 Minseo Ahn
Minseo Ahn Akhtar Ali
Akhtar Ali Jae Ho Seo
Jae Ho Seo- 1Department of Biochemistry, Wonkwang University School of Medicine, Iksan, Republic of Korea
- 2Sarcopenia Total Solution Center, Wonkwang University School of Medicine, Iksan, Republic of Korea
- 3Institute of Wonkwang Medical Science, Wonkwang University, Iksan, Republic of Korea
Mitochondrial regulation plays a crucial role in cancer immunity in the tumor microenvironment (TME). Infiltrating immune cells, including T cells, natural killer (NK) cells, and macrophages, undergo mitochondrial metabolic reprogramming to survive the harsh conditions of the TME and enhance their antitumor activity. On the other hand, immunosuppressive cells like myeloid-derived suppressor cells (MDSCs), regulatory T cells (Tregs), mast cells, and tumor-associated macrophages (TAMs) rely on mitochondrial regulation to maintain their function as well. Additionally, mitochondrial regulation of cancer cells facilitates immune evasion and even hijacks mitochondria from immune cells to enhance their function. Recent studies suggest that targeting mitochondria can synergistically reduce cancer progression, especially when combined with traditional cancer therapies and immune checkpoint inhibitors. Many mitochondrial-targeting drugs are currently in clinical trials and have the potential to enhance the efficacy of immunotherapy. This mini review highlights the critical role of mitochondrial regulation in cancer immunity and provides lists of mitochondrial targeting drugs that have potential to enhance the efficacy of cancer immunotherapy.
1 Introduction
The tumor microenvironment (TME) is a complex and dynamic environment that plays a crucial role in cancer development, progression, and therapeutic response (1). From the perspective of cancer immunity, the TME is not only occupied by cancer cells but also consists of a variety of cell types, including both immunosuppressive cells and immune cells (2). Among the many factors within this environment, mitochondria stand out as critical regulators, not just serving as the powerhouse of the cell but also playing key roles in metabolic pathways, apoptosis, and cellular differentiation (3–5). These mitochondrial functions can significantly impact the behavior of cells within the TME. Immunotherapy has emerged as a new type of anticancer treatment that aims to strengthen immune cell function to fight cancer. Immune checkpoint inhibitors that block immune checkpoints, such as CTLA-4, PD-1, and PD-L1, are the most widely used immunotherapy. However, immune checkpoint inhibitors are not always effective because some cancer cells do not respond to the treatments (6, 7). Many studies have shown that mitochondrial-targeting drugs synergistically enhance the efficacy of immune checkpoint inhibitors while improving immune cell function and reducing the activity of immunosuppressive cells. Therefore, understanding the mechanisms of mitochondrial metabolic reprogramming is crucial for developing strategies to enhance immune cell function in the tumor microenvironment, which could lead to more effective cancer treatments through immunotherapy. In this mini-review, we briefly explain the role of mitochondrial regulation in cancer immunity within the TME and mitochondrial-targeting drugs that might serve as novel immunotherapeutic treatments.
2 Mitochondrial metabolic reprogramming in immune cells of tumor microenvironment
Mitochondrial metabolic reprogramming refers to the process by which the function and metabolic pathways of mitochondria are altered, often in response to changes in the cell’s environment or state (8). Within the TME, stress conditions, such as reduced oxygen consumption, elevated reactive oxygen species (ROS) generation, depolarized membrane potential, and impaired biogenesis disrupt the mitochondrial metabolism of immune cells and impair their function (9–11). Therefore, mitochondrial metabolic reprogramming plays a key role in the antitumor activities of immune cells that infiltrate the TME (12).
T cells are among the most important immune cells that are responsible for identifying and destroying cancer cells. When T cells are activated, their metabolism shifts from oxidative phosphorylation (OXPHOS) to glycolysis, supporting their rapid proliferation and effector functions (13). However, T cells show a persistent loss of mitochondrial function and mass when infiltrating tumors (14). Furthermore, continuous T cell stimulation in hypoxic environments leads to Blimp-1-mediated suppression of PGC-1α-dependent mitochondrial reprogramming (14). Several studies showed that enhancing mitochondrial metabolism improves T cell function. For instance, overexpression of PGC-1α enhanced mitochondrial biogenesis and metabolic capacity, thus improving CD8+ T cell antitumor effects (15). Stimulating T cell surface receptor 4-1BB enhances mitochondrial fusion and biogenesis in CD8+ tumor-infiltrating lymphocytes, independently of PGC-1α and p38-MAPK signaling (16).
Natural killer (NK) cells are crucial cytotoxic lymphocytes that are involved in the innate immune response against infected or transformed cells (17). In their inactive state, NK cells generate ATP primarily via mitochondrial OXPHOS, and upon activation, both glycolysis and OXPHOS increase, boosting ATP production (18, 19). A decreased level of PGC-1α, a master regulator of mitochondrial biogenesis, significantly impaired the ability of NK cells to control B16F10 melanoma growth in vivo, showing that mitochondria play a key role in NK cells (20). Upon infiltration of the TME, NK cells exhibit significant disadvantages. NK cells isolated from liver tumors exhibit fragmented mitochondria and impaired metabolism characterized by suppressed glycolysis and mitochondrial dysfunction (21). Furthermore, TGF-β induces metabolic dysfunction in NK cells from patients with metastatic breast cancer, leading to reduced glycolysis and OXPHOS and increased mitochondrial fragmentation (22). Blocking TGF-β and/or GARP can improve NK cell metabolism and function (22). NK cells showed a significant enhancement in immune activity and cytotoxicity when functional allogeneic mitochondria were transferred, indicating that mitochondrial improvement may restore NK cell function in the TME (23).
In the context of macrophage differentiation and function, mitochondria play a vital role in the regulation of metabolic reprogramming, signaling pathways, and immune responses (24). Among the two macrophage phenotypes, M1 and M2, M1 macrophages promote antitumor immunity by producing proinflammatory cytokines, such as IL-1β, IL-6, and IL-12 (25). M1 macrophages rely primarily on glycolysis for ATP production, favoring glycolysis even in the presence of oxygen, similar to the Warburg effect observed in cancer cells (25). Most macrophages in the TME exhibit the M2-like phenotype (25). This is due to the low glucose, high lactate levels, and hypoxic conditions of the TME, which drive macrophages to undergo M2 polarization (26, 27). Furthermore, tumors secrete cytokines and chemokines, such as IL-10, TGF-β, CCL2, and CSF-1, which recruit and polarize macrophages to the M2 phenotype (27).
3 Mitochondrial metabolic regulation in immunosuppressive cells
The TME comprises diverse immunosuppressive cells that promote tumorigenesis and immune evasion (Figure 1). These include myeloid-derived suppressor cells (MDSCs), regulatory T cells (Tregs), mast cells, and tumor-associated macrophages (TAMs) (28, 29). Many studies have highlighted the essential roles of mitochondria and mitochondrial metabolism in immunosuppressive cells, indicating their potential as therapeutic targets.
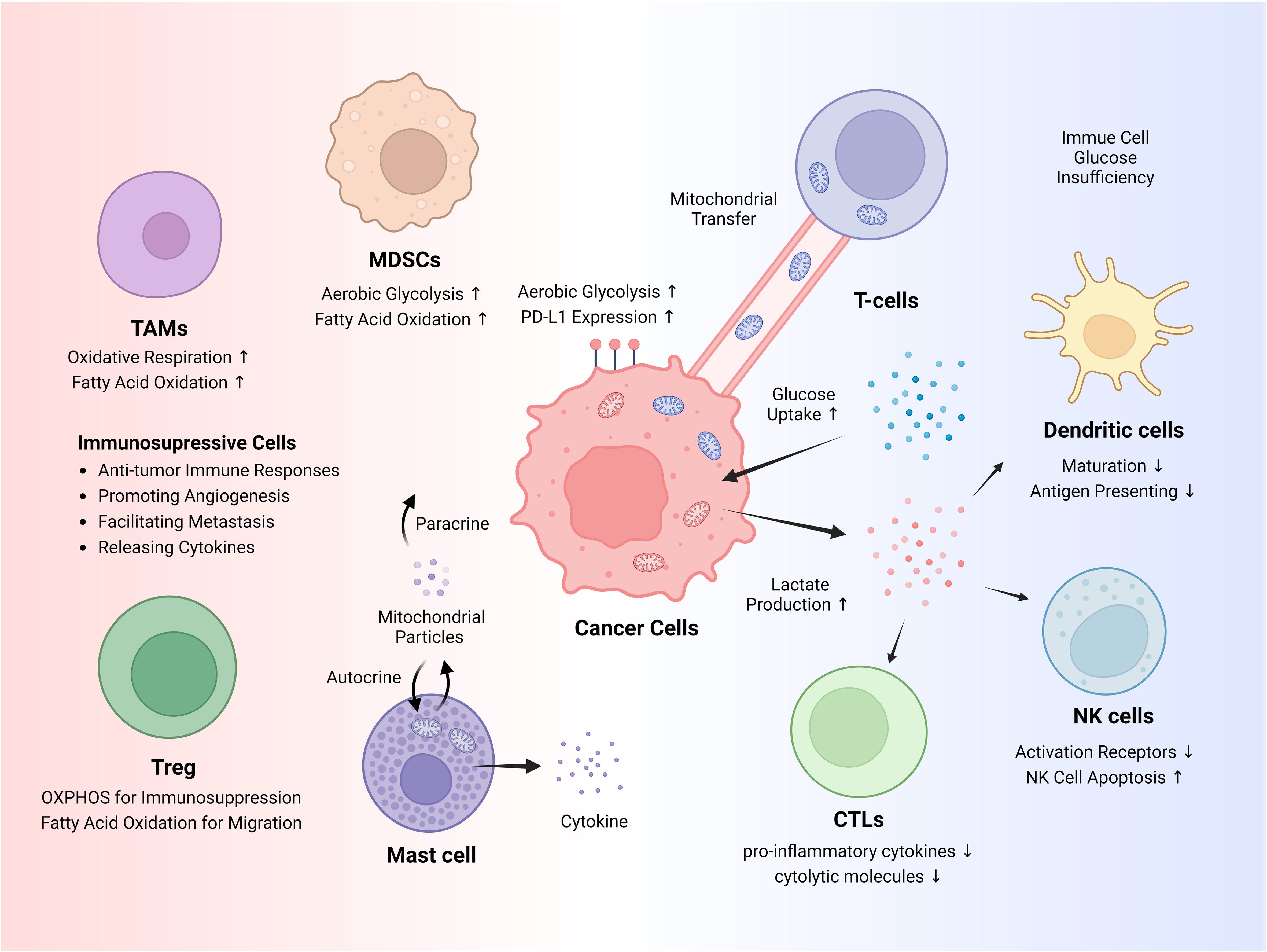
Figure 1. Cancer immune evasion and immunosuppressive cells. Cancer cells exhibit increased glucose uptake and heavily rely on glycolysis through a phenomenon known as the Warburg effect, a form of metabolic reprogramming. This heightened glucose uptake restricts glucose availability for immune cells, while lactate itself suppresses the antitumor activity of immune cells. Cancer cells can hijack mitochondria from T cells via mitochondrial transfer. Various immunosuppressive cells aid in tumorigenesis, and metabolic pathways, such as fatty acid oxidation (FAO) and oxidative phosphorylation (OXPHOS), adapt depending on the circumstances. NK, natural killer; Tregs, regulatory T cells; MDSCs, myeloid-derived suppressor cells; TAMs, tumor-associated macrophages.
MDSCs are immunosuppressive monocytes and neutrophils that rely heavily on glucose metabolism (30). For instance, β-adrenergic receptor signaling activates the STAT3 pathway, enhancing OXPHOS and glutamine utilization through the TCA cycle in MDSCs, reducing mitochondrial ROS via the NRF2 pathway, thereby improving MDSC survival (31). Decreased glucose availability, and consequently lactate production, results in smaller tumors, fewer MDSCs, and improved antitumor immune responses (32). Lipid metabolism is also crucial for MDSCs function. Tumor-derived cytokines activate STAT3 and STAT5 pathways and upregulate lipid transport receptors of MDSCs (33). This increases lipid uptake and enhances oxidative metabolism and immunosuppressive functions of MDSCs (33). Blocking fatty acid oxidation (FAO) in MDSCs, where fatty acid uptake and oxidation are crucial, reduces their immunosuppressive functions (34).
Tregs are another type of immunosuppressive cell that suppress the function of various immune cells, including CD4+ T helper cells, CD8+ cytotoxic T cells, NK cells, and NK T cells (35). Tregs rely heavily on OXPHOS and FAO for their energy needs (36). In the TME, where glucose is scarce and lactate is abundant, expression of Foxp3 increases, which in turn suppresses Myc and glycolysis, promotes OXPHOS, and helps Tregs adapt to their environment (37). HIF-1α serves as a metabolic switch in TME, oscillating between glycolysis-driven migration and OXPHOS-driven immunosuppression in Tregs (38). Tregs absorb fatty acids, store them as lipid droplets, and utilize fatty acid synthesis to aid their functional maturation (39). Blocking fatty acid-binding protein 5 in Tregs leads to the release of mitochondrial DNA, which activates cGAS-STING-dependent type I interferon signaling. This process increases the production of the regulatory cytokine IL-10 and suppresses the function of Tregs (40).
TAMs are predominantly of the M2 phenotype and support tumor growth and metastasis by releasing cytokines, chemokines, and growth factors (41). TAMs shift their metabolism towards OXPHOS and FAO while reducing glycolysis and the pentose phosphate pathway in the TME (42). This metabolic reprogramming promotes immunosuppressive signaling, which is favorable for tumor growth in the TME (43). Mast cells induce inflammation by releasing various chemokines and cytokines upon activation by a stimulus, which rapidly degranulates and releases their granule contents into the extracellular space through exocytosis (44). They contain extracellular mitochondrial particles and DNA, which can trigger cytokine release and exacerbate inflammation via autocrine or paracrine signaling (45). Overall, these findings illustrate the complex interplay between mitochondrial metabolic processes and immunosuppressive mechanisms within the TME and highlight potential targets for therapeutic interventions to enhance antitumor immune responses.
4 Mitochondrial regulation of cancer cells for immune evasion
Cancer cells exhibit considerable alterations in their metabolic processes, profoundly impacting cancer immunity in TME (Figure 1). One of the most prominent mechanisms of metabolic reprogramming is the Warburg effect, wherein glucose uptake and lactate production increase significantly in tumor cells, even in the presence of oxygen (46). Glycolytic activity and elevated glucose uptake by cancer cells can restrict glucose availability to tumor-infiltrating immune cells, thereby promoting immune escape (47). Furthermore, increased glycolytic activity in tumor cells correlates positively with elevated PD-L1 expression (48). Lactate, a primary metabolite generated by glycolysis, plays a direct role in facilitating the immune evasion of cancer cells (49). Lactate inhibits the maturation of dendritic cells and impairs their ability to present antigens by downregulating the expression of major histocompatibility complex class II molecules (50). Lactate impairs the effectiveness of cytotoxic T lymphocytes (CTLs) by lowering the production of proinflammatory cytokines, such as IFN-γ, which are essential for CTL-mediated antitumor activity, and by impairing the secretion pathways of cytolytic molecules (51). Tumor-derived lactate reduces NK cell activity by directly inhibiting their cytolytic function, decreasing the expression of activation receptors, such as NKp46, triggering apoptosis in NK cells, and indirectly increasing the number of MDSCs that suppress NK cytotoxicity (32, 52).
Other studies have demonstrated that mitochondrial function positively correlates with the immune evasion ability of cancer cells. In N-acetyltransferase 1-depleted breast cancer cells, a reduction in OXPHOS and mitochondrial biogenesis proteins was observed along with an elevation in antigen presentation proteins (53). The elevated expression of CD147, which is upregulated in various malignant tumors, is associated with increased GLUT1 and MCT1 levels (54). This enhanced glycolytic metabolism in hepatocellular carcinoma (HCC) cell lines correlated with immunosuppressive lymphocyte infiltration in HCC tissues (55). Furthermore, deletion of Complex II, one of the key components of the electron transport chain, inhibits melanoma tumor growth by enhancing antigen presentation and T cell-mediated death (56).
Cancer cells also disrupt mitochondrial metabolism in immune cells. For example, HCC secretes α-fetoprotein, which reduces SREBP-1 and PGC1-α in dendritic cells, leading to lower lipogenesis, oxygen consumption rate, and ATP synthesis of the cell (57). Interestingly, cancer cells can directly hijack mitochondria from immune cells in the TME to evade immune detection and destruction. The transfer of mitochondria from immune cells to cancer cells enhances cancer cell metabolism (58). Furthermore, mitochondria from T cells can transfer to cancer cells, leading to the upregulation of genes involved in cytoskeleton remodeling, energy production, and TNF-α signaling pathways, along with increasing cell cycle activity (59). In conclusion, cancer cells reprogram their metabolism, notably via the Warburg effect, to enhance their growth and evade immune responses. They also disrupt the mitochondrial metabolism and steal mitochondria from immune cells to evade immune detection.
5 Targeting mitochondria in cancer immunity
Since mitochondrial regulation plays a key role in cancer immunity, mitochondria-targeting drugs have the potential to boost immune cell function and immunotherapy for cancer. Many drugs targeting mitochondria are being investigated from an immunotherapy perspective, with some currently in clinical trials (Table 1).
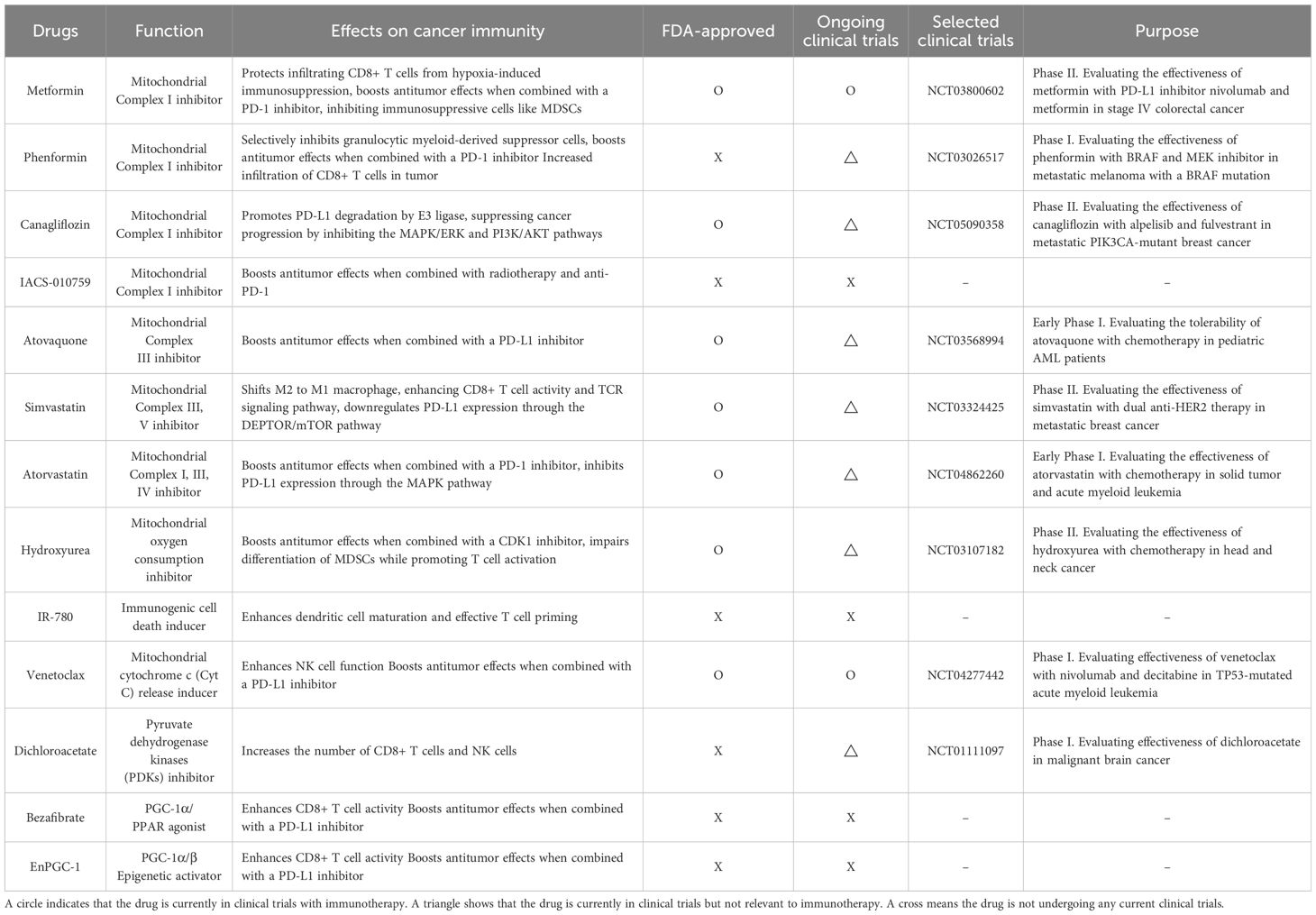
Table 1. Impact of mitochondria-targeting drugs on immunotherapy.
Metformin is one of the most extensively studied mitochondria-targeting drugs that enhances immune responses in cancer therapy, which inhibits Complex I of the mitochondrial respiratory chain (60, 61). Metformin improves cancer immunotherapy by directly protecting tumor-infiltrating CD8+ T cells from hypoxia-induced immunosuppression, potentially by reducing ROS production and preventing apoptosis (62). Combining metformin with an anti-PD-L1 antibody has been shown to induce tumor necrosis by enhancing CD8+ T cell infiltration and increasing IFN-γ expression (63). Furthermore, in STK11 mutant lung cancer, the combination of metformin with a PD-1 inhibitor boosts antitumor effects by inhibiting STING ubiquitination in an AXIN-1-dependent manner (64). Additionally, metformin activates AMPK phosphorylation in various cancer cells, which bolsters antitumor immunity by inhibiting immunosuppressive cells like MDSCs (65, 66). Phenformin, a drug structurally similar to metformin but with a stronger effect on Complex I inhibition, selectively targets MDSCs in vivo (67). Furthermore, combining phenformin with anti-PD-1 antibody therapy reduced tumor growth with greater infiltration of CD8+ T cells into a melanoma mouse model (67). Canagliflozin, a medication used to treat type 2 diabetes, is another drug that has been discovered to inhibit mitochondrial complex I activity. On the plasma membrane, SGLT2 (sodium/glucose cotransporter 2) physically interacts with PD-L1, preventing its proteasome-mediated degradation. Canagliflozin disrupts this interaction, leading to PD-L1 degradation by E3 ligase and consequently enhancing the activity of antitumor cytotoxic T cells (68, 69). Additionally, canagliflozin inhibits both the MAPK/ERK and PI3K/AKT signaling pathways, thereby suppressing cancer progression (69). A small molecule that inhibits mitochondrial complex I activity named IACS-010759 decreases radiation-induced Tregs, increases activated CD8+ T cells, and, when combined with radiotherapy and anti-PD-1, promotes abscopal responses and prolongs survival in a non-small cell lung cancer (NSCLC) xenograft mouse model (70). Atovaquone is a mitochondrial complex III inhibitor and alleviates hypoxia in TME in colorectal cancer in vivo. Furthermore, combination of atovaquone with anti-PD-L1 antibody greatly enhances tumor eradication in the CT26 colorectal cancer model by establishing a tumor-specific memory immune response (71).
Statins, which inhibit the enzyme HMG-CoA reductase involved in cholesterol synthesis, impair mitochondrial function by inhibiting mitochondrial respiratory chain (72). Seven statin drugs were tested for their ability to inhibit tumor cell proliferation using an ex vivo co-culture assay with murine cancer cells and tumor-infiltrating lymphocytes. Among these, simvastatin and lovastatin enhance T cell-mediated tumor cell killing and shift M2 to M1 macrophages in this model (73).
Simvastatin also boosts antitumor immunity by enhancing CD8+ T cell activity and T cell antigen receptor (TCR) signaling pathway (74). Simvastatin also reduced ILF3 expression by lowering H3K14 acetylation levels, and ILF3, in turn, downregulated PD-L1 expression through the DEPTOR/mTOR pathway (75). Another type of statin called atorvastatin significantly enhanced antitumor efficacy by promoting T cell activation in combination with an anti-PD-L1 antibody. Additionally, atorvastatin inhibited the MAPK pathway, leading to decreased PD-L1 expression (76). Hydroxyurea is an FDA-approved drug for treating both sickle cell disease and cancer, and it can be used alone or in combination with conventional chemotherapy or radiation therapy (77). Combining a checkpoint kinase 1 inhibitor with low-dose hydroxyurea reduced the tumor size of melanomas that are resistant to BRAF and immune checkpoint inhibitors in vivo (78). Mito-HU, a modified form of hydroxyurea designed to target mitochondria, disturbed differentiation of MDSCs while promoting T cell activation in vitro (77). IR-780 boosts cancer immunity by targeting mitochondria to induce immunogenic cell death, which exposes tumor-associated antigens, leading to enhanced dendritic cell maturation, effective T cell priming, and improved immune responses against tumors (79).
NK cells effectively drive cancer cells toward mitochondrial apoptosis, and when combined with the BCL-2 inhibitor venetoclax, they synergistically enhance the killing of cancer cells both in vitro and in vivo while pre-activated NK cells have been shown to become resistant to venetoclax (80). Furthermore, in combination with immune checkpoint blockade, venetoclax boosts infiltrating effector T cells and strengthens antitumor efficacy (81). Lactate produced by cancer cells promotes the IL-23/IL-17 inflammatory pathway and increases arginase I (ARG1) expression in macrophages, leading to the inhibition of T cell proliferation and activation (82). Dichloroacetate (DCA) reduces the IL-23/IL-17 inflammatory pathway and ARG1 expression in macrophages, while increasing the number of IFN-γ-producing CD8+ T cells and NK cells in vivo (82). Bezafibrate, a PGC-1α/PPAR agonist, promotes mitochondrial biogenesis and fatty acid oxidation (FAO) in T cells and increases the accumulation and activation of CD8+ T cells within tumors (83). In a lung carcinoma xenograft model, bezafibrate also enhanced the antitumor effects of PD-1 blockade (83). EnPGC-1, a DNA-based epigenetic activator that induces targeted expression of PGC-1α/β, enhances the mitochondrial activation, energy metabolism, and proliferation of CD8+ T cells in vitro. In a mouse model, EnPGC-1 synergizes with PD-1 blockade, leading to enhanced tumor inhibition (84). In summary, research into mitochondria-targeting drugs suggests that these agents can provide synergistic effects when combined with immunotherapy, making them promising candidates for clinical use. However, further investigation is needed to fully understand the underlying mechanisms and optimize their efficacy in clinical settings.
6 Conclusion
Despite the emergence of immunotherapy as a new strategy to treat cancer, some cancers are non-responsive to this treatment. Immune checkpoint inhibitors help the immune system recognize and attack cancer cells, but their effectiveness may be limited when used alone due to cancer’s immune evasion mechanisms (85, 86). Even cancer cells that respond to immune checkpoint inhibitors have been reported to develop resistance to immune checkpoint inhibitors over time (87, 88). Mitochondrial-targeted drugs can be used in combination with the treatment to regulate cellular energy metabolism and inhibit the survival and proliferation of cancer cells, thereby reducing the number of cancer cells and creating an environment in which immune cells can function more effectively (65). In addition, changes in the tumor microenvironment can promote the infiltration and activation of immune cells (89). This is expected to further increase the effectiveness of immune checkpoint inhibitors.
Numerous studies already have demonstrated that mitochondrial regulation plays a crucial role in cancer immunity and that mitochondria-targeting drugs enhance the efficiency of immunotherapy. Therefore, targeting mitochondria has the potential to restore and boost immune cell function within the TME and synergistically increase the effectiveness of immunotherapy. However, although many mitochondria-targeting drugs are currently in clinical trial, the primary focus is not directly connected to immunotherapy. In addition, the molecular mechanisms by which mitochondria-targeting drugs enhance the effectiveness of immunotherapy are not yet well understood. Therefore, further research is required to elucidate these mechanisms and their potential as immunotherapy adjuvants.
Author contributions
MA: Writing – original draft, Writing – review & editing. AA: Writing – original draft, Writing – review & editing. JS: Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by Wonkwang University in 2022.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Arneth B. Tumor microenvironment. Medicina (B Aires). (2019) 56:15. doi: 10.3390/medicina56010015
2. Lei X, Lei Y, Li JK, Du WX, Li RG, Yang J, et al. Immune cells within the tumor microenvironment: Biological functions and roles in cancer immunotherapy. Cancer Lett. (2020) 470:126–33. doi: 10.1016/j.canlet.2019.11.009
3. Li X, Jiang O, Chen M, Wang S. Mitochondrial homeostasis: shaping health and disease. Curr Med. (2024) 3:5. doi: 10.1007/s44194-024-00032-x
4. McBride HM, Neuspiel M, Wasiak S. Mitochondria: more than just a powerhouse. Curr Biol. (2006) 16:R551–60. doi: 10.1016/j.cub.2006.06.054
5. Anderson AJ, Jackson TD, Stroud DA, Stojanovski D. Mitochondria—hubs for regulating cellular biochemistry: emerging concepts and networks. Open Biol. (2019) 9:190126. doi: 10.1098/rsob.190126
6. Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer. (2018) 118:9–16. doi: 10.1038/bjc.2017.434
7. Yin Q, Wu L, Han L, Zheng X, Tong R, Li L, et al. Immune-related adverse events of immune checkpoint inhibitors: a review. Front Immunol. (2023) 14. doi: 10.3389/fimmu.2023.1167975
8. Fan X, Yang M, Lang Y, Lu S, Kong Z, Gao Y, et al. Mitochondrial metabolic reprogramming in diabetic kidney disease. Cell Death Dis. (2024) 15:442. doi: 10.1038/s41419-024-06833-0
9. Tian W, Liu Y, Cao C, Zeng Y, Pan Y, Liu X, et al. Chronic stress: impacts on tumor microenvironment and implications for anti-cancer treatments. Front Cell Dev Biol. (2021) 9. doi: 10.3389/fcell.2021.777018
10. Lei Y, Liao F, Tian Y, Wang Y, Xia F, Wang J. Investigating the crosstalk between chronic stress and immune cells: implications for enhanced cancer therapy. Front Neurosci. (2023) 17. doi: 10.3389/fnins.2023.1321176
11. Liu Y, Tian S, Ning B, Huang T, Li Y, Wei Y. Stress and cancer: The mechanisms of immune dysregulation and management. Front Immunol. (2022) 13. doi: 10.3389/fimmu.2022.1032294
12. Sukumar M, Kishton RJ, Restifo NP. Metabolic reprograming of anti-tumor immunity. Curr Opin Immunol. (2017) 46:14–22. doi: 10.1016/j.coi.2017.03.011
13. Kouidhi S, Elgaaied AB, Chouaib S. Impact of metabolism in on T-cell differentiation and function and cross talk with tumor microenvironment. Front Immunol. (2017) 8. doi: 10.3389/fimmu.2017.00270
14. Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, et al. The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity. (2016) 45:374–88. doi: 10.1016/j.immuni.2016.07.009
15. Dumauthioz N, Tschumi B, Wenes M, Marti B, Wang H, Franco F, et al. Enforced PGC-1α expression promotes CD8 T cell fitness, memory formation and antitumor immunity. Cell Mol Immunol. (2021) 18:1761–71. doi: 10.1038/s41423-020-0365-3
16. Menk AV, Scharping NE, Rivadeneira DB, Calderon MJ, Watson MJ, Dunstane D, et al. 4-1BB costimulation induces T cell mitochondrial function and biogenesis enabling cancer immunotherapeutic responses. J Exp Med. (2018) 215:1091–100. doi: 10.1084/jem.20171068
17. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. (2008) 9:503–10. doi: 10.1038/ni1582
18. Cong J. Metabolism of natural killer cells and other innate lymphoid cells. Front Immunol. (2020) 11. doi: 10.3389/fimmu.2020.01989
19. Wang Z, Guan D, Wang S, Chai LYA, Xu S, Lam KP. Glycolysis and oxidative phosphorylation play critical roles in natural killer cell receptor-mediated natural killer cell functions. Front Immunol. (2020) 11. doi: 10.3389/fimmu.2020.00202
20. Gerbec ZJ, Hashemi E, Nanbakhsh A, Holzhauer S, Yang C, Mei A, et al. Conditional deletion of PGC-1α Results in energetic and functional defects in NK cells. iScience. (2020) 23:101454. doi: 10.1016/j.isci.2020.101454
21. Zheng X, Qian Y, Fu B, Jiao D, Jiang Y, Chen P, et al. Mitochondrial fragmentation limits NK cell-based tumor immunosurveillance. Nat Immunol. (2019) 20:1656–67. doi: 10.1038/s41590-019-0511-1
22. Slattery K, Woods E, Zaiatz-Bittencourt V, Marks S, Chew S, Conroy M, et al. TGFβ drives NK cell metabolic dysfunction in human metastatic breast cancer. J Immunother Cancer. (2021) 9:e002044. doi: 10.1136/jitc-2020-002044
23. Kim SH, Kim MJ, Lim M, Kim J, Kim H, Yun CK, et al. Enhancement of the anticancer ability of natural killer cells through allogeneic mitochondrial transfer. Cancers (Basel). (2023) 15:3225. doi: 10.3390/cancers15123225
24. Hickman E, Smyth T, Cobos-Uribe C, Immormino R, Rebuli ME, Moran T, et al. Expanded characterization of in vitro polarized M0, M1, and M2 human monocyte-derived macrophages: Bioenergetic and secreted mediator profiles. PloS One. (2023) 18:e0279037. doi: 10.1371/journal.pone.0279037
25. Viola A, Munari F, Sánchez-Rodríguez R, Scolaro T, Castegna A. The metabolic signature of macrophage responses. Front Immunol. (2019) 10. doi: 10.3389/fimmu.2019.01462
26. Li M, He L, Zhu J, Zhang P, Liang S. Targeting tumor-associated macrophages for cancer treatment. Cell Biosci. (2022) 12:85. doi: 10.1186/s13578-022-00823-5
27. Boutilier AJ, Elsawa SF. Macrophage polarization states in the tumor microenvironment. Int J Mol Sci. (2021) 22:6995. doi: 10.3390/ijms22136995
28. Liu Y, Cao X. Immunosuppressive cells in tumor immune escape and metastasis. J Mol Med. (2016) 94:509–22. doi: 10.1007/s00109-015-1376-x
29. Tie Y, Tang F, Wei Yq, Wei Xw. Immunosuppressive cells in cancer: mechanisms and potential therapeutic targets. J Hematol Oncol. (2022) 15:61. doi: 10.1186/s13045-022-01282-8
30. Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol. (2021) 21:485–98. doi: 10.1038/s41577-020-00490-y
31. Daneshmandi S, Choi JE, Yan Q, MacDonald CR, Pandey M, Goruganthu M, et al. Myeloid-derived suppressor cell mitochondrial fitness governs chemotherapeutic efficacy in hematologic Malignancies. Nat Commun. (2024) 15:2803. doi: 10.1038/s41467-024-47096-9
32. Husain Z, Huang Y, Seth P, Sukhatme VP. Tumor-derived lactate modifies antitumor immune response: effect on myeloid-derived suppressor cells and NK cells. J Immunol. (2013) 191:1486–95. doi: 10.4049/jimmunol.1202702
33. Al-Khami AA, Zheng L, Del Valle L, Hossain F, Wyczechowska D, Zabaleta J, et al. Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. Oncoimmunology. (2017) 6:e1344804. doi: 10.1080/2162402X.2017.1344804
34. Hossain F, Al-Khami AA, Wyczechowska D, Hernandez C, Zheng L, Reiss K, et al. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol Res. (2015) 3:1236–47. doi: 10.1158/2326-6066.CIR-15-0036
35. Grover P, Goel PN, Greene MI. Regulatory T cells: regulation of identity and function. Front Immunol. (2021) 12. doi: 10.3389/fimmu.2021.750542
36. Blagih J, Hennequart M, Zani F. Tissue nutrient environments and their effect on regulatory T cell biology. Front Immunol. (2021) 12. doi: 10.3389/fimmu.2021.637960
37. Angelin A, Gil-de-Gómez L, Dahiya S, Jiao J, Guo L, Levine MH, et al. Foxp3 reprograms T cell metabolism to function in low-glucose, high-lactate environments. Cell Metab. (2017) 25:1282–1293.e7. doi: 10.1016/j.cmet.2016.12.018
38. Miska J, Lee-Chang C, Rashidi A, Muroski ME, Chang AL, Lopez-Rosas A, et al. HIF-1α Is a metabolic switch between glycolytic-driven migration and oxidative phosphorylation-driven immunosuppression of tregs in glioblastoma. Cell Rep. (2019) 27:226–237.e4. doi: 10.1016/j.celrep.2019.03.029
39. Zhang S, Lv K, Liu Z, Zhao R, Li F. Fatty acid metabolism of immune cells: a new target of tumour immunotherapy. Cell Death Discov. (2024) 10:39. doi: 10.1038/s41420-024-01807-9
40. Field CS, Baixauli F, Kyle RL, Puleston DJ, Cameron AM, Sanin DE, et al. Mitochondrial integrity regulated by lipid metabolism is a cell-intrinsic checkpoint for treg suppressive function. Cell Metab. (2020) 31:422–437.e5. doi: 10.1016/j.cmet.2019.11.021
41. Basak U, Sarkar T, Mukherjee S, Chakraborty S, Dutta A, Dutta S, et al. Tumor-associated macrophages: an effective player of the tumor microenvironment. Front Immunol. (2023) 14. doi: 10.3389/fimmu.2023.1295257
42. Hasan MN, Capuk O, Patel SM, Sun D. The role of metabolic plasticity of tumor-associated macrophages in shaping the tumor microenvironment immunity. Cancers (Basel). (2022) 14:3331. doi: 10.3390/cancers14143331
43. Dubey S, Ghosh S, Goswami D, Ghatak D, De R. Immunometabolic attributes and mitochondria-associated signaling of Tumor-Associated Macrophages in tumor microenvironment modulate cancer progression. Biochem Pharmacol. (2023) 208:115369. doi: 10.1016/j.bcp.2022.115369
44. Krystel-Whittemore M, Dileepan KN, Wood JG. Mast cell: A multi-functional master cell. Front Immunol. (2016) 6. doi: 10.3389/fimmu.2015.00620
45. Zhang B, Asadi S, Weng Z, Sismanopoulos N, Theoharides TC. Stimulated human mast cells secrete mitochondrial components that have autocrine and paracrine inflammatory actions. PloS One. (2012) 7:e49767. doi: 10.1371/journal.pone.0049767
46. Vaupel P, Schmidberger H, Mayer A. The Warburg effect: essential part of metabolic reprogramming and central contributor to cancer progression. Int J Radiat Biol. (2019) 95:912–9. doi: 10.1080/09553002.2019.1589653
47. Xia L, Oyang L, Lin J, Tan S, Han Y, Wu N, et al. The cancer metabolic reprogramming and immune response. Mol Cancer. (2021) 20:28. doi: 10.1186/s12943-021-01316-8
48. Jiang Z, Liu Z, Li M, Chen C, Wang X. Increased glycolysis correlates with elevated immune activity in tumor immune microenvironment. EBioMedicine. (2019) 42:431–42. doi: 10.1016/j.ebiom.2019.03.068
49. Caslin HL, Abebayehu D, Pinette JA, Ryan JJ. Lactate is a metabolic mediator that shapes immune cell fate and function. Front Physiol. (2021) 12. doi: 10.3389/fphys.2021.688485
50. Sangsuwan R, Thuamsang B, Pacifici N, Allen R, Han H, Miakicheva S, et al. Lactate exposure promotes immunosuppressive phenotypes in innate immune cells. Cell Mol Bioeng. (2020) 13:541–57. doi: 10.1007/s12195-020-00652-x
51. Haas R, Smith J, Rocher-Ros V, Nadkarni S, Montero-Melendez T, D’Acquisto F, et al. Lactate regulates metabolic and pro-inflammatory circuits in control of T cell migration and effector functions. PloS Biol. (2015) 13:e1002202. doi: 10.1371/journal.pbio.1002202
52. Jedlička M, Feglarová T, Janstová L, Hortová-Kohoutková M, Frič J. Lactate from the tumor microenvironment - A key obstacle in NK cell-based immunotherapies. Front Immunol. (2022) 13. doi: 10.3389/fimmu.2022.932055
53. Hong KU, Gardner JQ, Doll MA, Stepp MW, Wilkey DW, Benz FW, et al. Proteomic analysis of arylamine N-acetyltransferase 1 knockout breast cancer cells: Implications in immune evasion and mitochondrial biogenesis. Toxicol Rep. (2022) 9:1566–73. doi: 10.1016/j.toxrep.2022.07.010
54. Xin X, Zeng X, Gu H, Li M, Tan H, Jin Z, et al. CD147/EMMPRIN overexpression and prognosis in cancer: A systematic review and meta-analysis. Sci Rep. (2016) 6:32804. doi: 10.1038/srep32804
55. Li X, Zhang Y, Ma W, Fu Q, Liu J, Yin G, et al. Enhanced glucose metabolism mediated by CD147 contributes to immunosuppression in hepatocellular carcinoma. Cancer Immunol Immunother. (2020) 69:535–48. doi: 10.1007/s00262-019-02457-y
56. Mangalhara KC, Varanasi SK, Johnson MA, Burns MJ, Rojas GR, Esparza Moltó PB, et al. Manipulating mitochondrial electron flow enhances tumor immunogenicity. Sci (1979). (2023) 381:1316–23. doi: 10.1126/science.abq1053
57. Santos PM, Menk AV, Shi J, Tsung A, Delgoffe GM, Butterfield LH. Tumor-derived α-fetoprotein suppresses fatty acid metabolism and oxidative phosphorylation in dendritic cells. Cancer Immunol Res. (2019) 7:1001–12. doi: 10.1158/2326-6066.CIR-18-0513
58. Saha T, Dash C, Jayabalan R, Khiste S, Kulkarni A, Kurmi K, et al. Intercellular nanotubes mediate mitochondrial trafficking between cancer and immune cells. Nat Nanotechnol. (2022) 17:98–106. doi: 10.1038/s41565-021-01000-4
59. Zhang H, Yu X, Ye J, Li H, Hu J, Tan Y, et al. Systematic investigation of mitochondrial transfer between cancer cells and T cells at single-cell resolution. Cancer Cell. (2023) 41:1788–1802.e10. doi: 10.1016/j.ccell.2023.09.003
60. Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. (2000) 348:607–14. doi: 10.1042/bj3480607
61. Rena G, Hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia. (2017) 60:1577–85. doi: 10.1007/s00125-017-4342-z
62. Finisguerra V, Dvorakova T, Formenti M, Van Meerbeeck P, Mignion L, Gallez B, et al. Metformin improves cancer immunotherapy by directly rescuing tumor-infiltrating CD8 T lymphocytes from hypoxia-induced immunosuppression. J Immunother Cancer. (2023) 11:e005719. doi: 10.1136/jitc-2022-005719
63. Abdelmoneim M, Aboalela MA, Naoe Y, Matsumura S, Eissa IR, Bustos-Villalobos I, et al. The impact of metformin on tumor-infiltrated immune cells: preclinical and clinical studies. Int J Mol Sci. (2023) 24:13353. doi: 10.3390/ijms241713353
64. Wang Z, Lu C, Zhang K, Lin C, Wu F, Tang X, et al. Metformin combining PD-1 inhibitor enhanced anti-tumor efficacy in STK11 mutant lung cancer through AXIN-1-dependent inhibition of STING ubiquitination. Front Mol Biosci. (2022) 9. doi: 10.3389/fmolb.2022.780200
65. Bai R, Cui J. Mitochondrial immune regulation and anti-tumor immunotherapy strategies targeting mitochondria. Cancer Lett. (2023) 564:216223. doi: 10.1016/j.canlet.2023.216223
66. Xu P, Yin K, Tang X, Tian J, Zhang Y, Ma J, et al. Metformin inhibits the function of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. Biomed Pharmacother. (2019) 120:109458. doi: 10.1016/j.biopha.2019.109458
67. Kim SH, Li M, Trousil S, Zhang Y, Pasca di Magliano M, Swanson KD, et al. Phenformin inhibits myeloid-derived suppressor cells and enhances the anti-tumor activity of PD-1 blockade in melanoma. J Invest Dermatol. (2017) 137:1740–8. doi: 10.1016/j.jid.2017.03.033
68. Ding L, Chen X, Zhang W, Dai X, Guo H, Pan X, et al. Canagliflozin primes antitumor immunity by triggering PD-L1 degradation in endocytic recycling. J Clin Invest. (2023) 133(1):e154754. doi: 10.1172/JCI154754
69. Wang S, Sui M, Chen Q, Guo J, Yang H, Zhou Y, et al. Engineering PD-L1 targeted liposomal canagliflozin achieves multimodal synergistic cancer therapy. Chem Eng J. (2024) 498:155074. doi: 10.1016/j.cej.2024.155074
70. Chen D, Barsoumian HB, Fischer G, Yang L, Verma V, Younes AI, et al. Combination treatment with radiotherapy and a novel oxidative phosphorylation inhibitor overcomes PD-1 resistance and enhances antitumor immunity. J Immunother Cancer. (2020) 8:e000289. doi: 10.1136/jitc-2019-000289
71. Rodriguez-Berriguete G, Puliyadi R, MaChado N, Barberis A, Prevo R, McLaughlin M, et al. Antitumour effect of the mitochondrial complex III inhibitor Atovaquone in combination with anti-PD-L1 therapy in mouse cancer models. Cell Death Dis. (2024) 15:32. doi: 10.1038/s41419-023-06405-8
72. Mollazadeh H, Tavana E, Fanni G, Bo S, Banach M, Pirro M, et al. Effects of statins on mitochondrial pathways. J Cachexia Sarcopenia Muscle. (2021) 12:237–51. doi: 10.1002/jcsm.12654
73. Kansal V, Burnham AJ, Kinney BLC, Saba NF, Paulos C, Lesinski GB, et al. Statin drugs enhance responses to immune checkpoint blockade in head and neck cancer models. J Immunother Cancer. (2023) 11:e005940. doi: 10.1136/jitc-2022-005940
74. Yuan W, Ren X, Zhu J, Huang J, Zhang W, Zhang C, et al. Single-intraosseous simvastatin injection suppresses cancers via activating CD8+ T cells. Biomed Pharmacother. (2022) 155:113665. doi: 10.1016/j.biopha.2022.113665
75. Sun D, Yu W, Cui X. Effect of simvastatin on PD-L1 via ILF3 to enhance CD8 + T cell-mediated ferroptosis in gastric cancer cells. J Clin Oncol. (2024) 42:e16056–6. doi: 10.1200/JCO.2024.42.16_suppl.e16056
76. Choe EJ, Lee CH, Bae JH, Park JM, Park SS, Baek MC. Atorvastatin enhances the efficacy of immune checkpoint therapy and suppresses the cellular and extracellular vesicle PD-L1. Pharmaceutics. (2022) 14:1660. doi: 10.3390/pharmaceutics14081660
77. Cheng G, Hardy M, Topchyan P, Zander R, Volberding P, Cui W, et al. Mitochondria-targeted hydroxyurea inhibits OXPHOS and induces antiproliferative and immunomodulatory effects. iScience. (2021) 24:102673. doi: 10.1016/j.isci.2021.102673
78. Zeng Z, Ngo HL, Proctor M, Rizos H, Dolcetti R, Cruz JG, et al. Checkpoint kinase 1 inhibitor + low-dose hydroxyurea efficiently kills BRAF inhibitor- and immune checkpoint inhibitor-resistant melanomas. Pigment Cell Melanoma Res. (2024) 37:45–50. doi: 10.1111/pcmr.13120
79. Jiang Q, Zhang C, Wang H, Peng T, Zhang L, Wang Y, et al. Mitochondria-targeting immunogenic cell death inducer improves the adoptive T-cell therapy against solid tumor. Front Oncol. (2019) 9. doi: 10.3389/fonc.2019.01196
80. Pan R, Ryan J, Pan D, Wucherpfennig KW, Letai A. Augmenting NK cell-based immunotherapy by targeting mitochondrial apoptosis. Cell. (2022) 185:1521–1538.e18. doi: 10.1016/j.cell.2022.03.030
81. Kohlhapp FJ, Haribhai D, Mathew R, Duggan R, Ellis PA, Wang R, et al. Venetoclax increases intratumoral effector T cells and antitumor efficacy in combination with immune checkpoint blockade. Cancer Discov. (2021) 11:68–79. doi: 10.1158/2159-8290.CD-19-0759
82. Ohashi T, Akazawa T, Aoki M, Kuze B, Mizuta K, Ito Y, et al. Dichloroacetate improves immune dysfunction caused by tumor-secreted lactic acid and increases antitumor immunoreactivity. Int J Cancer. (2013) 133:1107–18. doi: 10.1002/ijc.v133.5
83. Wan H, Xu B, Zhu N, Ren B. PGC-1α activator–induced fatty acid oxidation in tumor-infiltrating CTLs enhances effects of PD-1 blockade therapy in lung cancer. Tumori J. (2020) 106:55–63. doi: 10.1177/0300891619868287
84. Malinee M, Pandian GN, Sugiyama H. Targeted epigenetic induction of mitochondrial biogenesis enhances antitumor immunity in mouse model. Cell Chem Biol. (2022) 29:463–475.e6. doi: 10.1016/j.chembiol.2021.08.001
85. Kim SK, Cho SW. The evasion mechanisms of cancer immunity and drug intervention in the tumor microenvironment. Front Pharmacol. (2022) 13. doi: 10.3389/fphar.2022.868695
86. Bagchi S, Yuan R, Engleman EG. Immune checkpoint inhibitors for the treatment of cancer: clinical impact and mechanisms of response and resistance. Annu Rev Pathol: Mech Dis. (2021) 16:223–49. doi: 10.1146/annurev-pathol-042020-042741
87. Barrueto L, Caminero F, Cash L, Makris C, Lamichhane P, Deshmukh RR. Resistance to checkpoint inhibition in cancer immunotherapy. Transl Oncol. (2020) 13:100738. doi: 10.1016/j.tranon.2019.12.010
88. Nagasaki J, Ishino T, Togashi Y. Mechanisms of resistance to immune checkpoint inhibitors. Cancer Sci. (2022) 113:3303–12. doi: 10.1111/cas.v113.10
Keywords: mitochondria, TME, immunotherapy, metabolism, immune evasion
Citation: Ahn M, Ali A and Seo JH (2024) Mitochondrial regulation in the tumor microenvironment: targeting mitochondria for immunotherapy. Front. Immunol. 15:1453886. doi: 10.3389/fimmu.2024.1453886
Received: 24 June 2024; Accepted: 27 September 2024;
Published: 11 October 2024.
Edited by:
Ren Zhao, Shanghai Jiao Tong University, ChinaReviewed by:
Jozsef Dudas, Innsbruck Medical University, AustriaMingxiao Feng, Johns Hopkins University, United States
Copyright © 2024 Ahn, Ali and Seo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jae Ho Seo, YmlvbmlhbjlAd2t1LmFjLmty
†These authors have contributed equally to this work