 Henry Smith
Henry Smith Edward Arbe-Barnes2
Edward Arbe-Barnes2 Shivan Sivakumar
Shivan Sivakumar- 1School of Medicine and Biomedical Sciences, University of Oxford, Oxford, United Kingdom
- 2Institute of Immunology and Transplantation, University College London, London, United Kingdom
- 3Sir William Dunn School of Pathology, University of Oxford, Oxford, United Kingdom
- 4Institute of Immunology and Immunotherapy, Birmingham Medical School, Birmingham, United Kingdom
- 5Birmingham Cancer Centre, Queen Elizabeth Hospital, Birmingham, United Kingdom
The five-year survival rates for pancreatic ductal adenocarcinoma (PDAC) have scarcely improved over the last half-century. It is inherently resistant to FDA-approved immunotherapies, which have transformed the outlook for patients with other advanced solid tumours. Accumulating evidence relates this resistance to its hallmark immunosuppressive milieu, which instils progressive dysfunction among tumour-infiltrating effector T cells. This milieu is established at the inception of neoplasia by immunosuppressive cellular populations, including regulatory T cells (Tregs), which accumulate in parallel with the progression to malignant PDAC. Thus, the therapeutic manipulation of Tregs has captured significant scientific and commercial attention, bolstered by the discovery that an abundance of tumour-infiltrating Tregs correlates with a poor prognosis in PDAC patients. Herein, we propose a mechanism for the resistance of PDAC to anti-PD-1 and CTLA-4 immunotherapies and re-assess the rationale for pursuing Treg-targeted therapies in light of recent studies that profiled the immune landscape of patient-derived tumour samples. We evaluate strategies that are emerging to limit Treg-mediated immunosuppression for the treatment of PDAC, and signpost early-stage trials that provide preliminary evidence of clinical activity. In this context, we find a compelling argument for investment in the ongoing development of Treg-targeted immunotherapies for PDAC.
1 Introduction
Since 1863 – when Rudolf Virchow first observed leukocyte infiltrates decorating neoplastic tissues – research has uncovered a dynamic interplay between the immune system and pre-malignant cells, which governs their progressive transformation to invasive derivatives (1). In parallel, efforts to leverage the immune system to treat malignancy have a long history; in 1868, Wilhelm Busch reported tumour regression after intentionally infecting patients with Streptococcus pyogenes (2). Today, immunotherapy has revolutionised clinical oncology: immune checkpoint inhibitors (ICIs; specifically anti-PD-1, -CTLA-4, and -PD-L1 antibodies) provide unprecedented rates of durable anti-tumour responses in patients with several types of cancer (3). However, ICIs, including the combination of anti-CTLA-4 and anti-PD-L1 antibodies, have yielded limited responses in pancreatic ductal adenocarcinoma (PDAC); a malignancy of the exocrine pancreas that constitutes 95% of pancreatic cancer cases (4, 5). Accordingly, PDAC carries a bleak prognosis: globally, the 5-year survival rate at the time of diagnosis is 9% (6).
Substantial research has sought to identify immunological mechanisms that render PDAC resistant to ICIs. Concomitantly, these studies have unearthed therapeutic targets that could feasibly be exploited to induce anti-tumour immunity in PDAC; indeed, strategies to restrain immunosuppressive regulatory T cells (Tregs), myeloid cells, and cancer-associated fibroblasts are currently under development (7, 8). The manipulation of CD4+ Tregs has gained considerable traction, stemming from the discovery that an abundance of intratumoral Tregs correlates with a poor prognosis in PDAC patients (9). Herein, we propose a mechanism for the intrinsic resistance of PDAC to ICIs; discuss the rationale for pursuing Treg-targeted therapies in the context of PDAC; and evaluate emerging strategies to limit Treg-mediated immunosuppression. Overall, we argue that Treg-targeted immunotherapies offer a valuable opportunity to improve clinical outcomes in PDAC.
2 Why have ICIs proved ineffective in the context of PDAC?
Any effective immunotherapy must induce lasting anti-tumour immunity, typically mediated by CD4+ and CD8+ effector T (Teff) cells and directed against tumour-associated antigens acquired during malignant progression (10, 11). Researchers have sought to identify immunological mechanisms that render PDAC resistant to ICIs. Initial efforts utilised autochthonous murine models of PDAC: KrasLSL-G12D/+;Pdx-1-Cre (KC) and KrasLSL-G12D/+;Trp53LSL-R172H/+;Pdx-1-Cre (KPC), which recapitulate features of the human disease (12, 13). More recent analyses have profiled the immune landscape of patient-derived tumour samples, facilitated by advances in single-cell multi-omic technologies (14–16).
It is well established that the baseline density of tumour-infiltrating Teff cells is a critical determinant of therapeutic responses to ICIs (17, 18). Thus, the immunologically ‘cold’ phenotype that characterises PDAC has often been attributed to the physical exclusion of Teff cells from the tumour microenvironment (TME) (19, 20). However, recent studies have challenged this paradigm, identifying heterogenous baseline infiltrates of CD4+ and CD8+ Teff cells that correlate with prolonged overall survival in PDAC patients (14, 15, 21–26). There is also evidence for ongoing anti-tumour immunity; Freed-Pastor et al. identified a population of HLA-DR+Ki67+CD57-CD8+ T cells – indicative of an activated, proliferative phenotype – that are present in the majority of patients (27). Altogether, these studies suggest that inducing Teff cell-mediated anti-tumour immunity in PDAC may not be as intractable as is widely considered (23).
In further support of this notion, a rare subset (~1.6.%) of PDAC patients with hypermutated mismatch repair deficient (dMMR) tumours exhibit marked therapeutic responses to anti-PD-1 antibodies (28). These tumours present a broad repertoire of neoantigens, which direct potent anti-tumour immune responses (29, 30). Indeed, in this patient cohort, sequencing of the TCR Vβ chain revealed that 94% of intratumoral T cell clonotypes were unique to tumours, implying the existence of a neoantigen-specific immune response (24). Overall, this highlights the importance of neoantigens as a substrate for Teff-mediated anti-tumour immunity – indeed, on the basis of this principle, pembrolizumab and nivolumab (anti-PD-1) were granted FDA-approval in 2017 for the treatment of dMMR tumours, irrespective of their tissue of origin (31). In this context, it is notable that recent studies have challenged the claim that MMR-proficient PDAC harbours a limited repertoire of neoantigens. Freed-Pastor et al. investigated a cohort of 57 advanced PDAC patients and discovered that they all possessed neoepitopes with predicted ability to bind MHC class-I molecules (27). Accordingly, studies have consistently identified intratumoral neoantigen-reactive CD8+ T cells in PDAC patients, indicating that these neoantigens are capable of directing anti-tumour immunity (27, 32, 33).
Nevertheless, it is evident that this population of intratumoral neoantigen-reactive CD8+ T cells is not sufficient to drive therapeutic responses to FDA-approved ICIs in MMR-proficient PDAC. Indeed, multi-omic profiling of the PDAC immune landscape in resectable patients has revealed that ‘dysfunctional’ and ‘senescent’ phenotypes – both hypofunctional states, defined by the expression of multiple inhibitory receptors: TIGIT, LAG-3, TIM-3, and CD39 – dominate the intratumoral Teff cell repertoire, leaving few activated T cells that are thus unable to control the tumour (15, 25). In addition, a more pronounced exhaustion signature has been observed in CD8+ T cells from fine-needle biopsy samples of advanced, unresectable PDAC (14).
This progressive dysfunction of intratumoral Teff cells can be attributed to the profoundly immunosuppressive TME. It is established by the progressive infiltration of immunosuppressive cells: Tregs, myeloid-derived suppressor cells, neutrophils, and tumour-associated macrophages (34). In the murine KC model, these populations dominate the immune landscape of pancreatic intraepithelial neoplasia (PanIN): precursor lesions that culminate in the development of PDAC (19). Other non-immune cellular populations also contribute to the immunosuppressive TME. For example, a subset of cancer-associated fibroblasts present antigenic peptides in association with MHC class-II molecules; however, they lack expression of classical co-stimulatory molecules and thus command CD4+ T cells to the Treg lineage (35). In summary, neoantigen-specific Teff responses are dampened by the gradual accumulation of immunosuppressive cells in the TME, which dictates the progression from PanIN to PDAC. Hence, the development of immunomodulatory therapies for PDAC must focus on surmounting the hallmark immunosuppressive TME (36). Importantly, the progressive nature of intratumoral Teff cell dysfunction promises to confer a broad window during which such therapies might be effective.
3 What is the phenotype of Tregs in PDAC?
To date, strategies targeting myeloid-derived suppressor cells or cancer-associated fibroblasts for the treatment of PDAC have generally failed to demonstrate therapeutic promise in clinical trials (37–40). However, one promising strategy – which has gained substantial traction in the context of PDAC – is combatting Treg-mediated immunosuppression. This originated from the discovery that an abundance of intratumoral Tregs correlates with a poorer prognosis in PDAC patients (9). Accordingly, the depletion of Tregs has been shown to delay tumour growth in orthotopically transplanted murine PDAC, albeit with conflicting results from other murine models (41, 42). However, recent single-cell analyses have uncovered extensive diversity among intratumoral Tregs; in this context, it is important to re-evaluate the rationale for the development of Treg-targeted therapies.
3.1 Effector Tregs are highly immunosuppressive
Classically, CD4+ Tregs have been defined according to expression of FOXP3, considered a lineage-specifying transcription factor (TF), or the interleukin (IL)-2 receptor α chain (CD25). In a seminal study, Hiraoka et al. discovered that the prevalence of FOXP3+ Tregs increases during the progression from PanIN to advanced PDAC – at this latter stage, they constitute 35% (± 11%) of the total intratumoral CD4+ population (9, 15). Further, it is estimated that 54% (± 19%) of intratumoral Tregs are effector Tregs (eTregs; CD45RA-FOXP3hiCD25hi) (15). These cells express high levels of TIGIT, CTLA-4, ICOS, CD39, and HLA-DR, which are indicative of functional activation and potent immunosuppressive capacity (14, 15). This activated state has been attributed to sustained TCR stimulation, provided by the plethora of self- and quasi-self-antigens present in the inflammatory TME (43). However, a stable eTreg phenotype is also dependent on the expression of Helios, a member of the Ikaros TF family. Indeed, intratumoral Helios+ Tregs exhibit significantly higher expression of FOXP3, compared to Helios- Tregs (44).
Intratumoral eTregs potently suppress CD8+ T cell-mediated immunity via the expression of co-inhibitory molecules e.g., CTLA-4, which prevents the functional maturation of dendritic cells (41); secretion of immunosuppressive cytokines e.g., IL-10, IL-35, and TGF-β; sequestration of IL-2, which hampers IL-2-dependent T cell activation; and the secretion of granzymes to lyse target CD8+ cells (45). In support of their immunosuppressive capacity in situ, spatial analyses reveal that 90% of Tregs reside in close proximity to a CD8+ T cell in the PDAC TME (15).
3.2 FOXP3+RORγt+ Tregs provide mitogenic signalling
FOXP3+ Tregs exhibit extensive heterogeneity in PDAC. Strikingly, studies have discovered populations of FOXP3+ Tregs that, in addition to IL-10, secrete high levels of pro-inflammatory cytokines. For example, Chellappa et al. identified Tregs that co-express FOXP3 and RORγt: a factor that specifies the type-17 T-helper cell lineage (TH17) (46). These cells retained markers associated with FOXP3+ Tregs e.g., CTLA-4, CD39, and ICOS, indicating an ability to robustly suppress anti-tumour immunity. However, through the simultaneous production of IL-17, these FOXP3+RORγt+ cells provide mitogenic signalling to transformed pancreatic epithelial cells, which upregulate the IL-17 receptor (47). Moreover, studies have identified populations of FOXP3- Treg-like cells that share expression of molecules classically associated with immunosuppressive Treg functions (e.g., IL-10, CCR8, TIGIT, ICOS, CTLA-4) (48, 49). Barilla et al. demonstrated that the gene expression profile of one such population, termed Tr1 cells (CD49b, CD73, and AHR), was associated with decreased overall survival in PDAC patients (49). Furthermore, Whiteside et al. suggest that intratumoral Teff cells may adopt this FOXP3- Treg-like phenotype following the ablation of FOXP3+ cells (48).
This profound heterogeneity likely explains conflicting reports regarding the overall contribution of Tregs in the pathophysiology of PDAC. One notable study reported an increased prevalence of Tregs in tumours of long-term PDAC survivors (24). Moreover, depletion of Tregs prior to the development of PanIN in KC mice has been shown to accelerate malignant progression (42). Conceivably, the use of different experimental systems, including varied methods for detecting and defining intratumoral Tregs, might accentuate specific Treg-associated functions and thereby explain these conflicting reports. Moreover, studies have suggested that, as part of normal immune homeostasis, intratumoral Tregs accompany CD8+ T cell infiltrates (21, 42, 49), which may further obscure any relationship between the prevalence of intratumoral Tregs and a poor prognosis. Nevertheless, harnessing the therapeutic manipulation of Tregs will require a targeted approach, based on a detailed understanding of the heterogeneous functions ascribed to Tregs, and their spatiotemporal dynamics in the PDAC TME (Figure 1). In addition, such an approach will reduce the systemic side-effects associated with Treg-targeted immunotherapies.
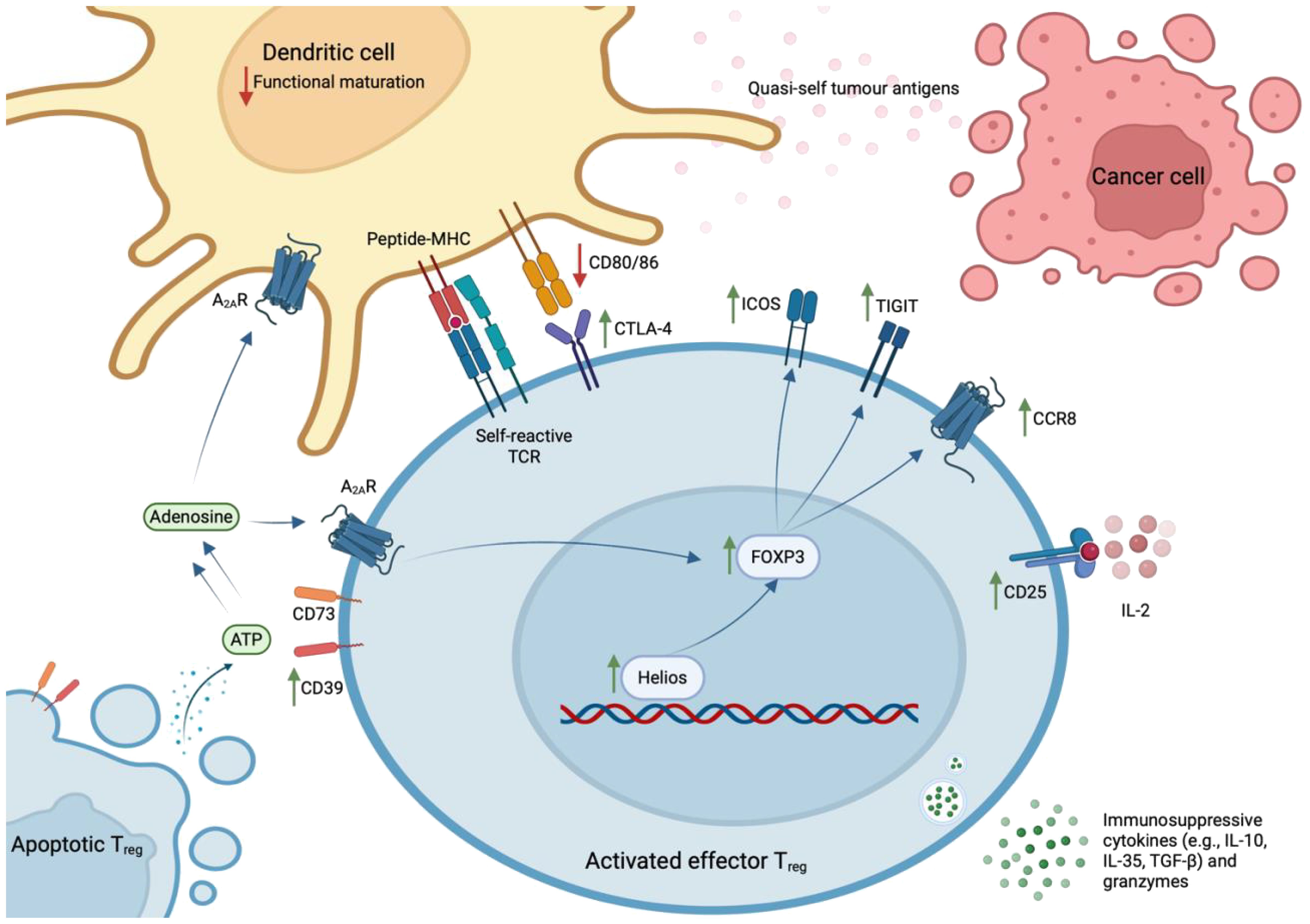
Figure 1 Phenotype of effector Treg cells in human PDAC. Effector Tregs – characterised by the expression of FOXP3, CD25, TIGIT, CTLA-4, ICOS, CD39, and CCR8 – are activated by sustained TCR stimulation with abundant self- and quasi-self-antigens and stabilised by expression of the Helios transcription factor. These cells exhibit potent immunosuppressive capacity within the PDAC TME, where they exist in close proximity to CD8+ T lymphocytes. Specifically, they express co-inhibitory molecules (e.g., CTLA-4, TIGIT, ICOS); convert ATP to immunosuppressive adenosine via ectoenzymes that remain catalytically active after cell-death (CD39 and CD73); secrete immunosuppressive cytokines (e.g., IL-10, IL-35, TGF-β) and granzymes that lyse CD8+ Teff cells; and sequester IL-2 that is required for Teff cell activation.
3.3 Apoptotic Tregs are paradoxically immunosuppressive
This hypothesis is fortified by the discovery that apoptotic Tregs, defined by increased expression of Ki67 and cleaved caspase-3, exert immunosuppressive effects in the oxidative TME. They release large quantities of ATP, which is converted into adenosine via CD39 and CD73 – ectoenzymes that are expressed by Tregs and remain catalytically active after cell-death (50). Through the A2A receptor, accumulating extracellular adenosine inhibits Teff cell proliferation; induces immunosuppressive dendritic cells; and stabilises surviving Tregs (51). Thus, CD39 and CD73 expression correlate with a poor prognosis in patients with various solid tumours (52, 53). Importantly, this paracrine signalling pathway is likely to be operating in human PDAC, as intratumoral Tregs express high levels of CD39.
4 What are the strategies to manipulate Tregs for the treatment of PDAC?
The manipulation of Tregs has captured significant attention from both scientific and commercial communities as a novel approach to the treatment of PDAC. The earliest attempts depleted Tregs by targeting CD25 with antibodies, daclizumab, or the IL-2-diphtheria toxin fusion protein, ONTAK (54, 55). However, IL-2 signalling via CD25 promotes the survival of activated Teff cells, conferring a limited therapeutic window to CD25-targeted interventions. Nevertheless, these efforts provided proof-of-concept for the therapeutic manipulation of Tregs. Today, numerous Treg-targeted therapies are under development for the treatment of advanced solid tumours, including PDAC (Table 1).
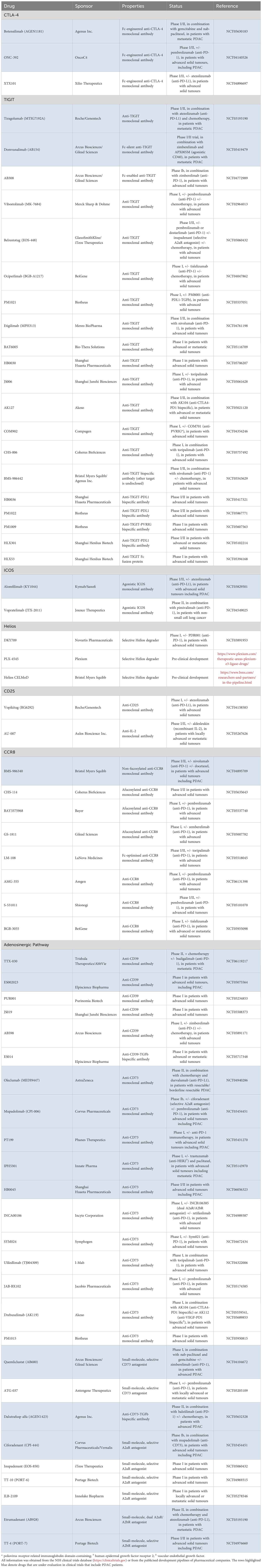
Table 1 Treg-targeted immunotherapies in current development (as of 01/05/2024).
4.1 Re-engineering next-generation ICIs
Allison and colleagues originally attributed the anti-tumour activity of anti-CTLA-4 monoclonal antibodies (mAbs) to the reinvigoration of dysfunctional Teff cells (56). However, accumulating evidence suggests that anti-CTLA-4 mAbs can preferentially deplete CTLA-4hi Tregs in vivo by antibody-dependent cellular cytotoxicity (ADCC) (57–60). Thus, in spite of the failure of prior clinical trials (4, 61), this novel mechanistic insight provides a rationale for the continued development of anti-CTLA-4 mAbs to treat PDAC. Clearly, however, this will necessitate re-engineering of existing anti-CTLA-4 mAbs; specifically, the fragment crystallisable (Fc) domain to enhance affinity for activatory Fcγ receptors and decrease affinity for inhibitory receptors, thereby promoting ADCC. This approach can be optimised with consideration of the relative abundance and distribution of specific FcγRs on local effector cells; indeed, the engineering of anti-CTLA-4 mAbs in this manner has been shown to increase therapeutic activity in tumour-bearing mice (59, 62). Therefore, it is important that studies have identified intratumoral populations of FcγRIIIA (CD16)-expressing natural killer and myeloid cells in human PDAC (14–16). Moreover, Agenus recently initiated a phase I/II trial to investigate botensilimab – an Fc-engineered anti-CTLA-4 mAb with enhanced affinity for FcγRIIIA – in metastatic PDAC patients (NCT05630183).
Further testament to the widespread interest in strategies to selectively deplete intratumoral Tregs, there is renewed attention on the development of anti-CD25 mAbs. For example, Solomon et al. developed an anti-CD25 mAb (RG6292) that selectively depletes CD25hi Tregs, whilst preserving CD25-STAT5 signalling required for Teff cell-mediated anti-tumour immunity (63). Indeed, a phase I trial of RG6292, conducted in patients with advanced/metastatic solid tumours, indicated a manageable safety profile and preliminary anti-tumour activity (64). However, multi-omic analysis of patient-derived tumour samples obtained during treatment with RG6292 is required to confirm this proposed mechanism of action in vivo.
4.2 Exploiting novel immune checkpoints
Since the discovery of CTLA-4 and PD-1, studies have identified a plethora of immune checkpoints – both inhibitory (e.g., TIGIT, LAG-3, TIM-3) and co-stimulatory (e.g., ICOS, OX40, GITR, 4–1BB) – that might be exploited therapeutically to augment anti-tumour immunity. In PDAC, TIGIT and ICOS are expressed at high levels on intratumoral eTregs (14, 15). TIGIT is also expressed, albeit at lower levels, by dysfunctional Teff cells, whereas ICOS is induced upon the activation of intratumoral Teff cells (14, 15, 27). Therefore, anti-TIGIT and agonistic ICOS mAbs might have a dual mechanism of action: the re-invigoration of dysfunctional Teff cells and selective depletion of activated Tregs (65). However, achieving the optimal balance between these mechanisms will require Fc engineering to effectively engage specific Fc receptors (66).
Tiragolumab (IgG1κ anti-TIGIT) has demonstrated tolerability and preliminary anti-tumour activity in patients with advanced solid tumours (67, 68). Consequently, two early-stage trials are investigating anti-TIGIT mAbs, incorporated into combinatorial regimens, for the treatment of metastatic PDAC (NCT03193190, NCT05419479). By contrast, a phase I/II trial, investigating vopratelimab (IgG1κ agonistic ICOS) for the treatment of advanced solid tumours, including three PDAC patients, reported limited efficacy (69). However, on-treatment emergence of ICOShi CD4+ Teff cells was associated with therapeutic responses, suggesting that vopratelimab might indeed re-invigorate dysfunctional Teff cells in patients through ICOS activation. More generally, this illustrates that multi-omic analyses of on-treatment patient-derived samples during clinical trials may further advance our understanding of the PDAC immune landscape.
4.3 De-stabilising activated Tregs
The development of strategies for selectively drugging Tregs has been the subject of considerable research. One potential target is Helios; in PDAC patients, Helios+ Tregs are significantly enriched in the TME (70). Moreover, Treg-intrinsic deletion of Helios has been shown to enhance anti-tumour immunity in tumour-bearing mice (71). Interestingly, Helios-deficient Tregs acquire a Teff phenotype including the production of pro-inflammatory cytokines (e.g., IFN-γ), which is attributed to downregulation of FOXP3 and de-repression of TH1/TH2 lineage determinants (43). In the absence of the stabilising influence of Helios, it appears that the inflammatory TME promotes the trans-differentiation of Tregs into activated Teff cells. Intriguingly, this novel Teff population is equipped with an inherently self-reactive TCR repertoire, which might be expected to direct a potent immune response against ‘quasi-self’ tumour antigens.
Transcription factors are traditionally considered difficult to drug. However, several recent studies have described small-molecules that selectively enhance the proteasomal degradation of Helios (72, 73). Future in vivo studies must determine whether these small-molecules can selectively destabilise activated intratumoral eTregs; one clinical trial is currently evaluating this approach in advanced solid tumours (NCT03891953).
4.4 Targeting chemokine receptors
The origin of intratumoral FOXP3+ Tregs is unclear – they may differentiate locally from Teff cells or be recruited from the circulation. For the latter, targeting chemokine signalling axes (e.g., CCL2-CCR4; CCL5-CCR5) that can recruit Tregs into the PDAC TME is of interest. However, this strategy has proved disappointing thus far; clinical trials investigating mogamulizumab (IgG1 anti-CCR4) reported off-target depletion of TH2/TH17 cells, reflecting heterogeneous expression of CCR4 (74, 75).
It is notable, therefore, that intratumoral eTregs uniquely express CCR8 (76). However, functional blockade of CCR8 does not affect Treg recruitment; they acquire CCR8 expression in the TME, perhaps suggesting that this axis mediates retention of intratumoral Tregs (77). Nevertheless, CCR8 constitutes a target for the selective depletion of intratumoral eTregs in PDAC. Pre-clinical studies have demonstrated that anti-CCR8 mAbs profoundly suppress tumour growth in tumour-bearing mice (76, 78). Further, this response coincided with the expansion of intratumoral CD4+ Teff cells and the preservation of systemic Treg populations, which may mitigate the risk of autoimmune-related adverse events. Currently, eight early-stage trials are investigating anti-CCR8 mAbs for the treatment of advanced solid tumours (NCT04895709, NCT06131398, NCT05635643, NCT05537740, NCT05007782, NCT05518045, NCT05101070, NCT05935098).
4.5 Combatting immunosuppressive adenosine
Apoptotic Tregs convert ATP to adenosine, an immunosuppressive metabolite, via ectoenzymes that remain catalytically active after cell-death. This raises the paradoxical possibility that the therapeutic depletion of Tregs might not limit Treg-cell-mediated immunosuppression. This discovery provided impetus to the development of immunotherapies that target the adenosinergic pathway: CD39, CD73, and the A2A/A2B receptors. It is hoped that these therapies will synergise with Treg-targeted approaches, or other immunotherapeutic modalities, to induce potent anti-tumour immunity. To date, however, attempts to target this pathway with anti-CD73 mAbs have demonstrated no clinical benefit for PDAC patients; a phase-II trial investigating the combination of anti-CD73, anti-PD-L1, and chemotherapy revealed comparable efficacy to chemotherapy alone (79).
5 Conclusions and future perspectives
The manipulation of intratumoral Tregs may prove a valuable addition to our currently limited armamentarium for the treatment of PDAC. This therapeutic strategy has the potential to re-invigorate anti-tumour immunity by reprogramming the immunosuppressive milieu that is first established in pre-malignant lesions. This notion is supported by promising early-stage clinical trials of Treg-targeted immunotherapies (68, 80). Moreover, data from trials investigating anti-CCR8 mAbs and selective Helios degraders, strategies to selectively target intratumoral effector Tregs, are eagerly awaited.
There are several outstanding questions, however, which threaten to hinder the effective therapeutic manipulation of intratumoral Tregs:
1. Given that intratumoral Tregs are present from early carcinogenesis to the development of metastatic disease, are Treg-targeted therapies effective in cohorts of patients from the full spectrum of the natural history of PDAC?
2. With novel Treg-targeted interventions, is there on-treatment emergence of immunosuppressive FOXP3- Treg-like cells (e.g., Tr1 cells) or other complementary immunosuppressive mechanisms?
3. How can we prevent immune-related adverse events, which so often necessitate treatment discontinuation, when targeting Tregs for the treatment of PDAC?
4. To what extent do Treg-targeted therapies synergise with anti-cancer agents from our existing repertoire, including immunotherapies and conventional chemotherapies?
Importantly, with preliminary clinical evidence for the efficacy of Treg-targeted therapies, there is a compelling argument for the allocation of resources to resolve these outstanding questions.
Author contributions
SS: Writing – original draft, Writing – review & editing. HS: Writing – original draft, Writing – review & editing. EA-B: Writing – original draft, Writing – review & editing. EA: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
SS has had a personal fellowship and funding from Bristol Myers Squibb. He has received payments for consultancy, speaker fees or attendance at meetings by Astrazeneca, Servier, Novartis and Momo biotech.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
2. Busch W. Aus der Sitzung der medicinischen Section vom 13 November 1867. Berl Klin Wochenschr. (1868) 5:137.
3. SChadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol. (2015) 33:1889–94. doi: 10.1200/JCO.2014.56.2736
4. O’Reilly EM, Oh D-Y, Dhani N, Renouf DJ, Lee MA, Sun W, et al. Durvalumab with or without tremelimumab for patients with metastatic pancreatic ductal adenocarcinoma: A phase 2 randomized clinical trial. JAMA Oncol. (2010) 5:1431–8. doi: 10.1001/jamaoncol.2019.1588
5. Mizrahi JD, Surana R, Valle JW, Shroff RT. Pancreatic cancer. Lancet. (2020) 395:2008–20. doi: 10.1016/S0140–6736(20)30974–0
6. International Agency for Research on Cancer. World Cancer Report 2020. Lyon, France: IARC Publications (2020). 367 p.
7. Bockorny B, Macarulla T, Semenisty V, Borazanci E, Feliu J, Ponz-Sarvise M, et al. Motixafortide and pembrolizumab combined to nanoliposomal irinotecan, fluorouracil, and folinic acid in metastatic pancreatic cancer: the COMBAT/KEYNOTE-202 trial. Clin Cancer Res. (2021) 27:5020–7. doi: 10.1158/1078–0432.CCR-21–0929
8. Bockorny B, Semenisty V, Macarulla T, Borazanci E, Wolpin BM, Stemmer SM, et al. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: the COMBAT trial. Nat Med. (2020) 26:878–85. doi: 10.1038/s41591–020-0880-x
9. Hiraoka N, Onozato K, Kosuge T, Hirohashi S. Prevalence of FOXP3+ Regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin Cancer Res. (2006) 12:5423–34. doi: 10.1158/1078–0432.CCR-06–0369
10. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. (2014) 515:568–71. doi: 10.1038/nature13954
11. Alspach E, Lussier DM, Miceli AP, Kizhvatov I, DuPage M, Luoma AM, et al. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature. (2019) 574:696–701. doi: 10.1038/s41586–019-1671–8
12. Hingorani SR, Petricoin EF III, Maitra A, Rajapakse V, King C, Jacobetz MA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. (2003) 4:437–50. doi: 10.1016/S1535–6108(03)00309-X
13. Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. (2005) 7:469–83. doi: 10.1016/j.ccr.2005.04.023
14. Steele NG, Carpenter ES, Kemp SB, Sirihorachai VR, The S, Delrosario L, et al. Multimodal mapping of the tumor and peripheral blood immune landscape in human pancreatic cancer. Nat Cancer. (2020) 1:1097–112. doi: 10.1038/s43018–020-00121–4
15. Sivakumar S, Abu-Shah E, Ahern DJ, Arbe-Barnes EH, Jainarayanan AK, Mangal N, et al. Activated regulatory T-cells, dysfunctional and senescent t-cells hinder the immunity in pancreatic cancer. Cancers (Basel). (2021) 13:1776. doi: 10.3390/cancers13081776
16. Sivakumar S, Jainarayanan A, Arbe-Barnes E, Sharma PK, Leathlobhair MN, Amin S, et al. Single-cell immune multi-omics and repertoire analyses in pancreatic ductal adenocarcinoma reveal differential immunosuppressive mechanisms within different tumour microenvironments. bioRxiv. (2023) 08.31.555730. doi: 10.1101/2023.08.31.555730
17. Chen P-L, Roh W, Reuben A, Cooper ZA, Spencer CN, Prieto PA, et al. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discovery. (2016) 6:827–37. doi: 10.1158/2159–8290.CD-15–1545
18. Spranger S, Koblish HK, Horton B, Scherle PA, Newton R, Gajewski TF. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8+ T cells directly within the tumor microenvironment. J Immunother Cancer. (2014) 2:3. doi: 10.1186/2051–1426-2–3
19. Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. (2007) 67:9518–27. doi: 10.1158/0008–5472.CAN-07–0175
20. Bear AS, Vonderheide RH, O’Hara MH. Challenges and opportunities for pancreatic cancer immunotherapy. Cancer Cell. (2020) 38:788–802. doi: 10.1016/j.ccell.2020.08.004
21. Carstens JL, Correa de Sampaio P, Yang D, Barua S, Wang H, Rao A, et al. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat Commun. (2017) 8:15095. doi: 10.1038/ncomms15095
22. Ene–Obong A, Clear AJ, Watt J, Wang J, Fatah R, Riches JC, et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology. (2013) 145:1121–32. doi: 10.1053/j.gastro.2013.07.025
23. Balli D, Rech AJ, Stanger BZ, Vonderheide RH. Immune cytolytic activity stratifies molecular subsets of human pancreatic cancer. Clin Cancer Res. (2017) 23:3129–38. doi: 10.1158/1078–0432.CCR-16–2128
24. Balachandran VP, Łuksza M, Zhao JN, Makarov V, Moral JA, Remark R, et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature. (2017) 551:512–6. doi: 10.1038/nature24462
25. Jainarayanan A, Mouroug-Anand N, Arbe-Barnes EH, Bush AJ, Bashford-Rogers R, Frampton A, et al. Pseudotime dynamics of T cells in pancreatic ductal adenocarcinoma inform distinct functional states within the regulatory and cytotoxic T cells. iScience. (2023) 26:106324. doi: 10.1016/j.isci.2023.106324
26. de Santiago I, Yau C, Heij L, Middleton MR, Markowetz F, Grabsch HI, et al. Immunophenotypes of pancreatic ductal adenocarcinoma: Meta-analysis of transcriptional subtypes. Int J Cancer. (2019) 145:1125–37. doi: 10.1002/ijc.32186
27. Freed-Pastor WA, Lambert LJ, Ely ZA, Pattada NB, Bhutkar A, Eng G, et al. The CD155/TIGIT axis promotes and maintains immune evasion in neoantigen-expressing pancreatic cancer. Cancer Cell. (2021) 39:1342–1360.e14. doi: 10.1016/j.ccell.2021.07.007
28. Lupinacci RM, Goloudina A, Buhard O, Bachet J-B, Maréchal R, Demetter P, et al. Prevalence of microsatellite instability in intraductal papillary mucinous neoplasms of the pancreas. Gastroenterology. (2018) 154:1061–5. doi: 10.1053/j.gastro.2017.11.009
29. McGranahan N, Furness AJS, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. (2016) 351:1463–9. doi: 10.1126/science.aaf1490
30. Li B, Li T, Pignon J-C, Wang B, Wang J, Shukla SA, et al. Landscape of tumor-infiltrating T cell repertoire of human cancers. Nat Genet. (2016) 48:725–32. doi: 10.1038/ng.3581
31. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. (2017) 357:409–13. doi: 10.1126/science.aan6733
32. Gros A, Tran E, Parkhurst MR, Ilyas S, Pasetto A, Groh EM, et al. Recognition of human gastrointestinal cancer neoantigens by circulating PD-1+ lymphocytes. J Clin Invest. (2019) 129:4992–5004. doi: 10.1172/JCI127967
33. Parkhurst MR, Robbins PF, Tran E, Prickett TD, Gartner JJ, Jia L, et al. Unique neoantigens arise from somatic mutations in patients with gastrointestinal cancers. Cancer Discovery. (2019) 9:1022–35. doi: 10.1158/2159–8290.CD-18–1494
34. Huber M, Brehm CU, Gress TM, Buchholz M, Alhamwe BA, von Strandmann EP, et al. The immune microenvironment in pancreatic cancer. Int J Mol Sci. (2020) 21:7307. doi: 10.3390/ijms21197307
35. Huang H, Wang Z, Zhang Y, Pradhan RN, Ganguly D, Chandra R, et al. Mesothelial cell-derived antigen-presenting cancer-associated fibroblasts induce expansion of regulatory T cells in pancreatic cancer. Cancer Cell. (2022) 40:656–73. doi: 10.1016/j.ccell.2022.04.011
36. Leinwand J, Miller G. Regulation and modulation of antitumor immunity in pancreatic cancer. Nat Immunol. (2020) 21:1152–9. doi: 10.1038/s41590–020-0761-y
37. Padrón LJ, Maurer DM, O’Hara MH, O’Reilly EM, Wolff RA, Wainberg ZA, et al. Sotigalimab and/or nivolumab with chemotherapy in first-line metastatic pancreatic cancer: clinical and immunologic analyses from the randomized phase 2 PRINCE trial. Nat Med. (2022) 28:1167–77. doi: 10.1038/s41591–022-01829–9
38. Kim EJ, Sahai V, Abel EV, Griffith KA, Greenson JK, Takebe N, et al. Pilot clinical trial of hedgehog pathway inhibitor GDC-0449 (vismodegib) in combination with gemcitabine in patients with metastatic pancreatic adenocarcinoma. Clin Cancer Res. (2014) 20:5937–45. doi: 10.1158/1078–0432.CCR-14–1269
39. Benson AB, Wainberg ZA, Hecht JR, Vyushkov D, Dong H, Bendell J, et al. A phase II randomized, double-blind, placebo-controlled study of simtuzumab or placebo in combination with gemcitabine for the first-line treatment of pancreatic adenocarcinoma. Oncologist. (2017) 22:241–e15. doi: 10.1634/theoncologist.2017–0024
40. Zhen DB, Whittle M, Ritch PS, Hochster HS, Coveler AL, George B, et al. Phase II study of PEGPH20 plus pembrolizumab for patients (pts) with hyaluronan (HA)-high refractory metastatic pancreatic adenocarcinoma (mPC). J Clin Oncol. (2022) 40:576. doi: 10.1200/JCO.2022.40.4-suppl.576
41. Jang J-E, Hajdu CH, Liot C, Miller G, Dustin ML, Bar-Sagi D. Crosstalk between regulatory T cells and tumor-associated dendritic cells negates anti-tumor immunity in pancreatic cancer. Cell Rep. (2017) 20:558–71. doi: 10.1016/j.celrep.2017.06.062
42. Zhang Y, Lazarus J, Steele NG, Yan W, Lee H-J, Nwosu ZC, et al. Regulatory T-cell depletion alters the tumor microenvironment and accelerates pancreatic carcinogenesis. Cancer Discovery. (2020) 10:422–39. doi: 10.1158/2159–8290.CD-19–0958
43. Yates K, Bi K, Haining WN, Cantor H, Kim H-J. Comparative transcriptome analysis reveals distinct genetic modules associated with Helios expression in intratumoral regulatory T cells. Proc Natl Acad Sci USA. (2018) 115:2162–7. doi: 10.1073/pnas.1720447115
44. Kim H-J, Barnitz RA, Kreslavsky T, Brown FD, Moffett H, Lemieux ME, et al. Stable inhibitory activity of regulatory T cells requires the transcription factor Helios. Science. (2015) 350:334–9. doi: 10.1126/science.aad0616
45. Sharabi A, Tsokos MG, Ding Y, Malek TR, Klatzmann D, Tsokos GC. Regulatory T cells in the treatment of disease. Nat Rev Drug Discovery. (2018) 17:823–44. doi: 10.1038/nrd.2018.148
46. Chellappa S, Hugenschmidt H, Hagness M, Line PD, Labori KJ, Wiedswang G, et al. Regulatory T cells that co-express RORγt and FOXP3 are pro-inflammatory and immunosuppressive and expand in human pancreatic cancer. Oncoimmunology. (2016) 5:e1102828. doi: 10.1080/2162402X.2015.1102828
47. McAllister F, Bailey JM, Alsina J, Nirschl CJ, Sharma R, Fan H, et al. Oncogenic kras activates a hematopoietic-to-epithelial IL-17 signaling axis in preinvasive pancreatic neoplasia. Cancer Cell. (2014) 25:621–37. doi: 10.1016/j.ccr.2014.03.014
48. Barilla RM, Diskin B, Caso RC, Lee KB, Mohan N, Buttar C, et al. Specialized dendritic cells induce tumor-promoting IL-10+IL-17+ FoxP3neg regulatory CD4+ T cells in pancreatic carcinoma. Nat Commun. (2019) 10:1424. doi: 10.1038/s41467–019-09416–2
49. Whiteside SK, Grant FM, Alvisi G, Clarke J, Tang L, Imianowski CJ, et al. Acquisition of suppressive function by conventional T cells limits antitumor immunity upon Treg depletion. Sci Immunol. (2023) 8:eabo5558. doi: 10.1126/sciimmunol.abo5558
50. Maj T, Wang W, Crespo J, Zhang H, Wang W, Wei S, et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat Immunol. (2017) 18:1332–41. doi: 10.1038/ni.3868
51. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. (2007) 204:1257–65. doi: 10.1084/jem.20062512
52. Loi S, Pommey S, Haibe-Kains B, Beavis PA, Darcy PK, Smyth MJ, et al. CD73 promotes anthracycline resistance and poor prognosis in triple negative breast cancer. Proc Natl Acad Sci USA. (2013) 110:11091–6. doi: 10.1073/pnas.1222251110
53. Wu X-R, He X-S, Chen Y-F, Yuan R-X, Zeng Y, Lian L, et al. High expression of CD73 as a poor prognostic biomarker in human colorectal cancer. J Surg Oncol. (2012) 106:130–7. doi: 10.1002/jso.23056
54. Onizuka S, Tawara I, Shimizu J, Sakaguchi S, Fujita T, Nakayama E. Tumor rejection by in vivo administration of anti-CD25 (Interleukin-2 receptor α) monoclonal antibody. Cancer Res. (1999) 59:3128–33.
55. Litzinger MT, Fernando R, Curiel TJ, Grosenbach DW, Schlom J, Palena C. IL-2 immunotoxin denileukin diftitox reduces regulatory T cells and enhances vaccine-mediated T-cell immunity. Blood. (2007) 110:3192–201. doi: 10.1182/blood-2007–06-094615
56. Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. (1995) 182:459–65. doi: 10.1084/jem.182.2.459
57. Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti–CTLA-4 therapy against melanoma. J Exp Med. (2013) 210:1695–710. doi: 10.1084/jem.20130579
58. Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, et al. Anti-CTLA-4 antibodies of igG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res. (2013) 1:32–42. doi: 10.1158/2326–6066.CIR-13–0013
59. Arce Vargas F, Furness AJS, Litchfield K, Joshi K, Rosenthal R, Ghorani E, et al. Fc effector function contributes to the activity of human anti-CTLA-4 antibodies. Cancer Cell. (2018) 33:649–63. doi: 10.1016/j.ccell.2018.02.010
60. Romano E, Kusio-Kobialka M, Foukas PG, Baumgaertner P, Meyer C, Ballabeni P, et al. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc Natl Acad Sci USA. (2015) 112:6140–5. doi: 10.1073/pnas.1417320112
61. Royal RE, Levy C, Turner K, Mathur A, Hughes M, Kammula US, et al. Phase 2 trial of single agent ipilimumab (Anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother. (2010) 33:828–33. doi: 10.1097/CJI.0b013e3181eec14c
62. Knorr DA, Blanchard L, Leidner RS, Jensen SM, Meng R, Jones A, et al. FcγRIIB is an immune checkpoint limiting the activity of treg-targeting antibodies in the tumor microenvironment. Cancer Immunol Res. (2024) 12:322–33. doi: 10.1158/2326–6066.CIR-23–0389
63. Solomon I, Amann M, Goubier A, Arce Vargas F, Zervas D, Qing C, et al. CD25-Treg-depleting antibodies preserving IL-2 signaling on effector T cells enhance effector activation and antitumor immunity. Nat Cancer. (2020) 1:1153–66. doi: 10.1038/s43018–020-00133–0
64. Gambardella V, Ong M, Ruiz MER, Machiels J-P, de Sanmamed MF, Tabernero JM, et al. Abstract CT110: Safety and anti-tumor activity of a novel Treg depleter RG6292, as a single agent and in combination with atezolizumab in patients with solid tumors. Cancer Res. (2023) 83:CT110. doi: 10.1158/1538–7445.am2023-ct110
65. Sainson RCA, Thotakura AK, Kosmac M, Borhis G, Parveen N, Kimber R, et al. An antibody targeting ICOS increases intratumoral cytotoxic to regulatory T-cell ratio and induces tumor regression. Cancer Immunol Res. (2020) 8:1568–82. doi: 10.1158/2326–6066.CIR-20–0034
66. Kraehenbuehl L, Weng C-H, Eghbali S, Wolchok JD, Merghoub T. Enhancing immunotherapy in cancer by targeting emerging immunomodulatory pathways. Nat Rev Clin Oncol. (2022) 19:37–50. doi: 10.1038/s41571–021-00552–7
67. Bendell JC, Bedard P, Bang Y-J, LoRusso P, Hodi S, Gordon M, et al. Abstract CT302: Phase Ia/Ib dose-escalation study of the anti-TIGIT antibody tiragolumab as a single agent and in combination with atezolizumab in patients with advanced solid tumors. Cancer Res. (2020) 80:302. doi: 10.1158/1538–7445.AM2020-CT302
68. Cho BC, Abreu DR, Hussein M, Cobo M, Patel AJ, Secen N, et al. Tiragolumab plus atezolizumab versus placebo plus atezolizumab as a first-line treatment for PD-L1-selected non-small-cell lung cancer (CITYSCAPE): primary and follow-up analyses of a randomised, double-blind, phase 2 study. Lancet Oncol. (2022) 23:781–92. doi: 10.1016/S1470–2045(22)00226–1
69. Yap TA, Gainor JF, Callahan MK, Falchook GS, Pachynski RK, LoRusso P, et al. First-in-human phase I/II ICONIC trial of the ICOS agonist vopratelimab alone and with nivolumab: ICOS-high CD4 T-cell populations and predictors of response. Clin Cancer Res. (2022) 28:3695–708. doi: 10.1158/1078–0432.CCR-21–4256
70. Liu M-F, Jin C, Wu T, Chen E-H, Lu M, Qin H-L. Helios serves as a suppression marker to reduce regulatory T cell function in pancreatic cancer patients. Immunol Res. (2021) 69:275–84. doi: 10.1007/s12026–021-09200–9
71. Nakagawa H, Sido JM, Reyes EE, Kiers V, Cantor H, Kim H-J. Instability of Helios-deficient Tregs is associated with conversion to a T-effector phenotype and enhanced antitumor immunity. Proc Natl Acad Sci USA. (2016) 113:6248–53. doi: 10.1073/pnas.1604765113
72. Wang ES, Verano AL, Nowak RP, Yuan JC, Donovan KA, Eleuteri NA, et al. Acute pharmacological degradation of Helios destabilizes regulatory T cells. Nat Chem Biol. (2021) 17:711–7. doi: 10.1038/s41589–021-00802-w
73. Bonazzi S, d’Hennezel E, Beckwith REJ, Xu L, Fazal A, Magracheva A, et al. Discovery and characterization of a selective IKZF2 glue degrader for cancer immunotherapy. Cell Chem Biol. (2023) 30:235–247.e12. doi: 10.1016/j.chembiol.2023.02.005
74. Kurose K, Ohue Y, Wada H, Iida S, Ishida T, Kojima T, et al. Phase ia study of foxP3+ CD4 treg depletion by infusion of a humanized anti-CCR4 antibody, KW-0761, in cancer patients. Clin Cancer Res. (2015) 21:4327–36. doi: 10.1158/1078–0432.CCR-15–0357
75. Doi T, Muro K, Ishii H, Kato T, Tsushima T, Takenoyama M, et al. A phase I study of the anti-CC chemokine receptor 4 antibody, mogamulizumab, in combination with nivolumab in patients with advanced or metastatic solid tumors. Clin Cancer Res. (2019) 25:6614–22. doi: 10.1158/1078–0432.CCR-19–1090
76. Kidani Y, Nogami W, Yasumizu Y, Kawashima A, Tanaka A, Sonoda Y, et al. CCR8-targeted specific depletion of clonally expanded Treg cells in tumor tissues evokes potent tumor immunity with long-lasting memory. Proc Natl Acad Sci USA. (2022) 119:e2114282119. doi: 10.1073/pnas.2114282119
77. Whiteside SK, Grant FM, Gyori DS, Conti AG, Imianowski CJ, Kuo P, et al. CCR8 marks highly suppressive Treg cells within tumours but is dispensable for their accumulation and suppressive function. Immunology. (2021) 163:512–20. doi: 10.1111/imm.13337
78. Campbell JR, McDonald BR, Mesko PB, Siemers NO, Singh PB, Selby M, et al. Fc-optimized anti-CCR8 antibody depletes regulatory T cells in human tumor models. Cancer Res. (2021) 81:2983–94. doi: 10.1158/0008–5472.CAN-20–3585
79. Coveler AL, Reilley M, Zalupski M, Macarulla T, Fountzilas C, Castanon Alvarez E, et al. Safety and clinical activity of oleclumab (O) ± durvalumab (D) + chemotherapy (CT) in patients (pts) with metastatic pancreatic ductal adenocarcinoma (mPDAC): A phase 1b/2 randomized study. J Clin Oncol. (2023) 41:4136. doi: 10.1200/JCO.2023.41.16_suppl.4136
80. Manji GA, Wainberg ZA, Krishnan K, Giafis N, Udyavar A, Quah CS, et al. ARC-8: Phase I/Ib study to evaluate safety and tolerability of AB680 + chemotherapy + zimberelimab (AB122) in patients with treatment-naive metastatic pancreatic adenocarcinoma (mPDAC). J Clin Oncol. (2021) 39:404. doi: 10.1200/JCO.2021.39.3_suppl.404
Keywords: immunotherapy, regulatory T cells, pancreatic ductal adenocarcinoma, TIGIT, CCR8, Helios, adenosine
Citation: Smith H, Arbe-Barnes E, Shah EA and Sivakumar S (2024) Manipulating regulatory T cells: is it the key to unlocking effective immunotherapy for pancreatic ductal adenocarcinoma? Front. Immunol. 15:1406250. doi: 10.3389/fimmu.2024.1406250
Received: 24 March 2024; Accepted: 14 May 2024;
Published: 30 May 2024.
Edited by:
Don J. Diamond, City of Hope National Medical Center, United StatesReviewed by:
Yvonne Samstag, Heidelberg University Hospital, GermanyCopyright © 2024 Smith, Arbe-Barnes, Shah and Sivakumar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shivan Sivakumar, cy5zaXZha3VtYXJAYmhhbS5hYy51aw==