 Beatriz Martín-Antonio1*
Beatriz Martín-Antonio1* Belén Blanco2*
Belén Blanco2* África González-Murillo3*
África González-Murillo3* Laura Hidalgo4*
Laura Hidalgo4* Jordi Minguillón5*
Jordi Minguillón5* Gema Pérez-Chacón6* and Next Generation CART MAD Consortium
Gema Pérez-Chacón6* and Next Generation CART MAD Consortium- 1Department of Experimental Hematology, Instituto de Investigación Sanitaria-Fundación Jiménez Diaz (IIS-FJD), Madrid, Spain
- 2Cancer Immunotherapy Unit (UNICA), Department of Immunology, Instituto de Investigación Sanitaria Hospital 12 de Octubre (imas12), Madrid, Spain
- 3Department of Pediatric Hematology and Oncology, Advanced Therapies Unit, Fundación Investigación Biomédica Hospital Infantil Universitario Niño Jesús, Madrid, Spain
- 4Cellular Biotechnology Unit, Instituto de Salud Carlos III (ISCIII), Madrid, Spain
- 5La Paz Hospital Institute for Health Research (IdiPAZ), Hospital Universitario La Paz. Universidad Autónoma de Madrid (UAM), Madrid, Spain
- 6Immunity, Immunopathology and Emergent Therapies Group. Instituto de Investigaciones Biomedicas Sols-Morreale. CSIC-UAM, Madrid, Spain
Adoptive T cellular immunotherapies have emerged as relevant approaches for treating cancer patients who have relapsed or become refractory (R/R) to traditional cancer treatments. Chimeric antigen receptor (CAR) T-cell therapy has improved survival in various hematological malignancies. However, significant limitations still impede the widespread adoption of these therapies in most cancers. To advance in this field, six research groups have created the “NEXT Generation CART MAD Consortium” (NEXT CART) in Madrid’s Community, which aims to develop novel cell-based immunotherapies for R/R and poor prognosis cancers. At NEXT CART, various basic and translational research groups and hospitals in Madrid concur to share and synergize their basic expertise in immunotherapy, gene therapy, and immunological synapse, and clinical expertise in pediatric and adult oncology. NEXT CART goal is to develop new cell engineering approaches and treatments for R/R adult and pediatric neoplasms to evaluate in multicenter clinical trials. Here, we discuss the current limitations of T cell-based therapies and introduce our perspective on future developments. Advancement opportunities include developing allogeneic products, optimizing CAR signaling domains, combining cellular immunotherapies, multi-targeting strategies, and improving tumor-infiltrating lymphocytes (TILs)/T cell receptor (TCR) therapy. Furthermore, basic studies aim to identify novel tumor targets, tumor molecules in the tumor microenvironment that impact CAR efficacy, and strategies to enhance the efficiency of the immunological synapse between immune and tumor cells. Our perspective of current cellular immunotherapy underscores the potential of these treatments while acknowledging the existing hurdles that demand innovative solutions to develop their potential for cancer treatment fully.
Introduction
In the last decade, CAR-T cell therapy has become 2nd line of treatment in relapsed/refractory hematological malignancies. Currently, there are four approved CAR-T cell products directed to CD19 (CART-19) for B cell malignancies (axi-cel, tisa-cel, liso-cel, and brexu-cel) and two products targeting B-cell maturation antigen (BCMA) (CART-BCMA) (ide-cel and cilta-cel) for multiple myeloma (MM) patients (1, 2). Despite this growing number of CAR-T cell products approved for hematological malignancies, no CAR-T cell-based treatments are available to treat solid tumors, evidencing distinct limitations that guide new lines of research to develop improved CAR-T cells. Clinical studies have demonstrated that limitations in the CAR treatment differ depending on the CAR and the malignancy and could arise at different stages of the treatment.
Among the most common complications in CAR-T therapy are failures in producing autologous CAR-T cells, loss of target antigen, the barriers imposed by the tumor microenvironment (TME), and the lack of efficacy and persistence of CAR-T cells. Of interest, the immunological synapse (IS) constitutes a platform that involves changes in the cortical F-actin to transfer cytotoxic proteins from immune T and B cells to tumor cells, and has been widely studied in physiological conditions (3). The IS between CAR-T cells and tumor cells is not entirely understood and research in this field might provide new insights and targets to tackle some of these problems. Here, we outline the ongoing strategies developed to avoid events that decrease the efficacy of CAR-T treatment. As a Consortium, we will provide our perspective on how these strategies will evolve in the coming years to solve the main current limitations of these therapies.
Off-the-shelf CAR therapies: developing alternatives to circumvent the failure of autologous CAR-T cell production
Tumor patients are often immunosuppressed and around 10% of patients enrolled in CAR-T therapy do not receive the CAR treatment due to a failure to produce autologous CAR-T cells (4, 5). The infusion of allogeneic universal CAR-T cells is a field in development to circumvent this problem. Current techniques knock out the T cell receptor α constant (TRAC) and CD52 genes/loci to reduce the risk of graft-versus-host disease (GvHD) and to avoid a host-versus-graft reaction, respectively. This option has been tested for CART-19 (6) and CART-BCMA cells (7), showing manageable safety. However, responses are still lower than those achieved with autologous CAR-T cells (5, 8). The previous depletion of the CD45RA+ fraction in CAR-T cells is another option to obtain a less alloreactive product enriched in memory T cells (9, 10). Indeed, our Consortium is administering allogenic memory CART-NKG2D cells in sarcoma patients (NCT06087341).
Other alternatives to circumvent the failure of autologous CAR-T cell production include allogeneic CAR-NK cells, which do not cause GVHD (11). Clinical trials have confirmed their safety (11), emerging as an alternative to producing allogeneic CAR cells (12). They are obtained from different sources, including cord blood (12), allogeneic induced pluripotent stem cells (iPSCs) (13), and peripheral blood. Despite the early expectations in this field, the intrinsic short half-life of NK cells might be responsible for the lower efficacy observed compared to CAR-T cells (14). Engineering CAR-NK cells with intrinsic NK receptor domains might improve their persistence. Indeed, various novel constructs that replace co-stimulatory and transmembrane domains of the CAR with more typical NK cell domains are being developed. Thus, NKG2D transmembrane, 2B4 co-stimulatory or DAP12 signaling domains (15) confer higher efficacy than CAR-NK cells containing CD28, 4-1BB, and CD3ζ; T cell signaling domains (16). Moreover, including specific cytokines in the construct should prolong CAR-NK cell persistence (17).
In addition, NK cells present additional advantages, such as CD16 expression that activates their antibody-dependent cell-mediated cytotoxicity (ADCC). Thus, combining CAR-NK cells with antibodies targeting tumor-associated antigens (TAAs) that bind to CD16 will trigger ADCC and could enhance their efficacy. Last but not least, CAR-NK treatment could also be combined with antibodies that block NK inhibitory receptors, such as NKG2A (18).
Another approach to explore in the clinic is exploiting the essential property of NK cells for activating other immune cells (15), including T cells and CAR-T cells (19). Indeed, a combined treatment based on CAR-T and NK cells enhances in vivo CAR-T efficacy (19), offering a therapeutic advantage.
Optimization of CAR domains to enhance the IS formation and function
Research on CAR domain modification to optimize IS formation and function between CAR cells and their tumor cell targets represents a field in development that might improve clinical results. Indeed, CAR-T cells’ IS differs from that of TCRs and T cells secreting bispecific T-cell engager antibodies (STAb-T cells) (20). CAR-T cells’ IS relies mainly on the CAR interaction with the tumor antigen, which limits the stability and strength of the IS. In contrast, STAb-T cells recapitulate key features of the TCR-mediated IS more closely (see below). The key characteristics of the distinct IS are shown in Figure 1 to illustrate differences that could guide new research lines to improve CAR-T cells’ IS formation and effector function.
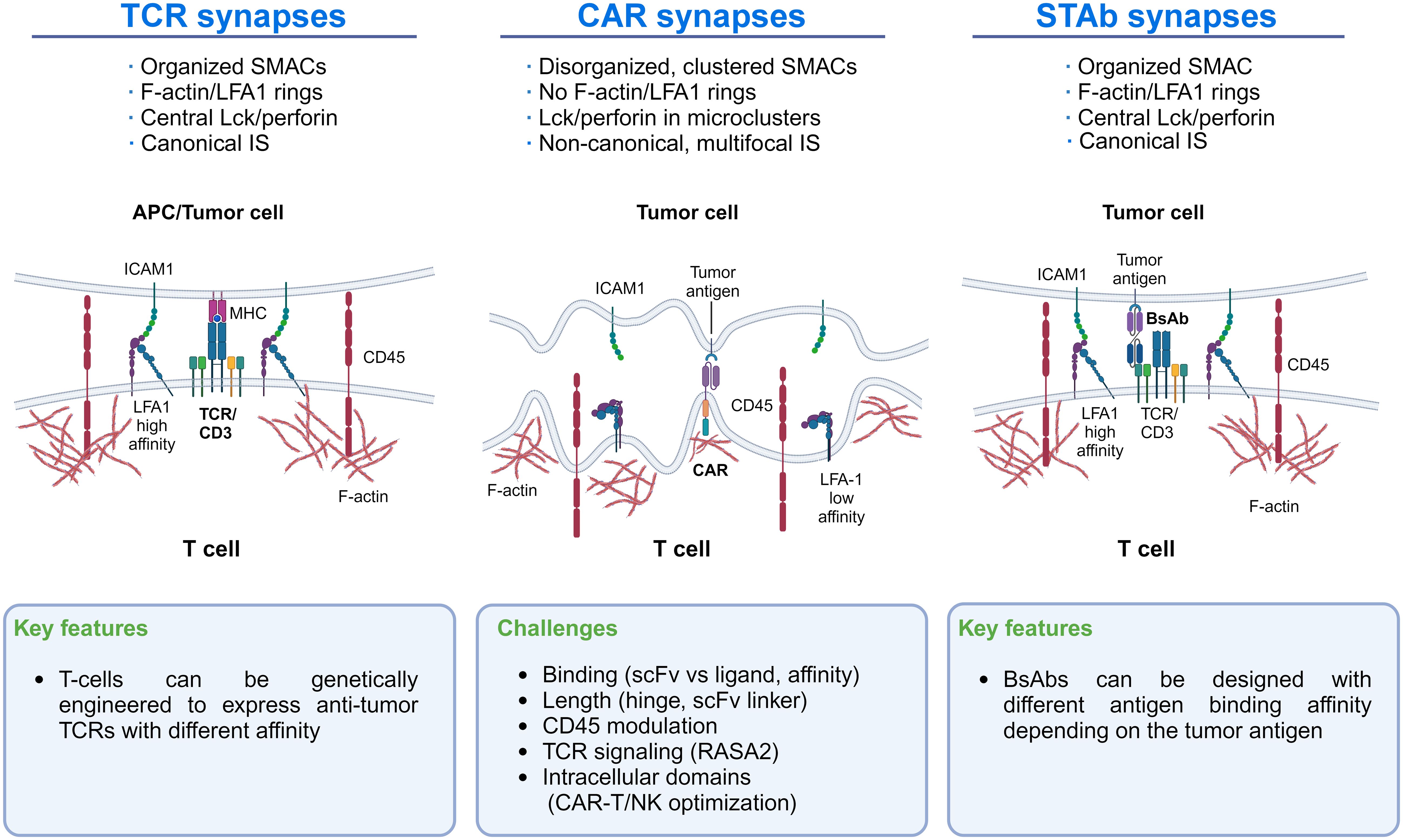
Figure 1 Immunological synapse features of TCRs, CAR-T cells, and STAb-T cells: The main differences in the IS of TCRs, CAR-T cells, and STAb-T cells are shown. These differences could guide new lines of research to improve CAR-T cells’ IS formation.
Increasing LFA-1/ICAM-1 interaction around CAR-T cell IS, as occurs with TCR-mediated IS, might improve the efficacy of CAR-T therapy. Indeed, recent studies highlight the significance of LFA-1 expression in forming the IS in T and NK cells (21, 22) and the importance of the LFA-1/ICAM-1 axis in CAR-T cell/tumor cell interactions in solid tumors (23, 24) (Figure 1). Initially, CAR-T cell activation strongly depends on the expression level of ICAM-1 on tumor cells (24), where CAR-T cells detach from malignant cells more quickly than normal T cells due to LFA-1 downregulation in the IS (25). Therefore, improving LFA-1/ICAM-1 interaction in the CAR-T synapse might help to establish more efficient IS, which could be achieved by enforcing in the CAR-T the production of cytokines that upregulate cell adhesion molecules (see below). Another suitable approach could be to express an anti-ICAM-1 scFv on the CAR-T that would interact with ICAM-1 in the tumor cell. However, limitations of this strategy would be the difficulty of restricting the scFv-ICAM-1 interaction to the IS and whether the strength and stability of this interaction would be counterproductive for CAR-T cell function. Other critical factors that modulate the IS formation and CAR-T activity include scFv/ligand conformation, the hinge length, and the presence of additional CAR domains (26, 27). For instance, in 4-1BB CARs targeting CD22, shortening the linker of the scFv drives antigen-independent signaling. This tonic signaling enhanced IS formation and conferred superior effector function, a finding not observed in CD28-based CARs, suggesting that shorter linkers may benefit 4-1BB CARs (28).
Furthermore, adding an intracellular scaffolding protein binding site, such as PDZ domain, enhances the IS formation and improves CAR-T and CAR-NK efficacy against solid tumors (29). Also, blocking TCR-signaling inhibitors, such as RASA2, improves the IS function, providing larger synaptic areas, increased lytic granules, and pZAP70 accumulation at the IS (30).
Studies analyzing CART-19 interactions have raised a “size exclusion” model whereby CAR-T activation depends on the size difference between the CAR-antigen pair and CD45 on the membrane. Precisely, CD19 engagement with CART-19 results in a narrow intermembrane space that excludes CD45, favoring CAR phosphorylation and CAR-T cell activation. Increasing the size of CAR extracellular domains attenuates CART-19 cell activation, and increasing the size of CD45 enhances CART-19 activation (31).
Several ex-vivo approaches based on artificial intelligence analyses of F-actin reorganization at the IS have assessed the IS quality made by CAR-T cells (32, 33). Indeed, in vitro learning-based IS quality based on F-actin measurements correlates with patient clinical outcomes upon CAR-T therapy (34). This strategy could provide guidelines for designing and optimizing CAR constructs and also optimize T cell-redirecting strategies such as STAb-T cells (see below) for potential clinical developments.
Improving CAR recognition domains to avoid early CAR-T disappearance
Most CARs used in the clinic are derived from murine single-chain variable fragments (scFvs), which induce the development of human anti-murine antibodies (HAMAs) by the patients, leading to early CAR-T cell disappearance, without time to achieve responses in the patient (35). Current strategies to avoid this event include humanizing murine CARs (5). Also, camelid heavy-chain variable domains (VHHs) present higher homology to human variable domains, being less immunogenic than murine scFv (2). These two options have shown higher rates of initial responses in MM than those derived from murine scFv (1, 2, 5). In addition, newer CAR constructs contain synthetic antigen-binding domains that are fully human, and have shown outstanding results, representing a promising alternative (36).
Multi-targeting to avoid relapses due to target antigen loss, tumor heterogeneity, and on-target, off-tumor toxicities
CART-19 therapy in pediatric B-ALL represents a curative option, with an overall remission rate of 82% after 38.8 months, and a median overall survival not reached (8). However, relapses still occur due to a target antigen loss on tumor cells. It happens more frequently in pediatric ALL patients (7) than in adult patients (13), with 68% vs. 13% of relapsed patients after CART-19, respectively. Moreover, some antigens are more prone to disappear than others. Thus, loss of BCMA in MM after CAR-T treatment is hardly observed or only suspected (5), whereas target antigen loss in solid tumors has been observed (37).
Multi-targeting CAR-T strategies, such as CARs in tandem/dual, bicistronic formats or co-administration/co-transduction and sequential approaches targeting CD19/CD22 or CD19/CD20, have been used in clinical studies in B-ALL as an alternative to avoid these relapses (38–43). Although results are encouraging, this strategy is hampered by the loss of the second target antigen (44), and, therefore, other approaches are being developed, such as CARs directed to antigens that do not undergo loss of expression or tri-specific approaches combining CD19/CD20/CD22 (45).
In other pediatric malignancies such as acute myeloid leukemia (AML), neuroblastoma, hepatoblastoma, or osteosarcoma, CAR-T cell therapy is still inefficient due to the heterogeneity of the tumor (46). Thus, in AML, dual/tandem CAR approaches targeting multiple antigens, including CD123/CLL1 (30), CD123/CD33, or CD33/CLL1/CD123 are being tested to fight tumor heterogeneity (47). In pediatric osteosarcoma, newer CAR designs, such as switchable CARs, are being evaluated in our Consortium (48) to solve tumor heterogeneity.
Moreover, finding tumor-specific antigens to avoid toxicities remains challenging for solid tumors. Dual gate-logic CARs, which require recognition of two antigens to become activated (49), and split, universal, and programmable (SUPRA) CARs (50) will help reduce on-target/off-tumor toxicities.
Shortening of CAR production and other approaches to avoid immunosenescence and improve CAR persistence
The most frequent relapses after achieving a response occur due to a lack of CAR-T cell persistence. The age of the patient (51), the number of previous lines of treatments (52), and the production of CAR-T cells (53) induce terminal T cell differentiation, where T cells become immunosenescent, presenting a sorter half-life and, therefore, a lower persistence in the patient. Indeed, MM patients, an elderly population, achieve 100% of objective responses after CAR-T treatment; however, a proportion of them end up relapsing before the 1st year with a lack of CAR-T cell persistence (5). Conversely, pediatric and young ALL patients treated with CAR-T cells have not relapsed after 38.8 months and still present B-cell aplasia, indicating CAR-T cell persistence (8). Different production techniques that shorten the time of manufacturing or adding drugs to avoid CAR-T cell differentiation (54) will be helpful strategies to prevent this issue.
TRUCKs to improve the quality of the IS and to fight the TME barriers
Additional factors that impact CAR-T persistence and efficacy are the barriers of the TME. Indeed, CAR-T cells can become anergic when they arrive at the TME. Cell-cell communication between tumor and CAR-T cells through the IS might lead to these events. It is well-known that the IS serves as a platform to transfer cytotoxic immune and tumor proteins that may impact the activity of immune cells. Indeed, the quality of the IS dictates the anti-tumor efficacy of T cells, NK cells, and CAR-T cells (33). Thus, an algorithm that measures the quality of the IS based on four parameters (F-actin, perforin, tumor antigen, and pZeta) demonstrated that the CAR-T IS correlates with the clinical response in patients treated with CART-Kappa cells (NCT00881920), supporting that this “ex vivo” synaptic parameter can predict the clinical response to CAR treatment (34).
Developing the so-called fourth-generation CAR-T cells or TRUCKs (T cells redirected for universal cytokine-mediated killing) could serve as a strategy to use molecules that stabilize and/or potentiate the IS between malignant and TRUCK-like cells. TRUCKs require CAR-T transduction with a transgene payload encoding various genes that may improve CAR-T efficacy in the TME (55, 56). In this regard, TME signals modulate the expression of some adhesion/costimulatory molecules that affect the quality of the CAR-T IS (57). In this regard, IFNγ produced by CAR-T cells upregulates ICAM-1 on tumor cells and strengthens CAR-T and tumor cell interaction, facilitating tumor killing in solid tumors (24, 58). Therefore, gene payloads encoding genes promoting cell adhesion, i.e. inducing the expression of LFA-1 (αLβ2) or other relevant integrins in the CAR-T cell, could be a valuable strategy to strengthen the IS of tumor cells and CAR-T cells.
In this line, our Consortium is evaluating the combination of CAR-T cells with other approaches targeting IFNγ and other cytokines to promote CAR-T/tumor cell IS. Indeed, radiotherapy, which increases IFNγ on the TME (59), or armed oncolytic viruses producing CXCR3 ligands, such as CXCL10, enhancing the avidity of LFA-1 on CAR-T cells (60), are being investigated by us.
Moreover, to fight the TME barriers, novel TRUCKs that secrete different molecules are approaches under investigation (61). The TRUCK concept is also suitable for other cells susceptible to carrying a CAR, such as NK cells and macrophages (62). New approaches include the use of payloads of a variety of immunomodulatory genes besides cytokines (63). For example, IL12 has been the subject of several studies addressing this issue, using distinct strategies to express IL12 in TILs or tumors (64, 65). Despite being a promising strategy, it has become evident that there is a need to adapt these strategies to the particularities of each TME. Thus, our Consortium is developing TRUCKs expressing transgenes under the control of promoters explicitly activated in the TME of each type of tumor. Identifying promoters activated by metabolites or signals overrepresented and/or specific to particular TMEs is crucial in restricting the activation of the gene payload to the tumor, minimizing toxicities.
TILs and TCRs to fight the heterogeneity of solid tumors
Our Consortium is also developing TILs, a group of lymphocytes that include T cells, NK cells, and other innate lymphocytes that infiltrate tumors and recognize and destroy tumor cells. The main advantages of TILs include their polyclonality, which enables targeting multiple tumor antigens and circumvents the problem of antigen heterogeneity in solid tumors. Moreover, by being isolated from tumors, they are already equipped with chemokine receptors, providing a higher capacity for homing into the tumor. Finally, most TILs present a TCR with high affinity to TAAs and neoantigens, which decreases the possible on-target, off-tumor toxicities. T cell activation is mediated through highly organized and dynamic IS where the TCRs and MHC-peptide complexes interact (66–68).
TIL therapy was first described as an adoptive cell therapy by Steven A. Rosenberg in 1988 (69), where they isolated and expanded TILs and re-infused them into patients with metastatic malignant melanoma, causing tumor regression. Afterwards, in 2015, they treated nine patients with metastatic cervical cancer, obtaining 33% of durable objective responses after a single infusion of TILs (NCT01585428) (70).
Despite their potential, TIL therapy still faces several challenges. The main one is the current procedure to obtain TILs, which requires two months of in vitro expansion to get the necessary number of TILs to treat patients. This long period may lead to a product with poor persistence (71). In addition, some patients with rapidly progressing diseases cannot wait long for the isolated TILs to expand. Recent strategies to improve TIL persistence and efficacy include the knockout of endogenous TCR (72) and PD1 to avoid T cell exhaustion and overcome the particularly immunosuppressive TME of solid tumors.
Another limitation is that not all tumors are suitable for isolating active TILs, and specific TAAs are lacking. Engineered TCR-based therapies have emerged to address this issue (73–76). This therapy arose with the discovery that TILs from different patients recognize the same antigens expressed in tumor cells, such as MART-1 and glycoprotein 100 (gp100) (77). Engineered TCRs require T cell transduction with antigen-specific TCRα and β chains to generate tumor-specific T cells, expansion, and reinfusion in the patient (76). They preserve many advantages inherent to natural TCRs, such as fully mediating TCR signaling. Moreover, TCRs are sensitive to small epitope densities (67, 78), and their activation depends on the antigen presented by the MHC of a large target antigen pool (79), which allows modulating TCR affinity, especially against low-density targets (80).
In January 2022, the FDA approved the first TCR-based therapeutic for treating uveal melanoma targeting gp100 presented on HLA-A2 (81). In February 2024, Iovance Biotherapeutics announced the FDA approval of AMTAGVI™ (lifileucel), a tumor-derived autologous T-cell immunotherapy for treating adult patients with unresectable or metastatic melanoma (82). Next, in 2024, the FDA is expected to approve an engineered TCR T cell therapy targeting MAGE-A4 to treat synovial sarcoma (83).
STAb-T cells, an optimal tool to enhance the efficacy of adoptive T cell therapy
An attractive and emerging approach in adoptive T cell therapy involves the infusion of T cells genetically modified to secrete T cell-redirecting bispecific Antibodies (STAb-T cells) directed to CD3 and to a tumor-associated antigen (84). Therefore, STAb-T strategy combines aspects of antibody- and cell-based therapies and might solve some of the limitations of the T cell-redirecting strategies currently used in the clinic. Indeed, unlike systemically administered bispecific antibodies (bsAbs), STAb-T cells have the ability to migrate to areas of inflammation and damage (as can be tumors) and actively extravasate to tissues by crossing the vascular-endothelial barrier. They also have a long half-life, and produce a constant release of the therapeutic bsAb, which eliminates the problems associated with the manufacture, storage, and administration of antibodies (84). Importantly, contrary to the membrane-anchored CAR, the soluble bsAbs secreted by STAb-T cells can recruit the entire pool of T lymphocytes, both engineered and unmodified bystander T cells, present at the tumor site (85–87), enhancing the anti-tumor response.
Moreover, when CAR-T cells encounter tumor cells, they form a disorganized synapse differing from the canonical “bull’s eye” structure initiated by TCR interactions with the MHC-peptide complex (25, 66, 85, 88). Unlike CARs, small-sized T cell-engaging bsAbs induce the formation of a seemingly canonical IS between T lymphocytes and tumor cells (Figure 1). Proper synapse formation plays an essential role in the T cell activation process triggered by the TCR and in the polarized secretion of lytic granules or cytokines to the synaptic cleft (89–91). Further studies will be necessary to determine whether such differences could represent an advantage of STAb-T cells over CAR-T cells in cancer immunotherapy. Recently, studies have demonstrated a higher efficacy of STAb-T cells vs. CAR-T cells targeting the same antigen in animal models of different hematological tumors (89–91). On the other hand, the generation of CAR-T cells that secrete bsAbs simultaneously with CAR expression (CAR-STAb-T cells) (92–95) and the generation of STAb-T cells (94) are attracting much interest. Our Consortium is leading research lines in these areas.
Adoptive T cell therapy to treat other diseases
Finally, all the strategies discussed above also offer a high potential to treat other pathologies than cancer, including autoimmune and infectious diseases (96). CART-19 therapy has already reached the clinic, with ongoing trials to analyze the safety and efficacy of CART-19 cells in treating systemic lupus erythematosus (97) (NCT05030779; NCT03030976). Other approaches for targeting autoreactive B cells are being developed (98, 99).
Regarding infectious diseases, opportunistic infections affecting immunocompromised patients remain a significant cause of morbidity and mortality (100). Since their immune system is impaired, adoptive T cell therapy in these patients would depend on allogeneic T cells. In this sense, our Consortium has conducted a study evaluating the treatment of immunocompromised patients suffering from viral or fungal infections with allogeneic CD45RA-T lymphocytes from healthy donors containing pathogen-specific memory T cells (101). Moreover, this procedure is being tested in a clinical trial against SARS-CoV-2 (NCT04578210) with promising results (102).
Concluding remarks
Adoptive cell-based immunotherapy has an enormous potential to treat refractory tumors and other diseases. In the coming years, we expect all the mentioned strategies will enable therapy optimization, especially for solid tumors and other diseases. Bringing together the efforts of basic/translational and clinical researchers will help to achieve this. To this end, our Consortium points at these hurdles that demand innovative solutions to fulfill their full potential in cancer therapy.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
Author contributions
BM-A: Conceptualization, Funding acquisition, Writing – original draft, Writing – review & editing. BB: Conceptualization, Funding acquisition, Writing – original draft, Writing – review & editing. ÁG-M: Conceptualization, Funding acquisition, Writing – original draft, Writing – review & editing. LH: Conceptualization, Funding acquisition, Writing – original draft, Writing – review & editing. JM: Conceptualization, Funding acquisition, Writing – original draft, Writing – review & editing. GP-C: Conceptualization, Funding acquisition, Writing – original draft, Writing – review & editing. NC: Writing – original draft, Writing – review & editing.
Group members of ‘Next Generation CART MAD Consortium’
Hospital Universitario la Paz, Universidad Autonoma de Madrid: Jordi Minguillón, Lucía Fernández, Halin Bareke, Andrés París-Muñoz, Adriana Mañas, Cristina Aguirre-Portolés, Alfonso Navarro-Zapata, Carmen Mestre-Durán, Marta Ibáñez-Navarro, Laura Clares-Villa, Sara Naharro and Antonio Perez-Martinez (PI),
Consejo Superior de Investigaciones Científicas (CSIC): Marcos Aldea, María Gaibar, Javier Ruiz-Navarro, Manuel Izquierdo and Juan M. Zapata (PI).
Fundación Jimenez Díaz: Esperanza Esquinas Tarifa, Juana Serrano-López, Pilar Llamas-Sillero and Beatriz Martin-Antonio (PI).
Hospital Universitario Infantil Niño Jesus: África González Murillo, Elena García Sánchez, Jorge García Martínez, Alba Rubio San Simón, Blanca Herrero, Gonzalo García Aguilera, Beatriz Horcajo Morera and Manuel Ramírez Orellana (PI).
Hospital Universitario 12 de Octubre: Belén Blanco-Durango, Anais Jiménez-Reinoso, Rodrigo Lázaro-Gorines, Ivana Zagorac, Antonio Tapia Galisteo, Ángel Ramirez-Fernández, Laura Díez-Alonso, Carmen Dominguez-Alonso, Ainhoa Erce-Llamazares, Oana Hangiu, Laura Rubio Pérez, Marina Gómez Rosel, Alejandro Segura Tudela, Javier Arroyo-Ródena and Luis Álvarez-Vallina (PI).
Instituto de Salud Carlos III (ISCIII): Miguel Ángel Rodríguez Milla, Patricia García Rodríguez, Beatriz Somovilla Crespo and Javier García-Castro (PI).
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by the “Comunidad de Madrid, Dirección General de Investigación e Innovación Tecnológica”, grant number: S2022/BMD-7225. Consorcio Next Generation CART MAD “Nueva Generación de Inmunoterapias STAb y CAR-T Multidianas”. BMA is supported by the Miguel Servet Program of ISCIII (CP21/00111).
Acknowledgments
The figure was made with Biorender.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Munshi NC, Anderson LD, Shah N, Madduri D, Berdeja J, Lonial S, et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N Engl J Med. (2021) 384:705–16. doi: 10.1056/NEJMoa2024850
2. Berdeja JG, Madduri D, Usmani SZ, Jakubowiak A, Agha M, Cohen AD, et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): a phase 1b/2 open-label study. Lancet. (2021) 398:314–24. doi: 10.1016/S0140-6736(21)00933-8
3. Calvo V, Izquierdo M. Role of actin cytoskeleton reorganization in polarized secretory traffic at the immunological synapse. Front Cell Dev Biol. (2021) 9:629097. doi: 10.3389/fcell.2021.629097
4. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. (2019) 380:45–56. doi: 10.1056/NEJMoa1804980
5. Oliver-Caldés A, González-Calle V, Cabañas V, Español-Rego M, Rodríguez-Otero P, Reguera JL, et al. Fractionated initial infusion and booster dose of ARI0002h, a humanized, BCMA-directed CAR T-cell therapy, for patients with relapsed or refractory multiple myeloma (CARTBCMA-HCB-01): a single-arm, multicenter, academic pilot study. Lancet Oncol. (2023) 24:913–24. doi: 10.1016/S1470-2045(23)00222-X
6. Benjamin R, Jain N, Maus MV, Boissel N, Graham C, Jozwik A, et al. UCART19, a first-in-class allogeneic anti-CD19 chimeric antigen receptor T-cell therapy for adults with relapsed or refractory B-cell acute lymphoblastic leukemia (CALM): a phase 1, dose-escalation trial. Lancet Haematol. (2022) 9:e833–43. doi: 10.1016/S2352-3026(22)00245-9
7. Mailankody S, Matous JV, Chhabra S, Liedtke M, Sidana S, Oluwole OO, et al. Allogeneic BCMA-targeting CAR T cells in relapsed/refractory multiple myeloma: phase 1 UNIVERSAL trial interim results. Nat Med. (2023) 29:422–9. doi: 10.1038/s41591-022-02182-7
8. Laetsch TW, Maude SL, Rives S, Hiramatsu H, Bittencourt H, Bader P, et al. Three-year update of tisagenlecleucel in pediatric and young adult patients with relapsed/refractory acute lymphoblastic leukemia in the ELIANA trial. J Clin Oncol. (2023) 41:1664–9. doi: 10.1200/JCO.22.00642
9. Kim-Hoehamer Y-I, Riberdy JM, Zheng F, Park JJ, Shang N, Métais J-Y, et al. Development of a cGMP-compliant process to manufacture donor-derived, CD45RA-depleted memory CD19-CAR T cells. Gene Ther. (2023) 30:222–31. doi: 10.1038/s41434-021-00307-0
10. Fernández L, Fernández A, Mirones I, Escudero A, Cardoso L, Vela M, et al. GMP-compliant manufacturing of NKG2D CAR memory T cells using cliniMACS prodigy. Front Immunol. (2019) 10:2361. doi: 10.3389/fimmu.2019.02361
11. Bachiller M, Battram AM, Perez-Amill L, Martín-Antonio B. Natural killer cells in immunotherapy: are we nearly there? Cancers (Basel). (2020) 12:3139. doi: 10.3390/cancers12113139
12. Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. (2020) 382:545–53. doi: 10.1056/NEJMoa1910607
13. Ueda T, Kumagai A, Iriguchi S, Yasui Y, Miyasaka T, Nakagoshi K, et al. Non-clinical efficacy, safety and stable clinical cell processing of induced pluripotent stem cell-derived anti-glypican-3 chimeric antigen receptor-expressing natural killer/innate lymphoid cells. Cancer Sci. (2020) 111:1478–90. doi: 10.1111/cas.14374
14. Marin D, Li Y, Basar R, Rafei H, Daher M, Dou J, et al. Safety, efficacy and determinants of response of allogeneic CD19-specific CAR-NK cells in CD19+ B cell tumors: a phase 1/2 trial. Nat Med. (2024) 30:772–84. doi: 10.1038/s41591-023-02785-8
15. Töpfer K, Cartellieri M, Michen S, Wiedemuth R, Müller N, Lindemann D, et al. DAP12-based activating chimeric antigen receptor for NK cell tumor immunotherapy. J Immunol. (2015) 194:3201–12. doi: 10.4049/jimmunol.1400330
16. Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell. (2018) 23:181–192.e5. doi: 10.1016/j.stem.2018.06.002
17. Li L, Mohanty V, Dou J, Huang Y, Banerjee PP, Miao Q, et al. Loss of metabolic fitness drives tumor resistance after CAR-NK cell therapy and can be overcome by cytokine engineering. Sci Adv. (2023) 9:eadd6997. doi: 10.1126/sciadv.add6997
18. Geurts VCM, Voorwerk L, Balduzzi S, Salgado R, Van de Vijver K, van Dongen MGJ, et al. Unleashing NK- and CD8 T cells by combining monalizumab and trastuzumab for metastatic HER2-positive breast cancer: Results of the MIMOSA trial. Breast. (2023) 70:76–81. doi: 10.1016/j.breast.2023.06.007
19. Bachiller M, Perez-Amill L, Battram AM, Carné SC, Najjar A, Verhoeyen E, et al. NK cells enhance CAR-T cell antitumor efficacy by enhancing immune/tumor cells cluster formation and improving CAR-T cell fitness. J Immunother Cancer. (2021) 9:e002866. doi: 10.1136/jitc-2021-002866
20. Ramírez-Fernández Á, Aguilar-Sopeña Ó, Díez-Alonso L, Segura-Tudela A, Domínguez-Alonso C, Roda-Navarro P, et al. Synapse topology and downmodulation events determine the functional outcome of anti-CD19 T cell-redirecting strategies. Oncoimmunology. (2020) 11:2054106. doi: 10.1080/2162402X.2022.2054106
21. Wang MS, Hu Y, Sanchez EE, Xie X, Roy NH, de Jesus M, et al. Mechanically active integrins target lytic secretion at the immune synapse to facilitate cellular cytotoxicity. Nat Commun. (2022) 13:3222. doi: 10.1038/s41467-022-30809-3
22. Pariani AP, Almada E, Hidalgo F, Borini-Etichetti C, Vena R, Marín L, et al. Identification of a novel mechanism for LFA-1 organization during NK cytolytic response. J Cell Physiol. (2023) 238:227–41. doi: 10.1002/jcp.30921
23. Ramkumar P, Abarientos AB, Tian R, Seyler M, Leong JT, Chen M, et al. CRISPR-based screens uncover determinants of immunotherapy response in multiple myeloma. Blood Adv. (2020) 4:2899–911. doi: 10.1182/bloodadvances.2019001346
24. Kantari-Mimoun C, Barrin S, Vimeux L, Haghiri S, Gervais C, Joaquina S, et al. CAR T-cell entry into tumor islets is a two-step process dependent on IFNγ and ICAM-1. Cancer Immunol Res. (2021) 9:1425–38. doi: 10.1158/2326-6066.CIR-20-0837
25. Davenport AJ, Cross RS, Watson KA, Liao Y, Shi W, Prince HM, et al. Chimeric antigen receptor T cells form nonclassical and potent immune synapses driving rapid cytotoxicity. Proc Natl Acad Sci U.S.A. (2018) 115:E2068–76. doi: 10.1073/pnas.1716266115
26. Xiong Y, Libby KA, Su X. The physical landscape of CAR-T synapse. Biophys J. (2023) S0006-3495:00581–7. doi: 10.1016/j.bpj.2023.09.004
27. Mazinani M, Rahbarizadeh F. CAR-T cell potency: from structural elements to vector backbone components. biomark Res. (2022) 10:70. doi: 10.1186/s40364-022-00417-w
28. Singh N, Frey NV, Engels B, Barrett DM, Shestova O, Ravikumar P, et al. Antigen-independent activation enhances the efficacy of 4-1BB-costimulated CD22 CAR T cells. Nat Med. (2021) 27:842–50. doi: 10.1038/s41591-021-01326-5
29. Chockley PJ, Ibanez-Vega J, Krenciute G, Talbot LJ, Gottschalk S. Synapse-tuned CARs enhance immune cell anti-tumor activity. Nat Biotechnol. (2023) 41:1434–45. doi: 10.1038/s41587-022-01650-2
30. Ibanez J, Houke H, Meehl M, Hebbar N, Thanekar U, Velasquez P, et al. DIPG-34. IMMUNE SYNAPSE OPTIMIZED GRP78-SPECIFIC CAR T CELLS ELICIT AN IMPROVED ANTITUMOR RESPONSE AGAINST DIPGS. Neuro-Oncology. (2023) 25:i20. doi: 10.1093/neuonc/noad073.081
31. Xiao Q, Zhang X, Tu L, Cao J, Hinrichs CS, Su X. Size-dependent activation of CAR-T cells. Sci Immunol. (2022) 7:eabl3995. doi: 10.1126/sciimmunol.abl3995
32. Lee M, Lee Y-H, Song J, Kim G, Jo Y, Min H, et al. Deep-learning-based three-dimensional label-free tracking and analysis of immunological synapses of CAR-T cells. Elife. (2020) 9:e49023. doi: 10.7554/eLife.49023
33. Xiong W, Chen Y, Kang X, Chen Z, Zheng P, Hsu Y-H, et al. Immunological synapse predicts effectiveness of chimeric antigen receptor cells. Mol Ther. (2018) 26:963–75. doi: 10.1016/j.ymthe.2018.01.020
34. Naghizadeh A, Tsao W-C, Hyun Cho J, Xu H, Mohamed M, Li D, et al. In vitro machine learning-based CAR T immunological synapse quality measurements correlate with patient clinical outcomes. PloS Comput Biol. (2022) 18:e1009883. doi: 10.1371/journal.pcbi.1009883
35. Ortíz-Maldonado V, Rives S, Castellà M, Alonso-Saladrigues A, Benítez-Ribas D, Caballero-Baños M, et al. CART19-BE-01: A multicenter trial of ARI-0001 cell therapy in patients with CD19+ Relapsed/refractory Malignancies. Mol Ther. (2021) 29:636–44. doi: 10.1016/j.ymthe.2020.09.027
36. Frigault MJ, Bishop MR, Rosenblatt J, O’Donnell EK, Raje N, Cook D, et al. Phase 1 study of CART-ddBCMA for the treatment of subjects with relapsed and refractory multiple myeloma. Blood Adv. (2023) 7:768–77. doi: 10.1182/bloodadvances.2022007210
37. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. New Engl J Med. (2016) 375:2561–9. doi: 10.1056/NEJMoa1610497
38. Cordoba S, Onuoha S, Thomas S, Pignataro DS, Hough R, Ghorashian S, et al. CAR T cells with dual targeting of CD19 and CD22 in pediatric and young adult patients with relapsed or refractory B cell acute lymphoblastic leukemia: a phase 1 trial. Nat Med. (2021) 27:1797–805. doi: 10.1038/s41591-021-01497-1
39. Shalabi H, Qin H, Su A, Yates B, Wolters PL, Steinberg SM, et al. CD19/22 CAR T cells in children and young adults with B-ALL: phase 1 results and development of a novel bicistronic CAR. Blood. (2022) 140:451–63. doi: 10.1182/blood.2022015795
40. Wang T, Tang Y, Cai J, Wan X, Hu S, Lu X, et al. Coadministration of CD19- and CD22-directed chimeric antigen receptor T-cell therapy in childhood B-cell acute lymphoblastic leukemia: A single-arm, multicenter, phase II trial. J Clin Oncol. (2023) 41:1670–83. doi: 10.1200/JCO.22.01214
41. Ghorashian S, Lucchini G, Richardson R, Nguyen K, Terris C, Guvenel A, et al. CD19/CD22 targeting with cotransduced CAR T cells to prevent antigen-negative relapse after CAR T-cell therapy for B-cell ALL. Blood. (2024) 143:118–23. doi: 10.1182/blood.2023020621
42. Cui W, Zhang X-Y, Li Z, Dai H-P, Yin J, Cui Q-Y, et al. Long-term follow-up of tandem CD19/CD22 CAR T-Cells in r/r B-ALL patients with high-risk features. Am J Hematol. (2023) 98:E338–40. doi: 10.1002/ajh.27076
43. Pan J, Tang K, Luo Y, Seery S, Tan Y, Deng B, et al. Sequential CD19 and CD22 chimeric antigen receptor T-cell therapy for childhood refractory or relapsed B-cell acute lymphocytic leukaemia: a single-arm, phase 2 study. Lancet Oncol. (2023) 24:1229–41. doi: 10.1016/S1470-2045(23)00436-9
44. Spiegel JY, Patel S, Muffly L, Hossain NM, Oak J, Baird JH, et al. CAR T cells with dual targeting of CD19 and CD22 in adult patients with recurrent or refractory B cell Malignancies: a phase 1 trial. Nat Med. (2021) 27:1419–31. doi: 10.1038/s41591-021-01436-0
45. Schneider D, Xiong Y, Wu D, Hu P, Alabanza L, Steimle B, et al. Trispecific CD19-CD20-CD22-targeting duoCAR-T cells eliminate antigen-heterogeneous B cell tumors in preclinical models. Sci Transl Med. (2021) 13:eabc6401. doi: 10.1126/scitranslmed.abc6401
46. Tosic N, Marjanovic I, Lazic J. Pediatric acute myeloid leukemia: Insight into genetic landscape and novel targeted approaches. Biochem Pharmacol. (2023) 215:115705. doi: 10.1016/j.bcp.2023.115705
47. Cummins K, Gill S. Chimeric antigen receptor T cells in acute myeloid leukemia. Hematol Oncol Clin North Am. (2023) 37:1125–47. doi: 10.1016/j.hoc.2023.06.004
48. Hidalgo L, Somovilla-Crespo B, Garcia-Rodriguez P, Morales-Molina A, Rodriguez-Milla MA, Garcia-Castro J. Switchable CAR T cell strategy against osteosarcoma. Cancer Immunol Immunother. (2023) 72:2623–33. doi: 10.1007/s00262-023-03437-z
49. Abbott RC, Hughes-Parry HE, Jenkins MR. To go or not to go? Biological logic gating engineered T cells. J Immunother Cancer. (2022) 10:e004185. doi: 10.1136/jitc-2021-004185
50. Cho JH, Collins JJ, Wong WW. Universal chimeric antigen receptors for multiplexed and logical control of T cell responses. Cell. (2018) 173:1426–1438.e11. doi: 10.1016/j.cell.2018.03.038
51. Thomas R, Wang W, Su D-M. Contributions of age-related thymic involution to immunosenescence and inflammaging. Immun Ageing. (2020) 17:2. doi: 10.1186/s12979-020-0173-8
52. Das RK, Vernau L, Grupp SA, Barrett DM. Naïve T-cell Deficits at Diagnosis and after Chemotherapy Impair Cell Therapy Potential in Pediatric Cancers. Cancer Discovery. (2019) 9:492–9. doi: 10.1158/2159-8290.CD-18-1314
53. Castella M, Caballero-Baños M, Ortiz-Maldonado V, González-Navarro EA, Suñé G, Antoñana-Vidósola A, et al. Point-of-care CAR T-cell production (ARI-0001) using a closed semi-automatic bioreactor: experience from an academic phase I clinical trial. Front Immunol. (2020) 11:482. doi: 10.3389/fimmu.2020.00482
54. Alsina M, Shah N, Raje NS, Jagannath S, Madduri D, Kaufman JL, et al. Updated results from the phase I CRB-402 study of anti-bcma CAR-T cell therapy bb21217 in patients with relapsed and refractory multiple myeloma: correlation of expansion and duration of response with T cell phenotypes. Blood. (2020) 136:25–6. doi: 10.1182/blood-2020-140410
55. Tang L, Pan S, Wei X, Xu X, Wei Q. Arming CAR-T cells with cytokines and more: Innovations in the fourth-generation CAR-T development. Mol Ther. (2023) 31:3146–62. doi: 10.1016/j.ymthe.2023.09.021
56. Tokarew N, Ogonek J, Endres S, von Bergwelt-Baildon M, Kobold S. Teaching an old dog new tricks: next-generation CAR T cells. Br J Cancer. (2019) 120:26–37. doi: 10.1038/s41416-018-0325-1
57. Harjunpää H, Llort Asens M, Guenther C, Fagerholm SC. Cell adhesion molecules and their roles and regulation in the immune and tumor microenvironment. Front Immunol. (2019) 10:1078. doi: 10.3389/fimmu.2019.01078
58. Larson RC, Kann MC, Bailey SR, Haradhvala NJ, Llopis PM, Bouffard AA, et al. CAR T cell killing requires the IFNγR pathway in solid but not liquid tumors. Nature. (2022) 604:563–70. doi: 10.1038/s41586-022-04585-5
59. Lugade AA, Sorensen EW, Gerber SA, Moran JP, Frelinger JG, Lord EM. Radiation-induced IFN-gamma production within the tumor microenvironment influences antitumor immunity. J Immunol. (2008) 180:3132–9. doi: 10.4049/jimmunol.180.5.3132
60. Espie D, Donnadieu E. New insights into CAR T cell-mediated killing of tumor cells. Front Immunol. (2022) 13:1016208. doi: 10.3389/fimmu.2022.1016208
61. Rodriguez-Garcia A, Palazon A, Noguera-Ortega E, Powell DJ, Guedan S. CAR-T cells hit the tumor microenvironment: strategies to overcome tumor escape. Front Immunol. (2020) 11:1109. doi: 10.3389/fimmu.2020.01109
62. Pan K, Farrukh H, Chittepu VCSR, Xu H, Pan C-X, Zhu Z. CAR race to cancer immunotherapy: from CAR T, CAR NK to CAR macrophage therapy. J Exp Clin Cancer Res. (2022) 41:119. doi: 10.1186/s13046-022-02327-z
63. Fowler D, Nattress C, Navarrete AS, Barisa M, Fisher J. Payload delivery: engineering immune cells to disrupt the tumor microenvironment. Cancers (Basel). (2021) 13:6000. doi: 10.3390/cancers13236000
64. Garaulet G, Pérez-Chacon G, Alarcón H, Alfranca A, Mulero F, Martínez-Torrecuadrada J, et al. Intratumoral expression using a NFkB-based promoter enhances IL12 antitumor efficacy. Cancer Gene Ther. (2019) 26:216–33. doi: 10.1038/s41417-018-0076-4
65. Cirella A, Luri-Rey C, Di Trani CA, Teijeira A, Olivera I, Bolaños E, et al. Novel strategies exploiting interleukin-12 in cancer immunotherapy. Pharmacol Ther. (2022) 239:108189. doi: 10.1016/j.pharmthera.2022.108189
66. Watanabe K, Kuramitsu S, Posey AD, June CH. Expanding the therapeutic window for CAR T cell therapy in solid tumors: the knowns and unknowns of CAR T cell biology. Front Immunol. (2018) 9:2486. doi: 10.3389/fimmu.2018.02486
67. Harris DT, Kranz DM. Adoptive T cell therapies: A comparison of T cell receptors and chimeric antigen receptors. Trends Pharmacol Sci. (2016) 37:220–30. doi: 10.1016/j.tips.2015.11.004
68. Finetti F, Baldari CT. The immunological synapse as a pharmacological target. Pharmacol Res. (2018) 134:118–33. doi: 10.1016/j.phrs.2018.06.009
69. Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med. (1988) 319:1676–80. doi: 10.1056/NEJM198812223192527
70. Stevanović S, Draper LM, Langhan MM, Campbell TE, Kwong ML, Wunderlich JR, et al. Complete regression of metastatic cervical cancer after treatment with human papillomavirus-targeted tumor-infiltrating T cells. J Clin Oncol. (2015) 33:1543–50. doi: 10.1200/JCO.2014.58.9093
71. Johnson LA, June CH. Driving gene-engineered T cell immunotherapy of cancer. Cell Res. (2017) 27:38–58. doi: 10.1038/cr.2016.154
72. Foy SP, Jacoby K, Bota DA, Hunter T, Pan Z, Stawiski E, et al. Non-viral precision T cell receptor replacement for personalized cell therapy. Nature. (2023) 615:687–96. doi: 10.1038/s41586-022-05531-1
73. Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. (2011) 29:917–24. doi: 10.1200/JCO.2010.32.2537
74. Zhang J, Wang L. The emerging world of TCR-T cell trials against cancer: A systematic review. Technol Cancer Res Treat. (2019) 18:1533033819831068. doi: 10.1177/1533033819831068
75. Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother. (2013) 36:133–51. doi: 10.1097/CJI.0b013e3182829903
76. Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. (2006) 314:126–9. doi: 10.1126/science.1129003
77. Wang RF, Rosenberg SA. Human tumor antigens recognized by T lymphocytes: implications for cancer therapy. J Leukoc Biol. (1996) 60:296–309. doi: 10.1002/jlb.60.3.296
78. Harris DT, Hager MV, Smith SN, Cai Q, Stone JD, Kruger P, et al. Comparison of T cell activities mediated by human TCRs and CARs that use the same recognition domains. J Immunol. (2018) 200:1088–100. doi: 10.4049/jimmunol.1700236
79. Esfahani K, Roudaia L, Buhlaiga N, Del Rincon SV, Papneja N, Miller WH. A review of cancer immunotherapy: from the past, to the present, to the future. Curr Oncol. (2020) 27:S87–97. doi: 10.3747/co.27.5223
80. Manfredi F, Cianciotti BC, Potenza A, Tassi E, Noviello M, Biondi A, et al. TCR redirected T cells for cancer treatment: achievements, hurdles, and goals. Front Immunol. (2020) 11:1689. doi: 10.3389/fimmu.2020.01689
81. Nathan P, Hassel JC, Rutkowski P, Baurain J-F, Butler MO, Schlaak M, et al. Overall survival benefit with tebentafusp in metastatic uveal melanoma. N Engl J Med. (2021) 385:1196–206. doi: 10.1056/NEJMoa2103485
82. Chesney J, Lewis KD, Kluger H, Hamid O, Whitman E, Thomas S, et al. Efficacy and safety of lifileucel, a one-time autologous tumor-infiltrating lymphocyte (TIL) cell therapy, in patients with advanced melanoma after progression on immune checkpoint inhibitors and targeted therapies: pooled analysis of consecutive cohorts of the C-144-01 study. J Immunother Cancer. (2022) 10:e005755. doi: 10.1136/jitc-2022-005755
83. Hong DS, Van Tine BA, Biswas S, McAlpine C, Johnson ML, Olszanski AJ, et al. Autologous T cell therapy for MAGE-A4+ solid cancers in HLA-A*02+ patients: a phase 1 trial. Nat Med. (2023) 29:104–14. doi: 10.1038/s41591-022-02128-z
84. Blanco B, Compte M, Lykkemark S, Sanz L, Alvarez-Vallina L. T cell-redirecting strategies to “STAb” Tumors: beyond CARs and bispecific antibodies. Trends Immunol. (2019) 40:243–57. doi: 10.1016/j.it.2019.01.008
85. Blanco B, Ramírez-Fernández Á, Bueno C, Argemí-Muntadas L, Fuentes P, Aguilar-Sopeña Ó, et al. Overcoming CAR-mediated CD19 downmodulation and leukemia relapse with T lymphocytes secreting anti-CD19 T-cell engagers. Cancer Immunol Res. (2022) 10:498–511. doi: 10.1158/2326-6066.CIR-21-0853
86. Jiménez-Reinoso A, Tirado N, Martinez-Moreno A, Díaz VM, García-Peydró M, Hangiu O, et al. Efficient preclinical treatment of cortical T cell acute lymphoblastic leukemia with T lymphocytes secreting anti-CD1a T cell engagers. J Immunother Cancer. (2022) 10:e005333. doi: 10.1136/jitc-2022-005333
87. Diez-Alonso L, Falgas A, Arroyo-Rodenas J, Romencin PA, Martinez A, Gomez-Rosel A, et al. Engineered T cells secreting anti-BCMA T cell engagers control multiple myeloma and promote immune memory in vivo. Sci Transl Med. (2024) 16:eadg7962. doi: 10.1126/scitranslmed.adg7962
88. Mukherjee M, Mace EM, Carisey AF, Ahmed N, Orange JS. Quantitative imaging approaches to study the CAR immunological synapse. Mol Ther. (2017) 25:1757–68. doi: 10.1016/j.ymthe.2017.06.003
89. Huehls AM, Coupet TA, Sentman CL. Bispecific T-cell engagers for cancer immunotherapy. Immunol Cell Biol. (2015) 93:290–6. doi: 10.1038/icb.2014.93
90. Offner S, Hofmeister R, Romaniuk A, Kufer P, Baeuerle PA. Induction of regular cytolytic T cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol Immunol. (2006) 43:763–71. doi: 10.1016/j.molimm.2005.03.007
91. Kouhestani D, Geis M, Alsouri S, Bumm TGP, Einsele H, Sauer M, et al. Variant signaling topology at the cancer cell-T-cell interface induced by a two-component T-cell engager. Cell Mol Immunol. (2021) 18:1568–70. doi: 10.1038/s41423-020-0507-7
92. Choi BD, Yu X, Castano AP, Bouffard AA, Schmidts A, Larson RC, et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat Biotechnol. (2019) 37:1049–58. doi: 10.1038/s41587-019-0192-1
93. Cao G, Zhang G, Liu M, Liu J, Wang Q, Zhu L, et al. GPC3-targeted CAR-T cells secreting B7H3-targeted BiTE exhibit potent cytotoxicity activity against hepatocellular carcinoma cell in the in vitro assay. Biochem Biophys Rep. (2022) 31:101324. doi: 10.1016/j.bbrep.2022.101324
94. Huang S-W, Pan C-M, Lin Y-C, Chen M-C, Chen Y, Jan C-I, et al. BiTE-secreting CAR-γδT as a dual targeting strategy for the treatment of solid tumors. Adv Sci (Weinh). (2023) 10:e2206856. doi: 10.1002/advs.202206856
95. Yin Y, Rodriguez JL, Li N, Thokala R, Nasrallah MP, Hu L, et al. Locally secreted BiTEs complement CAR T cells by enhancing killing of antigen heterogeneous solid tumors. Mol Ther. (2022) 30:2537–53. doi: 10.1016/j.ymthe.2022.05.011
96. O’Leary K. Obesity treatments gather momentum. Nat Med. (2022) 28:2450. doi: 10.1038/s41591-022-02149-8
97. Lee DSW, Rojas OL, Gommerman JL. B cell depletion therapies in autoimmune disease: advances and mechanistic insights. Nat Rev Drug Discovery. (2021) 20:179–99. doi: 10.1038/s41573-020-00092-2
98. Ellebrecht CT, Bhoj VG, Nace A, Choi EJ, Mao X, Cho MJ, et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science. (2016) 353:179–84. doi: 10.1126/science.aaf6756
99. Lee J, Lundgren DK, Mao X, Manfredo-Vieira S, Nunez-Cruz S, Williams EF, et al. Antigen-specific B cell depletion for precision therapy of mucosal pemphigus vulgaris. J Clin Invest. (2020) 130:6317–24. doi: 10.1172/JCI138416
100. Janssen NAF, Brüggemann RJM, Reijers MH, Henriet SSV, Ten Oever J, de Mast Q, et al. A multidisciplinary approach to fungal infections: one-year experiences of a center of expertise in mycology. J Fungi (Basel). (2020) 6:274. doi: 10.3390/jof6040274
101. Al-Akioui Sanz K, Echecopar Parente C, Ferreras C, Menéndez Ribes M, Navarro A, Mestre C, et al. Familial CD45RA- T cells to treat severe refractory infections in immunocompromised patients. Front Med (Lausanne). (2023) 10:1083215. doi: 10.3389/fmed.2023.1083215
102. Pérez-Martínez A, Mora-Rillo M, Ferreras C, Guerra-García P, Pascual-Miguel B, Mestre-Durán C, et al. Phase I dose-escalation single center clinical trial to evaluate the safety of infusion of memory T cells as adoptive therapy in COVID-19 (RELEASE). EClinicalMedicine. (2021) 39:101086. doi: 10.1016/j.eclinm.2021.101086
Keywords: CAR-T cells, NK cells, TILs, TCR therapy, immunological synapse, cancer
Citation: Martín-Antonio B, Blanco B, González-Murillo Á, Hidalgo L, Minguillón J, Pérez-Chacón G and Next Generation CART MAD Consortium (2024) Newer generations of multi-target CAR and STAb-T immunotherapeutics: NEXT CART Consortium as a cooperative effort to overcome current limitations. Front. Immunol. 15:1386856. doi: 10.3389/fimmu.2024.1386856
Received: 16 February 2024; Accepted: 23 April 2024;
Published: 08 May 2024.
Edited by:
Francisco Martin, University of Granada, SpainReviewed by:
David John Coe, University of Bristol, United KingdomZhitao Ying, Peking University, China
Copyright © 2024 Martín-Antonio, Blanco, González-Murillo, Hidalgo, Minguillón, Pérez-Chacón and Next Generation CART MAD Consortium. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Beatriz Martín-Antonio, YmVhdHJpei5hbnRvbmlvQHF1aXJvbnNhbHVkLmVz; Belén Blanco, YmJsYW5jby5pbWFzMTJAaDEyby5lcw==; África González-Murillo, YWZyaWNhLmdvbnphbGV6QHNhbHVkLm1hZHJpZC5vcmc=; Laura Hidalgo, bGhpZGFsZ29AaXNjaWlpLmVz; Jordi Minguillón, am1pbmd1aWxsb25AZXh0LmNuaW8uZXM=; Gema Pérez-Chacón, Z3BjaGFjb25AaWliLnVhbS5lcw==