 Ana Carolina Leão1,2*
Ana Carolina Leão1,2* Maria Jose Villar1,2Rakesh Adhikari1,2Cristina Poveda1,2,3
Maria Jose Villar1,2Rakesh Adhikari1,2Cristina Poveda1,2,3 Leroy Versteeg1,2,4Gregório Almeida5
Leroy Versteeg1,2,4Gregório Almeida5 Peter J. Hotez1,2,3,6
Peter J. Hotez1,2,3,6 Maria Elena Bottazzi1,2,3,6
Maria Elena Bottazzi1,2,3,6 Kathryn M. Jones1,2,3
Kathryn M. Jones1,2,3- 1Department of Pediatrics, Division of Tropical Medicine, Baylor College of Medicine, Houston, TX, United States
- 2Texas Children’s Hospital Center for Vaccine Development, Baylor College of Medicine, Houston, TX, United States
- 3Department of Molecular Virology and Microbiology, Baylor College of Medicine, Houston, TX, United States
- 4Cell Biology and Immunology Group, Wageningen University & Research, Wageningen, Netherlands
- 5Centro de Tecnologia em Vacinas, Universidade Federal de Minas Gerais, Belo Horizonte, Brazil
- 6Department of Biology, Baylor University, Waco, TX, United States
Introduction: Chagas disease (CD), caused by the parasite Trypanosoma cruzi, affects millions globally. Despite treatment options in the acute phase, most infections progress to a chronic indeterminate form or develop severe cardiac/gastrointestinal complications. Understanding the immune response is crucial for the development of vaccines and more efficient drugs for the disease control.
Methods: This work investigates the immune response to T. cruzi H1 K68 strain infection in female BALB/c and C57BL/6 mice to characterize differences in Tfh and B cell responses that may be involved in the poor parasite-specific antibody production during acute infection. For this, mice were euthanized 14, 28, and 49 days after infection, and splenic T and B cell populations were evaluated by flow cytometry.
Results: BALB/c mice exhibited a strong Th2-biased response with a massive expansion of classic Tfh cells and GC B cells, potentially linked with polyclonal B cell activation and hypergammaglobulinemia, but not with efficient parasite clearance. C57BL/6 mice displayed a Th1-skewed response with a population of "Th1-like Tfh" cells expressing IFN-γ and CXCR5 associated with lower parasite burden and more focused antibody response, including parasitespecific IgG2c during early acute infection.
Discussion: These findings suggest that these mouse models develop different immune responses mediated by Tfh cells, which are crucial for B cell activation and antibody production. The massive expansion of Tfh cells in BALB/c mice might lead to unspecific antibody production due to excessive B cell activation. Conversely, C57BL/6 mice exhibit a "Th1-like Tfh" response lacking classic Tfh cells, potentially explaining their weak parasite-specific antibody production throughout the acute infection. Overall, this study provides for the first time insights into the complex interplay between Tfh cells and antibody production during T. cruzi infection, suggesting potential targets for therapeutic intervention in CD.
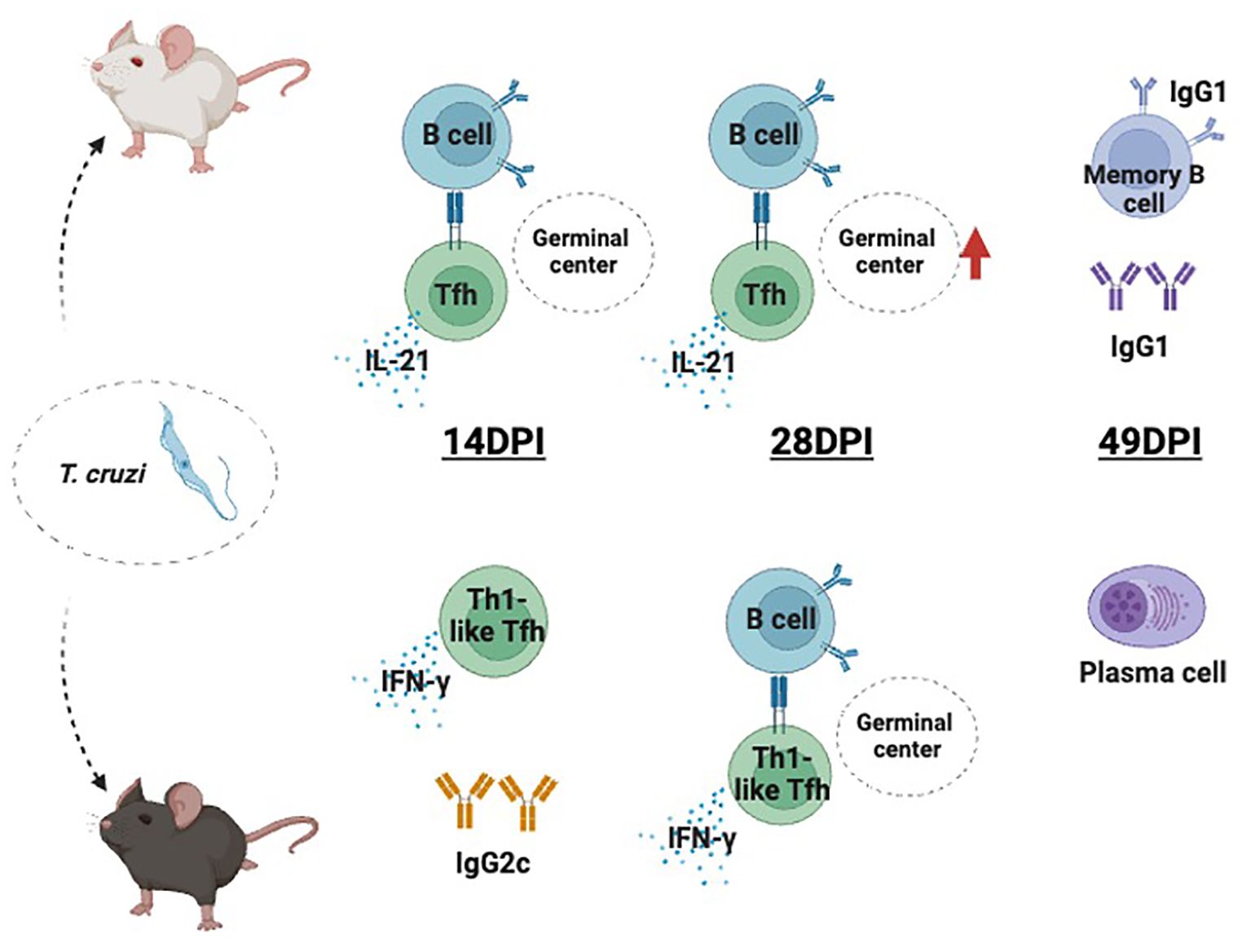
Graphical Abstract. Different responses involving Tfh during early T. cruzi H1 K68 strain infection. Classic Tfh cell production is extensively induced in BALB/c mice, which accordingly present a massive GC B cell expansion, that can be correlated to hypergammaglobulinemia. C57BL/6 mice presented a “Th1-like Tfh” response with IgG2c parasite-specific antibody production and generation of plasma cells. Created with BioRender.com.
1 Introduction
Chagas disease (CD) is a neglected tropical infectious disease caused by the protozoan parasite Trypanosoma cruzi (1, 2). About 6–7 million people worldwide are estimated to be infected with T. cruzi, and 75 million are at risk of infection (1). CD remains endemic to Latin American countries despite some decreases in infection due to vector control and societal poverty reduction, and urbanization (1, 3). In addition, CD has an increasing global presence derived from human migrations, especially to Southern Europe and Australia (1, 4), and expanding evidence for autochthonous transmission in Texas and elsewhere in the Southern United States (5). Furthermore, there are expanding modes of transmission beyond vector-borne illness, including oral and congenital infections (1, 3, 6).
The clinical outcomes of CD range from asymptomatic to severe chronic cardiovascular or gastrointestinal involvement (7, 8). The acute phase is characterized by high parasitemia, often accompanied by systemic symptoms, such as fever, headache, and diarrhea, among others (9). Occasionally hepatomegaly, splenomegaly, myocarditis, and meningoencephalitis (2, 10). Much less frequently people bitten by a triatomine bug show the characteristic first visible signs, which can be a skin lesion or a purplish swelling of the lids of one eye (inoculation chagoma or unilateral bi-palpebral edema) (1, 2). The acute stage is when the only two available anti-parasitic drugs, Benznidazole and Nifurtimox, exhibit reliable effectiveness (1, 7). However, the symptoms are generally mild, contributing to missed or late diagnosis that leads to the chronic phase of the infection (1, 7). When the parasitemia decreases, most individuals remain asymptomatic or in the indeterminate form, with no clinical symptoms (7, 8). Yet approximately 30% of these patients will develop cardiac and/or gastrointestinal symptoms that may be determinant of the morbidity of the disease (1, 7).
T. cruzi infection control depends on both innate and acquired immune mechanisms, including T and B cell responses (11, 12). Macrophages, NK cells, antibodies, CD8+ (cytotoxic), and CD4+ (helper) T lymphocytes producing high levels of IFN-γ are required to control the parasitemia (13–15). Studies on CD acute experimental models have shown the role of proinflammatory Th1 cytokines, such as IFN-γ and TNF-α, in resistance to T. cruzi infection (16, 17), while the Th2 profile is related to disease susceptibility with the production of IL-4 (18, 19). However, in murine experimental studies, over-expression of Th1 immunity can also lead to severe cardiac immunopathology associated with severe inflammation and morbidity. Therefore, effective control of the T. cruzi in the mammalian host may rely on balanced Th1/Th2 responses (20). Humoral immunity is also important for parasite control during T. cruzi infection; previous studies have shown that B cell depletion increases parasitemia and decreases mice survival in non-lethal infection (15). Further, the adoptive transfer of antibodies from late-stage T. cruzi-infected mice to naïve rapidly clears the parasite from the blood (21). Nonetheless, evidence indicates that most B cells are not parasite-specific during early T. cruzi infection (19). Also, reports of T. cruzi B-cell superantigens, like Tc24 protein, facilitate immune escape by interfering with antibody-mediated responses, eliminating catalytic activity from host innate antibodies (22).
T. cruzi has evolved an arsenal of strategies to evade and subvert host immunity, leading to life-long lasting infections. One of these strategies consists of modulating the B cell compartment, attributed to the high variability of parasite surface antigens and the presence of parasite-derived B cell mitogens that cause polyclonal B cell activation and hypergammaglobulinemia (23–26).
Follicular helper T (Tfh) cells are a specialized subset of CD4+ T cells that play a crucial role in assisting B cells during the T-dependent germinal center (GC) response, leading to the production of antigen-specific memory B and plasma cells (27). Tfh cells are distinguished from non-Tfh effector cell, like Th1, Th2, or Th17 cell, by their expression of the chemokine receptor CXCR5, costimulatory receptor ICOS, and transcription factor (TF) Bcl6 (27–29). Tfh cells support the GC reaction through their expression of CD40L that engages CD40 on GC B cells, and with the key help of the cytokine IL-21, promote GC B cell proliferation and maintenance and the generation of long-lived Plasma cells (28).
Mouse models have been informative for analyzing immune responses to T. cruzi infection. Yet mouse strains show variable disease progression and severity that also differ depending on the parasite strain (29, 30). In general, BALB/c mice are more susceptible to T. cruzi infection, compared to relatively resistant C57BL/6 mice, presenting increased parasitemia and mortality given a similar parasite challenge (12, 31). Resistance vs. susceptibility has been linked to C57BL/6 mice Th1 response, in contrast to BALB/c Th2 profile in T. cruzi infection, but also Leishmania, a related kinetoplastid protozoan species (18, 32). In a study comparing Leishmania infantum infection in three murine strains, BALB/c susceptible, C57BL/6 intermediate, and SV/129 resistant, results supported the development of a prevalent Th2-like response in BALB/c, but not in C57BL/6 or SV/129 mice. Also, results showed that both BALB/c and SV/129 presented higher levels of infection-induced splenic Tfh cells than C57BL/6 mice, with parasite-specific antibody significantly higher in SV/129 mice early after infection in comparison with the other two murine strains (33).
CD8+ T cell immunity has been extensively studied, given the critical importance of parasite-specific CD8+ T cells for host resistance throughout the infection (34, 35). In this work, we compare the immune response to T. cruzi H1 K68 strain infection in BALB/c and C57BL/6 mice models to describe for the first time differences in Tfh cell response that may be involved in the lack of parasite-specific antibody production during the initial stages of the infection, which may contribute to relative susceptibility and resistance.
Here, we report the Tfh cells response during T. cruzi acute infection by comparing two different mice models. BALB/c and C57BL/6 mice infected with T. cruzi H1 K68 strain present distinct responses. While BALB/c has a massive expansion of classic Tfh cells and GC B cells, that could be related to the polyclonal B cell activation and hypergammaglobulinemia. C57BL/6 mice present a “Th1-like Tfh” response (36), whereas the lack of classic Tfh cells may contribute to the poor production of parasite-specific antibodies during acute infection. Tfh different responses in T. cruzi infection may help to explain the diversity of CD progressions that range from indeterminate to severe cardiac involvement. A deeper understanding of CD immunopathogenesis is essential for the development of alternatives for treatment and control. Additionally, Tfh immune response presents a key role in vaccine development.
2 Materials and methods
2.1 Mice and parasite
Female C57BL/6 mice (Jackson Laboratories #000664) and female BALB/c mice (Jackson Laboratories #000651), aged 5–8 weeks, were used for the study. Animal experiments were performed in full compliance with the Guide for the Care and Use of Laboratory Animals, 8th edition (37), under a protocol approved by Baylor College of Medicine’s Institutional Animal Care and Use Committee (IACUC) under assurance number D16-00475. The T. cruzi H1 strain (TcI), transfected with the pTRIX2-RE9h plasmid containing the thermostable, red-shifted firefly luciferase gene PpyRE9h (38–40), designated H1 K68 was grown on monolayers of the C2C12 mouse myocyte cell line (ATCC CRL-1772) in RPMI media supplemented with 5% fetal bovine serum and 1X Penicillin/Streptomycin (cRPMI) to propagate tissue culture trypomastigotes (TCT) (41). Culture media containing TCT was collected, parasites were pelleted by centrifugation, washed once with sterile medical grade saline, and resuspended in sterile medical grade saline.
2.2 Study design
T. cruzi H1 K68 strain (TcI) is a mouse model for Chronic Chagasic cardiomyopathy (CCC) and was selected to ensure the survival of the animals during the study (41, 42). Ninety-six mice (48 BALB/c and 48 C57BL/6) were randomly assigned to form infected or control groups (Table 1). The infected groups (24 BALB/and 24 C57BL/6) received by intraperitoneal route 100μL of saline solution containing 5000 trypomastigotes of T. cruzi H1 K68, generated in our laboratory (Table 1) (41). The control groups (24 BALB/c and 24 C57BL/6) received the same volume of saline solution intraperitoneally (Table 1). Blood was collected by tail vein microsampling from all infected mice every week beginning at 7 days post-infection (DPI) to follow parasitemia by quantitative PCR. Mice were monitored daily for morbidity; no clinical signs or mortality were observed during the study. At the study endpoints, 14, 28, and 49 DPI, all mice were humanely euthanized, and hearts, whole blood, femur, and spleens were collected for analysis (Figure 1).
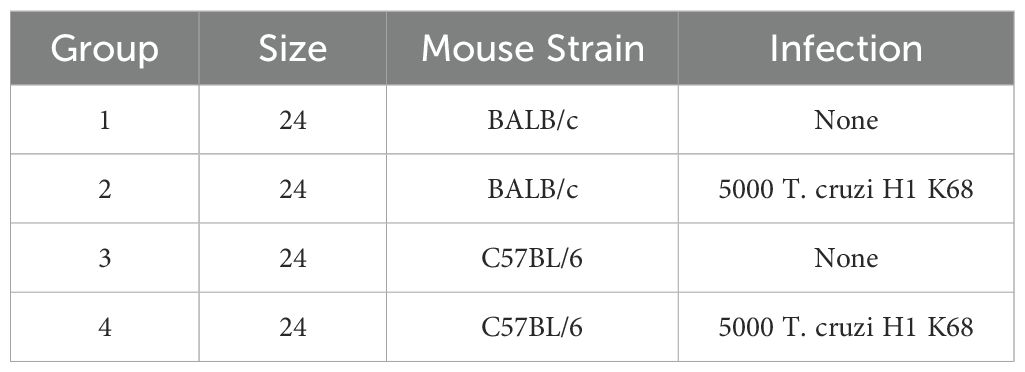
Table 1. Group scheme.
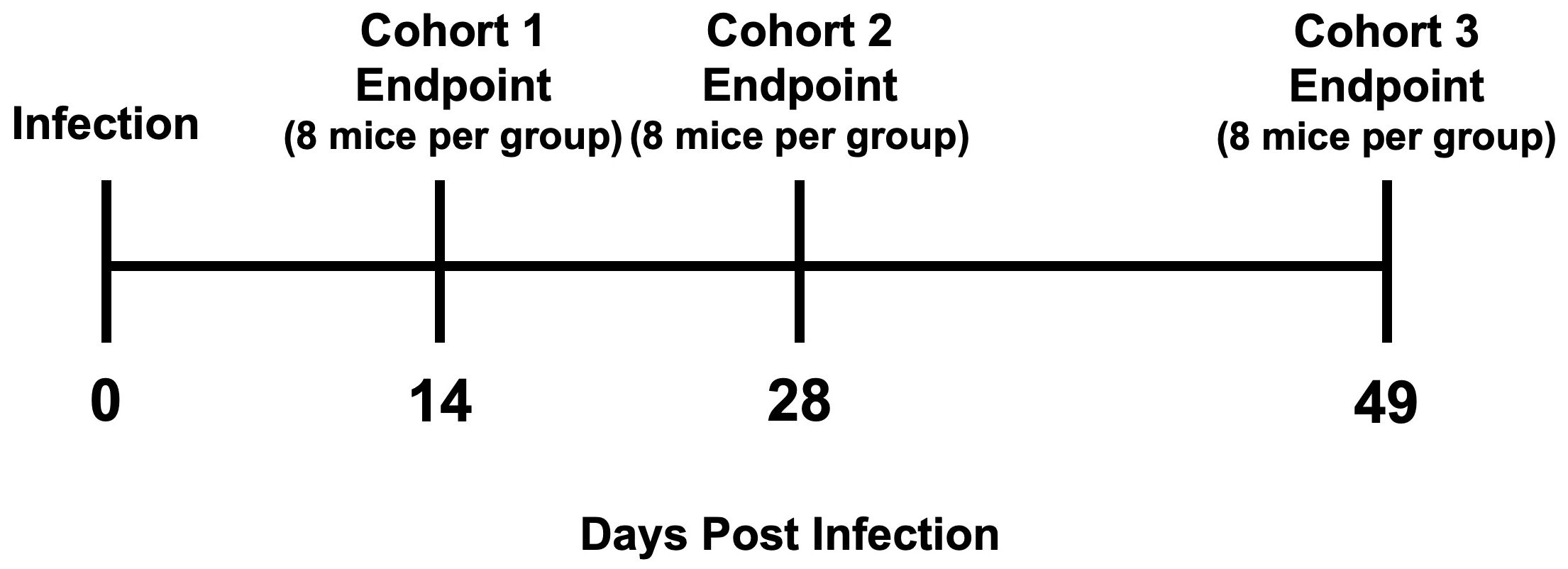
Figure 1. Study design scheme. Timeline with study endpoints.
2.3 QRT-PCR for parasite burden
According to the manufacturer’s guidelines, DNA was extracted from whole blood and frozen heart tissue (20mg) using a PDQeX Nucleic Acid Extractor (MicroGEM). To measure blood and tissue parasite burden, quantitative real-time PCR was performed using TaqMan Fast Advanced master mix (Life Technologies) and oligonucleotides specific for the satellite region of T. cruzi nuclear DNA (primers 5′-ASTCGGCTGATCGTTTTCGA-3′ and 5′-AATTCCTCCAAGCAGCGGATA-3′ and probe 5′-6-FAM-CACACACTGGACACCAA-MGB-3′. T. cruzi data were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH)using the following primers: 5’ CAATGTGTCCGTCGTGGATCT 3’ and 5’ GTCCTCAGTGTAGCCCAAGATG 3’, probe 5’ 6-FAM CGTGCCGCCTGGAGAAACCTGCC MGB 3’; (Life Technologies, CA, USA). After normalization, parasite burden was calculated based on the standard curve of known parasite concentrations. GraphPad Prism software was used to plot parasite equivalents per milliliter of blood over time, and the area under the curve (AUC) was calculated for each animal to determine overall parasitemia. Cardiac parasite burdens were calculated based on a standard curve and expressed as the number of parasites per milligram of tissue (43).
2.4 Splenocyte preparation
Spleens were rinsed in sterile 1X PBS, then transferred to a gentleMACS C Tube containing 3 mL of sterile 1X PBS. The tissue was homogenized using a gentleMACS Dissociator (Miltenyi Biotech, Surrey, UK). Red blood cells in the spleen homogenates were subsequently lysed with ACK lysis buffer (Lonza, 10-548E). The lysis solution was diluted 5-fold with RPMI medium supplemented with 10% fetal bovine serum FBS, 1× penicillin-streptomycin (Pen-Strep), and l-glutamine (cRPMI medium). Then splenocytes were pelleted by centrifugation for 5 min at 300 × g. Splenocytes were resuspended in 5 mL of cRPMI medium and passed through 40μm strainers (BD Biosciences, 352340). Cells were counted using acridine orange-propidium iodide (AOPI) live/dead dye and a Cellometer Auto 2000 automated cell counter. Then, 1 × 106 live splenocytes were incubated for each sample in a 96-well non-tissue culture plate. For intracellular staining, splenocytes were incubated with 50 ng/mL phorbol 12-myristate 13-acetate (PMA)–500 ng/mL ionomycin, or medium only for 5 h at 37°C in 5% CO2. These cells were then analyzed by flow cytometry and Luminex.
2.5 Bone marrow preparation
Bone marrow was extracted from the femurs of each mouse using a 25 G x 5/8 in. needle and 10 mL syringe into RPMI-1640 media supplemented with 10% fetal bovine serum FBS, 1× penicillin-streptomycin (Pen-Strep), and l-glutamine (cRPMI medium). The suspension was filtered through 40μm strainers (BD Biosciences, 352340). Cells were counted using acridine orange-propidium iodide (AOPI) live/dead dye and a Cellometer Auto 2000 automated cell counter. Then, 1×106 live cells were incubated for each sample in a 96-well non-tissue culture plate. These cells were then analyzed by flow cytometry.
2.6 Flow cytometry analysis
To measure CD4- and CD8- responses, splenocytes were collected 5 hours post restimulation for flow cytometry labeling, washed with PBS, and stained with Live/Dead NIR fixable viability dye, anti-CD3e PE (phycoerythrin)/Dazzle 594, anti-CD4 Alexa Fluor 700, and anti-CD8a peridinin chlorophyll protein (PerCP)-Cy5.5. Tfh was evaluated with anti-CD44 fluorescein isothiocyanate (FITC), anti-CD185 (CXCR5) BV421 Brilliant Violet 421 (BV-421) and anti-CD279 (PD-1) PE (phycoerythrin)-Cy7 (PE-Cy7) (Supplementary Table S1). To evaluate intracellular cytokine production, 4.1 μg/ml brefeldin A was added to splenocytes during the 5 h of restimulation. Splenocytes were stained for surface markers as described above, fixed with BD Cytofix/Cytoperm, and permeabilized according to the manufacturer’s instructions. Permeabilized splenocytes were stained with anti-IFN-γ Alexa Fluor 647, anti-TNF Brilliant Violet 605 (BV-605), IL-4 Brilliant Violet 711 (BV-711) and IL-21 phycoerythrin (PE) (Supplementary Table S1). To evaluated B cells, splenocytes or bone marrow cells were collected and stained with Live/Dead Aqua fixable viability dye, anti-CD4 FITC, anti-CD8 FITC, anti-Ly-6G/Ly-6C (Gr-1) FITC, anti-F4/80 FITC and anti-CD11c FITC, anti-CD19 BV-421 or BV-711, anti-CD38 PerCP-Cy5.5 or BV-711, anti-CD86 allophycocyanin (APC), anti- CD184 (CXCR4) PE, anti-mouse CD95 (Fas) BV-605, anti-CD138 APC, anti-CD45R (B220) PE-eFluor 610, anti-IgD Alexa Fluor 700, anti-IgM PE-Cy7 and anti-IgG1 PerCP-Cy5.5 (Supplementary Table S1). Samples were acquired on an Attune instrument, and at least 100,000 total events in a live gate were analyzed using FlowJo software. Cells were gated on forward and side scatter for lymphocytes, exclusion of viability dye, and singlet populations. T cells (Supplementary Figure S1), Tfh cells (Supplementary Figure S2), CXCR5+CD8+ cells (Supplementary Figure S3), B cells in the spleen (Supplementary Figure S4), and B cells in bone marrow (Supplementary Figure S5) were determined based on FMO. Absolute numbers were calculated by multiplying the frequency of live cells from the population of interest by the total number of cells collected from the bone marrow. Data were plotted using GraphPad Prism software.
2.7 Multiplex analysis of cytokines by Luminex
Cell-free supernatant from re-stimulated splenocytes was collected after 5 h and frozen at -80°C until Luminex analysis. Cytokines levels of IL-2, IL-4, IL-6, IL-10, IFN-γ, and TNF-α in supernatants were measured by Luminex-based assay using the Milliplex MAP Mouse kit (Millipore) as previously described (44).
2.8 ELISA parasite-specific IgM, IgG, IgG1, and IgG2a/c
Indirect ELISAs were conducted to measure T. cruzi parasite antibody titers from serum. 96-well NUNC ELISA plates were coated overnight at 4°C with 2 μg/mL of T. cruzi H1 K68 strain parasite lysate (45) in 1x coating buffer (KPL). The coating buffer was discarded the following day, and the plates were blocked with assay buffer (0.1% BSA/1X PBS/0.05%Tween 20) for 2 h at room temperature. Mouse serum was serially diluted two-fold in assay buffer, starting at a 1:200 dilution. For the negative control, pooled naïve mouse sera were also diluted to 1:200. ELISA plates were washed using a BioTek 405 TS plate washer and PBST (1X PBS/0.05%Tween 20). Diluted mouse serum and the controls were added to the washed ELISA plates in duplicate at 100 μL/well and incubated for 2 h at room temperature. Following incubation, the plates were washed four times. Then, 100 μL/well of the appropriate secondary antibodies were added: 1:6000 diluted goat anti-mouse IgM HRP (Lifespan Bioscience), goat anti-mouse IgG1 HRP (Lifespan Bioscience), goat anti-mouse IgG2a HRP (Lifespan Bioscience), or 1:40000 diluted goat anti-mouse Ig2c HRP (Lifespan Bioscience). After 1 h of incubation at room temperature, the ELISA plates were washed five times. Then, 100 μL of TMB substrate (KPL) was added to each well and incubated for 15 minutes. The reaction was stopped by adding 100 μL/well 1M HCl, and the absorbance at 450 nm was measured using an Epoch 2 spectrophotometer (Biotek). Duplicate values of the measured O.D. at 450 nm were averaged for data analysis. The titer cutoff value was calculated as follows: titer cutoff = average negative control + 3 x standard deviation of the negative control. The titer was determined for each mouse serum sample by taking the corresponding dilution factor of the highest dilution with an average O.D. value above the titer cutoff. If a sample did not show an average O.D. value above the titer cutoff at 1:200, an arbitrary titer value of 67 was assigned (baseline).
2.9 Statistical analysis
Statistical analysis was performed using GraphPad Prism software version 9.3.1 for Windows (GraphPad Software, San Diego, California, USA). Significance was calculated by the Normality test followed by the parametric Unpaired t-test or nonparametric Mann-Whitney test. P values ≤0.05 were considered significant. The figures’ P-values ≤0.05, ≤0.01, ≤0.001, and ≤0.0001 are represented as one, two, three, and four symbol characters, respectively.
3 Results
3.1 BALB/c but not C57BL/6 mice infected with T. cruzi present a classic Tfh cell response
BALB/c mice exhibit a classical Tfh cell response, including expansion of IL-21-producing Tfh cells, compared to C57BL/6 mice during H1 K68 T. cruzi infection. Tfh cell response was evaluated by flow cytometry. Results showed that BALB/c infected mice compared to naïve show an increase in CD4+CD44+CXCR5hiPD-1+ Tfh cells at 14, 28, and 49 DPI, which is not present in the C57BL/6 model (Figure 2A). In the same direction, when IL-21 Tfh-producing cells are evaluated, an expansion of CD4+CD44+CXCR5+IL-21+ cells are observed in BALB/c infected mice compared to naïve in all time points but not in C57BL/6 (Figure 2B). In addition, these cells are expanded in BALB/c infected mice compared to C57BL/6 infected mice at 28 and 49 DPI (Figure 2B). Otherwise, when CD4+CD44+CXCR5+IFN-γ+ cells are evaluated, we observe an expansion in C57BL/6 infected mice compared to naïve at 14, 28, and 49 DPI, while in BALB/c mice this increase is present only at 28 and 49 DPI (Figure 2D). Analyzing the infected group from both mouse models, there is an expansion of these cells in C57BL/6 compared to BALB/c mice in all time points (Figure 2D). Interestingly, evaluating CD4+CD44+CXCR5+ cells that are both IFN-γ+ and IL-21+, C57BL/6 infected mice show an increase of these cells compared to naïve and to BALB/c infected mice at 14 DPI (Figure 2E). However, at 28 DPI this population is only expanded in BALB/c infected mice compared to naïve (Figure 2E). Yet at 49 DPI, this polyfunctional cells are expanded in both infected mouse models compared to naïve, and in BALB/c infected mice compared to C57BL/6 infected mice (Figure 2E).
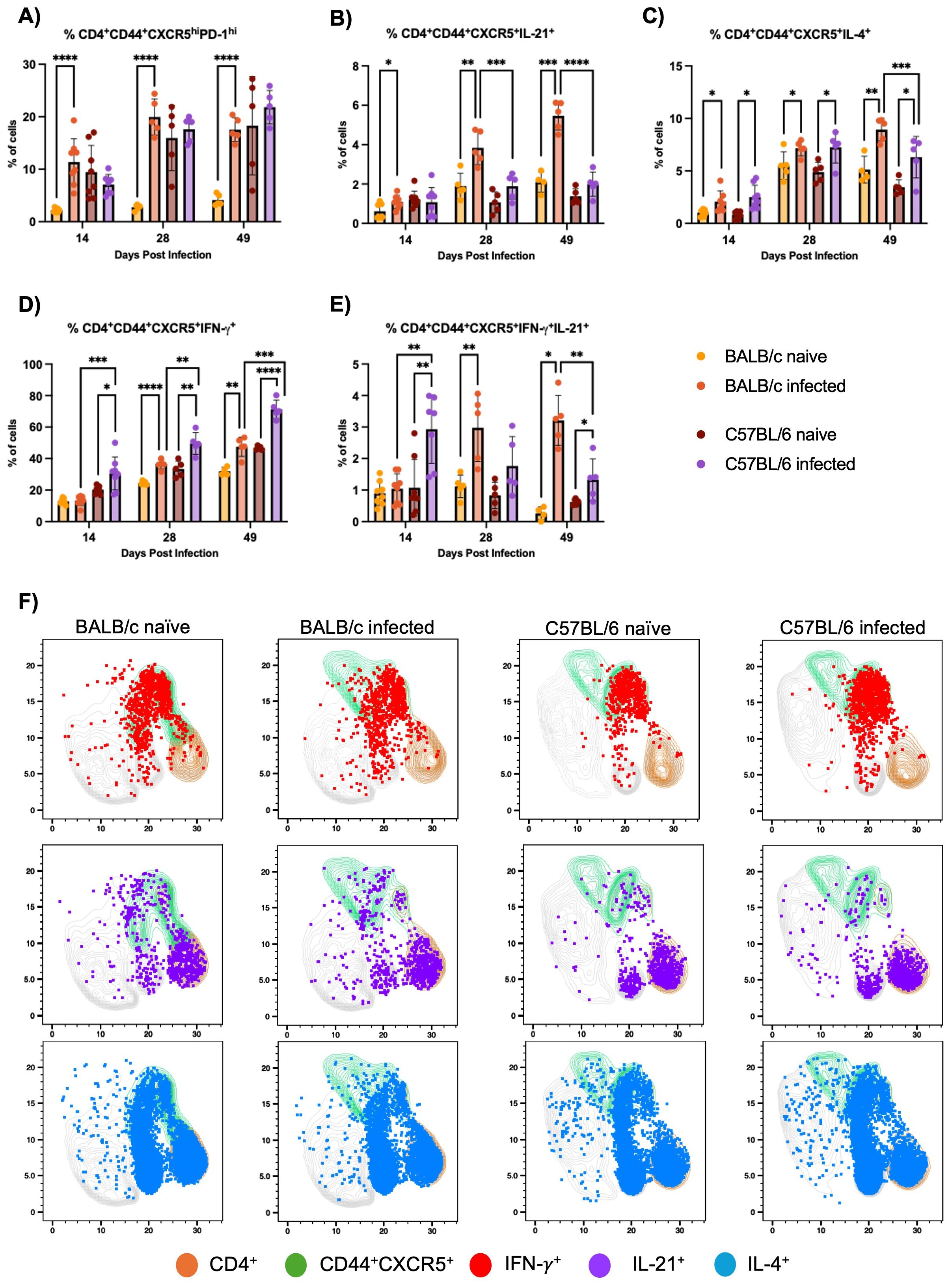
Figure 2. Tfh cell response evaluation. Flow cytometry analysis of splenocytes restimulated with PMA/I for 5 h from BALB/c and C57BL/6 mice naïve or infected with 5000 trypomastigotes of H1 K68 strain of T. cruzi. (A) Analysis of CD4+CD44+CXCR5hiPD-1hi (B) CD4+CD44+CXCR5+IL-21+ (C) CD4+CD44+CXCR5+IL-4+ (D) CD4+CD44+CXCR5+IFN-γ+ cells and (E) UMAP projection of IFN-γ, IL-21 and IL-4 production by CD4+ and CD4+CD44+CXCR5+T lymphocytes compartments at 14 DPI. Each point represents an individual mouse; 14 DPI n = 8, 28 DPI n=5 and 49 DPI n=5. Significance was calculated by the Normality test followed by the parametric Unpaired t-test or nonparametric Mann-Whitney test. P-values of ≤0.05, ≤0.01, ≤0.001, and ≤0.0001 are represented as one (*), two (**), three (***), and four (****) symbol characters, respectively.
Finally, the evaluation of CD4+CD44+CXCR5+IL-4+ cells showed that in the infected group from both models, these cells are expanded at 14, 28, and 49 DPI compared to naïve (Figure 2C). But at 49DPI BALB/c infected mice also present an expansion of these cells compared to C57BL/6 infected group (Figure 2C). A Uniform Manifold Approximation and Projection (UMAP) was generated to analyze cytokine production by CD4+CD44+CXCR5+ T cells at 14 DPI (Figure 2F). Results showed that most IFN-γ+ cells overlap with CD44+CXCR5+, contrarily to IL-21+ cells. In contrast, IL-4+ cells are spread in CD4+ regions that are CD44+CXCR5+ and not positive for these markers (Figure 2F).
3.2 Expansion of CD8+CXCR5+ cells in mice infected with T. cruzi
CD8+CXCR5+ T cells, including IL-21-producing subsets, are expanded in both BALB/c and C57BL/6 mice during H1 K68 T. cruzi infection, with C57BL/6 mice showing a more pronounced expansion at 14 DPI. Recent work in different models and diseases has identified unique subsets of CD8+ T cells expressing the chemokine receptor CXCR5, which directs these cells into secondary lymphoid follicles (47). Comparing H1 K68 T. cruzi infection using BALB/c and C57BL/6 mice, flow cytometry results showed that IL-21 CD8+CXCR5+ producing cells, that have been described as antibody-enhancement (B cell “helper”) subset (48, 49), are increased in infected mice compared to naïve in both models only at 14 DPI (Figure 3A). CD8+CXCR5+IL-4+ population is expanded exclusively in C57BL/6 infected mice compared to naïve at 14 and 49 DPI (Figure 3B). IL-21 or IL-4 CD8+CXCR5+ producing cells are also expanded in C57BL/6 infected mice compared to BALB/c infected mice at 14DPI (Figures 3A, B). Another CD8+CXCR5+ subset that has been reported as antibody-suppressor (50, 51) and described as CD8+CD44+CXCR5+PD-1-CXCR5+IFN-γ+ cells was also evaluated. Results showed that these cells are increased in the infected group compared to naïve in both models at 14, 28, and 49 DPI (Figure 3C).
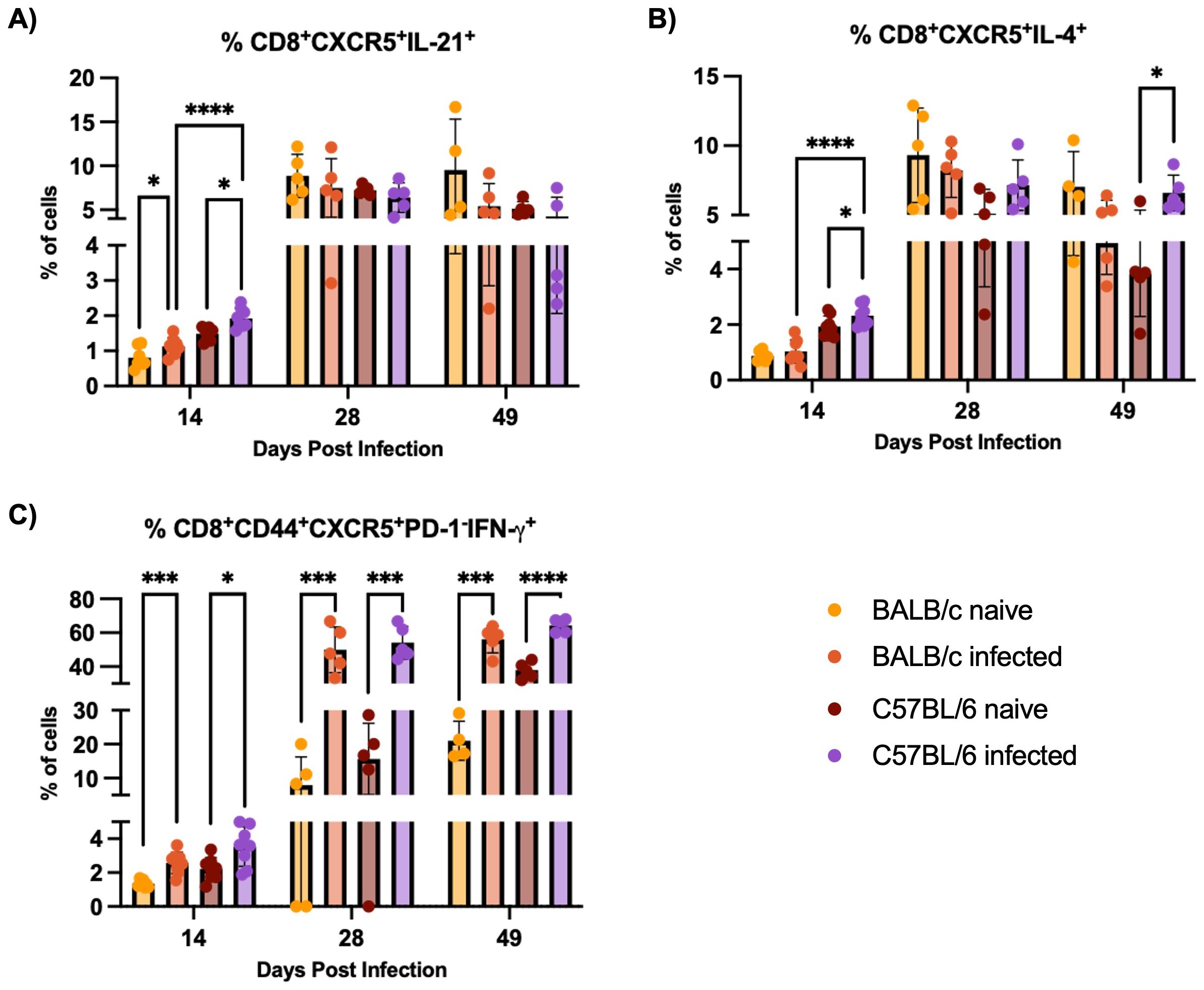
Figure 3. CD8+CXCR5+ cells response evaluation. Flow cytometry analysis of splenocytes restimulated with PMA/I for 5 h from BALB/c and C57BL/6 mice naïve or infected with 5000 trypomastigotes of H1 K68 strain of T. cruzi. (A) Analysis of CD8+CXCR5+IL-21+ (B) CD8+CXCR5+IL-4+ and (C) CD8+CD44+CXCR5+PD-1-IFN-γ+ cells. Each point represents an individual infected mouse: 14 DPI n = 8, 28 DPI n=5, and 49 DPI n=5. Significance was calculated by the Normality test followed by the parametric Unpaired t-test or nonparametric Mann-Whitney test. P-values of ≤0.05, ≤0.001, and ≤0.0001 are represented as one (*), three (***) and four (****) symbol characters, respectively.
3.3 Characterization of the B cell response in BALB/c and C57BL/6 mice infected with T. cruzi
BALB/c mice exhibit an earlier and stronger expansion of germinal center B cells compared to C57BL/6 mice during H1 K68 T. cruzi infection, with BALB/c mice producing higher levels of parasite-specific IgG1. To start the B cell immune response characterization, germinal center (GC) B cells were evaluated by flow cytometry. Results showed that in BALB/c infected mice, the expansion of CD19+CD38-FAS+ GC B cells starts at 14 DPI, massively expands at 28 DPI, and continues at 49 DPI (Figure 4A). In C57BL/6 mice, the expansion of GC B cells in infected mice compared to naïve only happens at 28 DPI and continues at 49 DPI (Figure 4A). Also, this population is expanded in BALB/c infected mice compared to C57BL/6 infected mice at 14 DPI (Figure 4A). When the dark zone (DZ), CD19+CD38-FAS+ CXCR4+CD86- cells, and the light zone (LZ), CD19+CD38-FAS+ CXCR4-CD86+ cells, components of the GC B cells were evaluated, we observe a significant increase in both populations in BALB/c infected mice at 14, 28 and 49 DPI compared to naïve (Figures 4B,C). Meanwhile, in C57BL/6 infected mice, only LZ GC B cells increase starting at 28 DPI and continuing at 49 DPI (Figures 4B, C).
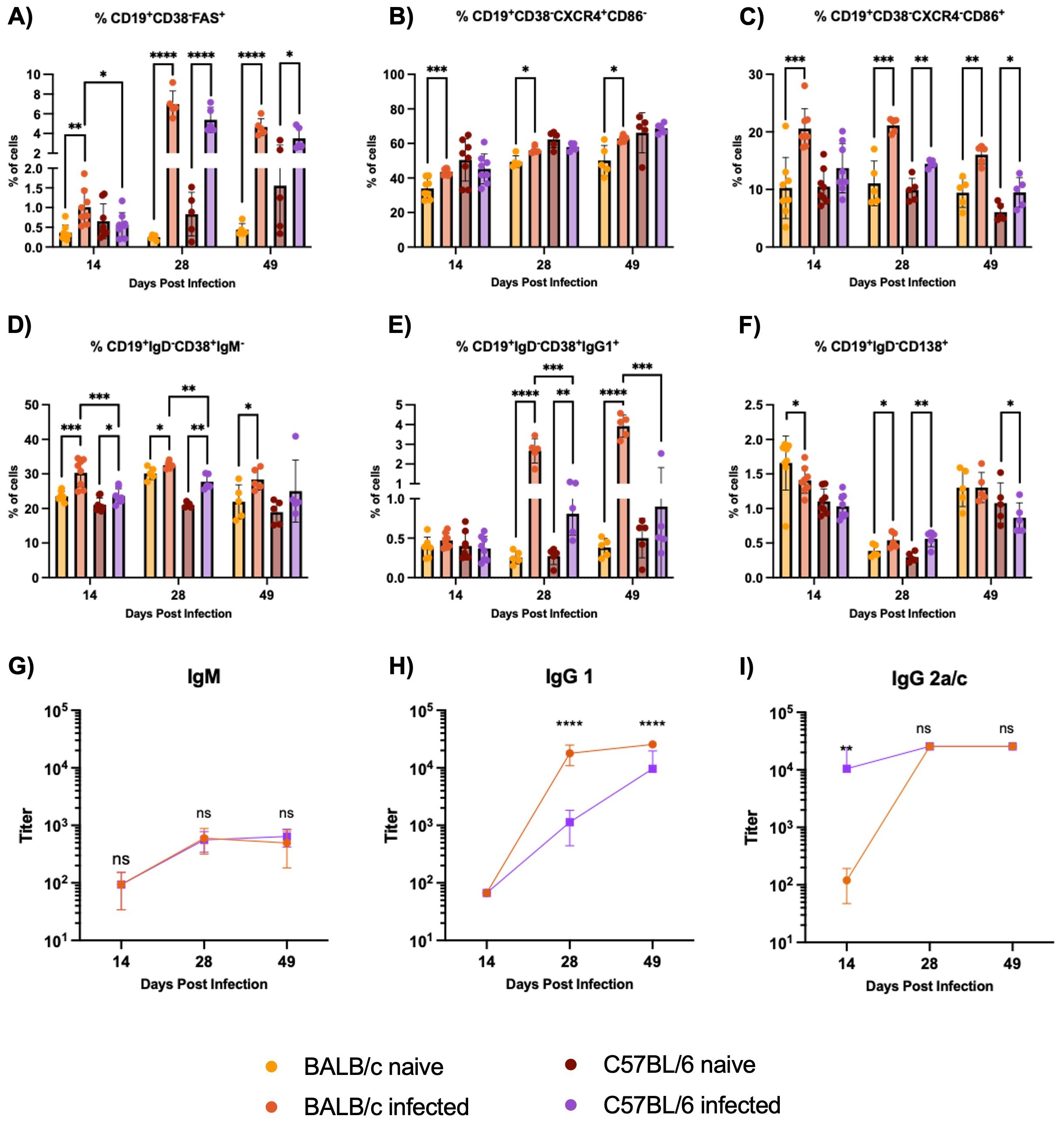
Figure 4. B cell immune response evaluation in the spleens. Flow cytometry analysis of B cells from non-stimulated splenocytes from BALB/c and C57BL/6 mice naïve or infected with 5000 trypomastigotes of H1 K68 strain of T. cruzi. (A) GC B cells CD19+CD38-FAS+ (B) Dark zone GC B cells CD19+CD38-FAS+CXCR4+CD86- (C) Light zone GC B cells CD19+CD38-FAS+CXCR4-CD86+ (D) Isotype switched memory B cells (MBC) CD19+CD38+ IgM- (E) IgG1+MBC CD19+CD38+IgG1+ (F) Antibody secreting cells (ASC) CD19+IgD-CD138+ (G) IgM T. cruzi-specific antibody titers (H) IgG1 T. cruzi-specific antibody titers (I) IgG2a/c T. cruzi-specific antibody titers. For the flow cytometry results, each point represents an individual mouse: 14 DPI n = 8, 28 DPI n=5, and 49 DPI n=5. Each point represents the average of 5 infected mice sera for the ELISA results. Significance was calculated by the Normality test followed by the parametric Unpaired t-test or nonparametric Mann-Whitney test. P-values of ≤0.05, ≤0.01, ≤0.001, and ≤0.0001 are represented as one (*), two (**), three (***), and four (****) symbol characters, respectively.
Following the B cell response characterization, memory B cells (MBC) and antibody-secreting cells (ASC) were evaluated by flow cytometry. Comparing naïve and infected groups, we observe an increase in the isotype switched C19+CD38+IgM- MBC cells in BALB/c infected mice at 14, 28, and 49 DPI. In C57BL/6 infected mice, this expansion is present only at 14 and 28 DPI (Figure 4D). In addition, this population is expanded in BALB/c infected mice compared to C57BL/6 infected mice at 14 and 28 DPI (Figure 4D). IgG1+MBC were also analyzed, results showed that these cells are highly expanded in BALB/c infected mice compared to naïve at 28 and 49 DPI, while in C57BL/6 infected mice, this population is increased only at 28 DPI (Figure 4E). These cells are also expanded in BALB/c infected mice compared to C57BL/6 infected mice at 28 and 49 DPI (Figure 4E). The percentage of CD19+IgD-CD138+ ASC is reduced in BALB/c infected mice compared to the naïve at 14 DPI (Figure 4F). However, at 28 DPI, these cells are expanded in BALB/c and C57BL/6 infected mice compared to the naïve groups (Figure 4F). Additionally, IgM, IgG1, and IgG2a for BALB/c mice, and IgG2c for C57BL/6 mice, T. cruzi-specific antibody titers were analyzed by ELISA. Results showed no significant differences in IgM production between BALB/c and C57BL/6 mice (Figure 4G). Also, higher titers of parasite-specific IgG1 are produced in BALB/c compared to C57BL/6 infected mice at 28 and 49 DPI (Figure 4H). However, C57BL/6 Ig2c titers were higher than BALB/c Ig2a at 14 DPI, and no differences were observed at 28 and 49 DPI (Figure 4I).
3.4 T. cruzi infection induces higher levels of antibody secreting cells (ASC) in C57BL/6 mice bone marrow
In response to H1 K68 T. cruzi infection, both BALB/c and C57BL/6 mice show a reduction in total B cell and antibody-secreting cell populations in the bone marrow at earlier time points (14 and 28 DPI). But by 49 DPI, there is an increase in B cell numbers in both strains, with C57BL/6 mice showing a rebound in ASC population. B cell immune response to H1 K68 T. cruzi infection was also evaluated in the bone marrow. Flow cytometry analysis showed that the percentage of B220+CD19+ total B cells decreases when we compare C57BL/6 naïve to infected mice at 14 and 28 DPI (Figure 5A). In addition, we observe a reduction in the number of these cells at 28 DPI (Figure 5B). BALB/c infected mice present a reduction in the frequency of total B cells at 28 DPI (Figure 5A). At 49 DPI, in both models, the infected mice have an increment in the number of these cells compared to the naïve (Figure 5B). ASC were also evaluated, the percentage of CD19+IgD-CD138+ cells decreased when we compared BALB/c naïve to infected mice at 14 and 28 DPI (Figure 5C). C57BL/6 infected mice present a reduction in ASC frequency at 28 DPI, yet an increment at 49 DPI compared to the naïve (Figure 5C). The number of this population decreased in BALB/c infected mice compared to naïve at 28 DPI and increased in C57BL/6 infected mice compared to the control at 49 DPI (Figure 5D).
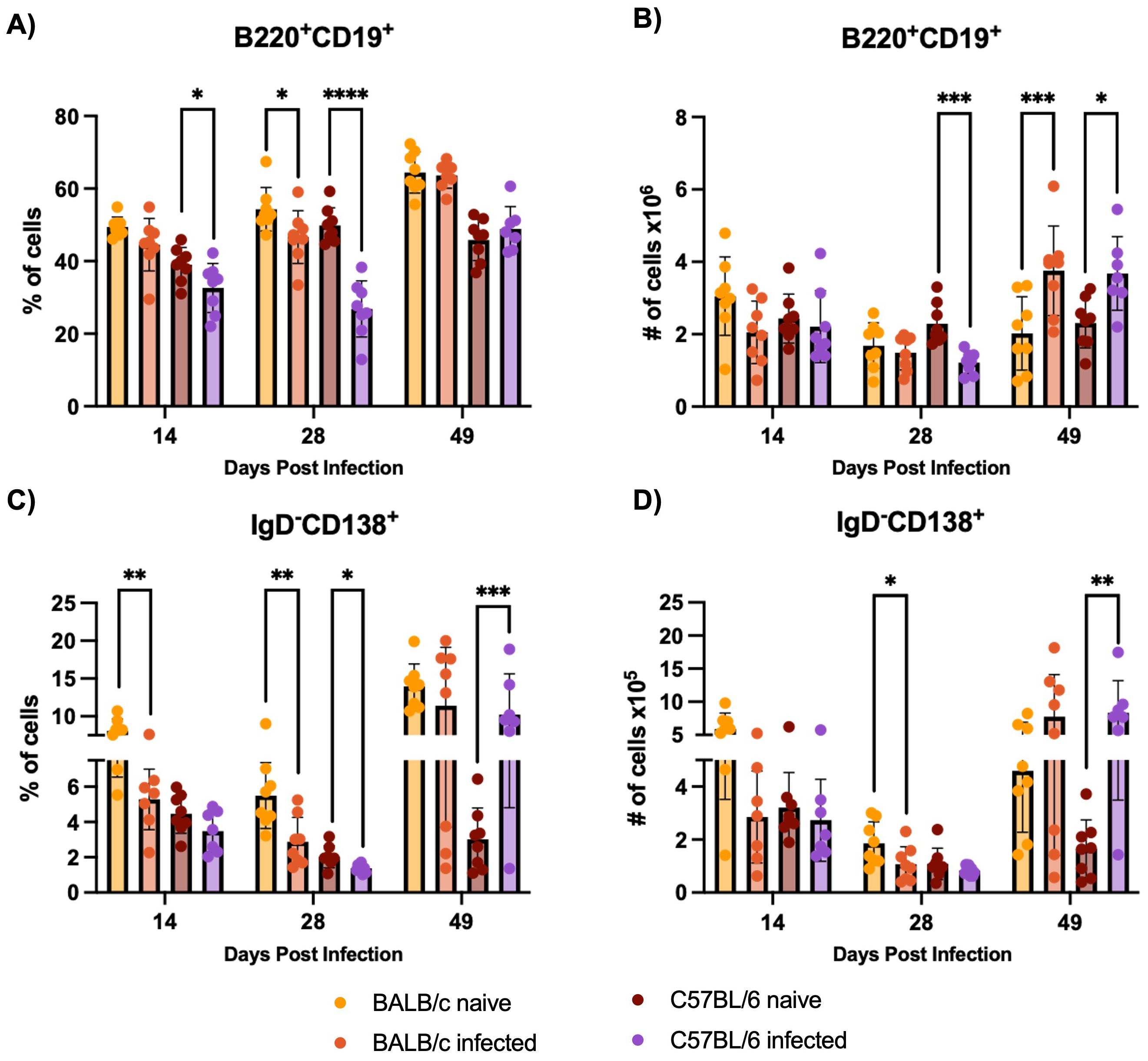
Figure 5. B cell immune response evaluation in the bone marrow. Flow cytometry analysis of bone marrow cells from BALB/c and C57BL/6 mice naïve or infected with 5000 trypomastigotes of H1 K68 strain of T. cruzi. (A) Percentage of B220+CD19+ Total B cells (B) Number of B220+CD19+ Total B cells (C) Percentage of CD19+IgD-CD138+ Antibody secreting cells (ASC) (D) Number of CD19+IgD-CD138+ ASC. Each point represents an individual mouse: 14 DPI n = 8, 28 DPI n=5, and 49 DPI n=5. Significance was calculated by the Normality test followed by the parametric Unpaired t-test or nonparametric Mann-Whitney test. P-values of ≤0.05, ≤0.01, ≤0.001, and ≤0.0001 are represented as one (*), two (**), three (***), and four (****) symbol characters, respectively.
3.5 Parasite burden is increased in BALB/c compared to C57BL/6 during early acute phase
BALB/c mice exhibit significantly higher parasitemia at 7 and 14 DPI and higher cardiac parasite burden at 28 DPI compared to C57BL/6 mice during H1 K68 T. cruzi infection. To compare T. cruzi infection between the two mouse models, BALB/c and C57BL/6 mice were infected with T. cruzi H1 K68, as described above. Parasites in blood were monitored weekly from 7 to 49 DPI (Figure 6A). Cardiac parasite burden was evaluated at the endpoints 14, 28, and 49 DPI (Figure 6B). BALB/c mice had significantly increased parasitemia at 7 and 14 DPI and cardiac parasite burden at 28 DPI compared to the C57BL/6 mice model (Figure 6A, B).
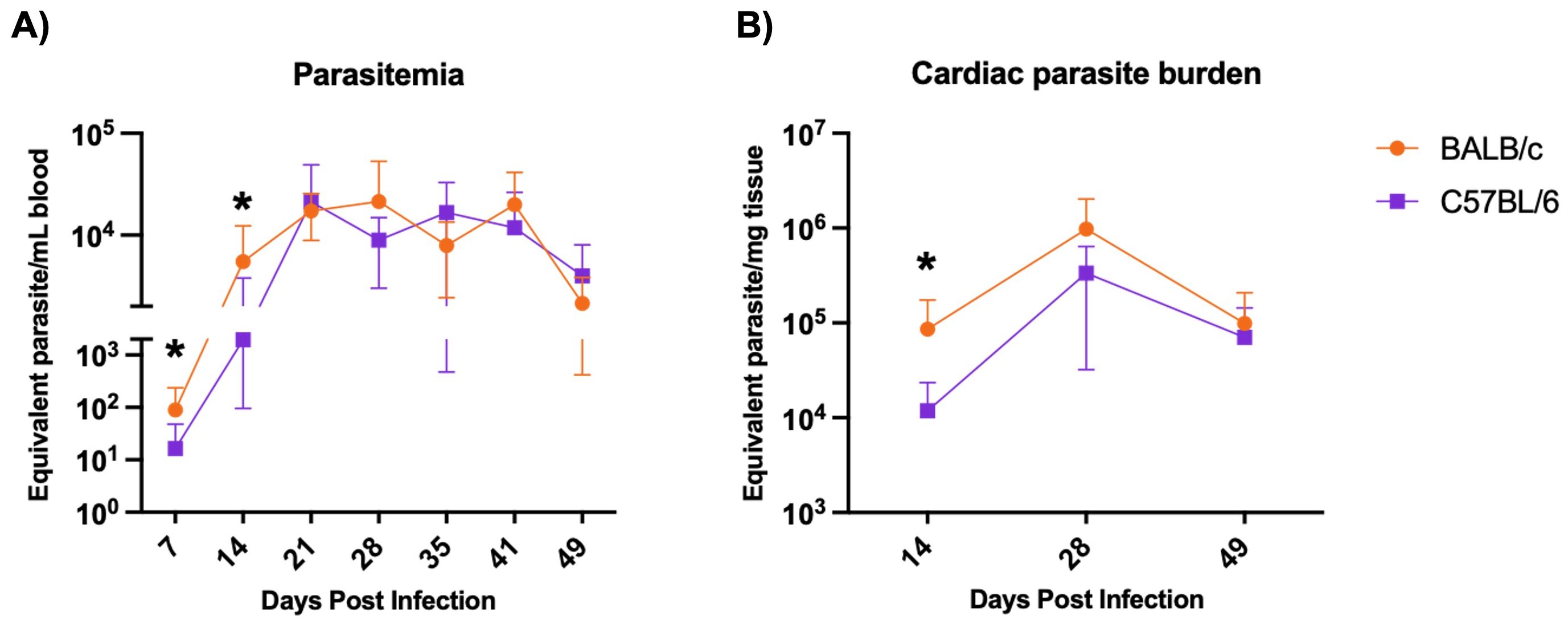
Figure 6. Parasite burden evaluation. Mice were infected with 5000 bft T.cruzi strain H1 – K68. (A) Infection was monitored by measuring parasitemia weekly. Each point represents mean values: 7 DPI n=24, 14 DPI n=24, 21 DPI n=16, 28 DPI n=16, 35 DPI n=8, 41 DPI n=8 and 49 DPI n=8. (B) Cardiac parasite burden was quantified by quantitative real-time PCR. Each point represents mean values n=8. Significance was calculated using the nonparametric Mann-Whitney test and the area under the curve (AUC). P-values of ≤0.05 are represented as one (*) symbol character. AUC from 7 and 14 DPI comparing BALB/c and C57BL/6 p value = 0.0172.
3.6 Secreted cytokines multiplex analysis and T cell response evaluation confirm C57BL/6 Th1 and BALB/c Th2 profiles
C57BL/6 mice exhibit a Th1 skewed immune response, with higher levels of IFN-γ and TNF-α while BALB/c mice show a Th2 response, characterized by higher IL-4 production, during H1 K68 T. cruzi infection. The characterization of the immune response by multiplex analysis of secreted cytokines was also performed. At 14 DPI, higher levels of IFN-γ and TNF- α were generated in C57BL/6 compared to BALB/c-infected mice, that continues at 28 DPI for IFN-γ (Figures 7A, F). Still, at 14 DPI, we have a significant increase of IL-4 in C57BL/6 compared to BALB/c, whereas, at 28 and 49 DPI, the concentration of IL-4 is higher in BALB/c than in C57BL/6 infected mice (Figure 7D). At 28 and 49 DPI, the levels of IL-2 and IL-10 are also higher in BALB/c compared to C57BL/6 infected mice (Figures 7B, E). Additionally, comparing infected to naïve mice, in C57BL/6 there are increased levels of IL-2 at 14 DPI and reduced levels at 28 and 49 DPI (Figure 7B). IL-6 is increased in C57BL/6 infected mice throughout the acute infection (Figure 7C). In BALB/c and C57BL/6 mice, IFN-γ and IL-4 are increased in the infected group compared to naïve throughout the acute infection (Figures 7A, D). While IL-2 is reduced at 28 and 49 DPI, and TNF-α only in BALB/c at 28 DPI (Figures 7B, F). Cytokine-producing CD4+ and CD8+ T-cells were also evaluated by flow cytometry to compare BALB/c and C57BL/6 mice responses to H1 K68 T. cruzi infection (Supplementary Figure S6).
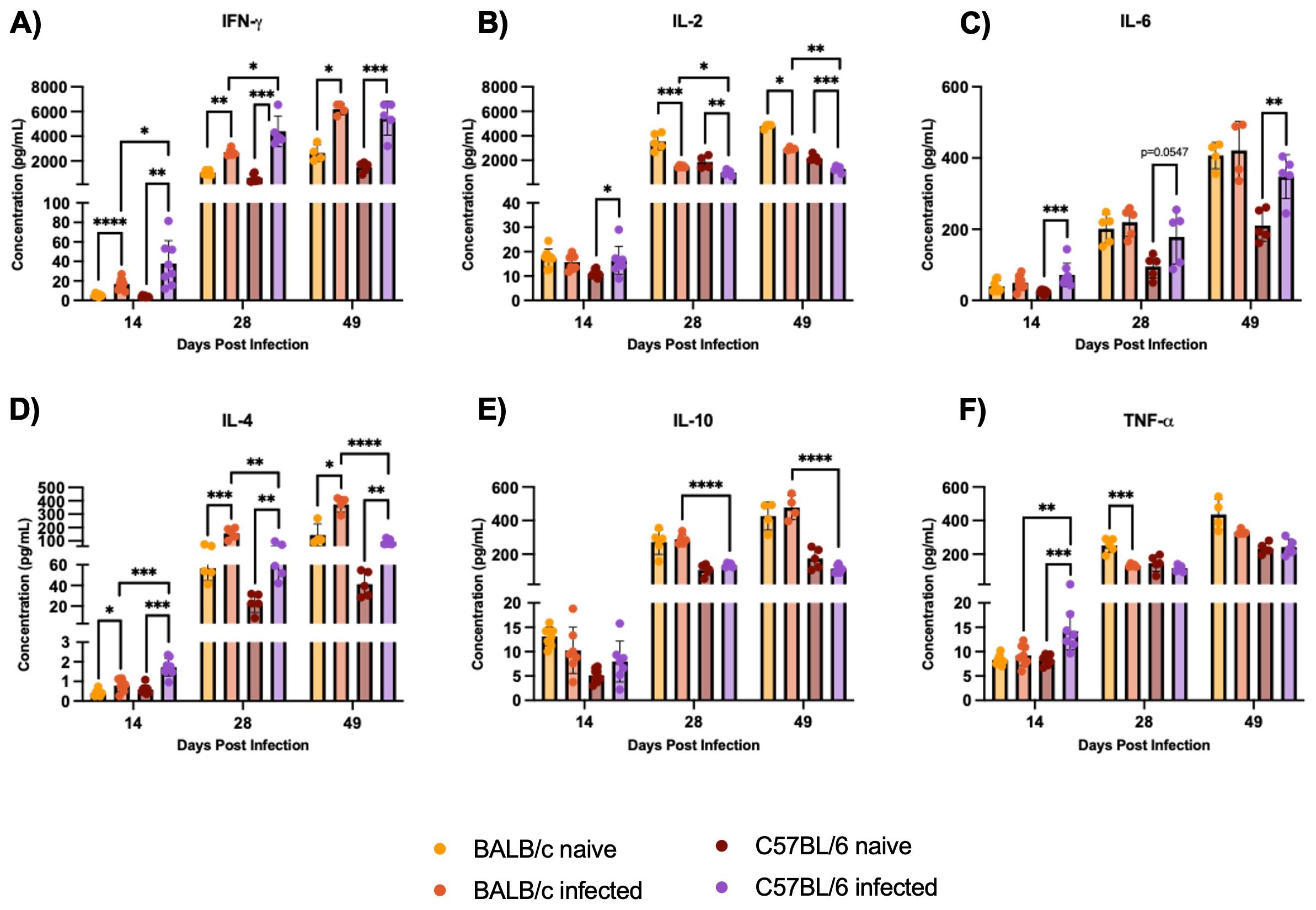
Figure 7. Multiplex analysis of secreted cytokines by Luminex. Splenocytes were restimulated with PMA/I for 5h. The multiplex analysis measured secreted cytokines from these splenocytes. (A) IFN-γ (B) IL-2 (C) IL-6 (D) IL-4 (E) IL-10 (F) TNF-α. Each point represents an individual mouse: 14 DPI n = 8, 28 DPI n=5, and 49 DPI n=5. Significance was calculated by the Normality test followed by the parametric Unpaired t-test or nonparametric Mann-Whitney test. P-values of ≤0.05, ≤0.01, ≤0.001, and ≤0.0001 are represented as one (*), two (**), three (***), and four (****) symbol characters, respectively.
4 Discussion
Induction of an appropriate immune response to T. cruzi during acute infection determines whether parasite burdens and tissue damage are quickly controlled to minimize organ dysfunction or if excessive inflammation leads to progressive damage and clinical disease. Here, we performed a thorough immune evaluation focused on elucidating the early kinetics of Tfh cell and B cell development, contributing to effective parasite control. Tfh cells are distinguishable from naive or other effector T cells by expression of the chemokine receptor CXCR5, which directs Tfh cells to the B cell follicle, and of the exhaustion marker PD-1 (28, 52). Poor Tfh response is associated with a defective GC reaction, while the overabundance can lead to pathogenic autoantibody production and autoimmune disease (53). We confirmed that our BALB/c infection model is relatively susceptible, with higher parasite burdens and a Th2-biased immune response, compared to the relatively resistant C57BL/6 infection model, with lower parasite burdens and a Th1-biased immune response. These data are in accordance with other acute CD (as well as other parasitic kinetoplastid) models that link proinflammatory Th1 cytokines like IFN-γ to resistance and Th2 response with IL-4 production to susceptibility to T. cruzi infection (17, 19).
To better understand the molecular and cellular mechanisms of this established observation, we showed that BALB/c and C57BL/6 mice infected with T. cruzi H1 K68 strain present different responses regarding Tfh cells. BALB/c infected mice exhibited massive expansion of CD4+CD44+CXCR5hiPD-1+ Tfh cells and increased IL-21-producing Tfh cells during acute infection. “Hybrid Th1/Tfh” or “Th1-like Tfh” have been used to describe any IFN-γ+ CD4 T cell also expressing IL-21 and/or CXCR5, functional markers of Tfh in Malaria models (36, 54). Here, we describe for the first time the production of CD4+CD44+CXCR5+IFN-γ+ cells during T. cruzi infection, with a high production of these cells in C57BL/6 infected mice. C57BL/6 mouse groups also showed frequencies of CD4+CD44+CXCR5hiPD-1+ Tfh cells as high as BALB/c infected mice. However, there is no difference between naive and infected C57BL/6 mice, suggesting that this high frequency is not caused by the infection. Further investigation is necessary to evaluate the specificity of this response to T. cruzi infection. In addition, the same is not observed when we analyzed IL-21 Tfh-producing cells, what corroborates that C57BL/6 mouse infected with T. cruzi H1 K68 do not present a classical Tfh cell response.
Recently, CXCR5+CD8 T cells have been described in several pathogenic conditions with varied functional capacity (55). In murine and human studies, antibody-enhancing CXCR5+CD8+ T cells have been reported to express IL-21 (48, 49). Interestingly, at 14 DPI C57BL/6 infected mice show higher frequencies of IL-21 CD8+CXCR5+ producing cells than BALB/c infected mice. What as well could be related to some protection observed in C57BL/6 mice. The role of IL-4-producing CD8+ T cells still hasn’t been well characterized; however, in the CD experimental model, the production of IFN-γ by CD8+ T cells in the absence of IL-4 has been linked to cardiac tissue damage (56). When CD8+CXCR5+IL-4+ is evaluated, we observe a significant increase in these cells in C57BL/6 mice compared to BALB/c. It remains to be investigated whether IL-4 produced by CD8+CXCR5+ would exert the same protective role in T. cruzi infection.
In contrast, antibody-suppressor CXCR5+CD8+ T cells have been identified in transplantation models as another subset by the absence of the co-inhibitory molecule PD-1, the lack of IL-21 production, and the expression of IFN-γ (47, 50). CD8+CD44+CXCR5+PD-1-IFN-γ+ evaluation showed that there is an expansion of this population in infected mice compared to the naïve in both models. Alloantibody suppression was described as mediated, partially, by CD8-mediated clearance of antibody-producing B cells through both FasL and perforin mechanisms (51). This cytotoxic clearance has been shown to be antigen-specific, as CD8+ T Ab-supp cells do not kill naïve or third-party primed IgG+ B cells in vitro or in vivo (47, 51). The Fas receptor/Fas ligand (FasR/FasL) pathway in T. cruzi infection has been described as one of the main mechanisms involved in B cell death, participating in the selectively elimination of IgG+ B cells reactive to parasite but not self-antigens (57, 58).
During the early stages of infection, T. cruzi causes polyclonal B cell activation, leading to hypergammaglobulinemia, yet most of the antibodies produced are not parasite-specific and unable to control infection (59–61). Correlated to the massive expansion of Tfh cells in BALB/c mice infected with H1 K68 T. cruzi strain, when we evaluated GC B cells, a vast expansion of this population was also observed in BALB/c mice. Despite not presenting a Tfh classic response, C57BL/6 mice also expanded GC B cells compared to naïve, but only at 28 and 49 DPI. Previous work has shown that C57BL/6 mice infected with T. cruzi Y strain presented lower polyclonal B cell activation than BALB/c mice, suggesting that polyclonal activation in T. cruzi infection highly depends on the host strain (30). This difference was associated with the Th1-focused C57BL/6 immune response, in contrast to the Th2-focused response developed by BALB/c mice (30). Nevertheless, it has been described that IL-4, predominantly secreted by Tfh and Th2 cells after antigen encounter, is crucial for the survival of GC B cells (62). Further, it has been suggested in Plasmodium spp. infection that the hybrid Th1/Tfh population producing IFN-γ, IL-21, and IL-10 are likely to concurrently provide cellular protection and limit the large humoral response that leads to hypergammaglobulinemia (36).
When the dark zone (DZ) and the light zone (LZ) GC B cells were evaluated, C57BL/6 infected mice only presented an expansion of the LZ compared to naïve mice, while BALB/c infected mice presented an expansion on both compartments. B cells in the DZ proliferate and undergo somatic hypermutation, producing cells that express antibodies that differ in their ability to bind antigen (28). Mutant GC B cells subsequently migrate to the LZ, where they test their newly mutated receptors and receive help from cognate Tfh cells, leading to positive selection of cells expressing higher-affinity antibodies (28). The polyclonal activation of the B cell compartment could restrict the size of the niche needed for optimal development of antigen-specific lymphocytes by increasing competition for activation and survival signals in the lymphoid tissues, resulting in a delayed parasite-specific antibody response (61, 63).
MBC and ASC were evaluated to assess the impact of the different Tfh responses in B cell development. IgG1+MBC is greatly expanded in BALB/c compared to C57BL/6 mice. Additionally, elevated titers of parasite-specific IgG1 are also found in sera from BALB/c-infected mice. IL-4 has been linked to class-switch recombination to IgG1 in mice; remarkably, IL-4 from Tfh cells rather than classical Th2 cells (62). In C57BL/6 mice, IgG2c and IgG2a in the BALB/c are more efficient than other subclasses in neutralizing viruses (64). C57BL/6 T. cruzi parasite-specific IgG2c antibody titers were higher than BALB/c IgG2a at 14 DPI. Although the GC was canonically thought to be the site of class-switch recombination, recent work suggests that switching occurs primarily before complete GC maturation in pre-GC B cells (28).
Another strategy to manipulate the B cell response by T. cruzi is the reduction in B cells in the bone marrow during the infection, enhancing B cell apoptosis and affecting B cell migration to the periphery (65, 66). After H1 K68 T. cruzi strain, B220+CD19+ total B cell decreases when we compare naïve to infected mice, but ASC increases at 49 DPI in the C57BL/6 model.
Studies have shown the role of cytokines that are signaled through signal transducer and activator of transcription 3 (STAT3), like IL-6 and IL-21, in promoting the Tfh cell phenotype (53, 62). STAT3 expression in CD4+ T cells is required for their differentiation into Tfh cells and promotion of GC B cell development and virus-specific antibody responses (53). In a lymphocytic choriomeningitis virus (LCMV) acute infection model, it was demonstrated that STAT3 downmodulates type I interferon (IFN) signaling, as STAT3-deficient Tfh cells display a marked increase in Th1 cell-associated and interferon-inducible transcripts (53). Also, antibody blockade of the IFNαβ receptor promoted Tfh cell differentiation in wild-type mice and mice containing STAT3-deficient CD4+ T cells (53). In a mouse model of Chronic Chagasic Cardiomyopathy (CCC), mice infected with T. cruzi H1 strain and then treated with TTI-101, a small molecule inhibitor of STAT3, showed that STAT3 inhibition eliminated cardiac fibrosis but increased cardiac inflammation. Also, upregulated cardiac gene expression of STAT1, IL-6 and Type I and Type II IFN responses (67).
Collectively, in this work, results suggest that different responses involving Tfh lead to poor and delayed antibody production during early T. cruzi H1 K68 strain infection. Classic Tfh cell production is extensively induced in BALB/c mice, which accordingly present a massive GC B cell expansion, that can be correlated to polyclonal B cell activation and hypergammaglobulinemia, generating unspecific antibodies. Contrarily, C57BL/6 mice presented a “Th1-like Tfh” response that restrained this hypergammaglobulinemia and could be connected to the IgG2c parasite-specific antibody production. This response may have contributed to the better performance of the C57BL/6 model, with less parasites in the blood and heart at 14 DPI. Concurrent, results suggest that H1 K68 T. cruzi infection doesn’t induce the expansion of classic Tfh cells in the C57BL/6 mice model, and it is possible that the lack of this population compromises a stronger specific antibody response during the infection.
Further investigation is needed to assess if the differences in Tfh response could be related to a higher activation of STAT3 over type I IFNs in BALB/c mice, with the opposite happening in the C57BL/6 model. As well to understand the role of Tfh and “Th1-like Tfh” response in T. cruzi infection and the impact of these cells in the generation of long-lived plasma cells in the bone marrow. Study evaluating circulating Tfh cell subsets (cTfh) from adult patients with different clinical forms of chronic Chagas disease showed phenotypic changes between asymptomatic patients and patients with chagasic dilated cardiomyopathy, suggesting that dysregulation of Tfh cells might contribute to Chagas disease progression (9). Moreover, clarifying the role of CD8+CXCR5+ cells in T. cruzi infection and elucidating whether a balanced Th1 and Tfh response would improve host resistance are also needed. The primary function of Tfh cells is to provide protection from infectious diseases, facilitating antibody responses to viral, bacterial, parasite, and fungal infections (46). Potentially, a better understanding of the Tfh response during T. cruzi infection could aid in the development of immunotherapies that generate an earlier and stronger parasite-specific antibody response to control disease progression.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was approved by Baylor College of Medicine’s Institutional Animal Care and Use Committee (IACUC). The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
AL: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Validation, Writing – original draft, Writing – review & editing. MV: Formal Analysis, Investigation, Methodology, Writing – review & editing. RA: Formal Analysis, Investigation, Methodology, Writing – review & editing. CP: Investigation, Methodology, Writing – review & editing. LV: Investigation, Methodology, Writing – review & editing. GA: Formal Analysis, Methodology, Writing – review & editing. PH: Funding acquisition, Resources, Writing – review & editing. MB: Funding acquisition, Resources, Writing – review & editing. KJ: Data curation, Funding acquisition, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. The authors declare financial support was received for the research, authorship, and/or publication of this article. This work was funded by the Robert J. Kleberg, Jr. and Helen C. Kleberg Foundation (PJH) and R01AI168038 (KMJ). This project was supported by the Cytometry and Cell Sorting Core at Baylor College of Medicine with funding from the NIH (NIAID P30AI036211, NCI P30CA125123, and NCRR S10RR024574) and the assistance of Joel M. Sederstrom.
Conflict of interest
All authors are involved in a Chagas Vaccine Development program. MB and PH are listed among the inventors on a Chagas Disease Vaccine patent submitted by Baylor College of Medicine.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1487317/full#supplementary-material
References
1. WHO. Chagas disease (2024). Available online at: https://www.who.int/news-room/fact-sheets/detail/chagas-disease-(american-trypanosomiasis) (Accessed June 13, 2024).
2. de Sousa AS, Vermeij D, Ramos AN Jr, Luquetti AO. Chagas disease. Lancet. (2024) 403(10422):203–18. doi: 10.1016/S0140-6736(23)01787-7
3. Rossi IV, de Souza DAS, Ramirez MI. The end justifies the means: Chagas disease from a perspective of the host-Trypanosoma cruzi interaction. Life (Basel). (2024) 14(4):488. doi: 10.3390/life14040488
4. Durães-Oliveira J, Palma-Marques J, Moreno C, Rodrigues A, Monteiro M, Alexandre-Pires G, et al. Chagas disease: A silent threat for dogs and humans. Int J Mol Sci. (2024) 25:3840. doi: 10.3390/ijms25073840
5. Hotez PJ. The rise of neglected tropical diseases in the “new Texas. PloS Negl Trop Dis. (2018) 12(1):e0005581. Public Library of Science. doi: 10.1371/journal.pntd.0005581
6. Agudelo Higuita NI, Beatty NL, Forsyth C, Henao-Martínez AF, Manne-Goehler J, US Chagas Research Consortium. Chagas disease in the United States: a call for increased investment and collaborative research. Lancet Regional Health Americas. (2024) 34:100768. doi: 10.1016/j.lana.2024.100768
7. Silvestrini MMA, Alessio GD, Frias BED, Sales Júnior PA, Araújo MSS, Silvestrini CMA, et al. New insights into Trypanosoma cruzi genetic diversity, and its influence on parasite biology and clinical outcomes. Front Immunol. (2024) 15. doi: 10.3389/fimmu.2024.1342431
8. Magalhães LMD, Gollob KJ, Zingales B, Dutra WO. Pathogen diversity, immunity, and the fate of infections: lessons learned from Trypanosoma cruzi human–host interactions. Lancet Microbe. (2022) 3:e711–22. Elsevier Ltd. doi: 10.1016/S2666-5247(21)00265-2
9. Quebrada Palacio LP, Fernández ER, Hernández-Vásquez Y, Petray PB, Postan M. Circulating T follicular helper cell abnormalities associated to different clinical forms of chronic chagas disease. Front Cell Infect Microbiol. (2020) 10:126. doi: 10.3389/fcimb.2020.00126
10. Martín-Escolano J, Marín C, Rosales MJ, Tsaousis AD, Medina-Carmona E, Martín-Escolano R. An updated view of the trypanosoma cruzi life cycle: intervention points for an effective treatment. ACS Infect Dis. (2022) 8(6):1107–15. doi: 10.1021/acsinfecdis.2c00123
11. Macaluso G, Grippi F, Di Bella S, Blanda V, Gucciardi F, Torina A, et al. A Review on the Immunological Response against Trypanosoma cruzi. Pathogens. (2023) 12(2):282. doi: 10.3390/pathogens12020282
12. Cristovão-Silva AC, Brelaz-de-Castro MCA, Hernandes MZ, Pereira VRA. Chagas disease: Immunology of the disease at a glance. Cytokine Growth Factor Rev. (2021) 62:15–22. Pergamon. doi: 10.1016/j.cytogfr.2021.10.001
13. Ferraz ML, Gazzinelli RT, Alves RO, Urbina JA, Romanha AJ. Absence of CD4+ T lymphocytes, CD8+ T lymphocytes, or B lymphocytes has different effects on the efficacy of posaconazole and benznidazole in treatment of experimental acute Trypanosoma cruzi infection. Antimicrob Agents Chemother. (2009) 53:174–9. doi: 10.1128/AAC.00779-08
14. Brener Z, Gazzinelli RT. Immnunological control of trypanosoma cruzi infection and pathogenesis of chagas’ Disease. Int Arch Allergy Immunol. (1997) 114:103–10. doi: 10.1159/000237653
15. Kumar S, Tarleton RL. The relative contribution of antibody production and CD8+ T cell function to immune control of Trypanosoma cruzi. Parasite Immunol. (1998) 20:207–16. doi: 10.1046/j.1365-3024.1998.00154.x
16. MaChado FS, Martins GA, Aliberti JCS, Mestriner FLAC, Cunha FQ, Silva JS. Trypanosoma cruzi –infected cardiomyocytes produce chemokines and cytokines that trigger potent nitric oxide–dependent trypanocidal activity. Circulation. (2000) 102:3003–8. doi: 10.1161/01.cir.102.24.3003
17. Hoft DF, Schnapp AR, Eickhoff CS, Roodman ST. Involvement of CD4+ Th1 cells in systemic immunity protective against primary and secondary challenges with Trypanosoma cruzi. Infect Immun. (2000) 68:197–204. doi: 10.1128/IAI.68.1.197-204.2000
18. Kumar S, Tarleton RL. Antigen-specific th1 but not th2 cells provide protection from lethal trypanosoma cruzi infection in mice. J Immunol. (2001) 166:4596–603. doi: 10.4049/jimmunol.166.7.4596
19. Hiyama K, Hamano S, Nakamura T, Nomoto K, Tada I. IL-4 reduces resistance of mice to Trypanosoma cruzi infection. Parasitol Res. (2001) 87:269–74. doi: 10.1007/pl00008577
20. Jones KM, Poveda C, Versteeg L, Bottazzi ME, Hotez PJ. Preclinical advances and the immunophysiology of a new therapeutic Chagas disease vaccine. Expert Rev Vaccines. (2022) 21:1185–203. doi: 10.1080/14760584.2022.2093721
21. Brodskyn CI, Silva AM, Takehara HA, Mota I. IgG subclasses responsible for immune clearance in mice infected with Trypanosoma cruzi. Immunol Cell Biol. (1989) 67:343–8. doi: 10.1038/icb.1989.50
22. Gunter SM, Jones KM, Zhan B, Essigmann HT, Murray KO, Garcia MN, et al. Identification and characterization of the trypanosoma cruzi B-cell superantigen Tc24. Am J Trop Med Hygiene. (2016) 94:114–21. doi: 10.4269/ajtmh.15-0438
23. Minoprio P. Parasite polyclonal activators: new targets for vaccination approaches? Int J Parasitol. (2001) 31:588–91. doi: 10.1016/s0020-7519(01)00171-0
24. Reina-San-Martín B, Degrave W, Rougeot C, Cosson A, Chamond N, Cordeiro-Da-Silva A, et al. A B-cell mitogen from a pathogenic trypanosome is a eukaryotic proline racemase. Nat Med. (2000) 6(8):890–7. doi: 10.1038/78651
25. Bermejo DA, Amezcua Vesely MC, Khan M, Acosta Rodriguez EV, Montes CL, Amezcua Vesely MC, et al. Trypanosoma cruzi infection induces a massive extrafollicular and follicular splenic B-cell response which is a high source of non-parasite-specific antibodies. Immunology. (2011) 132:123–33. doi: 10.1111/j.1365-2567.2010.03347.x
26. Cardoso MS, Reis-Cunha JL, Bartholomeu DC. Evasion of the immune response by trypanosoma cruzi during acute infection. Front Immunol. (2016) 6:1. doi: 10.3389/fimmu.2015.00659
27. Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol. (2011) 29:621–63. doi: 10.1146/annurev-immunol-031210-101400
28. Victora GD, Nussenzweig MC. Germinal Centers. Annu Rev Immunol. (2022) 40:413–42. doi: 10.1146/annurev-immunol-120419-022408.
29. Haolla FA, Claser C, de Alencar BCG, Tzelepis F, de Vasconcelos JR, de Oliveira G, et al. Strain-specific protective immunity following vaccination against experimental Trypanosoma cruzi infection. Vaccine. (2009) 27:5644–53. doi: 10.1016/j.vaccine.2009.07.013
30. Bryan MA, Guyach SE, Norris KA. Specific humoral immunity versus polyclonal B Cell activation in trypanosoma cruzi infection of susceptible and resistant mice. PloS Negl Trop Dis. (2010) 4(7):e733. doi: 10.1371/journal.pntd.0000733
31. Pellegrini A, Carrera-Silva EA, Arocena A, Cano RC, Aoki MP, Gea S. Trypanosoma cruzi antigen immunization induces a higher B cell survival in BALB/c mice, a susceptible strain, compared to C57BL/6 B lymphocytes, a resistant strain to cardiac autoimmunity. Med Microbiol Immunol. (2011) 200:209–18. doi: 10.1007/s00430-011-0192-3
32. Sacks D, Noben-Trauth N. The immunology of susceptibility and resistance to Leishmania major in mice. Nat Rev Immunol. (2002) 2:845–58. doi: 10.1038/nri933
33. Pérez-Cabezas B, Cecílio P, Gaspar TB, Gärtner F, Vasconcellos R, Cordeiro-Da-Silva A. Understanding resistance vs. susceptibility in visceral leishmaniasis using mouse models of Leishmania infantumInfection. Front Cell Infect Microbiol. (2019) 9. doi: 10.3389/fcimb.2019.00030
34. Tarleton RL. CD8+ T cells in trypanosoma cruzi infection. Semin Immunopathol. (2015) 37:233–8. doi: 10.1007/s00281-015-0481-9
35. Acosta Rodríguez EV, Araujo Furlan CL, Fiocca Vernengo F, Montes CL, Gruppi A. Understanding CD8+ T cell immunity to trypanosoma cruzi and how to improve it. Trends Parasitol. (2019) 35:899–917. Elsevier Ltd. doi: 10.1016/j.pt.2019.08.006
36. Carpio VH, Aussenac F, Puebla-Clark L, Wilson KD, Villarino AV, Dent AL, et al. T helper plasticity is orchestrated by STAT3, bcl6, and blimp-1 balancing pathology and protection in malaria. iScience. (2020) 23:101310. doi: 10.1016/j.isci.2020.101310
37. Garber JC, Barbee RW, Bielitzki JT, Clayton LA, Donovan JC, Hendriksen CFM. Guide for the Care and Use of Laboratory Animals. Washington, D.C: National Academies Press (2011). doi: 10.17226/12910
38. Lewis MD, Fortes Francisco A, Taylor MC, Burrell-Saward H, McLatchie AP, Miles MA, et al. Bioluminescence imaging of chronic Trypanosoma cruzi infections reveals tissue-specific parasite dynamics and heart disease in the absence of locally persistent infection. Cell Microbiol. (2014) 16:1285–300. doi: 10.1111/cmi.2014.16.issue-9
39. Branchini BR, Ablamsky DM, Davis AL, Southworth TL, Butler B, Fan F, et al. Red-emitting luciferases for bioluminescence reporter and imaging applications. Anal Biochem. (2010) 396:290–7. doi: 10.1016/j.ab.2009.09.009
40. Ruíz-Sánchez R, de León MP, Matta V, Reyes PA, López R, Jay D, et al. Trypanosoma cruzi isolates from Mexican and Guatemalan acute and chronic chagasic cardiopathy patients belong to Trypanosoma cruzi I. Mem Inst Oswaldo Cruz. (2005) 100:281–3. doi: 10.1590/S0074-02762005000300012
41. Liu Z, Ulrich vonBargen R, Kendricks AL, Wheeler K, Leão AC, Sankaranarayanan K, et al. Localized cardiac small molecule trajectories and persistent chemical sequelae in experimental Chagas disease. Nat Commun. (2023) 14:6769. doi: 10.1038/s41467-023-42247-w
42. Mancino C, Pollet J, Zinger A, Jones KM, Villar MJ, Leao AC, et al. Harnessing RNA technology to advance therapeutic vaccine antigens against chagas disease. ACS Appl Mater Interfaces. (2023) 16(13):15832–46. doi: 10.1021/acsami.3c18830
43. Jones K, Versteeg L, Damania A, Keegan B, Kendricks A, Pollet J, et al. Vaccine-linked chemotherapy improves benznidazole efficacy for acute chagas disease. Infect Immun. (2018) 86:1–15. doi: 10.1128/IAI.00876-17
44. Versteeg L, Le Guezennec X, Zhan B, Liu Z, Angagaw M, Woodhouse JD, et al. Transferring Luminex® cytokine assays to a wall-less plate technology: Validation and comparison study with plasma and cell culture supernatants. J Immunol Methods. (2017) 440:74–82. doi: 10.1016/j.jim.2016.11.003
45. Jones KM, Mangin EN, Reynolds CL, Villanueva LE, Cruz JV, Versteeg L, et al. Vaccine-linked chemotherapy improves cardiac structure and function in a mouse model of chronic Chagas disease. Front Cell Infect Microbiol. (2023) 13:1106315. doi: 10.3389/fcimb.2023.1106315
46. Crotty S. T follicular helper cell biology: A decade of discovery and diseases. Immunity. (2019) 50:1132–48. Cell Press. doi: 10.1016/j.immuni.2019.04.011
47. Elzein SM, Zimmerer JM, Han JL, Ringwald BA, Bumgardner GL. CXCR5+CD8+ T cells: A review of their antibody regulatory functions and clinical correlations. J Immunol. (2021) 206:2775–83. doi: 10.4049/jimmunol.2100082
48. Shen J, Luo X, Wu Q, Huang J, Xiao G, Wang L, et al. A subset of CXCR5+CD8+ T cells in the germinal centers from human tonsils and lymph nodes help B cells produce immunoglobulins. Front Immunol. (2018) 9:2287. doi: 10.3389/fimmu.2018.02287
49. Li Y, Tang L, Guo L, Chen C, Gu S, Zhou Y, et al. CXCL13-mediated recruitment of intrahepatic CXCR5+CD8+ T cells favors viral control in chronic HBV infection. J Hepatol. (2020) 72:420–30. doi: 10.1016/j.jhep.2019.09.031
50. Zimmerer JM, Basinger MW, Ringwald BA, Abdel-Rasoul M, Pelletier RP, Rajab A, et al. Inverse association between the quantity of human peripheral blood CXCR5+IFN-γ+CD8+ T cells with de novo DSA production in the first year after kidney transplant. Transplantation. (2020) 104:2424–34. doi: 10.1097/TP.0000000000003151
51. Zimmerer JM, Pham TA, Wright CL, Tobin KJ, Sanghavi PB, Elzein SM, et al. Alloprimed CD8(+) T cells regulate alloantibody and eliminate alloprimed B cells through perforin- and FasL-dependent mechanisms. Am J Transplant. (2014) 14:295–304. doi: 10.1111/ajt.12565
52. Arnold CN, Campbell DJ, Lipp M, Butcher EC. The germinal center response is impaired in the absence of T cell-expressed CXCR5. Eur J Immunol. (2007) 37:100–9. doi: 10.1002/eji.200636486
53. Ray JP, Marshall HD, Laidlaw BJ, Staron MM, Kaech SM, Craft J. Transcription factor STAT3 and type I interferons are corepressive insulators for differentiation of follicular helper and T helper 1 cells. Immunity. (2014) 40:367–77. doi: 10.1016/j.immuni.2014.02.005
54. Obeng-Adjei N, Portugal S, Tran TM, Yazew TB, Skinner J, Li S, et al. Circulating th1-cell-type tfh cells that exhibit impaired B cell help are preferentially activated during acute malaria in children. Cell Rep. (2015) 13:425–39. doi: 10.1016/j.celrep.2015.09.004
55. Valentine KM, Hoyer KK. CXCR5+ CD8 T cells: protective or pathogenic? Front Immunol. (2019) 10:1322. doi: 10.3389/fimmu.2019.01322
56. Soares MB, Silva-Mota KN, Lima RS, Bellintani MC, Pontes-de-Carvalho L, Ribeiro-dos-Santos R. Modulation of chagasic cardiomyopathy by interleukin-4: dissociation between inflammation and tissue parasitism. Am J Pathol. (2001) 159:703–9. doi: 10.1016/S0002-9440(10)61741-5
57. Silva-Barrios S, Charpentier T, Stäger S. The deadly dance of B cells with trypanosomatids. Trends Parasitol. (2018) 34:155–71. doi: 10.1016/j.pt.2017.10.001
58. Zuñiga E, Motran CC, Montes CL, Yagita H, Gruppi A. Trypanosoma cruzi infection selectively renders parasite-specific igG + B lymphocytes susceptible to fas/fas ligand-mediated fratricide. J Immunol. (2002) 168:3965–73. doi: 10.4049/jimmunol.168.8.3965
59. Minoprio P, Burlen O, Pereira P, Guilbert B, Andrade L, Hontebeyrie-Joskowicz M, et al. Most B cells in acute trypanosoma cruzi infection lack parasite specificity. Scand J Immunol. (1988) 28:553–61. doi: 10.1111/j.1365-3083.1988.tb01487.x
60. Montes CL, Zuñiga EI, Vazquez J, Arce C, Gruppi A. Trypanosoma cruzi mitochondrial malate dehydrogenase triggers polyclonal B-cell activation. Clin Exp Immunol. (2002) 127:27–36. doi: 10.1046/j.1365-2249.2002.01746.x
61. Flávia Nardy A, Freire-De-Lima CG, Morrot A. Immune evasion strategies of trypanosoma cruzi. J Immunol Res. (2015) 2015:178947. doi: 10.1155/2015/178947
62. Choi J, Crotty S, Choi YS. Cytokines in follicular helper T cell biology in physiologic and pathologic conditions. Immune Netw. (2024) 24(1):e8. Korean Association of Immunologists. doi: 10.4110/in.2024.24.e8
63. Freitas AA, Rocha B. Population biology of lymphocytes: the flight for survival. Annu Rev Immunol. (2000) 18:83–111. doi: 10.1146/annurev.immunol.18.1.83
64. Dahlgren MW, Plumb AW, Niss K, Lahl K, Brunak S, Johansson-Lindbom B. Type I interferons promote germinal centers through B cell intrinsic signaling and dendritic cell dependent th1 and tfh cell lineages. Front Immunol. (2022) 13. doi: 10.3389/fimmu.2022.932388
65. Müller U, Schaub GA, Mossmann H, Köhler G, Carsetti R, Hölscher C. Immunosuppression in experimental chagas disease is mediated by an alteration of bone marrow stromal cell function during the acute phase of infection. Front Immunol. (2018) 9:2794. doi: 10.3389/fimmu.2018.02794
66. Zuniga E, Acosta-Rodriguez E, Merino MC, Montes C, Gruppi A. Depletion of immature B cells during Trypanosoma cruzi infection: Involvement of myeloid cells and the cyclooxygenase pathway. Eur J Immunol. (2005) 35:1849–58. doi: 10.1002/(ISSN)1521-4141
Keywords: Chagas disease, Tfh, Th1, B cells, antibody, T. cruzi
Citation: Leão AC, Villar MJ, Adhikari R, Poveda C, Versteeg L, Almeida G, Hotez PJ, Bottazzi ME and Jones KM (2025) Different responses involving Tfh cells delay parasite-specific antibody production in Trypanosoma cruzi acute experimental models. Front. Immunol. 16:1487317. doi: 10.3389/fimmu.2025.1487317
Received: 27 August 2024; Accepted: 04 April 2025;
Published: 28 April 2025.
Edited by:
Alisa Gruden-Movsesijan, Institute for the Application of Nuclear Energy (INEP), SerbiaReviewed by:
Isabela Resende Pereira, Fluminense Federal University, BrazilFausto Edmundo Lima Pereira, Vila Velha University, Brazil
Maria Da Gloria Bonecini De Almeida, Oswaldo Cruz Foundation (Fiocruz), Brazil
Copyright © 2025 Leão, Villar, Adhikari, Poveda, Versteeg, Almeida, Hotez, Bottazzi and Jones. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ana Carolina Leão, YW5hLmRlYXJhdWpvbGVhb0BiY20uZWR1