 Yijin Cai1†
Yijin Cai1† Zhongyu Han1*†
Zhongyu Han1*† Hong Cheng2†
Hong Cheng2† Hongpeng Li1†
Hongpeng Li1† Ke Wang3
Ke Wang3 Jia Chen4
Jia Chen4 Zhi-Xiang Liu4Yulong Xie4Yumeng Lin3Shuwei Zhou5
Zhi-Xiang Liu4Yulong Xie4Yumeng Lin3Shuwei Zhou5 Siyu Wang6Xiao Zhou7Song Jin8*
Siyu Wang6Xiao Zhou7Song Jin8*- 1School of Medical and Life Sciences, Chengdu University of Traditional Chinese Medicine, Chengdu, China
- 2School of Automation Engineering, University of Electronic Science and Technology, Chengdu, China
- 3Eye School of Chengdu University of Traditional Chinese Medicine, Chengdu, China
- 4School of Health Preservation and Rehabilitation, Chengdu University of Traditional Chinese Medicine, Chengdu, China
- 5Jiangsu Key Laboratory of Molecular and Functional Imaging, Department of Radiology, Zhongda Hospital, School of Medicine, Southeast University, Nanjing, China
- 6Department of Gastroenterology, The First Hospital of Hunan University of Chinese Medicine, Changsha, China
- 7Second Clinical Medical College, Heilongjiang University of Chinese Medicine, Heilongjiang, China
- 8Department of Rehabilitation, Hospital of Chengdu University of Traditional Chinese Medicine, Chengdu, China
Ageing is an inevitable process that affects various tissues and organs of the human body, leading to a series of physiological and pathological changes. Mechanisms such as telomere depletion, stem cell depletion, macrophage dysfunction, and cellular senescence gradually manifest in the body, significantly increasing the incidence of diseases in elderly individuals. These mechanisms interact with each other, profoundly impacting the quality of life of older adults. As the ageing population continues to grow, the burden on the public health system is expected to intensify. Globally, the prevalence of musculoskeletal system diseases in elderly individuals is increasing, resulting in reduced limb mobility and prolonged suffering. This review aims to elucidate the mechanisms of ageing and their interplay while exploring their impact on diseases such as osteoarthritis, osteoporosis, and sarcopenia. By delving into the mechanisms of ageing, further research can be conducted to prevent and mitigate its effects, with the ultimate goal of alleviating the suffering of elderly patients in the future.
1 Introduction
Ageing occurs as a result of the interaction of countless factors, and it has extremely complex biological behaviors. Cellular ageing, telomere loss, genomic changes, and dysregulation of mitochondrial function drive the progression of ageing and promote the development of related diseases (1). The organs of the human body are altered in various ways by ageing (Figure 1). With the development of the ageing population, the study of ageing has become a hot topic of clinical research. Investigations into the molecular mechanisms of ageing have also greatly advanced.
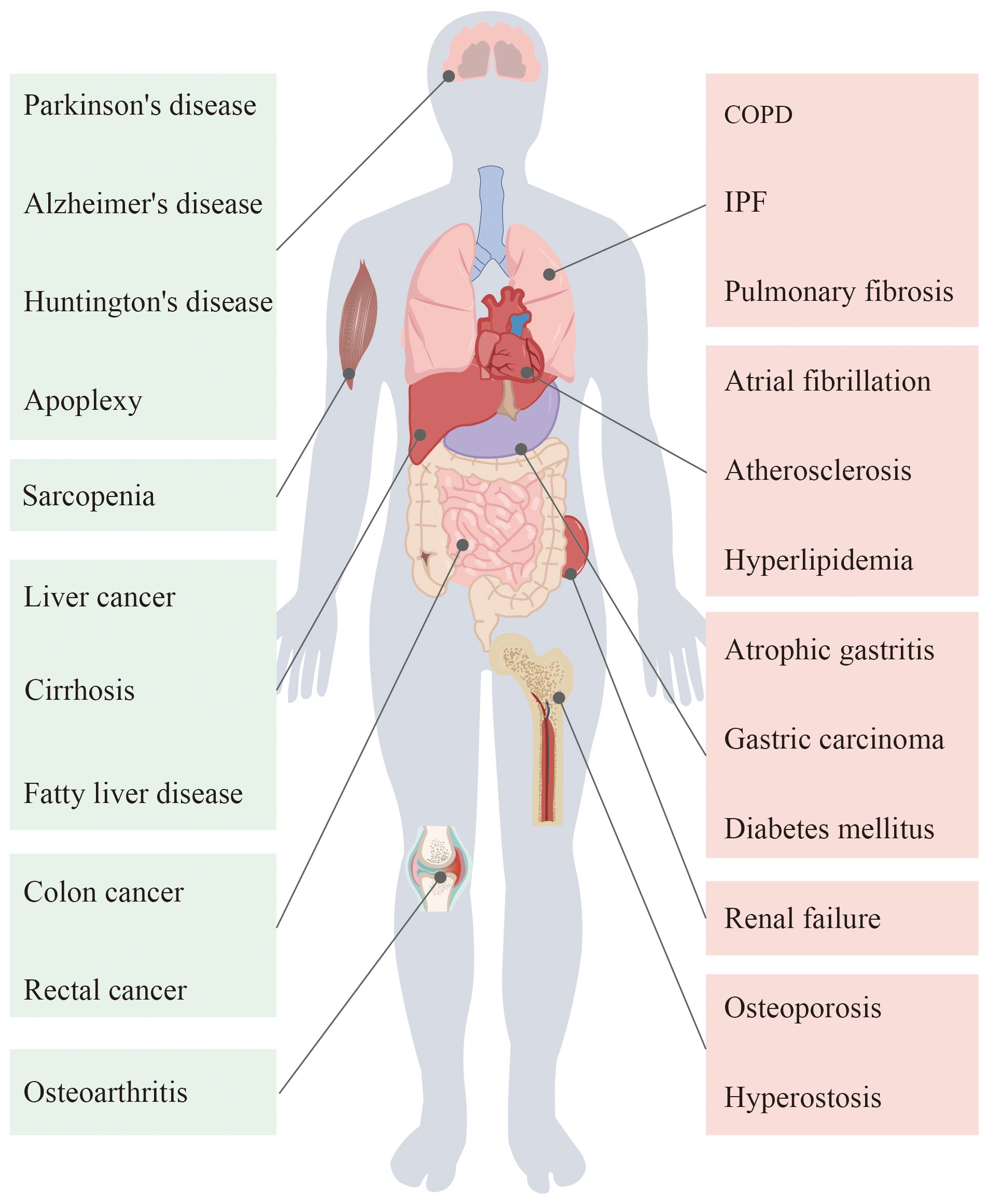
Figure 1 Ageing in the human body organs will appear different degrees of pathological changes, thus causing a variety of diseases.
Progressive declines in tissue and organ function are major features of ageing and represent a significant manifestation of musculoskeletal system disease. In people over 65 years of age, chronic musculoskeletal system disorders are the most common conditions leading to impaired physical frailty mobility (2). A large number of studies have shown that ageing is a clear cause of a series of musculoskeletal system diseases, such as osteoarthritis, osteoporosis, and sarcopenia (3). Some studies suggest that the cellular senescence secretory phenotype (SASP) involves a variety of cellular immune factors and enzymes caused by the occurrence of “ageing inflammation”, resulting in changes in the local microenvironment of the body and leading to cartilage and other connective tissue damage, thus causing a wide range of musculoskeletal diseases in elderly people.
Physiological ageing of the musculoskeletal system is characterized by loss of muscle strength and muscle mass, known as sarcopenia, bone loss or osteoporosis, and destruction of cartilage or osteoarthritis (OA) (4–6). The occurrence and development of musculoskeletal system-related diseases have become the most common disabling factors, resulting in a serious burden on social health care. In this paper, we will focus on the role of cellular senescence in musculoskeletal-related diseases and provide a basis for further research.
2 Mechanisms of ageing
The impact of ageing on the human body is comprehensive and complex, affecting various systems of the human body and leading to functional deterioration and even the occurrence of disease. With the deepening of ageing research, the mechanism of ageing has gradually surfaced. The mechanism of ageing is intricate and closely interrelated, affecting the state of the entire body (Figure 2).
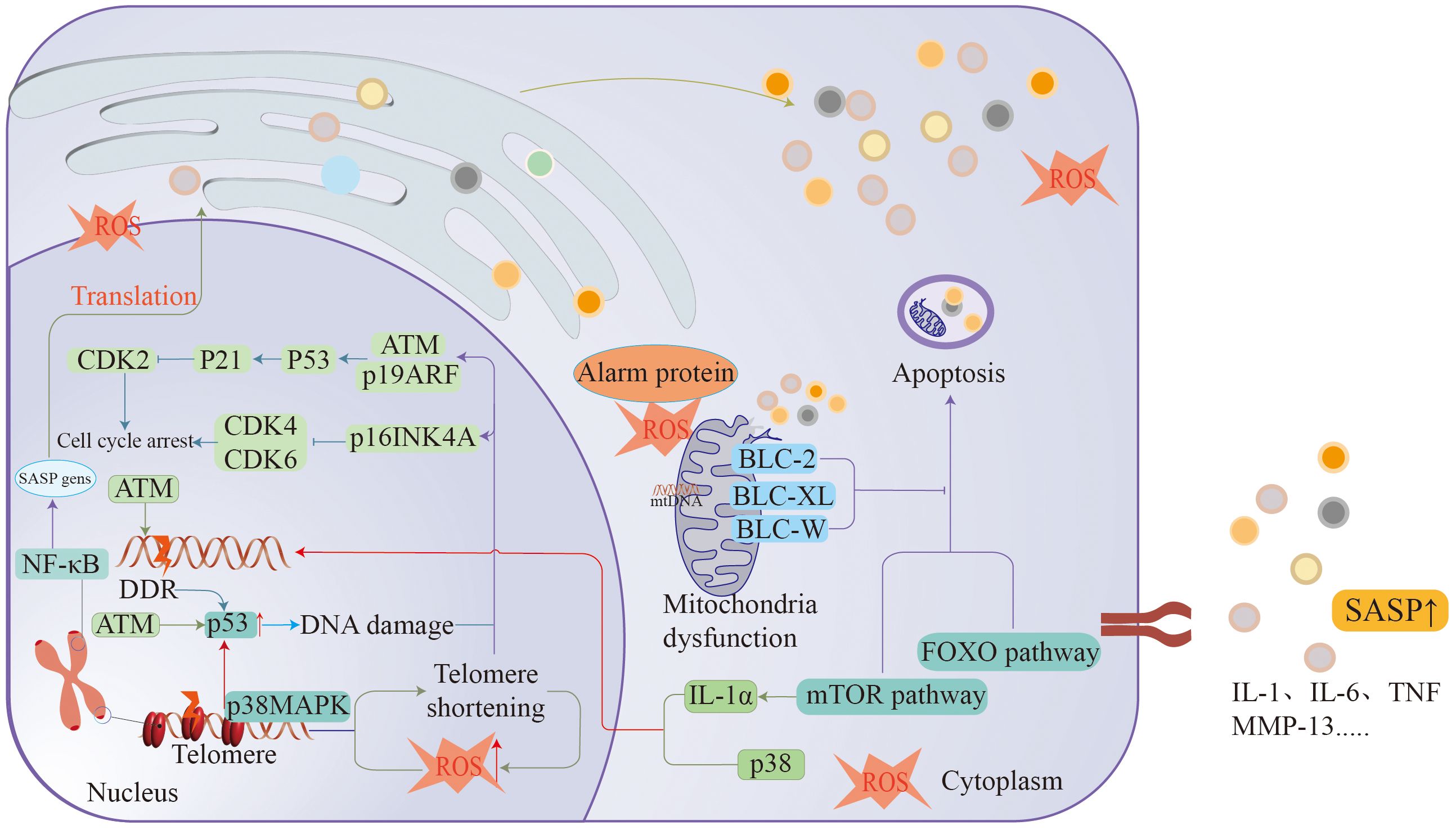
Figure 2 Ageing is associated with many mechanisms, such as DDR, cellular senescence, macrophage dysfunction, telomere loss, etc., all of which cause arrest of the cell division cycle to promote the formation of ageing.
2.1 Cellular senescence
Cellular senescence was first proposed by Hayflick and Moorhead in 1961 (7). Senescent cells undergo irreversible division arrest and gradually accumulate over time to cause dysfunction in the body. Shortening of telomeres, induction of oncogenes, and impairment of mitochondrial function can lead to the development of cellular senescence (8). Despite the arrest of ageing growth, the effects of cellular senescence on the body are clearly correlated with the altered performance of ageing cells (9). In some cases, cellular senescence plays an active role in the normal physiological activities of the body, and induced recovery of cytokines secreted by senescent cells can be observed during wound recovery. Ageing cells are able to exert anticancer effects by limiting the replication of precancerous cells (10–12). Moreover, the occurrence of cellular senescence also plays an important role in the physiopathological mechanisms of many age-related diseases.
Cellular senescence is a stable state of growth arrest, but it is a dynamic process during which cellular senescence occurs and is induced by many molecular pathways (13, 14). Cellular senescence involves complex mechanisms. DDR, telomere shortening and damage, activation of the INK4/ARF locus, induction of the p53/p21CIP1 pathway, and chromatin reorganization have all been shown to be associated with the development of cellular senescence (14–16). The DNA damage response (DDR) is considered to be a common factor in the development of cellular senescence. When DNA is damaged, DDR factors accumulate at the sites of damage, and cellular senescence is caused by entrapment of the cell division cycle into an arrested state through DDR signaling kinases (ATM, ATR, CHK1 and CHK2) (17–19). Moreover, the activation and expression of the related tumor suppressors p53, p21, and p16 also play important roles in the process of cellular senescence (20–22). As a means of fighting cancer, oncogene expression also induces cellular senescence. Over proliferation triggered by the expression of oncogenes is associated with altered DNA replication status and induces cellular senescence through participation in the DDR pathway (23, 24). Activation of the p53 and p16INK4a/Rb tumor suppressor networks is also a major factor in cell cycle arrest, reducing CDK2 activity through transcription of p53-inducible (CDKi) p21CIP1, allowing cells to exit the cell growth cycle (15, 25, 26). These activated networks intervene in the development of cellular senescence by hindering p53 expression (27, 28). The occurrence of cellular senescence is a mechanism by which the body inhibits the development of cancer. The activation of the INK4/ARF locus is another mechanism by which cells undergo senescence; the INK4/ARF locus encodes three tumor suppressors: ARF, p16INK4a, and p15INK4b. These pathways can hinder the degradation of p53 and affect cellular senescence (29, 30). P16INK4a is also often considered the most important cellular marker of cellular senescence (31, 32). Changes in some organelles can also cause ageing. Impaired ribosome biogenesis is an important factor in cell cycle arrest, excessive rRNA transcription in ageing cells or rRNA processing inhibition in replicative ageing can produce nuclear stress, and the accumulation of the ribosomal 40S subunit protein RPS14 can allow the cell cycle to fall into a state of arrest through the CDK4/rb pathway (33, 34). The RPL22/eL22 protein in ribosomes can then prevent the phosphorylation of RB and promote cell cycle arrest through the inhibition of CDK4-Cyclin D1 (35).
Cellular senescence also exerts biological effects in the local cellular environment or on the entire body, and the sum of various cytokines produced by senescent cells is called the SASP (36). Although the SASP is different during cellular senescence in different tissues, a large number of in vitro cellular senescence assays have reported that the SASP is composed mainly of IL-6, IL-8, and MCP1 and contains a variety of proinflammatory factors and various ECM remodeling-related enzymes (37–39). In recent years, proteomic analysis of the SASP has revealed a number of different SASP effectors in the human body that can act as soluble molecules or be released into the extracellular space via exosomes; moreover, many age-related diseases are associated with enrichment of the SASP in plasma (40). The SASP is regulated at multiple levels, and sustained signal transduction by the DDR is key to SASP-related inflammatory molecules, with ATM, NBS1, and CHK2 being the main regulators (41, 42). The SASP is regulated by many pathways and depends on various secretion pathways to regulate cell and tissue ageing. The characteristics of the SASP are also very different under different signaling pathways. The DDR and cGAS/STING pathways are related to SASP regulation. The SASP affects the development of cellular senescence through the regulation of various cascade reactions (43, 44).
2.2 Telomere attrition
Telomere shortening is a classic mechanism of ageing. In telomere attrition, as cells replicate and proliferate, telomeres shorten as the number of divisions increases, leading to the development of senescence (45, 46). Telomere shortening and dysfunction have been shown to be significantly associated with age-related diseases in many studies. In many tissues and organs with high proliferative activity, the continuous division of cells leads to gradual shortening of telomeres and gradually leads to abnormal changes in DNA. These changes can lead to a series of physiological changes, such as ageing, apoptosis, and blocked cell differentiation, to induce disease. However, in tissues and organs with a low proliferative tendency, telomeres can also be affected by factors such as ROS, leading to telomere damage. Oxidative stress can affect skeletal muscle, cardiac muscle and other cells to accelerate ageing and cause related diseases (47, 48). Telomere shortening is also strongly associated with ageing-related inflammation, and the development of ageing and chronic inflammation can lead to the development of diseases such as osteoarthritis, atherosclerosis, and type II diabetes. Inflammatory ageing caused by telomere loss and dysfunction is mainly driven by the secretion of high ROS, and high levels of ROS and an inflammatory response will continue to aggravate the loss and imbalance of telomeres, resulting in a malignant cycle (49, 50). Therefore, inhibiting the increase in ROS by maintaining mitochondrial activity is the main treatment for ageing-related diseases (49).
2.3 Inflammation
Inflammation often runs through aging as an important clue. Inflammation can occur widely in aging tissue cells and induce immune responses, leading to the development of diseases. The development of chronic inflammation is also considered one of the hallmarks of the development of aging (51, 52).
Chronic inflammation and aging are mutually influential processes. senescence-associated secretory phenotype (SASP) induces senescence in normal cells through the development of chronic inflammation. At the same time, chronic inflammation promotes aging of related immune cells resulting in the inability of people to timely remove aging cells and related inflammatory factors. Thus forming a malignant cycle causes further development of aging. At the cellular level, mitochondrial dysfunction and DNA damage and oxidative stress can cause chronic inflammation. These senescent cells secrete SASP that includes most of the inflammatory mediators such as IL-1β and IL-6 as key factors that promote the development of inflammation (53, 54). Aging-related chronic inflammatory responses are mainly endogenous inflammatory responses formed by endogenous cellular molecular junk stimulation (55, 56). Oxidative stress can induce systemic inflammatory response through signaling pathway by inducing the damage of intracellular biological factors. It also promotes oxidative stress through increased secretion of inflammatory mediators. Damage to the genome by ROS, prompting telomere shortening DNA fragmentation has been identified as a key factor in the development of cellular senescence These damaged DNA fragments leak into the cytosol directly causing inflammation (57).
2.4 Disruption of macroautophagy
Autophagy is a metabolic process in which various cellular components are degraded and recovered, and the stable operation of cells is maintained by the phagocytosis and decomposition of abnormal macromolecules and organelles (49). A large body of evidence suggests that a decline in autophagy is associated with the development of ageing and related diseases. With increasing age, autophagy naturally decreases. Simultaneously, stalled autophagic vesicles can accumulate with age, resulting in a systemic decline in autophagic activity (58, 59). The upstream regulatory function of autophagy disorder is mainly realized by the insulin/IGF-1 signaling pathway, which affects the level of autophagy activity through the combination of FOXO and the sensing regulation of mTOR trophic factors and growth factors. The activity of the transcription factors TFEB and FOXO is also affected when the insulin/IGF-1 signaling pathway is dysregulated, and these changes reduce autophagy and lysosome-related expression (60, 61). These regulatory pathways are vulnerable to age factors. Changes in mTOR activity can have a significant impact on autophagic flux, and increased mTOR conduction can be significantly observed in the muscle tissues of aged mice and humans (62). Dysregulation of FOXO activity induces musculoskeletal-related diseases, and autophagic gene expression is significantly lower in the cartilage of elderly osteoarthritis patients, demonstrating that reduced autophagic activity in cartilage tissue leads to the development of cartilage degeneration (63, 64).
2.5 Mitochondrial dysfunction
The mitochondria play a role as power providers in the normal physiological activity of cells, but mitochondria also play roles in the development of inflammation and cell death (65). Mutations in mtDNA can accumulate within mitochondria with age, resulting in the dysregulation of mitochondrial oxidative phosphorylation, increasing ROS production and thereby causing cellular senescence (66). According to related experiments, the accumulation of mtDNA mutations and deletions results in significant ageing phenotypes, including sarcopenia, osteoporosis, shortened lifespan, and decreased fertility (67, 68). Instability of mtDNA within mitochondria can also contribute to the development of ageing. mtDNA tends to replicate more frequently during physiological activities and is present in an oxidative environment with mitochondria. These conditions place mtDNA at a greater risk of mutation and deletion (69). Increased mtDNA mutations can be observed in various ageing biological tissue cell systems. Recent studies have shown that mtDNA is also exported only extracellularly and is detected in cerebrospinal fluid as a biological signal interconnected between mitochondria in distal tissues. Decreased secretion of melatonin in elderly individuals prompts mtDNA release from mitochondria and activates the cGAS/STING/IRF3 signaling pathway to produce inflammatory cytokines involved in the development of cellular senescence (70). Increased mtDNA mutations can be observed in various aging biological tissue cells. Recent studies have found that mtDNA also exports only extracellularly and is detected in cerebrospinal fluid as a biological signal interconnected between mitochondria in distal tissues. Decreased secretion of melatonin in the elderly prompts mtDNA release from mitochondria and activates the cGAS/STING/IRF3 signaling pathway to produce inflammatory cytokines involved in the development of cellular senescence (71–73). Mitochondrial dysfunction also leads to the development of chronic inflammation. Some immune factors are released in response to cell damage during the development of immune responses, and the related factors are called alarm proteins (74, 75). The immune factors include mitochondria-associated molecules, such as mtDNA, ATP, succinate, and ROS, and these mitochondria-derived molecules induce inflammation through different pathways (76). Among them, mtDNA has already been confirmed to be the strongest immune activator. Several PRRs bind mtDNA to trigger innate immune pathways to induce inflammation; for example, TLR9 binds mtDNA to stimulate NF-κB signaling pathways to stimulate inflammation development (77, 78). During organ transplantation, older donor organs can lead to aging of the transplanted person. After aging organ transplantation, the expression of SASP in recipients will be significantly increased to induce the aging of young cell cells, and even the aging cell population. Accumulation of mtDNA also greatly reduces the survival rate of transplanted organs (79, 80). In the experiment of Jasper Iske et al., senolytics therapy was effective in improving the survival probability of transplanted organs in recipient mice after treatment of aging donor mice. It also reduces the level of inflammation in recipient mice after transplantation (81). Mitochondrial dysfunction leading to chronic inflammation is a causative factor in many diseases, and targeted mitochondrial therapy via pharmacological intervention can effectively treat age-related diseases (66).
2.6 Protein homeostasis
Most of the physiological functions of humans are inseparable from proteins, and the damage and inactivation of normal proteins in the human body can lead to the occurrence of a series of related diseases (82). Many neurological diseases are mainly associated with protein damage. To prevent protein denaturation, cells prevent protein misfolding by producing chaperone proteins. When the structural homeostasis of proteins changes, autophagy and the unfolded protein response (UPR) together constitute a clearance system for abnormal proteins (83). The transcription factors ATF3 and ATF4, which regulate UPR activation, therefore play important roles in the processing of abnormal proteins. Abnormal expression levels of ATF3 and ATF4 lead to failure of the ubiquitination of abnormal proteins that accumulate protease targets to accelerate ageing and the development of related diseases; moreover, it has been shown that reducing the concentration of ubiquitinated targets can prolong lifespan (84–86). A newly discovered ubiquitin-like molecule, SUMO, can improve stability by modifying proteins. Silencing of SUMO-encoding genes has been found to lead to a reduced lifespan in C. elegans (87, 88). In addition, insulin-like growth factor-1 (IGF-1) inhibits the degradation of abnormal proteins by regulating the autophagy–lysosome system and inhibiting lysosomal protein degradation, such as that of the proteasome. IGF-1, a well-established ageing factor, can significantly inhibit ageing by inhibiting the IGF-1 signaling pathway (89). However, in the musculoskeletal system, certain levels of IGF-1 can promote muscle cell synthesis of proteins through the PI3K/Akt/mTOR and PI3K/Akt/GSK3β pathways and lead to a decrease in muscle strength when IGF-1 levels are too low (90, 91).
2.7 Genome instability
Genomes are frequently affected by various exogenous and endogenous factors. These effects often cause damage to a range of genetic materials. In response to these injuries, organisms have evolved a series of damage repair mechanisms. Damage to DNA activates the DNA damage response, preventing the transmission of damaged genetic information to offspring cells through the replication of damaged DNA in tissues. However, with age, these repair systems gradually fail, and damaged genes continue to accumulate, leading to ageing of the body (92, 93). Tabula et al. confirmed a positive correlation between the generation of genetic mutations and the development of ageing through the analysis of a large number of mouse cells of all ages (94). Improving the stability of the genome can effectively delay the occurrence of ageing. In an experiment, overexpression of SIRT6 in mice effectively improved the stability of the genome and prolonged the lifespan of the mice (95).
2.8 Epigenetic changes
Epigenetic changes such as DNA methylation, abnormal histone modification, and abnormal chromatin remodeling promote ageing. These changes can lead to a range of ageing-related diseases, including various cancers, metabolic diseases, neurological diseases, and musculoskeletal system diseases. DNA methylation in the human body mainly refers to the regulation of gene expression by the binding of DNA to proteins that inhibit transcription (96). Methylation, a stable DNA modification, can be used as an epigenetic marker (97). According to related studies, there are fewer DNA methylation modifications in senescent cells than in permanent and proliferating cells, and DNA methylation gradually decreases during cellular senescence. Steve Horvath proposed the concept of the epigenetic clock through methylation and age, and Levine proposed the theory of DNAm PhenoAge. These studies demonstrate a relationship between the epigenetic clock and ageing and indicate that ageing-related alterations can be deduced by DNA methylation levels (98, 99). Histone modifications are additional major forms of epigenetic alterations. Loss of histones and modification of histones can also be clearly observed when ageing occurs, with the greatest changes being methylation and acetylations (100). Histone modifications can be involved in the regulation of transcriptional function. In related studies, patients with Hutchinson-Gilford syndrome and Werner syndrome exhibited significant chromatin abnormalities and significant decreases in H3K9me3 or SUV39H (H3K9me3 histone methyltransferase) levels (101). Reducing the activity of histone acetyltransferases prolongs the lifespan of progeria mice (102).
2.9 Deregulated nutrient sensing
Nutrient sensing is the ability of human cells to recognize nutrients such as glucose and is regulated mainly by the insulin/IGF-1 signaling, mTOR, and AMPK pathways. Nutrient-sensing pathways can regulate the absorption and distribution of nutrients through the state of the cell (103). Various pathway regulatory networks interact with each other to form feedback loops to regulate metabolic processes in the body, such as protein and nucleotide synthesis. These regulatory networks mainly promote anabolic progression at young ages and ageing at old ages (104). The microbiota in the human body is also involved in nutrient sensing to some extent, and the gut–brain axis (GBA) demonstrates that there is communication between the gut microbiota and the brain (105, 106). The intestinal microbiota plays important roles in metabolism and the immune response, and ageing-related microbiota can promote the development of systemic inflammation, leading to the occurrence of ageing-related diseases. During ageing, glial cells undergo a series of disorders leading to neurodegenerative diseases in the brain. In experiments, microglial function was effectively improved by resupplementing mice with relevant gut microbiota metabolites (56, 107).
2.10 Stem cell exhaustion
Stem cells have the potential to regenerate and differentiate and are associated with cell turnover and the repair of damaged tissues in vivo. Stem cells, like ordinary cells, are ageing cells. Autophagy is an important mechanism for maintaining stem cell activity and differentiation ability. Autophagy activity in MSCs can effectively improve the ageing of stem cells. However, with ageing, decreased autophagic activity naturally causes the differentiation and proliferation of MSCs (108, 109). Ageing triggers a series of processes of asymmetric cell division and differentiation, resulting in stem cell depletion or abnormal proliferation and differentiation. According to related mouse experiments, ageing can significantly affect the division and differentiation of T cells, resulting in premature ageing and impaired T-cell function (110).
2.11 Changes in intercellular communication
The normal physiological activities of the human body are inseparable from the transmission of information, and the body’s complex and large communication network links various organs, tissues and even cells to form a whole. Intercellular communication occurs mainly through the exocytosis of signaling factors and other means to affect adjacent cells (111). The SASP causes inflammation and senescence in normal cells through its effects on adjacent cellular functions. Senescent cells in tissues mainly communicate between cells through juxtacrine signaling, cell fusion, cytoplasmic bridges, and extracellular vesicles, causing the “spread” of ageing in the local or even systemic circulation (112, 113).
3 Role of ageing in musculoskeletal system diseases
Diseases of the musculoskeletal system occur widely in the elderly population, with approximately 1.71 billion people worldwide suffering from musculoskeletal disorders. Diseases of the musculoskeletal system have a great impact on the quality of life of the population, severely limiting the patient’s mobility and the ability of the labor force to engage substantially in social activities. As the population ages, the number of diseases will continue to increase in the future (114) (Figure 3).
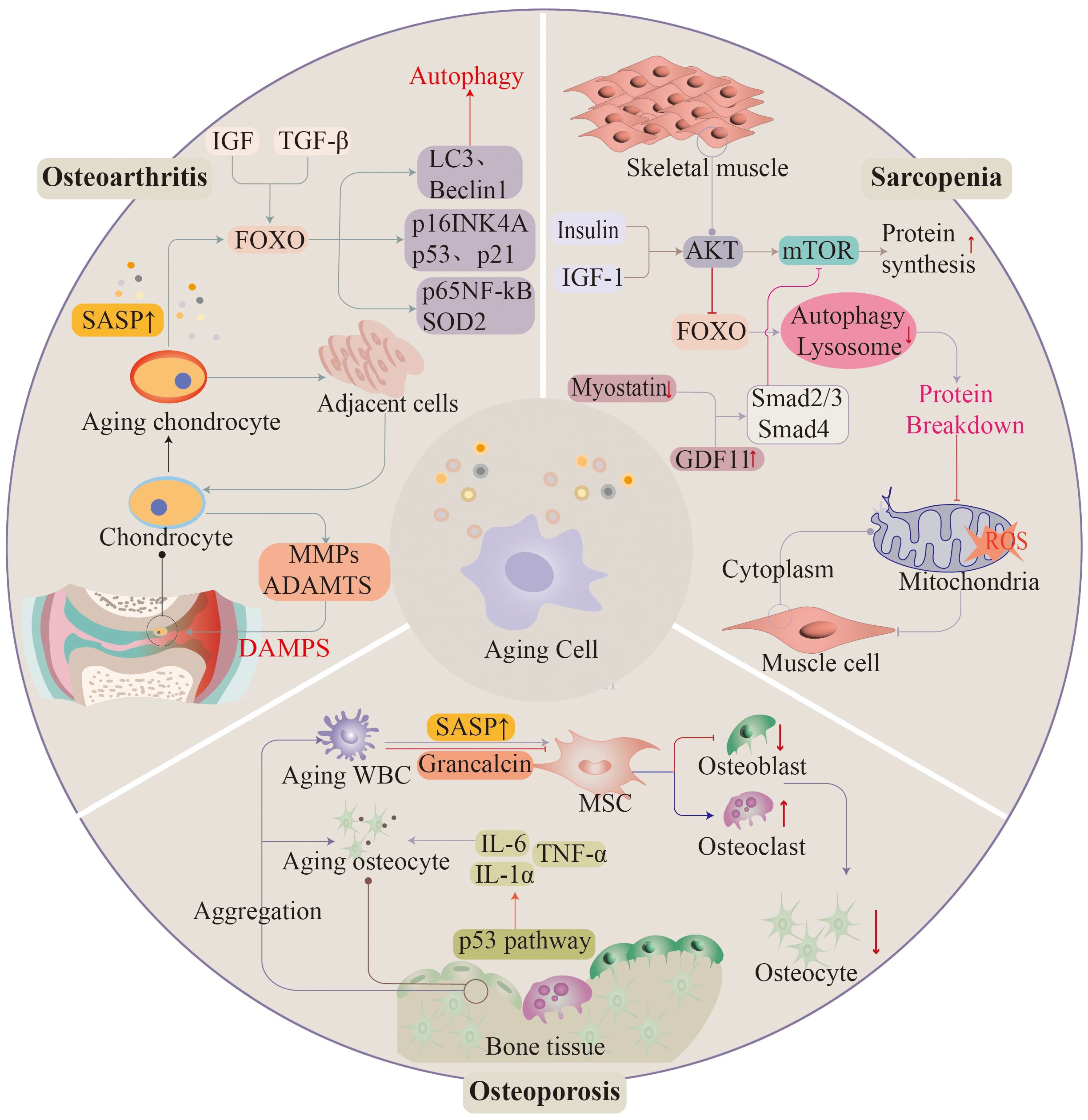
Figure 3 As ageing progresses, the musculoskeletal system often appears in significant functional deterioration. Osteoarthritis, osteoporosis, and sarcopenia are associated with ageing. Ageing of histiocytes promotes the development of these diseases.
4 Ageing and osteoarthritis
Osteoarthritis (OA) is a chronic degenerative disease that occurs within joints and is mainly characterized by cartilage degeneration and osteophyte formation in patients, often accompanied by joint pain, difficulty in movement and other symptoms, mostly in elderly patients (5, 83). As the most common joint disease, osteoarthritis causes not only physical pain but also mental pain. There are many risk factors for the development of osteoarthritis; obesity, a history of joint injury, sex, and genetics are all causative factors for its development, though the most important risk factor is age (115).
The development of osteoarthritis is closely related to the development of cellular senescence. Cellular senescence in articular cartilage is not only age-related, but mechanical damage or mechanical stress stimulation at the joint can also lead to the occurrence of chondrocyte senescence. The most common pathogenesis of osteoarthritis suggests that the occurrence of osteoarthritis is associated with the occurrence of subchondral sclerosis (116, 117). With respect to cartilage destruction, progressive loss of articular cartilage is a hallmark injury in OA. Cartilage damage is almost unavoidable with age, which in turn progresses to the bones and joints. Although chondrocytes have always been recognized as nonregenerable, according to some studies at the cellular level, chondrocytes can regenerate after articular cartilage injury, and the cartilage matrix proliferates in clusters during the early stages of OA (118). At the cellular level, cartilage organizers include a group of cells with stem cell functions, namely, CSPCs, which have a certain positive effect on cartilage injury. Cellular ageing has a certain positive effect on cartilage mechanisms. In addition to the ageing damage of chondrocytes themselves, the ageing of chondrocytes in OA causes changes in the secretory phenotype of other cells in the joint, that is, the effect of “SASP” paracrine signaling (119). Transplantation of senescent fibroblasts into the articular cavity in a mouse model resulted in cartilage damage, leading to changes in the cellular environment in the synovial membrane of the articular cavity and inducing OA (120). Elevated levels of IL-1α, IL-6, MCP-1 and other factors in synovial fluid can also be caused by cells other than chondrocytes (121). For example, premature senescence can be induced in normal human fibroblasts by IL-6–STAT3 signaling (122). Obviously, osteoarthritis development is not influenced by only one cytokine. However, there is relevant evidence that the secretion of ageing phenotypes associated with chondrocyte injury (e.g., IL-6, IL-1α) has an impact on the development of OA. However, because these cytokines are also present in osteoarthritic chondrocytes, the types of cells that cause the abnormal expression of these mediators has not been determined (123). Gene expression patterns can also change in articular chondrocytes as a result of increased SASP in osteoarthritic tissue cells with ageing. Growth factors are released directly or indirectly by chondrocytes during osteoarthritis, and high levels of synthesis and release promote downstream BMP-2 expression and stimulate extracellular matrix turnover (124). The ageing-induced release of cytokines from chondrocytes or other cells in the joint cavity induces the upregulation of MMP expression, leading to the degradation of ECM cartilage, thereby driving the development of osteoarthritis. MMP-13 can cut type II collagen, and in cartilage injury, cartilage breakdown products stimulate the secretion of MMP13 by related cells to further loss of cartilage and accelerate the development of OA (125). Through the transformation of related cellular ageing phenotypes, the chondrocyte ageing pathway, the upregulation of β-galactosidase production, the increase in p16 expression, the irreversible growth arrest of related cells, and the increase in extracellular vesicle secretion are all involved in the transformation of cell phenotypes that are strongly related to the development of osteoarthritis (126). Currently, cartilage damage caused by cellular senescence remains a major topic of research. Osteoarthritis is often treated clinically by repairing articular cartilage or injecting matrix into the joint cavity to relieve the progression of osteoarthritis, such as with intra-articular stem cell injection. The important role of cell senescence injury-related factors in the development of osteoarthritis has been confirmed. Studying the mechanism underlying the relationship between cellular senescence and the occurrence of osteoarthritis can provide more options for the treatment of cartilage injury in osteoarthritis or for delaying the progression of osteoarthritis and improving the quality of life of affected patients. Analyzing the process by which arthritic chondrocytes accumulate damage from the perspective of cellular ageing under the mechanism of SASP and identifying the targets of cytokine signaling pathways in the ageing process can reveal more effective methods for the treatment of osteoarthritis.
As SASP research progresses, a major goal of ageing research is to better understand common mechanisms that can delay loss of function in multiple systems, which will lead to improved “healthy longevity” rather than simply increasing longevity. Heterochronous symbiosis experiments combining the circulatory systems of juvenile and aged animals have revealed systemic factors that alter the ageing phenotype, including growth and differentiation factor 11 (GDF11), oxytocin, and IL-15, which may reduce ageing in multiple organ systems. An important aim of future studies will be to determine whether these ageing-related factors affect OA development.
5 Ageing and osteoporosis
OP is a disease characterized by bone loss and microstructural changes, and its occurrence is strongly correlated with age. With the progress of research, the occurrence of OP is also closely related to the occurrence of ageing. When ageing occurs, the release of large amounts of SASP products and other effects will lead to disruption of the balance of bone homeostasis, and changes in the local microenvironment or systemic factors will lead to the occurrence of osteoporosis (127, 128). Treatment of ageing cells has been shown to significantly improve bone loss in mice (129).
Osteocytes account for 90% of all bone-related cells and play important roles in the normal physiological structure and activity of bone. An increasing number of related studies have shown that osteocyte ageing is significantly associated with bone metabolism disorders (130). Senile osteoporosis is mainly characterized by the loss of cortical bone and trabeculae, which are mainly reflected in the loss of cortical bone during ageing (131). Osteocyte and osteoblast dysfunctions are considered to be the main causes of bone loss in elderly patients with OP (127). Ageing occurs in osteocytes for a variety of reasons: in addition to replicative ageing associated with telomere shortening during the cell cycle and body ageing, cytotoxic stress is induced; oncogenes are stimulated; free radical accumulation occurs; reactive oxygen species are generated; DNA damage, telomere damage, protein arrest, and mitochondrial dysfunction occur; and prosurvival pathways are activated (132–134). Changes in the expression levels of some genes, such as WNT10B, RUNX2, RANKL, Osterix, osteocalcin, osteoprotegerin(OPG), and Sclerosteosis(SOST), have revealed these effects, which sometimes differed in male patients with idiopathic osteoporosis, and their expression levels tended to decrease (135). Studies using RNA sequencing technology have assessed genes that change with age-related osteoporosis and revealed approximately 700 DEGs as well as 12 cellular pathways related to skeletal ageing (136).
With increasing age, ageing osteocytes and bone marrow cells upregulate the expression of multiple SASP factors in the bone microenvironment of aged mice, and the expression of the key ageing marker p16INK4a doubles with the ageing of bone-derived B cells and bone marrow T cells, osteoprogenitor cells, osteoblasts, and osteocytes (137, 138). The upregulation of several SASP-related mRNAs, such as p16INK4a and p21, was also evident from the comparison of the expression of related factors in elderly women, and the expression levels of SASP effectors were significantly greater in elderly women than in young women (139). The bone microenvironment of aged mice shows obvious ageing characteristics, and the occurrence of OP is strongly associated with the balance of osteoblasts and osteoclasts. Osteoblasts and leptin receptor-positive cells, which are the main source of osteoblasts, also senesce with increasing age in mice (140–142). This process leads to changes in osteoblast differentiation to adipocyte differentiation, which triggers senile osteoporosis (143, 144). Some studies have shown that p6 and p21 mRNA levels are significantly increased in osteocytes of aged mice (145). When ageing occurs, the levels of two autophagy marker genes (Atg7 and Map1lc3a) in the osteocytes of aged mice decrease, which indicates a reduction in mitophagy in mice, and the damaged mitochondria cannot be removed in time, leading to the generation of more reactive oxygen species (ROS) and subsequently contributing to ageing (139, 146). Although senescent osteocytes account for only a very small proportion of the total number of osteocytes, SASP release by these senescent osteocytes induces an inflammatory microenvironment in bone, resulting in disturbances in bone metabolism (145). Some specific SASP factors, such as TNF-α, IL-1 and IL-6, promote the ageing of nearby healthy cells and lead to bone resorption and the inhibition of bone formation; the resulting bone loss then induces OP. Treatment of mouse bone marrow cells with senescent cell culture medium has revealed that osteoclast differentiation is significantly increased in mice (129). Increased SASP-related senescence can significantly affect normal cells. There are also factors related to the SASP that can regulate gene expression in osteocytes and inhibit osteogenic differentiation and mineralization. Previous studies have shown that blocking the SASP and the secretion of MLO-Y4 cells can relieve the inhibition of bone mesenchymal stem cell (BMSC) differentiation and improve the occurrence of OP (147, 148). In clinical practice, cortical bone ageing loss is more severe than trabecular bone loss in patients with primary OP, suggesting that bone loss in ageing osteocytes may involve different mechanisms at different bone sites (149, 150).
Another important player in bone homeostasis is the immune system. According to Farr et al., immune cells also play an important role in the development of senile OP (137, 151). Neutrophils and macrophages undergo senescence in aged mice, and senescent macrophages also release SASP factors to promote M1 polarization and ageing in other macrophages (137, 139, 151). Large amounts of grancalcin are accumulated and secreted from the bone marrow, which bind and inhibit the signal transduction of the b2 receptor of MSCs, reducing osteogenesis (152). The ageing of immune cells is a potential factor in the development of senile OP. In alveolar bone, repeated stimulation of LSP triggers the activation of p53 and increases the expression levels of IL-1α, IL-6 and TNF-α, which in turn causes DNA damage and induces cellular senescence in alveolar bone, resulting in the secretion of local SASP to form OP (41, 153, 154).
Thus, the ageing of a variety of cells in the bone microenvironment can cause bone loss, and although the ageing of myeloid cells and osteocytes are predominant, the ageing of other cells still has a certain impact on the occurrence of OP (139).
Estrogen has a considerable effect on the metabolism of osteocytes and osteoblasts and can alleviate the ageing of osteocytes and osteoclasts to a certain extent (155). Therefore, changes in estrogen levels in the human body can have a certain degree of impact on the metabolism of osteocytes and osteoclasts (156). After menopause, a large decrease in estrogen levels leads to accelerated proliferation and metabolism of osteoclasts and stimulates bone resorption on trabecular bone surfaces. This process stimulates the activity of osteoclasts, causing the upregulation of SOST. The upregulation of SOST inhibits WNT signaling, increases osteoblast number, and decreases osteoblast activity (127). This causes an imbalance in bone metabolism to form OP. With the ageing of the body, bone cell ageing can lead to OP. Estrogen plays an important role in bone cell ageing. In OVX mice, the proportions of p16 and β-galactoside 127 in bone cells are high, and they produce effectors such as MMP-3, MMP-13, IL-6, IL-8, IL-1α and IL-1β. Other SASP component-related experiments have also demonstrated that exogenous estrogen supplementation is quite effective for bone loss in OVX mice; related studies have shown that exogenous estrogen supplementation can effectively alleviate osteocyte senescence in OVX mice when exogenous estrogen supplementation is available. By hindering p16 and other related signaling pathways, bone loss in OVX mice is substantially attenuated or prevented (157, 158). However, at present, the specific mechanism by which estrogen delays osteocyte senescence is not clear, and relevant studies have shown that this effect may be related to the inhibition of Usp10 by estrogen (159). A related study from 2019 showed that a decrease in estrogen levels did not have an adequate effect on bone cell SASP concentrations (160). Bone loss caused by estrogen deficiency may not be caused by decreased estrogen levels, which directly affect the cellular ageing process. It is more likely that estrogen deficiency leads to changes in cellular microenvironment-related factors that affect the ageing process of osteocytes, such as promoting their ageing by inducing oxidative stress.
Age-related bone loss can be prevented by eliminating senescent cells or blocking cellular senescence-associated pathways (129). Aged mice clearly showed improvements in skeletal microarchitecture and strength after 4 months of treatment with anti-ageing drugs (eliminating senescent cells) or 2 months of treatment with JAK inhibitors (blocking the proinflammatory secretome of senescent cells) (161, 162). However, IL-1a and Tnfsf11 mRNA could significantly decrease osteoclast numbers and increase osteoblast numbers via the effective clearance of senescent osteocytes (163, 164).
6 Ageing and sarcopenia
Sarcopenia is a disease characterized by the loss of muscle strength and mass (165). The musculoskeletal system allows movement and protects vital organs; over time, its erosion leads to an increased risk of fractures and physical weakness, which are hallmarks of ageing (166).
Cellular senescence, to some extent, can be regarded as inflammation at the molecular level in the human body, causing a range of age-related diseases as a major consequence of ageing due to DNA damage (167). In ageing mice, symptoms of sarcopenia are often observed, and elevated expression levels of SASP factors such as Igfbp2, MMP13, and Pai1 are clearly detected in their bones (168). Ageing can have a significant effect on the number of skeletal muscle cells, and SASP-related factors were significantly increased in the skeletal muscle cells of older rhesus monkeys (169, 170). The artificial induction of ectopic p16Ink4a expression in aged mice inhibited the proliferation of damaged skeletal muscle satellite cells. However, inhibiting the expression of p16Ink4a can significantly alleviate sarcopenia in ageing mice (168, 171). The number of skeletal muscle satellite cells decreases with increasing age. Moreover, skeletal muscle satellite cells in aged mice develop fibrosis by activating the Wnt signaling pathway, which in turn leads to fibrosis of skeletal muscle tissue (172). Increased cellular senescence and inflammatory responses can also lead to muscle loss by damaging stem cells. Inflammatory responses occurring throughout the body are also associated with decreased function of associated progenitor cells (161, 173). OPA1 regulates the metabolic level of muscle stem cells through FGF21, and muscle loss occurs when OPA1 is reduced. Cultivation of senescent mesenchymal progenitor cells with myoblasts in the experiment resulted in accelerated muscle loss. The expression of proinflammatory factors in the SASP is an important factor in sarcopenia, and the overexpression of IL-6 can significantly lead to muscle atrophy in mice (174, 175).
In recent years, researchers have determined that skeletal muscle is not only the motor unit of the human body but also has a certain immunoregulatory function, Skeletal muscle cells can regulate human immune function by releasing signals such as related cytokines (176). Muscle tissue can release more than 300 cytokines, one of which was previously identified as IL-6 (177, 178). IL-6 also acts as a member of the SASP, causing sarcopenia through proinflammatory effects (179, 180). IL-6 induces muscle atrophy by reducing muscle synthesis and may directly mediate muscle catabolism. IL-6 can also induce muscle atrophy by stimulating abnormal STAT3–IL-6 signaling (181, 182). When IL-6 is administered to mice with the corresponding antibody, muscle conditions can be effectively improved (183). Another apparent correlate is IL-15, a cytokine that is expressed in a variety of tissues, including muscle, and is a part of SASP (178). When synergized with IGF-1, IL-15 induces the synthesis of myosin heavy chain and promotes muscle recovery by inhibiting the differentiation of FAPs into adipocytes (184). The overexpression of FoxO also increases muscle protein breakdown and promotes ageing- and obesity-related muscle degeneration. IL-5 mRNA levels were substantially increased after recovery in subjects trained for high-intensity muscle resistance (185, 186). As an energy level sensor within skeletal muscle cells, AMPK is important for IL-15 production following exercise (187, 188). With increasing age, AMPK activity decreases in vivo, causing muscle loss in IL-15 signaling pathways (189).
7 Targeted ageing therapy for musculoskeletal system diseases
Ageing is inevitable. The incidence of various diseases will also increase with age. The main research ideas currently aim at treating and preventing some diseases by inhibiting the occurrence of cellular senescence. In many age-related diseases, curbing the growth of senescent cells is an effective treatment (190).
Current major ageing inhibition strategies are divided into senolytics and senomorphics. Senolytics are a strategy to promote apoptosis in senescent cells by intervening in various signaling regulatory pathways within senescent cells via the use of related drugs. Normally, senescent cells upregulate the expression of factors that negatively regulate apoptosis (e.g., BCL-2, BCL-W, BCL-X), thereby maintaining a stable state of senescence arrest (13, 191, 192). Senomorphics inhibit the damage of senescent cells to other normal tissue cells mainly by inhibiting the expression of SASP factors in senescent cells. The expression of SASP components secreted by senescent cells is reduced by related drugs, such as rapamycin (mTOR), which inhibits the occurrence of ageing (193–195). The characteristic of Senomorphics is that it can reduce SASP secretion while ensuring cell survival.
Senolytic therapy often has high requirements for the timing of administration during treatment. Inhibition of BCL-2 and BCL-Xl by Navitoclax at the initial stage of inflammation effectively removed senescent p16 stem cells, promoting histiocyte regeneration. The combined use of quercetin and dasatinib eliminated senescent cells in bone tissue and cartilage by stimulating senescence-associated tyrosine kinase targets (116, 196, 197). In addition, intra-articular injection of UBX0101 (an anti-ageing agent) for the treatment of osteoarthritis resulted in the local clearance of ageing cells to alleviate the progression of arthritis by blocking p53/MDM2-induced apoptosis of ageing cells (198). In a recent mouse study, there was a significant decrease in the number of hydroxylated proteins in synovial fluid and cartilage of articular cavities treated with UBX0101, which could reduce oxidative damage to related proteins. The progression of osteoarthritis was greatly slowed in these mice (199). Laccarin has shown benefits in the treatment of musculoskeletal disorders such as osteoporosis and osteoarthritis in relevant animal and clinical drug experiments. Laccarin can hinder the development of osteoporosis through osteoblast differentiation and inhibition of bone resorption (200, 201).
In the SASP, MMP13 has been identified as the most important factor contributing to the development of osteoarthritis. Inhibition of MMP13 by CL82198 effectively reduced the degradation of type II collagen and reduced the number of osteoarthritis-related lesions (202, 203). AP20187 is a cell-permeable molecule used to dimerize the FK506-binding protein (FKBP) fusion protein, initiate biological signaling cascades and gene expression or disrupt protein-protein interactions, and treatment with AP20187 can reduce the expression of p16Ink4a, thereby improving bone loss (204, 205). The number of osteoblasts and the rate of bone formation increased in mice treated with AP20187, and the amount of adipose tissue in the bone marrow decreased (129).
8 Discussion
Aging is a natural process that affects all systems and organs of the human body. It is an inevitable topic that requires careful consideration and understanding. Aging mechanisms are complex, and different factors interact and promote each other. Continued research on aging is important to prevent aging and its related diseases and improve the quality of life of the elderly.
This review summarizes the main mechanisms of aging and their effects in the musculoskeletal system. Musculoskeletal system diseases have a particularly wide impact on the quality of life, and severe cases can lead to loss of mobility causing serious harm to the psychophysiological health of patients. With increasing age, the number of patients involved is expected to continue to increase, placing a serious burden on the public healthcare system. Musculoskeletal system disorders have a large impact on the quality of prognosis by intervening early. In particular, OA, due to the particularity of cartilage tissue, the injured cartilage can hardly be recovered. Therefore, timely relevant treatment in the early stage of OA is the most suitable therapy.
Many current studies are limited to animal and cellular assays. In these studies, only a small part has been implicated in the promotion of aging by interactions between different mechanisms interacting with each other. As a whole, there are more or less mutual influence and interaction between various systems and organs of the human body. Many mechanistic studies of aging, especially the interactions between various aging mechanisms, are not clear. These issues remain to be further investigated in depth. There is still a long way to go before it is truly used in clinical practice. The problem of social labor can be greatly optimized if major breakthroughs are made in aging of the musculoskeletal system. In the future, Through the early intervention of aging, we can help middle-aged and elderly people effectively delay the development of musculoskeletal diseases with OA as the main disease. Reduce the public health care pressure caused by aging.
Author contributions
YC: Writing – original draft. ZH: Writing – original draft, Writing – review & editing. HC: Writing – original draft. HL: Writing – original draft. KW: Writing – original draft. JC: Writing – original draft. ZL: Writing – original draft. YX: Writing – original draft. YL: Writing – original draft. SZ: Writing – original draft. SW: Writing – original draft. XZ: Writing – original draft. SJ: Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Open Project Program of Engineering Research Center of Human-Robot Intelligent Technologies and Systems, MOE of China (under Grant Nos. KP2024PY015).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. Hallmarks of aging: An expanding universe. Cell. (2023) 186:243–78. doi: 10.1016/j.cell.2022.11.001
2. Roberts S, Colombier P, Sowman A, Mennan C, Rölfing JH, Guicheux J, et al. Ageing in the musculoskeletal system. Acta orthopaedica. (2016) 87:15–25. doi: 10.1080/17453674.2016.1244750
3. Song S, Guo Y, Yang Y, Fu D. Advances in pathogenesis and therapeutic strategies for osteoporosis. Pharmacol Ther. (2022) 237:108168. doi: 10.1016/j.pharmthera.2022.108168
4. Nedergaard A, Henriksen K, Karsdal MA, Christiansen C. Musculoskeletal ageing and primary prevention. Best Pract Res Clin obstetrics gynaecology. (2013) 27:673–88. doi: 10.1016/j.bpobgyn.2013.06.001
5. Martel-Pelletier J, Barr AJ, Cicuttini FM, Conaghan PG, Cooper C, Goldring MB, et al. Osteoarthritis. Nat Rev Dis Primers. (2016) 2:16072. doi: 10.1038/nrdp.2016.72
6. Cruz-Jentoft AJ, Baeyens JP, Bauer JM, Boirie Y, Cederholm T, Landi F, et al. Sarcopenia: European consensus on definition and diagnosis: Report of the European Working Group on Sarcopenia in Older People. Age Ageing. (2010) 39:412–23. doi: 10.1093/ageing/afq034
7. Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. (1961) 25:585–621. doi: 10.1016/0014-4827(61)90192-6
8. Di Micco R, Krizhanovsky V, Baker D, d'Adda di Fagagna F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol. (2021) 22:75–95. doi: 10.1038/s41580-020-00314-w
9. Salama R, Sadaie M, Hoare M, Narita M. Cellular senescence and its effector programs. Genes Dev. (2014) 28:99–114. doi: 10.1101/gad.235184.113
10. Jun JI, Lau LF. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat Cell Biol. (2010) 12:676–85. doi: 10.1038/ncb2070
11. Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell. (2014) 31:722–33. doi: 10.1016/j.devcel.2014.11.012
12. Collado M, Serrano M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer. (2010) 10:51–7. doi: 10.1038/nrc2772
13. Yosef R, Pilpel N, Tokarsky-Amiel R, Biran A, Ovadya Y, Cohen S, et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat Commun. (2016) 7:11190. doi: 10.1038/ncomms11190
14. Herranz N, Gil J. Mechanisms and functions of cellular senescence. J Clin Invest. (2018) 128:1238–46. doi: 10.1172/JCI95148
15. Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature. (2004) 432:307–15. doi: 10.1038/nature03098
16. Gil J, Peters G. Regulation of the INK4b-ARF-INK4a tumour suppressor locus: all for one or one for all. Nat Rev Mol Cell Biol. (2006) 7:667–77. doi: 10.1038/nrm1987
17. Fumagalli M, Rossiello F, Mondello C, d'Adda di Fagagna F. Stable cellular senescence is associated with persistent DDR activation. PloS One. (2014) 9:e110969. doi: 10.1371/journal.pone.0110969
18. D'adda Di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature. (2003) 426:194–8. doi: 10.1038/nature02118
19. Mallette FA, Ferbeyre G. The DNA damage signaling pathway connects oncogenic stress to cellular senescence. Cell Cycle (Georgetown Tex). (2007) 6:1831–6. doi: 10.4161/cc.6.15.4516
20. Beauséjour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, et al. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J. (2003) 22:4212–22. doi: 10.1093/emboj/cdg417
21. Dulić V, Beney GE, Frebourg G, Drullinger LF, Stein GH. Uncoupling between phenotypic senescence and cell cycle arrest in aging p21-deficient fibroblasts. Mol Cell Biol. (2000) 20:6741–54. doi: 10.1128/MCB.20.18.6741-6754.2000
22. Kamijo T, Zindy F, Roussel MF, Quelle DE, Downing JR, Ashmun RA, et al. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell. (1997) 91:649–59. doi: 10.1016/S0092-8674(00)80452-3
23. Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. (2006) 444:638–42. doi: 10.1038/nature05327
24. Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Sci (New York NY). (2008) 319:1352–5. doi: 10.1126/science.1140735
25. Kastenhuber ER, Lowe SW. Putting p53 in context. Cell. (2017) 170:1062–78. doi: 10.1016/j.cell.2017.08.028
26. d'Adda di Fagagna F. Living on a break: cellular senescence as a DNA-damage response. Nat Rev Cancer. (2008) 8:512–22. doi: 10.1038/nrc2440
27. Stewart SA, Ben-Porath I, Carey VJ, O'Connor BF, Hahn WC, Weinberg RA. Erosion of the telomeric single-strand overhang at replicative senescence. Nat Genet. (2003) 33:492–6. doi: 10.1038/ng1127
28. Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. (2005) 120:513–22. doi: 10.1016/j.cell.2005.02.003
29. Sharpless NE. INK4a/ARF: a multifunctional tumor suppressor locus. Mutat Res. (2005) 576:22–38. doi: 10.1016/j.mrfmmm.2004.08.021
30. Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers [J]. Nature. (2010) 463:899–905. doi: 10.1038/nature08822
31. Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. (1999) 397:164–8. doi: 10.1038/16476
32. Bracken AP, Kleine-Kohlbrecher D, Dietrich N, Pasini D, Gargiulo G, Beekman C, et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. (2007) 21:525–30. doi: 10.1101/gad.415507
33. Nishimura K, Kumazawa T, Kuroda T, Katagiri N, Tsuchiya M, Goto N, et al. Perturbation of ribosome biogenesis drives cells into senescence through 5S RNP-mediated p53 activation. Cell Rep. (2015) 10:1310–23. doi: 10.1016/j.celrep.2015.01.055
34. Lessard F, Igelmann S, Trahan C, Huot G, Saint-Germain E, Mignacca L, et al. Senescence-associated ribosome biogenesis defects contributes to cell cycle arrest through the Rb pathway. Nat Cell Biol. (2018) 20:789–99. doi: 10.1038/s41556-018-0127-y
35. Del Toro N, Fernandez-Ruiz A, Mignacca L, Kalegari P, Rowell MC, Igelmann S, et al. Ribosomal protein RPL22/eL22 regulates the cell cycle by acting as an inhibitor of the CDK4-cyclin D complex. Cell Cycle (Georgetown Tex). (2019) 18:759–70. doi: 10.1080/15384101.2019.1593708
36. Kuilman T, Peeper DS. Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer. (2009) 9:81–94. doi: 10.1038/nrc2560
37. Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PloS Biol. (2008) 6:2853–68. doi: 10.1371/journal.pbio.0060301
38. Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. (2010) 5:99–118. doi: 10.1146/annurev-pathol-121808-102144
39. Coppé JP, Patil CK, Rodier F, Krtolica A, Beauséjour CM, Parrinello S, et al. A human-like senescence-associated secretory phenotype is conserved in mouse cells dependent on physiological oxygen. PloS One. (2010) 5:e9188. doi: 10.1371/journal.pone.0009188
40. Tanaka T, Biancotto A, Moaddel R, Moore AZ, Gonzalez-Freire M, Aon MA, et al. Plasma proteomic signature of age in healthy humans. Aging Cell. (2018) 17:e12799. doi: 10.1111/acel.12799
41. Coppé JP, Rodier F, Patil CK, Freund A, Desprez PY, Campisi J.. Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J Biol Chem. (2011) 286:36396–403. doi: 10.1074/jbc.M111.257071
42. Rodier F, Coppé JP, Patil CK, Hoeijmakers WA, Muñoz DP, Raza SR, et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. (2009) 11:973–9. doi: 10.1038/ncb1909
43. Freund A, Patil CK, Campisi J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. (2011) 30:1536–48. doi: 10.1038/emboj.2011.69
44. Glück S, Guey B, Gulen MF, Wolter K, Kang TW, Schmacke NA, et al. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence [J]. Nat Cell Biol. (2017) 19:1061–70. doi: 10.1038/ncb3586
45. Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature. (1990) 345:458–60. doi: 10.1038/345458a0
46. Levy MZ, Allsopp RC, Futcher AB, Greider CW, Harley CB. Telomere end-replication problem and cell aging. J Mol Biol. (1992) 225:951–60. doi: 10.1016/0022-2836(92)90096-3
47. Puente BN, Kimura W, Muralidhar SA, Moon J, Amatruda JF, Phelps KL, et al. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell. (2014) 157:565–79. doi: 10.1016/j.cell.2014.03.032
48. González-Guardia L, Yubero-Serrano EM, Rangel-Zuñiga O, Marin C, Camargo A, Pérez-Martínez P, et al. Influence of endothelial dysfunction on telomere length in subjects with metabolic syndrome: LIPGENE study. Age (Dordrecht Netherlands). (2014) 36:9681. doi: 10.1007/s11357-014-9681-9
49. Dillon LM, Williams SL, Hida A, Peacock JD, Prolla TA, Lincoln J, et al. Increased mitochondrial biogenesis in muscle improves aging phenotypes in the mtDNA mutator mouse. Hum Mol Genet. (2012) 21:2288–97. doi: 10.1093/hmg/dds049
50. Risques RA, Lai LA, Himmetoglu C, Ebaee A, Li L, Feng Z, et al. Ulcerative colitis-associated colorectal cancer arises in a field of short telomeres, senescence, and inflammation. Cancer Res. (2011) 71:1669–79. doi: 10.1158/0008-5472.CAN-10-1966
51. Baechle JJ, Chen N, Makhijani P, Winer S, Furman D, Winer DA. Chronic inflammation and the hallmarks of aging. Mol Metab. (2023) 74:101755. doi: 10.1016/j.molmet.2023.101755
52. Li X, Li C, Zhang W, Wang Y, Qian P, Huang H. Inflammation and aging: signaling pathways and intervention therapies. Signal transduction targeted Ther. (2023) 8:239. doi: 10.1038/s41392-023-01502-8
53. Li X, Zhang Z, Wang Z, Gutiérrez-Castrellón P, Shi H. Cell deaths: Involvement in the pathogenesis and intervention therapy of COVID-19. Signal transduction targeted Ther. (2022) 7:186. doi: 10.1038/s41392-022-01043-6
54. Laing AG, Lorenc A, Del Molino Del Barrio I, Das A, Fish M, Monin L, et al. A dynamic COVID-19 immune signature includes associations with poor prognosis. Nat Med. (2020) 26:1623–35. doi: 10.1038/s41591-020-1038-6
55. Franceschi C, Garagnani P, Vitale G, Capri M, Salvioli S. Inflammaging and 'Garb-aging'. Trends Endocrinol metabolism: TEM. (2017) 28:199–212. doi: 10.1016/j.tem.2016.09.005
56. Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A. Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat Rev Endocrinol. (2018) 14:576–90. doi: 10.1038/s41574-018-0059-4
57. De La Fuente M, Miquel J. An update of the oxidation-inflammation theory of aging: the involvement of the immune system in oxi-inflamm-aging. Curr Pharm design. (2009) 15:3003–26. doi: 10.2174/138161209789058110
58. Yim WW, Mizushima N. Lysosome biology in autophagy. Cell Discovery. (2020) 6:6. doi: 10.1038/s41421-020-0141-7
59. Chang JT, Kumsta C, Hellman AB, Adams LM, Hansen M. Spatiotemporal regulation of autophagy during Caenorhabditis elegans aging. eLife. (2017) 6:e18459. doi: 10.7554/eLife.18459
60. Nieto-Torres JL, Hansen M. Macroautophagy and aging: The impact of cellular recycling on health and longevity. Mol aspects Med. (2021) 82:101020. doi: 10.1016/j.mam.2021.101020
61. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. (2013) 153:1194–217. doi: 10.1016/j.cell.2013.05.039
62. Carroll B, Nelson G, Rabanal-Ruiz Y, Kucheryavenko O, Dunhill-Turner NA, Chesterman CC, et al. Persistent mTORC1 signaling in cell senescence results from defects in amino acid and growth factor sensing. J Cell Biol. (2017) 216:1949–57. doi: 10.1083/jcb.201610113
63. Lotz MK, Caramés B. Autophagy and cartilage homeostasis mechanisms in joint health, aging and OA. Nat Rev Rheumatol. (2011) 7:579–87. doi: 10.1038/nrrheum.2011.109
64. Rached MT, Kode A, Silva BC, Jung DY, Gray S, Ong H, et al. FoxO1 expression in osteoblasts regulates glucose homeostasis through regulation of osteocalcin in mice. J Clin Invest. (2010) 120:357–68. doi: 10.1172/JCI39901
65. Hetz C, Zhang K, Kaufman RJ. Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol. (2020) 21:421–38. doi: 10.1038/s41580-020-0250-z
66. Amorim JA, Coppotelli G, Rolo AP, Palmeira CM, Ross JM, Sinclair DA. Mitochondrial and metabolic dysfunction in ageing and age-related diseases. Nat Rev Endocrinol. (2022) 18:243–58. doi: 10.1038/s41574-021-00626-7
67. Ross JM, Öberg J, BRENé S, Coppotelli G, Terzioglu M, Pernold K, et al. High brain lactate is a hallmark of aging and caused by a shift in the lactate dehydrogenase A/B ratio. Proc Natl Acad Sci United States America. (2010) 107:20087–92. doi: 10.1073/pnas.1008189107
68. Ross JM, Stewart JB, Hagström E, Brené S, Mourier A, Coppotelli G, et al. Germline mitochondrial DNA mutations aggravate ageing and can impair brain development. Nature. (2013) 501:412–5. doi: 10.1038/nature12474
69. Greaves LC, Nooteboom M, Elson JL, Tuppen HA, Taylor GA, Commane DM, et al. Clonal expansion of early to mid-life mitochondrial DNA point mutations drives mitochondrial dysfunction during human ageing. PloS Genet. (2014) 10:e1004620. doi: 10.1371/journal.pgen.1004620
70. Scheibye-Knudsen M, Fang EF, Croteau DL, Bohr VA. Contribution of defective mitophagy to the neurodegeneration in DNA repair-deficient disorders. Autophagy. (2014) 10:1468–9. doi: 10.4161/auto.29321
71. Yu CH, Davidson S, Harapas CR, Hilton JB, Mlodzianoski MJ, Laohamonthonkul P, et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell. (2020) 183:636–49.e18. doi: 10.1016/j.cell.2020.09.020
72. Son JM, Lee C. Aging: All roads lead to mitochondria. Semin Cell Dev Biol. (2021) 116:160–8. doi: 10.1016/j.semcdb.2021.02.006
73. Jauhari A, Baranov SV, Suofu Y, Kim J, Singh T, Yablonska S, et al. Melatonin inhibits cytosolic mitochondrial DNA-induced neuroinflammatory signaling in accelerated aging and neurodegeneration. J Clin Invest. (2020) 130:3124–36. doi: 10.1172/JCI135026
74. Dela Cruz CS, Kang MJ. Mitochondrial dysfunction and damage associated molecular patterns (DAMPs) in chronic inflammatory diseases. Mitochondrion. (2018) 41:37–44. doi: 10.1016/j.mito.2017.12.001
75. Gong T, Liu L, Jiang W, Zhou R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol. (2020) 20:95–112. doi: 10.1038/s41577-019-0215-7
76. Chandel NS. Evolution of mitochondria as signaling organelles. Cell Metab. (2015) 22:204–6. doi: 10.1016/j.cmet.2015.05.013
77. Roger AJ, Muñoz-Gómez SA, Kamikawa R. The origin and diversification of mitochondria. Curr Biol CB. (2017) 27:R1177–r92. doi: 10.1016/j.cub.2017.09.015
78. West AP, Shadel GS. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat Rev Immunol. (2017) 17:363–75. doi: 10.1038/nri.2017.21
79. Iske J, Roesel MJ, Martin F, Schroeter A, Matsunaga T, Maenosono R, et al. Transplanting old organs promotes senescence in young recipients. Am J Transplant Off J Am Soc Transplant Am Soc Transplant Surgeons. (2024) 24:391–405. doi: 10.1016/j.ajt.2023.10.013
80. Iske J, Matsunaga T, Zhou H, Tullius SG. Donor and recipient age-mismatches: the potential of transferring senescence. Front Immunol. (2021) 12:671479. doi: 10.3389/fimmu.2021.671479
81. Iske J, Seyda M, Heinbokel T, Maenosono R, Minami K, Nian Y, et al. Senolytics prevent mt-DNA-induced inflammation and promote the survival of aged organs following transplantation. Nat Commun. (2020) 11:4289. doi: 10.1038/s41467-020-18039-x
82. Hipp MS, Kasturi P, Hartl FU. The proteostasis network and its decline in ageing. Nat Rev Mol Cell Biol. (2019) 20:421–35. doi: 10.1038/s41580-019-0101-y
83. Zhou D, Borsa M, Simon AK. Hallmarks and detection techniques of cellular senescence and cellular ageing in immune cells. Aging Cell. (2021) 20:e13316. doi: 10.1111/acel.13316
84. Zhang C, Zhang X, Huang L, Guan Y, Huang X, Tian XL, et al. ATF3 drives senescence by reconstructing accessible chromatin profiles. Aging Cell. (2021) 20:e13315. doi: 10.1111/acel.13315
85. Koyuncu S, Loureiro R, Lee HJ, Wagle P, Krueger M, Vilchez D. Rewiring of the ubiquitinated proteome determines ageing in C. elegans. Nature. (2021) 596:285–90. doi: 10.1038/s41586-021-03781-z
86. Sun Y, Lin X, Liu B, Zhang Y, Li W, Zhang S, et al. Loss of ATF4 leads to functional aging-like attrition of adult hematopoietic stem cells. Sci Adv. (2021) 7:eabj6877. doi: 10.1126/sciadv.abj6877
87. Gao K, Li Y, Hu S, Liu Y. SUMO peptidase ULP-4 regulates mitochondrial UPR-mediated innate immunity and lifespan extension. eLife. (2019) 8:e41792. doi: 10.7554/eLife.41792
88. Larrick JW, Larrick JW, Mendelsohn AR. SUMO wrestles with mitophagy to extend lifespan. Rejuvenation Res. (2020) 23:527–32. doi: 10.1089/rej.2020.2406
89. Timmer LT, Hoogaars WMH, Jaspers RT. The role of IGF-1 signaling in skeletal muscle atrophy. Adv Exp Med Biol. (2018) 1088:109–37. doi: 10.1007/978-981-13-1435-3_6
90. Ascenzi F, Barberi L, Dobrowolny G, Villa Nova Bacurau A, Nicoletti C, Rizzuto E, et al. Effects of IGF-1 isoforms on muscle growth and sarcopenia. Aging Cell. (2019) 18:e12954. doi: 10.1111/acel.12954
91. Yoshida T, Delafontaine P. Mechanisms of IGF-1-mediated regulation of skeletal muscle hypertrophy and atrophy. Cells. (2020) 9(9):1970. doi: 10.3390/cells9091970
92. Vijg J, Dong X. Pathogenic mechanisms of somatic mutation and genome mosaicism in aging. Cell. (2020) 182:12–23. doi: 10.1016/j.cell.2020.06.024
93. Miller KN, Victorelli SG, Salmonowicz H, Dasgupta N, Liu T, Passos JF, et al. Cytoplasmic DNA: sources, sensing, and role in aging and disease. Cell. (2021) 184:5506–26. doi: 10.1016/j.cell.2021.09.034
94. Almanzar N, Antony J, Baghel AS, Bakerman I, Bansal I, Barres BA, et al. A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature. (2020) 583:590–5. doi: 10.1038/s41586-020-2496-1
95. Tian X, Firsanov D, Zhang Z, Cheng Y, Luo L, Tombline G, et al. SIRT6 is responsible for more efficient DNA double-strand break repair in long-lived species. Cell. (2019) 177:622–38.e22. doi: 10.1016/j.cell.2019.03.043
96. Sen P, Shah PP, Nativio R, Berger SL. Epigenetic mechanisms of longevity and aging. Cell. (2016) 166:822–39. doi: 10.1016/j.cell.2016.07.050
97. López V, Fernández AF, Fraga MF. The role of 5-hydroxymethylcytosine in development, aging and age-related diseases. Ageing Res Rev. (2017) 37:28–38. doi: 10.1016/j.arr.2017.05.002
98. Levine ME, Lu AT, Quach A, Chen BH, Assimes TL, Bandinelli S, et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging. (2018) 10:573–91. doi: 10.18632/aging.v10i4
99. Shireby GL, Davies JP, Francis PT, Burrage J, Walker EM, Neilson GWA, et al. Recalibrating the epigenetic clock: implications for assessing biological age in the human cortex. Brain J Neurol. (2020) 143:3763–75. doi: 10.1093/brain/awaa334
100. Paluvai H, Di Giorgio E, Brancolini C. The histone code of senescence. Cells. (2020) 9(2):466. doi: 10.3390/cells9020466
101. Yi SJ, Kim K. New insights into the role of histone changes in aging. Int J Mol Sci. (2020) 21(21):8241. doi: 10.3390/ijms21218241
102. Bonkowski MS, Sinclair DA. Slowing ageing by design: the rise of NAD(+) and sirtuin-activating compounds. Nat Rev Mol Cell Biol. (2016) 17:679–90. doi: 10.1038/nrm.2016.93
103. Shannon OM, Ashor AW, Scialo F, Saretzki G, Martin-Ruiz C, Lara J, et al. Mediterranean diet and the hallmarks of ageing. Eur J Clin Nutr. (2021) 75:1176–92. doi: 10.1038/s41430-020-00841-x
104. Kabacik S, Lowe D, Fransen L, Leonard M, Ang SL, Whiteman C, et al. The relationship between epigenetic age and the hallmarks of aging in human cells. Nat Aging. (2022) 2:484–93. doi: 10.1038/s43587-022-00220-0
105. Aman Y, Schmauck-Medina T, Hansen M, Morimoto RI, Simon AK, Bjedov I, et al. Autophagy in healthy aging and disease. Nat Aging. (2021) 1:634–50. doi: 10.1038/s43587-021-00098-4
106. Kowalski K, Mulak A. Brain-gut-microbiota axis in alzheimer's disease. J Neurogastroenterol Motil. (2019) 25:48–60. doi: 10.5056/jnm18087
107. Cryan JF, O'riordan KJ, Cowan CSM, Sandhu KV, Bastiaanssen TFS, Boehme M, et al. The microbiota-gut-brain axis. Physiol Rev. (2019) 99:1877–2013. doi: 10.1152/physrev.00018.2018
108. Revuelta M, Matheu A. Autophagy in stem cell aging. Aging Cell. (2017) 16:912–5. doi: 10.1111/acel.12655
109. Zhang D, Chen Y, Xu X, Xiang H, Shi Y, Gao Y, et al. Autophagy inhibits the mesenchymal stem cell aging induced by D-galactose through ROS/JNK/p38 signalling. Clin Exp Pharmacol Physiol. (2020) 47:466–77. doi: 10.1111/1440-1681.13207
110. Borsa M, Barandun N, Gräbnitz F, Barnstorf I, Baumann NS, Pallmer K, et al. Asymmetric cell division shapes naive and virtual memory T-cell immunity during ageing. Nat Commun. (2021) 12:2715. doi: 10.1038/s41467-021-22954-y
111. Fafián-Labora JA, O'loghlen A. Classical and nonclassical intercellular communication in senescence and ageing. Trends Cell Biol. (2020) 30:628–39. doi: 10.1016/j.tcb.2020.05.003
112. Narita M. Juxtacrine regulation of cellular senescence. BMB Rep. (2019) 52:3–4. doi: 10.5483/BMBRep.2019.52.1.289
113. Schaum N, Lehallier B, Hahn O, Pálovics R, Hosseinzadeh S, Lee SE, et al. Ageing hallmarks exhibit organ-specific temporal signatures. Nature. (2020) 583:596–602. doi: 10.1530/ey.18.14.11
114. Cieza A, Causey K, Kamenov K, Hanson SW, Chatterji S, Vos T. Global estimates of the need for rehabilitation based on the Global Burden of Disease study 2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet (London England). (2021) 396:2006–17. doi: 10.1016/S0140-6736(20)32340-0
115. Michael JW, Schlüter-Brust KU, Eysel P. The epidemiology, etiology, diagnosis, and treatment of osteoarthritis of the knee. Deutsches Arzteblatt Int. (2010) 107:152–62. doi: 10.3238/arztebl.2010.0152
116. Jeon OH, Kim C, Laberge RM, Demaria M, Rathod S, Vasserot AP, et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat Med. (2017) 23:775–81. doi: 10.1038/nm.4324
117. Burr DB, Gallant MA. Bone remodelling in osteoarthritis. Nat Rev Rheumatol. (2012) 8:665–73. doi: 10.1038/nrrheum.2012.130
118. Lotz MK, Otsuki S, Grogan SP, Sah R, Terkeltaub R, D'Lima D. Cartilage cell clusters. Arthritis rheumatism. (2010) 62:2206–18. doi: 10.1002/art.27528
119. Jiang Y, Tuan RS. Origin and function of cartilage stem/progenitor cells in osteoarthritis. Nat Rev Rheumatol. (2015) 11:206–12. doi: 10.1038/nrrheum.2014.200
120. Xu M, Bradley EW, Weivoda MM, Hwang SM, Pirtskhalava T, Decklever T, et al. Transplanted senescent cells induce an osteoarthritis-like condition in mice. The journals of gerontology Series A, Biol Sci Med Sci. (2017) 72:780–5. doi: 10.1093/gerona/glw154
121. Tsuchida AI, Beekhuizen M, t Hart MC, Radstake TR, Dhert WJ, Saris DB, et al. Cytokine profiles in the joint depend on pathology, but are different between synovial fluid, cartilage tissue and cultured chondrocytes. Arthritis Res Ther. (2014) 16:441. doi: 10.1186/s13075-014-0441-0
122. Pearson MJ, Herndler-Brandstetter D, Tariq MA, Nicholson TA, Philp AM, Smith HL, et al. IL-6 secretion in osteoarthritis patients is mediated by chondrocyte-synovial fibroblast cross-talk and is enhanced by obesity. Sci Rep. (2017) 7:3451. doi: 10.1038/s41598-017-03759-w
123. Demidenko ZN, Blagosklonny MV. Growth stimulation leads to cellular senescence when the cell cycle is blocked. Cell Cycle (Georgetown Tex). (2008) 7:3355–61. doi: 10.4161/cc.7.21.6919
124. Blaney Davidson EN, Vitters EL, Van Lent PL, van de Loo FA, van den Berg WB, van der Kraan PM. Elevated extracellular matrix production and degradation upon bone morphogenetic protein-2 (BMP-2) stimulation point toward a role for BMP-2 in cartilage repair and remodeling. Arthritis Res Ther. (2007) 9:R102. doi: 10.1186/ar2305
125. Loeser RF, Goldring SR, Scanzello CR, Goldring MB. Osteoarthritis: a disease of the joint as an organ. Arthritis rheumatism. (2012) 64:1697–707. doi: 10.1002/art.34453
126. Coryell PR, Diekman BO, Loeser RF. Mechanisms and therapeutic implications of cellular senescence in osteoarthritis. Nat Rev Rheumatol. (2021) 17:47–57. doi: 10.1038/s41584-020-00533-7
127. Li X, Xu J, Dai B, Wang X, Guo Q, Qin L. Targeting autophagy in osteoporosis: From pathophysiology to potential therapy. Ageing Res Rev. (2020) 62:101098. doi: 10.1016/j.arr.2020.101098
128. Watanabe K, Hishiya A. Mouse models of senile osteoporosis. Mol aspects Med. (2005) 26:221–31. doi: 10.1016/j.mam.2005.01.006
129. Farr JN, Xu M, Weivoda MM, Monroe DG, Fraser DG, Onken JL, et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat Med. (2017) 23:1072–9. doi: 10.1038/nm.4385
130. Cui J, Shibata Y, Zhu T, Zhou J, Zhang J. Osteocytes in bone aging: Advances, challenges, and future perspectives. Ageing Res Rev. (2022) 77:101608. doi: 10.1016/j.arr.2022.101608
131. Chen H, Zhou X, Fujita H, Onozuka M, Kubo KY. Age-related changes in trabecular and cortical bone microstructure. Int J Endocrinol. (2013) 2013:213234. doi: 10.1155/2013/213234
132. Serrano M, Lin AW, Mccurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. (1997) 88:593–602. doi: 10.1016/S0092-8674(00)81902-9
133. Rajarajacholan UK, Riabowol K. Aging with ING: a comparative study of different forms of stress induced premature senescence. Oncotarget. (2015) 6:34118–27. doi: 10.18632/oncotarget.v6i33
134. Kural KC, Tandon N, Skoblov M, Kel-Margoulis OV, Baranova AV. Pathways of aging: comparative analysis of gene signatures in replicative senescence and stress induced premature senescence. BMC Genomics. (2016) 17:1030. doi: 10.1186/s12864-016-3352-4
135. Patsch JM, Kohler T, Berzlanovich A, Muschitz C, Bieglmayr C, Roschger P, et al. Trabecular bone microstructure and local gene expression in iliac crest biopsies of men with idiopathic osteoporosis. J Bone mineral Res Off J Am Soc Bone Mineral Res. (2011) 26:1584–92. doi: 10.1002/jbmr.344
136. Farr JN, Roforth MM, Fujita K, Nicks KM, Cunningham JM, Atkinson EJ, et al. Effects of age and estrogen on skeletal gene expression in humans as assessed by RNA sequencing. PloS One. (2015) 10:e0138347. doi: 10.1371/journal.pone.0138347
137. Vicente R, Mausset-Bonnefont AL, Jorgensen C, Louis-Plence P, Brondello JM. Cellular senescence impact on immune cell fate and function [J]. Aging Cell. (2016) 15:400–6. doi: 10.1111/acel.2016.15.issue-3
138. Khosla S, Farr JN, Tchkonia T, Kirkland JL. The role of cellular senescence in ageing and endocrine disease. Nat Rev Endocrinol. (2020) 16:263–75. doi: 10.1038/s41574-020-0335-y
139. Farr JN, Fraser DG, Wang H, Jaehn K, Ogrodnik MB, Weivoda MM, et al. Identification of senescent cells in the bone microenvironment. J Bone mineral Res Off J Am Soc Bone Mineral Res. (2016) 31:1920–9. doi: 10.1002/jbmr.2892
140. Yue R, Zhou BO, Shimada IS, Zhao Z, Morrison SJ. Leptin receptor promotes adipogenesis and reduces osteogenesis by regulating mesenchymal stromal cells in adult bone marrow. Cell Stem Cell. (2016) 18:782–96. doi: 10.1016/j.stem.2016.02.015
141. Gao B, Lin X, Jing H, Fan J, Ji C, Jie Q, et al. Local delivery of tetramethylpyrazine eliminates the senescent phenotype of bone marrow mesenchymal stromal cells and creates an anti-inflammatory and angiogenic environment in aging mice. Aging Cell. (2018) 17:e12741. doi: 10.1111/acel.12741
142. An Y, Wang B, Wang X, Dong G, Jia J, Yang Q, et al. SIRT1 inhibits chemoresistance and cancer stemness of gastric cancer by initiating an AMPK/FOXO3 positive feedback loop. Cell Death Dis. (2020) 11:115. doi: 10.1038/s41419-020-2308-4
143. Pierce JL, Begun DL, Westendorf JJ, McGee-Lawrence ME. Defining osteoblast and adipocyte lineages in the bone marrow. Bone. (2019) 118:2–7. doi: 10.1016/j.bone.2018.05.019
144. Infante A, Rodríguez CI. Osteogenesis and aging: lessons from mesenchymal stem -cells. Stem Cell Res Ther. (2018) 9:244. doi: 10.1186/s13287-018-0995-x
145. Kenyon J, Gerson SL. The role of DNA damage repair in aging of adult stem cells. Nucleic Acids Res. (2007) 35:7557–65. doi: 10.1093/nar/gkm1064
146. Dalle Pezze P, Nelson G, Otten EG, Korolchuk VI, Kirkwood TB, von Zglinicki T, et al. Dynamic modelling of pathways to cellular senescence reveals strategies for targeted interventions. PloS Comput Biol. (2014) 10:e1003728. doi: 10.1371/journal.pcbi.1003728
147. Aquino-Martinez R, Eckhardt BA, Rowsey JL, Fraser DG, Khosla S, Farr JN, et al. Senescent cells exacerbate chronic inflammation and contribute to periodontal disease progression in old mice [J]. J periodontology. (2021) 92:1483–95. doi: 10.1002/JPER.20-0529
148. Xu L, Wang Y, Wang J, Zhai J, Ren L, Zhu G. Radiation-induced osteocyte senescence alters bone marrow mesenchymal stem cell differentiation potential via paracrine signaling. Int J Mol Sci. (2021) 22(17):9323. doi: 10.3390/ijms22179323
149. Piemontese M, Almeida M, Robling AG, Kim HN, Xiong J, Thostenson JD, et al. Old age causes de novo intracortical bone remodeling and porosity in mice. JCI Insight. (2017) 2(17):e93771. doi: 10.1172/jci.insight.93771
150. Almeida M, Han L, Martin-Millan M, Plotkin LI, Stewart SA, Roberson PK, et al. Skeletal involution by age-associated oxidative stress and its acceleration by loss of sex steroids. J Biol Chem. (2007) 282:27285–97. doi: 10.1074/jbc.M702810200
151. Khosla S, Farr JN, Monroe DG. Cellular senescence and the skeleton: pathophysiology and therapeutic implications. J Clin Invest. (2022) 132(3):e154888. doi: 10.1172/JCI154888
152. Li CJ, Xiao Y, Sun YC, He WZ, Liu L, Huang M, et al. Senescent immune cells release grancalcin to promote skeletal aging. Cell Metab. (2022) 34:184–5. doi: 10.1016/j.cmet.2021.12.003
153. Aquino-Martinez R, Rowsey JL, Fraser DG, Eckhardt BA, Khosla S, Farr JN, et al. LPS-induced premature osteocyte senescence: Implications in inflammatory alveolar bone loss and periodontal disease pathogenesis. Bone. (2020) 132:115220. doi: 10.1016/j.bone.2019.115220
154. Liu J, Zeng J, Wang X, Zheng M, Luan Q. P53 mediates lipopolysaccharide-induced inflammation in human gingival fibroblasts. J periodontology. (2018) 89:1142–51. doi: 10.1002/JPER.18-0026
155. Chen JR, Lazarenko OP, Haley RL, Blackburn ML, Badger TM, Ronis MJ. Ethanol impairs estrogen receptor signaling resulting in accelerated activation of senescence pathways, whereas estradiol attenuates the effects of ethanol in osteoblasts. J Bone mineral Res Off J Am Soc Bone Mineral Res. (2009) 24:221–30. doi: 10.1359/jbmr.081011
156. DEL Río JP, Alliende MI, Molina N, Serrano FG, Molina S, Vigil P. Steroid hormones and their action in women's brains: the importance of hormonal balance. Front Public Health. (2018) 6:141. doi: 10.3389/fpubh.2018.00141
157. Geng Q, Gao H, Yang R, Guo K, Miao D. Pyrroloquinoline quinone prevents estrogen deficiency-induced osteoporosis by inhibiting oxidative stress and osteocyte senescence. Int J Biol Sci. (2019) 15:58–68. doi: 10.7150/ijbs.25783
158. Li J, Karim MA, Che H, Geng Q, Miao D. Deletion of p16 prevents estrogen deficiency-induced osteoporosis by inhibiting oxidative stress and osteocyte senescence. Am J Trans Res. (2020) 12:672–83.
159. Wei Y, Fu J, Wu W, Ma P, Ren L, Wu J. Estrogen prevents cellular senescence and bone loss through Usp10-dependent p53 degradation in osteocytes and osteoblasts: the role of estrogen in bone cell senescence. Cell Tissue Res. (2021) 386:297–308. doi: 10.1007/s00441-021-03496-7
160. Farr JN, Rowsey JL, Eckhardt BA, Thicke BS, Fraser DG, Tchkonia T, et al. Independent roles of estrogen deficiency and cellular senescence in the pathogenesis of osteoporosis: evidence in young adult mice and older humans. J Bone mineral Res Off J Am Soc Bone Mineral Res. (2019) 34:1407–18. doi: 10.1002/jbmr.3729
161. Xu M, Tchkonia T, Ding H, Ogrodnik M, Lubbers ER, Pirtskhalava T, et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc Natl Acad Sci United States America. (2015) 112:E6301–10. doi: 10.1073/pnas.1515386112
162. Colavitti R, Finkel T. Reactive oxygen species as mediators of cellular senescence. IUBMB Life. (2005) 57:277–81. doi: 10.1080/15216540500091890
163. Kim HN, Xiong J, Macleod RS, Iyer S, Fujiwara Y, Cawley KM, et al. Osteocyte RANKL is required for cortical bone loss with age and is induced by senescence. JCI Insight. (2020) 5(19):e138815. doi: 10.1172/jci.insight.138815
164. Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O'Brien CA. Matrix-embedded cells control osteoclast formation. Nat Med. (2011) 17:1235–41. doi: 10.1038/nm.2448
165. Liu F, Yuan L, Li L, Yang J, Liu J, Chen Y, et al. S-sulfhydration of SIRT3 combats BMSC senescence and ameliorates osteoporosis via stabilizing heterochromatic and mitochondrial homeostasis. Pharmacol Res. (2023) 192:106788. doi: 10.1016/j.phrs.2023.106788
166. Janssen I, Shepard DS, Katzmarzyk PT, Roubenoff R. The healthcare costs of sarcopenia in the United States. J Am Geriatrics Soc. (2004) 52:80–5. doi: 10.1111/j.1532-5415.2004.52014.x
167. Zhao Y, Simon M, Seluanov A, Gorbunova V. DNA damage and repair in age-related inflammation. Nat Rev Immunol. (2023) 23:75–89. doi: 10.1038/s41577-022-00751-y
168. Baker DJ, Perez-Terzic C, Jin F, Pitel KS, Niederländer NJ, Jeganathan K, et al. Opposing roles for p16Ink4a and p19Arf in senescence and ageing caused by BubR1 insufficiency. Nat Cell Biol. (2008) 10:825–36. doi: 10.1038/ncb1744
169. Welle S, Brooks AI, Delehanty JM, Needler N, Bhatt K, Shah B, et al. Skeletal muscle gene expression profiles in 20-29 year old and 65-71 year old women. Exp gerontology. (2004) 39:369–77. doi: 10.1016/j.exger.2003.11.011
170. Kayo T, Allison DB, Weindruch R, Prolla TA. Influences of aging and caloric restriction on the transcriptional profile of skeletal muscle from rhesus monkeys. Proc Natl Acad Sci United States America. (2001) 98:5093–8. doi: 10.1073/pnas.081061898
171. Sousa-Victor P, Gutarra S, García-Prat L, Rodriguez-Ubreva J, Ortet L, Ruiz-Bonilla V, et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature. (2014) 506:316–21. doi: 10.1038/nature13013
172. Brack AS, Conboy MJ, Roy S, Lee M, Kuo CJ, Keller C, et al. Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Sci (New York NY). (2007) 317:807–10. doi: 10.1126/science.1144090
173. Conboy IM, Conboy MJ, Wagers AJ, Girma ER, Weissman IL, Rando TA. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature. (2005) 433:760–4. doi: 10.1038/nature03260
174. Tezze C, Romanello V, Desbats MA, Fadini GP, Albiero M, Favaro G, et al. Age-associated loss of OPA1 in muscle impacts muscle mass, metabolic homeostasis, systemic inflammation, and epithelial senescence. Cell Metab. (2017) 25:1374–89.e6. doi: 10.1016/j.cmet.2017.04.021
175. Tsujinaka T, Ebisui C, Fujita J, Kishibuchi M, Morimoto T, Ogawa A, et al. Muscle undergoes atrophy in association with increase of lysosomal cathepsin activity in interleukin-6 transgenic mouse. Biochem Biophys Res Commun. (1995) 207:168–74. doi: 10.1006/bbrc.1995.1168
176. Afzali AM, Müntefering T, Wiendl H, Meuth SG, Ruck T. Skeletal muscle cells actively shape (auto)immune responses. Autoimmun Rev. (2018) 17:518–29. doi: 10.1016/j.autrev.2017.12.005
177. Giudice J, Taylor JM. Muscle as a paracrine and endocrine organ. Curr Opin Pharmacol. (2017) 34:49–55. doi: 10.1016/j.coph.2017.05.005
178. Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, et al. Cellular senescence: defining a path forward. Cell. (2019) 179:813–27. doi: 10.1016/j.cell.2019.10.005
179. Dalle S, Rossmeislova L, Koppo K. The role of inflammation in age-related sarcopenia. Front Physiol. (2017) 8:1045. doi: 10.3389/fphys.2017.01045
180. Alemán H, Esparza J, Ramirez FA, Astiazaran H, Payette H. Longitudinal evidence on the association between interleukin-6 and C-reactive protein with the loss of total appendicular skeletal muscle in free-living older men and women. Age Ageing. (2011) 40:469–75. doi: 10.1093/ageing/afr040
181. Harder-Lauridsen NM, Krogh-Madsen R, Holst JJ, Plomgaard P, Leick L, Pedersen BK, et al. Effect of IL-6 on the insulin sensitivity in patients with type 2 diabetes. Am J Physiol Endocrinol Metab. (2014) 306:E769–78. doi: 10.1152/ajpendo.00571.2013
182. Madaro L, Passafaro M, Sala D, Etxaniz U, Lugarini F, Proietti D, et al. Denervation-activated STAT3-IL-6 signalling in fibro-adipogenic progenitors promotes myofibres atrophy and fibrosis. Nat Cell Biol. (2018) 20:917–27. doi: 10.1038/s41556-018-0151-y
183. Tsujinaka T, Fujita J, Ebisui C, Yano M, Kominami E, Suzuki K, et al. Interleukin 6 receptor antibody inhibits muscle atrophy and modulates proteolytic systems in interleukin 6 transgenic mice. J Clin Invest. (1996) 97:244–9. doi: 10.1172/JCI118398
184. Kang X, Yang MY, Shi YX, Xie MM, Zhu M, Zheng XL, et al. Interleukin-15 facilitates muscle regeneration through modulation of fibro/adipogenic progenitors. Cell communication Signaling CCS. (2018) 16:42. doi: 10.1186/s12964-018-0251-0
185. Yang H, Chang J, Chen W, Zhao L, Qu B, Tang C, et al. Treadmill exercise promotes interleukin 15 expression in skeletal muscle and interleukin 15 receptor alpha expression in adipose tissue of high-fat diet rats. Endocrine. (2013) 43:579–85. doi: 10.1007/s12020-012-9809-6
186. Kalinkovich A, Livshits G. Sarcopenic obesity or obese sarcopenia: A cross talk between age-associated adipose tissue and skeletal muscle inflammation as a main mechanism of the pathogenesis. Ageing Res Rev. (2017) 35:200–21. doi: 10.1016/j.arr.2016.09.008
187. Crane JD, Macneil LG, Lally JS, Ford RJ, Bujak AL, Brar IK, et al. Exercise-stimulated interleukin-15 is controlled by AMPK and regulates skin metabolism and aging. Aging Cell. (2015) 14:625–34. doi: 10.1111/acel.12341
188. Kjøbsted R, Hingst JR, Fentz J, Foretz M, Sanz MN, Pehmøller C, et al. AMPK in skeletal muscle function and metabolism. FASEB J Off Publ Fed Am Societies Exp Biol. (2018) 32:1741–77. doi: 10.1096/fj.201700442R
189. Salminen A, Kaarniranta K, Kauppinen A. Age-related changes in AMPK activation: Role for AMPK phosphatases and inhibitory phosphorylation by upstream signaling pathways. Ageing Res Rev. (2016) 28:15–26. doi: 10.1016/j.arr.2016.04.003
190. Childs BG, Gluscevic M, Baker DJ, Laberge RM, Marquess D, Dananberg J, et al. Senescent cells: an emerging target for diseases of ageing. Nat Rev Drug Discovery. (2017) 16:718–35. doi: 10.1038/nrd.2017.116
191. Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J, et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med. (2016) 22:78–83. doi: 10.1038/nm.4010
192. Ozsvari B, Nuttall JR, Sotgia F, Lisanti MP. Azithromycin and Roxithromycin define a new family of "senolytic" drugs that target senescent human fibroblasts. Aging. (2018) 10:3294–307. doi: 10.18632/aging.v10i11
193. Herranz N, Gallage S, Mellone M, Wuestefeld T, Klotz S, Hanley CJ, et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat Cell Biol. (2015) 17:1205–17. doi: 10.1038/ncb3225
194. Zhang B, Fu D, Xu Q, Cong X, Wu C, Zhong X, et al. The senescence-associated secretory phenotype is potentiated by feedforward regulatory mechanisms involving Zscan4 and TAK1. Nat Commun. (2018) 9:1723. doi: 10.1038/s41467-018-04010-4
195. Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. (2009) 460:392–5. doi: 10.1038/nature08221
196. Hickson LJ, Langhi Prata LGP, Bobart SA, Evans TK, Giorgadze N, Hashmi SK, et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine. (2019) 47:446–56. doi: 10.1016/j.ebiom.2019.08.069
197. Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, et al. Senolytics improve physical function and increase lifespan in old age. Nat Med. (2018) 24:1246–56. doi: 10.1038/s41591-018-0092-9
198. Faust HJ, Zhang H, Han J, Wolf MT, Jeon OH, Sadtler K, et al. IL-17 and immunologically induced senescence regulate response to injury in osteoarthritis. J Clin Invest. (2020) 130:5493–507. doi: 10.1172/JCI134091
199. Chin AF, Han J, Clement CC, Choi Y, Zhang H, Browne M, et al. Senolytic treatment reduces oxidative protein stress in an aging male murine model of post-traumatic osteoarthritis. Aging Cell. (2023) 22:e13979. doi: 10.1111/acel.13979
200. Yamaura K, Nelson AL, Nishimura H, Rutledge JC, Ravuri SK, Bahney C, et al. The effects of fisetin on bone and cartilage: A systematic review. Pharmacol Res. (2022) 185:106504. doi: 10.1016/j.phrs.2022.106504
201. Derakhshanian H, Djalali M, Djazayery A, Nourijelyani K, Ghadbeigi S, Pishva H, et al. Quercetin prevents experimental glucocorticoid-induced osteoporosis: a comparative study with alendronate. Can J Physiol Pharmacol. (2013) 91:380–5. doi: 10.1139/cjpp-2012-0190
202. Neuhold LA, Killar L, Zhao W, Sung ML, Warner L, Kulik J, et al. Postnatal expression in hyaline cartilage of constitutively active human collagenase-3 (MMP-13) induces osteoarthritis in mice. J Clin Invest. (2001) 107:35–44. doi: 10.1172/JCI10564
203. Mehana EE, Khafaga AF, El-Blehi SS. The role of matrix metalloproteinases in osteoarthritis pathogenesis: An updated review. Life Sci. (2019) 234:116786. doi: 10.1016/j.lfs.2019.116786
204. Ahmed S, Xie J, Horne D, Williams JC. Photocleavable dimerizer for the rapid reversal of molecular trap antagonists. J Biol Chem. (2014) 289:4546–52. doi: 10.1074/jbc.C113.513622
Keywords: ageing, cell senescence, SASP, osteoarthritis, osteoporosis, sarcopenia
Citation: Cai Y, Han Z, Cheng H, Li H, Wang K, Chen J, Liu Z-X, Xie Y, Lin Y, Zhou S, Wang S, Zhou X and Jin S (2024) The impact of ageing mechanisms on musculoskeletal system diseases in the elderly. Front. Immunol. 15:1405621. doi: 10.3389/fimmu.2024.1405621
Received: 23 March 2024; Accepted: 22 April 2024;
Published: 07 May 2024.
Edited by:
Marjorie Pion, Instituto de Investigación Sanitaria Gregorio Marañón, SpainReviewed by:
Jasper Iske, Brigham and Women’s Hospital and Harvard Medical School, United StatesCarlos Manuel Martinez, Biomedical Research Institute of Murcia (IMIB), Spain
Copyright © 2024 Cai, Han, Cheng, Li, Wang, Chen, Liu, Xie, Lin, Zhou, Wang, Zhou and Jin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhongyu Han, aHp5Y3p5MTk5N0AxNjMuY29t; Song Jin, MTA0OTE0NzAwMEBxcS5jb20=
†These authors have contributed equally to this work