 Quanren Pan
Quanren Pan Xuemei Huang
Xuemei Huang Chaobin Liu
Chaobin Liu Qingjun Pan
Qingjun Pan Shian Huang
Shian Huang- 1Guangdong Provincial Key Laboratory of Autophagy and Major Chronic Non-communicable Diseases, Clinical Research and Experimental Center, Department of Nephrology, Affiliated Hospital of Guangdong Medical University, Zhanjiang, China
- 2Laboratory of Cardiovascular Diseases, Cardiovascular Medicine Center, Affiliated Hospital of Guangdong Medical University, Zhanjiang, China
- 3Department of Clinical Laboratory, State Key Laboratory of Respiratory Disease, The First Affiliated Hospital of Guangzhou Medical University, Guangzhou, China
Patients with Systemic Lupus Erythematosus (SLE) are significantly more susceptible to atherosclerosis, which may elevate their mortality risk. The review explores recent understandings of the origins and remedies for atherosclerosis associated with SLE. Our focus is particularly on the consequences of immune system disparities, interruptions in intestinal bacteria, and metabolic complications. The influence of SLE on atherosclerosis extends past usual risk elements, including processes specific to the disease. The list encompasses excessive immune cell activity, production of autoantibodies, inflammatory responses. A variety of therapies for atherosclerosis linked to SLE encompass cholesterol-lowering medications, anti-inflammatory drugs, immune suppressors, antimalarials, interferon treatments, NET inhibitors, and methods aimed at T and B-cells. However, existing research has its shortcomings, necessitating additional clinical trials to ascertain the efficacy and security of these therapies. The direct interactions among SLE, gut microbiota, metabolism, and atherosclerosis is underexplored, presenting innovation opportunities. Research into specific gut microbial strains and metabolites’ effects on immune responses and atherosclerosis progression in SLE patients is needed. Such research could uncover novel therapeutic targets and biomarkers, advancing prevention and treatment strategies for SLE cardiovascular complications.
1 Introduction
Patients with systemic lupus erythematosus (SLE) face an elevated risk of cardiovascular diseases (CVDs), particularly atherosclerosis (AS), driven by both traditional and SLE-specific factors. Traditional risk factors such as dyslipidemia, hypertension, and smoking interact with SLE-related pathological mechanisms, including chronic inflammation, immune dysregulation (1, 2).
Macrophages contribute to vascular injury through oxidized low-density lipoprotein (ox-LDL) uptake and foam cell formation (3). Type I interferons (IFNs) enhance neutrophil extracellular traps (NETs) release, exacerbating endothelial dysfunction (4, 5). CXCR3+ T helper 1 (Th1) cells migrate to arterial walls, promoting vascular inflammation, while B cells drive atherosclerosis via autoantibody production (e.g., anti-endothelial cell antibodies) and immune complex deposition (6–8).
Pro-inflammatory gut microbiota elevate systemic inflammation (9, 10), and dietary L-carnitine conversion to trimethylamine N-oxide (TMAO) by Emergencia timonensis accelerates atherosclerosis in mice and humans (11). SLE patients exhibit gut dysbiosis characterized by reduced beneficial bacteria and increased pathogenic species (12, 13). However, direct evidence linking gut microbiota or metabolites to SLE-associated atherosclerosis remains sparse, highlighting the need for further research.
We have focused on discussing immune-targeted therapies (e.g., interferon inhibitors (14), T-cell (15), and B-cell therapies (16)) and inflammatory pathways. Emerging approaches modulating gut microbiota or metabolites (e.g., TMAO inhibitors) show preclinical potential to attenuate plaque inflammation, though clinical validation in SLE remains scarce (17). This underexplored axis offers novel therapeutic opportunities.
This review aims to offer novel insights and new research directions for atherosclerosis associated with SLE. Current therapeutic strategies for SLE-related atherosclerosis prioritize immune balance restoration (e.g., interferon inhibition and T/B-cell modulation). Simultaneously, targeting gut microbiota dysbiosis and metabolites like TMAO emerge as promising adjunct approaches to ameliorate both autoimmune dysregulation and atherosclerosis in SLE. Integrating these dual pathways may pioneer synergistic therapies to reduce cardiovascular burden in SLE patients.
2 The role of immune imbalance in the pathogenesis of SLE-related atherosclerosis
2.1 Pro-inflammatory environment, oxidative stress, and macrophage dysfunction
The pro-inflammatory environment, oxidative stress, and macrophage dysfunction constitute key mechanisms driving SLE-related atherosclerosis (Figure 1). SLE, characterized by chronic inflammation, induces endothelial cell damage and dysfunction (18). Pro-inflammatory cytokines (e.g., TNF-α, IFN-γ) upregulate endothelial adhesion molecules, facilitating monocyte-laden leukocyte migration into the vascular intima (19). Patients with SLE exhibit a distinct “lupus lipoprotein pattern” with reduced high-density lipoprotein (HDL) and elevated low-density lipoprotein (LDL), total cholesterol, and triglycerides (20). This lipid profile synergizes with chronic inflammation to promote vascular injury and plaque initiation (21). Oxidative stress, defined by reactive oxygen species (ROS) overproduction, critically accelerates AS pathogenesis (22). ROS increase vascular permeability (23), and convert LDL to oxidized LDL (oxLDL) that accumulates subendothelially (3). Key enzymes like myeloperoxidase (MPO) and NADPH oxidase (NOX) are involved in this oxidative process. Oxidized LDL (oxLDL) is particularly critical in atherosclerosis, inducing inflammation through scavenger and Toll-like receptors (TLR) (24). OxLDL promotes the migration of monocytes towards the vascular endothelium and facilitating monocyte chemotaxis and adhesion through interactions between surface molecules like CD36 on monocytes and endothelial adhesion molecules such as vascular Cell Adhesion Molecule-1 (VCAM-1) and Intercellular Adhesion Molecule-1 (ICAM-1) (3).
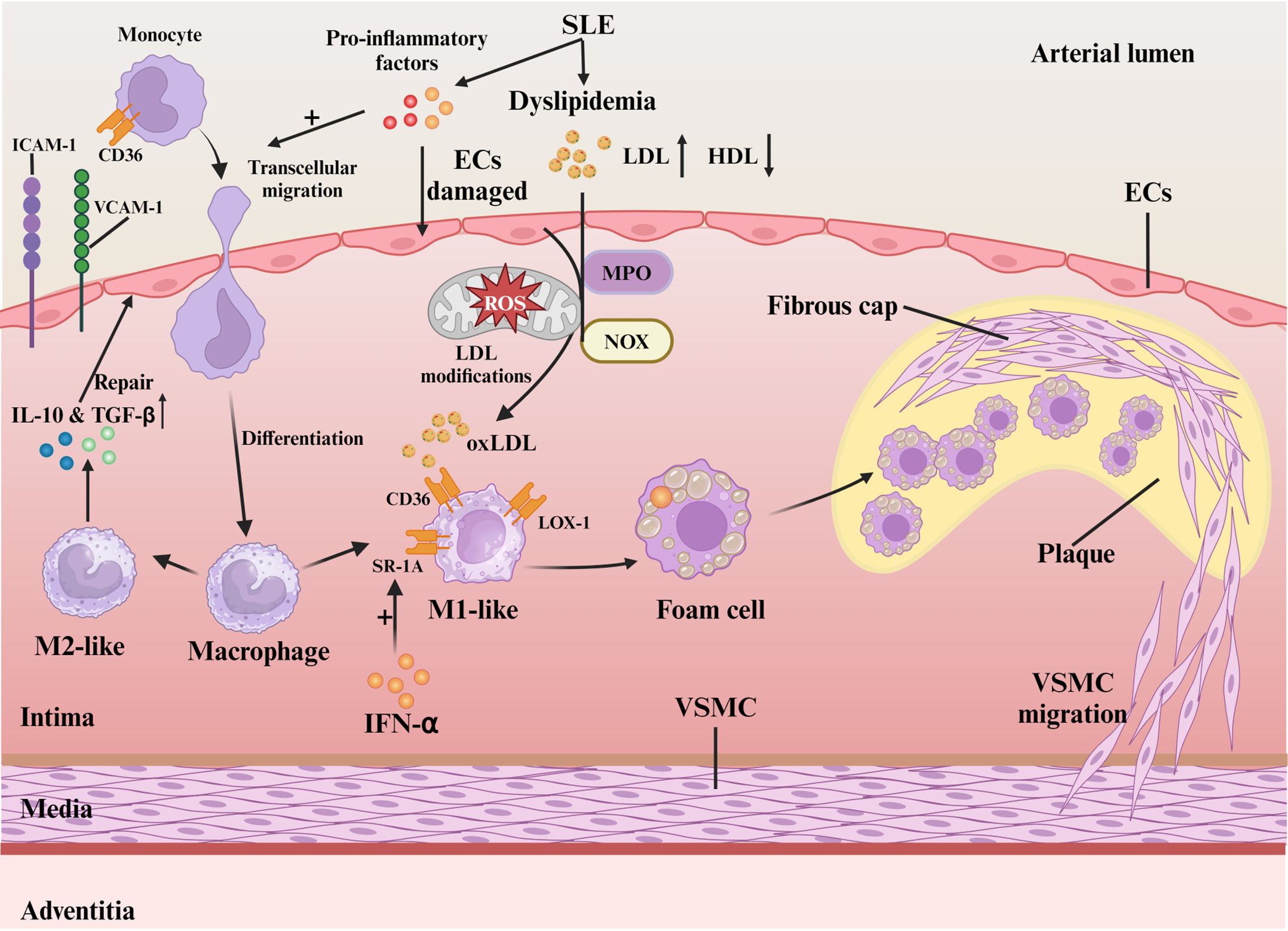
Figure 1. Mechanisms of oxidative stress and macrophage in SLE-associated atherosclerosis. Activated pro-inflammatory cytokines in SLE blood induce endothelial cell damage and monocyte migration via ICAM-1/VCAM-1. Differentiated macrophages polarize into M1/M2 based on local cues. LDL oxidation by myeloperoxidase and NADPH oxidase forms oxLDL, which M1 macrophages internalize via CD36, SR-A1, and LOX-1, leading to foam cell formation and plaque progression. VSMCs reinforce plaque stability by forming a fibrous cap.
Macrophage SRs (CD36, SR-A1, LOX-1) mediate oxLDL uptake, forming foam cells essential for AS progression (3, 25). M1 macrophages with high CD36/SR-A1 expression dominate early lesions (26), whereas M2 counterparts with reduced LOX-1 and elevated IL-10/TGF-β secretion exert protective effects (26). In SLE, IFN-α-induced SR-A1 overexpression (27) combined with elevated LDL (28) exacerbates foam cell formation. Patient-specific SR expression patterns modulate oxLDL clearance efficiency and inflammatory responses (25). Macrophage-T cell crosstalk in plaques sustains cytokine-driven inflammation (26). Polarization dynamics determine plaque fate: M1-derived pro-inflammatory mediators destabilize plaques (29), while M2 macrophages promote stabilization (29). oxLDL-driven M1 polarization predominance underlies SLE-accelerated AS (30).
The buildup of lipids, immune cells, and debris along with extracellular matrix (ECM) degradation and rising inflammation promotes unstable plaque development (31). vascular smooth muscle cell (VSMC)-mediated fibrous cap formation stabilizes plaques through ECM reinforcement (32). Neutrophil-induced endothelial injury disrupts this process, increasing thrombosis risk (33). Chronic SLE inflammation synergizes with traditional risk factors to create self-perpetuating cycles of plaque expansion/rupture, culminating in myocardial infarction or stroke (31). Therefore, targeting the oxidative stress-macrophage polarization axis may break this vicious cycle in SLE-associated AS.
2.2 Type I IFN response
Type I interferons (IFN-I) are crucial in the development of SLE-Related AS, as depicted in Figure 2. In SLE, IFN-I production originates from dendritic cells, neutrophils, and monocytes/macrophages (21), encompassing thirteen IFN-α subtypes, IFN-β, and rare variants (IFN-ϵ, IFN-κ, IFN-ω) (34). Over 60% of SLE patients demonstrate an “IFN signature” characterized by elevated IFN-regulated gene activity, with nearly 50% exhibiting sustained high IFN-I levels linked to genetic predisposition and enhanced disease risk (35). Defective apoptosis in SLE increases extracellular DNA/RNA that complexes with anti-DNA/RNA antibodies to form immune complexes (ICs) (36). ICs activate plasmacytoid dendritic cells (pDCs), which are primary IFN-I producers in SLE (37). pDCs recognize DNA and RNA in immune complexes via TLR9 and TLR7, triggering myeloid differentiation factor 88 (MyD88) and downstream signaling through nuclear factor kappa B (NF-κB) and Mitogen-activated protein kinases (MAPKs) (37). Subsequent nuclear translocation of interferon regulatory factor 7 (IRF7) triggers IFN-I transcription via binding to interferon-stimulated response element (ISRE) promoters (37, 38). Autocrine amplification occurs through IFNAR-mediated JAK kinase (TYK2/JAK1) activation, inducing STAT1/2 phosphorylation and ISGF3 complex formation that enhances interferon-stimulated genes (ISGs) expression (21).
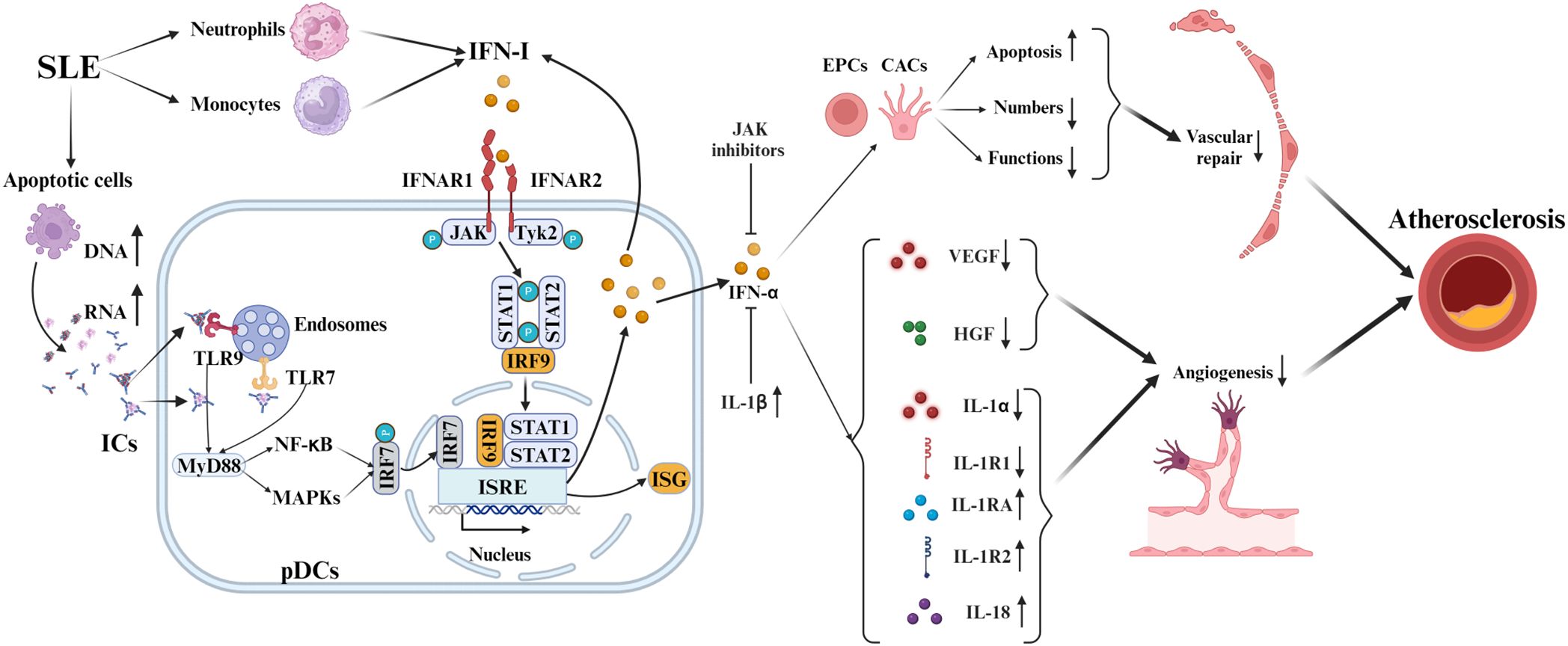
Figure 2. Mechanisms of IFN-I in SLE-associated atherosclerosis. In SLE, pDCs produce IFN-I via TLR7/9 sensing of immune complex nucleic acids, activating IRF7 and increasing IFN-I. IFN-I binding to IFNAR triggers JAK-STAT, enhancing ISG expression via IRF9. It causes endothelial injury and curbs angiogenesis by inhibiting VEGF and HGF, and modulates cytokines like IL-1 and IL-18, advancing SLE pathology. IFN-α also promotes EPC and CAC apoptosis and dysfunction. Counteracting IFN-α’s anti-angiogenic effects involves JAK inhibitors and IL-1β.
IFN-I-induced endothelial cell damage and repair-imbalance accelerate atherosclerosis (39). In SLE, IFN-α significantly exacerbates atherosclerosis by modulating endothelial progenitor cells (EPCs) and bone marrow-derived circulating angiogenic cells (CACs) (40). This modulation includes inducing EPC apoptosis, which negatively impacts their number, structure, and function, thus impairing vascular repair (41). Furthermore, IFN-α inhibits vascular endothelial growth factor (VEGF) and hepatocyte growth factor (HGF) activities, hindering neovascularization and repair (39). Inhibiting the IFNAR reverses EPC/CAC dysfunction in SLE, restoring angiogenic potential (39). In NZM-2328 mice, IFN-α worsens reduced endothelial function and EPC/CAC levels, which IFNAR knockout prevents (42).
Concurrently, IFN-α creates anti-angiogenic microenvironments via IL-1 pathway dysregulation (reduced IL-1α/1β/1R1; elevated IL-1Ra/1R2) that JAK inhibitors effectively counteract (40). IFN-I further induces pro-atherogenic cytokines like IL-18, exacerbating vascular injury and AS progression in SLE (43). In conclusion, Type I interferons play a crucial role in SLE, influencing immune responses and vascular damage.
2.3 Neutrophils and neutrophil extracellular traps
Neutrophils play a key role in SLE-related AS development by enhancing the IFN-I signature and NET formation, as shown in Figure 3. NETs are characterized by large, web-like structures composed of chromatin, histones, and proteins (44). The formation of these structures depends on ROS produced by NOX and/or mitochondrial ROS. Additionally, the activation of peptidyl arginine deiminase (PAD) and MPO converts arginine to citrulline, leading to chromatin decondensation (44).
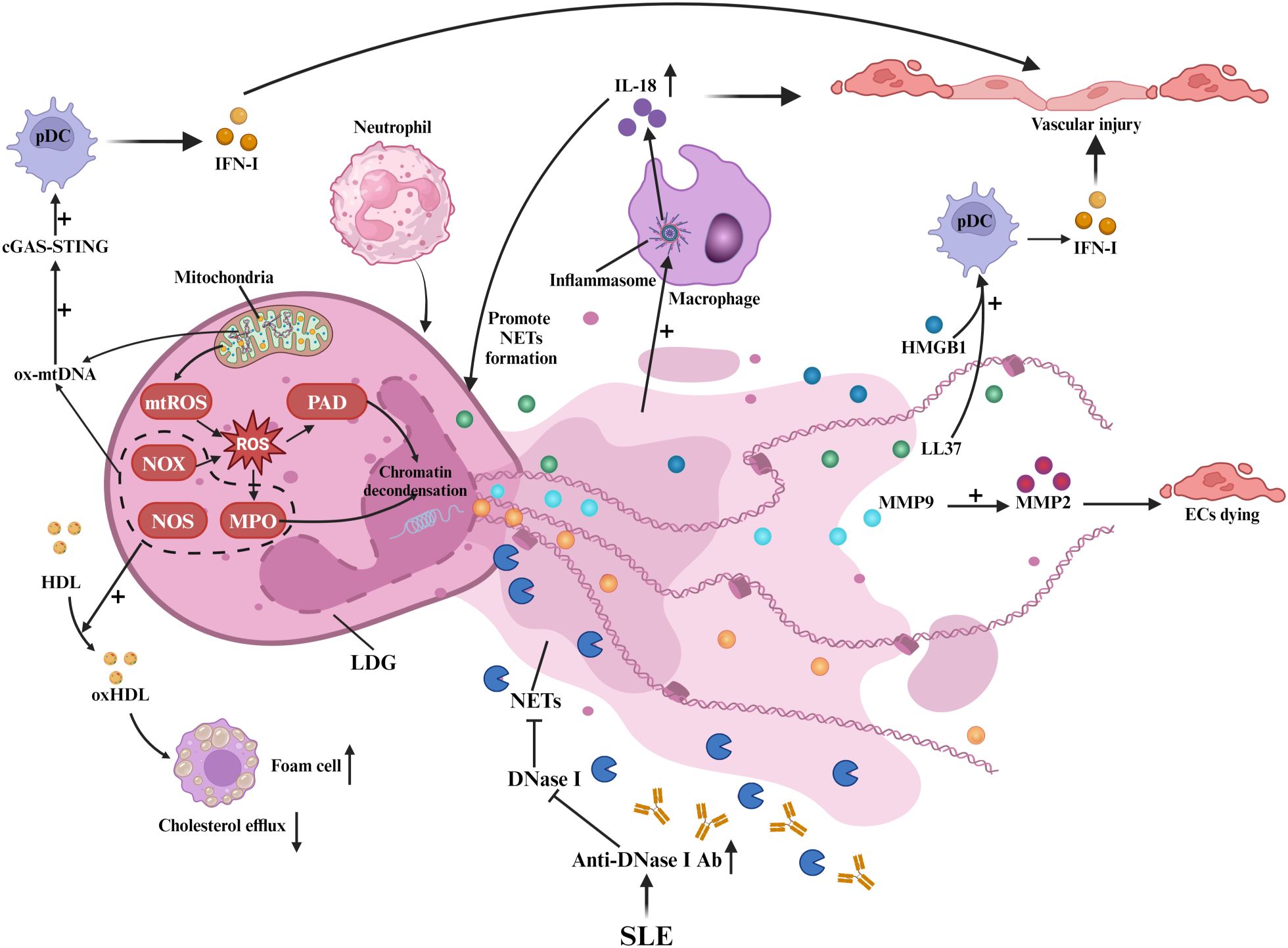
Figure 3. Neutrophils and NETs in SLE-associated atherosclerosis: Neutrophils and NETs. In SLE, LDGs release NETs, damaging ECs and triggering inflammation. NET formation involves ROS from NOX and mitochondria, with PAD and MPO inducing chromatin decondensation. Ox mtDNA activates cGAS-STING in pDCs, boosting IFN-I. NETs’ MMP9 activates MMP2, causing EC death. LL-37 and HMGB1 enhance pDC IFN-α, while inflammasome activity promotes IL-18. NOX, NOS, and MPO in NETs oxidize HDL, impairing cholesterol efflux. Anti-DNase I antibodies hinder NET clearance.
Low-density granulocytes (LDGs), a pro-inflammatory neutrophil subset, drive spontaneous NET release that induces endothelial dysfunction and vascular injury in SLE-associated AS (45). These cells overproduce TNF-α and IFN-I (45), while their NETs deliver autoantigens that perpetuate autoimmune responses (46). LDG-derived NETs activate T-cells (47) and transfer matrix metalloproteinase 9 (MMP9) to endothelial cells, triggering MMP2-mediated apoptosis (48). NET-associated small RNAs promote endothelial inflammation through TLR signaling (47, 49), whereas LL-37 (a cathelicidin peptide) stabilizes NET-DNA complexes and enhances plasmacytoid dendritic cell (pDC)-mediated IFN-α production (4, 50). Concurrently, oxidized mitochondrial DNA (ox-mtDNA) in NETs activates the cyclic GMP-AMP synthase - stimulator of interferon genes (cGAS-STING) pathway, amplifying IFN-I responses (51, 52). Impaired NET clearance in SLE, attributed to anti-DNase I antibodies (53), allows persistent High Mobility Group Box 1 (HMGB1) release that exacerbates chemotaxis and cytokine storms (4, 54).
NETs further sustain vascular inflammation via macrophage inflammasome activation and IL-18-driven NETosis (55), establishing an IFN-I-NET positive feedback loop that accelerates AS (4, 5). NOX, nitric oxide synthase (NOS), and MPO within NETs oxidize HDL, impairing cholesterol efflux and promoting foam cell formation (56). Collectively, neutrophil-derived NETs emerge as central mediators of SLE-related AS through multifaceted immune activation and vascular injury mechanisms.
2.4 T-cell dysfunction
T-cell dysfunction is essential in the development of AS in SLE patients, as shown in Figure 4. In SLE, CD4+ T-cells exhibit hyperactivation with reduced activation thresholds (57), where pDC-derived IFN-I induces endothelial cell production of CXCR3 ligands (CXCL9), promoting CD4+CXCR3+ T-cell arterial infiltration (58). Additionally, CD4+CCR5+ T-cells accumulate in SLE patients with carotid AS, demonstrating CCR5-mediated plaque homing (59). These CXCR3+ (tissue migration) and CCR5+ (plaque recruitment) subsets synergistically accelerate AS through complementary pathways (60, 61), independent of traditional risk factors like dyslipidemia (62).
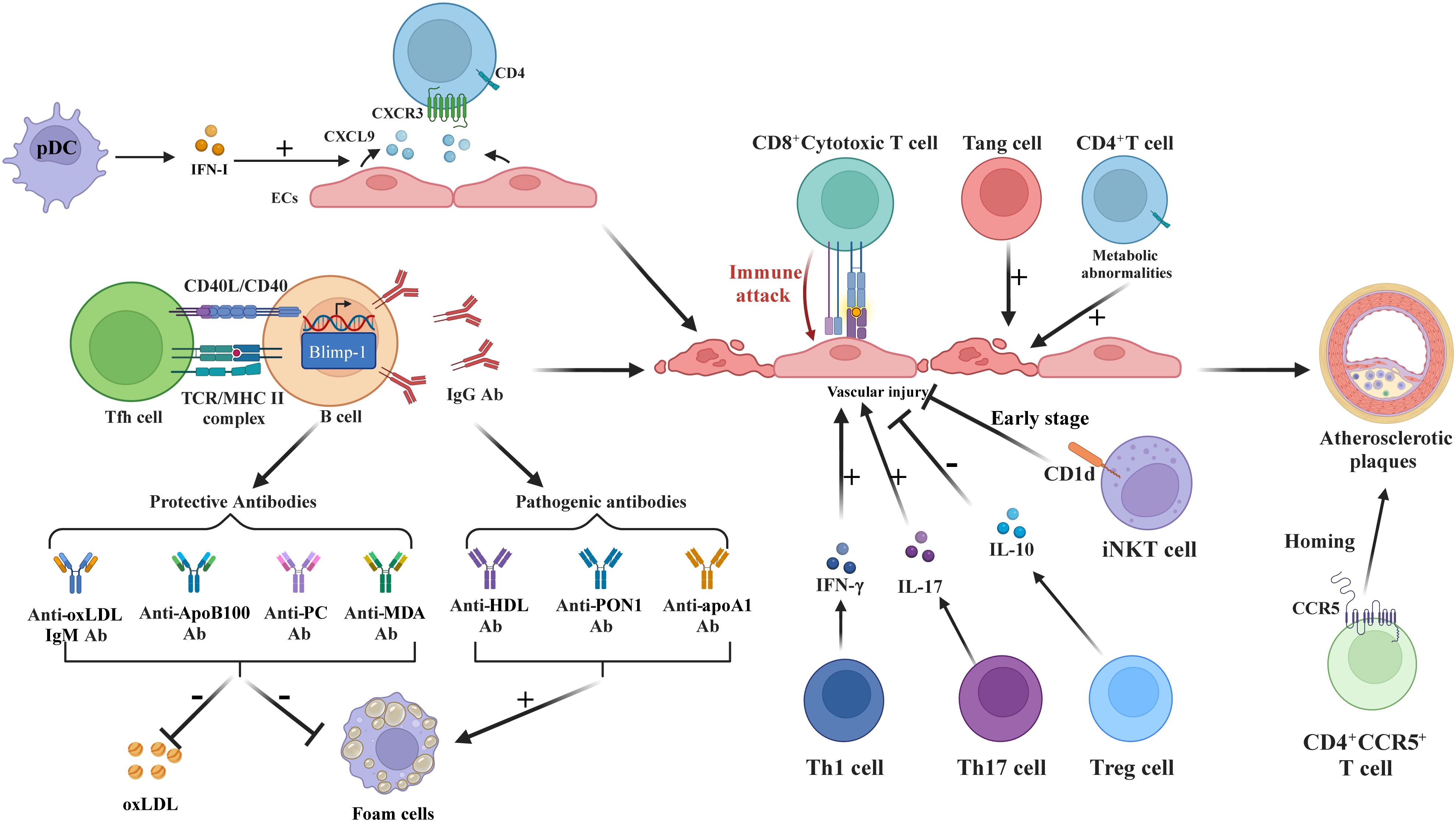
Figure 4. T and B Cells in SLE-associated atherosclerosis. IFN-I stimulates ECs to produce CXCR3 ligands, recruiting CD4+CXCR3+T-cells. Tfh enhances B-cell differentiation into IgG-secreting plasma cells. Th1/Th17 cells increase EC damage via IFN-γ and IL-17. CD8+T-cells disrupt ECs; Treg reduces inflammation via IL-10 and FoxP3. Tang cells aid repair but may worsen inflammation. iNKT initially protects but loses efficacy over time. CCR5 is essential for T-cell homing to atherosclerotic plaques. B-cells produce pathogenic antibodies (anti-HDL, anti-PON1, anti-apoA1) fostering foam cells, while protective antibodies (anti-oxLDL-IgM, anti-PC) lower oxLDL. SLE-related CD4+T-cell metabolic dysfunction exacerbates inflammation.
Follicular helper T-cells (Tfh) exacerbate AS via major histocompatibility complex II (MHCII)-dependent B-cell differentiation into IgG-secreting plasma cells (63, 64), while cTfh1/cTfh2/cTfh17 subpopulations interact with neutrophils to amplify vascular inflammation (65). Cholesterol-rich environments enhance splenic B-cell control over Tfh expansion through programmed death ligand 1 (PD-L1) regulation (63).
Imbalanced regulatory T-cell (Treg)/effector T-cell (Teff) ratios correlate with AS progression (62, 66). Dyslipidemia and elevated T-cell Pbx1d levels can intensify autoimmune reactions and atherosclerosis by disrupting Treg stability (62). Dyslipidemia-induced Pbx1d overexpression destabilizes Tregs, diminishing IL-10 and FoxP3 expression (66). Conversely, expanded Th17 cells secrete IL-17 to enhance plaque inflammation (66, 67), while Th1 and cytotoxic CD8+ T-cells directly damage endothelium via cytokine release (21, 68). Circulating IFN-Ihi CD4+ and exhausted/activated CD8+ T-cells infiltrate SLE vascular lesions (21, 69).
Other T-cell subsets, such as CD3+CD31+CXCR4+angiogenic T-cells (Tang), aid vascular endothelium repair during AS (70). However, in SLE patients, especially those positive for anti-dsDNA, elevated CD8+Tang cell levels in blood may signal endothelial damage and cardiovascular risks (71). The expanded CD28(null)Tang subset, associated with aging, intensifies endothelial inflammation via inflammatory factor release, leading to atherosclerosis and linked to SLE disease activity (72, 73). Invariant natural killer T (iNKT) cells lose early anti-AS capacity due to impaired CD1d-mediated lipid presentation (74, 75).
Metabolically reprogrammed CD4+ T-cells exhibit enhanced glycolysis via pyruvate kinase M2 (PKM2) activation, releasing pro-inflammatory cytokines (76). IFN-I disrupts T-cell bioenergetics (77), creating self-perpetuating inflammatory loops that pharmacological inhibition can reverse (78). Collectively, T-cell-mediated immunity constitutes a central axis in SLE-associated AS pathogenesis.
2.5 B-cell dysfunction
B-cells and their autoantibodies are key in SLE-induced AS, as shown in Figure 4. Elevated anti- HDL, paraoxonase 1 (PON1), and apolipoprotein A1 (apoA1) antibodies serve as progression markers (79, 80), with anti-HDL predicting cardiovascular risk and anti-PON1 correlating with intimal thickening (80). Patients with SLE exhibit reduced IgM/IgG anti-phosphocholine (PC) levels inversely associated with AS severity (81–85), where IgM anti-PC mitigates disease activity while IgG anti-PC inhibits endothelial adhesion molecules (83–85). Hyperlipidemia-driven B-cell activation promotes oxLDL generation (86, 87), further amplified by ICs containing ribonucleoproteins and anti-Ro/La antibodies that enhance anti-oxLDL production (88). OxLDL-β2-glycoprotein I complexes stimulate foam cell formation and autoantibody responses (89).
B-cell-activating factor (BAFF) elevation correlates with subclinical AS (90), while B1 cells paradoxically exhibit both protective (autoimmunity suppression) and pathogenic (class-switched IgG production) roles (91, 92). Lupus-prone mice demonstrate marginal zone B cell-driven pathogenic antibody secretion (93) and abnormal plasma cell differentiation involving somatic hypermutation (94). Pathogenic autoantibodies include anti-dsDNA (endothelial dysfunction/thrombosis induction) (95, 96), anti-endothelial cell antibodies (IgA-mediated vascular injury) (97, 98), and antiphospholipid antibodies (plaque progression in 20-30% patients) (99).
Protective autoantibodies counterbalance these effects: anti-oxLDL-IgM, anti-ApoB100, anti-PC, and anti-malondialdehyde (MDA) antibodies suppress oxLDL uptake and foam cell formation (100, 101). Reduced anti-ApoB100 levels in SLE imply impaired AS protection (101), while anti-C1q antibodies promote M2 macrophage polarization (102). Anti-PC-IgM modulates Treg/Th17 equilibrium, synergizing with anti-MDA-IgM for cardiovascular protection (84, 103). Nevertheless, dietary/metabolic constraints limit their therapeutic potential in SLE (104). Collectively, B-cell-derived autoantibody networks constitute pivotal drivers of SLE-associated AS.
3 The role of gut microbiota dysbiosis and metabolic disorders in the pathogenesis of atherosclerosis
Gut microbiota dysbiosis and metabolites (e.g., TMAO) are well - known to promote atherosclerosis via inflammatory pathways and immune dysregulation in various diseases, although their direct role in SLE associated atherosclerosis remains unclear. There are numerous studies exploring the relationships between SLE and gut microbiota, or between atherosclerosis and gut microbiota, and also between metabolic disorders and both of them. However, research comprehensively integrating SLE, atherosclerosis, gut microbiota, and metabolic disorders remains unexplored. While Cabana-Puig et al. (105) demonstrated high-fat diet (HFD) exacerbates atherosclerosis via Ly6C+ monocyte pathways and Lactobacillus improves glomerulonephritis, these interventions were examined independently. Thus, this study does not establish causal relationships between gut microbiota and SLE-associated atherosclerosis. This underscores the critical need for integrated investigations examining microbiome-atherosclerosis interactions specifically in SLE contexts.
In this section, considering the current state of research, we mainly focus on the general mechanisms by which gut microbiota dysbiosis and metabolic disorders lead to atherosclerosis (including in diseases other than SLE), as depicted in Figure 5.
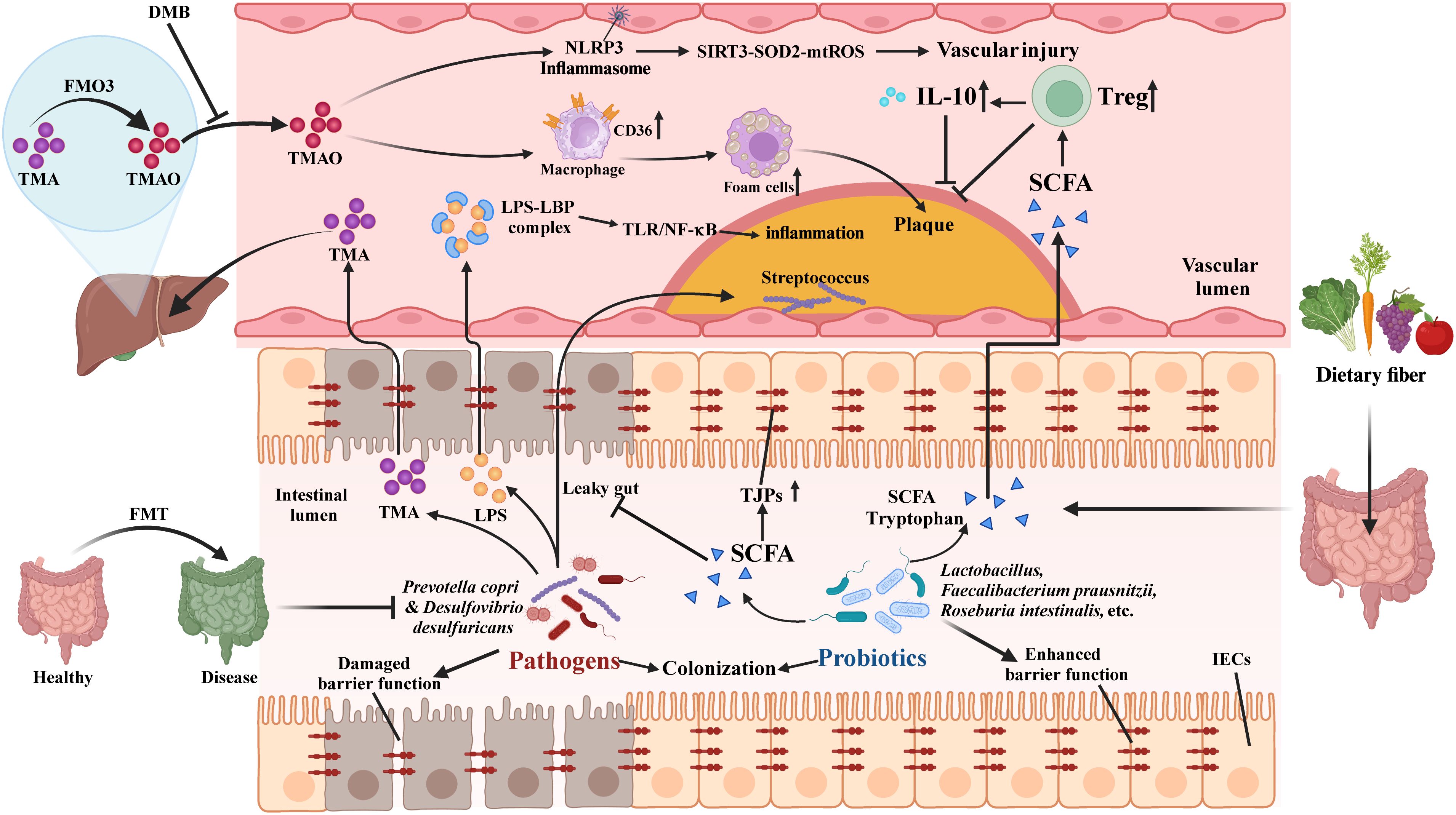
Figure 5. Potential mechanisms underlying gut microbiota dysbiosis in atherosclerosis. Pathogenic bacteria like Prevotella copri weaken the intestinal barrier, causing a “leaky gut” and allowing LPS into the bloodstream, which activates the LBP-TLR4/NF-κB pathway, triggering inflammation and promoting plaque formation. Streptococcus spp. can migrate to plaques, worsening the condition. Probiotics (e.g., Lactobacillus, Faecalibacterium prausnitzii) upregulate TJPs, reducing LPS-induced inflammation. Pathogenic bacteria produce TMA, converted to TMAO by FMO3, promoting atherosclerosis. DMB lowers TMAO and improves vascular function in lupus mice. SCFAs from probiotics enhance Tregs, and IL-10, and reverse cholesterol transport, preventing atherosclerosis. Probiotics, FMT, and dietary fiber modulate gut microbiota, lower TMA/TMAO and LPS, and reduce inflammation.
3.1 Genetic factors
The gut microbiota significantly influences the development of atherosclerosis, with genetic factors playing an important role. A study showed that proinflammatory gut microbiota from Caspase1-/- mice accelerated atherosclerosis in Ldlr-/- mice, as evidenced by increased levels of circulating monocytes, neutrophils, and proinflammatory cytokines. Fecal microbiota transplantation reduced short-chain fatty acid-producing taxa, such as Akkermansia and Clostridium, leading to decreased anti-inflammatory short-chain fatty acids in the cecum (9). This study highlights the multifaceted interplay between gut microbiota, genetic factors, and atherosclerosis.
3.2 Specific microbiota promotes atherosclerosis progression
Bacterial DNA in atherosclerotic plaques suggests intestinal bacteria can migrate to the vascular system, as seen with Streptococcus spp. moving to coronary artery plaques, leading to elevated pro-inflammatory cytokines and altered vascular tension, which contribute to coronary atherosclerosis (106, 107). Mechanistically, Desulfovibrio desulfuricans compromises intestinal barrier integrity, enabling lipopolysaccharide (LPS) translocation that activates the TLR4/NF-κB inflammatory axis (108). Circulating LPS subsequently recruits inflammatory cells to lesions through LPS-binding protein (LBP)/TLR4 signaling (109). Conversely, Faecalibacterium prausnitzii fortifies gut barriers and suppresses LPS synthesis, mitigating coronary artery disease (CAD) risk (110). Comparative studies reveal divergent microbial impacts: Porphyromonas gingivalis oral infection exacerbates atherosclerosis in ApoE-/- mice via gut dysbiosis and lipid abnormalities (reduced HDL/triglycerides) (111), while Bacteroides fragilis-induced dysbiosis (decreased Lactobacillaceae; increased Desulfovibrionaceae) promotes plaque formation through metabolic inflammation (10). Transplant experiments using Caspase1-/- mouse microbiota demonstrate accelerated atherosclerosis in Ldlr-/- recipients through systemic inflammation and microbial community shifts (9). These findings establish the gut-vascular axis as a multidimensional crosstalk involving barrier regulation, immunometabolic reprogramming, and microbial ecosystem remodeling.
3.3 TMAO as a promoter of atherosclerosis
TMAO accelerates atherosclerosis via multiple mechanisms. Gut microbes process dietary choline/carnitine/betaine into trimethylamine (TMA) using cut/cnt enzymes, with liver FMO3 converting TMA to TMAO (112). TMAO activates NF-κB to increase inflammatory cytokines for plaque formation (113), triggers NLRP3 inflammasome via SIRT3-SOD2-mtROS in endothelial cells (114), and enhances macrophage CD36/SR-A1-mediated ox-LDL uptake causing foam cell formation (115). It also suppresses bile acid synthesis through FXR/SHP receptors, impairing cholesterol excretion (116).
Gut microbiota directly modulates TMAO’s atherogenic effects. Lachnoclostridium saccharolyticum converts choline to TMA, correlating with plaque growth (117). Emergencia timonensis uniquely processes l-carnitine-derived γ-butyrobetaine into TMA (11). PSRC1 deficiency elevates TMAO and worsens atherosclerosis in ApoE-/- mice through gut dysbiosis (112).
Current research on TMAO in SLE remains limited. While TMAO’s pro-atherogenic mechanisms are well-characterized in other diseases (112–118), its specific role in SLE-related cardiovascular complications requires cautious interpretation given the limited disease-specific evidence. Clinical studies have demonstrated elevated plasma TMAO levels in lupus patients compared to healthy controls (119). González-Correa et al. (17) demonstrated that TMAO exacerbates vascular endothelial dysfunction via oxidative stress and impaired antioxidant defense in TLR7-activated lupus mice. Their intervention with 3,3-dimethyl-1-butanol (DMB), a TMA production inhibitor, effectively reduced circulating TMAO levels and alleviated hypertension in this lupus mouse model (17).
In conclusion, gut microbiota promotes TMAO production, which critically drives atherosclerosis through inflammatory and metabolic mechanisms (112–118). In lupus mice, TMAO causes vascular dysfunction that may accelerate atherosclerosis in SLE, but this connection requires further investigation. It should be emphasized that González-Correa et al. (17) specifically demonstrated TMAO’s contribution to vascular endothelial dysfunction in TLR7-activated SLE mice, but did not investigate atherosclerosis endpoints. To our knowledge, no published studies have directly examined TMAO’s effects on atherogenesis in lupus models or other systemic autoimmune diseases.
3.4 Tryptophan metabolism disruption
Recent studies have highlighted the role of tryptophan metabolism disruption in promoting atherosclerosis. A study showed that indoleamine 2,3-dioxygenase 1 (IDO1) promotes atherosclerosis by inhibiting IL-10 production, which activates a cAMP-dependent pathway and suppresses Erk1/2 phosphorylation, leading to increased plaque instability (120). Similarly, Laurans et al. found that IDO1 activity in obese mice disrupts the intestinal mucosal barrier, triggers chronic inflammation, and interferes with lipid metabolism, worsening obesity-related metabolic diseases (121). Additionally, Xue et al. identified a protective role for the gut microbial metabolite indole-3-propionic acid (IPA), which reduces plaque development by promoting reverse cholesterol transport in macrophages via the miR-142-5p/ABCA1 signaling pathway (122). These findings highlight the complex relationship between tryptophan metabolism and atherosclerosis.
3.5 Immune dysregulation
Recent research has highlighted the complex interactions between immune responses and gut microbiota in atherosclerosis development. One study found that under hyperlipidemic conditions, gut microbiota can activate B2 cells in perivascular adipose tissue, leading to increased IgG production and disease progression. Disrupting the gut microbiota with broad-spectrum antibiotics or depleting B2 cells pharmacologically mitigated atherosclerosis, emphasizing the role of B2 cells beyond lipid metabolism (123). Another study showed that immunization against gut bacterial outer membrane proteins reduced systemic inflammation and improved metabolic markers in mice fed a Western diet, mediated by apoE, suggesting a potential therapeutic approach for immune-metabolic diseases (124). Dietary modulation critically impacts this axis: high-fat/low-fiber diets induce microbial dysbiosis that amplifies mesenteric lymph node lymphocyte proliferation and accelerates plaque formation (125, 126).These findings underscore the importance of gut microbiota-immune interactions in atherosclerosis, offering insights into therapeutic strategies targeting the gut microbiome.
4 Therapeutic strategies for SLE-related atherosclerosis
4.1 Statins
Statins, cornerstone therapeutics for hyperlipidemia management, have garnered significant attention for their putative role in SLE-associated AS. Through HMG-CoA reductase inhibition, these agents inhibit cholesterol synthesis and ameliorate SLE-associated dyslipidemia—a pivotal atherosclerosis driver (5, 127). Preclinical studies demonstrate statins’ dual capacity to attenuate autoimmune activation and atherosclerotic progression (127), exerting anti-atherogenic effects through IL-6/IL-8/TNF-α suppression (5, 128). Clinical applications in SLE remain contentious (129): while observational data suggest cardiovascular risk reduction (130), the Lupus Atherosclerosis Prevention Study revealed no significant coronary artery or endothelial improvement after 24-month intervention (131). Current evidence thus indicates that despite plausible mechanistic benefits, statins’ clinical efficacy in SLE-related AS lacks robust validation, necessitating mechanistic investigations to delineate disease-specific pharmacodynamics and optimize therapeutic strategies.
4.2 Glucocorticoids
Glucocorticoids, cornerstone therapeutics for SLE, suppress inflammation and modulate atherogenesis through cytokine reduction (132, 133). Low-dose regimens (e.g., dexamethasone) demonstrate plaque-stabilizing effects (132, 133). However, dose-dependent cardiovascular risks emerge with prolonged high-dose administration, paradoxically accelerating plaque formation (133, 134). This bidirectional relationship necessitates strict adherence to minimal effective dosing with timely tapering to optimize risk-benefit ratios. Notably, chronic high-dose exposure elevates cardiovascular mortality in long-term treated cohorts, demanding vigilant monitoring (134). In conclusion, glucocorticoids should be administered with caution in SLE treatment, alongside other therapies, to balance their anti-inflammatory effects with the need to reduce cardiovascular hazards.
4.3 Immunosuppressant
Mycophenolate mofetil (MMF) and cyclophosphamide (CPA) demonstrate dual therapeutic potential in SLE-atherosclerosis pathogenic nexus. Preclinical evidence reveals MMF alleviates SLE manifestations and atherogenesis via inosine monophosphate dehydrogenase inhibition, curtailing lymphocyte-mediated inflammation (135). CPA exerts immunomodulatory effects through DNA synthesis disruption and lymphocyte clonal expansion blockade, synergistically attenuating plaque development (136). Clinical observations indicate MMF improves cardiovascular risk profiles in SLE cohorts (137, 138), though disease-specific clinical validation remains pending (139). While these immunosuppressants show mechanistic promise in atherosclerotic lesion regression, translating preclinical efficacy into optimized human SLE therapeutics requires systematic investigation of tissue-specific pharmacodynamics and long-term vascular outcomes.
4.4 Antimalarial drugs
The antimalarial agent hydroxychloroquine (HCQ) serves as a cornerstone therapy for SLE and its atherosclerotic complications. HCQ modulates SLE-associated atherogenesis through AP-1-mediated T-cell suppression (140) and endothelial stabilization via dual mechanisms: attenuation of TLR signaling and ROS generation, and AMPK-dependent VSMC cycle arrest (140, 141). Intriguingly, HCQ synergizes with high-dose statins to inhibit cholesterol biosynthesis through precursor regulation without altering serum cholesterol profiles (142). Multicenter clinical data validate its cardioprotective efficacy, demonstrating reductions in cholesterol levels, thrombotic events, and mortality rates in SLE cohorts (143). Extended analyses reveal 30% lower cardiovascular disease risk with HCQ use, particularly regarding CAD and cerebrovascular events (144, 145). These findings position HCQ as a dual-purpose therapeutic agent that concurrently addresses autoimmune dysregulation and vascular pathology in SLE management.
4.5 Therapeutic potential of interferon and NET inhibitors
IFN-I and NETs play a significant role in SLE-related AS, making them potential therapeutic targets. Anifrolumab, a monoclonal antibody, effectively treats moderate to severe SLE by blocking the type I interferon receptor, reducing NET formation, and lowering inflammatory cytokines (146). PADs—key enzymatic drivers of NET formation—show therapeutic promise by downregulating IFN-I signaling cascades in SLE-related AS (147). Janus kinase inhibitors (JAKi) including tofacitinib and baricitinib modulate SLE pathogenesis via JAK/STAT pathway inhibition, with preclinical evidence revealing tofacitinib’s dual capacity to reduce NET burden and restore endothelial homeostasis in MRL/lpr models (148–150). Early-phase clinical trials validate JAKi safety profiles and cardiometabolic benefits (149), while ruxolitinib extends this therapeutic spectrum in murine lupus (148). The TYK2 allosteric inhibitor deucravacitinib achieves comparable IFN-I suppression, with Phase II trials confirming both safety and clinical improvement in SLE cohorts (14, 21). Collectively, IFN-α antibodies and signaling inhibitors represent mechanistically grounded strategies for SLE-associated AS management, though definitive clinical validation requires large-scale trials to establish risk-benefit ratios across diverse patient populations.
4.6 Targeted T-cell therapy
The abnormal activation of T-cells plays an important role in atherosclerosis associated with SLE (67). For patients with SLE, Th17 cells exacerbate atherosclerosis advancement by generating inflammatory substances like IL-17 (66). Simultaneously, Tregs slow down atherosclerosis development by inhibiting effector T-cells and fostering anti-inflammatory reactions (151).
Notably, low-dose IL-2 selectively modulates the balance among Treg, Tfh, and Th17 cells, effectively reducing SLE disease activity and improving clinical outcomes while showing high tolerability and safety (15, 152). Consequently, a reduced dosage of IL-2 could decelerate atherosclerosis by controlling T-cells. To sum up, specific T-cell treatment methods may hold promise for atherosclerosis linked to SLE, yet additional studies are required to confirm their effectiveness and safety.
4.7 Targeted B cell therapy
B-cell-directed therapies emerge as viable interventions for SLE-linked AS. BAFF serves as a crucial modulator of B-cell homeostasis and SLE pathogenesis (153). The BAFF-neutralizing monoclonal antibody belimumab—an approved SLE biologic—demonstrates dual therapeutic capacity by achieving disease activity control and renal protection in lupus nephritis (153). Its anti-atherogenic effects exhibit context-dependent effects contingent upon lipidomic profiles and B-cell functional states, underscoring the necessity for precision targeting strategies (154). Clinical data reveal belimumab’s concomitant HDL elevation and Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) score reduction, suggesting synergistic metabolic-immune benefits in AS mitigation (155). Complementary approaches employ CD20-directed agents like rituximab that mediate pathogenic B-cell depletion while preserving protective autoantibody pools, concurrently suppressing pathogenic T-cell cross-activation to impede AS progression (16, 156). Notably, rituximab ameliorates dyslipidemia in refractory SLE cases and attenuates myocardial infarction risk through pleiotropic immunometabolic mechanisms (16, 157). These findings position B-cell modulation as a strategic axis in SLE-related AS management, though future investigations must delineate molecular stratification biomarkers and establish dynamic monitoring protocols to optimize risk-adapted therapeutic strategies.
4.8 Potential therapies of targeting gut microbiota and metabolism in treating SLE-related atherosclerosis
Atherosclerosis is a significant cardiovascular complication in patients with SLE. Targeting gut microbiota and metabolic disorders has emerged as a novel approach to treat SLE-related atherosclerosis. Despite limited preclinical evidence, this approach holds significant promise for modulating both autoimmunity and vascular inflammation. Potential therapies include gene intervention, probiotic therapy, fecal microbiota transplantation, TMAO and tryptophan metabolism regulation, SCFA therapy and pharmacological or nutritional therapies. Relevant studies are listed in Supplementary Table 1.
4.8.1 Gene intervention
Gene intervention may improve gut microbiota dysbiosis and metabolic abnormalities to benefit atherosclerosis. NOD1/NOD2-deficient mice had less atherosclerosis than Ldlr-/- mice after a 12-week high-fat diet, due to gut microbiota changes, increased intestinal cholesterol and coprostanol levels correlating with more Eubacterium coprostanoligenes, and lower plasma lipid levels and reduced foam cell formation from upregulated Abca1 and Abcg1 in macrophages (158). PSRC1 deficiency in ApoE-/- mice accelerated atherosclerosis by boosting TMAO, increased TMA-producing bacteria and plasma betaine and TMAO levels. These mice also showed colonic inflammation linked to gut microbiota dysregulation and PSRC1 deficiency increased liver flavin monooxygenase 3 (FMO3) expression, while PSRC1 overexpression suppressed FMO3 (112). These results highlight gene intervention’s potential to modulate gut microbiota and metabolic pathways to combat atherosclerosis, though more clinical research is needed for human validation.
4.8.2 Probiotic therapy
A population study and in vivo experiments using ApoE-/- mice revealed that Faecalibacterium prausnitzii presence is associated with the lowest CAD incidence. Faecalibacterium prausnitzii reduces intestinal LPS synthesis, enhances mechanical and mucosal barriers, decreases plasma LPS levels, and alleviates atherosclerosis in ApoE-/- mice (110). A human study and in vivo experiments showed Bacteroides vulgatus and Bacteroides dorei abundance is reduced in CAD patients, while administering these bacteria alleviates atherosclerotic lesion formation in mice and reduces gut microbiota LPS production (159). Another study demonstrated that Roseburia intestinalis interacts with dietary plant polysaccharides to improve gut metabolism, reduce systemic inflammation, and significantly alleviate atherosclerosis. Intestinal butyrate administration can also reduce endotoxemia and atherosclerosis development (160). The research investigated a high-fat diet and interventions with LGG or telmisartan on atherosclerosis in ApoE-/- mice, finding both LGG and telmisartan significantly reduced atherosclerotic plaque size and improved various biomarkers (161). However, further clinical research is needed to validate these therapeutic effects in human patients.
4.8.3 Fecal microbiota transplantation
FMT may be an effective strategy for treating atherosclerosis by modulating the gut microbiota. One study showed that Z-Ligustilide significantly alleviates atherosclerosis in ApoE-/- mice by reconstructing gut microbiota and maintaining intestinal barrier integrity via cannabinoid receptor 2 activation. Fecal bacteria from Z-Ligustilide-treated mice induced similar beneficial effects, and 16S RNA gene sequencing revealed a significant increase in Rikenella abundance in both Z-Ligustilide-treated and FMT mice (162). Another study showed Empagliflozin alleviates atherosclerosis, increases fecal probiotics abundance, reduces inflammatory responses, and alters gut microbiota metabolism; FMT from empagliflozin-treated mice to control mice also reduced atherosclerosis and systemic inflammatory responses (163). Furthermore, research using a C1q/TNF-related protein 9 gene-deficient (CTRP9-KO) mouse model found transplanting gut microbiota from wild-type mice into CTRP9-KO mice alleviated the development of atherosclerosis (164). However, further clinical research is needed to validate the efficacy and safety of FMT in human patients.
4.8.4 TMAO regulation therapy
Targeting TMAO biosynthesis emerges as a novel therapeutic frontier for SLE-associated atherosclerosis. Pharmacological inhibition of TMA lyase by 3,3-dimethyl-1-butanol (DMB) in imiquimod-induced lupus models demonstrates multi-organ protection through TMAO suppression, including endothelial function restoration and macrophage foam cell formation inhibition independent of cholesterol modulation (17).
Complementary studies reveal diverse TMAO modulation strategies: Puerarin exerts atheroprotection via Prevotella copri depletion and TMA precursor reduction, while berberine’s vitamin-mimetic metabolite dihydroberberine disrupts gut microbial TMA biosynthesis (165, 166). Dietary interventions further expand this paradigm – aged garlic oligosaccharides reshape gut ecology toward Akkermansia-dominant communities with concomitant TMAO reduction in hyperlipidemic ApoE-/- models, whereas hickory polyphenols attenuate atherosclerosis through hepatic FMO3 transcriptional downregulation in high-choline-fed C57BL/6J mice (167, 168). Further clinical trials are necessary to confirm the efficacy and safety of these interventions in human patients.
4.8.5 Tryptophan metabolism regulation therapy
Recent studies highlight tryptophan metabolism’s potential therapeutic role in atherosclerosis. A clinical trial and in vivo experiment in ApoE-/- mice revealed that IPA was significantly downregulated in CAD patients, correlating with atherosclerotic cardiovascular disease (ASCVD) risk and severity. Dietary IPA supplementation in ApoE-/- mice alleviated plaque development via the miR-142-5p/ABCA1 pathway by promoting cholesterol efflux. This study first uncovered the link between gut microbiota-derived tryptophan metabolites and ASCVD, providing a potential target (122). Another study showed that Aucubin improves atherosclerosis in ApoE-/- mice by modulating gut microbiota, particularly by increasing indole-3-acrylic acid (IAA). IAA alleviates atherosclerosis by activating the Aryl hydrocarbon receptor (AhR) and inhibiting the TGF-β/Smad pathway (169). Similarly, Indole-3-carbinol (I3C) inhibits atherosclerosis in ApoE-/- mice. I3C improves lipid profile, enhances gut microbiota diversity, and increases Verrucomicrobia abundance. Integrated analysis shows that 1-methyladenosine is a key modulator of I3C’s protective effect (170). These findings indicate that modulating tryptophan metabolism and gut microbiota could be promising therapeutic strategies for atherosclerosis, but more large-scale studies are needed to confirm their efficacy and safety in humans.
4.8.6 Short-chain fatty acids regulation therapy
Short-chain fatty acids (SCFAs) show potential in treating atherosclerosis. Propionate (PA) reduces intestinal cholesterol absorption and lowers plasma LDL cholesterol via immune mechanisms, increasing regulatory T cells and IL-10 while inhibiting Npc1l1 expression. Oral PA supplementation in humans significantly decreases cholesterol levels, offering a new therapeutic target (171). In ApoE-/- mice, Roseburia intestinalis interacts with dietary plant polysaccharides to improve gut metabolism and reduce atherosclerosis, revealing gut microbiota and diet interaction mechanisms (160). These studies suggest SCFAs’ role in combating atherosclerosis through gut microbiota and immune modulation, though human validation is still required.
4.8.7 Pharmacological or nutritional therapies
Recent studies have illuminated the potential of various treatments in improving atherosclerosis via gut microbiota modulation. One study found gut microbiota affects atherosclerosis development by regulating vascular microRNA-204 expression, which impacts vascular endothelial function by targeting Sirtuin1 (172). Bicyclol improves high-fat diet-induced atherosclerosis in ApoE-/- mice by restoring gut health and alleviating endothelial activation, macrophage infiltration, and cholesterol ester accumulation (173). Hydroxyurea effectively mitigates atherosclerosis in ApoE-/- mice by modulating gut microbiota and cholesterol metabolism, reducing plaque and liver lipid accumulation (174). Palmatine significantly mitigates atherosclerosis by modulating gut microbiota and phenylalanine metabolism, reshaping gut microbiota composition and reducing harmful bacteria abundance (175). Peanut Skin Extract significantly mitigates atherosclerosis in ApoE-/- mice by regulating lipid metabolism, exerting anti-inflammatory effects, and altering gut microbiota composition (176). Ginsenosides protect Ldlr-/- mice from atherosclerosis by enhancing bile salt hydrolase activity and protecting the intestinal barrier (177). ALW-II-41–27 significantly alleviates atherosclerosis in ApoE-/- mice by reshaping gut microbiota and modulating bile acid metabolism, reducing plaques and increasing collagen and smooth muscle cell content (178). Disulfiram reduces atherosclerosis in ApoE-/- mice by inhibiting GsdmD, inducing autophagy, and modulating gut microbiota (179). Dietary fiber also benefits SLE mice by modulating gut microbiota and improving vascular and renal health (180). These findings highlight the potential of targeting gut microbiota as a therapeutic strategy for atherosclerosis, with further human validation needed.
5 Future perspectives
SLE, a complex autoimmune disorder, increases the risk of atherosclerosis. Despite recent progress, challenges remain. This review connects SLE with atherosclerosis through immune imbalance, gut microbiota disturbances, and metabolic differences, and suggests future research and treatment strategies.
The paper revisits atherosclerosis risk factors in SLE, including both traditional cardiovascular risks and unique SLE pathophysiology. Recent studies highlight immune cell dysfunction, particularly macrophage metabolism imbalance and the role of LNGs and CD4+T-cells under type I interferon conditions, leading to mitochondrial damage and oxidative stress, which promote atherosclerosis. Lipid metabolism irregularities, disease activity also accelerate the condition.
Current therapies including statins, corticosteroids, immunosuppressants, antimalarials, and targeted molecular agents aim to modulate lipid profiles, inflammatory responses, immune activation, and gut microbiota composition to mitigate cardiovascular risks. Future investigations should prioritize uncovering novel atherogenic mechanisms in SLE, particularly immune-metabolic interactions in specialized cell populations.
Study limitations include predominant reliance on animal models and in vitro systems that may not accurately reflect human SLE pathophysiology. Disease heterogeneity necessitates rigorously designed clinical studies. Research priorities should focus on developing personalized therapies, elucidating immune cell contributions to vascular pathology, investigating gut microbiota-metabolism crosstalk, and identifying predictive biomarkers. The underexplored relationship between specific gut microbial species/metabolites and oxidative atherogenesis in SLE warrants particular attention, as this may reveal novel therapeutic targets and diagnostic tools.
Advances in SLE-related atherosclerosis research could inform cardiovascular management across autoimmune disorders. Multicenter clinical trials, interdisciplinary collaborations, and data-sharing platforms are critical for accelerating progress. Although challenges remain, emerging technologies offer optimism for developing precision therapies that improve SLE patients’ cardiovascular outcomes and overall quality of life. This synthesis enhances understanding of SLE-associated vascular pathogenesis and provides translational insights applicable to broader autoimmune research.
Author contributions
QRP: Writing – original draft, Writing – review & editing. XH: Writing – original draft, Writing – review & editing. CL: Writing – review & editing. QJP: Writing – review & editing. SH: Funding acquisition, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by the Construction of Zhanjiang Vascular remodeling and arrhythmia Medical Engineering Research Center (no. 2020A06003) (SH).
Acknowledgments
Figures were created using BioRender (BioRender, Canada; BioRender.com; Agreement numbers: HU284B7TAI; KH284B7UXZ; IL284B7WHC; HD284B7ZJG; ET284B82K5).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1492726/full#supplementary-material
Glossary
apoA1: Apolipoprotein A1
LDGs: Low-Density Granulocytes
AS: atherosclerosis
LOX-1: Lectin-like oxidized low-density lipoprotein receptor-1
ASCVD: atherosclerotic cardiovascular disease
LPS: Lipopolysaccharide
BAFF: B-cell-activating factor
MAPKs: Mitogen-activated protein kinases
CACs: circumferential artery cells
MDA: malondialdehyde
CAD: coronary artery disease
MMF: Mycophenolate mofetil
cGAS-STING: cyclic GMP-AMP synthase - stimulator of interferon genes pathway
MMP: Matrix Metalloproteinase
CPA: cyclophosphamide
MPO: Myeloperoxidase
CVDs: cardiovascular diseases
MyD88: myeloid differentiation factor 88
DMB: 3,3-dimethyl-1-butanol
NETs: Neutrophil Extracellular Traps
DNase I: Deoxyribonuclease I
NF-κB: Nuclear Factor Kappa B
ECM: extracellular matrix
NOX: NADPH Oxidase
EPCs: endothelial progenitor cells
NOS: Nitric Oxide Synthase
FMO3: flavin monooxygenase 3
Ox mtDNA: Oxidized Mitochondrial DNA
FMT: Fecal Microbiota Transplantation
oxLDL: Oxidized Low-Density Lipoprotein
HCQ: hydroxychloroquine
PAD: Peptidylarginine Deiminase
HDL: High-Density Lipoprotein
PC: Phosphatidylcholine
HFD: high-fat diet
pDCs: Plasmacytoid Dendritic Cells
HGF: hepatocyte growth factor
PD-L1: programmed death ligand 1
HMGB1: High Mobility Group Box 1
PKM2: pyruvate kinase M2
IAA: indole-3-acrylic acid
PON1: Paraoxonase 1
I3C: Indole-3-carbinol
ROS: Reactive Oxygen Species
ICAM-1: Intercellular Adhesion Molecule-1
SCFAs: Short-Chain Fatty Acids
ICs: immune complexes
SLE: systemic lupus erythematosus
IDO1: indoleamine 2,3-dioxygenase 1
SR-A1: scavenger receptor A1
IFN-I: Type I interferons
Tang cells: CD3+CD31+CXCR4+ angiogenic T-cells
TYK2/JAK1: IFNAR-mediated JAK kinase
Tfh: Follicular helper T-cells
iNKT: Invariant Natural Killer T-cells
Th1: CXCR3+ T helper 1 cells
IPA: indole-3-propionic acid
TJPs: Tight Junction Proteins
IRF: Interferon regulatory factor
TLR: Toll-like receptors
ISGs: interferon-stimulated genes
TMA: Trimethylamine
ISRE: interferon-stimulated response element
TMAO: Trimethylamine-N-Oxide
JAKi: Janus kinase inhibitors
VCAM-1: Vascular Cell Adhesion Molecule-1
LBP: Lipopolysaccharide Binding Protein
VEGF: vascular endothelial growth factor
LDL: low-density lipoprotein
VSMC: vascular smooth muscle cell.
References
1. Oliveira CB, Kaplan MJ. Cardiovascular disease risk and pathogenesis in systemic lupus erythematosus. Semin Immunopathol. (2022) 44:309–24. doi: 10.1007/s00281-022-00922-y
2. Jin L, Shi X, Yang J, Zhao Y, Xue L, Xu L, et al. Gut microbes in cardiovascular diseases and their potential therapeutic applications. Protein Cell. (2021) 12:346–59. doi: 10.1007/s13238-020-00785-9
3. Chen C, Khismatullin DB. Oxidized low-density lipoprotein contributes to atherogenesis via co-activation of macrophages and mast cells. PLoS One. (2015) 10:e0123088. doi: 10.1371/journal.pone.0123088
4. Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med. (2011) 3:73ra20. doi: 10.1126/scitranslmed.3001201
5. Teixeira V, Tam LS. Novel insights in systemic lupus erythematosus and atherosclerosis. Front Med (Lausanne). (2017) 4:262. doi: 10.3389/fmed.2017.00262
6. Schwartz DM, Burma AM, Kitakule MM, Luo Y, Mehta NN. T cells in autoimmunity-associated cardiovascular diseases. Front Immunol. (2020) 11:588776. doi: 10.3389/fimmu.2020.588776
7. Srikakulapu P, McNamara CA. B cells and atherosclerosis. Am J Physiol Heart Circ Physiol. (2017) 312:H1060–7. doi: 10.1152/ajpheart.00859.2016
8. da Silva FF, Levy RA, de Carvalho JF. Cardiovascular risk factors in the antiphospholipid syndrome. J Immunol Res. (2014) 2014:621270. doi: 10.1155/2014/621270
9. Brandsma E, Kloosterhuis NJ, Koster M, Dekker DC, Gijbels MJJ, van der Velden S, et al. A proinflammatory gut microbiota increases systemic inflammation and accelerates atherosclerosis. Circ Res. (2019) 124:94–100. doi: 10.1161/CIRCRESAHA.118.313234
10. Shi G, Lin Y, Wu Y, Zhou J, Cao L, Chen J, et al. Bacteroides fragilis supplementation deteriorated metabolic dysfunction, inflammation, and aorta atherosclerosis by inducing gut microbiota dysbiosis in animal model. Nutrients. (2022) 14:2199. doi: 10.3390/nu14112199
11. Koeth RA, Lam-Galvez BR, Kirsop J, Wang Z, Levison BS, Gu X, et al. l-Carnitine in omnivorous diets induces an atherogenic gut microbial pathway in humans. J Clin Invest. (2019) 129:373–87. doi: 10.1172/JCI94601
12. Pan Q, Guo F, Huang Y, Li A, Chen S, Chen J, et al. Gut microbiota dysbiosis in systemic lupus erythematosus: novel insights into mechanisms and promising therapeutic strategies. Front Immunol. (2021) 12:799788. doi: 10.3389/fimmu.2021.799788
13. Chen BD, Jia XM, Xu JY, Zhao LD, Ji JY, Wu BX, et al. An autoimmunogenic and proinflammatory profile defined by the gut microbiota of patients with untreated systemic lupus erythematosus. Arthritis Rheumatol. (2021) 73:232–43. doi: 10.1002/art.41511
14. Morand E, Pike M, Merrill JT, van Vollenhoven R, Werth VP, Hobar C, et al. Deucravacitinib, a tyrosine kinase 2 inhibitor, in systemic lupus erythematosus: A phase II, randomized, double-blind, placebo-controlled trial. Arthritis Rheumatol. (2023) 75:242–52. doi: 10.1002/art.42391
15. He J, Zhang R, Shao M, Zhao X, Miao M, Chen J, et al. Efficacy and safety of low-dose IL-2 in the treatment of systemic lupus erythematosus: a randomised, double-blind, placebo-controlled trial. Ann Rheum Dis. (2020) 79:141–9. doi: 10.1136/annrheumdis-2019-215396
16. Pego-Reigosa JM, Lu TY, Fontanillo MF, del Campo-Perez V, Rahman A, Isenberg DA. Long-term improvement of lipid profile in patients with refractory systemic lupus erythematosus treated with B-cell depletion therapy: a retrospective observational study. Rheumatol (Oxford). (2010) 49:691–6. doi: 10.1093/rheumatology/kep446
17. Gonzalez-Correa C, Moleon J, Minano S, Visitacion N, Robles-Vera I, Gomez-Guzman M, et al. Trimethylamine N-oxide promotes autoimmunity and a loss of vascular function in toll-like receptor 7-driven lupus mice. Antioxidants (Basel). (2021) 11:84. doi: 10.3390/antiox11010084
18. Sircana MC, Erre GL, Castagna F, Manetti R. Crosstalk between inflammation and atherosclerosis in rheumatoid arthritis and systemic lupus erythematosus: is there a common basis? Life (Basel). (2024) 14:716. doi: 10.3390/life14060716
19. Mehta NN, Teague HL, Swindell WR, Baumer Y, Ward NL, Xing X, et al. IFN-gamma and TNF-alpha synergism may provide a link between psoriasis and inflammatory atherogenesis. Sci Rep. (2017) 7:13831. doi: 10.1038/s41598-017-14365-1
20. Kim SY, Yu M, Morin EE, Kang J, Kaplan MJ, Schwendeman A. High-density lipoprotein in lupus: disease biomarkers and potential therapeutic strategy. Arthritis Rheumatol. (2020) 72:20–30. doi: 10.1002/art.41059
21. Ambler WG, Kaplan MJ. Vascular damage in systemic lupus erythematosus. Nat Rev Nephrol. (2024) 20:251–65. doi: 10.1038/s41581-023-00797-8
22. Perl A. Oxidative stress in the pathology and treatment of systemic lupus erythematosus. Nat Rev Rheumatol. (2013) 9:674–86. doi: 10.1038/nrrheum.2013.147
23. Lopez-Pedrera C, Barbarroja N, Jimenez-Gomez Y, Collantes-Estevez E, Aguirre MA, Cuadrado MJ. Oxidative stress in the pathogenesis of atherothrombosis associated with anti-phospholipid syndrome and systemic lupus erythematosus: new therapeutic approaches. Rheumatol (Oxford). (2016) 55:2096–108. doi: 10.1093/rheumatology/kew054
24. Boren J, Chapman MJ, Krauss RM, Packard CJ, Bentzon JF, Binder CJ, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. (2020) 41:2313–30. doi: 10.1093/eurheartj/ehz962
25. Martin-Fuentes P, Civeira F, Recalde D, Garcia-Otin AL, Jarauta E, Marzo I, et al. Individual variation of scavenger receptor expression in human macrophages with oxidized low-density lipoprotein is associated with a differential inflammatory response. J Immunol. (2007) 179:3242–8. doi: 10.4049/jimmunol.179.5.3242
26. Mushenkova NV, Bezsonov EE, Orekhova VA, Popkova TV, Starodubova AV, Orekhov AN. Recognition of oxidized lipids by macrophages and its role in atherosclerosis development. Biomedicines. (2021) 9:915. doi: 10.3390/biomedicines9080915
27. Li J, Fu Q, Cui H, Qu B, Pan W, Shen N, et al. Interferon-alpha priming promotes lipid uptake and macrophage-derived foam cell formation: a novel link between interferon-alpha and atherosclerosis in lupus. Arthritis Rheum. (2011) 63:492–502. doi: 10.1002/art.30165
28. Robinson GA, Wilkinson MGL, Wincup C. The role of immunometabolism in the pathogenesis of systemic lupus erythematosus. Front Immunol. (2021) 12:806560. doi: 10.3389/fimmu.2021.806560
29. Barrett TJ. Macrophages in atherosclerosis regression. Arterioscler Thromb Vasc Biol. (2020) 40:20–33. doi: 10.1161/ATVBAHA.119.312802
30. Wang Y, Su W, Li Y, Yuan J, Yao M, Su X, et al. Analyzing the pathogenesis of systemic lupus erythematosus complicated by atherosclerosis using transcriptome data. Front Immunol. (2022) 13:935545. doi: 10.3389/fimmu.2022.935545
31. Appleton BD, Major AS. The latest in systemic lupus erythematosus-accelerated atherosclerosis: related mechanisms inform assessment and therapy. Curr Opin Rheumatol. (2021) 33:211–8. doi: 10.1097/BOR.0000000000000773
32. Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res. (2016) 118:692–702. doi: 10.1161/CIRCRESAHA.115.306361
33. Quillard T, Araujo HA, Franck G, Shvartz E, Sukhova G, Libby P. TLR2 and neutrophils potentiate endothelial stress, apoptosis and detachment: implications for superficial erosion. Eur Heart J. (2015) 36:1394–404. doi: 10.1093/eurheartj/ehv044
34. McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I interferons in infectious disease. Nat Rev Immunol. (2015) 15:87–103. doi: 10.1038/nri3787
35. Postal M, Vivaldo JF, Fernandez-Ruiz R, Paredes JL, Appenzeller S, Niewold TB. Type I interferon in the pathogenesis of systemic lupus erythematosus. Curr Opin Immunol. (2020) 67:87–94. doi: 10.1016/j.coi.2020.10.014
36. O’Neill LA. Immunology. Sensing the dark side of DNA. Science. (2013) 339:763–4. doi: 10.1126/science.1234724
37. Ding X, Xiang W, He X. IFN-I mediates dysfunction of endothelial progenitor cells in atherosclerosis of systemic lupus erythematosus. Front Immunol. (2020) 11:581385. doi: 10.3389/fimmu.2020.581385
38. Ahad A, Smita S, Mishra GP, Biswas VK, Sen K, Gupta B, et al. NCoR1 fine-tunes type-I IFN response in cDC1 dendritic cells by directly regulating Myd88-IRF7 axis under TLR9. Eur J Immunol. (2020) 50:1959–75. doi: 10.1002/eji.202048566
39. Denny MF, Thacker S, Mehta H, Somers EC, Dodick T, Barrat FJ, et al. Interferon-alpha promotes abnormal vasculogenesis in lupus: a potential pathway for premature atherosclerosis. Blood. (2007) 110:2907–15. doi: 10.1182/blood-2007-05-089086
40. Thacker SG, Berthier CC, Mattinzoli D, Rastaldi MP, Kretzler M, Kaplan MJ. The detrimental effects of IFN-alpha on vasculogenesis in lupus are mediated by repression of IL-1 pathways: potential role in atherogenesis and renal vascular rarefaction. J Immunol. (2010) 185:4457–69. doi: 10.4049/jimmunol.1001782
41. Rajagopalan S, Somers EC, Brook RD, Kehrer C, Pfenninger D, Lewis E, et al. Endothelial cell apoptosis in systemic lupus erythematosus: a common pathway for abnormal vascular function and thrombosis propensity. Blood. (2004) 103:3677–83. doi: 10.1182/blood-2003-09-3198
42. Thacker SG, Zhao W, Smith CK, Luo W, Wang H, Vivekanandan-Giri A, et al. Type I interferons modulate vascular function, repair, thrombosis, and plaque progression in murine models of lupus and atherosclerosis. Arthritis Rheum. (2012) 64:2975–85. doi: 10.1002/art.34504
43. Kahlenberg JM, Thacker SG, Berthier CC, Cohen CD, Kretzler M, Kaplan MJ. Inflammasome activation of IL-18 results in endothelial progenitor cell dysfunction in systemic lupus erythematosus. J Immunol. (2011) 187:6143–56. doi: 10.4049/jimmunol.1101284
44. Azzouz D, Khan MA, Palaniyar N. ROS induces NETosis by oxidizing DNA and initiating DNA repair. Cell Death Discov. (2021) 7:113. doi: 10.1038/s41420-021-00491-3
45. Rahman S, Sagar D, Hanna RN, Lightfoot YL, Mistry P, Smith CK, et al. Low-density granulocytes activate T cells and demonstrate a non-suppressive role in systemic lupus erythematosus. Ann Rheum Dis. (2019) 78:957–66. doi: 10.1136/annrheumdis-2018-214620
46. Wang H, Li T, Chen S, Gu Y, Ye S. Neutrophil extracellular trap mitochondrial DNA and its autoantibody in systemic lupus erythematosus and a proof-of-concept trial of metformin. Arthritis Rheumatol. (2015) 67:3190–200. doi: 10.1002/art.39296
47. Wigerblad G, Kaplan MJ. Neutrophil extracellular traps in systemic autoimmune and autoinflammatory diseases. Nat Rev Immunol. (2023) 23:274–88. doi: 10.1038/s41577-022-00787-0
48. Carmona-Rivera C, Zhao W, Yalavarthi S, Kaplan MJ. Neutrophil extracellular traps induce endothelial dysfunction in systemic lupus erythematosus through the activation of matrix metalloproteinase-2. Ann Rheum Dis. (2015) 74:1417–24. doi: 10.1136/annrheumdis-2013-204837
49. Blanco LP, Wang X, Carlucci PM, Torres-Ruiz JJ, Romo-Tena J, Sun HW, et al. RNA externalized by neutrophil extracellular traps promotes inflammatory pathways in endothelial cells. Arthritis Rheumatol. (2021) 73:2282–92. doi: 10.1002/art.41796
50. Kahlenberg JM, Kaplan MJ. Little peptide, big effects: the role of LL-37 in inflammation and autoimmune disease. J Immunol. (2013) 191:4895–901. doi: 10.4049/jimmunol.1302005
51. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. (2016) 22:146–53. doi: 10.1038/nm.4027
52. Caielli S, Athale S, Domic B, Murat E, Chandra M, Banchereau R, et al. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J Exp Med. (2016) 213:697–713. doi: 10.1084/jem.20151876
53. Hakkim A, Furnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, et al. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci U S A. (2010) 107:9813–8. doi: 10.1073/pnas.0909927107
54. Magna M, Pisetsky DS. The role of HMGB1 in the pathogenesis of inflammatory and autoimmune diseases. Mol Med. (2014) 20:138–46. doi: 10.2119/molmed.2013.00164
55. Kahlenberg JM, Carmona-Rivera C, Smith CK, Kaplan MJ. Neutrophil extracellular trap-associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. J Immunol. (2013) 190:1217–26. doi: 10.4049/jimmunol.1202388
56. Smith CK, Vivekanandan-Giri A, Tang C, Knight JS, Mathew A, Padilla RL, et al. Neutrophil extracellular trap-derived enzymes oxidize high-density lipoprotein: an additional proatherogenic mechanism in systemic lupus erythematosus. Arthritis Rheumatol. (2014) 66:2532–44. doi: 10.1002/art.38703
57. Conti F, Spinelli FR, Alessandri C, Pacelli M, Ceccarelli F, Marocchi E, et al. Subclinical atherosclerosis in systemic lupus erythematosus and antiphospholipid syndrome: focus on beta2GPI-specific T cell response. Arterioscler Thromb Vasc Biol. (2014) 34:661–8. doi: 10.1161/ATVBAHA.113.302680
58. Clement M, Charles N, Escoubet B, Guedj K, Chauveheid MP, Caligiuri G, et al. CD4+CXCR3+ T cells and plasmacytoid dendritic cells drive accelerated atherosclerosis associated with systemic lupus erythematosus. J Autoimmun. (2015) 63:59–67. doi: 10.1016/j.jaut.2015.07.001
59. Baragetti A, Ramirez GA, Magnoni M, Garlaschelli K, Grigore L, Berteotti M, et al. Disease trends over time and CD4(+)CCR5(+) T-cells expansion predict carotid atherosclerosis development in patients with systemic lupus erythematosus. Nutr Metab Cardiovasc Dis. (2018) 28:53–63. doi: 10.1016/j.numecd.2017.09.001
60. Qin S, Rottman JB, Myers P, Kassam N, Weinblatt M, Loetscher M, et al. The chemokine receptors CXCR3 and CCR5 mark subsets of T cells associated with certain inflammatory reactions. J Clin Invest. (1998) 101:746–54. doi: 10.1172/JCI1422
61. Li J, McArdle S, Gholami A, Kimura T, Wolf D, Gerhardt T, et al. CCR5+T-bet+FoxP3+ Effector CD4 T cells drive atherosclerosis. Circ Res. (2016) 118:1540–52. doi: 10.1161/CIRCRESAHA.116.308648
62. Li W, Elshikha AS, Cornaby C, Teng X, Abboud G, Brown J, et al. T cells expressing the lupus susceptibility allele Pbx1d enhance autoimmunity and atherosclerosis in dyslipidemic mice. JCI Insight. (2020) 5:e138274. doi: 10.1172/jci.insight.138274
63. Gaddis DE, Padgett LE, Wu R, McSkimming C, Romines V, Taylor AM, et al. Apolipoprotein AI prevents regulatory to follicular helper T cell switching during atherosclerosis. Nat Commun. (2018) 9:1095. doi: 10.1038/s41467-018-03493-5
64. Tay C, Liu YH, Kanellakis P, Kallies A, Li Y, Cao A, et al. Follicular B cells promote atherosclerosis via T cell-mediated differentiation into plasma cells and secreting pathogenic immunoglobulin G. Arterioscler Thromb Vasc Biol. (2018) 38:e71–84. doi: 10.1161/ATVBAHA.117.310678
65. Ghamar Talepoor A, Khosropanah S, Doroudchi M. Functional subsets of circulating follicular helper T cells in patients with atherosclerosis. Physiol Rep. (2020) 8:e14637. doi: 10.14814/phy2.14637
66. Zhu M, Mo H, Li D, Luo X, Zhang L. Th17/Treg imbalance induced by increased incidence of atherosclerosis in patients with systemic lupus erythematosus (SLE). Clin Rheumatol. (2013) 32:1045–52. doi: 10.1007/s10067-013-2237-z
67. Wilhelm AJ, Rhoads JP, Wade NS, Major AS. Dysregulated CD4+ T cells from SLE-susceptible mice are sufficient to accelerate atherosclerosis in LDLr-/- mice. Ann Rheum Dis. (2015) 74:778–85. doi: 10.1136/annrheumdis-2013-203759
68. Tellides G, Tereb DA, Kirkiles-Smith NC, Kim RW, Wilson JH, Schechner JS, et al. Interferon-gamma elicits arteriosclerosis in the absence of leukocytes. Nature. (2000) 403:207–11. doi: 10.1038/35003221
69. Perez RK, Gordon MG, Subramaniam M, Kim MC, Hartoularos GC, Targ S, et al. Single-cell RNA-seq reveals cell type-specific molecular and genetic associations to lupus. Science. (2022) 376:eabf1970. doi: 10.1126/science.abf1970
70. Hur J, Yang HM, Yoon CH, Lee CS, Park KW, Kim JH, et al. Identification of a novel role of T cells in postnatal vasculogenesis: characterization of endothelial progenitor cell colonies. Circulation. (2007) 116:1671–82. doi: 10.1161/CIRCULATIONAHA.107.694778
71. Miao J, Qiu F, Li T, Zhao P, Zhang K, Lv M, et al. Circulating angiogenic T cells and their subpopulations in patients with systemic lupus erythematosus. Mediators Inflammation. (2016) 2016:2842143. doi: 10.1155/2016/2842143
72. Lopez P, Rodriguez-Carrio J, Martinez-Zapico A, Caminal-Montero L, Suarez A. Senescent profile of angiogenic T cells from systemic lupus erythematosus patients. J Leukoc Biol. (2016) 99:405–12. doi: 10.1189/jlb.5HI0215-042R
73. Bortoluzzi A, Chighizola CB, Fredi M, Raschi E, Bodio C, Privitera D, et al. The IMMENSE study: the interplay between iMMune and ENdothelial cells in mediating cardiovascular risk in systemic lupus erythematosus. Front Immunol. (2020) 11:572876. doi: 10.3389/fimmu.2020.572876
74. Cho YN, Kee SJ, Lee SJ, Seo SR, Kim TJ, Lee SS, et al. Numerical and functional deficiencies of natural killer T cells in systemic lupus erythematosus: their deficiency related to disease activity. Rheumatol (Oxford). (2011) 50:1054–63. doi: 10.1093/rheumatology/keq457
75. Bondarenko S, Catapano AL, Norata GD. The CD1d-natural killer T cell axis in atherosclerosis. J Innate Immun. (2014) 6:3–12. doi: 10.1159/000351034
76. Lu S, Deng J, Liu H, Liu B, Yang J, Miao Y, et al. PKM2-dependent metabolic reprogramming in CD4(+) T cells is crucial for hyperhomocysteinemia-accelerated atherosclerosis. J Mol Med (Berl). (2018) 96:585–600. doi: 10.1007/s00109-018-1645-6
77. Yennemadi AS, Keane J, Leisching G. Mitochondrial bioenergetic changes in systemic lupus erythematosus immune cell subsets: Contributions to pathogenesis and clinical applications. Lupus. (2023) 32:603–11. doi: 10.1177/09612033231164635
78. Yin Y, Choi SC, Xu Z, Perry DJ, Seay H, Croker BP, et al. Normalization of CD4+ T cell metabolism reverses lupus. Sci Transl Med. (2015) 7:274ra18. doi: 10.1126/scitranslmed.aaa0835
79. Croca S, Bassett P, Chambers S, Davari M, Alber KF, Leach O, et al. IgG anti-apolipoprotein A-1 antibodies in patients with systemic lupus erythematosus are associated with disease activity and corticosteroid therapy: an observational study. Arthritis Res Ther. (2015) 17:26. doi: 10.1186/s13075-015-0539-z
80. Lopez P, Rodriguez-Carrio J, Martinez-Zapico A, Perez-Alvarez AI, Lopez-Mejias R, Benavente L, et al. Serum levels of anti-PON1 and anti-HDL antibodies as potential biomarkers of premature atherosclerosis in systemic lupus erythematosus. Thromb Haemost. (2017) 117:2194–206. doi: 10.1160/TH17-03-0221
81. Gronwall C, Reynolds H, Kim JK, Buyon J, Goldberg JD, Clancy RM, et al. Relation of carotid plaque with natural IgM antibodies in patients with systemic lupus erythematosus. Clin Immunol. (2014) 153:1–7. doi: 10.1016/j.clim.2014.03.017
82. Anania C, Gustafsson T, Hua X, Su J, Vikstrom M, de Faire U, et al. Increased prevalence of vulnerable atherosclerotic plaques and low levels of natural IgM antibodies against phosphorylcholine in patients with systemic lupus erythematosus. Arthritis Res Ther. (2010) 12:R214. doi: 10.1186/ar3193
83. Gronwall C, Akhter E, Oh C, Burlingame RW, Petri M, Silverman GJ. IgM autoantibodies to distinct apoptosis-associated antigens correlate with protection from cardiovascular events and renal disease in patients with SLE. Clin Immunol. (2012) 142:390–8. doi: 10.1016/j.clim.2012.01.002
84. Rahman M, Sing S, Golabkesh Z, Fiskesund R, Gustafsson T, Jogestrand T, et al. IgM antibodies against malondialdehyde and phosphorylcholine are together strong protection markers for atherosclerosis in systemic lupus erythematosus: Regulation and underlying mechanisms. Clin Immunol. (2016) 166-167:27–37. doi: 10.1016/j.clim.2016.04.007
85. Su J, Hua X, Concha H, Svenungsson E, Cederholm A, Frostegard J. Natural antibodies against phosphorylcholine as potential protective factors in SLE. Rheumatol (Oxford). (2008) 47:1144–50. doi: 10.1093/rheumatology/ken120
86. Braun N, Wade NS, Wakeland EK, Major AS. Accelerated atherosclerosis is independent of feeding high fat diet in systemic lupus erythematosus-susceptible LDLr(-/-) mice. Lupus. (2008) 17:1070–8. doi: 10.1177/0961203308093551
87. Stanic AK, Stein CM, Morgan AC, Fazio S, Linton MF, Wakeland EK, et al. Immune dysregulation accelerates atherosclerosis and modulates plaque composition in systemic lupus erythematosus. Proc Natl Acad Sci U S A. (2006) 103:7018–23. doi: 10.1073/pnas.0602311103
88. Kurien BT, Fesmire J, Anderson CJ, Scofield RH. Anti-ro and concomitant anti-la autoantibodies strongly associated with anti-oxLDL or anti-phospholipid antibody in systemic lupus erythematosus. J Clin Rheumatol. (2016) 22:418–25. doi: 10.1097/RHU.0000000000000429
89. Zhang X, Xie Y, Zhou H, Xu Y, Liu J, Xie H, et al. Involvement of TLR4 in oxidized LDL/beta2GPI/anti-beta2GPI-induced transformation of macrophages to foam cells. J Atheroscler Thromb. (2014) 21:1140–51. doi: 10.5551/jat.24372
90. Theodorou E, Nezos A, Antypa E, Ioakeimidis D, Koutsilieris M, Tektonidou M, et al. B-cell activating factor and related genetic variants in lupus related atherosclerosis. J Autoimmun. (2018) 92:87–92. doi: 10.1016/j.jaut.2018.05.002
91. Nguyen TT, Baumgarth N. Natural igM and the development of B cell-mediated autoimmune diseases. Crit Rev Immunol. (2016) 36:163–77. doi: 10.1615/CritRevImmunol.2016018175
92. Enghard P, Humrich JY, Chu VT, Grussie E, Hiepe F, Burmester GR, et al. Class switching and consecutive loss of dsDNA-reactive B1a B cells from the peritoneal cavity during murine lupus development. Eur J Immunol. (2010) 40:1809–18. doi: 10.1002/eji.200940050
93. Wither JE, Roy V, Brennan LA. Activated B cells express increased levels of costimulatory molecules in young autoimmune NZB and (NZB x NZW)F(1) mice. Clin Immunol. (2000) 94:51–63. doi: 10.1006/clim.1999.4806
94. Malkiel S, Barlev AN, Atisha-Fregoso Y, Suurmond J, Diamond B. Plasma cell differentiation pathways in systemic lupus erythematosus. Front Immunol. (2018) 9:427. doi: 10.3389/fimmu.2018.00427
95. Doring Y, Manthey HD, Drechsler M, Lievens D, Megens RT, Soehnlein O, et al. Auto-antigenic protein-DNA complexes stimulate plasmacytoid dendritic cells to promote atherosclerosis. Circulation. (2012) 125:1673–83. doi: 10.1161/CIRCULATIONAHA.111.046755
96. Patino-Trives AM, Perez-Sanchez C, Perez-Sanchez L, Luque-Tevar M, Abalos-Aguilera MC, Alcaide-Ruggiero L, et al. Anti-dsDNA antibodies increase the cardiovascular risk in systemic lupus erythematosus promoting a distinctive immune and vascular activation. Arterioscler Thromb Vasc Biol. (2021) 41:2417–30. doi: 10.1161/ATVBAHA.121.315928
97. Cieslik P, Semik-Grabarczyk E, Hrycek A, Holecki M. The impact of anti-endothelial cell antibodies (AECAs) on the development of blood vessel damage in patients with systemic lupus erythematosus: the preliminary study. Rheumatol Int. (2022) 42:791–801. doi: 10.1007/s00296-022-05104-5
98. Kondo A, Takahashi K, Mizuno T, Kato A, Hirano D, Yamamoto N, et al. The level of igA antibodies to endothelial cells correlates with histological evidence of disease activity in patients with lupus nephritis. PLoS One. (2016) 11:e0163085. doi: 10.1371/journal.pone.0163085
99. Perez-Sanchez C, Barbarroja N, Messineo S, Ruiz-Limon P, Rodriguez-Ariza A, Jimenez-Gomez Y, et al. Gene profiling reveals specific molecular pathways in the pathogenesis of atherosclerosis and cardiovascular disease in antiphospholipid syndrome, systemic lupus erythematosus and antiphospholipid syndrome with lupus. Ann Rheum Dis. (2015) 74:1441–9. doi: 10.1136/annrheumdis-2013-204600
100. de Faire U, Frostegard J. Natural antibodies against phosphorylcholine in cardiovascular disease. Ann N Y Acad Sci. (2009) 1173:292–300. doi: 10.1111/j.1749-6632.2009.04748.x
101. Svenungsson E, Engelbertsen D, Wigren M, Gustafsson JT, Gunnarsson I, Elvin K, et al. Decreased levels of autoantibodies against apolipoprotein B-100 antigens are associated with cardiovascular disease in systemic lupus erythematosus. Clin Exp Immunol. (2015) 181:417–26. doi: 10.1111/cei.12651
102. Ho MM, Manughian-Peter A, Spivia WR, Taylor A, Fraser DA. Macrophage molecular signaling and inflammatory responses during ingestion of atherogenic lipoproteins are modulated by complement protein C1q. Atherosclerosis. (2016) 253:38–46. doi: 10.1016/j.atherosclerosis.2016.08.019
103. Sun J, Lundstrom SL, Zhang B, Zubarev RA, Steuer J, Gillgren P, et al. IgM antibodies against phosphorylcholine promote polarization of T regulatory cells from patients with atherosclerotic plaques, systemic lupus erythematosus and healthy donors. Atherosclerosis. (2018) 268:36–48. doi: 10.1016/j.atherosclerosis.2017.11.010
104. Lourdudoss C, Ajeganova S, Frostegard J. Association between dietary and metabolic factors and IgM antibodies to phosphorylcholine and malondialdehyde in patients with systemic lupus erythematosus and population-based matched controls. Clin Exp Rheumatol. (2018) 36:428–33.
105. Cabana-Puig X, Lu R, Geng S, Michaelis JS, Oakes V, Armstrong C, et al. CX(3)CR1 modulates SLE-associated glomerulonephritis and cardiovascular disease in MRL/lpr mice. Inflammation Res. (2023) 72:1083–97. doi: 10.1007/s00011-023-01731-1
106. Sanchez-Rodriguez E, Egea-Zorrilla A, Plaza-Diaz J, Aragon-Vela J, Munoz-Quezada S, Tercedor-Sanchez L, et al. The gut microbiota and its implication in the development of atherosclerosis and related cardiovascular diseases. Nutrients. (2020) 12:605. doi: 10.3390/nu12030605
107. Tuomisto S, Huhtala H, Martiskainen M, Goebeler S, Lehtimaki T, Karhunen PJ. Age-dependent association of gut bacteria with coronary atherosclerosis: Tampere Sudden Death Study. PLoS One. (2019) 14:e0221345. doi: 10.1371/journal.pone.0221345
108. Zhang K, Qin X, Qiu J, Sun T, Qu K, Din AU, et al. Desulfovibrio desulfuricans aggravates atherosclerosis by enhancing intestinal permeability and endothelial TLR4/NF-kappaB pathway in Apoe (-/-) mice. Genes Dis. (2023) 10:239–53. doi: 10.1016/j.gendis.2021.09.007
109. Shen X, Li L, Sun Z, Zang G, Zhang L, Shao C, et al. Gut microbiota and atherosclerosis-focusing on the plaque stability. Front Cardiovasc Med. (2021) 8:668532. doi: 10.3389/fcvm.2021.668532
110. Yang HT, Jiang ZH, Yang Y, Wu TT, Zheng YY, Ma YT, et al. Faecalibacterium prausnitzii as a potential Antiatherosclerotic microbe. Cell Commun Signal. (2024) 22:54. doi: 10.1186/s12964-023-01464-y
111. Park S, Kim I, Han SJ, Kwon S, Min EJ, Cho W, et al. Oral Porphyromonas gingivalis infection affects intestinal microbiota and promotes atherosclerosis. J Clin Periodontol. (2023) 50:1553–67. doi: 10.1111/jcpe.13864
112. Luo T, Guo Z, Liu D, Guo Z, Wu Q, Li Q, et al. Deficiency of PSRC1 accelerates atherosclerosis by increasing TMAO production via manipulating gut microbiota and flavin monooxygenase 3. Gut Microbes. (2022) 14:2077602. doi: 10.1080/19490976.2022.2077602
113. Seldin MM, Meng Y, Qi H, Zhu W, Wang Z, Hazen SL, et al. Trimethylamine N-oxide promotes vascular inflammation through signaling of mitogen-activated protein kinase and nuclear factor-kappaB. J Am Heart Assoc. (2016) 5:e002767. doi: 10.1161/JAHA.115.002767
114. Chen ML, Zhu XH, Ran L, Lang HD, Yi L, Mi MT. Trimethylamine-N-oxide induces vascular inflammation by activating the NLRP3 inflammasome through the SIRT3-SOD2-mtROS signaling pathway. J Am Heart Assoc. (2017) 6:e006347. doi: 10.1161/JAHA.117.006347
115. Geng J, Yang C, Wang B, Zhang X, Hu T, Gu Y, et al. Trimethylamine N-oxide promotes atherosclerosis via CD36-dependent MAPK/JNK pathway. BioMed Pharmacother. (2018) 97:941–7. doi: 10.1016/j.biopha.2017.11.016
116. Ding L, Chang M, Guo Y, Zhang L, Xue C, Yanagita T, et al. Trimethylamine-N-oxide (TMAO)-induced atherosclerosis is associated with bile acid metabolism. Lipids Health Dis. (2018) 17:286. doi: 10.1186/s12944-018-0939-6
117. Cai YY, Huang FQ, Lao X, Lu Y, Gao X, Alolga RN, et al. Integrated metagenomics identifies a crucial role for trimethylamine-producing Lachnoclostridium in promoting atherosclerosis. NPJ Biofilms Microbiomes. (2022) 8:11. doi: 10.1038/s41522-022-00273-4
118. Zhu W, Gregory JC, Org E, Buffa JA, Gupta N, Wang Z, et al. Gut microbial metabolite TMAO enhances platelet hyperreactivity and thrombosis risk. Cell. (2016) 165:111–24. doi: 10.1016/j.cell.2016.02.011
119. Li Y, Liang L, Deng X, Zhong L. Lipidomic and metabolomic profiling reveals novel candidate biomarkers in active systemic lupus erythematosus. Int J Clin Exp Pathol. (2019) 12:857–66.
120. Metghalchi S, Ponnuswamy P, Simon T, Haddad Y, Laurans L, Clement M, et al. Indoleamine 2,3-dioxygenase fine-tunes immune homeostasis in atherosclerosis and colitis through repression of interleukin-10 production. Cell Metab. (2015) 22:460–71. doi: 10.1016/j.cmet.2015.07.004
121. Laurans L, Venteclef N, Haddad Y, Chajadine M, Alzaid F, Metghalchi S, et al. Genetic deficiency of indoleamine 2,3-dioxygenase promotes gut microbiota-mediated metabolic health. Nat Med. (2018) 24:1113–20. doi: 10.1038/s41591-018-0060-4
122. Xue H, Chen X, Yu C, Deng Y, Zhang Y, Chen S, et al. Gut microbially produced indole-3-propionic acid inhibits atherosclerosis by promoting reverse cholesterol transport and its deficiency is causally related to atherosclerotic cardiovascular disease. Circ Res. (2022) 131:404–20. doi: 10.1161/CIRCRESAHA.122.321253
123. Chen L, Ishigami T, Nakashima-Sasaki R, Kino T, Doi H, Minegishi S, et al. Commensal microbe-specific activation of B2 cell subsets contributes to atherosclerosis development independently of lipid metabolism. EBioMedicine. (2016) 13:237–47. doi: 10.1016/j.ebiom.2016.10.030
124. Saita D, Ferrarese R, Foglieni C, Esposito A, Canu T, Perani L, et al. Adaptive immunity against gut microbiota enhances apoE-mediated immune regulation and reduces atherosclerosis and western-diet-related inflammation. Sci Rep. (2016) 6:29353. doi: 10.1038/srep29353
125. Pindjakova J, Sartini C, Lo Re O, Rappa F, Coupe B, Lelouvier B, et al. Gut dysbiosis and adaptive immune response in diet-induced obesity vs. Systemic Inflammation. Front Microbiol. (2017) 8:1157. doi: 10.3389/fmicb.2017.01157
126. Laurans L, Mouttoulingam N, Chajadine M, Lavelle A, Diedisheim M, Bacquer E, et al. An obesogenic diet increases atherosclerosis through promoting microbiota dysbiosis-induced gut lymphocyte trafficking into the periphery. Cell Rep. (2023) 42:113350. doi: 10.1016/j.celrep.2023.113350
127. Wilhelm AJ, Major AS. Accelerated atherosclerosis in SLE: mechanisms and prevention approaches. Int J Clin Rheumtol. (2012) 7:527–39. doi: 10.2217/ijr.12.46
128. Tu H, Li Q, Xiang S, Jiang H, Mao Y, Shou Z, et al. Dual effects of statins therapy in systemic lupus erythematosus and SLE-related atherosclerosis: the potential role for regulatory T cells. Atherosclerosis. (2012) 222:29–33. doi: 10.1016/j.atherosclerosis.2012.02.023
129. de Jong HJ, Tervaert JW, Saldi SR, Vandebriel RJ, Souverein PC, Meyboom RH, et al. Association between statin use and lupus-like syndrome using spontaneous reports. Semin Arthritis Rheum. (2011) 41:373–81. doi: 10.1016/j.semarthrit.2011.06.002
130. De Jong HJI, van Staa TP, Lalmohamed A, de Vries F, Vandebriel RJ, Van Loveren H, et al. Pattern of risks of systemic lupus erythematosus among statin users: a population-based cohort study. Ann Rheum Dis. (2017) 76:1723–30. doi: 10.1136/annrheumdis-2016-210936
131. Petri MA, Kiani AN, Post W, Christopher-Stine L, Magder LS. Lupus atherosclerosis prevention study (LAPS). Ann Rheum Dis. (2011) 70:760–5. doi: 10.1136/ard.2010.136762
132. MacLeod C, Hadoke PWF, Nixon M. Glucocorticoids: fuelling the fire of atherosclerosis or therapeutic extinguishers? Int J Mol Sci. (2021) 22:7622. doi: 10.3390/ijms22147622
133. Blachut D, Przywara-Chowaniec B, Harpula J, Tomasik A, Nowalany-Kozielska E, Morawiec B. The effects of glucocorticoid treatment on cardiovascular system in patients with systemic lupus erythematosus. Arch Rheumatol. (2022) 37:495–503. doi: 10.46497/ArchRheumatol.2022.9255
134. Mejia-Vilet JM, Ayoub I. The use of glucocorticoids in lupus nephritis: new pathways for an old drug. Front Med (Lausanne). (2021) 8:622225. doi: 10.3389/fmed.2021.622225
135. Richez C, Richards RJ, Duffau P, Weitzner Z, Andry CD, Rifkin IR, et al. The effect of mycophenolate mofetil on disease development in the gld.apoE (-/-) mouse model of accelerated atherosclerosis and systemic lupus erythematosus. PLoS One. (2013) 8:e61042. doi: 10.1371/journal.pone.0061042
136. Sato-Okabayashi Y, Isoda K, Heissig B, Kadoguchi T, Akita K, Kitamura K, et al. Low-dose oral cyclophosphamide therapy reduces atherosclerosis progression by decreasing inflammatory cells in a murine model of atherosclerosis. Int J Cardiol Heart Vasc. (2020) 28:100529. doi: 10.1016/j.ijcha.2020.100529
137. McMahon M, Skaggs B, Grossman J, Wong WK, Sahakian L, Chen W, et al. Comparison of PREDICTS atherosclerosis biomarker changes after initiation of new treatments in patients with SLE. Lupus Sci Med. (2019) 6:e000321. doi: 10.1136/lupus-2019-000321
138. van Leuven SI, van Wijk DF, Volger OL, de Vries JP, van der Loos CM, de Kleijn DV, et al. Mycophenolate mofetil attenuates plaque inflammation in patients with symptomatic carotid artery stenosis. Atherosclerosis. (2010) 211:231–6. doi: 10.1016/j.atherosclerosis.2010.01.043
139. Kiani AN, Magder LS, Petri M. Mycophenolate mofetil (MMF) does not slow the progression of subclinical atherosclerosis in SLE over 2 years. Rheumatol Int. (2012) 32:2701–5. doi: 10.1007/s00296-011-2048-y
140. Floris A, Piga M, Mangoni AA, Bortoluzzi A, Erre GL, Cauli A. Protective effects of hydroxychloroquine against accelerated atherosclerosis in systemic lupus erythematosus. Mediators Inflammation. (2018) 2018:3424136. doi: 10.1155/2018/3424136
141. Lee H, Han JH, Kim S, Kim S, Cho DH, Woo CH. Anti-malarial drugs reduce vascular smooth muscle cell proliferation via activation of AMPK and inhibition of smad3 signaling. J Lipid Atheroscler. (2019) 8:267–76. doi: 10.12997/jla.2019.8.2.267
142. Ulander L, Simonen P, Tolppanen H, Hartman O, Rissanen TT, Eklund KK, et al. The effect of hydroxychloroquine on cholesterol metabolism in statin treated patients after myocardial infarction. Atheroscler Plus. (2023) 53:26–32. doi: 10.1016/j.athplu.2023.06.003
143. Rua-Figueroa I, Rua-Figueroa D, Perez-Veiga N, Anzola AM, Galindo-Izquierdo M, Calvo-Alen J, et al. Antimalarials exert a cardioprotective effect in lupus patients: Insights from the Spanish Society of Rheumatology Lupus Register (RELESSER) analysis of factors associated with heart failure. Semin Arthritis Rheum. (2022) 52:151946. doi: 10.1016/j.semarthrit.2021.11.012
144. Yang DH, Leong PY, Sia SK, Wang YH, Wei JC. Long-term hydroxychloroquine therapy and risk of coronary artery disease in patients with systemic lupus erythematosus. J Clin Med. (2019) 8:796. doi: 10.3390/jcm8060796
145. Liu D, Li X, Zhang Y, Kwong JS, Li L, Zhang Y, et al. Chloroquine and hydroxychloroquine are associated with reduced cardiovascular risk: a systematic review and meta-analysis. Drug Des Devel Ther. (2018) 12:1685–95. doi: 10.2147/DDDT.S166893
146. Furie R, Khamashta M, Merrill JT, Werth VP, Kalunian K, Brohawn P, et al. Anifrolumab, an anti-interferon-alpha receptor monoclonal antibody, in moderate-to-severe systemic lupus erythematosus. Arthritis Rheumatol. (2017) 69:376–86. doi: 10.1002/art.39962
147. Knight JS, Subramanian V, O’Dell AA, Yalavarthi S, Zhao W, Smith CK, et al. Peptidylarginine deiminase inhibition disrupts NET formation and protects against kidney, skin and vascular disease in lupus-prone MRL/lpr mice. Ann Rheum Dis. (2015) 74:2199–206. doi: 10.1136/annrheumdis-2014-205365
148. Richter P, Cardoneanu A, Burlui AM, Macovei LA, Bratoiu I, Buliga-Finis ON, et al. Why do we need JAK inhibitors in systemic lupus erythematosus? Int J Mol Sci. (2022) 23:11788. doi: 10.3390/ijms231911788
149. Hasni SA, Gupta S, Davis M, Poncio E, Temesgen-Oyelakin Y, Carlucci PM, et al. Phase 1 double-blind randomized safety trial of the Janus kinase inhibitor tofacitinib in systemic lupus erythematosus. Nat Commun. (2021) 12:3391. doi: 10.1038/s41467-021-23361-z
150. Furumoto Y, Smith CK, Blanco L, Zhao W, Brooks SR, Thacker SG, et al. Tofacitinib ameliorates murine lupus and its associated vascular dysfunction. Arthritis Rheumatol. (2017) 69:148–60. doi: 10.1002/art.39818
151. Foks AC, Lichtman AH, Kuiper J. Treating atherosclerosis with regulatory T cells. Arterioscler Thromb Vasc Biol. (2015) 35:280–7. doi: 10.1161/ATVBAHA.114.303568
152. He J, Zhang X, Wei Y, Sun X, Chen Y, Deng J, et al. Low-dose interleukin-2 treatment selectively modulates CD4(+) T cell subsets in patients with systemic lupus erythematosus. Nat Med. (2016) 22:991–3. doi: 10.1038/nm.4148
153. Mockel T, Basta F, Weinmann-Menke J, Schwarting A. B cell activating factor (BAFF): Structure, functions, autoimmunity and clinical implications in Systemic Lupus Erythematosus (SLE). Autoimmun Rev. (2021) 20:102736. doi: 10.1016/j.autrev.2020.102736
154. Saidoune F, Even G, Lamri Y, Chezel J, Gaston AT, Escoubet B, et al. Effects of BAFF neutralization on atherosclerosis associated with systemic lupus erythematosus. Arthritis Rheumatol. (2021) 73:255–64. doi: 10.1002/art.41485
155. Liang D, Huang S, Ding R. Effects of belimumab on the lipid profile in systemic lupus erythematosus patients: an observational study. Clin Rheumatol. (2024) 43:2513–20. doi: 10.1007/s10067-024-07029-9
156. Ait-Oufella H, Herbin O, Bouaziz JD, Binder CJ, Uyttenhove C, Laurans L, et al. B cell depletion reduces the development of atherosclerosis in mice. J Exp Med. (2010) 207:1579–87. doi: 10.1084/jem.20100155
157. Kridin K, Mruwat N, Ludwig RJ. Association of rituximab with risk of long-term cardiovascular and metabolic outcomes in patients with pemphigus. JAMA Dermatol. (2023) 159:56–61. doi: 10.1001/jamadermatol.2022.5182
158. Vlacil AK, Schuett J, Ruppert V, Soufi M, Oberoi R, Shahin K, et al. Deficiency of Nucleotide-binding oligomerization domain-containing proteins (NOD) 1 and 2 reduces atherosclerosis. Basic Res Cardiol. (2020) 115:47. doi: 10.1007/s00395-020-0806-2
159. Yoshida N, Emoto T, Yamashita T, Watanabe H, Hayashi T, Tabata T, et al. Bacteroides vulgatus and Bacteroides dorei Reduce Gut Microbial Lipopolysaccharide Production and Inhibit Atherosclerosis. Circulation. (2018) 138:2486–98. doi: 10.1161/CIRCULATIONAHA.118.033714
160. Kasahara K, Krautkramer KA, Org E, Romano KA, Kerby RL, Vivas EI, et al. Interactions between Roseburia intestinalis and diet modulate atherogenesis in a murine model. Nat Microbiol. (2018) 3:1461–71. doi: 10.1038/s41564-018-0272-x
161. Chan YK, Brar MS, Kirjavainen PV, Chen Y, Peng J, Li D, et al. High fat diet induced atherosclerosis is accompanied with low colonic bacterial diversity and altered abundances that correlates with plaque size, plasma A-FABP and cholesterol: a pilot study of high fat diet and its intervention with Lactobacillus rhamnosus GG (LGG) or telmisartan in ApoE(-/-) mice. BMC Microbiol. (2016) 16:264. doi: 10.1186/s12866-016-0883-4
162. Liu SJ, Fu JJ, Liao ZY, Liu YX, He J, He LY, et al. Z-ligustilide alleviates atherosclerosis by reconstructing gut microbiota and sustaining gut barrier integrity through activation of cannabinoid receptor 2. Phytomedicine. (2024) 135:156117. doi: 10.1016/j.phymed.2024.156117
163. Hao H, Li Z, Qiao SY, Qi Y, Xu XY, Si JY, et al. Empagliflozin ameliorates atherosclerosis via regulating the intestinal flora. Atherosclerosis. (2023) 371:32–40. doi: 10.1016/j.atherosclerosis.2023.03.011
164. Kim ES, Yoon BH, Lee SM, Choi M, Kim EH, Lee BW, et al. Fecal microbiota transplantation ameliorates atherosclerosis in mice with C1q/TNF-related protein 9 genetic deficiency. Exp Mol Med. (2022) 54:103–14. doi: 10.1038/s12276-022-00728-w
165. Li ZH, Weng J, Yan J, Zeng YH, Hao QY, Sheng HF, et al. Puerarin alleviates atherosclerosis via the inhibition of Prevotella copri and its trimethylamine production. Gut. (2024) 73:1934–43. doi: 10.1136/gutjnl-2024-331880
166. Ma SR, Tong Q, Lin Y, Pan LB, Fu J, Peng R, et al. Berberine treats atherosclerosis via a vitamine-like effect down-regulating Choline-TMA-TMAO production pathway in gut microbiota. Signal Transduct Target Ther. (2022) 7:207. doi: 10.1038/s41392-022-01027-6
167. Wang X, Cui J, Gu Z, Guo L, Liu R, Guo Y, et al. Aged garlic oligosaccharides modulate host metabolism and gut microbiota to alleviate high-fat and high-cholesterol diet-induced atherosclerosis in ApoE(-/-) mice. Food Chem. (2025) 463:141409. doi: 10.1016/j.foodchem.2024.141409
168. Jiang C, Wang S, Wang Y, Wang K, Huang C, Gao F, et al. Polyphenols from hickory nut reduce the occurrence of atherosclerosis in mice by improving intestinal microbiota and inhibiting trimethylamine N-oxide production. Phytomedicine. (2024) 128:155349. doi: 10.1016/j.phymed.2024.155349
169. Luo Z, Yang L, Zhu T, Fan F, Wang X, Liu Y, et al. Aucubin ameliorates atherosclerosis by modulating tryptophan metabolism and inhibiting endothelial-mesenchymal transitions via gut microbiota regulation. Phytomedicine. (2024) 135:156122. doi: 10.1016/j.phymed.2024.156122
170. He Y, Zhu Y, Shui X, Huang Z, Li K, Lei W. Gut microbiome and metabolomic profiles reveal the antiatherosclerotic effect of indole-3-carbinol in high-choline-fed ApoE(-/-) mice. Phytomedicine. (2024) 129:155621. doi: 10.1016/j.phymed.2024.155621
171. Haghikia A, Zimmermann F, Schumann P, Jasina A, Roessler J, Schmidt D, et al. Propionate attenuates atherosclerosis by immune-dependent regulation of intestinal cholesterol metabolism. Eur Heart J. (2022) 43:518–33. doi: 10.1093/eurheartj/ehab644
172. Vikram A, Kim YR, Kumar S, Li Q, Kassan M, Jacobs JS, et al. Vascular microRNA-204 is remotely governed by the microbiome and impairs endothelium-dependent vasorelaxation by downregulating Sirtuin1. Nat Commun. (2016) 7:12565. doi: 10.1038/ncomms12565
173. Li XL, Cui JJ, Zheng WS, Zhang JL, Li R, Ma XL, et al. Bicyclol alleviates atherosclerosis by manipulating gut microbiota. Small. (2022) 18:e2105021. doi: 10.1002/smll.202105021
174. Yang XY, Yu H, Fu J, Guo HH, Han P, Ma SR, et al. Hydroxyurea ameliorates atherosclerosis in ApoE(-/-) mice by potentially modulating Niemann-Pick C1-like 1 protein through the gut microbiota. Theranostics. (2022) 12:7775–87. doi: 10.7150/thno.76805
175. Wang A, Guan B, Yu L, Liu Q, Hou Y, Li Z, et al. Palmatine protects against atherosclerosis by gut microbiota and phenylalanine metabolism. Pharmacol Res. (2024) 209:107413. doi: 10.1016/j.phrs.2024.107413
176. Xu M, Lv C, Wang H, Lu Q, Ye M, Zhu X, et al. Peanut skin extract ameliorates high-fat diet-induced atherosclerosis by regulating lipid metabolism, inflammation reaction and gut microbiota in ApoE(-/-) mice. Food Res Int. (2022) 154:111014. doi: 10.1016/j.foodres.2022.111014
177. Wang Y, Wu J, Hong Y, Zhu J, Zhang Y, Zhang J, et al. Ginsenosides retard atherogenesis via remodelling host-microbiome metabolic homeostasis. Br J Pharmacol. (2024) 181:1768–92. doi: 10.1111/bph.16320
178. Lu C, Liu D, Wu Q, Zeng J, Xiong Y, Luo T. EphA2 blockage ALW-II-41–27 alleviates atherosclerosis by remodeling gut microbiota to regulate bile acid metabolism. NPJ Biofilms Microbiomes. (2024) 10:108. doi: 10.1038/s41522-024-00585-7
179. Traughber CA, Timinski K, Prince A, Bhandari N, Neupane K, Khan MR, et al. Disulfiram reduces atherosclerosis and enhances efferocytosis, autophagy, and atheroprotective gut microbiota in hyperlipidemic mice. J Am Heart Assoc. (2024) 13:e033881. doi: 10.1161/JAHA.123.033881
180. Moleon J, Gonzalez-Correa C, Robles-Vera I, Minano S, de la Visitacion N, Barranco AM, et al. Targeting the gut microbiota with dietary fibers: a novel approach to prevent the development cardiovascular complications linked to systemic lupus erythematosus in a preclinical study. Gut Microbes. (2023) 15:2247053. doi: 10.1080/19490976.2023.2247053
Keywords: systemic lupus erythematosus, atherosclerosis, immune dysregulation, gut microbiome imbalance, metabolic disorders
Citation: Pan Q, Huang X, Liu C, Pan Q and Huang S (2025) Systemic lupus erythematosus and atherosclerosis: immune pathways and the uncharted territory of gut microbiota and metabolism. Front. Immunol. 16:1492726. doi: 10.3389/fimmu.2025.1492726
Received: 07 September 2024; Accepted: 07 April 2025;
Published: 28 April 2025.
Edited by:
Kim Maree O’Sullivan, Monash University, AustraliaReviewed by:
Daming Zuo, Southern Medical University, ChinaSimona Rolla, University of Turin, Italy
Patricia López, University of Oviedo, Spain
Copyright © 2025 Pan, Huang, Liu, Pan and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qingjun Pan, c3RpbHdhcGFuQGdtYWlsLmNvbQ==; Shian Huang, aHVhbmdzaGlhbkBnZG11LmVkdS5jbg==
†These authors have contributed equally to this work