 Agnieszka Piekarska
Agnieszka Piekarska Katarzyna Pawelec2
Katarzyna Pawelec2 Anna Szmigielska-Kapłon
Anna Szmigielska-Kapłon Marek Ussowicz
Marek Ussowicz- 1Department of Hematology and Transplantology, Medical University of Gdansk, Gdansk, Poland
- 2Department of Oncology, Pediatric Hematology, Clinical Transplantology and Pediatrics, Medical University of Warsaw, Warsaw, Poland
- 3Department of Hematology, Medical University of Lodz, Lodz, Poland
- 4Department of Pediatric Bone Marrow Transplantation, Oncology and Hematology, Wroclaw Medical University, Wroclaw, Poland
Acquired aplastic anemia (AA) is an immune-mediated bone marrow (BM) failure where marrow disruption is driven by a cytotoxic T-cell–mediated autoimmune attack against hematopoietic stem cells. The key diagnostic challenge in children, but also in adults, is to exclude the possible underlying congenital condition and myelodysplasia. The choice of treatment options, either allogeneic hematopoietic cell transplantation (alloHCT) or immunosuppressive therapy (IST), depends on the patient’s age, comorbidities, and access to a suitable donor and effective therapeutic agents. Since 2022, horse antithymocyte globulin (hATG) has been available again in Europe and is recommended for IST as a more effective option than rabbit ATG. Therefore, an update on immunosuppressive strategies is warranted. Despite an improved response to the new immunosuppression protocols with hATG and eltrombopag, some patients are not cured or remain at risk of aplasia relapse or clonal evolution and require postponed alloHCT. The transplantation field has evolved, becoming safer and more accessible. Upfront alloHCT from unrelated donors is becoming a tempting option. With the use of posttransplant cyclophosphamide, haploidentical HCT offers promising outcomes also in AA. In this paper, we present the state of the art in the management of severe AA for pediatric and adult patients based on the available guidelines and recently published studies.
1 Introduction
Bone marrow failure (BMF) syndromes are a consequence of inherited or acquired conditions, resulting in hematopoiesis failure. The hematopoietic stem cell (HSC) compartment in BMF is disrupted either by constitutional mutations of genes involved in hematopoiesis, or through a direct destruction of HSCs by cytotoxic agents or an immune-mediated attack.
Aplastic anemia (AA) is a hematological entity associated with bone marrow (BM) aplasia and hematopoiesis failure, which was first described at the turn of the 19th and 20th centuries (1). The historical term is slightly confusing, as most patients experience pancytopenia in addition to anemia, while “aplastic” defines the inability of the BM to effectively produce blood cells as a result of various mechanisms. The key diagnostic challenge is to identify the underlying condition involved in aplasia in individual patients to choose the optimal treatment option: either allogeneic hematopoietic cell transplantation (alloHCT) or immunosuppressive therapy (IST).
The diagnostic hallmark of AA is the presence of at least two-lineage cytopenia with hemoglobin levels lower than 10 g/dL, platelet count lower than 50 × 109/L, or neutrophil count lower than 1.5 × 109/L (2). The modified Camitta criteria classified AA into three categories on the basis of BM cellularity, an absolute neutrophil count (ANC), platelet count, and reticulocyte count (3). Severe AA (SAA) is characterized by pancytopenia with ANC lower than 0.5 × 109/L, platelet count lower than 20 × 109/L, reticulocyte count lower than 20 × 109/L (manual count) or 60 × 109/L (using an automated analyzer), and less than 25% of normal cellularity in BM biopsy. Very severe AA (vSAA) is defined by the same criteria except ANC of less than 0.2 × 109/L. Patients not fulfilling the SAA/vSAA criteria with BM cellularity are classified as having nonsevere AA, but many patients in this category have other BMF syndromes that require a diagnosis.
2 Pathophysiology and clonal evolution
In short, AA is an acquired immune-mediated BMF syndrome where marrow disruption is driven by a cytotoxic T-cell–mediated autoimmune attack against HSCs sustained by type I interferons that polarize the immune system toward T-helper 1 responses in early phases and then toward T-helper 17 and effector memory CD8+ T-cell in late stage and severe disease (4). The immune-mediated factors play a central role in the pathogenesis of hepatitis-associated AA – a distinct variant of AA with a prevalence of about 5%, in which pancytopenia appears 2 to 3 months after acute hepatitis and shows quite good response to IST of 55% to 70% in patients who lack an optimal HSC donor (5–8).
Among multiple studied factors, the dysfunction of BM environment and mesenchymal stromal cell dysregulation might contribute to the damage in the stem-cell niche (9). Immune pressure from T-cell–mediated mechanisms leads to somatic gene alterations in HSCs, resulting in immune evasion (10). The telomere length in AA patients was reported to be significantly shorter than in age-matched controls (11, 12). Approximately 35% of patients with AA show telomere length shortening in granulocytes and mononuclear cells, which results in HSC exhaustion and is associated with the risk of relapse, clonal evolution, and worse overall survival (OS) (13, 14). In turn, telomere shortening due to mitotic demand in the HSC compartment can destabilize the genome by chromosome rearrangement and aneuploidy, which negatively affects therapy response and survival (14–16).
Fattizzo et al. reported a high prevalence of paroxysmal nocturnal hemoglobinuria (PNH) clones caused by somatic PIGA mutations in patients with AA and other hematological disorders (17). Next-generation sequencing (NGS) allowed the identification of somatic mutations responsible for clonality in AA even in small subclones. Moreover, different mutations in the same PIGA gene in a single patient supports the hypothesis that the mutations are acquired independently in different clones, promoting their expansion and evolution. The carriers of somatic PIGA and BCOR/BCORL1 mutations show a lower risk of AA transformation to myelodysplastic syndrome (MDS), whereas myeloid driver lesions such as DNMT3A and ASXL1 mutations characterize the group with a higher risk of malignancy (18). While most patients with AA have normal BM karyotype, trisomy 8 and del(13q) were reported and were linked to a more favorable response to therapy (19, 20). Uniparental disomy (UPD) of chromosome 6p (a locus containing the human leukocyte [HLA] cluster) can be identified in up to 11% of AA patients and is highly specific for AA, but not for other BMF syndromes (21, 22). In contrast, the presence of monosomy 7 in BMF/SAA suggests the diagnosis of MDS and a higher probability of constitutional disorders, such as GATA2 deficiency, SAMD9/SAMD9L-related disorders, or telomer biology disorders (23, 24). In adults with AA, cytogenetic abnormalities at diagnosis are observed in 12% to 15% of patients, with the following findings reported in the literature: trisomies (+8, +6, +15), aberrations of chromosomes 7 and 13, UPD(6p), and del(5q) (25, 26).
Poor-risk mutation carriers with AA have a 40% higher risk of clonal evolution to MDS or acute myeloid leukemia (AML) compared with patients with PIGA, BCOR, and BCORL1 mutations (27–30). Apart from the risk of clonal evolution, adverse effects in terms of response to IST, risk of relapse, and inferior OS were observed in patients without a PNH clone, with poor-risk mutations (ASXL1, TP53, RUNX1, DNMT3A), and shortened telomeres (31, 32). However, a detailed NGS-based analysis of the patients participating in the RACE study showed fluctuations in the variant allele frequency of detected mutations, and no mutation correlated with response to IST or OS (33).
3 Diagnosis
The diagnosis of AA must include the differentiation between the various pathologies resulting in pancytopenia and BMF syndromes. In histopathological BM assessment, hypoplastic MDS, acute leukemia, T-cell large granular lymphocyte leukemia, cancer metastases, and reticulin fibrosis must be excluded and BM cellularity should be clearly described. Diagnostic BM workup includes cytogenetic examination with fluorescent in situ hybridization (FISH) (1, 30, 34, 35). Immunophenotyping from peripheral blood (PB) directed to detect PNH is obligatory as PNH clone is observed in almost half of the patients with AA and has clinical implications (31, 36, 37). Evaluation in the direction of hepatitis-derived marrow failure including classic hepatitis viruses but also parvovirus B19, CMV, and EBV is also recommended at diagnosis (7).
In the differential diagnosis of AA, other conditions leading to BMF must be considered. Direct BM damage can be caused by radiation, chemotherapy, or exposure to toxic compounds such as benzene, and various drugs including chloramphenicol, gold compounds, nonsteroid anti-inflammatory drugs, and anticonvulsants (38, 39).
There is a high prevalence of inherited BMF syndromes in children and young adults. Constitutional BMF can be a manifestation of germline defect resulting from a loss of function of one or more of the genes involved in DNA repair (Fanconi anemia), telomere maintenance (dyskeratosis congenita and other telomeropathies), and deficiencies leading to hematopoiesis regulation (GATA2, CTLA4) (1). Genomic screening for constitutional syndromes and telomere length measurement (if available) should be done, and the presence of somatic mutation should be evaluated (40).
Hypocellular MDS can mimic the presentation of AA and evolve from various BMF syndromes, and the differentiation between these entities can pose a practical challenge. In a recent study from the United States, including 732 children and adult HCT recipients clinically diagnosed with acquired AA, a comprehensive genetic analysis using NGS identified 48 patients (6.6%) with unrecognized inherited BMF syndromes who had 62 germline pathogenic variants in 22 genes associated with inherited BMF syndromes (41). Moreover, patients with unrecognized inherited BMF syndromes had worse survival because of treatment-related mortality from organ failure.
Adults are less likely than children to have constitutional BMF, but the possibility should not be neglected because incomplete gene penetrance and variable expressivity do not always lead to physical abnormalities and might manifest solely with BMF. However, usually a family history is suggestive. Considering better access to NGS, a panel including ASXL1, DNMT3A, BCOR, BCORL1, PIGA, RUNX1, TP53 and GATA2 genes can be done.
To sum up, the diagnostic workup recommendations for SAA were presented by the Severe Aplastic Anemia Working Party of the European Society for Blood and Marrow Transplantation (EBMT) in 2013 and the North American Pediatric Aplastic Anemia Consortium in 2021 (35, 42). Due to a wide range of diagnostic recommendations, only key points should be mentioned. In addition to standard laboratory testing, as well as bone marrow aspirate and biopsy, patients should be evaluated for bone marrow karyotype, FISH for abnormalities of chromosomes 7, 8, 5, and del20q, presence of the PNH clone, chromosomal breakage and/or genetic Fanconi anemia testing, and telomere length (with flow FISH techniques). The availability of NGS testing prompts early evaluation for germline mutations in the GATA2, SAMD9/9L, ERCC6L2, and EVI1 genes, telomere biology disorders/dyskeratosis congenita, Fanconi anemia, ribosomopathies (Shwachman-Diamond syndrome, Diamond-Blackfan anemia) when appropriate, but especially in children and young adults or patients with atypical or nonhematological manifestations. Although not used in current clinical algorithms, the NGS panel to detect the presence of somatic variants (consisting typically of PIGA, DNMT3A, ASXL2, TET2, BCOR, BCORL, NPM1, ETV6, TP53, but not limited to this list) might be integrated in the nearest future but their clinical value has to be validated (43). The growing knowledge on monogenic defects resulting in one- or multilineage cytopenias and rapid progress in this field warrant clinical alertness and unhindered referral for genetic testing (44). Patients should undergo the work-up for autoimmune cytopenias with ANA and infectious screening.
4 History of AA therapy
Initially, the treatment of patients with AA consisted of supportive transfusions and the administration of steroids and vitamins. Historically, the first effective pharmacotherapy of AA was developed in the 1960s and was based on androgen administration (45). Thus, androgen therapy has established its place in the therapy of AA, but was associated with multiple adverse effects, such as virilization, jaundice and hepatotoxicity, hyperlipidemia, and behavioral effects with no improvement in mortality (46, 47). Although alloHCT and IST are highly effective in AA, androgens continue to be an option, even in the frontline, for transplant ineligible patients with no access to modern IST (46).
In 1970s, alloHCT was proved to be more effective than conventional therapies, thus becoming the cornerstone of the curative treatment of AA (48, 49). Currently, alloHCT is the widely accepted and recommended first-line therapy in children with matched sibling donors (MSDs), and fit young adults. The increasing availability of matched unrelated donors (MUDs) has led to the concept of upfront alloHCT from MUDs, but the time to arranging transplantation in this situation is longer and can reach 2 to 3 months.
In the early 1980s, another therapeutical option was offered for patients with AA. The phenomenon of alloHCT rejection with autologous reconstitution resulting in hematological remission of AA warranted studies of immunosuppression. The antilymphocyte globulin (ALG) was studied in different clinical settings (with or without alloHCT or androgens). For ALG alone, the treatment showed the response rates ranging from 30% to 40% (50, 51). Considering the immune origin of the disorder, intensive IST with antithymocyte globulin (ATG) was also evaluated (52, 53). Antithymocyte globulin contains polyclonal antibodies directed against different T-cell antigens including CD2, CD3, CD4, CD8, CD11, CD18, CD25, and others. Treatment with ATG results in the anergy of T-lymphocyte cells and induces their depletion via antibody-dependent cell-mediated cytotoxicity. Complement-dependent lysis is an additional mechanism resulting in T-cell lysis. The mechanisms of action of ATG were reviewed by Mohty et al. (54). Since 2022, horse ATG (hATG) has been available again in Europe and is recommended for IST. Therefore, the update of immunosuppressive strategies is warranted.
The next step towards improvement of the outcomes was made with implementation of eltrombopag (ELT), an oral thrombopoietin receptor agonist (TPO-RA) to the IST protocols. It is licensed in the therapy of immune thrombocytopenia due to megakaryocyte stimulating properties. It can also lead to clinically significant improvements in blood counts in nearly half of the patients with AA refractory to IST, increasing BM cellularity, CD34+ count, and progenitor cells through a direct effect on marrow cells (55–57). The mechanism of ELT action in AA is improving signaling via TPO receptor (c-mpl) expressed on the surface of HSCs and progenitor cells irrespective of interferon-γ inflammatory microenvironment in BM, by bypassing inhibition caused by interferon-γ competitively binding to the same receptor (58). In fact, about 45% of patients experienced a hematological response to ELT alone, having not only higher platelet counts but also significant improvements in hemoglobin levels and ANC. Townsley et al. evaluated the efficacy of hATG and CsA with addition of ELT in 92 patients in open-label pivotal study (55). Data were compared with results from a historical group of patients receiving solely hATG with CsA. Overall response at 3 months was 74% and 80% at 6 months in comparison with results obtained in a historical cohort of patients (66% at 6 months, P <0.001).
Current recommendations on the treatment of SAA in children and adolescents were published by the European Working Group of Myelodysplastic Syndrome (MDS) and Severe Aplastic Anemia (SAA) (EWOG-MDS/SAA) in 2023 and are available online (59). For adult patients, the recommendations of the EBMT are most often used (60). There are also national UK guidelines available (61).
5 Nontransplant therapeutic options in AA
5.1 Evaluation of pediatric recommendations
Thanks to IST combined with hATG and cyclosporine A (CsA), the 5-year OS of patients under 16 years of age increased from 50% before 1990 to approximately 80% today (62–64). In a Polish study, the 10-year OS rate reached 78% in children treated mainly with hATG (65). According to a German study, children with vSAA had a higher rate of complete response (CR) than children with SAA (68% vs 45%; P = 0.009). They also showed better survival (93% vs 81%; P < 0.001) (66).
Different ATG preparations were used in IST with various biological effects. Lymphoglobulin® (hATG, Genzyme) had been widely used for IST in Europe, but it was withdrawn from the market in 2007. In the absence of licensed hATG, rabbit ATG (rATG) sera were used. However, the effectiveness of rATG treatment was worse compared with hATG. In a randomized study by Scheinberg et al., the 3-year OS after rATG was inferior to that of hATG (76% vs 96%) (67). Similarly, the EBMT Severe Aplastic Anemia Working Party (EBMT-SAA-WP) reported that the 2-year OS after r-ATG was only 68% vs 86% for hATG (68). Some studies (mainly retrospective ones) showed similar effectiveness of hATG and rATG (68–74). Chang et al. reported the 4-year OS of 75% without statistical difference between patients treated with hATG (n = 29) and rATG (n = 33) (75). In contrast, the EWOG-SAA-2010 interim analysis showed a lower overall response rate (ORR) to IST in patients treated with rATG compared with hATG (22% vs 42%, P = 0.03) but without differences in OS (88% vs 93%) (76). Similarly, Pawelec et al. demonstrated the 3-year OS of 78% and the 10-year survival of 67% for hATG and rATG, respectively (77). However, in a meta-analysis by Hayakawa et al., the ORR was significantly higher for hATG vs rATG (risk ratio [RR], 1.27; 95% CI, 1.05-1.54; P = 0.015) (78). In another meta-analysis by Yang et al., the beneficial effect of hATG on the ORR at 6 months in treatment-naïve patients with AA was also shown (RR, 1.86) with comparable safety profiles (79). A summarized comparison of studies with hATG and rATG in IST of AA patients is shown in Table 1.

Table 1 Studies comparing hATG and rATG for the treatment of aplastic anemia.
A third source of ATG – porcine serum (pATG) – was studied in a few Chinese trials. In a study by Bing et al., the ORR was 83.3% (CR, 54.2%; partial response [PR], 29.2%) with a median time of 90 days (range, 23-380), and 10.4% of patients died of infection within 30 days (80). A pediatric study by Zhu et al. revealed a similar therapeutic effect and safety of pATG or rATG combined with CsA (24-month ORR, 81.6% vs 78.6%) as first-line therapy for pediatric patients with AA (81). In retrospective studies by Liu et al. and Chen et al., no significant differences in OS between pATG and rATG were observed (82, 83).
Factors increasing the ORR to IST in children include severity (ORR better in vSAA than in SAA), younger age, higher pretreatment reticulocyte count and lymphocyte count, male sex, and leukocyte count (84, 85). In a meta-analysis by Gu et al., the earlier start of IST, higher neutrophil count at IST, and rate of lymphocyte clearance during IST were associated with improved response rates (86).
The addition of granulocyte-colony-stimulating factor (G-CSF) may reduce the rate of early infectious episodes and days of hospitalization in vSAA patients, but has no effect on the response rate to IST and survival (87, 88). The safety of G-CSF after IST might be compromised. Japanese studies from 1998 and 2002 suggested a close relationship between the long-term use of G-CSF and secondary MDS in nonresponders to IST (89, 90). In a study on 802 SAA patients, a multivariate analysis revealed increasing age, higher disease severity, and increasing number of G-CSF therapy days to be risk factors for MDS/AML transformation. However, the prolonged G-CSF administration in these studies was typical for patients with suboptimal response, and this factor can be associated with other, nonimmune mediated mechanisms of SAA pathogenesis and, in turn, with a higher risk of clonal disease (91). A meta-analysis by Ding et al. and a follow-up study of a randomized, prospective trial by Tichelli et al. did not confirm any impact of G-CSF on the risk of clonal evolution in patients with SAA (92, 93). Regardless of the use of G-CSF, SAA patients treated with IST are at risk of several late malignant and nonmalignant complications. There is a significant risk of relapse (10% at 10 years) and 8%–16% risk of clonal disease (MDS/AML), which does not reach plateau (64, 85, 93).
In the pediatric EWOG strategy for SAA treatment if hATG is not available, rATG may be used as an alternative therapy. Currently, G-CSF should only be used in patients with severe neutropenia (ANC <0.5 × 109/L) and prolonged administration of G-CSF should be avoided. Children not responding to the IST therapy or relapsing afterwards must seek alloHCT from MUDs or alternative donors (ADs) (59). According to the Pediatric Haemato-Oncology Italian Association, patients without MUD can receive the second course of IST (94). A switch of serotherapy from hATG to rATG and vice-versa should be considered, but the expected ORR is only 30% after failure of the first IST and 60% after relapse (95). The remaining options include transplantation from ADs and immunotherapy with alemtuzumab and ELT. The overview of the SAA management in the pediatric population based on the current recommendations is presented in Figure 1.
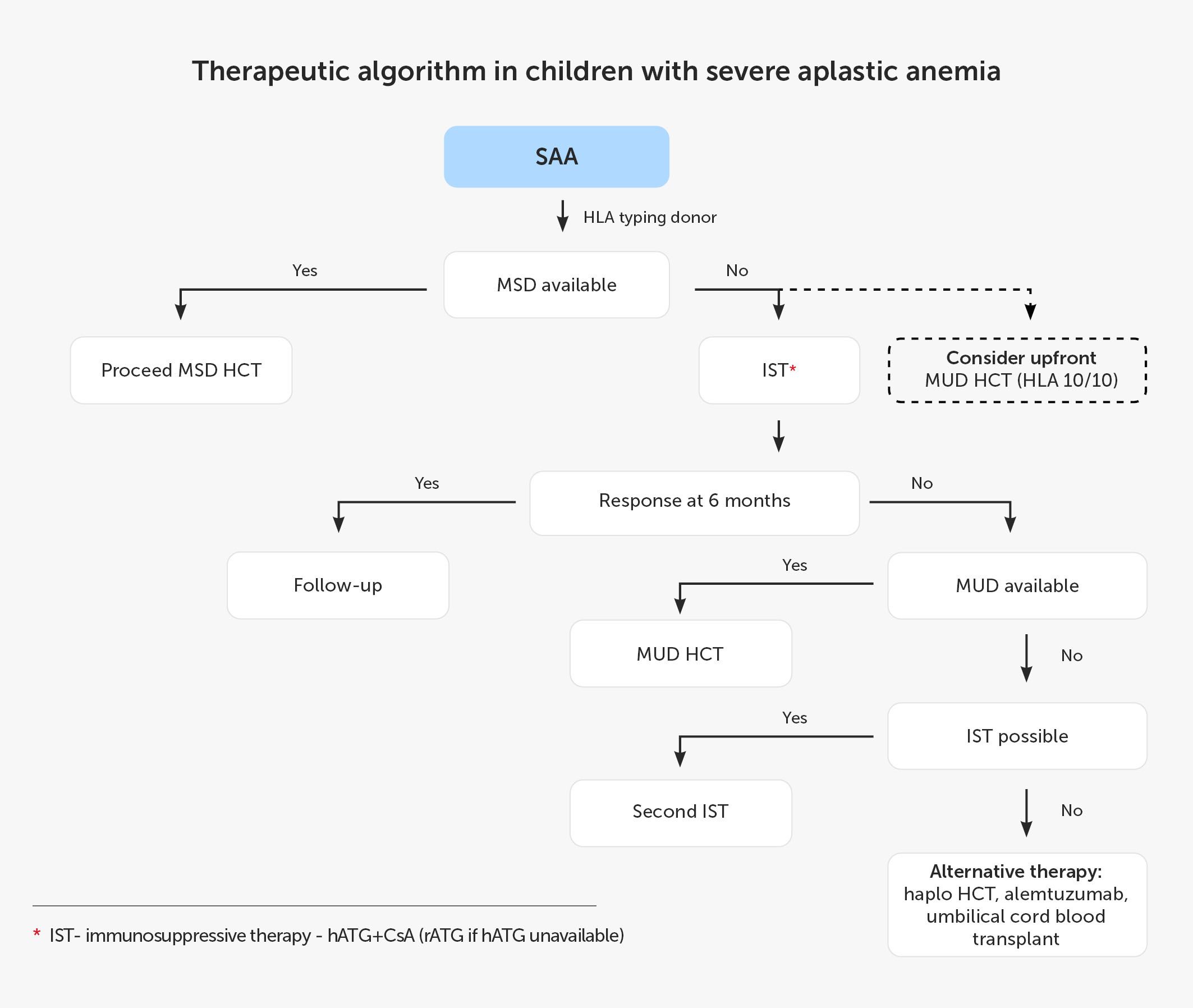
Figure 1 Therapeutic algorithm in children with severe aplastic anemia. CsA, cyclosporin A; hATG, horse anti-thymocyte globulin; HCT, hematopoietic cell transplantation; Haplo, haploidentical donor; HLA, human leukocyte antigen; IST, immunosuppressive treatment; MSD, 8/8 matched sibling donor; MUD, 10/10 matched unrelated donor; rATG, rabbit anti-thymocyte globulin; SAA, severe aplastic anemia.
5.2 Evaluation of adult recommendations
According to EBMT recommendations, patients younger than 40 years should undergo alloHCT from MSD. Patients above this age limit and patients without MSD are candidates for IST (60). Best outcomes are reported with hATG combined with CsA and ELT, and this triple combination is currently recommended in adult patients. The current treatment schedule for patients with SAA based on EBMT recommendations and data from the available literature is shown in Figure 2.
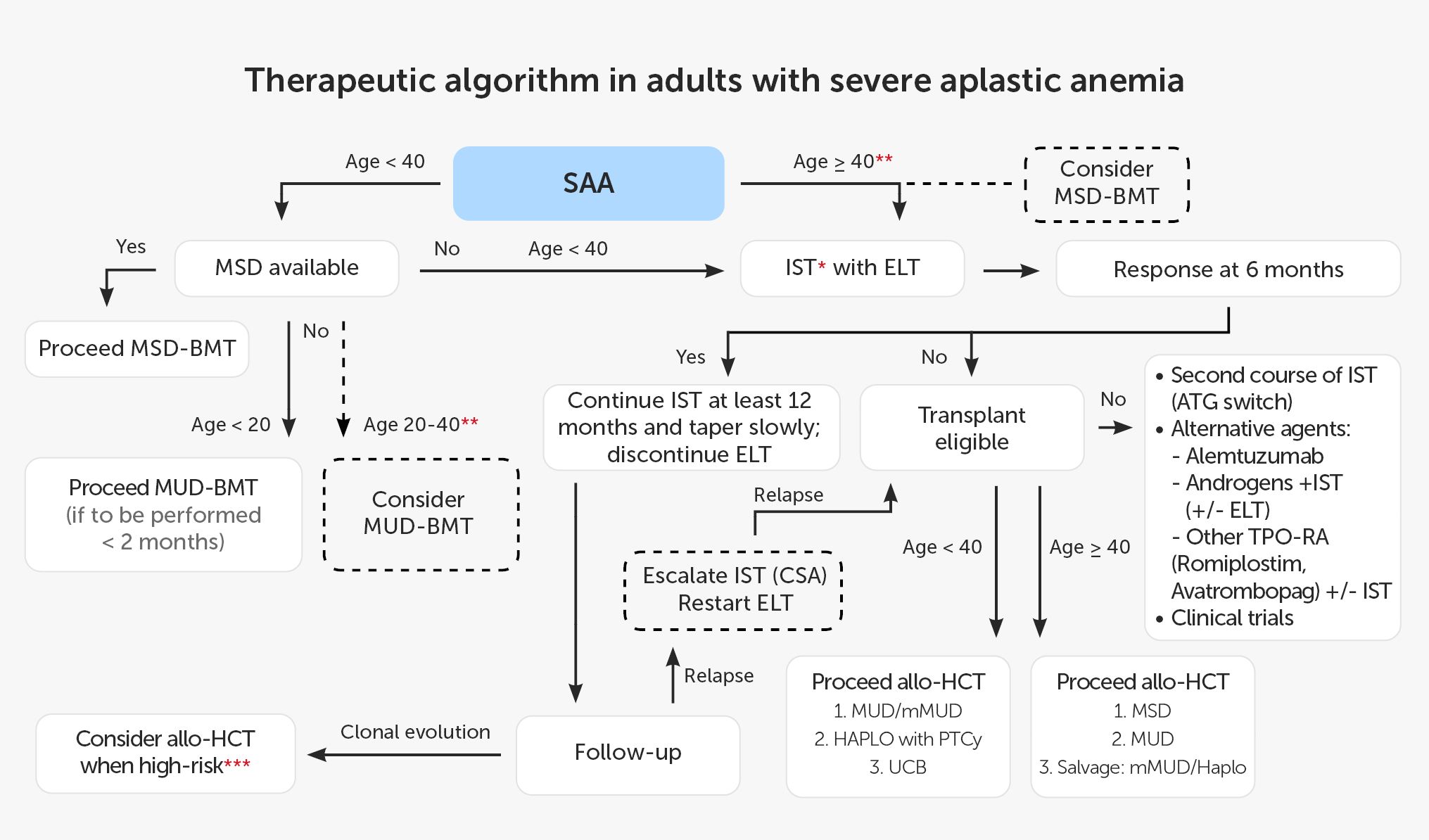
Figure 2 Therapeutic algorithm in adults with severe aplastic anemia. * IST: hATG + CsA (rATG if hATG not available; without ELT if unavailable). ** present predictors of poor response to IST (e.g. age, low reticulocyte count, low lymphocyte count, vSAA, no PNH clone). *** e.g. del7, complex karyotype, dysplasia, increased percentage of blasts). allo-HCT, allogeneic hematopoietic cell transplantation; ATG, anti-thymocyte globulin; BMT, bone marrow transplantation; CsA, cyclosporin A; ELT,eltrombopag; HAPLO, haploidentical donor; hATG, horse anti-thymocyte globulin; IST, immunosuppressive treatment; MSD, 8/8 matched sibling donor; MUD, 10/10 matched unrelated donor; mMUD, 9/10 mismatched unrelated donor; PNH, paroxysmal nocturnal hemoglobinuria; PTCy, post-transplant cyclophosphamide; rATG, rabbit anti-thymocyte globulin; SAA, severe aplastic anemia; TPO-RA, thrombopoietin receptor agonist; UCB, umbilical cord blood;vSAA, very SAA.
A standard total dose of hATG (ATGAM®) is 160 mg/kg. Most studies evaluated the dose given in a 4-day schedule (40 mg/kg daily for 4 days). The dose was evaluated in 2 prospective randomized studies by Scheinberg et al.: one study evaluating the use of hATG plus CsA with or without sirolimus and the other comparing hATG with rATG (67, 96). Cyclosporine A was given orally at an initial dose of 5 mg/kg for several months (34, 61). In earlier studies, ATGAM® was given at lower daily doses for 8 or 10 days, but at the same total dose of 160 mg/kg. Champlin et al. evaluated hATG given for 8 days (20 mg/kg daily) in monotherapy or with androgens (50). Doney et al. used hATG for 10 days in combination with oxymetholone given orally for 3 months (97). In both studies, CsA was not incorporated in IST protocols, so it is impossible to compare the outcomes with those obtained in modern studies. A lower total dose of hATG was evaluated in a retrospective trial by Scott et al. Two schedules of IST based on CsA and hATG were compared: one group (n = 14) received hATG at a standard dose of 40 mg/kg daily for 4 days, and the other group (n = 17) received 15 mg/kg daily for 5 days (98). The efficacy of both treatment regimens was comparable, although a low number of patients and the retrospective trial design should be considered.
Best outcomes were obtained when hATG was combined with CsA, resulting in 60%-65% of responses, including 10% of CR (96, 99). The addition of other immunosuppressive agents, including sirolimus and mycophenolate mofetil (MMF), did not improve the response rate in prospective randomized trials (52, 96, 100). A retrospective multicenter real-life study included all patients diagnosed with AA between 2000 and 2011 in Sweden and treated in the first line with ATG plus CsA. The study evaluated 158 patients, 128 of whom received rATG. Response to treatment was reported in 47% of patients. The 5-year OS was significantly higher in patients achieving PR or CR as compared with nonresponders (89% vs 52%, P <0.001). The 5-year OS was 77.3% and 58.5% for SAA and vSAA, respectively (P = 0.002). The age of patients did not influence the efficacy of treatment (74). The EBMT performed a retrospective study evaluating the efficacy of rATG in Europe and Asia. The study included 955 patients receiving the first-line treatment with ATG and CsA between 2001 and 2012. The ORR was 37% at 3 months and 52% at 6 months, with a subsequent increase up to 65% at 12 months after treatment. The OS at 10 years was 70% (101).
The prospective randomized trial by Scheinberg et al. revealed superior efficacy of hATG vs rATG (67). A total of 120 patients were evaluated, 60 patients per group. The ORR at 3 months of IST was 62% in the hATG group and 33% in the rATG group (P = 0.0017). The response evaluated at 6 months was also superior for hATG compared with rATG (68% vs 37%; P <0.001). The 3-year OS was also significantly higher for hATG than for rATG (96% vs 76%, P = 0.04). Real-world data from France concerning the efficacy of hATG in SAA were also encouraging (102).
The safety issues concerning the infusion of either hATG or rATG are similar, as the most common complications include skin reactions, headache, chills, and fever. In rare cases, anaphylactic reactions or systemic inflammatory reactions due to cytokine release may occur such as cytokine release syndrome or systemic inflammatory response syndrome. To reduce the incidence of immediate adverse reactions, corticosteroids, antipyretics, and antihistamines are recommended (60). To reduce the risk of infusion-related side effects, the preinfusion skin testing should be considered, especially in patients with an increased risk of anaphylaxis (e.g. patients with atopic eczema). However, the negative results of the skin testing do not exclude anaphylactic reactions or development of serum sickness. To reduce the risk of thrombophlebitis, ATG administration via central line is recommended. The prolonged infusion time (minimum 12 hours) is linked to better infusion tolerance (60). Several days after ATG infusion, serum sickness may occur including rash, arthralgia, and fever. The symptoms respond well to steroid treatment. A temporary increase in liver function tests may be observed after ATG administration. The most frequent severe adverse events were infections and hemorrhage (67, 96, 102).
Best response to IST should be evaluated 4 to 6 months after hATG administration (1, 60, 61, 103). The criteria of CR used in recent clinical trials are as follows: ANC >1 × 109/L, hemoglobin >10 g/dL, and platelet count >100 × 109/L (96). Most patients achieve PR when they no longer meet criteria for severe type of AA and become transfusion independent but do not meet CR criteria (ANC >0.5 × 109/L, Hb >8 g/dL, platelet count >20 × 109/L). Long-term responses to IST are observed in 60% of patients. However, some patients require more prolonged CsA treatment exceeding the minimal recommended 12 months and continuation of CsA for a longer time with subsequent slow tapering to minimize the risk of relapse (34). In 10% to 30% of patients, SAA may recur after initial response (1, 60, 61, 103). In such cases, alloHCT should be performed if possible or an alternative strategy with repeating IST might be considered, depending on the individual patient risk assessment and donor access. The second IST pathway should incorporate the option of the change of ATG origin or the use of alemtuzumab and addition of TPO-RA; however, insufficient stem cell reserve might limit the efficacy of IST (34, 52, 60, 61, 95, 104).
5.3 Thrombopoietin-mimetic therapy and immunosuppression
In adult patients, ELT proved to be an effective agent in patients with AA refractory to previous IST. Desmond et al. evaluated 43 patients unresponsive to IST and observed long-lasting responses in 40% of cases (56). In the prospective randomized RACE trial conducted by the EBMT-SAA-WP, 101 patients in the hATG + CsA arm and 96 patients in the hATG + CsA + ELT arm were evaluated (33). The median patient age was 53 years, and only 9 patients were children. The dose of ELT was 150 mg daily from day 14. The results were in favor of ELT addition in the first-line treatment with a significantly higher CR in the group receiving ELT at 3 months (22% vs 10%; P = 0.01) and a higher ORR at 3 months (59% vs 31%) and at 6 months (68% vs 41%). The ELT group responded faster to treatment and had no additional toxicity, including hepatotoxicity. The 2-year OS was comparable in both arms. The incidence of somatic mutations was similar in both groups and increased at 6 months after the administration of hATG, with clonal evolution observed in 10% to 15% of cases (33). In a Chinese study, the beneficial effect of prolonged ELT administration was demonstrated in patients without CR at 6 months (105). Based on data from these pivotal studies, the current standard approach to the therapy of SAA in adults includes the combination of IST with hATG with addition of ELT.
However, the effects of ELT on pediatric patients with SAA are less well documented and remain controversial, and the evidence is conflicting. Among the pivotal study population, 21% of patients were children aged 3-18 years, but in contrast to adults, in the pediatric sub-cohort analysis by Groarke et al., in children treated with ELT plus IST there was no significant difference in either the ORR or CR rate at 6 months (ORR: 70% in the ELT group and 72% in the historical group; P = 0.78) (57). In a study by Fang et al., better responses at 6 months were seen in children treated with ELT plus IST (CR: 50% vs 17.9%, P < 0.05; ORR: 94.4% vs 69.2%; P < 0.05) (106). Goronkova et al. reported a similar ORR in the ELT + IST and IST groups (65% vs 53%; P = 0.218). However, the CR rate was significantly higher in the ELT + IST group (31% vs 12%; P = 0.027) (107). Of note, the greatest benefit from ELT combined with IST was observed in patients with SAA with ORR 89% vs 57% (P = .028) but not in those with vSAA (52% vs 50%; P = .902). Other studies conducted in a small number of patients confirmed more rapid hematological responses and CR rates but failed to show ORR and survival advantage in the pediatric population (108). As there are no sufficient data to support the advantage of combined treatment of IST with ELT, the EWOG does not generally recommend this therapy in children (59).
Apart from ELT, also other TPO-RA are investigated in the SAA setting (109). Romiplostim, an Fc-peptide fusion protein administered subcutaneously, activates intracellular transcriptional pathways via the TPO receptor (110). In the dose-finding and long-term treatment phase 2 trial in SAA patients refractory to IST, 10 patients (30%) maintained a platelet response at 2 and 3 years, including 9 and 5 patients with erythroid and neutrophil response, respectively, without significant toxicity. A romiplostim dose of 10 μg/kg once weekly was suggested as a recommended starting dose (111). In a phase 2/3 study in 31 patients with SAA refractory to IST, a romiplostim dose was titrated by platelet response from 10 μg/kg once weekly up to a maximum of 20 μg/kg, and any hematological response at week 27 and 53 was 84% and 81%, respectively (112). In a retrospective study of 10 patients switched to romiplostim (20 μg/kg/week) after failure of the maximum ELT dose, 7 patients achieved neutrophil, erythroid, or platelet response, including one CR at 12 months (113). There is an ongoing phase 2 clinical trial in patients with SAA (either treatment-naïve or relapsed/refractory) with avatrombopag, a second-generation oral TPO-RA, administered from day 1 until day 180 at a dose of 60 mg/day, with dose adjustment according to platelet count, in combination with IST (hATG and CsA) or without IST (114).
5.4 Other nontransplant options in AA
Androgens have been used in AA for decades. Their mechanism of action in BMF is complex. Androgens induce erythropoiesis by increasing erythropoietin secretion and response to erythropoietin; they also stimulate the expansion of hematopoietic progenitors (50). Androgens also act by ameliorating telomerase activity in HSCs, resulting the in prevention of telomere attrition both in acquired SAA and in constitutional BMF syndromes (15, 115, 116). Some androgens have additional immunosuppressive mechanisms. For example, danazol inhibits interleukin-1 and TNF-α secretion by human monocytes in a dose-dependent manner (117). Several studies, including one randomized trial, showed the benefit of adding androgens to IST (34, 51, 118). Combined therapy with an immunosuppressive agent, ELT, and androgens in refractory SAA was also reported, with an ORR of 42% (119). The recommended doses for androgens in SAA are 2.5 mg/kg/d for oxymetholone and methenolone and 1 mg/kg/d for methandrostenolone and norethandrolone (46, 120).
Alemtuzumab (a humanized monoclonal antibody directed against CD52 protein) is one of the lymphocytotoxic serotherapeutic options for the treatment of autoimmune diseases. However, in SAA the best results for alemtuzumab were obtained in the relapse and refractory settings, with an ORR of 56% and a 3-year OS of 86% (121).
6 Hematopoietic cell transplantation
6.1 Challenges in pediatric HCT
AlloHCT in AA is associated with several challenges that have been overcome during the last 40 years. The major concern in AA was the high risk of graft rejection, which was related to the presence of stem cell–eliminating T-lymphocytes and high sensitization towards HLA antigens in heavily transfused patients. The benefit of nonmyeloablative but intensive immunosuppressive transplant protocols with hATG and cyclophosphamide (Cy) was proved in transplantations from MSD in the 1990s (122, 123). Similarly, Cy with rATG-based serotherapy showed very good outcomes with high engraftment rates and mortality associated mostly with invasive fungal infections (124). Improved survival after alloHCT from MUDs was associated with improvements in donor typing, less toxic conditioning regimens with omission of total body irradiation (TBI), and use of leukodepleted blood products (125–127). Advances in supportive care with the availability of new antifungals and effective management of viral infections were pivotal in the reduction of transplant-related mortality.
One of the unresolved controversies in the field of pediatric AA is the problem of upfront HCT from MUDs. Some of the previous evidence supports the benefit of an upfront MUD-HCT in SAA, but randomized clinical trials are needed to confirm such an approach. The analysis of studies conducted between 1998 and 2019 that compared the outcomes of alloHCT and IST as an upfront therapy in SAA showed improved OS in upfront transplant recipients (128). In another study, the outcomes of upfront MUD-HCT in pediatric SAA were similar to those observed for MSD-HCT and superior to those for IST and MUD-HCT performed after IST failure (129). Conducting a randomized controlled trial in the setting of SAA poses a significant challenge. The North American Pediatric Aplastic Anemia Consortium and the Pediatric Transplantation and Cellular Therapy Consortium presented the results of a pilot study randomizing patients to upfront MUD-HCT or IST (130). In this study, 23 of the 57 patients with confirmed SAA underwent randomization and received therapy with a median follow-up of 18 months. This can be seen as a promising herald of high-quality evidence for the upfront therapy of SAA.
6.2 Challenges in adult HCT
Transplantation options in adult patients with AA include “upfront” or “delayed” alloHCT after failed IST. The outcomes of alloHCT in AA improved in the last decade with long-lasting OS reaching 70% to 90% (131). On the other hand, despite the improved response to the new immunosuppression protocols with hATG and ELT, some patients are not cured or remain at risk of aplasia relapse or clonal evolution (33). About 3 months after successful IST, in about 19% of patients, chromosomal aberrations, including chromosome 7, were detected. There is also a significant risk of progression to MDS or AML within a median time of 4 to 6 years (27, 31, 55, 103, 132). Therefore, according to the available evidence, the decision to proceed to alloHCT should be based on the benefit-risk analysis of IST vs alloHCT. Accessible factors increasing the probability of response to IST and the risk of clonal evolution should be considered in decision-making.
Evaluated biomarkers potentially predictive of better response to IST are as follows: baseline absolute lymphocyte count ≥1 × 109/L; absolute reticulocyte count ≥25 × 109/L; PIGA, BCOR, and BCORL mutations. There are also potential biomarker candidates requiring clinical validation: predominance of memory/activated phenotype of T regulatory cells; increased intracellular IFN-γ levels in circulating T cells; IFN-γ +874T/A gene polymorphism; bone marrow enrichment of CD8+ T cells and T cell–activating intercellular interactions; and miR-150-5p expression (31, 133–138). Poor response to IST might be expected with an undetectable PNH clone and a shorter telomere length, and detection of adverse genetic mutations: ASXL1, DNMT3A, RUNX1, TP53, but also with TPO plasma levels of >1,796.7 pg/ml (32, 139, 140). Telomere shortening and the above adverse mutations have also unfavorable impact on relapse incidence and clonal evolution (14, 140). If germline mutations were detected, the family history should be carefully collected to determine the malignant hematologic disease germline predisposition because for these patients alloHCT is usually needed.
Factors determining the choice of the therapeutic pathway include the patient’s age, comorbidities, availability of an optimal donor, AA severity at diagnosis as well as access to key medical agents effective in nontransplant therapies: hATG and ELT (26). In a study published by the EBMT, the identified risk factors for alloHCT that should be considered were as follows: PB as the source of hematopoietic cells, time interval to alloHCT exceeding 6 months from diagnosis, patient’s age ≥20 years, no ATG use in conditioning regimen, and the cytomegalovirus (CMV) donor/recipient serostatus other than negative. The presence of risk factors determined inferior survival assessed as 77% and 67% in the intermediate and high-risk groups, respectively, compared with 90% in the low-risk group (141). However, the last risk factor is of a lower value due to a broader access to letermovir prophylaxis in CMV seropositive patients. In another study conducted in patients who underwent transplantation with the use of fludarabine (Flu), the risk factors leading to worse OS included age ≥20 years, shorter time interval (3 months), transfusion burden (>20 units of packed red cells, >50 platelet concentrates), and previous alloHCT. However, in a multivariate analysis, all factors but age were associated with poor survival prognosis (142). Therefore, with the use of Flu, age as a sole risk factor should not be treated as an absolute contraindication to alloHCT. In the study by Sheth et al., the outcome of patients transplanted with FCC conditioning protocol (Flu, Cy, and alemtuzumab) was summarized, and no significant differences were found between patients younger and older than 50 years, but alloHCT outcomes were much worse for patients with a HCT–comorbidity index score (HCT-CI) higher than 3 (143).
For adult patients at the age of 40 years or younger without significant comorbidities, alloHCT from MSDs remains the best option. For older patients, due to higher mortality rates, IST is recommended (60, 144, 145). Nevertheless, some recently published papers showed no significant differences in OS between the age groups when conditioning regimens are based on Flu (143, 146). However, it should be noted that the risk of complications related to graft-versus-host disease (GVHD) is much higher in elderly patients; therefore, a decision to perform upfront alloHCT from MSD in selected patients should be individualized (147). The transplant pathway in the older age group may be recommended for patients with a good performance status and a low HCT-CI in the case of SAA or vSAA and a lack of factors increasing the probability of response to IST (26). Considering disease severity, according to the published data, patients with SAA and vSAA more rarely respond to IST compared with those with nonsevere (moderate) AA. Moreover, they more often experience life-threatening infections and bleeding complications (133, 148).
In the last decade, we observe improved outcomes of alloHCT from MUDs, also in patients with AA (149). It may be explained by better high-resolution donor typing, improved supportive care, and the use of Flu and rATG or alemtuzumab in conditioning protocols with the avoidance of higher doses of TBI and PB as a source of hematopoietic cells (26, 34). However, transplant centers without extensive experience with alloHCT from MUDs and ADs should transfer patients to more experienced centers if such a procedure is planned.
For patients younger than 40 years without MSD, IST is the preferred frontline option; however, in young adults (<20 years) with urgent indications to alloHCT (e.g. recurrent infections) and an available MUD within 8 to 12 weeks, serious consideration should be given to this option (26). In retrospective analyses, the results of alloHCT from MSDs and MUDs were comparable in the population of children and teenagers (129). Another factor to determine opting for upfront alloHCT from MUDs would be the detection of a genetically unfavorable clone increasing the risk of progression to MDS or AML. Early alloHCT from MUDs is the option in patients older than 40 years with failed IST, especially in the case of clonal evolution. However, in the older age group, worse outcomes can be expected, including lower OS and disease-free survival resulting from transplant-related mortality, GVHD, and graft failure (GF) (1, 60, 61). The studies on transplantation techniques and alloHCT outcomes for the treatment of SAA are summarized in Table 2.
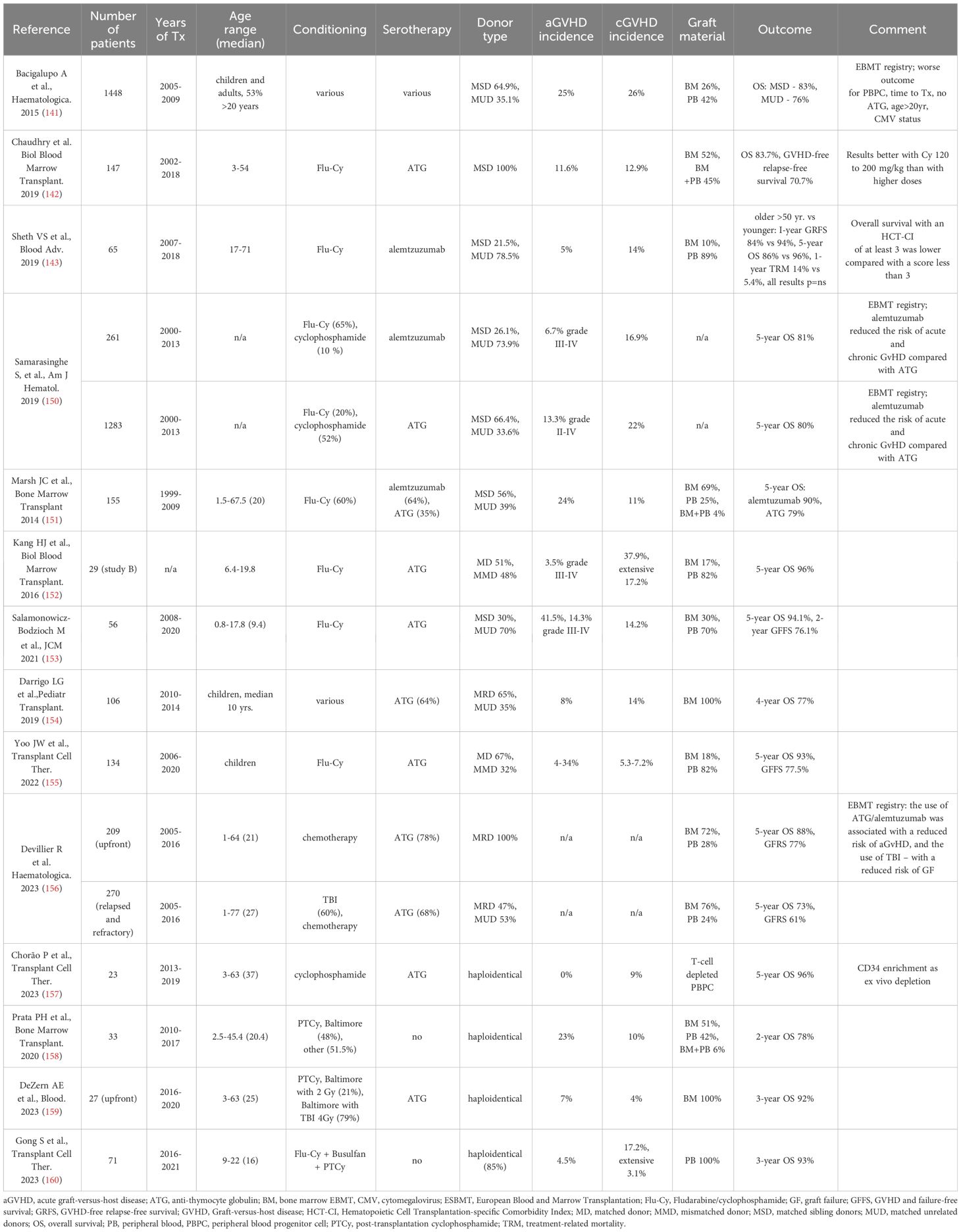
Table 2 Key studies on transplantation techniques and alloHCT outcomes for the treatment of aplastic anemia.
6.3 Pretransplant management
The patient’s workup before alloHCT should include the assessment of the HCT-CI score, collection of data on previous and current infections (including tuberculosis, viral and fungal infections), and laboratory screening for latent infections. For transfusions, blood products must be irradiated and leukodepleted to avoid alloimmunization against HLA antigens. In patients with significant transfusion burden, serum ferritin should be evaluated. To decrease the risk of organ toxicity, infections, and GF, iron chelation therapy should be considered if the ferritin concentration is higher than 1000 ng/ml.
Fertility preservation issues should be discussed with patients when potentially gonadotoxic conditioning regimen is planned (26, 161–165). The severity of conditioning toxicities can be calculated using a cyclophosphamide equivalent dose (CED) (166). The risk of infertility depends on age and summarized exposure to gonadotoxic chemotherapeutics and radiotherapy (162, 163). The risk of irreversible gonadotoxicity is very high in men receiving a CED higher than 4000 mg/m2 and in women receiving a CED higher than 6000 mg/m2 (163–165, 167),.
6.4 Conditioning regimens
The choice of conditioning regimen in pediatric SAA has evolved over the last 40 years. Among other factors, the omission of initially popular irradiation techniques was associated with progress of HCT and reduced complication rates in this population (168, 169).
Another pivotal step forward in the conditioning was related to the addition of Flu to the MUD-HCT (170, 171). The introduction of Flu into the conditioning regimens of adults with SAA in the last 2 decades was associated with improved outcomes (172). The reduction in the intensity of the conditioning protocol is associated with improved survival but graft-versus-host-free/relapse-free survival (GRFS) might not exceed 80% (152, 153). This issue is especially important in the pediatric population due to long life expectancy of posttransplant children, and the risk of GVHD-associated organ complications and metabolic consequences of life-long steroid immunosuppression that can arise (153).
Twenty years ago, Flu-Cy-ATG greatly improved the outcome of MUD-HCT in children with SAA (173, 174). In the case of MUD-HCT, the optimal conditioning protocol was not evident, but the 5-year OS probability did not exceed 80%, which was inferior to the newer results observed in children. In a retrospective study by the Pediatric Hematopoietic Stem Cell Transplant Working Group of the Brazilian Bone Marrow Transplantation Society and the Brazil-Seattle Consortium (Gedeco), the 4-year OS for MRD-HCT was lower than expected and did not differ significantly from that for MUD-HCT (82% vs. 69%, respectively; P = 0.08). The authors highlighted the need for standardized pediatric protocols (154).
Despite the existing variability of transplant protocols in SAA, in 2023 most studies agree that the Flu and Cy chemotherapy backbone is the standard of care for pediatric population. The original Cy dose of 200 mg/kg used in SAA for MSD-HCT can be safely decreased to 100 mg/kg (or 3000 mg/m2) in combination with Flu, which results in improved survival, as showed by numerous studies (152, 153, 175). However, at the lower end of the Cy dosing range, in children receiving the dose of 60 mg/kg, an increased risk of graft rejection was observed (176).
A retrospective cohort of 56 children with SAA after Flu-Cy-ATG conditioning showed favorable outcomes (153). The overall incidence of acute GVHD (aGVHD) was 41.5%, and that of grade III-IV aGVHD – 14.3%. Chronic GVHD (cGVHD) was diagnosed in 14.2% of children. The probability of the 2-year GVHD-free survival was 76.1%. In the univariate analysis, a higher dose of Cy and previous IST were significant risk factors for worse OS. Episodes of viral replication occurred in 33 of 56 patients (58.9%), but it did not influence OS. The study showed improvement over the previous treosulfan-based protocol for MUD-HCT (177).
In a Korean study, Yoo et al. described a low rate of secondary GF, higher failure-free survival (FFS), and manageable GVHD regardless of HLA compatibility (155). In 134 children with SAA transplanted with HLA-matched BM transplantation (BMT; n = 24), HLA-matched PB stem cell transplantation (PBSCT; n = 66), and HLA-mismatched PBSCT (n = 44), the estimated 5-year OS, FFS, and GVHD-free FFS of the total cohort were 93.0%, 89.5%, and 77.5%, respectively (155). The incidence of grade II-IV aGVHD was significantly higher in PBSCT than in BMT, but the incidence of grade III-IV aGVHD and cGVHD was similar across all the 3 groups.
In 2023, Vissers et al. described a single-center experience with 228 HCT procedures in pediatric BMF (n = 103) and SAA (n = 125) (178). Patients with SAA transplanted after 2017 had a superior 5-year OS and event-free survival (EFS) of 97% and 85%, respectively, compared with 68% and 59%, respectively, in the cohort transplanted before 2017 (P = 0.0011 and P = 0.017). In the case of BMF, OS and EFS were 89% for those transplanted after 2017 compared with 62% and 59%, respectively, for the other cohort (P = nonsignificant). The authors noticed the typical evolution of the conditioning regimen in SAA, with a change from more toxic to an immune-ablative regimen consisting of Flu/Cy without radiation.
A recent analysis of GRFS by the EBMT-SAA-WP included 479 patients with idiopathic SAA. The probabilities of 5-year GFRS in patients transplanted upfront or in relapsed/refractory cohorts were 77% and 61%, respectively (156). Approximately one-third of all patients were below the age of 20 years, and the group consisted mainly of children. Age was the main risk factor of death (hazard ratio [HR], 1.04; 95% CI, 1.02-1.06; P <0.001), aGvHD (HR, 1.03; 95% CI, 1.00-1.07; P = 0.041), and cGvHD (HR, 1.04; 95% CI, 1.01-1.08; P = 0.032) as the causes of inferior GRFS. The use of ATG/alemtuzumab was associated with a reduced risk of aGvHD (yes vs no: 4% vs 16%; P = 0.006), and the use of TBI – with a reduced risk of GF (yes vs no: 4% vs 11%; P = 0.039). There were no significant differences in GRFS and in the causes of GRFS failure according to graft source, donor type, or time from diagnosis to alloHCT. These results suggest that although cured in terms of hematopoiesis recovery, many patients can suffer from detrimental health issues due to long-term sequelae.
The use of MSD-HCT after conditioning with low-dose Cy (80 mg/kg), Flu (175 mg/m2), and Thymoglobulin (10 mg/kg) was studied in 32 children with BMF below 14 years of age. While the treatment led to primary engraftment in all patients, 2 patients later developed GF or MDS and only 7 patients sustained donor chimerism above 99% (179). The study was heterogenous and contained 9 patients with constitutional BMF (two ERCC6L2, two ANKRD26, two TINF2, one LZTFL1, one RTEL1, and one DNAJC21), for which Flu/Cy–based conditioning is not optimal.
The chemotherapy backbone recommended by the EWOG for alloHCT in children with SAA consists of Flu (30 mg/m2/day, × 4 days) and Cy (25 mg/kg/day, × 4 days) (59). Some small studies indicated that other reduced toxicity protocols, such as Flu with melphalan ± thiotepa, might be used in BMF syndromes (180, 181).
In adult patients, the choice of conditioning regimen is based on the patient’s age and comorbidities, a donor type, evaluation of the risk for graft rejection, access to alemtuzumab, and local experience (26). In the latest evaluation of 955 SAA patients transplanted from HLA-MSD, Flu/Cy/ATG, and Cy/ATG conditioning regimens were associated with the highest survival rates, as compared with Cy ± Flu and busulfan/Cy, with the 5-year probabilities of survival equal to 91%, 91%, 80%, and 84%, respectively (P = 0.001). In the case of 409 recipients of 8/8 and 7/8 HLA allele-matched unrelated donor transplantation (n = 409), the 5-year probabilities of survival with Cy/ATG/TBI-2Gy, Flu/Cy/ATG/TBI-2Gy, Flu/Cy/ATG, and Cy/ATG were 77%, 80%, 75%, and 72%, respectively (P = 0.61) (182). In the MSD setting, Cy-based protocols (200 mg/kg) are recommended in patients younger than 30 years, while in older age groups, Flu (120-150 mg/m2) plus Cy (120 mg/kg) are preferred, with the addition of serotherapy in both cases (26, 142, 151, 182, 183). However, in young adult patients at risk of cardiotoxicity, a Flu/Cy-based regimens should be considered. In the 10/10 MUD and 9/10 mismatched unrelated donor settings, low-dose TBI of 2 Gy should be added to the Flu/Cy/serotherapy backbone to decrease the risk of graft rejection (26, 61). In the haploidentical (haploHCT) setting, an increase in the TBI dose to 4-6 Gy may be considered to achieve sustained engraftment (184–186).
6.5 Transplantation techniques
The choice of transplant material in alloHCT for SAA is under debate, and resource availability can affect the choice of stem cell source. Bone marrow is considered as the stem cell source for the first alloHCT in SAA because of higher survival probability and lower GVHD incidence (141, 187, 188). In SAA patients, the use of PBSC in MSD was associated with a high incidence of cGVHD of up to 43% (189). The retrospective analysis of 1886 patients with SAA by Bacigalupo et al. showed that BM was superior to PBSC in all age groups and was associated with a lower incidence of aGVHD and cGVHD as well as lower mortality rates (190). However, for technical reasons, the popularity of alloHCT from PB has increased in the recent years (129, 191). In a study by Kumar et al., a multivariate analysis showed the highest OS with BM as graft source in high-income countries compared with PBSCs in all countries or BM in not-high-income countries (P <0.001) (192). The authors concluded that PBSCs may be an acceptable alternative in countries with limited resources when treating patients at high risk of GF and infective complications (193). The transplant outcome in MSD and MUD-HCT for SAA is also improved by the shorter interval from diagnosis to transplantation (127, 128, 141, 190, 194, 195).
To reduce the complication rates, there is a search for ways to improve GVHD prophylaxis. The ex-vivo immunomagnetic T-cell depletion of MSD PBPC is challenging and requires a specialized graft-engineering facility, but it is associated with faster neutrophil engraftment, lower aGVHD incidence, and similar OS as the standard BM (157). However, the T-cell depletion techniques in haploHCT for SAA are associated with an increased risk of GF (196). With the increased data on favorable outcomes with posttransplant Cy (PTCy) as GVHD prophylaxis not only in haploHCT, but also in the MUD and mMUD settings, this option should also be considered (197).
In adult patients, BM is a recommended source of hematopoietic cells in the MSD and MUD settings, but PB might be accepted when alemtuzumab is used (141, 143, 187, 190). The cellularity of the BM graft should be higher than in hematologic malignancies, with the minimal quantity established as 3 × 106 CD34/kg (3 × 108 total nucleated cells [TNC]/kg) (61). In haploHCT, there are no clear recommendations concerning the source and an optimal dose of cells, with a suggestion for a higher dose to diminish the risk of GF. There are different protocols including the preferential use of BM or G-CSF-mobilized BM and PB (186, 198). It must be noted that in the EBMT study no difference in GVHD-free survival was reported when PB was used in haploHCT, with superior platelet engraftment and lower GF rates with a medium graft cellularity of 3.5 × 108 TNC/kg for BM and 7.1 × 106 CD34/kg for PB (158). An option to perform syngeneic HCT should be used irrespective of the patient’s age because of the very good outcomes (long-term survival of about 90%), and in this case, PB is the preferable HSC source (199).
6.6 Serotherapy in HCT
The conditioning regimen including serotherapy with rATG, hATG, or alemtuzumab in SAA was shown to be associated with a survival advantage (150, 200). The inclusion of serotherapy with rATG is associated with both reduced risk of cGVHD and increased risk of opportunistic infections in alloHCT recipients (201). In some studies, the benefit of ATG in MSD-HCT was unclear. A multicenter prospective trial by the Center for International Blood & Marrow Transplant Research reported a cGVHD incidence of 21% and 32% in patients conditioned with either Cy alone or Cy/ATG, respectively, while the OS was 74% and 80%, respectively (200). Compared with hATG, rATG was associated with a lower risk of grade II to IV aGVHD (HR, 0.39; P <0.001) but not of cGVHD (182). In a study by Atta et al., rATG was protective against aGVHD and cGVHD, but a higher incidence of invasive fungal disease and mixed chimerism was noted in the rATG vs hATG cohorts (202). In another study, 21 patients with SAA, including children, underwent BMT from MSDs following a standard conditioning regimen with Cy (200 mg/kg) and hATG (30 mg/kg/day × 3 days) (203). None of the patients rejected the graft, all had sustained engraftment, and cGVHD developed in 24% of patients. The currently recommended cumulative doses of rATG in children undergoing HCT are 40 to 60 mg/kg for Grafalon and 8 to 10 mg/kg for Thymoglobulin, depending on the donor and grafting material (59).
A strategy to reduce the incidence of GVHD in SAA is the replacement of ATG with alemtuzumab (150, 204, 205). The protocol containing Flu (120 mg/m2) and Cy (120 mg/m2) with alemtuzumab (40-100 mg) was studied in 50 SAA patients transplanted from MSDs (n = 21) or MUDs (n = 29) (206). Acute GVHD was observed only in 13.5% of patients (all grade I-II), and only 2 patients (4%) developed cGVHD, but GF was present in 9.5% of patients after MSD and 14.5% of those after MUD.
In adult patients, the standard total doses of rATG for SAA in MSD and MUD-HCT are 5 to 10 mg/kg (Thymoglobulin) or 60 mg/kg (Grafalon), but in patients at high risk of graft rejection, a higher Thymoglobulin dose of 10-20 mg/kg might be considered (26, 142). A total dose of alemtuzumab ranges from 40 to 100 mg, and usually 0.2 mg/kg/day is administered for 5 consecutive days (151, 206). In patients referred for alloHCT with alemtuzumab with a high PNH clone associated with SAA, a lowered CD52 expression is possible on the PNH clone-derived lymphocytes as CD52 is a glycosylphosphatidylinositol-anchored protein. Despite theoretical concerns there have been no reports of increased graft rejection or graft failure rates in AA patients with PNH clones using alemtuzumab-containing regimens (207).
6.7 Haploidentical HCT
Over the last 20 years, developments in haploHCT with ex-vivo depletion techniques or PTCy gained a lot of attention in multiple indications, including SAA. Nevertheless, SAA HCT protocols should acknowledge the fact that Cy induces cardiotoxicity in 7% to 28% of patients with a mortality rate of 11% to 43% at the therapeutic dose of 170 to 180 mg/kg intravenously (IV). Therefore, caution should be exerted when increasing the cumulative (pre-HCT and post-HCT) Cy dose (208). In patients with a significant risk of cardiotoxicity, a Flu/Cy-based conditioning regimen should be considered. There are also data on the reduction of a standard total PTCy dose of 100 mg/kg to 80 mg/kg (209).
Initial reports on haploHCT showed an engraftment rate of 67% and a 2-year OS of 78% in 33 patients (39% children) transplanted between 2011 and 2017 with PTCy (158). The outcomes of related haploidentical donors after reduced-intensity conditioning with PTCy were presented in 37 patients with SAA: 20 relapsed/refractory and 17 treatment-naïve (186). The 2-year OS probability was 94%, and the cumulative incidence of grade II-IV aGVHD at day 100 and cGVHD at 2 years was 11% and 8%, respectively. The GF incidence was 11% including 1 case in a relapsed/refractory group and 3 in a treatment-naïve patients receiving 2 Gy of TBI while no GF was observed in patients who received 4 Gy of TBI. The role of irradiation cannot be generalized because low-dose TBI/total lymphatic irradiation reduces the risk of GF in adult SAA and is deemed safe, but in children the irradiation techniques, even at a low dose, are not recommended due to excellent transplant outcomes in pediatric SAA and historic reports showing the risk of malignancies in the radiation field (59, 127, 156, 210, 211).
Haploidentical HCT with CD45RA (4 patients), TCRαβ-CD19 depletion or PTCy (5 patients) was evaluated in 11 patients with SAA or MDS-refractory cytopenia of childhood (212). The conditioning regimens were heterogenous and consisted of Flu combined with irradiation techniques (9), Cy (9), busulfan, treosulfan, or thiotepa. Despite engraftment, 2 patients died, and 4 of 9 long-term survivors developed cGVHD.
The clinical protocols for alloHCT in SAA in some countries show fundamental differences, especially by including a high number of partially matched donors and more myeloablative conditioning protocols. Therefore, a direct comparison is not feasible. Older studies that did not implement PTCy after haploHCT revealed an elevated incidence of aGVHD and worse OS probability (213–215). In a study by Qin X et al., in an analysis of 115 patients (mostly children), a conditioning regimen without low-dose irradiation or busulfan was associated with an inferior FFS in a multivariate analysis, but 60% of donors were partially matched in HLA (216). In an Indian study, children and young adults (aged 21 years or younger) with SAA underwent haploHCT after Flu-Cy-ATG/TBI with or without thiotepa/melphalan and PTCy (217). At a median follow-up of 48 months, the OS and EFS were 61.6% and 58.1%, respectively. Chinese protocols in pediatric SAA contain a more intensive chemotherapy backbone, such as Flu (200 mg/m2), Cy (30-100 mg/kg), and busulfan (6.4 mg/kg) (160). In a study by Gong et al., 61 of the 71 patients with SAA below the age of 20 years were transplanted from haploidentical donors (160). At a median follow-up of 42 months (95% CI, 33-50 months), the OS and EFS were 93.0% (95% CI, 85.3%-97.3%) and 81.7% (95% CI, 71.5-89.3%), respectively. A more intensive myeloablative protocol resulted in complete donor chimerism in all long-term surviving HCT recipients. In addition, the frequencies of grade II–IV aGVHD, overall cGVHD, and moderate-severe cGVHD were 4.5% (95% CI, 1.3-11.6), 17.2% (95% CI, 9.5-27.8), and 3.1% (95% CI, 0.7-9.6), respectively.
In adult patients, retrospective analyses show minor differences between the outcomes of alloHCT from MUDs and MSDs compared with those of haploidentical donors with the use of PTCy as GVHD prophylaxis. It was suggested that haploHCT should be reserved for younger patients without MSD and MUD after an unsuccessful outcome of IST (34, 104, 218). However, the recent study in mixed age cohort showed encouraging outcomes with haploBMT approach in SAA refractory to IST or relapsed after initial response with 1-year OS 81% and the PTCy-based GVHD prophylaxis. The authors concluded that haploBMT with PTCy could now be considered a standard approach for salvage treatment of SAA (219). Moreover, another prospective study by DeZern et al. enrolling 37 SAA patients with median age 25 (range, 3-63 years) to haploBMT as an upfront approach with PTCy showed OS of 92% at 1, 2 and 3 years with minimal GVHD, while in last 20 consecutive patients receiving a TBI dose increased from 2 Gy to 4 Gy, OS of 100% was achieved (159).
Based on increasing available data, the haploBMT-PTCy platform as the salvage or even initial treatment is of special interest in pediatric and adult patients originating from the ethnical minorities but should be also considered as an alternative option to the MUD/mMUD platform when beneficial outcomes over IST are expected and a delayed alloHCT procedure poses a risk of life-threatening complications.
When donor-specific antibodies (DSA) are identified in pretransplant evaluation of the patient, the preferred haploidentical donor is without antigens corresponding to the detected DSA in the recipient. In the case of a lack of a donor choice and DSA above 1000 MFI, there are 2 options to prevent GF: the search for AD or desensitization of the recipient (220).
6.8 Cord blood transplantations
The role of cord blood (CB) in HCT for SAA remains to be determined, although the number of CB transplantations has been on the wane in recent years (221). Increased experience with haploHCT and suboptimal historic outcomes of CB transplantation in SAA suggest that this tendency is becoming stronger (222–224). According to the Chinese study, Haplo-BMT (35 patients) or haploidentical CB (56 patients) HCT based on the Beijing Protocol, with Flu at a dose of 45 mg/m2/day on days −7 to −4, busulfan at a dose of 3.2 mg/kg/day IV on days −7 to −6, Cy at a dose of 40 mg/kg/day on days −5 to −2, and rATG (Thymoglobulin) at a dose of 2.5 mg/kg/day IV on days −5 to −2, and GVHD prophylaxis with CsA/tacrolimus with MMF and methotrexate (MTX) was feasible in 91 children (225, 226). The Haplo and Haplo-CB groups had similar 3-year OS (92.8% ± 4.9% vs 94.3% ± 3.2, P = nonsignificant), with a lower incidence of aGVHD and cGVHD in the Haplo-CB HCT recipients. In a retrospective analysis of 240 children with SAA transplanted at single center, CB recipients had inferior FFS and GRFS compared with Haplo and MUD cohorts (227). Additionally, CB was associated with a higher GF rate and inferior platelet engraftment. In a study from 2021, HCT in 60 pediatric patients with SAA used unmanipulated haploHCT with third-party 1-5/6 HLA antigen–matched umbilical CB after myeloablative conditioning with busulfan, Cy, and Flu (228). The 5-year transplant-related mortality, OS, and FFS were 2.8%, 97.2%, and 96.2%, respectively. Considering that patients with SAA constituted only a subgroup of 109 patients in the study cohort, the detailed analysis was not provided. It is of note, however, that at +30 days after transplantation, the engrafted HSCs were derived from CB in 5 of the 109 patients. In unique cases, a successful autologous CB transplantation was reported in SAA, but there were differences regarding the use of conditioning therapy before the transplantation (229–232).
The use of CB for transplantation in adults is limited by graft cellularity. Longer time to engraftment is another concern. However, the published outcomes of CB transplantation (CBT) are promising, with an OS of 88% and a low rate of aGVHD and cGVHD of about 15% (233). The APCORD protocol (FluCyATG+2GyTBI) also showed very good results in a prospective trial (222). The recommended minimum TNC dose is 4 × 107 TNC/kg. In a retrospective study of the Japanese Society for Transplantation and Cellular Therapy comparing haploHCT (n = 24) with PTCy vs CBT (n = 59) in adult SAA patients, no significant differences were noted in OS, the cumulative incidence of grade II-IV aGVHD, and cGVHD. However, patients after PTCy-haploHCT experienced significantly higher rates of engraftment in neutrophils (95.8% vs 78%; P <0.001) and platelets (83.3% vs 72.9%; P = 0.025). Moreover, a higher 1-year FFS rate was observed in patients younger than 40 years after haploHCT with PTCy compared with the CBT group (92.9% vs 63.9%; P = 0.047) (218).
6.9 Posttransplant management and sequelae
The advantages of MUD-HCT include a short time to neutrophil engraftment and high survival rates, but they are counterbalanced by the need for donor search and arrangement of graft donation, which is associated with an up to 4-month delay in therapy. The waiting time for an HSC donor can be used as a window for pharmacotherapy involving IST with or without additional drugs (e.g. TPO-RA or androgens). In addition, HCT can result in treatment-related organ toxicities, infections, GF, poor graft function, acute and chronic GVHD, infertility, and secondary neoplasms, supporting the need for a judicious referral for HCT (234, 235).
The factor with the highest influence on the quality of life is GVHD, and GFRS is the best composite endpoint measuring the success of alloHCT. A risk of GF is another key issue after alloHCT in AA (236). Therefore, the suitable and often individualized posttransplant IST has the most important impact on alloHCT outcome.
Posttransplant GVHD prophylaxis in children typically consists of MTX and CsA, since Locatelli et al. proved this combination to be superior to CsA alone in a randomized controlled trial (237). Due to increased popularity and low cost, PTCy as the sole GVHD prophylaxis after PBSCT from MSDs can be considered, as it is associated with low rates of aGVHD and cGVHD (238).
In adult patients, in the MSD and MUD alloHCT platforms with rATG use, calcineurin inhibitors (CNI) with a short course of MTX is recommended, while alemtuzumab administration is followed by sole CNI-based IST. CNI should be continued for 9 to 12 months with CNI concentrations maintained on higher levels than in hematological malignancy to avoid GF (CsA, 200-300 ng/ml; tacrolimus, 10-15 ng/ml) and very careful tapering under the control of donor chimerism and evidence of GF (61). In haploHCT, there are protocols based on PTCy ± rATG with MMF and tacrolimus given after PTCy administration and the Chinese protocol based on rATG with CsA+MMF starting on day+1 and MTX administrations on days +1, +3, +6, and +11 (186, 239, 240). The APCORD protocol for umbilical CBT comprises CsA at a dose of 200-400 ng/ml for 3 months, with gradual tapering up to 1 year after alloHCT (222).
Donor chimerism is monitored at least 1, 3, 6, and 12 months after alloHCT for the assessment of engraftment kinetics and prediction of GF with progressing mixed chimerism that requires intensification of post-HCT IST (26). Transient mixed chimerism is a common finding after rATG and in the FCC protocol with alemtuzumab, stable mixed chimerism in T lymphocytes indicates that immunotolerance has been achieved (241). In the case of poor graft function with complete donor chimerism, ELT administration or an additional boost of hematopoietic cells without additional conditioning should be considered (242, 243).
In case of alloHCT failure in SAA, second transplants are associated with very high success rates, even with the retransplantation from an original donor (244). In 55 children who experienced GF after the first alloHCT, the 5-year OS and FFS were 82.9% and 81.2%, respectively (245).
The long-term survival of children with SAA after HCT is very high, and the OS probability at 5 and 10 years is 96% and 94%, respectively (246). Changes in the conditioning protocols and improved genetic testing over the last 40 years positively affect the survival outcomes, but caution is warranted. Despite excellent survival probability, the freedom from long-term complications in HCT survivors have not been determined (247). According to Buchbinder et al., the cumulative incidence of late effects among survivors of MSD-HCT was less than 3% and did not increase over time, but after MUD-HCT, the incidence of late effects exceeded 3% by 5 years: gonadal dysfunction, 10.5% (95% CI, 7.3-14.3); growth disturbance, 7.2% (95% CI, 4.4-10.7); avascular necrosis, 6.3% (95% CI, 3.6-9.7); hypothyroidism, 5.5% (95% CI, 2.8-9.0); and cataracts, 5.1% (95% CI, 2.9-8.0). However, the majority of these transplantations included irradiation as part of conditioning regimen (248).
Among the consequences of HCT, the incidence of subsequent malignancies should be considered, estimated at 1.1% among the SAA patients (249). Improved differential diagnosis and genetic testing reduced the misdiagnosis of clonal disorders as SAA and, consequently, the occurrence of myeloid malignancies after HCT (84, 250). It can be suggested that HCT results would further improve in the future due to eliminated or reduced exposure to TBI or alkylating agents. However, long-term surveillance is necessary, because the majority of nonleukemic malignancies developed at least 5 years after transplantation.
7 Conclusions
Although the body of evidence on the treatment outcomes in patients with AA is mostly based on retrospective studies and nonrandomized trials, the up-to-date recommendations can be summarized as follows:
1. The diagnosis of patients with AA must include testing for constitutional defects associated with BMF syndromes. Potential related transplant donors must always be tested for constitutional defects if such are found in a patient (proband).
2. Pretransplant care should include transfusions of irradiated leukodepleted blood products as well as anti-infective screening and prophylaxis.
3. Upfront alloHCT from MSD is recommended in children and young adults (age <40 years).
4. Upfront IST with hATG is recommended in elderly patients and children and young adults without MSD and should be performed without delay.
5. Addition of ELT to IST in adults should be considered as a standard of care incorporated in the IST protocols.
6. The option of upfront MUD-HCT can be considered in patients lacking MSD if the donor search and transplantation procedures would be completed within 2 months and the patient is eligible for HCT. However, further studies are necessary to confirm this.
7. Bone marrow is preferred as the source of stem cells due to better outcomes in most transplantation settings.
8. Conditioning regimen for alloHCT should be based on Flu, Cy, and rATG in children; in adults on Cy (age <30 years) and on Cy with Flu (age >30 years), while serotherapy options include rATG or alemtuzumab. In the MUD and Haplo setting in adults, a low dose of TBI should be added to reduce the risk of GF.
9. GVHD prophylaxis after alloHCT should consist of CNI and a short course of MTX or CNI alone when alemtuzumab is used in conditioning.
10. HaploHCT and CBT should not be considered a standard therapy, unless as a salvage after IST failure and the unavailability of MSD/MUD. In the haploHCT setting, PTCy as the GVHD prophylaxis is recommended. Based on increasing available data, the haploBMT-PTCy platform as the salvage or initial treatment might be considered as an alternative option to MUD/mMUD, especially in patients originating from the ethnical minorities.
11. Androgens can be considered as a supportive treatment in patients not responding to IST and ineligible for other therapies.
12. Fertility preservation strategies should be discussed and implemented in patients eligible for alloHCT with potentially gonadotoxic protocols.
13. Long-term surveillance in alloHCT survivors is necessary.
Author contributions
AP: Conceptualization, Investigation, Methodology, Project administration, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. KP: Investigation, Validation, Visualization, Writing – original draft. AS: Investigation, Validation, Writing – original draft. MU: Conceptualization, Investigation, Methodology, Supervision, Validation, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study received funding from Pfizer. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication. All authors declare no other competing interests.
Acknowledgments
The Authors would like to thank Proper Medical Writing, Poland for the editorial and language assistance in the preparation of this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
2. Peslak SA, Olson T, Babushok DV. Diagnosis and treatment of aplastic anemia. Curr Treat Options Oncol. (2017) 18:70. doi: 10.1007/s11864-017-0511-z
3. Camitta BM, Rappeport JM, Parkman R, Nathan DG. Selection of patients for bone marrow transplantation in severe aplastic anemia. Blood. (1975) 45:355–63. doi: 10.1182/blood.V45.3.355.bloodjournal453355
4. Giudice V, Selleri C. Aplastic anemia: pathophysiology. Semin Hematol. (2022) 59:13–20. doi: 10.1053/j.seminhematol.2021.12.002
5. Locasciulli A, Bacigalupo A, Bruno B, Montante B, Marsh J, Tichelli A, et al. Hepatitis-associated aplastic anaemia: epidemiology and treatment results obtained in Europe. A report of The EBMT aplastic anaemia working party. Br J Haematol. (2010) 149:890–5. doi: 10.1111/j.1365-2141.2010.08194.x
6. Osugi Y, Yagasaki H, Sako M, Kosaka Y, Taga T, Ito T, et al. Antithymocyte globulin and cyclosporine for treatment of 44 children with hepatitis associated aplastic anemia. Haematologica. (2007) 92:1687–90. doi: 10.3324/haematol.11359
7. Rauff B, Idrees M, Shah SA, Butt S, Butt AM, Ali L, et al. Hepatitis associated aplastic anemia: a review. Virol J. (2011) 8:87. doi: 10.1186/1743-422X-8-87
8. Zhang X, Yang W, Yang D, Wei J, Zhang P, Feng S, et al. Comparison of hematopoietic stem cell transplantation and immunosuppressive therapy as the first-line treatment option for patients with severe hepatitis-associated aplastic anemia. Front Immunol. (2023) 14:1146997. doi: 10.3389/fimmu.2023.1146997
9. Atmar K, Tulling AJ, Lankester AC, Bartels M, Smiers FJ, van der Burg M, et al. Functional and immune modulatory characteristics of bone marrow mesenchymal stromal cells in patients with aplastic anemia: A systematic review. Front Immunol. (2022) 13:859668. doi: 10.3389/fimmu.2022.859668
10. Pagliuca S, Gurnari C, Hercus C, Hergalant S, Nadarajah N, Wahida A, et al. Molecular landscape of immune pressure and escape in aplastic anemia. Leukemia. (2023) 37:202–11. doi: 10.1038/s41375-022-01723-w
11. Ball SE, Gibson FM, Rizzo S, Tooze JA, Marsh JC, Gordon-Smith EC. Progressive telomere shortening in aplastic anemia. Blood. (1998) 91:3582–92. doi: 10.1182/blood.V91.10.3582
12. Barade A, Aboobacker F, Korula A, Lakshmi K, Devasia A, Abraham A, et al. Impact of donor telomere length on survival in patients undergoing matched sibling donor transplantation for aplastic anaemia. Br J Haematol. (2022) 196:724–34. doi: 10.1111/bjh.17880
13. Brummendorf TH, Maciejewski JP, Mak J, Young NS, Lansdorp PM. Telomere length in leukocyte subpopulations of patients with aplastic anemia. Blood. (2001) 97:895–900. doi: 10.1182/blood.V97.4.895
14. Scheinberg P, Cooper JN, Sloand EM, Wu CO, Calado RT, Young NS. Association of telomere length of peripheral blood leukocytes with hematopoietic relapse, Malignant transformation, and survival in severe aplastic anemia. JAMA. (2010) 304:1358–64. doi: 10.1001/jama.2010.1376
15. Calado RT, Cooper JN, Padilla-Nash HM, Sloand EM, Wu CO, Scheinberg P, et al. Short telomeres result in chromosomal instability in hematopoietic cells and precede Malignant evolution in human aplastic anemia. Leukemia. (2012) 26:700–7. doi: 10.1038/leu.2011.272
16. Townsley DM, Dumitriu B, Young NS. Bone marrow failure and the telomeropathies. Blood. (2014) 124:2775–83. doi: 10.1182/blood-2014-05-526285
17. Fattizzo B, Ireland R, Dunlop A, Yallop D, Kassam S, Large J, et al. Clinical and prognostic significance of small paroxysmal nocturnal hemoglobinuria clones in myelodysplastic syndrome and aplastic anemia. Leukemia. (2021) 35:3223–31. doi: 10.1038/s41375-021-01190-9
18. Gurnari C, Pagliuca S, Prata PH, Galimard JE, Catto LFB, Larcher L, et al. Clinical and molecular determinants of clonal evolution in aplastic anemia and paroxysmal nocturnal hemoglobinuria. J Clin Oncol. (2023) 41:132–42. doi: 10.1200/JCO.22.00710
19. Li J, Liu X, Zhao X, Yang W, Zhang L, Jing L, et al. Long-term outcomes of aplastic anemia with cytogenetic abnormalities at diagnosis. Eur J Haematol. (2023) 110:379–85. doi: 10.1111/ejh.13913
20. Hosokawa K, Katagiri T, Sugimori N, Ishiyama K, Sasaki Y, Seiki Y, et al. Favorable outcome of patients who have 13q deletion: a suggestion for revision of the WHO ‘MDS-U’ designation. Haematologica. (2012) 97:1845–9. doi: 10.3324/haematol.2011.061127
21. Betensky M, Babushok D, Roth JJ, Mason PJ, Biegel JA, Busse TM, et al. Clonal evolution and clinical significance of copy number neutral loss of heterozygosity of chromosome arm 6p in acquired aplastic anemia. Cancer Genet. (2016) 209:1–10. doi: 10.1016/j.cancergen.2015.10.002
22. Shah YB, Priore SF, Li Y, Tang CN, Nicholas P, Kurre P, et al. The predictive value of PNH clones, 6p CN-LOH, and clonal TCR gene rearrangement for aplastic anemia diagnosis. Blood Adv. (2021) 5:3216–26. doi: 10.1182/bloodadvances.2021004201
23. Olson TS, Dickerson KE, Nakano TA, Wlodarski M. Monosomy 7 Predisposition Syndromes Overview. Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, editors. Seattle (WA: GeneReviews (1993).
24. Wlodarski MW, Collin M, Horwitz MS. GATA2 deficiency and related myeloid neoplasms. Semin Hematol. (2017) 54:81–6. doi: 10.1053/j.seminhematol.2017.05.002
25. Gupta V, Brooker C, Tooze JA, Yi QL, Sage D, Turner D, et al. Clinical relevance of cytogenetic abnormalities at diagnosis of acquired aplastic anaemia in adults. Br J Haematol. (2006) 134:95–9. doi: 10.1111/j.1365-2141.2006.06105.x
26. Iftikhar R, Chaudhry QUN, Anwer F, Neupane K, Rafae A, Mahmood SK, et al. Allogeneic hematopoietic stem cell transplantation in aplastic anemia: current indications and transplant strategies. Blood Rev. (2021) 47:100772. doi: 10.1016/j.blre.2020.100772
27. Kulasekararaj AG, Jiang J, Smith AE, Mohamedali AM, Mian S, Gandhi S, et al. Somatic mutations identify a subgroup of aplastic anemia patients who progress to myelodysplastic syndrome. Blood. (2014) 124:2698–704. doi: 10.1182/blood-2014-05-574889
28. Negoro E, Nagata Y, Clemente MJ, Hosono N, Shen W, Nazha A, et al. Origins of myelodysplastic syndromes after aplastic anemia. Blood. (2017) 130:1953–7. doi: 10.1182/blood-2017-02-767731
29. Visconte V, Maciejewski JP. Clonal dynamics of hematopoietic stem cell compartment in aplastic anemia. Semin Hematol. (2022) 59:47–53. doi: 10.1053/j.seminhematol.2021.12.003
30. Bonadies N, Rovo A, Porret N, Bacher U. When should we think of myelodysplasia or bone marrow failure in a thrombocytopenic patient? A practical approach to diagnosis. J Clin Med. (2021) 10(5):1026. doi: 10.3390/jcm10051026
31. Yoshizato T, Dumitriu B, Hosokawa K, Makishima H, Yoshida K, Townsley D, et al. Somatic mutations and clonal hematopoiesis in aplastic anemia. N Engl J Med. (2015) 373:35–47. doi: 10.1056/NEJMoa1414799
32. Narita A, Muramatsu H, Sekiya Y, Okuno Y, Sakaguchi H, Nishio N, et al. Paroxysmal nocturnal hemoglobinuria and telomere length predicts response to immunosuppressive therapy in pediatric aplastic anemia. Haematologica. (2015) 100:1546–52. doi: 10.3324/haematol.2015.132530
33. Peffault de Latour R, Kulasekararaj A, Iacobelli S, Terwel SR, Cook R, Griffin M, et al. Eltrombopag added to immunosuppression in severe aplastic anemia. N Engl J Med. (2022) 386:11–23. doi: 10.1056/NEJMoa2109965
34. Bacigalupo A. How I treat acquired aplastic anemia. Blood. (2017) 129:1428–36. doi: 10.1182/blood-2016-08-693481
35. Rovo A, Tichelli A, Dufour C, Saa-Wp E. Diagnosis of acquired aplastic anemia. Bone Marrow Transplant. (2013) 48:162–7. doi: 10.1038/bmt.2012.230
36. Sugimori C, Mochizuki K, Qi Z, Sugimori N, Ishiyama K, Kondo Y, et al. Origin and fate of blood cells deficient in glycosylphosphatidylinositol-anchored protein among patients with bone marrow failure. Br J Haematol. (2009) 147:102–12. doi: 10.1111/j.1365-2141.2009.07822.x
37. Scheinberg P, Marte M, Nunez O, Young NS. Paroxysmal nocturnal hemoglobinuria clones in severe aplastic anemia patients treated with horse anti-thymocyte globulin plus cyclosporine. Haematologica. (2010) 95:1075–80. doi: 10.3324/haematol.2009.017889
38. Aksoy M, Erdem S, Dincol G, Bakioglu I, Kutlar A. Aplastic anemia due to chemicals and drugs: a study of 108 patients. Sex Transm Dis. (1984) 11:347–50. doi: 10.1097/00007435-198410001-00007
39. Rich ML, Ritterhoff RJ, Hoffmann RJ. A fatal case of aplastic anemia following chloramphenicol (chloromycetin) therapy. Ann Intern Med. (1950) 33:1459–67. doi: 10.7326/0003-4819-33-6-1459
40. Solimando AG, Palumbo C, Pragnell MV, Bittrich M, Argentiero A, Krebs M. Aplastic anemia as a roadmap for bone marrow failure: an overview and a clinical workflow. Int J Mol Sci. (2022) 23(19):11765. doi: 10.3390/ijms231911765
41. McReynolds LJ, Rafati M, Wang Y, Ballew BJ, Kim J, Williams VV, et al. Genetic testing in severe aplastic anemia is required for optimal hematopoietic cell transplant outcomes. Blood. (2022) 140:909–21. doi: 10.1182/blood.2022016508
42. Shimano KA, Narla A, Rose MJ, Gloude NJ, Allen SW, Bergstrom K, et al. Diagnostic work-up for severe aplastic anemia in children: Consensus of the North American Pediatric Aplastic Anemia Consortium. Am J Hematol. (2021) 96:1491–504. doi: 10.1002/ajh.26310
43. Schoettler ML, Nathan DG. The pathophysiology of acquired aplastic anemia: current concepts revisited. Hematol Oncol Clin North Am. (2018) 32:581–94. doi: 10.1016/j.hoc.2018.03.001
44. Oved JH, Babushok DV, Lambert MP, Wolfset N, Kowalska MA, Poncz M, et al. Human mutational constraint as a tool to understand biology of rare and emerging bone marrow failure syndromes. Blood Adv. (2020) 4:5232–45. doi: 10.1182/bloodadvances.2020002687
45. Shahidi NT, Diamond LK. Testosterone-induced remission in aplastic anemia of both acquired and congenital types. Further observations in 24 cases. N Engl J Med. (1961) 264:953–67. doi: 10.1056/NEJM196105112641901
46. Nassani M, Fakih RE, Passweg J, Cesaro S, Alzahrani H, Alahmari A, et al. The role of androgen therapy in acquired aplastic anemia and other bone marrow failure syndromes. Front Oncol. (2023) 13:1135160. doi: 10.3389/fonc.2023.1135160
47. Li FP, Alter BP, Nathan DG. The mortality of acquired aplastic anemia in children. Blood. (1972) 40:153–62. doi: 10.1182/blood.V40.2.153.153
48. Camitta BM, Thomas ED. Severe aplastic anaemia: a prospective study of the effect of androgens or transplantation on haematological recovery and survival. Clin Haematol. (1978) 7:587–95. doi: 10.1016/S0308-2261(21)00051-5
49. Camitta BM, Thomas ED, Nathan DG, Santos G, Gordon-Smith EC, Gale RP, et al. Severe aplastic anemia: a prospective study of the effect of early marrow transplantation on acute mortality. Blood. (1976) 48:63–70. doi: 10.1182/blood.V48.1.63.bloodjournal48163
50. Champlin RE, Ho WG, Feig SA, Winston DJ, Lenarsky C, Gale RP. Do androgens enhance the response to antithymocyte globulin in patients with aplastic anemia? A prospective randomized trial. Blood. (1985) 66:184–8. doi: 10.1182/blood.V66.1.184.184
51. Bacigalupo A, Chaple M, Hows J, Van Lint MT, McCann S, Milligan D, et al. Treatment of aplastic anaemia (AA) with antilymphocyte globulin (ALG) and methylprednisolone (MPred) with or without androgens: a randomized trial from the EBMT SAA working party. Br J Haematol. (1993) 83:145–51. doi: 10.1111/j.1365-2141.1993.tb04645.x
52. Scheinberg P. Acquired severe aplastic anaemia: how medical therapy evolved in the 20th and 21st centuries. Br J Haematol. (2021) 194:954–69. doi: 10.1111/bjh.17403
53. Miller WJ, Branda RF, Flynn PJ, Howe RB, Ramsay NK, Condie RM, et al. Antithymocyte globulin treatment of severe aplastic anaemia. Br J Haematol. (1983) 55:17–25. doi: 10.1111/j.1365-2141.1983.tb01220.x
54. Mohty M. Mechanisms of action of antithymocyte globulin: T-cell depletion and beyond. Leukemia. (2007) 21:1387–94. doi: 10.1038/sj.leu.2404683
55. Townsley DM, Scheinberg P, Winkler T, Desmond R, Dumitriu B, Rios O, et al. Eltrombopag added to standard immunosuppression for aplastic anemia. N Engl J Med. (2017) 376:1540–50. doi: 10.1056/NEJMoa1613878
56. Desmond R, Townsley DM, Dumitriu B, Olnes MJ, Scheinberg P, Bevans M, et al. Eltrombopag restores trilineage hematopoiesis in refractory severe aplastic anemia that can be sustained on discontinuation of drug. Blood. (2014) 123:1818–25. doi: 10.1182/blood-2013-10-534743
57. Groarke EM, Patel BA, Gutierrez-Rodrigues F, Rios O, Lotter J, Baldoni D, et al. Eltrombopag added to immunosuppression for children with treatment-naive severe aplastic anaemia. Br J Haematol. (2021) 192:605–14. doi: 10.1111/bjh.17232
58. Alvarado LJ, Huntsman HD, Cheng H, Townsley DM, Winkler T, Feng X, et al. Eltrombopag maintains human hematopoietic stem and progenitor cells under inflammatory conditions mediated by IFN-gamma. Blood. (2019) 133:2043–55. doi: 10.1182/blood-2018-11-884486
59. Bierings MKK, Masmas T, Schmugge M, Sedlacek P, Sevilla J, Strahm B, et al. EWOG-SAA: consensus for the treatment of SAA, version 1.7., 21.03.2023. EWOG-MDS/SAA European Working Group of Myelodysplastic Syndrome (MDS) and Severe Aplastic Anemia (SAA) in children and adolescents. (2023).
60. Carreras ED C, Mohty M, Kroger N. The EBMT Handbook. Carreras ED C, Mohty M, Kroger N, editors (2019) Cham, Switzerland: Springer Nature Switzerland AG Gewerbesstrasse, p. 185.
61. Killick SB, Bown N, Cavenagh J, Dokal I, Foukaneli T, Hill A, et al. Guidelines for the diagnosis and management of adult aplastic anaemia. Br J Haematol. (2016) 172:187–207. doi: 10.1111/bjh.13853
62. Passweg JR, Marsh JC. Aplastic anemia: first-line treatment by immunosuppression and sibling marrow transplantation. Hematol Am Soc Hematol Educ Program. (2010) 2010:36–42. doi: 10.1182/asheducation-2010.1.36
63. Pulsipher MA, Young NS, Tolar J, Risitano AM, Deeg HJ, Anderlini P, et al. Optimization of therapy for severe aplastic anemia based on clinical, biologic, and treatment response parameters: conclusions of an international working group on severe aplastic anemia convened by the Blood and Marrow Transplant Clinical Trials Network, March 2010. Biol Blood Marrow Transplant. (2011) 17:291–9. doi: 10.1016/j.bbmt.2010.10.028
64. Saracco P, Quarello P, Iori AP, Zecca M, Longoni D, Svahn J, et al. Cyclosporin A response and dependence in children with acquired aplastic anaemia: a multicentre retrospective study with long-term observation follow-up. Br J Haematol. (2008) 140:197–205. doi: 10.1111/j.1365-2141.2007.06903.x
65. Pawelec K, Matysiak M, Niewiadomska E, Rokicka-Milewska R, Kowalczyk J, Stefaniak J, et al. [Results of immunosupressive therapy in children with severe aplastic anaemia. Report of the Polish Paediatric Haematology Group]. Med Wieku Rozwoj. (2008) 12:1092–7.
66. Fuhrer M, Rampf U, Baumann I, Faldum A, Niemeyer C, Janka-Schaub G, et al. Immunosuppressive therapy for aplastic anemia in children: a more severe disease predicts better survival. Blood. (2005) 106:2102–4. doi: 10.1182/blood-2005-03-0874
67. Scheinberg P, Nunez O, Weinstein B, Scheinberg P, Biancotto A, Wu CO, et al. Horse versus rabbit antithymocyte globulin in acquired aplastic anemia. N Engl J Med. (2011) 365:430–8. doi: 10.1056/NEJMoa1103975
68. Marsh JC, Bacigalupo A, Schrezenmeier H, Tichelli A, Risitano AM, Passweg JR, et al. Prospective study of rabbit antithymocyte globulin and cyclosporine for aplastic anemia from the EBMT Severe Aplastic Anaemia Working Party. Blood. (2012) 119:5391–6. doi: 10.1182/blood-2012-02-407684
69. Afable MG 2nd, Shaik M, Sugimoto Y, Elson P, Clemente M, Makishima H, et al. Efficacy of rabbit anti-thymocyte globulin in severe aplastic anemia. Haematologica. (2011) 96:1269–75. doi: 10.3324/haematol.2011.042622
70. Shin SH, Yoon JH, Yahng SA, Lee SE, Cho BS, Eom KS, et al. The efficacy of rabbit antithymocyte globulin with cyclosporine in comparison to horse antithymocyte globulin as a first-line treatment in adult patients with severe aplastic anemia: a single-center retrospective study. Ann Hematol. (2013) 92:817–24. doi: 10.1007/s00277-013-1674-8
71. Jeong DC, Chung NG, Cho B, Zou Y, Ruan M, Takahashi Y, et al. Long-term outcome after immunosuppressive therapy with horse or rabbit antithymocyte globulin and cyclosporine for severe aplastic anemia in children. Haematologica. (2014) 99:664–71. doi: 10.3324/haematol.2013.089268
72. Vallejo C, Montesinos P, Polo M, Cuevas B, Morado M, Rosell A, et al. Rabbit antithymocyte globulin versus horse antithymocyte globulin for treatment of acquired aplastic anemia: a retrospective analysis. Ann Hematol. (2015) 94:947–54. doi: 10.1007/s00277-015-2305-3
73. Suzuki T, Kobayashi H, Kawasaki Y, Okazuka K, Hatano K, Fujiwara S, et al. Efficacy of combination therapy with anti-thymocyte globulin and cyclosporine A as a first-line treatment in adult patients with aplastic anemia: a comparison of rabbit and horse formulations. Int J Hematol. (2016) 104:446–53. doi: 10.1007/s12185-016-2046-7
74. Vaht K, Goransson M, Carlson K, Isaksson C, Lenhoff S, Sandstedt A, et al. Low response rate to ATG-based immunosuppressive therapy in very severe aplastic anaemia - A Swedish nationwide cohort study. Eur J Haematol. (2018) 100:613–20. doi: 10.1111/ejh.13057
75. Chang MH, Kim KH, Kim HS, Jun HJ, Kim DH, Jang JH, et al. Predictors of response to immunosuppressive therapy with antithymocyte globulin and cyclosporine and prognostic factors for survival in patients with severe aplastic anemia. Eur J Haematol. (2010) 84:154–9. doi: 10.1111/j.1600-0609.2009.01378.x
76. Yoshimi A, Niemeyer CM, Fuhrer MM, Strahm B. Comparison of the efficacy of rabbit and horse antithymocyte globulin for the treatment of severe aplastic anemia in children. Blood. (2013) 121:860–1. doi: 10.1182/blood-2012-10-461509
77. Pawelec K, Salamonowicz M, Panasiuk A, Demkow U, Kowalczyk J, Balwierz W, et al. First-line immunosuppressive treatment in children with aplastic anemia: rabbit antithymocyte globulin. Adv Exp Med Biol. (2015) 836:55–62. doi: 10.1007/5584_2014_38
78. Hayakawa J, Kanda J, Akahoshi Y, Harada N, Kameda K, Ugai T, et al. Meta-analysis of treatment with rabbit and horse antithymocyte globulin for aplastic anemia. Int J Hematol. (2017) 105:578–86. doi: 10.1007/s12185-017-2179-3
79. Yang N, Chen J, Zhang H, Dai Z, Yao H, Ma X, et al. Horse versus rabbit antithymocyte globulin in immunosuppressive therapy of treatment-naive aplastic anemia: a systematic review and meta-analysis. Ann Hematol. (2017) 96:2031–43. doi: 10.1007/s00277-017-3136-1
80. Bing H, Siyi Y, Wei Z, Jian L, Minghui D, Li J, et al. The use of anti-human T lymphocyte porcine immunoglobulin and cyclosporine a to treat patients with acquired severe aplastic anemia. Acta Haematol. (2010) 124:245–50. doi: 10.1159/000321790
81. Zhu Y, Yang Y, Yang W, Song L, Li Y, Fan H, et al. Efficacy and safety of porcine ALG compared to rabbit ATG as first-line treatment for children with acquired aplastic anemia. Eur J Haematol. (2020) 104:562–70. doi: 10.1111/ejh.13398
82. Chen M, Liu C, Qiao X, Zhou D, Zhuang J, Han B. Comparative study of porcine anti-human lymphocyte immunoglobulin and rabbit anti-human thymocyte immunoglobulin as a first-line treatment of acquired severe aplastic anemia. Leuk Res. (2018) 65:55–60. doi: 10.1016/j.leukres.2018.01.001
83. Liu L, Ding L, Hao L, Zhang X, Li X, Zhang L, et al. Efficacy of porcine antihuman lymphocyte immunoglobulin compared to rabbit antithymocyte immunoglobulin as a first-line treatment against acquired severe aplastic anemia. Ann Hematol. (2015) 94:729–37. doi: 10.1007/s00277-014-2279-6
84. Fuhrer M, Burdach S, Ebell W, Gadner H, Haas R, Harbott J, et al. Relapse and clonal disease in children with aplastic anemia (AA) after immunosuppressive therapy (IST): the SAA 94 experience. German/Austrian Pediatric Aplastic Anemia Working Group. Klin Padiatr. (1998) 210:173–9. doi: 10.1055/s-2008-1043875
85. Scheinberg P, Wu CO, Nunez O, Young NS. Long-term outcome of pediatric patients with severe aplastic anemia treated with antithymocyte globulin and cyclosporine. J Pediatr. (2008) 153:814–9. doi: 10.1016/j.jpeds.2008.06.004
86. Gu C, Zhu X, Qiao X, Zhai X, Shi W, Xie X. Multivariate logistic analysis of predictors of response to immunosuppressive therapy in children with aplastic anemia: a double-center study. Hematology. (2019) 24:282–9. doi: 10.1080/16078454.2019.1565149
87. Locasciulli A, Bruno B, Rambaldi A, Saracco P, Dufour C, Finelli C, et al. Treatment of severe aplastic anemia with antilymphocyte globulin, cyclosporine and two different granulocyte colony-stimulating factor regimens: a GITMO prospective randomized study. Haematologica. (2004) 89:1054–61.
88. Tichelli A, Schrezenmeier H, Socie G, Marsh J, Bacigalupo A, Duhrsen U, et al. A randomized controlled study in patients with newly diagnosed severe aplastic anemia receiving antithymocyte globulin (ATG), cyclosporine, with or without G-CSF: a study of the SAA Working Party of the European Group for Blood and Marrow Transplantation. Blood. (2011) 117:4434–41. doi: 10.1182/blood-2010-08-304071
89. Kaito K, Kobayashi M, Katayama T, Masuoka H, Shimada T, Nishiwaki K, et al. Long-term administration of G-CSF for aplastic anaemia is closely related to the early evolution of monosomy 7 MDS in adults. Br J Haematol. (1998) 103:297–303. doi: 10.1046/j.1365-2141.1998.01014.x
90. Kojima S, Ohara A, Tsuchida M, Kudoh T, Hanada R, Okimoto Y, et al. Risk factors for evolution of acquired aplastic anemia into myelodysplastic syndrome and acute myeloid leukemia after immunosuppressive therapy in children. Blood. (2002) 100:786–90. doi: 10.1182/blood.V100.3.786
91. Li Y, Li X, Ge M, Shi J, Qian L, Zheng Y, et al. Long-term follow-up of clonal evolutions in 802 aplastic anemia patients: a single-center experience. Ann Hematol. (2011) 90:529–37. doi: 10.1007/s00277-010-1140-9
92. Ding SX, Chen T, Wang T, Liu CY, Lu WL, Fu R. The risk of clonal evolution of granulocyte colony-stimulating factor for acquired aplastic anemia: A systematic review and meta-analysis. Acta Haematol. (2018) 140:141–5. doi: 10.1159/000491816
93. Tichelli A, de Latour RP, Passweg J, Knol-Bout C, Socie G, Marsh J, et al. Long-term outcome of a randomized controlled study in patients with newly diagnosed severe aplastic anemia treated with antithymocyte globulin and cyclosporine, with or without granulocyte colony-stimulating factor: a Severe Aplastic Anemia Working Party Trial from the European Group of Blood and Marrow Transplantation. Haematologica. (2020) 105:1223–31. doi: 10.3324/haematol.2019.222562
94. Barone A, Lucarelli A, Onofrillo D, Verzegnassi F, Bonanomi S, Cesaro S, et al. Diagnosis and management of acquired aplastic anemia in childhood. Guidelines from the Marrow Failure Study Group of the Pediatric Haemato-Oncology Italian Association (AIEOP). Blood Cells Mol Dis. (2015) 55:40–7. doi: 10.1016/j.bcmd.2015.03.007
95. Scheinberg P, Nunez O, Young NS. Retreatment with rabbit anti-thymocyte globulin and ciclosporin for patients with relapsed or refractory severe aplastic anaemia. Br J Haematol. (2006) 133:622–7. doi: 10.1111/j.1365-2141.2006.06098.x
96. Scheinberg P, Wu CO, Nunez O, Scheinberg P, Boss C, Sloand EM, et al. Treatment of severe aplastic anemia with a combination of horse antithymocyte globulin and cyclosporine, with or without sirolimus: a prospective randomized study. Haematologica. (2009) 94:348–54. doi: 10.3324/haematol.13829
97. Doney K, Dahlberg SJ, Monroe D, Storb R, Buckner CD, Thomas ED. Therapy of severe aplastic anemia with anti-human thymocyte globulin and androgens: the effect of HLA-haploidentical marrow infusion. Blood. (1984) 63:342–8. doi: 10.1182/blood.V63.2.342.bloodjournal632342
98. Scott A, Morris K, Butler J, Mills AK, Kennedy GA. Treatment of aplastic anaemia with lower-dose anti-thymocyte globulin produces similar response rates and survival as per standard dose anti-thymocyte globulin schedules. Intern Med J. (2016) 46:1198–203. doi: 10.1111/imj.13175
99. Rosenfeld SJ, Kimball J, Vining D, Young NS. Intensive immunosuppression with antithymocyte globulin and cyclosporine as treatment for severe acquired aplastic anemia. Blood. (1995) 85:3058–65. doi: 10.1182/blood.V85.11.3058.bloodjournal85113058
100. Scheinberg P, Nunez O, Wu C, Young NS. Treatment of severe aplastic anaemia with combined immunosuppression: anti-thymocyte globulin, ciclosporin and mycophenolate mofetil. Br J Haematol. (2006) 133:606–11. doi: 10.1111/j.1365-2141.2006.06085.x
101. Bacigalupo A, Oneto R, Schrezenmeier H, Hochsmann B, Dufour C, Kojima S, et al. First line treatment of aplastic anemia with thymoglobuline in Europe and Asia: Outcome of 955 patients treated 2001-2012. Am J Hematol. (2018) 93:643–8. doi: 10.1002/ajh.25081
102. Peffault de Latour R, Tabrizi R, Marcais A, Leblanc T, Lamy T, Mohty M, et al. Nationwide survey on the use of horse antithymocyte globulins (ATGAM) in patients with acquired aplastic anemia: A report on behalf of the French Reference Center for Aplastic Anemia. Am J Hematol. (2018) 93:635–42. doi: 10.1002/ajh.25050
103. Scheinberg P, Young NS. How I treat acquired aplastic anemia. Blood. (2012) 120:1185–96. doi: 10.1182/blood-2011-12-274019
104. Marsh JC, Kulasekararaj AG. Management of the refractory aplastic anemia patient: what are the options? Blood. (2013) 122:3561–7. doi: 10.1182/blood-2013-05-498279
105. Li R, Wang N, Chai X, Yang L, Liu K, He H, et al. Prolonged use of eltrombopag in patients with severe aplastic anemia in the real world. Clin Exp Med. (2023) 23:2619–27. doi: 10.1007/s10238-023-00989-3
106. Fang M, Song H, Zhang J, Li S, Shen D, Tang Y. Efficacy and safety of immunosuppressive therapy with or without eltrombopag in pediatric patients with acquired aplastic anemia: A Chinese retrospective study. Pediatr Hematol Oncol. (2021) 38:633–46. doi: 10.1080/08880018.2021.1895924
107. Goronkova O, Novichkova G, Salimova T, Kalinina I, Baidildina D, Petrova U, et al. Efficacy of combined immunosuppression with or without eltrombopag in children with newly diagnosed aplastic anemia. Blood Adv. (2023) 7:953–62. doi: 10.1182/bloodadvances.2021006716
108. Zhao Y, Yang W, Zhao X, Hu X, Hu J, Liu X, et al. Efficacy of eltrombopag with immunosuppressive therapy for children with acquired aplastic anemia. Front Pediatr. (2022) 10:1095143. doi: 10.3389/fped.2022.1095143
109. Zhao LP, Sicre De Fontbrune F, Contejean A, Abraham J, Terriou L, Chabrot C, et al. Nationwide survey in France on the use of romiplostim in patients with refractory severe aplastic anemia. Bone Marrow Transplant. (2019) 54:1161–3. doi: 10.1038/s41409-019-0452-1
110. Bussel JB, Soff G, Balduzzi A, Cooper N, Lawrence T, Semple JW. A review of romiplostim mechanism of action and clinical applicability. Drug Des Devel Ther. (2021) 15:2243–68. doi: 10.2147/DDDT.S299591
111. Lee JW, Lee SE, Jung CW, Park S, Keta H, Park SK, et al. Romiplostim in patients with refractory aplastic anaemia previously treated with immunosuppressive therapy: a dose-finding and long-term treatment phase 2 trial. Lancet Haematol. (2019) 6:e562–e72. doi: 10.1016/S2352-3026(19)30153-X
112. Jang JH, Tomiyama Y, Miyazaki K, Nagafuji K, Usuki K, Uoshima N, et al. Efficacy and safety of romiplostim in refractory aplastic anaemia: a Phase II/III, multicentre, open-label study. Br J Haematol. (2021) 192:190–9. doi: 10.1111/bjh.17190
113. Ise M, Iizuka H, Kamoda Y, Hirao M, Kida M, Usuki K. Romiplostim is effective for eltrombopag-refractory aplastic anemia: results of a retrospective study. Int J Hematol. (2020) 112:787–94. doi: 10.1007/s12185-020-02971-1
114. McQuilten Z, Heritier S, Fox L, Fox V, Young L, Blombery P, et al. Efficacy and safety of avatrombopag in combination with immunosuppressive therapy in treatment-naive and relapsed/refractory severe aplastic anaemia: protocol for the DIAAMOND-Ava-FIRST and DIAAMOND-Ava-NEXT Bayesian Optimal Phase II trials. BMJ Open. (2024) 14:e076246. doi: 10.1136/bmjopen-2023-076246
115. Calado RT, Yewdell WT, Wilkerson KL, Regal JA, Kajigaya S, Stratakis CA, et al. Sex hormones, acting on the TERT gene, increase telomerase activity in human primary hematopoietic cells. Blood. (2009) 114:2236–43. doi: 10.1182/blood-2008-09-178871
116. Townsley DM, Dumitriu B, Young NS. Danazol treatment for telomere diseases. N Engl J Med. (2016) 375:1095–6. doi: 10.1056/NEJMc1607752
117. Mori H, Nakagawa M, Itoh N, Wada K, Tamaya T. Danazol suppresses the production of interleukin-1 beta and tumor necrosis factor by human monocytes. Am J Reprod Immunol. (1990) 24:45–50. doi: 10.1111/j.1600-0897.1990.tb01037.x
118. Leleu X, Terriou L, Duhamel A, Moreau AS, Andrieux J, Dupire S, et al. Long-term outcome in acquired aplastic anemia treated with an intensified dose schedule of horse antilymphocyte globulin in combination with androgens. Ann Hematol. (2006) 85:711–6. doi: 10.1007/s00277-006-0152-y
119. Gao Q, Zhang L, Zhao X, Zhu Y, Peng G, Li Y, et al. Eltrombopag, oral immunosuppressant and androgen combination therapy in twelve patients with refractory severe aplastic anemia. Hematology. (2020) 25:341–7. doi: 10.1080/16078454.2020.1815129
120. Sanchez-Medal L, Gomez-Leal A, Duarte L, Guadalupe Rico M. Anabolic androgenic steroids in the treatment of acquired aplastic anemia. Blood. (1969) 34:283–300. doi: 10.1182/blood.V34.3.283.283
121. Scheinberg P, Nunez O, Weinstein B, Scheinberg P, Wu CO, Young NS. Activity of alemtuzumab monotherapy in treatment-naive, relapsed, and refractory severe acquired aplastic anemia. Blood. (2012) 119:345–54. doi: 10.1182/blood-2011-05-352328
122. Storb R, Etzioni R, Anasetti C, Appelbaum FR, Buckner CD, Bensinger W, et al. Cyclophosphamide combined with antithymocyte globulin in preparation for allogeneic marrow transplants in patients with aplastic anemia. Blood. (1994) 84:941–9. doi: 10.1182/blood.V84.3.941.bloodjournal843941
123. Storb R, Weiden PL, Sullivan KM, Appelbaum FR, Beatty P, Buckner CD, et al. Second marrow transplants in patients with aplastic anemia rejecting the first graft: use of a conditioning regimen including cyclophosphamide and antithymocyte globulin. Blood. (1987) 70:116–21. doi: 10.1182/blood.V70.1.116.bloodjournal701116
124. Kroger N, Zabelina T, Renges H, Kruger W, Kordes U, Rischewski J, et al. Long-term follow-up of allogeneic stem cell transplantation in patients with severe aplastic anemia after conditioning with cyclophosphamide plus antithymocyte globulin. Ann Hematol. (2002) 81:627–31. doi: 10.1007/s00277-002-0566-0
125. Deeg HJ, O’Donnell M, Tolar J, Agarwal R, Harris RE, Feig SA, et al. Optimization of conditioning for marrow transplantation from unrelated donors for patients with aplastic anemia after failure of immunosuppressive therapy. Blood. (2006) 108:1485–91. doi: 10.1182/blood-2006-03-005041
126. Perez-Albuerne ED, Eapen M, Klein J, Gross TJ, Lipton JM, Baker KS, et al. Outcome of unrelated donor stem cell transplantation for children with severe aplastic anemia. Br J Haematol. (2008) 141:216–23. doi: 10.1111/j.1365-2141.2008.07030.x
127. Bacigalupo A, Socie G, Lanino E, Prete A, Locatelli F, Locasciulli A, et al. Fludarabine, cyclophosphamide, antithymocyte globulin, with or without low dose total body irradiation, for alternative donor transplants, in acquired severe aplastic anemia: a retrospective study from the EBMT-SAA Working Party. Haematologica. (2010) 95:976–82. doi: 10.3324/haematol.2009.018267
128. Alotaibi H, Aljurf M, de Latour R, Alfayez M, Bacigalupo A, Fakih RE, et al. Upfront alternative donor transplant versus immunosuppressive therapy in patients with severe aplastic anemia who lack a fully HLA-matched related donor: systematic review and meta-analysis of retrospective studies, on behalf of the severe aplastic anemia working party of the european group for blood and marrow transplantation. Transplant Cell Ther. (2022) 28:105 e1– e7. doi: 10.1016/j.jtct.2021.10.006
129. Dufour C, Veys P, Carraro E, Bhatnagar N, Pillon M, Wynn R, et al. Similar outcome of upfront-unrelated and matched sibling stem cell transplantation in idiopathic paediatric aplastic anaemia. A study on behalf of the UK Paediatric BMT Working Party, Paediatric Diseases Working Party and Severe Aplastic Anaemia Working Party of EBMT. Br J Haematol. (2015) 171:585–94. doi: 10.1111/bjh.13614
130. Pulsipher MA, Lehmann LE, Bertuch AA, Sasa G, Olson T, Nakano T, et al. A study assessing the feasibility of randomization of pediatric and young adult patients between matched unrelated donor bone marrow transplantation and immune-suppressive therapy for newly diagnosed severe aplastic anemia: A joint pilot trial of the North American Pediatric Aplastic Anemia Consortium and the Pediatric Transplantation and Cellular Therapy Consortium. Pediatr Blood Cancer. (2020) 67:e28444. doi: 10.1002/pbc.28444
131. Georges GE, Doney K, Storb R. Severe aplastic anemia: allogeneic bone marrow transplantation as first-line treatment. Blood Adv. (2018) 2:2020–8. doi: 10.1182/bloodadvances.2018021162
132. Moore MA, Castro-Malaspina H. Immunosuppression in aplastic anemia–postponing the inevitable? N Engl J Med. (1991) 324:1358–60. doi: 10.1056/NEJM199105093241909
133. Scheinberg P, Wu CO, Nunez O, Young NS. Predicting response to immunosuppressive therapy and survival in severe aplastic anaemia. Br J Haematol. (2009) 144:206–16. doi: 10.1111/j.1365-2141.2008.07450.x
134. Kordasti S, Costantini B, Seidl T, Perez Abellan P, Martinez Llordella M, McLornan D, et al. Deep phenotyping of Tregs identifies an immune signature for idiopathic aplastic anemia and predicts response to treatment. Blood. (2016) 128:1193–205. doi: 10.1182/blood-2016-03-703702
135. Sloand E, Kim S, Maciejewski JP, Tisdale J, Follmann D, Young NS. Intracellular interferon-gamma in circulating and marrow T cells detected by flow cytometry and the response to immunosuppressive therapy in patients with aplastic anemia. Blood. (2002) 100:1185–91. doi: 10.1182/blood-2002-01-0035
136. Chang H, Zeng F, Zhang JY, Mu XY, Meng WT, Ma HB, et al. Association of the interferon-gamma single nucleotide polymorphism +874(T/A) with response to immunosuppressive therapy in patients with severe aplastic anemia. Blood Cells Mol Dis. (2010) 45:313–6. doi: 10.1016/j.bcmd.2010.09.003
137. Jang J, Kim H, Park SS, Kim M, Min YK, Jeong HO, et al. Single-cell RNA sequencing reveals novel cellular factors for response to immunosuppressive therapy in aplastic anemia. Hemasphere. (2023) 7:e977. doi: 10.1097/HS9.0000000000000977
138. Hosokawa K, Kajigaya S, Feng X, Desierto MJ, Fernandez Ibanez MD, Rios O, et al. A plasma microRNA signature as a biomarker for acquired aplastic anemia. Haematologica. (2017) 102:69–78. doi: 10.3324/haematol.2016.151076
139. Elmahdi S, Muramatsu H, Narita A, Ismael O, Hama A, Nishio N, et al. Markedly high plasma thrombopoietin (TPO) level is a predictor of poor response to immunosuppressive therapy in children with acquired severe aplastic anemia. Pediatr Blood Cancer. (2016) 63:659–64. doi: 10.1002/pbc.25820
140. Park HS, Park SN, Im K, Kim SM, Kim JA, Hwang SM, et al. Telomere length and somatic mutations in correlation with response to immunosuppressive treatment in aplastic anaemia. Br J Haematol. (2017) 178:603–15. doi: 10.1111/bjh.14691
141. Bacigalupo A, Socie G, Hamladji RM, Aljurf M, Maschan A, Kyrcz-Krzemien S, et al. Current outcome of HLA identical sibling versus unrelated donor transplants in severe aplastic anemia: an EBMT analysis. Haematologica. (2015) 100:696–702. doi: 10.3324/haematol.2014.115345
142. Chaudhry QUN, Iftikhar R, Satti TM, Mahmood SK, Ghafoor T, Shamshad GU, et al. Outcome of fludarabine-based conditioning in high-risk aplastic anemia patients undergoing matched related donor transplantation: A single-center study from Pakistan. Biol Blood Marrow Transplant. (2019) 25:2375–82. doi: 10.1016/j.bbmt.2019.07.029
143. Sheth VS, Potter V, Gandhi SA, Kulasekararaj AG, de Lavallade H, Muus P, et al. Similar outcomes of alemtuzumab-based hematopoietic cell transplantation for SAA patients older or younger than 50 years. Blood Adv. (2019) 3:3070–9. doi: 10.1182/bloodadvances.2019000480
144. Bacigalupo A, Brand R, Oneto R, Bruno B, Socie G, Passweg J, et al. Treatment of acquired severe aplastic anemia: bone marrow transplantation compared with immunosuppressive therapy–The European Group for Blood and Marrow Transplantation experience. Semin Hematol. (2000) 37:69–80. doi: 10.1053/shem.2000.0370069
145. Yoshida N, Kobayashi R, Yabe H, Kosaka Y, Yagasaki H, Watanabe K, et al. First-line treatment for severe aplastic anemia in children: bone marrow transplantation from a matched family donor versus immunosuppressive therapy. Haematologica. (2014) 99:1784–91. doi: 10.3324/haematol.2014.109355
146. Shin SH, Jeon YW, Yoon JH, Yahng SA, Lee SE, Cho BS, et al. Comparable outcomes between younger (≦̸40 years) and older (>40 years) adult patients with severe aplastic anemia after HLA-matched sibling stem cell transplantation using fludarabine-based conditioning. Bone Marrow Transplant. (2016) 51:1456–63. doi: 10.1038/bmt.2016.171
147. Rice C, Eikema DJ, Marsh JCW, Knol C, Hebert K, Putter H, et al. Allogeneic hematopoietic cell transplantation in patients aged 50Years or older with severe aplastic anemia. Biol Blood Marrow Transplant. (2019) 25:488–95. doi: 10.1016/j.bbmt.2018.08.029
148. Jalaeikhoo H, Khajeh-Mehrizi A. Immunosuppressive therapy in patients with aplastic anemia: a single-center retrospective study. PloS One. (2015) 10:e0126925. doi: 10.1371/journal.pone.0126925
149. Marsh JCW, Risitano AM, Mufti GJ. The case for upfront HLA-matched unrelated donor hematopoietic stem cell transplantation as a curative option for adult acquired severe aplastic anemia. Biol Blood Marrow Transplant. (2019) 25:e277–e84. doi: 10.1016/j.bbmt.2019.05.012
150. Samarasinghe S, Clesham K, Iacobelli S, Sbianchi G, Knol C, Hamladji RM, et al. Impact of T-cell depletion strategies on outcomes following hematopoietic stem cell transplantation for idiopathic aplastic anemia: A study on behalf of the European blood and marrow transplant severe aplastic anemia working party. Am J Hematol. (2019) 94:80–6. doi: 10.1002/ajh.25314
151. Marsh JC, Pearce RM, Koh MB, Lim Z, Pagliuca A, Mufti GJ, et al. Retrospective study of alemtuzumab vs ATG-based conditioning without irradiation for unrelated and matched sibling donor transplants in acquired severe aplastic anemia: a study from the British Society for Blood and Marrow Transplantation. Bone Marrow Transplant. (2014) 49:42–8. doi: 10.1038/bmt.2013.115
152. Kang HJ, Hong KT, Lee JW, Kim H, Park KD, Shin HY, et al. Improved outcome of a reduced toxicity-fludarabine, cyclophosphamide, plus antithymocyte globulin conditioning regimen for unrelated donor transplantation in severe aplastic anemia: comparison of 2 multicenter prospective studies. Biol Blood Marrow Transplant. (2016) 22:1455–9. doi: 10.1016/j.bbmt.2016.04.003
153. Salamonowicz-Bodzioch M, Rosa M, Fraczkiewicz J, Gorczynska E, Gul K, Janeczko-Czarnecka M, et al. Fludarabine-cyclophosphamide-based conditioning with antithymocyte globulin serotherapy is associated with durable engraftment and manageable infections in children with severe aplastic anemia. J Clin Med. (2021) 10(19):4416. doi: 10.3390/jcm10194416
154. Darrigo LG Jr., Colturato V, de Souza MP, Loth G, Calixto R, Seber A, et al. Allogeneic bone marrow transplants for pediatric severe aplastic anemia: real-world data comparing matched related and unrelated donors in a developing country. retrospective study on behalf of the pediatric hematopoietic stem cell transplant working group of the brazilian bone marrow transplantation society (sbtmo) and the brazil-seattle consortium (gedeco). Pediatr Transplant. (2019) 23:e13552. doi: 10.1016/j.ejca.2022.05.013
155. Yoo JW, Kim S, Lee JW, Jang PS, Jeong DC, Cho B, et al. High failure-free survival after unrelated donor peripheral blood stem cell transplantation in pediatric severe aplastic anemia. Transplant Cell Ther. (2022) 28:103 e1– e8. doi: 10.1016/j.jtct.2021.11.008
156. Devillier R, Eikema DJ, Dufour C, Aljurf M, Wu D, Maschan A, et al. Graft-versus-host disease and relapse/rejection-free survival after allogeneic transplantation for idiopathic severe aplastic anemia: a comprehensive analysis from the SAAWP of the EBMT. Haematologica. (2023) 108:2305–15. doi: 10.3324/haematol.2022.281876
157. Chorao P, Montoro J, Balaguer-Rosello A, Guerreiro M, Villalba M, Facal A, et al. T cell-depleted peripheral blood versus unmanipulated bone marrow in matched sibling transplantation for aplastic anemia. Transplant Cell Ther. (2023) 29:322.e1–e5. doi: 10.1016/j.jtct.2023.01.016
158. Prata PH, Eikema DJ, Afansyev B, Bosman P, Smiers F, Diez-Martin JL, et al. Haploidentical transplantation and posttransplant cyclophosphamide for treating aplastic anemia patients: a report from the EBMT Severe Aplastic Anemia Working Party. Bone Marrow Transplant. (2020) 55:1050–8. doi: 10.1038/s41409-019-0773-0
159. DeZern AE, Zahurak M, Symons HJ, Cooke KR, Huff CA, Jain T, et al. Alternative donor BMT with posttransplant cyclophosphamide as initial therapy for acquired severe aplastic anemia. Blood. (2023) 141:3031–8. doi: 10.1182/blood.2023020435
160. Gong S, Chen C, Chen K, Yang R, Wang L, Yang K, et al. Alternative transplantation with post-transplantation cyclophosphamide in aplastic anemia: A retrospective report from the BMF-WG of Hunan province, China. Transplant Cell Ther. (2023) 29:48.e1– e7. doi: 10.1016/j.jtct.2022.10.006
161. Bedoschi G, Navarro PA, Oktay K. Chemotherapy-induced damage to ovary: mechanisms and clinical impact. Future Oncol. (2016) 12:2333–44. doi: 10.2217/fon-2016-0176
162. Loren AW, Senapati S. Fertility preservation in patients with hematologic Malignancies and recipients of hematopoietic cell transplants. Blood. (2019) 134:746–60. doi: 10.1182/blood.2018846790
163. Mulder RL, Font-Gonzalez A, Green DM, Loeffen EAH, Hudson MM, Loonen J, et al. Fertility preservation for male patients with childhood, adolescent, and young adult cancer: recommendations from the PanCareLIFE Consortium and the International Late Effects of Childhood Cancer Guideline Harmonization Group. Lancet Oncol. (2021) 22:e57–67. doi: 10.1016/S1470-2045(20)30582-9
164. Mulder RL, Font-Gonzalez A, Hudson MM, van Santen HM, Loeffen EAH, Burns KC, et al. Fertility preservation for female patients with childhood, adolescent, and young adult cancer: recommendations from the PanCareLIFE Consortium and the International Late Effects of Childhood Cancer Guideline Harmonization Group. Lancet Oncol. (2021) 22:e45–56. doi: 10.1016/S1470-2045(20)30594-5
165. Rives N, Courbiere B, Almont T, Kassab D, Berger C, Grynberg M, et al. What should be done in terms of fertility preservation for patients with cancer? The French 2021 guidelines. Eur J Cancer. (2022) 173:146–66. doi: 10.1016/j.ejca.2022.05.013
166. Green DM, Nolan VG, Goodman PJ, Whitton JA, Srivastava D, Leisenring WM, et al. The cyclophosphamide equivalent dose as an approach for quantifying alkylating agent exposure: a report from the Childhood Cancer Survivor Study. Pediatr Blood Cancer. (2014) 61:53–67. doi: 10.1002/pbc.24679
167. Green DM, Liu W, Kutteh WH, Ke RW, Shelton KC, Sklar CA, et al. Cumulative alkylating agent exposure and semen parameters in adult survivors of childhood cancer: a report from the St Jude Lifetime Cohort Study. Lancet Oncol. (2014) 15:1215–23. doi: 10.1016/S1470-2045(14)70408-5
168. Ades L, Mary JY, Robin M, Ferry C, Porcher R, Esperou H, et al. Long-term outcome after bone marrow transplantation for severe aplastic anemia. Blood. (2004) 103:2490–7. doi: 10.1182/blood-2003-07-2546
169. Gluckman E, Esperou H, Devergie A, Traineau R, Leverger G, Schaison G. Pediatric bone marrow transplantation for leukemia and aplastic anemia. Report of 222 cases transplanted in a single center. Nouv Rev Fr Hematol (1978). (1989) 31:111–4. doi: 10.1007/s00005-012-0174-1
170. Bacigalupo A, Locatelli F, Lanino E, Marsh J, Socie G, Maury S, et al. Fludarabine, cyclophosphamide and anti-thymocyte globulin for alternative donor transplants in acquired severe aplastic anemia: a report from the EBMT-SAA Working Party. Bone Marrow Transplant. (2005) 36:947–50. doi: 10.1038/sj.bmt.1705165
171. Resnick IB, Aker M, Shapira MY, Tsirigotis PD, Bitan M, Abdul-Hai A, et al. Allogeneic stem cell transplantation for severe acquired aplastic anaemia using a fludarabine-based preparative regimen. Br J Haematol. (2006) 133:649–54. doi: 10.1111/j.1365-2141.2006.06084.x
172. Viollier R, Socie G, Tichelli A, Bacigalupo A, Korthof ET, Marsh J, et al. Recent improvement in outcome of unrelated donor transplantation for aplastic anemia. Bone Marrow Transplant. (2008) 41:45–50. doi: 10.1038/sj.bmt.1705894
173. George B, Mathews V, Viswabandya A, Kavitha ML, Srivastava A, Chandy M. Fludarabine based reduced intensity conditioning regimens in children undergoing allogeneic stem cell transplantation for severe aplastic anemia. Pediatr Transplant. (2008) 12:14–9. doi: 10.1111/j.1399-3046.2007.00825.x
174. Kang HJ, Shin HY, Choi HS, Ahn HS. Fludarabine, cyclophosphamide plus thymoglobulin conditioning regimen for unrelated bone marrow transplantation in severe aplastic anemia. Bone Marrow Transplant. (2004) 34:939–43. doi: 10.1038/sj.bmt.1704720
175. Chung NG, Lee JW, Jang PS, Jeong DC, Cho B, Kim HK. Reduced dose cyclophosphamide, fludarabine and antithymocyte globulin for sibling and unrelated transplant of children with severe and very severe aplastic anemia. Pediatr Transplant. (2013) 17:387–93. doi: 10.1111/petr.12073
176. McGuinn C, Geyer MB, Jin Z, Garvin JH, Satwani P, Bradley MB, et al. Pilot trial of risk-adapted cyclophosphamide intensity based conditioning and HLA matched sibling and unrelated cord blood stem cell transplantation in newly diagnosed pediatric and adolescent recipients with acquired severe aplastic anemia. Pediatr Blood Cancer. (2014) 61:1289–94. doi: 10.1002/pbc.24976
177. Szpecht D, Gorczynska E, Kalwak K, Owoc-Lempach J, Choma M, Styczynski J, et al. Matched sibling versus matched unrelated allogeneic hematopoietic stem cell transplantation in children with severe acquired aplastic anemia: experience of the polish pediatric group for hematopoietic stem cell transplantation. Arch Immunol Ther Exp (Warsz). (2012) 60:225–33. doi: 10.1007/s00005-012-0174-1
178. Vissers L, van der Burg M, Lankester A, Smiers F, Mohseny A. Optimizing diagnostic methods and stem cell transplantation outcomes in pediatric bone marrow failure: a 50-year single center experience. Eur J Pediatr. (2023) 182:4195–203. doi: 10.1007/s00431-023-05093-y
179. Alsultan A, Abujoub R, Alsudairy R, Memon S, Jarrar MS, Alafghani S, et al. Human leucocyte antigen-matched related haematopoietic stem cell transplantation using low-dose cyclophosphamide, fludarabine and thymoglobulin in children with severe aplastic anaemia. Br J Haematol. (2023) 203:255–63. doi: 10.1111/bjh.19004
180. Ngwube A, Hayashi RJ, Murray L, Loechelt B, Dalal J, Jaroscak J, et al. Alemtuzumab based reduced intensity transplantation for pediatric severe aplastic anemia. Pediatr Blood Cancer. (2015) 62:1270–6. doi: 10.1002/pbc.25458
181. Wang YM, Anderson E, Gloude N, Marion A, King C, Leung W, et al. Irradiation-free RIC HSCT has a tolerable safety profile and is effective therapy for pediatric bone marrow failure syndromes. Pediatr Transplant. (2021) 25:e13855. doi: 10.1111/petr.13855
182. Bejanyan N, Kim S, Hebert KM, Kekre N, Abdel-Azim H, Ahmed I, et al. Choice of conditioning regimens for bone marrow transplantation in severe aplastic anemia. Blood Adv. (2019) 3:3123–31. doi: 10.1182/bloodadvances.2019000722
183. Aljurf M, Al-Zahrani H, Van Lint MT, Passweg JR. Standard treatment of acquired SAA in adult patients 18-40 years old with an HLA-identical sibling donor. Bone Marrow Transplant. (2013) 48:178–9. doi: 10.1038/bmt.2012.223
184. ElGohary G, El Fakih R, de Latour R, Risitano A, Marsh J, Schrezenmeier H, et al. Haploidentical hematopoietic stem cell transplantation in aplastic anemia: a systematic review and meta-analysis of clinical outcome on behalf of the severe aplastic anemia working party of the European group for blood and marrow transplantation (SAAWP of EBMT). Bone Marrow Transplant. (2020) 55:1906–17. doi: 10.1038/s41409-020-0897-2
185. Lee SE, Park SS, Jeon YW, Yoon JH, Cho BS, Eom KS, et al. Optimal conditioning regimen for haplo-identical stem cell transplantation in adult patients with acquired severe aplastic anemia: Prospective de-escalation study of TBI and ATG dose. Am J Hematol. (2018) 93:1368–75. doi: 10.1002/ajh.25257
186. DeZern AE, Zahurak ML, Symons HJ, Cooke KR, Rosner GL, Gladstone DE, et al. Haploidentical BMT for severe aplastic anemia with intensive GVHD prophylaxis including posttransplant cyclophosphamide. Blood Adv. (2020) 4:1770–9. doi: 10.1182/bloodadvances.2020001729
187. Schrezenmeier H, Passweg JR, Marsh JC, Bacigalupo A, Bredeson CN, Bullorsky E, et al. Worse outcome and more chronic GVHD with peripheral blood progenitor cells than bone marrow in HLA-matched sibling donor transplants for young patients with severe acquired aplastic anemia. Blood. (2007) 110:1397–400. doi: 10.1182/blood-2007-03-081596
188. Eapen M, Le Rademacher J, Antin JH, Champlin RE, Carreras J, Fay J, et al. Effect of stem cell source on outcomes after unrelated donor transplantation in severe aplastic anemia. Blood. (2011) 118:2618–21. doi: 10.1182/blood-2011-05-354001
189. Chu R, Brazauskas R, Kan F, Bashey A, Bredeson C, Camitta B, et al. Comparison of outcomes after transplantation of G-CSF-stimulated bone marrow grafts versus bone marrow or peripheral blood grafts from HLA-matched sibling donors for patients with severe aplastic anemia. Biol Blood Marrow Transplant. (2011) 17:1018–24. doi: 10.1016/j.bbmt.2010.10.029
190. Bacigalupo A, Socie G, Schrezenmeier H, Tichelli A, Locasciulli A, Fuehrer M, et al. Bone marrow versus peripheral blood as the stem cell source for sibling transplants in acquired aplastic anemia: survival advantage for bone marrow in all age groups. Haematologica. (2012) 97:1142–8. doi: 10.3324/haematol.2011.054841
191. Nakamura Y, Mori T, Kako S, Yamazaki H, Kanda Y, Uchida N, et al. Outcome of peripheral blood stem cell transplantation from HLA-identical sibling donors for adult patients with aplastic anemia. Int J Hematol. (2023) 117:356–65. doi: 10.1007/s12185-022-03487-6
192. Kumar R, Kimura F, Ahn KW, Hu ZH, Kuwatsuka Y, Klein JP, et al. Comparing outcomes with bone marrow or peripheral blood stem cells as graft source for matched sibling transplants in severe aplastic anemia across different economic regions. Biol Blood Marrow Transplant. (2016) 22:932–40. doi: 10.1016/j.bbmt.2016.01.012
193. Hamidieh AA, Mozafari M, Noshad S, Alimoghaddam K, Behfar M, Ghavamzadeh A. Matched related donor hematopoietic stem cell transplantation results in a long-term follow-up of a pediatric acquired severe aplastic anemia subset: A stem cell source perspective. Pediatr Transplant. (2015) 19:399–407. doi: 10.1111/petr.12458
194. Gupta V, Eapen M, Brazauskas R, Carreras J, Aljurf M, Gale RP, et al. Impact of age on outcomes after bone marrow transplantation for acquired aplastic anemia using HLA-matched sibling donors. Haematologica. (2010) 95:2119–25. doi: 10.3324/haematol.2010.026682
195. Devillier R, Dalle JH, Kulasekararaj A, D’Aveni M, Clement L, Chybicka A, et al. Unrelated alternative donor transplantation for severe acquired aplastic anemia: a study from the French Society of Bone Marrow Transplantation and Cell Therapies and the EBMT Severe Aplastic Anemia Working Party. Haematologica. (2016) 101:884–90. doi: 10.3324/haematol.2015.138727
196. Im HJ, Koh KN, Choi ES, Jang S, Kwon SW, Park CJ, et al. Excellent outcome of haploidentical hematopoietic stem cell transplantation in children and adolescents with acquired severe aplastic anemia. Biol Blood Marrow Transplant. (2013) 19:754–9. doi: 10.1016/j.bbmt.2013.01.023
197. Pedraza A, Jorge S, Suarez-Lledo M, Pereira A, Gutierrez-Garcia G, Fernandez-Aviles F, et al. High-dose cyclophosphamide and tacrolimus as graft-versus-host disease prophylaxis for matched and mismatched unrelated donor transplantation. Transplant Cell Ther. (2021) 27:619 e1– e8. doi: 10.1016/j.jtct.2021.03.022
198. Xu LP, Wang SQ, Wu DP, Wang JM, Gao SJ, Jiang M, et al. Haplo-identical transplantation for acquired severe aplastic anaemia in a multicentre prospective study. Br J Haematol. (2016) 175:265–74. doi: 10.1111/bjh.14225
199. Gerull S, Stern M, Apperley J, Beelen D, Brinch L, Bunjes D, et al. Syngeneic transplantation in aplastic anemia: pre-transplant conditioning and peripheral blood are associated with improved engraftment: an observational study on behalf of the Severe Aplastic Anemia and Pediatric Diseases Working Parties of the European Group for Blood and Marrow Transplantation. Haematologica. (2013) 98:1804–9. doi: 10.3324/haematol.2013.091074
200. Champlin RE, Perez WS, Passweg JR, Klein JP, Camitta BM, Gluckman E, et al. Bone marrow transplantation for severe aplastic anemia: a randomized controlled study of conditioning regimens. Blood. (2007) 109:4582–5. doi: 10.1182/blood-2006-10-052308
201. Baron F, Mohty M, Blaise D, Socie G, Labopin M, Esteve J, et al. Anti-thymocyte globulin as graft-versus-host disease prevention in the setting of allogeneic peripheral blood stem cell transplantation: a review from the Acute Leukemia Working Party of the European Society for Blood and Marrow Transplantation. Haematologica. (2017) 102:224–34. doi: 10.3324/haematol.2016.148510
202. Atta EH, de Sousa AM, Schirmer MR, Bouzas LF, Nucci M, Abdelhay E. Different outcomes between cyclophosphamide plus horse or rabbit antithymocyte globulin for HLA-identical sibling bone marrow transplant in severe aplastic anemia. Biol Blood Marrow Transplant. (2012) 18:1876–82. doi: 10.1016/j.bbmt.2012.07.004
203. Gallo S, Woolfrey AE, Burroughs LM, Storer BE, Flowers ME, Hari P, et al. Marrow grafts from HLA-identical siblings for severe aplastic anemia: does limiting the number of transplanted marrow cells reduce the risk of chronic GvHD? Bone Marrow Transplant. (2016) 51:1573–8. doi: 10.1038/bmt.2016.198
204. Kennedy-Nasser AA, Leung KS, Mahajan A, Weiss HL, Arce JA, Gottschalk S, et al. Comparable outcomes of matched-related and alternative donor stem cell transplantation for pediatric severe aplastic anemia. Biol Blood Marrow Transplant. (2006) 12:1277–84. doi: 10.1016/j.bbmt.2006.07.011
205. Gupta V, Ball SE, Yi QL, Sage D, McCann SR, Lawler M, et al. Favorable effect on acute and chronic graft-versus-host disease with cyclophosphamide and in vivo anti-CD52 monoclonal antibodies for marrow transplantation from HLA-identical sibling donors for acquired aplastic anemia. Biol Blood Marrow Transplant. (2004) 10:867–76. doi: 10.1016/j.bbmt.2004.09.001
206. Marsh JC, Gupta V, Lim Z, Ho AY, Ireland RM, Hayden J, et al. Alemtuzumab with fludarabine and cyclophosphamide reduces chronic graft-versus-host disease after allogeneic stem cell transplantation for acquired aplastic anemia. Blood. (2011) 118:2351–7. doi: 10.1182/blood-2010-12-327536
207. Garland RJ, Groves SJ, Diamanti P, West SE, Winship KL, Virgo PF, et al. Early emergence of PNH-like T cells after allogeneic stem cell transplants utilising CAMPATH-1H for T cell depletion. Bone Marrow Transplant. (2005) 36:237–44. doi: 10.1038/sj.bmt.1705049
208. Iqubal A, Iqubal MK, Sharma S, Ansari MA, Najmi AK, Ali SM, et al. Molecular mechanism involved in cyclophosphamide-induced cardiotoxicity: Old drug with a new vision. Life Sci. (2019) 218:112–31. doi: 10.1016/j.lfs.2018.12.018
209. Fuji S, Sugita J, Najima Y, Konishi T, Tanaka T, Ohigashi H, et al. Low- versus standard-dose post-transplant cyclophosphamide as GVHD prophylaxis for haploidentical transplantation. Br J Haematol. (2023) 67(2):544–51. doi: 10.1016/j.ijrobp.2006.08.049
210. Pierga JY, Socie G, Gluckman E, Devergie A, Henry-Amar M, Bridier A, et al. Secondary solid Malignant tumors occurring after bone marrow transplantation for severe aplastic anemia given thoraco-abdominal irradiation. Radiother Oncol. (1994) 30:55–8. doi: 10.1016/0167-8140(94)90009-4
211. Belkacemi Y, Labopin M, Hennequin C, Hoffstetter S, Mungai R, Wygoda M, et al. Reduced-intensity conditioning regimen using low-dose total body irradiation before allogeneic transplant for hematologic malignancies: Experience from the European Group for Blood and Marrow Transplantation. Int J Radiat Oncol Biol Phys. (2007) 67(2):544–51. doi: 10.1016/j.ijrobp.2006.08.049
212. Quintero V, Bueno-Sanchez D, Mozo-Del-Castillo Y, Urtasun-Erburu A, Sisinni L, Lopez-Duarte M, et al. Haploidentical hematopoietic stem cell transplantation in pediatric patients with acquired hypocellular bone marrow failure. Transplant Cell Ther. (2023) 29:621.e1–e6. doi: 10.1016/j.jtct.2023.07.011
213. Xu LP, Zhang XH, Wang FR, Mo XD, Han TT, Han W, et al. Haploidentical transplantation for pediatric patients with acquired severe aplastic anemia. Bone Marrow Transplant. (2017) 52:381–7. doi: 10.1038/bmt.2016.281
214. Li S, Wang B, Fu L, Pang Y, Zhu G, Zhou X, et al. Hematopoietic stem cell transplantation without in vivo T-cell depletion for pediatric aplastic anemia: A single-center experience. Pediatr Transplant. (2018) 22:e13204. doi: 10.1111/petr.13204
215. Zhang Y, Guo Z, Liu XD, He XP, Yang K, Chen P, et al. Comparison of haploidentical hematopoietic stem cell transplantation and immunosuppressive therapy for the treatment of acquired severe aplastic anemia in pediatric patients. Am J Ther. (2017) 24:e196–201. doi: 10.1097/MJT.0000000000000366
216. Qin X, Zhu YP, Luo CJ, Zhou M, Huang K, Chen C, et al. Optimizing conditioning regimen with low-dose irradiation or busulfan enables the outcome of transplantation from a 6-7/8 HLA-matched donor comparable to that from an 8/8 HLA-matched unrelated donor in severe aplastic anemia patients under 40 years. Ann Hematol. (2021) 100:2363–73. doi: 10.1007/s00277-021-04540-w
217. Kharya G, Jaiswal SR, Bhat S, Raj R, Yadav SP, Dua V, et al. Impact of conditioning regimen and graft-versus-host disease prophylaxis on the outcome of haploidentical peripheral blood stem cell transplantation for high-risk severe aplastic anemia in children and young adults: A report from the pediatric severe aplastic anemia consortium of India. Transplant Cell Ther. (2023) 29:199.e1– e10. doi: 10.1016/j.jtct.2023.09.009
218. Onishi Y, Mori T, Yamazaki H, Hiramoto N, Zaimoku Y, Kanaya M, et al. Comparison of haploidentical stem cell transplantation with post-transplantation cyclophosphamide versus umbilical cord blood transplantation in adult patients with aplastic anemia. Transplant Cell Ther. (2023) 9(9):e660–e669. doi: 10.1016/j.jtct.2023.09.009
219. DeZern AE, Eapen M, Wu J, Talano JA, Solh M, Davila Saldana BJ, et al. Haploidentical bone marrow transplantation in patients with relapsed or refractory severe aplastic anaemia in the USA (BMT CTN 1502): a multicentre, single-arm, phase 2 trial. Lancet Haematol. (2022) 9(9):e660–e9. doi: 10.1016/S2352-3026(22)00206-X
220. Xu LP, Wang SQ, Ma YR, Gao SJ, Cheng YF, Zhang YY, et al. Who is the best haploidentical donor for acquired severe aplastic anemia? Experience from a multicenter study. J Hematol Oncol. (2019) 12:87. doi: 10.1186/s13045-019-0775-9
221. Pagliuca S, Ruggeri A, Peffault de Latour R. Cord blood transplantation for bone marrow failure syndromes: state of art. Stem Cell Investig. (2019) 6:39. doi: 10.21037/sci
222. Peffault de Latour R, Chevret S, Jubert C, Sirvent A, Galambrun C, Ruggeri A, et al. Unrelated cord blood transplantation in patients with idiopathic refractory severe aplastic anemia: a nationwide phase 2 study. Blood. (2018) 132:750–4. doi: 10.1182/blood-2018-01-829630
223. Park M, Lee YH, Kang HR, Lee JW, Kang HJ, Park KD, et al. Unrelated donor cord blood transplantation for non-malignant disorders in children and adolescents. Pediatr Transplant. (2014) 18:221–9. doi: 10.1111/petr.12213
224. Chan KW, McDonald L, Lim D, Grimley MS, Grayson G, Wall DA. Unrelated cord blood transplantation in children with idiopathic severe aplastic anemia. Bone Marrow Transplant. (2008) 42:589–95. doi: 10.1038/bmt.2008.227
225. Chang YJ, Zhao XY, Huang XJ. Granulocyte colony-stimulating factor-primed unmanipulated haploidentical blood and marrow transplantation. Front Immunol. (2019) 10:2516. doi: 10.3389/fimmu.2019.02516
226. Yao D, Tian Y, Li J, Li B, Lu J, Ling J, et al. Association between haploidentical hematopoietic stem cell transplantation combined with an umbilical cord blood unit and graft-versus-host disease in pediatric patients with acquired severe aplastic anemia. Ther Adv Hematol. (2022) 13:20406207221134409. doi: 10.1177/20406207221134409
227. Lu Y, Xiong M, Sun RJ, Zhang JP, Zhao YL, Wei ZJ, et al. Comparisons of unmanipulated haploidentical donor, unrelated cord blood donor and matched unrelated donor hematopoietic stem cell transplantation in pediatric acquired severe aplastic anemia: a single center study. Leuk Lymphoma. (2022) 63:3307–16. doi: 10.1080/10428194.2022.2118527
228. Tang X, Yu Z, Ping L, Lu W, Jing Y, Cao X. Improved outcomes using unmanipulated haploidentical hematopoietic stem cells combined with third-party umbilical cord blood transplantation for non-malignant diseases in children: The experience of a single center. Pediatr Transplant. (2021) 25:e13995. doi: 10.1111/petr.13995
229. Sun Y, Liu Z, Xiao J, Liu X, Jiang F, Fan S, et al. Autologous cord blood transplantation in children with acquired severe aplastic anemia. Pediatr Transplant. (2019) 23:e13325. doi: 10.1111/petr.13325
230. Rosa M, Gajek K, Salamonowicz-Bodzioch M, Mielcarek-Siedziuk M, Fraczkiewicz J, Jarmolinski T, et al. Successful bone marrow recovery after an immunoablative regimen with autologous cord blood transplant in a child with idiopathic severe aplastic anemia: A case report. Transplant Proc. (2020) 52:653–6. doi: 10.1016/j.transproceed.2019.12.006
231. Avgerinou G, Oikonomopoulou C, Kaisari A, Ioannidou E, Komitopoulou A, Paisou A, et al. Successful long-term hematological and immunological reconstitution by autologous cord blood transplantation combined with post-transplant immunosuppression in two children with severe aplastic anemia. Pediatr Transplant. (2019) 23:e13320. doi: 10.1111/petr.13320
232. Buchbinder D, Hsieh L, Puthenveetil G, Soni A, Stites J, Huynh V, et al. Successful autologous cord blood transplantation in a child with acquired severe aplastic anemia. Pediatr Transplant. (2013) 17:E104–7. doi: 10.1111/petr.12068
233. Pagliuca S, Peffault de Latour R, Volt F, Locatelli F, Zecca M, Dalle JH, et al. Long-term outcomes of cord blood transplantation from an HLA-identical sibling for patients with bone marrow failure syndromes: A report from eurocord, cord blood committee and severe aplastic anemia working party of the european society for blood and marrow transplantation. Biol Blood Marrow Transplant. (2017) 23:1939–48. doi: 10.1016/j.bbmt.2017.08.004
234. Majhail NS. Long-term complications after hematopoietic cell transplantation. Hematol Oncol Stem Cell Ther. (2017) 10:220–7. doi: 10.1016/j.hemonc.2017.05.009
235. Klasa L, Sadowska-Klasa A, Piekarska A, Wydra D, Zaucha JM. The management of gynecological complications in long-term survivors after allogeneic hematopoietic cell transplantation-a single-center real-life experience. Ann Hematol. (2020) 99:1361–8. doi: 10.1007/s00277-020-04034-1
236. Kanda Y, Usuki K, Inagaki M, Ohta A, Ogasawara Y, Obara N, et al. Decision analysis of allogeneic bone marrow transplantation versus immunosuppressive therapy for young adult patients with aplastic anemia. Int J Hematol. (2023) 117:660–8. doi: 10.1007/s12185-022-03530-6
237. Locatelli F, Bruno B, Zecca M, Van-Lint MT, McCann S, Arcese W, et al. Cyclosporin A and short-term methotrexate versus cyclosporin A as graft versus host disease prophylaxis in patients with severe aplastic anemia given allogeneic bone marrow transplantation from an HLA-identical sibling: results of a GITMO/EBMT randomized trial. Blood. (2000) 96:1690–7.
238. George B, Pn N, Devasia AJ, Kulkarni U, Korula A, Lakshmi KM, et al. Post-transplant cyclophosphamide as sole graft-versus-host disease prophylaxis is feasible in patients undergoing peripheral blood stem cell transplantation for severe aplastic anemia using matched sibling donors. Biol Blood Marrow Transplant. (2018) 24:494–500. doi: 10.1016/j.bbmt.2017.10.034
239. Clay J, Kulasekararaj AG, Potter V, Grimaldi F, McLornan D, Raj K, et al. Nonmyeloablative peripheral blood haploidentical stem cell transplantation for refractory severe aplastic anemia. Biol Blood Marrow Transplant. (2014) 20:1711–6. doi: 10.1016/j.bbmt.2014.06.028
240. Xu LP, Jin S, Wang SQ, Xia LH, Bai H, Gao SJ, et al. Upfront haploidentical transplant for acquired severe aplastic anemia: registry-based comparison with matched related transplant. J Hematol Oncol. (2017) 10:25. doi: 10.1186/s13045-017-0398-y
241. Grimaldi F, Potter V, Perez-Abellan P, Veluchamy JP, Atif M, Grain R, et al. Mixed T cell chimerism after allogeneic hematopoietic stem cell transplantation for severe aplastic anemia using an alemtuzumab-containing regimen is shaped by persistence of recipient CD8 T cells. Biol Blood Marrow Transplant. (2017) 23:293–9. doi: 10.1016/j.bbmt.2016.11.003
242. Marotta S, Marano L, Ricci P, Cacace F, Frieri C, Simeone L, et al. Eltrombopag for post-transplant cytopenias due to poor graft function. Bone Marrow Transplant. (2019) 54:1346–53. doi: 10.1038/s41409-019-0442-3
243. Kircali E, Cengiz Seval G, Ozturk C, Yilmaz H, Koyun D, Civriz Bozdag S, et al. Eltrombopag for the treatment of poor graft function following haematopoietic cell transplantation: real-life data. Balkan Med J. (2023) 40:51–6. doi: 10.4274/balkanmedj
244. Fuhrer M, Claviez A, Klein B, Humpe A, Schrauder A. Re-transplantation from the same unrelated donor in three adolescents with severe aplastic anemia after graft rejection. Klin Padiatr. (2009) 221:358–61. doi: 10.1055/s-0029-1239530
245. Kudo K, Muramatsu H, Yoshida N, Kobayashi R, Yabe H, Tabuchi K, et al. Second allogeneic hematopoietic stem cell transplantation in children with severe aplastic anemia. Bone Marrow Transplant. (2015) 50:1312–5. doi: 10.1038/bmt.2015.153
246. Bhatia S, Francisco L, Carter A, Sun CL, Baker KS, Gurney JG, et al. Late mortality after allogeneic hematopoietic cell transplantation and functional status of long-term survivors: report from the Bone Marrow Transplant Survivor Study. Blood. (2007) 110:3784–92. doi: 10.1182/blood-2007-03-082933
247. Samarasinghe S, Steward C, Hiwarkar P, Saif MA, Hough R, Webb D, et al. Excellent outcome of matched unrelated donor transplantation in paediatric aplastic anaemia following failure with immunosuppressive therapy: a United Kingdom multicentre retrospective experience. Br J Haematol. (2012) 157:339–46. doi: 10.1111/j.1365-2141.2012.09066.x
248. Buchbinder D, Nugent DJ, Brazauskas R, Wang Z, Aljurf MD, Cairo MS, et al. Late effects in hematopoietic cell transplant recipients with acquired severe aplastic anemia: a report from the late effects working committee of the center for international blood and marrow transplant research. Biol Blood Marrow Transplant. (2012) 18:1776–84. doi: 10.1016/j.bbmt.2012.06.018
249. Kahn JM, Brazauskas R, Tecca HR, Bo-Subait S, Buchbinder D, Battiwala M, et al. Subsequent neoplasms and late mortality in children undergoing allogeneic transplantation for nonmalignant diseases. Blood Adv. (2020) 4:2084–94. doi: 10.1182/bloodadvances.2019000839
250. Baumann I, Fuhrer M, Behrendt S, Campr V, Csomor J, Furlan I, et al. Morphological differentiation of severe aplastic anaemia from hypocellular refractory cytopenia of childhood: reproducibility of histopathological diagnostic criteria. Histopathology. (2012) 61:10–7. doi: 10.1111/j.1365-2559.2011.04156.x
Keywords: aplastic anemia, antithymocyte globulin, eltrombopag, hATG, hematopoietic cell transplantation, immunosuppression, rATG
Citation: Piekarska A, Pawelec K, Szmigielska-Kapłon A and Ussowicz M (2024) The state of the art in the treatment of severe aplastic anemia: immunotherapy and hematopoietic cell transplantation in children and adults. Front. Immunol. 15:1378432. doi: 10.3389/fimmu.2024.1378432
Received: 01 February 2024; Accepted: 22 March 2024;
Published: 05 April 2024.
Edited by:
Giuseppe Milone, University Hospital Polyclinic Vittorio Emanuele, ItalyReviewed by:
Daria Babushok, University of Pennsylvania, United StatesYanzhi Song, Beijing Boren Hospital, China
Copyright © 2024 Piekarska, Pawelec, Szmigielska-Kapłon and Ussowicz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Agnieszka Piekarska, YWduaWVzemthLnBpZWthcnNrYUBndW1lZC5lZHUucGw=