, Oncology, and Blood and Marrow Transplantation, and Holden Comprehensive Cancer Center, University of Iowa.
Conflict of interest
The authors declare that this work was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer (2004) 4(2):143–53. doi: 10.1038/nrc1279
2. Jones PA, Baylin SB. The epigenomics of cancer. Cell (2007) 128(4):683–92. doi: 10.1016/j.cell.2007.01.029
3. Stearns V, Zhou Q, Davidson NE. Epigenetic regulation as a new target for breast cancer therapy. Cancer Invest (2007) 25(8):659–65. doi: 10.1080/07357900701719234
4. Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet (2002) 3(6):415–28. doi: 10.1038/nrg816
5. Kouzarides T. Chromatin modifications and their function. Cell (2007) 128(4):693–705. doi: 10.1016/j.cell.2007.02.005
6. Huang Y, Nayak S, Jankowitz R, Davidson NE, Oesterreich S. Epigenetics in breast cancer: what’s new? Breast Cancer Res (2011) 13(6). doi: 10.1186/bcr2925
7. Katz TA, Huang Y, Davidson NE, Jankowitz RC. Epigenetic reprogramming in breast cancer: from new targets to new therapies. Ann Med (2014) 46(6):397–408. doi: 10.3109/07853890.2014.923740
8. Bao X, Anastasov N, Wang Y, Rosemann M. A novel epigenetic signature for overall survival prediction in patients with breast cancer. J Transl Med (2019) 17(1):380. doi: 10.1186/s12967-019-2126-6
9. Terry MB, McDonald JA, Wu HC, Eng S, Santella RM. Epigenetic biomarkers of breast cancer risk: across the breast cancer prevention continuum. Adv Exp Med Biol (2016) 882:33–68. doi: 10.1007/978-3-319-22909-6_2
10. Rentscher KE, Bethea TN, Zhai W, Small BJ, Zhou X, Ahles TA, et al. Epigenetic aging in older breast cancer survivors and noncancer controls: preliminary findings from the Thinking and Living with Cancer Study. Cancer (2023) 129(17):2741–53. doi: 10.1002/cncr.34818
11. Sher G, Salman NA, Khan AQ, Prabhu KS, Raza A, Kulinski M, et al. Epigenetic and breast cancer therapy: Promising diagnostic and therapeutic applications. Semin Cancer Biol (2020). 82:152–65. doi: 10.1016/j.semcancer.2020.08.009
12. . Available at: https://clinicaltrials.gov/.
13. Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol (2020) 20(11):651–68. doi: 10.1038/s41577-020-0306-5
14. Kraehenbuehl L, Weng CH, Eghbali S, Wolchok JD, Merghoub T. Enhancing immunotherapy in cancer by targeting emerging immunomodulatory pathways. Nat Rev Clin Oncol (2022) 19(1):37–50. doi: 10.1038/s41571-021-00552-7
15. Kim R, Kin T. Current and future therapies for immunogenic cell death and related molecules to potentially cure primary breast cancer. Cancers (Basel) (2021) 13(19). doi: 10.3390/cancers13194756
16. Emens LA. Breast cancer immunotherapy: facts and hopes. Clin Cancer Res (2018) 24(3):511–20. doi: 10.1158/1078-0432.CCR-16-3001
17. Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med (2018) 379(22):2108–21. doi: 10.1056/NEJMoa1809615
18. Schmid P, Cortes J, Dent R, Pusztai L, McArthur H, Kummel S, et al. Event-free survival with pembrolizumab in early triple-negative breast cancer. N Engl J Med (2022) 386(6):556–67. doi: 10.1056/NEJMoa2112651
19. Gatti-Mays ME, Balko JM, Gameiro SR, Bear HD, Prabhakaran S, Fukui J, et al. If we build it they will come: targeting the immune response to breast cancer. NPJ Breast Cancer (2019) 5:37. doi: 10.1038/s41523-019-0133-7
20. Solinas C, Gombos A, Latifyan S, Piccart-Gebhart M, Kok M, Buisseret L. Targeting immune checkpoints in breast cancer: an update of early results. ESMO Open (2017) 2(5):e000255. doi: 10.1136/esmoopen-2017-000255
21. Bird AP. CpG-rich islands and the function of DNA methylation. Nature (1986) 321(6067):209–13. doi: 10.1038/321209a0
22. Yen RW, Vertino PM, Nelkin BD, Yu JJ, el-Deiry W, Cumaraswamy A, et al. Isolation and characterization of the cDNA encoding human DNA methyltransferase. Nucleic Acids Res (1992) 20(9):2287–91. doi: 10.1093/nar/20.9.2287
23. Lo PK, Sukumar S. Epigenomics and breast cancer. Pharmacogenomics (2008) 9(12):1879–902. doi: 10.2217/14622416.9.12.1879
24. Chatterji S, Krzoska E, Thoroughgood CW, Saganty J, Liu P, Elsberger B, et al. Defining genomic, transcriptomic, proteomic, epigenetic, and phenotypic biomarkers with prognostic capability in male breast cancer: a systematic review. Lancet Oncol (2023) 24(2):e74–85. doi: 10.1016/S1470-2045(22)00633-7
25. Garcia-Recio S, Hinoue T, Wheeler GL, Kelly BJ, Garrido-Castro AC, Pascual T, et al. Multiomics in primary and metastatic breast tumors from the AURORA US network finds microenvironment and epigenetic drivers of metastasis. Nat Cancer (2023) 4(1):128–47. doi: 10.1038/s43018-022-00491-x
26. Stefansson OA, Esteller M. Epigenetic modifications in breast cancer and their role in personalized medicine. Am J Pathol (2013) 183(4):1052–63. doi: 10.1016/j.ajpath.2013.04.033
27. Lin LH, Tran I, Yang Y, Shen G, Miah P, Cotzia P, et al. DNA methylation identifies epigenetic subtypes of triple-negative breast cancers with distinct clinicopathologic and molecular features. Mod Pathol (2023) 36(11):100306. doi: 10.1016/j.modpat.2023.100306
28. Yoo CB, Jeong S, Egger G, Liang G, Phiasivongsa P, Tang C, et al. Delivery of 5-aza-2’-deoxycytidine to cells using oligodeoxynucleotides. Cancer Res (2007) 67(13):6400–8. doi: 10.1158/0008-5472.CAN-07-0251
29. Yoo CB, Chuang JC, Byun HM, Egger G, Yang AS, Dubeau L, et al. Long-term epigenetic therapy with oral zebularine has minimal side effects and prevents intestinal tumors in mice. Cancer Prev Res (Phila) (2008) 1(4):233–40. doi: 10.1158/1940-6207.CAPR-07-0008
30. Yang X, Ferguson AT, Nass SJ, Phillips DL, Butash KA, Wang SM, et al. Transcriptional activation of estrogen receptor alpha in human breast cancer cells by histone deacetylase inhibition. Cancer Res (2000) 60(24):6890–4.
31. Nam JS, Ino Y, Kanai Y, Sakamoto M, Hirohashi S. 5-aza-2’-deoxycytidine restores the E-cadherin system in E-cadherin-silenced cancer cells and reduces cancer metastasis. Clin Exp Metastasis (2004) 21(1):49–56. doi: 10.1023/B:CLIN.0000017180.19881.c1
32. Suzuki H, Toyota M, Carraway H, Gabrielson E, Ohmura T, Fujikane T, et al. Frequent epigenetic inactivation of Wnt antagonist genes in breast cancer. Br J Cancer (2008) 98(6):1147–56. doi: 10.1038/sj.bjc.6604259
33. Mirza S, Sharma G, Pandya P, Ralhan R. Demethylating agent 5-aza-2-deoxycytidine enhances susceptibility of breast cancer cells to anticancer agents. Mol Cell Biochem (2010) 342(1-2):101–9. doi: 10.1007/s11010-010-0473-y
34. Sandhu R, Rivenbark AG, Coleman WB. Enhancement of chemotherapeutic efficacy in hypermethylator breast cancer cells through targeted and pharmacologic inhibition of DNMT3b. Breast Cancer Res Treat (2012) 131(2):385–99. doi: 10.1007/s10549-011-1409-2
35. Buocikova V, Longhin EM, Pilalis E, Mastrokalou C, Miklikova S, Cihova M, et al. Decitabine potentiates efficacy of doxorubicin in a preclinical trastuzumab-resistant HER2-positive breast cancer models. BioMed Pharmacother (2022) 147:112662. doi: 10.1016/j.biopha.2022.112662
36. Shan W, Jiang Y, Yu H, Huang Q, Liu L, Guo X, et al. HDAC2 overexpression correlates with aggressive clinicopathological features and DNA-damage response pathway of breast cancer. Am J Cancer Res (2017) 7(5):1213–26.
37. Li Y, Seto E. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb Perspect Med (2016) 6(10). doi: 10.1101/cshperspect.a026831
38. Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet (2009) 10(1):32–42. doi: 10.1038/nrg2485
39. Keen JC, Yan L, Mack KM, Pettit C, Smith D, Sharma D, et al. A novel histone deacetylase inhibitor, scriptaid, enhances expression of functional estrogen receptor alpha (ER) in ER negative human breast cancer cells in combination with 5-aza 2’-deoxycytidine. Breast Cancer Res Treat (2003) 81(3):177–86. doi: 10.1023/A:1026146524737
40. Zhou Q, Atadja P, Davidson NE. Histone deacetylase inhibitor LBH589 reactivates silenced estrogen receptor alpha (ER) gene expression without loss of DNA hypermethylation. Cancer Biol Ther (2007) 6(1):64–9. doi: 10.4161/cbt.6.1.3549
41. Pruitt K, Zinn RL, Ohm JE, McGarvey KM, Kang SH, Watkins DN, et al. Inhibition of SIRT1 reactivates silenced cancer genes without loss of promoter DNA hypermethylation. PloS Genet (2006) 2(3):e40. doi: 10.1371/journal.pgen.0020040
42. Stearns V, Jacobs LK, Fackler M, Tsangaris TN, Rudek MA, Higgins M, et al. Biomarker modulation following short-term vorinostat in women with newly diagnosed primary breast cancer. Clin Cancer Res (2013) 19(14):4008–16. doi: 10.1158/1078-0432.CCR-13-0033
43. Luu TH, Morgan RJ, Leong L, Lim D, McNamara M, Portnow J, et al. A phase II trial of vorinostat (suberoylanilide hydroxamic acid) in metastatic breast cancer: a California Cancer Consortium study. Clin Cancer Res (2008) 14(21):7138–42. doi: 10.1158/1078-0432.CCR-08-0122
44. Connolly RM, Zhao F, Miller KD, Lee MJ, Piekarz RL, Smith KL, et al. E2112: randomized phase III trial of endocrine therapy plus entinostat or placebo in hormone receptor-positive advanced breast cancer. A trial of the ECOG-ACRIN cancer research group. J Clin Oncol (2021). 39(28):3171–81. doi: 10.1200/JCO.21.00944
45. Michalak EM, Visvader JE. Dysregulation of histone methyltransferases in breast cancer - Opportunities for new targeted therapies? Mol Oncol (2016) 10(10):1497–515. doi: 10.1016/j.molonc.2016.09.003
46. Ferrari KJ, Scelfo A, Jammula S, Cuomo A, Barozzi I, Stutzer A, et al. Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity. Mol Cell (2014) 53(1):49–62. doi: 10.1016/j.molcel.2013.10.030
47. Bachmann IM, Halvorsen OJ, Collett K, Stefansson IM, Straume O, Haukaas SA, et al. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J Clin Oncol (2006) 24(2):268–73. doi: 10.1200/JCO.2005.01.5180
48. Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci U S A. (2003) 100(20):11606–11. doi: 10.1073/pnas.1933744100
49. Jones B, Su H, Bhat A, Lei H, Bajko J, Hevi S, et al. The histone H3K79 methyltransferase Dot1L is essential for mammalian development and heterochromatin structure. PloS Genet (2008) 4(9):e1000190. doi: 10.1371/journal.pgen.1000190
50. Oktyabri D, Ishimura A, Tange S, Terashima M, Suzuki T. DOT1L histone methyltransferase regulates the expression of BCAT1 and is involved in sphere formation and cell migration of breast cancer cell lines. Biochimie (2016) 123:20–31. doi: 10.1016/j.biochi.2016.01.005
51. Nassa G, Salvati A, Tarallo R, Gigantino V, Alexandrova E, Memoli D, et al. Inhibition of histone methyltransferase DOT1L silences ERalpha gene and blocks proliferation of antiestrogen-resistant breast cancer cells. Sci Adv (2019) 5(2):eaav5590. doi: 10.1126/sciadv.aav5590
52. Zhang L, Deng L, Chen F, Yao Y, Wu B, Wei L, et al. Inhibition of histone H3K79 methylation selectively inhibits proliferation, self-renewal and metastatic potential of breast cancer. Oncotarget (2014) 5(21):10665–77. doi: 10.18632/oncotarget.2496
53. Byun WS, Kim WK, Han HJ, Chung HJ, Jang K, Kim HS, et al. Targeting histone methyltransferase DOT1L by a novel psammaplin A analog inhibits growth and metastasis of triple-negative breast cancer. Mol Ther Oncol (2019) 15:140–52. doi: 10.1016/j.omto.2019.09.005
54. Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell (2004) 119(7):941–53. doi: 10.1016/j.cell.2004.12.012
55. Lee MG, Wynder C, Cooch N, Shiekhattar R. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature (2005) 437(7057):432–5. doi: 10.1038/nature04021
56. Huang Y, Marton LJ, Woster PM, Casero RA. Polyamine analogues targeting epigenetic gene regulation. Essays Biochem (2009) 46:95–110. doi: 10.1042/bse0460007
57. Amente S, Lania L, Majello B. The histone LSD1 demethylase in stemness and cancer transcription programs. Biochim Biophys Acta (2013) 1829(10):981–6. doi: 10.1016/j.bbagrm.2013.05.002
58. Lan F, Nottke AC, Shi Y. Mechanisms involved in the regulation of histone lysine demethylases. Curr Opin Cell Biol (2008) 20(3):316–25. doi: 10.1016/j.ceb.2008.03.004
59. Serce N, Gnatzy A, Steiner S, Lorenzen H, Kirfel J, Buettner R. Elevated expression of LSD1 (Lysine-specific demethylase 1) during tumour progression from pre-invasive to invasive ductal carcinoma of the breast. BMC Clin Pathol (2012) 12:13. doi: 10.1186/1472-6890-12-13
60. Lim S, Janzer A, Becker A, Zimmer A, Schule R, Buettner R. Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis (2010) 31:512–20. doi: 10.1093/carcin/bgp324
61. Qin Y, Vasilatos SN, Chen L, Wu H, Cao Z, Fu Y, et al. Inhibition of histone lysine-specific demethylase 1 elicits breast tumor immunity and enhances antitumor efficacy of immune checkpoint blockade. Oncogene (2019) 38(3):390–405. doi: 10.1038/s41388-018-0451-5
62. Nagasawa S, Sedukhina AS, Nakagawa Y, Maeda I, Kubota M, Ohnuma S, et al. LSD1 overexpression is associated with poor prognosis in basal-like breast cancer, and sensitivity to PARP inhibition. PloS One (2015) 10(2):e0118002. doi: 10.1371/journal.pone.0118002
63. Karytinos A, Forneris F, Profumo A, Ciossani G, Battaglioli E, Binda C, et al. A novel mammalian flavin-dependent histone demethylase. J Biol Chem (2009) 284(26):17775–82. doi: 10.1074/jbc.M109.003087
64. Huang Y, Yin Y, Sun M. Targeting LSD2 in breast cancer. Aging (Albany NY). (2018) 10(1):11–2. doi: 10.18632/aging.101371
65. Chen L, Vasilatos SN, Qin Y, Katz TA, Cao C, Wu H, et al. Functional characterization of lysine-specific demethylase 2 (LSD2/KDM1B) in breast cancer progression. Oncotarget (2017) 8(47):81737–53. doi: 10.18632/oncotarget.19387
66. Katz TA, Vasilatos SN, Harrington E, Oesterreich S, Davidson NE, Huang Y. Inhibition of histone demethylase, LSD2 (KDM1B), attenuates DNA methylation and increases sensitivity to DNMT inhibitor-induced apoptosis in breast cancer cells. Breast Cancer Res Treat (2014) 146(1):99–108. doi: 10.1007/s10549-014-3012-9
67. Dimitrova E, Turberfield AH, Klose RJ. Histone demethylases in chromatin biology and beyond. EMBO Rep (2015) 16(12):1620–39. doi: 10.15252/embr.201541113
68. Accari SL, Fisher PR. Emerging roles of jmjC domain-containing proteins. Int Rev Cell Mol Biol (2015) 319:165–220. doi: 10.1016/bs.ircmb.2015.07.003
69. Klose RJ, Kallin EM, Zhang Y. JmjC-domain-containing proteins and histone demethylation. Nat Rev Genet (2006) 7(9):715–27. doi: 10.1038/nrg1945
70. Xie G, Liu X, Zhang Y, Li W, Liu S, Chen Z, et al. UTX promotes hormonally responsive breast carcinogenesis through feed-forward transcription regulation with estrogen receptor. Oncogene (2017) 36(39):5497–511. doi: 10.1038/onc.2017.157
71. Yamane K, Tateishi K, Klose RJ, Fang J, Fabrizio LA, Erdjument-Bromage H, et al. PLU-1 is an H3K4 demethylase involved in transcriptional repression and breast cancer cell proliferation. Mol Cell (2007) 25(6):801–12. doi: 10.1016/j.molcel.2007.03.001
72. Varghese B, Del Gaudio N, Cobellis G, Altucci L, Nebbioso A. KDM4 involvement in breast cancer and possible therapeutic approaches. Front Oncol (2021) 11:750315. doi: 10.3389/fonc.2021.750315
73. Benedetti R, Dell’Aversana C, De Marchi T, Rotili D, Liu NQ, Novakovic B, et al. Inhibition of histone demethylases LSD1 and UTX regulates ERalpha signaling in breast cancer. Cancers (Basel) (2019) 11(12). doi: 10.3390/cancers11122027
74. Wu SY, Chiang CM. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J Biol Chem (2007) 282(18):13141–5. doi: 10.1074/jbc.R700001200
75. Belkina AC, Denis GV. BET domain co-regulators in obesity, inflammation and cancer. Nat Rev Cancer (2012) 12(7):465–77. doi: 10.1038/nrc3256
76. Andrikopoulou A, Liontos M, Koutsoukos K, Dimopoulos MA, Zagouri F. The emerging role of BET inhibitors in breast cancer. Breast (2020) 53:152–63. doi: 10.1016/j.breast.2020.08.005
77. Perez-Pena J, Paez R, Nieto-Jimenez C, Sanchez VC, Galan-Moya EM, Pandiella A, et al. Mapping Bromodomains in breast cancer and association with clinical outcome. Sci Rep (2019) 9(1):5734. doi: 10.1038/s41598-019-41934-3
78. Khandekar D, Tiriveedhi V. Role of BET inhibitors in triple negative breast cancers. Cancers (Basel) (2020) 12(4). doi: 10.3390/cancers12040784
79. Li Z, Liu P, Chen W, Liu X, Tong F, Sun J, et al. Hypoxia-cleavable and specific targeted nanomedicine delivers epigenetic drugs for enhanced treatment of breast cancer and bone metastasis. J Nanobiotechnol (2023) 21(1):221. doi: 10.1186/s12951-023-01939-7
80. Hayes J, Peruzzi PP, Lawler S. MicroRNAs in cancer: biomarkers, functions and therapy. Trends Mol Med (2014) 20(8):460–9. doi: 10.1016/j.molmed.2014.06.005
81. Yan LX, Huang XF, Shao Q, Huang MY, Deng L, Wu QL, et al. MicroRNA miR-21 overexpression in human breast cancer is associated with advanced clinical stage, lymph node metastasis and patient poor prognosis. RNA (2008) 14(11):2348–60. doi: 10.1261/rna.1034808
82. Chen J, Wang BC, Tang JH. Clinical significance of microRNA-155 expression in human breast cancer. J Surg Oncol (2012) 106(3):260–6. doi: 10.1002/jso.22153
83. Zhang J, Yang J, Zhang X, Xu J, Sun Y, Zhang P. MicroRNA-10b expression in breast cancer and its clinical association. PloS One (2018) 13(2):e0192509. doi: 10.1371/journal.pone.0192509
84. Wu Y, Shi W, Tang T, Wang Y, Yin X, Chen Y, et al. miR-29a contributes to breast cancer cells epithelial-mesenchymal transition, migration, and invasion via down-regulating histone H4K20 trimethylation through directly targeting SUV420H2. Cell Death Dis (2019) 10(3):176. doi: 10.1038/s41419-019-1437-0
85. Maryam M, Naemi M, Hasani SS. A comprehensive review on oncogenic miRNAs in breast cancer. J Genet (2021) 100. doi: 10.1007/s12041-021-01265-7
86. Yu N, Huangyang P, Yang X, Han X, Yan R, Jia H, et al. microRNA-7 suppresses the invasive potential of breast cancer cells and sensitizes cells to DNA damages by targeting histone methyltransferase SET8. J Biol Chem (2013) 288(27):19633–42. doi: 10.1074/jbc.M113.475657
87. Uhlmann S, Zhang JD, Schwager A, Mannsperger H, Riazalhosseini Y, Burmester S, et al. miR-200bc/429 cluster targets PLCgamma1 and differentially regulates proliferation and EGF-driven invasion than miR-200a/141 in breast cancer. Oncogene (2010) 29(30):4297–306. doi: 10.1038/onc.2010.201
88. Kawaguchi T, Yan L, Qi Q, Peng X, Gabriel EM, Young J, et al. Overexpression of suppressive microRNAs, miR-30a and miR-200c are associated with improved survival of breast cancer patients. Sci Rep (2017) 7(1):15945. doi: 10.1038/s41598-017-16112-y
89. Wang W, Luo YP. MicroRNAs in breast cancer: oncogene and tumor suppressors with clinical potential. J Zhejiang Univ Sci B (2015) 16(1):18–31. doi: 10.1631/jzus.B1400184
90. Si ML, Zhu S, Wu H, Lu Z, Wu F, Mo YY. miR-21-mediated tumor growth. Oncogene (2007) 26(19):2799–803. doi: 10.1038/sj.onc.1210083
91. Saito Y, Liang G, Egger G, Friedman JM, Chuang JC, Coetzee GA, et al. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell (2006) 9(6):435–43. doi: 10.1016/j.ccr.2006.04.020
92. Tutar L, Tutar E, Tutar Y. MicroRNAs and cancer; an overview. Curr Pharm Biotechnol (2014) 15(5):430–7. doi: 10.2174/1389201015666140519095304
93. Zhang T, Hu H, Yan G, Wu T, Liu S, Chen W, et al. Long non-coding RNA and breast cancer. Technol Cancer Res Treat (2019) 18:1533033819843889. doi: 10.1177/1533033819843889
94. Yang F, Shen Y, Zhang W, Jin J, Huang D, Fang H, et al. An androgen receptor negatively induced long non-coding RNA ARNILA binding to miR-204 promotes the invasion and metastasis of triple-negative breast cancer. Cell Death Differ (2018) 25(12):2209–20. doi: 10.1038/s41418-018-0123-6
95. Wang Z, Yang B, Zhang M, Guo W, Wu Z, Wang Y, et al. lncRNA epigenetic landscape analysis identifies EPIC1 as an oncogenic lncRNA that interacts with MYC and promotes cell-cycle progression in cancer. Cancer Cell (2018) 33(4):706–20 e9. doi: 10.1016/j.ccell.2018.03.006
96. Li W, Zhang Z, Liu X, Cheng X, Zhang Y, Han X, et al. The FOXN3-NEAT1-SIN3A repressor complex promotes progression of hormonally responsive breast cancer. J Clin Invest (2017) 127(9):3421–40. doi: 10.1172/JCI94233
97. Huang X, Xie X, Liu P, Yang L, Chen B, Song C, et al. Adam12 and lnc015192 act as ceRNAs in breast cancer by regulating miR-34a. Oncogene (2018) 37(49):6316–26. doi: 10.1038/s41388-018-0410-1
98. Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, et al. Long noncoding RNA as modular scaffold of histone modification complexes. Science (2010) 329(5992):689–93. doi: 10.1126/science.1192002
99. Padua Alves C, Fonseca AS, Muys BR, de Barros ELBR, Burger MC, de Souza JE, et al. Brief report: The lincRNA Hotair is required for epithelial-to-mesenchymal transition and stemness maintenance of cancer cell lines. Stem Cells (2013) 31(12):2827–32. doi: 10.1002/stem.1547
100. Xu Y, Wang Y, Yuan C, Sheng X, Sha R, Dai H, et al. Predictive and prognostic value of EPIC1 in patients with breast cancer receiving neoadjuvant chemotherapy. Ther Adv Med Oncol (2020) 12:1758835920940886. doi: 10.1177/1758835920940886
101. Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, et al. Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell (2018) 174(5):1293–308 e36. doi: 10.1016/j.cell.2018.05.060
102. Degnim AC, Brahmbhatt RD, Radisky DC, Hoskin TL, Stallings-Mann M, Laudenschlager M, et al. Immune cell quantitation in normal breast tissue lobules with and without lobulitis. Breast Cancer Res Treat (2014) 144(3):539–49. doi: 10.1007/s10549-014-2896-8
103. Reed JR, Schwertfeger KL. Immune cell location and function during post-natal mammary gland development. J Mammary Gland Biol Neoplasia (2010) 15(3):329–39. doi: 10.1007/s10911-010-9188-7
104. Henning AN, Roychoudhuri R, Restifo NP. Epigenetic control of CD8(+) T cell differentiation. Nat Rev Immunol (2018) 18(5):340–56. doi: 10.1038/nri.2017.146
105. Busslinger M, Tarakhovsky A. Epigenetic control of immunity. Cold Spring Harb Perspect Biol (2014) 6(6). doi: 10.1101/cshperspect.a019307
106. Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature (2015) 527(7577):249–53. doi: 10.1038/nature15520
107. Llinas-Arias P, Iniguez-Munoz S, McCann K, Voorwerk L, Orozco JIJ, Ensenyat-Mendez M, et al. Epigenetic regulation of immunotherapy response in triple-negative breast cancer. Cancers (Basel) (2021) 13(16). doi: 10.3390/cancers13164139
108. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity (2013) 39(1):1–10. doi: 10.1016/j.immuni.2013.07.012
109. Cao J, Yan Q. Cancer epigenetics, tumor immunity, and immunotherapy. Trends Cancer (2020) 6(7):580–92. doi: 10.1016/j.trecan.2020.02.003
110. Lam RA, Tien TZ, Joseph CR, Lim JX, Thike AA, Iqbal J, et al. Cancer-testis antigens in triple-negative breast cancer: role and potential utility in clinical practice. Cancers (Basel) (2021) 13(15). doi: 10.3390/cancers13153875
111. Li Y, Li J, Wang Y, Zhang Y, Chu J, Sun C, et al. Roles of cancer/testis antigens (CTAs) in breast cancer. Cancer Lett (2017) 399:64–73. doi: 10.1016/j.canlet.2017.02.031
112. Jakobsen MK, Traynor S, Staehr M, Duijf PG, Nielsen AY, Terp MG, et al. The cancer/testis antigen gene VCX2 is rarely expressed in Malignancies but can be epigenetically activated using DNA methyltransferase and histone deacetylase inhibitors. Front Oncol (2020) 10:584024. doi: 10.3389/fonc.2020.584024
113. Cornel AM, Mimpen IL, Nierkens S. MHC class I downregulation in cancer: underlying mechanisms and potential targets for cancer immunotherapy. Cancers (Basel) (2020) 12(7). doi: 10.3390/cancers12071760
114. DhatChinamoorthy K, Colbert JD, Rock KL. Cancer immune evasion through loss of MHC class I antigen presentation. Front Immunol (2021) 12:636568. doi: 10.3389/fimmu.2021.636568
115. Taylor BC, Balko JM. Mechanisms of MHC-I downregulation and role in immunotherapy response. Front Immunol (2022) 13:844866. doi: 10.3389/fimmu.2022.844866
116. Luo N, Nixon MJ, Gonzalez-Ericsson PI, Sanchez V, Opalenik SR, Li H, et al. DNA methyltransferase inhibition upregulates MHC-I to potentiate cytotoxic T lymphocyte responses in breast cancer. Nat Commun (2018) 9(1):248. doi: 10.1038/s41467-017-02630-w
117. Zhou Z, van der Jeught K, Fang Y, Yu T, Li Y, Ao Z, et al. An organoid-based screen for epigenetic inhibitors that stimulate antigen presentation and potentiate T-cell-mediated cytotoxicity. Nat BioMed Eng (2021) 5(11):1320–35. doi: 10.1038/s41551-021-00805-x
118. Vergara Bermejo A, Ragonnaud E, Daradoumis J, Holst P. Cancer associated endogenous retroviruses: ideal immune targets for adenovirus-based immunotherapy. Int J Mol Sci (2020) 21(14). doi: 10.3390/ijms21144843
119. Deblois G, Tonekaboni SAM, Grillo G, Martinez C, Kao YI, Tai F, et al. Epigenetic switch-induced viral mimicry evasion in chemotherapy-resistant breast cancer. Cancer Discovery (2020) 10(9):1312–29. doi: 10.1158/2159-8290.CD-19-1493
120. Sheng W, LaFleur MW, Nguyen TH, Chen S, Chakravarthy A, Conway JR, et al. LSD1 ablation stimulates anti-tumor immunity and enables checkpoint blockade. Cell (2018) 174(3):549–63 e19. doi: 10.1016/j.cell.2018.05.052
121. Zhu Y, An X, Zhang X, Qiao Y, Zheng T, Li X. STING: a master regulator in the cancer-immunity cycle. Mol Cancer (2019) 18(1):152. doi: 10.1186/s12943-019-1087-y
122. Kwon J, Bakhoum SF. The cytosolic DNA-sensing cGAS-STING pathway in cancer. Cancer Discovery (2020) 10(1):26–39. doi: 10.1158/2159-8290.CD-19-0761
123. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature (2008) 455(7213):674–8. doi: 10.1038/nature07317
124. Chen Q, Sun L, Chen ZJ. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol (2016) 17(10):1142–9. doi: 10.1038/ni.3558
125. Gaston J, Cheradame L, Yvonnet V, Deas O, Poupon MF, Judde JG, et al. Intracellular STING inactivation sensitizes breast cancer cells to genotoxic agents. Oncotarget (2016) 7(47):77205–24. doi: 10.18632/oncotarget.12858
126. Wu SY, Xiao Y, Wei JL, Xu XE, Jin X, Hu X, et al. MYC suppresses STING-dependent innate immunity by transcriptionally upregulating DNMT1 in triple-negative breast cancer. J Immunother Cancer (2021) 9(7). doi: 10.1136/jitc-2021-002528
127. Wu L, Cao J, Cai WL, Lang SM, Horton JR, Jansen DJ, et al. KDM5 histone demethylases repress immune response via suppression of STING. PloS Biol (2018) 16(8):e2006134. doi: 10.1371/journal.pbio.2006134
128. Ong LT, Lee WC, Ma S, Oguz G, Niu Z, Bao Y, et al. IFI16-dependent STING signaling is a crucial regulator of anti-HER2 immune response in HER2+ breast cancer. Proc Natl Acad Sci U S A. (2022) 119(31):e2201376119. doi: 10.1073/pnas.2201376119
129. Li X, Gruosso T, Zuo D, Omeroglu A, Meterissian S, Guiot MC, et al. Infiltration of CD8(+) T cells into tumor cell clusters in triple-negative breast cancer. Proc Natl Acad Sci U S A. (2019) 116(9):3678–87. doi: 10.1073/pnas.1817652116
130. Mahmoud SM, Paish EC, Powe DG, Macmillan RD, Grainge MJ, Lee AH, et al. Tumor-infiltrating CD8+ lymphocytes predict clinical outcome in breast cancer. J Clin Oncol (2011) 29(15):1949–55. doi: 10.1200/JCO.2010.30.5037
131. Tu WJ, McCuaig RD, Tan AHY, Hardy K, Seddiki N, Ali S, et al. Targeting nuclear LSD1 to reprogram cancer cells and reinvigorate exhausted T cells via a novel LSD1-EOMES switch. Front Immunol (2020) 11:1228. doi: 10.3389/fimmu.2020.01228
132. Lee DY, Salahuddin T, Iqbal J. Lysine-specific demethylase 1 (LSD1)-mediated epigenetic modification of immunogenicity and immunomodulatory effects in breast cancers. Curr Oncol (2023) 30(2):2127–43. doi: 10.3390/curroncol30020164
133. Li Q, Shi L, Gui B, Yu W, Wang J, Zhang D, et al. Binding of the JmjC demethylase JARID1B to LSD1/NuRD suppresses angiogenesis and metastasis in breast cancer cells by repressing chemokine CCL14. Cancer Res (2011) 71(21):6899–908. doi: 10.1158/0008-5472.CAN-11-1523
134. Van Damme J, Proost P, Lenaerts JP, Opdenakker G. Structural and functional identification of two human, tumor-derived monocyte chemotactic proteins (MCP-2 and MCP-3) belonging to the chemokine family. J Exp Med (1992) 176(1):59–65. doi: 10.1084/jem.176.1.59
135. Zhang M, Yang W, Wang P, Deng Y, Dong YT, Liu FF, et al. CCL7 recruits cDC1 to promote antitumor immunity and facilitate checkpoint immunotherapy to non-small cell lung cancer. Nat Commun (2020) 11(1):6119. doi: 10.1038/s41467-020-19973-6
136. Janzer A, Stamm K, Becker A, Zimmer A, Buettner R, Kirfel J. The H3K4me3 histone demethylase Fbxl10 is a regulator of chemokine expression, cellular morphology, and the metabolome of fibroblasts. J Biol Chem (2012) 287(37):30984–92. doi: 10.1074/jbc.M112.341040
137. Deng Y, Li Z, Pan M, Wu H, Ni B, Han X. Implications of inflammatory cell death-related IFNG and co-expressed RNAs (AC006369.1 and CCR7) in breast carcinoma prognosis, and anti-tumor immunity. Front Genet (2023) 14:1112251. doi: 10.3389/fgene.2023.1112251
138. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science (2018) 359(6382):1350–5. doi: 10.1126/science.aar4060
139. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi: 10.1038/nrc3239
140. Solinas C, Garaud S, De Silva P, Boisson A, Van den Eynden G, de Wind A, et al. Immune checkpoint molecules on tumor-infiltrating lymphocytes and their association with tertiary lymphoid structures in human breast cancer. Front Immunol (2017) 8:1412. doi: 10.3389/fimmu.2017.01412
141. Sasidharan Nair V, El Salhat H, Taha RZ, John A, Ali BR, Elkord E. DNA methylation and repressive H3K9 and H3K27 trimethylation in the promoter regions of PD-1, CTLA-4, TIM-3, LAG-3, TIGIT, and PD-L1 genes in human primary breast cancer. Clin Epigenetics (2018) 10:78. doi: 10.1186/s13148-018-0512-1
142. Cha JH, Chan LC, Li CW, Hsu JL, Hung MC. Mechanisms controlling PD-L1 expression in cancer. Mol Cell (2019) 76(3):359–70. doi: 10.1016/j.molcel.2019.09.030
143. Elashi AA, Sasidharan Nair V, Taha RZ, Shaath H, Elkord E. DNA methylation of immune checkpoints in the peripheral blood of breast and colorectal cancer patients. Oncoimmunology (2019) 8(2):e1542918. doi: 10.1080/2162402X.2018.1542918
144. Darvin P, Sasidharan Nair V, Elkord E. PD-L1 expression in human breast cancer stem cells is epigenetically regulated through posttranslational histone modifications. J Oncol (2019) 2019:3958908. doi: 10.1155/2019/3958908
145. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med (2012) 366(26):2443–54. doi: 10.1056/NEJMoa1200690
146. Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med (2015) 372(21):2018–28. doi: 10.1056/NEJMoa1501824
147. Almand B, Clark JI, Nikitina E, van Beynen J, English NR, Knight SC, et al. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol (2001) 166(1):678–89. doi: 10.4049/jimmunol.166.1.678
148. Wang HF, Ning F, Liu ZC, Wu L, Li ZQ, Qi YF, et al. Histone deacetylase inhibitors deplete myeloid-derived suppressor cells induced by 4T1 mammary tumors in vivo and in vitro. Cancer Immunol Immunother (2017) 66(3):355–66. doi: 10.1007/s00262-016-1935-1
149. Yu B, Luo F, Sun B, Liu W, Shi Q, Cheng SY, et al. KAT6A acetylation of SMAD3 regulates myeloid-derived suppressor cell recruitment, metastasis, and immunotherapy in triple-negative breast cancer. Adv Sci (Weinh) (2021) 8(20):e2100014. doi: 10.1002/advs.202100014
150. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol (2008) 9(5):503–10. doi: 10.1038/ni1582
151. Chan IS, Knutsdottir H, Ramakrishnan G, Padmanaban V, Warrier M, Ramirez JC, et al. Cancer cells educate natural killer cells to a metastasis-promoting cell state. J Cell Biol (2020) 219(9). doi: 10.1083/jcb.202001134
152. Yu Y, Liu Y, Zong C, Yu Q, Yang X, Liang L, et al. Mesenchymal stem cells with Sirt1 overexpression suppress breast tumor growth via chemokine-dependent natural killer cells recruitment. Sci Rep (2016) 6:35998. doi: 10.1038/srep35998
153. Shen J, Pan J, Du C, Si W, Yao M, Xu L, et al. Silencing NKG2D ligand-targeting miRNAs enhances natural killer cell-mediated cytotoxicity in breast cancer. Cell Death Dis (2017) 8(4):e2740. doi: 10.1038/cddis.2017.158
154. Mantovani A, Locati M. Tumor-associated macrophages as a paradigm of macrophage plasticity, diversity, and polarization: lessons and open questions. Arterioscler Thromb Vasc Biol (2013) 33(7):1478–83. doi: 10.1161/ATVBAHA.113.300168
155. Li Z, Wang P, Cui W, Yong H, Wang D, Zhao T, et al. Tumour-associated macrophages enhance breast cancer Malignancy via inducing ZEB1-mediated DNMT1 transcriptional activation. Cell Biosci (2022) 12(1):176. doi: 10.1186/s13578-022-00913-4
156. Ma C, He D, Tian P, Wang Y, He Y, Wu Q, et al. miR-182 targeting reprograms tumor-associated macrophages and limits breast cancer progression. Proc Natl Acad Sci U.S.A. (2022) 119(6). doi: 10.1073/pnas.2114006119
157. Meng Z, Zhang R, Wang Y, Zhu G, Jin T, Li C, et al. miR-200c/PAI-2 promotes the progression of triple negative breast cancer via M1/M2 polarization induction of macrophage. Int Immunopharmacol (2020) 81:106028. doi: 10.1016/j.intimp.2019.106028
158. Hey J, Halperin C, Hartmann M, Mayer S, Schonung M, Lipka DB, et al. DNA methylation landscape of tumor-associated macrophages reveals pathways, transcription factors and prognostic value relevant to triple-negative breast cancer patients. Int J Cancer (2023) 152(6):1226–42. doi: 10.1002/ijc.34364
159. Tan AHY, Tu W, McCuaig R, Hardy K, Donovan T, Tsimbalyuk S, et al. Lysine-specific histone demethylase 1A regulates macrophage polarization and checkpoint molecules in the tumor microenvironment of triple-negative breast cancer. Front Immunol (2019) 10:1351. doi: 10.3389/fimmu.2019.01351
160. Emens LA, Cruz C, Eder JP, Braiteh F, Chung C, Tolaney SM, et al. Long-term clinical outcomes and biomarker analyses of atezolizumab therapy for patients with metastatic triple-negative breast cancer: A phase 1 study. JAMA Oncol (2019) 5(1):74–82. doi: 10.1001/jamaoncol.2018.4224
161. Mina LA, Lim S, Bahadur SW, Firoz AT. Immunotherapy for the treatment of breast cancer: emerging new data. Breast Cancer (Dove Med Press) (2019) 11:321–8. doi: 10.2147/BCTT.S184710
162. Kim K, Skora AD, Li Z, Liu Q, Tam AJ, Blosser RL, et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci U S A. (2014) 111(32):11774–9. doi: 10.1073/pnas.1410626111
163. Li W, Sang M, Hao X, Wu Y, Shan B. CTLA−4 blockade combined with 5−aza−2’−deoxycytidine enhances the killing effect of MAGE−A family common antigen peptide−specific cytotoxic T cells on breast cancer. Oncol Rep (2020) 44(4):1758–70. doi: 10.3892/or.2020.7701
164. Taylor K, Loo Yau H, Chakravarthy A, Wang B, Shen SY, Ettayebi I, et al. An open-label, phase II multicohort study of an oral hypomethylating agent CC-486 and durvalumab in advanced solid tumors. J Immunother Cancer (2020) 8(2). doi: 10.1136/jitc-2020-000883
165. Wang C, Liu Y, Dong L, Li X, Yang Q, Brock MV, et al. Efficacy of Decitabine plus Anti-PD-1 Camrelizumab in Patients with Hodgkin Lymphoma Who Progressed or Relapsed after PD-1 Blockade Monotherapy. Clin Cancer Res (2021) 27(10):2782–91. doi: 10.1158/1078-0432.CCR-21-0133
166. Terranova-Barberio M, Thomas S, Ali N, Pawlowska N, Park J, Krings G, et al. HDAC inhibition potentiates immunotherapy in triple negative breast cancer. Oncotarget (2017) 8(69):114156–72. doi: 10.18632/oncotarget.23169
167. Christmas BJ, Rafie CI, Hopkins AC, Scott BA, Ma HS, Cruz KA, et al. Entinostat converts immune-resistant breast and pancreatic cancers into checkpoint-responsive tumors by reprogramming tumor-infiltrating MDSCs. Cancer Immunol Res (2018) 6(12):1561–77. doi: 10.1158/2326-6066.CIR-18-0070
168. Roussos Torres ET, Rafie C, Wang C, Lim D, Brufsky A, LoRusso P, et al. Phase I study of entinostat and nivolumab with or without ipilimumab in advanced solid tumors (ETCTN-9844). Clin Cancer Res (2021) 27(21):5828–37. doi: 10.1158/1078-0432.CCR-20-5017
169. Shi J, Wang Y, Zeng L, Wu Y, Deng J, Zhang Q, et al. Disrupting the interaction of BRD4 with diacetylated Twist suppresses tumorigenesis in basal-like breast cancer. Cancer Cell (2014) 25(2):210–25. doi: 10.1016/j.ccr.2014.01.028
170. Jing X, Shao S, Zhang Y, Luo A, Zhao L, Zhang L, et al. BRD4 inhibition suppresses PD-L1 expression in triple-negative breast cancer. Exp Cell Res (2020) 392(2):112034. doi: 10.1016/j.yexcr.2020.112034
171. Lai X, Stiff A, Duggan M, Wesolowski R, Carson WE 3rd, Friedman A. Modeling combination therapy for breast cancer with BET and immune checkpoint inhibitors. Proc Natl Acad Sci U S A. (2018) 115(21):5534–9. doi: 10.1073/pnas.1721559115
Keywords: breast cancer, epigenetic alterations, DNA methylation, histone modifications, non-coding RNA, epigenetic therapy, immunotherapy, combination therapy
Citation: Yin J, Gu T, Chaudhry N, Davidson NE and Huang Y (2024) Epigenetic modulation of antitumor immunity and immunotherapy response in breast cancer: biological mechanisms and clinical implications. Front. Immunol. 14:1325615. doi: 10.3389/fimmu.2023.1325615
Received: 21 October 2023; Accepted: 22 December 2023;
Published: 10 January 2024.
Edited by:
Peter Hamar, Semmelweis University, HungaryReviewed by:
Ming Yi, Zhejiang University, ChinaAlessandro Poggi, San Martino Hospital (IRCCS), Italy
Umar Mehraj, Duke University, United States
Copyright © 2024 Yin, Gu, Chaudhry, Davidson and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi Huang, eWktaHVhbmdAdWlvd2EuZWR1