 Ana Sami
Ana Sami Afsheen Raza
Afsheen Raza- 1Department of Medicine and Dentistry, Barts Cancer Institute, Queen Mary University of London, London, United Kingdom
- 2Department of Biomedical Sciences, College of Health Sciences, Abu Dhabi University, Abu Dhabi, United Arab Emirates
Breast cancer has the highest global incidence among all cancers, affecting more than 2 million individuals annually. Despite the availability of new drugs and novel treatment combinations, it is postulated that the incidence and mortality of breast cancer will rise by 40.8% and 51.9% respectively by 2040. Such dire statistics are associated with the clonal evolution of cancer cells that leads to therapeutic resistance and consequent relapse in breast cancer patients. On the other hand, the tumor microenvironment (TME) comprising of tumor cells, cancer-associated immune cells, re-programmed stromal cells, and the extracellular matrix (ECM) creates an immunosuppressive niche facilitating immune evasion. This review focuses on a critical cellular component of the tumor microenvironment, the tumor-associated macrophages (TAMs) in breast cancer immunotherapy. Macrophages are inherently plastic and can convert from an anti-tumor M1 phenotype to a pro-tumor M2 phenotype based on microenvironmental cues. Cancer cells facilitate these cues, allowing the tumor-associated macrophages to gain M2 phenotype and mediate immune evasion. Therefore, knowledge of the distinct role of tumor-associated macrophages in immune evasion can help design therapeutics such as engineered macrophages, M2 targeting drugs, and novel macrophage-mediated drug delivery strategies for long-term survival in breast cancer.
1 Introduction
Breast Cancer (BC) is a leading cause of morbidity and mortality in females affecting more than 2 million individuals annually (1). This is due to multiple factors, the most important being tumoral heterogeneity.
Breast cancer heterogeneity, facilitated by clonal evolution of the tumor or by cancer stem cells (2) is associated with therapeutics that lead to the selection and propagation of drug-resistant clones, causing resistance to treatment modalities (3–5). At least 20% of patients receiving treatment for breast cancer relapse within 5 years, resulting in metastatic disease with poor outcomes (6, 7).
To facilitate cancer progression and resistance to treatment modalities, tumor cells interact with various cellular components within the microenvironment to create a highly favorable tumor microenvironment (TME). The TME components consist of tumor cells, immune cells, re-programmed stromal cells, and immunosuppressive cells such as the Myeloid-Derived Suppressor Cells (MDSCs), regulatory T cells (Tregs), Tumor-Associated Macrophages (TAMs), cytokines and chemokines, etc. These components not only facilitate the generation of a hypoxic environment and epithelial to mesenchymal transition (EMT), they also play a significant role in the suppression of innate immune response and exhaustion of cytotoxic T and Natural Killer (NK) cells (8), thus promoting a vicious cycle that facilitates uncontrolled proliferation and tumor growth (9–11).
Though all components of the TME impact immune evasion and therapeutic resistance, tumor-associated macrophages are key modulators of these events. This is due to their ability to undergo cellular plasticity that catalyzes their phenotypic change from M1 pro-inflammatory to M2 anti-inflammatory phenotype in response to external and internal stimuli (12). In the TME, the dynamic interaction between the tumor cells and macrophages stimulates cellular plasticity and the M2 phenotype. M2 macrophages or TAMs, due to their immunosuppressive nature, promote drug resistance, making these cells attractive targets for cancer therapeutics (9, 13).
In this review, we aim to focus on the impact of TAM cellular plasticity on breast cancer immunotherapy. Novel approaches to TAM targeting that enhance immunotherapy efficacy and counter therapeutic resistance have been discussed to provide a broad understanding of the role of TAMs in improving patient outcomes in breast cancer.
1.1 Classically activated macrophages (M-1 phenotype)
Macrophages activate and modulate the innate immune response through phagocytosis, cellular cytotoxicity, secretion of pro-inflammatory cytokines such as Interleukin-6 (IL-6), IL-12, tumor necrosis factor (TNF), and antigen presentation to cytotoxic T cells through the major histocompatibility complex II (MHC-II) molecules (14). Their phenotype is significantly influenced by the tumors they infiltrate (15). In breast cancer, macrophages constitute greater than 50% of the tumor-infiltrating cells indicating their significance in the pathogenesis of BC (16).
Macrophages are classified into the M1-anti-tumor phenotype and the M2-pro-tumor phenotype (17). M1 Macrophages overexpress cell surface markers CD80, CD86, and CD16/32 (18) as shown in Supplementary Figure 1, leading to the activation of T cells. Furthermore, signaling through Toll-like receptors (TLRs) in M1 macrophages stimulates the activation of various transcription factors such as nuclear factor kappa-light-chain-enhancer (NF-κB) which in turn initiates the production of pro-inflammatory cytokines such as Interferon-γ (IFN-γ) (19, 20). In addition to this, the IFN-γ produced by T Helper Cell 1 (Th1) promotes M1 macrophage polarization and upregulation of nitric oxide synthase-2 (NOS2) production (21). NOS2 is a major activator of cytotoxic activity against tumor cells via oxidation of L-arginine to L-citrulline and cytotoxic nitric oxide (22, 23). The role of M1 macrophages in eradicating tumor cell proliferation and growth in BC has been evidenced in several studies. For example, a survey of forty HER2+ breast cancer patients, treated with trastuzumab reported that patients with elevated levels of M1-like macrophages (iNOS+) exhibited significantly improved survival. In contrast, high levels of M2-like macrophages (CD163+) were associated with poor prognosis (24).
1.2 M2 phenotype or tumor-associated macrophages
Pro-tumor macrophages or anti-inflammatory Tumor-Associated Macrophages (TAMs) are derived from circulating monocytes and tissue-resident macrophages. Tumor cells initiate recruitment of monocyte-derived circulating macrophages by production of Colony Stimulating Factor 1 (CSF1) and CC chemokine Ligand 2 (CCL2), leading to their binding to CSF1 receptor (CSF1R) and CCL2 receptor (CCR2) on the macrophage cell surface. This binding initiates the conversion of M1 anti-tumoral macrophage into a tumor-associated M2 macrophage (25, 26). On the other hand, Tissue-resident macrophages (TRMs), upon tumoral stimulation, activate Tregs thus initiating epithelial-mesenchymal transition (EMT), tumor invasion, and metastasis via the TAM phenotype (27).
Bone-marrow-derived monocytes can also convert to M2 phenotype when stimulated with immunosuppressive cytokines such as IL-10, or TGF-β in the TME, via phosphatidylinositol 3 kinase (PI3K)/protein kinase B (Akt) signaling pathway (28, 29). Studies on mouse models of breast cancer have documented that IL-10 secreted by TAMs prevents CD8+ T cell cytotoxic activity by inhibiting IL-12 expression on dendritic cells required for anti-tumoral differentiation of T cells into T helper-1 (Th1) cells (30). Moreover, IL-4 and IL-13 secreted by M2 macrophages stimulate T helper-2 (Th2) to secrete anti-inflammatory cytokines and chemokines that inhibit M1 polarization (31), and promote tumor cell growth and proliferation.
Hypoxia within the tumor microenvironment induces the TAM/M2 phenotype. Rapid tumor growth leads to hypoxic conditions and lactic acid accumulation within the TME leading to Vascular Endothelial Growth Factor (VEGF) and Arginase I (ARG-1) expression by macrophages (32). VEGF is a critical factor associated with the promotion of tumor angiogenesis and support for sustained tumor growth. On the other hand, ARG-1 hydrolyzes L-arginine (an essential amino acid required for T-cells and NK cell activation) to urea and L-ornithine (Supplementary Figure 1) leading to the inhibition of T and NK cell activation and proliferation (33, 34)
Studies on pre-clinical BC models have reported that Granulocyte-macrophage colony-stimulating factor (GM-CSF) secreted by tumor cells promotes programmed death ligand-1 (PD-L1) overexpression on TAM cell surface, thereby deactivating CD8+ cytotoxic T cells via the immunosuppressive PD-1/PD L1 interaction (35). Thus, it is evident that TAMs contribute, via multiple mechanisms, to promoting the uncontrolled proliferation of tumor cells and supporting immune evasion, leading to the invasion and metastasis of breast cancer.
1.3 TAM-targeted therapies for breast cancer
Strategies to reduce M2-macrophages in breast cancer tumor microenvironment include inhibition of macrophage recruitment, TAM depletion, and repolarization of pro-tumoral M2- -macrophages to the anti-tumoral M1 phenotype. Keeping these strategies in perspective, several novel TAM-targeting drugs are being tested in clinical trials and pre-clinical studies to improve the efficacy of BC immunotherapy, as shown in Table 1. Some of these drugs are discussed below:
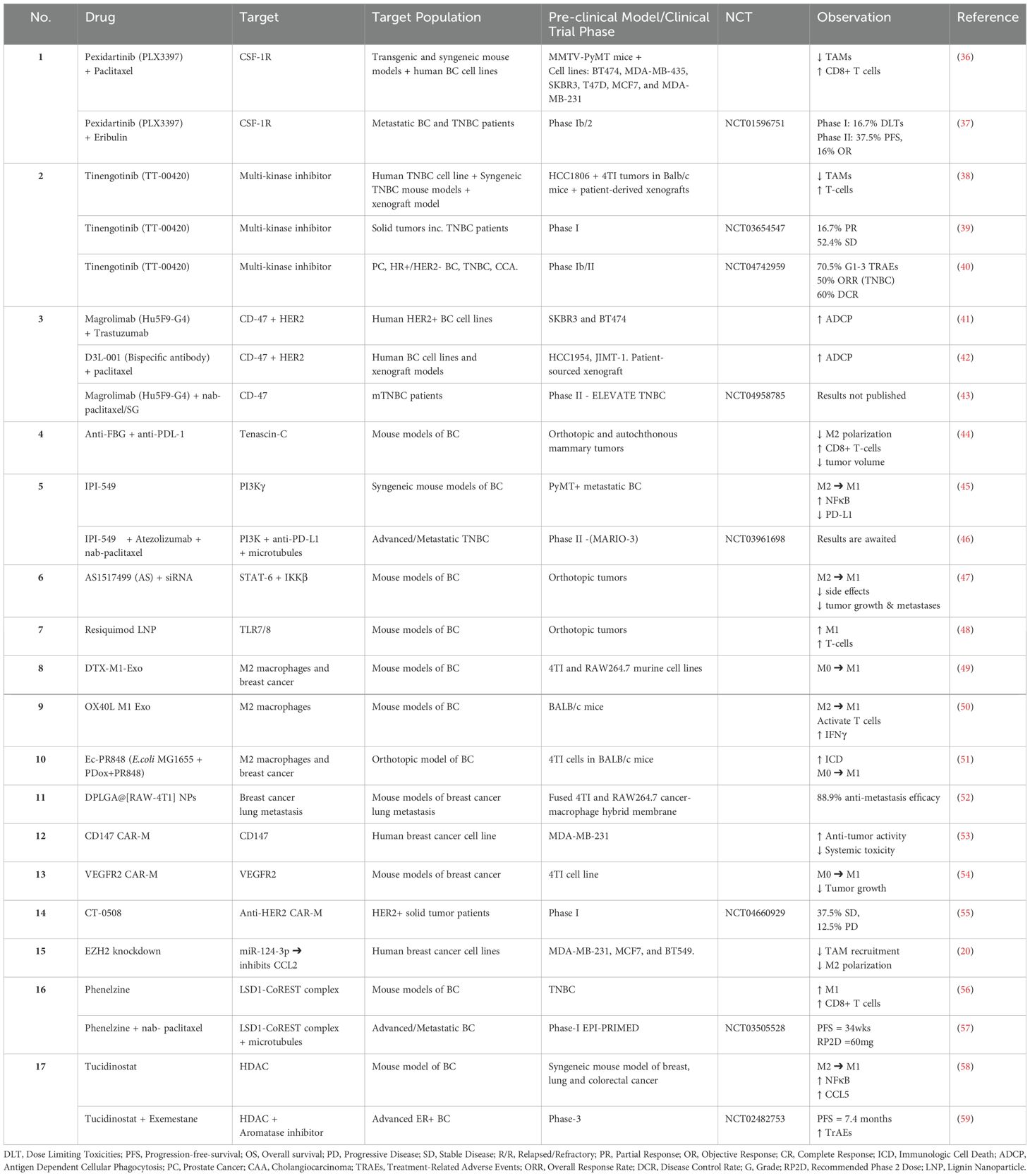
Table 1. Pre-clinical studies and clinical trials targeting TAMs in breast cancer.
1.3.1 Pexidartinib; anti-Colony Stimulating Factor-1 Receptor
Pexidartinib (PLX3397) is an oral, small-molecule inhibitor of Colony Stimulating Factor-1 Receptor (CSF1R). CSF1R is implicated in the recruitment of macrophages to the TME, and their polarization to an anti-tumor M2 phenotype. Mechanistically, CSF-1 produced by tumor cells, binds to CSF1-receptor (CSF1-R) on the macrophage cell surface as shown in Figure 1 (25). This binding alters macrophage metabolism via the downstream PI3K/Akt signaling pathway and promotes the expression of rapamycin complex -2 (mTORC-2), and interferon regulatory factor 4 (IRF4), leading to M2 macrophage polarization (60). Therefore, Pexidartinib, a CSF1-R inhibitor, reduces TAM recruitment within the TME and enhances the cytotoxic activity of immune cells.
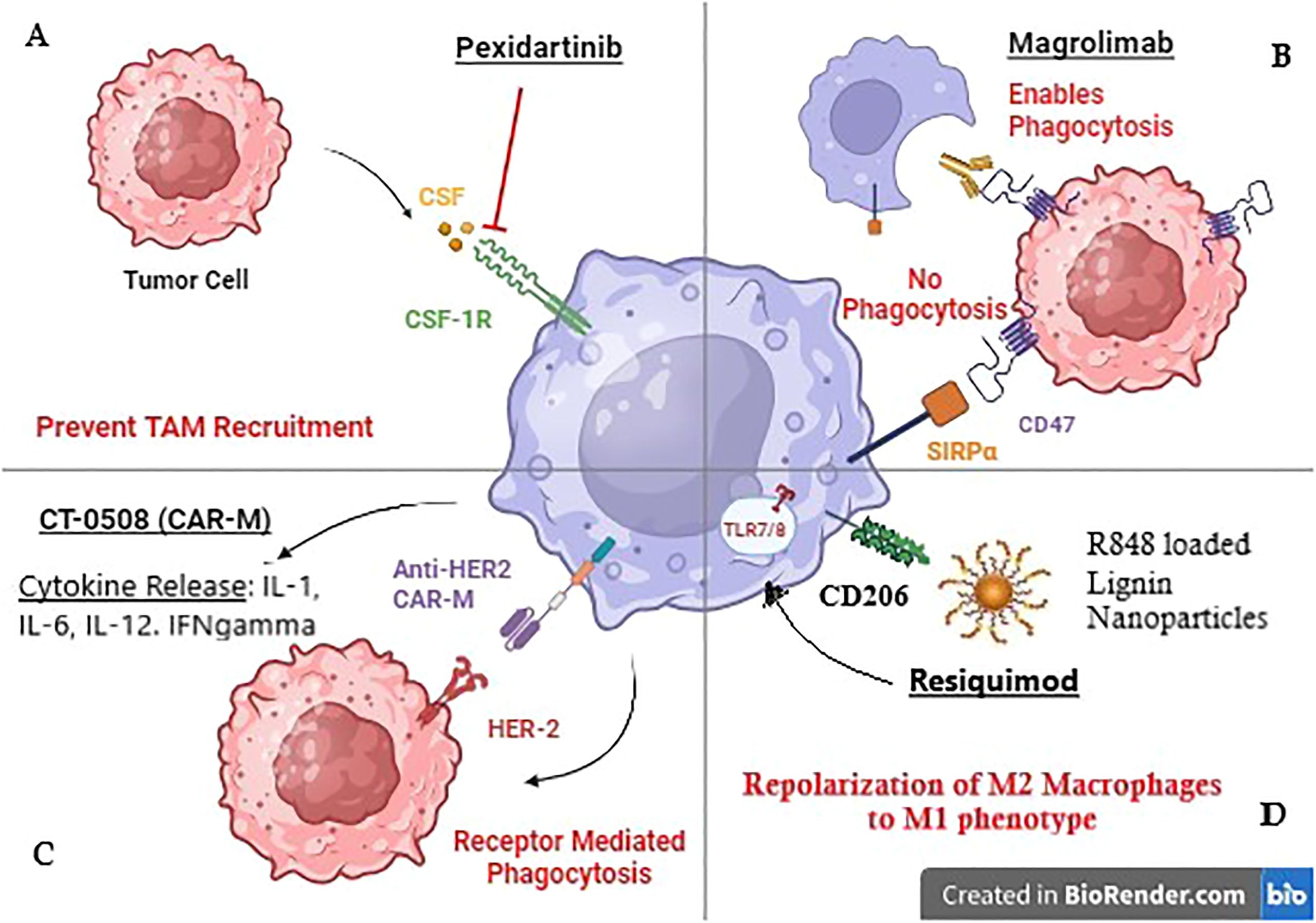
Figure 1. Mechanism of action of TAM-targeted therapies in Breast cancer. (A) Pexidartinib inhibits CSF-1R receptor on TAMS to prevent their recruitment to the tumor site. (B) Magrolimab blocks CD47 receptor on tumor cells to prevent its binding with Macrophage receptor SIRPa promoting phagocytosis. (C) CT-0508 is a HER-2 targeting CAR-M that has shown promising results in clinical trials. (D) Resiquimod is an R848 (TLR 7/8 agonist) loaded lignin nanoparticle targeting CD206 Mannose receptor on macrophages.
In pre-clinical mouse models, Pexidartinib has been shown to revert immune suppression, reduce tumor growth, and improve survival (36). A Phase Ib/II Clinical trial (NCT01596751) tested Pexidartinib (PLX3397) in combination with non-taxane chemotherapy Eribulin in metastatic breast cancer patients. Promising results from Phase Ib reported dose-limiting toxicities (at 800mg/day and 1000mg/day dose) in 16.7% of patients, while Phase II results showed 3 months of progression-free survival in 35.7% of TNBC patients and objective response in 16% of patients (61).
1.3.2 Tinengotinib (multi-kinase inhibitor)
Tinengotinib (TT-00420) is an oral, multi-kinase inhibitor that targets kinases Aurora A/B, Fibroblast Growth Factor Receptor (FGFR1/2/3), Vascular Endothelial Growth Factor Receptors (VEGFRs), Janus Kinases (JAK1/2), and CSF1R (38).
In pre-clinical studies on syngeneic mouse models of TNBC, Tinengotinib treatment resulted in up-regulation of chemokines CXCL10 and 11 (ligands for T-cell receptor CXCR3) leading to increased infiltration of T cells and reduced numbers of tumor-associated macrophages (TAMs). Inhibition of tumor proliferation, decreased angiogenesis, and an amplified immune response were observed in this study (38). The study provided evidence for a ‘first-in-human’ phase I clinical trial of Tinengotinib, for treating TNBC (NCT03654547) (62). Promising results were reported with 7/42 (16.7%) patients exhibiting partial response (PR) and 22/42 (52.4%) showing stable disease (SD). Additionally, the drug was well tolerated with a limited number of patients experiencing dose-limiting toxicities (39).
1.3.3 Magrolimab (Anti-CD47)
Tumor cell surface molecule CD47 binds to its ligand, the signal regulatory protein α (SIRPα) on the macrophage cell surface, and provides a safety signal, blocking phagocytosis (63). CD47 is overexpressed in HER2+ tumors. Macrophage checkpoint blockade, using the anti-CD47 antibody Magrolimab has been investigated in HER2+ breast cancer cells in combination with the anti-HER2 antibody Trastuzumab (41). Results showed upregulation of macrophage-mediated, Fc-dependent antibody-dependent cellular phagocytosis (ADCP).
A phase II clinical trial ELEVATE TNBC (NCT04958785) is underway in patients with locally advanced or metastatic TNBC. The trial combines CD47 inhibition through Magrolimab (Hu5F9-G4) with taxane chemotherapy nab-paclitaxel/paclitaxel or sacituzumab govitecan-hziy (SG) (44, 49, 52). SG is an antibody-drug conjugate that combines the anti-trophoblast cell-surface antigen 2 (Trop-2) antibody hRS7 IgG1, with a topoisomerase1 inhibitor SN-38, inhibiting DNA replication. Trop-2 is overexpressed in up to 90% of TNBC with poor prognoses (64, 65). The trial aims to investigate the potential of macrophage-mediated ADCP due to the amplification of phagocytic signals on the tumor cell surface after taxane treatment (43).
D3L-001, a bispecific antibody against HER2 and CD47 was investigated in HER-2+ breast cancer cell lines HCC1954, JIMT-1, and patient-sourced xenograft models of breast cancer. Results showed significant anti-tumor activity due to the synergistic effect of simultaneous dual targeting of HER2 and CD47 leading to antibody-dependent cellular phagocytosis. D3L-001 also showed improved efficacy when combined with chemotherapy such as paclitaxel (42).
1.3.4 Anti-FBG (Fibrinogen-like globe) antibody
Tenascin-C (TNC) is an extracellular matrix (ECM) glycoprotein, overexpressed in solid tumors. Tumor-derived Tenascin-C facilitates immunosuppression and promotes pro-tumor (M2) macrophage phenotype via activation of macrophage toll-like receptor-4 (TLR-4) through the binding of the TNC protein C-terminal Fibrinogen-like globe (FBG) domain (66). This is a paradoxical situation as TLR-4 classically activates the pro-inflammatory M1 macrophage phenotype.
Pre-clinical studies on murine models of breast cancer have documented that, anti-FGB treatment, when combined with anti-PDL-1 immunotherapy, led to reduced polarization of macrophages to TAMs, increased infiltration of CD8+ cytotoxic T lymphocytes, and significant reduction of tumor volume and lung metastases (44).
1.4 Macrophage re-polarization therapy
Macrophages constitute 50% of the cells infiltrating the TME, thus repolarizing anti-inflammatory M2 TAMs to pro-inflammatory M1 phenotype can greatly turn the odds in our favor (16). Multiple strategies have been evaluated in this regard, including but not limited to, Phosphatidylinositol 3-kinases (PI3Ks) inhibitors, STAT inhibitors, and Toll-like receptors agonists (TLR-3/4/7&8/9 agonists) (67).
PI3Ks promote cell survival, proliferation, and differentiation (68). PI3Kγ, via Akt and mTOR signaling, blocks NF-κB activation, and stimulates transcription factor CCAAT/enhancer binding protein β (C/EBPβ). C/EBPβ, in turn, promotes the M2-associated genes (including Arg1, Il10, and Mrc1), causing immunosuppression (69, 70). In syngeneic mouse models of breast cancer, PI3K-γ inhibitor IPI-549 repolarized TAMs from M2 to M1, stimulated macrophage NF-κB expression, and reduced PD-L1 expression, thus promoting CD8 + T cell cytotoxicity (45). A phase II clinical trial MARIO-3 (Macrophage Reprogramming in Immuno-Oncology) (NCT03961698) is currently underway to evaluate IPI-549 in combination with Atezolizumab and nab-paclitaxel as first-line therapy in advanced/metastatic TNBC patients. The results of the trial are awaited (46).
The JAK/STAT signaling pathway is integral to M1/M2 macrophage polarization (69). The STAT6 transcription factor regulates genes such as arginase 1 (Arg1), and macrophage mannose receptor 1 (Mrc1) required for IL-4-mediated M2 macrophage polarization (71). Transcription factor NF-κB activates a pro-inflammatory TME (72). Upstream inhibitors of NF-κB activation, Inhibitory kappa B kinases (IKKs), prevent inflammatory response and M1 macrophage polarization (73). An M2 macrophage targeting, Ph-sensitive, PEG-coated nanodrug encapsulating STAT6 inhibitor AS1517499 (AS) and IKKβ siRNA was tested in orthotopic mouse models of breast cancer. The therapy reduced tumor growth and metastasis and caused M2 to M1 macrophage polarization. Targeted drug delivery to TAMs also reduced immune side effects (47).
Toll-like receptors (TLRs) are transmembrane or intracellular pattern recognition receptors on antigen-presenting cells such as macrophages (74). Endosomal TLR 7/8 recognizes single-stranded RNA to initiate an inflammatory response (75). In pre-clinical orthotopic mouse models of TNBC, TLR 7/8 agonist Resiquimod was tested through lignin nanoparticles (LNPs). These LNPs targeted CD206+ M2 TAMs via the mUNO peptide, reducing off-target effects, as shown in Figure 1. Results showed increased numbers of M1 macrophages and cytotoxic T cells. The therapy had a synergistic effect with chemotherapy Vinblastine (48).
These studies testify to the impact of Macrophage polarization on tumor growth, proliferation, and drug resistance.
1.5 Engineered macrophages for drug delivery
Macrophages are the most abundant immune cells in the tumor microenvironment, making them attractive agents for drug delivery. They also have a prolonged half-life and an inherent ability to migrate to the tissue microenvironment, thus reducing off-target effects and associated toxicities. The applications of engineered macrophages in cancer immunotherapy include macrophage-mediated drug delivery, chimeric antigen receptor macrophage (CAR-M) therapy, and combined treatment approaches deploying bacterial drug delivery and macrophage exosomes (76).
1.5.1 Docetaxel-M1; DTX-M1-exosomes
Immune cell-derived exosomes can be used as vehicles to deliver chemotherapeutic drugs such as docetaxel (DTX). A pre-clinical study investigated anti-tumoral M1 macrophage-derived exosomes (M1-Exo) loaded with Docetaxel to create the DTX-M1-Exo drug delivery system. The experiment utilized the 4T1 murine breast cancer cell line and RAW264.7 murine macrophage cell line for in-vivo mouse models of breast cancer. The therapy induced polarization of M0 macrophages to the M1 phenotype in the immunosuppressive TME, while inhibiting repolarization to the pro-tumoral M2 phenotype. Thus, DTX-M1-Exo is a novel treatment combining immunotherapy through macrophage polarization with docetaxel chemotherapy to achieve substantial antitumor therapeutic efficacy (49).
1.5.2 OX40L M1 exosomes
The OX40 receptor is expressed on the surface of immune cells, such as CD8+ T cells while its ligand OX40L is expressed on antigen-presenting cells, such as macrophages (77). In an in-vivo study conducted on mouse models of breast cancer, OX40L M1-exosomes accumulated in tumor tissues, repolarized M2-macrophages into M1-macrophages and promoted phagocytosis. OX40L M1-exos activated T-cells by binding to OX40 on their cell surface, promoting IFN-γ secretion. Thus, the dual impact of innate and adaptive immunity successfully blocked the growth and metastasis of mouse breast cancer (50).
1.5.3 Bacterial therapy
Bacteria have tumor-homing ability and can survive in hypoxic conditions in the TME. They are thus being deployed as therapeutic carriers for anti-tumor drugs. Ec-PR848 comprises E.coli MG1655 loaded with 2 types of nanoparticles. The PDOX nanoparticles (NP) consist of chemotherapy doxorubicin (Dox)-loaded onto poly lactic-co-glycolic acid (PLGA) core, while PR848 consists of toll-like receptor 7/8 (TLR7/8) agonist Resiquimod (R848) loaded onto PLGA. R8484 binds to macrophage endosomes to promote an anti-tumor M1 phenotype (51). This combines macrophage re-polarization therapy with chemotherapy-induced immunologic cell death (ICD), which activates cytotoxic T cells and promotes their infiltration into the TME.
1.5.4 Cell membrane-based nanoparticles
In a pre-clinical study on mouse models of breast cancer lung metastasis, cell membranes from the mouse macrophage RAW264.7 cells and mouse breast cancer 4T1 cells were fused to create a macrophage-cancer hybrid membrane. This was then coated onto doxorubicin (Dox)-loaded poly (lactic-co-glycolic acid) (PLGA) NPs (DPLGA@[RAW-4T1] NPs) for treating breast cancer lung metastasis in-vivo. The results exhibited 88.9% anti-metastasis efficacy due to metastasis targeting through the α4 and β1 integrin expression, on the macrophage membrane thus showing potential as a future drug delivery strategy in metastatic disease (52).
1.5.5 CAR-M cellular therapy
Adoptive Cellular therapy utilizing Chimeric Antigen Receptor T cells (CAR-T) cells has evolved to incorporate genetically engineered Macrophages (CAR-Ms). CAR-Ms offer the potential benefit of producing Matrix Metalloproteases (MMPs) that degrade the ECM allowing greater access to the tumor site (78). This enables CAR-Ms to phagocytose tumor cells, present tumor antigens to T cells, and activate the TME as shown in Supplementary Figure 2 (55). Macrophages express CD46, the docking protein for group B adenoviruses such as Ad35, making Adenoviruses reliable vectors for macrophage engineering (79).
CD-147 is overexpressed in multiple tumors including breast cancer. In a preclinical study on MDA-MB-231 cell lines CD147CAR-Ms were investigated. Enhanced antitumor activity and reduced systemic toxicity were observed indicating the role of CAR-M as a therapeutic tool to target cancer cells in vitro (53).
Vascular Endothelial Growth Factor Receptor (VEGFR2) is overexpressed in multiple tumors. Targeting of VEGFR2 via CAR-Ms was evaluated in 4T1 breast cancer mouse models. CAR-Ms were activated through TLR-4 and/or IFN-γ receptors leading to M1 macrophage polarization. This is due to the TLR intracellular domain of CAR-M. Enhanced macrophage infiltration into the tumors was observed resulting in reduced tumor size. This indicates the utility of CAR-Ms as inhibitors of BC growth and proliferation (54)
A first-in-human clinical trial (NCT04660929) is currently underway in HER2 overexpressing solid tumors including breast cancer. The trial is investigating CT-0508, an anti-HER2 CAR Macrophage, and Phase-1 results have shown that CT-0508 is well tolerated without any dose-limiting toxicities, adverse events, or on-target off-tumor activity. The trial reported 37.5% (3/8 patients) with stable disease and only 1 out of 8 patients exhibited progressive disease. Mechanistically, CT-0508 modulated the TME causing T cell activation, proliferation, and infiltration into the TME, making it a novel therapeutic agent in BC (55).
1.6 Epigenetic control of macrophage polarization
Epigenetic alterations induce phenotype changes by mediating the expression of transcription factors through histone modification, DNA methylation, histone acetylation and deacetylation, Micro RNA, etc.
These epigenetic alterations are also capable of influencing macrophage plasticity in the TME (76) due to their unique epigenomic memory leading to rapid phenotypic changes from recurrent environmental signals (80).
Enhancer of Zeste Homolog 2 (EZH2) causes histone H3 tri-methylation and is implicated in TAM polarization (81). Studies on CRISPR Cas9-mediated EZH2 knockdown in breast cancer cell lines MDA-MB-231, MCF7, and BT549, reported DNA demethylation and consequent upregulation of miR-124-3p and inhibition of its target gene CCL2, which helps recruit macrophages to the tumor site (20). This reduced TAM infiltration into the TME and inhibited M2 polarization (Qian et al., 2011).
Lysine-specific histone demethylase 1A (LSD1), is differentially expressed in M1 and M2 macrophages and is implicated in the epigenetic regulation of EMT, and cancer stem cell (CSC) genes (82). In murine models of TNBC, LSD1 inhibitor Phenelzine targets LSD1 binding domains nuclear REST corepressor 1 (CoREST) and flavin adenine dinucleotide (FAD) to disintegrate the LSD1-CoREST complex (56). This induces the expression of M1 macrophage genes, establishes the M1 phenotype, increases CD8+ T-cell infiltration into the TME, and improves the efficacy of anti-PD-1 immunotherapy (83, 84). A phase I clinical trial EPI-PRIMED (NCT03505528) evaluated the impact of Phenelzine Sulfate in combination with nab-paclitaxel in patients with advanced/metastatic breast cancer. Results showed median PFS at 34 weeks and established 60mg Phenelzine as the recommended Phase 2 dose (57).
Histone deacetylase (HDAC) is a molecular eraser that causes transcriptional repression (85). HDAC inhibitors stop cancer cell proliferation and upregulate MHC class I genes (86). In syngeneic murine breast, lung, and colorectal cancer models, HDAC inhibitor Tucidinostat polarized M2 macrophages to the M1 phenotype by activating the NF-κB signaling pathway, promoted the expression of chemoattractant CCL5 leading to the infiltration of CD8 + T cells, and decreased tumor resistance to anti-PD-L1 immunotherapy (58). A Phase 3 clinical trial NCT02482753 assessed the safety and efficacy of Tucidnostat (Chidamide) in combination with aromatase inhibitor Exemestane in patients with advanced Estrogen Receptor + (ER+) BC. Results showed improved PFS with a higher incidence of grade 3/4 hematological adverse events (59).
Alternatively, the transcription factor Zinc-finger E-box-binding homeobox 1 (ZEB1) facilitates EMT and is crucial for breast cancer invasion and metastasis. It causes the acetylation of DNA Methyl Transferase 1 (DNTM1), through histone acetyltransferase p300, resulting in increased expression. TAMs support DNMT1 overexpression through the IL-6-pSTAT3-ZEB1-DNMT1 pathway, that in turn supports breast cancer invasion (87). This elucidates the significance of epigenetics in promoting or halting cancer growth and metastasis.
1.6.1 Micro RNA therapy
Micro RNAs are small non-coding RNAs implicated in macrophage activation, polarization, and cytokine secretion. MicroRNAs known to impact macrophage polarization include miR-147 and miR-223. These miRNAs when activated by TLR stimulation, downregulate macrophage production of pro-inflammatory cytokines IL-6 and IL-1β, indicating a pro-tumoral effect (88, 89). Similarly, miR-125a-5p when stimulated by TLR2/4, promotes M2 macrophage polarization via IL-4 and downregulates the M1 macrophage phenotype, (90) while miR-21 induces M2 macrophage polarization via blocking the JAK2/STAT1 signaling pathway (91). Knowledge of the critical signaling pathways associated with these micro-RNAs can facilitate miRNA-targeted drug development, and stimulate an anti-tumor TME for breast cancer immunotherapy.
For M1 macrophage polarization, miR-16 has been shown to influence polarization in mouse peritoneal macrophages via overexpression of M1 marker CD16/32, cytokine IL-12, and nitric oxide. Moreover, it also induces depletion of M2 marker CD206 indicating TAM inhibition. In addition to this, miR-16 also downregulates macrophage expression of PD-L1 (92), creating a conducive environment for immune cell infiltration. Similarly, miR-17, miR-20a, and miR-106a, when activated by lipopolysaccharide (LPS), have been reported to cause reduced signal regulatory protein α (SIRPα) expression, and promote pro-inflammatory cytokine secretion, (93) thus initiating an anti-tumor immune response.
2 Conclusion
Immunotherapy heralded as the miracle cure, has seen limited success in solid tumors, this is mainly due to the fibrotic and immunosuppressive TME. Utilizing immune cells abundantly found in the TME such as macrophages, has the potential to specifically target tumor cells, limiting systemic toxicities such as inflammation.
Therapies targeted towards the anti-tumor M2 Macrophages have shown positive results in pre-clinical studies. Deploying targeted therapies to revert the immunosuppressive nature of the TME can help eliminate tumors and prevent recurrence. Macrophage therapy despite showing promising results in pre-clinical studies, is still in its infancy and needs further characterization for safety and reliability. Emerging results from the first-in-human clinical trials shall light the path for the future of this innovative therapy.
Engineered macrophages hold great promise as drug delivery vehicles and as agents to influence an anti-tumor TME. Further research into lowering costs and creating a standardized version of this personalized therapy can help bring these therapies to the clinic and benefit patients with metastatic, relapsed/refractory breast tumors.
Author contributions
AS: Conceptualization, Investigation, Writing – original draft, Writing – review & editing. AR: Conceptualization, Formal Analysis, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1457491/full#supplementary-material
Supplementary Figure 1 | Differences between M1 and M2 Macrophage Phenotype. Specific differences in activating molecules, cell surface receptors, secreted molecules, and Arginine metabolism.
Supplementary Figure 1 | Properties of CAR-M cells. 1. CAR-M can recognize tumor-associated antigens (TAA) to initiate phagocytosis. 2. They secrete pro-inflammatory cytokines to facilitate immune cell infiltration. 3. They secrete Matrix Metalloproteinases (MMPs) to degrade extracellular matrix (EMC) and facilitate T-cell infiltration and drug access. 4. CAR-M present TAA to CD8+ T-cells via MHC-II molecules to activate cytotoxic immune response.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer J Clin. (2021) 71:209–49. doi: 10.3322/CAAC.21660
2. Nowell PC. The clonal evolution of tumor cell populations. Science. (1976) 194:23–8. doi: 10.1126/SCIENCE.959840
3. Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol. (2017) 15:81–94. doi: 10.1038/nrclinonc.2017.166
4. Sivakumar S, Jin DX, Tukachinsky H, Murugesan K, McGregor K, Danziger N, et al. Tissue and liquid biopsy profiling reveal convergent tumor evolution and therapy evasion in breast cancer. Nat Commun. (2022) 13:1–13. doi: 10.1038/s41467-022-35245-x
5. Venizelos A, Engebrethsen C, Deng W, Geisler J, Geisler S, Iversen GT, et al. Clonal evolution in primary breast cancers under sequential epirubicin and docetaxel monotherapy. Genome Med. (2022) 14:1–18. doi: 10.1186/S13073-022-01090-2/FIGURES/5
6. Kennecke H, Yerushalmi R, Woods R, Cheang MCU, Voduc D, Speers CH, et al. Metastatic behavior of breast cancer subtypes. J Clin Oncology : Off J Am Soc Clin Oncol. (2010) 28:3271–7. doi: 10.1200/JCO.2009.25.9820
7. Wu Q, Li J, Zhu S, Wu J, Chen C, Liu Q, et al. Breast cancer subtypes predict the preferential site of distant metastases: a SEER based study. Oncotarget. (2017) 8:27990. doi: 10.18632/ONCOTARGET.15856
8. Chung W, Eum HH, Lee HO, Lee KM, Lee HB, Kim KT, et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat Commun. (2017) 8:1–12. doi: 10.1038/ncomms15081
9. Deepak KGK, Vempati R, Nagaraju GP, Dasari VR, Nagini S, Rao DN, et al. Tumor microenvironment: Challenges and opportunities in targeting metastasis of triple negative breast cancer. Pharmacol Res. (2020) 153:104683. doi: 10.1016/J.PHRS.2020.104683
10. Zhao H, Yin X, Wang L, Liu K, Liu W, Bo L, et al. Identifying tumour microenvironment-related signature that correlates with prognosis and immunotherapy response in breast cancer. Sci Data. (2023) 10:1–14. doi: 10.1038/s41597-023-02032-2
11. Salemme V, Centonze G, Cavallo F, Defilippi P, Conti L. The crosstalk between tumor cells and the immune microenvironment in breast cancer: implications for immunotherapy. Front Oncol. (2021) 11:610303/BIBTEX. doi: 10.3389/FONC.2021.610303/BIBTEX
12. Lüönd F, Tiede S, Christofori G. Breast cancer as an example of tumour heterogeneity and tumour cell plasticity during Malignant progression. Br J Cancer. (2021) 125:164–75. doi: 10.1038/s41416-021-01328-7
13. Yu T, Di G. Role of tumor microenvironment in triple-negative breast cancer and its prognostic significance. Chin J Cancer Res. (2017) 29:237. doi: 10.21147/J.ISSN.1000-9604.2017.03.10
14. Franken L, Schiwon M, Kurts C. Macrophages: sentinels and regulators of the immune system. Cell Microbiol. (2016) 18:475–87. doi: 10.1111/CMI.12580
15. Niu Y, Chen J, Qiao Y. Epigenetic modifications in tumor-associated macrophages: A new perspective for an old foe. Front Immunol. (2022) 13:836223/BIBTEX. doi: 10.3389/FIMMU.2022.836223/BIBTEX
16. Hill BS, Sarnella A, D’Avino G, Zannetti A. Recruitment of stromal cells into tumour microenvironment promote the metastatic spread of breast cancer. Semin Cancer Biol. (2020) 60:202–13. doi: 10.1016/J.SEMCANCER.2019.07.028
17. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. (2002) 23:549–55. doi: 10.1016/S1471-4906(02)02302-5
18. Yunna C, Mengru H, Lei W, Weidong C. Macrophage M1/M2 polarization. Eur J Pharmacol. (2020) 877:173090. doi: 10.1016/J.EJPHAR.2020.173090
19. Rodriguez PC, Ochoa AC, Al-Khami AA. Arginine metabolism in myeloid cells shapes innate and adaptive immunity. Front Immunol. (2017) 8:93/BIBTEX. doi: 10.3389/FIMMU.2017.00093/BIBTEX
20. Wang YF, Yu L, Hu ZL, Fang YF, Shen YY, Song MF, et al. Regulation of CCL2 by EZH2 affects tumor-associated macrophages polarization and infiltration in breast cancer. Cell Death Dis. (2022) 13:1–15. doi: 10.1038/s41419-022-05169-x
21. Modolell M, Corraliza IM, Link F, Soler G, Eichmann K. Reciprocal regulation of the nitric oxide synthase/arginase balance in mouse bone marrow-derived macrophages by TH 1 and TH 2 cytokines. Eur J Immunol. (1995) 25:1101–4. doi: 10.1002/EJI.1830250436
22. MacMicking J, Xie QW, Nathan C. Nitric oxide and macrophage function. Annu Rev Immunol. (1997) 15:323–50. doi: 10.1146/ANNUREV.IMMUNOL.15.1.323/CITE/REFWORKS
23. Geeraerts X, Bolli E, Fendt SM, Van Ginderachter JA. Macrophage metabolism as therapeutic target for cancer, atherosclerosis, and obesity. Front Immunol. (2017) 8:289/BIBTEX. doi: 10.3389/FIMMU.2017.00289/BIBTEX
24. Honkanen TJ, Tikkanen A, Karihtala P, Mäkinen M, Väyrynen JP, Koivunen JP. Prognostic and predictive role of tumour-associated macrophages in HER2 positive breast cancer. Sci Rep. (2019) 9:1–9. doi: 10.1038/s41598-019-47375-2
25. Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. (2014) 74:5057–69. doi: 10.1158/0008-5472.CAN-13-3723/657718/AM/CSF1-CSF1R-BLOCKADE-REPROGRAMS-TUMOR-INFILTRATING
26. Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nat 2011. (2011) 475:222–5. doi: 10.1038/nature10138
27. Casanova-Acebes M, Dalla E, Leader AM, LeBerichel J, Nikolic J, Morales BM, et al. Tissue-resident macrophages provide a pro-tumorigenic niche to early NSCLC cells. Nature. (2021) 595:578–84. doi: 10.1038/s41586-021-03651-8
28. Cassetta L, Fragkogianni S, Sims AH, Swierczak A, Forrester LM, Zhang H, et al. Human tumor-associated macrophage and monocyte transcriptional landscapes reveal cancer-specific reprogramming, biomarkers, and therapeutic targets. Cancer Cell. (2019) 35:588–602.e10. doi: 10.1016/J.CCELL.2019.02.009
29. Vergadi E, Ieronymaki E, Lyroni K, Vaporidi K, Tsatsanis C. Akt signaling pathway in macrophage activation and M1/M2 polarization. J Immunol. (2017) 198:1006–14. doi: 10.4049/JIMMUNOL.1601515
30. Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho CMT, Pryer N, et al. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell. (2014) 26:623–37. doi: 10.1016/J.CCELL.2014.09.006/ATTACHMENT/32966374-2A6B-400A-80EB-941328B1EAE4/MMC2.PDF
31. Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol. (2010) 22:231–7. doi: 10.1016/J.COI.2010.01.009
32. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. (2014) 513:559–63. doi: 10.1038/nature13490
33. Steggerda SM, Bennett MK, Chen J, Emberley E, Huang T, Janes JR, et al. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J ImmunoTherapy Cancer. (2017) 5:1–18. doi: 10.1186/S40425-017-0308-4/FIGURES/8
34. Rath M, Müller I, Kropf P, Closs EI, Munder M, Li G, et al. ‘Metabolism via arginase or nitric oxide synthase: Two competing arginine pathways in macrophages’, Front Immunol. (2014) 5:120091. doi: 10.3389/FIMMU.2014.00532/BIBTEX
35. Yonemitsu K, Pan C, Fujiwara Y, Miyasato Y, Shiota T, Yano H, et al. GM-CSF derived from the inflammatory microenvironment potentially enhanced PD-L1 expression on tumor-associated macrophages in human breast cancer. Sci Rep. (2022) 12:1–12. doi: 10.1038/s41598-022-16080-y
36. DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discovery. (2011) 1:54–67. doi: 10.1158/2159-8274.CD-10-0028
37. Phase Ib/II Study of PLX 3397 and Eribulin in Patients With Metastatic Breast Cancer - Full Text View - ClinicalTrials.gov (n.). Available online at: https://classic.clinicaltrials.gov/ct2/show/NCT01596751 (Accessed August 5, 2023).
38. Peng P, Qiang X, Li G, Li L, Ni S, Yu Q, et al. Tinengotinib (TT-00420), a novel spectrum-selective small-molecule kinase inhibitor, is highly active against triple-negative breast cancer. Mol Cancer Ther. (2023) 22:205–14. doi: 10.1158/1535-7163.MCT-22-0012
39. Piha-Paul SA, Xu B, Raghav KPS, Meric-Bernstam F, Janku F, Dumbrava EE, et al. First-in-human, phase I study of TT-00420, a multiple kinase inhibitor, as a single agent in advanced solid tumors. J Clin Oncol. (2022) 40(16_suppl):3013–3. doi: 10.1200/JCO.2022.40.16_suppl.3013
40. Piha-Paul SA, Goel S, Liao C-Y, Gabrail NY, Dayyani F, Kazmi SMA, et al. Preliminary safety and efficacy of tinengotinib tablets as monotherapy and combination therapy in advanced solid tumors: A phase Ib/II clinical trial. J Clin Oncol. (2023) 41:3019–9. doi: 10.1200/JCO.2023.41.16_SUPPL.3019
41. Upton R, Banuelos A, Feng D, Biswas T, Kao K, Mckenna K, et al. Combining CD47 blockade with trastuzumab eliminates HER2-positive breast cancer cells and overcomes trastuzumab tolerance. Proc Natl Acad Sci U S A. (2021) 118. doi: 10.1073/pnas.2026849118/-/DCSupplemental
42. Rui H, Yang X, Chen J, Wang J, Zheng Z, Chen Z, et al. Abstract 1873: D3L-001, a novel bispecific antibody targeting HER2 and CD47, demonstrates potent preclinical efficacy in solid tumors. Cancer Res. (2023) 83:1873–3. doi: 10.1158/1538-7445.AM2023-1873
43. Rainey N, Joshi R, Chiu JWY, Chen A, Wang H, Odegard J, et al. Abstract OT2-10-01: A phase 2, randomized study of magrolimab combination therapy in adult patients with unresectable locally advanced or metastatic triple-negative breast cancer. Cancer Res. (2023) 83:OT2-10-01. doi: 10.1158/1538-7445.SABCS22-OT2-10-01
44. Deligne C, Murdamoothoo D, Gammage AN, Gschwandtner M, Erne W, Loustau T, et al. Matrix-targeting immunotherapy controls tumor growth and spread by switching macrophage phenotype. Cancer Immunol Res. (2020) 8:368–82. doi: 10.1158/2326-6066.CIR-19-0276/470872/AM/MATRIX-TARGETING-IMMUNOTHERAPY-CONTROLS-TUMOR
45. Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, et al. PI3Kγ is a molecular switch that controls immune suppression. Nature. (2016) 539:437–42. doi: 10.1038/nature19834
46. Study details | Evaluation of IPI-549 Combined With Front-line Treatments in Pts. With Triple-Negative Breast Cancer or Renal Cell Carcinoma | ClinicalTrials.gov. (n.). Available online at: https://clinicaltrials.gov/study/NCT03961698?cond=Breast%20Cancer&intr=IPI549&rank=2 (Accessed October 26, 2024).
47. Xiao H, Guo Y, Li B, Li X, Wang Y, Han S, et al. M2-like tumor-associated macrophage-targeted codelivery of STAT6 inhibitor and IKKβ siRNA induces M2-to-M1 repolarization for cancer immunotherapy with low immune side effects. ACS Cent Sci. (2020) 6:1208–22. doi: 10.1021/ACSCENTSCI.9B01235/ASSET/IMAGES/LARGE/OC9B01235_0007.JPEG
48. Figueiredo P, Lepland A, Scodeller P, Fontana F, Torrieri G, Tiboni M, et al. Peptide-guided resiquimod-loaded lignin nanoparticles convert tumor-associated macrophages from M2 to M1 phenotype for enhanced chemotherapy. Acta Biomaterialia. (2021) 133:231–43. doi: 10.1016/J.ACTBIO.2020.09.038
49. Zhao Y, Zheng Y, Zhu Y, Li H, Zhu H, Liu T. Docetaxel-loaded M1 macrophage-derived exosomes for a safe and efficient chemoimmunotherapy of breast cancer. J Nanobiotechnology. (2022) 20:1–12. doi: 10.1186/S12951-022-01526-2/FIGURES/9
50. Yu Y, Li T, Ou M, Luo R, Chen H, Ren H, et al. OX40L-expressing M1-like macrophage exosomes for cancer immunotherapy. J Controlled Release. (2024) 365:469–79. doi: 10.1016/J.JCONREL.2023.11.051
51. Wei B, Pan J, Yuan R, Shao B, Wang Y, Guo X, et al. Polarization of tumor-associated macrophages by nanoparticle-loaded escherichia coli combined with immunogenic cell death for cancer immunotherapy. Nano Lett. (2021) 21:4231–40. doi: 10.1021/ACS.NANOLETT.1C00209/ASSET/IMAGES/LARGE/NL1C00209_0005.JPEG
52. Gong C, Yu X, You B, Wu Y, Wang R, Han L, et al. Macrophage-cancer hybrid membrane-coated nanoparticles for targeting lung metastasis in breast cancer therapy. J Nanobiotechnology. (2020) 18:1–17. doi: 10.1186/S12951-020-00649-8/FIGURES/5
53. Chupradit K, Muneekaew S, Wattanapanitch M. Engineered CD147-CAR macrophages for enhanced phagocytosis of cancers. Cancer Immunology Immunotherapy. (2024) 73:1–15. doi: 10.1007/S00262-024-03759-6/FIGURES/6
54. Duan Z, Li Z, Wang Z, Chen C, Luo Y. Chimeric antigen receptor macrophages activated through TLR4 or IFN-γ receptors suppress breast cancer growth by targeting VEGFR2. Cancer Immunology Immunotherapy. (2023) 72:3243–57. doi: 10.1007/S00262-023-03490-8/METRICS
55. Reiss KA, Yuan Y, Ueno NT, Johnson ML, Gill S, Dees EC, et al. A phase 1, first-in-human (FIH) study of the anti-HER2 CAR macrophage CT-0508 in subjects with HER2 overexpressing solid tumors. J Clin Oncol. (2022) 40:2533–3. doi: 10.1200/JCO.2022.40.16_SUPPL.2533
56. Tan AHY, Tu WJ, McCuaig R, Hardy K, Donovan T, Tsimbalyuk S, et al. Lysine-specific histone demethylase 1A regulates macrophage polarization and checkpoint molecules in the tumor microenvironment of triple-negative breast cancer. Front Immunol. (2019) 10:1351/BIBTEX. doi: 10.3389/FIMMU.2019.01351/BIBTEX
57. Prasanna T, Malik L, McCuaig RD, Tu WJ, Wu F, Lim PS, et al. A phase 1 proof of concept study evaluating the addition of an LSD1 inhibitor to nab-paclitaxel in advanced or metastatic breast cancer (EPI-PRIMED). Front Oncol. (2022) 12:862427. doi: 10.3389/FONC.2022.862427
58. Zhang P, Du Y, Bai H, Wang Z, Duan J, Wang X, et al. Optimized dose selective HDAC inhibitor tucidinostat overcomes anti-PD-L1 antibody resistance in experimental solid tumors. BMC Med. (2022) 20:1–19. doi: 10.1186/S12916-022-02598-5/FIGURES/7
59. Jiang Z, Li W, Hu X, Zhang Q, Sun T, Cui S, et al. Tucidinostat plus exemestane for postmenopausal patients with advanced, hormone receptor-positive breast cancer (ACE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. (2019) 20:806–15. doi: 10.1016/S1470-2045(19)30164-0
60. Huang SCC, Smith AM, Everts B, Colonna M, Pearce EL, Schilling JD, et al. Metabolic reprogramming mediated by the mTORC2-IRF4 signaling axis is essential for macrophage alternative activation. Immunity. (2016) 45:817–30. doi: 10.1016/J.IMMUNI.2016.09.016/ATTACHMENT/4A8BDC05-9D16-482D-81AF-7878681C2E69/MMC2.PDF
61. Study Results | Phase Ib/II Study of PLX 3397 and Eribulin in Patients With Metastatic Breast Cancer | ClinicalTrials.gov. (n.). Available online at: https://clinicaltrials.gov/study/NCT01596751?cond=Breast%20Cancer&intr=PLX3397&rank=1&tab=resultsoutcome-measures (Accessed September 25, 2024).
62. Study Record | ClinicalTrials.gov. (n.d.-). Available online at: https://clinicaltrials.gov/study/NCT03654547?cond=Breast%20Cancer&term=STAT6%20inhibitor&rank=6 (Accessed August 4, 2023).
63. Willingham SB, Volkmer JP, Gentles AJ, Sahoo D, Dalerba P, Mitra SS, et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci United States America. (2012) 109:6662–7. doi: 10.1073/PNAS.1121623109/SUPPL_FILE/SM02.AVI
64. Bardia A, Mayer IA, Vahdat LT, Tolaney SM, Isakoff SJ, Diamond JR, et al. Sacituzumab govitecan-hziy in refractory metastatic triple-negative breast cancer. New Engl J Med. (2019) 380:741–51. doi: 10.1056/NEJMOA1814213/SUPPL_FILE/NEJMOA1814213_DATA-SHARING.PDF
65. Study Record | ClinicalTrials.gov. (n.d.-). Available online at: https://clinicaltrials.gov/study/NCT04958785?cond=Breast%20Cancer&intr=Magrolimab&rank=2 (Accessed August 10, 2023).
66. Swanson L, Katkar GD, Tam J, Pranadinata RF, Chareddy Y, Coates J, et al. TLR4 signaling and macrophage inflammatory responses are dampened by GIV/Girdin. Proc Natl Acad Sci United States America. (2020) 117:26895–906. doi: 10.1073/PNAS.2011667117/SUPPL_FILE/PNAS.2011667117.SAPP.PDF
67. Wang S, Wang J, Chen Z, Luo J, Guo W, Sun L, et al. Targeting M2-like tumor-associated macrophages is a potential therapeutic approach to overcome antitumor drug resistance. NPJ Precis Oncol. (2024) 8:1–19. doi: 10.1038/s41698-024-00522-z
68. Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet 2006 7:8. (2006) 7:606–19. doi: 10.1038/nrg1879
69. Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol. (2011) 11:750–61. doi: 10.1038/nri3088
70. Ruffell D, Mourkioti F, Gambardella A, Kirstetter P, Lopez RG, Rosenthal N, et al. A CREB-C/EBPβ cascade induces M2 macrophage-specific gene expression and promotes muscle injury repair. Proc Natl Acad Sci United States America. (2009) 106:17475–80. doi: 10.1073/PNAS.0908641106/SUPPL_FILE/0908641106SI.PDF
71. Takeda H, Tanaka T, Shi W, Matsumoto M, Minami M, Kashiwamura SI, et al. Essential role of Stat6 in IL-4 signalling. Nature. (1996) 380:627–30. doi: 10.1038/380627a0
72. Hagemann T, Lawrence T, McNeish I, Charles KA, Kulbe H, Thompson RG, et al. Re-educating” tumor-associated macrophages by targeting NF-κB. J Exp Med. (2008) 205:1261. doi: 10.1084/JEM.20080108
73. Gamble C, McIntosh K, Scott R, Ho KH, Plevin R, Paul A. Inhibitory kappa B kinases as targets for pharmacological regulation. Br J Pharmacol. (2012) 165:802. doi: 10.1111/J.1476-5381.2011.01608.X
74. Tran TH, Tran TTP, Truong DH, Nguyen HT, Pham TT, Yong CS, et al. Toll-like receptor-targeted particles: A paradigm to manipulate the tumor microenvironment for cancer immunotherapy. Acta Biomaterialia. (2019) 94:82–96. doi: 10.1016/J.ACTBIO.2019.05.043
75. Vizcaino Castro A, Daemen T, Oyarce C. Strategies to reprogram anti-inflammatory macrophages towards pro-inflammatory macrophages to support cancer immunotherapies. Immunol Lett. (2024) 267:106864. doi: 10.1016/J.IMLET.2024.106864
76. Yang S, Wang Y, Jia J, Fang Y, Yang Y, Yuan W, et al. Advances in engineered macrophages: A new frontier in cancer immunotherapy. Cell Death Dis. (2024) 15:1–12. doi: 10.1038/s41419-024-06616-7
77. Webb GJ, Hirschfield GM, Lane PJL. OX40, OX40L and autoimmunity: a comprehensive review. Clin Rev Allergy Immunol. (2015) 50:312–32. doi: 10.1007/S12016-015-8498-3
78. Chen Y, Yu Z, Tan X, Jiang H, Xu Z, Fang Y, et al. CAR-macrophage: A new immunotherapy candidate against solid tumors. Biomedicine Pharmacotherapy. (2021) 139:111605. doi: 10.1016/J.BIOPHA.2021.111605
79. Gaggar A, Shayakhmetov DM, Lieber A. CD46 is a cellular receptor for group B adenoviruses. Nat Med. (2003) 9:1408–12. doi: 10.1038/nm952
80. Ostuni R, Piccolo V, Barozzi I, Polletti S, Termanini A, Bonifacio S, et al. Latent enhancers activated by stimulation in differentiated cells. Cell. (2013) 152:157–71. doi: 10.1016/j.cell.2012.12.018
81. Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, et al. Role of histone H3 lysine 27 methylation in polycomb-group silencing. Science. (2002) 298:1039–43. doi: 10.1126/SCIENCE.1076997/SUPPL_FILE/CAOR1076997.SUP.PDF
82. Boulding T, McCuaig RD, Tan A, Hardy K, Wu F, Dunn J, et al. LSD1 activation promotes inducible EMT programs and modulates the tumour microenvironment in breast cancer. Sci Rep. (2018) 8:1–18. doi: 10.1038/s41598-017-17913-x
83. Qin Y, Vasilatos SN, Chen L, Wu H, Cao Z, Fu Y, et al. Inhibition of histone lysine-specific demethylase 1 elicits breast tumor immunity and enhances antitumor efficacy of immune checkpoint blockade. Oncogene 2018 38:3. (2018) 38:390–405. doi: 10.1038/s41388-018-0451-5
84. Zhou Z, van der Jeught K, Fang Y, Yu T, Li Y, Ao Z, et al. An organoid-based screen for epigenetic inhibitors that stimulate antigen presentation and potentiate T-cell-mediated cytotoxicity. Nat Biomed Eng. (2021) 5:1320–35. doi: 10.1038/s41551-021-00805-x
85. Teicher BA. Combinations of PARP, hedgehog and HDAC inhibitors with standard drugs. Curr Opin Pharmacol. (2010) 10:397–404. doi: 10.1016/j.coph.2010.04.014
86. Mohammad HP, Barbash O, Creasy CL. Targeting epigenetic modifications in cancer therapy: erasing the roadmap to cancer. Nat Med. (2019) 25:403–18. doi: 10.1038/s41591-019-0376-8
87. Li Z, Wang P, Cui W, Yong H, Wang D, Zhao T, et al. Tumour-associated macrophages enhance breast cancer Malignancy via inducing ZEB1-mediated DNMT1 transcriptional activation. Cell Bioscience. (2022) 12:1–13. doi: 10.1186/S13578-022-00913-4/FIGURES/6
88. Chen Q, Wang H, Liu Y, Song Y, Lai L, Han Q, et al. Inducible microRNA-223 down-regulation promotes TLR-triggered IL-6 and IL-1β Production in macrophages by targeting STAT3. PloS One. (2012) 7:e42971. doi: 10.1371/JOURNAL.PONE.0042971
89. Liu G, Friggeri A, Yang Y, Park YJ, Tsuruta Y, Abraham E. miR-147, a microRNA that is induced upon toll-like receptor stimulation, regulates murine macrophage inflammatory responses. Proc Natl Acad Sci United States America. (2009) 106:15819–24. doi: 10.1073/PNAS.0901216106/SUPPL_FILE/0901216106SI.PDF
90. Banerjee S, Cui H, Xie N, Tan Z, Yang S, Icyuz M, et al. MiR-125a-5p regulates differential activation of macrophages and inflammation. J Biol Chem. (2013) 288:35428–36. doi: 10.1074/JBC.M112.426866/ASSET/D95F10B3-9115-4ADB-96F3-AEB216DB6523/MAIN.ASSETS/GR7.JPG
91. Xi J, Huang Q, Wang L, Ma X, Deng Q, Kumar M, et al. miR-21 depletion in macrophages promotes tumoricidal polarization and enhances PD-1 immunotherapy. Oncogene. (2018) 37:3151–65. doi: 10.1038/s41388-018-0178-3
92. Jia X, Li X, Shen Y, Miao J, Liu H, Li G, et al. MiR-16 regulates mouse peritoneal macrophage polarization and affects T-cell activation. J Cell Mol Med. (2016) 20:1898–907. doi: 10.1111/JCMM.12882
Keywords: immunotherapy, breast cancer, tumor microenvironment, tumor-associated macrophages (TAMs), nanoparticle, epigenetics
Citation: Sami A and Raza A (2024) Reprogramming the tumor microenvironment – macrophages emerge as key players in breast cancer immunotherapy. Front. Immunol. 15:1457491. doi: 10.3389/fimmu.2024.1457491
Received: 30 June 2024; Accepted: 07 November 2024;
Published: 26 November 2024.
Edited by:
Reem Amr Assal, Heliopolis University, EgyptReviewed by:
Alessandro Poggi, San Martino Hospital (IRCCS), ItalyHaitao Wang, National Cancer Institute (NIH), United States
Byungheon Lee, Kyungpook National University, Republic of Korea
Copyright © 2024 Sami and Raza. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Afsheen Raza, UmF6YS5hZnNoZWVuQGFkdS5hYy5hZQ==