 Víctor Albarrán1*
Víctor Albarrán1* María San Román1Javier Pozas2Jesús Chamorro1Diana Isabel Rosero1Patricia Guerrero1Juan Carlos Calvo1Carlos González1Coral García de Quevedo1Patricia Pérez de Aguado1Jaime Moreno1Alfonso Cortés1Ainara Soria1
María San Román1Javier Pozas2Jesús Chamorro1Diana Isabel Rosero1Patricia Guerrero1Juan Carlos Calvo1Carlos González1Coral García de Quevedo1Patricia Pérez de Aguado1Jaime Moreno1Alfonso Cortés1Ainara Soria1- 1Department of Medical Oncology, Ramon y Cajal University Hospital, Madrid, Spain
- 2Department of Medical Oncology, The Royal Marsden Hospital, London, United Kingdom
Adoptive cell therapy (ACT) comprises different strategies to enhance the activity of T lymphocytes and other effector cells that orchestrate the antitumor immune response, including chimeric antigen receptor (CAR) T-cell therapy, T-cell receptor (TCR) gene-modified T cells, and therapy with tumor-infiltrating lymphocytes (TILs). The outstanding results of CAR-T cells in some hematologic malignancies have launched the investigation of ACT in patients with refractory solid malignancies. However, certain characteristics of solid tumors, such as their antigenic heterogeneity and immunosuppressive microenvironment, hamper the efficacy of antigen-targeted treatments. Other ACT modalities, such as TIL therapy, have emerged as promising new strategies. TIL therapy has shown safety and promising activity in certain immunogenic cancers, mainly advanced melanoma, with an exciting rationale for its combination with immune checkpoint inhibitors. However, the implementation of TIL therapy in clinical practice is hindered by several biological, logistic, and economic challenges. In this review, we aim to summarize the current knowledge, available clinical results, and potential areas of future research regarding the use of T cell therapy in patients with solid tumors
1 Introduction
1.1 T cells, antitumor response, and immune evasion
The antitumor activity of our immune system is a highly sophisticated process with several regulatory and negative feedback pathways. When malignant cells are identified and attacked by macrophages and natural killer (NK) cells -components of the innate immunity-, aberrant proteins derived from the cumulative occurrence of mutations are released and phagocytosed by dendritic and other antigen-presenting cells (APC) (1). In the peripheral lymph nodes, these tumor-associated antigens (TAA) are exposed by APC through major histocompatibility complex type I (MHC-I) molecules to the T cell receptor (TCR) of naïve CD8+ T cells, leading to their activation. For this ‘immune synapsis’ to be successful, other co-stimulating receptors on the T cell membrane (such as B7) should be activated (2). At the same time, the interaction between MHC type II (MHC-II) molecules and the TCR of CD4+ T helper lymphocytes leads to the activation of B cells and subsequent production of antitumor antibodies (3), and unleashes additional mechanisms that elicit CD8+ T cells function and differentiation, including dendritic cell licensing and cytokine production (4). Once CD8+ T cells are activated, they travel to the tumor site and recognize TAA presented by MHC-II molecules on the surface of malignant cells, unleashing the effector phase of adaptive immunity, and ultimately leading to tumor cell death (4). The quantity and phenotype of these tumor-infiltrating lymphocytes (TILs) have been widely associated with the biological behavior, prognosis, and response to anti-cancer therapies in virtually all subtypes of solid cancers (5–7). The success of the effector phase is compromised by the inhibition of T-cell response by immunosuppressive cells from the tumor microenvironment (TME), including myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), and regulatory T lymphocytes (T-regs) (8). Tumor cells are able to modulate their function and differentiation through the activation of NF-κB and STAT3 signaling pathways, inducing the release of immunosuppressive cytokines (IL-6, IL-10, TGF-β) that inhibit TILs antitumor activity (9).
The antitumor response is controlled by negative feedback mechanisms performed by molecules known as ‘immune checkpoints’, both in the priming phase -including CTLA-4, LAG-3 and TIM-3- and in the effector phase -mainly programmed cell death protein 1 (PD-1), activated by ligands (PD-L1) expressed both by cells from the tumor and TME- (10). The mechanisms of the antitumor response and potential immune biomarkers are shown in Figure 1.
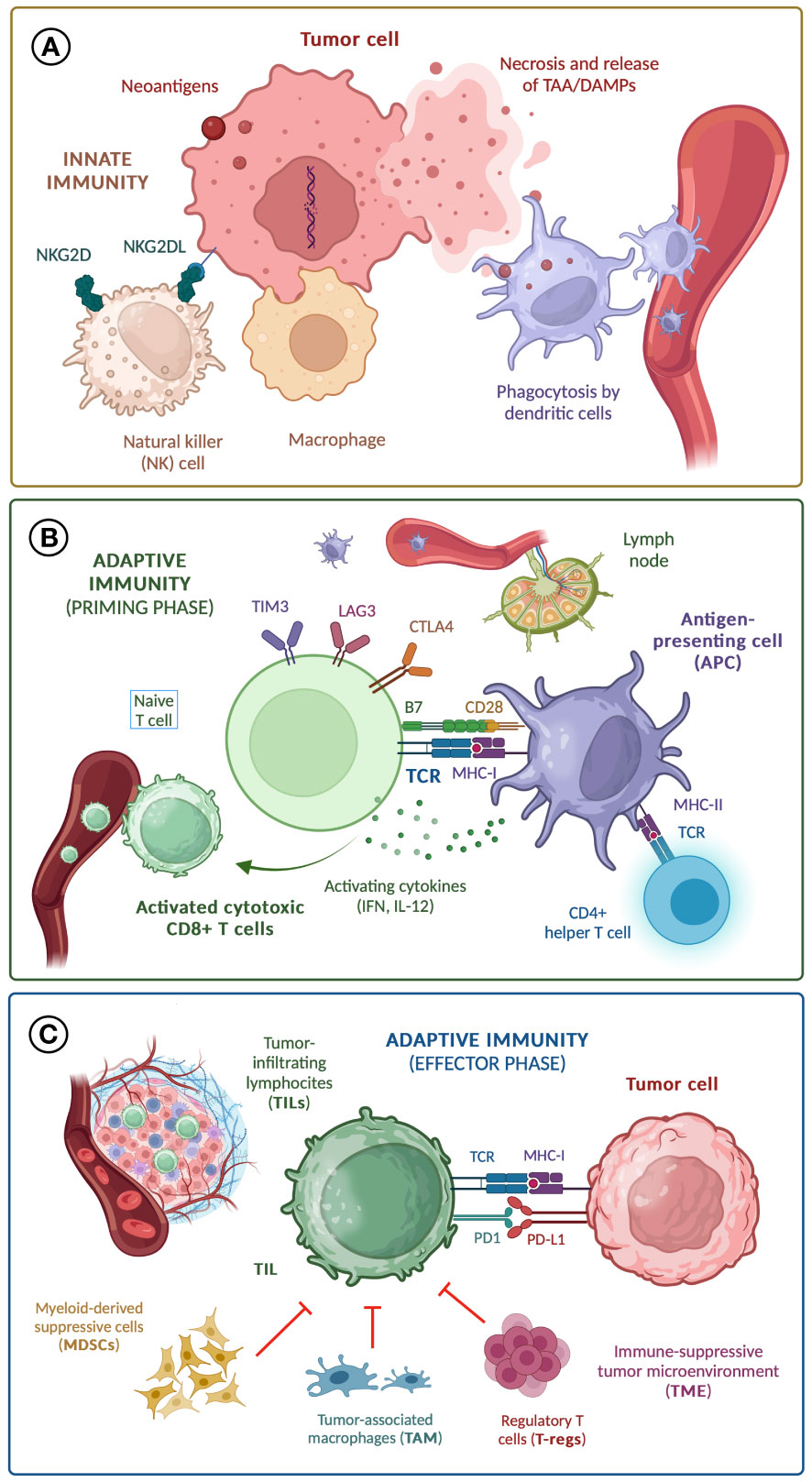
Figure 1 Mechanisms of anti-tumor response; (A) Innate immunity; (B) Adaptive immunity (priming phase); (C) Adaptive immunity (effector phase). TAA, tumor-associated antigen; DAMPs, damage-associated molecular patterns; TCR, T cell receptor; MHC, major histocompatibility complex; IL-12, interleukin-12; IFN, interferons.
The ability to avoid the immune system is a hallmark of malignant cells (11). Some of the most relevant mechanisms of immune evasion in solid tumors are the upregulation of immune checkpoints (12) -which sets the rationale for the use of immune checkpoint inhibitors (ICI)-, the loss of MHC-I or other molecules with a key role in antigen presentation (13), the production of cytokines (IL-6, IL-10, TGF-β) that lead to an immunosuppressive TME (14), and the activation of oncogenic routes that promote T-regs infiltration -such as the indoleamine 2,3-dioxygenase (IDO) pathway (15)- or inhibit CD8+ T-cell trafficking to the tumor site -such as the Wnt/β-catenin pathway (16)-. Recent data suggest that remote transference of biomolecular cargoes from malignant to healthy cells, mediated by exosomes, may also play a crucial role in misleading the mechanisms of antigenic recognition, thus contributing to immune tumor evasion (17).
Since the FDA approval of anti-CTLA4 and anti-PD1 therapy for advanced melanoma in 2011 and 2015, respectively, ICI alone or in combination with other therapies have transformed the therapeutic landscape of nearly all solid tumors (18). However, there are several other emerging strategies to enhance the immune antitumor response, some of them with promising results in ICI-refractory tumors, which will surely increase the relevance of immunotherapy in cancer treatment throughout the following years.
1.2 Basis of adoptive cell therapy
Adoptive cell therapy (ACT) encompasses several techniques that use the T lymphocytes themselves, after a process of artificial modification or genetic engineering, to improve their antitumor activity (19). This implies the extraction of autologous T lymphocytes from the patient, and their manipulation and amplification in vitro. Meanwhile, the patient undergoes treatment with lymphodepleting chemotherapy (CT) -usually fludarabine plus cyclophosphamide- to annihilate ineffective and immune-suppressing lymphocytes. After this process, the improved cell product is reinfused into the patient.
The difference between different ACT modalities lies in the characteristics of T cell modification in vitro: CAR-T and TCR gene-modified T cells are genetically engineered to incorporate modified membrane receptors with a high affinity for selected tumor antigens. However, in contrast to hematologic malignancies, solid tumors are composed of polyclonal cell populations with huge antigenic heterogeneity, which hampers the efficacy of antigen-targeted therapies. TIL therapy is based on the activation and expansion of infiltrating T cells extracted from the tumor itself, which are intrinsically reactive against tumor antigens, setting an interesting rationale for its use against the changing and heterogeneous cell population of solid cancers.
2 CAR-T cells, a role in solid cancers?
2.1 Introduction
CAR-T cells are genetically modified lymphocytes that incorporate a chimeric antigen receptor (CAR) composed by three parts: an extracellular domain with a single-chain fragment variable (scFv) that allows antigen recognition, a transmembrane domain -linked to the extracellular part through a spacer-, and an intracellular domain. This includes a CD3 complex -which activates the downstream signaling pathways- and several costimulatory domains (usually CD28 and/or 4-1BB) that intensify the cytoplasmatic activity of T cells unchained by antigenic recognition (20). In recent years, innovations in the structure and manufacturing of CAR-T cells have led to significant improvements in their clinical efficacy, especially with the development of fourth-generation CAR-T cells (21). Fifth generation CARs equipped with three costimulatory domains and able to secrete anti-PDL1 scFv blockade molecules, targeted against B cell maturation antigen (BCMA) have shown heightened antitumor efficacy and decrease of T cell exhaustion in patients with multiple myeloma (22).
Nanobodies or single domain antibodies (VHH) have recently been exploited as an alternative to scFvs for antigen-targeting domains on T cell surface, based on numerous advantages including their small size, high affinity, specificity and stability (23). VHH-based CD19-redirected CAR-T cells have shown similar expansion rate, cytotoxicity and anti-tumor reactions when compared with their scFv-based counterparts (24).
Due to their molecular structure, CAR-T cells only recognize extracellular antigens, and are particularly efficient when their scFv has a high affinity for the targeted protein -and it is homogeneously expressed by tumor cells-. This explains the efficacy of CAR-T cells in patients with leukemia and lymphoma, which comprise a clonal population of cells that uniformly express certain antigens -such as CD19- on their membrane (25). Since 2018, tisagenlecleucel and axicabtagene-citoleucel have EMA approval for the treatment of B-cell LLA and refractory non-Hodgkin lymphoma, and in 2020, brexucabtagene-autoleucel was approved by the EMA for mantle lymphoma, achieving complete response rates of over 50% in heavily pre-treated patients (26).
The outstanding results of CAR-T cells in hematologic malignancies have led to their investigation in solid tumors, mainly using overexpressed epithelial antigens as targets. The epithelial growth factor receptor (EGFR), HER2, carcinoembryonic antigen (CEA), mesothelin and soluble antigen GD2 have been frequent targets of CAR-T therapies, although many other antigens have been the object of preclinical studies (27).
2.2 Clinical outcomes
Nearly 500 clinical trials evaluating CAR-T cells in solid tumors have been registered, most of them in Asian population, and many still ongoing (28). Most completed studies are phase I/II trials that have reported modest results, with only occasional and generally brief clinical responses. Clinical research on CAR-T cells has mainly focused on glioblastoma (GBM), sarcoma, neuroblastoma, and gastrointestinal cancer.
In 2016, Brown et al. (29) reported the case of a GBM patient with an 8-month complete response (CR) after IL13-targeted CAR-T therapy, although further research has failed to confirm these results (30). Her2 may be another interesting target for CAR-T cells in GBM; in a clinical trial with 17 patients, 1 partial response (PR) (lasting 9 months) and 7 cases of stable disease (SD) (ranging from 2 to 29 months) were reported (31). EGFR-targeted CAR-T cells have been evaluated in GBM in two clinical trials, with negative results (32, 33).
Her2-targeted CAR-T cells have also been tested in sarcoma patients. In a phase I/II study including 19 patients with Her2+ recurrent or refractory sarcoma of several histological subtypes, 4 SD were observed [3 osteosarcoma and 1 small round cell desmoplastic tumor) (34). In neuroblastoma, at least three clinical trials have evaluated the efficacy of GD2-targeted CAR-T cells -based on the efficacy of anti-GD2 monoclonal antibodies such as dinutuximab- (35), with promising results (3 CR among 19 patients (36), 4 PR among 8 patients (37), and 5 SD among 11 patients (38)].
As for gastrointestinal (GI) cancer, several antigens have been evaluated as potential targets of CAR-T therapy. In a phase I trial including 23 patients with several GI tumors treated with CD133-targeted CAR-T cells, 3 PR (2 pancreatic and 1 hepatocellular carcinoma [HCC]) and 14 SD were observed (39). Zhan et al. (40) evaluated CAR-T therapy targeting Claudin 18.2 (CLDN 18.2) in 11 patients with CLDN 18.2-positive gastric or pancreatic carcinoma, reporting 1 CR, 3 PR and 5 SD. EGFR-CAR-T therapy has mainly been evaluated in biliopancreatic tumors, with promising results. Liu et al. (41) conducted a phase I study including 14 patients with refractory advanced pancreatic carcinoma, reporting 4 PR and 8 SD; in a phase I study by Guo et al. (42) with 19 patients (14 cholangiocarcinoma, 5 gallbladder carcinoma), 1 CR and 10 SD were observed. Glypican-3 (GPC3)-targeted (43) and CEA-targeted (44) CAR-T cells have shown modest activity in HCC and colorectal cancer, respectively.
Mesothelin-targeted intrapleural CAR-T cells have been evaluated -in combination with ICI- in 14 patients with malignant mesothelioma and non-small cell lung cancer (NSCLC), with promising results (2 CR, 5 PR, and 4 SD) (45). In NSCLC, a phase I study showed clinical activity of EGFR-CAR-T cells, with 2 PR and 5 SD among 11 patients (46), and ROR1-directed CAR-T cells showed preliminary positive results in ROR1+ tumors (4 PR among 6 patients) (47). CAR-T therapy has obtained modest results for metastatic castration-resistant prostate cancer (mCRPC) -targeting prostate specific membrane antigen (PSMA)- (48). Evidence for CAR-T therapy in other solid tumors is even scarcer and mainly comes from preclinical studies (49).
The outcomes of the most relevant clinical trials that evaluated CAR-T cells in solid tumors are summarized in Figure 2.
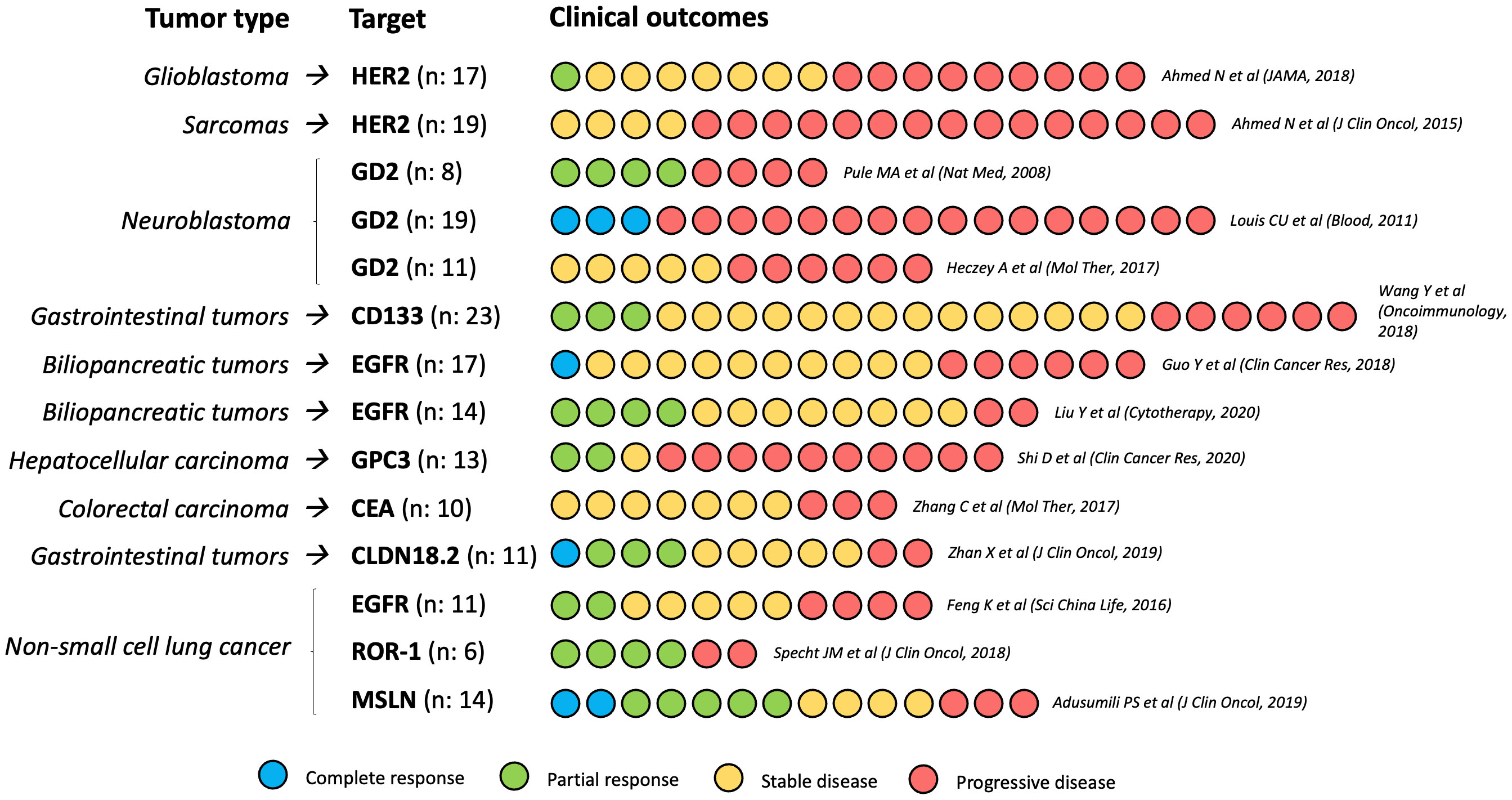
Figure 2 Clinical outcomes of the most relevant CAR-T cells phase I/II trials in solid tumors. N, number of participants in each study.
3 TCR-engineered T cells, more of the same?
3.1 Introduction
T cells expressing an engineered T-cell receptor (TCR-T-cells) represent an alternative modality of ACT, with several advantages compared to CAR T therapy (50). By using engineered TCR -instead of CAR-, T cells can recognize not only membrane proteins but also intracellular antigens presented by MHC molecules, covering a wider repertoire of tumor neoantigens and therapeutic targets. Intrinsic features of T cells, such as high antigen sensitivity and the use of physiological signaling pathways, improve TCR-T-cells antitumor functions and reduce the risk of on-target off-tumor (OTOT) toxicity.
However, TCR-T-cells also have some disadvantages compared to CAR-T cells, such as their weaker avidity for target antigens and their limitation to a certain human leukocyte antigen (HLA) haplotype, which restricts the number of patients that potentially benefit from each modified TCR. In addition, as antigen-targeted modified T lymphocytes, all the limitations of CAR-T therapy in solid tumors -antigenic heterogeneity, difficulty of T cell trafficking, and detrimental effects of immunosuppressive TME- are also applicable to TCR-T-cells (51).
3.2 Clinical outcomes
In 2006, Morgan et al. (52) published the results of the first trial with TCR-T-cells in solid tumors, targeting MART-1 in 17 patients with metastatic melanoma (with 2 PR). Since then, at least three other phase I/II clinical trials have evaluated MART-1-targeted TCR-T-cells in advanced melanoma -all using retrovirus as vectors-, and have reported variable results, with ORR ranging from 0% to 30% (53–55). In one of these trials (53), protein gp100 was also evaluated as a target in 16 patients, observing 1 sustained CR (>14 months) and 2 PR (ORR: 18.8%). TCR-T-cell-related toxicity was similar in all these studies, with a predominance of skin toxicity (23-94%) and a low incidence of CRS (<15%).
The New York esophageal squamous cell carcinoma (NY-ESO)-1 antigen is a promising target both in melanoma and in some sarcoma immunogenic subtypes -particularly synovial sarcoma, with NY-ESO-1 overexpression in 80% of patients (56)-. In a phase II trial with 38 melanoma and synovial sarcoma patients treated with NY-ESO-1 TCR-T-cells, there was an ORR of 57.9%, including 5 maintained CR and several long-term PR, with no severe toxicity related to TCR-T-cells (57). Similar results were obtained in two other phase I studies, with an incidence of CRS of 10% in one of them (58, 59). Two other phase I/II trials evaluated NY-ESO-1 TCR-T-cells in patients with synovial sarcoma -using lentivirus as a vector instead of retrovirus-, obtaining similar results (ORR 30% to 50%), with an incidence of CRS of 41.7% in one of them (60, 61). NY-ESO-1 was also used as TCR-T-cells target in a small phase I trial including 3 patients with different solid tumors, with negative results (62).
Other cancer-tesis antigens, such as MAGE family proteins, have been evaluated as TCR-T-cell targets in multi-tumor phase I/II trials. A phase I trial evaluated MAGE-A3-targeted TCR-T-cells in 17 patients with solid tumors and observed an ORR of 23.5% (including 1 sustained CR) (63). Morgan et al. (64) treated 9 solid tumors patients (including 7 synovial sarcomas) with MAGE-A3-targeted cells, obtaining an ORR of 56% (including 1 sustained CR), but with severe toxicity (ICANS in 3 patients and 2 treatment-related deaths). In a phase I trial with 38 patients treated with MAGE-A4-TCR-T-cells, 50% of CRS was observed (65), and another trial with MAGE-A3-TCR-T-cells was suspended after 2 treatment-related deaths (66). Cross-reactions against proteins normally expressed by nervous system cells -such as EPS8L2- have been hypothesized as the source of severe neurotoxicity and toxic deaths reported by MAGE-TCR-T-cells trials (67).
MAGE-targeted TCR-T-cells have obtained negative or modest results in NSCLC (68) and esophageal cancer (69). Similarly, significant toxicity has been observed in studies targeting other proteins that are not exclusively expressed by tumor cells. For example, a phase I trial evaluating CEA-targeted TCR-T-cells in patients with colorectal cancer was suspended due to severe colitis in 100% of the patients (70).
The treatment of virus-related tumors is an interesting approach for TCR-T-cells, since targeting specific viral antigens, which are expressed by infected tumor cells but not by normal tissues, should avoid the problem of cross-reactions and OTOT toxicity. In fact, two trials have evaluated TCR-T-cells targeted against human papillomavirus (HPV) carcinogenesis-related proteins (E6 and E7) in HPV+ tumors, without any relevant toxicity and interesting clinical outcomes (ORR of 16.7% and 50%, respectively) (71, 72). Meng et al. evaluated TCR-T-cells against hepatitis B virus (HBV) in 8 patients with HBV+ HCC, observing 1 prolonged PR and only one case of liver toxicity (73). Veatch et al. (74) tested TCR-T-cells against Merkel carcinoma polyomavirus (MCPyV) in 5 patients with refractory Merkel cell carcinoma, reporting 1 PR and no significant adverse events. However, given that virus-related tumors represent a small proportion of advanced solid cancers, other strategies are needed to overcome TCR-T-cells cross-reactions and OTOT toxicity.
In relation to this, an exciting hypothesis is the use of neoantigens -byproduct of tumor somatic mutations, not present in non-malignant tissues- as tumor-restricted and immunogenic targets of TCR-T-cells. In fact, some studies suggest that long-term responses to immune checkpoint inhibitors are mediated by neoantigen-reactive effector T cells (75), providing a rationale for combining TCR-T-cells and ICI. Unfortunately, a phase I trial exploring the effectiveness of personalized neoantigen-targeted TCR-T-cells in 16 patients with advanced solid tumors has obtained disappointing results (51).
Occasional responses to TCR-T-cells have been observed in other tumors. Leidner et al. (76) reported a partial response with KRAS G12D-targeted TCR-T-cells (lasting >6 months) in a patient with advanced pancreatic adenocarcinoma. Kim et al. (77) reported a partial response to TP53-targeted TCR-T-cells in a patient with metastatic breast cancer.
The outcomes of published TCR-T-cells clinical trials for solid tumors have been summarized in Table 1.
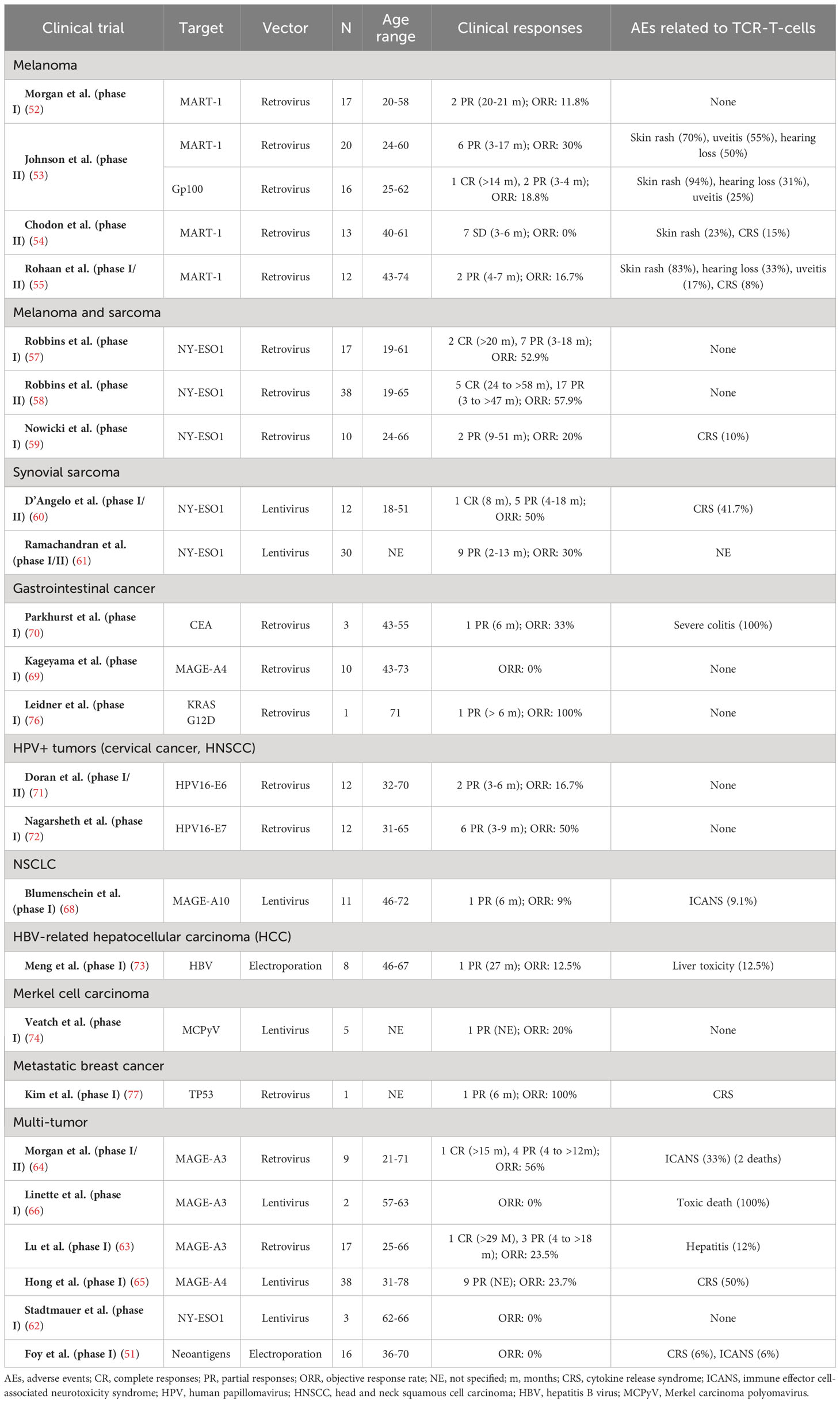
Table 1 Clinical trials with published results of TCR-T-cells in solid tumors.
4 Challenges and future strategies
4.1 Challenges and future strategies
Both CAR-T and TCR-T cells harbor inherent limitations for the treatment of patients with solid tumors, which may explain the significantly worse clinical outcomes than those obtained with CAR-T therapy in hematologic malignancies.
4.1.1 Antigenic heterogeneity
In contrast to the clonal nature of lymphomas and leukemias, solid tumors -particularly in the context of metastatic disease- are characterized by the progressive acquisition of somatic mutations that lead to polyclonal expansion of different cellular lineages, giving rise to genomic instability and antigenic heterogeneity.
Significant intratumoral heterogeneity in neo-epitope expression and clonal expansion of the adaptive immune system in distant regions of the same disease have also been demonstrated in hepatitis B virus (HBV)-related liver cancer (78). In Her2+ breast cancer, spatial transcriptomic studies have shown intra-patient heterogeneity in the expression of gene signatures that determine cellular interactions with T lymphocytes and other immune cells (79). O’Rourke et al. (33) studied the expression of several antigens in tumor cells from 7 GBM patients, before and after a single infusion of EGFR-targeted CAR-T cells, observing in all of them a significant decrease in EGFR expression, but an important increase in several other antigens with known immunosuppressor functions (CD8, GRZMB, CD25, IDO1, PDL1, and FoxP3). These results shed light on how antigen-specific T cell therapies might quickly promote the selection of resistant cellular subclones with immunosuppressive activity.
T cells with multi-antigenic recognition have been proposed as a potential strategy to overcome this problem. Some preclinical studies have suggested that CAR-T cells with bispecific adapters can facilitate the eradication of antigenically different tumors (80). Moving a step forward, CAR-T cells equipped with synthetic Notch (synNotch) receptors might be able to induce CAR expression only after the recognition of tumor-specific antigens, creating precise prime-and-kill recognition circuits (81). Although these are promising strategies, they have not yet been evaluated in clinical trials, and further research is required to assess their feasibility.
4.1.2 On-target off-tumor toxicity
Most antigens targeted by CAR-T or TCR-T cells in solid tumors are not specific to cancer cells and are also expressed on non-malignant tissues, leading to potential OTOT toxicity. In addition to the usual adverse events observed in patients with hematologic malignancies, such as cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS), which are generally manageable (82), the OTOT effects of these cell therapies add significant toxicity and are usually dose-limiting in patients with solid tumors.
The release of perforin and granzymes following T cell activation is assumed to play a key role in OTOT cytotoxicity, although the upregulation of T cell-surface pro-apoptotic molecules (such as FAS ligands) might also contribute to tissue destruction (83). Acute respiratory distress, digestive hemorrhage, and severe mucocutaneous toxicity have been reported as OTOT effects in several clinical trials, particularly with Her2 (84)-, CLDN18.2 (85)- and EGFR (41, 42)-targeted CAR-T cells. Interestingly, no OTOT toxicity has been reported for anti-GD2 CAR-T cells in patients with diffuse midline gliomas (86) despite the fact that GD2 is expressed in healthy brain tissue (87). Although little is known about the threshold for antigen recognition (88), this suggests that CAR-T therapy may be feasible without significant OTOT effects in cases with different levels of antigen expression between tumor and healthy cells.
Some additional theoretical strategies to overcome OTOT toxicity include the modulation of scFv affinity and/or CAR architecture, the locoregional administration of CAR-T cells to avoid systemic effects, the development of engineering approaches to exogenously control CAR-T cell activity, and the design of protein-based logic-circuit strategies to restrict CAR-T cell activation (89). Further research is needed to assess the clinical feasibility of these approaches and discover predictive biomarkers of severe toxicity.
4.1.3 T cells trafficking
Ensuring contact between CAR-T cells and tumor cells is not a problem in leukemias and lymphomas, since malignant cells concentrate within the blood and lymph nodes; however, it is a significant obstacle in solid metastatic tumors, particularly those with infiltration of immune-privileged organs -such as the central nervous system-. Several studies have shown that metastatic lesions from solid cancers have significantly lower lymphocytic infiltration than primary tumors, suggesting that the loss of T cell trafficking to the tumor site is a relevant mechanism for immune escape and may facilitate tumor progression (90).
In patients with metastatic melanoma, Harlin et al. (91) showed that the expression of certain cytokines (CCL2, CCL3, CCL4, CCL5, CXCL9, and CXCL10) is significantly higher in lesions enriched with TILs than in those with poor lymphocytic infiltration, suggesting that these molecules play an important role in T cell recruitment. Targeting the tumor vasculature and microenvironment to modulate the chemotactic response is an exciting research topic to improve CAR-T cell trafficking to solid tumors (92).
4.1.4 Immunosuppressive TME
In addition to hampering T cell trafficking, the TME has inhibitory effects on the lymphocytes that get to infiltrate the tumor site, which is an obstacle for CAR T cell function. In addition, sustained exposure to tumor antigens and inflammatory signals is thought to progressively mitigate the function and proliferation of modified T cells, leading to CAR T cell ‘exhaustion’. Targeting T cell intrinsic pathways (PD-1/PD-L1 axis, TOX/NR4A, TGF-β, CBL-B), using CRISPR technology to modulate the surface expression of CAR, and uncoupling antigen recognition from CAR activation signaling, are exciting approaches under research to overcome exhaustion and improve CAR T cells clinical outcomes in solid tumors (93).
5 TIL therapy, the hope for ACT in solid tumors?
5.1 Introduction
Ex vivo expanded tumor-infiltrating lymphocytes (TILs) from different solid cancers share a composition of oligoclonal effector T cells that are reactive against a heterogeneous repertoire of tumor-associated antigens (94). This establishes the rationale for TILs artificial expansion and activation in vitro (out of the TME detrimental influence) and their subsequent reinfusion -together with stimulating cytokine IL-2- into a more favorable environment, after chemical depletion of immunosuppressive cells.
Not being a specific antigen-targeted therapy and using naturally ‘selected’ tumor-reactive lymphocytes, TIL therapy may theoretically overcome the problems of antigenic heterogeneity, tumor trafficking, and on target off tumor toxicity that limit the effectiveness of CAR-T and TCR-T therapies against advanced solid tumors.
5.2 Clinical outcomes in melanoma
TIL therapy has mainly been evaluated in advanced melanoma. Since the first positive studies conducted by Rosenberg (95–97), several phase I/II trials have obtained clinical responses with expanded TILs -alone or in combination with total body irradiation- with a significant variability in the IL2 dosage, the number of infused cells, and the intensity of lymphodepletion (98–101). All these studies were performed before the large-scale expansion of ICI and targeted therapy as the standard of care for advanced melanoma.
In ASCO 2020, Sarnaik et al. (102) communicated the results of a phase II trial evaluating cryopreserved autologous TIL therapy lifileucel (LN-144) in patients with metastatic melanoma in progression to anti-PD1 +/- anti-CTLA4 therapy (and BRAF/MEK inhibitors in BRAF-mutant tumors). Among the 66 evaluable patients, there were 2 CR and 22 PR (ORR 36.4%), with the median duration of response not reached at 18.7 months. A reduction in tumor burden was observed in 81% of patients. Responses were demonstrated regardless of the BRAF mutational status, PD-L1 expression, and tumor location. Objective responses were observed in patients with brain and liver metastases, baseline bulky disease, elevated lactate dehydrogenase (LDH) levels, and prior anti-PD1 treatment. The safety profile was consistent with the known toxicities of the lymphodepletion and IL-2 regimens.
Two years later, Rohaan et al. (103) published the first phase III trial of ACT for solid tumors, comparing TIL therapy with ipilimumab in patients with stage IIIC/IV melanoma. A total of 168 patients were randomly assigned in a 1:1 ratio to receive TILs (at least 5 x 109 cells, preceded by lymphodepleting CT, followed by IL2 at high-doses of 600.000 IU/kg) or ipilimumab (84 patients in each group). 89% of the patients had received previous systemic therapy, most of them adjuvant or first-line anti-PD1 antibodies. Median progression-free survival (mPFS) was significantly higher in the TIL group (7.2 vs 3.1 months; hazard ratio [HR] 0.50; p <0.001), as well as the ORR (49% vs 21%). The median overall survival was 25.8 months (vs 18.9 months in the ipilimumab group).
All the above-mentioned studies have been conducted on patients with cutaneous melanoma. A phase II trial evaluated TILs in 21 patients with metastatic uveal melanoma, with 1 sustained CR and 6 PR (ORR of 35%) (104). Interestingly, 3 of these responders (43%) had previously received immunotherapy with anti-PD1 and/or anti-CTLA4 agents, without any clinical benefit. These results demonstrate that TIL therapy merits further research in non-cutaneous melanoma, especially considering its refractory nature to ICI and other systemic treatments.
The outcomes of the published clinical trials of TIL therapy have been summarized in Table 2.
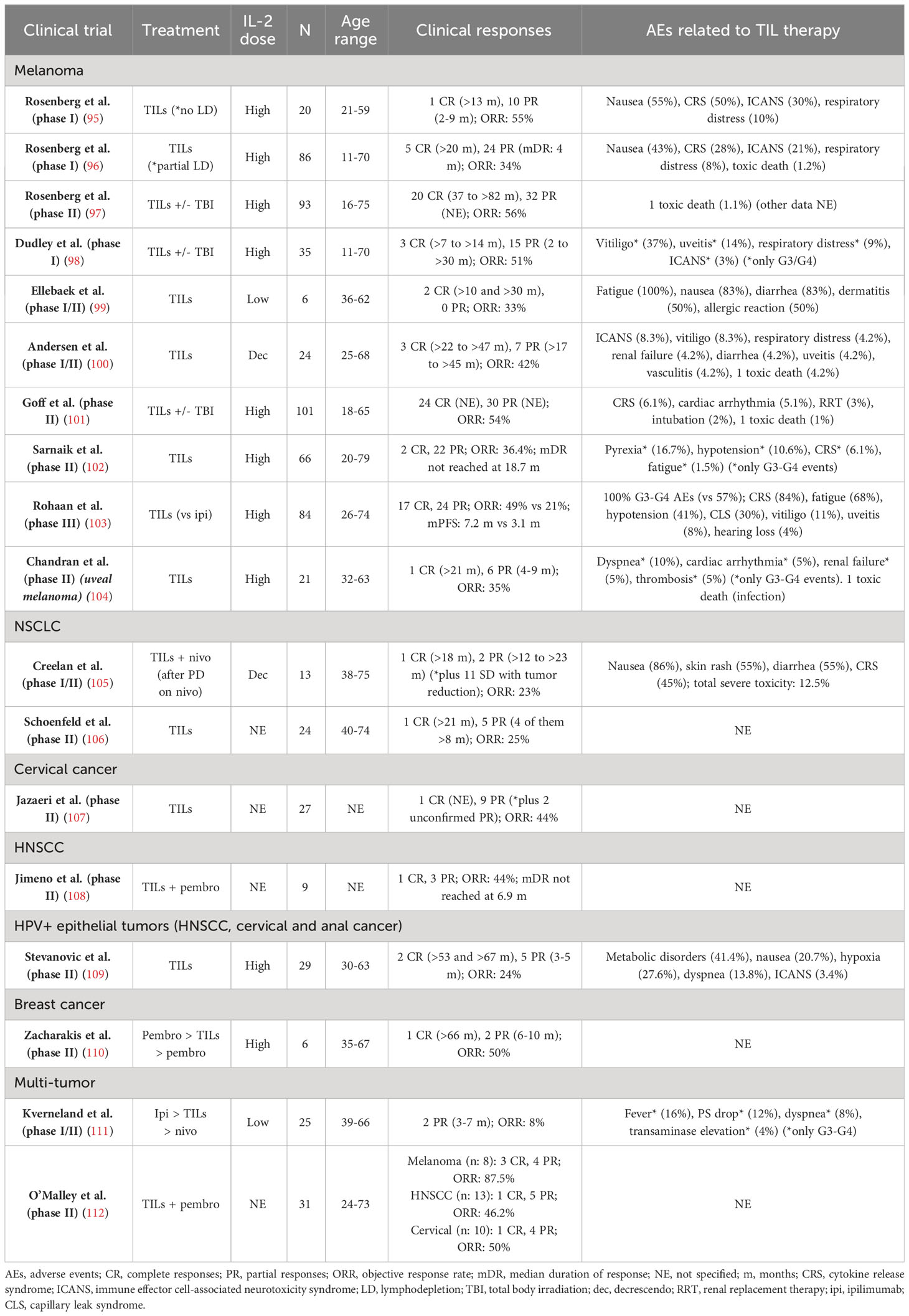
Table 2 Clinical trials with published results of TIL therapy in solid tumors.
5.3 Clinical outcomes in other solid tumors
Although TIL therapy has not yet achieved robust results in non-melanoma tumors, some phase I/II clinical trials have shown promising data in other immunogenic malignancies, such as NSCLC, cervical cancer, and HNSCC.
In a phase I/II trial, Creelan et al. (105) evaluated the efficacy of autologous TILs in combination with anti-PD1 nivolumab in 20 patients with advanced NSCLC following progression to nivolumab monotherapy. Among the 13 evaluable patients, there were 3 objective responses (2 CR -both ongoing >18 months-, 1 PR, and 8 SD with tumor reduction). Interestingly, one of the patients with sustained CR was EGFR-mutant (exon 19 deletion), which is a known predictor of anti-PD1 failure (113), suggesting that NSCLC subtypes that are commonly refractory to ICI might not be resistant to TIL therapy. Further research is required to assess whether TIL therapy could have a re-sensitizing effect for ICI use.
A phase II trial evaluated TIL monotherapy (LN-145) in 28 patients with advanced pretreated NSCLC, observing 1 CR (>21 months) and 5 PR (4 of them >8 months) among 24 evaluable patients (ORR 25%) (106).
TIL therapy also has promising results in a phase II trial with patients with advanced cervical cancer, observing 1 CR and 11 PR (2 of them unconfirmed by study criteria) (ORR: 44%) (107). Another study evaluated TILs in 29 patients with HPV+ epithelial tumors (HNSCC, cervical and anal cancer), with an ORR of 24% (including 2 CR, ongoing after 53 and 67 months) (109).
O’Malley et al. (112) conducted a multi-tumor trial with TIL therapy plus pembrolizumab in 31 patients (13 HNSCC, 10 cervical cancers, and 8 melanoma), with positive results (ORR 46.2%, 50%, and 87.5%, respectively). This TIL+ICI combination has also been evaluated in small phase II trials in HNSCC (108) and metastatic breast cancer (110), with promising results (ORR 44% and 50%, respectively). Interestingly, the second study included a patient with hormone-positive breast cancer who achieved a sustained CR (>5 years). However, a limitation of these trials is that some patients, especially those with ICI-responding tumors such as melanoma and HNSCC, might have responded to anti-PD1 blockade itself, making the role of TIL therapy difficult to assess. The sequential use of ipilimumab, TIL therapy, and nivolumab in different solid tumors was evaluated in a phase I/II trial of 25 patients, with modest results (2 PR ranging from 3 to 7 months) (111).
5.4 Challenges for clinical practice
Despite the promising results of TIL therapy in clinical trials, many practical and economic challenges have limited its large-scale implementation (114).
TIL manufacturing begins with a surgical resection of tumor tissue, preferably from metastases accessible with minimally invasive surgery. To date, the generation of TILs has been equally successful regardless of the resected lesion site (115). Enzymatic digestion and/or mechanical fragmentation of the surgical sample is followed by culture of the fragments in IL-2 containing media, a process that might take 2-6 weeks. This minimally expanded or “young” TILs are massively expanded using high doses of IL-2, anti-CD3, and irradiated peripheral blood mononuclear cells (PBMCs) or ‘feeder cells’, within a 2-weeks rapid expansion protocol (REP). The expanded TILs are eventually reinfused into the lymphodepleted patients, followed by high-dose bolus IL2 (114).
This is an expensive and logistically complex process that requires highly specialized facilities, protocolized procedures, and qualified technical staff. These are important limitations for the widespread application of TIL therapy, particularly in developing countries. Grade 3-4 adverse events, which appear in virtually all patients treated with TIL therapy, mainly following lymphodepletion and IL2 administration, also require highly specialized management. The significant treatment toxicity, together with the long duration of the manufacturing process, are substantial obstacles to the successful application of TIL therapy in patients with refractory metastatic tumors that often lead to rapid clinical deterioration.
To date, TIL therapy has been evaluated in young, fit patients with an ECOG performance status (PS) of 0-1. Even in this selected population from clinical trials, it is often found that only a small proportion of the screened patients can finally receive treatment. For example, in a phase II trial of TIL therapy in advanced breast cancer, only 6 of the 46 screened patients (13%) received TILs infusion (110). No residual or evaluable disease after resection, inadequate material for screening, negative or weak isolation of lymphocytes in the resected samples, clinical progression, PS deterioration, and lymphodepletion-related severe toxicity were common causes of treatment failure.
Lastly, not only the economic expenses but also the individualized nature of TIL therapy, which differs from conventional commercial drugs, are important challenges for TIL therapy regulatory approval. Previous experience with CAR-T cells in hematologic malignancies will surely smooth the way for dealing with the health-economic aspects of TIL therapy and its implementation in clinical practice.
6 Conclusion
The clinical outcomes of adoptive cell therapy (ACT) in solid tumors are conditioned by biological aspects that substantially differ from those of hematologic malignancies, such as their antigenic heterogeneity, immunosuppressive microenvironment, and immune scape ability. Dozens of phase I/II trials with CAR-T cells have only led to sporadic, usually short-term clinical responses, although novel strategies, such as multiantigenic recognition, SynNotch receptors, and CAR modulation through CRISPR technology, are promising approaches to improve their efficacy. Despite the inherent limitations of antigen-targeted treatments, TCR-engineered T-cells have several advantages over CAR-T cells and have shown promising results in certain tumors, such as melanoma and synovial sarcoma with NY-ESO1 overexpression. In the opinion of the authors, the treatment of HPV+ and other virus-related malignancies, as well as the use of neoantigens as targets, are exciting fields of TCR-T-cells research.
Conceptually different from other ACT modalities, TIL therapy is based on the extraction, ex vivo expansion, and stimulation of naturally reactive tumor-infiltrating lymphocytes, followed by their reinfusion into lymphodepleted patients. TILs have shown positive results in advanced melanoma, with a recent positive phase III trial that has proved their superiority to ipilimumab in anti-PD1 refractory disease. The efficacy of TIL therapy, alone or in combination with checkpoint inhibitors, has also been demonstrated in other solid tumors including NSCLC, cervical cancer, and HNSCC. Strategies to selectively expand neoantigen-reactive TILs or genetically modify the expanding cells through CRISPR technology are exciting lines of research, to improve the efficacy of TIL therapy in patients with melanoma and extend its clinical benefit to other malignancies. However, numerous clinical and practical limitations that currently hinder its large-scale implementation still need to be overcome.
Author contributions
VA: Writing – original draft, Writing – review & editing. MS: Writing – review & editing. JP: Writing – review & editing. JC: Writing – review & editing. DR: Writing – review & editing. PG: Writing – review & editing. JCC: Writing – review & editing. CG: Writing – review & editing. CGQ: Writing – review & editing. PP: Writing – review & editing. JM: Writing – review & editing. AC: Writing – review & editing. AS: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
BioRender.com was used for the design of Figure 1.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. (2013) 39:1–10. doi: 10.1016/j.immuni.2013.07.012
2. Dustin ML. The immunological synapse. Cancer Immunol Res. (2014) 2:1023–33. doi: 10.1158/2326-6066.CIR-14-0161
3. Mills DM, Cambier JC. B lymphocyte activation during cognate interactions with CD4+ T lymphocytes: molecular dynamics and immunologic consequences. Semin Immunol. (2003) 15:325–9. doi: 10.1016/j.smim.2003.09.004
4. Slaney CY, Kershaw MH, Darcy PK. Trafficking of T cells into tumors. Cancer Res. (2014) 74:7168–74. doi: 10.1158/0008-5472.CAN-14-2458
5. Chen B, Li H, Liu C, Xiang X, Wang S, Wu A, et al. Prognostic value of the common tumour-infiltrating lymphocyte subtypes for patients with non-small cell lung cancer: A meta-analysis. PloS One. (2020) 15:e0242173. doi: 10.1371/journal.pone.0242173
6. Bi Q, Liu Y, Yuan T, Wang H, Li B, Jiang Y, et al. Predicted CD4+ T cell infiltration levels could indicate better overall survival in sarcoma patients. J Int Med Res. (2021) 49:300060520981539. doi: 10.1177/0300060520981539
7. Fridman WH, Pagès F, Sautès-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. (2012) 12:298–306. doi: 10.1038/nrc3245
8. Hadrup S, Donia M, Thor Straten P. Effector CD4 and CD8 T cells and their role in the tumor microenvironment. Cancer Microenviron. (2013) 6:123–33. doi: 10.1007/s12307-012-0127-6
9. Li L, Yu R, Cai T, Chen Z, Lan M, Zou T, et al. Effects of immune cells and cytokines on inflammation and immunosuppression in the tumor microenvironment. Int Immunopharmacol. (2020) 88:106939. doi: 10.1016/j.intimp.2020.106939
10. Kong X. Discovery of new immune checkpoints: family grows up. Adv Exp Med Biol. (2020) 1248:61–82. doi: 10.1007/978-981-15-3266-5_4
11. Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discovery. (2022) 12:31–46. doi: 10.1158/2159-8290.CD-21-1059
12. Dubrot J, Du PP, Lane-Reticker SK, Kessler EA, Muscato AJ, Mehta A, et al. In vivo CRISPR screens reveal the landscape of immune evasion pathways across cancer. Nat Immunol. (2022) 23:1495–506. doi: 10.1038/s41590-022-01315-x
13. DhatChinamoorthy K, Colbert JD, Rock KL. Cancer immune evasion through loss of MHC class I antigen presentation. Front Immunol. (2021) 12:636568. doi: 10.3389/fimmu.2021.636568
14. Batlle E, Massagué J. Transforming growth factor-β Signaling in immunity and cancer. Immunity. (2019) 50:924–40. doi: 10.1016/j.immuni.2019.03.024
15. Zhai L, Ladomersky E, Lenzen A, Nguyen B, Patel R, Lauing KL, et al. IDO1 in cancer: a Gemini of immune checkpoints. Cell Mol Immunol. (2018) 15:447–57. doi: 10.1038/cmi.2017.143
16. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature. (2015) 523:231–5. doi: 10.1038/nature14404
17. Essola JM, Zhang M, Yang H, Li F, Xia B, Mavoungou JF, et al. Exosome regulation of immune response mechanism: Pros and cons in immunotherapy. Bioact Mater. (2024) 32:124–46. doi: 10.1016/j.bioactmat.2023.09.018
18. Chang E, Pelosof L, Lemery S, Gong Y, Goldberg KB, Farrell AT, et al. Systematic review of PD-1/PD-L1 inhibitors in oncology: from personalized medicine to public health. Oncologist. (2021) 26:e1786–99. doi: 10.1002/onco.13887
19. Morotti M, Albukhari A, Alsaadi A, Artibani M, Brenton JD, Curbishley SM, et al. Promises and challenges of adoptive T-cell therapies for solid tumours. Br J Cancer. (2021) 124:1759–76. doi: 10.1038/s41416-021-01353-6
20. Zhang C, Liu J, Zhong JF, Zhang X. Engineering CAR-T cells. biomark Res. (2017) 5:22. doi: 10.1186/s40364-017-0102-y
21. Huang R, Li X, He Y, Zhu W, Gao L, Liu Y, et al. Recent advances in CAR-T cell engineering. J Hematol Oncol. (2020) 13:86. doi: 10.1186/s13045-020-00910-5
22. Yuti P, Sawasdee N, Natungnuy K, Rujirachaivej P, Luangwattananun P, Sujjitjoon J, et al. Enhanced antitumor efficacy, proliferative capacity, and alleviation of T cell exhaustion by fifth-generation chimeric antigen receptor T cells targeting B cell maturation antigen in multiple myeloma. Biomed Pharmacother. (2023) 168:115691. doi: 10.1016/j.biopha.2023.115691
23. Safarzadeh Kozani P, Naseri A, Mirarefin SMJ, Salem F, Nikbakht M, Evazi Bakhshi S, et al. Nanobody-based CAR-T cells for cancer immunotherapy. biomark Res. (2022) 10:24. doi: 10.1186/s40364-022-00371-7
24. Nasiri F, Safarzadeh Kozani P, Rahbarizadeh F. T-cells engineered with a novel VHH-based chimeric antigen receptor against CD19 exhibit comparable tumoricidal efficacy to their FMC63-based counterparts. Front Immunol. (2023) 14:1063838. doi: 10.3389/fimmu.2023.1063838
25. Shank BR, Do B, Sevin A, Chen SE, Neelapu SS, Horowitz SB. Chimeric antigen receptor T cells in hematologic Malignancies. Pharmacotherapy. (2017) 37:334–45. doi: 10.1002/phar.1900
26. Sermer D, Brentjens R. CAR T-cell therapy: Full speed ahead. Hematological Oncol. (2019) 37:95–100. doi: 10.1002/hon.2591
27. Marofi F, Motavalli R, Safonov VA, Thangavelu L, Yumashev AV, Alexander M, et al. CAR T cells in solid tumors: challenges and opportunities. Stem Cell Res Ther. (2021) 12:81. doi: 10.1186/s13287-020-02128-1
28. Wang V, Gauthier M, Decot V, Reppel L, Bensoussan D. Systematic review on CAR-T cell clinical trials up to 2022: academic center input. Cancers. (2023) 15:1003. doi: 10.3390/cancers15041003
29. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. (2016) 375:2561–9. doi: 10.1056/NEJMoa1610497
30. Bagley SJ, Desai AS, Linette GP, June CH, O’Rourke DM. CAR T-cell therapy for glioblastoma: recent clinical advances and future challenges. Neuro-Oncology. (2018) 20:1429–38. doi: 10.1093/neuonc/noy032
31. Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. HER2-specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: A phase 1 dose-escalation trial. JAMA Oncol. (2017) 3:1094–101. doi: 10.1001/jamaoncol.2017.0184
32. Goff SL, Morgan RA, Yang JC, Sherry RM, Robbins PF, Restifo NP, et al. Pilot trial of adoptive transfer of chimeric antigen receptor–transduced T cells targeting EGFRvIII in patients with glioblastoma. J Immunother. (2019) 42:126–35. doi: 10.1097/CJI.0000000000000260
33. O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. (2017) 9:eaaa0984. doi: 10.1126/scitranslmed.aaa0984
34. Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, et al. Human epidermal growth factor receptor 2 (HER2) -specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol. (2015) 33:1688–96. doi: 10.1200/JCO.2014.58.0225
35. Yu AL, Gilman AL, Ozkaynak MF, Naranjo A, Diccianni MB, Gan J, et al. Long-term follow-up of a phase III study of ch14.18 (Dinutuximab) + Cytokine immunotherapy in children with high-risk neuroblastoma: COG study ANBL0032. Clin Cancer Res. (2021) 27:2179–89. doi: 10.1158/1078-0432.CCR-20-3909
36. Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. (2011) 118:6050–6. doi: 10.1182/blood-2011-05-354449
37. Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. (2008) 14:1264–70. doi: 10.1038/nm.1882
38. Heczey A, Louis CU, Savoldo B, Dakhova O, Durett A, Grilley B, et al. CAR T cells administered in combination with lymphodepletion and PD-1 inhibition to patients with neuroblastoma. Mol Ther. (2017) 25:2214–24. doi: 10.1016/j.ymthe.2017.05.012
39. Wang Y, Chen M, Wu Z, Tong C, Dai H, Guo Y, et al. CD133-directed CAR T cells for advanced metastasis Malignancies: A phase I trial. Oncoimmunology. (2018) 7:e1440169. doi: 10.1080/2162402X.2018.1440169
40. Zhan X, Wang B, Li Z, Li J, Wang H, Chen L, et al. Phase I trial of Claudin 18.2-specific chimeric antigen receptor T cells for advanced gastric and pancreatic adenocarcinoma. JCO. (2019) 37:2509. doi: 10.1200/JCO.2019.37.15_suppl.2509
41. Liu Y, Guo Y, Wu Z, Feng K, Tong C, Wang Y, et al. Anti-EGFR chimeric antigen receptor-modified T cells in metastatic pancreatic carcinoma: A phase I clinical trial. Cytotherapy. (2020) 22:573–80. doi: 10.1016/j.jcyt.2020.04.088
42. Guo Y, Feng K, Liu Y, Wu Z, Dai H, Yang Q, et al. Phase I study of chimeric antigen receptor–modified T cells in patients with EGFR-positive advanced biliary tract cancers. Clin Cancer Res. (2018) 24:1277–86. doi: 10.1158/1078-0432.CCR-17-0432
43. Shi D, Shi Y, Kaseb AO, Qi X, Zhang Y, Chi J, et al. Chimeric antigen receptor-glypican-3 T-cell therapy for advanced hepatocellular carcinoma: results of phase I trials. Clin Cancer Res. (2020) 26:3979–89. doi: 10.1158/1078-0432.CCR-19-3259
44. Zhang C, Wang Z, Yang Z, Wang M, Li S, Li Y, et al. Phase I escalating-dose trial of CAR-T therapy targeting CEA+ Metastatic colorectal cancers. Mol Ther. (2017) 25:1248–58. doi: 10.1016/j.ymthe.2017.03.010
45. Adusumilli PS, Zauderer MG, Rusch VW, O’Cearbhaill R, Zhu A, Ngai D, et al. Regional delivery of mesothelin-targeted CAR T cells for pleural cancers: Safety and preliminary efficacy in combination with anti-PD-1 agent. JCO. (2019) 37:2511. doi: 10.1200/JCO.2019.37.15_suppl.2511
46. Feng K, Guo Y, Dai H, Wang Y, Li X, Jia H, et al. Chimeric antigen receptor-modified T cells for the immunotherapy of patients with EGFR-expressing advanced relapsed/refractory non-small cell lung cancer. Sci China Life Sci. (2016) 59:468–79. doi: 10.1007/s11427-016-5023-8
47. Specht JM, Lee S, Turtle C, Berger C, Veatch J, Gooley T, et al. Phase I study of immunotherapy for advanced ROR1+ Malignancies with autologous ROR1-specific chimeric antigen receptor-modified (CAR)-T cells. JCO. (2018) 36:TPS79. doi: 10.1200/JCO.2018.36.5_suppl.TPS79
48. Narayan V, Barber-Rotenberg JS, Jung I-Y, Lacey SF, Rech AJ, Davis MM, et al. PSMA-targeting TGFβ-insensitive armored CAR T cells in metastatic castration-resistant prostate cancer: a phase 1 trial. Nat Med. (2022) 28:724–34. doi: 10.1038/s41591-022-01726-1
49. Maalej KM, Merhi M, Inchakalody VP, Mestiri S, Alam M, Maccalli C, et al. CAR-cell therapy in the era of solid tumor treatment: current challenges and emerging therapeutic advances. Mol Cancer. (2023) 22:20. doi: 10.1186/s12943-023-01723-z
50. Baulu E, Gardet C, Chuvin N, Depil S. TCR-engineered T cell therapy in solid tumors: State of the art and perspectives. Sci Adv. (2023) 9:eadf3700. doi: 10.1126/sciadv.adf3700
51. Foy SP, Jacoby K, Bota DA, Hunter T, Pan Z, Stawiski E, et al. Non-viral precision T cell receptor replacement for personalized cell therapy. Nature. (2023) 615:687–96. doi: 10.1038/s41586-022-05531-1
52. Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. (2006) 314:126–9. doi: 10.1126/science.1129003
53. Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. (2009) 114:535–46. doi: 10.1182/blood-2009-03-211714
54. Chodon T, Comin-Anduix B, Chmielowski B, Koya RC, Wu Z, Auerbach M, et al. Adoptive transfer of MART-1 T-cell receptor transgenic lymphocytes and dendritic cell vaccination in patients with metastatic melanoma. Clin Cancer Res. (2014) 20:2457–65. doi: 10.1158/1078-0432.CCR-13-3017
55. Rohaan MW, Gomez-Eerland R, van den Berg JH, Geukes Foppen MH, van Zon M, Raud B, et al. MART-1 TCR gene-modified peripheral blood T cells for the treatment of metastatic melanoma: a phase I/IIa clinical trial. Immunooncol Technol. (2022) 15:100089. doi: 10.1016/j.iotech.2022.100089
56. Iura K, Maekawa A, Kohashi K, Ishii T, Bekki H, Otsuka H, et al. Cancer-testis antigen expression in synovial sarcoma: NY-ESO-1, PRAME, MAGEA4, and MAGEA1. Hum Pathol. (2017) 61:130–9. doi: 10.1016/j.humpath.2016.12.006
57. Robbins PF, Kassim SH, Tran TLN, Crystal JS, Morgan RA, Feldman SA, et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clin Cancer Res. (2015) 21:1019–27. doi: 10.1158/1078-0432.CCR-14-2708
58. Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. (2011) 29:917–24. doi: 10.1200/JCO.2010.32.2537
59. Nowicki TS, Berent-Maoz B, Cheung-Lau G, Huang RR, Wang X, Tsoi J, et al. A pilot trial of the combination of transgenic NY-ESO-1-reactive adoptive cellular therapy with dendritic cell vaccination with or without ipilimumab. Clin Cancer Res. (2019) 25:2096–108. doi: 10.1158/1078-0432.CCR-18-3496
60. D’Angelo SP, Melchiori L, Merchant MS, Bernstein D, Glod J, Kaplan R, et al. Antitumor activity associated with prolonged persistence of adoptively transferred NY-ESO-1 c259T cells in synovial sarcoma. Cancer Discovery. (2018) 8:944–57. doi: 10.1158/2159-8290.CD-17-1417
61. Ramachandran I, Lowther DE, Dryer-Minnerly R, Wang R, Fayngerts S, Nunez D, et al. Systemic and local immunity following adoptive transfer of NY-ESO-1 SPEAR T cells in synovial sarcoma. J Immunother Cancer. (2019) 7:276. doi: 10.1186/s40425-019-0762-2
62. Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, et al. CRISPR-engineered T cells in patients with refractory cancer. Science. (2020) 367:eaba7365. doi: 10.1126/science.aba7365
63. Lu Y-C, Parker LL, Lu T, Zheng Z, Toomey MA, White DE, et al. Treatment of patients with metastatic cancer using a major histocompatibility complex class II-restricted T-cell receptor targeting the cancer germline antigen MAGE-A3. J Clin Oncol. (2017) 35:3322–9. doi: 10.1200/JCO.2017.74.5463
64. Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother. (2013) 36:133–51. doi: 10.1097/CJI.0b013e3182829903
65. Hong DS, Van Tine BA, Olszanski AJ, Johnson ML, Liebner DA, Trivedi T, et al. Phase I dose escalation and expansion trial to assess the safety and efficacy of ADP-A2M4 SPEAR T cells in advanced solid tumors. JCO. (2020) 38:102. doi: 10.1200/JCO.2020.38.15_suppl.102
66. Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. (2013) 122:863–71. doi: 10.1182/blood-2013-03-490565
67. Martin AD, Wang X, Sandberg ML, Negri KR, Wu ML, Toledo Warshaviak D, et al. Re-examination of MAGE-A3 as a T-cell therapeutic target. J Immunother. (2021) 44:95–105. doi: 10.1097/CJI.0000000000000348
68. Blumenschein GR, Devarakonda S, Johnson M, Moreno V, Gainor J, Edelman MJ, et al. Phase I clinical trial evaluating the safety and efficacy of ADP-A2M10 SPEAR T cells in patients with MAGE-A10+ advanced non-small cell lung cancer. J Immunother Cancer. (2022) 10:e003581. doi: 10.1136/jitc-2021-003581
69. Kageyama S, Ikeda H, Miyahara Y, Imai N, Ishihara M, Saito K, et al. Adoptive transfer of MAGE-A4 T-cell receptor gene-transduced lymphocytes in patients with recurrent esophageal cancer. Clin Cancer Res. (2015) 21:2268–77. doi: 10.1158/1078-0432.CCR-14-1559
70. Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan D-AN, Feldman SA, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. (2011) 19:620–6. doi: 10.1038/mt.2010.272
71. Doran SL, Stevanović S, Adhikary S, Gartner JJ, Jia L, Kwong MLM, et al. T-cell receptor gene therapy for human papillomavirus-associated epithelial cancers: A first-in-human, phase I/II study. J Clin Oncol. (2019) 37:2759–68. doi: 10.1200/JCO.18.02424
72. Nagarsheth NB, Norberg SM, Sinkoe AL, Adhikary S, Meyer TJ, Lack JB, et al. TCR-engineered T cells targeting E7 for patients with metastatic HPV-associated epithelial cancers. Nat Med. (2021) 27:419–25. doi: 10.1038/s41591-020-01225-1
73. Meng F, Zhao J, Tan AT, Hu W, Wang S-Y, Jin J, et al. Immunotherapy of HBV-related advanced hepatocellular carcinoma with short-term HBV-specific TCR expressed T cells: results of dose escalation, phase I trial. Hepatol Int. (2021) 15:1402–12. doi: 10.1007/s12072-021-10250-2
74. Veatch J, Paulson K, Asano Y, Martin L, Lee B, Hall ET, et al. Merkel polyoma virus specific T-cell receptor transgenic T-cell therapy in PD-1 inhibitor refractory Merkel cell carcinoma. JCO. (2022) 40:9549. doi: 10.1200/JCO.2022.40.16_suppl.9549
75. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. (2015) 348:124–8. doi: 10.1126/science.aaa1348
76. Leidner R, Sanjuan Silva N, Huang H, Sprott D, Zheng C, Shih Y-P, et al. Neoantigen T-cell receptor gene therapy in pancreatic cancer. N Engl J Med. (2022) 386:2112–9. doi: 10.1056/NEJMoa2119662
77. Kim SP, Vale NR, Zacharakis N, Krishna S, Yu Z, Gasmi B, et al. Adoptive cellular therapy with autologous tumor-infiltrating lymphocytes and T-cell receptor-engineered T cells targeting common p53 neoantigens in human solid tumors. Cancer Immunol Res. (2022) 10:932–46. doi: 10.1158/2326-6066.CIR-22-0040
78. Losic B, Craig AJ, Villacorta-Martin C, Martins-Filho SN, Akers N, Chen X, et al. Intratumoral heterogeneity and clonal evolution in liver cancer. Nat Commun. (2020) 11:291. doi: 10.1038/s41467-019-14050-z
79. Andersson A, Larsson L, Stenbeck L, Salmén F, Ehinger A, Wu SZ, et al. Spatial deconvolution of HER2-positive breast cancer delineates tumor-associated cell type interactions. Nat Commun. (2021) 12:6012. doi: 10.1038/s41467-021-26271-2
80. Lee YG, Marks I, Srinivasarao M, Kanduluru AK, Mahalingam SM, Liu X, et al. Use of a single CAR T cell and several bispecific adapters facilitates eradication of multiple antigenically different solid tumors. Cancer Res. (2019) 79:387–96. doi: 10.1158/0008-5472.CAN-18-1834
81. Yu S, Yi M, Qin S, Wu K. Next generation chimeric antigen receptor T cells: safety strategies to overcome toxicity. Mol Cancer. (2019) 18:125. doi: 10.1186/s12943-019-1057-4
82. Sheth VS, Gauthier J. Taming the beast: CRS and ICANS after CAR T-cell therapy for ALL. Bone Marrow Transplant. (2021) 56:552–66. doi: 10.1038/s41409-020-01134-4
83. Benmebarek M-R, Karches CH, Cadilha BL, Lesch S, Endres S, Kobold S. Killing mechanisms of chimeric antigen receptor (CAR) T cells. Int J Mol Sci. (2019) 20:1283. doi: 10.3390/ijms20061283
84. Feng K, Liu Y, Guo Y, Qiu J, Wu Z, Dai H, et al. Phase I study of chimeric antigen receptor modified T cells in treating HER2-positive advanced biliary tract cancers and pancreatic cancers. Protein Cell. (2018) 9:838–47. doi: 10.1007/s13238-017-0440-4
85. Qi C, Gong J, Li J, Liu D, Qin Y, Ge S, et al. Claudin18.2-specific CAR T cells in gastrointestinal cancers: phase 1 trial interim results. Nat Med. (2022) 28:1189–98. doi: 10.1038/s41591-022-01800-8
86. Majzner RG, Ramakrishna S, Yeom KW, Patel S, Chinnasamy H, Schultz LM, et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature. (2022) 603:934–41. doi: 10.1038/s41586-022-04489-4
87. Mount CW, Majzner RG, Sundaresh S, Arnold EP, Kadapakkam M, Haile S, et al. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M+ diffuse midline gliomas. Nat Med. (2018) 24:572–9. doi: 10.1038/s41591-018-0006-x
88. Majzner RG, Rietberg SP, Sotillo E, Dong R, Vachharajani VT, Labanieh L, et al. Tuning the antigen density requirement for CAR T-cell activity. Cancer Discovery. (2020) 10:702–23. doi: 10.1158/2159-8290.CD-19-0945
89. Flugel CL, Majzner RG, Krenciute G, Dotti G, Riddell SR, Wagner DL, et al. Overcoming on-target, off-tumour toxicity of CAR T cell therapy for solid tumours. Nat Rev Clin Oncol. (2023) 20:49–62. doi: 10.1038/s41571-022-00704-3
90. Ogiya R, Niikura N, Kumaki N, Bianchini G, Kitano S, Iwamoto T, et al. Comparison of tumor-infiltrating lymphocytes between primary and metastatic tumors in breast cancer patients. Cancer Sci. (2016) 107:1730–5. doi: 10.1111/cas.13101
91. Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, et al. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. (2009) 69:3077–85. doi: 10.1158/0008-5472.CAN-08-2281
92. Donnadieu E, Dupré L, Pinho LG, Cotta-de-Almeida V. Surmounting the obstacles that impede effective CAR T cell trafficking to solid tumors. J Leukoc Biol. (2020) 108:1067–79. doi: 10.1002/JLB.1MR0520-746R
93. Gumber D, Wang LD. Improving CAR-T immunotherapy: Overcoming the challenges of T cell exhaustion. eBioMedicine. (2022) 77:103941. doi: 10.1016/j.ebiom.2022.103941
94. Junker N, Kvistborg P, Køllgaard T, Straten PT, Andersen MH, Svane IM. Tumor associated antigen specific T-cell populations identified in ex vivo expanded TIL cultures. Cell Immunol. (2012) 273:1–9. doi: 10.1016/j.cellimm.2011.12.004
95. Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. N Engl J Med. (1988) 319:1676–80. doi: 10.1056/NEJM198812223192527
96. Rosenberg SA, Yannelli JR, Yang JC, Topalian SL, Schwartzentruber DJ, Weber JS, et al. Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2. J Natl Cancer Inst. (1994) 86:1159–66. doi: 10.1093/jnci/86.15.1159
97. Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. (2011) 17:4550–7. doi: 10.1158/1078-0432.CCR-11-0116
98. Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. (2005) 23:2346–57. doi: 10.1200/JCO.2005.00.240
99. Ellebaek E, Iversen TZ, Junker N, Donia M, Engell-Noerregaard L, Met Ö, et al. Adoptive cell therapy with autologous tumor infiltrating lymphocytes and low-dose Interleukin-2 in metastatic melanoma patients. J Transl Med. (2012) 10:169. doi: 10.1186/1479-5876-10-169
100. Andersen R, Donia M, Ellebaek E, Borch TH, Kongsted P, Iversen TZ, et al. Long-lasting complete responses in patients with metastatic melanoma after adoptive cell therapy with tumor-infiltrating lymphocytes and an attenuated IL2 regimen. Clin Cancer Res. (2016) 22:3734–45. doi: 10.1158/1078-0432.CCR-15-1879
101. Goff SL, Dudley ME, Citrin DE, Somerville RP, Wunderlich JR, Danforth DN, et al. Randomized, prospective evaluation comparing intensity of lymphodepletion before adoptive transfer of tumor-infiltrating lymphocytes for patients with metastatic melanoma. J Clin Oncol. (2016) 34:2389–97. doi: 10.1200/JCO.2016.66.7220
102. Sarnaik AA, Hamid O, Khushalani NI, Lewis KD, Medina T, Kluger HM, et al. Lifileucel, a tumor-infiltrating lymphocyte therapy, in metastatic melanoma. J Clin Oncol. (2021) 39:2656–66. doi: 10.1200/JCO.21.00612
103. Rohaan MW, Borch TH, van den Berg JH, Met Ö, Kessels R, Geukes Foppen MH, et al. Tumor-infiltrating lymphocyte therapy or ipilimumab in advanced melanoma. N Engl J Med. (2022) 387:2113–25. doi: 10.1056/NEJMoa2210233
104. Chandran SS, Somerville RPT, Yang JC, Sherry RM, Klebanoff CA, Goff SL, et al. Treatment of metastatic uveal melanoma with adoptive transfer of tumour-infiltrating lymphocytes: a single-centre, two-stage, single-arm, phase 2 study. Lancet Oncol. (2017) 18:792–802. doi: 10.1016/S1470-2045(17)30251-6
105. Creelan BC, Wang C, Teer JK, Toloza EM, Yao J, Kim S, et al. Tumor-infiltrating lymphocyte treatment for anti-PD-1-resistant metastatic lung cancer: a phase 1 trial. Nat Med. (2021) 27:1410–8. doi: 10.1038/s41591-021-01462-y
106. Schoenfeld A, Lee S, Paz-Ares L, Doger B, Gettinger S, Haefliger S, et al. 458 First phase 2 results of autologous tumor-infiltrating lymphocyte (TIL; LN-145) monotherapy in patients with advanced, immune checkpoint inhibitor-treated, non-small cell lung cancer (NSCLC). J Immunother Cancer. (2021) 9:A486–7. doi: 10.1136/jitc-2021-SITC2021.458
107. Jazaeri AA, Zsiros E, Amaria RN, Artz AS, Edwards RP, Wenham RM, et al. Safety and efficacy of adoptive cell transfer using autologous tumor infiltrating lymphocytes (LN-145) for treatment of recurrent, metastatic, or persistent cervical carcinoma. JCO. (2019) 37:2538. doi: 10.1200/JCO.2019.37.15_suppl.2538
108. Jimeno A, Papa S, Haigentz M, Rodríguez-Moreno J, Schardt J, Fardis M, et al. 353 Safety and efficacy of tumor infiltrating lymphocytes (TIL, LN-145) in combination with pembrolizumab for advanced, recurrent or metastatic HNSCC. J ImmunoTher Cancer. (2020) 8. doi: 10.1136/jitc-2020-SITC2020.0353
109. Stevanović S, Helman SR, Wunderlich JR, Langhan MM, Doran SL, Kwong MLM, et al. A phase II study of tumor-infiltrating lymphocyte therapy for human papillomavirus-associated epithelial cancers. Clin Cancer Res. (2019) 25:1486–93. doi: 10.1158/1078-0432.CCR-18-2722
110. Zacharakis N, Huq LM, Seitter SJ, Kim SP, Gartner JJ, Sindiri S, et al. Breast cancers are immunogenic: immunologic analyses and a phase II pilot clinical trial using mutation-reactive autologous lymphocytes. J Clin Oncol. (2022) 40:1741–54. doi: 10.1200/JCO.21.02170
111. Kverneland AH, Chamberlain CA, Borch TH, Nielsen M, Mørk SK, Kjeldsen JW, et al. Adoptive cell therapy with tumor-infiltrating lymphocytes supported by checkpoint inhibition across multiple solid cancer types. J Immunother Cancer. (2021) 9:e003499. doi: 10.1136/jitc-2021-003499
112. O’Malley D, Lee S, Psyrri A, Sukari A, Thomas S, Wenham R, et al. 492 Phase 2 efficacy and safety of autologous tumor-infiltrating lymphocyte (TIL) cell therapy in combination with pembrolizumab in immune checkpoint inhibitor-naïve patients with advanced cancers. J Immunother Cancer. (2021) 9:A523–4. doi: 10.1136/jitc-2021-SITC2021.492
113. Yang F, Wang Y, Tang L, Mansfield AS, Adjei AA, Leventakos K, et al. Efficacy of immune checkpoint inhibitors in non-small cell lung cancer: A systematic review and meta-analysis. Front Oncol. (2022) 12:955440. doi: 10.3389/fonc.2022.955440
114. Monberg TJ, Borch TH, Svane IM, Donia M. TIL therapy: facts and hopes. Clin Cancer Res. (2023) 29(17):3275–83. doi: 10.1158/1078-0432.CCR-22-2428
115. Besser MJ, Shapira-Frommer R, Treves AJ, Zippel D, Itzhaki O, Hershkovitz L, et al. Clinical responses in a phase II study using adoptive transfer of short-term cultured tumor infiltration lymphocytes in metastatic melanoma patients. Clin Cancer Res. (2010) 16:2646–55. doi: 10.1158/1078-0432.CCR-10-0041
Keywords: immunotherapy, adoptive cell therapy, T cells, car-t, TCR-modified cells, TIL therapy, melanoma
Citation: Albarrán V, San Román M, Pozas J, Chamorro J, Rosero DI, Guerrero P, Calvo JC, González C, García de Quevedo C, Pérez de Aguado P, Moreno J, Cortés A and Soria A (2024) Adoptive T cell therapy for solid tumors: current landscape and future challenges. Front. Immunol. 15:1352805. doi: 10.3389/fimmu.2024.1352805
Received: 08 December 2023; Accepted: 04 March 2024;
Published: 14 March 2024.
Edited by:
MIchael E. Hurwitz, Yale University, United StatesReviewed by:
Jorge Ibanez-Vega, St. Jude Children’s Research Hospital, United StatesPhilippe Lewalle, Université libre de Bruxelles, Belgium
Pouya Safarzadeh Kozani, Tarbiat Modares University, Iran
Copyright © 2024 Albarrán, San Román, Pozas, Chamorro, Rosero, Guerrero, Calvo, González, García de Quevedo, Pérez de Aguado, Moreno, Cortés and Soria. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Víctor Albarrán, dmljYWxiYXJyYW5mZXJuYW5kZXpAZ21haWwuY29t