 Yiran Qin
Yiran Qin Mingxuan Li
Mingxuan Li Haibo Liu
Haibo Liu- 1Department of Cardiology, Qingpu Hospital Affiliated to Fudan University, Shanghai, China
- 2Department of Cardiology, Huadong Hospital, Fudan University, Shanghai, China
Myocardial infarction (MI) is one of the leading causes of death worldwide. It is triggered by thrombosis or vascular occlusion. After MI, damaged cardiomyocytes are replaced by scar tissue, leading to systolic and diastolic dysfunction, followed by adverse remodeling. Regulatory T cells (Tregs), as major immune cells, play a crucial role in post-MI inflammation and immunomodulation. Tregs improve cardiac remodeling after MI through various mechanisms, including inhibiting inflammatory cell infiltration, inducing anti-inflammatory macrophages, suppressing cell apoptosis, regulating fibroblast function, and promoting angiogenesis. The modulation of Tregs number or function may provide novel methods for improving post-MI remodeling. This review describes the immunoregulatory roles of Tregs, their regulatory mechanisms in post-MI ventricular remodeling, and the prospects and challenges for clinical application. However, the exact molecular mechanisms of Tregs in ventricular remodeling remain to be investigated. Although most of the current studies are at the preclinical stage, they hold great potential for further application in the future.
1 Introduction
Myocardial infarction (MI) is a global health problem with a serious economic burden, which has been considered the main cause of cardiovascular morbidity and mortality (1). It is an irreversible consequence of coronary artery ischemia. Patients are prone to heart failure after MI, which directly affects their quality of life and prognosis (2). Ventricular remodeling plays a pivotal role in the pathological process of ventricular dysfunction after infarction. It is clinically characterized by ventricular dilation, with other changes including collagen deposition, scar formation, fibrosis, and hypertrophy (3). A variety of drugs have been used to inhibit left ventricular remodeling, such as angiotensin-converting enzyme inhibitors and beta-blockers (4). However, current therapeutic strategies for preventing adverse clinical outcomes remain limited. Therefore, exploring the pathological mechanisms of post-MI ventricular remodeling and developing more effective prevention and treatment strategies are receiving increasing emphasis.
Regulatory T cells (Tregs) are a major type of immune cells involved in MI (5). In recent years, with the deepening of research in immunology and cardiology, the role of Tregs in ventricular remodeling has gradually garnered attention (6–8). Activation of the inflammatory immune system post-MI affects myocardial damage repair. Excessive inflammation results in an increased infarct size and aggravates cardiac remodeling (9). Tregs are a crucial type of lymphocytes that exhibit anti-inflammatory effects (10). They have immunomodulatory and recovery-promoting roles in atherosclerosis (11), acute coronary syndrome (ACS) (12), and chronic heart failure (13). The current review focuses on the role and mechanisms of Tregs in immune-mediated cardiac remodeling following MI, aiming to provide novel perspectives for the treatment and prognosis of MI.
2 Mechanisms of ventricular remodeling after MI
Post-MI ventricular remodeling refers to changes in the morphology, size, and tissue structure of the ventricle following myocardial ischemia and hypoxia caused by acute occlusion of the coronary artery. It is a process in which the heart initiates a series of adaptive responses for self-repair and compensation. Long-term and sustained pathological remodeling contributes to heart failure (4). At the histological level, the pathological changes manifest as cardiomyocyte hypertrophy, apoptosis, myofibroblast proliferation, and interstitial fibrosis in the non-infarcted area, leading to the progressive alteration in left ventricular myofibrillar structure (3, 14). The macroscopic manifestations include thinning of the infarcted myocardium, left ventricular dilation, and compensatory myocardial hypertrophy in the infarct border zone. This leads to an increase in both left ventricular end-diastolic and end-systolic volumes over time. The left ventricle gradually transitions from a normal ellipsoidal shape to a spherical one, with abnormal regional wall motion and decreased left ventricular ejection fraction (LVEF) (15).
The mechanisms underlying ventricular remodeling involve several aspects, including metabolic alterations in cardiomyocytes under ischemic and hypoxic conditions, activation of the neuroendocrine system, and changes in the extracellular matrix (16). In the hypoxic environment caused by MI, normal aerobic oxidation in cardiomyocytes is inhibited, and anaerobic glycolysis serves as the main source of energy. Although this metabolic switch can provide energy in a short period of time, the amount of ATP produced is much less than that from aerobic oxidation. Additionally, the accumulation of metabolites such as lactate may lead to intracellular acidosis and impaired cell function (17). After MI, the body rapidly initiates the activation of the sympathetic nervous system and the renin-angiotensin-aldosterone system. These systems collectively promote the development of ventricular remodeling by regulating extracellular matrix deposition and promoting cardiomyocyte hypertrophy and interstitial fibrosis (18). Changes in collagen, fibronectin, and other components of the extracellular matrix lead to disruption of connections between cardiomyocytes and enlargement of the ventricular cavity. Meanwhile, the imbalance between matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs) further exacerbates ventricular remodeling (2, 16, 19).
Immune response plays a significant role in post-MI healing and remodeling (20). Hypoxia and nutrients deficiency result in the death of cardiomyocytes, thereby triggering inflammation (21). Immune cells participate in all stages of MI progression, where they can both exacerbate the death of cardiomyocytes and promote the regeneration of damaged myocardium. Additionally, they regulate the dynamic and complex inflammatory responses (20, 22). Monocytes and macrophages in the infarct area are activated and polarized at different times. They produce pro-inflammatory or anti-inflammatory cytokines, regulate cardiomyocyte proliferation and apoptosis, and influence cardiac remodeling (23). T lymphocytes and B lymphocytes are recruited to the infarct area to remove and repair damaged cells and tissues (24, 25). Understanding the role of Tregs, as key inflammatory suppressive lymphocytes, in post-MI ventricular remodeling is helpful to develop effective therapeutic targets (26).
3 Characteristics and immunoregulatory roles of Tregs
Tregs are a subset of CD4+ T cells, accounting for 5%-10% of circulating CD4+ T cells. The features of Tregs include high expression of CD25 (IL-2 receptor alpha chain), high expression of transcription factor Foxp3, and low expression of CD127 (IL-7 receptor) (27). Foxp3 is the core molecule responsible for the function of Tregs, and its expression level directly determines the immunosuppressive capacity (28). Foxp3 deficiency leads to severe systemic inflammatory diseases (29). Tregs can be categorized into two types: natural Tregs derived from the thymus and induced Tregs generated from peripheral effector T cells (Table 1). They play different immunoregulatory roles under various physiological and pathological conditions (30).
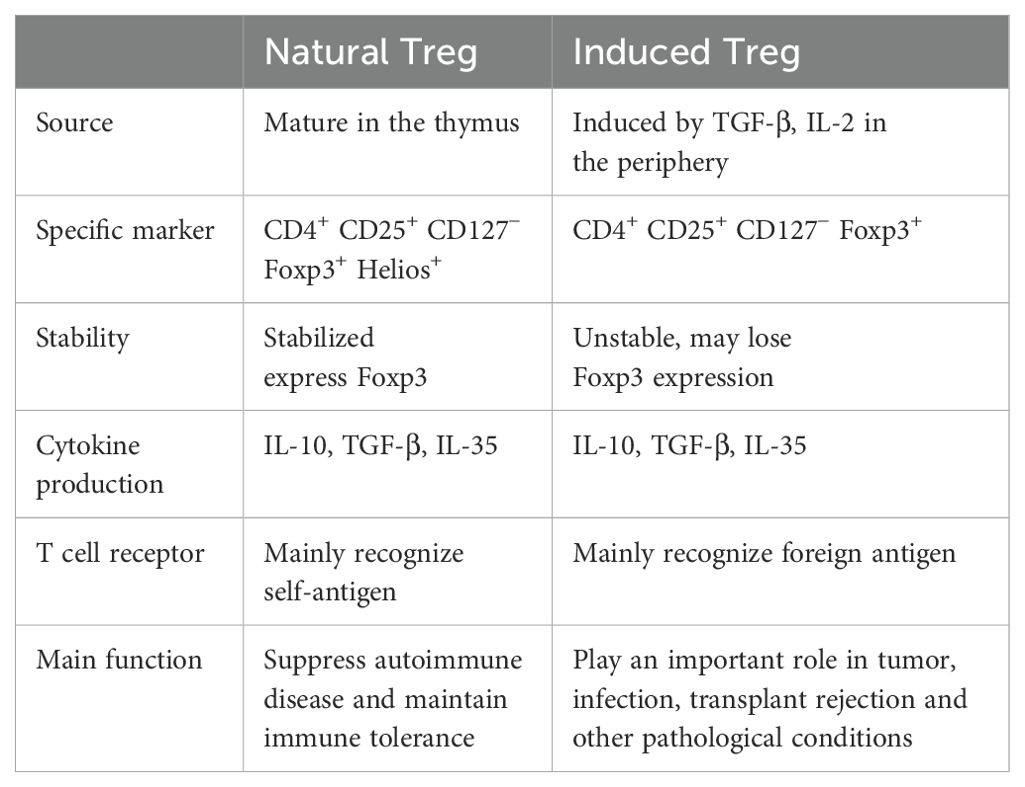
Table 1. Characterization of Treg subsets.
The immunomodulatory function of Tregs is primarily characterized by the suppression of over-activated immune response. Tregs exert immunosuppressive function on effector cells of innate and adaptive immunity, such as macrophages, neutrophils, dendritic cells (DCs), and T cells, through the utilization of multiple mechanisms (31). There are three main mechanisms by which Tregs exert their suppressive effects, including cell-to-cell interactions, cytokine release, and interference with target cell metabolism (Figure 1).
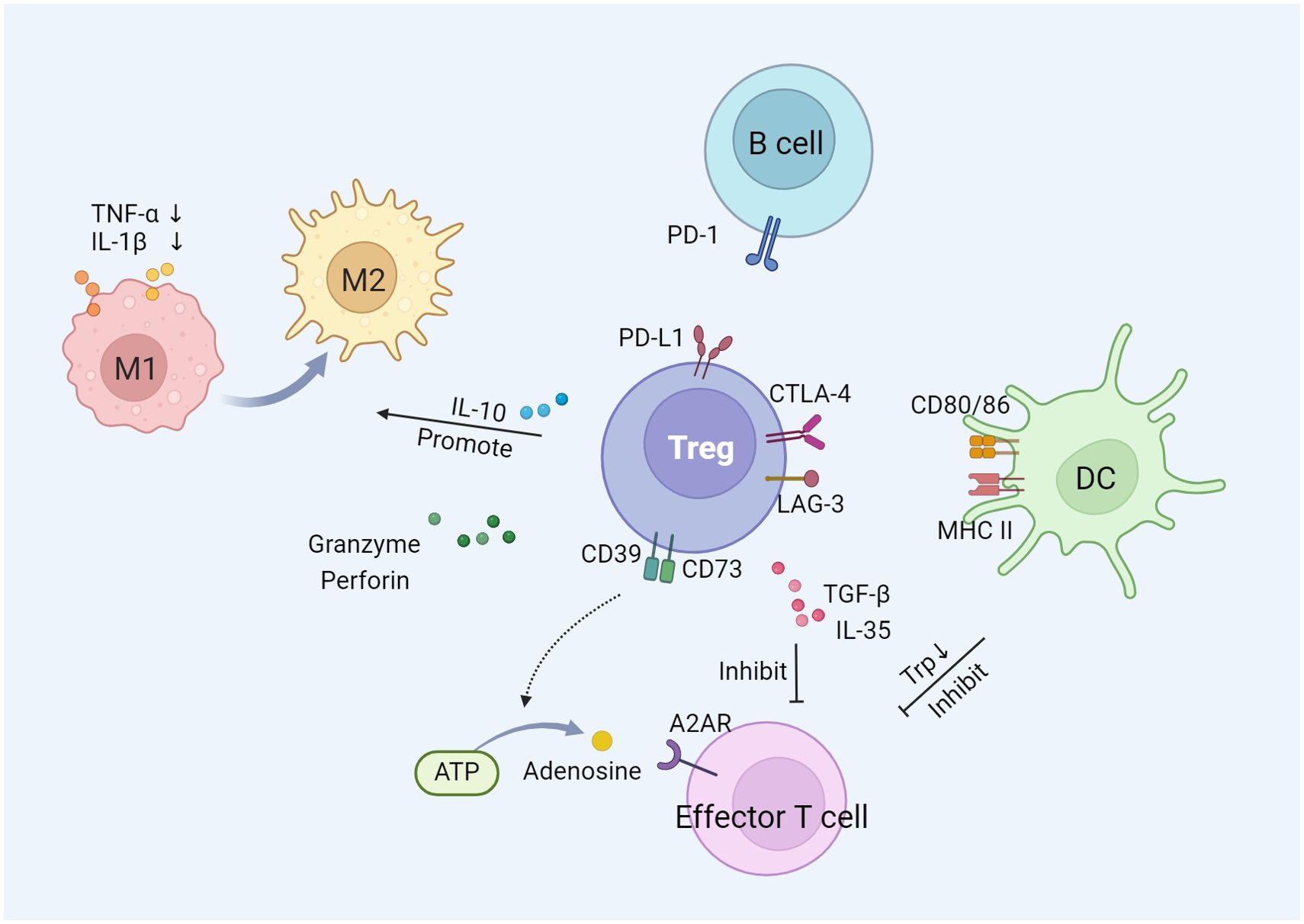
Figure 1. The immunosuppression mechanism of Tregs.
Tregs communicate directly with effector T cells or other immune cells through intercellular contact mechanisms. The surface of Tregs exhibits high expression of co-inhibitory receptors, including cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), lymphocyte activation gene 3 (LAG-3), and programmed death-ligand 1 (PD-L1) (32). CTLA-4 competitively binds to B7 molecules (CD80, CD86) on the surface of DCs, preventing the interaction between CD28 and B7, thereby delivering inhibitory signals and suppressing the activation and proliferation of T cells (33). The binding of CTLA-4 to B7 mediates the expression of indoleamine 2,3-dioxygenase by DCs, which degrades tryptophan essential for T cell proliferation (34, 35). LAG-3 binds to MHC class II molecules on the cell membrane of DCs and inhibits the maturation of DCs through cytoplasmic signaling. PD-L1 binds to programmed death receptor 1 (PD-1) on B cells, inhibiting their proliferation and activation, and inducing B cell apoptosis (36).
Tregs release a variety of inhibitory cytokines, including interleukin (IL)-10, transforming growth factor-β (TGF-β), and IL-35, to regulate the immune response of target cells (37). IL-10 inhibits the secretion of two pro-inflammatory cytokines, tumor necrosis factor-α (TNF-α) and IL-1β, by monocytes and macrophages (38). It also induces the transformation of pro-inflammatory M1 macrophages into anti-inflammatory and pro-repair M2 macrophages (39). Anti-IL-10 neutralizing antibodies block the inhibition of effector T cells mediated by Tregs (40). Intracellular TGF-β induces an increase in Foxp3 expression, promoting the generation of Tregs (41). Blocking TGF-β signaling reduces Tregs-mediated immunosuppression and exerts anti-tumor therapeutic effects (42). Similar to TGF-β and IL-10, IL-35 plays an important role in immunomodulation and immune homeostasis by regulating the development of Tregs (43).
Tregs exert their suppressive function by interfering with the metabolic pathways of target cells. Specifically, Tregs are able to deliver a large amount of cyclic adenosine monophosphate (cAMP) to effector T cells, interfering with their glycolysis and oxidative phosphorylation processes, thereby reducing the energy supply and proliferative capacity of effector T cells (44). Furthermore, Tregs compete with effector T cells to consume IL-2, which is a crucial cytokine necessary for the proliferation and survival of effector T cells. By limiting the availability of IL-2, Tregs induce effector T cell death and immunosuppression (45, 46). Tregs express the exonucleases CD39 and CD73 on their surface, which catalyze the production of adenosine from ATP. The adenosine activates adenosine receptor A2 on effector T cells, exerting an inhibitory effect (37). In addition, Tregs can inhibit effector T cell-mediated tumor clearance through cytolysis. Tregs induce apoptosis in effector T cells by producing granzyme and perforin, thereby reducing the number of effector cells and mediating immune suppression (47).
4 Tregs improve ventricular remodeling after MI
Tang et al. amplified Tregs in MI rats by using adoptive transfer and CD28 superagonist antibody. They confirmed that increasing Tregs number in infarcted hearts prevented adverse ventricular remodeling and improved cardiac function (48). This study demonstrates for the first time that Tregs have an important regulatory role in post-MI remodeling. Lin et al. provided evidence that the level of Tregs in peripheral blood is significantly reduced in patients with ACS, compared to those with stable angina pectoris and individuals with normal coronary arteries (49). The migration of circulating Tregs to the site of cardiac inflammation leads to a decrease in the number of peripheral Tregs and an increase in the peri-infarct area. Furthermore, the frequency of Tregs is positively correlated with LVEF in ACS patients. Prospective studies on the relationship between Tregs and cardiovascular risk showed that low level of circulating Tregs were associated with a higher risk of MI (50).
A recent study showed that the number of circulating Tregs in patients with acute MI (AMI) at 72h was higher than that in healthy controls and patients with cardiovascular risks such as type 2 diabetes, dyslipidemia, and smoking. And the IL-10 secreted by Tregs was increased in AMI (7). The reason for this phenomenon may be that Tregs counteracted the inflammatory response following acute ischemia. Further analysis revealed that the percentage of Tregs in AMI patients remained relatively unchanged at 3 months but significantly decreased after 6 months. At the 3-month point after AMI, patients with LVEF greater than 50% have higher levels of Tregs. These studies suggest that the cardiac function seems to be influenced by the number of Tregs. Tregs may play a pivotal role as a potential therapeutic target in the progression and prognosis of MI.
5 The regulatory mechanism of Tregs in post-MI ventricular remodeling
5.1 Suppression of excessive inflammation
The inflammatory response triggered by AMI is an important factor in myocardial injury (9). Tregs limit the inflammatory response by inhibiting the infiltration of inflammatory cells at the site of myocardial injury. Within hours after AMI, there is an increase in the number of neutrophils in the infarct area, followed by monocytes/macrophages infiltration. This triggers an excessive inflammatory response (51). Expanding Tregs in the infarcted heart can reduce the infiltration of neutrophils, macrophages, and T lymphocytes, reduce the production of pro-inflammatory cytokines, inhibit the cytotoxic effects of CD8+ T lymphocytes, and mitigate ventricular remodeling (48). In contrast, Tregs depletion leads to an increased density of CD45+ inflammatory cells, significantly exacerbating both dilated and hypertrophic remodeling (52, 53). Reperfusion injury is a key pathophysiological process of revascularization in AMI. Selective depletion of Tregs exacerbated myocardial ischemia/reperfusion injury (MIRI), while injection of Tregs activated in vitro inhibited MIRI and alleviated ventricular remodeling (54). Studies on their mechanisms have shown that Tregs exert cardioprotective effects in a CD39-dependent manner by activating the AKT/ERK pathway, inhibiting cardiomyocyte apoptosis, and reducing neutrophil infiltration. When CD39 is deficient, this protective effect disappears, and the ability of Tregs to inhibit neutrophils is impaired. The IL-2/anti-IL-2 complex (IL-2C) can significantly expand Tregs in the spleen and heart of mice. Helper T cells (Th) 1 and Th17 secrete interferon-γ and IL-17 respectively, exacerbating the immune-inflammatory response after MI. IL-2C pretreatment before MIRI reduced the number of Th1 and Th17, decreased inflammatory cell infiltration after MIRI, and improved cardiac function. Moreover, IL-2C decreased the expression of pro-inflammatory cytokines TNF-α, interferon-γ, IL-12, and IL-17A in the heart (55).
Tregs improve adverse post-MI remodeling by modulating the ratio of pro-inflammatory/anti-inflammatory macrophages. Tregs promote the polarization of macrophages towards the M2 phenotype, which has anti-inflammatory and tissue repair properties, while inhibiting the M1-type polarization of pro-inflammatory macrophages, thereby facilitating the repair of damaged myocardial tissue (56). The research by Saha et al. supported this conclusion. They used intravenous infusion of neonatal mesenchymal stromal cells to enhance the proliferation of Tregs mediated by CD44, which promoted the differentiation of monocytes into M2 macrophage and improved the cardiac structure and function in MI rats (57). Zhang et al. also showed that exosomes secreted by DCs activated Tregs to transform macrophages into M2 type. This resulted in a significant increase of Tregs and M2 macrophages infiltration in the border zone of AMI mice, thereby improving cardiac function (58). IL-2C inhibited the expression of pro-inflammatory cytokine genes and macrophage infiltration in the MI region, and induced the differentiation of macrophages from M1 to M2 phenotype in the border zone. IL-2C therapy may be a potential way to improve ischemic heart disease (59). All of these results suggest that Tregs promote macrophage differentiation toward an anti-inflammatory phenotype. In a mouse model of Tregs depleted by anti-CD25 antibody, the proportion of pro-inflammatory macrophages in the MI region was increased (56). Tregs inhibited the activation of pro-inflammatory macrophages. In MIRI mice, CXC chemokine receptor 4 antagonists reduced the expression of inflammatory genes in monocytes and macrophages by mobilizing splenic Tregs to the infarct zone (60).
Jia et al. investigated the mechanism by which Tregs promote the survival of anti-inflammatory macrophages. IL-35 is mainly expressed in Tregs. Ly6Clow is a monocyte/macrophage subtype with anti-inflammatory effects. IL-35 promoted the survival of Ly6Clow macrophages by activating the expression of CX3CR1 and TGF-β1 in macrophages through the phosphorylation of STAT1 and STAT4. IL-35 inhibition reduced the survival of Ly6Clow macrophages in infarcted heart. This resulted in impaired healing of the infarct area and aggravated cardiac remodeling (61). Feng et al. showed that chemokine ligand 17 (CCL17) was expressed in CCR2+ macrophages and DCs infiltrating the heart after myocardial injury. CCL17 inhibited Tregs recruitment through activation of CCR4 and exacerbated ventricular remodeling in MIRI mice. The absence of CCL17 led to an increase in the number of Tregs in the myocardium, reduced left ventricular remodeling, and improved systolic function. Increased Tregs further inhibited the expression of pro-inflammatory cytokines and chemokines in macrophages (6). This study reveals a potential link between Tregs and macrophages in post-MI ventricular remodeling. The interaction between Tregs and macrophages may be conducive to the formation of an anti-inflammatory microenvironment. Additionally, Tregs may influence macrophage polarization through the release of exosomes. Hu et al. found that exosomes secreted by Tregs after MI promoted anti-inflammatory macrophage polarization, reduced infarct size and inhibited cardiomyocyte apoptosis (62).
In summary, Tregs improve post-MI ventricular remodeling by inhibiting the infiltration of inflammatory cells, regulating macrophage differentiation, and reducing the expression of inflammatory cytokines.
5.2 Inhibition of cardiomyocyte apoptosis
Tregs play a beneficial role in adverse post-MI ventricular remodeling by modulating cardiomyocyte proliferation and apoptosis. In a lipopolysaccharide-induced inflammation model in vitro, Tregs reduced apoptosis of neonatal rat cardiomyocytes through direct intercellular contact and the secretion of IL-10 (48). Another study found that Tregs increased the phosphorylation levels of Akt and ERK1/2 in cardiomyocytes of MIRI mice. This confirmed that Tregs inhibited cardiomyocyte apoptosis by activating the ERK/Akt signaling pathway (54). Furthermore, Tregs can promote cardiomyocyte proliferation. Li et al. performed single-cell RNA sequencing on Tregs. They found that Tregs could promote neonatal cardiomyocyte proliferation by paracrine secretion of factors such as CCL24, GAS6, or AREG (63). Zacchigna et al. detected proliferative cardiomyocytes in the hearts of pregnant mice, and Tregs depletion during pregnancy reduced the proliferation of maternal and fetal cardiomyocytes. The researchers also injected Tregs into the area surrounding ischemia, which resulted in an increase in cardiomyocyte proliferation and a decrease in infarct size. Further studies have shown that Tregs promote cardiomyocyte proliferation and improve post-MI remodeling through paracrine secretion of cytokines such as Cst7 (53).
Apoptosis is closely related to inflammatory response. Anti-inflammatory effects help protect cardiomyocytes from attack by excessive immune response, which in turn reduce cardiomyocyte apoptosis. Recently, Wang et al. constructed a drug-loaded microgel system that responds to reactive oxygen species stimulation, effectively converting pro-inflammatory Th17 into anti-inflammatory Tregs. This system reduced early cardiomyocyte apoptosis post-MI, decreased inflammation in vivo, and protected cardiac function (64). The microgel system opens a new avenue for immunotherapy to improve ventricular remodeling.
5.3 Regulation of cardiac fibroblast function
Tregs participate in regulating fibroblast phenotype, inhibiting myocardial fibrosis and ventricular remodeling, and play a positive role in recovery after MI. Ramjee et al. observed that loss of IFN-γ in the myocardium led to reduced Tregs recruitment and enhanced fibrotic response in damaged myocardium post-MI (65). The number of Tregs in the left ventricular free wall of mice treated with IFN-γ doubled, and the area of myocardial fibrosis was relatively reduced by 34.4%. It suggested that IFN-γ recruited Tregs to the infarct area, limiting excessive myocardial fibrosis. In vitro co-culture experiment of Tregs with cardiac fibroblasts showed that Tregs regulated the function of cardiac fibroblasts. Tregs decreased the expression of α-smooth muscle actin and MMP-3, and attenuated collagen contraction (52).
However, Tregs may have a dual role in regulating myocardial fibrosis. Tregs facilitate the formation of new extracellular matrix and promote myocardial fibrosis after AMI by enhancing collagen deposition. After MI, Tregs were activated with superagonistic CD28-specific monoclonal antibody, which increased the level of TGF-β1 in myocardium (56). TGF-β1 promoted collagen synthesis in myofibroblasts. Increased collagen content and maturation in the infarct area of mice resulted in the formation of stable scar tissue, which prevented post-infarction left ventricular dilatation and rupture, as well as undesirable remodeling of the surviving myocardium.
Although the role of Tregs in cardiac fibrosis remains controversial, the above findings confirm the importance of Tregs in ventricular remodeling. Future studies may provide insight into the complex balance between the pro-fibrotic and anti-fibrotic effects of Tregs. It could help to develop new strategies for the treatment of MI-related ventricular remodeling.
5.4 Promoting angiogenesis
Angiogenesis is a critical process in heart repair, helping to restore blood supply to ischemic areas, reduce cardiomyocyte death, and promote recovery of cardiac function. Administering exogenous Tregs to MI mice resulted in a significant increase in the number of small capillaries within the infarcted heart, as demonstrated by CD31 staining (66). While ablation of Tregs led to a decrease in the number of small capillaries. Xiao et al. also observed an increase in small vessel density following Tregs amplification in the MIRI model (55). These results indicate that Tregs play an important role in microangiogenesis.
Tregs may promote angiogenesis by affecting macrophage polarization. Tregs are able to regulate monocytes/macrophages from a pro-inflammatory to a pro-repair state. This transformation involves an increase in the secretion of angiogenic factors by macrophages, such as vascular endothelial growth factor and fibroblast growth factor, which contribute to angiogenesis (67, 68).
IL-10 secreted by Tregs is a mediator that induces angiogenesis after MI. On the one hand, IL-10 can inhibit the production of pro-inflammatory cytokines, reducing their inhibitory effects on angiogenesis. On the other hand, IL-10 can also directly act on vascular endothelial cells, promoting their proliferation and migration, thus accelerating the formation of new blood vessels (30). Tregs can also promote endothelial cell proliferation by increasing the expression of Apelin, a cardiovascular active peptide (69). It follows that Tregs may promote angiogenesis through multiple mechanisms.
6 Clinical application prospects of Tregs
Although there is plenty of evidence supporting the benefits of Tregs in animal models of MI, it often takes a long time to translate these benefits into practical clinical applications for patients. Conventional tools for Tregs amplification include antibodies such as anti-CD3/CD28, and cytokines such as IL-2. As early as 2006, research institutions attempted to develop immunotherapies that activate endogenous Tregs. They conducted a Phase 1 trial using Tregs agonists in six healthy young volunteers (70). Within 90 minutes after receiving intravenous injection of the super-agonist anti-CD28 monoclonal antibody (TGN1412), all subjects experienced systemic inflammatory reactions, including headache, myalgia, nausea, diarrhea, and hypotension. Twelve hours later, patients were in critical condition and received cardiopulmonary support in the intensive care unit. Although all six subjects ultimately survived, this case nevertheless reveals the complexity and high risk of Tregs-activated therapy. A study conducted by Tian et al. evaluated the safety and efficacy of low-dose IL-2 in patients with ischemic heart disease (71). They demonstrated that low-dose subcutaneous injections of IL-2 successfully elevated Tregs levels without significant adverse effects. This indicates that low-dose IL-2 may have potential to treat ischemic heart disease. In addition to the activation of endogenous Tregs, the adoptive transfer of exogenous Tregs has also shown significant promise in improving post-MI ventricular remodeling (46, 48). It can be divided into autologous Tregs reinfusion and allogeneic Tregs transplantation. Autologous Tregs reinfusion has a lower risk of immune rejection. However, its preparation process is time-consuming and is influenced by the quantity and functionality of the patient’s own Tregs, which limits its application in AMI. In contrast, off-the-shelf allogeneic Tregs derived from healthy donors can be prepared and stored in advance, making them suitable for rapid treatment. Nevertheless, allogeneic Tregs may trigger host immune rejection, and their long-term survival and functional stability still require further optimization (26). Therefore, autologous Tregs may be more suitable for the regulation of chronic inflammation, while allogeneic Tregs may be more appropriate for rapid intervention during the acute phase of MI. In the future, large-scale studies will be needed to confirm its safety and efficacy.
Tregs play a crucial role in maintaining immune homeostasis through their antigen specificity. Antigen-specific Tregs suppress immune responses against specific antigens, demonstrating significant therapeutic potential in autoimmune diseases and transplant tolerance (72). However, the pathological process of MI involves multiple antigens and extensive inflammatory responses. Polyclonal Tregs possess a broader antigen recognition capability, enabling them to suppress multiple immune responses simultaneously, which may confer an advantage in the treatment of MI. Nonetheless, their non-specific suppression may increase the risk of infections or tumorigenesis. Thus, careful consideration of the balance between efficacy and safety remains essential for their clinical application.
In recent years, the clinical application of chimeric antigen receptor T (CAR-T) cell therapy is rapidly developing. CAR is a modified receptor that directs T cells to recognize and eliminate cells expressing the target antigen. Excessive myocardial fibrosis is known to be a cause of adverse ventricular remodeling after AMI. CD5-targeted lipid nanoparticles encapsulating modified mRNA can transiently generate CAR-T cells targeting fibroblast activation protein, reduce myocardial fibrosis and improve cardiac systolic and diastolic function in mouse models of heart failure (73). Targeted regulation of fibroblast activation protein provides new research insights for CAR-Treg cells to improve ventricular remodeling after MI.
Heart transplantation is an effective treatment for patients with end-stage heart failure, but it is limited by allograft rejection. Increasing the number of Tregs or enhancing their suppressive function can help prevent allograft rejection. In a mouse model of allogeneic heart transplant rejection, low-dose IL-2 increased Tregs infiltration in the spleen and graft, prolonged graft survival, and prevented chronic rejection (74). Studies have shown that the combination of matrine and tacrolimus can alleviate acute rejection in mouse allogeneic heart transplantation by inhibiting the maturation of DCs and increasing the proportion of Tregs, thereby mediating immune tolerance post-heart transplantation (75).
Adoptive transfer of exogenous Tregs or amplification of endogenous Tregs has demonstrated protective effects on post-MI ventricular remodeling. Extensive basic experimental research and rigorous evaluations of efficacy and safety are essential before conducting clinical studies. Future research should also address how to optimize the Tregs amplification pathway, how to maintain their stability, and how to direct them to target the infarct area, in order to successfully translate this new strategy into an effective clinical approach for the treatment of MI.
7 Conclusions
Increasing the function or number of Tregs can ameliorate post-MI ventricular remodeling, with the primary mechanisms encompassing the suppression of inflammatory response, reduction of cardiomyocyte apoptosis, modulation of cardiac fibroblast function, and promotion of angiogenesis (Figure 2). Tregs are considered as potential therapeutic targets. In recent years, a growing number of studies have focused on facilitating the transition of Tregs from basic research to clinical applications. The development of biomimetic nanoparticles and engineered Tregs has also brought promises in this therapeutic field (76, 77). Future research needs to further delve into the role and mechanisms of Tregs after MI, and carry out necessary clinical trials. Effective utilization of Tregs may provide a new potential avenue for the treatment of MI.
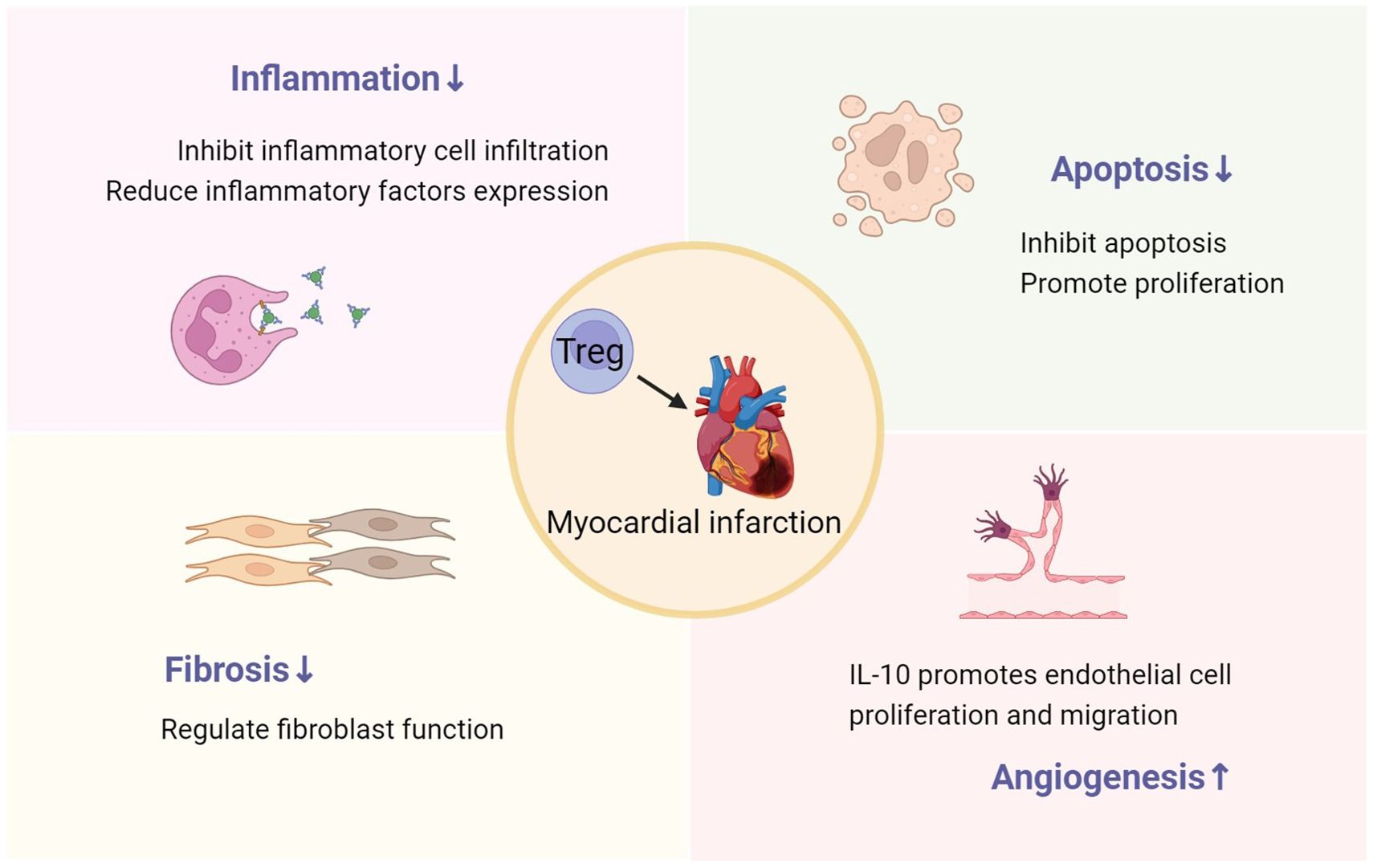
Figure 2. The role of Tregs in post-MI ventricular remodeling, including inhibiting inflammation, reducing cardiomyocyte apoptosis and fibrosis, and promoting angiogenesis.
Author contributions
YQ: Writing – original draft. ML: Writing – review & editing. HL: Conceptualization, Funding acquisition, Supervision, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by National Natural Science Foundation of China (81770350) and Natural Science Foundation of Shanghai (23ZR1411600).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, et al. Heart disease and stroke statistics-2020 update: A report from the american heart association. Circulation. (2020) 141:e139–596. doi: 10.1161/CIR.0000000000000757
2. Hilgendorf I, Frantz S, Frangogiannis NG. Repair of the infarcted heart: cellular effectors, molecular mechanisms and therapeutic opportunities. Circ Res. (2024) 134:1718–51. doi: 10.1161/CIRCRESAHA.124.323658
3. Gissler MC, Antiochos P, Ge Y, Heydari B, Grani C, Kwong RY. Cardiac magnetic resonance evaluation of LV remodeling post-myocardial infarction: prognosis, monitoring and trial endpoints. JACC Cardiovasc Imaging. (2024) 7:1366–80. doi: 10.1016/j.jcmg.2024.03.012
4. Frantz S, Hundertmark MJ, Schulz-Menger J, Bengel FM, Bauersachs J. Left ventricular remodelling post-myocardial infarction: pathophysiology, imaging, and novel therapies. Eur Heart J. (2022) 43:2549–61. doi: 10.1093/eurheartj/ehac223
5. DeBerge M, Chaudhary R, Schroth S, Thorp EB. Immunometabolism at the heart of cardiovascular disease. JACC Basic Transl Sci. (2023) 8:884–904. doi: 10.1016/j.jacbts.2022.12.010
6. Feng G, Bajpai G, Ma P, Koenig A, Bredemeyer A, Lokshina I, et al. CCL17 aggravates myocardial injury by suppressing recruitment of regulatory T cells. Circulation. (2022) 145:765–82. doi: 10.1161/CIRCULATIONAHA.121.055888
7. Martinez-Shio EB, Marin-Jauregui LS, Rodriguez-Ortega AC, Doniz-Padilla LM, Gonzalez-Amaro R, Escobedo-Uribe CD, et al. Regulatory T-cell frequency and function in acute myocardial infarction patients and its correlation with ventricular dysfunction. Clin Exp Immunol. (2024) 216:262–71. doi: 10.1093/cei/uxae014
8. Kumar V, Prabhu SD, Bansal SS. CD4(+) T-lymphocytes exhibit biphasic kinetics post-myocardial infarction. Front Cardiovasc Med. (2022) 9:992653. doi: 10.3389/fcvm.2022.992653
9. Ong SB, Hernandez-Resendiz S, Crespo-Avilan GE, Mukhametshina RT, Kwek XY, Cabrera-Fuentes HA, et al. Inflammation following acute myocardial infarction: Multiple players, dynamic roles, and novel therapeutic opportunities. Pharmacol Ther. (2018) 186:73–87. doi: 10.1016/j.pharmthera.2018.01.001
10. Zhuang R, Meng Q, Ma X, Shi S, Gong S, Liu J, et al. CD4(+)FoxP3(+)CD73(+) regulatory T cell promotes cardiac healing post-myocardial infarction. Theranostics. (2022) 12:2707–21. doi: 10.7150/thno.68437
11. Saigusa R, Winkels H, Ley K. T cell subsets and functions in atherosclerosis. Nat Rev Cardiol. (2020) 17:387–401. doi: 10.1038/s41569-020-0352-5
12. Zhu L, Jia L, Liu Z, Zhang Y, Wang J, Yuan Z, et al. Elevated methylation of FOXP3 (Forkhead box P3)-TSDR (Regulatory T-cell-specific demethylated region) is associated with increased risk for adverse outcomes in patients with acute coronary syndrome. Hypertension. (2019) 74:581–89. doi: 10.1161/HYPERTENSIONAHA.119.12852
13. Bansal SS, Ismahil MA, Goel M, Zhou G, Rokosh G, Hamid T, et al. Dysfunctional and proinflammatory regulatory T-lymphocytes are essential for adverse cardiac remodeling in ischemic cardiomyopathy. Circulation. (2019) 139:206–21. doi: 10.1161/CIRCULATIONAHA.118.036065
14. Konstam MA, Kramer DG, Patel AR, Maron MS, Udelson JE. Left ventricular remodeling in heart failure: current concepts in clinical significance and assessment. JACC Cardiovasc Imaging. (2011) 4:98–108. doi: 10.1016/j.jcmg.2010.10.008
15. Burchfield JS, Xie M, Hill JA. Pathological ventricular remodeling: mechanisms: part 1 of 2. Circulation. (2013) 128:388–400. doi: 10.1161/CIRCULATIONAHA.113.001878
16. Leanca SA, Crisu D, Petris AO, Afrasanie I, Genes A, Costache AD, et al. Left ventricular remodeling after myocardial infarction: from physiopathology to treatment. Life (Basel). (2022) 12:1111. doi: 10.3390/life12081111
17. Jiang M, Xie X, Cao F, Wang Y. Mitochondrial metabolism in myocardial remodeling and mechanical unloading: implications for ischemic heart disease. Front Cardiovasc Med. (2021) 8:789267. doi: 10.3389/fcvm.2021.789267
18. Gajarsa JJ, Kloner RA. Left ventricular remodeling in the post-infarction heart: a review of cellular, molecular mechanisms, and therapeutic modalities. Heart Fail Rev. (2011) 16:13–21. doi: 10.1007/s10741-010-9181-7
19. Nielsen SH, Mouton AJ, DeLeon-Pennell KY, Genovese F, Karsdal M, Lindsey ML. Understanding cardiac extracellular matrix remodeling to develop biomarkers of myocardial infarction outcomes. Matrix Biol. (2019) 75-76:43–57. doi: 10.1016/j.matbio.2017.12.001
20. Feng Q, Li Q, Zhou H, Sun L, Lin C, Jin Y, et al. The role of major immune cells in myocardial infarction. Front Immunol. (2022) 13:1084460. doi: 10.3389/fimmu.2022.1084460
21. Saparov A, Ogay V, Nurgozhin T, Chen W, Mansurov N, Issabekova A, et al. Role of the immune system in cardiac tissue damage and repair following myocardial infarction. Inflammation Res. (2017) 66:739–51. doi: 10.1007/s00011-017-1060-4
22. Prabhu SD, Frangogiannis NG. The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circ Res. (2016) 119:91–112. doi: 10.1161/CIRCRESAHA.116.303577
23. Hasan AS, Luo L, Yan C, Zhang TX, Urata Y, Goto S, et al. Cardiosphere-derived cells facilitate heart repair by modulating M1/M2 macrophage polarization and neutrophil recruitment. PloS One. (2016) 11:e0165255. doi: 10.1371/journal.pone.0165255
24. Zouggari Y, Ait-Oufella H, Bonnin P, Simon T, Sage AP, Guerin C, et al. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat Med. (2013) 19:1273–80. doi: 10.1038/nm.3284
25. Wang J, Xu T, Xu M. Roles and mechanisms of TGR5 in the modulation of CD4(+) T cell functions in myocardial infarction. J Cardiovasc Transl Res. (2022) 15:350–59. doi: 10.1007/s12265-021-10164-2
26. Wu Q, Wu M, Zhang K, Sun R, Li H, Tong J, et al. Regulatory T cells as a therapeutic target in acute myocardial infarction. Mol Immunol. (2024) 172:17–22. doi: 10.1016/j.molimm.2024.06.003
27. Schmetterer KG, Neunkirchner A, Pickl WF. Naturally occurring regulatory T cells: markers, mechanisms, and manipulation. FASEB J. (2012) 26:2253–76. doi: 10.1096/fj.11-193672
28. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. (2003) 299:1057–61. doi: 10.1126/science.1079490
29. Hu W, Wang ZM, Feng Y, Schizas M, Hoyos BE, van der Veeken J, et al. Regulatory T cells function in established systemic inflammation and reverse fatal autoimmunity. Nat Immunol. (2021) 22:1163–74. doi: 10.1038/s41590-021-01001-4
30. Fung T, Yang KY, Lui KO. An emerging role of regulatory T-cells in cardiovascular repair and regeneration. Theranostics. (2020) 10:8924–38. doi: 10.7150/thno.47118
31. Okeke EB, Uzonna JE. The pivotal role of regulatory T cells in the regulation of innate immune cells. Front Immunol. (2019) 10:680. doi: 10.3389/fimmu.2019.00680
32. Hu W, Li J, Cheng X. Regulatory T cells and cardiovascular diseases. Chin Med J (Engl). (2023) 136:2812–23. doi: 10.1097/CM9.0000000000002875
33. Onishi Y, Fehervari Z, Yamaguchi T, Sakaguchi S. Foxp3+ natural regulatory T cells preferentially form aggregates on dendritic cells in vitro and actively inhibit their maturation. Proc Natl Acad Sci U S A. (2008) 105:10113–18. doi: 10.1073/pnas.0711106105
34. Munn DH, Sharma MD, Lee JR, Jhaver KG, Johnson TS, Keskin DB, et al. Potential regulatory function of human dendritic cells expressing indoleamine 2,3-dioxygenase. Science. (2002) 297:1867–70. doi: 10.1126/science.1073514
35. Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. (2003) 9:1269–74. doi: 10.1038/nm934
36. Gotot J, Dhana E, Yagita H, Kaiser R, Ludwig-Portugall I, Kurts C. Antigen-specific Helios(-), Neuropilin-1(-) Tregs induce apoptosis of autoreactive B cells via PD-L1. Immunol Cell Biol. (2018) 96:852–62. doi: 10.1111/imcb.12053
37. Albany CJ, Trevelin SC, Giganti G, Lombardi G, Scottà C. Getting to the heart of the matter: the role of regulatory T-cells (Tregs) in cardiovascular disease (CVD) and atherosclerosis. Front Immunol. (2019) 10:2795. doi: 10.3389/fimmu.2019.02795
38. Armstrong L, Jordan N, Millar A. Interleukin 10 (IL-10) regulation of tumour necrosis factor alpha (TNF-alpha) from human alveolar macrophages and peripheral blood monocytes. Thorax. (1996) 51:143–49. doi: 10.1136/thx.51.2.143
39. Tiemessen MM, Jagger AL, Evans HG, van Herwijnen MJ, John S, Taams LS. CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proc Natl Acad Sci U S A. (2007) 104:19446–51. doi: 10.1073/pnas.0706832104
40. Rivas JR, Liu Y, Alhakeem SS, Eckenrode JM, Marti F, Collard JP, et al. Interleukin-10 suppression enhances T-cell antitumor immunity and responses to checkpoint blockade in chronic lymphocytic leukemia. Leukemia. (2021) 35:3188–200. doi: 10.1038/s41375-021-01217-1
41. Hata A, Chen YG. TGF-beta signaling from receptors to smads. Cold Spring Harb Perspect Biol. (2016) 8:a022061. doi: 10.1101/cshperspect.a022061
42. Chen SY, Mamai O, Akhurst RJ. TGFbeta: signaling blockade for cancer immunotherapy. Annu Rev Cancer Biol. (2022) 6:123–46. doi: 10.1146/annurev-cancerbio-070620-103554
43. Zong Y, Deng K, Chong WP. Regulation of Treg cells by cytokine signaling and co-stimulatory molecules. Front Immunol. (2024) 15:1387975. doi: 10.3389/fimmu.2024.1387975
44. Dou AX, Feng LL, Liu XQ, Wang X. Cyclic adenosine monophosphate involvement in low-dose cyclophosphamide-reversed immune evasion in a mouse lymphoma model. Cell Mol Immunol. (2012) 9:482–88. doi: 10.1038/cmi.2012.34
45. Thornton AM, Shevach EM. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J Exp Med. (1998) 188:287–96. doi: 10.1084/jem.188.2.287
46. Zhuang R, Feinberg MW. Regulatory T cells in ischemic cardiovascular injury and repair. J Mol Cell Cardiol. (2020) 147:1–11. doi: 10.1016/j.yjmcc.2020.08.004
47. Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica-Worms DR, et al. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity. (2007) 27:635–46. doi: 10.1016/j.immuni.2007.08.014
48. Tang TT, Yuan J, Zhu ZF, Zhang WC, Xiao H, Xia N, et al. Regulatory T cells ameliorate cardiac remodeling after myocardial infarction. Basic Res Cardiol. (2012) 107:232. doi: 10.1007/s00395-011-0232-6
49. Lin YZ, Lu SH, Lu ZD, Huang Y, Shi Y, Liu L, et al. Downregulation of CD4+LAP+ and CD4+CD25+ regulatory T cells in acute coronary syndromes. Mediators Inflamm. (2013) 2013:764082. doi: 10.1155/2013/764082
50. Wigren M, Bjorkbacka H, Andersson L, Ljungcrantz I, Fredrikson GN, Persson M, et al. Low levels of circulating CD4+FoxP3+ T cells are associated with an increased risk for development of myocardial infarction but not for stroke. Arterioscler Thromb Vasc Biol. (2012) 32:2000–04. doi: 10.1161/ATVBAHA.112.251579
51. Yan X, Anzai A, Katsumata Y, Matsuhashi T, Ito K, Endo J, et al. Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J Mol Cell Cardiol. (2013) 62:24–35. doi: 10.1016/j.yjmcc.2013.04.023
52. Saxena A, Dobaczewski M, Rai V, Haque Z, Chen W, Li N, et al. Regulatory T cells are recruited in the infarcted mouse myocardium and may modulate fibroblast phenotype and function. Am J Physiol Heart Circ Physiol. (2014) 307:H1233–42. doi: 10.1152/ajpheart.00328.2014
53. Zacchigna S, Martinelli V, Moimas S, Colliva A, Anzini M, Nordio A, et al. Paracrine effect of regulatory T cells promotes cardiomyocyte proliferation during pregnancy and after myocardial infarction. Nat Commun. (2018) 9:2432. doi: 10.1038/s41467-018-04908-z
54. Xia N, Jiao J, Tang TT, Lv BJ, Lu YZ, Wang KJ, et al. Activated regulatory T-cells attenuate myocardial ischaemia/reperfusion injury through a CD39-dependent mechanism. Clin Sci (Lond). (2015) 128:679–93. doi: 10.1042/CS20140672
55. Xiao J, Yu K, Li M, Xiong C, Wei Y, Zeng Q. The IL-2/anti-IL-2 complex attenuates cardiac ischaemia-reperfusion injury through expansion of regulatory T cells. Cell Physiol Biochem. (2017) 44:1810–27. doi: 10.1159/000485818
56. Weirather J, Hofmann UD, Beyersdorf N, Ramos GC, Vogel B, Frey A, et al. Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ Res. (2014) 115:55–67. doi: 10.1161/CIRCRESAHA.115.303895
57. Saha P, Guru SA, Ge ZD, Simms L, Chen L, Bolli R, et al. Neonatal Cardiac Mesenchymal Stromal Cells Promote Recovery of Infarcted Myocardium through CD44 Mediated FoxP3(+) T-Regulatory Cells after Vascular Infusion. Stem Cell Rev Rep. (2024) 20:1843–53. doi: 10.1007/s12015-024-10750-2
58. Zhang Y, Cai Z, Shen Y, Lu Q, Gao W, Zhong X, et al. Hydrogel-load exosomes derived from dendritic cells improve cardiac function via Treg cells and the polarization of macrophages following myocardial infarction. J Nanobiotechnology. (2021) 19:271. doi: 10.1186/s12951-021-01016-x
59. Zeng Z, Yu K, Chen L, Li W, Xiao H, Huang Z. Interleukin-2/Anti-Interleukin-2 Immune Complex Attenuates Cardiac Remodeling after Myocardial Infarction through Expansion of Regulatory T Cells. J Immunol Res. (2016) 2016:8493767. doi: 10.1155/2016/8493767
60. Wang Y, Dembowsky K, Chevalier E, Stuve P, Korf-Klingebiel M, Lochner M, et al. C-X-C motif chemokine receptor 4 blockade promotes tissue repair after myocardial infarction by enhancing regulatory T cell mobilization and immune-regulatory function. Circulation. (2019) 139:1798–812. doi: 10.1161/CIRCULATIONAHA.118.036053
61. Jia D, Jiang H, Weng X, Wu J, Bai P, Yang W, et al. Interleukin-35 promotes macrophage survival and improves wound healing after myocardial infarction in mice. Circ Res. (2019) 124:1323–36. doi: 10.1161/CIRCRESAHA.118.314569
62. Hu H, Wu J, Cao C, Ma L. Exosomes derived from regulatory T cells ameliorate acute myocardial infarction by promoting macrophage M2 polarization. IUBMB Life. (2020) 72:2409–19. doi: 10.1002/iub.2364
63. Li J, Yang KY, Tam R, Chan VW, Lan HY, Hori S, et al. Regulatory T-cells regulate neonatal heart regeneration by potentiating cardiomyocyte proliferation in a paracrine manner. Theranostics. (2019) 9:4324–41. doi: 10.7150/thno.32734
64. Wang S, Yao Y, Song L, Zhai Z, Li S, Wang Z, et al. ROS-responsive drug-releasing injectable microgels for ameliorating myocardial infarction. Biomaterials. (2024) 307:122534. doi: 10.1016/j.biomaterials.2024.122534
65. Ramjee V, Li D, Manderfield LJ, Liu F, Engleka KA, Aghajanian H, et al. Epicardial YAP/TAZ orchestrate an immunosuppressive response following myocardial infarction. J Clin Invest. (2017) 127:899–911. doi: 10.1172/JCI88759
66. Sharir R, Semo J, Shimoni S, Ben-Mordechai T, Landa-Rouben N, Maysel-Auslender S, et al. Experimental myocardial infarction induces altered regulatory T cell hemostasis, and adoptive transfer attenuates subsequent remodeling. PloS One. (2014) 9:e113653. doi: 10.1371/journal.pone.0113653
67. Wang N, Liu C, Wang X, He T, Li L, Liang X, et al. Hyaluronic acid oligosaccharides improve myocardial function reconstruction and angiogenesis against myocardial infarction by regulation of macrophages. Theranostics. (2019) 9:1980–92. doi: 10.7150/thno.31073
68. Tang J, Shen Y, Chen G, Wan Q, Wang K, Zhang J, et al. Activation of E-prostanoid 3 receptor in macrophages facilitates cardiac healing after myocardial infarction. Nat Commun. (2017) 8:14656. doi: 10.1038/ncomms14656
69. Tempel D, de Boer M, van Deel ED, Haasdijk RA, Duncker DJ, Cheng C, et al. Apelin enhances cardiac neovascularization after myocardial infarction by recruiting aplnr+ circulating cells. Circ Res. (2012) 111:585–98. doi: 10.1161/CIRCRESAHA.111.262097
70. Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. (2006) 355:1018–28. doi: 10.1056/NEJMoa063842
71. Zhao TX, Sriranjan RS, Tuong ZK, Lu Y, Sage AP, Nus M, et al. Regulatory T-cell response to low-dose interleukin-2 in ischemic heart disease. NEJM Evid. (2022) 1:EVIDoa2100009. doi: 10.1056/EVIDoa2100009
72. Imura Y, Ando M, Kondo T, Ito M, Yoshimura A. CD19-targeted CAR regulatory T cells suppress B cell pathology without GvHD. JCI Insight. (2020) 5:e136185. doi: 10.1172/jci.insight.136185
73. Rurik JG, Tombacz I, Yadegari A, Mendez FP, Shewale SV, Li L, et al. CAR T cells produced in vivo to treat cardiac injury. Science. (2022) 375:91–6. doi: 10.1126/science.abm0594
74. Ravichandran R, Itabashi Y, Fleming T, Bansal S, Bowen S, Poulson C, et al. Low-dose IL-2 prevents murine chronic cardiac allograft rejection: Role for IL-2-induced T regulatory cells and exosomes with PD-L1 and CD73. Am J Transplant. (2022) 22:2180–94. doi: 10.1111/ajt.17101
75. Zheng S, Chen Y, Wang Z, Che Y, Wu Q, Yuan S, et al. Combination of matrine and tacrolimus alleviates acute rejection in murine heart transplantation by inhibiting DCs maturation through ROS/ERK/NF-kappaB pathway. Int Immunopharmacol. (2021) 101:108218. doi: 10.1016/j.intimp.2021.108218
76. Li F, Liu D, Liu M, Ji Q, Zhang B, Mei Q, et al. Tregs biomimetic nanoparticle to reprogram inflammatory and redox microenvironment in infarct tissue to treat myocardial ischemia reperfusion injury in mice. J Nanobiotechnology. (2022) 20:251. doi: 10.1186/s12951-022-01445-2
Keywords: regulatory T cells, myocardial infarction, ventricular remodeling, immune regulation, inflammation
Citation: Qin Y, Li M and Liu H (2025) Regulatory T cells: a promising new therapeutic target in ventricular remodeling after myocardial infarction. Front. Immunol. 16:1514335. doi: 10.3389/fimmu.2025.1514335
Received: 20 October 2024; Accepted: 24 March 2025;
Published: 07 April 2025.
Edited by:
Yibei Zhu, Soochow University, ChinaReviewed by:
Bernhard Fidelis Maier, Indiana University-Purdue University Indianapolis, United StatesSudipta Tripathi, University of Massachusetts Medical School, United States
Copyright © 2025 Qin, Li and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haibo Liu, aGFpYm9saXUxM0BmdWRhbi5lZHUuY24=