 Guillaume Kellermann
Guillaume Kellermann Nicolas Leulliot2‡
Nicolas Leulliot2‡ Julien Cherfils-Vicini
Julien Cherfils-Vicini Patrick Brest
Patrick Brest- 1Telomium, SAS, Ivry-sur-Seine, France
- 2Université Paris Cité, Centre national de la recherche scientifique (CNRS), Cibles Thérapeutiques et Conception de Médicaments (CiTCoM), Paris, France
- 3Université Côte d’Azur, Institute for Research on Cancer and Aging, Nice (IRCAN), Centre national de la recherche scientifique (CNRS), Institut national de la santé et de la recherche médicale (INSERM), Centre Antoine Lacassagne, Institut Hospitalo-Universitaire (IHU), RESPIRera, Fédérations Hospitalo-Universitaires (FHU)OncoAge, Nice, France
Immune checkpoint therapies (ICT) have transformed the treatment of cancer over the past decade. However, many patients do not respond or suffer relapses. Successful immunotherapy requires epitope spreading, but the slow or inefficient induction of functional antitumoral immunity delays the benefit to patients or causes resistances. Therefore, understanding the key mechanisms that support epitope spreading is essential to improve immunotherapy. In this review, we highlight the major role played by B-cells in breaking immune tolerance by epitope spreading. Activated B-cells are key Antigen-Presenting Cells (APC) that diversify the T-cell response against self-antigens, such as ribonucleoproteins, in autoimmunity but also during successful cancer immunotherapy. This has important implications for the design of future cancer vaccines.
1 Introduction
The breakthrough in cancer immunotherapy was awarded by the Nobel Prize in Physiology or Medicine to James Allison and Tasuku Honjo in 2018 (1). Immune checkpoints Therapies (ICT) have fundamentally changed cancer treatment approaches over the past decade. ICT have become a robust method to reinvigorate exhausted T-cells targeting neoepitopes on cancer cells. Neoepitopes occurrence in tumors correlates with tumor mutation burden (TMB). Some neoepitopes are recognized as foreign antigens by T-cells and become major drivers of antitumoral immunity when their activity is prolonged by ICT [1].
However, the delay between treatment initiation and response is a weakness. Indeed, the benefit of ICT is not apparent until 2-3 months, and sometimes not until 6-12 months after treatment initiation, comparing unfavorably to chemotherapy (2, 3). Therefore, ICT is often less effective for very aggressive tumors. Combining ICT with intra-tumoral injections of the replicating virus T-VEC (FDA-approved), failed to accelerate therapeutic responses: the mean time to response was 8.4 months (4). In a HNSCC trial combining T-VEC and ICT, 27.8% of patients died before the first evaluation at week 9 (5), highlighting the need to accelerate the induction of functional antitumor immune responses. Moreover, the majority of patients do not respond at all to ICT, because most tumors do not present enough neoepitopes. Persistent clonal neoepitopes, required for long term remissions, are often missing (6, 7), and inefficient epitope spreading to non-mutated tumor associated-antigens (TAA), explains the primary, but also secondary resistance to ICT (8).
Developing diversified immune responses is critical to overcome the limitations of current immunotherapies. Understanding why and how epitope spreading occurs holds the key. In this review, we take an evolutionary approach to provide a unifying explanation for this phenomenon and describe the key pathways driving it. By leveraging these insights, future immunotherapies can be designed to activate these pathways and unleash the full potential of the immune system against cancer.
1.1 Break of tolerance during immunotherapy
Neoepitopes and chronic inflammation in some tumors break immune tolerance to tissue-specific, non-mutated, self-antigens (8), leading to serious adverse events when these tumor antigens are expressed in other normal tissues (9). Nevertheless, these autoimmune side effects often correlate with survival in patients treated with ICT (10). This suggests that these reactions are not only adverse events but also real contributors to the therapeutic effect (11). They however remain difficult to predict. Self-tolerance does not impede the generation of activated T-cells against many self-antigens, but may strongly impair their functional activity in tissues (12). Consequently, spontaneous immune responses occurring naturally against self-associated tumor antigens, or T-cell responses elicited by cancer vaccines do not consistently correlate with therapeutic outcomes observed in vivo and can’t be used to prove or rule out a break of immune tolerance, not even for neoepitopes (13).
Rather than relying on unpredictable, rare, delayed, poorly targeted, and potentially dangerous autoimmune reactions, a more rational immunotherapy should directly break immune tolerance to specific cancer targets. This would allow safer, faster, and more reliable tumor regression regardless of TMB or neoepitope load. This was the original goal of cancer vaccines, but their inability to reliably break immune tolerance, in clinical conditions, has so far limited their efficacy, except in the rare patients experiencing epitope spreading (14).
Epitope spreading is a diversification of the initial T-cell response against novel epitopes that are different from, and not cross-reactive with the original epitope(s) targeted in the primary immunization (Figure 1). Epitope spreading can occur to cryptic epitopes of the same targeted antigen (intra-molecular spreading) or against other antigens (inter-molecular spreading or antigen spreading) (15). Antigen spreading against other immunodominant epitopes readily occurs through bystander activation during many immune responses, but this is usually unrelated to a break of immune tolerance. On the contrary, intra-molecular diversification of the T-cell response reported for self-antigens such as MAGE Family Member A3 (MAGEA3) (16) or Telomerase (TERT) (17) provided stronger evidence that a true break of immune tolerance occurred in the few patients successfully reacting to cancer vaccines. Epitope spreading occurred in these patients several months after the initial vaccination, and this latter diversification of the T-cell response was correlated with the appearance of objective therapeutic responses, as if the vaccine-induced T-cells were not the direct mediators of tumor regression, but rather initiators of a broader T-cell response after several cycles of epitope spreading (16–18). It is therefore crucial to identify which cell populations are promoting such epitope diversification.
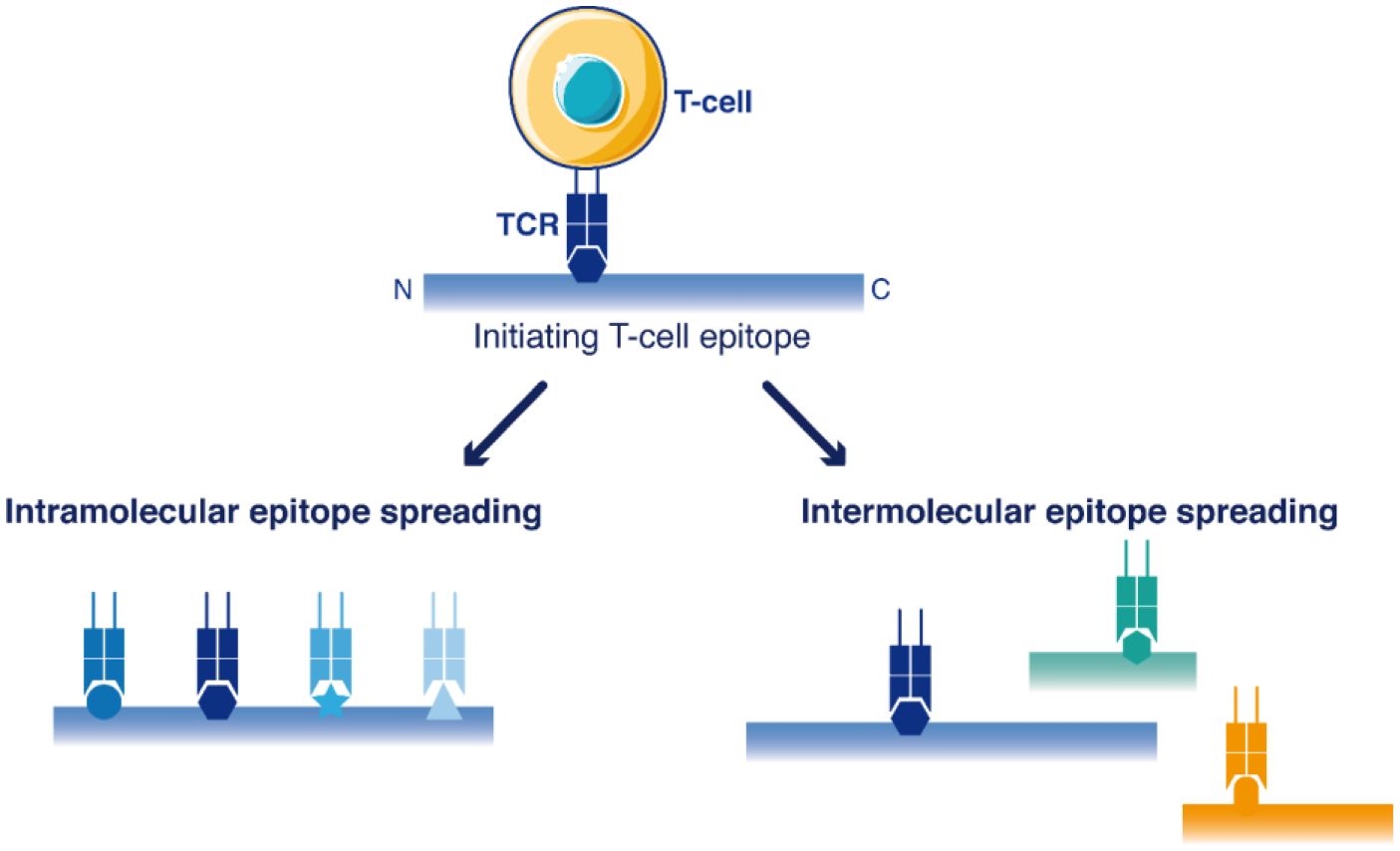
Figure 1 Schematic view of T-cell epitope spreading into the sequences of antigens. The primary immune response targets dominant initiating T-cell epitope(s). Then, the response may be further extended to other T-cell epitopes within the same antigen (intramolecular epitope spreading) or among other antigens (antigen spreading/intermolecular epitope spreading).
1.2 Activated B-Cells promote epitope spreading
As early as 1993, Charles Janeway hypothesized that B-cells are the key antigen-presenting cells (APC) behind the diversification of T-cell responses (19). In the last three decades, many studies have supported the key involvement of B-cells as critical APC for T-cell activation in many autoimmune diseases (20–25). The fundamental role of auto-reactive B-cells as a key antigen presenting cell (APC) to activate T-cells is widely established in autoimmune diseases (20–24), where B-cells have even become a therapeutic target (24, 26). B-cells diversify CD4 T-cell responses (22), cross-present antigens during autoimmunity (27, 28) and also help CD8 T-cell independently of antigen presentation (29). However, in cancer immunotherapy, epitope spreading has been mostly attributed to dendritic cells (DC) (30). The anti-tumoral role of B-cells has long been neglected, because tumor mouse models have mainly predicted a pro-tumoral role of B-cells, in contrast to the situation in humans (31, 32). However, recent data has strengthened the evidence for a strong correlation between B-cell activation and successful immunotherapy in humans (33–38). This suggests that B-cells play a key role in tumor antigen presentation (39) and are critical for successful cancer immunotherapy (32, 40) and also to the associated-autoimmune reactions (11, 25).
1.3 The viral escape theory: a potential fundamental reason for epitope spreading
During a primary viral infection, DC capture and present foreign antigens to activate high-avidity T-cells that kill infected cells. Later, serum antibodies rise to suppress viral dissemination and prevent recurrence. However, when the virus escapes through random mutations at protective T-cell epitopes, B-cells still capture virus particles released from infected cells resistant to T-cell killing (41). In this situation, B-cells are continuously activated via their BCR (B-Cell Receptor) and present antigens to T-cells. Thus, persistent antigen presentation by memory B-cells means that an immune response has been induced, but also that this immune response has failed to completely remove the pathogen. Therefore, these conditions may have been evolutionarily selected to provide special signaling to recruit a new wave of effectors, that are more likely to cause autoimmune damage, but are able to remove escape variants that cannot be cured with the primary response (42–45).
High-avidity T-cells reacting against self-antigens are deleted by central tolerance, but low avidity-cells T-cells are not deleted. They are held in reserve in the case the primary response fails (42, 46–48). With the help of B-cells, T-cells with gradually lower TCR affinity are progressively recruited (45), which may ultimately break immune tolerance and cause autoimmune damage. During stepwise expansion of the immune response, the hierarchical order of epitope spreading starts with the most immunodominant epitope and then extends to the least dominant (15). Activated T-cell reaching subdominant epitopes and cryptic epitopes that have not been tolerized break immune tolerance (49, 50).
B-cells are highly efficient at reconcentrating diluted antigens released from dead cells by capture through their BCR which allows the selective presentation of a specific antigen to activate T-cells (19). On the contrary, DCs capture and present antigens non-specifically. Therefore, DCs need to remain under the strict control of T regulatory cells (Treg) (51), whereas B-cells are less stringently controlled by Treg (52, 53). Indeed, B-cells have a unique ability to activate T-cells and support the differentiation of low-avidity T-cells (44, 45) which are normally suppressed by T-regulatory cells (Treg) (54, 55). DC play a crucial role in the initial T-cell activation, but B-cells, not DC, uniquely support CD4 T-cells cooperation and their further activation (22, 56) through the OX40-OX40L pathway (22, 57). Memory B-cells act as specific APC that overcome the suppressive activity of Treg in many autoimmune disorders (23, 58, 59).
Activated B-cells also provide additional APC numbers to bypass T-cell competition, reduce immunodominance and diversify the T-cell response. Indeed, during viral infections, T-cells compete for APC and costimulatory molecules, establishing a state where only a few epitopes dominate the T-cell response. Dominant epitopes usually display a high affinity for HLA and provide the strongest T-cell activation (60), but immunodominance can be bypassed by artificially increasing the number of APC (61–63). During an infection, if the antigen is only presented by dendritic cells (DCs), the number of activated APC is limiting, therefore creating a competition between T-cells for the few activated APCs and favoring the dominance of a T-cell response against only a few epitopes. On the contrary, expanded B-cells can outnumber DC and provide an increased number of APC that relief the competition for APC. This bypasses the immunodominance and support the extreme diversification of the immune response to multiple new epitopes: epitope spreading (45). Moreover, plasma B cells secrete high levels of antibodies that bind the antigen and facilitate the subsequent mobilization of other non-conventional APC (64), further enhancing antigen presentation and cross-presentation which favors the break of tolerance (65).
Broad immune responses targeting multiple epitopes are more efficient than restricted responses, but carry a higher risk of autoimmunity (66). While the risk-benefit ratio of inducing a broader T-cell activation may not always be positive during a chronic infection, a live pathogen that acutely escape immune control and becomes detected as a threat out-of-control, provides a stronger signal than a chronic infection (67). Acute reinfection with a pathogen expressing a self-antigen, but not a standard DC vaccine, can directly activate low avidity T-cells and induce autoimmunity after only one week (47), demonstrating that epitope spreading is not necessarily a slow process but can be accelerated. However, this also suggests that standard vaccine technologies and current adjuvants are suboptimal for inducing epitope spreading. It is therefore crucial to identify the most suitable signaling pathways that, can efficiently drive epitope spreading.
1.4 The unique role of TLR7 in B-cells to break tolerance
Toll-like receptor (TLR) -9 or TLR3 agonists (CpG and PolyI:C) are usually scored as the more powerful adjuvants for standard antigens in mice, but a TLR7 agonist was reported as the most effective adjuvant for a self-antigen (68). TLR7, a sensor for viral RNA, has a superior ability to induce IL-6 dependent resistance to the suppressive activity of Treg (69, 70). TLR7 agonists are more effective in bypassing Treg than agonists for TLR4, TLR5 (71) and even TLR3, RIG-I, MDA-5 (68, 72).
TLR7 plays an essential role in B-cells unlike any other TLR, including TLR9, despite both receptors rely on MyD88 signaling (73). TLR7, but not TLR2, TLR3, TLR4 or TLR9 is required for the formation of spontaneous germinal centers in vivo (74). TLR7 agonists, but not TLR3, TLR4 or TLR9 agonists, induce the accumulation of atypical memory CD11c+ B cells (ABCs), associated with autoimmunity (75).
The major importance of TLR7 in the break of immune tolerance is strengthened by the independent discovery that several genetic/epigenetic variations in TLR7 leads to autoimmunity (76–78). Indeed, at the genome level, a single gene duplication in TLR7 induces autoimmune pathology, demonstrating that TLR7 levels must remain tightly regulated in vivo (79). At the transcription level, type II interferon potentiates the TLR7 pathway (80, 81) and type I interferon up-regulates TLR7 expression 40-fold in B-cells, without changing the levels of other TLR, thereby increasing the sensitivity of B-cells specifically against RNA viruses (82). Of note, RNA viruses have a higher mutation rate than any other pathogen. They pose the risk of escaping the immune response by random mutation (41). As a result, TLR7 signaling in B-cells seems to have been evolutionarily selected to respond efficiently against viral escape variants by facilitating epitope spreading.
1.5 RNP: optimal inducer of the dual BCR/TLR7 signaling
Ribonucleoproteins (RNPs) are RNA-protein complexes that can be mistaken as RNA virus. They constitute a major class of self-antigens targeted in autoimmune diseases (83). Autoantibodies against different RNPs antigens are frequently found in various autoimmune diseases. However, mice genetically deficient in secreting antibody showed that serum antibodies are not essential to the autoimmune pathology. Rather, it was found that the presence of non-secreted auto-antibodies at the surface of B-cells (the BCR) is critical, suggesting that it is the APC function of B-cells which is key for autoimmunity (21, 22, 84). Genome-wide association studies have also identified the BCR signaling among the most frequent genetic polymorphisms predisposing to autoimmunity (20).
Noticeably, RNPs induce a dual BCR/TLR signaling (Figure 2), by the sequential engagement of the antigen signaling at the cell-surface (BCR or FcR through their protein part), followed then by TLR7 signaling in endosomes (through their RNA part) (78). This acute or prolonged dual signaling by RNPs better mimics a RNA virus escaping immune control than any other self-antigen, explaining why RNP often break tolerance by epitope spreading (83).
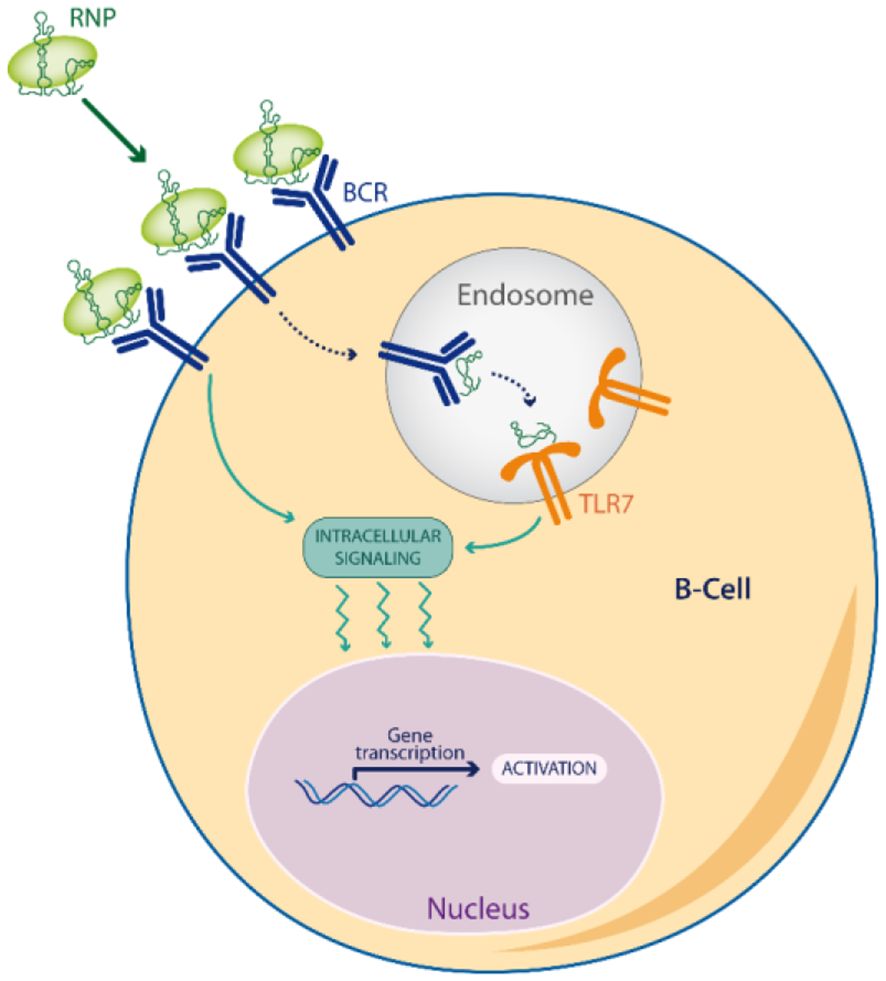
Figure 2 Dual BCR/TLR signaling. Viral Escape Mimetics (VEM), like RNPs antigens, induce a dual BCR/TLR signaling in memory B-cells which enhances antigen presentation to T-cells, and favors a break of tolerance by epitope spreading.
The co-stimulation of the BCR is required to unleash the full potential of TLR7 signaling. Otherwise, TLR7 activation alone is rapidly restrained to hypo-responsiveness (85). Antigens that activate the dual BCR/TLR7 signaling, but not dual BCR/TLR9 or TLR7 alone, promote B-cell differentiation into plasma cells (86), found to correlate with antitumor responses to anti-PD-L1 (37). Noticeably, the dual BCR/TLR7 signaling has also been experimentally found as the optimal combination for vaccination with human B-cells (87). Current studies support a model where the dual activation of the BCR/TLR7 signaling by a foreign RNP antigen in B-cells is the first event that orchestrates the break of tolerance in several autoimmune diseases (83, 88), suggesting a cooperation between the three main autoimmune mechanisms: molecular mimicry, bystander help and epitope spreading.
Ribonucleoprotein antigens are also the major class of self-antigens targeted by B-cells in tumors (89). Among the most studied TAA, telomerase is a RNP absent in most normal adult tissues but reactivated in most cancers and essential for prolonged tumor growth and metastasis. After vaccination with a single peptide from TERT, extensive epitope spreading into the entire TERT protein has been observed in the few patients achieving tumor regression, but not in non-responders (17). In these responders, the T-cell response against TERT was so widespread that T-cell reactivity was detected at almost half of the positions tested along the protein, providing evidence of profound epitope spreading (17). Interestingly, the long-term survival of these patients without autoimmune extension to other tissues (90), illustrates the concept of beneficial autoimmunity (11). This supports the development of immune activators capable of directly eliciting such extensive and diversified immune responses in the majority of patients.
2 Conclusions and perspectives
No cancer vaccine has been specifically designed to break immune tolerance against a tumor antigen by inducing intramolecular epitope spreading through the specific activation of the dual BCR/TLR7 signaling in B-cells. All mRNA/DNA/viral cancer vaccines suffer from a de-coupling of timing and localization between early TLR/IFN stimulation in endosomes and the delayed antigen expression in the cytoplasm (91). Current cancer vaccines do not sequentially activate the BCR at the cell-surface, followed by connected TLR7 activation and MHC antigen presentation in the resulting endosome (28). Neither unassociated adjuvants commonly co-administrated in protein/peptide vaccine formulations, nor antigens delivered by mRNA/DNA/viral vectors appropriately reproduce the temporospatial trafficking of a viral particle captured by an APC.
Vaccines based on antigen-adjuvant conjugates should provide better Viral Escape Mimetics (VEM) to induce epitope spreading. Recently, peptides linked to the synthetic TLR7 agonists (imidazoquinoline) have been tested (92). However, small peptides do not contain enough B and T epitopes linked on the same antigen to support direct epitope spreading, while the in vivo adjuvancy of imidazoquinolines is vastly inferior to RNAs, because of abnormal TLR7 trafficking when TLR7 is activated by these small synthetic agonists instead of an RNA (93). Larger protein-RNA complexes should be preferred, as antigens close to a viral size display an optimal trafficking into lymph nodes, contrary to small antigens (94). Finally, many of the current cancer vaccines still use oil adjuvants which induce antibody production by B-cells that prevent infectious diseases, but abrogate the antitumoral activity of B-cells (95) and the response to ICTs (96). Therefore, future cancer vaccine must activate the most potent type of B-cells for ICT.
Vaccines against neoepitopes can induce broader immune response, but they have nevertheless mostly failed to solve the primary resistance to ICTs (97), possibly because a standard immunization approach doesn’t reverse self-tolerance (12, 47). Many neoepitopes do not display enough foreignness and several work as suppressive regulatory T cell epitopes (98). Therefore, in most patients, ICTs can only become efficient with a break of immune tolerance induced by profound epitope spreading against native non-mutated epitopes, a process lead by activated B-cells, as evidence in autoimmunity.
Author contributions
GK: Funding acquisition, Visualization, Writing – original draft, Writing – review & editing. NL: Funding acquisition, Visualization, Writing – original draft, Writing – review & editing. JC-V: Writing – original draft, Writing – review & editing. MB: Funding acquisition, Visualization, Writing – original draft, Writing – review & editing. PB: Funding acquisition, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Funding is acknowledged from the French Government (Agence Nationale de Recherche, ANR) through the ‘Investments for the Future’ LABEX SIGNALIFE (ANR-11-LABX-0028-01 and IDEX UCAJedi ANR-15-IDEX-01) and RNPVAX (ANR-23-CE17-0011-01, ANR-23-CE17-0011-02, ANR-23-CE17-0011-03), Canceropole PACA; Région Sud, CNRS, Université Paris Cité, INSERM cancer; INCA Plan Cancer; ITMO Cancer.
Acknowledgments
The authors apologize for citations that are not included in the bibliography due to limited numbers. The authors would like to thank Dr. Joost Louwagie and Dr Nicolas Cuburu for proofreading the manuscript.
Conflict of interest
GK is shareholder of Telomium, a company developing vaccines to break immune tolerance.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be constructed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
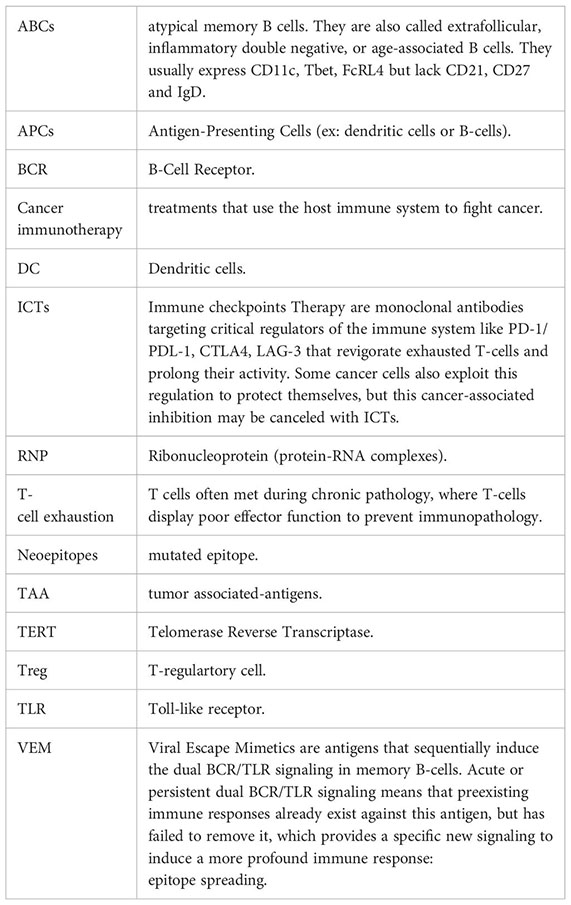
Glossary
References
1. Ledford H, Else H, Warren M. Cancer immunologists scoop medicine Nobel prize. Nature. (2018) 562:20–1. doi: 10.1038/d41586-018-06751-0
2. Herbst RS, Giaccone G, de Marinis F, Reinmuth N, Vergnenegre A, Barrios CH, et al. Atezolizumab for first-line treatment of PD-L1-selected patients with NSCLC. N Engl J Med. (2020) 383:1328–39. doi: 10.1056/NEJMoa1917346
3. Hellmann MD, Paz-Ares L, Bernabe Caro R, Zurawski B, Kim S-W, Carcereny Costa E, et al. Nivolumab plus ipilimumab in advanced non-small-cell lung cancer. N Engl J Med. (2019) 381:2020–31. doi: 10.1056/NEJMoa1910231
4. Chesney JA, Puzanov I, Collichio FA, Singh P, Milhem MM, Glaspy J, et al. Talimogene laherparepvec in combination with ipilimumab versus ipilimumab alone for advanced melanoma: 5-year final analysis of a multicenter, randomized, open-label, phase II trial. J Immunother Cancer. (2023) 11:e006270. doi: 10.1136/jitc-2022-006270
5. Harrington KJ, Kong A, Mach N, Chesney JA, Fernandez BC, Rischin D, et al. Talimogene laherparepvec and pembrolizumab in recurrent or metastatic squamous cell carcinoma of the head and neck (MASTERKEY-232): A multicenter, phase 1b study. Clin Cancer Res Off J Am Assoc Cancer Res. (2020) 26:5153–61. doi: 10.1158/1078-0432.CCR-20-1170
6. Rosenthal R, Cadieux EL, Salgado R, Bakir MA, Moore DA, Hiley CT, et al. Neoantigen-directed immune escape in lung cancer evolution. Nature. (2019) 567:479–85. doi: 10.1038/s41586-019-1032-7
7. Niknafs N, Balan A, Cherry C, Hummelink K, Monkhorst K, Shao XM, et al. Persistent mutation burden drives sustained anti-tumor immune responses. Nat Med. (2023) 29:440–9. doi: 10.1038/s41591-022-02163-w
8. Lo JA, Kawakubo M, Juneja VR, Su MY, Erlich TH, LaFleur MW, et al. Epitope spreading toward wild-type melanocyte-lineage antigens rescues suboptimal immune checkpoint blockade responses. Sci Transl Med. (2021) 13:eabd8636. doi: 10.1126/scitranslmed.abd8636
9. Kråkenes T, Herdlevaer I, Raspotnig M, Haugen M, Schubert M, Vedeler CA. CDR2L is the major yo antibody target in paraneoplastic cerebellar degeneration. Ann Neurol. (2019) 86:316–21. doi: 10.1002/ana.25511
10. Petrelli F, Grizzi G, Ghidini M, Ghidini A, Ratti M, Panni S, et al. Immune-related adverse events and survival in solid tumors treated with immune checkpoint inhibitors: A systematic review and meta-analysis. J Immunother Hagerstown Md 1997. (2020) 43:1–7. doi: 10.1097/CJI.0000000000000300
11. Zitvogel L, Perreault C, Finn OJ, Kroemer G. Beneficial autoimmunity improves cancer prognosis. Nat Rev Clin Oncol. (2021) 18:591–602. doi: 10.1038/s41571-021-00508-x
12. Truckenbrod EN, Burrack KS, Knutson TP, Borges da Silva H, Block KE, O’Flanagan SD, et al. CD8+ T cell self-tolerance permits responsiveness but limits tissue damage. eLife. (2021) 10:e65615. doi: 10.7554/eLife.65615
13. Brennick CA, George MM, Moussa MM, Hagymasi AT, Seesi SA, Shcheglova TV, et al. An unbiased approach to defining bona fide cancer neoepitopes that elicit immune-mediated cancer rejection. J Clin Invest. (2021) 131:e142823. doi: 10.1172/JCI142823
14. Lin MJ, Svensson-Arvelund J, Lubitz GS, Marabelle A, Melero I, Brown BD, et al. Cancer vaccines: the next immunotherapy frontier. Nat Cancer. (2022) 3:911–26. doi: 10.1038/s43018-022-00418-6
15. Vanderlugt CL, Miller SD. Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nat Rev Immunol. (2002) 2:85–95. doi: 10.1038/nri724
16. Sahin U, Oehm P, Derhovanessian E, Jabulowsky RA, Vormehr M, Gold M, et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature. (2020) 585:107–12. doi: 10.1038/s41586-020-2537-9
17. Inderberg-Suso E-M, Trachsel S, Lislerud K, Rasmussen A-M, Gaudernack G. Widespread CD4+ T-cell reactivity to novel hTERT epitopes following vaccination of cancer patients with a single hTERT peptide GV1001. Oncoimmunology. (2012) 1:670–86. doi: 10.4161/onci.20426
18. Türeci Ö, Löwer M, Schrörs B, Lang M, Tadmor A, Sahin U. Challenges towards the realization of individualized cancer vaccines. Nat BioMed Eng. (2018) 2:566–9. doi: 10.1038/s41551-018-0266-2
19. Mamula MJ, Janeway CA. Do B cells drive the diversification of immune responses? Immunol Today. (1993) 14:151–152; discussion 153-154. doi: 10.1016/0167-5699(93)90274-O
20. Corneth OBJ, Neys SFH, Hendriks RW. Aberrant B cell signaling in autoimmune diseases. Cells. (2022) 11:3391. doi: 10.3390/cells11213391
21. Chan OT, Hannum LG, Haberman AM, Madaio MP, Shlomchik MJ. A novel mouse with B cells but lacking serum antibody reveals an antibody-independent role for B cells in murine lupus. J Exp Med. (1999) 189:1639–48. doi: 10.1084/jem.189.10.1639
22. Kroeger DR, Rudulier CD, Bretscher PA. Antigen presenting B cells facilitate CD4 T cell cooperation resulting in enhanced generation of effector and memory CD4 T cells. PloS One. (2013) 8:e77346. doi: 10.1371/journal.pone.0077346
23. Jelcic I, Nimer FA, Wang J, Lentsch V, Planas R, Jelcic I, et al. Memory B cells activate brain-homing, autoreactive CD4+ T cells in multiple sclerosis. Cell. (2018) 175:85–100. doi: 10.1016/j.cell.2018.08.011
24. Kansal R, Richardson N, Neeli I, Khawaja S, Chamberlain D, Ghani M, et al. Sustained B cell depletion by CD19-targeted CAR T cells is a highly effective treatment for murine lupus. Sci Transl Med. (2019) 11:eaav1648. doi: 10.1126/scitranslmed.aav1648
25. Das R, Bar N, Ferreira M, Newman AM, Zhang L, Bailur JK, et al. Early B cell changes predict autoimmunity following combination immune checkpoint blockade. J Clin Invest. (2018) 128:715–20. doi: 10.1172/JCI96798
26. Mackensen A, Müller F, Mougiakakos D, Böltz S, Wilhelm A, Aigner M, et al. Anti-CD19 CAR T cell therapy for refractory systemic lupus erythematosus. Nat Med. (2022) 28:2124–32. doi: 10.1038/s41591-022-02017-5
27. Mariño E, Tan B, Binge L, Mackay CR, Grey ST. B-cell cross-presentation of autologous antigen precipitates diabetes. Diabetes. (2012) 61:2893–905. doi: 10.2337/db12-0006
28. Tohme M, Maisonneuve L, Achour K, Dussiot M, Maschalidi S, Manoury B. TLR7 trafficking and signaling in B cells is regulated by the MHCII-associated invariant chain. J Cell Sci. (2020) 133:jcs236711. doi: 10.1242/jcs.236711
29. Klarquist J, Cross EW, Thompson SB, Willett B, Aldridge DL, Caffrey-Carr AK, et al. B cells promote CD8 T cell primary and memory responses to subunit vaccines. Cell Rep. (2021) 36:109591. doi: 10.1016/j.celrep.2021.109591
30. Brossart P. The role of antigen spreading in the efficacy of immunotherapies. Clin Cancer Res Off J Am Assoc Cancer Res. (2020) 26:4442–7. doi: 10.1158/1078-0432.CCR-20-0305
31. Guy TV, Terry AM, Bolton HA, Hancock DG, Shklovskaya E, Fazekas de St. Groth B. Pro- and anti-tumour effects of B cells and antibodies in cancer: a comparison of clinical studies and preclinical models. Cancer Immunol Immunother CII. (2016) 65:885–96. doi: 10.1007/s00262-016-1848-z
32. Kinker GS, Vitiello GAF, Ferreira WAS, Chaves AS, Cordeiro de Lima VC, Medina TdaS. B cell orchestration of anti-tumor immune responses: A matter of cell localization and communication. Front Cell Dev Biol. (2021) 9:678127. doi: 10.3389/fcell.2021.678127
33. Fridman WH, Meylan M, Petitprez F, Sun C-M, Italiano A, Sautès-Fridman C. B cells and tertiary lymphoid structures as determinants of tumour immune contexture and clinical outcome. Nat Rev Clin Oncol. (2022) 19:441–57. doi: 10.1038/s41571-022-00619-z
34. Ye J, Lee PP. B cell receptor signaling strength modulates cancer immunity. J Clin Invest. (2022) 132:e157665. doi: 10.1172/JCI157665
35. Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature. (2020) 577:549–55. doi: 10.1038/s41586-019-1922-8
36. Vanhersecke L, Brunet M, Guégan J-P, Rey C, Bougouin A, Cousin S, et al. Mature tertiary lymphoid structures predict immune checkpoint inhibitor efficacy in solid tumors independently of PD-L1 expression. Nat Cancer. (2021) 2:794–802. doi: 10.1038/s43018-021-00232-6
37. Patil NS, Nabet BY, Müller S, Koeppen H, Zou W, Giltnane J, et al. Intratumoral plasma cells predict outcomes to PD-L1 blockade in non-small cell lung cancer. Cancer Cell. (2022) 40:289–300.e4. doi: 10.1016/j.ccell.2022.02.002
38. Petitprez F, de Reyniès A, Keung EZ, Chen TW-W, Sun C-M, Calderaro J, et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature. (2020) 577:556–60. doi: 10.1038/s41586-019-1906-8
39. Cui C, Wang J, Fagerberg E, Chen P-M, Connolly KA, Damo M, et al. Neoantigen-driven B cell and CD4 T follicular helper cell collaboration promotes anti-tumor CD8 T cell responses. Cell. (2021) 184:6101–6118.e13. doi: 10.1016/j.cell.2021.11.007
40. Wennhold K, Thelen M, Lehmann J, Schran S, Preugszat E, Garcia-Marquez M, et al. CD86+ Antigen-presenting B cells are increased in cancer, localize in tertiary lymphoid structures, and induce specific T-cell responses. Cancer Immunol Res. (2021) 9:1098–108. doi: 10.1158/2326-6066.CIR-20-0949
41. Agerer B, Koblischke M, Gudipati V, Montaño-Gutierrez LF, Smyth M, Popa A, et al. SARS-CoV-2 mutations in MHC-I-restricted epitopes evade CD8+ T cell responses. Sci Immunol. (2021) 6:eabg6461. doi: 10.1126/sciimmunol.abg6461
42. Yu W, Jiang N, Ebert PJR, Kidd BA, Müller S, Lund PJ, et al. Clonal deletion prunes but does not eliminate self-specific αβ CD8(+) T lymphocytes. Immunity. (2015) 42:929–41. doi: 10.1016/j.immuni.2015.05.001
43. Straub A, Grassmann S, Jarosch S, Richter L, Hilgendorf P, Hammel M, et al. Recruitment of epitope-specific T cell clones with a low-avidity threshold supports efficacy against mutational escape upon re-infection. Immunity. (2023) 56:1269–1284.e6. doi: 10.1016/j.immuni.2023.04.010
44. Keck S, Schmaler M, Ganter S, Wyss L, Oberle S, Huseby ES, et al. Antigen affinity and antigen dose exert distinct influences on CD4 T-cell differentiation. Proc Natl Acad Sci U.S.A. (2014) 111:14852–7. doi: 10.1073/pnas.1403271111
45. Merkenschlager J, Ploquin MJ, Eksmond U, Andargachew R, Thorborn G, Filby A, et al. Stepwise B-cell-dependent expansion of T helper clonotypes diversifies the T-cell response. Nat Commun. (2016) 7:10281. doi: 10.1038/ncomms10281
46. Sandberg JK, Franksson L, Sundbäck J, Michaelsson J, Petersson M, Achour A, et al. T cell tolerance based on avidity thresholds rather than complete deletion allows maintenance of maximal repertoire diversity. J Immunol Baltim Md 1950. (2000) 165:25–33. doi: 10.4049/jimmunol.165.1.25
47. Zehn D, Bevan MJ. T cells with low avidity for a tissue-restricted antigen routinely evade central and peripheral tolerance and cause autoimmunity. Immunity. (2006) 25:261–70. doi: 10.1016/j.immuni.2006.06.009
48. Cebula A, Kuczma M, Szurek E, Pietrzak M, Savage N, Elhefnawy WR, et al. Dormant pathogenic CD4+ T cells are prevalent in the peripheral repertoire of healthy mice. Nat Commun. (2019) 10:4882. doi: 10.1038/s41467-019-12820-3
49. Cibotti R, Kanellopoulos JM, Cabaniols JP, Halle-Panenko O, Kosmatopoulos K, Sercarz E, et al. Tolerance to a self-protein involves its immunodominant but does not involve its subdominant determinants. Proc Natl Acad Sci U.S.A. (1992) 89:416–20. doi: 10.1073/pnas.89.1.416
50. Slifka MK, Blattman JN, Sourdive DJD, Liu F, Huffman DL, Wolfe T, et al. Preferential escape of subdominant CD8+ T cells during negative selection results in an altered antiviral T cell hierarchy. J Immunol Baltim Md 1950. (2003) 170:1231–9. doi: 10.4049/jimmunol.170.3.1231
51. Houot R, Perrot I, Garcia E, Durand I, Lebecque S. Human CD4+CD25high regulatory T cells modulate myeloid but not plasmacytoid dendritic cells activation. J Immunol Baltim Md 1950. (2006) 176:5293–8. doi: 10.4049/jimmunol.176.9.5293
52. Onishi Y, Fehervari Z, Yamaguchi T, Sakaguchi S. Foxp3+ natural regulatory T cells preferentially form aggregates on dendritic cells in vitro and actively inhibit their maturation. Proc Natl Acad Sci U.S.A. (2008) 105:10113–8. doi: 10.1073/pnas.0711106105
53. Hong S, Zhang Z, Liu H, Tian M, Zhu X, Zhang Z, et al. B cells are the dominant antigen-presenting cells that activate naive CD4+ T cells upon immunization with a virus-derived nanoparticle antigen. Immunity. (2018) 49:695–708. doi: 10.1016/j.immuni.2018.08.012
54. Pace L, Tempez A, Arnold-Schrauf C, Lemaitre F, Bousso P, Fetler L, et al. Regulatory T cells increase the avidity of primary CD8+ T cell responses and promote memory. Science. (2012) 338:532–6. doi: 10.1126/science.1227049
55. Fontaine M, Vogel I, Van Eycke Y-R, Galuppo A, Ajouaou Y, Decaestecker C, et al. Regulatory T cells constrain the TCR repertoire of antigen-stimulated conventional CD4 T cells. EMBO J. (2018) 37:398–412. doi: 10.15252/embj.201796881
56. Kleindienst P, Brocker T. Concerted antigen presentation by dendritic cells and B cells is necessary for optimal CD4 T-cell immunity in vivo. Immunology. (2005) 115:556–64. doi: 10.1111/j.1365-2567.2005.02196.x
57. Gerloni M, Xiong S, Mukerjee S, Schoenberger SP, Croft M, Zanetti M. Functional cooperation between T helper cell determinants. Proc Natl Acad Sci U.S.A. (2000) 97:13269–74. doi: 10.1073/pnas.230429197
58. Olson TS, Bamias G, Naganuma M, Rivera-Nieves J, Burcin TL, Ross W, et al. Expanded B cell population blocks regulatory T cells and exacerbates ileitis in a murine model of Crohn disease. J Clin Invest. (2004) 114:389–98. doi: 10.1172/JCI20855
59. Ellis JS, Braley-Mullen H. Mechanisms by which B cells and regulatory T cells influence development of murine organ-specific autoimmune diseases. J Clin Med. (2017) 6:13. doi: 10.3390/jcm6020013
60. Kaseke C, Park RJ, Singh NK, Koundakjian D, Bashirova A, Garcia Beltran WF, et al. HLA class-I-peptide stability mediates CD8+ T cell immunodominance hierarchies and facilitates HLA-associated immune control of HIV. Cell Rep. (2021) 36:109378. doi: 10.1016/j.celrep.2021.109378
61. Kedl RM, Rees WA, Hildeman DA, Schaefer B, Mitchell T, Kappler J, et al. T cells compete for access to antigen-bearing antigen-presenting cells. J Exp Med. (2000) 192:1105–13. doi: 10.1084/jem.192.8.1105
62. Farrington LA, Smith TA, Grey F, Hill AB, Snyder CM. Competition for antigen at the level of the APC is a major determinant of immunodominance during memory inflation in murine cytomegalovirus infection. J Immunol Baltim Md 1950. (2013) 190:3410–6. doi: 10.4049/jimmunol.1203151
63. Burchill MA, Tamburini BA, Kedl RM. T cells compete by cleaving cell surface CD27 and blocking access to CD70-bearing APCs. Eur J Immunol. (2015) 45:3140–9. doi: 10.1002/eji.201545749
64. Mysore V, Cullere X, Mears J, Rosetti F, Okubo K, Liew PX, et al. FcγR engagement reprograms neutrophils into antigen cross-presenting cells that elicit acquired anti-tumor immunity. Nat Commun. (2021) 12:4791. doi: 10.1038/s41467-021-24591-x
65. Harbers SO, Crocker A, Catalano G, D’Agati V, Jung S, Desai DD, et al. Antibody-enhanced cross-presentation of self antigen breaks T cell tolerance. J Clin Invest. (2007) 117:1361–9. doi: 10.1172/JCI29470
66. Kotliarov Y, Sparks R, Martins AJ, Mulè MP, Lu Y, Goswami M, et al. Broad immune activation underlies shared set point signatures for vaccine responsiveness in healthy individuals and disease activity in patients with lupus. Nat Med. (2020) 26:618–29. doi: 10.1038/s41591-020-0769-8
67. Nelson CE, Thompson EA, Quarnstrom CF, Fraser KA, Seelig DM, Bhela S, et al. Robust iterative stimulation with self-antigens overcomes CD8+ T cell tolerance to self- and tumor antigens. Cell Rep. (2019) 28:3092–3104.e5. doi: 10.1016/j.celrep.2019.08.038
68. Kumai T, Lee S, Cho H-I, Sultan H, Kobayashi H, Harabuchi Y, et al. Optimization of peptide vaccines to induce robust antitumor CD4 T-cell responses. Cancer Immunol Res. (2017) 5:72–83. doi: 10.1158/2326-6066.CIR-16-0194
69. Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. (2003) 299:1033–6. doi: 10.1126/science.1078231
70. Vanden Bush TJ, Buchta CM, Claudio J, Bishop GA. Cutting Edge: Importance of IL-6 and cooperation between innate and adaptive immune receptors in cellular vaccination with B lymphocytes. J Immunol Baltim Md 1950. (2009) 183:4833–7. doi: 10.4049/jimmunol.0900968
71. Hackl D, Loschko J, Sparwasser T, Reindl W, Krug AB. Activation of dendritic cells via TLR7 reduces Foxp3 expression and suppressive function in induced Tregs. Eur J Immunol. (2011) 41:1334–43. doi: 10.1002/eji.201041014
72. Anz D, Koelzer VH, Moder S, Thaler R, Schwerd T, Lahl K, et al. Immunostimulatory RNA blocks suppression by regulatory T cells. J Immunol Baltim Md 1950. (2010) 184:939–46. doi: 10.4049/jimmunol.0901245
73. Leibler C, John S, Elsner RA, Thomas KB, Smita S, Joachim S, et al. Genetic dissection of TLR9 reveals complex regulatory and cryptic proinflammatory roles in mouse lupus. Nat Immunol. (2022) 23:1457–69. doi: 10.1038/s41590-022-01310-2
74. Soni C, Wong EB, Domeier PP, Khan TN, Satoh T, Akira S, et al. B cell-intrinsic TLR7 signaling is essential for the development of spontaneous germinal centers. J Immunol Baltim Md 1950. (2014) 193:4400–14. doi: 10.4049/jimmunol.1401720
75. Rubtsov AV, Rubtsova K, Fischer A, Meehan RT, Gillis JZ, Kappler JW, et al. Toll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c+ B-cell population is important for the development of autoimmunity. Blood. (2011) 118:1305–15. doi: 10.1182/blood-2011-01-331462
76. Brown GJ, Cañete PF, Wang H, Medhavy A, Bones J, Roco JA, et al. TLR7 gain-of-function genetic variation causes human lupus. Nature. (2022) 605:349–56. doi: 10.1038/s41586-022-04642-z
77. Fillatreau S, Manfroi B, Dörner T. Toll-like receptor signalling in B cells during systemic lupus erythematosus. Nat Rev Rheumatol. (2021) 17:98–108. doi: 10.1038/s41584-020-00544-4
78. Jackson SW, Kolhatkar NS, Rawlings DJ. B cells take the front seat: dysregulated B cell signals orchestrate loss of tolerance and autoantibody production. Curr Opin Immunol. (2015) 33:70–7. doi: 10.1016/j.coi.2015.01.018
79. Deane JA, Pisitkun P, Barrett RS, Feigenbaum L, Town T, Ward JM, et al. Control of toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity. (2007) 27:801–10. doi: 10.1016/j.immuni.2007.09.009
80. Zumaquero E, Stone SL, Scharer CD, Jenks SA, Nellore A, Mousseau B, et al. IFNγ induces epigenetic programming of human T-bethi B cells and promotes TLR7/8 and IL-21 induced differentiation. eLife. (2019) 8:e41641. doi: 10.7554/eLife.41641
81. Chodisetti SB, Fike AJ, Domeier PP, Singh H, Choi NM, Corradetti C, et al. Type II but not type I IFN signaling is indispensable for TLR7-promoted development of autoreactive B cells and systemic autoimmunity. J Immunol Baltim Md 1950. (2020) 204:796–809. doi: 10.4049/jimmunol.1901175
82. Bekeredjian-Ding IB, Wagner M, Hornung V, Giese T, Schnurr M, Endres S, et al. Plasmacytoid dendritic cells control TLR7 sensitivity of naive B cells via type I IFN. J Immunol Baltim Md 1950. (2005) 174:4043–50. doi: 10.4049/jimmunol.174.7.4043
83. Wolin S. RNPs and autoimmunity: 20 years later. RNA N Y N. (2015) 21:548–9. doi: 10.1261/rna.050385.115
84. Molnarfi N, Schulze-Topphoff U, Weber MS, Patarroyo JC, Prod’homme T, Varrin-Doyer M, et al. MHC class II-dependent B cell APC function is required for induction of CNS autoimmunity independent of myelin-specific antibodies. J Exp Med. (2013) 210:2921–37. doi: 10.1084/jem.20130699
85. Poovassery JS, Bishop GA. Type I IFN receptor and the B cell antigen receptor regulate TLR7 responses via distinct molecular mechanisms. J Immunol Baltim Md 1950. (2012) 189:1757–64. doi: 10.4049/jimmunol.1200624
86. Nündel K, Green NM, Shaffer AL, Moody KL, Busto P, Eilat D, et al. Cell-intrinsic expression of TLR9 in autoreactive B cells constrains BCR/TLR7-dependent responses. J Immunol Baltim Md 1950. (2015) 194:2504–12. doi: 10.4049/jimmunol.1402425
87. Buchta CM, Bishop GA. Toll-like receptors and B cells: functions and mechanisms. Immunol Res. (2014) 59:12–22. doi: 10.1007/s12026-014-8523-2
88. Greiling TM, Dehner C, Chen X, Hughes K, Iñiguez AJ, Boccitto M, et al. Commensal orthologs of the human autoantigen Ro60 as triggers of autoimmunity in lupus. Sci Transl Med. (2018) 10:eaan2306. doi: 10.1126/scitranslmed.aan2306
89. Atsumi S, Katoh H, Komura D, Hashimoto I, Furuya G, Koda H, et al. Focal adhesion ribonucleoprotein complex proteins are major humoral cancer antigens and targets in autoimmune diseases. Commun Biol. (2020) 3:588. doi: 10.1038/s42003-020-01305-5
90. Hansen GL, Gaudernack G, Brunsvig PF, Cvancarova M, Kyte JA. Immunological factors influencing clinical outcome in lung cancer patients after telomerase peptide vaccination. Cancer Immunol Immunother CII. (2015) 64:1609–21. doi: 10.1007/s00262-015-1766-5
91. Verbeke R, Lentacker I, De Smedt SC, Dewitte H. Three decades of messenger RNA vaccine development. Nano Today. (2019) 28:100766. doi: 10.1016/j.nantod.2019.100766
92. Lynn GM, Sedlik C, Baharom F, Zhu Y, Ramirez-Valdez RA, Coble VL, et al. Peptide-TLR-7/8a conjugate vaccines chemically programmed for nanoparticle self-assembly enhance CD8 T-cell immunity to tumor antigens. Nat Biotechnol. (2020) 38:320–32. doi: 10.1038/s41587-019-0390-x
93. Rajagopal D, Paturel C, Morel Y, Uematsu S, Akira S, Diebold SS. Plasmacytoid dendritic cell-derived type I interferon is crucial for the adjuvant activity of Toll-like receptor 7 agonists. Blood. (2010) 115:1949–57. doi: 10.1182/blood-2009-08-238543
94. Mehta NK, Pradhan RV, Soleimany AP, Moynihan KD, Rothschilds AM, Momin N, et al. Pharmacokinetic tuning of protein-antigen fusions enhances the immunogenicity of T-cell vaccines. Nat BioMed Eng. (2020) 4:636–48. doi: 10.1038/s41551-020-0563-4
95. Newman JH, Chesson CB, Herzog NL, Bommareddy PK, Aspromonte SM, Pepe R, et al. Intratumoral injection of the seasonal flu shot converts immunologically cold tumors to hot and serves as an immunotherapy for cancer. Proc Natl Acad Sci U.S.A. (2020) 117:1119–28. doi: 10.1073/pnas.1904022116
96. Hailemichael Y, Woods A, Fu T, He Q, Nielsen MC, Hasan F, et al. Cancer vaccine formulation dictates synergy with CTLA-4 and PD-L1 checkpoint blockade therapy. J Clin Invest. (2018) 128:1338–54. doi: 10.1172/JCI93303
97. Blass E, Ott PA. Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat Rev Clin Oncol. (2021) 18:215–29. doi: 10.1038/s41571-020-00460-2
Keywords: B-cell, tolerance, immunotherapy, epitope spreading, RNA binding protein
Citation: Kellermann G, Leulliot N, Cherfils-Vicini J, Blaud M and Brest P (2024) Activated B-Cells enhance epitope spreading to support successful cancer immunotherapy. Front. Immunol. 15:1382236. doi: 10.3389/fimmu.2024.1382236
Received: 05 February 2024; Accepted: 26 February 2024;
Published: 19 March 2024.
Edited by:
Kevin M. Mcbride, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
Tahereh Soltantoye, Tehran University of Medical Sciences, IranCopyright © 2024 Kellermann, Leulliot, Cherfils-Vicini, Blaud and Brest. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guillaume Kellermann, dGVsb21pdW0uYmlvdGVjaEBnbWFpbC5jb20=; Patrick Brest, cGF0cmljay5icmVzdEB1bml2LWNvdGVkYXp1ci5mcg==
†These authors have contributed equally to this work and share last authorship
‡ORCID: Guillaume Kellermann, orcid.org/0009-0003-4047-6476
Nicolas Leulliot, orcid.org/0000-0003-0283-4298
Julien Cherfils-Vicini, orcid.org/0000-0001-7813-1527
Magali Blaud, orcid.org/0000-0001-8425-9754
Patrick Brest, orcid.org/0000-0002-1252-4747