 Lu Yang
Lu Yang Sijia Li
Sijia Li- Department of Oncology, Suining Central Hospital, Suining, Sichuan, China
Glioblastoma(GBM) is a highly malignant primary central nervous system tumor that poses a significant threat to patient survival due to its treatment resistance and rapid recurrence.Current treatment options, including maximal safe surgical resection, radiotherapy, and temozolomide (TMZ) chemotherapy, have limited efficacy.In recent years, the role of glycolytic metabolic reprogramming in GBM has garnered increasing attention. This review delves into the pivotal role of glycolytic metabolic reprogramming in GBM, with a particular focus on the multifaceted roles of lactate, a key metabolic product, within the tumor microenvironment (TME). Lactate has been implicated in promoting tumor cell proliferation, invasion, and immune evasion. Additionally, this review systematically analyzes potential therapeutic strategies targeting key molecules within the glycolytic pathway, such as Glucose Transporters (GLUTs), Monocarboxylate Transporters(MCTs), Hexokinase 2 (HK2), 6-Phosphofructo-2-Kinase/Fructose-2,6-Biphosphatase 3 (PFKFB3), Pyruvate Kinase Isozyme Type M2 (PKM2), and the Lactate Dehydrogenase A (LDHA). These studies provide a novel perspective for GBM treatment. Despite progress made in existing research, challenges remain, including drug penetration across the blood-brain barrier, side effects, and resistance. Future research will aim to address these challenges by improving drug delivery, minimizing side effects, and exploring combination therapies with radiotherapy, chemotherapy, and immunotherapy to develop more precise and effective personalized treatment strategies for GBM.
1 Introduction
Glioblastoma (GBM) is a highly malignant and prognostically poor primary brain tumor, classified as Grade IV glioma by the World Health Organization (WHO), accounting for approximately 15% of all primary intracranial malignant tumors (1, 2). GBM is characterized by its aggressive nature, high recurrence rate, and treatment resistance, resulting in a dismal prognosis with a median survival of less than 15 months despite maximal safe surgical resection, radiotherapy, and temozolomide (TMZ) chemotherapy (3, 4). The primary challenges in GBM treatment stem from its high heterogeneity and complex microenvironment. Extensive research has implicated glycolytic metabolic reprogramming as a potential initiating event in GBM development and as a regulator of the plasticity that contributes to glioma heterogeneity (5, 6). Beyond providing essential energy and biosynthetic precursors for tumor cells, glycolytic metabolic reprogramming profoundly impacts the TME (7). The shift from mitochondrial oxidative phosphorylation (OXPHOS) to aerobic glycolysis (Warburg effect) is a hallmark of tumor cell growth and metastasis. Consequently, targeting aerobic glycolysis in tumor cells holds great promise for anti-tumor therapy. Studies have shown that reversing the energy production pathway of OXPHOS can induce the differentiation of GBM cells into astrocytes (8). Therefore, interventions targeting the abnormal glycolytic characteristics of GBM have emerged as a crucial direction in modern cancer therapy research. Significant progress has been made in understanding tumor metabolic reprogramming in GBM; however, the roles of lactate, the main product of glycolysis, in tumor development remain incompletely elucidated.
This study aims to explore the critical role of glycolytic metabolic reprogramming in GBM, with a specific focus on the mechanisms of lactate’s action within the TME. By thoroughly analyzing GBM glycolysis, this study seeks to provide a theoretical basis for developing novel therapeutic targets and improving clinical outcomes for GBM patients.
2 The role of lactate, a glycolytic metabolic product, in shaping the GBM tumor microenvironment and immune response
In physiological conditions, cells exposed to ample oxygen typically utilize the highly efficient and energy-rich OXPHOS system for energy production, maximizing energy extraction from glucose molecules and converting it into adenosine triphosphate (ATP) for cellular use (9). However, in malignant tumors like GBM, cancer cells exhibit a pathological metabolic shift, preferentially relying on glycolysis, an inefficient energy production pathway, even in the presence of sufficient oxygen (10). Although glycolysis produces fewer ATP molecules per glucose unit compared to OXPHOS, its rapid rate allows for the swift accumulation of ATP, fulfilling the energy demands of rapidly proliferating and invasive tumor cells (11, 12). This phenomenon, first observed by Otto Warburg in the 20th century, is widely known as the Warburg effect or aerobic glycolysis and is a common metabolic feature in numerous malignancies (Figure 1).
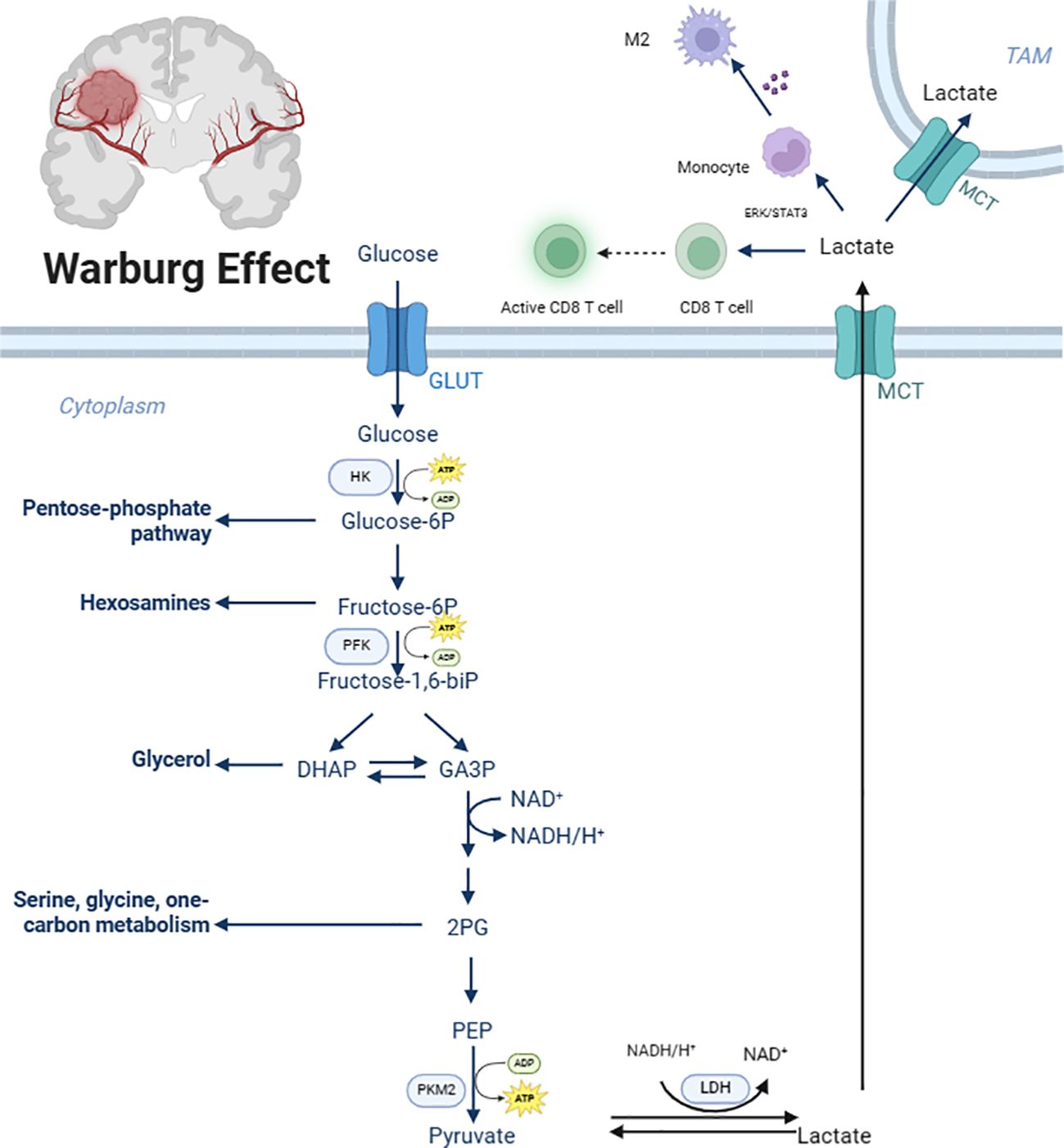
Figure 1. Glycolytic metabolic reprogramming in glioblastoma. GLUT, Glucose transporter; HK, Hexokinase 2; Glucose-6P, Glucose-6 phosphate; Fructose-6P, fructose-6-phosphate; PFK, Phosphofructokinase; fructose-1,6-bip, Fructose-1,6-biphosphate; GA3P, glyceraldehyde-3-phosphate; 2PG, 2-phosphoglycerate; PEP, Phosphoenolpyruvate; PKM2, Pyruvate Kinase 2; MCT, Monocarboxylate transporter. Created with BioRender.com.
2.1 Physiological roles of lactic acid
The excessive activation of glycolytic metabolic reprogramming plays a crucial role in GBM development, progression, and immune resistance. It not only provides a rapid means of ATP generation to meet the immense energy requirements of tumor cells but also influences the TME. The product of glycolysis, lactate, has been shown to play significant roles in GBM proliferation, differentiation, and progression (13). Lactate serves as an internal energy source for GBM tumor cells, converting within the mitochondria via the tricarboxylic acid (TCA) cycle to produce ATP, partially uncoupling energy production from glycolysis and allowing glucose to be primarily utilized for cell proliferation and metastasis. Additionally, the accumulation of lactate within the GBM TME leads to a decrease in environmental pH, creating an acidic TME (14). The acidic TME generates an inwardly directed proton gradient across the cancer cell plasma membrane, providing a driving force for proton-coupled transporters to enhance the supply of select nutrients. Research by Colen CB et al. demonstrated that inhibition of lactate efflux in rats with intracranially implanted glioma cells significantly impaired tumor invasion, potentially through the promotion of Epithelial-Mesenchymal Transition (EMT), tumor angiogenesis, metastasis, and colonization of distant organs (15–17). Besides, clinical studies have confirmed that elevated lactate levels are associated with GBM grading, higher Ki-67 index, larger tumor volume, and poorer prognosis (18, 19).
2.2 Regulation of lactate on the activation status and functions of macrophages
Macrophages are the most abundant immune cells within the GBM TME, comprising over 30% of infiltrating cells and reaching up to 70% in certain cases (20, 21). Studies have shown that microglia, a type of macrophage, play a crucial role in GBM progression (22). The production of lactate by cancer cells in GBM has been reported to promote M2 polarization and tumor angiogenesis of tumor-associated macrophages (TAMs) (23), the possible mechanism is that lactate can upregulate the expression of Vascular Endothelial Growth Factor (VEGF) and ARG1 genes through HIF1a-mediated mechanisms, promoting macrophage M2 polarization (24). In vitro experiments by Noe JT et al. revealed that supplementation of lactate to glucose-starved mouse bone marrow-derived macrophages increased histone H3 Lysine 9 (H3K9) acetylation in the ARG1 and Retnla promoter regions, indicating that lactate can regulate macrophage gene expression through epigenetic mechanisms (25). Lonhitano et al. explored the interaction between lactate and Lnsulin-like Growth Factor-binding Protein 6 (IGFBP6) in GBM cells, revealing that lactate can regulate IGFBP6 expression, further modulating microglial polarization and contributing to tumor development and therapeutic resistance (26). Similarly, Yan C et al. demonstrated that the proton-sensing G Protein-coupled Receptor (GPCRs) GPR65 can respond to lactate stimulation via the cAMP/PKA/CREB signaling pathway, triggering HMGB1 release and promoting M2 polarization of TAMs, subsequently enhancing glioma cell proliferation, migration, invasion, and mesenchymal transition (27). Li Y et al. showed that lactate depletion and SIRPα signaling blockade could potentially re-educate TAMs, reversing the immunosuppressive TME, enhancing macrophage anti-tumor activity, and effectively inhibiting tumor growth (28). Furthermore, research conducted by Ye Z et al. has shown that when macrophages in GBM exhibit high oxidative stress and reduced antigen presentation, it leads to a significant decrease in the number and function of CD8+ T cells, ultimately promoting an immunosuppressive microenvironment and the growth and progression of GBM (29)
2.3 Regulation of lactate on the activation status and functions of T cells
Lactate is not only a byproduct of tumor cell metabolism but also an important immunoregulatory factor that can influence the activation state and function of T cells (30), T cells are diverse and can be primarily divided into two types: CD8+ T cells and CD4+ T cells. On one hand, lactate acts as a physiological carbon source, facilitating effective CD8+ T cell activation even when glucose is abundant (31). Research has shown that lactate reduces the binding of Glyceraldehyde-3-phosphate Dehydrogenase (GAPDH) to cytokine mRNA, thereby releasing more free cytokine mRNA and enhancing cytokine production by regulatory T cells (32–34). The main mechanism involves lactate inhibiting histone deacetylase activity, increasing H3K27 acetylation in the TCF7 super enhancer region, and upregulating TCF7 gene expression, thereby enhancing CD8 T cell stemness and anti-tumor efficacy, Feng Q et al. demonstrated that lactate-pretreated CD8 T cells effectively suppressed tumor growth when transferred to tumor-bearing mice, providing evidence for the intrinsic role of lactate in anti-tumor immunity, independent of its pH-dependent effects, which may advance the development of cancer immunotherapy (35).
On the other hand, lactate-induced TME acidification inhibits CD8 T cell proliferation, cytokine production, and cytotoxic function. This effect can be reversed by adjusting the pH of lactate-containing cell culture media to 7.4, restoring the cytokine production ability of human CD8 T cells and highlighting the importance of the acidic environment for T cell function (36).
Wang Z et al. conducted an RNA-seq analysis to explore the role of lactate in the progression of GBM, uncovering that elevated lactate levels in the TME impact the migration and infiltration rate of CD8+ T cells in GBM. Specifically, in a high-lactate TME, the migratory capacity of CD8+ T cells is inhibited, resulting in decreased infiltration of these cells within the tumor tissue. This effect may be mediated through the binding of lactate to specific receptors on the surface of CD8+ T cells, such as GPR65, which in turn influences intracellular signaling pathways, including MAPK and WNT/β-catenin signaling (30).
Furthermore, lactate has been shown to regulate Foxp3-dependent RNA splicing through the Cytotoxic T-lymphocyte-Associated Protein 4 (CTLA-4) pathway, maintaining the stable phenotype and function of tumor-infiltrating regulatory T cells (Tregs) (37). The balance between Programmed Cell Death Protein 1(PD-1)-expressing CD8 T cells and Tregs within the TME determines the clinical efficacy of PD-1 blockade therapy. Kumagai S et al. demonstrated that lactate in highly glycolytic TMEs upregulates PD-1 expression on Treg cells while suppressing PD-1 expression on effector T cells, facilitating tumor cell evasion of the immune system and promoting tumor progression (38). Moreover, Fischer K’s research indicates that high extracellular lactate levels block the efflux of lactate from T cells (36). Lactate enters CD4+ T cells through MCT1 and is converted to pyruvate by LDHB, thereby driving the TCA cycle and reducing glycolysis within these cells. This metabolic shift not only hinders the proliferation of CD4+ T cells but also enhances the expansion of Tregs (39). Building on this, Zhang YT et al. further investigated the mechanisms by which lactate regulates CD4+ T cells. Their study revealed that lactate promotes the conversion of α-ketoglutarate (α-KG) to 2-hydroxyglutarate (2HG) by increasing the expression and activity of Lactate Dehydrogenase A (LDHA). This alteration affects mTOR phosphorylation and HIF-1α synthesis, ultimately leading to an increased proportion of Treg cells (40).
CD47 plays a prominent role in the immune evasion pathways of GBM and other cancers by delivering a “do not eat me” signal through binding to SIRPα expressed on macrophages and microglia (41), Similarly, research by Wang S et al. has shown that lactic acid modulates both adaptive and innate immune responses by inducing CD47 expression and STAT3 activation. Furthermore, compared to either treatment alone, combination therapy with DCA and anti-CD47 increased the infiltration of CD4+ and CD8+ T cells (42).
In summary, lactate remodels the TME through complex signaling networks, inducing an immunosuppressive environment and contributing to the establishment of resistance.
3 Targeting the glycolytic metabolic reprogramming pathway in GBM
3.1 Targeting transporter
3.1.1 Glucose transporters
The initial step in glycolysis involves the transport of glucose molecules from the extracellular environment into the cytoplasmic matrix. This crucial transport action is primarily mediated by the GLUT family members, characterized by their high affinity for glucose and efficient import of glucose into cells to sustain the survival, proliferation, and growth of cancer cells (43, 44). In GBM, the expression of GLUT-1 and GLUT-3 is particularly prominent, and their levels correlate with reduced patient survival (45). Extensive evidence suggests that GLUT1 expression is upregulated in various malignancies and is associated with poor prognosis (45, 46). Studies have demonstrated that silencing GLUT1 expression effectively reduces glioma stem cell (GSC) tumor sphere formation, decreases self-renewal and proliferation in vitro, indicating its potential as a therapeutic target in GBM (46). Zhang Z et al. showed that mutating the CYS207 residue of GLUT1 to serine abolished its palmitoylation and membrane localization, resulting in decreased glycolysis, reduced cancer cell proliferation, and suppression of GBM tumorigenic potential (47). The research by Y. Li et al. found that HSP90B1 is significantly upregulated in radioresistant GBM cell lines, and that HSP90B1 promotes the localization of GLUT1 on the plasma membrane, enhances glycolytic activity, and thereby enhances tumor growth and radioresistance in GBM cells. In vivo experiments showed that HSP90B1 knockdown combined with radiotherapy can improve the survival rate of mice with GBM (48). Furthermore, natural products from plants and fungi, such as quercetin, can specifically block the function of GLUT-1 transporters. Both in vivo and in vitro experiments have confirmed that quercetin can significantly inhibit the growth of GBM and extend the survival time of mice (49). These results indicate that quercetin is a potential therapeutic drug for GBM.
Similar to GLUT-1, GLUT-3, also known as the neuronal glucose transporter, is highly expressed in brain tumor-initiating cells (BTICs) and exhibits a higher affinity for glucose (44). The expression of GLUT-3 is significantly higher in Grade 3 and 4 gliomas compared to lower-grade gliomas, suggesting its crucial role in glucose transport in high-grade gliomas (50). Libby et al. further demonstrated that overexpression of GLUT-3 in GBM is associated with increased invasiveness (51). Indeed, experiments have shown that knocking down GLUT-3 can more effectively regulate anaerobic pyruvate utilization and significantly slow down cancer cell proliferation (52). GLUTs enhance cancer cell glucose uptake, driving glycolytic metabolism and providing the necessary energy support for cancer cell survival, proliferation, and growth. Therefore, targeting GLUT1/GLUT3 is considered an ideal strategy to delay GBM tumor cell proliferation and overcome treatment resistance. Kwak S et al. confirmed the efficacy of GBM cell death induction by HDAC2 knockdown, targeting GLUT3 expression, using an in vivo orthotopic xenograft tumor model (53). Pucci G et al. demonstrated that GLUT-3 gene knockdown provides a better opportunity to control anaerobic pyruvate utilization and significantly reduces GBM tumor proliferation, suggesting GLUT-3 as a suitable silencing target for overcoming radiation resistance (52).
3.1.2 Monocarboxylate transporters
Excessive lactate generated during glycolysis accumulates, leading to the accumulation of MCTs. MCTs utilize a proton symport mechanism to extrude lactate from cells, which is crucial for maintaining high glycolytic rates in cancer cells and promoting extracellular TME acidification (54–56). Among MCTs, MCT1 is the primary plasma membrane transporter responsible for lactate efflux, particularly in Isocitrate Dehydrogenase (IDH)-wildtype gliomas (57). Consequently, MCT1 is considered a reliable target for targeting glycolytic metabolic reprogramming. Miranda-Gonçalves V et al. showed that inhibiting MCT1 significantly improved the survival rate of brain tumor-bearing mice, reduced lactate efflux and cell invasiveness, and enhanced the survival rate of mice treated with orthotopic gliomas, especially when combined with TMZ therapy, MCT1 knockdown significantly increased the sensitivity of GBM cells to TMZ, thereby significantly prolonging the survival of treated mice (58). Based on these findings, MCT1 inhibitors like AZD3965 have entered Phase I/II clinical trials for the treatment of small cell lung cancer (59) and lymphoma (60).
3.2 Targeting key enzymes of glycolytic metabolic reprogramming
3.2.1 Hexokinase 2
HK is a key enzyme in the first step of glycolysis, catalyzing the conversion of glucose to Glucose-6-Phosphate (G-6-P). HK2 is the most well-characterized isoform of the HK family and is significantly overexpressed in GBM tumor cells compared to adjacent normal tissues, correlating with GBM prognosis (61). Wolf A et al. demonstrated that transient inhibition of HK2 in GBM cells suppresses tumor cell proliferation and enhances sensitivity to apoptosis-inducing stimuli such as radiotherapy and chemotherapeutic alkylating agent TMZ. Furthermore, stable HK2 depletion inhibits aerobic glycolysis and promotes normal oxidative glucose metabolism, including decreased extracellular lactate, increased OXPHOS protein expression, and increased oxygen consumption. Interestingly, they found that supplementing HK1 in HK2-deficient cells restored overall HK activity but failed to rescue the aerobic glycolysis phenotype, strongly indicating the unique role of HK2 in GBM progression beyond that of HK1 (62). The role of HK2 in inhibiting tumor cell growth has been confirmed in breast cancer (63), oral cancer (64), and cervical cancer (65). The main therapeutic approaches targeting HK2 include reducing its catalytic activity by inhibiting its expression, directly inhibiting its enzymatic activity to block glucose phosphorylation, and disrupting its interaction with mitochondria to cut off an important energy pathway for tumor cells. Natural products like resveratrol and ginsenosides have been shown to effectively inhibit HK2 activity and exhibit potential in suppressing glycolysis and inducing apoptosis in various cancer experimental models (66, 67). Resveratrol has been shown to significantly inhibit the proliferation of U87 glioma cells and multiple patient-derived GSC cell lines in GBM (68). Additionally, antifungal azole drugs have been identified as a class of potential tumor metabolism inhibitors. Studies have revealed that ketoconazole and posaconazole can effectively inhibit the growth of GBM in vitro and in vivo by regulating HK2-related genes and signaling pathways. These drugs improve survival rates in GBM mouse models, reduce tumor cell proliferation, and decrease overall tumor metabolism levels (69).
The development of small-molecule HK2 inhibitors is also actively pursued and shows promising prospects. 2-DG is a small-molecule HK2 inhibitor that competitively inhibits HK2-mediated glucose phosphorylation, weakens the immunosuppressive network and macrophage polarization, enhances anti-tumor immunity, and contributes to local tumor control in mouse models (69). The research by Pająk et al. confirms that novel 2-DG analogs, such as WP1122, and HDAC inhibitors (like sodium butyrate and sodium valproate) exhibit significant synergistic anticancer effects, inhibiting the proliferation of GBM cells and inducing apoptosis. In vivo experimental results demonstrate that, in mouse models, the combination therapy significantly suppressed tumor growth, improved the survival rate of mice, and no significant toxicity was observed (70). This suggests that 2-DG, as a glycolysis inhibitor, can enhance the sensitivity of GBM cells to radiotherapy and other chemotherapeutic drugs, providing a new strategy for the treatment of GBM. In addition to combination with chemotherapy and radiotherapy, the combination of HK inhibitors and anti-PD-1 antibodies has also shown promising efficacy. HK2 can act as a protein kinase and phosphorylate IκBα at T291, leading to NF-κB-dependent upregulation of PD-L1 expression, which suppresses CD8 T cell activation and infiltration into tumor tissues, accelerating brain tumor growth and increasing resistance to immune checkpoint blockade therapy. Therefore, the combination of HK inhibitors and anti-PD-1 antibodies exhibits superior anti-tumor effects compared to monotherapy with either drug (71).
It is noteworthy that disrupting the interaction between HK2 and mitochondria is another promising strategy. Li R et al. found that Gomisin J, a lignan derivative, can inhibit the proliferation of glioma cells, induce apoptosis, and suppress glycolysis regulated by HK2, thereby inhibiting the progression of gliomas. Gomisin J may represent a potential therapeutic strategy for the treatment of gliomas with relatively low toxicity (72). Similarly, Shteinfer-Kuzmine A et al. focused on cell-penetrating peptides based on Voltage-Dependent Anion Channel 1 (VDAC1), which can perturb the energy and metabolic homeostasis of GBM cells, leading to apoptosis of GBM cells and derived stem cell lines. These peptides also demonstrated inhibitory
effects on tumor growth, invasion, metabolism, stemness, and promoted apoptosis in subcutaneous and intracranial orthotopic GBM xenograft mouse models, suggesting that peptide compounds based on VDAC1 may become a novel candidate for GBM treatment (73). Additionally, the use of VDAC1-specific short interfering RNA (si-VDAC1) to treat GBM cell lines and subcutaneous or intracranial orthotopic GBM xenograft mouse models has been shown to significantly inhibit tumor growth. The remaining tumor tissue exhibited reversed oncogenic characteristics, including metabolic reprogramming, proliferation inhibition, reduced angiogenesis, weakened EMT, decreased invasiveness, and loss of stemness, with some tumor cells differentiating into neuron-like and astrocyte-like cells (74).
3.2.2 Phosphofructokinase-2/fructose-2,6-bisphosphatase 3
Phosphofructokinase-1 (PFK1) is a key regulatory and rate-limiting step in glycolysis, catalyzing the conversion of fructose-6-phosphate and ATP to fructose-1,6-bisphosphate (F-1,6-BP) and ADP (75). PFKFB3 is one of the isoforms of PFK-2 and has been confirmed to be the most highly expressed enzyme in GBM (76). Inhibition of PFKFB3 expression or activity can decrease the level of F-2,6-BP, thereby inhibiting the activity of PFK1 and the rate of glycolysis, ultimately suppressing cell proliferation (77, 78). Clem BF et al. demonstrated that chemical inhibitors targeting PFKFB3, a key enzyme in glycolysis, can effectively reduce glycolysis rate and inhibit tumor cell proliferation (79). This key finding not only confirms the crucial role of PFKFB3 in tumor metabolism regulation but also opens up new strategies for cancer treatment. Li FL et al. further demonstrated the impact of PFKFB3 acetylation status on its activity, showing that acetylated PFKFB3 can significantly promote glycolysis and protect cells from cisplatin-induced apoptosis, revealing the potential role of PFKFB3 modification status in tumor resistance mechanisms (80). Current research trends also lean towards combining PFKFB3 inhibitors with other existing therapies to achieve more comprehensive and effective treatment outcomes. Zhang J’s research team found that simultaneous inhibition of VEGF and glycolysis activator PFKFB3 can significantly prolong the survival of GBM patients and slow down tumor growth. This dual therapy not only inhibited cell proliferation but also promoted apoptosis and enhanced tumor vessel normalization, improving tumor hypoxia, reducing lactate accumulation, and increasing the efficacy and delivery efficiency of chemotherapeutic drugs like doxorubicin (81). This indicates that by integrating the synergistic effects of VEGF and PFKFB3 blockade, it is possible to overcome the current limitations of bevacizumab monotherapy. Nakada M et al. explored the possibility of using PFKFB3 inhibitors to address TKI (tyrosine kinase inhibitor) resistance in GBM, suggesting that the combination of PFKFB3 inhibitors and TKIs may bring breakthroughs in clinical practice (82). Additionally, a series of small-molecule inhibitors targeting PFKFB3 have been extensively investigated, including KAN0438757, developed by Gustafsson NMS et al. Experimental data show that KAN0438757 significantly reduces glioblastoma cell viability and migration ability, with a time- and dose-dependent effect (83). Other inhibitors like 3-PO, PFK15, PFK158, YN1, and N4A have also shown strong antitumor proliferation efficacy in preclinical studies (84). 3-(3-Pyridinyl)-1-(4-pyridinyl)-2-propen-1-one (3-PO) is a novel compound widely reported to inhibit glycolysis flux by competitively inhibiting PFKFB3 (85). Li J’s research team compared the differences in glycolysis flux (analyzed by glucose uptake and lactate secretion) and cell viability (assessed by CCK8 assay) between experimental groups with and without 3-PO addition, providing strong evidence for 3-PO’s role in significantly inhibiting tumor cell glycolytic activity and proliferation (86). The combined application of 3-PO and bevacizumab (a monoclonal antibody targeting VEGF) has been confirmed to reduce GBM cell proliferation and increase apoptosis, thereby delaying tumor growth and improving patient survival (81). PFK15, as a derivative of 3-PO, has demonstrated approximately 100 times higher PFKFB3 inhibitory activity than 3-PO in preclinical studies. Although its anti-GBM growth activity is slightly inferior to that of TMZ, preclinical studies still indicate that PFK15 and its closely related structural derivatives have the potential to exhibit potent cytotoxic activity against various human cancers in future clinical trials. However, it is noteworthy that these compounds have not yet entered the clinical trial phase (79). The combination of PFK158 with CTLA-4 antibody has demonstrated synergistic inhibition of cancer growth, providing a promising perspective for the integration of immunotherapy and targeted glycolytic metabolism therapy (88).
3.2.3 Pyruvate kinase M2
Pyruvate kinase (PK) has two main isoforms: PKM1 and PKM2. It catalyzes the transfer of inorganic phosphate from Phosphoenolpyruvate (PEP) to ADP, generating pyruvate and the energy-rich molecule ATP (82). PKM2 is minimally expressed in healthy brains but highly expressed in GBM cells. Studies have shown that GBM cells undergo a switch from PKM1 to PKM2 isoforms (87). Further experiments have revealed that replacing PKM2 with PKM1 variants in GBM cells leads to restricted biosynthetic pathways and inhibits tumor growth (88). These studies collectively indicate that PKM2 is an important biomarker of glycolytic reprogramming. PKM2 primarily exists in the forms of tetramers and dimers. Dimeric PKM2 is predominantly located in the nuclei of tumor cells and plays a pivotal role in tumor cell proliferation, invasion, and metastasis. Specifically, when the expression of dimeric PKM2 is preferred, it can regulate aerobic glycolysis through metabolic reprogramming of cancer cell metabolic pathways, leading to increased macromolecular biosynthesis in cancer cells and accelerated cancer cell growth. Conversely, activating PKM2 can promote the formation of tetramers, which can inhibit the nuclear import of PKM2 and subsequently suppress the STAT3 signaling pathway, thereby inhibiting the proliferation and metastasis of GBM cells (88, 89). This dynamic equilibrium mechanism between the dimeric and tetrameric forms of PKM2 enables proliferating cancer cells to regulate their anabolic and catabolic demands (90). Liang J et al. demonstrated that activation of the epidermal growth factor receptor (EGFR) induces PKM2 nuclear translocation, upregulating cyclin D1 expression in GBM U87 cell lines and other human cancer cells, accelerating tumor formation (91). On the other hand, the naphthoquinone compound Shikonin, extracted from the traditional Chinese herb Zicao, has been found to inhibit PKM2 activity. In vitro experiments have demonstrated its inhibitory effect on the proliferation, migration, and invasion of glioblastoma cell lines U87 and U251. Furthermore, studies have shown that Shikonin can also inhibit the migration and invasion of GBM cells by targeting phosphorylated β-Catenin and phosphorylated PI3K/Akt signaling pathways (92). Ding Y et al. extensively investigated the effects of a series of parthenolide dimers as PKM2 activators and found that compound 5 exhibited high activity. It promotes PKM2 tetramer formation in GBM cells, reduces PKM2 nuclear translocation without affecting total PKM2 expression, and thereby inhibits the STAT3 signaling pathway in vitro and in vivo, as a result, it suppresses the proliferation and metastasis of GBM cells and induces apoptosis (93). These findings provide potential drug targets and therapeutic approaches for the development of new GBM treatment strategies. Metformin and vitamin K, as PKM2 inhibitors, have shown inhibitory effects on glycolysis, cell proliferation, and drug resistance in various cancers such as osteosarcoma (94), non-small cell lung cancer (95), and hepatocellular carcinoma (96). Metformin has been evaluated for its potential therapeutic effect on glioblastoma in several clinical trials. For example, a cohort study by Adeberg et al. involving 276 patients with primary GBM suggested that the use of metformin in diabetic GBM patients may help slow disease progression and improve patient survival (97),This study provides evidence for metformin as a potential therapeutic agent and lays the foundation for future clinical trials. Additionally, research by Qiao X et al. suggests that metformin can penetrate the blood-brain barrier (BBB) and enter brain tissue, inhibiting the malignant progression of gliomas. The study showed that metformin alone can induce apoptosis in glioma cells (98). However, the specific role of vitamin K in gliomas has not been reported as of now, and further research is needed to elucidate it in the future. Notably, a small molecule compound called dimethylaminochlorocycline (DMAMCL) can modulate glycolysis pathways by activating PKM2, reducing GBM cell proliferation. DMAMCL has been applied in clinical trials for recurrent GBM. This compound selectively binds to the monomeric form of PKM2, promoting PKM2 tetramerization and increasing its pyruvate kinase activity. The inhibitory effect of DMAMCL in GBM cells is weakened when PKM2 is depleted, further demonstrating the critical role of PKM2 in mediating the sensitivity of GBM cells to DMAMCL (99). Furthermore, research by Gao M et al. has shown that Trametinib inhibits the growth and aerobic glycolysis of glioma cells by targeting the PKM2/c-Myc axis. Trametinib suppresses the expression of PKM2 in glioma cells and inhibits the translocation of PKM2 into the nucleus, thereby affecting the expression of c-Myc (100). These findings not only reveal the potential mechanism of Trametinib in antitumor activity but also provide new insights into the clinical drug treatment of gliomas.
3.2.4 Lactate dehydrogenase
Lactate dehydrogenase (LDH) is a core component of the Warburg effect. In hypoxic or oxidative stress conditions, tumor cells adaptively upregulate LDH activity to promote the rapid conversion of pyruvate to lactate during glycolysis, rather than entering the tricarboxylic acid cycle (TCA cycle) for complete oxidation (101, 102). In GBM, the Lactate Dehydrogenase A (LDHA) isoform is typically overexpressed, promoting the flux of substrates towards lactate production during glycolysis, enhancing tumor cell survival and proliferation (103). Studies on various cell lines have shown that inhibiting LDHA function leads to decreased cancer cell proliferation, increased apoptosis, and weakened migration and invasion, further confirming the importance of LDHA as a potential therapeutic target. Galloflavin, a synthesized LDHA and LDHB inhibitor, exhibits high levels of cell penetration (104, 105). Research has shown that Galloflavin inhibits the conversion of pyruvate to lactate, directing pyruvate into the TCA cycle, prompting tumor cells that rely on glycolysis to switch to oxidative phosphoryation (OXPHOS) for energy production, this metabolic shift is detrimental to tumor cells as it increases oxygen consumption and reactive oxygen species (ROS) generation, leading to mitochondrial oxidative damage (106). Gossypol, a derivative of cottonseed oil, has been found to inhibit LDHA. Research by T. Coyle et al. has demonstrated that gossypol exhibits significant in vitro and in vivo cytotoxicity against central nervous system tumor cells. In vitro, gossypol displays concentration- and time-dependent cytotoxicity against various glioma cell lines. In vivo, gossypol significantly inhibits tumor growth in a xenograft model in nude mice, suggesting its potential as a therapeutic agent for the treatment of primary malignancies of the central nervous system (107). Gossypol has been shown to be well-tolerated in clinical trials and has shown promise in trials for recurrent malignant gliomas (NCT00540722 and NCT00390403). FX11, a novel gossypol-derived selective LDHA small-molecule inhibitor, has been developed and has shown promise in preclinical studies. It inhibits LDHA activity, decreases intracellular ATP levels, and induces oxidative stress and cell death, which can be partially reversed by the antioxidant N-acetylcysteine (108). Recently, FX11 has been commercialized, and studies in human lymphoma and pancreatic cancer xenografts have shown that it inhibits tumor progression and induces significant oxidative stress and necrosis (108) Daniele S et al. found that NHI-1 and NHI-2 (LDH-A inhibitors) can reduce GBM cell proliferation, trigger apoptosis, and block the cell cycle (103). Additionally, oxalate has been considered to have LDH-inhibitory effects (109). The study of SunT et al. confirmed that Oxamate can reprogram glucose metabolism of cancer stem cells, and can also alleviate immunosuppression of TME and reduce tumor-infiltrating CAR-Treg cells, which may be a potential strategy to enhance CAR-T function in glioblastoma therapy (110). Moorhouse AD et al. designed a novel dual-functional ligand that inhibits LDHA, with activity 9 times higher than sodium oxalate,however, subsequent reports on detailed molecular modeling studies and biological tests targeting cancer cell lines have not been forthcoming (111). The specific application of targeting glycolytic metabolic reprogramming pathways in GBM is demonstrated in Table 1.
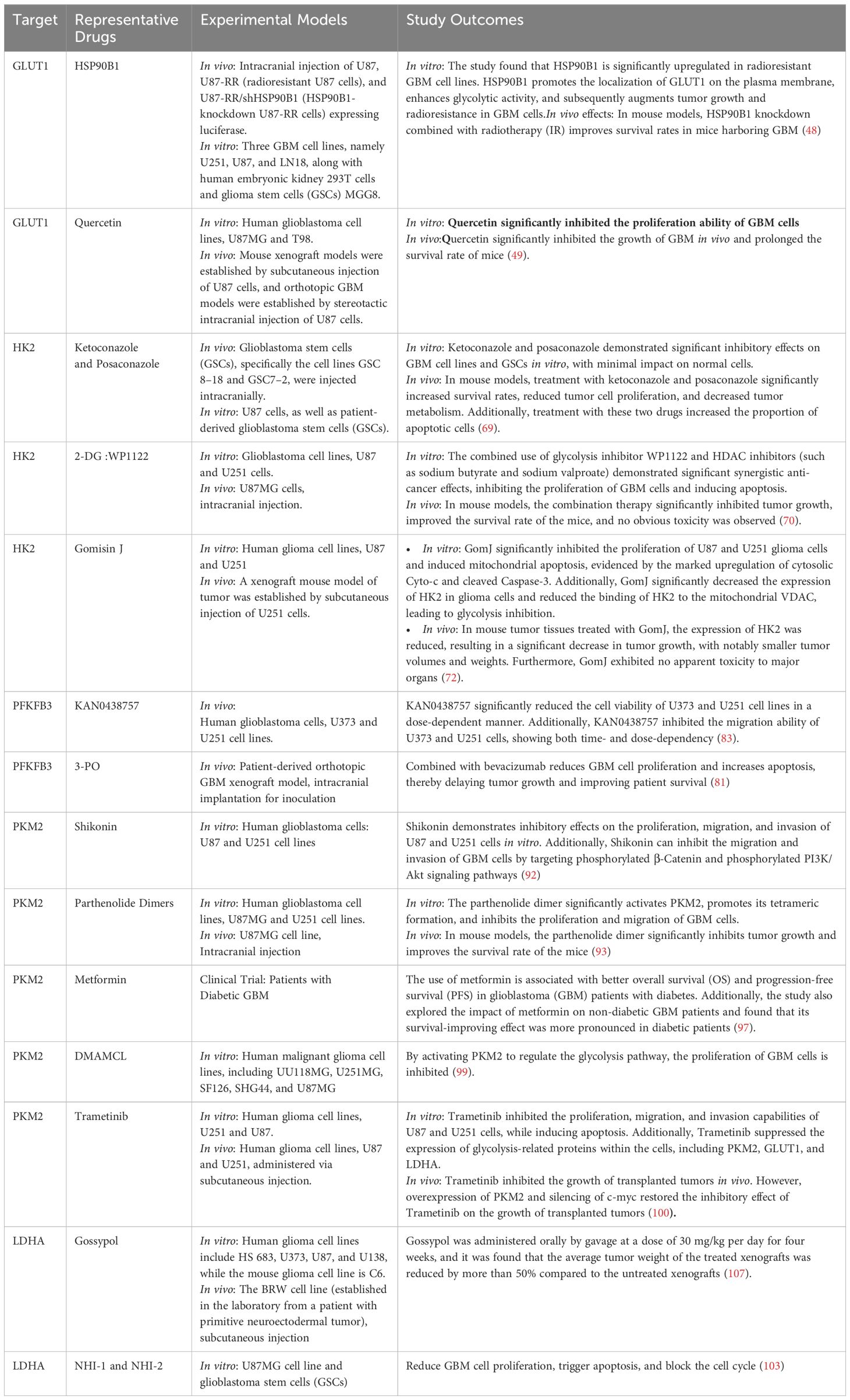
Table 1. Targeted glycolysis inhibitors in the treatment of GBM.
4 Current challenges and the future perspectives
Glycolytic metabolic reprogramming plays a pivotal role in the development, invasion, and treatment resistance of GBM. It not only provides essential energy and biosynthetic precursors for rapidly proliferating tumor cells but also profoundly shapes the GBM microenvironment, influencing angiogenesis, inflammation, and the dynamic balance of the immune microenvironment. As researchers gain deeper insights into the functions of key glycolytic enzymes and their roles in tumor metabolism regulation, targeting the glycolytic pathway has emerged as a promising therapeutic strategy. Currently, a series of small-molecule inhibitors targeting glycolytic metabolic reprogramming pathways have shown therapeutic effects in preclinical and clinical studies. However, challenges remain, including optimizing drug design to enhance specificity and efficacy, addressing issues related to drug penetration across the blood-brain barrier, potential side effects, and resistance. Combining these inhibitors with existing therapies such as radiotherapy, chemotherapy, targeted therapy, and emerging immunotherapies holds promise for effectively controlling GBM.
In this review, we systematically elucidated the key role of glycolytic metabolic product - lactate, in the occurrence and development of GBM. By thoroughly analyzing existing literature and delving into empirical research, we revealed that abnormal activation of glycolysis is a core feature of GBM metabolic reprogramming, not only providing necessary energy and biomass for the rapid proliferation of tumor cells but also significantly shaping the TME, conferring GBM with enhanced invasiveness and resistance.
Given the central role of glycolytic metabolic reprogramming in GBM, we propose several directions for future research and clinical treatment. Firstly, further elucidation of the molecular mechanisms of glycolysis and their differences in various stages and subtypes of GBM will aid in the development of more precise diagnostic tools and prognostic indicators. For example, monitoring the expression levels, activity, and metabolic product content of key glycolytic enzymes in blood or tumor tissue may serve as powerful biomarkers for assessing disease progression, predicting treatment responses, and prognosis. Secondly, the design and optimization of targeted therapeutic strategies against the glycolytic pathway are crucial. Existing research has shown the potential of drugs inhibiting glycolytic enzyme activity in GBM treatment. However, challenges remain in addressing issues related to drug delivery across the blood-brain barrier, potential side effects, and resistance. Combining these inhibitors with radiotherapy, chemotherapy, and other emerging immunotherapies holds promise for achieving more effective and sustained control of GBM. Finally, given the role of glycolytic products in regulating the TME and immune evasion, future research should focus on the specific effects of glycolytic metabolism on the tumor immune microenvironment, seeking and verifying strategies that can enhance anti-tumor immune responses by regulating glycolysis and altering TME pH. For instance, targeting lactate transporters or interfering with lactate metabolism may help improve immune cell function, enabling better recognition and attack of cancer cells and ultimately enhancing the efficacy of immunotherapy.
5 Conclusion
Gaining a deeper insight into and implementing targeted interventions for abnormal glycolysis in GBM offers a promising new strategy to combat this difficult-to-treat tumor. Future research endeavors should concentrate on adapting these fundamental discoveries into clinical applications, thereby furnishing GBM patients with more potent and tailored treatment options.
Author contributions
LY: Conceptualization, Data curation, Formal analysis, Investigation, Resources, Visualization, Writing – original draft. SL: Conceptualization, Formal analysis, Methodology, Writing – review & editing. LY: Conceptualization, Formal analysis, Methodology, Resources, Writing – review & editing. JL: Conceptualization, Data curation, Formal analysis, Writing – original draft. NL: Conceptualization, Resources, Supervision, Validation, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, et al. The 2007 WHO classification of tumours of the central nervous system. Acta neuropathologica. (2007) 114:97–109. doi: 10.1007/s00401-007-0243-4
2. Ostrom QT, Cioffi G, Gittleman H, Patil N, Waite K, Kruchko C, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2012-2016. Neuro-oncology. (2019) 21:v1–v100. doi: 10.1093/neuonc/noz150
3. Stupp R, Taillibert S, Kanner AA, Kesari S, Steinberg DM, Toms SA, et al. Maintenance therapy with tumor-treating fields plus temozolomide vs temozolomide alone for glioblastoma: A randomized clinical trial. Jama. (2015) 314:2535–43. doi: 10.1001/jama.2015.16669
4. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. New Engl J Med. (2005) 352:987–96. doi: 10.1056/NEJMoa043330
5. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. IDH1 and IDH2 mutations in gliomas. New Engl J Med. (2009) 360:765–73. doi: 10.1056/NEJMoa0808710
6. Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. (2012) 21:297–308. doi: 10.1016/j.ccr.2012.02.014
7. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Sci. (2009) 324:1029–33. doi: 10.1126/science.1160809
8. Xing F, Luan Y, Cai J, Wu S, Mai J, Gu J, et al. The Anti-Warburg Effect Elicited by the cAMP-PGC1α Pathway Drives Differentiation of Glioblastoma Cells into Astrocytes. Cell Rep. (2017) 18:468–81. doi: 10.1016/j.celrep.2016.12.037
9. Tretter V. Special issue: cellular oxygen homeostasis. Int J Mol Sci. (2022) 23(9):4505. doi: 10.3390/ijms23094505
10. Liberti MV, Locasale JW. The warburg effect: how does it benefit cancer cells? Trends Biochem Sci. (2016) 41:211–8. doi: 10.1016/j.tibs.2015.12.001
11. Zhou Y, Tozzi F, Chen J, Fan F, Xia L, Wang J, et al. Intracellular ATP levels are a pivotal determinant of chemoresistance in colon cancer cells. Cancer Res. (2012) 72:304–14. doi: 10.1158/0008-5472.CAN-11-1674
12. Locasale JW, Cantley LC. Altered metabolism in cancer. BMC Biol. (2010) 8:88. doi: 10.1186/1741-7007-8-88
13. Pavlova NN, Zhu J, Thompson CB. The hallmarks of cancer metabolism: Still emerging. Cell Metab. (2022) 34:355–77. doi: 10.1016/j.cmet.2022.01.007
14. Colen CB, Shen Y, Ghoddoussi F, Yu P, Francis TB, Koch BJ, et al. Metabolic targeting of lactate efflux by Malignant glioma inhibits invasiveness and induces necrosis: an in vivo study. Neoplasia (New York N.Y.). (2011) 13:620–32. doi: 10.1593/neo.11134
15. Goetze K, Walenta S, Ksiazkiewicz M, Kunz-Schughart LA, Mueller-Klieser W. Lactate enhances motility of tumor cells and inhibits monocyte migration and cytokine release. Int J Oncol. (2011) 39:453–63. doi: 10.3892/ijo.2011.1055
16. Yang J, Ren B, Yang G, Wang H, Chen G, You L, et al. The enhancement of glycolysis regulates pancreatic cancer metastasis. Cell Mol Life sciences: CMLS. (2020) 77:305–21. doi: 10.1007/s00018-019-03278-z
17. Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. (2003) 3:721–32. doi: 10.1038/nrc1187
18. Sitter B, Forsmark A, Solheim O. Elevated serum lactate in glioma patients: associated factors. Front Oncol. (2022) 12:831079. doi: 10.3389/fonc.2022.831079
19. Maldonado F, Fábregas N, Aldecoa I, González J, García-Orellana M, Belda I, et al. Association between preoperative serum lactate concentrate with tumor cell proliferative index in primary brain tumor. J neurosurgical Sci. (2022) 66:91–5. doi: 10.23736/S0390-5616.19.04715-5
20. Wang G, Zhong K, Wang Z, Zhang Z, Tang X, Tong A, et al. Tumor-associated microglia and macrophages in glioblastoma: From basic insights to therapeutic opportunities. Front Immunol. (2022) 13:964898. doi: 10.3389/fimmu.2022.964898
21. Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell. (2019) 178:835–849.e21. doi: 10.1016/j.cell.2019.06.024
22. Fan D, Yue Q, Chen J, Wang C, Yu R, Jin Z, et al. Reprogramming the immunosuppressive microenvironment of IDH1 wild-type glioblastoma by blocking Wnt signaling between microglia and cancer cells. Oncoimmunology. (2021) 10:1932061. doi: 10.1080/2162402X.2021.1932061
23. Wang F, Liao W, Li C, Zhu L. Silencing BMAL1 promotes M1/M2 polarization through the LDHA/lactate axis to promote GBM sensitivity to bevacizumab. Int Immunopharmacol. (2024) 134:112187. doi: 10.1016/j.intimp.2024.112187
24. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. (2014) 513:559–63. doi: 10.1038/nature13490
25. Noe JT, Rendon BE, Geller AE, Conroy LR, Morrissey SM, Young LEA, et al. Lactate supports a metabolic-epigenetic link in macrophage polarization. Sci Adv. (2021) 7:eabi8602. doi: 10.1126/sciadv.abi8602
26. Longhitano L, Vicario N, Forte S, Giallongo C, Broggi G, Caltabiano R, et al. Lactate modulates microglia polarization via IGFBP6 expression and remodels tumor microenvironment in glioblastoma. Cancer immunology immunotherapy: CII. (2023) 72:1–20. doi: 10.1007/s00262-022-03215-3
27. Yan C, Yang Z, Chen P, Yeh Y, Sun C, Xie T, et al. GPR65 sensing tumor-derived lactate induces HMGB1 release from TAM via the cAMP/PKA/CREB pathway to promote glioma progression. J Exp Clin Cancer research: CR. (2024) 43:105. doi: 10.1186/s13046-024-03025-8
28. Li Y, Wei Y, Huang Y, Qin G, Zhao C, Ren J, et al. Lactate-responsive gene editing to synergistically enhance macrophage-mediated cancer immunotherapy. Small (Weinheim an der Bergstrasse Germany). (2023) 19:e2301519. doi: 10.1002/smll.202301519
29. Ye Z, Ai X, Yang K, Yang Z, Fei F, Liao X, et al. Targeting microglial metabolic rewiring synergizes with immune-checkpoint blockade therapy for glioblastoma. Cancer Discovery. (2023) 13:974–1001. doi: 10.1158/2159-8290.CD-22-0455
30. Wang Z, Dai Z, Zhang H, Liang X, Zhang X, Wen Z, et al. Tumor-secreted lactate contributes to an immunosuppressive microenvironment and affects CD8 T-cell infiltration in glioblastoma. Front Immunol. (2023) 14:894853. doi: 10.3389/fimmu.2023.894853
31. Kaymak I, Luda KM, Duimstra LR, Ma EH, Longo J, Dahabieh MS, et al. Carbon source availability drives nutrient utilization in CD8(+) T cells. Cell Metab. (2022) 34:1298–1311.e6. doi: 10.1016/j.cmet.2022.07.012
32. Millet P, Vachharajani V, McPhail L, Yoza B, McCall CE. GAPDH binding to TNF-α mRNA contributes to posttranscriptional repression in monocytes: A novel mechanism of communication between inflammation and metabolism. J Immunol (Baltimore Md.: 1950). (2016) 196:2541–51. doi: 10.4049/jimmunol.1501345
33. Wen J, Cheng S, Zhang Y, Wang R, Xu J, Ling Z, et al. Lactate anions participate in T cell cytokine production and function. Sci China. Life Sci. (2021) 64:1895–905. doi: 10.1007/s11427-020-1887-7
34. Chang CH, Curtis JD, Maggi LB Jr., Faubert B, Villarino AV, O'Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. (2013) 153:1239–51. doi: 10.1016/j.cell.2013.05.016
35. Feng Q, Liu Z, Yu X, Huang T, Chen J, Wang J, et al. Lactate increases stemness of CD8 + T cells to augment anti-tumor immunity. Nat Commun. (2022) 13:4981. doi: 10.1038/s41467-022-32521-8
36. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. (2007) 109:3812–9. doi: 10.1182/blood-2006-07-035972
37. Ding R, Yu X, Hu Z, Dong Y, Huang H, Zhang Y, et al. Lactate modulates RNA splicing to promote CTLA-4 expression in tumor-infiltrating regulatory T cells. Immunity. (2024) 57:528–540.e6. doi: 10.1016/j.immuni.2024.01.019
38. Kumagai S, Koyama S, Itahashi K, Tanegashima T, Lin YT, Togashi Y, et al. Lactic acid promotes PD-1 expression in regulatory T cells in highly glycolytic tumor microenvironments. Cancer Cell. (2022) 40:201–218.e9. doi: 10.1016/j.ccell.2022.01.001
39. Kaushik DK, Bhattacharya A, Mirzaei R, Rawji KS, Ahn Y, Rho JM, et al. Enhanced glycolytic metabolism supports transmigration of brain-infiltrating macrophages in multiple sclerosis. J Clin Invest. (2019) 129:3277–92. doi: 10.1172/JCI124012
40. Zhang YT, Xing ML, Fang HH, Li WD, Wu L, Chen ZP. Effects of lactate on metabolism and differentiation of CD4(+)T cells. Mol Immunol. (2023) 154:96–107. doi: 10.1016/j.molimm.2022.12.015
41. Hutter G, Theruvath J, Graef CM, Zhang M, Schoen MK, Manz EM, et al. Microglia are effector cells of CD47-SIRPα antiphagocytic axis disruption against glioblastoma. Proc Natl Acad Sci USA. (2019) 116:997–1006. doi: 10.1073/pnas.1721434116
42. Wang S, Huang T, Wu Q, Yuan H, Wu X, Yuan F, et al. Lactate reprograms glioblastoma immunity through CBX3-regulated histone lactylation. J Clin Invest 134. (2024) 134(22):e176851. doi: 10.1172/JCI176851
43. Labak CM, Wang PY, Arora R, Guda MR, Asuthkar S, Tsung AJ, et al. Glucose transport: meeting the metabolic demands of cancer, and applications in glioblastoma treatment. Am J Cancer Res. (2016) 6:1599–608.
44. Flavahan WA, Wu Q, Hitomi M, Rahim N, Kim Y, Sloan AE, et al. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat Neurosci. (2013) 16:1373–82. doi: 10.1038/nn.3510
45. Chamarthy S, Mekala JR. Functional importance of glucose transporters and chromatin epigenetic factors in Glioblastoma Multiforme (GBM): possible therapeutics. Metab Brain Dis. (2023) 38:1441–69. doi: 10.1007/s11011-023-01207-5
46. Guda MR, Labak CM, Omar SI, Asuthkar S, Airala S, Tuszynski J, et al. GLUT1 and TUBB4 in glioblastoma could be efficacious targets. Cancers. (2019) 11(9):1308. doi: 10.3390/cancers11091308
47. Zhang Z, Li X, Yang F, Chen C, Liu P, Ren Y, et al. DHHC9-mediated GLUT1 S-palmitoylation promotes glioblastoma glycolysis and tumorigenesis. Nat Commun. (2021) 12:5872. doi: 10.1038/s41467-021-26180-4
48. Li Y, Ge Y, Zhao M, Ding F, Wang X, Shi Z, et al. HSP90B1-mediated plasma membrane localization of GLUT1 promotes radioresistance of glioblastomas. J Biomed Res. (2023) 37:326–39. doi: 10.7555/JBR.37.20220234
49. Wang L, Ji S, Liu Z, Zhao JJC. Quercetin inhibits glioblastoma growth and prolongs survival rate through inhibiting glycolytic metabolism. Chemotherapy. (2022) 67(3):132–41. doi: 10.1159/000523905
50. Liu Y, Li YM, Tian RF, Liu WP, Fei Z, Long QF, et al. The expression and significance of HIF-1alpha and GLUT-3 in glioma. Brain Res. (2009) 1304:149–54. doi: 10.1016/j.brainres.2009.09.083
51. Libby CJ, Gc S, Benavides GA, Fisher JL, Williford SE, Zhang S, et al. A role for GLUT3 in glioblastoma cell invasion that is not recapitulated by GLUT1. Cell adhesion migration. (2021) 15:101–15. doi: 10.1080/19336918.2021.1903684
52. Pucci G, Minafra L, Bravatà V, Calvaruso M, Turturici G, Cammarata FP, et al. Glut-3 gene knockdown as a potential strategy to overcome glioblastoma radioresistance. Int J Mol Sci. (2024) 25(4):2079. doi: 10.3390/ijms25042079
53. Kwak S, Park SH, Kim SH, Sung GJ, Song JH, Jeong JH, et al. miR-3189-targeted GLUT3 repression by HDAC2 knockdown inhibits glioblastoma tumorigenesis through regulating glucose metabolism and proliferation. J Exp Clin Cancer research: CR. (2022) 41:87. doi: 10.1186/s13046-022-02305-5
54. Ortega AD, Sánchez-Aragó M, Giner-Sánchez D, Sánchez-Cenizo L, Willers I, Cuezva JM. Glucose avidity of carcinomas. Cancer Lett. (2009) 276:125–35. doi: 10.1016/j.canlet.2008.08.007
55. Vanhove K, Graulus GJ, Mesotten L, Thomeer M, Derveaux E, Noben JP, et al. The metabolic landscape of lung cancer: new insights in a disturbed glucose metabolism. Front Oncol. (2019) 9:1215. doi: 10.3389/fonc.2019.01215
56. Miranda-Gonçalves V, Reis RM, Baltazar F. Lactate transporters and pH regulation: potential therapeutic targets in glioblastomas. Curr Cancer Drug Targets. (2016) 16:388–99. doi: 10.2174/1568009616666151222150543
57. Miranda-Gonçalves V, Honavar M, Pinheiro C, Martinho O, Pires MM, Pinheiro C, et al. Monocarboxylate transporters (MCTs) in gliomas: expression and exploitation as therapeutic targets. Neuro-oncology. (2013) 15:172–88. doi: 10.1093/neuonc/nos298
58. Miranda-Gonçalves V, Gonçalves CS, Granja S, Vieira de Castro J, Reis RM, Costa BM, et al. MCT1 is a new prognostic biomarker and its therapeutic inhibition boosts response to temozolomide in human glioblastoma. Cancers. (2021) 13(14):3468. doi: 10.3390/cancers13143468
59. Polański R, Hodgkinson CL, Fusi A, Nonaka D, Priest L, Kelly P, et al. Activity of the monocarboxylate transporter 1 inhibitor AZD3965 in small cell lung cancer. Clin Cancer research: an Off J Am Assoc Cancer Res. (2014) 20:926–37. doi: 10.1158/1078-0432.CCR-13-2270
60. Noble RA, Bell N, Blair H, Sikka A, Thomas H, Phillips N, et al. Inhibition of monocarboxyate transporter 1 by AZD3965 as a novel therapeutic approach for diffuse large B-cell lymphoma and Burkitt lymphoma. Haematologica. (2017) 102:1247–57. doi: 10.3324/haematol.2016.163030
61. Huang Y, Ouyang F, Yang F, Zhang N, Zhao W, Xu H, et al. The expression of Hexokinase 2 and its hub genes are correlated with the prognosis in glioma. BMC Cancer. (2022) 22:900. doi: 10.1186/s12885-022-10001-y
62. Wolf A, Agnihotri S, Micallef J, Mukherjee J, Sabha N, Cairns R, et al. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J Exp Med. (2011) 208:313–26. doi: 10.1084/jem.20101470
63. Aft RL, Zhang FW, Gius D. Evaluation of 2-deoxy-D-glucose as a chemotherapeutic agent: mechanism of cell death. Br J Cancer. (2002) 87:805–12. doi: 10.1038/sj.bjc.6600547
64. Chakraborty P, Lubna S, Bhuin S, K D, Chakravarty M, Jamma T, et al. Targeting hexokinase 2 for oral cancer therapy: structure-based design and validation of lead compounds. Front Pharmacol. (2024) 15:1346270. doi: 10.3389/fphar.2024.1346270
65. Heslop KA, Milesi V, Maldonado EN. VDAC modulation of cancer metabolism: advances and therapeutic challenges. Front Physiol. (2021) 12:742839. doi: 10.3389/fphys.2021.742839
66. Zhou Y, Zheng X, Lu J, Chen W, Li X, Zhao L. Ginsenoside 20(S)-rg3 inhibits the warburg effect via modulating DNMT3A/ miR-532-3p/HK2 pathway in ovarian cancer cells. Cell Physiol biochemistry: Int J Exp Cell physiology biochemistry Pharmacol. (2018) 45:2548–59. doi: 10.1159/000488273
67. Li W, Ma X, Li N, Liu H, Dong Q, Zhang J, et al. Resveratrol inhibits Hexokinases II mediated glycolysis in non-small cell lung cancer via targeting Akt signaling pathway. Exp Cell Res. (2016) 349:320–7. doi: 10.1016/j.yexcr.2016.11.002
68. Clark PA, Bhattacharya S, Elmayan A, Darjatmoko SR, Thuro BA, Yan MB, et al. Resveratrol targeting of AKT and p53 in glioblastoma and glioblastoma stem-like cells to suppress growth and infiltration. J Neurosurg. (2017) 126:1448–60. doi: 10.3171/2016.1.JNS152077
69. Agnihotri S, Mansouri S, Burrell K, Li M, Mamatjan Y, Liu J, et al. Ketoconazole and posaconazole selectively target HK2-expressing glioblastoma cells. Clin Cancer research: an Off J Am Assoc Cancer Res. (2019) 25:844–55. doi: 10.1158/1078-0432.CCR-18-1854
70. Pająk B, Siwiak-Niedbalska E, Jaśkiewicz A, Sołtyka M, Zieliński R, Domoradzki T, et al. Synergistic anticancer effect of glycolysis and histone deacetylases inhibitors in a glioblastoma model. Biomedicines. (2021) 9(12):1749. doi: 10.3390/biomedicines9121749
71. Guo D, Tong Y, Jiang X, Meng Y, Jiang H, Du L, et al. Aerobic glycolysis promotes tumor immune evasion by hexokinase2-mediated phosphorylation of IκBα. Cell Metab. (2022) 34:1312–1324.e6. doi: 10.1016/j.cmet.2022.08.002
72. Li R, Yang W. Gomisin J inhibits the glioma progression by inducing apoptosis and reducing HKII-regulated glycolysis. Biochem Biophys Res Commun. (2020) 529:15–22. doi: 10.1016/j.bbrc.2020.05.109
73. Shteinfer-Kuzmine A, Arif T, Krelin Y, Tripathi SS, Paul A, Shoshan-Barmatz V. Mitochondrial VDAC1-based peptides: Attacking oncogenic properties in glioblastoma. Oncotarget. (2017) 8:31329–46. doi: 10.18632/oncotarget.15455
74. Arif T, Krelin Y, Nakdimon I, Benharroch D, Paul A, Dadon-Klein D, et al. VDAC1 is a molecular target in glioblastoma, with its depletion leading to reprogrammed metabolism and reversed oncogenic properties. Neuro-oncology. (2017) 19:951–64. doi: 10.1093/neuonc/now297
75. Mor I, Cheung EC, Vousden KH. Control of glycolysis through regulation of PFK1: old friends and recent additions. Cold Spring Harbor Symp quantitative Biol. (2011) 76:211–6. doi: 10.1101/sqb.2011.76.010868
76. Blum R, Jacob-Hirsch J, Amariglio N, Rechavi G, Kloog Y. Ras inhibition in glioblastoma down-regulates hypoxia-inducible factor-1alpha, causing glycolysis shutdown and cell death. Cancer Res. (2005) 65:999–1006. doi: 10.1158/0008-5472.999.65.3
77. Hers HG, Van Schaftingen E. Fructose 2,6-bisphosphate 2 years after its discovery. Biochem J. (1982) 206:1–12. doi: 10.1042/bj2060001
78. Pilkis SJ, Claus TH, Kurland IJ, Lange AJ. 6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase: a metabolic signaling enzyme. Annu Rev Biochem. (1995) 64:799–835. doi: 10.1146/annurev.bi.64.070195.004055
79. Clem BF, O'Neal J, Tapolsky G, Clem AL, Imbert-Fernandez Y, , Kerr DA 2nd, et al. Targeting 6-phosphofructo-2-kinase (PFKFB3) as a therapeutic strategy against cancer. Mol Cancer Ther. (2013) 12:1461–70. doi: 10.1158/1535-7163.MCT-13-0097
80. Li FL, Liu JP, Bao RX, Yan G, Feng X, Xu YP, et al. Acetylation accumulates PFKFB3 in cytoplasm to promote glycolysis and protects cells from cisplatin-induced apoptosis. Nat Commun. (2018) 9:508. doi: 10.1038/s41467-018-02950-5
81. Zhang J, Xue W, Xu K, Yi L, Guo Y, Xie T, et al. Dual inhibition of PFKFB3 and VEGF normalizes tumor vasculature, reduces lactate production, and improves chemotherapy in glioblastoma: insights from protein expression profiling and MRI. Theranostics. (2020) 10:7245–59. doi: 10.7150/thno.44427
82. Nakada M, Kita D, Watanabe T, Hayashi Y, Hamada J. Mechanism of chemoresistance against tyrosine kinase inhibitors in Malignant glioma. Brain tumor Pathol. (2014) 31:198–207. doi: 10.1007/s10014-013-0174-9
83. Saruhan S, Özdemir D, Safa R, Agca CA. Investigation of the effects of PFKFB3 small molecule inhibitor KAN0438757 on cell migration and expression level of N-cadherin protein in glioblastoma cell lines. Türk doğa ve fen dergisi. (2024) 13(1):47–53. doi: 10.46810/tdfd.1385118
84. Zhang Y, Li Q, Huang Z, Li B, Nice EC, Huang C, et al. Targeting glucose metabolism enzymes in cancer treatment: current and emerging strategies. Cancers. (2022) 14(19):4568. doi: 10.3390/cancers14194568
85. Clem B, Telang S, Clem A, Yalcin A, Meier J, Simmons A, et al. Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flux and tumor growth. Mol Cancer Ther. (2008) 7:110–20. doi: 10.1158/1535-7163.MCT-07-0482
86. Li J, Zhang S, Liao D, Zhang Q, Chen C, Yang X, et al. Overexpression of PFKFB3 promotes cell glycolysis and proliferation in renal cell carcinoma. BMC Cancer. (2022) 22:83. doi: 10.1186/s12885-022-09183-2
87. Desai S, Ding M, Wang B, Lu Z, Zhao Q, Shaw K, et al. Tissue-specific isoform switch and DNA hypomethylation of the pyruvate kinase PKM gene in human cancers. Oncotarget. (2014) 5:8202–10. doi: 10.18632/oncotarget.v5i18
88. Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. (2008) 452:230–3. doi: 10.1038/nature06734
89. Israelsen WJ, Dayton TL, Davidson SM, Fiske BP, Hosios AM, Bellinger G, et al. PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell. (2013) 155:397–409. doi: 10.1016/j.cell.2013.09.025
90. Zahra K, Dey T, Ashish, Mishra SP, Pandey U. Pyruvate kinase M2 and cancer: the role of PKM2 in promoting tumorigenesis. Front Oncol. (2020) 10:159. doi: 10.3389/fonc.2020.00159
91. Liang J, Cao R, Zhang Y, Xia Y, Zheng Y, Li X, et al. PKM2 dephosphorylation by Cdc25A promotes the Warburg effect and tumorigenesis. Nat Commun. (2016) 7:12431. doi: 10.1038/ncomms12431
92. Zhang F-Y, Hu Y, Que Z-Y, Wang P, Liu Y-H, Wang Z-H, et al. Shikonin inhibits the migration and invasion of human glioblastoma cells by targeting phosphorylated β-catenin and phosphorylated PI3K/Akt: a potential mechanism for the anti-glioma efficacy of a traditional Chinese herbal medicine. Int J Mol Sci. (2015) 16:23823–48. doi: 10.3390/ijms161023823
93. Ding Y, Xue Q, Liu S, Hu K, Wang D, Wang T, et al. Identification of parthenolide dimers as activators of pyruvate kinase M2 in xenografts of glioblastoma multiforme in vivo. J medicinal Chem. (2020) 63:1597–611. doi: 10.1021/acs.jmedchem.9b01328
94. Shang D, Wu J, Guo L, Xu Y, Liu L, Lu J. Metformin increases sensitivity of osteosarcoma stem cells to cisplatin by inhibiting expression of PKM2. Int J Oncol. (2017) 50:1848–56. doi: 10.3892/ijo.2017.3950
95. Liu M, Zhang Z, Wang H, Chen X, Jin C. Activation of AMPK by metformin promotes renal cancer cell proliferation under glucose deprivation through its interaction with PKM2. Int J Biol Sci. (2019) 15:617–27. doi: 10.7150/ijbs.29689
96. Chen J, Jiang Z, Wang B, Wang Y, Hu X. Vitamin K(3) and K(5) are inhibitors of tumor pyruvate kinase M2. Cancer Lett. (2012) 316:204–10. doi: 10.1016/j.canlet.2011.10.039
97. Adeberg S, Bernhardt D, Ben Harrabi S, Bostel T, Mohr A, Koelsche C, et al. Metformin influences progression in diabetic glioblastoma patients. Strahlentherapie und Onkologie: Organ der Deutschen Rontgengesellschaft … [et al]. (2015) 191:928–35. doi: 10.1007/s00066-015-0884-5
98. Qiao X, Wang Z, Chen Y, Peng N, Zhang H, Niu C, et al. Combined metformin and simvastatin therapy inhibits SREBP2 maturation and alters energy metabolism in glioma. Cell Death Dis. (2024) 15:809. doi: 10.1038/s41419-024-07169-5
99. Guo J, Xue Q, Liu K, Ge W, Liu W, Wang J, et al. Dimethylaminomicheliolide (DMAMCL) suppresses the proliferation of glioblastoma cells via targeting pyruvate kinase 2 (PKM2) and rewiring aerobic glycolysis. Front Oncol. (2019) 9:993. doi: 10.3389/fonc.2019.00993
100. Gao M, Yang J, Gong H, Lin Y, Liu J. Trametinib inhibits the growth and aerobic glycolysis of glioma cells by targeting the PKM2/c-myc axis. Front Pharmacol. (2021) 12:760055. doi: 10.3389/fphar.2021.760055
101. Sharma D, Singh M, Rani R. Role of LDH in tumor glycolysis: Regulation of LDHA by small molecules for cancer therapeutics. Semin Cancer Biol. (2022) 87:184–95. doi: 10.1016/j.semcancer.2022.11.007
102. Urbańska K, Orzechowski A. Unappreciated role of LDHA and LDHB to control apoptosis and autophagy in tumor cells. Int J Mol Sci. (2019) 20(9):2085. doi: 10.3390/ijms20092085
103. Daniele S, Giacomelli C, Zappelli E, Granchi C, Trincavelli ML, Minutolo F, et al. Lactate dehydrogenase-A inhibition induces human glioblastoma multiforme stem cell differentiation and death. Sci Rep. (2015) 5:15556. doi: 10.1038/srep15556
104. Wang Z, Wang D, Han S, Wang N, Mo F, Loo TY, et al. Bioactivity-guided identification and cell signaling technology to delineate the lactate dehydrogenase A inhibition effects of Spatholobus suberectus on breast cancer. PLoS One. (2013) 8:e56631. doi: 10.1371/journal.pone.0056631
105. Han X, Sheng X, Jones HM, Jackson AL, Kilgore J, Stine JE, et al. Evaluation of the anti-tumor effects of lactate dehydrogenase inhibitor galloflavin in endometrial cancer cells. J Hematol Oncol. (2015) 8:2. doi: 10.1186/s13045-014-0097-x
106. Manerba M, Vettraino M, Fiume L, Di Stefano G, Sartini A, Giacomini E, et al. Galloflavin (CAS 568-80-9): a novel inhibitor of lactate dehydrogenase. ChemMedChem. (2012) 7:311–7. doi: 10.1002/cmdc.201100471
107. Coyle T, Levante S, Shetler M, Winfield J. In vitro and in vivo cytotoxicity of gossypol against central nervous system tumor cell lines. J neuro-oncology. (1994) 19:25–35. doi: 10.1007/BF01051046
108. Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, et al. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci USA. (2010) 107:2037–42. doi: 10.1073/pnas.0914433107
109. Tennant DA, Durán RV, Gottlieb E. Targeting metabolic transformation for cancer therapy. Nat Rev Cancer. (2010) 10:267–77. doi: 10.1038/nrc2817
110. Sun T, Liu B, Li Y, Wu J, Cao Y, Yang S, et al. Oxamate enhances the efficacy of CAR-T therapy against glioblastoma via suppressing ectonucleotidases and CCR8 lactylation. J Exp Clin Cancer research: CR. (2023) 42:253. doi: 10.1186/s13046-023-02815-w
Keywords: glioblastoma, glycolytic metabolic reprogramming, lactate, tumor microenvironment, targeted therapy
Citation: Yang L, Li S, Yu L, Leng J and Li N (2025) Targeting glycolysis: exploring a new frontier in glioblastoma therapy. Front. Immunol. 15:1522392. doi: 10.3389/fimmu.2024.1522392
Received: 04 November 2024; Accepted: 23 December 2024;
Published: 14 January 2025.
Edited by:
Ekene Emmanuel Nweke, University of the Witwatersrand, South AfricaReviewed by:
Sylvester Omoruyi, University of the Witwatersrand, South AfricaJitcy Joseph, National Health Laboratory Service (NHLS), South Africa
Copyright © 2025 Yang, Li, Yu, Leng and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Na Li, MTUwMDkyMTI0NUBxcS5jb20=