 Patrick Nylund
Patrick Nylund Anna Nikkarinen1
Anna Nikkarinen1 Sara Ek
Sara Ek Ingrid Glimelius
Ingrid Glimelius- 1Department of Immunology, Genetics and Pathology, Cancer Precision Medicine Unit, Uppsala University, Uppsala, Sweden
- 2Department of Immunotechnology, Lund University, Lund, Sweden
- 3Division of Clinical Epidemiology, Department of Medicine, Karolinska Institute, Stockholm, Sweden
In Mantle Cell Lymphoma (MCL), the role of macrophages within the tumour microenvironment (TME) has recently gained attention due to their impact on prognosis and response to therapy. Despite their low absolute number in MCL tumour tissue, recent findings reveal an association between the levels of macrophages and prognosis, consistent with trends observed in other lymphoma subtypes. M2-like macrophages, identified by markers such as CD163, contribute to angiogenesis and suppression of the immune response. Clinical trials with MCL patients treated with chemoimmunotherapy and targeted treatments underscore the adverse impact of high levels of M2-like macrophages. Immunomodulatory drugs like lenalidomide reduce the levels of MCL-associated CD163+ macrophages and enhance macrophage phagocytic activity. Similarly, clinical approaches targeting the CD47 “don’t eat me” signalling, in combination with the anti-CD20-antibody rituximab, demonstrate increased macrophage activity and phagocytosis of MCL tumour cells. Cell-based therapies such as chimeric antigen receptor (CAR) T-cell have shown promise but various challenges persist, leading to a potential interest in CAR-macrophages (CAR-M). When macrophages are recruited to the TME, they offer advantages including phagocytic function and responsiveness to microenvironment alterations, suggesting their potential as a manipulable and inducible alternative when CAR T-cell therapies fails in the complex landscape of MCL treatment.
Introduction
Mantle cell lymphoma (MCL) corresponds to about 5% of all non-Hodgkin’s lymphomas (NHL) and originates from the malignant transformation of B-cells within the mantle zones of the lymph nodes. MCL can also arise from antigen-experienced B-cells that have undergone immunoglobulin heavy-chain variable (IGVH) somatic hypermutations elsewhere in the lymphatic tissue (1). Due to disease heterogeneity, MCL is challenging to treat and remains mostly incurable, with patients exhibiting a median overall survival of 1.8 to 9.4 years depending on their individual genetic and pathological risk factors (2–4). Furthermore, patients often relapse as a result of drug resistance, leading to an overall poor prognosis (5, 6). Interestingly, recently developed immunotherapies including anti-CD19 chimeric antigen receptor T (CAR-T) cell therapy (7) and bispecific antibodies (8) have shown promising treatment outcomes at relapse. Thus, in the present review we aim to elucidate the current knowledge of the MCL tumour microenvironment (TME) and discuss how targeting these systems could improve patient response and survival even further.
Established prognostic factors in mantle cell lymphoma
The primary oncogenic event that takes place in MCL cells is t(11;14)(q13;q32), but this genetic aberration alone is not enough to drive lymphoma pathogenesis (9, 10). Instead, MCL pathogenesis is underpinned by increased genomic instability by mutations in genes that regulate cell cycle progression, such as ATM, TP53 and RB1, as well as genes that regulate DNA repair, NF-κB signalling and apoptosis. As such, detection of TP53 mutations in the tumour cells is one of the strongest negative prognostic markers in patients. In the absence of these specific gene mutations, transcriptional dysregulation can also cause aberrant protein function, as exemplified by the poor prognosis for patients with MYC overexpression (11, 12).
Markers for poor prognosis that are used in the clinic include a high mantle cell lymphoma international prognostic index (MIPI), high proliferation rate measured with Ki-67, blastoid and/or pleomorphic histology, as well as an abnormal karyotype (13–17). However, it has become clear that patient outcome cannot be determined by studying only the disease-specific cell types, but must include the much wider system of supporting and interacting cells that constitute the TME (18–20). The tumour architecture, presence and composition of inflammatory cells in the tumour tissue, and soluble biomarkers are important aspects of the TME. However, markers reflecting the TME are not yet being used as prognostic factors in MCL care. Despite this, the TME is highly involved in the established treatments, and the prognostic factors of the TME are becoming increasingly important to consider with the future expansion of immunotherapy in MCL treatment.
Current therapeutic approaches in mantle cell lymphoma
In younger MCL patients, the standard therapy has consisted of monoclonal anti-CD20 antibody rituximab together with intensive chemotherapy, i.e., cyclophosphamide, doxorubicin, vincristine prednisone (CHOP) and cytarabine or oxaliplatin, followed by an autologous stem cell transplantation (21). However, the introduction of novel targeted drugs, i.e., the Bruton tyrosine kinase inhibitor (BTKi) ibrutinib as maintenance after induction chemotherapy, has proven to be as effective with our without autologous stem cell transplantation (22). On the other hand, for elderly patients there is no general agreement on treatment internationally (23), but most patients have received chemotherapy (CHOP or bendamustine) in combination with rituximab.
A new era utilizing targeted drugs and potentially omitting chemotherapy is emerging in MCL care (24–26). The covalent BTKi:s ibrutinib, acalabrutinib and zanubrutinib have shown efficacy in relapsed patients (27). However, the survival benefit when using BTKi as a first-line therapy compared to chemotherapy is unknown, and we are eagerly awaiting the results for the randomized ENRICH trial comparing chemotherapy to first-line BTKi in elderly patients. A key question is whether BTKi can replace chemotherapy or add to its efficacy. Furthermore, the non-covalent BTKis pirtobrutinib and nemtabrutinib have also proven effective when used in conjunction with covalent BTKis at disease progression for various B-cell malignancies (28, 29).
The novel field of CAR-T cell therapy and bispecific antibodies is the next step in treatment. The CAR-T cell-based therapies tisagenlecleucel (Kymriah), axicabtagene ciloleucel (Yescarta), brexucabtagene autoleucel (Tecartus) and lisocabtagene maraleucel (Breyanzi) have been FDA approved for treatment of various hematological malignancies. In addition, the bispecific antibody-based therapy epcoritamab has also recently been FDA approved, but is not yet readily available in many countries due to reimbursement regulations. Immunotherapy has revolutionized the care of MCL patients relapsing after chemoimmunotherapy and BTKi (7, 30, 31). Summarized in Tables 1, 2 are a selection of ongoing and actively-recruiting clinical trials on the next generation of CAR-T cell therapy in MCL (Table 1) as well as a table of ongoing trials on different bispecific antibodies in MCL (Table 2). Resistance mechanisms of BTKi have been attributed to mutations in the BTK binding site as well as to various somatic mutations. However, for novel drugs such as the new generation of BTKi, CAR-T cell therapy and bispecific antibodies, specific resistance mechanisms are largely underexplored. The most described cause is loss of the target molecule, usually CD19, but other resistance mechanisms can include exhaustion of the CAR-T cells. It now remains to be determined which local factors in the TME and more general factors of the individual’s immune response and immune state can influence the response to treatment (Table 3).
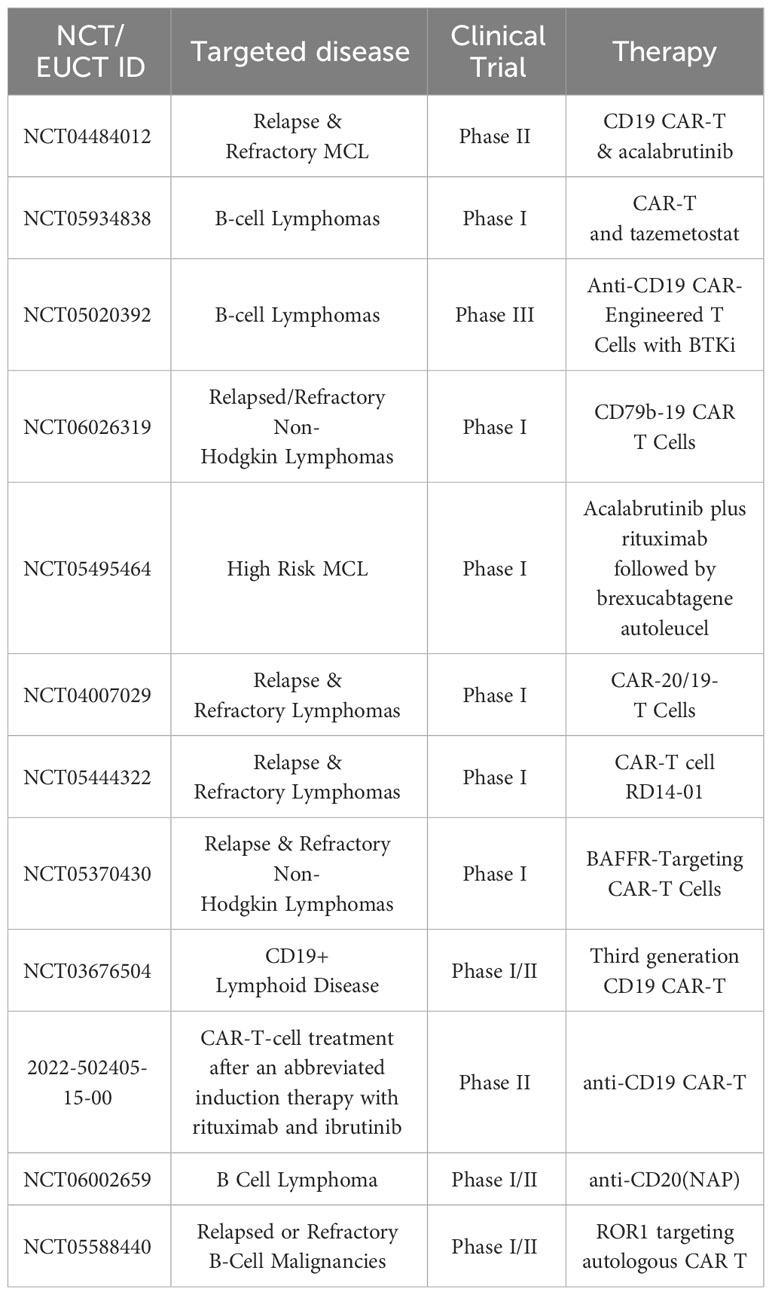
Table 1 Selected planned, ongoing actively recruiting CAR-T cell clinical trials in MCL on clinicaltrials.gov February 2024.
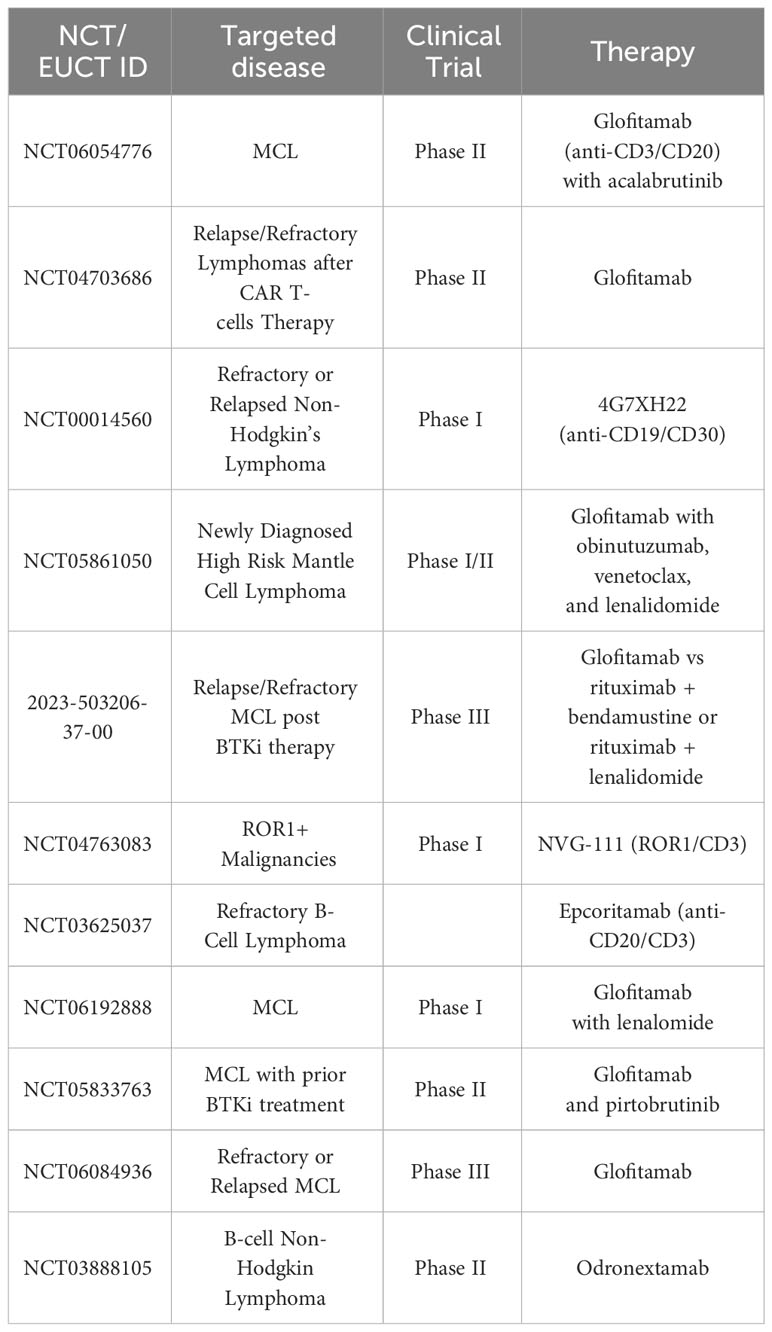
Table 2 Selected planned, ongoing and actively recruiting bispecific antibody clinical trials in MCL on clinicaltrials.gov February 2024.
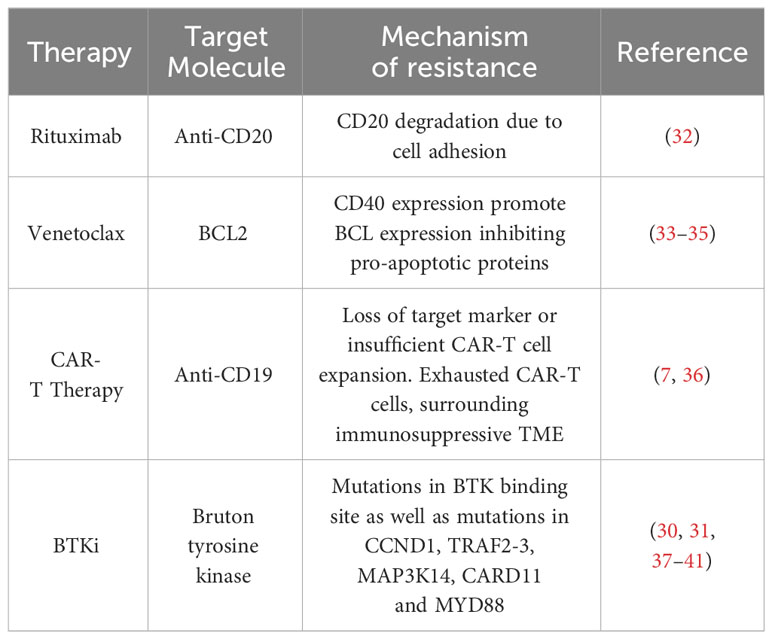
Table 3 Example of drug resistance mechanisms in mantle cell lymphoma.
The tumour microenvironment causing resistance to therapy
Drug resistance has previously been attributed to cellular processes within the TME. MCL cells are dependent on the TME for increased proliferative capacity, cellular survival and immune system evasion. As such, there is a rationale for targeting the TME in combination with conventional and novel immunotherapeutic drugs to improve patient response and outcome (Figure 1). Recent studies have shown that MCL cells invading the bone marrow became resistant to rituximab as a result of losing CD20 expression, while the tumour cells in the lymph node tissue maintained their extensive CD20 expression (32). Here, the process of CD20 degradation was attributed to bone marrow stromal cell adhesion (32). Combinatorial inhibition of focal adhesion kinase and ibrutinib has been shown to overcome resistance to ibrutinib in MCL cells (47) (Table 3).
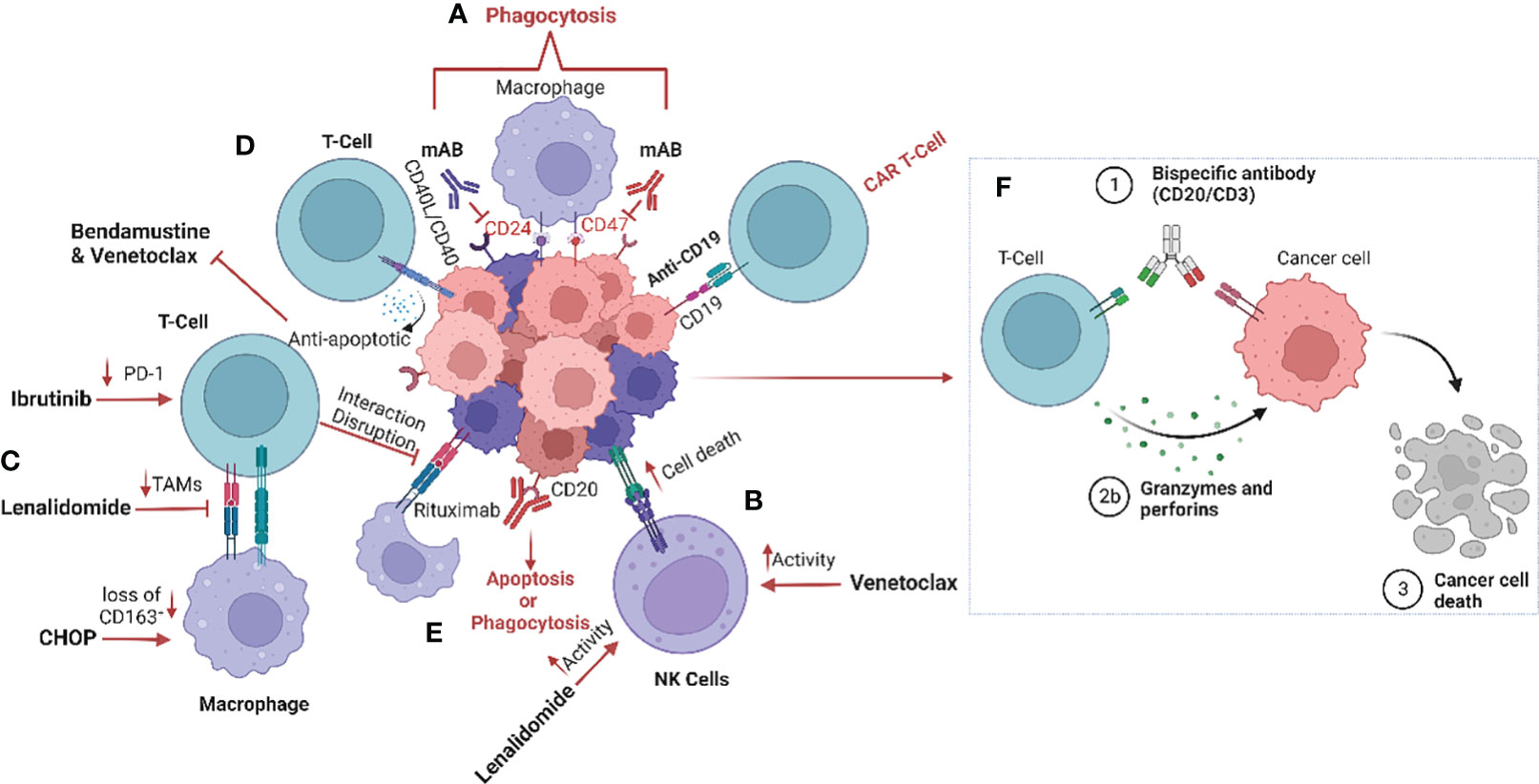
Figure 1 Schematic representation of the impact of treatment regimens on the MCL tumour microenvironment (TME). (A) The current strategies targeting the tumour microenvironment (TME), especially macrophages, include anti-CD24 and anti-CD47 monoclonal antibodies to enhance phagocytosis by inhibiting the “don’t eat me’ signalling (42). (B) Furthermore, treatment with lenalidomide and venetoclax increase natural killer (NK) cell activity and (C) reduce tumour-associated macrophages (TAMs) within the TME (35, 43–45). (D) CD40/CD40L interaction induces venetoclax and bendamustine resistance by promoting release of anti-apoptotic signalling such as BCL family proteins (33–35). (E) CD20 monoclonal antibody therapy results in the induction of phagocytosis, and more recently (F) bispecific antibodies promote the interaction between T-cells and tumour cells, causing T-cell induced cytotoxicity (46). Image was created with biorender.com.
It has previously been established that MCL cells are dependent on the CD40-CD40L interaction between the tumour cells and T-cells, and that this interaction plays an important role in promoting MCL cell survival (Figure 1). MCL cells that express CD40 induce the expression of the B-cell lymphoma (BCL) genes, a family of anti-apoptotic proteins which inhibit mitochondrial apoptotic priming and counteract pro-apoptotic proteins (33, 34) (Table 3), leading to resistance to the BCL2 inhibitor venetoclax (35) (Figure 1). However, treatment with the anti-CD20 antibody obinutuzumab can overcome venetoclax resistance by counteracting NF-κB-induced Bcl-XL expression (33, 35) (Figure 1). Interestingly, obinutuzumab treatment of MCL cells impaired p52 expression and reduced the expression of NF-κB target genes in cells with high expression of CD20 (33). Venetoclax also reduces the number of T regulatory cells (Tregs), lowers PD-1 expression in T-cells and increases natural killer (NK) cell function (Figure 1). Many MCL cells overexpress the transcription factor SOX11 which promotes expression of another co-stimulatory molecule, such as CD70 (48). CD70 bind to CD27 on T-cells in non-Hodgins lymphoma, resulting in T-cell exhaustion (49).
Different T-cell-engaging treatments such as CAR-T cells and bispecific antibodies function by targeting the TME directly or indirectly. In CAR-T cell treatment, T-cells are harvested from the patient, modified, and returned to take part in targeting the lymphoma cells and inducing anti-tumoral immune responses. Anti-CD19 CAR-T cell therapy has been approved for treatment of MCL patients that relapse after BTK treatment, who typically have a poor prognosis (7) (Figure 1). However, CAR-T cell treatment failures are frequent due to loss of target marker or insufficient CAR T-cell expansion (Table 3). Different armed CAR-T cells, the addition of co-stimulatory factors, production of CAR-T cells from T-cells harvested earlier in the disease course and dual binding sites are ways to improve the efficacy of CAR-T cells (36). Another way of improving efficacy is to use combinatorial therapies. Bispecific antibody therapy after CAR-T treatment is also currently undergoing evaluation of efficacy when loss of target for the CAR-T cells occurs and in abrogating T-cell exhaustion. The bispecific antibodies (anti-CD20/CD3) bind T-cells to lymphoma cells, facilitating T-cell engagement and activation of antibody-dependent cellular cytotoxicity (ADCC) against the lymphoma cells (50) (Figure 1). However, resistance is also observed in treatments with bispecific antibodies, particularly over time.
Macrophages in MCL therapy
Macrophages in MCL
Macrophages are a crucial component of the immune infiltrate in MCL. These immune cells are often referred to as tumour-associated macrophages (TAMs) when found within the TME. TAMs are highly plastic and can exhibit distinct functional states, with the two primary polarization states being M1-like (pro-inflammatory and anti-tumoral) and M2-like (anti-inflammatory and pro-tumoral). This division is likely an over-simplification but still useful. CD32, CD64, CD68, CD80 and CD86 have all been reported as surface markers for M1-like macrophages (51). M2-like macrophages are most commonly assessed using CD163, a scavenger receptor on the macrophage surface that can be released into the surrounding tissue or bloodstream upon activation (52). Another cell surface marker for identifying M2-like macrophages that are induced as a result of inflammation is CD206 (53). In addition, M1 and M2 cellular differentiation is highly dependent on several TME conditions such as hypoxia and hypoglycaemia. TAM utilize lipid metabolic pathways to sustain energy needs which can promote or inhibit tumour pathogenesis (54). Furthermore, other studies suggests that TAMs are subjected to the Warburg effect by utilizing oxidative phosphorylation rather than aerobic glycolysis for ATP production, which promotes the differentiation of M2-like macrophages. This provides a rationale to target the lipid metabolic pathway and/or mechanism of the Warburg effect to promote M1-like phenotypic differentiation of TAM which could promote response to conventional immunotherapies and reduce resistance (55, 56).
Although absolute numbers of macrophages are low in MCL tumour tissue, their presence was recently shown to be associated with poorer prognosis (19). This is not surprising, since it is known from the literature that macrophages correlate with poorer prognosis in many lymphoma subtypes (46, 57–60). However, this knowledge has only recently begun being utilized in predicting prognosis (19), identifying soluble markers (18) and enhancing the efficacy of novel immunotherapy treatments in the MCL setting (61).
Targeting macrophages in MCL
Macrophages play an important role in regulating the response to classical chemotherapy (62). The underlying biological mechanisms of CHOP response, which has been the backbone in MCL treatment, are also dependant on alteration of the microenvironment. In mice with diffuse large B-cell lymphoma (DLBCL) that were treated with CHOP and R-CHOP (addition of rituximab to standard CHOP therapy), the proportion of CD163+ macrophages were lower after both treatments, indicating a repolarization of M2-like macrophages to M1-like with these regimens (Figure 1) (63). It has however not yet been investigated how much of this effect was due to the glucocorticoid prednisolone, which itself has a modulatory effect on macrophages. In a wound repair model, macrophages treated with glucocorticoids upregulated CD163 and CD206 expression, thus demonstrating polarization towards anti-inflammatory M2-like macrophages which also showed a decreased migration ability (64). Although glucocorticoids are widely used in oncology, the effects on the TME are not yet fully understood and the elucidation of their impact will require further investigation.
In MCL, M2-polarization of macrophages is common, contributing to angiogenesis and inhibition of the immune response (65). Co-culture of monocytes with MCL cells isolated from newly diagnosed patient biopsies promotes M2-like macrophage differentiation with increased expression of CD163, which is associated with poor patient outcome and blastoid morphology, independent of T-cell infiltration (19). Clinical trials of NHL (including MCL patients treated with rituximab, intensive chemotherapy and autologous stem cell transplantation) showed a poorer outcome for patients exhibiting a high presence of M2-like macrophages, and it was suggested that infiltration of these macrophages could have an impact on the response to anti-CD20 therapy (19, 57, 60, 66). Furthermore, MCL cells secrete colony stimulating factor 1(CSF1), which promotes the differentiation of monocytes into M2-like CD163+ macrophages and thereby stimulates MCL proliferation and survival (67). Depletion of macrophages by blocking the CSF1/CSF1R axis in vivo has been demonstrated to reduce differentiation and survival of M2-type macrophages (68). Other drugs such as the BTKi ibrutinib also impacts the TME by exerting an immunomodulatory effect through regulation of tumour-infiltrating macrophages (69). In addition, ibrutinib downregulates PD-1 expression on T-cells, disrupts communication between MCL cells and macrophages, and impairs macrophage phagocytic function (70) (Figure 1).
We have previously confirmed the prognostic value of CD163 by measuring soluble CD163 (sCD163) in the serum of patients with MCL. High sCD163 was associated with shorter progression-free survival and poor outcome in patients treated with rituximab, ibrutinib and lenalidomide in a phase 2 clinical trial (18). Although sCD163 levels were higher in patients with p53-deficient tumours, the prognostic value of sCD163 was independent of TP53 mutations and other clinical factors such as MIPI, Ki-67 or blastoid morphology, while low sCD163 levels could identify patients with a favourable prognosis (18). Various clinical approaches have been investigated to target M2-like macrophages and disrupt MCL proliferative advantage. One such approach is lenalidomide, which is an immunomodulatory drug targeting cereblon (CRBN), a highly conserved gene that promotes ubiquitination. Lenalidomide is currently approved for treatment of multiple myeloma (MM) and MCL (71), and has been found to affect many cells of the TME, including macrophages. Accordingly, lenalidomide reduced the abundance of TAMs (43), enhanced macrophage phagocytic activity and increased macrophage communication with CD8+ T-cells (44) (Figure 1). In addition, treatment with lenalidomide has a clinical benefit in relapsed MCL patients, defined by increased NK cell activity (45). Just recently, the combination of lenalidomide, venetoclax and rituximab was shown to be feasible and efficacious in relapsed MCL patients, using a minimal residual disease (MRD)-driven treatment design (72).
To be able to study the MCL lymph node signalling, heterogeneity and TME, 3D models of MCL-spheroids have been created (70). These spheroids caused monocytes to differentiate into M2-like macrophages, which were interestingly reprogrammed into a more immunogenic phenotype upon blockade of the chemokine receptor CCR1 (53, 70). Prior studies have shown that co-culturing MCL cells with TAMs assisted the tumour cells to avoid phagocytosis. However, dual inhibition of CD24 and CD47, two molecules enhancing the “don’t eat me” signal, in combination with rituximab induced increased macrophage activity and promoted phagocytosis in MCL cells (42) (Figure 1). Interestingly, other “don’t eat me” signalling pathways have been identified and include the PD-1/PD-L1, MHC-I/LILLRB1/2 and CD24/SIGLEC-10 axes. Targeting these mechanisms have been shown to induce activation of macrophages and increase phagocytosis (73–75). In addition, monoclonal antibodies targeting CD47 on the tumour cells have been demonstrated to activate macrophages and induce phagocytosis of the tumour cells. Furthermore, ongoing clinical trials in MCL are exploring the targeting of “don’t eat me” signalling by utilizing anti-CD24 therapy. CD24-expressing cells promotes immune response escape by reprogramming macrophages not to target them for phagocytosis (NCT05888701). Clinical studies have demonstrated the importance of blocking the “don’t eat me” signalling pathways in various haematological malignancies including MCL by specifically targeting the CD47/SIRPα axis (76). Thus, utilizing CD47-targeting bispecific antibodies to enhance the response may be a promising therapeutic intervention to increase treatment efficiency. Preclinical evaluation of CD47/PD-1, CD47/CD33 CD47/PD-L1, CD47/CD20, CD47/CD19 and CD47/CD20 are currently ongoing (77–82).
CAR-macrophage therapy
Despite the relatively high success rate of CAR T-cell therapies in haematological malignancies, numerous challenges remain, including a lack of specific targets. Furthermore, CAR T-cell trafficking and infiltration to tumour sites is difficult due to the abnormal vasculature and dysregulated adhesion matrices (83). Another limitation in CAR T-cell therapies is that the TME includes Tregs, TAMs, tumour-associated fibroblasts and myeloid-derived suppressor cells that create a hostile environment by secreting immunosuppressive cytokines that impair CAR T-cell function. Antigen escape and lack of CAR T-cell expansion and persistence during treatment can also limit the effectiveness of the therapy (84). Direct TAM-targeted therapies have been reported but are highly dependent on the presence of either activating or suppressive macrophage markers, and can cause adverse effects. Clinical data of these therapies is currently lacking for MCL.
Recently, the development of CAR-macrophages (CAR-M) has become increasingly interesting as a potential solution to the challenges observed with TAM-targeting and CAR T-cell therapies. As of now, CAR-M is not yet in clinical trials for MCL, but since macrophages are recruited to the TME as a result of the production of cancer-associated cytokines and hypoxic conditions, it has been suggested that CAR-M could be utilized to overcome the problem of poor capacity for trafficking and infiltration (85). Early precursors of CAR-M were the simultaneous blocking of CD47/SIRPα which have been shown to promote phagocytotic activity with high specificity against the tumour antigen (86). In fact, next generation CAR-M therapies targeting CD19 promoted phagocytosis in an antigen-specific manner and converted M2-like macrophages to M1-like. Interestingly, these CAR-M also secreted proinflammatory cytokines and chemokines, upregulated antigen presentation, promoted activation of cytotoxic T-cells and overcame the immunosuppressive microenvironment in vivo (87, 88). Additional preclinical studies of CAR-M include the target antigens CD5, CD22, HER2, CCR7 and ALK (89, 90). Another potential advantage of this therapy is that, even when in the immunosuppressive M2-like state, macrophages still maintain the function of phagocytosis. It has also been suggested that macrophages are more responsive to alterations in the TME, making them easier to manipulate and induce than CAR T-cells (91) (Figure 2). However, several challenges remain on this front, including finding an appropriate source of macrophages for modification, since immortalized cells cannot be applicable for clinical utility while blood and bone marrow samples cannot be modified efficiently (Figure 2). Interestingly, induced pluripotent stem cells (iPSCs)-derived macrophages have been demonstrated to recapitulate similar anti-tumour function described by Klichinsky et al. (87), in both solid and haematological malignancies (92). Currently, there are two ongoing phase I clinical trials evaluating the potential of CAR-M in HER2-overexpressing solid tumours and advanced gastric cancer with peritoneal metastases (NCT04660929 & NCT06224738), but no clinical trials have been performed in MCL to date. To conclude, the strengths of CAR-M include increased tumour infiltration, reduction of TAMs and alteration of TAM phenotypes within the TME. CAR-M can also stimulate increased phagocytosis and promote antigen presentation, resulting in improved cytotoxic T-cell-mediated anti-tumour effects with limited off-target effects. As such, the potential of CAR-M should be further explored.
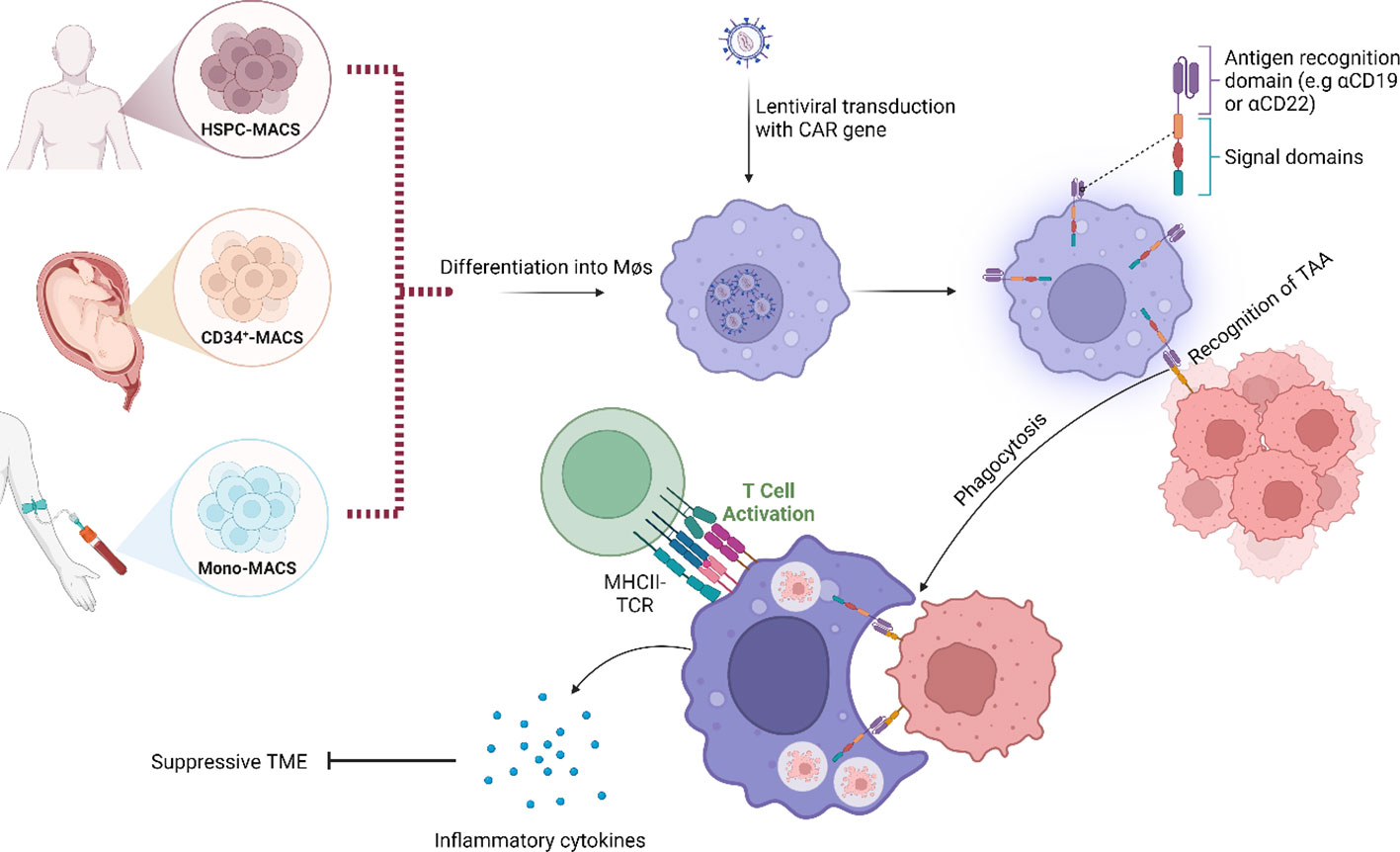
Figure 2 Schematic representation of the modes of action of chimeric antigen receptor–macrophages (CAR-M). Instead of using a T-cell as carrier for the CAR construct, a macrophage is used. Binding of tumour-associated antigen by CAR-M induces phagocytosis, accompanied by the release of inflammatory cytokines that activate additional immune responses within the tumour microenvironment (TME). Examples of downstream effects include T-cell-mediated anti-tumour immune responses. Image was created with biorender.com.
Discussion
Herein, we comprehensively explore the intricate relationship between the TME and its profound impact on treatment response, resistance mechanisms, and ultimately patient outcomes, with a particular focus on macrophages. We highlight the significance of macrophages in treatment dynamics, and their dual role in either promoting or inhibiting therapeutic responses. TAMs are highly plastic and have the ability to adopt distinct functional states, notably M1-like with pro-inflammatory and anti-tumoral characteristics, or M2-like with anti-inflammatory and pro-tumoral features. This polarization dynamic could be essential as a determinant in treatment outcomes.
We have summarised the current treatment strategies and stipulated the many challenges associated with choice of drug. The next objective in immunotherapy is to investigate if, and how, a patient’s individual immune state would impact the degree of side effects caused by different immunotherapies. We are constantly learning more about effective side effect management, but patients are often still hospitalized for safety reasons. For example, high-dose steroids are required for prophylaxis, and patients can suffer from severe complications such as cytokine release syndrome and immune effector cell-associated neurotoxicity syndrome. While the exact mechanisms of these immune-mediated toxicities are not clearly understood, emerging pre-clinical and clinical studies have revealed the pivotal role of myeloid cells, particularly macrophages, as contributors to the efficacy of treatments but also as crucial mediators of toxicity (93).
Immunotherapy takes advantage of the TME to defeat the malignant cells, as opposed to focusing on the disruption of such interactions. CAR-T cell therapies and bispecific antibodies have already paved the way in relapsed/refractory disease in various haematological malignancies and the next step is testing them as first-line treatment combinations for high-risk patients. M2-like macrophages are involved in CAR-T cell therapy response both in vivo and in vitro, leading to CAR-T cell therapy failure and disease progression in DLBCL (94), likely having similar activities in MCL. Targeting macrophages with bispecific antibodies is a therapeutic possibility, but has currently not been evaluated in a clinical setting (95). Furthermore, reprogramming of pro-tumoral macrophages to anti-tumoral macrophages, as well as utilization of CAR-M are potential novel approaches in MCL therapy (95). The balance between achieving efficacy and the potential toxicity of macrophage-targeted therapies will however be challenging, since they exhibit a high degree of diversity and plasticity and can adopt different functional states in response to the TME. Modulating macrophage activity may trigger inflammatory responses, leading to cytokine release syndrome or other immune-related adverse events, and as such balancing therapeutic efficacy with the risk of systemic inflammation is crucial. Similarly, macrophages can play both pro-tumor and anti-tumor roles, which in combination with a lack of target specificity can lead to unintended effects on other tissues or immune cells.
In summary, the TME plays a large role in determining the response to both conventional but also next generation therapies for MCL, where a high presence of CD163+ macrophages have a negative prognostic impact. It is evident that by combining the therapies now in clinical utility with strategies directly targeting the TME, patient outcome could be improved, but additional clinical evaluation is required.
Author contributions
PN: Conceptualization, Project administration, Visualization, Writing – original draft, Writing – review & editing. AN: Project administration, Writing – original draft, Writing – review & editing. SE: Writing – original draft, Writing – review & editing. IG: Conceptualization, Funding acquisition, Project administration, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The project was supported by grants from the Swedish Research Council (Dnr: 2022-00801), the Swedish Cancer Society (CAN 222167Pj) and The Sjöberg Foundation (2023-01-03:3).
Acknowledgments
We are grateful to Tiarne van de Walle for her excellent assistance, advice and input on this review.
Conflict of interest
IG has received support to the department from Jansen Cilag and Abbvie for educational purposes. IG participated in a real-world data project supported by Takeda with reimbursement to the department.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Navarro A, Bea S, Jares P, Campo E. Molecular pathogenesis of mantle cell lymphoma. Hematol Oncol Clin North Am. (2020) 34:795–807. doi: 10.1016/j.hoc.2020.05.002
2. Wang ML, Rule S, Martin P, Goy A, Auer R, Kahl BS, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med. (2013) 369:507–16. doi: 10.1056/NEJMoa1306220
3. Gonzalez-Santamarta M, Quinet G, Reyes-Garau D, Sola B, Roue G, Rodriguez MS. Resistance to the proteasome inhibitors: lessons from multiple myeloma and mantle cell lymphoma. Adv Exp Med Biol. (2020) 1233:153–74.
4. Perez-Galan P, Mora-Jensen H, Weniger MA, Shaffer AL 3rd, Rizzatti EG, Chapman CM, et al. Bortezomib resistance in mantle cell lymphoma is associated with plasmacytic differentiation. Blood. (2011) 117:542–52. doi: 10.1182/blood-2010-02-269514
5. Nadeu F, Martin-Garcia D, Clot G, Diaz-Navarro A, Duran-Ferrer M, Navarro A, et al. Genomic and epigenomic insights into the origin, pathogenesis, and clinical behavior of mantle cell lymphoma subtypes. Blood. (2020) 136:1419–32. doi: 10.1182/blood.2020005289
6. Campo E, Jaffe ES, Cook JR, Quintanilla-Martinez L, Swerdlow SH, Anderson KC, et al. The international consensus classification of mature lymphoid neoplasms: a report from the clinical advisory committee. Blood. (2022) 140:1229–53. doi: 10.1182/blood.2022015851
7. Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, et al. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. N Engl J Med. (2020) 382:1331–42. doi: 10.1056/NEJMoa1914347
8. Dickinson M, Carlo-Stella C, Morschhauser F, Patel K, Khan C, Bartlett NL, et al. Glofitamab monotherapy provides durable responses after fixed-length dosing in relapsed/refractory (R/R) non-Hodgkin lymphoma (NHL) patients (pts). Blood. (2021) 138:2478. doi: 10.1182/blood-2021-146845
9. Bodrug SE, Warner BJ, Bath ML, Lindeman GJ, Harris AW, Adams JM. Cyclin D1 transgene impedes lymphocyte maturation and collaborates in lymphomagenesis with the myc gene. EMBO J. (1994) 13:2124–30. doi: 10.1002/embj.1994.13.issue-9
10. Lovec H, Grzeschiczek A, Kowalski MB, Moroy T. Cyclin D1/bcl-1 cooperates with myc genes in the generation of B-cell lymphoma in transgenic mice. EMBO J. (1994) 13:3487–95. doi: 10.1002/embj.1994.13.issue-15
11. Kumar A. MYC overexpression: adding another piece to the puzzle of high-risk mantle cell lymphoma. Haematologica. (2023). doi: 10.3324/haematol.2023.284105
12. Rodrigues JM, Hollander P, Schmidt L, Gkika E, Razmara M, Kumar D, et al. MYC protein is a high-risk factor in mantle cell lymphoma and identifies cases beyond morphology, proliferation and TP53/p53 - a Nordic Lymphoma Group study. Haematologica. (2023). doi: 10.3324/haematol.2023.283352
13. Rodrigues JM, Hassan M, Freiburghaus C, Eskelund CW, Geisler C, Raty R, et al. p53 is associated with high-risk and pinpoints TP53 missense mutations in mantle cell lymphoma. Br J Haematol. (2020) 191(5):796–805. doi: 10.1111/bjh.17023
14. Jain P, Zhang S, Kanagal-Shamanna R, Ok CY, Nomie K, Gonzalez GN, et al. Genomic profiles and clinical outcomes of de novo blastoid/pleomorphic MCL are distinct from those of transformed MCL. Blood Adv. (2020) 4:1038–50. doi: 10.1182/bloodadvances.2019001396
15. Scheubeck G, Jiang L, Hermine O, Kluin-Nelemans HC, Schmidt C, Unterhalt M, et al. Clinical outcome of Mantle Cell Lymphoma patients with high-risk disease (high-risk MIPI-c or high p53 expression). Leukemia. (2023) 37:1887–94. doi: 10.1038/s41375-023-01977-y
16. Silkenstedt E, Dreyling M. Mantle cell lymphoma-Update on molecular biology, prognostication and treatment approaches. Hematol Oncol. (2023) 41 Suppl 1:36–42. doi: 10.1002/hon.3149
17. Zhang S, Jiang VC, Han G, Hao D, Lian J, Liu Y, et al. Longitudinal single-cell profiling reveals molecular heterogeneity and tumor-immune evolution in refractory mantle cell lymphoma. Nat Commun. (2021) 12:2877. doi: 10.1038/s41467-021-22872-z
18. Nikkarinen A, Lokhande L, Amini RM, Jerkeman M, Porwit A, Molin D, et al. Soluble CD163 predicts outcome in both chemoimmunotherapy and targeted therapy-treated mantle cell lymphoma. Blood Adv. (2023) 7:5304–13. doi: 10.1182/bloodadvances.2023010052
19. Rodrigues JM, Nikkarinen A, Hollander P, Weibull CE, Raty R, Kolstad A, et al. Infiltration of CD163-, PD-L1- and FoxP3-positive cells adversely affects outcome in patients with mantle cell lymphoma independent of established risk factors. Br J Haematol. (2021) 193:520–31. doi: 10.1111/bjh.17366
20. Nygren L, Wasik AM, Baumgartner-Wennerholm S, Jeppsson-Ahlberg A, Klimkowska M, Andersson P, et al. T-cell levels are prognostic in mantle cell lymphoma. Clin Cancer Res. (2014) 20:6096–104. doi: 10.1158/1078-0432.CCR-14-0889
21. Glimelius I, Smedby KE, Albertsson-Lindblad A, Crowther MJ, Eloranta S, Jerkeman M, et al. Unmarried or less-educated patients with mantle cell lymphoma are less likely to undergo a transplant, leading to lower survival. Blood Adv. (2021) 5:1638–47. doi: 10.1182/bloodadvances.2020003645
22. Dreyling M, Doorduijn JK, Gine E, Jerkeman M, Walewski J, Hutchings M, et al. Efficacy and safety of ibrutinib combined with standard first-line treatment or as substitute for autologous stem cell transplantation in younger patients with mantle cell lymphoma: results from the randomized triangle trial by the European MCL network. Blood. (2022) 140:1–3. doi: 10.1182/blood-2022-163018
23. Dreyling M, Geisler C, Hermine O, Kluin-Nelemans HC, Le Gouill S, Rule S, et al. Newly diagnosed and relapsed mantle cell lymphoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. (2014) 25 Suppl 3:iii83–92. doi: 10.1093/annonc/mdu264
24. Ye H, Desai A, Zeng D, Romaguera J, Wang ML. Frontline treatment for older patients with mantle cell lymphoma. Oncologist. (2018) 23(11): 1337–1348. doi: 10.1634/theoncologist.2017-0470
25. Martin P, Ruan J, Leonard JP. The potential for chemotherapy-free strategies in mantle cell lymphoma. Blood. (2017) 130(17):1881–1888. doi: 10.1182/blood-2017-05-737510
26. Gine E, de la Cruz F, Jimenez Ubieto A, Lopez Jimenez J, Martin Garcia-Sancho A, Terol MJ, et al. Ibrutinib in combination with rituximab for indolent clinical forms of mantle cell lymphoma (IMCL-2015): A multicenter, open-label, single-arm, phase II trial. J Clin Oncol. (2022), 40:JCO2102321. doi: 10.1200/JCO.21.02321
27. Rule S, Dreyling M, Goy A, Hess G, Auer R, Kahl B, et al. Ibrutinib for the treatment of relapsed/refractory mantle cell lymphoma: extended 3.5-year follow up from a pooled analysis. Haematologica. (2019) 104:e211–e4. doi: 10.3324/haematol.2018.205229
28. Mato AR, Shah NN, Jurczak W, Cheah CY, Pagel JM, Woyach JA, et al. Pirtobrutinib in relapsed or refractory B-cell Malignancies (BRUIN): a phase 1/2 study. Lancet. (2021) 397:892–901. doi: 10.1016/S0140-6736(21)00224-5
29. Reiff SD, Mantel R, Smith LL, Greene JT, Muhowski EM, Fabian CA, et al. The BTK inhibitor ARQ 531 targets ibrutinib-resistant CLL and richter transformation. Cancer Discovery. (2018) 8:1300–15. doi: 10.1158/2159-8290.CD-17-1409
30. Wang Y, Jain P, Locke FL, Maurer MJ, Frank MJ, Munoz JL, et al. Brexucabtagene autoleucel for relapsed or refractory mantle cell lymphoma in standard-of-care practice: results from the US lymphoma CAR T consortium. J Clin Oncol. (2023) 41:2594–606. doi: 10.1200/JCO.22.01797
31. Hutchings M, Morschhauser F, Iacoboni G, Carlo-Stella C, Offner FC, Sureda A, et al. Glofitamab, a novel, bivalent CD20-targeting T-cell-engaging bispecific antibody, induces durable complete remissions in relapsed or refractory B-cell lymphoma: A phase I trial. J Clin Oncol. (2021) 39:1959–70. doi: 10.1200/JCO.20.03175
32. Kuroda Y, Yashima-Abo A, Koyama D, Kikuchi J, Mori S, Ito S, et al. Bone marrow stromal cell-mediated degradation of CD20 leads to primary rituximab resistance in mantle cell lymphoma. Leukemia. (2021) 35:1506–10. doi: 10.1038/s41375-020-01035-x
33. Chiron D, Bellanger C, Papin A, Tessoulin B, Dousset C, Maiga S, et al. Rational targeted therapies to overcome microenvironment-dependent expansion of mantle cell lymphoma. Blood. (2016) 128:2808–18. doi: 10.1182/blood-2016-06-720490
34. Decombis S, Papin A, Bellanger C, Sortais C, Dousset C, Le Bris Y, et al. The IL32/BAFF axis supports prosurvival dialogs in the lymphoma ecosystem and is disrupted by NIK inhibition. Haematologica. (2022) 107:2905–17. doi: 10.3324/haematol.2021.279800
35. Agarwal R, Chan YC, Tam CS, Hunter T, Vassiliadis D, Teh CE, et al. Dynamic molecular monitoring reveals that SWI-SNF mutations mediate resistance to ibrutinib plus venetoclax in mantle cell lymphoma. Nat Med. (2019) 25:119–29. doi: 10.1038/s41591-018-0243-z
36. Ruella M, Korell F, Porazzi P, Maus MV. Mechanisms of resistance to chimeric antigen receptor-T cells in haematological Malignancies. Nat Rev Drug Discovery. (2023) 22:976–95. doi: 10.1038/s41573-023-00807-1
37. Chiron D, Di Liberto M, Martin P, Huang X, Sharman J, Blecua P, et al. Cell-cycle reprogramming for PI3K inhibition overrides a relapse-specific C481S BTK mutation revealed by longitudinal functional genomics in mantle cell lymphoma. Cancer Discovery. (2014) 4:1022–35. doi: 10.1158/2159-8290.CD-14-0098
38. Rahal R, Frick M, Romero R, Korn JM, Kridel R, Chan FC, et al. Pharmacological and genomic profiling identifies NF-kappaB-targeted treatment strategies for mantle cell lymphoma. Nat Med. (2014) 20:87–92. doi: 10.1038/nm.3435
39. Wu C, de Miranda NF, Chen L, Wasik AM, Mansouri L, Jurczak W, et al. Genetic heterogeneity in primary and relapsed mantle cell lymphomas: Impact of recurrent CARD11 mutations. Oncotarget. (2016) 7:38180–90. doi: 10.18632/oncotarget.v7i25
40. Mohanty A, Sandoval N, Das M, Pillai R, Chen L, Chen RW, et al. CCND1 mutations increase protein stability and promote ibrutinib resistance in mantle cell lymphoma. Oncotarget. (2016) 7:73558–72. doi: 10.18632/oncotarget.v7i45
41. Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. (2011) 470:115–9. doi: 10.1038/nature09671
42. Aroldi A, Mauri M, Ramazzotti D, Villa M, Malighetti F, Crippa V, et al. Effects of blocking CD24 and CD47 'don't eat me' signals in combination with rituximab in mantle-cell lymphoma and chronic lymphocytic leukaemia. J Cell Mol Med. (2023) 27:3053–64. doi: 10.1111/jcmm.17868
43. Song K, Herzog BH, Sheng M, Fu J, McDaniel JM, Chen H, et al. Lenalidomide inhibits lymphangiogenesis in preclinical models of mantle cell lymphoma. Cancer Res. (2013) 73:7254–64. doi: 10.1158/0008-5472.CAN-13-0750
44. Svanberg R, Janum S, Patten PEM, Ramsay AG, Niemann CU. Targeting the tumor microenvironment in chronic lymphocytic leukemia. Haematologica. (2021) 106:2312–24. doi: 10.3324/haematol.2020.268037
45. Hagner PR, Chiu H, Ortiz M, Apollonio B, Wang M, Couto S, et al. Activity of lenalidomide in mantle cell lymphoma can be explained by NK cell-mediated cytotoxicity. Br J Haematol. (2017) 179:399–409. doi: 10.1111/bjh.14866
46. Touati M, Delage-Corre M, Monteil J, Abraham J, Moreau S, Remenieras L, et al. CD68-positive tumor-associated macrophages predict unfavorable treatment outcomes in classical Hodgkin lymphoma in correlation with interim fluorodeoxyglucose-positron emission tomography assessment. Leuk Lymphoma. (2015) 56:332–41. doi: 10.3109/10428194.2014.917636
47. Rudelius M, Rosenfeldt MT, Leich E, Rauert-Wunderlich H, Solimando AG, Beilhack A, et al. Inhibition of focal adhesion kinase overcomes resistance of mantle cell lymphoma to ibrutinib in the bone marrow microenvironment. Haematologica. (2018) 103:116–25. doi: 10.3324/haematol.2017.177162
48. Balsas P, Veloza L, Clot G, Sureda-Gomez M, Rodriguez ML, Masaoutis C, et al. SOX11, CD70, and Treg cells configure the tumor-immune microenvironment of aggressive mantle cell lymphoma. Blood. (2021) 138:2202–15. doi: 10.1182/blood.2020010527
49. Yang ZZ, Novak AJ, Ziesmer SC, Witzig TE, Ansell SM. CD70+ non-Hodgkin lymphoma B cells induce Foxp3 expression and regulatory function in intratumoral CD4+CD25 T cells. Blood. (2007) 110:2537–44. doi: 10.1182/blood-2007-03-082578
50. Salvaris R, Ong J, Gregory GP. Bispecific antibodies: A review of development, clinical efficacy and toxicity in B-cell lymphomas. J Pers Med. (2021) 11:355. doi: 10.3390/jpm11050355
51. Oshi M, Tokumaru Y, Asaoka M, Yan L, Satyananda V, Matsuyama R, et al. M1 Macrophage and M1/M2 ratio defined by transcriptomic signatures resemble only part of their conventional clinical characteristics in breast cancer. Sci Rep. (2020) 10:16554. doi: 10.1038/s41598-020-73624-w
52. Møller HJ. Soluble CD163. Scand J Clin Lab Invest. (2012) 72:1–13. doi: 10.3109/00365513.2011.626868
53. Le K, Sun J, Ghaemmaghami J, Smith MR, Ip WKE, Phillips T, et al. Blockade of CCR1 induces a phenotypic shift in macrophages and triggers a favorable antilymphoma activity. Blood Adv. (2023) 7:3952–67. doi: 10.1182/bloodadvances.2022008722
54. Qiao X, Hu Z, Xiong F, Yang Y, Peng C, Wang D, et al. Lipid metabolism reprogramming in tumor-associated macrophages and implications for therapy. Lipids Health Dis. (2023) 22:45. doi: 10.1186/s12944-023-01807-1
55. Zhu L, Yang T, Li L, Sun L, Hou Y, Hu X, et al. TSC1 controls macrophage polarization to prevent inflammatory disease. Nat Commun. (2014) 5:4696. doi: 10.1038/ncomms5696
56. Kim J, Bae JS. Metabolic regulation of macrophages in tumor microenvironment. Curr Opin Hematol. (2018) 25:52–9. doi: 10.1097/MOH.0000000000000390
57. Kridel R, Xerri L, Gelas-Dore B, Tan K, Feugier P, Vawda A, et al. The prognostic impact of CD163-positive macrophages in follicular lymphoma: A study from the BC cancer agency and the lymphoma study association. Clin Cancer Res. (2015) 21:3428–35. doi: 10.1158/1078-0432.CCR-14-3253
58. Scott DW, Gascoyne RD. The tumour microenvironment in B cell lymphomas. Nat Rev Cancer. (2014) 14:517–34. doi: 10.1038/nrc3774
59. Wada N, Zaki MA, Hori Y, Hashimoto K, Tsukaguchi M, Tatsumi Y, et al. Tumour-associated macrophages in diffuse large B-cell lymphoma: a study of the Osaka Lymphoma Study Group. Histopathology. (2012) 60:313–9. doi: 10.1111/j.1365-2559.2011.04096.x
60. Canioni D, Salles G, Mounier N, Brousse N, Keuppens M, Morchhauser F, et al. High numbers of tumor-associated macrophages have an adverse prognostic value that can be circumvented by rituximab in patients with follicular lymphoma enrolled onto the GELA-GOELAMS FL-2000 trial. J Clin Oncol. (2008) 26:440–6. doi: 10.1200/JCO.2007.12.8298
61. Qiu Y, Liao P, Wang H, Chen J, Hu Y, Hu R, et al. Enhanced tumor immunotherapy by polyfunctional CD19-CAR T cells engineered to secrete anti-CD47 single-chain variable fragment. Int J Biol Sci. (2023) 19:4948–66. doi: 10.7150/ijbs.86632
62. Lossos C, Liu Y, Kolb KE, Christie AL, Van Scoyk A, Prakadan SM, et al. Mechanisms of lymphoma clearance induced by high-dose alkylating agents. Cancer Discovery. (2019) 9:944–61. doi: 10.1158/2159-8290.CD-18-1393
63. Zhang Y, Xiang J, Sheng X, Zhu N, Deng S, Chen J, et al. GM-CSF enhanced the effect of CHOP and R-CHOP on inhibiting diffuse large B-cell lymphoma progression via influencing the macrophage polarization. Cancer Cell Int. (2021) 21:141. doi: 10.1186/s12935-021-01838-7
64. Desgeorges T, Caratti G, Mounier R, Tuckermann J, Chazaud B. Glucocorticoids shape macrophage phenotype for tissue repair. Front Immunol. (2019) 10:1591. doi: 10.3389/fimmu.2019.01591
65. Le K, Sun J, Khawaja H, Shibata M, Maggirwar SB, Smith MR, et al. Mantle cell lymphoma polarizes tumor-associated macrophages into M2-like macrophages, which in turn promote tumorigenesis. Blood Adv. (2021) 5:2863–78. doi: 10.1182/bloodadvances.2020003871
66. Kridel R, Steidl C, Gascoyne RD. Tumor-associated macrophages in diffuse large B-cell lymphoma. Haematologica. (2015) 100:143–5. doi: 10.3324/haematol.2015.124008
67. Papin A, Tessoulin B, Bellanger C, Moreau A, Le Bris Y, Maisonneuve H, et al. CSF1R and BTK inhibitions as novel strategies to disrupt the dialog between mantle cell lymphoma and macrophages. Leukemia. (2019) 33:2442–53. doi: 10.1038/s41375-019-0463-3
68. Rojo R, Raper A, Ozdemir DD, Lefevre L, Grabert K, Wollscheid-Lengeling E, et al. Deletion of a Csf1r enhancer selectively impacts CSF1R expression and development of tissue macrophage populations. Nat Commun. (2019) 10:3215. doi: 10.1038/s41467-019-11053-8
69. Ping L, Ding N, Shi Y, Feng L, Li J, Liu Y, et al. The Bruton's tyrosine kinase inhibitor ibrutinib exerts immunomodulatory effects through regulation of tumor-infiltrating macrophages. Oncotarget. (2017) 8:39218–29. doi: 10.18632/oncotarget.v8i24
70. Araujo-Ayala F, Dobano-Lopez C, Valero JG, Nadeu F, Gava F, Faria C, et al. A novel patient-derived 3D model recapitulates mantle cell lymphoma lymph node signaling, immune profile and in vivo ibrutinib responses. Leukemia. (2023) 37:1311–23. doi: 10.1038/s41375-023-01885-1
71. Morschhauser F, Fowler NH, Feugier P, Bouabdallah R, Tilly H, Palomba ML, et al. Rituximab plus lenalidomide in advanced untreated follicular lymphoma. N Engl J Med. (2018) 379:934–47. doi: 10.1056/NEJMoa1805104
72. Jerkeman M, Kolstad A, Hutchings M, Pasanen A, Meriranta L, Niemann CU, et al. MRD-driven treatment with venetoclax-R2 in mantle cell lymphoma: the Nordic Lymphoma Group MCL7 VALERIA trial. Blood Adv. (2023) 8(2):407–415. doi: 10.1182/bloodadvances.2023011920
73. Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. (2017) 545:495–9. doi: 10.1038/nature22396
74. Barkal AA, Weiskopf K, Kao KS, Gordon SR, Rosental B, Yiu YY, et al. Engagement of MHC class I by the inhibitory receptor LILRB1 suppresses macrophages and is a target of cancer immunotherapy. Nat Immunol. (2018) 19:76–84. doi: 10.1038/s41590-017-0004-z
75. Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, Zaro BW, et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature. (2019) 572:392–6. doi: 10.1038/s41586-019-1456-0
76. Eladl E, Tremblay-LeMay R, Rastgoo N, Musani R, Chen W, Liu A, et al. Role of CD47 in hematological Malignancies. J Hematol Oncol. (2020) 13:96. doi: 10.1186/s13045-020-00930-1
77. Piccione EC, Juarez S, Liu J, Tseng S, Ryan CE, Narayanan C, et al. A bispecific antibody targeting CD47 and CD20 selectively binds and eliminates dual antigen expressing lymphoma cells. MAbs. (2015) 7:946–56. doi: 10.1080/19420862.2015.1062192
78. van Bommel PE, He Y, Schepel I, Hendriks M, Wiersma VR, van Ginkel RJ, et al. CD20-selective inhibition of CD47-SIRPalpha "don't eat me" signaling with a bispecific antibody-derivative enhances the anticancer activity of daratumumab, alemtuzumab and obinutuzumab. Oncoimmunology. (2018) 7:e1386361. doi: 10.1080/2162402X.2017.1386361
79. Ma L, Zhu M, Gai J, Li G, Chang Q, Qiao P, et al. Preclinical development of a novel CD47 nanobody with less toxicity and enhanced anti-cancer therapeutic potential. J Nanobiotechnology. (2020) 18:12. doi: 10.1186/s12951-020-0571-2
80. Buatois V, Johnson Z, Salgado-Pires S, Papaioannou A, Hatterer E, Chauchet X, et al. Preclinical development of a bispecific antibody that safely and effectively targets CD19 and CD47 for the treatment of B-cell lymphoma and leukemia. Mol Cancer Ther. (2018) 17:1739–51. doi: 10.1158/1535-7163.MCT-17-1095
81. Hatterer E, Barba L, Noraz N, Daubeuf B, Aubry-Lachainaye JP, von der Weid B, et al. Co-engaging CD47 and CD19 with a bispecific antibody abrogates B-cell receptor/CD19 association leading to impaired B-cell proliferation. MAbs. (2019) 11:322–34. doi: 10.1080/19420862.2018.1558698
82. Wang Y, Ni H, Zhou S, He K, Gao Y, Wu W, et al. Tumor-selective blockade of CD47 signaling with a CD47/PD-L1 bispecific antibody for enhanced anti-tumor activity and limited toxicity. Cancer Immunol Immunother. (2021) 70:365–76. doi: 10.1007/s00262-020-02679-5
83. Wang LC, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res. (2014) 2:154–66. doi: 10.1158/2326-6066.CIR-13-0027
84. Pan K, Farrukh H, Chittepu V, Xu H, Pan CX, Zhu Z. CAR race to cancer immunotherapy: from CAR T, CAR NK to CAR macrophage therapy. J Exp Clin Cancer Res. (2022) 41:119. doi: 10.1186/s13046-022-02327-z
85. Henze AT, Mazzone M. The impact of hypoxia on tumor-associated macrophages. J Clin Invest. (2016) 126:3672–9. doi: 10.1172/JCI84427
86. Morrissey MA, Williamson AP, Steinbach AM, Roberts EW, Kern N, Headley MB, et al. Chimeric antigen receptors that trigger phagocytosis. Elife. (2018) 7:e36688. doi: 10.7554/eLife.36688
87. Klichinsky M, Ruella M, Shestova O, Lu XM, Best A, Zeeman M, et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat Biotechnol. (2020) 38:947–53. doi: 10.1038/s41587-020-0462-y
88. Chen Y, Yu Z, Tan X, Jiang H, Xu Z, Fang Y, et al. CAR-macrophage: A new immunotherapy candidate against solid tumors. BioMed Pharmacother. (2021) 139:111605. doi: 10.1016/j.biopha.2021.111605
89. Zhang W, Liu L, Su H, Liu Q, Shen J, Dai H, et al. Chimeric antigen receptor macrophage therapy for breast tumours mediated by targeting the tumour extracellular matrix. Br J Cancer. (2019) 121:837–45. doi: 10.1038/s41416-019-0578-3
90. Kang M, Lee SH, Kwon M, Byun J, Kim D, Kim C, et al. Nanocomplex-mediated in vivo programming to chimeric antigen receptor-M1 macrophages for cancer therapy. Adv Mater. (2021) 33:e2103258. doi: 10.1002/adma.202103258
91. Schulz D, Severin Y, Zanotelli VRT, Bodenmiller B. In-depth characterization of monocyte-derived macrophages using a mass cytometry-based phagocytosis assay. Sci Rep. (2019) 9:1925. doi: 10.1038/s41598-018-38127-9
92. Zhang L, Tian L, Dai X, Yu H, Wang J, Lei A, et al. Pluripotent stem cell-derived CAR-macrophage cells with antigen-dependent anti-cancer cell functions. J Hematol Oncol. (2020) 13:153. doi: 10.1186/s13045-020-00983-2
93. Al Zaki A, McCurry D, Strati P. CAR T-cells and macrophages in large B-cell lymphoma: impact on toxicity and efficacy. Leuk Lymphoma. (2023) 64:808–15. doi: 10.1080/10428194.2023.2185090
94. Yan Z, Li L, Fu D, Wu W, Qiao N, Huang Y, et al. Immunosuppressive tumor microenvironment contributes to tumor progression in diffuse large B-cell lymphoma upon anti-CD19 chimeric antigen receptor T therapy. Front Med. (2023) 17:699–713. doi: 10.1007/s11684-022-0972-8
Keywords: mantle cell lymphoma, macrophages, tumor microenvironment, immunotherapy, car-t, CAR-M, drug resistance
Citation: Nylund P, Nikkarinen A, Ek S and Glimelius I (2024) Empowering macrophages: the cancer fighters within the tumour microenvironment in mantle cell lymphoma. Front. Immunol. 15:1373269. doi: 10.3389/fimmu.2024.1373269
Received: 19 January 2024; Accepted: 29 February 2024;
Published: 19 March 2024.
Edited by:
Jeanfrancois Rossi, University of Montpellier 1, FranceReviewed by:
Walter Hanel, The Ohio State University, United StatesCopyright © 2024 Nylund, Nikkarinen, Ek and Glimelius. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ingrid Glimelius, aW5ncmlkLmdsaW1lbGl1c0BpZ3AudXUuc2U=