 Haiyan Xu
Haiyan Xu Ying Yang2†
Ying Yang2† PingLi Wang
PingLi Wang Zhiyong Xu
Zhiyong Xu- 1Department of Biobank, The Second Affiliated Hospital of Zhejiang University School of Medicine, Hangzhou, China
- 2Department of Respiratory and Critical Care Medicine, The Fourth Affiliated Hospital of Zhejiang University School of Medicine, Yiwu, China
- 3Department of Respiratory and Critical Care Medicine, The Second Affiliated Hospital of Zhejiang University School of Medicine, Hangzhou, China
- 4Breast Cancer Center, Zhejiang Cancer Hospital, Hangzhou Institute of Medicine, Chinese Academy of Sciences, Hangzhou, China
Adenocarcinoma-to-squamous cell carcinoma transformation (AST) induces drug resistance in patients with lung adenocarcinoma (LUAD), often resulting in unfavorable clinical outcomes. In recent years, it has been found that alterations in the tumor immune microenvironment (TIME) during adenosquamous carcinoma trans-differentiation also influence the efficacy of immunotherapy. Moreover, the aberrant expression and activation of several driver genes for AST lead to abnormal infiltration and function of immune cell by remodeling the cellular inflammatory phenotype. In this review, we will systematically present the changes in the TIME and molecular regulatory mechanisms during adenosquamous carcinoma differentiation, aiming to gain a better understand of the function of immune cells during this process and the potential value of combining immunotherapy to enhance the treatment of non-small cell lung cancer (NSCLC).
1 Introduction
Non-small cell lung cancer (NSCLC) accounts for approximately 85% of all lung cancers, with lung adenocarcinoma (LUAD) and squamous cell carcinoma (LUSC) being the two most common subtypes. The phenomenon of adenocarcinoma-to-squamous cell carcinoma transformation (AST) has gained attention due to its association with drug resistance and poor prognosis (1, 2). Notably, lung adenosquamous carcinoma (LUAS), a rare but aggressive biphasic subtype of NSCLC, accounts for 0.7%–11.4% of all NSCLC cases and uniquely exhibits geographically distinct glandular and squamous components (each ≥10% of tumor volume), representing a transitional entity bridging LUAD and LUSC (3). Despite significant advances in understanding cancer biology, the underlying mechanisms driving AST and its impact on immunotherapy remain largely unexplored (1, 2).
Lineage plasticity describes the phenomenon where cancer cells undergo dynamic transformation of their phenotypes during the onset and progression of cancer (2). Through lineage plasticity, tumor cells can shift to different histological subtypes, thereby increasing the heterogeneity of tumors (1) and facilitating immune escape, allowing adaptation to the tumor microenvironment (TME) post-treatment. In clinical practice, following treatment with epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs) or KRAS glycine-to-cysteine substitution at codon 12 (G12C) inhibitors (adagrasib and sotorasib), the transformation of lung AST is frequently observed (4–6). Similarly, LUAD patients also exhibit transformation of lung AST after receiving immunotherapy and chemotherapy (Table 1).
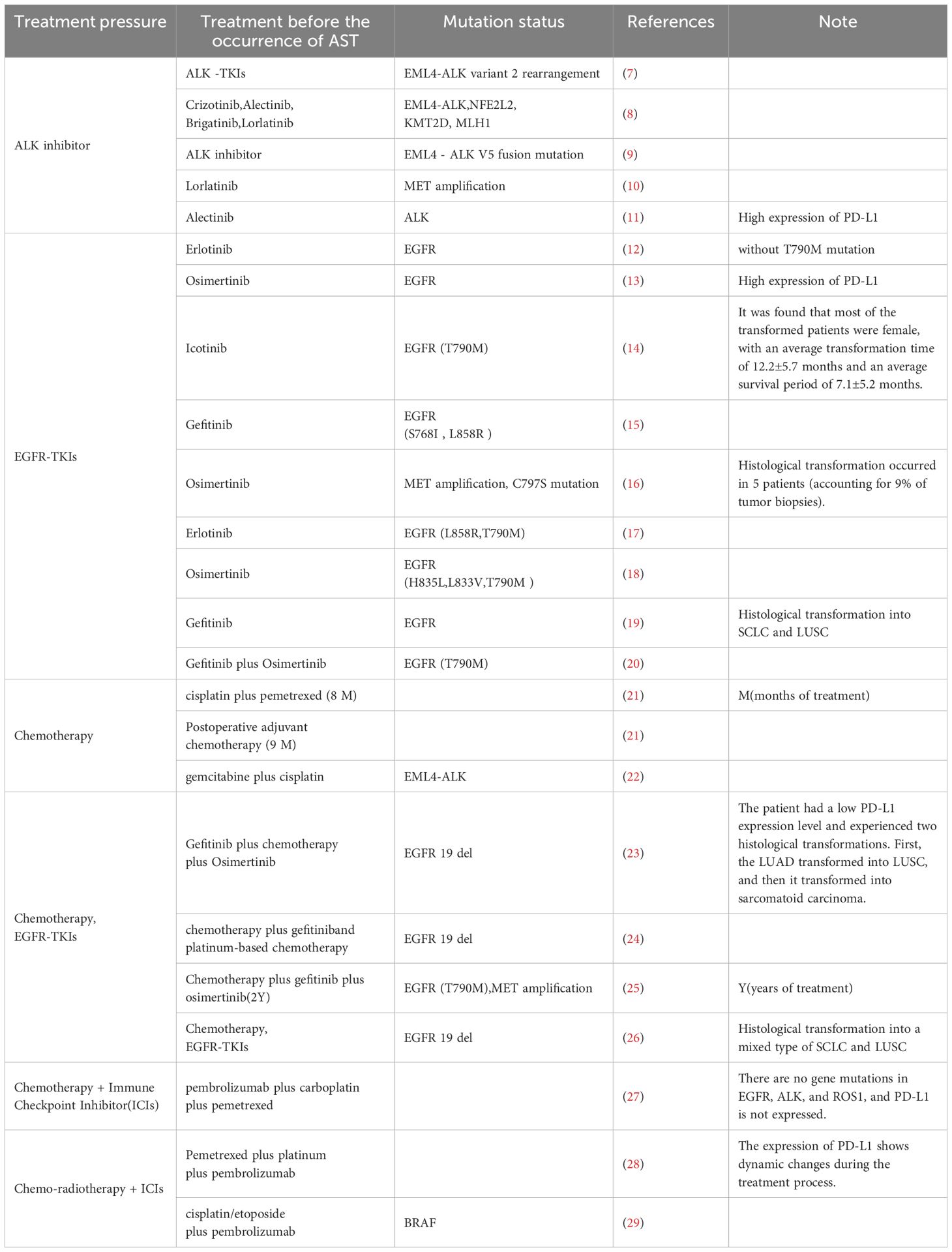
Table 1. Clinical cases of AST following different treatment regimens.
Histologic transformation, such as AST, is a critical resistance mechanism in LUAD. Epidemiologically, AST occurs in up to 9% of EGFR-mutant patients relapsing on osimertinib and contributes to markedly poor prognosis (median survival <6 months post-relapse) (30). Similarly, KRAS G12C-mutant LUAD (13% of all LUAD) treated with adagrasib exhibits AST as a resistance pathway alongside secondary KRAS mutations and RTK-RAS reactivation (5). These transformations are characterized by lineage marker switching (e.g., TTF-1 loss, p63 gain) and activation of pro-squamous transcriptional programs, rendering tumors refractory to lineage-specific therapies. This underscores the need for early detection and adaptive therapeutic strategies, and this transformation is closely linked to immune responses. Interestingly, a study described a 69-year-old never-smoking male NSCLC patient with EGFR/ALK wild-type adenocarcinoma, who developed sequential histological transformations—adenocarcinoma with sarcomatoid change, squamous cell carcinoma with sarcomatoid change, and pure squamous cell carcinoma—during chemotherapy, radiotherapy, and pembrolizumab treatment. PD-L1 expression shifted from positive to negative, highlighting dynamic phenotypic evolution under therapeutic pressure (28). These findings suggest that AST is a key mechanism underlying drug resistance in LUAD treatment.
Recent studies have begun to unravel how AST influences the immune landscape of NSCLC (13), highlighting the role of immune checkpoint inhibitors in modulating the inflammatory response during trans-differentiation. The trans-differentiation from adenocarcinoma to squamous cell carcinoma is not unique to lung cancer but is also prevalent in other organs (31–37). There is a significant correlation between AST triggered by the inactivation of Liver kinase B1 (LKB1), also known as serine-threonine kinase 11 (STK 11), and targeted-therapy resistance (38). It is widely acknowledged that lineage plasticity and immune escape are common mechanisms resulting to acquired drug resistance and subsequent treatment failure.
As two different subtypes of NSCLC, LUAD and LUSC differ in histopathological features, gene expression profiles, and responses to drug treatments (39, 40). Pathological confirmation of LUAS requires strict WHO criteria: (1) morphologically distinct glandular/squamous components via hematoxylin-eosin staining; (2) immunohistochemical validation (e.g., TTF-1/napsin A for adenocarcinoma, p40/p63 for squamous carcinoma); and (3) spatial segregation of components (each ≥10% tumor volume). While small biopsy samples pose diagnostic challenges due to sampling bias, identification of biphenotypic differentiation should prompt molecular or surgical validation (3). Interestingly, approximately 4% to 9% of human NSCLC tumors contain mixed adenomatous and squamous pathologies in a single lesion, clinically termed adenosquamous lung carcinoma (ASLC) (41, 42). While LUAD and LUSC share certain genomic alterations (e.g., TP53 mutations), their overall somatic single-nucleotide variant (SNV) and insertion/deletion (InDel) landscapes exhibit significant divergence, particularly in driver oncogenes and tumor-specific pathways (43). Recent studies have shown that during the AST, an immunosuppressive state is observed in either LUAD or LUSC, whereas their transformed intermediate state exhibits an inflammatory state characterized by increased immune infiltration (3). This implies that the TIME undergoes significant changes during AST, and the understanding of the relevant regulatory mechanisms can help guide the combined application of immunotherapy in clinical practice.
Moreover, common genomic alterations include TP53 and EGFR mutations, as well as gene deletions in the 9q21 chromosomal region, which strongly indicate a common clonal origin for both subtypes (30). This phenomenon implies that there is a potential phenotypic transformation in NSCLC, fully demonstrating that NSCLC has strong cancer plasticity (this plasticity is not limited to lineage plasticity). Additionally, Patients with ASLC usually have poor treatment effects and a poor prognosis (44).
Moving forward, we will delve into the intricacies of the TIME and the molecular mechanisms that drive adenosquamous carcinoma differentiation. We aim to elucidate the role of immune cells in this transformative process and to explore the potential benefits of integrating immunotherapy to improve treatment outcomes for patients with NSCLC.
2 Immunotherapy challenges in AST: genetic and immunological factors
KRAS stands out as one of the most prevalent mutated genes in lung cancer (45). Among these mutations, KRAS G12C inhibitors, such as adagrasib and sotorasib, have presented certain clinical efficacy in the treatment of KRAS G12C-mutated lung cancer (46). However, most patients will eventually develop drug resistance during subsequent treatment. For individuals harboring STK11/LKB1 mutations, tumors exhibiting high-expression of LUSC gene features often respond poorly to adagrasib treatment. STK11/LKB1 mutations may facilitate the occurrence of AST in tumors by inducing epigenetic plasticity, thereby contributing to resistance to KRAS inhibitors (47). Whole-genome sequencing (WGS) results show that in mixed-histology tumors, LUAD and LUSC have a common clonal origin. In addition, changes in TBX3, MET, RBM10, etc. may be related to trans-differentiation, thus affecting the occurrence of AST (30). It’s worth noting that adenocarcinoma subclones might initially emerge and subsequently undergo trans-differentiation into squamous lesions (3). The main driving factor behind the transformation from LUAD to LUSC appears to be transcriptional reprogramming rather than mutational events. During this transformation process, genes related to the PI3K/AKT, MYC, and PRC2 pathways are consistently up-regulated (30). Moreover, the combined activation of PI3K/AKT and MYC can induce squamous features in pre-clinical models of EGFR-mutated LUAD, including mouse models and patient-derived xenograft (PDX) models. Additionally, inhibition of EZH2 or the PI3K/AKT signaling pathway can restore sensitivity to osimertinib in those drug-resistant squamous-like tumors (30).
The Tang team performed WGS and RNA-seq on surgical specimens from 109 LUAS patients (71 males [65.1%], 38 females [34.9%]; median age: 62 years, range: 32–84). The cohort comprised 46.8% non-smokers (lower than Asian LUAD cohorts [58–62.8%]) and predominantly early-stage tumors (Stage I: 50.5%, II: 17.4%, III: 31.2%, IV: 0.9%). Mutational analysis revealed TP53 (59%) and EGFR (43%) as the most frequent alterations, with ALK fusions occurring in 8% of cases. This demographic and clinical profiling underscores the cohort’s relevance to LUAS biology in resectable disease contexts (3). This team identified several potential oncogenic drivers during the development of LUAS, including RAC1, ALK, and AKT1 (3). Through multidimensional scaling analysis of the RNA-seq data of LUAS and LUSC data, LUAS was categorized into three mRNA subtypes: terminal respiratory unit-like (TRU-like), inflammatory, and Basal-like. Among these, the inflammatory subtype features enhanced immune infiltration and serves as an intermediate stage of AST. This implies that cancer cells gradually progress from an adenocarcinoma state to an inflammatory state and ultimately evolve into a squamous state (3). Pathological analysis demonstrates that most tumors exhibit the classic histological pattern of LUAD, while some tumors possess squamous pathological features or mixed pathology. Over time, tumor development transitions from LUAD-dominant LUAS to LUSC-dominant LUAS and finally to typical LUSC, with LUSC tumors being larger in volume (41). The analysis of the mRNA subtype of LUAS identified a dysregulated upstream transcription factor (TFs) network centered around NKX2-1, FOXA2, SOX2, and TP63, which has a regulatory effect on the development of LUAS (3). Furthermore, the research of the Tong team further discovered that the dynamic dysregulation of lineage-specific TFs, including LUAD-related TFs (NKX2–1 and FOXA2) and LUSC-related TFs (ΔNp63 and SOX2), finely tunes the AST process (4). Notably, TTF1 + ΔNp63 + serves as an important marker for AST. The analysis of human ALK-rearranged lung cancer samples also found that some LUADs present squamous features, indicating a tendency towards squamous transformation (41). Clinical analysis revealed that the progression-free survival (PFS) of LUSC patients receiving ALK-TKI treatment is significantly shorter than that of LUAD patients. Moreover, the expression of squamous biomarkers was detected in biopsy samples of recurrent patients, which means that AST is associated with drug resistance (41). Besides, the gene set variation analysis (GSVA) scores of immune cells (such as neutrophils, T-cells, and B-cells) in the inflammatory subtype are significantly higher.
The above mechanisms, including KRAS gene mutations, genetic alterations in mixed-histology tumors, characteristic gene mutations in LUAS, dysregulation of lineage-specific transcription factor expression, and remodeling of the immune microenvironment, collectively induce resistance through a cascade of “genetic variations (e.g., STK11/LKB1, TP53, EGFR mutations)-transcriptional reprogramming (disruption of lineage-specific transcription factor networks and abnormal activation of PI3K/AKT and MYC pathways)-immune microenvironment remodeling (dysfunctional immune infiltration in the inflammatory subtype and the paradox of immune cell accumulation)”. This multi-dimensional resistance is manifested as: 1) intrinsic tumor cell resistance, where cancer cells directly reduce sensitivity to immune checkpoint inhibitors (ICI) through epigenetic plasticity and lineage transdifferentiation; 2) immune microenvironment-mediated resistance, where the “quality” of immune infiltration (e.g., enrichment of immunosuppressive cells or functional exhaustion of effector T cells), rather than just the “quantity” of immune cells, becomes a critical bottleneck for immune therapy response; and 3) unresolved cross-resistance mechanisms between targeted therapies (such as KRAS G12C inhibitors or EGFR tyrosine kinase inhibitors) and ICI, with their interactive roles during AST still requiring in-depth clarification. The convergence of these multiple resistance mechanisms constitutes the complex challenges facing immunotherapy for AST-related lung cancer.
3 TFs Networks and signaling pathway alterations in AST
3.1 Role of TFs in KRAS-driven ASLC
In KRAS-driven LUAD, the TFs networks of LUAD and LUSC can be visualized as a complex seesaw. These networks are intricately balanced, mutually inhibiting each other to maintain a delicate dynamic equilibrium. Disruption of this balance tilts the seesaw towards one side, leading to squamous cell differentiation when the influence of LUSC-related TFs (NKX2–1 and FOXA2) outweighs that of LUAD (p63 and SOX2), ultimately resulting in ASLC (3, 48)
Knockout of NKX2–1 and FOXA2 can drive the occurrence of LUAS, and SOX2 plays a key role in driving squamous trans-differentiation (3). DNp63, the predominant p63 transcript, is overexpressed in tumors compared with normal tissues (49). Through organoid culture of LUAD from KRASLSL-G12D/+/LKB1flox/flox (KL) mice, research has revealed that E74-like factor 5 (ELF5) is crucial for maintaining ADC lineage characteristics and maintaining sensitivity to KRAS inhibitors, while ΔNp63 promotes squamous transformation and resistance to KRAS inhibitors (4). Elf5 serves as an inhibitor of Vezf1. Upon reducing Vezf1 expression, ΔNp63 expression decreased, and NKX2–1 expression increased. This finding suggests that during the AST process, Elf5 can regulate ΔNp63 expression by modulating Vezf1, potentially inhibiting squamous transformation. Moreover, these alterations in the expression of DNp63 and related genes not only drive AST but also maintain SCC characteristics and lead to resistance to KRAS inhibition (47).
In KRAS-mutant lung cancer with LKB1 deletion, Wnt signaling plays a role in maintaining the adenocarcinoma state by activating NKX2-1. However, in the KL mouse model, Wnt signaling is inactivated due to oxidative stress (50). Its downstream effector FOXO3A can be inactivated by reactive oxygen species (ROS). ROS mediates the shutdown of Wnt/β-catenin signaling through FOXO3A, disrupting the balance of the glandular and squamous lineage TF networks, thereby promoting squamous trans-differentiation (51).
In the Rosa26LSL-Sox2-IRES-GFP; LKB1fl/fl (SL) mouse model, with overexpression of the TFs Sox2 and loss of the tumor suppressor LKB1, the tumors exhibit LUSC characteristics, such as expressing markers like KRT5 and DNp63, and are similar to human LUSC in gene expression and immune infiltration characteristics, including an abundance of tumor-associated neutrophils (TANs), low expression of NKX2-1, and activation of the mTOR pathway (high expression of p4EBP1). In this model, SOX2 inhibits the activity of NKX2-1, which in turn leads to the deletion of NKX2-1 (3, 48). SOX2 can promote the recruitment and infiltration of TANs mediated by CXCL3 and CXCL5 (48), and this phenomenon is independent of the tumor tissue type (48). NKX2–1 functions as a tumor suppressor in LUSC, and its deletion may promote SOX2-driven transformation by inhibiting SOX2 targeted genes, thereby accelerating the development of LUSC (48).
In SNL (deletion of Nkx2–1 in SL mice) mice, the deletion of NKX2–1 significantly accelerates the tumorigenesis driven by the deletion of SOX2 and LKB1. The induced tumors are mainly mucinous adenocarcinomas. Over time, these tumors undergo trans-differentiation into LUSC, with an increasing number of ΔNp63+ tumor cells (41). In the KL mouse model, the level of TET-mediated DNA demethylation increases during AST. Knockout of the Tet2 gene indicates its indispensability for squamous carcinoma transformation (36). TET2 promotes lipid transfer of neutrophils through STAT3-mediated CXCL5 expression, thereby promoting the AST process (36). Changes in P63 and its related genes maintain the characteristics of LUSC (46). Targeting STAT3 can significantly reduce the expression of P63 and inhibit the AST process (47). Therefore, targeting STAT3 can enhance the anti-cancer immune response of tumors, rescue the suppressed TIME (52, 53), and simultaneously target the connection between STAT3-CXCL5 to reduce neutrophil infiltration and effectively inhibit squamous transformation (36).
3.2 ALK rearrangement and JAK-STAT pathway activation in ASLC
Research findings reveal that anaplastic lymphoma kinase (ALK) rearrangement can be detected in 5.1% ~ 7.5% of ASLC. The EML4-ALK G1202R mutation drives Epithelial-Mesenchymal Transition (EMT) via constitutive activation of the STAT3/Slug signaling axis. Mechanistically, G1202R-enhanced ALK kinase activity phosphorylates STAT3, which directly upregulates Slug (SNAI2) expression. Slug orchestrates EMT by repressing epithelial markers (e.g., E-cadherin) and inducing mesenchymal markers (e.g., vimentin, N-cadherin), thereby augmenting tumor cell migration, invasion, and metastatic dissemination (PMID: 35085771). This molecular cascade links ALK mutational activation to both EMT phenotype acquisition and clinically observed aggressiveness in ALK-rearranged tumors (54), indicating that this mutation promotes tumor progression and augments its metastatic potential. Further research has shown that EML4-ALK initially promotes the formation of LUAD and drives squamous transformation in the late stage, altering the morphology and characteristics of the tumor (46, 55). Simultaneously, it has also been found that the JAK-STAT signal activated by EML4-ALK promotes AST, thereby resulting in resistance to ALK inhibitors (46). In addition, research indicates that the EML4-ALK fusion protein may activate the PI3K/AKT signaling pathway to promote the proliferation, survival, and migration of lung cancer cells, thus driving tumor progression. The dysregulation of the PI3K/AKT signaling pathway is relatively common in ASLC, and this dysregulation may be closely associated with tumorigenesis, development, and poor prognosis (56). The well-known inhibitors of the JAK-STAT pathway are ruxolitinib (JAK1/2 inhibitor) and fedratinib (JAK2 inhibitor) (57). Moreover, the combination of JAK1/2 inhibitors with TKI therapy and the combined use of ALK and STAT3 inhibitors can regulate the immune response and restore the sensitivity of tumor cells to treatment (58), thereby overcoming the resistance driven by AST and improving the treatment outcome. In conclusion, the JAK/STAT signaling pathway plays a significant promoting role in the process of LUAD transforming into AST.
3.3 The Hippo pathway’s role in mediating AST
The Hippo pathway, along with its downstream effectors, the transcriptional co-activator Yes-associated protein (YAP) and the transcriptional co-activator with PDZ-binding motif (TAZ), can bind to TEADs to regulate gene expression (59). These proteins critical regulatory roles in organ growth, cell plasticity, proliferation, and regeneration (60). In particular, the YAP/TAZ-TEAD transcriptional complex has emerged as a promising target for cancer therapy, with strategies aimed at inhibiting the Hippo pathway by disrupting the YAP/TAZ-TEAD interface (61).
Experimental manipulations of YAP in lung cancer cell lines have revealed that overexpression of YAP results in a marked down-regulation of S100A7 and ΔNp63, alongside a substantial up-regulation of TTF1 and napsin A, effectively inhibiting squamous trans-differentiation. Conversely, knockdown of YAP alone facilitates this trans-differentiation process (59). LSD1 can mediate a large number of genes down-regulated by YAP/TAZ, including tumor suppressors in YAP/TAZ-activated cells, which confirms that YAP/TAZ drives cell proliferation and tumor growth through the polyamine-eIF5A oligomerization-LSD1 axis. In addition, the deletion of LKB1 leads to YAP activation, causing an up-regulation of ZEB2 expression and inhibiting DNp63 transcription. YAP inhibits LKB1-deleted ASLC by inhibiting ZEB2-mediated DNp63 (62). In summary, we have compiled the key factors influencing the balance of TFs in LUAD and LUSC during AST (Figure 1).
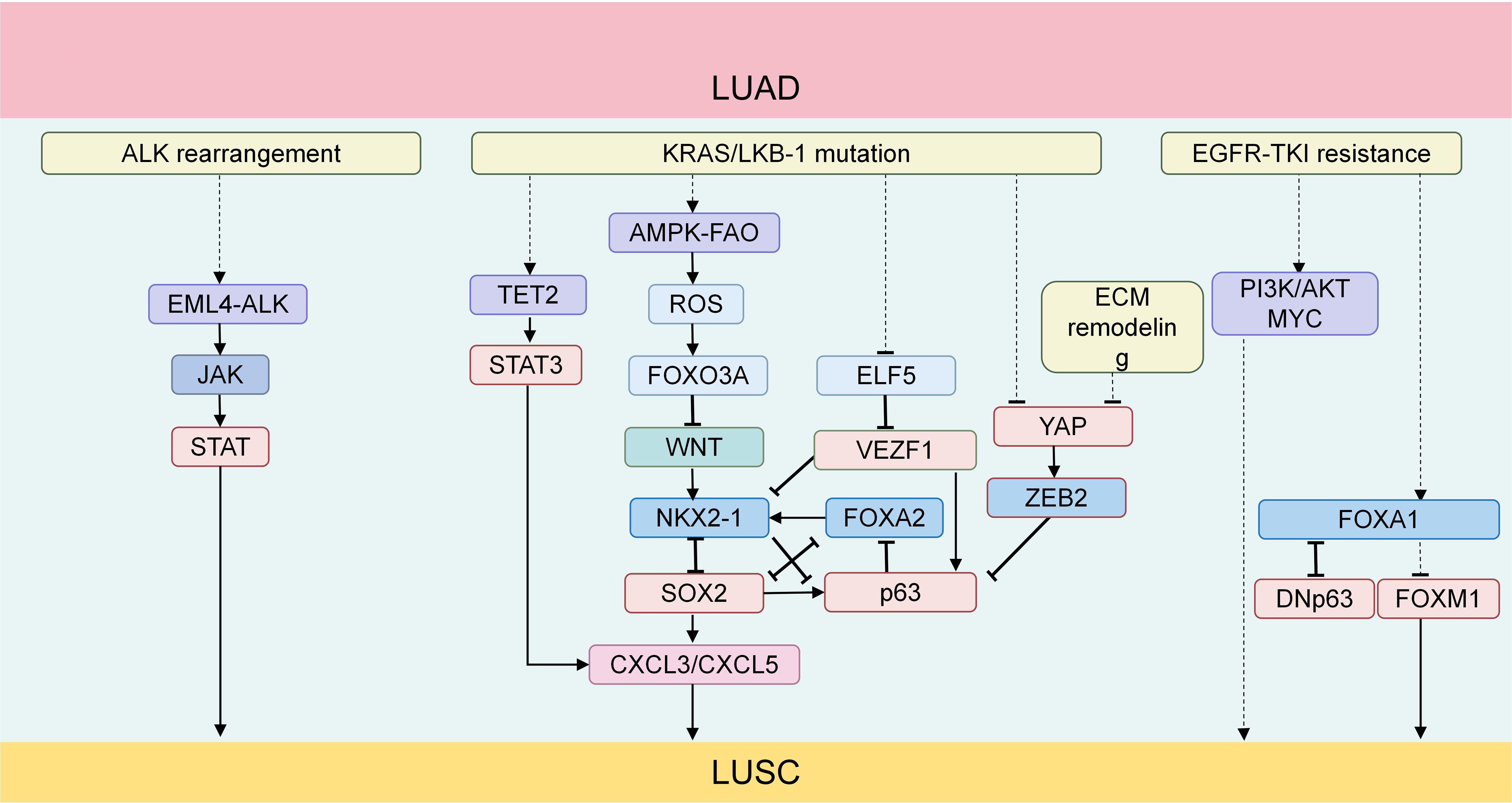
Figure 1. (1) The EML4-ALK fusion protein produced by ALK rearrangement can activate the JAK-STAT signaling pathway. The activated JAK-STAT signaling plays an important promoting role in the process of AST; (2) TET2 promotes neutrophil lipid transfer through STAT3-mediated CXCL5 expression, thus facilitating the process of AST; (3) In KRAS-mutated lung cancer with LKB1 deficiency, the AMPK-FAO pathway is disrupted by the excessively accumulated ROS. Meanwhile, ROS mediates the shutdown of the Wnt/β-catenin signaling pathway through FOXO3A, disrupting the balance of the TF network,thus promoting the development of AST; (4) The upstream TFs of ASLC form a regulatory network centered around NKX2-1, FOXA2, SOX2 and TP63; (5) In KRAS-driven LUAD, SOX2 promotes the recruitment and infiltration of TANs through CXCL3 and CXCL5; (6) YAP inhibits ASLC with LKB1 deficiency by suppressing DNp63 mediated by ZEB2; (7) Activation of the PI3K/AKT pathway can induce squamous characteristics in an EGFR-mutated LUAD model; (8) The deletion of FOXA1and the overexpression of FOXM1synergistically drive AST (63); (9) The knockout of FOXA1 significantly upregulates the transcription of DNp63 in tumor cells. The protein level of FOXA1 is downregulated in tumors with DNp63 overexpression, while it is upregulated in tumors with DNp63 knockout. TFs, transcription factors; LUSC, lung squamous cell carcinoma; LUAD, lung adenocarcinoma; ASLC, adeno-squamous lung carcinoma; TANs, tumor-associated neutrophils; AST, adeno-squamous transformation.
4 LKB1 inactivation: a driver of adenosquamous transformation
LKB1, a tumor-suppressor gene, is commonly found to be inactivated in a wide variety of tumor types, with this phenomenon being particularly prominent in LUAD (about 30% of cases) (64). Additionally, LKB1 serves as a central regulator of chromatin accessibility. LKB1 mutations account for around 17% in LUAD, and the mutation rate is even higher in ASLC, averaging 39.66%. LKB1 inactivation can drive AST in lung cancer (49, 65, 66).
Research indicates that in LUAD with LKB1 deficiency, the reduction in lysyl oxidase (Lox) leads to a decrease in collagen distribution, triggering extracellular matrix remodeling. Simultaneously, the up-regulation of p63 expression prompts LUAD to gradually trans-differentiate into LUSC through pathologically mixed ASLC intermediates (67). Moreover, KRAS-mutated lung cancer with LKB1 deletion exhibits high plasticity (49). Furthermore, LKB1 deletion results in severe metabolic imbalance and excessive accumulation of ROS.ROS can drive the AST process by disrupting fatty acid oxidation (FAO) mediated by adenosine monophosphate-activated protein kinase (AMPK) (66). ROS can act on various stromal cells, providing metabolic support for tumors, ensuring blood supply, and influencing the tumor’s immune response (68). LKB1 deficiency also promotes the formation of an immunosuppressive microenvironment and may be associated with primary resistance to anti-PD-1/anti-PD-L1 (64).
Both the oncogene KRAS and the tumor-suppressor gene STK11 play regulatory roles in metabolism, and they are frequently mutated in NSCLC (69). KRAS-driven lung cancer often leads to the inactivation of TP53 and/or STK11/LKB1 (70). In LUAD patients, STK11/LKB1 mutations are significantly associated with squamous cell characteristics (44). Recent studies have revealed that in LUAD, tumors with KRAS/TP53 co-mutations often display significantly elevated PD-L1 expression and an accumulation of cytotoxic T cells, whereas tumors with KRAS/LKB1 (K/L) co-mutations are usually negative for PD-L1 expression and exhibit minimal cytotoxic immune infiltration (71). Lung cancer with K/L deletion is highly invasive (69), lacks PD-L1, and has a poor response to immune checkpoint blockade (ICB) (70).
A comprehensive analysis of factors associated with LKB1 deletion is pivotal in elucidating the intricate mechanisms underlying the initiation, progression, and heterogeneity of ASLC. This line of inquiry not only deepens our understanding of the molecular foundations of this malignancy but also serves as a cornerstone for the development of targeted therapeutic interventions. Leveraging these insights, we have formulated innovative strategies to address these complexities, as delineated in Table 2.
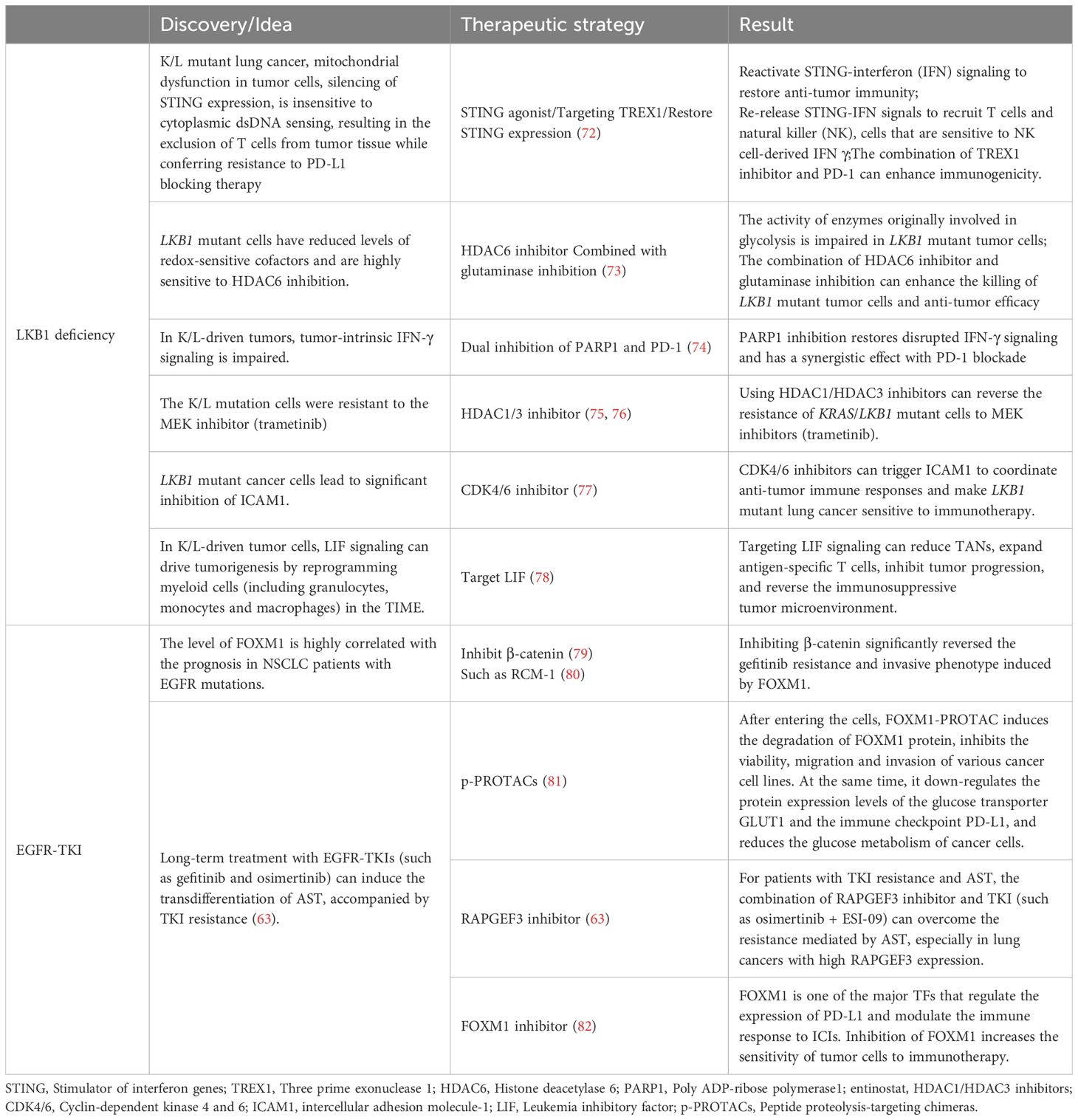
Table 2. Immunotherapy strategies for the AST process.
5 Immune microenvironment dynamics in AST
5.1 The role of TANs in AST
The efficacy of immunotherapy is shaped by the dynamic interplay between immune cells and cancer cells within the TIME (83). TIME heterogeneity contributes significantly to tumor metastasis, recurrence, and drug resistance (38, 84). TANs in lung cancer exhibit an activated phenotype, secreting various cytokines and chemokines, and some possess antigen-presenting-cell-like capabilities, stimulating anti-tumor T-cell responses (85). They can also exert anti-tumor effects by directly killing tumor cells and participating in networks that mediates drug resistance. Conversely, TANs can promote tumor growth by driving angiogenesis, remodeling the extracellular matrix, facilitating metastasis, and inducing immunosuppression (86). For example, LUAD cells can activate TANs, upregulating the expression of Notch3 in cancer cells and thereby promoting their own invasion and migration (87). Tumor cells can also induce neutrophils through TGFβ1, activate the Smad2/3 signaling pathway, leading to increased FAM3C production. FAM3C promotes the EMT of tumor cells through the JNK-ZEB1/Snail signaling pathway (88). This interaction enhances the affinity between neutrophils and tumor cells, making TANs an important component of TIME. Locally aggregated TANs may be triggered by external stimuli in the TME and switch between anti-tumor and pro-tumor phenotypes (86). In NSCLC, TANs are more abundant in LUSC than in LUAD and are associated with a poor prognosis (46). In the process of AST, there are often reports of TANs recruitment and infiltration (33, 46, 49, 51, 53). Therefore, the infiltration density of TANs may be a new marker for a poor prognosis in AST. TANs have pro-tumor characteristics and preferentially promote the development of squamous tumors. They may affect the fate of tumor cells by creating a favorable TIME or accelerating adenosquamous transdifferentiation (46).
In KRAS-driven adenocarcinoma, SOX2 promotes the recruitment and infiltration of TANs through CXCL3 and CXCL5 (46) (Figure 2). SOX2 is a TFs related to LUSC and it inhibits the activity of NKX2-1, a TFs related to LUAD. The absence of NKX2–1 significantly accelerates the occurrence of LUSC (41, 46). Consequently, TAN recruitment and infiltration are partially driven by TFs imbalance, facilitating AST. TANs are rich in triglycerides and can transfer lipids to tumor cells, promoting cell proliferation and squamous transformation (33). Inhibiting the STAT3-CXCL5 axis can reduce the infiltration of TANs and thus inhibit squamous cell carcinoma transformation (33). TANs can significantly inhibit T-cell proliferation and reduce the secretion of interferon (IFN-γ) and tumor necrosis factor-α (TNF-α), adversely affecting the TIME (89). Disrupting the balance of TFs in adenocarcinoma and squamous cell carcinoma to promote squamous cell differentiation will have an impact on the infiltration and function of immune cells. The immunosuppressive effect of TANs further worsens this adverse TIME, creating conditions for AST. Intrinsically, the tumor-driving factors SOX2 and NKX2–1 have opposing regulatory effects on TAN accumulation, suggesting that cancer cell intrinsic factors also shape TIME (35), challenging the notion that tissue type alone determines TIME.
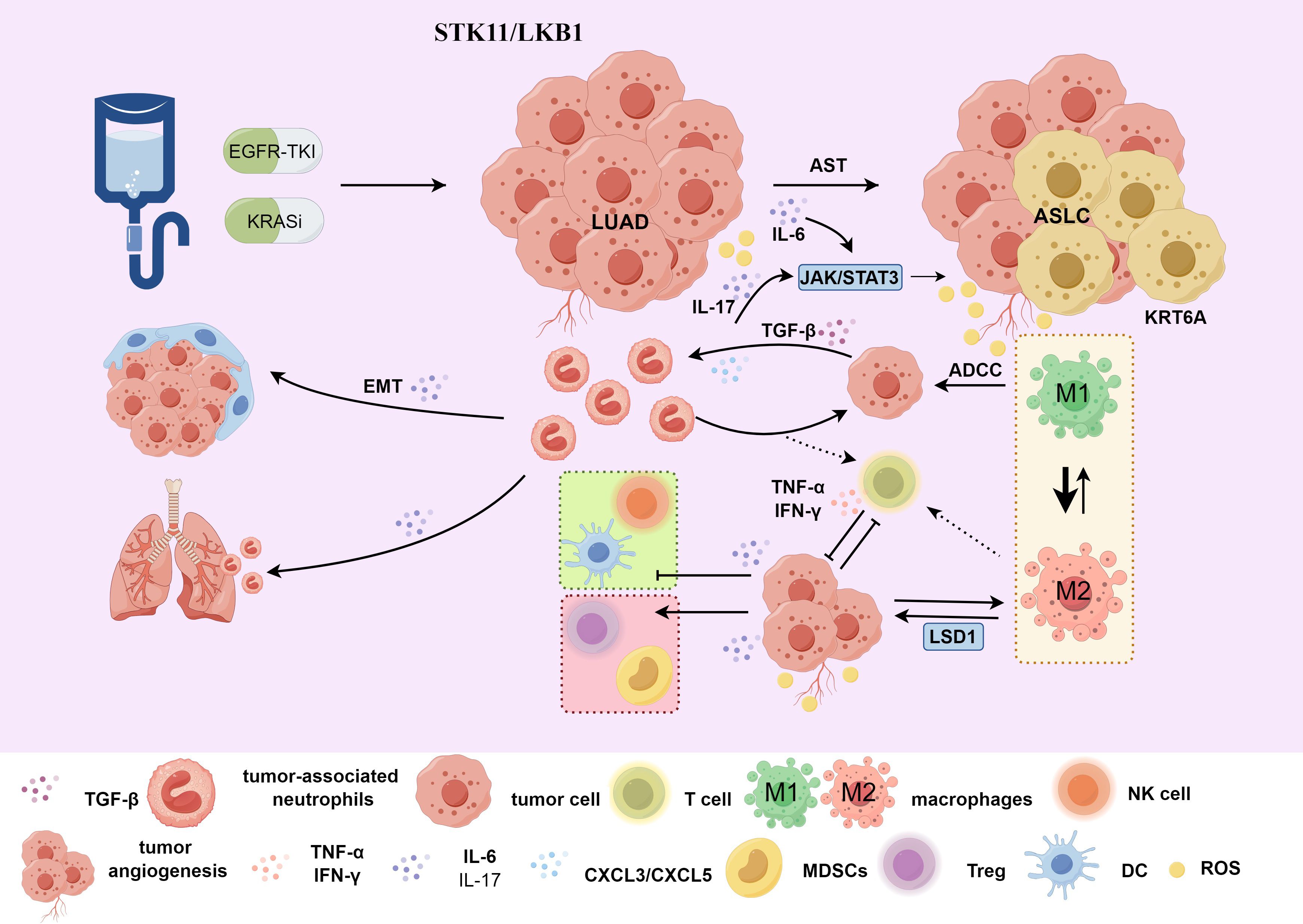
Figure 2. (1) Treatment with EGFR-TKI or KRAS inhibitors may lead to AST. For patients with STK11/LKB1 mutations, the KRT6A gene is highly expressed in the AST transitional state. Under the influence of the local TME, M2-TAMs affects LSD1 and indirectly promotes the tumor cell proliferation and invasion process mediated by KRT6A, affecting treatment prognosis; (2) Tumor cells can induce neutrophils through TGFβ1 to promote EMT. In KRAS-driven LUAD, there is recruitment and infiltration of TANs mediated by CXCL3 and CXCL5. TET2 promotes neutrophil lipid transfer via STAT3-mediated CXCL5 expression. TANs inhibit T cell proliferation and reduce the secretion of IFN-γ and TNF-α, affecting the TIME and promoting the AST process; (3) IL-6/IL-17 indirectly influences the TIME of AST through the JAK-STAT signaling pathway, negatively regulating neutrophils, natural killer cells, effector T cells and dendritic cells, and positively regulating regulatory T cells and MDSCs. AST, adenosquamous transformation; M2-TAMs, M2 macrophages; TANs, tumor-associated neutrophils; TIME, tumor immune microenvironment; ADCC, antibody-dependent cell-mediated cytotoxicity; ASLC, adenosquamous lung carcinoma; LUAD, lung adenocarcinoma; EMT, Epithelial-Mesenchymal Transition. Use the website "https://www.figdraw.com/#/" for drawing.
In conclusion, AST-driving genes promote the infiltration of TANs, which contributes to oncogenesis through promoting EMT, angiogenesis, inhibiting T-cell activation and killing, and facilitating the formation of an immunosuppressive TIME. Therefore, we hypothesize that the extensive infiltration of TANs may play a crucial role in the transformation of AST from an intermediate state to squamous cell carcinoma. Thus, targeting TANs can effectively inhibit the process of AST.
5.2 The promoting role of IL-6/IL-17 pro-inflammatory cytokines on the transformation of TIME
Interleukin-6 (IL-6), produced by various cells within the TME, is a pleiotropic pro-inflammatory cytokine (90). The classic signaling pathway of IL-6 (mIL-6R) is associated with anti-inflammatory functions, while the trans-signaling pathway (sIL-6R) is related to pro-inflammatory responses. The sIL-6R must simultaneously activate the JAK/STAT3 and PI3K/AKT pathways to induce human vascular endothelial cells to release the pro-inflammatory chemokine monocyte chemoattractant protein-1 (MCP-1, also known as CCL2) (91). CCL2 has a dual role: it regulates lymphocyte and NK cell homing, migration, activation, differentiation, and development positively, while also attracting and enhancing other inflammatory factors, promoting monocyte and macrophage infiltration (92–94). CCL2, through its interaction with CCR2, facilitates cancer cell migration and recruits immunosuppressive cells into the TME, fostering cancer progression (95). Therefore, early detection of CCL2 changes can provide insights into the progression of LUAD, and the state of the inflammatory response, particularly in the inflammatory subtype in ASLC. This is helpful for timely adjusting the treatment plan for adenocarcinoma-squamous transformation and further exploring the specific mechanism of CCL2 in AST.
In the TME, the IL-6/JAK/STAT3 signaling can drive the proliferation, survival, invasion, and metastasis of tumor cells and strongly suppress the anti-tumor immune response (96). STAT3 is often over-activated in tumor-infiltrating immune cells, negatively regulating neutrophils, natural killer cells, effector T-cells, and dendritic cells, while positively regulating regulatory T-cells and myeloid-derived suppressor cell (MDSC) populations (96), thereby affecting immune function. The up-regulation of CD47 expression in drug-resistant cells enhances the escape ability of cancer cells. STAT3 is associated with CD47 expression. Inhibiting STAT3 can enhance the phagocytic activity of tumor-associated macrophages (TAMs) and alleviate drug resistance. The combined use of gefitinib, a STAT3 inhibitor, and an anti-CD47 monoclonal antibody can address drug resistance (97). IL-6 activates the JAK/STAT3 signaling in tumor cells and tumor-infiltrating immune cells, participating in the construction of an adverse TIME (91) (Figure 2), potentially facilitating AST. Inhibitors targeting IL-6 production, IL-6R, and IL-6 signaling (90) can be used to impede the AST process.
In addition, studies have shown that during acute inflammation, IL-17 can mediate the migration of neutrophils to the lungs, resulting in lung tissue damage (98). The expression of cytokines mediated by IL-17 and the recruitment of neutrophils can trigger tumor proliferation (99) and resistance to immunotherapy (100) in lung cancer. TANs can produce IL-17a and promote the AST process through JAK2/STAT3 signaling (Figure 2). Blocking the IL-17a signaling with neutralizing antibodies can inhibit the activity of TANs-stimulated tumor cells (101).
5.3 LSD1-mediated TAM polarization in ASLC
Currently, squamous transformation is considered an escape mechanism for adenocarcinoma to evade anti-cancer therapies. However, its key driving factors and molecular alterations have not been fully explored. A clinical trial study showed that squamous transformation occurs in LUAD patients after they develop resistance to the KRAS inhibitor adagrasib (48). After analyzing the transcriptomic features and clinical outcomes, it was found that only in LUAD patients, STK11/LKB1 mutations are significantly associated with squamous cell features and adagrasib resistance, and the keratin type II cytoskeleton 6A (KRT6A) gene is highly expressed during the AST transition state (2, 44). KRT6A expression inversely correlates with treatment duration, predicting poor prognosis in NSCLC patients treated with adagrasib alone, as high KRT6A levels are associated with shorter survival, higher KEAP1 and STK11/LKB1 mutation rates, and promote NSCLC cell proliferation and invasion (44, 102, 103).
Mechanistically, KRT6A promotes tumor progression through the pentose phosphate pathway regulated by MYC, and LSD1 can promote KRT6A gene expression (104). The Che team’s research found that KRT6A acts downstream of LSD1 to promote the proliferation and invasion of NSCLC cells (105) In addition, highly expressed KRT6A in LUAD can promote the proliferation and metastasis of lung cancer through EMT and cancer stem cell transformation (106), and can also promote the radioresistance, invasion, and metastasis of lung cancer through the p53 signaling pathway (107).
In summary, KRT6A plays a promoting role in the transformation of LUAD to squamous type, and the promoting effect of LSD1 on KRT6A may directly affect the malignant progression of the tumor. Detecting elevated KRT6A expression early in LUAD can identify AST and adagrasib resistance, aiding in the timely adjustment of treatment strategies to address potential drug resistance.
Tumors and the TME can affect the pro-metastatic ability of TANs. TAMs, among others, can promote neutrophil-mediated tumor metastasis (46). It is known that inflammatory stimuli promoted by TNF-α can induce an increase in LSD1 expression in M2-TAMs (108). LSD1 promotes pro-inflammatory polarization of human macrophages (M1-TAM polarization) by inhibiting the transcription of catalase (109). During this process, hydrogen peroxide (H2O2) is produced as a by-product (110). Under normal circumstances, this by-product inhibits catalase, thereby maintaining the M1 polarization state. When an LSD1 inhibitor is used, the catalase level can be maintained (109), preventing LSD1 from promoting M1-TAM polarization. Meanwhile, research has found that M1-TAM polarization, in turn, reduces LSD1 expression (111), forming a feedback-regulation mechanism. Therefore, LSD1 plays a key regulatory role in the TIME of ASLC (Figure 3).
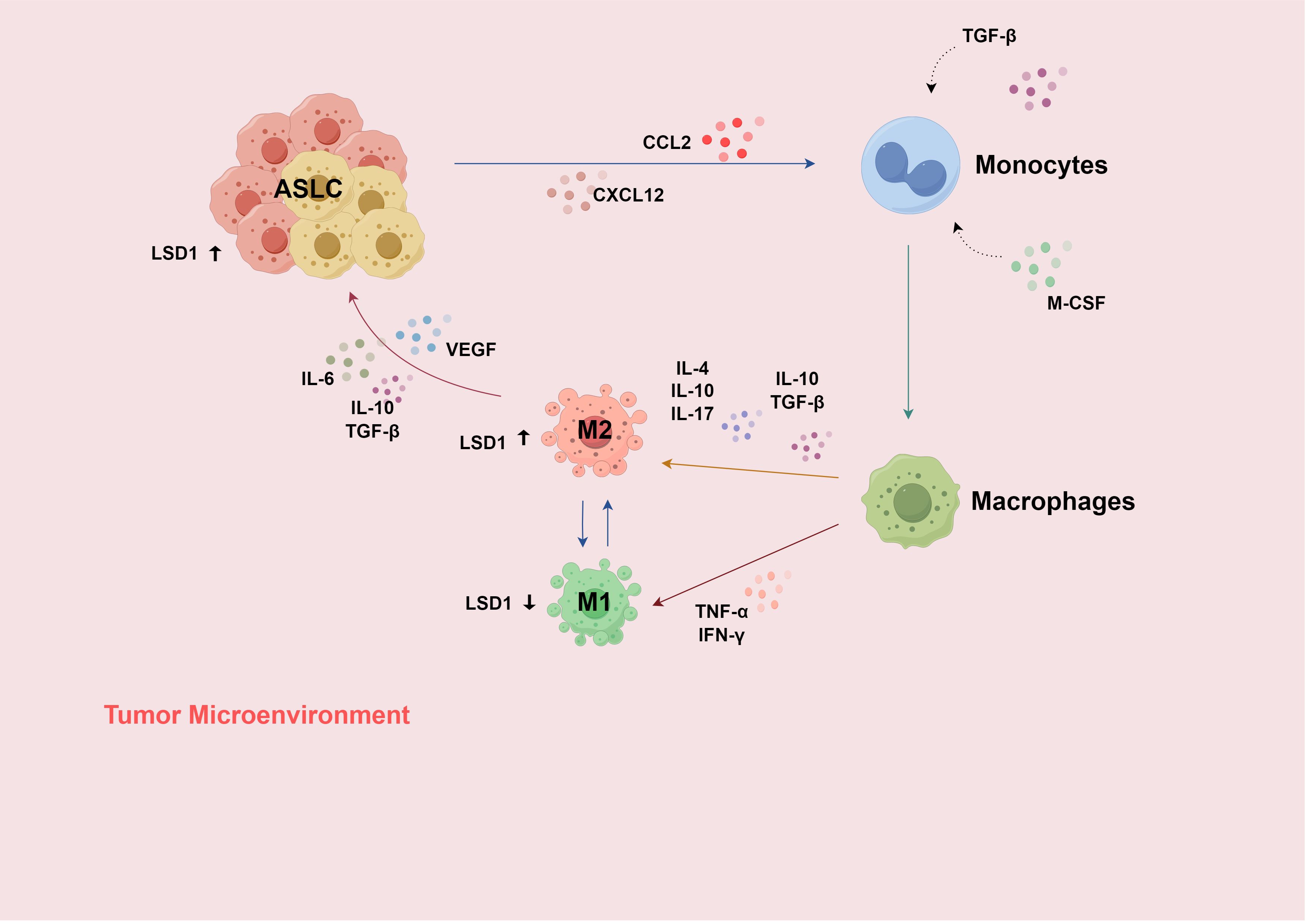
Figure 3. (1) Recruited by the chemokine signals of CCL-2 and CXCL-12, monocytes enter the TME and differentiate into macrophages under the influence of cytokines such as M-CSF and TGF-β; (2) Macrophages are subsequently stimulated by more cytokines in the TME and differentiate into two phenotypes, M1 and M2; (3) When the TME changes or under therapeutic intervention, TAMs can transform into each other; (4) Inflammatory stimulation induced by TNF-α can increase the expression of LSD1 in M2-TAM; LSD1 generates H2O2 and inhibits CAT, thereby promoting M1 polarization and reducing the expression of LSD1; (5) In the inflammatory subtype of ASLC, immune infiltration increases, and CCL2 promotes the migration and infiltration of macrophages at the site of inflammation. H2O2, hydrogen peroxide; CAT, catalase; TAMs, tumor-associated macrophages. Use the website "https://www.figdraw.com/#/" for drawing.
Under the influence of the local TME, TAMs exhibit great phenotypic heterogeneity and diverse functional capabilities (112), and can transform into each other when the TME changes or during treatment intervention. The interaction between the TME and cancer cells plays an important role in acquired resistance to EGFR-TKIs (97). Among them, M2-like reprogramming of TAMs and the reduction in macrophage phagocytosis are related to drug resistance (97). TAMs mainly have two functional subtypes. Classically activated M1-TAMs (pro-inflammatory) can directly mediate cytotoxicity and antibody-dependent cell-mediated cytotoxicity (ADCC) to kill tumor cells (113), while M2-TAMs (anti-inflammatory) can promote tumor cell occurrence, metastasis (114), inhibit T-cell-mediated anti-tumor immune responses, and promote tumor angiogenesis, thereby leading to tumor progression (113, 115). (Figure 2)
The expression of LSD1 in TAMs is regulated by inflammatory stimuli (57, 116, 117). In turn, LSD1 can regulate the polarization state of TAMs. This forms a complex interaction network impacting the progression and development of ASLC. In summary, LSD1 and TAMs interact through multiple mechanisms in ASLC, having an important impact on tumor progression, the TIME, etc., providing potential targets and research directions for the treatment of ASLC (Figure 3).
USP7, highly expressed in M2 macrophages compared to M1, can be silenced with a specific inhibitor, altering M2-TAM phenotype and function. This promotes CD8+ T-cell proliferation and increases PD-L1 expression in tumor cells in vitro. The combined use of a USP7 inhibitor and an anti-PD-1 antibody can produce a synergistic anti-tumor effect (118). Manipulating macrophage polarization has emerged as a potential strategy for treating ASLC, either by inhibiting M2-type polarization signals or promoting M1-type polarization to enhance anti-tumor immune responses.
5.4 LSD1 inhibition: synergistic potential with immunotherapy in ASLC treatment
Dysregulation of various cell types within the TME can trigger immunosuppressive functions and result in the growth of aggressive tumors (119). LSD1 (lysine-specific histone demethylase 1, also known as KDM1A) is crucial in tumorigenesis and development and has a close connection with immunity (120–122). LSD1 is widely expressed in multiple cancer types. It can block normal cell differentiation, promote the proliferation, migration, and invasion of tumor cells (123), and drive the formation of cancer stem cell phenotypes (124–129). Previous research in ASLC has shown that LSD1, upstream of the highly-expressed KRT6A, promotes KRT6A gene expression, thus facilitating lung cancer cell growth and invasion (105). Depletion of LSD1 enhances the immunogenicity of poorly immunogenic tumors and T-cell infiltration (130), suggesting that inhibiting LSD1 can improve the anti-tumor effect of immunotherapy. Similarly, the Tang group found in the KL model that LSD1 deletion completely blocks the AST process. In this model, only LUAD pathology is present, and the incidence of AST and tumor burden are significantly reduced (3).
H2O2 is important in the interaction between LSD1 and TAMs in ASLC (57, 116, 117). Superoxide dismutase 2 (SOD2), a mitochondrial superoxide scavenger and H2O2 regulator, may affect tumor development in the TME by regulating oxidative stress levels. For example, using mitochondrial antioxidants or other methods to regulate SOD2 function can inhibit tumor-associated inflammatory responses and reduce the tumor-promoting effect of inflammation (69). SOD2 promotes the immunosuppressive function of mesenchymal stem cells at the expense of adipocyte differentiation. In an inflammatory microenvironment, it helps regulate the immune response and reduce inflammatory damage (131). The lack of LSD1 activity and the maintenance of PARP1 on the SOD2 promoter significantly increase SOD2 expression. Although LSD1 is considered tumor-promoting, it can also promote M1-TAM polarization. LSD1 and SOD2 may jointly participate in TME regulation. Further study of their interaction mechanism could lead to more effective treatment strategies.
Tolperisone can induce a tumor-suppressive response mediated by the unfolded protein response (UPR) (132). As a potential target for LSD1, it may alter the gene expression profile and biological behavior of tumor cells by regulating LSD1 activity or its demethylation of specific genes. It can also act synergistically with proteasome inhibitors like MG132 to enhance the inhibitory effect on tumor cells. Tolperisone can reprogram M2-TAMs to M1-TAMs, enhancing the immune response in the tumor microenvironment and making the tumor more sensitive to immunotherapy (133). This provides a good basis for further research on combining tolperisone with ASLC treatment.
6 Therapeutic strategies targeting key drivers in AST
6.1 Targeting TAN in immune microenvironment remodeling
Enhanced immune cell infiltration into the TME may be facilitated by chemokines. Knocking out CXCL3 and CXCL5 can inhibit AST (41). Macropinocytosis (MP) enables tumor cells to extract nutrients from extracellular sources and use them to generate energy (134). Pharmacological inhibition of MP can significantly inhibit the lipid transfer from TANs to cancer cells and block squamous transformation (33). The selective CXCR2 inhibitor SB225002 shows good therapeutic effects by reducing neutrophil infiltration and boosting anti-tumor T-cell activity through CD8+ T cell activation (135). It has been found that granulocyte-macrophage colony-stimulating factor (GM-CSF) derived from tumor cells triggers the expression of the anti-apoptotic Bcl-xL protein and enhances the survival of neutrophils through JAK/STAT signaling. Oral administration of a specific BH3 mimetic (A-1331852) can reduce the survival rate and abundance of TANs and inhibit lung tumor growth without causing neutropenia (136).
6.2 Targeting LSD1-mediated epigenetic reprogramming in lineage transdifferentiation
GSK2879552, an irreversible LSD1 inhibitor, has been reported to effectively suppress small-cell lung cancer (SCLC) (137). Treatment of the KL model with GSK2879552 significantly inhibits KL tumor progression and neutrophil infiltration. Most mice show ADC pathology, and the incidence of AST and tumor burden are also greatly decreased (127), highlighting LSD1 as a potential therapeutic target for STK11-mutated ASLC.
Interestingly, inhibiting LSD1 upregulates PD-L1 expression on tumor cells, affecting the response to immune checkpoint inhibitors (117). In related studies, PD-L1-mediated in-vivo T-cell immunity in exosomes can regulate tumor cell proliferation. LSD1 deletion reduces PD-L1 expression in exosomes and restores T-cell responses (127). Similarly, in the NSCLC xenograft model, the combination of an LSD1 inhibitor and a ferroptosis inducer has a stronger anti-tumor effect than either drug alone (116). The Mamun team’s research also confirms the potential of combining LSD1 inhibitors with ICB therapy in future cancer research (119), providing new directions for ASLC treatment.
7 Conclusion
In-depth exploration of the changes in key signaling molecules and multiple signaling pathways during the transformation process of ASLC holds irreplaceably significant implications for further unveiling the mystery of the lung cancer development mechanism and for mining potential therapeutic targets. This review has systematically presented the molecular and immunological landscape of AST, highlighting the intricate interplay between lineage plasticity, immune escape, and treatment efficacy.
Our analysis revealed that LKB1 inactivation in tumors, particularly in LUAD and ASLC, significantly drives AST through mechanisms involving extracellular matrix remodeling, metabolic imbalance, and the formation of an immunosuppressive microenvironment. These findings underscore the pivotal role of LKB1 in maintaining lung cancer lineage and suggest that targeting LKB1-related pathways could be a promising therapeutic strategy for ASLC. The imbalance between TFs like SOX2 and NKX2–1 was identified as a central driver of AST. The dynamic dysregulation of lineage-specific TFs finely tunes the AST process, indicating that targeting these networks could potentially halt or reverse squamous transformation. The inflammatory subtype of ASLC, serving as an intermediate stage of AST, exhibits increased immune cell infiltration. This suggests that the TIME undergoes significant changes during AST, potentially offering a window for immunotherapy intervention. The role of pro-inflammatory cytokines like IL-6 and IL-17 in promoting the transformation of the TME was also highlighted, suggesting a dual role in both inflammation and immune modulation.
The phenomenon of AST profoundly impacts cancer treatment efficacy, revealing the complexity and challenges in managing lung cancer. Our findings emphasize the urgent need to explore immunotherapy strategies that account for lineage plasticity and immune escape. For instance, the observed association between STK11/LKB1 mutations and resistance to KRAS inhibitors in LUAD patients with squamous features provides a critical clue for understanding drug resistance mechanisms. While previous studies have noted the association between lineage plasticity and drug resistance, our review provides a comprehensive overview of how AST influences the immune landscape of NSCLC. We link specific genetic alterations, like ALK rearrangements and the JAK-STAT pathway, with AST and resistance to targeted therapies, offering a more nuanced understanding than previously reported.
One limitation of this review is the reliance on preclinical models, which may not fully capture the heterogeneity of human tumors. The complexity of the TME and the dynamic nature of immune cell interactions within it pose challenges in translating findings into effective clinical interventions. Furthermore, while we propose several potential therapeutic strategies, their efficacy and safety in clinical settings require further validation. Future research should focus on comprehensive molecular profiling to identify predictive biomarkers for AST transitions, further investigation into the molecular mechanisms driving lineage plasticity, particularly the roles of TFs and their regulatory networks, development of combination therapies that target both the tumor cells and the TIME to enhance treatment efficacy and overcome resistance, exploration of targeted therapies based on key signaling pathways identified in this review, including LSD1 inhibitors and other epigenetic modifiers, and employing single-cell technologies to dissect tumor heterogeneity and immune cell dynamics within ASLC.
The evolving understanding of lung cancer heterogeneity demands focused efforts on LUAS and AST. Future research should prioritize: (1) single-cell multi-omics (scRNA/ATAC-seq) to resolve LUAS plasticity and immune-microenvironment crosstalk; (2) AST-focused trials targeting high-risk subgroups (e.g., KRAS/EGFR-mutant LUAD with LKB1 loss) with ctDNA-guided monitoring; (3) multi-omics harmonization bridging genomics (TP53/EGFR), proteomics, and preclinical models.
In summary, the interplay between signaling pathways, immune cell dynamics, and the TME in ASLC, particularly during the AST process, requires a nuanced understanding. This review underscores the need for personalized treatment strategies that address not only the tumor cells but also the supportive microenvironment, thereby improving treatment outcomes and overcoming resistance. By integrating these insights, we can move towards more effective and tailored therapeutic interventions for ASLC.
Author contributions
HX: Writing – original draft, Writing – review & editing. YY: Resources, Writing – review & editing. PW: Funding acquisition, Writing – review & editing. SL: Writing – review & editing. XZ: Writing – review & editing. HN: Writing – review & editing. ZX: Writing – review & editing, Resources, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by grants from the National Natural Science Foundation of China (No. 82371837; No. 82102852).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Liu T, Han C, Fang P, Zhu H, Wang S, Ma Z, et al. Long non-coding RNAs in lung cancer: implications for lineage plasticity-mediated TKI resistance. Cell Mol Life Sci. (2021) 78:1983–2000. doi: 10.1007/s00018-020-03691-9
2. Quintanal-Villalonga A, Chan JM, Yu HA, Pe’Er D, Sawyers CL, Sen T, et al. Lineage plasticity in cancer: a shared pathway of therapeutic resistance. Nat Rev Clin Oncol. (2020) 17:360–71. doi: 10.1038/s41571-020-0340-z
3. Tang S, Xue Y, Qin Z, Fang Z, Sun Y, Yuan C, et al. Counteracting lineage-specific transcription factor network finely tunes lung adeno-to-squamous transdifferentiation through remodeling tumor immune microenvironment. Natl Sci Rev. (2023) 10:d28. doi: 10.1093/nsr/nwad028
4. Tong X, Zhang N, Xue Y, and Ji H. Comments on ‘Adeno-to-squamous transition drives resistance to KRAS inhibition in LKB1 mutant lung cancer. J Mol Cell Biol. (2024) 16:mjae013. doi: 10.1093/jmcb/mjae013
5. Awad MM, Liu S, Rybkin II, Arbour KC, Dilly J, Zhu VW, et al. Acquired Resistance to KRAS(G12C) Inhibition in Cancer. N Engl J Med. (2021) 384:2382–93. doi: 10.1056/NEJMoa2105281
6. Chen Y, Tang WY, Tong X, and Ji H. Pathological transition as the arising mechanism for drug resistance in lung cancer. Cancer Commun (Lond). (2019) 39:53. doi: 10.1186/s40880-019-0402-8
7. Park S, Han J, and Sun JM. Histologic transformation of ALK-rearranged adenocarcinoma to squamous cell carcinoma after treatment with ALK inhibitor. Lung Cancer. (2019) 127:66–8. doi: 10.1016/j.lungcan.2018.11.027
8. Ball M, Christopoulos P, Kirchner M, Allgauer M, Brandt R, Winter H, et al. Histological and molecular plasticity of ALK-positive non-small-cell lung cancer under targeted therapy: a case report. Cold Spring Harb Mol Case Stud. (2022) 8:a006156. doi: 10.1101/mcs.a006156
9. Gong J, Gregg JP, Ma W, Yoneda K, Moore EH, Daly ME, et al. Squamous Cell Transformation of Primary Lung Adenocarcinoma in a Patient With EML4-ALK Fusion Variant 5 Refractory to ALK Inhibitors. J Natl Compr Canc Netw. (2019) 17:297–301. doi: 10.6004/jnccn.2019.7291
10. Ueda S, Shukuya T, Hayashi T, Suzuki M, Kondo A, Arai Y, et al. Transformation from adenocarcinoma to squamous cell lung carcinoma with MET amplification after lorlatinib resistance: A case report. Thorac Cancer. (2021) 12:715–19. doi: 10.1111/1759-7714.13829
11. Zhang Y, Qin Y, Xu H, Yao Q, Gao Y, Feng Y, et al. Case Report: A Case Report of a Histological Transformation of ALK-Rearranged Adenocarcinoma With High Expression of PD-L1 to Squamous Cell Carcinoma After Treatment With Alectinib. Pathol Oncol Res. (2021) 27:637745. doi: 10.3389/pore.2021.637745
12. Hsieh MS, Jhuang JY, Hua SF, and Chou YH. Histologic evolution from adenocarcinoma to squamous cell carcinoma after gefitinib treatment. Ann Thorac Surg. (2015) 99:316–19. doi: 10.1016/j.athoracsur.2014.02.075
13. Takahashi S, Sato Y, Sato Y, Hirabayashi R, Hara S, Takahashi Y, et al. Long-Term Efficacy of Immune Checkpoint Inhibitor for Squamous Cell Carcinoma Lesion Transformed From EGFR-Mutated Adenocarcinoma After Osimertinib Treatment: A Case Report. JTO Clin Res Rep. (2024) 5:100639. doi: 10.1016/j.jtocrr.2024.100639
14. Xi YZ, Xie L, Tan XW, and Zeng SL. Transformation of adenocarcinoma to squamous cell carcinoma as a source of EGFR-TKI resistance: A case report and literature review. Front Oncol. (2022) 12:942084. doi: 10.3389/fonc.2022.942084
15. Longo L, Mengoli MC, Bertolini F, Bettelli S, Manfredini S, and Rossi G. Synchronous occurrence of squamous-cell carcinoma “transformation” and EGFR exon 20 S768I mutation as a novel mechanism of resistance in EGFR-mutated lung adenocarcinoma. Lung Cancer. (2017) 103:24–6. doi: 10.1016/j.lungcan.2016.11.012
16. Mehlman C, Cadranel J, Rousseau-Bussac G, Lacave R, Pujals A, Girard N, et al. Resistance mechanisms to osimertinib in EGFR-mutated advanced non-small-cell lung cancer: A multicentric retrospective French study. Lung Cancer. (2019) 137:149–56. doi: 10.1016/j.lungcan.2019.09.019
17. Izumi H, Yamasaki A, Ueda Y, Sumikawa T, Maeta H, Nakamoto S, et al. Squamous Cell Carcinoma Transformation from EGFR-mutated Lung Adenocarcinoma: A Case Report and Literature Review. Clin Lung Cancer. (2018) 19:e63–66. doi: 10.1016/j.cllc.2017.10.005
18. Chiang CL, Yeh YC, Chou TY, and Chiu CH. Squamous cell carcinoma transformation after acquired resistance to osimertinib in a patient with lung adenocarcinoma harboring uncommon EGFR mutation. J Formos Med Assoc. (2020) 119:1439–41. doi: 10.1016/j.jfma.2019.12.017
19. Yao Y, Zhu Z, Wu Y, and Chai Y. Histologic transformation from adenocarcinoma to both small cell lung cancer and squamous cell carcinoma after treatment with gefitinib: A case report. Med (Baltimore). (2018) 97:e650. doi: 10.1097/MD.0000000000010650
20. Roca E, Pozzari M, Vermi W, Tovazzi V, Baggi A, Amoroso V, et al. Outcome of EGFR-mutated adenocarcinoma NSCLC patients with changed phenotype to squamous cell carcinoma after tyrosine kinase inhibitors: A pooled analysis with an additional case. Lung Cancer. (2019) 127:12–8. doi: 10.1016/j.lungcan.2018.11.016
21. Shao Y, Zhong DS, and Guan SS. Histologic transformation of lung adenocarcinoma to squamous cell carcinoma after chemotherapy: two case reports. Transl Cancer Res. (2020) 9:388–93. doi: 10.21037/tcr.2019.11.34
22. Wang F, Qin J, Xie F, Wu Q, and Lu H. Transformation of EML4-ALK fusion-positive adenocarcinoma into squamous cell carcinoma in association with acquired resistance to crizotinib. Lung Cancer. (2020) 140:118–20. doi: 10.1016/j.lungcan.2020.01.001
23. Lee PH and Chang GC. Transformations First Into Squamous-Cell Carcinoma and Later Into Sarcomatoid Carcinoma After Acquired Resistance to Osimertinib in a Patient With EGFR-Mutant Lung Adenocarcinoma: Case Report. Clin Lung Cancer. (2021) 22:e536–41. doi: 10.1016/j.cllc.2020.06.026
24. Haratani K, Hayashi H, Watanabe S, Kaneda H, Yoshida T, Takeda M, et al. Two cases of EGFR mutation-positive lung adenocarcinoma that transformed into squamous cell carcinoma: successful treatment of one case with rociletinib. Ann Oncol. (2016) 27:200–02. doi: 10.1093/annonc/mdv495
25. Haruki T, Nakanishi A, Matsui S, Kidokoro Y, Kubouchi Y, Takagi Y, et al. Transformation from adenocarcinoma to squamous cell carcinoma associated with long-term administration of EGFR-TKIs. Mol Clin Oncol. (2020) 13:82. doi: 10.3892/mco.2020.2152
26. Hakozaki T, Kitazono M, Takamori M, and Kiriu T. Combined Small and Squamous Transformation in EGFR-mutated Lung Adenocarcinoma. Intern Med. (2020) 59:1291–94. doi: 10.2169/internalmedicine.3542-19
27. Kozuma Y, Toyokawa G, Shoji F, Yamazaki K, Kawauchi S, Momosaki S, et al. Squamous Cell Carcinoma Transformation as a Possible Resistant Mechanism Against Pembrolizumab Plus Chemotherapy. J Thorac Oncol. (2019) 14:e238–40. doi: 10.1016/j.jtho.2019.05.039
28. Hsu CL, Chen KY, Kuo SW, and Chang YL. Histologic Transformation in a Patient with Lung Cancer Treated with Chemotherapy and Pembrolizumab. J Thorac Oncol. (2017) 12:e75–76. doi: 10.1016/j.jtho.2017.02.006
29. Mariniello A, Righi L, Morrone A, Carnio S, and Bironzo P. Squamous cell histological transformation in a lung adenocarcinoma patient (hyper) progressing upon immunotherapy. Tumori. (2022) 108:P15–19. doi: 10.1177/03008916221080487
30. Quintanal-Villalonga A, Taniguchi H, Zhan YA, Hasan MM, Chavan SS, Meng F, et al. Comprehensive molecular characterization of lung tumors implicates AKT and MYC signaling in adenocarcinoma to squamous cell transdifferentiation. J Hematol Oncol. (2021) 14:170. doi: 10.1186/s13045-021-01186-z
31. Stolnicu S, Hoang L, Zhou Q, Iasonos A, Terinte C, Pesci A, et al. Cervical Adenosquamous Carcinoma: Detailed Analysis of Morphology, Immunohistochemical Profile, and Outcome in 59 Cases. Int J Gynecol Pathol. (2023) 42:259–69. doi: 10.1097/PGP.0000000000000921
32. Liu CY. Understanding gastric adenosquamous cell carcinoma: Insights from immunoprofiling. J Chin Med Assoc. (2023) 86:780. doi: 10.1097/JCMA.0000000000000967
33. Lu SB, Ge FS, Liu C, and Hua YW. Adenosquamous carcinoma of sigmoid colon in an adolescent: A case report and literature review. Asian J Surg. (2022) 45:1055–56. doi: 10.1016/j.asjsur.2022.01.055
34. Gao R, Zhang X, and Jin L. Adenosquamous carcinoma-like mesonephric adenocarcinoma. Pathology. (2022) 54:828–30. doi: 10.1016/j.pathol.2021.11.012
35. Xiong Q, Zhang Z, Xu Y, and Zhu Q. Pancreatic Adenosquamous Carcinoma: A Rare Pathological Subtype of Pancreatic Cancer. J Clin Med. (2022) 11:7401. doi: 10.3390/jcm11247401
36. Romanucci G, Mercogliano S, Carucci E, Lunardi M, Caneva A, Benassuti C, et al. Low-grade adenosquamous carcinoma of the breast: a review with focus on imaging and management. Acta Radiol Open. (2021) 10:217675395. doi: 10.1177/20584601211013501
37. Ichaoui H, Nasr SB, Gargouri F, Zribi A, Hermi A, Fendri S, et al. Transformation of a prostatic adenocarcinoma into squamous cell carcinoma after luteinizing hormone-releasing hormone (LHRH) agonist and radiotherapy treatment. Pan Afr Med J. (2019) 34:125. doi: 10.11604/pamj.2019.34.125.19421
38. Xue Y, Chen Y, Sun S, Tong X, Chen Y, Tang S, et al. TET2-STAT3-CXCL5 nexus promotes neutrophil lipid transfer to fuel lung adeno-to-squamous transition. J Exp Med. (2024) 221:e20240111. doi: 10.1084/jem.20240111
39. Campbell JD, Alexandrov A, Kim J, Wala J, Berger AH, Pedamallu CS, et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet. (2016) 48:607–16. doi: 10.1038/ng.3564
40. Langer CJ, Obasaju C, Bunn P, Bonomi P, Gandara D, Hirsch FR, et al. Incremental Innovation and Progress in Advanced Squamous Cell Lung Cancer: Current Status and Future Impact of Treatment. J Thorac Oncol. (2016) 11:2066–81. doi: 10.1016/j.jtho.2016.08.138
41. Qin Z, Yue M, Tang S, Wu F, Sun H, Li Y, et al. EML4-ALK fusions drive lung adeno-to-squamous transition through JAK-STAT activation. J Exp Med. (2024) 221:e20232028. doi: 10.1084/jem.20232028
42. Hou S, Zhou S, Qin Z, Yang L, Han X, Yao S, et al. Evidence, Mechanism, and Clinical Relevance of the Transdifferentiation from Lung Adenocarcinoma to Squamous Cell Carcinoma. Am J Pathol. (2017) 187:954–62. doi: 10.1016/j.ajpath.2017.01.009
43. Ding Y, Zhang L, Guo L, Wu C, Zhou J, Zhou Y, et al. Comparative study on the mutational profile of adenocarcinoma and squamous cell carcinoma predominant histologic subtypes in Chinese non-small cell lung cancer patients. Thorac Cancer. (2020) 11:103–12. doi: 10.1111/1759-7714.13208
44. Schoenfeld AJ, Chan JM, Kubota D, Sato H, Rizvi H, Daneshbod Y, et al. Tumor Analyses Reveal Squamous Transformation and Off-Target Alterations As Early Resistance Mechanisms to First-line Osimertinib in EGFR-Mutant Lung Cancer. Clin Cancer Res. (2020) 26:2654–63. doi: 10.1158/1078-0432.CCR-19-3563
45. Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. (2014) 511:543. doi: 10.1038/nature13385
46. Ryan MB and Corcoran RB. Therapeutic strategies to target RAS-mutant cancers. Nat Rev Clin Oncol. (2018) 15:709–20. doi: 10.1038/s41571-018-0105-0
47. Tong X, Patel AS, Kim E, Li H, Chen Y, Li S, et al. Adeno-to-squamous transition drives resistance to KRAS inhibition in LKB1 mutant lung cancer. Cancer Cell. (2024) 42:413–28. doi: 10.1016/j.ccell.2024.01.012
48. Mollaoglu G, Jones A, Wait SJ, Mukhopadhyay A, Jeong S, Arya R, et al. The Lineage-Defining Transcription Factors SOX2 and NKX2–1 Determine Lung Cancer Cell Fate and Shape the Tumor Immune Microenvironment. Immunity. (2018) 49:764–79. doi: 10.1016/j.immuni.2018.09.020
49. Redon R, Muller D, Caulee K, Wanherdrick K, Abecassis J, and du Manoir S. A simple specific pattern of chromosomal aberrations at early stages of head and neck squamous cell carcinomas: PIK3CA but not p63 gene as a likely target of 3q26-qter gains. Cancer Res. (2001) 61:4122–29.
50. Ebrahimi KB, Cano M, Rhee J, Datta S, Wang L, and Handa JT. Oxidative Stress Induces an Interactive Decline in Wnt and Nrf2 Signaling in Degenerating Retinal Pigment Epithelium. Antioxid Redox Signal. (2018) 29:389–407. doi: 10.1089/ars.2017.7084
51. Fang Z, Han X, Chen Y, Tong X, Xue Y, Yao S, et al. Oxidative stress-triggered Wnt signaling perturbation characterizes the tipping point of lung adeno-to-squamous transdifferentiation. Signal Transduct Target Ther. (2023) 8:16. doi: 10.1038/s41392-022-01227-0
52. Dong Y, Chen J, Chen Y, and Liu S. Targeting the STAT3 oncogenic pathway: Cancer immunotherapy and drug repurposing. BioMed Pharmacother. (2023) 167:115513. doi: 10.1016/j.biopha.2023.115513
53. Zou S, Tong Q, Liu B, Huang W, Tian Y, and Fu X. Targeting STAT3 in Cancer Immunotherapy. Mol Cancer. (2020) 19:145. doi: 10.1186/s12943-020-01258-7
54. Lin JJ, Zhu VW, Yoda S, Yeap BY, Schrock AB, Dagogo-Jack I, et al. Impact of EML4-ALK Variant on Resistance Mechanisms and Clinical Outcomes in ALK-Positive Lung Cancer. J Clin Oncol. (2018) 36:1199–206. doi: 10.1200/JCO.2017.76.2294
55. Qin Z, Sun H, Yue M, Pan X, Chen L, Feng X, et al. Phase separation of EML4-ALK in firing downstream signaling and promoting lung tumorigenesis. Cell Discov. (2021) 7:33. doi: 10.1038/s41421-021-00270-5
56. Shen J, Meng Y, Wang K, Gao M, Du J, Wang J, et al. EML4-ALK G1202R mutation induces EMT and confers resistance to ceritinib in NSCLC cells via activation of STAT3/Slug signaling. Cell Signal. (2022) 92:110264. doi: 10.1016/j.cellsig.2022.110264
57. Beckman JD, DaSilva A, Aronovich E, Nguyen A, Nguyen J, Hargis G, et al. JAK-STAT inhibition reduces endothelial prothrombotic activation and leukocyte-endothelial proadhesive interactions. J Thromb Haemost. (2023) 21:1366–80. doi: 10.1016/j.jtha.2023.01.027
58. Elshatlawy M, Sampson J, Clarke K, and Bayliss R. EML4-ALK biology and drug resistance in non-small cell lung cancer: a new phase of discoveries. Mol Oncol. (2023) 17:950–63. doi: 10.1002/1878-0261.13446
59. Wang R, Li Y, Hu E, Kong F, Wang J, Liu J, et al. S100A7 promotes lung adenocarcinoma to squamous carcinoma transdifferentiation, and its expression is differentially regulated by the Hippo-YAP pathway in lung cancer cells. Oncotarget. (2017) 8:24804–14. doi: 10.18632/oncotarget.15063
60. Moya IM and Halder G. Hippo-YAP/TAZ signalling in organ regeneration and regenerative medicine. Nat Rev Mol Cell Biol. (2019) 20:211–26. doi: 10.1038/s41580-018-0086-y
61. Crawford JJ, Bronner SM, and Zbieg JR. Hippo pathway inhibition by blocking the YAP/TAZ-TEAD interface: a patent review. Expert Opin Ther Pat. (2018) 28:867–73. doi: 10.1080/13543776.2018.1549226
62. Gao Y, Zhang W, Han X, Li F, Wang X, Wang R, et al. YAP inhibits squamous transdifferentiation of Lkb1-deficient lung adenocarcinoma through ZEB2-dependent DNp63 repression. Nat Commun. (2014) 5:4629. doi: 10.1038/ncomms5629
63. Wang H, Tang S, Wu Q, He Y, Zhu W, Xie X, et al. Integrative study of lung cancer adeno-to-squamous transition in EGFR TKI resistance identifies RAPGEF3 as a therapeutic target. Natl Sci Rev. (2024) 11:e392. doi: 10.1093/nsr/nwae392
64. Bonanno L, Zulato E, Pavan A, Attili I, Pasello G, Conte P, et al. LKB1 and Tumor Metabolism: The Interplay of Immune and Angiogenic Microenvironment in Lung Cancer. Int J Mol Sci. (2019) 20:1874. doi: 10.3390/ijms20081874
65. Zhang H, Fillmore BC, Koyama S, Redig AJ, Chen T, Li S, et al. Lkb1 inactivation drives lung cancer lineage switching governed by Polycomb Repressive Complex 2. Nat Commun. (2017) 8:14922. doi: 10.1038/ncomms14922
66. Li F, Han X, Li F, Wang R, Wang H, Gao Y, et al. LKB1 Inactivation Elicits a Redox Imbalance to Modulate Non-small Cell Lung Cancer Plasticity and Therapeutic Response. Cancer Cell. (2015) 27:698–711. doi: 10.1016/j.ccell.2015.04.001
67. Han X, Li F, Fang Z, Gao Y, Li F, Fang R, et al. Transdifferentiation of lung adenocarcinoma in mice with Lkb1 deficiency to squamous cell carcinoma. Nat Commun. (2014) 5:3261. doi: 10.1038/ncomms4261
68. Cheung EC and Vousden KH. The role of ROS in tumour development and progression. Nat Rev Cancer. (2022) 22:280–97. doi: 10.1038/s41568-021-00435-0
69. Kim J, Hu Z, Cai L, Li K, Choi E, Faubert B, et al. CPS1 maintains pyrimidine pools and DNA synthesis in KRAS/LKB1-mutant lung cancer cells. Nature. (2017) 546:168–72. doi: 10.1038/nature22359
70. Kitajima S, Ivanova E, Guo S, Yoshida R, Campisi M, Sundararaman SK, et al. Suppression of STING Associated with LKB1 Loss in KRAS-Driven Lung Cancer. Cancer Discov. (2019) 9:34–45. doi: 10.1158/2159-8290.CD-18-0689
71. Gu M, Xu T, and Chang P. KRAS/LKB1 and KRAS/TP53 co-mutations create divergent immune signatures in lung adenocarcinomas. Ther Adv Med Oncol. (2021) 13:17548314. doi: 10.1177/17588359211006950
72. Tani T, Mathsyaraja H, Campisi M, Li ZH, Haratani K, Fahey CG, et al. TREX1 Inactivation Unleashes Cancer Cell STING-Interferon Signaling and Promotes Antitumor Immunity. Cancer Discov. (2024) 14:752–65. doi: 10.1158/2159-8290.CD-23-0700
73. Zhang H, Nabel CS, Li D, O’Connor RI, Crosby CR, Chang SM, et al. Histone Deacetylase 6 Inhibition Exploits Selective Metabolic Vulnerabilities in LKB1 Mutant, KRAS Driven NSCLC. J Thorac Oncol. (2023) 18:882–95. doi: 10.1016/j.jtho.2023.03.014
74. Long LL, Ma SC, Guo ZQ, Zhang YP, Fan Z, Liu LJ, et al. PARP Inhibition Induces Synthetic Lethality and Adaptive Immunity in LKB1-Mutant Lung Cancer. Cancer Res. (2023) 83:568–81. doi: 10.1158/0008-5472.CAN-22-1740
75. McGuire CK, Meehan AS, Couser E, Bull L, Minor AC, Kuhlmann-Hogan A, et al. Transcriptional repression by HDAC3 mediates T cell exclusion from Kras mutant lung tumors. Proc Natl Acad Sci U S A. (2024) 121:e1977273175. doi: 10.1073/pnas.2317694121
76. Eichner LJ, Curtis SD, Brun SN, McGuire CK, Gushterova I, Baumgart JT, et al. HDAC3 is critical in tumor development and therapeutic resistance in Kras-mutant non-small cell lung cancer. Sci Adv. (2023) 9:d3243. doi: 10.1126/sciadv.add3243
77. Bai X, Guo ZQ, Zhang YP, Fan ZZ, Liu LJ, Liu L, et al. CDK4/6 inhibition triggers ICAM1-driven immune response and sensitizes LKB1 mutant lung cancer to immunotherapy. Nat Commun. (2023) 14:1247. doi: 10.1038/s41467-023-36892-4
78. Rashidfarrokhi A, Pillai R, Hao Y, Wu WL, Karadal-Ferrena B, Dimitriadoy SG, et al. Tumor-intrinsic LKB1-LIF signaling axis establishes a myeloid niche to promote immune evasion and tumor growth. bioRxiv. (2023). doi: 10.1101/2023.07.15.549147
79. Guan S, Chen X, Chen Y, Xie W, Liang H, Zhu X, et al. FOXM1 Variant Contributes to Gefitinib Resistance via Activating Wnt/beta-Catenin Signal Pathway in Patients with Non-Small Cell Lung Cancer. Clin Cancer Res. (2022) 28:3770–84. doi: 10.1158/1078-0432.CCR-22-0791
80. Shukla S, Milewski D, Pradhan A, Rama N, Rice K, Le T, et al. The FOXM1 Inhibitor RCM-1 Decreases Carcinogenesis and Nuclear beta-Catenin. Mol Cancer Ther. (2019) 18:1217–29. doi: 10.1158/1535-7163.MCT-18-0709
81. Wang K, Dai X, Yu A, Feng C, Liu K, and Huang L. Peptide-based PROTAC degrader of FOXM1 suppresses cancer and decreases GLUT1 and PD-L1 expression. J Exp Clin Cancer Res. (2022) 41:289. doi: 10.1186/s13046-022-02483-2
82. Madhi H, Lee JS, Choi YE, Li Y, Kim MH, Choi Y, et al. FOXM1 Inhibition Enhances the Therapeutic Outcome of Lung Cancer Immunotherapy by Modulating PD-L1 Expression and Cell Proliferation. Adv Sci (Weinh). (2022) 9:e2202702. doi: 10.1002/advs.202202702
83. Lv B, Wang Y, Ma D, Cheng W, Liu J, Yong T, et al. Immunotherapy: Reshape the Tumor Immune Microenvironment. Front Immunol. (2022) 13:844142. doi: 10.3389/fimmu.2022.844142
84. Xue R, Zhang Q, Cao Q, Kong R, Xiang X, Liu H, et al. Liver tumour immune microenvironment subtypes and neutrophil heterogeneity. Nature. (2022) 612:141–47. doi: 10.1038/s41586-022-05400-x
85. Shaul ME and Fridlender ZG. Tumour-associated neutrophils in patients with cancer. Nat Rev Clin Oncol. (2019) 16:601–20. doi: 10.1038/s41571-019-0222-4
86. Jaillon S, Ponzetta A, Di Mitri D, Santoni A, Bonecchi R, and Mantovani A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat Rev Cancer. (2020) 20:485–503. doi: 10.1038/s41568-020-0281-y
87. Peng W, Sheng Y, Xiao H, Ye Y, Kwantwi LB, Cheng L, et al. Lung Adenocarcinoma Cells Promote Self-Migration and Self-Invasion by Activating Neutrophils to Upregulate Notch3 Expression of Cancer Cells. Front Mol Biosci. (2021) 8:762729. doi: 10.3389/fmolb.2021.762729
88. Wang Y, Li X, Zhang T, Li F, Shen Y, He Y, et al. Neutrophils promote tumor invasion via FAM3C-mediated epithelial-to-mesenchymal transition in gastric cancer. Int J Biol Sci. (2023) 19:1352–68. doi: 10.7150/ijbs.79022
89. Zhang D, Zhou J, Tang D, Zhou L, Chou L, Chou KY, et al. Neutrophil infiltration mediated by CXCL5 accumulation in the laryngeal squamous cell carcinoma microenvironment: A mechanism by which tumour cells escape immune surveillance. Clin Immunol. (2017) 175:34–40. doi: 10.1016/j.clim.2016.11.009
90. Kaur S, Bansal Y, Kumar R, and Bansal G. A panoramic review of IL-6: Structure, pathophysiological roles and inhibitors. Bioorg Med Chem. (2020) 28:115327. doi: 10.1016/j.bmc.2020.115327
91. Zegeye MM, Lindkvist M, Falker K, Kumawat AK, Paramel G, Grenegard M, et al. Activation of the JAK/STAT3 and PI3K/AKT pathways are crucial for IL-6 trans-signaling-mediated pro-inflammatory response in human vascular endothelial cells. Cell Commun Signal. (2018) 16:55. doi: 10.1186/s12964-018-0268-4
92. Liu Y, Xu K, Xiang Y, Ma B, Li H, Li Y, et al. Role of MCP-1 as an inflammatory biomarker in nephropathy. Front Immunol. (2023) 14:1303076. doi: 10.3389/fimmu.2023.1303076
93. Singh S, Anshita D, and Ravichandiran V. MCP-1: Function, regulation, and involvement in disease. Int Immunopharmacol. (2021) 101:107598. doi: 10.1016/j.intimp.2021.107598
94. Yoshimura T. The chemokine MCP-1 (CCL2) in the host interaction with cancer: a foe or ally? Cell Mol Immunol. (2018) 15:335–45. doi: 10.1038/cmi.2017.135
95. Xu M, Wang Y, Xia R, Wei Y, and Wei X. Role of the CCL2-CCR2 signalling axis in cancer: Mechanisms and therapeutic targeting. Cell Prolif. (2021) 54:e13115. doi: 10.1111/cpr.13115
96. Johnson DE, O’Keefe RA, and Grandis JR. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol. (2018) 15:234–48. doi: 10.1038/nrclinonc.2018.8
97. Lu J, Li J, Lin Z, Li H, Lou L, Ding W, et al. Reprogramming of TAMs via the STAT3/CD47-SIRPalpha axis promotes acquired resistance to EGFR-TKIs in lung cancer. Cancer Lett. (2023) 564:216205. doi: 10.1016/j.canlet.2023.216205
98. Chuammitri P, Wongsawan K, Pringproa K, and Thanawongnuwech R. Interleukin 17 (IL-17) manipulates mouse bone marrow- derived neutrophils in response to acute lung inflammation. Comp Immunol Microbiol Infect Dis. (2019) 67:101356. doi: 10.1016/j.cimid.2019.101356
99. Que H, Fu Q, Lan T, Tian X, and Wei X. Tumor-associated neutrophils and neutrophil-targeted cancer therapies. Biochim Biophys Acta Rev Cancer. (2022) 1877:188762. doi: 10.1016/j.bbcan.2022.188762
100. Ritzmann F, Lunding LP, Bals R, Wegmann M, and Beisswenger C. IL-17 Cytokines and Chronic Lung Diseases. Cells. (2022) 11:2132. doi: 10.3390/cells11142132
101. Li S, Cong X, Gao H, Lan X, Li Z, Wang W, et al. Tumor-associated neutrophils induce EMT by IL-17a to promote migration and invasion in gastric cancer cells. J Exp Clin Cancer Res. (2019) 38:6. doi: 10.1186/s13046-018-1003-0
102. Wang Y, Huang X, Fan H, Xu Y, Qi Z, Zhang Y, et al. Identification of fatty acid-related subtypes, the establishment of a prognostic signature, and immune infiltration characteristics in lung adenocarcinoma. Aging (Albany NY). (2023) 15:4202–35. doi: 10.18632/aging.204725
103. Zhou J, Jiang G, Xu E, Zhou J, Liu L, and Yang Q. Identification of SRXN1 and KRT6A as Key Genes in Smoking-Related Non-Small-Cell Lung Cancer Through Bioinformatics and Functional Analyses. Front Oncol. (2021) 11:810301. doi: 10.3389/fonc.2021.810301
104. Augert A, Eastwood E, Ibrahim AH, Wu N, Grunblatt E, Basom R, et al. Targeting NOTCH activation in small cell lung cancer through LSD1 inhibition. Sci Signal. (2019) 12:eaau2922. doi: 10.1126/scisignal.aau2922
105. Che D, Wang M, Sun J, Li B, Xu T, Lu Y, et al. KRT6A Promotes Lung Cancer Cell Growth and Invasion Through MYC-Regulated Pentose Phosphate Pathway. Front Cell Dev Biol. (2021) 9:694071. doi: 10.3389/fcell.2021.694071
106. Yang B, Zhang W, Zhang M, Wang X, Peng S, and Zhang R. KRT6A Promotes EMT and Cancer Stem Cell Transformation in Lung Adenocarcinoma. Technol Cancer Res Treat. (2020) 19:1079188896. doi: 10.1177/1533033820921248
107. Xu Q, Yu Z, Mei Q, Shi K, Shen J, Gao G, et al. Keratin 6A (KRT6A) promotes radioresistance, invasion, and metastasis in lung cancer via p53 signaling pathway. Aging (Albany NY). (2024) 16:7060–72. doi: 10.18632/aging.205742
108. Soldani C, De Simone G, Polidoro MA, Morabito A, Franceschini B, Colombo FS, et al. Riboflavin-LSD1 axis participates in the in vivo tumor-associated macrophage morphology in human colorectal liver metastases. Cancer Immunol Immunother. (2024) 73:63. doi: 10.1007/s00262-024-03645-1
109. Sobczak M, Strachowska M, Gronkowska K, Karwaciak I, Pulaski L, and Robaszkiewicz A. LSD1 Facilitates Pro-Inflammatory Polarization of Macrophages by Repressing Catalase. Cells. (2021) 10:2465. doi: 10.3390/cells10092465
110. Seifermann M and Epe B. Oxidatively generated base modifications in DNA: Not only carcinogenic risk factor but also regulatory mark? Free Radic Biol Med. (2017) 107:258–65. doi: 10.1016/j.freeradbiomed.2016.11.018
111. Tan A, Tu W, McCuaig R, Hardy K, Donovan T, Tsimbalyuk S, et al. Lysine-Specific Histone Demethylase 1A Regulates Macrophage Polarization and Checkpoint Molecules in the Tumor Microenvironment of Triple-Negative Breast Cancer. Front Immunol. (2019) 10:1351. doi: 10.3389/fimmu.2019.01351
112. Petty AJ and Yang Y. Tumor-associated macrophages: implications in cancer immunotherapy. Immunotherapy. (2017) 9:289–302. doi: 10.2217/imt-2016-0135
113. Gao J, Liang Y, and Wang L. Shaping Polarization Of Tumor-Associated Macrophages In Cancer Immunotherapy. Front Immunol. (2022) 13:888713. doi: 10.3389/fimmu.2022.888713
114. Strizova Z, Benesova I, Bartolini R, Novysedlak R, Cecrdlova E, Foley LK, et al. M1/M2 macrophages and their overlaps - myth or reality? Clin Sci (Lond). (2023) 137:1067–93. doi: 10.1042/CS20220531
115. Pan Y, Yu Y, Wang X, and Zhang T. Tumor-Associated Macrophages in Tumor Immunity. Front Immunol. (2020) 11:583084. doi: 10.3389/fimmu.2020.583084
116. Du L, Yang H, Ren Y, Ding Y, Xu Y, Zi X, et al. Inhibition of LSD1 induces ferroptosis through the ATF4-xCT pathway and shows enhanced anti-tumor effects with ferroptosis inducers in NSCLC. Cell Death Dis. (2023) 14:716. doi: 10.1038/s41419-023-06238-5
117. Alhousami T, Diny M, Ali F, Shin J, Kumar G, Kumar V, et al. Inhibition of LSD1 Attenuates Oral Cancer Development and Promotes Therapeutic Efficacy of Immune Checkpoint Blockade and YAP/TAZ Inhibition. Mol Cancer Res. (2022) 20:712–21. doi: 10.1158/1541-7786.MCR-21-0310
118. Dai X, Lu L, Deng S, Meng J, Wan C, Huang J, et al. USP7 targeting modulates anti-tumor immune response by reprogramming Tumor-associated Macrophages in Lung Cancer. Theranostics. (2020) 10:9332–47. doi: 10.7150/thno.47137
119. Mamun M, Zhang Y, Zhao JY, Shen DD, Guo T, Zheng YC, et al. LSD1: an emerging face in altering the tumor microenvironment and enhancing immune checkpoint therapy. J BioMed Sci. (2023) 30:60. doi: 10.1186/s12929-023-00952-0
120. Li H, Wu BK, Kanchwala M, Cai J, Wang L, Xing C, et al. YAP/TAZ drives cell proliferation and tumour growth via a polyamine-eIF5A hypusination-LSD1 axis. Nat Cell Biol. (2022) 24:373–83. doi: 10.1038/s41556-022-00848-5
121. Yang XF, Zhou SY, Wang C, Huang W, Li N, He F, et al. Inhibition of LSD1 promotes the differentiation of human induced pluripotent stem cells into insulin-producing cells. Stem Cell Res Ther. (2020) 11:185. doi: 10.1186/s13287-020-01694-8
122. Cuyas E, Gumuzio J, Verdura S, Brunet J, Bosch-Barrera J, Martin-Castillo B, et al. The LSD1 inhibitor iadademstat (ORY-1001) targets SOX2-driven breast cancer stem cells: a potential epigenetic therapy in luminal-B and HER2-positive breast cancer subtypes. Aging (Albany NY). (2020) 12:4794–814. doi: 10.18632/aging.102887
123. Noce B, Di Bello E, Fioravanti R, and Mai A. LSD1 inhibitors for cancer treatment: Focus on multi-target agents and compounds in clinical trials. Front Pharmacol. (2023) 14:1120911. doi: 10.3389/fphar.2023.1120911
124. Li M, Liu M, Han W, Wang Z, Han D, Patalano S, et al. LSD1 Inhibition Disrupts Super-Enhancer-Driven Oncogenic Transcriptional Programs in Castration-Resistant Prostate Cancer. Cancer Res. (2023) 83:1684–98. doi: 10.1158/0008-5472.CAN-22-2433
125. Sang N, Zhong X, Gou K, Liu H, Xu J, Zhou Y, et al. Pharmacological inhibition of LSD1 suppresses growth of hepatocellular carcinoma by inducing GADD45B. Medcomm (2020). (2023) 4:e269. doi: 10.1002/mco2.269
126. Liu B, Liu X, Han L, Chen X, Wu X, Wu J, et al. BRD4-directed super-enhancer organization of transcription repression programs links to chemotherapeutic efficacy in breast cancer. Proc Natl Acad Sci U S A. (2022) 119:e2109133119. doi: 10.1073/pnas.2109133119
127. Shen DD, Pang JR, Bi YP, Zhao LF, Li YR, Zhao LJ, et al. LSD1 deletion decreases exosomal PD-L1 and restores T-cell response in gastric cancer. Mol Cancer. (2022) 21:75. doi: 10.1186/s12943-022-01557-1
128. Wang H, Song Z, Xie E, Chen J, Tang B, Wang F, et al. Targeting the LSD1-G9a-ER Stress Pathway as a Novel Therapeutic Strategy for Esophageal Squamous Cell Carcinoma. Res (Wash D C). (2022) 2022:9814652. doi: 10.34133/2022/9814652
129. Ravasio R, Ceccacci E, Nicosia L, Hosseini A, Rossi PL, Barozzi I, et al. Targeting the scaffolding role of LSD1 (KDM1A) poises acute myeloid leukemia cells for retinoic acid-induced differentiation. Sci Adv. (2020) 6:x2746. doi: 10.1126/sciadv.aax2746
130. Sheng W, LaFleur MW, Nguyen TH, Chen S, Chakravarthy A, Conway JR, et al. LSD1 Ablation Stimulates Anti-tumor Immunity and Enables Checkpoint Blockade. Cell. (2018) 174:549–63. doi: 10.1016/j.cell.2018.05.052
131. Li Y, Wang T, Li X, Li W, Lei Y, Shang Q, et al. SOD2 promotes the immunosuppressive function of mesenchymal stem cells at the expense of adipocyte differentiation. Mol Ther. (2024) 32:1144–57. doi: 10.1016/j.ymthe.2024.01.031
132. Zanetti M, Xian S, Dosset M, and Carter H. The Unfolded Protein Response at the Tumor-Immune Interface. Front Immunol. (2022) 13:823157. doi: 10.3389/fimmu.2022.823157
133. Jiang W, Yang Z, Chen P, Zhao M, Wang Y, Wang J, et al. Tolperisone induces UPR-mediated tumor inhibition and synergizes with proteasome inhibitor and immunotherapy by targeting LSD1. Expert Opin Ther Targets. (2023) 27:879–95. doi: 10.1080/14728222.2023.2259097
134. Su H, Yang F, Fu R, Li X, French R, Mose E, et al. Cancer cells escape autophagy inhibition via NRF2-induced macropinocytosis. Cancer Cell. (2021) 39:678–93. doi: 10.1016/j.ccell.2021.02.016
135. Cheng Y, Mo F, Li Q, Han X, Shi H, Chen S, et al. Targeting CXCR2 inhibits the progression of lung cancer and promotes therapeutic effect of cisplatin. Mol Cancer. (2021) 20:62. doi: 10.1186/s12943-021-01355-1
136. Bodac A, Mayet A, Rana S, Pascual J, Bowler AD, Roh V, et al. Bcl-xL targeting eliminates ageing tumor-promoting neutrophils and inhibits lung tumor growth. EMBO Mol Med. (2024) 16:158–84. doi: 10.1038/s44321-023-00013-x
Keywords: adenosquamous lung carcinoma (ASLC), adenocarcinoma-to-squamous cell carcinoma transformation (AST), tumor immune microenvironment (TIME), tumor-associated macrophages (TAMs), tumor-associated neutrophils (TANs)
Citation: Xu H, Yang Y, Wang P, Lin S, Zhang X, Ni H and Xu Z (2025) Unraveling the immune mechanisms and therapeutic targets in lung adenosquamous transformation. Front. Immunol. 16:1542526. doi: 10.3389/fimmu.2025.1542526
Received: 10 December 2024; Accepted: 14 May 2025;
Published: 03 June 2025.
Edited by:
Yongqiang Zhang, Henan Academy of Innovations in Medical Science, ChinaReviewed by:
Zhimin Zeng, Second Affiliated Hospital of Nanchang University, ChinaYuqin Tang, Henan Provincial People’s Hospital, China
Xin-Yu Shi, Capital Medical University, China
Yanjie Huang, Karolinska Institutet (KI), Sweden
Copyright © 2025 Xu, Yang, Wang, Lin, Zhang, Ni and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhiyong Xu, eHV6aGl5b25nQHpqdS5lZHUuY29t
†These authors have contributed equally to this work