 Dan Yuan
Dan Yuan Na Liang2
Na Liang2- 1Department of Pathology, Affiliated Hospital of Zunyi Medical University, Zunyi, China
- 2Department of Histology and Embryology, School of Basic Medicine, Zunyi Medical University, Zunyi, China
- 3Department of Pathology, Jilin Municipal People’s Hospital, Jilin, China
- 4Department of Pathology, Peking Union Medical College Hospital, Beijing, China
Objective: This study aimed to investigate the clinicopathological features of indolent T-cell lymphoproliferative disease of the gastrointestinal tract (ITLPD-GI) and to improve its diagnostic and therapeutic approaches.
Methods: A retrospective analysis was conducted on eight ITLPD-GI patients treated between January 2018 and January 2024. Clinical data, pathological features, immunophenotypes, molecular testing results, and follow-up records were reviewed.
Results: Clinical characteristics: Male-to-female ratio 3:5; mean age at onset 42 years. Symptoms: Predominantly diarrhea and abdominal pain. Endoscopic findings: Erosions, multiple shallow ulcers, and small polypoid lesions. Pathological features: Histology: Atrophy of gastric/intestinal glands with diffuse infiltration of small lymphocytes (round/irregular nuclei, dense chromatin) in the lamina propria; rare mitoses; absence of angioinvasion or necrosis. Notably, two cases showed prominent plasma cell infiltration in the superficial mucosa. Immunophenotype: Pan-T-cell markers positive (5/8); CD4−/CD8+ (5/8), CD4+/CD8+ (2/8), CD4+/CD8− (1/8); aberrant CD20 expression (2/8); low Ki-67 index. TCR rearrangement: Monoclonal in all four tested cases. Treatment and prognosis: Supportive therapy (five cases): Dietary modification, immunosuppression, immunomodulation, and anti-infective agents. Symptoms resolved in one case but persisted in four. Targeted therapy (one CD20+ case): Rituximab added, with no improvement after 14 months of follow-up. Chemotherapy (two cases): Prednisone + thalidomide; one achieved significant remission at 9 months, while the other showed no response (persistent diarrhea/anxiety) at 35 months. No disease progression was observed during follow-up.
Conclusion: ITLPD-GI is a rare indolent monoclonal T-cell proliferation with non-specific clinical/endoscopic features, necessitating differentiation from aggressive lymphomas to avoid misdiagnosis and overtreatment. Diagnosis relies on histomorphology, immunohistochemistry, and TCR clonality assessment (critical for atypical cases, e.g., CD20+). The majority of patients have favorable outcomes with conservative management. Enhanced clinical awareness and novel therapeutic targets warrant further exploration.
1 Introduction
Indolent T-cell lymphoproliferative disease of the gastrointestinal tract (ITLPD-G) is a rare subtype of intestinal T-cell lymphoma that was first characterized by Perry et al. (1) in 2013 and subsequently incorporated into the WHO (2017) classification of hematopoietic and lymphoid neoplasms. This entity follows an indolent clinical course with a favorable prognosis and rarely progresses to aggressive lymphoma. While its exact pathogenesis remains unclear, persistent antigen stimulation (e.g., from immune dysregulation or chronic infection) has been implicated (1–3). Currently, no standardized treatment guidelines exist for ITLPD-G. The disease frequently poses diagnostic challenges due to its clinical and pathological similarities to inflammatory bowel disease and aggressive T-cell lymphomas, often leading to misdiagnosis and unnecessary overtreatment. To enhance the understanding of this condition, we present a comprehensive analysis of clinicopathological features, immunophenotypic characteristics, molecular genetic alterations, and therapeutic approaches through the retrospective evaluation of eight cases and a literature review.
2 Materials and methods
2.1 Clinical data
We retrospectively analyzed eight cases of ITLPD-GI diagnosed between January 2018 and January 2024, including six in-house biopsy specimens and two consultation cases. All pathology slides were independently reviewed by two senior hematopathologists specializing in lymphoma, with the final diagnosis established according to the 2017 WHO classification criteria for hematopoietic and lymphoid neoplasms.
2.2 Methods
Histopathological examination: Tissue specimens were fixed in 10% neutral buffered formalin, routinely processed for paraffin embedding, and sectioned at 4 μm thickness for hematoxylin-eosin (HE) staining and light microscopic evaluation.
Immunohistochemistry (IHC): The EnVision two-step method was employed using primary antibodies against CD2, CD3, CD5, CD7, CD4, CD8, CD56, CD20, CD79α, TIA-1, granzyme B, and Ki-67 (all purchased from Zhongshan Golden Bridge Biotechnology, Beijing, China). Staining procedures followed the manufacturer’s protocols.
T-cell receptor (TCR) gene rearrangement analysis: Genomic DNA was analyzed using the BIOMED-2 multiplex PCR protocol (Europe) on an ABI 3500DX Genetic Analyzer. The assay covered TCRβ (Vβ-Jβ and Dβ-Jβ segments), TCRγ (Vγ1-11-Jγ segments), and TCRδ (Vδ-Dδ-Jδ segments). All procedures were performed in strict accordance with the manufacturer’s instructions.
3 Results
Detailed clinicopathological characteristics are summarized in Table 1.
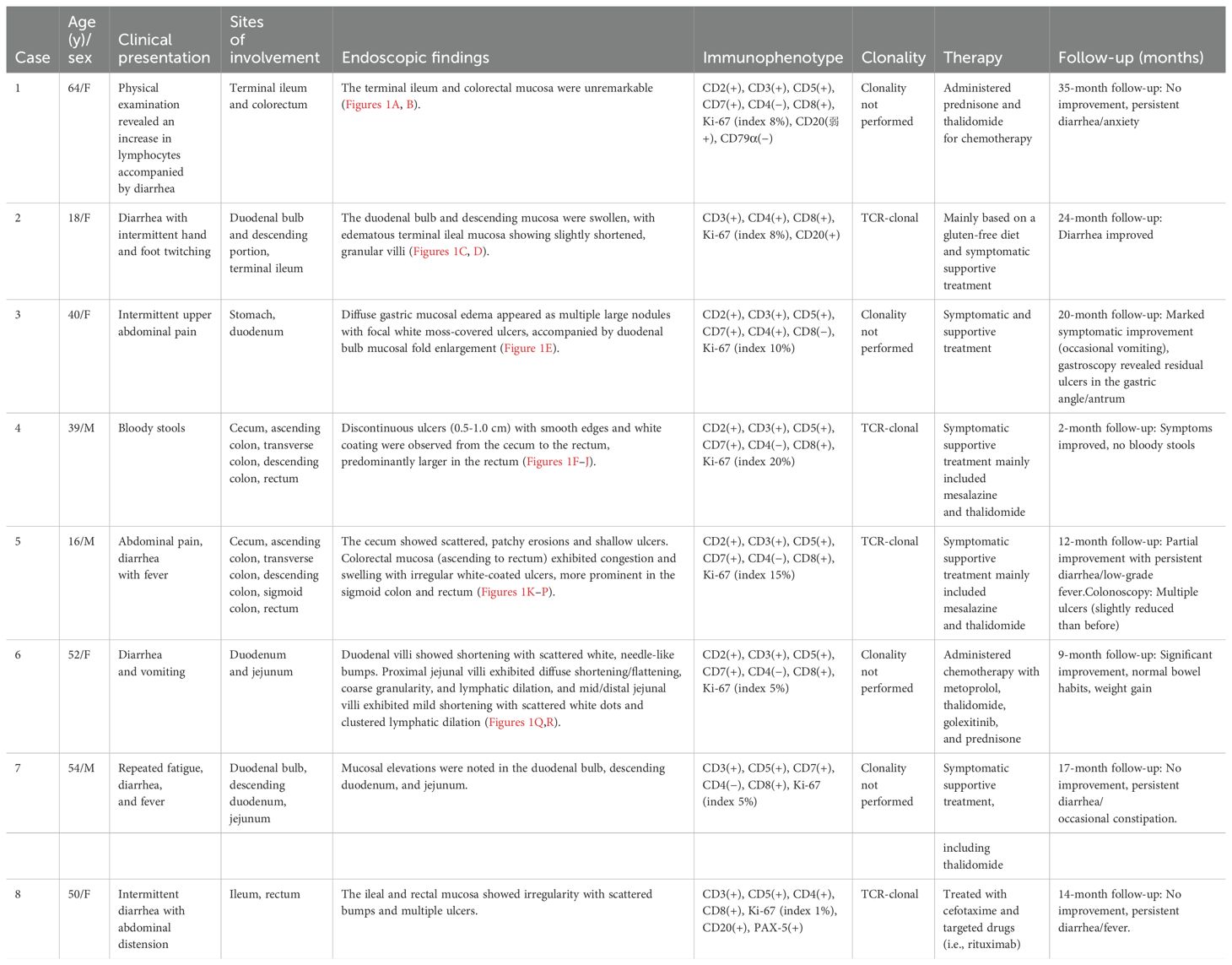
Table 1. Clinical and pathological characteristics of eight cases of indolent T-cell lymphoproliferative disease of the gastrointestinal tract.
3.1 Clinical characteristics
The cohort comprised eight patients (five women, three men) with a mean age of 42 years (median: 45 years), demonstrating a female predominance (F:M ratio 1.7:1). All patients presented with chronic diarrhea and abdominal pain, while concurrent symptoms included fever (n=3), vomiting (n=2), and hematochezia (n=1). Notably, one patient was initially identified by laboratory findings of lymphocytosis (WBC 12-15×109/L, lymphocyte percentage 80%) before developing diarrhea.
3.2 Endoscopic findings
Endoscopic evaluations revealed erosive changes with multiple ulcers and mucosal hyperemia/edema (n=3); multiple small protrusions with diffuse villous shortening, flattening, and granular appearance (n=3); combined ulcerative and protrusive lesions (n=1); and normal endoscopic findings (n=1). All cases had multi-site gastrointestinal involvement, with six cases having small intestine involvement (n=6), including two cases with concurrent large intestine involvement and one with additional gastric involvement. Two cases had isolated large intestine involvement (Figure 1).
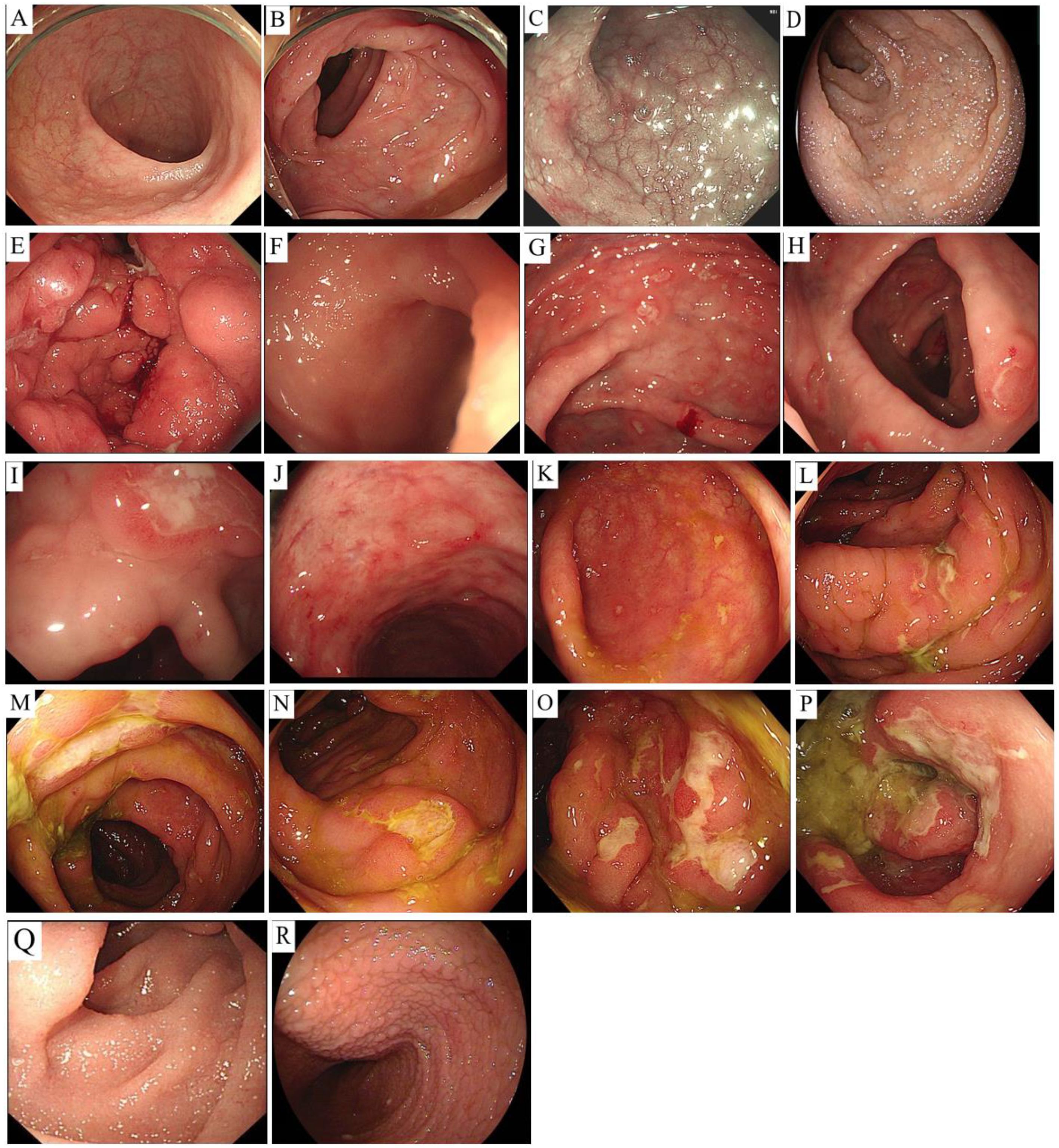
Figure 1. Endoscopic findings. Case 1: Terminal ileum and colorectal mucosa are unremarkable (A, B). Case 2: Duodenal bulb/descending mucosa swelling (C); terminal ileum edema with granular, shortened villi (D). Case 3: Diffuse gastric edema with nodular changes and focal white-coated ulcers (E). Case 4: Non-continuous ulcers were observed in the cecum (F), ascending colon (G), transverse colon (H), descending colon (I), and rectum (J), with smooth edges and a white coating, ranging in size from 0.5-1.0 cm. Larger lesions were more common in the rectum (F-J). Case 5: Scattered patchy erosions and shallow ulcers were observed in the cecum (K), while the mucosa of the ascending colon (L), transverse colon (M), descending colon (N), sigmoid colon (O), and rectum (P) were congested and swollen. Multiple irregular, patchy ulcers were scattered and covered with a white coating, with larger and more obvious ulcers in the sigmoid colon and rectum (K–P). Case 6: Duodenal villi were shortened with white and white needle-like protrusions (Q); proximal jejunal villi were diffusely flattened/granular with lymphatic dilation, and mid/distal jejunal villi were mildly shortened with white dots (R).
3.3 Treatment outcomes
Therapeutic interventions yielded variable responses:
1. Supportive care group (n=5; dietary modification, immunomodulation, anti-infectives): This treatment led to partial symptom improvement with residual diarrhea/vomiting/fever in four cases and complete resolution in one.
2. Targeted therapy (n=1; CD20+ case with rituximab): This treatment led to no clinical improvement after 14 months of follow-up (persistent diarrhea/fever).
3. Chemotherapy group (n=2; prednisone + thalidomide): This treatment led to significant improvement at 9 months of follow-up in one case and treatment failure at 35 months of follow-up (ongoing diarrhea/anxiety) in another.
3.4 Histopathological features
Gross examination: All specimens consisted of 2–5 endoscopic biopsy fragments (diameter: 2–3 mm).
Microscopic findings: All eight cases exhibited identical histological features. Glandular architecture: Atrophy of gastric/intestinal mucosal glands. Infiltrate pattern: Dense, monotonous lymphoid infiltration confined to the lamina propria (Figures 2A, B). Cytomorphology: Small lymphocytes with scant cytoplasm; round to mildly irregular nuclei with condensed chromatin; inconspicuous nucleoli; rare mitotic figures or apoptosis. Key negative findings: No angioinvasion, necrosis, or lymphoepithelial lesions; infiltration strictly limited to the mucosal layer (Figures 2C, D). Additional observations: Mild scattered plasma cells and eosinophils in the superficial mucosa (six cases); prominent plasma cell infiltration in the superficial mucosa (two cases) (Figure 3A).
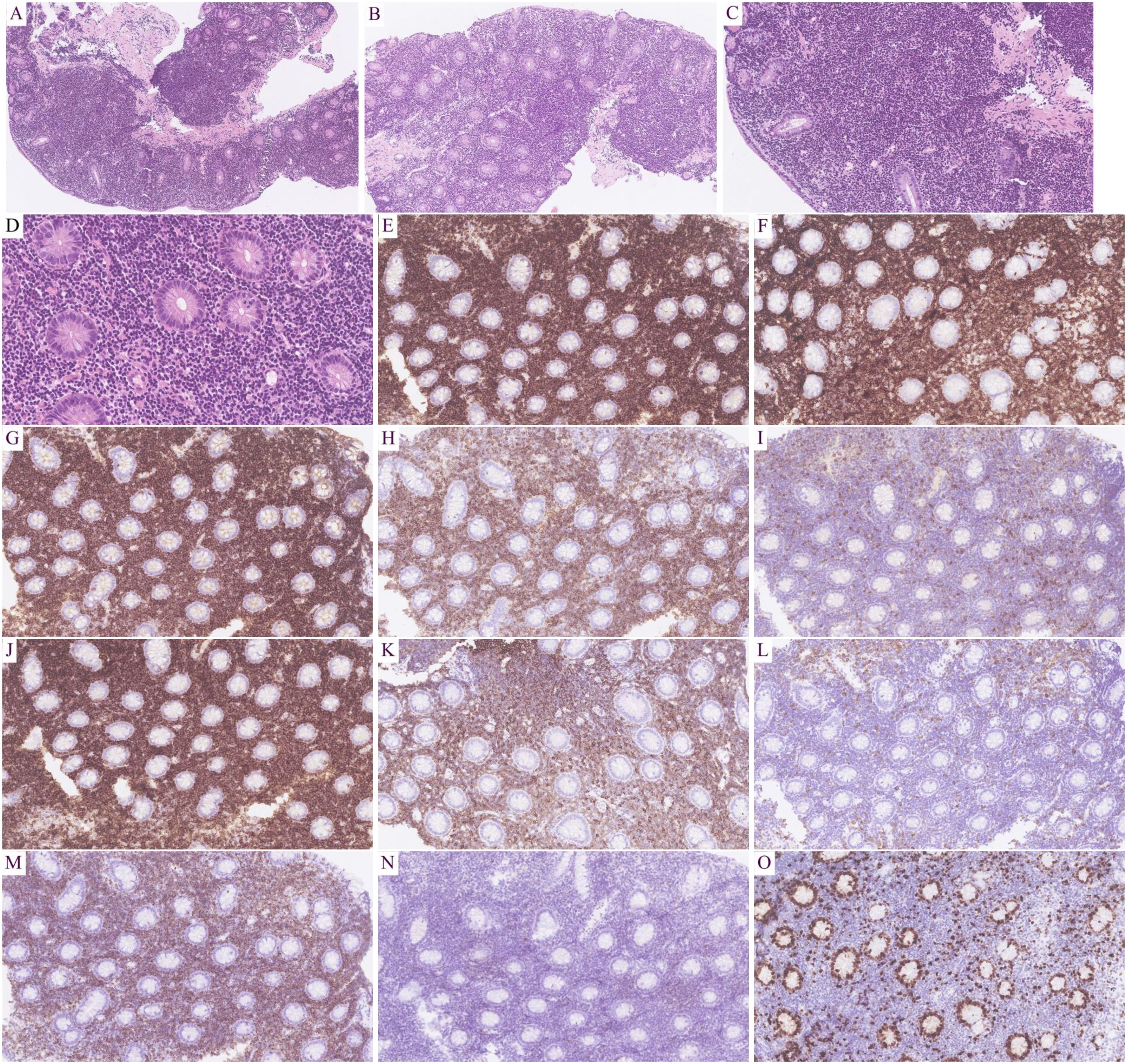
Figure 2. Results of hematoxylin-eosin (HE) staining and immunohistochemistry (IHC) assays. HE: Intestinal gland atrophy with diffuse lymphoid infiltration of the lamina propria (A, B; 100×).Uniform small cells: scant cytoplasm, round/irregular nuclei, dense chromatin, inconspicuous nucleoli, rare mitosis (C, 200×; D, 400×).IHC: T-cell markers: CD2+(E; 200×), CD3+(F; 200×), CD5+(G; 200×), CD7+(H; 200×); Subset: CD4−(I; 200×), CD8+ (J; 200×); Aberrant: CD20+ (K; 200×), CD79α− (L; 200×); Cytotoxic: TIA1+ (M; 200×), Granzyme B− (N; 200×); Proliferation: Ki-67 ~10% (O; 200×).
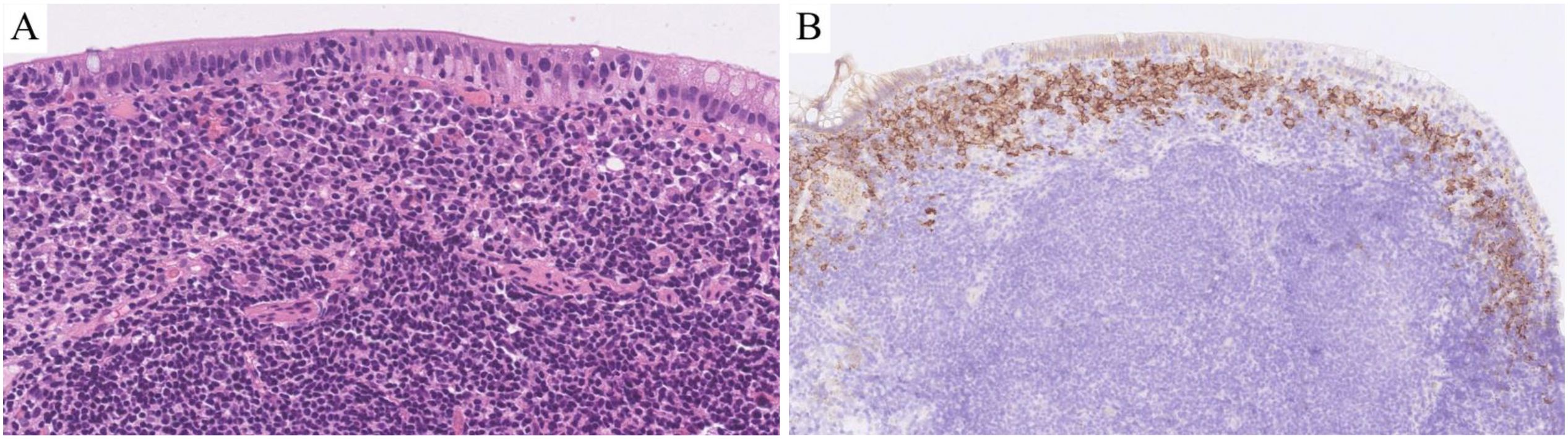
Figure 3. Results of hematoxylin-eosin (HE) staining and immunohistochemistry (IHC) assays. HE: There was a large amount of plasma cell infiltration in the superficial mucosa (A; HE 400×).IHC: There was a large amount of CD138-positive plasma cell infiltration in the superficial mucosa (B; IHC 200×).
3.5 Immunophenotypic and TCR gene rearrangement characteristics
T-cell markers: Pan-T markers [CD2(Figure 2E), CD3 (Figure 2F), CD5 (Figure 2G), CD7 (Figure 2H)] were positive in five cases. One case with limited panel: CD3+ only. Two consultation cases: Case 1: CD3+/CD5+/CD7+; Case 2: CD3+/CD5+. CD4/CD8 subsets: CD4-/CD8+ (five cases, Figures 2I, J): TIA1+ (Figure 2M) /Granzyme B- (Figure 2N) (three cases), TIA1+/Granzyme B+ (one case), not tested (one case); CD4+/CD8+ (two cases): TIA1+/Granzyme B- (one case), not tested (one case); CD4+/CD8- (one case, not tested). Aberrant markers: CD20+ (two cases, Figure 2K), one co-expressed PAX5 (but CD79α-, Figure 2L). CD56- in all cases. Proliferation index: Low Ki-67 (<15%, Figure 2O). Plasma cell infiltration: CD138+ plasma cell clusters in the superficial mucosa (two cases, Figure 3B). TCR gene rearrangement: Monoclonal TCR rearrangement confirmed in 4/4 tested cases; four cases not tested (see Table 1).
4 Discussion and conclusion
4.1 Clinical characteristics
ITLPD-GI remains exceptionally rare, with less than 80 cases reported in the literature as of 2021, and a total of less than 90 documented cases (4–9). Our cohort of eight patients (F:M ratio 1.7:1, mean age 42 years) showed a slight female predominance compared to previous reports (M:F ≈1.5:1) (4), which may reflect the limited sample size.
4.2 Etiology and pathogenesis
The exact etiology is unknown, but persistent antigen stimulation—potentially driven by immune-mediated or infectious conditions (e.g., IBD, viral infections, autoimmune disorders, or Helicobacter pylori infection)—is hypothesized to play a role (1–3). In our series, three cases arose from long-standing inflammatory bowel disease (IBD; Crohn’s disease/ulcerative colitis), supporting the theory of chronic immune stimulation leading to monoclonal T-cell proliferation (9). Notably, TNF-α inhibitors have been implicated in triggering ITLPD-GI, with documented regression upon discontinuation (10).
4.3 Clinical manifestations
Typical symptoms: Diarrhea, abdominal pain, fever, vomiting, and hematochezia, consistent with previous reports (4–9).
Novel findings: One case presented with peripheral lymphocytosis (WBC 12–15×109/L, LY% 80%), possibly linked to sustained antigen exposure, although further studies are needed.
Other rare manifestations: Our cohort included tetany while the literature also describes refractory oral ulcers, anal fistulae (8, 9, 11), metabolic disturbances (hypocalcemia/hypokalemia/hypomagnesemia/hypozincemia), and neurological symptoms (paresthesia, confusion) (1). Asymptomatic cases are occasionally detected incidentally during endoscopy or lymph node biopsy (12).
4.4 Disease distribution
Gastrointestinal involvement: All cases exhibited multi-site involvement (six cases: small intestine ± large intestine/stomach; two cases: large intestine only), aligning with published data (4–9, 13). Lesions predominantly affected the small bowel/colon, with sporadic gastric/esophageal involvement, typically showing discontinuous focal distribution.
Extraintestinal spread: In patients with this condition, rare cases involve the liver, bone marrow, peripheral blood, or lungs (4, 6, 9, 14, 15), with isolated reports of vitreoretinal infiltration (16).
4.5 Endoscopic features
None of the eight cases displayed pathognomonic endoscopic findings. Observations included erosions, shallow ulcers, mucosal hyperemia/edema, and villous shortening (one case appeared normal), mimicking IBD. Notably, mass formation or perforation is uncommon (4–9, 13).
4.6 Pathological features
4.6.1 Histomorphology
The diagnosis of ITLPD-G relies on histopathological examination, as both clinical and endoscopic findings lack specificity.
Characteristic features: Mucosal glandular atrophy with dense, monotonous infiltration of small lymphocytes in the lamina propria. Cytomorphology: Round to mildly irregular nuclei, condensed chromatin, inconspicuous nucleoli. Low proliferative activity: Rare apoptosis or mitotic figures. Absence of angioinvasion, necrosis, or lymphoepithelial lesions. Superficial epithelium may show erosions.
Depth of infiltration: Typically confined to the lamina propria, with occasional extension to the muscularis mucosae/submucosa. Transmural involvement is exceedingly rare (only 1 reported case by Huang et al. (7)).
Microenvironment: Six of eight cases exhibited mild plasma cell and eosinophil infiltration in the superficial mucosa. Two of eight cases showed prominent plasma cell infiltration. This finding may serve as a diagnostic clue or prognostic indicator, although larger studies are needed for validation. Lucioni et al. (17) reported that dense superficial plasma cell infiltration in ITLPD-G may help differentiate it from other GI T-cell lymphomas, suggesting microenvironmental immune cells (e.g., plasma cells) as potential diagnostic markers.
4.6.2 Immunophenotypic and molecular characteristics
T-cell markers: The majority of cases express pan-T markers (CD2/CD3/CD5/CD7), though partial loss may occur. Predominant subsets: CD4+/CD8- or CD4-/CD8+, with fewer CD4+/CD8+ or CD4-/CD8- cases (4, 9, 12, 13, 15). Our cohort: five cases expressed full T-cell markers. CD4-/CD8+ subset (n=5): three cases TIA1+/Granzyme B-; one case TIA1+/Granzyme B+; one case not tested. CD4+/CD8- (n=1): Not tested.CD4+/CD8+ (n=2): one case TIA1+/Granzyme B-; one case not tested. These results align with previous reports, wherein CD4−/CD8+ cases typically express TIA-1 but rarely Granzyme B, while CD4+/CD8− cases are usually negative for both markers (9, 18). There is a consistently low Ki-67 index.
Aberrant CD20 expression: Two cases demonstrated CD20 positivity, including one case with concurrent PAX5 expression. This is a potential diagnostic pitfall that may lead to misclassification as B-cell lymphoma. In such scenarios, TCR gene rearrangement analysis serves as a crucial confirmatory test. The underlying mechanism may involve either neoplastic transformation of specific T-cell subsets or aberrant activation-induced phenotypic alterations in T lymphocytes (14, 19).
Molecular findings: Monoclonal TCR rearrangement was diagnostic, as 4/4 tested cases were positive (including one CD20+ case); four cases were not tested.
T-helper cell polarization: CD4+ or CD4+/CD8+ cases may show Th1/Th2 hybrid phenotypes; CD8+ or CD4-/CD8- cases often exhibit Th2 polarization, and some demonstrate lineage plasticity (Soderquist et al. (12)).
4.7 Molecular genetic characteristics
4.7.1 Key findings
Approximately 80% of ITLPD-GI cases harbor pathogenic or likely pathogenic genetic alterations (12), predominantly affecting the following pathways.
4.7.1.1 JAK-STAT pathway abnormalities
STAT3-JAK2 fusions were highly prevalent in CD4+ cases (4/5 cases, Sharma et al. (20)) and may drive oncogenesis through STAT5 phosphorylation (Hu et al. (21)). STAT family phosphorylation was common in CD4+/CD8+ and double-negative cases (STAT1/2/5 phosphorylation; Soderquist et al. (12)).
4.7.1.2 Epigenetic regulator mutations
There were recurrent mutations in TET2, DNMT3A, and KMT2D (4/10 cases (12)) and potential early events in lymphomagenesis (22). Co-occurring alterations included CDKN2A missense mutations and TNFAIP3 nonsense mutations (12).
4.7.1.3 CD8+ case signature
In total, 50% of cases exhibit chromosomal structural alterations in the IL-2 gene 3’UTR region (12). IDH1/2 or SETD2 mutations were notably absent (12, 23).
4.7.1.4 Transformation-associated mutations
TP53 and POLE mutations were observed in cases progressing to aggressive lymphoma (12).
4.8 Differential diagnoses of ITLPD-GI
4.8.1 Diagnostic challenges
ITLPD-GI poses significant diagnostic difficulties due to its rarity and non-specific clinical manifestations, frequently leading to misdiagnosis as IBD or aggressive T-cell lymphomas. Accurate diagnosis requires a comprehensive evaluation incorporating histopathology, immunohistochemistry, and molecular testing (Table 2).
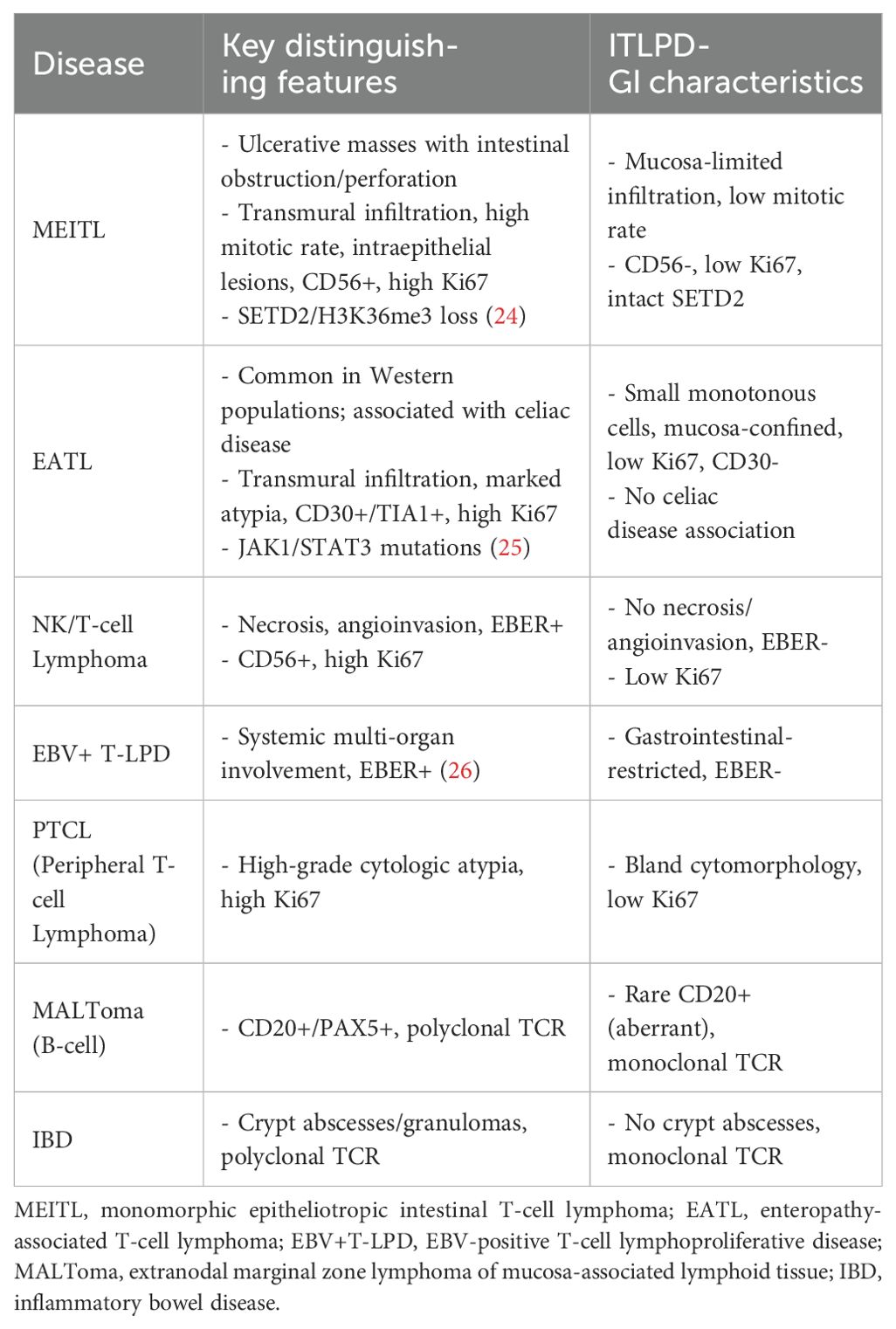
Table 2. Key differential diagnoses and distinguishing features.
4.8.2 Key diagnostic considerations
4.8.2.1 Endoscopic/histopathological features
ITLPD-GI: Superficial mucosal involvement without deep ulceration/perforation; small, monotonous lymphocytes with bland cytomorphology; rare mitotic figures (<5/10 HPF); absence of necrosis/angioinvasion; low proliferative index (Ki67 <15%).
Aggressive lymphomas: Transmural infiltration; marked cytologic atypia; frequent mitoses (>10/10 HPF); common necrosis/angiodestruction; high Ki67 (>50%).
4.8.2.2 Molecular diagnostics
Monoclonal TCR rearrangement serves as the diagnostic gold standard and is particularly crucial for atypical presentations (e.g., CD20+ cases).
4.8.2.3 Clinical caveats
Diagnostic pitfalls in our series included two cases that were initially misdiagnosed (as MALToma and EATL/MEITL). These were corrected through repeat biopsy with TCR clonality assessment. Thus, we recommend maintaining high suspicion for ITLPD-GI in cases with chronic GI symptoms refractory to IBD therapy, with discordant histological and immunophenotypic findings.
4.9 Treatment and prognosis
Currently, there is no consensus on the treatment of ITLPD-GI, with “watchful waiting” being the primary recommended strategy (4, 9). Conventional chemotherapy typically shows limited efficacy (4), although some alternative therapies have demonstrated certain therapeutic effects. Low-dose radiotherapy (30 Gy/20 fractions) successfully treated a gastric case with 1-year recurrence-free survival (27). Furthermore, corticosteroids or anti-CD52 monoclonal antibodies can improve symptoms (3, 5). Metronomic chemotherapy (prednisone + cyclophosphamide + thalidomide) achieved 1-year clinical remission in patients with severe symptoms (5).
In our case series, of the two patients treated with prednisone plus thalidomide, one showed improvement after 9 months, while the other still experienced diarrhea and anxiety after 35 months, suggesting the need for psychological intervention. Of the five patients receiving symptomatic treatment, four showed varying degrees of symptom improvement (although with residual diarrhea, vomiting, or fever), while one achieved complete resolution. Notably, one CD20-positive patient with ITLPD-GI showed no response to rituximab after 14 months of treatment, indicating that its efficacy in T-cell lymphomas requires further validation (19).
Although ITLPD-GI typically follows an indolent course (median survival >10 years) (4), there is a risk of progression. ITLPD-GI can transform into aggressive T-cell lymphomas (e.g., ALCL), often with hepatic/bone marrow involvement and an extremely poor prognosis (2, 11, 20). The transformation mechanism may correlate with the CD4-positive phenotype (accounting for 60% of progressive cases) or genetic mutations, while CD8-positive cases may have a better prognosis (11, 13, 20). Additionally, ITLPD-GI may coexist with other lymphomas (e.g., diffuse large B-cell lymphoma, Hodgkin’s lymphoma) (28, 29) or autoimmune diseases (this series is the first to report co-occurrence with Castleman disease and celiac disease, the latter improved with a gluten-free diet). Notably, one case initially misdiagnosed as EATL showed a transient response to a PD-1 inhibitor before progressing to confirmed ITLPD-GI, highlighting the diagnostic complexity. However, this study has limitations. Given the rarity of ITLPD-GI, only eight cases were included. Future research should expand the cohort and investigate molecular mechanisms to refine treatment strategies.
In conclusion, ITLPD-GI is a rare indolent monoclonal T-cell proliferative disorder with non-specific clinical and endoscopic manifestations, requiring differentiation from aggressive T-cell lymphomas (e.g., MEITL, EATL) and IBD to avoid misdiagnosis and unnecessary treatment. Diagnosis primarily relies on histopathology (mucosa-limited small lymphocyte infiltration) and immunohistochemistry, while TCR gene monoclonal rearrangement testing is crucial for confirmation in cases with atypical immunophenotypes (e.g., aberrant CD20 expression). The disease typically follows an indolent course, potentially associated with chronic antigen stimulation (e.g., IBD, infection), with the majority of patients having a favorable prognosis, and “watchful waiting” being recommended. However, a minority of cases may progress to aggressive T-cell lymphoma with a significantly worse prognosis, although the exact transformation mechanisms remain unclear. Therefore, enhancing awareness among clinicians and pathologists is critical, and future research should further explore the molecular mechanisms to optimize treatment strategies.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by the Biomedical Research Ethics Committee of the Affiliated Hospital of Zunyi Medical University(approval number: KLL-2024-535).
Author contributions
DY: Conceptualization, Data curation, Formal Analysis, Writing – original draft, Writing – review & editing. NL: Data curation, Formal Analysis, Writing – review & editing. D-YW: Data curation, Methodology, Writing – review & editing. J-JW: Writing – review & editing. C-WJ: Conceptualization, Data curation, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Science and Technology Plan Project of Zunyi City, Zunshi Kehe HZ (2023) 234, the Guizhou Provincial Health Commission Science and Technology Fund Project for 2024 (Grant No. D596), and the Zunyi Medical University Affiliated Hospital Excellent Youth Talent Training Program Project (Grant No. rc220240423).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Perry AM, Warnke RA, Hu Q, Gaulard P, Copie-Bergman C, Alkan S, et al. Indolent T cell lymphoproliferative disease of the gastrointestinal tract. Blood. (2013) 122:3599–606. doi: 10.1182/blood-2013-07-512830
2. Margolskee E, Jobanputra V, Lewis SK, Alobeid B, Green PH, and Bhagat G. Indolent small intestinal CD4+ T cell lymphoma is a distinct entity with unique biologic and clinical features. PLoS One. (2013) 8:e68343. doi: 10.1371/journal.pone.0068343
3. Malamut G, Meresse B, Kaltenbach S, Derrieux C, Verkarre V, Macintyre E, et al. Small intestinal CD4+ T cell lymphoma is a heterogenous entity with common pathology features;. Clin Gastroenterol Hepatol. (2014) 12:599–608. doi: 10.1016/j.cgh.2013.11.028
4. Sanguedolce F, Zanelli M, Zizzo M, Luminari S, Martino G, Soriano A, et al. Indolent T cell lymphoproliferative disorders of the gastrointestinal tract (iTLPD-GI): A review. Cancers (Basel). (2021) 13:2790. doi: 10.3390/cancers13112790
5. Liu H, Cao L, Zhao X, Miao Y, Wu W, Shi X, et al. Metronomic chemotherapy for indolent T cell lymphoproliferative disorder of the gastrointestinal tract. Cancer Sci. (2023) 114:3793–6. doi: 10.1111/cas.15880
6. David R, Mishra K, Gilbert ER, Mirza KM, and Hendler S. Indolent T cell lymphoproliferative disease: A rare case of a benign lymphoma of the gastrointestinal tract with extra-gastrointestinal involvement. ACG Case Rep J. (2022) 9:e00879. doi: 10.14309/crj.0000000000000879
7. Huang WY, Zeng L, Liao SS, Zhang W, Liu FR, Li LX, et al. Indolent T cell lymphoproliferative disorder of the gastrointestinal tract with the whole wall involvement: report of a case. Zhonghua Bing Li Xue Za Zhi. (2022) 51:1051–3. doi: 10.3760/cma.j.cn112151-20220314-00178
8. Lu J, Song S, Hou W, Hou W, Shen M, Duan X, et al. Clinical and pathological characteristics analysis of gastrointestinal inert T cell lymphoid tissue proliferative diseases. Chin J Med. (2020) 100:2537–40. doi: 10.3760/cma.jcn112137-20191206-02669
9. Shao S, Gu H, Lin D, Shi H, Zhang Y, and Li Y. Clinical and pathological characteristics of five cases of gastrointestinal inert T cell lymphoid tissue proliferative diseases. Chin J Pathol. (2019) 48:762–6. doi: 10.3760/cma.jissen.0529-5807.2019.10.003
10. Edison N, Belhanes-Peled H, Eitan Y, Guthmann Y, Yeremenko Y, Raffeld M, et al. Indolent T cell lymphoproliferative disease of the gastrointestinal tract after treatment with adalimumab in resistant Crohn’s colitis. Hum Pathol. (2016) 57:45–50. doi: 10.1016/j.humpath.2016.06.021
11. Perry AM, Bailey NG, Bonnett M, Jaffe ES, and Chan WC. Disease progression in a patient with indolent T cell lymphoproliferative disease of the gastrointestinal tract. Int J Surg Pathol. (2019) 27:102–7. doi: 10.1177/1066896918785985
12. Soderquist CR, Patel N, Murty VV, Betman S, Aggarwal N, Young KH, et al. Genetic and phenotypic characterization of indolent T cell lymphoproliferative disorders of the gastrointestinal tract. Haematologica. (2020) 105:1895–906. doi: 10.3324/haematol.2019.230961
13. Soderquist CR and Bhagat G. Gastrointestinal T- and NK-cell lymphomas and indolent lymphoproliferative disorders. Semin Diagn Pathol. (2020) 37:11–23. doi: 10.1053/j.semdp.2019.08.001
14. Wang X, Ng CS, Chen C, Yu G, and Yin W. An unusual case report of indolent T cell lymphoproliferative disorder with aberrant CD20 expression involving the gastrointestinal tract and bone marrow. Diagn Pathol. (2018) 13:82. doi: 10.1186/s13000-018-0762-4
15. Lewis SK, Green PH, Hsiao S, Mansukhani MM, Hsi ED, de Leval L, et al. Genetic and phenotypic characterization of indolent T cell lymphoproliferative disorders of the gastrointestinal tract. Haematologica. (2020) 105:1895–906. doi: 10.3324/haematol.2019.230961
16. Salman AR, Ferenchak K, Olsen TW, Smith WM, Ketterling RP, McPhail ED, et al. Vitreoretinal involvement by indolent t cell lymphoproliferative disorder of the gastrointestinal tract diagnosed by fluorescence in situ hybridization. Retin cases Brief Rep. (2023) 17:572–6. doi: 10.1097/ICB.0000000000001263
17. Lucioni M, Fraticelli S, Santacroce G, Bonometti A, Aronico N, Sciarra R, et al. Clinical and histopathological features of an italian monocentric series of primary small bowel T-cell lymphomas. Cancers (Basel). (2023) 15:2743. doi: 10.3390/cancers15102743
18. Li W, Zhu Z, Chen Z, Zhao P, Li X, and Zhang G. Research progress on gastrointestinal inert T cell lymphoid tissue proliferative diseases. Chin J Gastroenterol. (2022) 42:418–21. doi: 10.3760/cma.j.cn311367-20211114-00621
19. Li YL, Wang HP, Zhang C, and Zhai ZM. CD20-positive primary nasal peripheral T cell lymphoma: An analysis of one case and review of the literature. Cytom B Clin Cytom. (2020) 98:348–54. doi: 10.1002/cyto.b.21852
20. Sharma A, Oishi N, Boddicker RL, Hu G, Benson HK, Ketterling RP, et al. Recurrent STAT3-JAK2 fusions in indolent T cell lymphoproliferative disorder of the gastrointestinal tract. Blood. (2018) 131:2262–6. doi: 10.1182/blood-2018-01-830968
21. Hu G, Phillips JL, Dasari S, Jacobs HK, Luchtel RA, Oishi N, et al. Targetability of STAT3-JAK2 fusions: implications for T cell lymphoproliferative disorders of the gastrointestinal tract. Leukemia. (2020) 34:1467–71. doi: 10.1038/s41375-019-0678-3
22. Van Arnam JS, Lim MS, and Elenitoba-Johnson KSJ. Novel insights into the pathogenesis of T cell lymphomas. Blood. (2018) 131:2320–30. doi: 10.1182/blood-2017-11-764357
23. Moffitt AB, Ondrejka SL, McKinney M, Rempel RE, Goodlad JR, Teh CH, et al. Enteropathy-associated T cell lymphoma subtypes are characterized by loss of function of SETD2. J Exp Med. (2017) 214:1371–86. doi: 10.1084/jem.20160894
24. KiiQiik C, Wei L, and You H. Indolent T cell lymphoproliferative disease of the GI tract: insights for better diagnosis, prognosis, and appropriate therapy. Front Oncol. (2020) 10:1276. doi: 10.3389/fonc.2020.01276
25. Nicolae A, Xi L, Pham TH, Pham TA, Navarro W, Meeker HG, et al. Mutations in the JAK/STAT and RAS signaling pathways are common in intestinal T cell lymphomas. Leukemia. (2016) 30:2245–7. doi: 10.1038/leu.2016.178
26. Wang Y, Li Y, Meng X, Duan X, Wang M, Chen W, et al. Epstein-barr virus-associated T cell lymphoproliferative disorder presenting as chronic diarrhea and intestinal bleeding: a case report. Front Immunol. (2018) 9:2583. doi: 10.3389/fimmu.2018.02583
27. Takahashi N, Tsukasaki K, Kohri M, Akuzawa Y, Saeki T, Okamura D, et al. Indolent T cell lymphoproliferative disorder of the stomach successfully treated by radiotherapy. J Clin Exp Hematop. (2020) 60:7–10. doi: 10.3960/jslrt.19022
28. Guo L, Wen Z, Su X, Xiao S, and Wang Y. Indolent T cell lymphoproliferative disease with synchronous diffuse large B-cell lymphoma: A case report. Med (Baltimore). (2019) 98:e15323. doi: 10.1097/MD.0000000000015323
Keywords: Indolent T-cell lymphoproliferative disease of the gastrointestinal tract (ITLPD-GT), clinical and pathological features, abnormal positive expression of CD20, TCR monoclonal rearrangement, treatment
Citation: Yuan D, Liang N, Wang D-Y, Wang J-J and Jia C-W (2025) Clinical and pathological analysis of indolent T-cell lymphoproliferative disease of the gastrointestinal tract. Front. Immunol. 16:1530149. doi: 10.3389/fimmu.2025.1530149
Received: 26 November 2024; Accepted: 30 April 2025;
Published: 21 May 2025.
Edited by:
David Robert Kroeger, Zymeworks Inc., CanadaReviewed by:
Arturo Bonometti, Humanitas University, ItalyVijaya Lakshmi Muram Reddy, Narayana Medical College and Hospital, India
Copyright © 2025 Yuan, Liang, Wang, Wang and Jia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cong-Wei Jia, ZGF2aWRfamlhMDgxNEAxNjMuY29t