 Hui Yang
Hui Yang Jinhong Li3†
Jinhong Li3† Richard Bucala
Richard Bucala Anping Xu
Anping Xu Hui-Yao Lan
Hui-Yao Lan- 1Department of Nephrology, Sun Yat‐Sen Memorial Hospital, Sun Yat‐Sen University, Guangzhou, China
- 2Department of Medicine and Therapeutics, Li Ka Shing Institute of Health Sciences, Lui Che Woo Institute of Innovative Medicine, The Chinese University of Hong Kong, Hong Kong, Hong Kong SAR, China
- 3Department of Nephrology, The Seventh Affiliated Hospital of Sun Yat‐sen University, SunYat‐sen University, Shenzhen, China
- 4Departments of Nephrology and Pathology, Guangdong Provincial Hospital, Southern Medical University, Guangzhou, China
- 5Department of Internal Medicine, Yale University School of Medicine, New Haven, CT, United States
Macrophages are a rich source of macrophage migration inhibitory factor (MIF). It is well established that macrophages and MIF play a pathogenic role in anti-glomerular basement membrane crescentic glomerulonephritis (anti-GBM CGN). However, whether macrophages mediate anti-GBM CGN via MIF-dependent mechanism remains unexplored, which was investigated in this study by specifically deleting MIF from macrophages in MIFf/f−lysM−cre mice. We found that compared to anti-GBM CGN induced in MIFf/f control mice, conditional ablation of MIF in macrophages significantly suppressed anti-GBM CGN by inhibiting glomerular crescent formation and reducing serum creatinine and proteinuria while improving creatine clearance. Mechanistically, selective MIF depletion in macrophages largely inhibited renal macrophage and T cell recruitment, promoted the polarization of macrophage from M1 towards M2 via the CD74/NF-κB/p38MAPK-dependent mechanism. Unexpectedly, selective depletion of macrophage MIF also significantly promoted Treg while inhibiting Th1 and Th17 immune responses. In summary, MIF produced by macrophages plays a pathogenic role in anti-GBM CGN. Targeting macrophage-derived MIF may represent a novel and promising therapeutic approach for the treatment of immune-mediated kidney diseases.
1 Introduction
Anti-glomerular basement membrane crescentic glomerulonephritis (anti-GBM CGN) is an autoimmune glomerular disease that progresses rapidly. It is characterized by glomerular crescentic formation and the presence of autoantibodies that target specific epitopes on the a3 chain of type IV collagen (1–3). The development of anti-GBM CGN involves different cellular components, such as macrophages, lymphocytes, intrinsic renal cells, and a complex network of cytokines (4, 5). Despite extensive research, the precise mechanisms underlying this disease are still not fully understood.
Macrophages play a crucial role in the development of anti-GBM GN by infiltrating the affected kidneys and contributing to inflammation and fibrosis (6). Recent studies have shown that depletion of macrophages or inhibiting the production of cytokines by macrophages can alleviate kidney injury in anti-GBM CGN (7–9). The severity of the disease is associated with the infiltration and activation of macrophages, which can exhibit different phenotypes depending on the local environment. Pro-inflammatory M1 macrophages promote renal injury, whereas anti-inflammatory M2 macrophages offer protection against kidney diseases (6, 10–14).
Macrophage migration inhibitory factor (MIF) is a versatile proinflammatory cytokine that plays a crucial role in triggering the release of multiple downstream cytokines and facilitating the recruitment of leukocytes to inflammatory organs by binding to CD74 (15, 16). In the pathogenesis of murine autoimmune glomerulonephritis (GN), including anti-GBM CGN, MIF has been identified as a key player, influencing both the inflammatory and adaptive immune responses (17–21). Recent research demonstrated that mice lacking the MIF gene were protected from renal injury in a murine CGN model. Additionally, studies involving bone marrow reconstitution revealed that the absence of MIF from both bone marrow-derived and non-myeloid-derived sources improves experimental anti-GBM GN (22). However, further investigation is required to establish the specific contribution of macrophage-derived MIF in the context of anti-GBM CGN.
In order to investigate the potential role of macrophage-derived MIF in anti-GBM CGN and to uncover the underlying mechanisms, we utilized genetic techniques to create a conditional knockout of MIF specifically in macrophages. Through comprehensive evaluations, we determined the role of macrophage-derived MIF in the development of anti-GBM CGN. In addition, we also aimed to elucidate the underlying mechanisms of macrophage-derived MIF in the pathogenesis of anti-GBM CGN.
2 Results
2.1 Characterization of macrophage-specific MIF deficient mouse
To evaluate the pathogenic role of macrophage-derived MIF, we employed a Cre-loxP strategy to generate mice with a specific deletion of MIF within their macrophages. To confirm the efficiency of MIF deletion, bone marrow was isolated from MIFf/f-lysM-cre mice, control MIFf/f mice, and MIF KO littermates. The isolated bone marrow cells were cultured and induced to differentiate into macrophages. As shown in Figure 1A, the deficiency of macrophage-derived MIF led to a notable reduction in MIF secretion by BMDM (Figure 1A). Additionally, MIF and CD74 mRNA expression were significantly lower in macrophages of MIFf/f-lysM-cre mice compared with MIFf/f mice under basal conditions (Figures 1B, C). Furthermore, TNF-a treatment for 24 hours significantly increased MIF protein expression in macrophages derived from MIFf/f mice but not in macrophages from MIFf/f-lysM-cre mice (Figure 1D). These results demonstrated the successful deletion of MIF from macrophages in MIFf/f-lysM-cre mice.
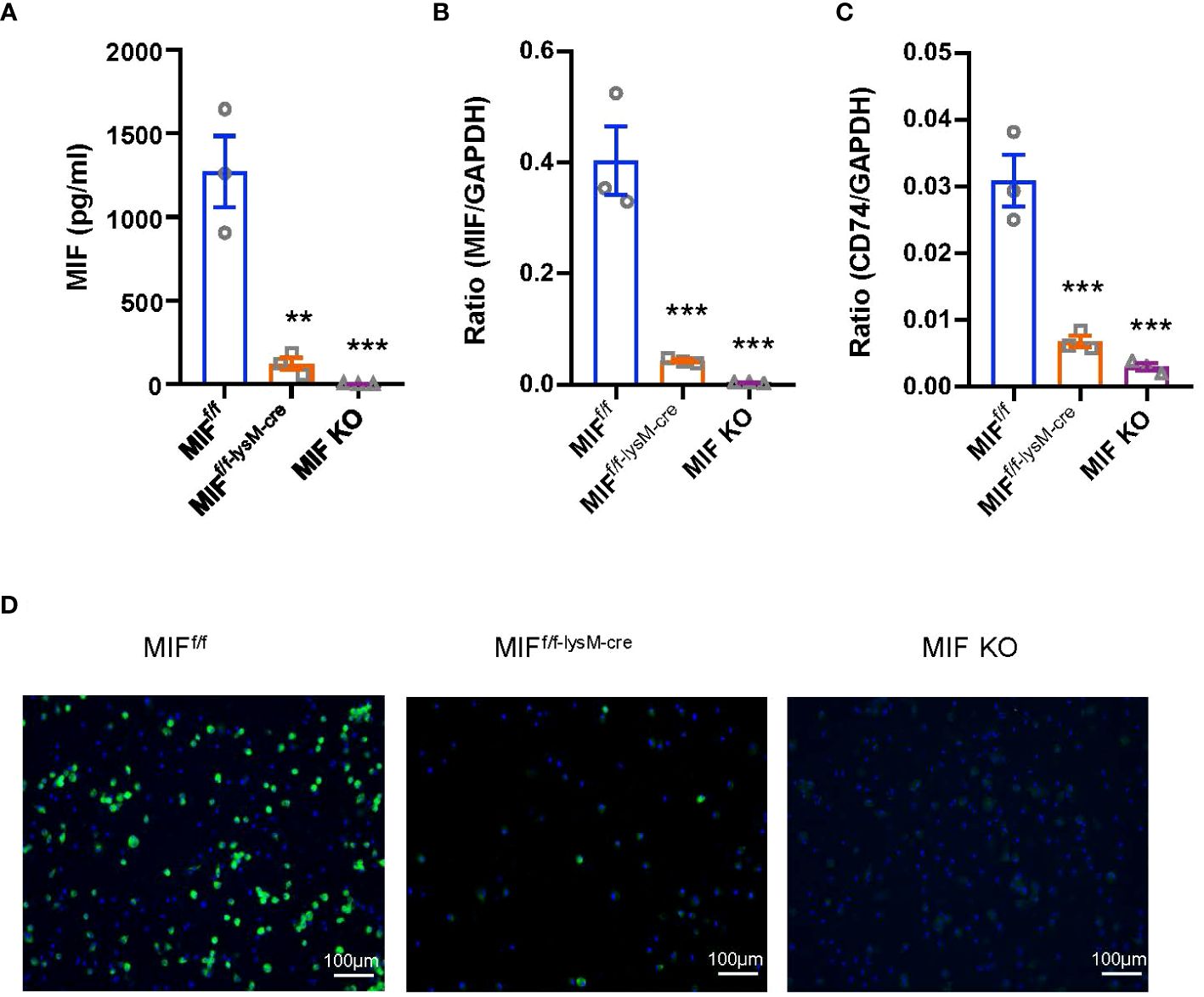
Figure 1 Characterization of MIFf/f-lysM-cre mice. (A) Bone marrow-derived macrophages (BMDMs) were isolated from MIFf/f, MIFf/f-lysM-cre and MIF KO mice. Enzymelinked immunosorbent assay (ELISA) show that MIFf/f-lysM-cre and MIF KO mice inhibit MIF expression by BMDMs. (B, C) RNA was isolated from cells for quantitative reverse transcription-PCR (qRT-PCR) quantitation of MIF and CD74. (D) Immunofluorescence staining for MIF(green) with nuclear DAPI (blue) counterstain in macrophages stimulated with TNF-a (10ng/ml for 24hours). Each bar represents mean±SEM. Each dot represents one mouse. **P < 0.01, ***P < 0.001 compared with MIFf/f mice; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
2.2 Selective MIF depletion in macrophages ameliorates experimental anti-GBM GN
To examine the involvement of macrophage-derived MIF in experimental anti-GBM CGN, we conducted experiments using both MIFf/f and MIFf/f-lysM-cre mice. The mice were induced to develop anti-GBM CGN, and the renal injuries were evaluated. Notably, the MIFf/f-lysM-cre mice showed a significant inhibition in renal injuries such as segmental glomerular capillary necrosis and crescent formation compared to the anti-GBM GN MIFf/f mice (Figures 2A, B). Renal dysfunction such as the urine albumin/creatinine ratio (Figure 2C), serum creatinine levels (Figure 2D), and creatinine clearance (Figure 2E) were also significantly improved in MIFf/f-lysM-cre mice compared to the anti-GBM CGN MIFf/f mice. These findings indicate that macrophage-derived MIF plays a pathogenic role in anti-GBM CGN.
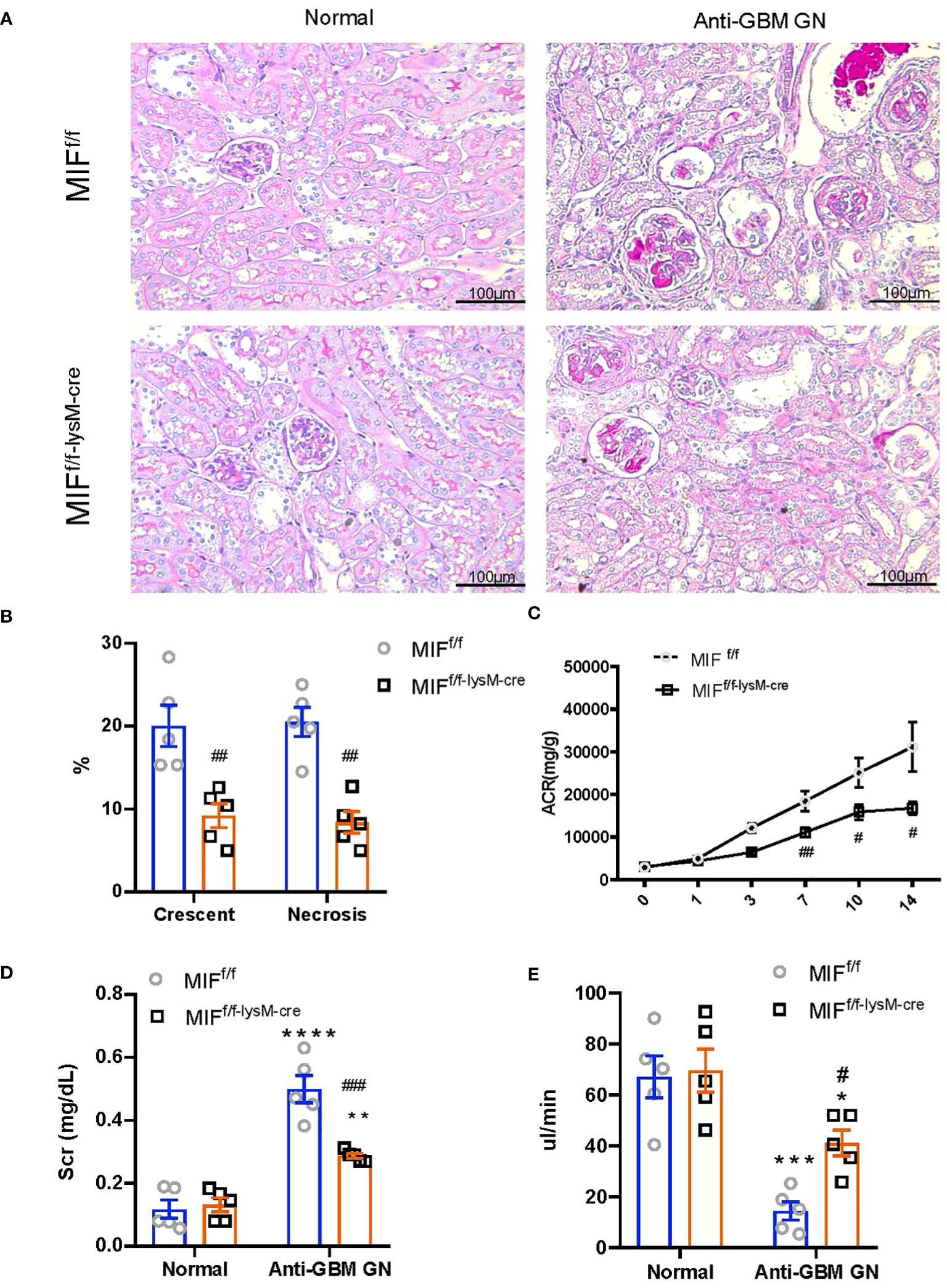
Figure 2 Selective MIF depletion in macrophages ameliorates experimental anti-GBM GN. (A) Representative images in PAS sections (magnification X200). (B) Semi-quantitative analysis of histology. (C) Urine albumin creatinine ration (ACR) over the disease course. (D) plasma creatinine. (E) Creatinine clearance. Each bar represents mean±SEM. Each dot represents one mouse. *p <0.05, **p <0.01, ***p< 0.001, ****p< 0.0001 versus corresponding control; #p<0.05, ##p < 0.01, ###p< 0.001 versus corresponding MIFf/f.
2.3 Deletion of macrophage MIF inhibits macrophage and T cell infiltration in a mouse model of anti-GBM CGN
We next examined macrophage-derived MIF on cellular immune response during anti-GBM CGN. Immunohistochemistry detected that a massive F4/80+ macrophages infiltrating the anti-GBM CGN in MIFf/f mice, which was largely inhibited in MIFf/f-lysM-cre mice (Figures 3A-C). Moreover, the infiltration of glomerular and interstitial CD3+ T cells in MIFf/f-lysM-cre GN mice was also significantly lower than in MIFf/f GN mice (Figures 3D-F). These findings demonstrate that selective depletion of MIF in macrophages suppresses the infiltration of macrophages and T cells in the kidney during anti-GBM GN.
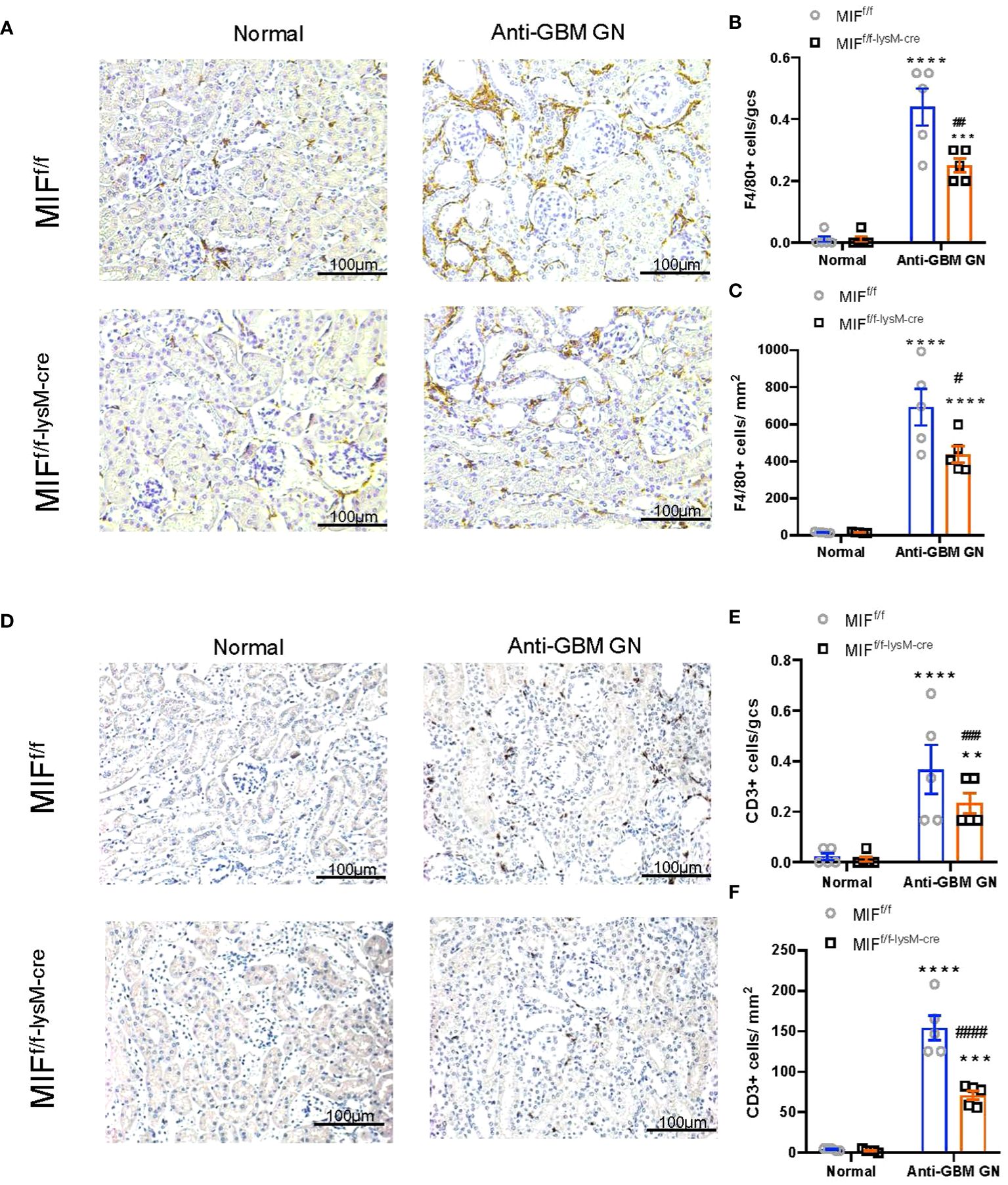
Figure 3 Selective MIF depletion in macrophages reduces macrophage and T cell recruitment in experimental anti-GBM GN. (A) Immunohistochemical staining for F4/80-positive macrophages in the kidney with anti-GBM crescentic GN on Day 14 after disease induction. (B, C) Summary data for macrophages in glomerulus and tubulointerstitium. (D) Immunohistochemical staining for CD3-positive T cells in the kidney with anti-GBM crescentic GN on Day 14 after disease induction. (E, F) Summary data for T cells in glomerulus and tubulointerstitium. Original magnification 200X. Each bar represents mean±SEM. Each dot represents one mouse. **p <0.01, ***p< 0.001, ****p< 0.0001 versus corresponding control; #p<0.05, ##p < 0.01, ###p< 0.001, ####p<0.0001 versus corresponding MIFf/f.
2.4 Deletion of macrophage MIF suppresses antigen-specific antibody production in a mouse model of anti-GBM CGN
Immunofluorescence was used to assess the glomerular deposition of sheep anti-mouse GBM antibody, mouse IgG, and complement component C3 in MIFf/f and MIFf/f-lysM-cre mice (Figure 4A). Interestingly, there was no significant difference in the glomerular deposition of these markers between the two groups, indicating that macrophage-specific MIF depletion did not affect immune complex deposition in inflamed glomeruli. However, as demonstrated in Figure 4B, the serum levels of mouse anti-sheep IgG antibodies were notably reduced in MIFf/f-lysM-cre GN mice compared to MIFf/f GN mice. This was associated with significant reduction in serum MIF levels in MIFf/f-lysM-cre GN mice compared to MIFf/f GN mice (Figure 4C). These results indicate that macrophage-specific MIF depletion reduced systemic MIF levels and selectively inhibited the antigen-specific antibody production without influencing the immune complex deposition in the inflamed glomeruli.
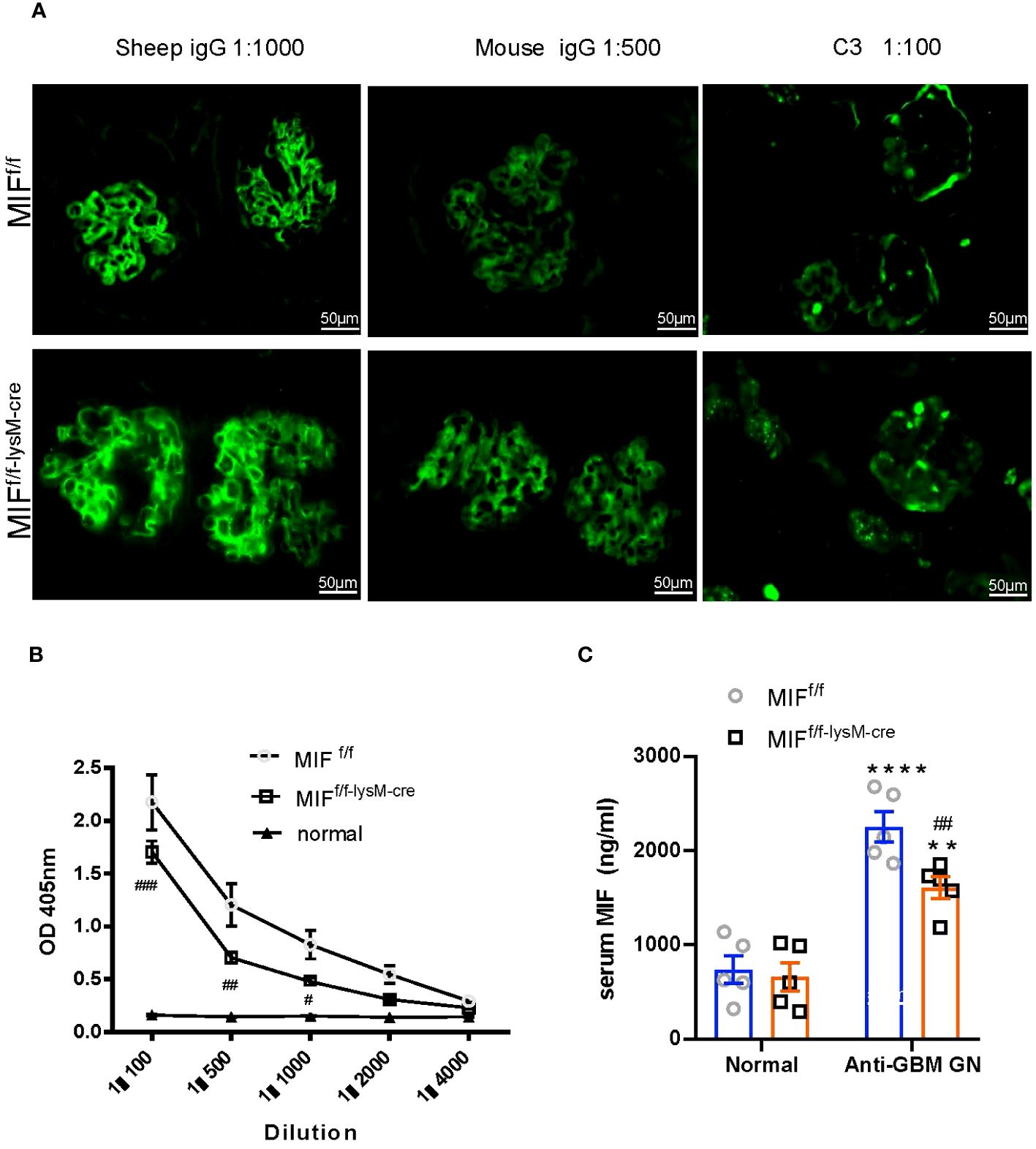
Figure 4 Selective MIF depletion in macrophages attenuates plasma levels of mouse anti-sheep IgG antibody and serum MIF production, which does not affect immune complex deposition in inflamed glomeruli. (A) Immunofluorescence staining for the deposition of sheep IgG, mouse IgG and mouse C3 in the kidney glomeruli with anti-GBM crescentic GN on Day 14 after disease induction. Plasma mouse anti-sheep IgG (B) and serum MIF (C) were determined using ELISA kits according to the manufacturer's protocol. Original magnification 200X. Each bar represents mean±SEM. Each dot represents one mouse. **p <0.01, ****p< 0.0001 versus corresponding control; #p<0.05, ##p < 0.01, ###p< 0.001 versus corresponding MIFf/f.
2.5 Deletion of macrophage MIF enhances macrophage polarization from M1 to M2 through in a mouse model of anti-GBM CGN
To investigate the impact of macrophage-derived MIF on macrophage polarization in the kidneys of mice with anti-GBM CGN, we utilized flow cytometry to determine the populations of M1 (F4/80+CD86+) and M2 (F4/80+CD206+) macrophages. Notably, MIFf/f-lysM-cre GN mice exhibited a significantly decreased M1 macrophages while increasing the M2 macrophages compared to MIFf/f GN mice (Figures 5A, B; Supplementary Figure S1). Furthermore, real-time PCR analysis revealed that selective MIF depletion in macrophages led to a significant inhibition of pro-inflammatory cytokines including MCP-1 and IL-1β while increasing the anti-inflammatory cytokine IL-10 in CGN mice (Figures 5C–E). These findings indicate that selective depletion of MIF in macrophages results in a shift in macrophage polarization from the pro-inflammatory M1 phenotype towards the anti-inflammatory M2 phenotype in experimental anti-GBM CGN. Moreover, this shift is associated with reduced pro-inflammatory cytokine expression and increased anti-inflammatory cytokine expression.
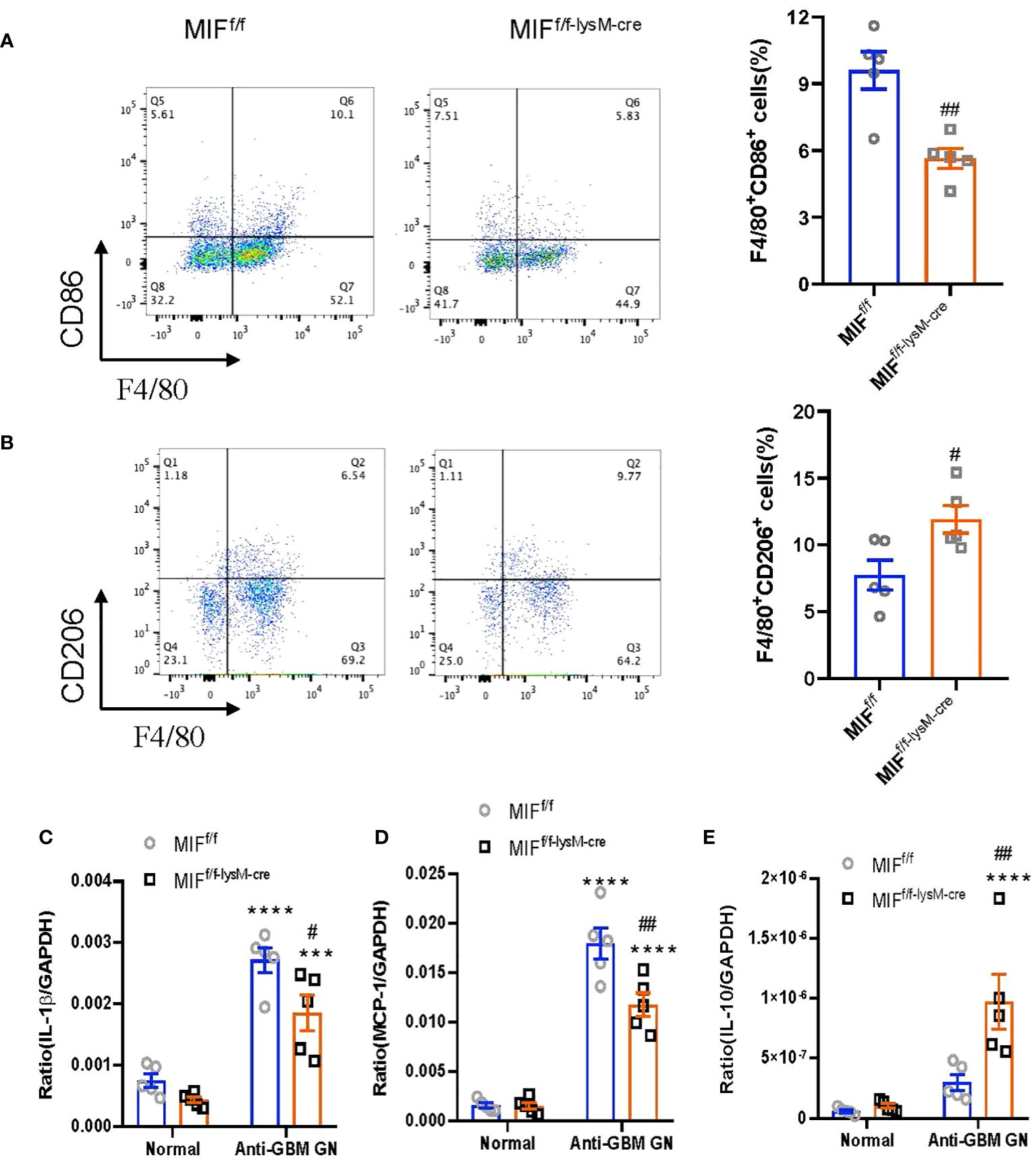
Figure 5 Selective MIF depletion in macrophages promotes macrophage polarization from M1 towards M2. (A) Representative flow cytometry plots and quantification of renal singlets analyzed for M1 (F4/80+CD86+). (B) Representative flow cytometry plots and quantification of renal singlets analyzed for M2 (F4/80+CD206+). Reverse transcription-PCR (RT-PCR) was performed on whole mouse kidney total RNA with anti-GBM crescentic GN on Day 14 after disease induction. (C–E) IL-1 β, monocyte chemoattractant protein-1 (MCP-1) and IL-10mRNA expression were normalized with GAPDH mRNA. Each bar represents mean±SEM. Each dot represents one mouse. ****p< 0.0001 versus corresponding control; #p<0.05, ##p < 0.01 versus corresponding MIFf/f.
2.6 Deletion of macrophage MIF promotes Treg but inhibits Th1 and Th17 immune responses in a mouse model of anti-GBM CGN
We next examined whether disrupted macrophage MIF influences T cell immunity as it is well-established that Th1 and Th17 are pathogenic whereas Treg is protective in anti-GBM CGN (23–26). In light of this, we investigated the impact of selective MIF depletion in macrophages on the immune differentiation of CD4+ T cells, specifically focusing on Th1 (CD4+IFNγ+), Th2 (CD4+IL-4+), Th17 (CD4+IL-17a+) and Treg (CD4+CD25+FoxP3+) subpopulations. Flow cytometry analysis revealed that selective MIF depletion in macrophages led to a significant reduction in Th1 (CD4+IFNγ+) and Th17 (CD4+IL-17a+) cells in the anti-GBM CGN kidney (Figures 6A, C; Supplementary Figures S2A, C). Conversely, there was an increase in Treg population (CD4+CD25+FoxP3+), however, the Th2 (CD4+IL-4+) immune response remained unaffected (Figures 6D, B; Supplementary Figure S3, Supplementary Figure S2B). These findings indicate that selective MIF depletion in macrophages enhances the Treg immune response while inhibiting the Th1 and Th17 immune responses in experimental anti-GBM CGN. This shift in immune cell differentiation may contribute to the amelioration of renal injury observed in this context.
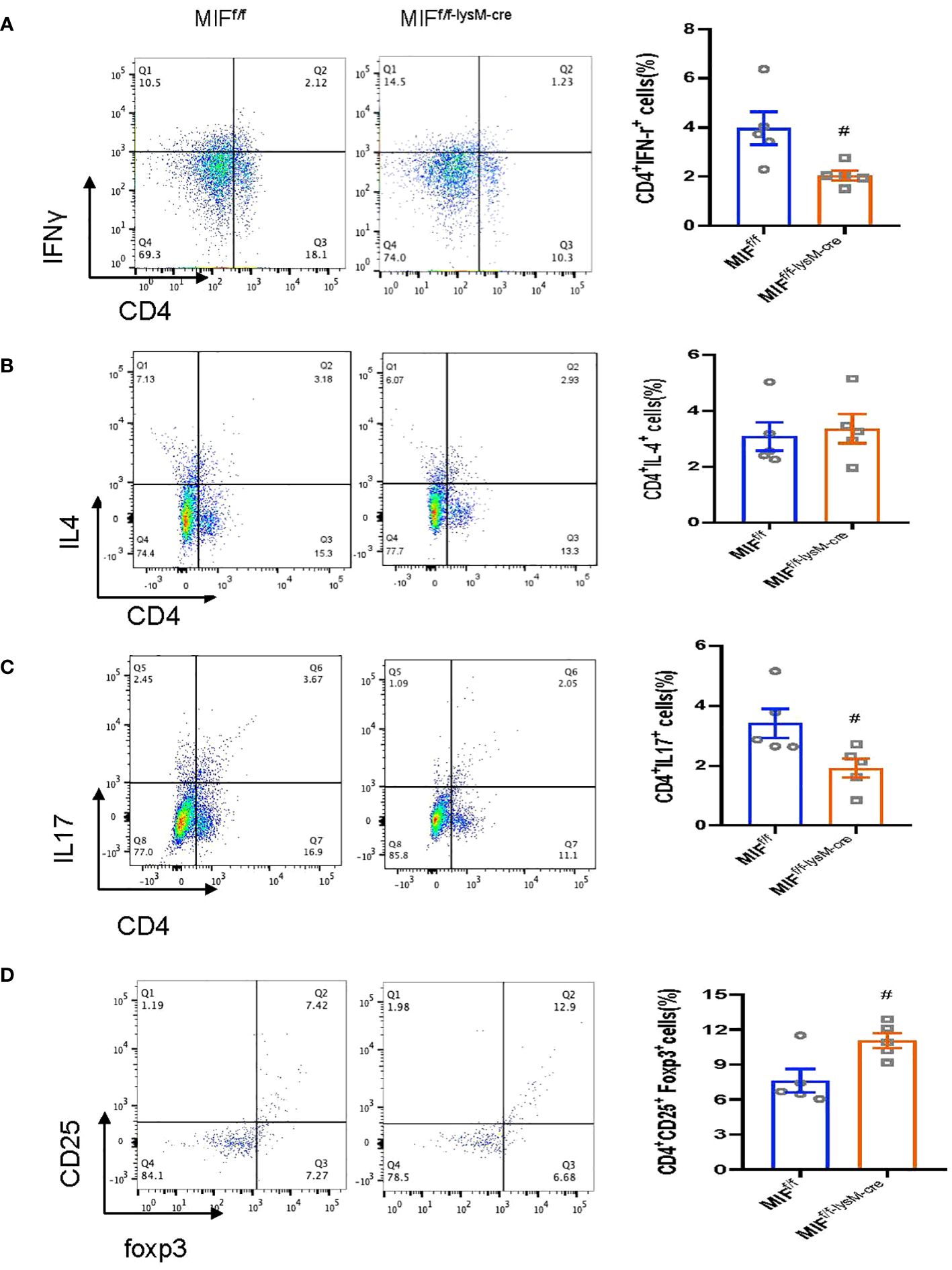
Figure 6 Selective MIF depletion in macrophages downregulates renal Th1/Th17 response and upregulates renal Treg response in experimental anti-GBM GN. Flow cytometry analysis of renal infiltrated CD4+IFNy+Th1 cells (A), CD4+ IL-4+Th2 cells (B), CD4+ IL-17+Th17 cells (C) and CD4+CD25+ Foxp3+Treg (D). CD4+CD25+ Foxp3+Treg is Gated on CD4+ T cells. Each bar represents mean±SEM. Each dot represents one mouse. #p<0.05 versus corresponding MIFf/f.
2.7 Deletion of macrophage MIF inhibits Anti-GBM GN by inactivating M1 macrophages via CD74/NF-κB and p38 MAPK-dependent mechanisms in vivo and in vitro
We next examined the mechanisms through which specific deletion of macrophage MIF inhibits anti-GBM GN induced in MIF f/f and MIF f/f-lysM-cre mice. Western blot analysis revealed that there was a marked upregulation of CD74 and activation of NF-κB/p65 and p38 MAPK signaling and expression of iNOS in the diseased kidney of MIF f/f mice (Figure 7). In contrast, selective MIF depletion from macrophages significantly inhibited the expression of CD74 and phosphorylation of NF-κB/p65 and p38 MAPK, as well as expression of iNOS in MIFf/f-lysM-cre GN mice (Figure 7). All of these findings indicated that MIF may promote anti-GBM GN by activating M1 macrophages through the CD74/NF-kB/p38 MAPK signaling. This was further demonstrated in vitro in cultured BMDM from MIF WT and MIF KO mice. We found that addition of TNF-α largely promoted iNOS-producing M1 macrophages in MIF WT BMDM by activating CD74/NF-κB/p38 MAPK signaling, which was blocked in BMDM lacking MIF (Figure 8). Thus, macrophage-derived MIF may mediate anti-GBM GN by promoting M1 macrophage activation via the CD74/NK-κB/p38 MAPK-dependent mechanism.
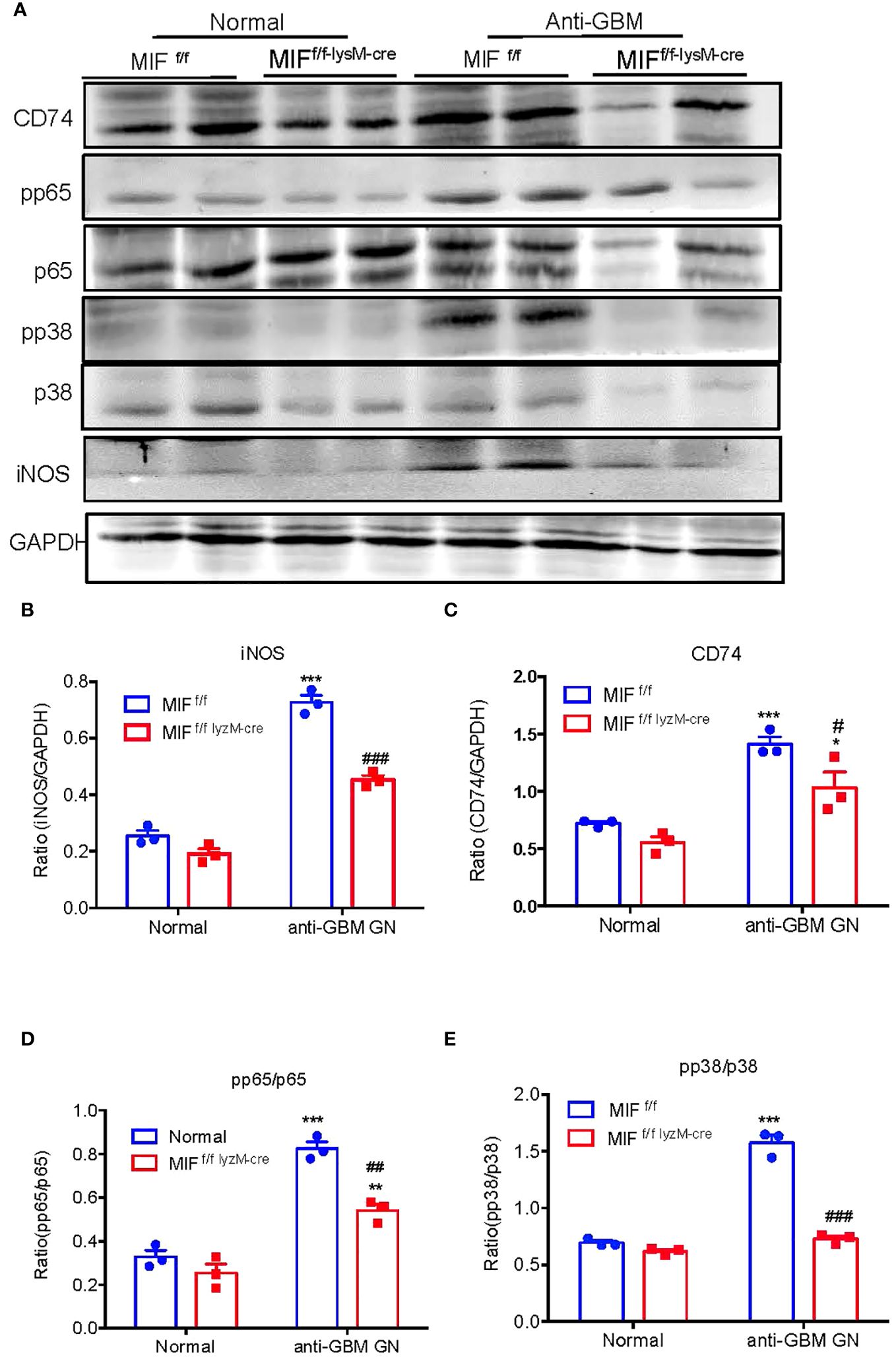
Figure 7 Selective MIF depletion in macrophages ameliorates experimental anti- GBM GN by inactivating NF-kB and p38/MAPK signaling and inhibiting M1 macrophages activation. (A) Western blots. (B–E) Statistics data of the protein expression (iNOS, CD74, pp65/p65 and pp38/p38). Each bar represents mean ± SEM. Each dot represents one mouse. *p <0.05, ***p<0.001 versus corresponding control; #p<0.05, ##p < 0.01, ###p< 0.001 versus corresponding MIFf/f.
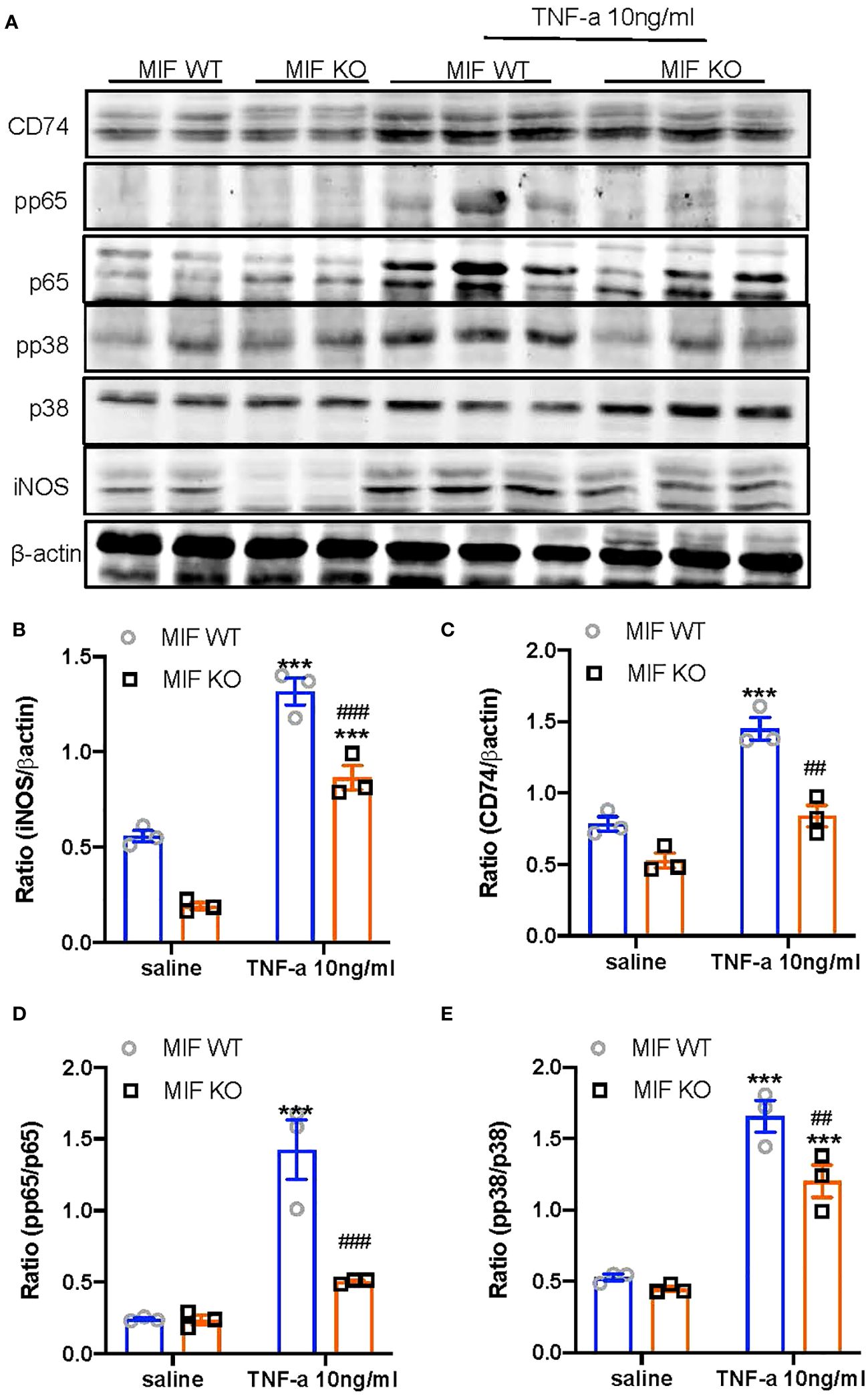
Figure 8 Bone marrow-derived macrophages (BMDM) lacking MIF show inhibition of MIF signaling and M1 macrophage activation in response to TNFa in vitro. (A) Western blots; (B–E) Statistics data of the protein expression (CD74, pp65/p65, pp38/p38, INOS). Results show that bone marrow-derived macrophages (BMDMs) isolated MIF KO mice inhibit TNF-α (10ng/ml)-induced upregulation of MIF signaling by downregulating CD74 expression and activation of NF-kB/p65 and p38/MAPK, there by inhibiting M1 macrophage activation by suppressing iNOS expression. Each bar represents mean±SEM. Each dot represents one mouse. ***P < 0.001 compared with MIF WT mice; ##P < 0.01, ###p< 0.001 compared with MIF WT mice treated with TNF-a 10ng/ml.
3 Discussion
MIF has been implicated in the pathogenesis of various diseases, including infectious diseases, inflammatory diseases, immune diseases such as rheumatoid arthritis, septic shock, and cardiovascular disease (20, 23–26). Previous studies have also demonstrated the importance of MIF in kidney-related conditions like acute kidney injury (AKI), chronic kidney disease (CKD), diabetic nephropathy, autosomal dominant polycystic kidney disease (ADPKD), and vasculitides (27–35).In glomerulonephritis, inhibition of MIF by neutralizing antibodies has shown renal protective in IgA nephritis and in rats with crescentic GN (36–38). Additionally, systemic MIF knockout (KO) can also suppress lupus nephritis and anti-GBM CGN (17, 19–22). However, it should be noted that systemic MIF KO models cannot distinguish the source of functional MIF since MIF is released by both macrophages and other intrinsic cells within the kidney. To address this, we generated mice with a macrophage-specific MIF KO and investigated the role and mechanisms of macrophage-derived MIF in a mouse model of anti-GBM GN. The results demonstrated that mice with macrophage-specific deletion of MIF were protected from the development of anti-GBM CGN. These findings highlight the crucial role of macrophage-derived MIF in the pathogenesis of anti-GBM CGN, shedding light on the specific contribution of macrophage-derived MIF in this disease context.
It is well-established that macrophages play a crucial role in the progressive renal injury associated with glomerular crescentic formation (6–8, 10, 11, 39). It is reported that MIF regulates macrophage activation via Toll-like receptor 4 (TLR4) (40). Thus, deletion of macrophage TLR4 inhibits anti-GBM cGN (39). It is also well defined that MIF is pathogenic in immunologically-mediated kidney disease as mice lacking MIF are protected against lupus mice and anti-GBM GN (17, 22). Similarly, systemic or bone marrow disruption of MIF also inhibits cardiac remodeling by suppressing myocardial leukocyte infiltration and the expression of inflammatory mediators (41). Findings from the present study added new information that macrophages may mediate anti-GBM CGN via MIF-dependent mechanism as selective depletion of macrophage MIF protected against anti-GBM CGN by inhibiting macrophage infiltration and promoting macrophage polarization from the pro-inflammatory M1 phenotype towards the anti-inflammatory M2 phenotype. These results suggest that macrophage-derived MIF may have a critical role in modulating macrophage activation and function in the pathogenesis of anti-GBM CGN.
Furthermore, an intriguing aspect of our findings is that the specific depletion of MIF in macrophages appears to confer kidney protection in crescentic GN by promoting renal Treg cells while suppressing Th1 and Th17 immune responses. The involvement of Th1 and Th17 immune responses in the pathogenesis of anti-GBM GN is well-established (42–44), whereas Treg cells are known to have a protective role (44, 45). In murine crescentic glomerulonephritis, Treg cells have been shown to regulate the Th1 immune response (45). Additionally, recent studies have highlighted the important role of cytokines and chemokines in the cross-regulation of Th1 and Th17 immune responses in experimental anti-GBM GN (46). Interestingly, MIF has been shown to enhance the acquisition of a Th17 cell-like phenotype in spondylarthritis (47). Based on these findings, it is reasonable to speculate that selective depletion of MIF in macrophages may protect against anti-GBM crescentic GN by suppressing Th1/Th17 while promoting Treg immune responses.
CD74 is identified as main receptor of MIF (48). The binding of MIF to CD74 triggers the activation of mitogen-activated protein kinase (MAPK) and NK-kB signaling (49, 50). It has been reported that M1 macrophage activation in anti-GBM GN is NF-κB-dependent (51) and M1-mediated NF-κB signaling can release cytokines including IL-1β, IL-6, TNF-α and granulocyte colony-stimulating factors (G-CSF) (52). The present study unraveled that deletion of macrophage MIF ameliorated anti-GBM GN by shifting the M1 macrophages to M2 macrophages via the CD74/NK-kB/p38 MAPK-dependent mechanism. Importantly, deletion of macrophage MIF inhibited the Th1 and Th17 immune responses, while increasing Treg. These findings also suggest that macrophage-derived MIF may play a regulatory role in T cell immunity during the development of anti-GBM GN, although the mechanisms remain largely unclear.
In summary, macrophage-derived MIF plays an important role in anti-GBM CGN. Mechanistically, as shown in Figure 9, macrophage-derived MIF may mediate anti-GBM CGN by promoting proinflammatory M1 macrophage infiltration and activation via the NK-κB and p38 MAPK pathways and promoting the Th1/Th17 immune responses while suppressing the Treg population. These findings are in line with our previous studies that T cell-mediated immunity plays a crucial role in anti-GBM disease, despite not necessarily impacting the glomerular deposition of immune complexes (44).
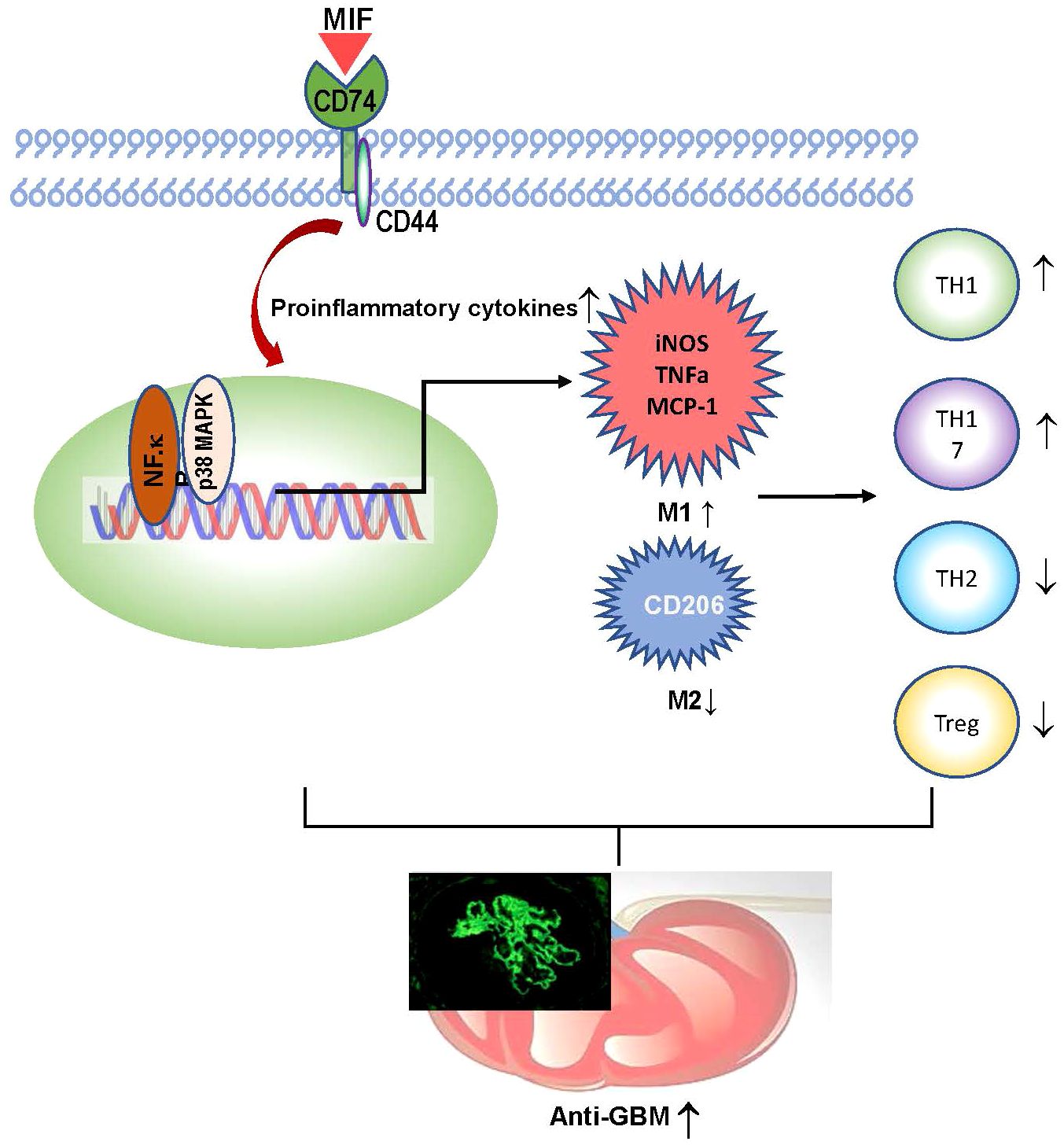
Figure 9 Mechanisms of MIF mediate experimental anti-GBM GN.
4 Materials and methods
4.1 Generation of macrophage-specific MIF deletion mice
To generate mice with myeloid-specific MIF deletion (MIFf/f−lysM−cre), we utilized C57BL/6 mice carrying MIF genes with homozygous loxP-flanked regions (MIFf/f), which were previously described (53). Lysozyme M promoter-driven cre (lysM-cre) mice were obtained from the Jackson Laboratory in the USA. The MIFf/f mice were crossed with lysM-cre mice to obtain the desired genotype. The genotypes of the resulting littermates were confirmed using PCR with specific primers recommended by the Jackson Laboratory. All mice used in the study were maintained under specific pathogen-free conditions at a temperature of 25°C and a 12-hour light-dark cycle. They were housed in our animal facility and provided with standard food and water ad libitum.
4.2 Isolation and culture of bone marrow-derived macrophages
To culture bone marrow-derived macrophages (BMDMs), bone marrow cells were isolated from the tibias and femurs of MIFf/f, MIFf/f−lysM−cre, and MIF knockout (KO) mice. These cells were then cultured in Dulbecco modified Eagle medium (DMEM) supplemented with 50 ng/ml macrophage colony-stimulating factor (M-CSF) for 7 days, following a previously established protocol (54). After the 7-day culture period, BMDMs were stimulated with TNF-α (10 ng/ml) for 24 hours and proteins were collected for western blot analysis.
4.3 Real-time PCR analysis
Total RNA was extracted from either diseased kidney tissue or cultured cells using the RNeasy Isolation Kit (Qiagen, Valencia, CA) following the manufacturer’s protocol. RT-PCR was conducted following established methods (44, 55). The mRNA expression levels of the target genes were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as an internal control. The primer sequences for mouse MIF, CD74, MCP-1, IL-1β, IL-10, and GAPDH were previously reported (44, 55, 56).
4.4 Induction of anti-GBM glomerulonephritis in mice
The mouse model of anti-glomerular basement membrane crescentic glomerulonephritis (anti-GBM cGN) was established using male MIFf/f and MIFf/f−lysM−cre mice (8–12 weeks old), following a well-established protocol (44). Herein, the procedure involved the following steps: Firstly, groups of MIFf/f and MIFf/f−lysM−cre mice received a flank subcutaneous immunization with normal sheep IgG mixed with Freund’s complete adjuvant (Sigma Aldrich, St. Louis, Missouri, USA) five days prior to the initiation of the experiment. Anti-GBM cGN was induced by administering sheep anti-mouse GBM IgG via tail vein injection at a dose of 60 μg/g of body weight (referred to as day 0). On day 14, the mice were sacrificed using a lethal dose of ketamine and xylazine mixture. Age-matched normal male MIFf/f and MIFf/f−lysM−cre mice (n = 5 per group) were included as normal control counterparts. All animal experiments were conducted in compliance with the guidelines approved by the Animal Experimentation Ethics Committee at the Chinese University of Hong Kong.
4.5 Measurement of proteinuria and creatinine
Analysis of proteinuria: Urinary samples were collected at various time points, including before and after the induction of anti-GBM disease on days 0, 1, 3, 7, and 14. Proteinuria analysis was performed following the manufacturer’s protocols, as described previously (44). Urinary albumin excretion was quantified as total urinary albumin/creatinine ratio (expressed as micrograms per milligram). Measurement of urinary and serum creatinine levels was conducted using an enzymatic kit (Stanbio Laboratory, Boerne, USA).
4.6 Renal pathology and immunohistochemistry
Renal pathology assessment was conducted on methyl Carnoy’s fixed, paraffin-embedded tissue sections (4μm thick). The sections were stained with periodic acid Schiff (PAS) to visualize the renal structures. Glomerular crescents and necrosis were quantified by examining 50 glomeruli per diseased kidney section and calculating the percentage of affected glomeruli. Immunohistochemistry was performed on paraffin sections stained with monoclonal an anti-F4/80 antibody (Serotec, Oxford, UK) to detect macrophages, and a rabbit anti-mouse polyclonal CD3+ antibody (SP7) (Abcam, Cambridge, UK) to identify total T cells. The number of positive cells for CD3 and F4/80 was counted in 20 glomeruli and expressed as cells per glomerular cross-section (gcs). In the tubulointerstitium, positive cells were counted under high-intensity fields (400× magnification) using a 0.0625 mm2 graticule fitted in the microscope eyepiece. The cell count was then reported as cells per square millimeter (mm2).
4.7 Immunofluorescence
To assess the presence of MIF-expressing macrophages in the kidney, immunofluorescence staining was performed. Acetone-fixed bone marrow-derived macrophages (BMDMs) were cultured with the MIF antibody (sc-20121; Santa Cruz) followed by incubation with a fluorescein isothiocyanate anti-rabbit secondary antibody, as previously described (54). To evaluate immune deposition in the glomeruli, direct immunofluorescence was conducted using FITC-conjugated polyclonal antibodies specific to sheep IgG, mouse IgG, and complement C3, following established protocols (38).
4.8 Enzyme-linked immunosorbent assay
Plasma levels of mouse anti-sheep IgG were quantified using a method previously described (56). The concentration of serum MIF was determined using ELISA kits (R&D Systems, Minneapolis, USA) according to the manufacturer’s instructions.
4.9 Flow cytometry analysis
Kidney single cells were prepared and subjected to flow cytometry analysis following a previously described protocol (44, 56). Single-cell suspensions were treated with IC Fixation Buffer and Permeabilization Buffer (eBioscience) to allow intracellular staining. The suspended kidney cells were then incubated with specific conjugated antibody cocktails in the dark for 30 minutes on ice. Negative controls included cells treated with irrelevant antibodies (isotype). Cells were also incubated with only one specific conjugated antibody. The antibodies used in this study were as follows: F4/80-Pacific blue (BioLegend, Catalog: 123124), CD86-APC (BioLegend, Catalog: 10512), CD206-Alexa 647 (Serotec, Catalog: MCA2235), CD4-FITC (eBioscience, Catalog: 11–0042-86), IFNγ-APC (eBioscience, Catalog: 17–7311-82), IL-4-PE (eBioscience, Catalog: 12–7041-82), IL-17a-PE (eBioscience, Catalog: 12–7177-81), CD25-PE (eBioscience, Catalog: 12–0251-83), and Foxp3-APC (eBioscience, Catalog: 12–0251-83). Flow cytometry analysis was performed using a FACS Calibar instrument and analyzed using the CellQuest Pro Analysis software (BD Biosciences, Franklin Lakes, New Jersey, USA).
4.10 Western blot analysis
Western blotting was performed as described previously (55, 57–59). Proteins from BMDMs and the kidney cortex were extracted with RIPA lysis buffer. After blocking nonspecific binding with 5% BSA, membranes were incubated overnight at 4 ° C with the primary antibodies against rabbit anti-iNOS (Abcam ab-15323), rabbit anti-iNOS (Abcam ab-178945), goat anti-CD74 (Santa Cruz, sc-5438), mouse anti-CD74 (Santa Cruz, sc-6267), goat anti-CD74 (Santa Cruz, sc-5438), rabbit anti-phosphorylated NF-kB p65 (Cell Signaling, #3031), rabbit anti-phosphorylated NF-kB p65 (Cell Signaling, #3033s), mouse anti-NF-kB p65 (Cell Signaling, #6965), rabbit anti-NF-kB p65 (Cell Signaling, # 8242S), rabbit anti-pp38 (Cell signaling, #9211), rabbit anti-pp38 (Cell signaling, # 4631L), rabbit anti-p38 (Cell signaling, #9212), rabbit anti-p38 (Cell signaling, # 8690S), mouse anti-ß-actin (Santa Cruz, sc-69879), mouse anti-GAPDH(proteintech,#60004–1-Ig).Then the membranes were incubated with IRDye800-conjugated secondary antibody (Rockland Immun- chemicals). Signals were scanned using the Odyssey IR imaging system (LI-COR Biosciences). Image J software (National Institutes of Health) was used for quantitative analysis of images.
4.11 Statistical analysis
Statistical analysis was conducted using Prism 9.0 GraphPad Software (GraphPad Software, La Jolla, California, USA). Data obtained from the study were presented as the mean ± standard error of the mean (SEM). Two-group comparisons were assessed using an independent sample t-test. Multiple group comparisons were performed using one-way analysis of variance (ANOVA) followed by Tukey’s post hoc tests. A p-value less than 0.05 was considered statistically significant in this experiment.
5 Conclusions
Our study demonstrates that selective MIF depletion in macrophages ameliorates experimental anti-GBM cGN by promoting macrophage polarization from M1 towards M2, enhancing Treg while inhibiting Th1 and Th17 immune responses via CD74/NF-κB/p38 MAPK signaling. Thus, targeting macrophage-derived MIF could be a novel therapy for anti-GBM cGN.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was approved by The Chinese University of Hong Kong. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
HY: Writing – original draft. JL: Writing – original draft. XH: Methodology, Writing – review & editing. RB: Methodology, Resources, Writing – review & editing. AX: Funding acquisition, Writing – review & editing. HL: Funding acquisition, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by the Research Grants Council of Hong Kong (GRF 14104019, 14101121, and R4012-18); the High-Level Hospital Construction Project from Guangdong Provincial People’s Hospital, Guangdong Academy of Medical Science (KJ012019108), the Guangdong-Hong Kong-Macao-Joint Labs Program (2019B121205005), the Lui Che Woo Institute of Innovative Medicine (CARE program), NIH 1R01-AR078334 (RB), Shenzhen Technology Project (JCYJ20190809120801655, JCYJ20180307150634856), the National Natural Science Funds of China (81870481), the Guangdong Provincial Natural Science Foundation (2021A1515011625) and the Guangdong Provincial Natural Science Foundation (2022A1515012308).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1361343/full#supplementary-material
References
1. Lan HY, Nikolic-Paterson DJ, Atkins RC. Involvement of activated periglomerular leukocytes in the rupture of Bowman's capsule and glomerular crescent progression in experimental glomerulonephritis. Lab Invest. (1992) 67:743–51.
2. McAdoo SP, Pusey CD. Anti-glomerular basement membrane disease. Clin J Am Soc Nephrol. (2017) 12:1162–72. doi: 10.2215/CJN.01380217
3. Shi Y, Jia XY, Gu QH, Wang M, Cui Z, Zhao MH. A modified peptide derived from goodpasture autoantigen arrested and attenuated kidney injuries in a rat model of anti-GBM glomerulonephritis. J Am Soc Nephrol. (2020) 31:40–53. doi: 10.1681/ASN.2019010067
4. Bolton WK, Innes DJ Jr, Sturgill BC, Kaiser DL. T-cells and macrophages in rapidly progressive glomerulonephritis: clinicopathologic correlations. Kidney Int. (1987) 32:869–76. doi: 10.1038/ki.1987.288
5. Tipping PG, Holdsworth SR. T cells in crescentic glomerulonephritis. J Am Soc Nephrol. (2006) 17:1253–63. doi: 10.1681/ASN.2005091013
6. Tang PM, Nikolic-Paterson DJ, Lan HY. Macrophages: versatile players in renal inflammation and fibrosis. Nat Rev Nephrol. (2019) 15:144–58. doi: 10.1038/s41581-019-0110-2
7. Fujinaka H, Yamamoto T, Takeya M, Feng L, Kawasaki K, Yaoita E, et al. Suppression of anti-glomerular basement membrane nephritis by administration of anti-monocyte chemoattractant protein-1 antibody in WKY rats. J Am Soc Nephrol. (1997) 8:1174–8. doi: 10.1681/ASN.V871174
8. Duffield JS, Tipping PG, Kipari T, Cailhier JF, Clay S, Lang R, et al. Conditional ablation of macrophages halts progression of crescentic glomerulonephritis. Am J Pathol. (2005) 167:1207–19. doi: 10.1016/S0002-9440(10)61209-6
9. Chalmers SA, Chitu V, Herlitz LC, Sahu R, Stanley ER, Putterman C. Macrophage depletion ameliorates nephritis induced by pathogenic antibodies. J Autoimmun. (2015) 57:42–52. doi: 10.1016/j.jaut.2014.11.007
10. Rogers NM, Ferenbach DA, Isenberg JS, Thomson AW, Hughes J. Dendritic cells and macrophages in the kidney: a spectrum of good and evil. Nat Rev Nephrol. (2014) 10:625–43. doi: 10.1038/nrneph.2014.170
11. Yang N, Isbel NM, Nikolic-Paterson DJ, Li Y, Ye R, Atkins RC, et al. Local macrophage proliferation in human glomerulonephritis. Kidney Int. (1998) 54:143–51. doi: 10.1046/j.1523-1755.1998.00978.x
12. Meng XM, Tang PM, Li J, Lan HY. Macrophage phenotype in kidney injury and repair. Kidney Dis (Basel). (2015) 1:138–46. doi: 10.1159/000431214
13. Chen T, Cao Q, Wang Y, Harris DCH. M2 macrophages in kidney disease: biology, therapies, and perspectives. Kidney Int. (2019) 95:760–73. doi: 10.1016/j.kint.2018.10.041
14. Lee H, Fessler MB, Qu P, Heymann J, Kopp JB. Macrophage polarization in innate immune responses contributing to pathogenesis of chronic kidney disease. BMC Nephrol. (2020) 21:270. doi: 10.1186/s12882-020-01921-7
15. Lin SG, Yu XY, Chen YX, Huang XR, Metz C, Bucala R, et al. De novo expression of macrophage migration inhibitory factor in atherogenesis in rabbits. Circ Res. (2000) 87:1202–8. doi: 10.1161/01.RES.87.12.1202
16. Chen Z, Sakuma M, Zago AC, Zhang X, Shi C, Leng L, et al. Evidence for a role of macrophage migration inhibitory factor in vascular disease. Arterioscler Thromb Vasc Biol. (2004) 24:709–14. doi: 10.1161/01.ATV.0000119356.35748.9e
17. Hoi AY, Hickey MJ, Hall P, Yamana J, O'Sullivan KM, Santos LL, et al. Macrophage migration inhibitory factor deficiency attenuates macrophage recruitment, glomerulonephritis, and lethality in MRL/lpr mice. J Immunol. (2006) 177:5687–96. doi: 10.4049/jimmunol.177.8.5687
18. Lan HY, Yang N, Nikolic-Paterson DJ, Yu XQ, Mu W, Isbel NM, et al. Expression of macrophage migration inhibitory factor in human glomerulonephritis. Kidney Int. (2000) 57:499–509. doi: 10.1046/j.1523-1755.2000.t01-1-00869.x
19. Wang FF, Zhu LA, Zou YQ, Zheng H, Wilson A, Yang CD, et al. New insights into the role and mechanism of macrophage migration inhibitory factor in steroid-resistant patients with systemic lupus erythematosus. Arthritis Res Ther. (2012) 14:R103. doi: 10.1186/ar3828
20. Bilsborrow JB, Doherty E, Tilstam PV, Bucala R. Macrophage migration inhibitory factor (MIF) as a therapeutic target for rheumatoid arthritis and systemic lupus erythematosus. Expert Opin Ther Targets. (2019) 23:733–44. doi: 10.1080/14728222.2019.1656718
21. Kang I, Bucala R. The immunobiology of MIF: function, genetics and prospects for precision medicine. Nat Rev Rheumatol. (2019) 15:427–37. doi: 10.1038/s41584-019-0238-2
22. Djudjaj S, Lue H, Rong S, Papasotiriou M, Klinkhammer BM, Zok S, et al. Macrophage migration inhibitory factor mediates proliferative GN via CD74. J Am Soc Nephrol. (2016) 27:1650–64. doi: 10.1681/ASN.2015020149
23. Bernhagen J, Krohn R, Lue H, Gregory JL, Zernecke A, Koenen RR, et al. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med. (2007) 13:587–96. doi: 10.1038/nm1567
24. Lehmann L, Novender U, Schroeder S, Pietsch T, von Spiegel T, Putensen C, et al. Plasma levels of macrophage migration inhibitory factor are elevated in patients with severe sepsis. Intensive Care Med. (2001) 27:1412–5. doi: 10.1007/s001340101022
25. Zernecke A, Bernhagen Jr., Weber C. Macrophage migration inhibitory factor in cardiovascular disease. Circulation. (2008) 117:1594–602. doi: 10.1161/CIRCULATIONAHA.107.729125
26. Averdunk LS, Goetzenich A, Bernhagen J, Bucala R, Stoppe C. The protective role of macrophage migration inhibitory factor (MIF) in acute kidney injury after cardiac surgery. Circulation. (2017) 136. doi: 10.26226/morressier.58f5b031d462b80296c9d416
27. Brown FG, Nikolic-Paterson DJ, Hill PA, Isbel NM, Dowling J, Metz CM, et al. Urine macrophage migration inhibitory factor reflects the severity of renal injury in human glomerulonephritis. J Am Soc Nephrol. (2002) 13 Suppl 1:S7–13. doi: 10.1681/ASN.V13suppl_1s7
28. Li JH, Tang Y, Lv J, Wang XH, Yang H, Tang PMK, et al. Macrophage migration inhibitory factor promotes renal injury induced by ischemic reperfusion. J Cell Mol Med. (2019) 23:3867–77. doi: 10.1111/jcmm.14234
29. Li J, Tang Y, Tang PMK, Lv J, Huang XR, Carlsson-Skwirut C, et al. Blocking macrophage migration inhibitory factor protects against cisplatin-induced acute kidney injury in mice. Mol Ther. (2018) 26:2523–32. doi: 10.1016/j.ymthe.2018.07.014
30. Bruchfeld A, Carrero JJ, Qureshi AR, Lindholm B, Barany P, Heimburger O, et al. Elevated serum macrophage migration inhibitory factor (MIF) concentrations in chronic kidney disease (CKD) are associated with markers of oxidative stress and endothelial activation. Mol Med. (2009) 15:70–5. doi: 10.2119/molmed.2008.00109
31. Rammos C, Hendgen-Cotta UB, Sobierajski J, Adamczyk S, Hetzel GR, Kleophas W, et al. Macrophage migration inhibitory factor is associated with vascular dysfunction in patients with end-stage renal disease. Int J Cardiol. (2013) 168:5249–56. doi: 10.1016/j.ijcard.2013.08.021
32. Watanabe T, Tomioka NH, Doshi M, Watanabe S, Tsuchiya M, Hosoyamada M. Macrophage migration inhibitory factor is a possible candidate for the induction of microalbuminuria in diabetic db/db mice. Biol Pharm Bull. (2013) 36:741–7. doi: 10.1248/bpb.b12-00741
33. Sanchez-Nino MD, Sanz AB, Ihalmo P, Lassila M, Holthofer H, Mezzano S, et al. The MIF receptor CD74 in diabetic podocyte injury. J Am Soc Nephrol. (2009) 20:353–62. doi: 10.1681/ASN.2008020194
34. Torres VE, Chapman AB, Devuyst O, Gansevoort RT, Grantham JJ, Higashihara E, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med. (2012) 367:2407–18. doi: 10.1056/NEJMoa1205511
35. Wendt M, Borjesson O, Avik A, Bratt J, Anderstam B, Qureshi AR, et al. Macrophage migration inhibitory factor (MIF) and thyroid hormone alterations in antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV). Mol Med. (2013) 19:109–14. doi: 10.2119/molmed.2012.00352
36. Leung JC, Chan LY, Tsang AW, Liu EW, Lam MF, Tang SC, et al. Anti-macrophage migration inhibitory factor reduces transforming growth factor-beta 1 expression in experimental IgA nephropathy. Nephrol Dial Transplant. (2004) 19:1976–85. doi: 10.1093/ndt/gfh323
37. Yang N, Nikolic-Paterson DJ, Ng YY, Mu W, Metz C, Bacher M, et al. Reversal of established rat crescentic glomerulonephritis by blockade of macrophage migration inhibitory factor (MIF): potential role of MIF in regulating glucocorticoid production. Mol Med. (1998) 4:413–24. doi: 10.1007/BF03401748
38. Lan HY, Bacher M, Yang N, Mu W, Nikolic-Paterson DJ, Metz C, et al. The pathogenic role of macrophage migration inhibitory factor in immunologically induced kidney disease in the rat. J Exp Med. (1997) 185:1455–65. doi: 10.1084/jem.185.8.1455
39. Yang F, Chen J, Huang XR, Yiu WH, Yu X, Tang SCW, et al. Regulatory role and mechanisms of myeloid TLR4 in anti-GBM glomerulonephritis. Cell Mol Life Sci. (2021) 78:6721–34. doi: 10.1007/s00018-021-03936-1
40. Roger T, David J, Glauser MP, Calandra T. MIF regulates innate immune responses through modulation of Toll-like receptor 4. Nature. (2001) 414:920–4. doi: 10.1038/414920a
41. White DA, Su Y, Kanellakis P, Kiriazis H, Morand EF, Bucala R, et al. Differential roles of cardiac and leukocyte derived macrophage migration inhibitory factor in inflammatory responses and cardiac remodelling post myocardial infarction. J Mol Cell Cardiol. (2014) 69:32–42. doi: 10.1016/j.yjmcc.2014.01.015
42. Summers SA, Steinmetz OM, Li M, Kausman JY, Semple T, Edgtton KL, et al. Th1 and Th17 cells induce proliferative glomerulonephritis. J Am Soc Nephrol. (2009) 20:2518–24. doi: 10.1681/ASN.2009030337
43. Kitching AR, Holdsworth SR. The emergence of th17 cells as effectors of renal injury. J Am Soc Nephrol. (2011) 22:235–8. doi: 10.1681/ASN.2010050536
44. Yang C, Huang XR, Fung E, Liu HF, Lan HY. The regulatory T-cell transcription factor foxp3 protects against crescentic glomerulonephritis. Sci Rep. (2017) 7:1481. doi: 10.1038/s41598-017-01515-8
45. Paust HJ, Ostmann A, Erhardt A, Turner JE, Velden J, Mittrucker HW, et al. Regulatory T cells control the Th1 immune response in murine crescentic glomerulonephritis. Kidney Int. (2011) 80:154–64. doi: 10.1038/ki.2011.108
46. Paust H-J, Turner J-E, Riedel J-H, Disteldorf E, Peters A, Schmidt T, et al. Chemokines play a critical role in the cross-regulation of Th1 and Th17 immune responses in murine crescentic glomerulonephritis. Kidney Int. (2012) 82:72–83. doi: 10.1038/ki.2012.101
47. Nakamura A, Zeng F, Nakamura S, Reid KT, Gracey E, Lim M, et al. Macrophage migration inhibitory factor drives pathology in a mouse model of spondyloarthritis and is associated with human disease. Sci Transl Med. (2021) 13:eabg1210. doi: 10.1126/scitranslmed.abg1210
48. Leng L, Metz CN, Fang Y, Xu J, Donnelly S, Baugh J, et al. MIF signal transduction initiated by binding to CD74. J Exp Med. (2003) 197:1467–76. doi: 10.1084/jem.20030286
49. Tillmann S, Bernhagen J, Noels H. Arrest functions of the MIF ligand/receptor axes in atherogenesis. Front Immunol. (2013) 4:115. doi: 10.3389/fimmu.2013.00115
50. Schulze-Osthoff K, Ferrari D, Riehemann K, Wesselborg S. Regulation of NF-kappa B activation by MAP kinase cascades. Immunobiology. (1997) 198:35–49. doi: 10.1016/S0171-2985(97)80025-3
51. Tomita N, Morishita R, Lan HY, Yamamoto K, Hashizume M, Notake M, et al. In vivo administration of a nuclear transcription factor-kappaB decoy suppresses experimental crescentic glomerulonephritis. J Am Soc Nephrol. (2000) 11:1244–52. doi: 10.1681/ASN.V1171244
52. Murray PJ. Macrophage polarization. Annu Rev Physiol. (2017) 79:541–66. doi: 10.1146/annurev-physiol-022516-034339
53. Fingerle-Rowson G, Petrenko O, Metz CN, Forsthuber TG, Mitchell R, Huss R, et al. The p53-dependent effects of macrophage migration inhibitory factor revealed by gene targeting. Proc Natl Acad Sci U.S.A. (2003) 100:9354–9. doi: 10.1073/pnas.1533295100
54. Wang S, Meng XM, Ng YY, Ma FY, Zhou S, Zhang Y, et al. TGF-beta/Smad3 signalling regulates the transition of bone marrow-derived macrophages into myofibroblasts during tissue fibrosis. Oncotarget. (2016) 7:8809–22. doi: 10.18632/oncotarget.v7i8
55. Lv J, Huang Xiao R, Klug J, Fröhlich S, Lacher P, Xu A, et al. Ribosomal protein S19 is a novel therapeutic agent in inflammatory kidney disease. Clin Sci. (2013) 124:627–37. doi: 10.1042/CS20120526
56. Wang YY, Jiang H, Wang YC, Huang XR, Pan J, Yang C, et al. Deletion of Smad3 improves cardiac allograft rejection in mice. Oncotarget. (2015) 6:17016–30. doi: 10.18632/oncotarget.v6i19
57. Qin W, Chung AC, Huang XR, Meng XM, Hui DS, Yu CM, et al. TGF-β/Smad3 signaling promotes renal fibrosis by inhibiting miR-29. J Am Soc Nephrol. (2011) 22:1462–74. doi: 10.1681/ASN.2010121308
58. Lv LL, Tang PM-K, Li CJ, You YK, Li J, Huang X-R, et al. The pattern recognition receptor, Mincle, is essential for maintaining the M1 macrophage phenotype in acute renal inflammation. Kidney Int. (2017) 91:587–602. doi: 10.1016/j.kint.2016.10.020
Keywords: macrophages, MIF, T cells, anti-GBM crescentic glomerulonephritis, inflammation
Citation: Yang H, Li J, Huang X-r, Bucala R, Xu A and Lan H-Y (2024) Macrophage-derived macrophage migration inhibitory factor mediates renal injury in anti-glomerular basement membrane glomerulonephritis. Front. Immunol. 15:1361343. doi: 10.3389/fimmu.2024.1361343
Received: 25 December 2023; Accepted: 30 April 2024;
Published: 23 May 2024.
Edited by:
Xu-jie Zhou, Peking University, ChinaCopyright © 2024 Yang, Li, Huang, Bucala, Xu and Lan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui-Yao Lan, aHlsYW5AY3Voay5lZHUuaGs=; Anping Xu, eHVhbnBpbmdAbWFpbC5zeXN1LmVkdS5jbg==
†These authors have contributed equally to this work