 Qianxin Zhang
Qianxin Zhang Lijuan Shen1†
Lijuan Shen1†- 1Department of Cardiology, The People’s Hospital of Yuhuan, Taizhou, Zhejiang, China
- 2Department of Cardiology, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, Zhejiang, China
- 3The Key Laboratory of Cardiovascular Disease of Wenzhou, Department of Cardiology, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, Zhejiang, China
Sterile inflammation, characterized by a persistent chronic inflammatory state, significantly contributes to the progression of various diseases such as autoimmune, metabolic, neurodegenerative, and cardiovascular disorders. Recent evidence has increasingly highlighted the intricate connection between inflammatory responses and cardiovascular diseases, underscoring the pivotal role of the Stimulator of Interferon Genes (STING). STING is crucial for the secretion of type I interferon (IFN) and proinflammatory cytokines in response to cytosolic nucleic acids, playing a vital role in the innate immune system. Specifically, research has underscored the STING pathway involvement in unregulated inflammations, where its aberrant activation leads to a surge in inflammatory events, enhanced IFN I responses, and cell death. The primary pathway triggering STING activation is the cyclic GMP-AMP synthase (cGAS) pathway. This review delves into recent findings on STING and the cGAS-STING pathways, focusing on their regulatory mechanisms and impact on cardiovascular diseases. It also discusses the latest advancements in identifying antagonists targeting cGAS and STING, and concludes by assessing the potential of cGAS or STING inhibitors as treatments for cardiovascular diseases.
Introduction
Cardiovascular diseases (CVDs) represent a wide array of complex conditions, including coronary artery diseases, thoracic aortic aneurysms, heart failure (HF), cardiomyopathies, and congenital cardiac anomalies (1). Their global prevalence highlights a significant impact on health worldwide, contributing to a substantial number of fatalities (2). In 2015, CVDs affected 11 million people in European Society of Cardiology member countries, with a total prevalence of approximately 83.5 million (3). These conditions lead to over 17 million deaths globally each year, including 4 million in Europe alone (4, 5). The interaction between immune responses and inflammation in CVDs’ pathophysiology has become a focal point of recent research. Despite advances in surgery and pharmacology improving clinical outcomes, the overall prognosis for CVD patients remains concerning (6). Immune responses and inflammation are pivotal in the pathogenesis of CVDs, as demonstrated by both innate and adaptive immune responses playing critical roles in atherosclerosis progression. A landmark clinical trial in 2017 showed that targeting inflammation could significantly reduce cardiovascular events in high-risk patients (7, 8). Under pathological conditions, cytoplasmic DNA accumulates through various mechanisms, including efflux from mitochondrial and nuclear DNA into the cytosol and release from dying cells (9, 10). This accumulation triggers the cGAS pathway upon binding with double-stranded DNA, leading to STING activation in the endoplasmic reticulum (11, 12). Activated STING initiates a cascade of immune responses, including the activation of interferon regulatory factor 3 (IRF3), TANK binding kinase 1 (TBK1), and nuclear factor-κB (NF-κB) (13–15). While this process upregulates interferons and inflammatory factors, excessive inflammation can lead to tissue damage and organ dysfunction, exacerbating various diseases (16–18). Inflammation is a key determinant in the onset, progression, and outcomes of CVDs, particularly in atherosclerosis and heart failure, where it significantly contributes to disease progression and adverse outcomes (5, 19). The cGAS-STING signaling pathway has been identified as critical in CVD pathogenesis with recent evidence suggesting that targeting cGAS-STING-mediated inflammation holds promise as a treatment for CVDs (20–22). This comprehensive review explores the impact of cGAS-STING-mediated sterile inflammation on CVDs, including the pathway’s intricate functions and its role in coordinating the IFN immune response. We also examine the landscape of suppressors targeting both cGAS and STING, highlighting potential therapeutic strategies to modulate this crucial pathway.
Overview of cGAS–STING pathway
DNA, recognized as a crucial damage-associated molecular pattern (DAMP), triggers the innate immune system by engaging corresponding receptors and initiating intracellular pathways (23). The sources of dsDNA include dying cells, DNA viruses, genomic instability, bacteria, damaged mitochondria, retroviruses, and DNA damage (24, 25). Various DNA sensors, such as absent in melanoma 2 (AIM2), Toll-like receptor (TLR) 9, and cyclic GMP-AMP synthase (cGAS), play pivotal roles in mediating DNA-stimulated innate immune responses (26–28). Among pattern recognition receptors (PRRs), the TLR family, particularly TLR9, is notable (29). Located in the endosomal membrane, TLR9 is highly sensitive to hypomethylated cytidine-phosphate-guanosine regions in DNA from bacteria or viruses, activating the innate immune response (30). Discovered in 2009, AIM2 acts as a cytoplasmic DNA receptor, forming an interaction with dsDNA and associating with the adaptor molecule apoptosis-associated speck-like protein, which activates and recruit caspases (31). This interaction leads to inflammasome/pyroptosome formation, activating both NF-κB and caspase-1 (32). In 2013, Chen et al. identified cGAS, a nucleotides-transferase, that catalyzes the production of the second messenger cGAMP, which binds to STING on the endoplasmic reticulum (ER) membrane, triggering the IFN-I pathway (28). cGAS recognizes both exogenous DNA from pathogens and endogenous DNA from various subcellular locations, such as cytoplasmic chromatin, micronuclei, and mitochondria (33–35). Upon dsDNA recognition, cGAS forms a complex, inducing conformational changes to synthesize cGAMP from ATP and GTP (36, 37). cGAMP binds to STING, causing a conformational change that activates the protein. STING then relocates from the ER to the ER-Golgi intermediate compartment and the Golgi apparatus, activating TBK1. TBK1 phosphorylates itself and STING, activating the IRF3 transcription factor. Dimerized IRF3 translocate into the nucleus, initiating IFN-I synthesis (38–40). Additionally, STING can activate IKK kinase, leading to the phosphorylation of NF-κB transcription factor inhibitors. This cascade ultimately stimulates the production of inflammatory cytokines, including TNF and IL-6 (41) (Figure 1).
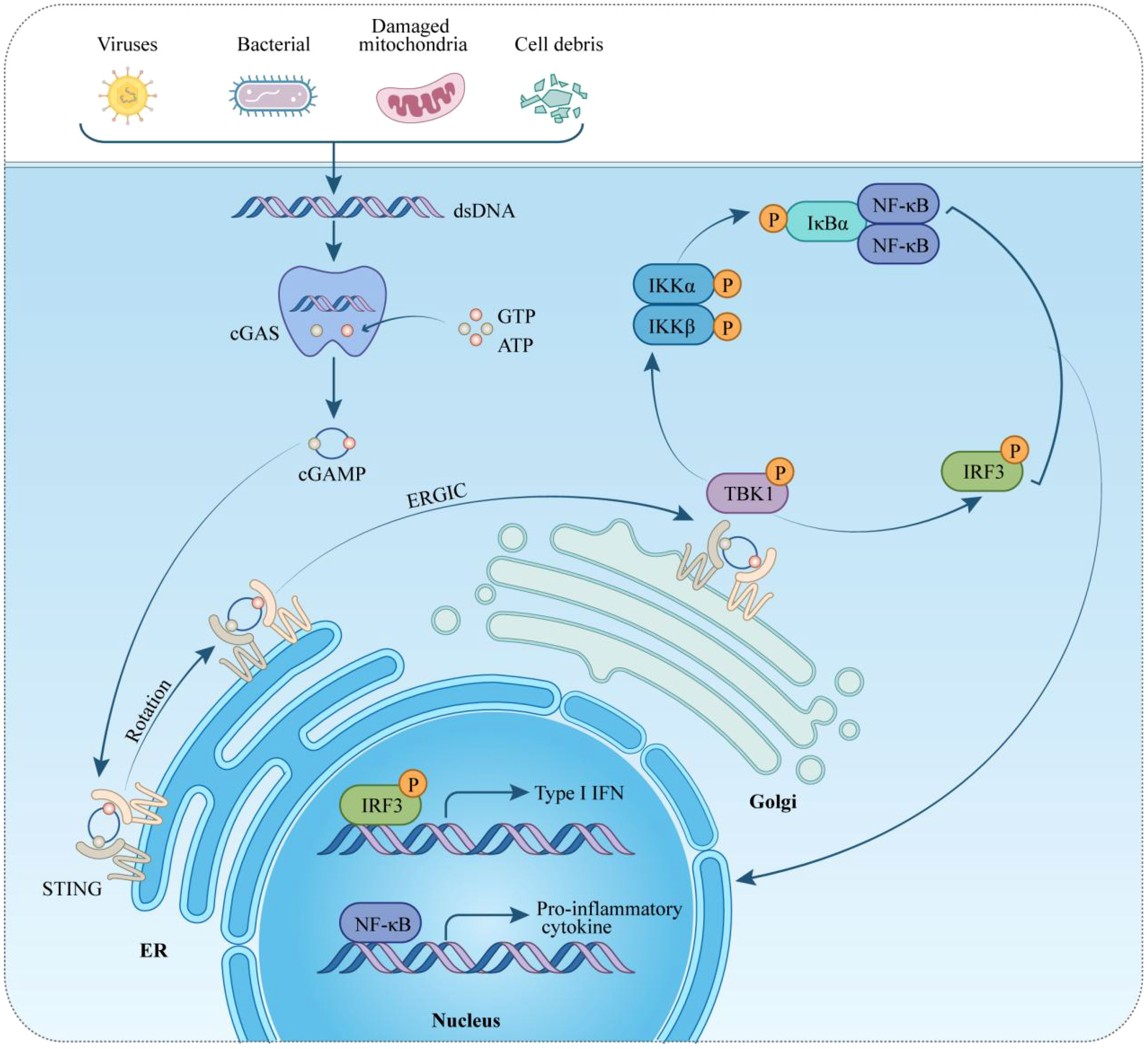
Figure 1 Overview of the cGAS-STING pathway in cardiovascular diseases. Initially, the cGAS-STING signaling pathway serves to protect host cells from pathogens. Further studies have highlighted its significance in the context of cardiovascular diseases, where cGAS detects DNA from both intracellular and extracellular origins. Upon DNA detection, cGAS converts its substrates, GTP and ATP, into cGAMP. STING, situated on the endoplasmic reticulum (ER), detects cGAMP and, when bound to it, becomes activated. This activation leads STING to recruit and phosphorylate TBK1 and IKKs. These kinases then phosphorylate IRF3 and IκBα, respectively. Phosphorylated IRF3 moves to the nucleus, initiating the transcription of type I interferon (IFN-I) genes. Concurrently, phosphorylated IκBα facilitates the recruitment of NF-κB to the nucleus, triggering the transcription of genes encoding proinflammatory cytokines. STING, stimulator of interferon genes; ER, endoplasmic reticulum; ERGIC, ER-Golgi intermediate compartment; TBK1, TANK-binding kinase 1; IRF-3, interferon regulatory factor-3; IκBα: inhibitors of transcription factor NF-κB; type-I IFN, type-I interferon; IL-6, interleukin 6; TNF-α, tumor necrosis factor-α; GTP, guanosine triphosphate; ATP, adenosine triphosphate.
The function of cGAS-STING pathway on cardiovascular diseases
Inflammation plays a pivotal role in the progression of cardiovascular diseases. Recent advancements in understanding the STING-mediated inflammatory response have made significant contributions to cardiovascular disease research. This focus has sharpened on the accumulation of cytoplasmic DNA and its role in activating the cGAS-STING-mediated inflammatory response within pathological cardiovascular environments. This section offers a comprehensive review of the pathological processes in cardiovascular diseases, highlighting the regulatory role of inflammation driven by the cGAS-STING pathway (Table 1).
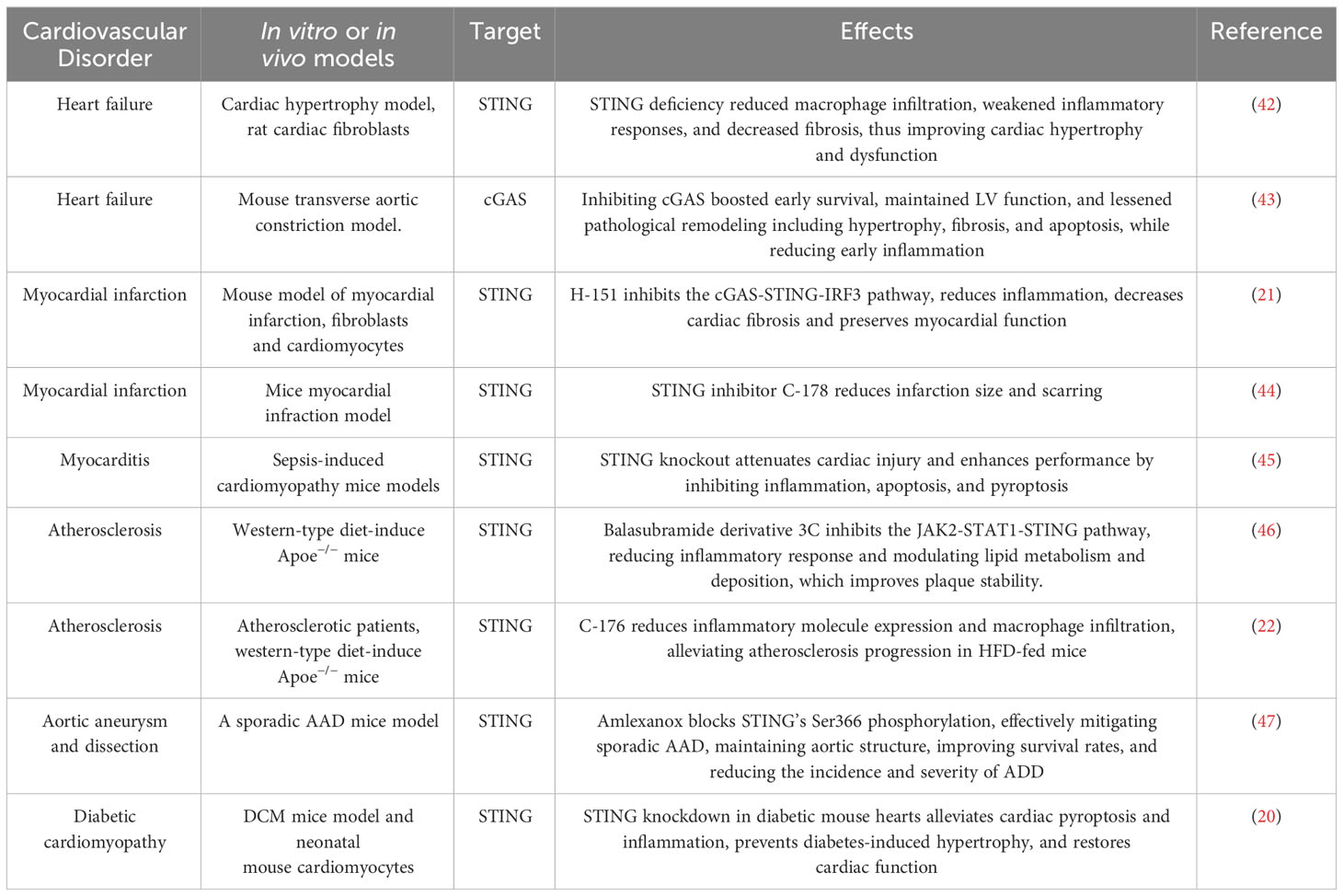
Table 1 The connection between cardiovascular diseases and the cGAS-STING pathway.
Heart failure
Heart failure (HF) manifests as a syndrome characterized by symptoms such as dyspnea, edema, and lower limb weakness, often accompanied by higher pulmonary edema, peripheral edema, and jugular vein strain (48). It results from impaired heart structure or function, significantly contributing to rising global mortality rates (49). There is mounting evidence linking inflammatory pathways’ activations to the progression of left ventricular remodeling and dysfunction in HF patients, suggesting a potential chronic inflammatory aspect to HF. However, the specific function of inflammation in HF remains incompletely understood. In a study investigating non-ischemic pressure-overload induced HF (transverse aortic constriction, TAC), characterized by cardiac dysfunction, hypertrophy, and fibrosis, elevated IFNα, IFNβ, and STING expressions were observed (42). Remarkably, STING knockout (STING-KO) mice exhibited a return to baseline levels of these indicators, indicating a direct association (42). Similarly, angiotensin II-treated neonatal rat cardiomyocytes displayed heightened STING, IFN-α/-β expressions. Inhibiting STING via siRNA in these cells resulted in a significant reduction in TNFα, IFNα/β, and IL-6/-1β levels. Elevated IFNα/β and STING expressions were also noted in dilative and hypertrophic cardiomyopathies specimens from humans (42). Complementary findings emerged from another study reporting substantial increases in IFN, STING, cGAS, as well as IFN-induced chemokines ISG15, IFIT3, CXCL10 expressions, three days post TAC (43). cGAS silence using adeno-associated virus 9 (AAV9) led to a notable drop in left ventricular remodeling and fibrosis, underscoring the potential therapeutic relevance of the cGAS-STING pathway in HF (43). While scientific investigations have initiated exploration into the relationship between HF and the cGAS-STING signaling pathway, further research is imperative to gain a comprehensive understanding of its precise role in disease progression.
Myocardial infarction
Myocardial infarction (MI), a severe manifestation of coronary heart disease, significantly impacts patient survival, with the potential for ventricular remodeling, progressive deterioration in cardiac function, and eventual development of chronic heart failure (HF), resulting in a 10-year survival rate typically below 50% (50). Following cardiac cell injury and death during MI, the release of damage-associated molecular patterns (DAMPs) initiates the innate immune response. In a mouse model of MI, activation of interferon regulatory factor 3 (IRF3), a molecule downstream of STING, was found to amplify the release of interferon type I (IFN-I) and inflammation significantly. Disrupting IRF3 signaling markedly reduced the production of inflammatory markers (CXCL10 and IFNB1) and cell infiltration, improving survival and cardiac function post-MI (51). This suggests that inhibiting IRF3 signaling could provide cardiac protection after MI. Rech et al. found that pharmacologic inhibition of STING reduces infarction size and scarring, restoring ventricular systolic function to near-normal levels three weeks post-reperfusion in MI mice, with a noted decrease in myocardial hypertrophy (44). They assessed the inflammatory response by examining expressions of pro-inflammatory genes, uncovering significant differences in IFI44 and CXCL10 expressions between the inhibition and control groups. Hu et al. further confirmed the protective effect of silencing STING through the development of H-151, a selective STING pathway suppressor, both in vivo and in vitro. STING suppression mitigated the impact of MI by reducing cardiomyocyte apoptosis and fibrosis in fibroblasts induced by the STING pathway in dsDNA-triggered infiltrated macrophages (21). Additionally, inhibiting the macrophage-derived IFN-I signaling pathway, using antibodies against IFN, STING, or a cGAS inhibitor, led to a notable reduction in infarct size in mice subjected to myocardial ischemia-reperfusion (52). These findings collectively suggest that DAMPs released during MI prompt a STING-mediated inflammatory response, contributing to the pathological process of MI.
Myocarditis
Myocarditis can arise from a variety of sources, including fungi, parasites, viruses, bacteria, autoimmune reactions, or as side effects of drugs (53). Viral infections play a crucial role in the initiation and progression of myocarditis, leading to inflammation that can cause cardiac cells to undergo necrosis or apoptosis, which are key aspects of the disease’s pathophysiological process (54, 55). Recent research has shown that the cGAS-STING pathway has a dual role in viral myocarditis. Following a viral infection, the presence of viral DNA within the cytoplasm can lead to two contrasting outcomes: on one hand, it activates the cGAS-STING pathway, promoting the production of IFN-I, which helps the body combat viruses; on the other hand, this same pathway can exacerbate myocardial damage by inducing inflammation (56, 57). In addition to its role in viral myocarditis, the cGAS-STING pathway is also significant in non-infectious myocarditis and microbial infections. An experimental study using a lipopolysaccharide-induced myocardial infarction mouse model with sepsis observed a notable increase in STING and p-IRF3 expression. Remarkably, STING knockout (STING-KO) effectively reduced inflammation, apoptosis, and cardiomyocyte scarring induced by lipopolysaccharide, while simultaneously improving cardiac function and extending mice survival (45). Myocarditis following Trypanosoma cruzi infection, which activates the poly-ADP ribose polymerase 1-cGAS-NF-κB pathway, facilitating the conversion of inflammatory macrophages, has also been observed. In addition, blocking cGAS activation has proven effective in mitigating this inflammatory response (58).
Atherosclerosis
Atherosclerosis (AS) is recognized as a chronic inflammatory condition and a primary cause of clinical cardiovascular events. The presence of DNA damage and extracellular vesicles containing DNA at vascular sites contributes to vascular disorders, especially atherosclerosis, by promoting the progression from calcification to plaque formation, and subsequently thrombosis (59). DNA damage is a notable feature in various vascular diseases, including genetic vascular degenerative syndromes (59). While the presence of microbial DNA in human atherosclerotic plaques has been noted, the exact pathophysiological connection to vascular disease progression is yet to be fully understood (60, 61). However, a correlation between the amount of bacterial DNA at plaque sites and the recruitment and accumulation of monocytes suggests a critical role for DNA-modulated monocyte recruitment in both vascular and systemic inflammation (62). Oxidized mitochondrial DNA from macrophages within atherosclerotic plaques is known to induce STING-dependent inflammation, exacerbating atherosclerosis (22). Pham PT et al. reported an accumulation of cytoplasmic DNA and increased cGAMP levels due to DNA damage in the atherosclerotic plaques of mice on a high-fat diet (HFD) (22). Further research indicated that activation of the cGAS-STING pathway in macrophages leads to persistent vascular inflammation and the induction of multiple inflammatory factors (22). Mechanistically, this process may be initiated by the release of mitochondrial DNA induced by the transactive response DNA-binding protein-43 kDa (TDP43) (63). Importantly, either genetic deletion of STING in macrophages or pharmacological blockade of STING has shown to reduce the expression of inflammatory molecules and macrophage infiltration, thereby alleviating the progression of atherosclerosis in mice on an HFD (22, 46).
Aortic aneurysms and dissections
Aortic aneurysm and dissection (AAD) are closely linked cardiovascular conditions that significantly increase morbidity and mortality risks (64). Currently, effective clinically proven medications to halt the progression of aortic degeneration are absent. AAD may manifest sporadically or in conjunction with genetic disorders. Sporadic AAD is characterized by a progressive loss of smooth muscle cells (SMCs), alongside fragmentation and depletion of the extracellular matrix (ECM), culminating in aneurysm development, dissection, and possible rupture (65). Luo et al. found that inducing sporadic AAD in Sting-deficient mice led to reduced aortic enlargement, decreased elastic fiber fragmentation, and a lower incidence of ADD compared to the wild-type group (47). This finding implies that the absence of STING contributes to the preservation of aortic structure and the smooth muscle layer, underscoring STING’s critical role in the progression of sporadic AAD and aortic degeneration. Further analysis through single-cell transcriptomics of the aorta, GO analyses, and in vitro experiments highlighted STING’s involvement in several biological processes, such as response to reactive oxygen species (ROS), DNA damage response, inflammatory response, and cell death in aortic smooth muscle (47). ROS-mediated DNA damage initiates the release of DNA fragments, activating the cGAS-STING-TBK1-IRF3 pathway, which in turn promotes the death of aortic SMCs. It was also discovered that tissue monocytes primarily detect and engulf dsDNA from damaged aortic SMCs, activating the STING-IRF3 pathway and increasing the expression of matrix metallopeptidase 9 (MMP-9), an enzyme that plays a role in ECM degradation. Importantly, the use of a pharmacological TBK1 suppressor to block phosphorylation at STING’s Ser366 was effective in mitigating sporadic AAD. This approach helped maintain the aortic structure, improved survival rates, and reduced the incidence and severity of ADD (47).
Diabetic cardiomyopathy
Diabetes, a chronic condition, can lead to heart failure due to prolonged cardiac pressure overload (66). Recent research has revealed STING’s role in diabetes-related processes, including cholesterol metabolism, liver inflammation, and islet cell damage (67, 68). Inflammation is a key factor in the development of diabetic cardiomyopathy. Yan et al. demonstrated that activation of the cGAS-STING pathway leads to NLRP3 inflammasome-induced pyroptosis, exacerbating diabetic cardiomyopathy (20). In diabetic mice, hyperlipidemia induces the release of DNA from myocardial cells, which activates the cGAS-STING pathway, resulting in pyroptosis and inflammation. This contributes to myocardial hypertrophy and remodeling (20). However, studies have shown that blocking the Sting gene with AAV9 or pharmacologically suppressing STING can significantly reduce diabetic cardiomyopathy and myocardial inflammation (20, 69). These findings highlight the critical role of cytosolic mtDNA-induced cGAS-STING activation in the pathogenesis of obesity-related diabetic cardiomyopathy (DCM) and provide preclinical evidence supporting the targeting of this pathway as a potential therapeutic strategy for DCM.
Cardiac aging
Inflammation is a fundamental hallmark of the cardiac aging process. Chronic low-grade inflammation, mediated through specific pathways, is a well-recognized precursor to the development of age-related CVDs (70). Central to this process is mitochondrial dysfunction, which plays a key role in the progression of senescence in CVD (71). One critical outcome of mitochondrial dysfunction is the release of mitochondrial DNA (mtDNA) into the cytoplasm (72). This event can activate various inflammatory pathways, including the cGAS pathway. cGAS, stimulated by the presence of cytosolic DNA, serves as a potent activator of the STING pathway. Upon activation, STING induces the transcription factor interferon-regulatory factor 3 (IRF3) to enter the nucleus, promoting the secretion of interferons and further enhancing the inflammatory response (73, 74). Recent research by Wang et al. reported that nanoplastics cause cardiomyocytes senescence by triggering a series of senescence-related marker molecules (75). Further experiments showed that nano-scaled microplastics may lead to ROS production by inducing calcium overload. The excessive accumulation of ROS leads to the release of mtDNA from mitochondria into the cytoplasm, which in turn activates the cGAS-STING signaling pathway, thus leading to cardiomyocyte senescence (75). In light of these findings, targeting the STING pathway may represent a novel therapeutic strategy in managing age-associated cardiovascular conditions by moderating the senescent inflammatory processes that significantly contribute to disease progression.
Discovery of cGAS-STING-targeting drugs
As previously emphasized, the cGAS-STING signaling pathway plays a pivotal role in the progression of various sterile cardiovascular diseases (CVDs). This pathway has emerged as a critical therapeutic target not only for the induction of cytokines, including IFN-I, but also in addressing broader health challenges. Several small-molecule agonists targeting this pathway have been developed and are actively being tested in clinical trials (NCT03172936, NCT04144140) (76). These efforts highlight the potential of modulating this pathway. Currently, treatments such as the JAK inhibitor baricitinib and steroids are used clinically to manage patients with STING activation (77, 78). However, it is necessary to develop novel drug interventions that more directly target cGAS and STING. This section will explore the use of cGAS and STING suppressors specifically in the context of cardiovascular diseases, analyzing their potential therapeutic benefits and implications. By targeting these molecular mechanisms, novel treatment strategies aim to offer more precise control over the inflammatory processes that contribute to the progression of CVDs, providing a promising avenue for therapeutic intervention (Figure 2) (Table 2).
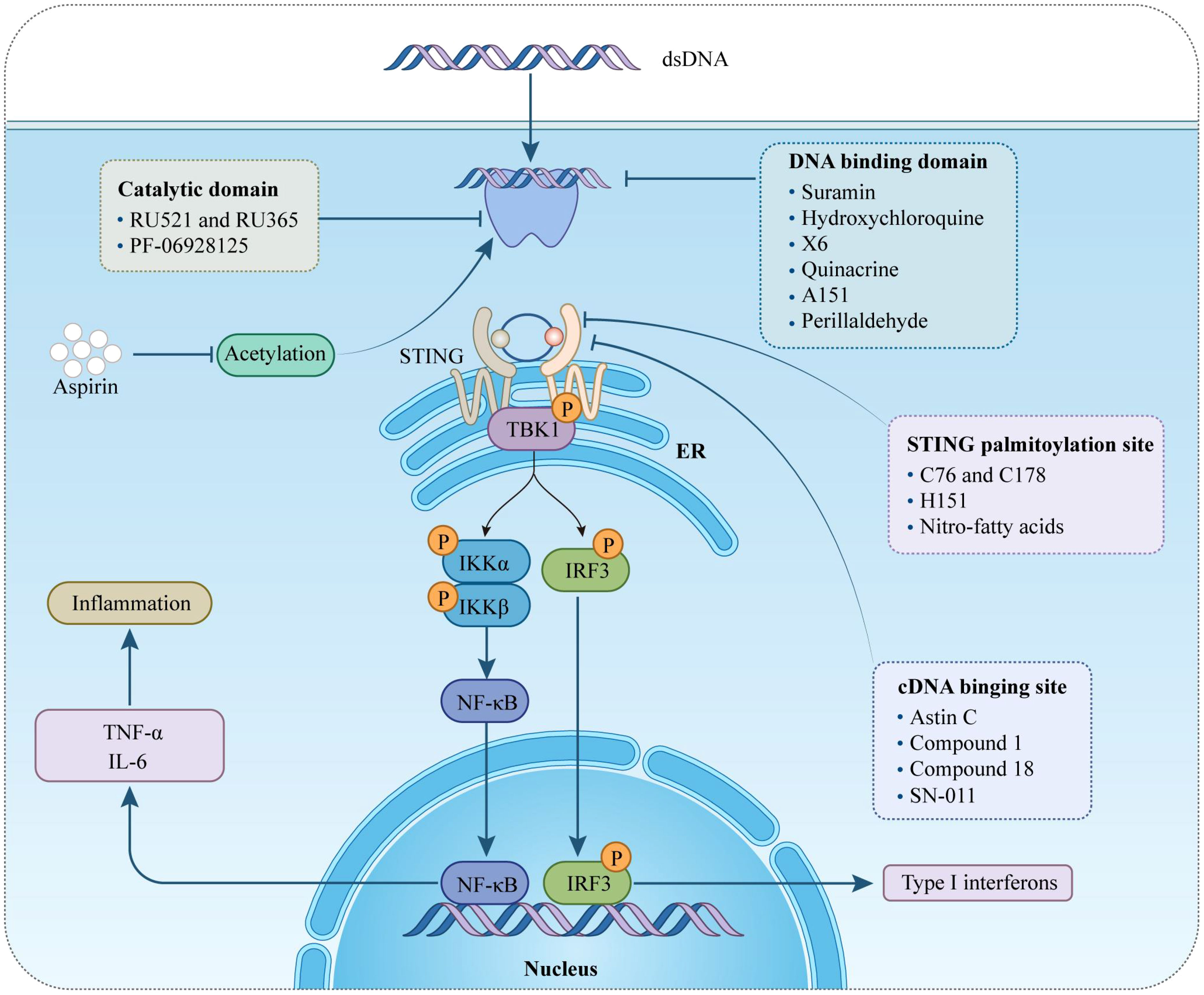
Figure 2 Therapeutical potential of cGAS-STING pathway targeting against inflammation in cardiovascular diseases. This figure provides an overview of different pharmacological agents to modulate the cGAS-STING signaling pathway by intervening at various targets. STING, stimulator of interferon genes; ER, endoplasmic reticulum; TBK1, TANK-binding kinase 1; IRF-3, interferon regulatory factor-3; IκBα, inhibitors of transcription factor NF-κB; type-I IFN, type-I interferon; IL-6, interleukin 6; TNF-α, tumor necrosis factor-α.
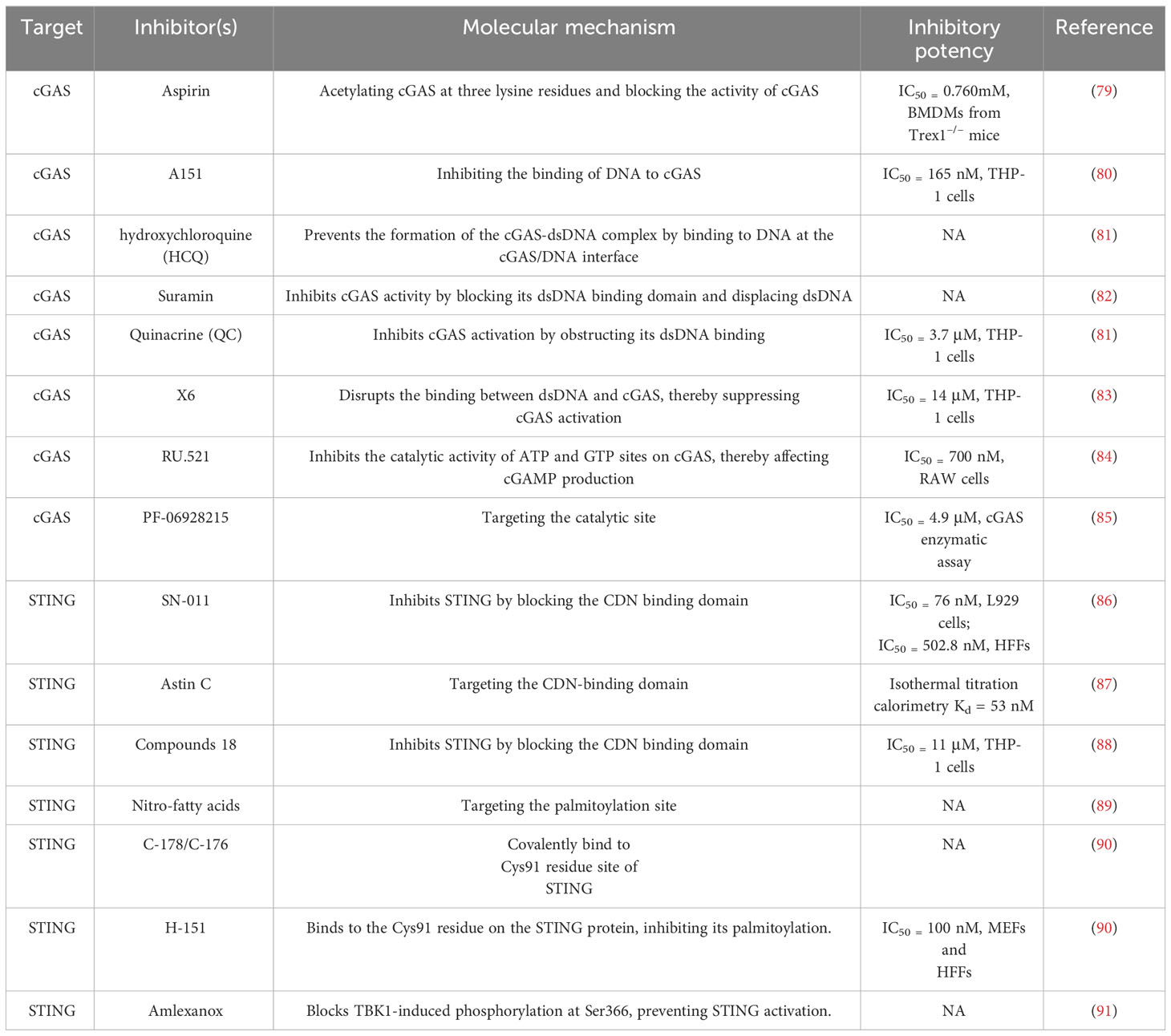
Table 2 Overview of cGAS-STING signaling pathway inhibitors.
cGAS inhibitors
cGAS acts as both a molecular alarm and an enzyme, catalyzing the production of cGAMP for STING signaling. The development of small molecule cGAS suppressors is based on three distinct mechanisms: The first is that mediating cGAS post-translational modification (92); The second is blocking the binding of DNA to cGAS (46); The third is targeting the catalytic pocket of cGAS (59). In the realm of cGAS inhibitors, the first mechanism focuses on the post-translational modifications of cGAS, with Aspirin being an example that targets cGAS acetylation at Lys 384/394/414 to effectively suppress autoimmunity caused by TREX1 genetic deletion (79). The second mechanism is aimed at interrupting the interaction between cGAS and dsDNA. Compounds such as A151, hydroxychloroquine (HCQ), Suramin, quinacrine (QC), and X6 prevent the formation of the cGAS-dsDNA complex by binding to the DNA minor groove at the cGAS/DNA interface (79–81, 83). Suramin, a nucleic acid mimic, competes with dsDNA for the cGAS binding site, reducing the migration and proliferation of vascular smooth muscle cells (VSMCs) and decreasing neointima hyperplasia (82, 93, 94). The third mechanism involves inhibitors that compete with ATP or GTP, the natural substrates of cGAS, or with cGAMP, the product. This category includes non-substrate-competitive suppressors like RU.521/365, which target essential residues in the catalytic site to reduce binding to ATP/GTP and, consequently, downregulate IFN expression. RU.521 specifically binds to the cGAS catalytic sites, inhibiting cGAMP synthesis and subsequent STING activation, and has been shown to protect against septic cardiomyopathy induced by LPS (84, 95). Meanwhile, substrate-competitive inhibitors, such as PF-06928215, directly inhibit the catalytic activity of cGAS and have been proven to alleviate palmitic acid-induced contractile dysfunction in cardiomyocytes (85, 96). Overall, cGAS inhibitors demonstrate protective effects in cardiovascular diseases.
Inhibitors of STING
STING is a critical signaling molecule in the cGAS–STING DNA-sensing pathway, with its activation essential for various intracellular signaling cascades. Development of STING inhibitors has focused on the palmitoylation site and ligand-binding pocket, utilizing computer-aided design (97, 98). This led to the identification of candidate molecules through high-throughput screening, with their effectiveness confirmed in mouse or human models. Suppressor designs aim to block STING activation by its endogenous ligand, cGAMP, with notable examples including SN-011, the natural cyclic peptide Astin C, and tetrahydroisoquinoline compounds (1 and 18) (86–88). Compound 18, targeting the CDN-binding domain (CBD) of STING, binds to the STING homodimer in an inactive ‘open’ conformation, as shown by crystallographic studies. Despite modest antagonistic effects in vitro, it offers favorable oral exposure (88). Astin C, from Aster tataricus, disrupts IRF3 recruitment to the STING signalosome while maintaining the STING-TBK1 interaction (87). Gong et al. highlighted Astin C’s potential to alleviate palmitic acid-induced cardiomyocyte contractile dysfunction by inhibiting the cGAS-STING pathway (96). STING palmitoylation at Cys88/91, crucial for signal transduction, is facilitated by palmitoyltransferases within the Golgi apparatus (15). Nitro-fatty acids accumulate during viral infections and inhibit STING activation through nitroalkylation at its palmitoylation site, showing protective effects against various cardiovascular diseases (89, 99–101). CXA-10, a well-tolerated nitro-fatty acid, is under phase II clinical trials for pulmonary hypertension (NCT04125745/NCT04053543/NCT03449524) (18). Nitrofuran derivatives like C-178, H-151, and C-176, identified via cell-based chemical screening, specifically target Cys91 on STING, blocking activation-induced palmitoylation (90). These compounds have shown efficacy in cardiovascular disease models, displaying anti-inflammatory effects. C-176 and H-151 protect against cardiomyopathy, including myocardial infarction, ischemia-reperfusion injury, and diabetic cardiomyopathy (21, 44, 69). C-176 also suppressed atherosclerosis and chronic kidney disease (CKD) development in HFD-induced Apoe−/− mice (22, 102). Notably, C-176/178 target mouse STING, whereas H-151 is effective against both mouse and human STING. H-151 disrupts STING activation’s necessary palmitoylation and prevents the assembly of the STING polymer complex, reducing infarct dilation and scar formation, thus restoring left ventricular systolic function and reducing myocardial hypertrophy in a myocardial infarction mouse model (44, 90). Additionally, targeting posttranslational modifications of STING and its associated kinases presents viable methods for inhibiting STING activity. For example, the FDA-approved drug amlexanox, known for its anti-inflammatory effects, is utilized in clinical settings to treat asthma and recurrent aphthous ulcers (103). Amlexanox effectively prevents complete activation of STING by blocking TBK1-induced phosphorylation at Ser366, due to its strong affinity and specificity for TBK1 (91). In summary, STING inhibitors have shown protective effects against cardiovascular diseases, presenting a novel strategy for treatment.
Conclusion and perspective
In the realm of cardiovascular diseases, the persistence of an inflammatory response significantly impacts initiation, development, and outcomes, often leading to adverse clinical consequences (19, 104). The cGAS-STING pathway, recognized for its role in mediating sterile inflammation, has increasingly become a focus of interest over the years (17). Pathological scenarios such as mitochondrial damage or cell death in immune cells, vascular endothelial cells, vascular smooth muscle cells, or cardiomyocytes can cause mitochondrial or nuclear DNA to be released into the cytoplasm (18). This cytoplasmic DNA then activates the cGAS-STING pathway, upregulating inflammatory factors, chemokines, and interferons, thereby exacerbating the inflammatory response (105). This review highlights the critical association between cardiac dysfunction and the cGAS-STING pathway, which is central to cell death and influences various physiological functions. The pathway is instrumental in the onset and progression of diverse cardiovascular disorders, such as MI, HF, and myocarditis, by mediating inflammation, regulating immunity, promoting autophagy, and contributing to aging. Animal studies have consistently shown that inhibiting this pathway can improve cardiac function in these diseases, also leading to increased survival rates. Given these promising findings, the cGAS-STING pathway is considered a potential therapeutic target for various cardiovascular disorders. Nonetheless, due to the complex and varied nature of cardiovascular diseases, further exploration and a deeper understanding of the cGAS-STING pathway’s role in different cardiovascular conditions are essential.
Author contributions
QZ: Conceptualization, Data curation, Resources, Writing – original draft. LS: Investigation, Methodology, Validation, Writing – review & editing. HR: Supervision, Validation, Visualization, Writing – review & editing. ZH: Conceptualization, Funding acquisition, Resources, Validation, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the Wenzhou Science and Technology Bureau, China (grant no. Y20210136), Traditional Chinese Medicine Administration of Zhejiang Province (grant no 2022ZA096).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Saeed S, Khan SU, Khan WU, Abdel-Maksoud MA, Mubarak AS, Aufy M, et al. Genome editing technology: A new frontier for the treatment and prevention of cardiovascular diseases. Curr Probl Cardiol. (2023) 48:101692. doi: 10.1016/j.cpcardiol.2023.101692
2. Abbas A, Raza A, Ullah M, Hendi AA, Akbar F, Khan SU, et al. A comprehensive review: epidemiological strategies, catheterization and biomarkers used as a bioweapon in diagnosis and management of cardio vascular diseases. Curr Probl Cardiol. (2023) 48:101661. doi: 10.1016/j.cpcardiol.2023.101661
3. Timmis A, Townsend N, Gale CP, Torbica A, Lettino M, Petersen SE, et al. European society of cardiology: cardiovascular disease statistics 2019. Eur Heart J. (2020) 41:12–85. doi: 10.1093/eurheartj/ehz859
4. Timmis A, Townsend N, Gale C, Grobbee R, Maniadakis N, Flather M, et al. European society of cardiology: cardiovascular disease statistics 2017. Eur Heart J. (2018) 39:508–79. doi: 10.1093/eurheartj/ehx628
5. Golia E, Limongelli G, Natale F, Fimiani F, Maddaloni V, Pariggiano I, et al. Inflammation and cardiovascular disease: from pathogenesis to therapeutic target. Curr Atheroscler Rep. (2014) 16:435. doi: 10.1007/s11883-014-0435-z
6. Ud Din SR, Saeed S, Khan SU, Kiani FA, Alsuhaibani AM, Zhong M. Bioactive compounds (BACs): A novel approach to treat and prevent cardiovascular diseases. Curr Probl Cardiol. (2023) 48:101664. doi: 10.1016/j.cpcardiol.2023.101664
7. Kong P, Cui ZY, Huang XF, Zhang DD, Guo RJ, Han M. Inflammation and atherosclerosis: signaling pathways and therapeutic intervention. Signal Transduct Target Ther. (2022) 7:131. doi: 10.1038/s41392-022-00955-7
8. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. (2017) 377:1119–31. doi: 10.1056/NEJMoa1707914
9. Zierhut C, Funabiki H. Regulation and consequences of cGAS activation by self-DNA. Trends Cell Biol. (2020) 30:594–605. doi: 10.1016/j.tcb.2020.05.006
10. Uggenti C, Lepelley A, Crow YJ. Self-awareness: nucleic acid-driven inflammation and the type I interferonopathies. Annu Rev Immunol. (2019) 37:247–67. doi: 10.1146/annurev-immunol-042718-041257
11. Gentili M, Lahaye X, Nadalin F, Nader GPF, Puig Lombardi E, Herve S, et al. The N-terminal domain of cGAS determines preferential association with centromeric DNA and innate immune activation in the nucleus. Cell Rep. (2019) 26:2377–2393.e13. doi: 10.1016/j.celrep.2019.01.105
12. Li Q, Cao Y, Dang C, Han B, Han R, Ma H, et al. Inhibition of double-strand DNA-sensing cGAS ameliorates brain injury after ischemic stroke. EMBO Mol Med. (2020) 12:e11002. doi: 10.15252/emmm.201911002
13. Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. (2009) 461:788–92. doi: 10.1038/nature08476
14. Fang R, Jiang Q, Guan Y, Gao P, Zhang R, Zhao Z, et al. Golgi apparatus-synthesized sulfated glycosaminoglycans mediate polymerization and activation of the cGAMP sensor STING. Immunity. (2021) 54:962–975.e8. doi: 10.1016/j.immuni.2021.03.011
15. Mukai K, Konno H, Akiba T, Uemura T, Waguri S, Kobayashi T, et al. Activation of STING requires palmitoylation at the Golgi. Nat Commun. (2016) 7:11932. doi: 10.1038/ncomms11932
16. Ablasser A, Chen ZJ. cGAS in action: Expanding roles in immunity and inflammation. Science. (2019) 363(6431). doi: 10.1126/science.aat8657
17. Bai J, Liu F. The cGAS-cGAMP-STING pathway: A molecular link between immunity and metabolism. Diabetes. (2019) 68:1099–108. doi: 10.2337/dbi18-0052
18. Oduro PK, Zheng X, Wei J, Yang Y, Wang Y, Zhang H, et al. The cGAS-STING signaling in cardiovascular and metabolic diseases: Future novel target option for pharmacotherapy. Acta Pharm Sin B. (2022) 12:50–75. doi: 10.1016/j.apsb.2021.05.011
19. Adamo L, Rocha-Resende C, Prabhu SD, Mann DL. Reappraising the role of inflammation in heart failure. Nat Rev Cardiol. (2020) 17:269–85. doi: 10.1038/s41569-019-0315-x
20. Yan M, Li Y, Luo Q, Zeng W, Shao X, Li L, et al. Mitochondrial damage and activation of the cytosolic DNA sensor cGAS-STING pathway lead to cardiac pyroptosis and hypertrophy in diabetic cardiomyopathy mice. Cell Death Discovery. (2022) 8:258. doi: 10.1038/s41420-022-01046-w
21. Hu S, Gao Y, Gao R, Wang Y, Qu Y, Yang J, et al. The selective STING inhibitor H-151 preserves myocardial function and ameliorates cardiac fibrosis in murine myocardial infarction. Int Immunopharmacol. (2022) 107:108658. doi: 10.1016/j.intimp.2022.108658
22. Pham PT, Fukuda D, Nishimoto S, Kim-Kaneyama JR, Lei XF, Takahashi Y, et al. STING, a cytosolic DNA sensor, plays a critical role in atherogenesis: a link between innate immunity and chronic inflammation caused by lifestyle-related diseases. Eur Heart J. (2021) 42:4336–48. doi: 10.1093/eurheartj/ehab249
23. Paludan SR. Activation and regulation of DNA-driven immune responses. Microbiol Mol Biol Rev. (2015) 79:225–41. doi: 10.1128/mmbr.00061-14
24. Riley JS, Tait SW. Mitochondrial DNA in inflammation and immunity. EMBO Rep. (2020) 21:e49799. doi: 10.15252/embr.201949799
25. Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature. (2017) 548:461–5. doi: 10.1038/nature23449
26. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. (2010) 11:373–84. doi: 10.1038/ni.1863
27. Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. (2009) 458:514–8. doi: 10.1038/nature07725
28. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. (2013) 339:786–91. doi: 10.1126/science.1232458
29. Vollmer J. TLR9 in health and disease. Int Rev Immunol. (2006) 25:155–81. doi: 10.1080/08830180600743107
30. Hayashi T, Raz E. TLR9-based immunotherapy for allergic disease. Am J Med. (2006) 119:897.e1–6. doi: 10.1016/j.amjmed.2005.12.028
31. Wang L, Sun L, Byrd KM, Ko CC, Zhao Z, Fang J. AIM2 inflammasome’s first decade of discovery: focus on oral diseases. Front Immunol. (2020) 11:1487. doi: 10.3389/fimmu.2020.01487
32. Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. (2009) 458:509–13. doi: 10.1038/nature07710
33. Heijink AM, Talens F, Jae LT, van Gijn SE, Fehrmann RSN, Brummelkamp TR, et al. BRCA2 deficiency instigates cGAS-mediated inflammatory signaling and confers sensitivity to tumor necrosis factor-alpha-mediated cytotoxicity. Nat Commun. (2019) 10:100. doi: 10.1038/s41467-018-07927-y
34. Glück S, Guey B, Gulen MF, Wolter K, Kang TW, Schmacke NA, et al. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol. (2017) 19:1061–70. doi: 10.1038/ncb3586
35. West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. (2015) 520:553–7. doi: 10.1038/nature14156
36. Li XD, Wu J, Gao D, Wang H, Sun L, Chen ZJ. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science. (2013) 341(6152):1390–4. doi: 10.1126/science.1244040
37. Gao P, Ascano M, Wu Y, Barchet W, Gaffney BL, Zillinger T, et al. Cyclic [G(2’,5’)pA(3’,5’)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell. (2013) 153:1094–107. doi: 10.1016/j.cell.2013.04.046
38. Shang G, Zhang C, Chen ZJ, Bai XC, Zhang X. Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP-AMP. Nature. (2019) 567:389–93. doi: 10.1038/s41586-019-0998-5
39. Zhao B, Du F, Xu P, Shu C, Sankaran B, Bell SL, et al. A conserved PLPLRT/SD motif of STING mediates the recruitment and activation of TBK1. Nature. (2019) 569:718–22. doi: 10.1038/s41586-019-1228-x
40. Zhang C, Shang G, Gui X, Zhang X, Bai XC, Chen ZJ. Structural basis of STING binding with and phosphorylation by TBK1. Nature. (2019) 567:394–8. doi: 10.1038/s41586-019-1000-2
41. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. (2008) 455:674–8. doi: 10.1038/nature07317
42. Zhang Y, Chen W, Wang Y. STING is an essential regulator of heart inflammation and fibrosis in mice with pathological cardiac hypertrophy via endoplasmic reticulum (ER) stress. BioMed Pharmacother. (2020) 125:110022. doi: 10.1016/j.biopha.2020.110022
43. Hu D, Cui YX, Wu MY, Li L, Su LN, Lian Z, et al. Cytosolic DNA sensor cGAS plays an essential pathogenetic role in pressure overload-induced heart failure. Am J Physiol Heart Circ Physiol. (2020) 318:H1525–h1537. doi: 10.1152/ajpheart.00097.2020
44. Rech L, Abdellatif M, Pöttler M, Stangl V, Mabotuwana N, Hardy S, et al. Small molecule STING inhibition improves myocardial infarction remodeling. Life Sci. (2022) 291:120263. doi: 10.1016/j.lfs.2021.120263
45. Li N, Zhou H, Wu H, Wu Q, Duan M, Deng W, et al. STING-IRF3 contributes to lipopolysaccharide-induced cardiac dysfunction, inflammation, apoptosis and pyroptosis by activating NLRP3. Redox Biol. (2019) 24:101215. doi: 10.1016/j.redox.2019.101215
46. Cai D, Liu H, Wang J, Hou Y, Pang T, Lin H, et al. Balasubramide derivative 3C attenuates atherosclerosis in apolipoprotein E-deficient mice: role of AMPK-STAT1-STING signaling pathway. Aging (Albany NY). (2021) 13:12160–78. doi: 10.18632/aging.202929
47. Luo W, Wang Y, Zhang L, Ren P, Zhang C, Li Y, et al. Critical role of cytosolic DNA and its sensing adaptor STING in aortic degeneration, dissection, and rupture. Circulation. (2020) 141:42–66. doi: 10.1161/circulationaha.119.041460
48. Tanai E, Frantz S. Pathophysiology of heart failure. Compr Physiol. (2015) 6:187–214. doi: 10.1002/cphy.c140055
49. Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, et al. Heart disease and stroke statistics-2019 update: A report from the American heart association. Circulation. (2019) 139:e56–e528. doi: 10.1161/cir.0000000000000659
50. Wang Y, Leifheit EC, Krumholz HM. Trends in 10-year outcomes among medicare beneficiaries who survived an acute myocardial infarction. JAMA Cardiol. (2022) 7:613–22. doi: 10.1001/jamacardio.2022.0662
51. King KR, Aguirre AD, Ye YX, Sun Y, Roh JD, Ng RP Jr., et al. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat Med. (2017) 23:1481–7. doi: 10.1038/nm.4428
52. Lai L, Zhang A, Yang B, Charles EJ, Kron IL, Yang Z. Plasmacytoid dendritic cells mediate myocardial ischemia/reperfusion injury by secreting type I interferons. J Am Heart Assoc. (2021) 10:e020754. doi: 10.1161/jaha.121.020754
53. Fung G, Luo H, Qiu Y, Yang D, McManus B. Myocarditis. Circ Res. (2016) 118:496–514. doi: 10.1161/circresaha.115.306573
54. Trachtenberg BH, Hare JM. Inflammatory cardiomyopathic syndromes. Circ Res. (2017) 121:803–18. doi: 10.1161/circresaha.117.310221
55. Tschöpe C, Cooper LT, Torre-Amione G, Van Linthout S. Management of myocarditis-related cardiomyopathy in adults. Circ Res. (2019) 124:1568–83. doi: 10.1161/circresaha.118.313578
56. Song JH, Ahn JH, Kim SR, Cho S, Hong EH, Kwon BE, et al. Manassantin B shows antiviral activity against coxsackievirus B3 infection by activation of the STING/TBK-1/IRF3 signalling pathway. Sci Rep. (2019) 9:9413. doi: 10.1038/s41598-019-45868-8
57. Ma Z, Damania B. The cGAS-STING defense pathway and its counteraction by viruses. Cell Host Microbe. (2016) 19:150–8. doi: 10.1016/j.chom.2016.01.010
58. Choudhuri S, Garg NJ. PARP1-cGAS-NF-κB pathway of proinflammatory macrophage activation by extracellular vesicles released during Trypanosoma cruzi infection and Chagas disease. PloS Pathog. (2020) 16:e1008474. doi: 10.1371/journal.ppat.1008474
59. Uryga A, Gray K, Bennett M. DNA damage and repair in vascular disease. Annu Rev Physiol. (2016) 78:45–66. doi: 10.1146/annurev-physiol-021115-105127
60. Hayashi C, Viereck J, Hua N, Phinikaridou A, Madrigal AG, Gibson FC 3rd, et al. Porphyromonas gingivalis accelerates inflammatory atherosclerosis in the innominate artery of ApoE deficient mice. Atherosclerosis. (2011) 215:52–9. doi: 10.1016/j.atherosclerosis.2010.12.009
61. Calandrini CA, Ribeiro AC, Gonnelli AC, Ota-Tsuzuki C, Rangel LP, Saba-Chujfi E, et al. Microbial composition of atherosclerotic plaques. Oral Dis. (2014) 20:e128–34. doi: 10.1111/odi.12205
62. Koren O, Spor A, Felin J, Fåk F, Stombaugh J, Tremaroli V, et al. Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc Natl Acad Sci U.S.A. (2011) 108 Suppl 1:4592–8. doi: 10.1073/pnas.1011383107
63. Huangfu N, Wang Y, Xu Z, Zheng W, Tao C, Li Z, et al. TDP43 exacerbates atherosclerosis progression by promoting inflammation and lipid uptake of macrophages. Front Cell Dev Biol. (2021) 9:687169. doi: 10.3389/fcell.2021.687169
64. Fradet G, Jamieson WR, Janusz MT, Ling H, Miyagishima RT, Munro AI. Aortic dissection: current expectations and treatment. Experience with 258 patients over 20 years. Can J Surg. (1990) 33:465–9.
65. Sakalihasan N, Limet R, Defawe OD. Abdominal aortic aneurysm. Lancet. (2005) 365:1577–89. doi: 10.1016/s0140-6736(05)66459-8
66. Zlobine I, Gopal K, Ussher JR. Lipotoxicity in obesity and diabetes-related cardiac dysfunction. Biochim Biophys Acta. (2016) 1861:1555–68. doi: 10.1016/j.bbalip.2016.02.011
67. Yu Y, Liu Y, An W, Song J, Zhang Y, Zhao X. STING-mediated inflammation in Kupffer cells contributes to progression of nonalcoholic steatohepatitis. J Clin Invest. (2019) 129:546–55. doi: 10.1172/jci121842
68. Qiao J, Zhang Z, Ji S, Liu T, Zhang X, Huang Y, et al. A distinct role of STING in regulating glucose homeostasis through insulin sensitivity and insulin secretion. Proc Natl Acad Sci USA. (2022) 119(7). doi: 10.1073/pnas.2101848119
69. Ma XM, Geng K, Law BY, Wang P, Pu YL, Chen Q, et al. Lipotoxicity-induced mtDNA release promotes diabetic cardiomyopathy by activating the cGAS-STING pathway in obesity-related diabetes. Cell Biol Toxicol. (2023) 39:277–99. doi: 10.1007/s10565-021-09692-z
70. Guzik TJ, Touyz RM. Oxidative stress, inflammation, and vascular aging in hypertension. Hypertension. (2017) 70:660–7. doi: 10.1161/hypertensionaha.117.07802
71. Picca A, Mankowski RT, Burman JL, Donisi L, Kim JS, Marzetti E, et al. Mitochondrial quality control mechanisms as molecular targets in cardiac ageing. Nat Rev Cardiol. (2018) 15:543–54. doi: 10.1038/s41569-018-0059-z
72. Lewis-McDougall FC, Ruchaya PJ, Domenjo-Vila E, Shin Teoh T, Prata L, Cottle BJ, et al. Aged-senescent cells contribute to impaired heart regeneration. Aging Cell. (2019) 18:e12931. doi: 10.1111/acel.12931
73. Li T, Chen ZJ. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med. (2018) 215:1287–99. doi: 10.1084/jem.20180139
74. Loo TM, Miyata K, Tanaka Y, Takahashi A. Cellular senescence and senescence-associated secretory phenotype via the cGAS-STING signaling pathway in cancer. Cancer Sci. (2020) 111:304–11. doi: 10.1111/cas.14266
75. Wang K, Du Y, Li P, Guan C, Zhou M, Wu L, et al. Nanoplastics causes heart aging/myocardial cell senescence through the Ca(2+)/mtDNA/cGAS-STING signaling cascade. J Nanobiotechnol. (2024) 22:96. doi: 10.1186/s12951-024-02375-x
76. Zhu HF, Li Y. Small-molecule targets in tumor immunotherapy. Nat Prod Bioprospect. (2018) 8:297–301. doi: 10.1007/s13659-018-0177-7
77. Lin B, Berard R, Al Rasheed A, Aladba B, Kranzusch PJ, Henderlight M, et al. A novel STING1 variant causes a recessive form of STING-associated vasculopathy with onset in infancy (SAVI). J Allergy Clin Immunol. (2020) 146:1204–1208.e6. doi: 10.1016/j.jaci.2020.06.032
78. Sanchez GAM, Reinhardt A, Ramsey S, Wittkowski H, Hashkes PJ, Berkun Y, et al. JAK1/2 inhibition with baricitinib in the treatment of autoinflammatory interferonopathies. J Clin Invest. (2018) 128:3041–52. doi: 10.1172/jci98814
79. Dai J, Huang YJ, He X, Zhao M, Wang X, Liu ZS, et al. Acetylation blocks cGAS activity and inhibits self-DNA-induced autoimmunity. Cell. (2019) 176:1447–1460.e14. doi: 10.1016/j.cell.2019.01.016
80. Steinhagen F, Zillinger T, Peukert K, Fox M, Thudium M, Barchet W, et al. Suppressive oligodeoxynucleotides containing TTAGGG motifs inhibit cGAS activation in human monocytes. Eur J Immunol. (2018) 48:605–11. doi: 10.1002/eji.201747338
81. An J, Woodward JJ, Sasaki T, Minie M, Elkon KB. Cutting edge: Antimalarial drugs inhibit IFN-β production through blockade of cyclic GMP-AMP synthase-DNA interaction. J Immunol. (2015) 194:4089–93. doi: 10.4049/jimmunol.1402793
82. Wang M, Sooreshjani MA, Mikek C, Opoku-Temeng C, Sintim HO. Suramin potently inhibits cGAMP synthase, cGAS, in THP1 cells to modulate IFN-β levels. Future Med Chem. (2018) 10:1301–17. doi: 10.4155/fmc-2017-0322
83. An J, Woodward JJ, Lai W, Minie M, Sun X, Tanaka L, et al. Inhibition of cyclic GMP-AMP synthase using a novel antimalarial drug derivative in trex1-deficient mice. Arthritis Rheumatol. (2018) 70:1807–19. doi: 10.1002/art.40559
84. Vincent J, Adura C, Gao P, Luz A, Lama L, Asano Y, et al. Small molecule inhibition of cGAS reduces interferon expression in primary macrophages from autoimmune mice. Nat Commun. (2017) 8:750. doi: 10.1038/s41467-017-00833-9
85. Hall J, Brault A, Vincent F, Weng S, Wang H, Dumlao D, et al. Discovery of PF-06928215 as a high affinity inhibitor of cGAS enabled by a novel fluorescence polarization assay. PloS One. (2017) 12:e0184843. doi: 10.1371/journal.pone.0184843
86. Hong Z, Mei J, Li C, Bai G, Maimaiti M, Hu H, et al. STING inhibitors target the cyclic dinucleotide binding pocket. Proc Natl Acad Sci USA. (2021) 118(24). doi: 10.1073/pnas.2105465118
87. Li S, Hong Z, Wang Z, Li F, Mei J, Huang L, et al. The cyclopeptide astin C specifically inhibits the innate immune CDN sensor STING. Cell Rep. (2018) 25:3405–3421.e7. doi: 10.1016/j.celrep.2018.11.097
88. Siu T, Altman MD, Baltus GA, Childers M, Ellis JM, Gunaydin H, et al. Discovery of a novel cGAMP competitive ligand of the inactive form of STING. ACS Med Chem Lett. (2019) 10:92–7. doi: 10.1021/acsmedchemlett.8b00466
89. Hansen AL, Buchan GJ, Rühl M, Mukai K, Salvatore SR, Ogawa E, et al. Nitro-fatty acids are formed in response to virus infection and are potent inhibitors of STING palmitoylation and signaling. Proc Natl Acad Sci U.S.A. (2018) 115:E7768–e7775. doi: 10.1073/pnas.1806239115
90. Haag SM, Gulen MF, Reymond L, Gibelin A, Abrami L, Decout A, et al. Targeting STING with covalent small-molecule inhibitors. Nature. (2018) 559:269–73. doi: 10.1038/s41586-018-0287-8
91. Reilly SM, Chiang SH, Decker SJ, Chang L, Uhm M, Larsen MJ, et al. An inhibitor of the protein kinases TBK1 and IKK-ε improves obesity-related metabolic dysfunctions in mice. Nat Med. (2013) 19:313–21. doi: 10.1038/nm.3082
92. Song B, Liu D, Greco TM, Cristea IM. Post-translational modification control of viral DNA sensors and innate immune signaling. Adv Virus Res. (2021) 109:163–99. doi: 10.1016/bs.aivir.2021.03.001
93. Hu Y, Zou Y, Dietrich H, Wick G, Xu Q. Inhibition of neointima hyperplasia of mouse vein grafts by locally applied suramin. Circulation. (1999) 100:861–8. doi: 10.1161/01.cir.100.8.861
94. Jia S, Chen Z, Li J, Chi Y, Wang J, Li S, et al. FAM3A promotes vascular smooth muscle cell proliferation and migration and exacerbates neointima formation in rat artery after balloon injury. J Mol Cell Cardiol. (2014) 74:173–82. doi: 10.1016/j.yjmcc.2014.05.011
95. Xu Q, Xiong H, Zhu W, Liu Y, Du Y. Small molecule inhibition of cyclic GMP-AMP synthase ameliorates sepsis-induced cardiac dysfunction in mice. Life Sci. (2020) 260:118315. doi: 10.1016/j.lfs.2020.118315
96. Gong Y, Li G, Tao J, Wu NN, Kandadi MR, Bi Y, et al. Double knockout of Akt2 and AMPK accentuates high fat diet-induced cardiac anomalies through a cGAS-STING-mediated mechanism. Biochim Biophys Acta Mol Basis Dis. (2020) 1866:165855. doi: 10.1016/j.bbadis.2020.165855
97. Hong Z, Mei J, Guo H, Zhu J, Wang C. Intervention of cGAS−STING signaling in sterile inflammatory diseases. J Mol Cell Biol. (2022) 14(2). doi: 10.1093/jmcb/mjac005
98. Chauhan C, Kaundal RK. The role of cGAS-STING signaling in ischemic stroke: From immune response to therapeutic targeting. Drug Discovery Today. (2023) 28:103792. doi: 10.1016/j.drudis.2023.103792
99. Mollenhauer M, Mehrkens D, Klinke A, Lange M, Remane L, Friedrichs K, et al. Nitro-fatty acids suppress ischemic ventricular arrhythmias by preserving calcium homeostasis. Sci Rep. (2020) 10:15319. doi: 10.1038/s41598-020-71870-6
100. Zhao Y, Chang Z, Zhao G, Lu H, Xiong W, Liang W, et al. Suppression of vascular macrophage activation by nitro-oleic acid and its implication for abdominal aortic aneurysm therapy. Cardiovasc Drugs Ther. (2021) 35:939–51. doi: 10.1007/s10557-020-07031-8
101. Klinke A, Möller A, Pekarova M, Ravekes T, Friedrichs K, Berlin M, et al. Protective effects of 10-nitro-oleic acid in a hypoxia-induced murine model of pulmonary hypertension. Am J Respir Cell Mol Biol. (2014) 51:155–62. doi: 10.1165/rcmb.2013-0063OC
102. Bi X, Du C, Wang X, Wang XY, Han W, Wang Y, et al. Mitochondrial damage-induced innate immune activation in vascular smooth muscle cells promotes chronic kidney disease-associated plaque vulnerability. Adv Sci (Weinh). (2021) 8:2002738. doi: 10.1002/advs.202002738
103. Ozasa K, Temizoz B, Kusakabe T, Kobari S, Momota M, Coban C, et al. Cyclic GMP-AMP triggers asthma in an IL-33-dependent manner that is blocked by amlexanox, a TBK1 inhibitor. Front Immunol. (2019) 10:2212. doi: 10.3389/fimmu.2019.02212
104. Silvestre-Roig C, Braster Q, Ortega-Gomez A, Soehnlein O. Neutrophils as regulators of cardiovascular inflammation. Nat Rev Cardiol. (2020) 17:327–40. doi: 10.1038/s41569-019-0326-7
Keywords: CGAS, STING, inflammation, cardiovascular diseases, IFN
Citation: Zhang Q, Shen L, Ruan H and Huang Z (2024) cGAS-STING signaling in cardiovascular diseases. Front. Immunol. 15:1402817. doi: 10.3389/fimmu.2024.1402817
Received: 18 March 2024; Accepted: 29 April 2024;
Published: 13 May 2024.
Edited by:
Bhesh Raj Sharma, St. Jude Children’s Research Hospital, United StatesReviewed by:
Yaqiu Wang, St. Jude Children’s Research Hospital, United StatesMorgan Salmon, University of Michigan, United States
Pinglong Xu, Zhejiang University, China
Copyright © 2024 Zhang, Shen, Ruan and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongbiao Ruan, cmhvbmdiaWFvQDEyNi5jb20=; Zhouqing Huang, c3VzaWVoenFAMTI2LmNvbQ==
†These authors have contributed equally to this work