 Elena Jechel1†
Elena Jechel1† Alin Horatiu Nedelcu2†
Alin Horatiu Nedelcu2† Felicia Dragan3*
Felicia Dragan3* Vasile Valeriu Lupu1*
Vasile Valeriu Lupu1* Iuliana Magdalena Starcea1
Iuliana Magdalena Starcea1 Adriana Mocanu1
Adriana Mocanu1 Solange Tamara Rosu2
Solange Tamara Rosu2 Violeta Streanga1
Violeta Streanga1 Ruxandra Russu2Ginel Baciu4†Ciprian Danielescu2†
Ruxandra Russu2Ginel Baciu4†Ciprian Danielescu2† Delia Lidia Salaru2Ionela Daniela Morariu5Olga Cirstea6Emil Anton2†
Delia Lidia Salaru2Ionela Daniela Morariu5Olga Cirstea6Emil Anton2† Ancuta Lupu1
Ancuta Lupu1- 1Pediatrics, Faculty of Medicine, “Grigore T. Popa” University of Medicine and Pharmacy, Iasi, Romania
- 2Faculty of Medicine, “Grigore T. Popa” University of Medicine and Pharmacy, Iasi, Romania
- 3Faculty of Medicine and Pharmacy, University of Oradea, Oradea, Romania
- 4Pediatrics, Faculty of Medicine and Pharmacy, “Dunarea de Jos” University of Galati, Galati, Romania
- 5Faculty of Pharmacy, “Grigore T. Popa” University of Medicine and Pharmacy, Iasi, Romania
- 6Pediatrics, “Nicolae Testemitanu” State University of Medicine and Pharmacy, Chisinau, Moldova
Pediatric nephrotic syndrome remains a complex clinical entity, with incompletely elucidated pathogenetic mechanisms, in which oxidative stress appears to have a substantial etiopathogenic role. Recent evidence supports the involvement of redox imbalance in podocyte damage, impaired glomerular function, and systemic decline. All this suggests that antioxidant interventions can favorably modulate the course of the disease. This narrative review aims to synthesize the most relevant data from the current literature on the interaction between oxidative stress and nephrotic syndrome in children, with a focus on the therapeutic potential of antioxidants. The analysis focuses on the molecular mechanisms by which oxidative stress contributes to the progression of renal dysfunction, the role of oxidative biomarkers in disease monitoring, and the ability of antioxidants to reduce the need for immunosuppressants and corticosteroids, thus contributing to the decrease in associated morbidity. The translational perspectives of antioxidant therapy are also discussed, in the context of the urgent need for effective adjuvant strategies with a safety profile superior to conventional therapies. By integrating these data, the paper supports the valorization of antioxidant interventions as an emerging direction in the management of pediatric nephrotic syndrome and substantiates the need for controlled clinical trials, with rigorous design, in this field.
1 Introduction
Nephrotic syndrome (NS) is a complex condition characterized by a chronic undulating evolution and is associated with increased morbidity and mortality. Particularly concerning pediatric patients, its individualized, multidisciplinary management is vital to mitigate systemic decline and improve both morbidity and psycho-social well-being (1, 2). Clinically, NS presents with proteinuria (urinary protein/creatinine ratio over 24 hours ≥ 200 mg/mmol or 3+ on urine strips), hypoalbuminemia (serum albumin < 3 g/dl), and edema. Additionally, patients with NS can also exhibit hyperlipidemia and an increased risk of thrombosis (3, 4). The incidence and prevalence of pediatric NS are approximately 3 per 100,000 and 16 per 100,000 children respectively, with a peak occurrence between ages 2 and 7. Genetic predisposition and geographic factors contribute significantly to disease distribution. Multiple genetic variations have been identified in intrafamilial aggregates and NS appears to be more common among young boys (5–7).
NS can be divided into primary and secondary forms. Secondary NS arises as a consequence of other systemic pathologies (e.g., infections, neoplasms, systemic lupus erythematosus, diabetes) or the improper use of nephrotoxic substances (e.g., medications) (8, 9). A rarer form, congenital NS is defined as the appearance of symptoms in newborns or infants younger than 3 months and is primarily linked to genetic variations, perinatal infections (e.g. syphilis, toxoplasmosis), or alloimmune diseases (10, 11). Among these, primary NS is the most prevalent in pediatrics. Depending on the histopathological characteristics of kidney damage, NS most commonly presents with minimal lesions or as focal segmental glomerulosclerosis (FSGS) (8). In addition to the microscopic aspect, the minimal lesions appear earlier (median age of 3 years versus 6 years in FSGS) and respond more favorably to corticosteroid treatment. In contrast, current data suggests that FSGS responds poorly to steroids and is therefore an important cause of progression to chronic kidney disease in children (6, 8, 12).
If a patient is unresponsive to steroid treatment, the current guidelines recommend escalating therapy by combining immunosuppressive drugs such as calcineurin inhibitors (Cyclosporin, Tacrolimus), mycophenolate mofetil, anti-CD20 monoclonal antibodies (Rituximab, Ofatumumab), and CTLA- 4 (Abatacept, Belatacept). The preference for a certain type of medication remains dictated by the particularities of the patient (12–15). To prevent and counteract the main comorbidities specific to NS, supportive therapy must include lifestyle modifications (e.g., diet) or the administration of pharmacological substances such as diuretics, lipid-lowering drugs, antihypertensives (renin-angiotensin axis inhibitors), and anticoagulants (8, 12, 16). Current global research highlights the decisive role of intestinal microbiota modulation through the intestinal axes - target organ system. Studies note that microbiome dynamics can negatively impact the prevalence and clinical course of various pathologies, including inflammatory, autoimmune, and atopic diseases. Among the physiological mechanisms affected in this process, we note the induction of chronic low-grade systemic inflammation and an imbalance in oxidative status, marked by increased oxidative stress (OS) and reduced total antioxidant capacity (17–20). The interrelationship between the two (increased OS, together with decreased antioxidant capacity) induces a systemic deficiency in endogenous antioxidant (EA) defenses (e.g., glutathione, superoxide dismutase, catalase) which aggravates cellular damage, promotes podocyte damage, and increases proteinuria. This sustains the loss of urinary albumin, a protein with antioxidant properties, while maintaining the vicious cycle of inflammation, glomerular injury, and endothelial dysfunction. The effect of proteinuria on urinary excretion of microelements in NS has been intensively studied, revealing significantly higher urinary losses in children experiencing relapse. This loss is accompanied by a blood deficiency of these microelements compared to children in remission and healthy controls. Over time, these factors contribute to the disruption of systemic and renal homeostasis, leading to NS-specific complications, progression to chronic kidney disease, and a diminished capacity for optimal treatment response. Additionally, the literature highlights a bidirectional correlation between oxidative status and therapeutic response/efficacy. Zinc has been identified as a promising adjuvant in the treatment of pediatric NS, with supplementation helping to reduce relapses and infection-related episodes associated with relapse, thereby prolonging the disease-free interval. A decrease in pro-inflammatory status parallels the development of remission, while oxidative and nitrative stress influence the response to steroid therapy. This relationship is reinforced by the strong link between selenium deficiency – resulting from urinary albumin-induced losses – and steroid resistance. The latter may occur possibly through the oxidation and inactivation of glucocorticoid receptors, coupled with reduced receptor expression at the cellular level. Selenium also intervenes in the dynamics of the malondialdehyde-glutathione balance and total antioxidant capacity. Consequently, we are confident that reducing OS markers in children with NS will enhance therapeutic response (21–30).
Based on these findings and current trends, this work intends to showcase the current understanding of how exogenous (ExA) and EAs influence the progression of pediatric NS. We will look at data on the absorption and secretion of antioxidants and antioxidant-related components, physiological metabolism, and the pathological pathways that arise when internal homeostasis is disrupted. To meet our goal, we performed a comprehensive literature review without setting strict inclusion criteria. We sourced the most recent available data from PubMed, Google Scholar, Web of Science (WoS), Scopus, and Embase, including subject encyclopedias. Search criteria were, therefore, defined by using terms and phrases like: “Nephrotic Syndrome”, “Renal Damage”, “Chronic Renal Progression”, combined with “Child”, “Oxidative Stress”, “Endogenous Antioxidants”, “Exogenous Antioxidants”, “Prognostic Biomarkers”, “alternative therapies”, and “Individualized management”. The study population included patients younger than 18 years at the time of reporting. To address gaps in the pediatric literature, we supplemented our analysis with relevant findings from adult studies and murine models where necessary. We minimized the risk of bias by not restricting our search based on language. Through this research, we aim to expand and integrate knowledge about oxidative balance in NS dynamics, considering major pathophysiological mechanisms and their implications. Ultimately, we hope our findings will contribute to advancing the individualized diagnosis and management of pediatric NS.
2 Main pathophysiological mechanisms involved in NS
At the level of the renal interstitium, the boundary between the glomerular capillaries and the urinary space is formed by the filtration membrane, which serves as a dual structural and electrochemical barrier. This membrane is made up of the capillary endothelium, the glomerular basement membrane, and podocyte extensions. Its main role is to stop the passage of substances with high molecular weight (e.g., proteins) into the urine. In NS, the glomerular basement membrane undergoes structural changes that alter its mechanical function and electrostatic balance. The main changes are reflected in the podocyte structures, the essential element in the glomerular filtration barrier. Notably, the disappearance of the slit diaphragm and the erasure of podocytic processes were observed (31–33). Following these changes, the injured glomerular filtration membrane allows the passage of albumin molecules (selective proteinuria) or high molecular weight plasma proteins (non-selective proteinuria) into the urine. Among the proteins with a functional role lost in the case of non-selective proteinuria, we note transferrin, ceruloplasmin, immunoglobulins, and fractions of the complement system, factors involved in the dynamics of coagulation, but also other enzymes and vitamins with a role in various metabolic processes (31, 34–36).
Persistent proteinuria contributes to progressive renal impairment, ultimately leading to the destruction of physiological architecture, tubular atrophy, and interstitial fibrosis (37, 38). Among the clinical-biological consequences of the pathological glomerular disturbances encountered in NS, we note malnutrition, anemia, nutritional deficiencies, procoagulant status, hypoalbuminemia, dyslipidemias, endocrinopathies, and increased susceptibility to infections due to immune deficiency. These, together with other such examples, are presented in a centralized manner in Table 1. The entire hemodynamic balance is deregulated by the activation of the renin-angiotensin system, the occurrence of hydro-electrolytic and acid-base imbalances, and peripheral edema, localized or generalized, which affect vital organs such as the nervous system, osteoarticular apparatus, liver, lung parenchyma, and cardiovascular system (38–42). This process perpetuates a vicious cycle of renal deterioration, characterized by hyperfiltration, reduced arteriolar resistance, compensatory glomerular hypertrophy, and progressive systemic decline (35, 36).
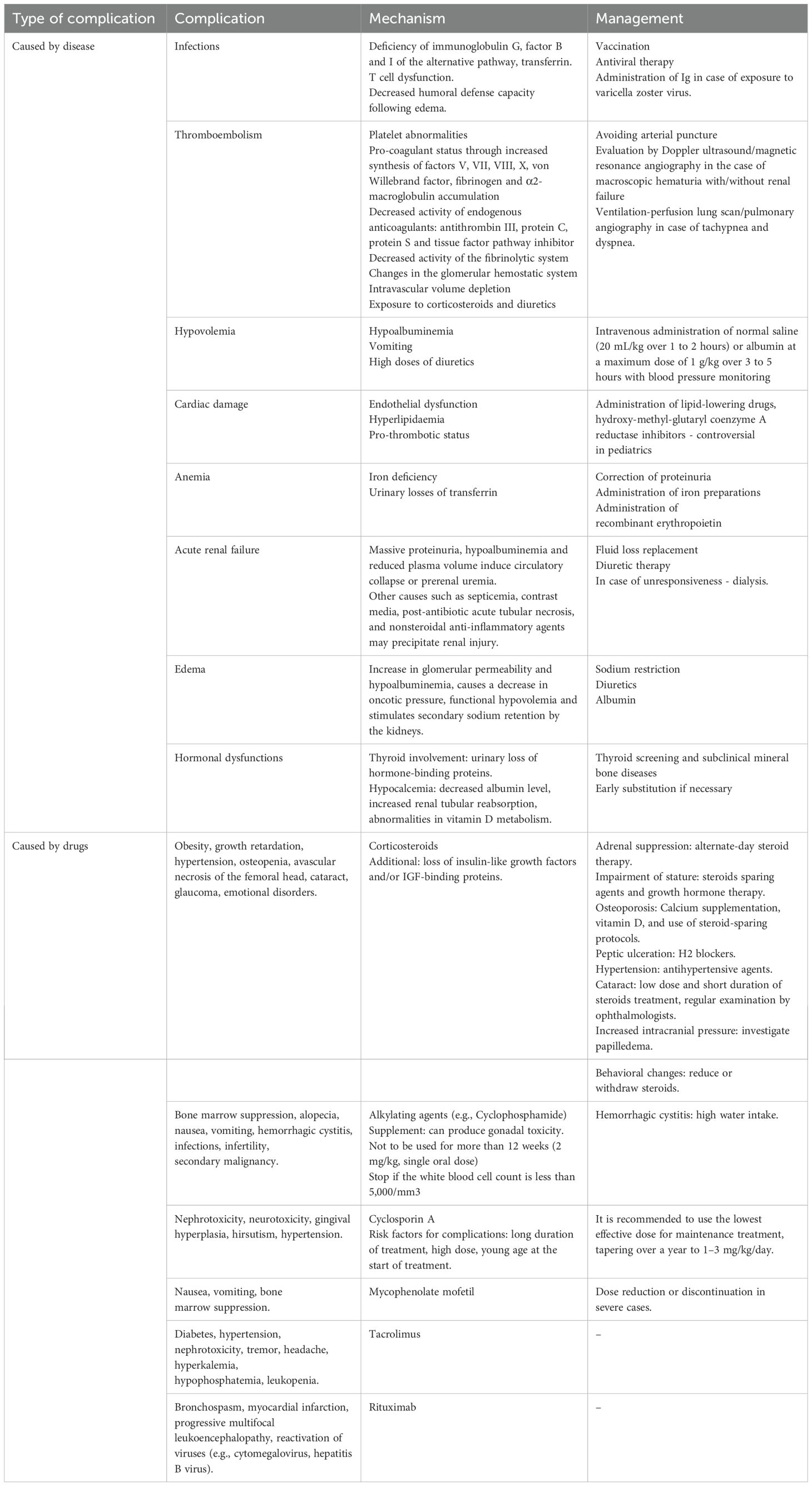
In the extensive process described previously, the systemic pro-oxidative status appears to be the central driver of the vicious cycle. The increase in free radicals together with the decrease in antioxidant capacity both play a pivotal role in potentiating renal damage by causing damage to the glomerular filtration barrier, cell damage, and endothelial dysfunction. Aside from this, they also promote inflammation, disrupt lipid metabolism, alter the optimal therapeutic response, or favor the development of comorbidities - aspects observed in the pediatric population, but also in adults. The pro-oxidant – antioxidant balance has also been shown to correlate with the evolutionary stages of NS (43–45). Over time, dyslipidemia and OS become critical factors in atherosclerosis development and the progression to cardiovascular disease and chronic renal failure. In support of this, Jabarpour M. et al. emphasize the importance of implementing therapies to mitigate these two variables (46). Furthermore, Bufna A. et al. report that corticosteroid induction therapy appeared to improve total antioxidant capacity (47)., However, caution is warranted, as other studies report that markers of OS remain elevated even after steroid therapy, highlighting its persistent role in NS pathophysiology (45). Consequently, its implications in NS dynamics remain indisputable, being detailed in what follows. Finally, Figure 1 schematically illustrates the pathophysiological cascade specific to the onset, progression, and complication of NS in pediatrics with respect to OS.
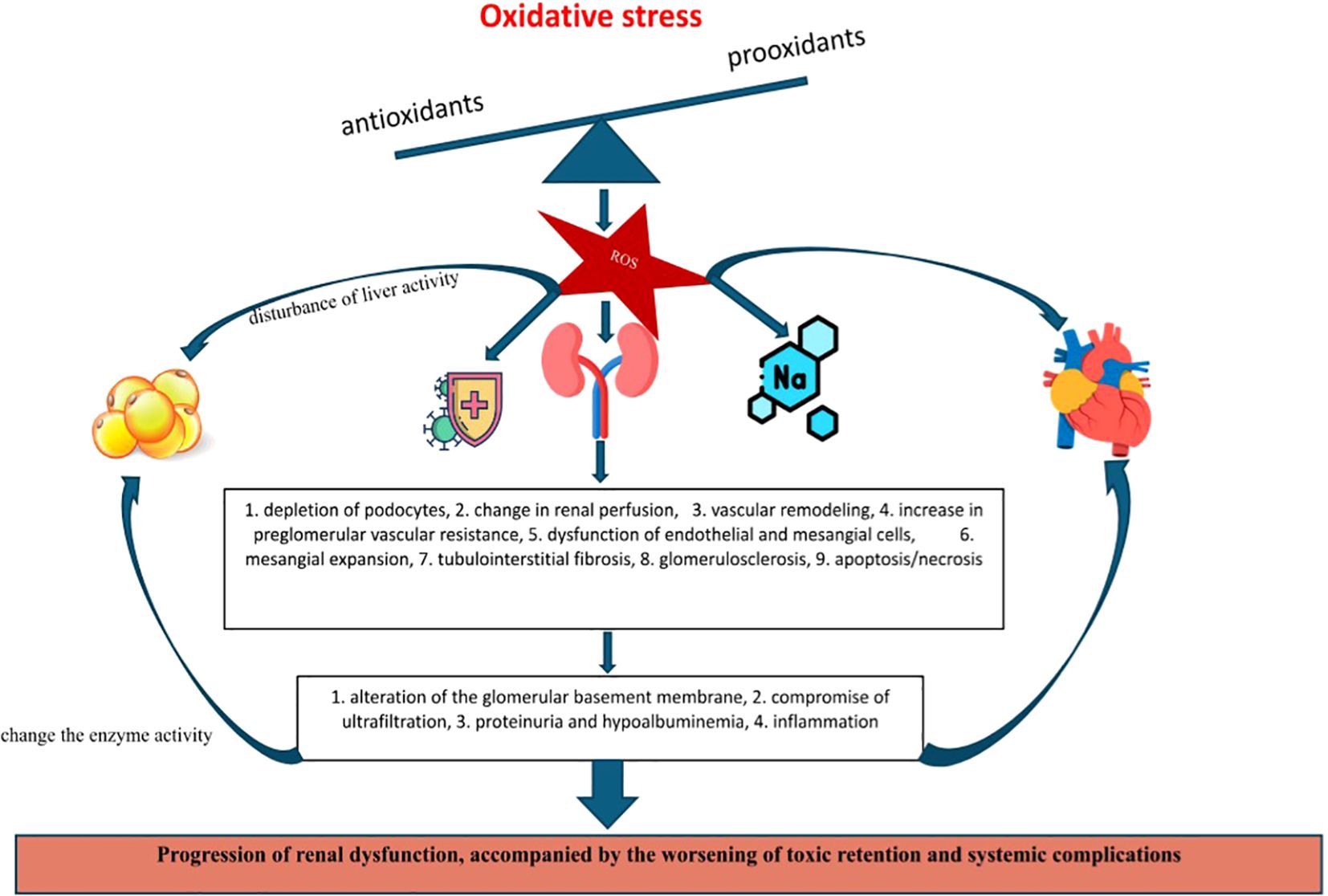
Figure 1. OS implications in the promotion of renal dysfunction and specific complications of pediatric NS.
3 Oxidative stress
OS is defined as a disturbance of the systemic pro-oxidative - anti-oxidative balance. The consequences of this imbalance are the excessive production of reactive oxygen species (ROS), reactive nitrogen species, and other species of free radicals. At the same time, there is a reduced antioxidant capacity to neutralize reactive species and counteract damage. Physiologically, OS has a role in cell signaling and immune defense. But, when its level exceeds the capacity of antioxidant defense, reactive species can induce the oxidation of lipids, proteins, and nucleic acids. These seem to be the mechanisms by which ROS participate in the induction of low-grade chronic inflammation, cellular dysfunction, and implicitly in the promotion and aggravation of chronic conditions (e.g., cardiovascular, pulmonary, renal, metabolic, neurological, malignancies) (48, 49). Following on from what has been discussed, Bissinger R. et al. note that OS acts as a bridge between chronic conditions and various complications such as anemia (50).
Regarding kidney damage, OS is correlated in the literature with both antioxidant depletion and excessive ROS production, serving as both a cause and an effect of renal degradation. The kidney’s vulnerability to OS appears to stem from its intense metabolic activity at the mitochondrial level. In relation to this, Daenen K. et al. reassert the link between OS and the progression of renal damage, further complicated by hypertension, atherosclerosis, inflammation, and anemia (51). In particular, cardiovascular damage is greatly accentuated in patients undergoing replacement therapy, such as hemodialysis, peritoneal dialysis, or renal transplantation, where persistent exposure to a pro-oxidative state intensifies systemic deterioration (52).
Given the complexity of this systemic condition, Table 2 summarizes the main exogenous and endogenous factors that interfere with the oxidative balance in kidney damage. To counteract their effects and restore homeostasis, the presence of enzymatic and non-enzymatic antioxidants in circulation is necessary. These antioxidants are currently recognized as important both in expanding diagnostic capabilities but also for their therapeutic role in mitigating systemic damage (53, 54).
4 Consequences of oxidative stress in renal and systemic decline
Summarizing the above, we note that OS (whether exogenous or endogenous) serves an important role in promoting and sustaining of systemic damage. This is mainly achieved by maintaining a chronic inflammatory status that disrupts physiological molecular processes. In the renal system, OS has been implicated both in renal injury and in increasing the risk of complications, including impaired therapeutic responses (e.g., corticosteroid resistance). The following sections provide a detailed exploration of these aspects and the pathophysiological mechanisms that amplify their impact.
4.1 Renal and glomerular injuries
Optimal renal functions – such as waste filtration, regulation of electrolyte balance, and bone integrity maintenance – are closely linked to adequate circulatory flow. It is estimated that the kidneys receive approximately one-fifth of cardiac output at rest, yet the oxygen pressure in the renal interstitium remains relatively low due to the arterio-venous mechanism. Notably, the partial pressure of O2 is higher in the renal vein than in the efferent arteriole or cortex. Renal blood flow is regulated by both intrinsic factors (e.g., myogenic response, tubulo-glomerular feedback) and extrinsic factors (e.g., neuro-hormonal signals). These self-regulatory changes in the renal plasma flow are intended to minimize the variations in the ultrafiltered volumes (65–68). Under conditions of oxidative stress, reactive species can alter renal perfusion, leading to vascular remodeling and increased preglomerular resistance – key factors in the development of hypertension as well as acute and chronic renal lesions. The primary target of this process is the tone of the renal afferent arteriole, followed by alterations in the myogenic response and tubulo-glomerular feedback (68, 69).
At the cellular level, OS represents a significant cause of endothelial and mesangial cell dysfunction, which in turn compromises vascular and glomerular integrity. Under normal conditions, endothelial cells promote vasodilation by facilitating the release of vasoactive substances. However, excess ROS interact with nitric oxide to form peroxynitrite, reducing nitric oxide bioavailability. Given nitric oxide’s critical role in vasodilation, anti-aggregation, and leukocyte adhesion prevention, its depletion leads to endothelial dysfunction. Subsequently, the activated endothelial cells produce vasoconstrictor agents, amplifying the inflammatory response. The resulting upregulation of adhesion molecules and secretion of chemokines establishes a vicious cycle that sustains inflammation (70).
The reference studies attest that endothelial dysfunction appears from the first stages of renal damage and correlates with disease activity, particularly the degree of proteinuria and serum albumin levels. Paraclinically, markers such as soluble thrombomodulin, tissue plasminogen activator, plasminogen activator inhibitor-1, and von-Willebrand factor help to assess this dysfunction. Endothelial damage not only contributes to systemic atherosclerosis but also accelerates kidney disease progression by increasing capillary permeability and aggravating proteinuria. However, evidence suggests that this process may be reversible, making it a potential therapeutic target for mitigating renal injury (71–73).
Further studies on chronic kidney damage models, such as diabetic kidney disease, reveal that podocyte depletion occurs due to the exposure of glomeruli to harmful stimuli, OS inducers such as hyperglycemia, hyperuremia, increased levels of fatty acids, growth factors, cytokines, and hormones (74–76). OS-induced podocyte damage manifests in multiple ways, primarily through the disruption of structural and functional integrity. This includes membrane dysfunction, cytoskeletal damage, mitochondrial impairments, inefficient energy production, as well as the activation of pro-apoptotic and pro-necrotic signaling pathways (e.g., caspases, mitogen-activated protein kinase). Other mechanisms involved in OS-induced podocyte damage include autophagy, pyroptosis, ferroptosis, and mitotic catastrophe.
In this sense, it is noted that targeting cellular pathways involved in OS dynamics (e.g., modulation of the TSP-1/TGF-β1/Smad pathway by overexpression of Sestrin2) protects podocytes from apoptosis. Conversely, exposure to harmful stimuli (e.g., hyperglycemia) has been associated with decreased protective autophagy. Thus, current efforts are focused on preserving basal autophagy levels in podocytes, a goal achievable through the administration of antioxidant compounds such as berberine, mangiferin, notoginsenoside R1, and geniposide (75, 77, 78). From a pathophysiological perspective, OS-induced podocyte apoptosis and necrosis compromise the glomerular filtration barrier, impairing ultrafiltration. This disruption allows plasma proteins to pass through the glomerular barrier, depending on their size and extent of the damage. The subsequent proteinuria perpetuates a vicious cycle by triggering inflammatory responses which further exacerbate OS, worsening podocyte damage and sustaining NS-specific symptomatology – possibly through the activation of the Wnt/β-catenin pathway (77–81).
Excessive OS exerts a dual impact on the integrity of the glomerular filtration barrier, both inducing dysfunction and exacerbating proteinuria and renal decline. Research highlights a bidirectional relationship between glomerular endothelial dysfunction following OS and podocyte damage, reflected in the dynamics of vascular endothelial growth factor A, angiopoietins, CXCL12/CXCR4/CXCR7, endothelin-1, interleukin (IL)-6, and extracellular vesicles at the paraclinical level. In practical terms, endothelial dysfunction disrupts glomerular pressure and blood flow, exacerbating local ischemia and the pro-oxidative status, accelerating podocyte damage. In turn, damaged podocytes lose their ability to maintain glomerular structure and trigger a local inflammatory reaction. It is also noted that counteracting mitochondrial OS by means of ExAs has led to the prevention of podocyte loss, albuminuria, and glomerulosclerosis, improving renal function. Findings from murine models suggest that early renal damage is characterized by endothelial cell injury, which precedes podocyte process deletion (74, 75, 77, 80, 82).
4.2 Inflammation and fibrosis
Following renal parenchymal damage due to OS, repair and regeneration processes are initiated. Physiologically, the repair of damaged tissue is characterized in the first phase by the appearance of myofibroblasts which contribute to the production of collagen and other components of the extracellular matrix. The success of parenchymal regeneration depends on the integrity of this process. Pathological variations occur when inflammatory and fibrogenic signaling is either incomplete or persistently activated. In the former case, repair remains latent and seems to re-initiate after an acute insult. In the latter, excessive extracellular matrix deposition leads to structural and functional impairment, culminating glomerular sclerosis (83).
Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) plays an important regulatory role in the expression of genes involved in inflammation, immune response, and cell survival. Under physiological conditions, NF-κB is located in the cytosol in an inactive state, bound to inhibitory proteins. Upon stimulation by viral or bacterial antigens, cytokines, or growth factors, NF-κB activation is transient, lasting approximately 30–60 minutes. It is noted that, under the conditions of a prolonged pro-inflammatory status, ROS can activate kinases such as the IKK complex (composed of two kinases, IKKα and IKKβ, and the regulatory subunit NEMO/IKKγ). In response to this interaction, IKK phosphorylates inhibitory κB proteins at the level of two critical serine residues. They are subsequently degraded in the proteasome and translocated into the nucleus where they bind to specific DNA sequences and initiate the transcription of target genes involved in the immune response, inflammation (e.g., tumor necrosis factor (TNF)-α, ILs 1 and 6, prostaglandins), chemotaxis (e.g., IL-8), cell adhesion (intercellular adhesion molecule-1, vascular adhesion molecule-1), growth regulation, and apoptosis resistance. In turn, cytokines and chemokines attract and activate inflammatory cells (neutrophils, macrophages, T lymphocytes) within the renal tissue, amplifying the chronic inflammatory response and perpetuating NF-κB activation (84–87). Thus, NF-κB inhibition through antioxidants has been proposed as a therapeutic target in chronic inflammatory conditions, including atherosclerosis and OS-induced nephrotoxicity (87, 88). In addition to stopping the chronic inflammatory feedback loop, Udwan K. notes that targeting Nf-κB appears to be involved in preventing changes in water permeability and protein reflection coefficient, thereby reducing ascites (89).
Furthermore, chronic inflammation leads to the progressive deterioration of renal function by promoting and maintaining fibrosis. This interplay is mediated primarily by transforming growth factor β1 (TGF-β1). By stimulating the TGF-β1/Smad pathway and interacting with surface receptors, numerous signaling pathways are activated that ultimately result in the activation of myofibroblasts, either directly from interstitial fibroblasts or through the trans-differentiation of endothelial cells or mesangial epithelial cells. Connective tissue growth factor enhances this process by improving the affinity of TGF-β for its receptors, thereby sustaining fibrogenic activity. Subsequently, the myofibroblasts excessively stimulate the production of collagen and components of the extracellular matrix, leading to tubulointerstitial fibrosis, mesangial expansion, and glomerulosclerosis. This pathological cascade has emerged as a therapeutic target for antioxidant-based interventions, particularly those based on activators of nuclear factor erythroid 2-related factor 2, which aim to reduce fibrosis, senescence, and apoptosis while enhancing regeneration capacity (90–93). Additional pathways implicated in tubulointerstitial injury and renal fibrosis include TNF-α and tissue inhibitor of metalloproteinase mRNA-1, which facilitate fibrosis by preventing the breakdown of collagen (93). Finally, the Jun N-terminal kinase signaling pathway interacts with the TGF-β1/Smad pathway, establishing its role in fibrotic progression (94). Additionally, we note the possibility of using pharmacologically active substances that target various pro-oxidative and pro-fibrotic pathways found in chronic renal damage including hyperglycemia, mitochondrial energy dysregulation, mineralocorticoid hormonal signaling, and hypoxia-inducible factors (95). Ultimately, persistent fibrosis, progressive inflammation, and maladaptive renal repair compromise glomerular function, exacerbating toxin retention, oxidative imbalance, and the self-perpetuating cycle of renal decline (93).
4.3 Apoptosis and cell necrosis
Regarding cell death induced by OS, it is noted that, depending on the intensity of the stimulus, it can manifest itself in the form of apoptosis (controlled self-destruction of the cell in response to moderate OS, without generating major inflammation) or necrosis (uncontrolled cell death, generative of significant inflammation, in response to major OS) (96, 97).
Continuing from what was previously discussed, the transcription factors and essential pathways involved in the initiation and promotion of apoptosis and necrosis are the p53 factor, the mitogen-activated protein kinase pathways, NF-κB, and caspases. The first three regulate OS responses and promote apoptosis by modulating the expression of pro-inflammatory and pro-apoptotic genes. Caspases, however, serve a dual role, both supporting inflammation and inducing cell death by degrading cellular components in a controlled manner (96, 97). Functionally, caspases are classified as “initiator caspases” and “effector/executioner caspases”. Apoptosis can occur via two primary pathways: intrinsic (mitochondrial) or extrinsic (death-receptor-mediated), depending on the triggering mechanisms. The intrinsic apoptotic pathway is triggered by mitochondrial membrane permeabilization and damage. This pathway is regulated by B-cell lymphoma 2 family proteins and ROS-induced stress. These events result in the release of cytochrome c, exacerbation of OS, and disruption of ATP synthesis. Cytochrome c binds to apoptotic protease-activating factor-1, forming the apoptosome, a quaternary protein complex. The apoptosome then facilitates the recruitment and activation of inhibitor caspase-9, which subsequently activates effector caspases, leading to the controlled dismantling of cellular structures (96–99). The extrinsic apoptotic pathway is mediated by the death-inducing signalling complex, initiated by interactions between extracellular death ligands (e.g., Fas/CD95) and their respective cell surface receptors. This interaction activates intracellular death domains, recruiting Fas-associated protein with death domain (FADD) through a death domain interaction. FADD, via its death effector domain activates initiator caspase-8, which subsequently activates effector caspases (caspase-3, -6, and -7), culminating in apoptotic cell death (96, 99–101).
In contrast, necrosis resulting from severe OS starts by damaging the cell membranes. Lipid peroxidation, following the actions of ROS, induces membrane permeabilization in the early stages. In response to cytosolic calcium, intracellular enzymes (e.g., phospholipases, proteases) activate and damage membrane integrity by breaking down membrane and intracellular components. Later, the damage becomes irreversible, eventually leading to the rupture of the membrane with the release of its contents into the extracellular space. Among the components released at the extracellular level, we especially note damage-associated molecular patterns (DAMPs). DAMPs act predominantly on the immune system, inducing a strong inflammatory response that perpetuates the vicious cycle described in subchapter 4.2. and they exacerbate renal injury and accelerate NS progression. Additionally, DAMPs contribute to post-injury renal tissue regeneration and scarring (97, 99, 102–105).
At the mitochondrial level, ROS reduces the production of ATP and this is an important determinant of apoptosis versus necrosis susceptibility, affecting cellular functions such as sodium ion pump activity. Necrosis is promoted through cellular swelling (edema) and membrane depolarization due to calcium and water influx (97, 103, 106). More recently, literature has described regulated necrosis (necroptosis) as an intermediary process between apoptosis and necrosis, exhibiting a more structured and programmed nature. In this situation, the prototypical pathway is based on the interaction of TNF family members with specific ligands, the recruitment of receptor-interacting protein kinase 1, the inhibition of caspase 8, the assembly of the RIP1/RIP3 signaling complex, and the formation of the necrosome. In the necrosome, the phosphorylation of the mixed line kinase domain-like protein (MLKL) takes place, with its subsequent translocation to the plasma membrane. Here phosphorylated MLKL interacts with phosphatidylinositol phosphates, inducing its oligomerization and insertion at the membrane level. This culminates in cell lysis and the extracellular release of DAMPs. Additionally, phosphorylated MLKL activates the pyrin domain of the NOD-like receptor (NLR) family, specifically/including/and by extension inflammasome-3, promoting the secretion of pro-inflammatory cytokines IL-1β and IL-18, thereby perpetuating inflammation (103, 106–108).
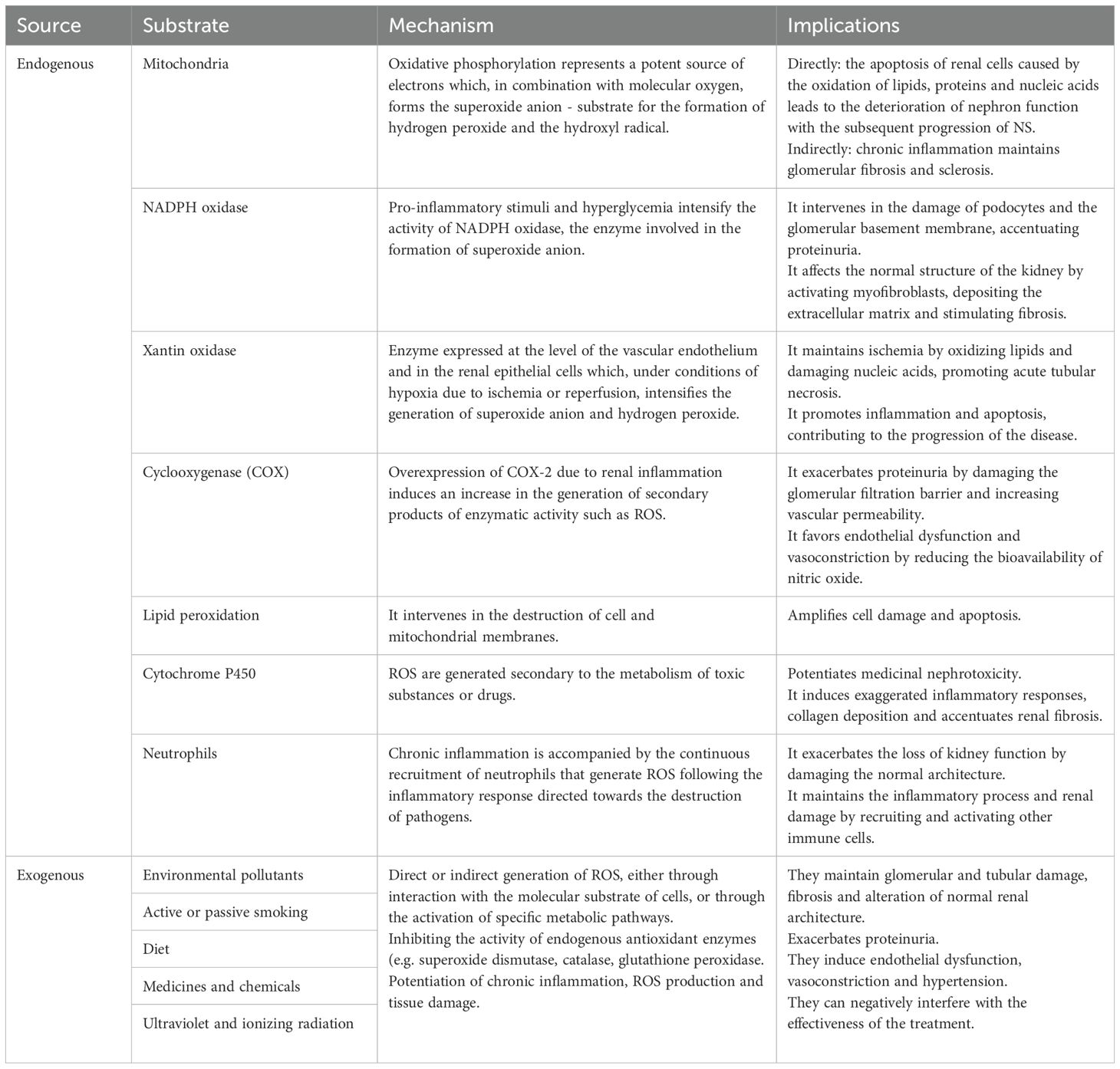
As a consequence, in the dynamics of NS, the mechanisms of cell death – apoptosis and necrosis – operate in a complex way, being induced depending on the strength of the pro-oxidative stresses. These processes result in either cell proliferation or destruction of the glomerular barriers with varying degrees of inflammation and renal injuries that further exacerbate the course of the disease. In this intricate process, antioxidants have proven useful, with resveratrol emerging as an effective prophylactic and therapeutic agent due to its renoprotective properties. It has demonstrated its ability to directly or indirectly counteract the effects of ROS, as well as modulating key pathways such as NF-κB and maintaining mitochondrial energy balance (109). Finally, Figure 2 illustrates how the increase in pro-oxidative status in NS initiates and maintains functional and structural renal decline.
Figure 2. Pathophysiological cascade of renal structural and functional damage induced by oxidative stress and chronic inflammation.
4.4 Hyperlipidemia
Alterations in lipid metabolism, characterized by an increase in cholesterol, triglycerides, and low-density lipoproteins (LDL), are one of the major complications of NS that frequently go under-recognized. Although dyslipidemia is reported at a lower frequency in children than in adults, it acts as a significant contributor in the progression of both cardiovascular (e.g., atherosclerosis, thrombosis) and renal damage in affected individuals (110). Regarding the pathogenesis of NS-related dyslipidemia, extensive reporting has revealed impairments in the synthesis, plasma catabolism, and clearing of lipoproteins. These reports correlate the severity of hyperlipidemia with the degree of proteinuria, disturbances in enzyme activity following chronic tissue damage, and relapse. Additionally, dysfunction in hepatic, muscular, and adipose tissue exacerbates the risk of dyslipidemia by reducing the activity of hepatic lipase and lipoprotein lipase, while increasing hepatic levels of proprotein convertase subtilisin/kexin type 9, an enzyme involved in LDL receptor degradation (110–112).
OS appears to play a central role in dyslipidemia related to NS or chronic kidney disease, contributing to the worsening of proteinuria, hypoalbuminemia, and systemic inflammation. By these mechanisms, OS reduce blood enzyme activity. Its role in dyslipidemia is related to the oxidation of LDL, which occurs in this context of OS, marked by elevated ROS. Oxidized LDL contributes to dyslipidemia in two ways; first, increased amounts worsen the general systemic proinflammatory status and endothelial dysfunction. Secondly, as clearance is impeded during this pro-oxidative state, the concentration of circulating LDL becomes persistently high, increasing atherogenic risk. Additionally, OS disrupts liver activity, promoting increased insulin resistance, which in turn inhibits lipolysis in adipose tissue. This increases the level of free fatty acids taken up by the liver, and therefore the excessive production of LDL., At the renal level, the accumulation of excess lipid molecules causes cellular and mitochondrial damage, perpetuating the vicious cycle of renal dysfunction (113–116). Another component of the lipid balance affected in NS are high-density lipoproteins (HDL). We know that HDL is a protector against cardiovascular diseases, inflammation, and OS. However, in NS, HDL appears to undergo structural and functional changes which are reflected in a decreased HDL cholesterol-to-total cholesterol ratio and a reduced capacity to mature through esterification. These changes contribute to the deterioration of HDL-mediated reverse cholesterol transport, potentiating the atherogenic effects of proteinuria (116, 117). The studies carried out on the adult population indicated that supplementation with small doses of vitamin E was effective in protecting LDL from lipid peroxidation, thus promoting renal health (118). Other antioxidants useful in counteracting OS renal damage are N-acetyl cysteine, ascorbic acid, L-carnitine, coenzyme Q10, curcumin, and resveratrol (119).
4.5 Cardiovascular complications
Given that cardiovascular damage is less severe among children with NS than in adults, we shall briefly explore its implications. We believe that patient education, prevention, and optimal management of OS, systemic inflammation, and NS are key elements in reducing cardiovascular risk in children over the medium and long term. In support of this belief, Tkaczyk M. et al. argue that a severe clinical evolution of NS can be accompanied by an increased risk of accelerated atherogenesis. Also, persistent abnormalities of lipid metabolism may increase early atherogenic risk. In this sense, the current research is focused on defining the risk-to-benefit ratio of using lipid-lowering drugs in the pediatric population (73, 120).
As presented previously, we reiterate that NS is characterized by a persistent vicious cycle that emerges from the effects of OS, chronic pro-inflammatory status, hyperlipidemia, and metabolic imbalances. These components modulate each other, promoting and maintaining systemic damage by dysregulating the prooxidant/antioxidant balance. OS is important both in renal injury and subsequent cardiovascular-renal remodeling. Alongside renal dysfunction, OS and the activation of the renin-angiotensin system (RAS) represent key factors in the promotion of endothelial damage, arterial stiffening, atherosclerosis, arterial hypertension, and ventricular dysfunction. These comorbidities together may significantly impact NS-related morbidity and mortality. For example, it is known that atherosclerosis is important in determining the need for dialysis, kidney transplant outcomes, and mortality risk (121–123).
The biological regulation of homocysteine, inflammatory and prothrombotic blood factors, and adhesion molecules is one of the most important determinants in the earliest stages of atherosclerosis. Many of these factors are present in children with NS, either at initial presentation or during disease relapses. Literature links their presence to selective proteinuria, OS, and steroid therapy. An additional risk factor is renal replacement therapy, which exacerbates OS by accentuating the loss of antioxidant substances. OS-induced endothelial injury accelerates atherosclerosis, thereby increasing the risk of thrombotic events. This triad forms a critical bridge between nephrotic and vascular damage, ultimately leading to cardiac and cerebral damage of the ischemic type (72, 123). Supporting this theory, a correlation has been observed between urinary OS biomarkers and the arteriosclerosis index (123).
Regarding cardiac damage from a hemodynamic and clinical point of view, Kamel A. et al. reported that approximately 30% of children with NS exhibit right ventricular dysfunction, while 20% have left ventricular dysfunction. These impairments correlate significantly with disease duration, treatment history, relapse patterns, steroid resistance, and response to immunosuppressive medication (124). Similarly, Tharanidharan SP. et al. note an increase in the average myocardial performance index which was directly correlated to the frequency of relapses, disease duration, and a history of hypertension and hypercholesterolemia (115).
In summary, a self-perpetuating cycle underlies NS-associated cardiovascular damage, with OS at its core. OS severity itself is determined by various factors; among those that potentiate OS are carbohydrate metabolism dysregulation leading to advanced glycation end products, lipid profile abnormalities resulting in plasma lipid oxidation, immune system activation triggering proinflammatory cytokine release, renal hypoxia, and the adverse effects of steroid or immunosuppressant therapy. Meanwhile, antioxidant reserves are severely depleted in NS due to massive losses of plasma proteins, leading over time to a reduction in the body’s capacity to neutralize ROS. OS further perpetuates proteinuria, weakens antioxidant defense, sustains inflammation, and disrupts homeostasis. Additionally, it triggers RAS activation, promoting endothelial damage, arterial stiffening, atherosclerosis, arterial hypertension, and thrombosis. The ultimate cardiovascular consequences of this cascade include pathological cardiovascular remodeling, ventricular dysfunction, and ischemia (72, 73, 115, 120–124).
In managing the salient effects of OS on microvascular and renal homeostasis, we present the results obtained by Hertiš Petek T. et al. who highlight the benefits of lifestyle modifications, including regular physical exercise, reduction of sedentary periods, and an adequate diet, in improving insulin sensitivity while also enhancing antioxidant and anti-inflammatory capacity. The authors note the importance of maintaining a constant level of antioxidants in the plasma. Dietary supplementation with antioxidants (e.g., vitamin C, E, zinc, L-arginine, carnosine, phytophenols) or probiotics remains under research regarding its adjuvant therapeutic effects. One approach involves omega-3 polyunsaturated fatty acid supplementation, which has shown effectiveness in reducing the level of cytokines, ROS, and adhesion molecules. Other dietary strategies include carbohydrate-restricted and methionine-restricted diets (123). Currently, pharmacological interventions targeting the consequences of OS dominate clinical management strategies for cardiovascular complications in NS. We further urge the concentration of efforts in identifying and implementing adjuvant therapeutic schemes based on diets rich in antioxidants and/or their exogenous supplementation. We are confident that administering well-established antioxidant compounds with cardiovascular benefits will have the effect of reducing systemic inflammation, OS, and by extension cardiac damage by targeting pathophysiological cascades at three important levels: increasing systemic antioxidant capacity, reducing renal injury, and preventing disease relapses.
4.6 Effects on the immune system
NS in children, primarily in those with active diseases, is frequently accompanied by an increased risk of infection compared to the general population (125–127). Kalra S. et al. found that early detection of recurrence significantly reduced the number of patients experiencing severe infections (128). With regards to OS, immune disorders in pediatric NS () are related to disruptions in immune cell proliferation and function, urinary loss of immunoglobulins, and an imbalance in immune responses – suppression of cellular immunity (low Th1) and an excessive stimulation of the humoral response (high Th2). This imbalance promotes the secretion of TNF-α, IL-6, IL-8, and IL-1β, complement activation, and neutrophil recruitment, which further increases the production of free radicals, amplifying the vicious cycle of proteinuria, podocyte damage, inflammation, and OS.
At the same time, corticosteroid therapy induces lymphocyte apoptosis and weakens the body’s capacity to combat viral and bacterial infections, as well as compromising the barrier function of immune cells following lipid peroxidation and cell membrane damage (121). While the urinary losses of functional proteins have been previously detailed, we turn our attention now to the damage inflicted on immune cells in NS. Once again, excessive concentrations of ROS are implicated among other reasons. Though physiological levels of ROS play a crucial role in immune function, excessive amounts have shown to impair the viability and function of immune cells, reducing the body’s ability to counteract infections. Furthermore, infections exacerbate inflammation, tissue damage, and metabolic disturbances, all of which sustain OS, Therefore, targeted OS management through antioxidant therapies and ExAs (e.g., α-tocopherol, ascorbic acid, coenzyme Q10, N-acetylcysteine, resveratrol, selenium, zinc, and curcumin) appears to be a reliable treatment approach to improving immune resistance in patients with NS. However, in order not to produce more harm than good, we must aim to restore ROS to physiological levels rather than eliminating them entirely (129–131).
Another mechanism contributing to immune dysregulation and increased relapse susceptibility in NS is intestinal dysbiosis. Alterations in intestinal microbiota have been shown to impair inflammatory cell function by disrupting the Th17/Treg axis following the depletion of butyrate- or short-chain fatty acid-producing bacteria. Furthermore, dysbiosis has also been shown to correlate with the stages of NS. Impaired microbiota function has clear implications in the pathogenesis of NS, linking intestinal dysbiosis with Treg cell dysfunction and increased relapse risk. In accordance with the above, clinical trials have demonstrated that modulation of the gut microbiota by administering probiotics based on butyrate-producing bacteria led to a longer relapse interval and a reduced need for immunosuppressive agents. Also, treatment with prebiotics and probiotics in addition to steroids demonstrated a significant increase in T-reg cells (CD4+/CD25+/FOXp3+) in peripheral blood and a higher level of Lactobacillus species in stool, along with a significant decrease in relapse rate (132–136).
4.7 Edema and electrolyte imbalances
OS encountered in pediatric NS triggers and sustains a pathophysiological cascade that progressively worsens glomerular lesions, proteinuria, and hypoalbuminemia. Additionally, studies in murine models have demonstrated the critical role of ROS in hydro-electrolyte disturbances associated with NS. OS primarily affects three key mechanisms: (1) up-regulation of Na/K-ATPase, (2) increases water permeability while simultaneously reducing the protein reflection coefficient through the peritoneal barrier, and (3) stimulation of angiotensin-induced aldosterone secretion (137). Also, salt overload (e.g., in the case of a high-sodium diet) has been shown to promote OS by modulating key molecules in immunity and inflammation, thereby perpetuating electrolyte imbalances, endothelial damage, and the progression of renal disease (138–140). The results of these systemic-related mechanisms contribute to the formation of edema characteristic of NS (141). However, some studies implicate the epithelial sodium channel activated by proteolytic enzymes or their precursors in sodium retention and subsequent edema, in particular cases (142–144). Therefore, the mechanism of edema production in renal diseases is complex, involving several intricate factors, among which OS plays a significant role.
4.8 Response to corticosteroids
OS, an important element in NS, may appear to modulate treatment response. The intensity and duration of OS exposure appear to correlate with corticosteroid responsiveness,. Assessing total antioxidant capacity can be useful in predicting the response to corticosteroids at diagnosis or relapse after the interruption of steroid treatment (145). In contrast to other chronic inflammatory diseases such as asthma or chronic lung disease, the mechanisms underlying steroid sensitivity variability in NS remain unclear. However, a disturbed redox environment (marked by decreased levels of enzymatic and non-enzymatic substances with antioxidant roles) affects glucocorticoid receptor sensitivity and immune cell function. There are many interesting areas of research that can be explored, such as the impact of OS on the structural conformation of corticosteroid receptors, intracellular signaling pathways, renal cell integrity, or hormonal dynamics,. Excessive ROS may interfere with glucocorticoid receptor degradation and translocation, affecting their activity. At the same time, persistent inflammation and chronic activation of NF-κB can lead over time to decreased sensitivity to steroids. Finally, ROS can activate mitogen-activated protein kinase which further interferes with glucocorticoid receptor signaling by phosphorylating and reducing its ability to bind to DNA and regulate anti-inflammatory genes. To mitigate this damage, antioxidants known to specifically target the oxidative pathways can be used such as N-acetylcysteine, glutathione precursors, vitamins C and E, curcumin and other NF-κB inhibitors, and mitochondria-targeted antioxidants such as MitoQ. Of these substances, the most used in practice will be detailed below (146–150).
5 Reliable biomarkers in the assessment of the oxidative status and prognosis of NS
Considering the multiple implications of OS in the dynamics of NS, we will discuss the main biomarkers at the systemic level, as well as the utility of renal prognosis. For an easier understanding, biomarkers can be categorized as enzymatic and non-enzymatic (stable ROS attack products). Their clinical value lies in enabling disease monitoring, guiding therapeutic adaptation, prevention of relapses, and improving medium and long-term prognosis. A notable advantage of oxidative biomarker assessment is its non-invasiveness. However, selecting relevant biomarkers requires a critical analysis of specificity, measurement site, and technique, as individual variations have been reported. The practical challenges of implementing biomarker-based monitoring strategies include being unable to reliably and clinically predict NS progression, lack of standardized measurement methods, and the need for advanced equipment and financial resources, which may limit accessibility in public healthcare systems. At the same time, we recognize that results must be interpreted in the context of genetic factors, diet, comorbidities, and concurrent treatments,. Large-scale clinical trials are needed to establish the real benefits of biomarker-based, personalized treatments in NS. Although there are challenges in achieving consensus within the medical community and updating treatment guidelines accordingly, personalizing treatment based on OS biomarkers could represent an important step in improving NS management, optimizing therapies, and reducing disease progression (151, 152).
Enzymatic biomarkers primarily include antioxidant enzymes such as superoxide dismutase, catalase (CAT), and glutathione peroxidase. These enzyme systems intervene in the dismutation of superoxide anion free radicals into molecular oxygen and hydrogen peroxide, the reduction of hydrogen peroxide and lipid hydroperoxides. In this way, antioxidant enzyme systems protect cells from damage induced by ROS attacks. During these processes, optimal enzyme activity depends on essential cofactors (e.g., copper, zinc), and their reduced activity signals increased OS, and compromised antioxidant defense (153–157). Additionally, it is possible to evaluate the degree of OS by measuring the ratio between reduced glutathione and oxidized glutathione (158). Non-enzymatic biomarkers of NS-specific OS include advanced protein oxidation products, malondialdehyde (a lipid peroxidation marker), 8-hydroxydeoxyguanosine (a marker of oxidative DNA damage), and total antioxidant capacity (TAC). In contrast to TAC, whose low level is associated with redox system dysfunction, the other biomarkers are associated with OS at increased values (159, 160).
From a prognostic perspective, patients with a lower-than-normal antioxidant status experience greater OS, such that they are more likely to suffer from unfavorable prognostic outcomes, for example, increased risk of relapse and quicker degeneration of kidney tissues. Hence, the integration of enzymatic and non-enzymatic biomarkers into NS treatment strategies may enable personalized therapy. Emerging research focuses on CAT gene polymorphism and their role in steroid resistance in idiopathic NS (161, 162). In practical terms, we have found studies that associate OS biomarkers and antioxidant potential with NS progression, relapse frequency, and renal failure (163–166).
6 Means of manipulating antioxidant capacity
Although the progression to chronic renal failure is rarer in children than in adults, the effective management of OS remains crucial, given its role in systemic decline,. Controlling the pro-oxidative status allows clinicians to reduce the morbidity associated with long-term complications of the underlying disease (e.g., cardiovascular and metabolic diseases in adult life) (167).
As previously discussed, antioxidants involved in combating NS-specific OS can be categorized as enzymatic and non-enzymatic. Enzymatic antioxidants rely heavily on endogenous production and functionality, necessitating optimal cofactor levels through exogenous intake (54, 83, 155). In contrast, non-enzymatic antioxidants, primarily obtained from external sources (e.g., vitamin C and E, coenzyme Q, carotenes, and trace elements), play roles in neutralizing ROS, oxidation prevention, and enzymatic function. However, certain non-enzymatic antioxidants – such as glutathione, ubiquinone, albumin, metallothioneins, and uric acid – are predominantly produced endogenously (28–30, 155, 168).
Antioxidant supplementation, whether dietary or pharmacological, is used as an adjuvant therapeutic method alongside standard steroid or immunosuppressive treatments. It can have various homeostatic benefits with regards to the systemic response to oxidative aggression, and these benefits depend on individual substance properties (e.g., bioavailability, synergy). Therefore, understanding the physicochemical characteristics, mechanisms of action, and interactions of ExAs is essential for optimizing bioavailability and bioactivity. Additionally, we especially emphasize the usefulness of antioxidants as adjunctive therapy, complementary to lifestyle changes (e.g., diet based on fruits, tea, vegetables or cereals) and targeted pharmacological interventions. The most effective approach integrates dietary changes, conventional pharmacological therapy, and antioxidant supplementation, as supplementation alone cannot counteract an unhealthy lifestyle (28–30, 155, 167, 168).
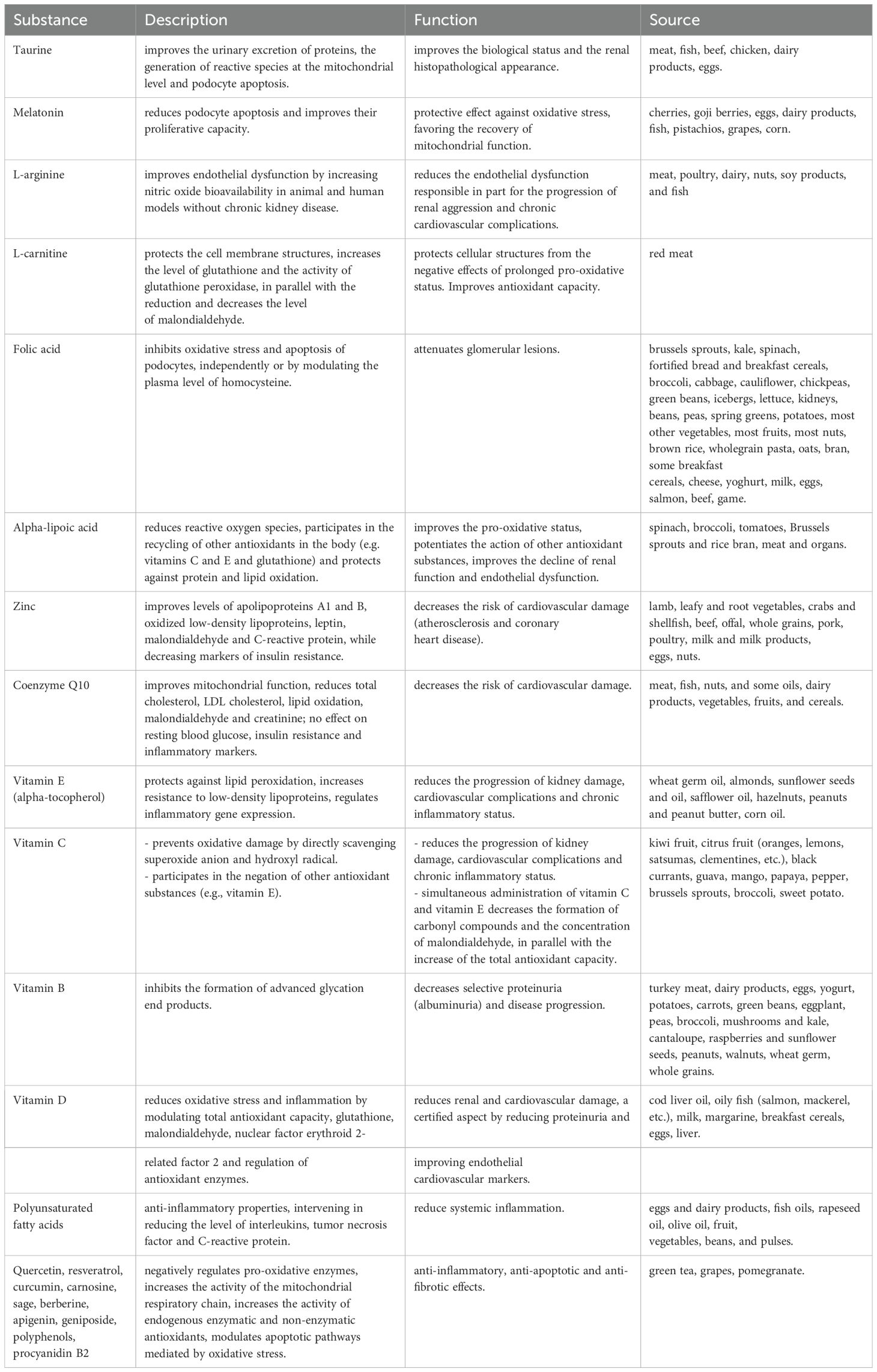
We reiterate that in the case of NS, chronic biological disturbances cause fluctuations in antioxidant levels, a phenomenon also observed in the case of infectious processes related to the activation of systemic inflammatory pathways. Since NS is a chronic pathology with a fluctuating evolution and multiple intercurrents, continuous OS management is important for reducing morbidity, slowing renal disease progression, minimizing relapses, and optimizing treatment response. To reduce OS, three levels must be targeted: decreasing exposure to environmental pollutants with oxidizing properties, increasing the administered level of EAs and ExAs, and reducing OS generation. Long-term lifestyle strategies must be carefully developed through cooperation within multidimensional teams that integrate both pharmacologists and dieticians. These strategies may include antioxidant supplementation, pharmacological agents with antioxidant properties (e.g., statins), and strict inflammation control. Importantly, care must be taken not to create a pro-oxidative environment through the excess administration of corticosteroids., Adopting an antioxidant-rich diet with a high content of polyphenols and flavonoids, reducing salt and unsaturated fat consumption, and ensuring adequate protein intake can mitigate OS, reduce cardiovascular complications, and prevent malnutrition. Psychological stress can act as a relapse trigger and should be managed with relaxation techniques and moderate physical exercise., Given their role in the pathophysiological cascade of NS, there is a growing interest in studying and integrating OS biomarkers into clinical practice for individual personalized therapy. Current research is focused on identifying and optimizing antioxidant supplementation strategies to reduce NS-related morbidity (134, 163, 169–171). The key related approaches are summarized in Table 3.
7 Conclusions
There is a strong association between systemic OS and the progression of pediatric NS,. The relationship is bidirectional; excess ROS participates through various pathophysiological mechanisms in the onset and persistence of renal damage and its complications – cardiovascular diseases, hydro-electrolytic and acid-base imbalances, immune dysfunction, dyslipidemia, and disease progression - while renal and systemic homeostasis disruption perpetuates a pro-inflammatory state, toxic substance retention, and a variety of metabolic abnormalities, in turn fueling OS. However, pharmacotherapy for NS, as well as other exogenous toxicants, are also implicated in OS etiology.
This review intended to clearly and comprehensively summarize the key pathophysiological mechanisms underlying renal and systemic deterioration in pediatric NS from an OS perspective. Our practical purpose is to demonstrate and encourage the use of antioxidant-rich diets and, non-enzymatic antioxidant supplementation in interrupting the pathophysiological cascade of NS. Implementing these strategies may help prevent or mitigate local and systemic complications over the medium and long term while reducing disease-specific morbidity at minimal cost and invasiveness.
We also emphasize the potential of enzymatic and non-enzymatic biomarkers as diagnostic and prognostic tools. Finally, we advocate for direct global efforts towards the development and patenting ofmethods for assessing oxidant and antioxidant status in pediatric clinical practice, particularly for chronic disorders characterized by low-grade chronic systemic inflammation. We believe that they will offer clinicians novel prospects for the personalized, diagnostic, and therapeutic management of pediatric NS, resulting in significant reduction of morbidity and mortality.
Author contributions
EJ: Conceptualization, Investigation, Writing – original draft. AN: Software, Visualization, Writing – original draft. VL: Conceptualization, Supervision, Writing – review & editing. IS: Investigation, Methodology, Writing – original draft. AM: Investigation, Writing – original draft. SR: Validation, Visualization, Writing – review & editing. VS: Investigation, Writing – original draft. RR: Investigation, Writing – original draft. GB: Validation, Visualization, Writing – review & editing. DS: Validation, Visualization, Writing – review & editing. IM: Validation, Visualization, Writing – review & editing. FD: Investigation, Writing – original draft. OC: Validation, Visualization, Writing – review & editing. AL: Investigation, Methodology, Writing – original draft. CD: Writing – review & editing, Validation. EA: Writing – review & editing, Validation.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Iorga M, Starcea IM, Munteanu M, Sztankovszky LZ. Psychological and social problems of children with chronic kidney disease. Eur J Sci Theology. (2014) 10:179–88.
2. Robinson RF, Nahata MC, Mahan JD, Batisky DL. Management of nephrotic syndrome in children. Pharmacotherapy. (2003) 23:1021–36. doi: 10.1592/phco.23.8.1021.32885
3. Trautmann A, Boyer O, Hodson E, Bagga A, Gipson DS, Samuel S, et al. International Pediatric Nephrology Association. IPNA clinical practice recommendations for the diagnosis and management of children with steroid-sensitive nephrotic syndrome. Pediatr Nephrol. (2023) 38:877–919. doi: 10.1007/s00467-022-05739-3
4. Kidney Disease: Improving Global Outcomes (KDIGO). KDIGO 2020 clinical practice guideline on glumerular diseases. Kidney Int. (2020), 153–6.
5. Mattoo TK, Sanjad S. Current understanding of nephrotic syndrome in children. Pediatr Clin North. Am. (2022) 69:1079–98. doi: 10.1016/j.pcl.2022.08.002
6. Wang CS, Greenbaum LA. Nephrotic syndrome. Pediatr Clin North Am. (2019) 66:73–85. doi: 10.1016/j.pcl.2018.08.006
7. Caridi G, Trivelli A, Sanna-Cherchi S, Perfumo F, Ghiggeri GM. Familial forms of nephrotic syndrome. Pediatr Nephrol. (2010) 25:241–52. doi: 10.1007/s00467-008-1051-3
8. Khanna R. Clinical presentation & management of glomerular diseases: hematuria, nephritic & nephrotic syndrome. Mo Med. (2011) 108:33–6.
10. Hölttä T, Jalanko H. Congenital nephrotic syndrome: is early aggressive treatment needed? Yes. Pediatr Nephrology. (2020) 35:1985–90. doi: 10.1007/s00467-020-04578-4
11. Boyer O, Schaefer F, Haffner D, Bockenhauer D, Hölttä T, Bérody S, et al. Management of congenital nephrotic syndrome: consensus recommendations of the ERKNet-ESPN Working Group. Nat Rev Nephrol. (2021) 17:277–89. doi: 10.1038/s41581-020-00384-1
12. Lee JM, Kronbichler A, Shin JI, Oh J. Current understandings in treating children with steroid-resistant nephrotic syndrome. Pediatr Nephrol. (2021) 36:747–61. doi: 10.1007/s00467-020-04476-9
13. Sinha A, Bagga A. Clinical practice guidelines for nephrotic syndrome: consensus is emerging. Pediatr Nephrol. (2022) 37:2975–84. doi: 10.1007/s00467-022-05639-6
14. Agrwal S, Mantan M, Dabas A, Batra VV. Childhood steroid-resistant nephrotic syndrome: long-term outcomes from a tertiary care center. Indian J Nephrol. (2022) 32:320–6. doi: 10.4103/ijn.ijn_258_21
15. Sachdeva S, Khan S, Davalos C, Avanthika C, Jhaveri S, Babu A, et al. Management of steroid-resistant nephrotic syndrome in children. Cureus. (2021) 13:e19363. doi: 10.7759/cureus.19363
16. Frățilă VG, Lupușoru G, Sorohan BM, Obrișcă B, Mocanu V, Lupușoru M, et al. Nephrotic syndrome: from pathophysiology to novel therapeutic approaches. Biomedicines. (2024) 12:569. doi: 10.3390/biomedicines12030569
17. Lupu VV, Miron I, Trandafir LM, Jechel E, Starcea IM, Ioniuc I, et al. Challenging directions in pediatric diabetes - the place of oxidative stress and antioxidants in systemic decline. Front Pharmacol. (2024) 15:1472670. doi: 10.3389/fphar.2024.1472670
18. Sun Y, Wang X, Li L, Zhong C, Zhang Y, Yang X, et al. The role of gut microbiota in intestinal disease: from an oxidative stress perspective. Front Microbiol. (2024) 15:1328324. doi: 10.3389/fmicb.2024.1328324
19. Kunst C, Schmid S, Michalski M, Tümen D, Buttenschön J, Müller M, et al. The influence of gut microbiota on oxidative stress and the immune system. Biomedicines. (2023) 11:1388. doi: 10.3390/biomedicines11051388
20. Lupu VV, Jechel E, Mihai CM, Mitrofan EC, Lupu A, Starcea IM, et al. Connection between celiac disease and systemic lupus erythematosus in children-A development model of autoimmune diseases starting from what we inherit to what we eat. Nutrients. (2023) 15:2535. doi: 10.3390/nu15112535
21. Parmar G, Mistry K, Gang S. Correlation of serum albumin and creatinine with oxidative stress markers in patients having nephrotic syndrome. Int J Pharm Pharmaceutical Sci. (2021) 13:20–4. doi: 10.22159/ijpps.2021v13i12.42931
22. Habriel Ruslie R, Rina Ramayani O, Darmadi D, Alamsyah Siregar G. Oxidative stress markers in initial therapy and remission of nephrotic syndrome and serum malondialdehyde level predictor from routine laboratory test. Med Glas (Zenica). (2021) 18:90–5. doi: 10.17392/1192-21
23. Barnes PJ. Mechanisms and resistance in glucocorticoid control of inflammation. J Steroid Biochem Mol Biol. (2010) 120:76–85. doi: 10.1016/j.jsbmb.2010.02.018
24. Zakeri N, Kelishadi MR, Asbaghi O, Naeini F, Afsharfar M, Mirzadeh E, et al. Selenium supplementation and oxidative stress: A review. PharmaNutrition. (2021) 17:100263. doi: 10.1016/j.phanu.2021.100263
25. Tashima Y, Terui M, Itoh H, Mizunuma H, Kobayashi R, Marumo F, et al. Effect of selenite on glucocorticoid receptor. J Biochem. (1989) 105(3):358–61. doi: 10.1093/oxfordjournals.jbchem.a122668
26. Abdallah ABE, El-Ghannam MA, Hasan AA, Mohammad LG, Noura MM, Alsayed EM, et al. Selenium nanoparticles modulate steroidogenesis-related genes and improve ovarian functions via regulating androgen receptors expression in polycystic ovary syndrome rat model. Biol Trace Elem Res. (2023) 201:5721–33. doi: 10.1007/s12011-023-03616-0
27. Daragó A, Klimczak M, Stragierowicz J, Stasikowska-Kanicka O, Kilanowicz A. The effect of zinc, selenium, and their combined supplementation on androgen receptor protein expression in the prostate lobes and serum steroid hormone concentrations of wistar rats. Nutrients. (2020) 12:153. doi: 10.3390/nu12010153
28. Bhatt GC, Jain S, Das RR. Zinc supplementation as an adjunct to standard therapy in childhood nephrotic syndrome - a systematic review. World J Clin Pediatr. (2016) 5:383–90. doi: 10.5409/wjcp.v5.i4.383
29. Mbanefo NR, Uwaezuoke SN, Eneh CI, Odimegwu CL, Chikani UN, Muoneke UV, et al. Can oral zinc supplementation reduce relapses in childhood steroid-sensitive nephrotic syndrome? A systematic review. Int J Nephrol Renovasc Dis. (2023) 16:143–53. doi: 10.2147/IJNRD.S403699
30. Stec J, Podracká L, Pavkovceková O, Kollár J. Zinc and copper metabolism in nephrotic syndrome. Nephron. (1990) 56:186–7. doi: 10.1159/000186130
31. D’Amico G, Bazzi C. Pathophysiology of proteinuria. Kidney Int. (2003) 63:809–25. doi: 10.1046/j.1523-1755.2003.00840.x
32. Toblli JE, Bevione P, Di Gennaro F, Madalena L, Cao G, Angerosa M. Understanding the mechanisms of proteinuria: therapeutic implications. Int J Nephrol. (2012) 2012:546039. doi: 10.1155/2012/546039
33. Dobrinskikh E, Blaine J. Glomerular mechanisms of proteinuria. In: Blaine J, editor. Proteinuria: Basic Mechanisms, Pathophysiology and Clinical Relevance. Springer, Cham (2016). doi: 10.1007/978-3-319-43359-2_2
34. Kasper DL, Fauci AS, Hauser SL, Longo DL, Jameson JL, Loscalzo J. Harrison’s Principles of Internal Medicine. 19th edition. McGraw-Hill Education / Medical (2015).
35. Kodner C. Diagnosis and management of nephrotic syndrome in adults: A practical review. Am Family Physician. (2016) 93:479–85.
36. Porth CM. Essentials of Pathophysiology: Concepts of Altered Health States. 4th edition. Lippincott Williams & Wilkins (2014), ISBN: ISBN: 978-1451190809.
37. Cravedi P, Remuzzi G. Pathophysiology of proteinuria and its value as an outcome measure in chronic kidney disease. Br J Clin Pharmacol. (2013) 76:516–23. doi: 10.1111/bcp.12104
38. Claudio P, Gabriella M. Nephrotic syndrome: pathophysiology and consequences. J Nephrol. (2023) 36:2179–90. doi: 10.1007/s40620-023-01697-7
39. Busuioc RM, Mircescu G. Nephrotic syndrome complications - new and old. Part 2. Maedica (Bucur). (2022) 17:404–14. doi: 10.26574/maedica.2022.17.2.395
40. Verma PR, Patil P. Nephrotic syndrome: A review. Cureus. (2024) 16:e53923. doi: 10.7759/cureus.53923
41. Park SJ, Shin JI. Complications of nephrotic syndrome. Korean J Pediatr. (2011) 54:322–8. doi: 10.3345/kjp.2011.54.8.322
42. Niaudet P. Nephrotic syndrome: classification and evaluation. In: Emma F, Goldstein S, Bagga A, Bates CM, Shroff R, editors. Pediatric Nephrology. Springer, Berlin, Heidelberg (2021). doi: 10.1007/978-3-642-27843-3_24-3
43. Kamireddy R, Kavuri S, Devi S, Vemula H, Chandana D, Harinarayanan S, et al. Oxidative stress in pediatric nephrotic syndrome. Clin Chim Acta. (2002) 325:147–50. doi: 10.1016/s0009-8981(02)00294-2
44. Bulucu F, Vural A, Aydin A, Sayal A. Oxidative stress status in adults with nephrotic syndrome. Clin Nephrol. (2000) 53:169–73.
45. Mulat SY, Mihajlović M, Antonić T, Miloševski-Lomić G, Peco-Antić A, Jovanović D, et al. Pediatric nephrotic syndrome: The interplay of oxidative stress and inflammation. J Med Biochem. (2024) 43:424–35. doi: 10.5937/jomb0-46526
46. Jabarpour M, Rashtchizadeh N, Argani H, Ghorbanihaghjo A, Ranjbarzadhag M, Sanajou D, et al. The impact of dyslipidemia and oxidative stress on vasoactive mediators in patients with renal dysfunction. Int Urol Nephrol. (2019) 51:2235–42. doi: 10.1007/s11255-019-02319-7
47. Bufna A, Sarkar PD. Oxidant stress in primary nephrotic syndrome in relation to dyslipidemia. Int J Biol Med Res. (2010) 1:144–8.
48. Reddy VP. Oxidative stress in health and disease. Biomedicines. (2023) 11(11):2925. doi: 10.3390/biomedicines11112925
49. Chatterjee S. Chapter two - oxidative stress, inflammation, and disease. Oxid Stress Biomaterials. (2016), 35–58. doi: 10.1016/B978-0-12-803269-5.00002-4
50. Bissinger R, Bhuyan AAM, Qadri SM, Lang F. Oxidative stress, eryptosis and anemia: a pivotal mechanistic nexus in systemic diseases. FEBS J. (2019) 286:826–54. doi: 10.1111/febs.14606
51. Daenen K, Andries A, Mekahli D, Van Schepdael A, Jouret F, Bammens B. Oxidative stress in chronic kidney disease. Pediatr Nephrol. (2019) 34:975–91. doi: 10.1007/s00467-018-4005-4
52. Coppolino G, Leonardi G, Andreucci M, Bolignano D. Oxidative stress and kidney function: A brief update. Curr Pharm Des. (2018) 24:4794–9. doi: 10.2174/1381612825666190112165206
53. Forman HJ, Zhang H. Targeting oxidative stress in disease: promise and limitations of antioxidant therapy. Nat Rev Drug Discov. (2021) 20:689–709. doi: 10.1038/s41573-021-00233-1
54. Krata N, Zagożdżon R, Foroncewicz B, Mucha K. Oxidative stress in kidney diseases: the cause or the consequence? Arch Immunol Ther Exp. (Warsz). (2018) 66(3):211–20. doi: 10.1007/s00005-017-0496-0
55. Johnson RJ, Feehally J, Floege J, Turner KJJ. Comprehensive Clinical Nephrology. Sixth Edition. Elsevier (2018).
56. Irazabal MV, Torres VE. Reactive oxygen species and redox signaling in chronic kidney disease. Cells. (2020) 9:1342. doi: 10.3390/cells9061342
57. Duni A, Liakopoulos V, Roumeliotis S, Peschos D, Dounousi E. Oxidative stress in the pathogenesis and evolution of chronic kidney disease: untangling ariadne’s thread. Int J Mol Sci. (2019) 20:3711. doi: 10.3390/ijms20153711
58. Cachofeiro V, Goicochea M, de Vinuesa SG, Oubiña P, Lahera V, Luño J. Oxidative stress and inflammation, a link between chronic kidney disease and cardiovascular disease. Kidney Int Suppl. (2008) 111:S4–9. doi: 10.1038/ki.2008.516
59. Doke T, Susztak K. The multifaceted role of kidney tubule mitochondrial dysfunction in kidney disease development. Trends Cell Biol. (2022) 32:841–53. doi: 10.1016/j.tcb.2022.03.012
60. Xu X, Nie S, Ding H, Hou FF. Environmental pollution and kidney diseases. Nat Rev Nephrol. (2018) 14:313–24. doi: 10.1038/nrneph.2018.11
61. Ria P, De Pascalis A, Zito A, Barbarini S, Napoli M, Gigante A, et al. Diet and proteinuria: state of art. Int J Mol Sci. (2022) 24:44. doi: 10.3390/ijms24010044
62. Seo YS, Park JM, Kim JH, Lee MY. Cigarette smoke-induced reactive oxygen species formation: A concise review. Antioxidants. (2023) 12:1732. doi: 10.3390/antiox12091732
63. Baliga R, Ueda N, Walker PD, Shah SV. Oxidant mechanisms in toxic acute renal failure. Drug Metab Rev. (1999) 31:971–97. doi: 10.1081/dmr-100101947
64. Klaus R, Niyazi M, Lange-Sperandio B. Radiation-induced kidney toxicity: molecular and cellular pathogenesis. Radiat Oncol. (2021) 16:43. doi: 10.1186/s13014-021-01764-y
65. Dalal R, Bruss ZS, Sehdev JS. Physiology, Renal Blood Flow and Filtration. Treasure Island (FL: StatPearls Publishing (2023). Available at: https://www.ncbi.nlm.nih.gov/books/NBK482248/.
66. Edwards A, Kurtcuoglu V. Renal blood flow and oxygenation. Pflugers Arch. (2022) 474:759–70. doi: 10.1007/s00424-022-02690-y
67. Leatherby RJ, Theodorou C, Dhanda R. Renal physiology: blood flow, glomerular filtration and plasma clearance. Anaesthesia Intensive Care Med. (2021) 22:439–42. doi: 10.1016/j.mpaic.2021.05.003
68. Honda T, Hirakawa Y, Nangaku M. The role of oxidative stress and hypoxia in renal disease. Kidney Res Clin Pract. (2019) 38:414–26. doi: 10.23876/j.krcp.19.063
69. Xu N, Jiang S, Persson PB, Persson EAG, Lai EY, Patzak A. Reactive oxygen species in renal vascular function. Acta Physiol (Oxf). (2020) 229:e13477. doi: 10.1111/apha.13477
70. Sena CM, Leandro A, Azul L, Seiça R, Perry G. Vascular oxidative stress: impact and therapeutic approaches. Front Physiol. (2018) 9:1668. doi: 10.3389/fphys.2018.01668
71. Shouman M, Abdallah N, Tablawy NE, Rashed LA. Clinical research Biochemical markers of endothelial dysfunction in pediatric nephrotic syndrome. Arch Med Sci. (2009) 5:415–21.
72. Sharma B, Saha A, Dubey NK, Kapoor K, Anubhuti, Batra VV, Upadhayay AD. Endothelial dysfuntion in children with idiopathic nephrotic syndrome. Atherosclerosis. (2014) 233:704–6. doi: 10.1016/j.atherosclerosis.2014.01.055
73. Tkaczyk M, Czupryniak A, Owczarek D, Lukamowicz J, Nowicki M. Markers of endothelial dysfunction in children with idiopathic nephrotic syndrome. Am J Nephrol. (2008) 28:197–202. doi: 10.1159/000110088
74. Daehn IS. Glomerular endothelial cell stress and cross-talk with podocytes in early diabetic kidney disease. Front Med (Lausanne). (2018) 5:113. doi: 10.3389/fmed.2018.00113
75. Zhu YT, Wan C, Lin JH, Hammes HP, Zhang C. Mitochondrial oxidative stress and cell death in podocytopathies. Biomolecules. (2022) 12:403. doi: 10.3390/biom12030403
76. Ao L, Xie Y. Research advance in the mechanism for oxidative stress-induced podocyte injury in diabetic kidney disease. Zhong Nan Da Xue Xue Bao Yi Xue Ban. (2021) 46:1403–8. doi: 10.11817/j.issn.1672-7347.2021.210199
77. Nagata M. Podocyte injury and its consequences. Kidney Int. (2016) 89:1221–30. doi: 10.1016/j.kint.2016.01.012
78. Yang C, Zhang Z, Liu J, Chen P, Li J, Shu H, et al. Research progress on multiple cell death pathways of podocytes in diabetic kidney disease. Mol Med. (2023) 29:135. doi: 10.1186/s10020-023-00732-4
79. Patrakka J, Tryggvason K. New insights into the role of podocytes in proteinuria. Nat Rev Nephrol. (2009) 5:463–8. doi: 10.1038/nrneph.2009.108
80. Barutta F, Bellini S, Gruden G. Mechanisms of podocyte injury and implications for diabetic nephropathy. Clin Sci (Lond). (2022) 136:493–520. doi: 10.1042/CS20210625
81. Zhou L, Chen X, Lu M, Wu Q, Yuan Q, Hu C, et al. Wnt/β-catenin links oxidative stress to podocyte injury and proteinuria. Kidney Int. (2019) 95:830–45. doi: 10.1016/j.kint.2018.10.032
82. Li M, Armelloni S, Mattinzoli D, Ikehata M, Chatziantoniou C, Alfieri C, et al. Crosstalk mechanisms between glomerular endothelial cells and podocytes in renal diseases and kidney transplantation. Kidney Res. Clin Pract. (2024) 43:47–62. doi: 10.23876/j.krcp.23.071
83. Black LM, Lever JM, Agarwal A. Renal inflammation and fibrosis: A double-edged sword. J Histochem Cytochem. (2019) 67:663–81. doi: 10.1369/0022155419852932
84. Zhang H, Sun SC. NF-κB in inflammation and renal diseases. Cell Biosci. (2015) 5:63. doi: 10.1186/s13578-015-0056-4
85. Lingappan K. NF-κB in oxidative stress. Curr Opin Toxicol. (2018) 7:81–6. doi: 10.1016/j.cotox.2017.11.002
86. Israël A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb Perspect Biol. (2010) 2:a000158. doi: 10.1101/cshperspect.a000158
87. Dąbek J, Kułach A, Gąsior Z. Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB): a new potential therapeutic target in atherosclerosis? Pharmacol Rep. (2010) 62:778–83. doi: 10.1016/s1734-1140(10)70338-8
88. Ranasinghe R, Mathai M, Zulli A. Cytoprotective remedies for ameliorating nephrotoxicity induced by renal oxidative stress. Life Sci. (2023) 318:121466. doi: 10.1016/j.lfs.2023.121466
89. Udwan K, Brideau G, Fila M, Edwards A, Vogt B, Doucet A. Oxidative stress and nuclear factor κB (NF-κB) increase peritoneal filtration and contribute to ascites formation in nephrotic syndrome. J Biol Chem. (2016) 291:11105–13. doi: 10.1074/jbc.M116.724690
90. Lv W, Booz GW, Fan F, Wang Y, Roman RJ. Oxidative stress and renal fibrosis: recent insights for the development of novel therapeutic strategies. Front Physiol. (2018) 9:105. doi: 10.3389/fphys.2018.00105
91. Su H, Wan C, Song A, Qiu Y, Xiong W, Zhang C. Oxidative stress and renal fibrosis: mechanisms and therapies. Adv Exp Med Biol. (2019) 1165:585–604. doi: 10.1007/978-981-13-8871-2_29
92. Stenvinkel P, Chertow GM, Devarajan P, Levin A, Andreoli SP, Bangalore S, et al. Chronic inflammation in chronic kidney disease progression: role of nrf2. Kidney Int Rep. (2021) 6:1775–87. doi: 10.1016/j.ekir.2021.04.023
93. Antar SA, Ashour NA, Marawan ME, Al-Karmalawy AA. Fibrosis: types, effects, markers, mechanisms for disease progression, and its relation with oxidative stress, immunity, and inflammation. Int J Mol Sci. (2023) 24:4004. doi: 10.3390/ijms24044004
94. Grynberg K, Ma FY, Nikolic-Paterson DJ. The JNK signaling pathway in renal fibrosis. Front Physiol. (2017) 8:829. doi: 10.3389/fphys.2017.00829
95. Patera F, Gatticchi L, Cellini B, Chiasserini D, Reboldi G. Kidney fibrosis and oxidative stress: from molecular pathways to new pharmacological opportunities. Biomolecules. (2024) 14:137. doi: 10.3390/biom14010137
96. Chen Q, Kang J, Fu C. The independence of and associations among apoptosis, autophagy, and necrosis. Sig Transduct Target Ther. (2018) 3:18. doi: 10.1038/s41392-018-0018-5
97. Padanilam BJ. Cell death induced by acute renal injury: a perspective on the contributions of apoptosis and necrosis. Am J Physiol Renal Physiol. (2003) 284:F608–27. doi: 10.1152/ajprenal.00284.2002
98. Edlich F. BCL-2 proteins and apoptosis: Recent insights and unknowns. Biochem Biophys Res Commun. (2018) 500:26–34. doi: 10.1016/j.bbrc.2017.06.190
99. Bertheloot D, Latz E, Franklin BS. Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cell Mol Immunol. (2021) 18:1106–21. doi: 10.1038/s41423-020-00630-3
100. Mahdizadeh SJ, Thomas M, Eriksson LA. Reconstruction of the fas-based death-inducing signaling complex (DISC) using a protein-protein docking meta-approach. J Chem Inf Model. (2021) 61:3543–58. doi: 10.1021/acs.jcim.1c00301
101. Tourneur L, Chiocchia G. FADD: a regulator of life and death. Trends Immunol. (2010) 31:260–9. doi: 10.1016/j.it.2010.05.005
102. Linkermann A, Chen G, Dong G, Kunzendorf U, Krautwald S, Dong Z. Regulated cell death in AKI. J Am Soc Nephrol. (2014) 25:2689–701. doi: 10.1681/ASN.2014030262
103. Khalid N, Azimpouran M. Necrosis. Treasure Island (FL: StatPearls Publishing (2024). Available at: https://www.ncbi.nlm.nih.gov/books/NBK557627/.
104. Mulay SR, Kumar SV, Lech M, Desai J, Anders HJ. How kidney cell death induces renal necroinflammation. Semin Nephrol. (2016) 36:162–73. doi: 10.1016/j.semnephrol.2016.03.004
105. Anders HJ, Schaefer L. Beyond tissue injury-damage-associated molecular patterns, toll-like receptors, and inflammasomes also drive regeneration and fibrosis. J Am Soc Nephrol. (2014) 25:1387–400. doi: 10.1681/ASN.2014010117
106. Noh MR, Padanilam BJ. Cell death induced by acute renal injury: a perspective on the contributions of accidental and programmed cell death. Am J Physiol Renal Physiol. (2024) 327:F4–F20. doi: 10.1152/ajprenal.00275.2023
107. Kers J, Leemans JC, Linkermann A. An overview of pathways of regulated necrosis in acute kidney injury. Semin Nephrol. (2016) 36:139–52. doi: 10.1016/j.semnephrol.2016.03.002
108. Sarhan M, Land WG, Tonnus W, Hugo CP, Linkermann A. Origin and consequences of necroinflammation. Physiol Rev. (2018) 98:727–80. doi: 10.1152/physrev.00041.2016
109. Rashid H, Jali A, Akhter MS, Abdi SAH. Molecular mechanisms of oxidative stress in acute kidney injury: targeting the loci by resveratrol. Int J Mol Sci. (2024) 25:3. doi: 10.3390/ijms25010003
110. Agrawal S, Zaritsky JJ, Fornoni A, Smoyer WE. Dyslipidaemia in nephrotic syndrome: mechanisms and treatment. Nat Rev Nephrol. (2018) 14:57–70. doi: 10.1038/nrneph.2017.155
111. Hassan Thabet MAE, Salcedo JR, Chan JCM. Hyperlipidemia in childhood nephrotic syndrome. Pediatr Nephrol. (1993) 7:559–66. doi: 10.1007/BF00852550
112. Querfeld U. Should hyperlipidemia in children with the nephrotic syndrome be treated? Pediatr Nephrol. (1999) 13:77–84. doi: 10.1007/s004670050568
113. Yuan Q, Tang B, Zhang C. Signaling pathways of chronic kidney diseases, implications for therapeutics. Sig Transduct Target Ther. (2022) 7:182. doi: 10.1038/s41392-022-01036-5
114. Mitrofanova A, Merscher S, Fornoni A. Kidney lipid dysmetabolism and lipid droplet accumulation in chronic kidney disease. Nat Rev Nephrol. (2023) 19:629–45. doi: 10.1038/s41581-023-00741-w
115. Ling XC, Kuo KL. Oxidative stress in chronic kidney disease. Ren Replace Ther. (2018) 4:53. doi: 10.1186/s41100-018-0195-2
116. Vaziri ND. Disorders of lipid metabolism in nephrotic syndrome: mechanisms and consequences. Kidney Int. (2016) 90:41–52. doi: 10.1016/j.kint.2016.02.026
117. Vaziri N. HDL abnormalities in nephrotic syndrome and chronic kidney disease. Nat Rev Nephrol. (2016) 12:37–47. doi: 10.1038/nrneph.2015.180
118. Princen HM, van Duyvenvoorde W, Buytenhek R, van der Laarse A, van Poppel G, Gevers Leuven JA, et al. Supplementation with low doses of vitamin E protects LDL from lipid peroxidation in men and women. Arterioscler Thromb Vasc Biol. (1995) 15:325–33. doi: 10.1161/01.atv.15.3.325
119. Verma S, Singh P, Khurana S, Ganguly NK, Kukreti R, Saso L, et al. Implications of oxidative stress in chronic kidney disease: a review on current concepts and therapies. Kidney Res Clin Pract. (2021) 40:183–93. doi: 10.23876/j.krcp.20.163
120. Hari P, Khandelwal P, Smoyer WE. Dyslipidemia and cardiovascular health in childhood nephrotic syndrome. Pediatr Nephrol. (2020) 35:1601–19. doi: 10.1007/s00467-019-04301-y
121. Ravarotto V, Bertoldi G, Innico G, Gobbi L, Calò LA. The pivotal role of oxidative stress in the pathophysiology of cardiovascular-renal remodeling in kidney disease. Antioxidants (Basel). (2021) 10:1041. doi: 10.3390/antiox10071041
122. Kamel A, Elnour SA, Ragaey M. Cardiac performance evaluation in children with nephrotic syndrome. Fayoum Univ Med J. (2020) 6:18–27. doi: 10.21608/fumj.2020.114316
123. Hertiš Petek T, Petek T, Močnik M, Marčun Varda N. Systemic inflammation, oxidative stress and cardiovascular health in children and adolescents: A systematic review. Antioxidants (Basel). (2022) 11:894. doi: 10.3390/antiox11050894
124. Hilmanto D, Mawardi F, Lestari AS, Widiasta A. Disease-associated systemic complications in childhood nephrotic syndrome: A systematic review. Int J Nephrol Renovasc Dis. (2022) 15:53–62. doi: 10.2147/IJNRD.S351053
125. Tharanidharan SP, Singh RK, Goel M, Shrivastava JS. A study on myocardial performance index in children with nephrotic syndrome in age group 1–12 years. J Clin Nephrol Ther. (2022) 6:106. doi: 10.35841/aacnt-6.2.106
126. Lebel A, Kropach N, Ashkenazi-Hoffnung L, Huber-Yaron A, Davidovits M. Infections in children with nephrotic syndrome: twenty years of experience. Clin Pediatr (Phila). (2020) 59:692–8. doi: 10.1177/0009922820908583
127. Moorani KN, Khan KM, Ramzan A. Infections in children with nephrotic syndrome. J Coll Physicians Surg Pak. (2003) 13:337–9.
128. Kalra S, Daryani H, Saxena A, Bhandari S, Sharma S. Infectious complications in children with nephrotic syndrome: Can they be prevented? Med J Armed Forces India. (2022) 78:170–4. doi: 10.1016/j.mjafi.2020.10.010
129. Pohanka M. Role of oxidative stress in infectious diseases. A review. Folia Microbiol. (2013) 58:503–13. doi: 10.1007/s12223-013-0239-5
130. Moro-García MA, Mayo JC, Sainz RM, Alonso-Arias R. Influence of inflammation in the process of T lymphocyte differentiation: proliferative, metabolic, and oxidative changes. Front Immunol. (2018) 9:339. doi: 10.3389/fimmu.2018.00339
131. Canton M, Sánchez-Rodríguez R, Spera I, Venegas FC, Favia M, Viola A, et al. Reactive oxygen species in macrophages: sources and targets. Front Immunol. (2021) 12:734229. doi: 10.3389/fimmu.2021.734229
132. Kaneko K, Tsuji S, Kimata T. Role of gut microbiota in idiopathic nephrotic syndrome in children. Med Hypotheses. (2017) 108:35–7. doi: 10.1016/j.mehy.2017.07.035
133. Ma X, Li T, Liu C, Ge H, Zheng D, Ma J, et al. Alterations of gut microbiota and metabolome are associated with primary nephrotic syndrome in children. BMC Microbiol. (2024) 24:519. doi: 10.1186/s12866-024-03667-w
134. Yamaguchi T, Tsuji S, Akagawa S, Akagawa Y, Kino J, Yamanouchi S, et al. Clinical significance of probiotics for children with idiopathic nephrotic syndrome. Nutrients. (2021) 13:365. doi: 10.3390/nu13020365
135. Kaneko K. Gut dysbiosis as a susceptibility factor in childhood idiopathic nephrotic syndrome. Pediatr Neonatol. (2025) 66:S2–7. doi: 10.1016/j.pedneo.2024.10.003
136. Mohammed RA, Hussein SK, Gaber SN, Abonaga F, Abdelfattah W, ES S, et al. The role of prebiotics and probiotics as an adjuvant therapy in children with idiopathic relapsing nephrotic syndrome: A prospective open-label clinical trial. Saudi J Kidney Dis Transpl. (2022) 33:S169–78. doi: 10.4103/1319-2442.384189
137. Khalil U. Role of oxidative stress in primary sodium retention and edema formation in nephrotic syndrome. In: Urology and Nephrology. Université Pierre et Marie Curie - Paris VI (2015). Available at: https://theses.hal.science/tel-01542593/document (Accessed November 12, 2024).
138. Sulyok E, Farkas B, Nagy B, Várnagy Á, Kovács K, Bódis J. Tissue sodium accumulation: pathophysiology and clinical implications. Antioxidants (Basel). (2022) 11:750. doi: 10.3390/antiox11040750
139. Patik JC, Lennon SL, Farquhar WB, Edwards DG. Mechanisms of dietary sodium-induced impairments in endothelial function and potential countermeasures. Nutrients. (2021) 13:270. doi: 10.3390/nu13010270
140. Soi V, Yee J. Sodium homeostasis in chronic kidney disease. Adv Chronic Kidney Dis. (2017) 24:325–31. doi: 10.1053/j.ackd.2017.08.001
141. Siddall EC, Radhakrishnan J. The pathophysiology of edema formation in the nephrotic syndrome. Kidney Int. (2012) 82:635–42. doi: 10.1038/ki.2012.180
142. Hinrichs GR, Jensen BL, Svenningsen P. Mechanisms of sodium retention in nephrotic syndrome. Curr Opin Nephrol Hypertens. (2020) 29:207–12. doi: 10.1097/MNH.0000000000000578
143. Deschênes G, Guigonis V, Doucet A. Molecular mechanism of edema formation in nephrotic syndrome. Arch Pediatr. (2004) 11:1084–94. doi: 10.1016/j.arcped.2004.03.029
144. Rondon-Berrios H. New insights into the pathophysiology of oedema in nephrotic syndrome. Nefrologia. (2011) 31:148–54. doi: 10.3265/Nefrologia.pre2010.Nov.10724
145. Bakr A, Abul Hassan S, Shoker M, Zaki M, Hassan R. Oxidant stress in primary nephrotic syndrome: does it modulate the response to corticosteroids? Pediatr Nephrol. (2009) 24:2375–80. doi: 10.1007/s00467-009-1246-2
146. Heijink I, van Oosterhout A, Kliphuis N, Jonker M, Hoffmann R, Telenga E, et al. Oxidant-induced corticosteroid unresponsiveness in human bronchial epithelial cells. Thorax. (2014) 69:5–13. doi: 10.1136/thoraxjnl-2013-203520
147. Barnes PJ. Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J Allergy Clin Immunol. (2013) 131:636–45. doi: 10.1016/j.jaci.2012.12.1564
148. Lewis BW, Ford ML, Rogers LK, Britt RD Jr. Oxidative stress promotes corticosteroid insensitivity in asthma and COPD. Antioxidants (Basel). (2021) 10:1335. doi: 10.3390/antiox10091335
149. Hapgood JP, Avenant C, Moliki JM. Glucocorticoid-independent modulation of GR activity: Implications for immunotherapy. Pharmacol Ther. (2016) 165:93–113. doi: 10.1016/j.pharmthera.2016.06.002
150. Ayroldi E, Cannarile L, Migliorati G, Nocentini G, Delfino DV, Riccardi C. Mechanisms of the anti-inflammatory effects of glucocorticoids: genomic and nongenomic interference with MAPK signaling pathways. FASEB J. (2021) 26:4805–20. doi: 10.1096/fj.12-216382
151. Tsukahara H. Biomarkers for oxidative stress: clinical application in pediatric medicine. Curr Med Chem. (2007) 14:339–51. doi: 10.2174/092986707779941177
152. Marrocco I, Altieri F, Peluso I. Measurement and clinical significance of biomarkers of oxidative stress in humans. Oxid Med Cell Longev. (2017) 2017:6501046. doi: 10.1155/2017/6501046
153. Halliwell B. Understanding mechanisms of antioxidant action in health and disease. Nat Rev Mol Cell Biol. (2024) 25:13–33. doi: 10.1038/s41580-023-00645-4
154. Ali SS, Ahsan H, Zia MK, Siddiqui T, Khan FH. Understanding oxidants and antioxidants: Classical team with new players. J Food Biochem. (2020) 44:e13145. doi: 10.1111/jfbc.13145
155. Losada-Barreiro S, Sezgin-Bayindir Z, Paiva-Martins F, Bravo-Díaz C. Biochemistry of antioxidants: mechanisms and pharmaceutical applications. Biomedicines. (2022) 10:3051. doi: 10.3390/biomedicines10123051
156. Younus H. Therapeutic potentials of superoxide dismutase. Int J Health Sci (Qassim). (2018) 12:88–93.
157. Vašková J, Kočan L, Vaško L, Perjési P. Glutathione-related enzymes and proteins: A review. Molecules. (2023) 28:1447. doi: 10.3390/molecules28031447
158. Nuhu F, Gordon A, Sturmey R, Seymour AM, Bhandari S. Measurement of glutathione as a tool for oxidative stress studies by high performance liquid chromatography. Molecules. (2020) 25:4196. doi: 10.3390/molecules25184196
159. Duni A, Liakopoulos V, Roumeliotis S, Peschos D, Dounousi E. Oxidative stress in the pathogenesis and evolution of chronic kidney disease: untangling ariadne’s thread. Int J Mol Sci. (2019) 20:3711. doi: 10.3390/ijms20153711
160. Liu Z, Wang JJ, Liu Q, Li J, Jiang S, Ma YQ, et al. Urinary 8-oxoGuo as a potential novel evaluation index for patients with nephrotic syndrome. Free Radic Res. (2022) 56:691–8. doi: 10.1080/10715762.2023.2166504
161. Ghodake SR, Suryakar AN, Ankush RD, Katkam RV, Shaikh K, Katta AV. Role of free radicals and antioxidant status in childhood nephrotic syndrome. Indian J Nephrol. (2011) 21:37–40. doi: 10.4103/0971-4065.78062
162. Shi J, Li W, Tao R, Zhou D, Guo Y, Fu H, et al. Association of catalase gene polymorphisms with idiopathic nephrotic syndrome in a Chinese pediatric population. Lab Med. (2023) 54:35–40. doi: 10.1093/labmed/lmac062
163. Mishra OP, Gupta AK, Prasad R, Ziledar A, Upadhyay RS, Mishra SP, et al. Antioxidant status of children with idiopathic nephrotic syndrome. Pediatr Nephrol. (2011) 26:251–6. doi: 10.1007/s00467-010-1696-6
164. Mulat SY, Mihajlović M, Antonić T, Miloševski-Lomić G, Peco-Antić A, Jovanović D, et al. Pediatric nephrotic syndrome: The interplay of oxidative stress and inflammation. J Med Biochem. (2024) 43:424–35. doi: 10.5937/jomb0-46526
165. Villalpando-Sánchez DC, Barajas-Medina CA, Alvarez-Aguilar C, López-Ortiz G, Romero-Henríquez LF, Gómez-García A. Advanced oxidative protein products had a diagnostic accuracy for identifying chronic kidney disease in adult population. Metabolites. (2024) 14:37. doi: 10.3390/metabo14010037
166. Arimura N, Watanabe H, Kato H, Imafuku T, Nakano T, Sueyoshi M, et al. Advanced oxidation protein products contribute to chronic-kidney-disease-induced adipose inflammation through macrophage activation. Toxins. (2023) 15:179. doi: 10.3390/toxins15030179
167. Sharifi-Rad M, Anil Kumar NV, Zucca P, Varoni EM, Dini L, Panzarini E, et al. Lifestyle, oxidative stress, and antioxidants: back and forth in the pathophysiology of chronic diseases. Front Physiol. (2020) 11:694. doi: 10.3389/fphys.2020.00694
168. Flieger J, Flieger W, Baj J, Maciejewski R. Antioxidants: classification, natural sources, activity/capacity measurements, and usefulness for the synthesis of nanoparticles. Materials (Basel). (2021) 14:4135. doi: 10.3390/ma14154135
169. García-Sánchez A, Miranda-Díaz AG, Cardona-Muñoz EG. The role of oxidative stress in physiopathology and pharmacological treatment with pro- and antioxidant properties in chronic diseases. Oxid Med Cell Longev. (2020) 2020:2082145. doi: 10.1155/2020/2082145
170. Krishnamurthy HK, Pereira M, Rajavelu I, Jayaraman V, Krishna K, Wang T, et al. Oxidative stress: fundamentals and advances in quantification techniques. Front Chem. (2024) 12:1470458. doi: 10.3389/fchem.2024.1470458
171. Poljsak B. Strategies for reducing or preventing the generation of oxidative stress. Oxid Med Cell Longev. (2011) 2011:194586. doi: 10.1155/2011/194586
172. Gutierrez-Mariscal FM, Arenas-de Larriva AP, Limia-Perez L, Romero-Cabrera JL, Yubero-Serrano EM, López-Miranda J. Coenzyme Q10 supplementation for the reduction of oxidative stress: clinical implications in the treatment of chronic diseases. Int J Mol Sci. (2020) 21:7870. doi: 10.3390/ijms21217870
173. Meng X, Li Y, Li S, Zhou Y, Gan RY, Xu DP, et al. Dietary sources and bioactivities of melatonin. Nutrients. (2017) 9:367. doi: 10.3390/nu9040367
174. Salehi B, Berkay Yılmaz Y, Antika G, Boyunegmez Tumer T, Fawzi Mahomoodally M, Lobine D, et al. Insights on the use of α-lipoic acid for therapeutic purposes. Biomolecules. (2019) 9:356. doi: 10.3390/biom9080356
175. Pravst I, Zmitek K, Zmitek J. Coenzyme Q10 contents in foods and fortification strategies. Crit Rev Food Sci Nutr. (2010) 50:269–80. doi: 10.1080/10408390902773037
Keywords: oxidative stress, child, kidney damage, systemic progression, nutritional management
Citation: Jechel E, Nedelcu AH, Dragan F, Lupu VV, Starcea IM, Mocanu A, Rosu ST, Streanga V, Russu R, Baciu G, Danielescu C, Salaru DL, Morariu ID, Cirstea O, Anton E and Lupu A (2025) Nutritional management of pediatric nephrotic syndrome regarding oxidative stress and antioxidant balance. Front. Immunol. 16:1542735. doi: 10.3389/fimmu.2025.1542735
Received: 10 December 2024; Accepted: 09 April 2025;
Published: 01 May 2025.
Edited by:
Ana Sanches Silva, National Institute for Agricultural and Veterinary Research (INIAV), PortugalReviewed by:
Brandon Marc Lane, Duke University, United StatesUjjawal Sharma, Central University of Punjab, India
Munkhtuya Tumurkhuu, Wake Forest Baptist Medical Center, United States
Copyright © 2025 Jechel, Nedelcu, Dragan, Lupu, Starcea, Mocanu, Rosu, Streanga, Russu, Baciu, Danielescu, Salaru, Morariu, Cirstea, Anton and Lupu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Felicia Dragan, ZmFybWFmZWxpQGdtYWlsLmNvbQ==; Vasile Valeriu Lupu, dmFsZXJpdWx1cHVAeWFob28uY29t
†These authors have contributed equally to this work